Способ получения дулоксетина и новые ключевые интермедиаты для его применения
Номер патента: 15828
Опубликовано: 30.12.2011
Авторы: Фараго Янош, Шиндлер Йожеф, Вукич Кристина, Гати Тамаш, Фогашши Эелемер, Цоке Каталин, Елеш Янош, Темешвари Кристина, Боди Йожеф






Формула / Реферат
1. Способ получения дулоксетина

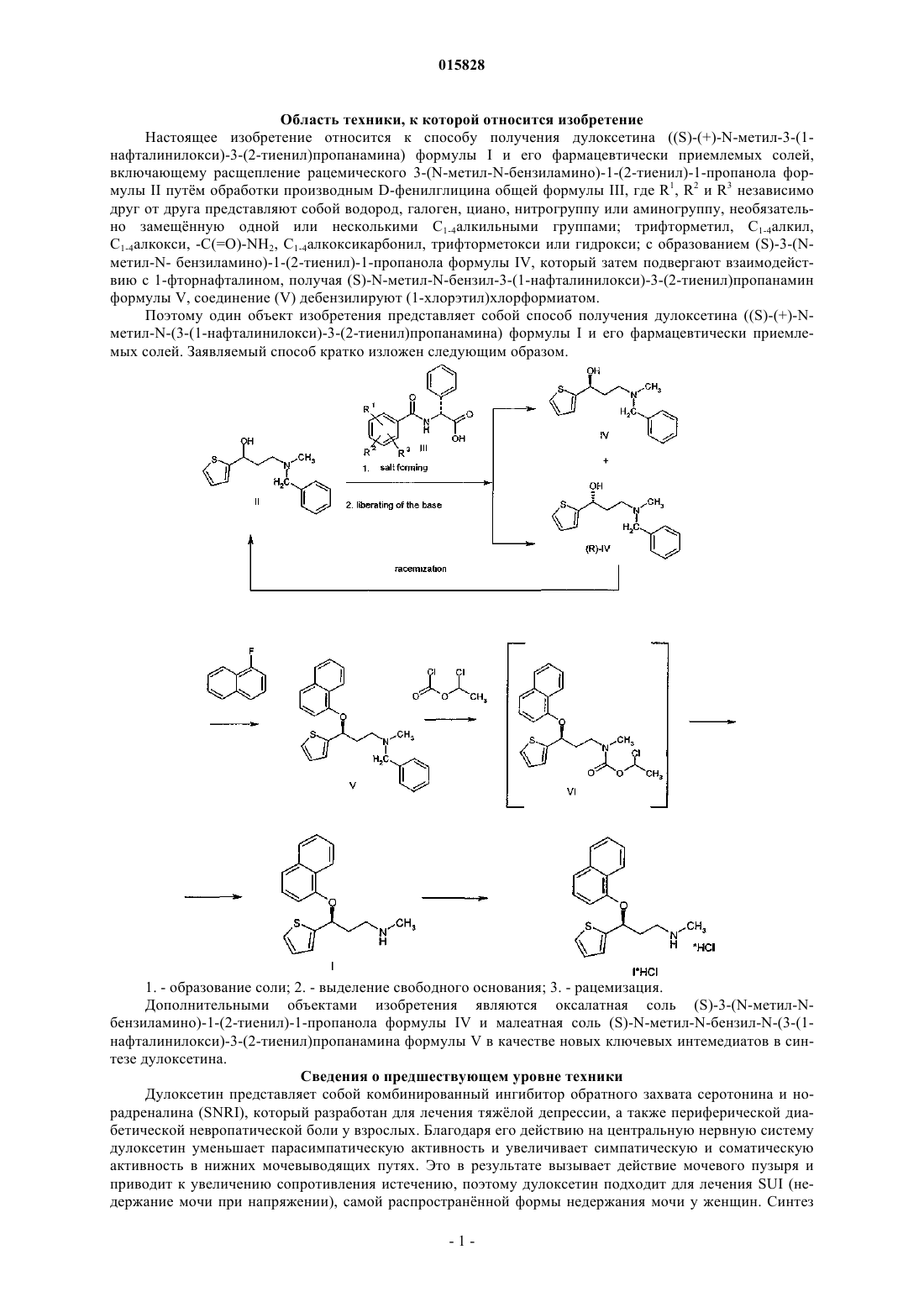
((S)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей, который включает следующие стадии:
расщепление рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы II

производным D-фенилглицина общей формулы III

где R1, R2 и R3представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещенную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси;
с образованием (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV

взаимодействие полученного соединения формулы IV с 1-фторнафталином с образованием (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V

взаимодействие полученного соединения формулы V с 1-хлорэтилхлорформиатом с образованием дулоксетина формулы I.
2. Способ по п.1, отличающийся тем, что расщепление проводят N-бензоил-D-фенилглицином.
3. Способ по п.1 или 2, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV очищают фракционной кристаллизацией.
4. Способ по п.3, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV.
5. Способ по п.1 или 2, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V очищают фракционной кристаллизацией.
6. Способ по п.5, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V.
7. Способ по п.1 или 2, отличающийся тем, что дулоксетин ((S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I очищают фракционной кристаллизацией.
8. Способ по п.7, отличающийся тем, что дулоксетин ((S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в дулоксетин ((S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I.
9. Способ по п.1 или 2, отличающийся тем, что ((S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I осаждают в форме его малеатной соли, которую затем в растворе выделяют в свободное основание и, не получая в чистом виде, подвергают его взаимодействию с безводным хлористым водородом и получают активное вещество гидрохлорид дулоксетина.
10. Способ по п.1 или 2, отличающийся тем, что (S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы I выделяют в форме его малеатной соли, затем, не выделяя в свободное основание, малеатную соль подвергают взаимодействию с концентрированной водной хлористо-водородной кислотой в органическом растворителе и получают активное вещество гидрохлорид дулоксетина.
11. Способ получения (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV

и его кислотно-аддитивных солей, отличающийся тем, что рацемический 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы II

расщепляют производным D-фенилглицина общей формулы III

где R1, R2 и R3представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещенную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; и
получают (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV и, если требуется, полученное соединение формулы IV осаждают в форме его кислотно-аддитивной соли.
12. Способ по п.11, отличающийся тем, что расщепление осуществляют N-бензоил-D-фенилглицином.
13. Способ по п.11 или 12, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV очищают фракционной кристаллизацией.
14. Способ по п.13, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV.
15. Способ получения (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V

и его кислотно-аддитивных солей, отличающийся тем, что

рацемический 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы II расщепляют производным D-фенилглицина общей формулы III

где R1, R2 и R3представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещенную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; и полученный (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV

подвергают взаимодействию с 1-фторнафталином и получают (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V и, если требуется, полученное соединение формулы V осаждают в форме его кислотно-аддитивной соли.
16. Способ по п.15, отличающийся тем, что расщепление осуществляют N-бензоил-D-фенилглицином.
17. Способ по п.15 или 16, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV очищают фракционной кристаллизацией.
18. Способ по п.17, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV.
19. Способ по п.15 или 16, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V очищают фракционной кристаллизацией.
20. Способ по п.19, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V.
Текст
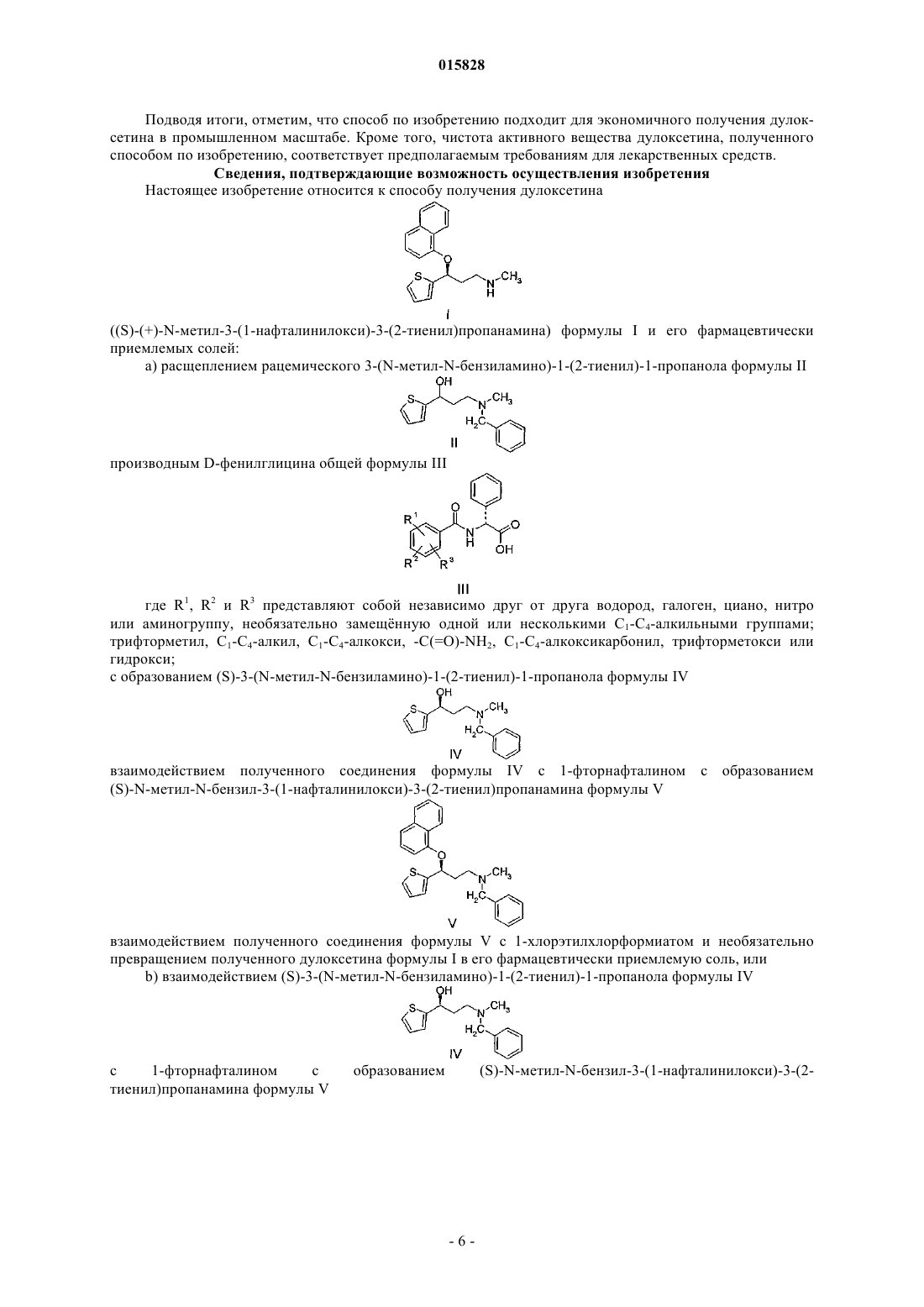
ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Дата публикации и выдачи патента СПОСОБ ПОЛУЧЕНИЯ ДУЛОКСЕТИНА И НОВЫЕ КЛЮЧЕВЫЕ ИНТЕРМЕДИАТЫ ДЛЯ ЕГО ПРИМЕНЕНИЯ Настоящее изобретение относится к способу получения дулоксетина S)-(+)-N-метил-N-(3-(1 нафталинилокси)-3-(2-тиенил)пропанамина) формулы (I) и его фармацевтически приемлемых солей, включающему стадии а) расщепления рацемического 3-(N-метил-N-бензиламино)-1-(2 тиенил)-1-пропанола формулы (II) производным D-фенилглицина с образованием соединения формулы (IV) и b) взаимодействия (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы (IV) с 1-фторнафталином с образованием (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3(2-тиенил)пропанамина формулы (V), взаимодействия полученного соединения формулы (V) с 1 хлорэтилхлорформиатом с образованием дулоксетина формулы (I) и, если требуется, превращения полученного дулоксетина формулы (I) в его кислотно-аддитивную соль. 015828 Область техники, к которой относится изобретение Настоящее изобретение относится к способу получения дулоксетина S)-(+)-N-метил-3-(1 нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей,включающему расщепление рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы II путм обработки производным D-фенилглицина общей формулы III, где R1, R2 и R3 независимо друг от друга представляют собой водород, галоген, циано, нитрогруппу или аминогруппу, необязательно замещнную одной или несколькими C1-4 алкильными группами; трифторметил, C1-4 алкил,C1-4 алкокси, -C(=O)-NH2, C1-4 алкоксикарбонил, трифторметокси или гидрокси; с образованием (S)-3-(Nметил-N- бензиламино)-1-(2-тиенил)-1-пропанола формулы IV, который затем подвергают взаимодействию с 1-фторнафталином, получая (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V, соединение (V) дебензилируют (1-хлорэтил)хлорформиатом. Поэтому один объект изобретения представляет собой способ получения дулоксетина S)-(+)-Nметил-N-(3-(1-нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей. Заявляемый способ кратко изложен следующим образом. 1. - образование соли; 2. - выделение свободного основания; 3. - рацемизация. Дополнительными объектами изобретения являются оксалатная соль (S)-3-(N-метил-Nбензиламино)-1-(2-тиенил)-1-пропанола формулы IV и малеатная соль (S)-N-метил-N-бензил-N-(3-(1 нафталинилокси)-3-(2-тиенил)пропанамина формулы V в качестве новых ключевых интемедиатов в синтезе дулоксетина. Сведения о предшествующем уровне техники Дулоксетин представляет собой комбинированный ингибитор обратного захвата серотонина и норадреналина (SNRI), который разработан для лечения тяжлой депрессии, а также периферической диабетической невропатической боли у взрослых. Благодаря его действию на центральную нервную систему дулоксетин уменьшает парасимпатическую активность и увеличивает симпатическую и соматическую активность в нижних мочевыводящих путях. Это в результате вызывает действие мочевого пузыря и приводит к увеличению сопротивления истечению, поэтому дулоксетин подходит для лечения SUI (недержание мочи при напряжении), самой распространнной формы недержания мочи у женщин. Синтез-1 015828 для получения дулоксетина и родственных соединений первоначально был описан в европейской патентной заявке ЕР 273658 (Eli Lilly и Co.). Согласно европейскому патенту ЕР 273658 гидрохлоридN,N-диметил-N-3-оксо-3-(2-тиенил)пропанамина получают реакцией конденсации Манниха, исходя из 2 ацетилтиофена, затем полученный кетен восстанавливают натрийборгидридом и получают 3-диметиламино-1-(2-тиенил)-1-пропанол. Арилирование вторичной гидроксильной группы проводят,подвергая его взаимодействию с 1-фторнафталином в присутствии основания гидрида натрия, и получают N,N-диметил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин, который затем деметилируют по двухстадийной методике. На первой стадии N,N-диметил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин подвергают взаимодействию с фенилхлорформиатом, получая соответствующий фенилкарбамат. Затем получают рацемический дулоксетин основным гидролизом, осуществляемым при высокой температуре, и выделяют его в форме оксалатной соли. После расщепления рацемического дулоксетина дибензоил-D-винной кислотой получают чистый S-энантиомер. Недостатком вышеописанного способа является серьзная угроза безопасности из-за применения гидрида натрия, который чрезвычайно пожароопасен. Кроме того, проведение стадии расщепления в конце последовательности операций увеличивает издержки производства. Согласно заявке на выдачу патента США 5362886 (Eli Lilly и Co.) стадию расщепления проводят на ранней фазе синтеза. При этом 3-диметиламино-1-(2-тиенил)-1-пропанол расщепляют(S)-(+)-миндальной кислотой, затем полученный S-(-)-3-диметиламино-1-(2-тиенил)-1-пропанол арилируют 1-фторнафталином в присутствии гидрида натрия и бензоата калия. Однако сообщается о частичной рацемизации и оптическая чистота полученного (S)-(+)-N,N-диметил-N-3-(1-нафталинилокси)-3-(2 тиенил)пропанамина составляет только 91% ее. Первый энантиоселективный способ получения производных 3-диалкиламино-1-арил-1-пропанона описан также Eli Lilly и Со. в заявке на европейский патент ЕР 457559 и в Tetrahedron Letters, 31(49),7101-04 (1990). В упомянутом способе в качестве восстановителя используют комплекс литийалюминийгидрид/тетрагидрофуран (LiAlH4/THF)2, который при применении в промышленном масштабе вызывает угрозу безопасности, как и при использовании гидрида натрия в способе, описанном в ЕР 273658. Чтобы обеспечить энантиоселективность, применяют большой избыток хирального катализатора(2R,3S)-4-диметиламино-1,2-дифенил-3-метил-2-бутанола. Энантиомерный избыток (S)-энантиомера составляет 80-88%, полученный 1,3-аминоспирт очищают кристаллизацией и получают целевой продукт с выходом около 50%. При повторении способа в увеличенном масштабе наблюдается значительное уменьшение энантиоселективности. Различные способы стереоселективного восстановления оксогруппы также опубликованы. Многочисленные патентные заявки касаются получения (S)-3-метиламино-1-(2-тиенил)-1-пропанола, где (S)-3 метиламино-1-(2-тиенил)-1-пропанол указан в качестве возможного интермедиата для синтеза дулоксетина, так как его можно нафтилировать одностадийным способом, получая целевое производное дулоксетина. В связи с этим разработан ряд различных химических способов энантиоселективного восстановления 3-метиламино-1-(2-тиенил)-1-пропанона с различной стереоселективностью, которые приводят к получению (S)-3-метиламино-1-(2-тиенил)-1-пропанола. Согласно международным заявкамWO 2004/020389 и WO 2004/031168 восстановление осуществляют гидрированием в присутствии Rh иRu катализаторов, содержащих хиральные лиганды, кроме того, описан биохимический способ восстановления 3-метиламино-1-(2-тиенил)-1-пропанона в международной заявке WO 2004/013123. ЗаявкиUS 2004/0249170 и WO 2004/005307 описывают расщепление рацемического 3-метиламино-1-(2-тиенил)1-пропанола. Однако взаимодействие (S)-3-метиламино-1-(2-тиенил)-1-пропанола с 1-фторнафталином в присутствии сильного основания приводит к образованию дулоксетина с умеренным выходом 10-45% из-за наличия вторичного амина. Более того, выделение конечного продукта приходится осуществлять хроматографически согласно Chirality 2000, 12, 26-29; J. Lab. Comp. Radiopharm, 1955, 36(3), 213-223. Изза вышеупомянутых недостатков (S)-3-метиламино-1-(2-тиенил)-1-пропанол не может рассматриваться в качестве интермедиата, применимого для экономичного промышленного способа. В способе, описанном в международной заявке WO 2004/056795, 3-диметиламино-1-(2-тиенил)-1 пропанол нафтилируют посредством основания гидроксида калия и катализатора межфазного переноса,18-краун-6. В заявке указано, что рацемизацию полученного (S)-N,N-диметил-N-3-(1-нафталинилокси)-3(2-тиенил)пропанамина, можно подавить, однако степень рацемизации и энантиомерная чистота продукта не установлены. Согласно ещ одной методике, описанной в международной заявке WO 2006/027798, можно не допустить нежелательной рацемизации, происходящей во время процесса нафтилирования, если проводить стадию расщепления после введения нафтильной группы в молекулу. Заявлено расщеплениеN,N-диметил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин, оставшийся в маточном растворе, подвергают рацемизации обработкой сильным основанием (трет-бутоксидом калия) в растворителе диметил-2 015828 сульфоксиде. Полученный с выходом 65% рацемат N,N-диметил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамина затем повторно используют на стадии расщепления, увеличивая, таким образом,выход нужного изомера. Способ по WO 2006/027798 устраняет нежелательную рацемизацию, происходящую во время нафтилирования, однако расщепление дорогим реагентом 1-фторнафталином после стадии нафтилирования делает этот способ слишком дорогостоящим. В свете недостатков способов, перечисленных выше, цель авторов настоящего изобретения состояла в создании способа получения дулоксетина, который безопасен при применении в промышленных масштабах и использует экономичные и удобные реакции на стадиях получения активного вещества с чистотой, требуемой для лекарственных средств. Ещ одна цель состояла в создании способа, согласно которому активное вещество высокой чистоты можно получать с хорошими выходами без применения усложннных стадий реакции или технологий, требующих жстких условий, а промежуточные соединения можно выделять, используя простые стадии реакции. Кроме того, цель авторов настоящего изобретения состояла в том, чтобы проводить дорогостоящие стадии реакции в конце процесса и регенерировать дорогие (т.е. оптически активные) ингредиенты. В ходе проведнных экспериментов авторы настоящего изобретения неожиданно обнаружили, что в способе по изобретению, исходя из 3-(N-метил-N-бензоламино)-1-(2-тиенил)-1-пропанола, получается кристаллический интермедиат, а именно малеатная соль (S)-N-метил-N-бензол-N-3-(1-нафталинилокси)3-(2-тиенил)пропанамина, которую можно очищать просто перекристаллизацией и получать превосходное вещество, чистое с химической и оптической точки зрения. Используя указанное промежуточное соединение, можно получать дулоксетин в промышленном масштабе синтезом, состоящим лишь из нескольких стадий. Сущность изобретения Способ по изобретению включает следующие стадии. Стадия 1. 3-(N-Метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы II (патентные заявки ЕР 273 658 и ЕР 457 559) подвергают взаимодействию с производным D-фенилглицина общей формулы III, где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу,необязательно замещнную одной или несколькими C1-4 алкильными группами; трифторметил, C1-4 алкил,C1-4 алкокси, -C(=O)-NH2, C1-4 алкоксикарбонил, трифторметокси или гидрокси, и получают (S)-3-(Nметил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV, который очищают в форме его оксалатной соли:(S)-3-(N-Метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV, полученный на стадии 1,подвергают взаимодействию с 1-фторнафталином и получают (S)-N-метил-N-бензил-N-3-(1 нафталинилокси)-3-(2-тиенил)пропанамин формулы V, который очищают в форме его малеатной соли:(S)-N-Метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V, полученный на стадии 2, подвергают взаимодействию с 1-хлорэтилхлорформиатом и получают дулоксетин S)-(+)-Nметил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I (через не выделенное промежуточное соединение формулы VI), который очищают в форме его малеатной соли: Стадия 4. Дулоксетин S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I, полученный на стадии 3, превращают в его гидрохлоридную соль в подходящем растворителе, например в этилацетате или ацетоне, и получают активное вещество гидрохлорид дулоксетина (IHCl). Альтернативно,гидрохлорид дулоксетина может быть получен непосредственно превращением малеатной соли (S)-(+)N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина в целевую гидрохлоридную соль: Сведения, подтверждающие возможность осуществления изобретения Стадия 1. 3-(N-Метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы II расщепляют, прибавляя 0,5-1 экв. производного D-фенилглицина общей формулы III, где R1, R2 и R3 независимо друг от друга представляют собой водород, галоген, циано, нитрогруппу или аминогруппу, необязательно замещнную одной или несколькими C1-4 алкильными группами; трифторметил, C1-4 алкил, C1-4 алкокси, -C(=O)-NH2,C1-4 алкоксикарбонил, трифторметокси или гидрокси предпочтительно, N-бензоил-D-фенилглицинаH2SO4. Полученную кристаллическую диастереоизомерную соль отфильтровывают. Затем диастереоизомерную соль обрабатывают смесью водного аммиака или карбоната калия и подходящего органического растворителя, предпочтительно н-гексана. Органический и водный слои разделяют, органический слой упаривают и получают свободное основание дулоксетина. Полученный 3-(N-метил-N-бензиламино)-1-(2 тиенил)-1-пропанол (оптическая чистота составляет около 80% ее для S-изомера) подвергают взаимодействию с щавелевой кислотой в метаноле и получают оксалатную соль. После прибавления третбутилметилового эфира и отфильтровывания кристаллического рацемата метанольный маточный раствор упаривают и получают кристаллическую оксалатную соль (S)-3-(N-метил-N-бензиламино)-1-(2 тиенил)-1-пропанола. Энантиомерная чистота полученного (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола составляет 98% ее. Водную фазу обрабатывают хлористо-водородной кислотой и выпавшее в осадок производное D-фенилглицина общей формулы III (расщепляющий агент) возвращают для дальнейшего ис-4 015828 пользования. Производное аминоспирта, обогащнное (R)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанолом, выделяют из ацетонового маточного раствора и подвергают рацемизации, нагревая его в водной серной кислоте, затем повторяют стадию расщепления. Стадия 2. В своих экспериментах авторы настоящего изобретения неожиданно обнаружили, что скорость реакции 1-фторнафталина с (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанолом в присутствии гидроксида натрия является одинаковой как в присутствии катализатора межфазного переноса, так и в его отсутствие, следовательно, наличие катализатора межфазного переноса не является необходимым. Согласно экспериментам авторов настоящего изобретения и известным литературным данным рацемизация является неизбежной в условиях реакции нафтилирования. Степень рацемизации можно уменьшить,применяя водный ДМСО в качестве растворителя и уменьшая температуру реакционной смеси. Поэтому реакцию нафтилирования проводят следующим образом. В смеси ДМСО и тетрагидрофурана, содержащей 20-40% ДМСО (предпочтительно 40% ДМСО), (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV подвергают взаимодействию с 1,1-1,5 мол.экв. 1-фторнафталина, предпочтительно 1,2 мол.экв. в присутствии 5-10 моль, предпочтительно 7 моль основания гидроксида калия при температуре 40-45 С. Степень ожидаемой рацемизации составляет 1-2%, следовательно, оптическая чистота продукта 94-96% ее. Продукт (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V - выделяют в форме малеатной соли после экстракционной обработки, затем полученную соль необязательно очищают кристаллизацией. Очистка в форме малеатной соли особенно предпочтительна потому,что кристаллизация(S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамина из перенасыщенного раствора дат продукт с высокой оптической чистотой, тогда как рацемат остатся в маточном растворе. Из малеатной соли получают основание для использования на следующей стадии 3. Полученный (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V имеет оптическую чистоту 99,8% ее. Стадия 3.(S)-N-Метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V в дихлорметане подвергают взаимодействию с 1-1,1 мол.экв. 1-хлорэтилформиата в присутствии 0,3-1 экв. диизопропилэтиламина при температуре 20-30 С. Полученный промежуточный карбамат формулы VI перемешивают в метаноле при температуре 20-30 С, затем экстрагируют реакционную смесь и полученный дулоксетин S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I осаждают из подходящего органического растворителя, например из этилацетата или трет-бутилметилового эфира,в форме оксалатной или малеатной соли, предпочтительно в форме малеатной соли. Затем из малеатной соли получают основание и превращают его в гидрохлоридную соль дулоксетина на следующей стадии. Стадия 4.(S)-(+)-N-Метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы I растворяют в безводном апротонном органическом растворителе, предпочтительно в этилацетате или ацетоне, затем прибавляют этилацетат, содержащий 0,95-0,98 экв. хлористого водорода или водной хлористо-водородной кислоты при температуре 0-25 С и получают гидрохлоридную соль. Оптическая чистота полученной гидрохлоридной соли дулоксетина (IHCl) после отфильтровывания и сушки составляет 99,8% ее. Альтернативно, малеатную соль (S)-(+)-N-метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы I,полученную на стадии 3, суспендируют в ацетоне, затем прибавляют конц. раствор хлористо-водородной кислоты, содержащий 0,95-0,98 экв. хлористого водорода, осаждают гидрохлорид дулоксетина. Способ по изобретению имеет следующие преимущества.a) Благодаря отличной способности кристаллизоваться новый ключевой интермедиат (S)-N-метилN-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V можно эффективно очищать простой кристаллизацией. Из нового ключевого интермедиата - соединения формулы V - можно получать активное вещество дулоксетин высокой химической и оптической чистоты без применения специальных реагентов и жестких условий реакции.b) В способе по изобретению расщепление проводят на ранней стадии пути синтеза, что позволяет существенно уменьшить производственные затраты. На указанной стадии получают новое промежуточное соединение (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV легко осуществляемым и экономичным способом.c) (R)-3-(N-Метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV, накопившийся в маточном растворе во время проведения расщепления, легко рацемизируют, затем полученный рацемический продукт расщепляют повторно.d) Степень рацемизации, происходящей во время реакции нафтилирования, минимизирована при использовании подходящей смеси растворителей.e) Нет необходимости получения интермедиатов с высокой степенью оптической чистоты ни на стадии расщепления, ни на стадии нафтилирования, так как благодаря превосходным кристаллизационным свойствам малеатную соль ключевого интермедиата (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3(2-тиенил)пропанамина можно эффективно очищать простой кристаллизацией.-5 015828 Подводя итоги, отметим, что способ по изобретению подходит для экономичного получения дулоксетина в промышленном масштабе. Кроме того, чистота активного вещества дулоксетина, полученного способом по изобретению, соответствует предполагаемым требованиям для лекарственных средств. Сведения, подтверждающие возможность осуществления изобретения Настоящее изобретение относится к способу получения дулоксетинаS)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей: а) расщеплением рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы II где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; с образованием (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV взаимодействием полученного соединения формулы IV с 1-фторнафталином с образованием взаимодействием полученного соединения формулы V с 1-хлорэтилхлорформиатом и необязательно превращением полученного дулоксетина формулы I в его фармацевтически приемлемую соль, или который затем реагирует с 1-хлорэтилхлорформиатом с образованием дулоксетина формулы I, и необязательно превращением полученного дулоксетина формулы I в его фармацевтически приемлемую соль,или с) взаимодействием (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V с 1-хлорэтилхлорформиатом с образованием дулоксетина формулы I и необязательно превращением полученного дулоксетина формулы I в его фармацевтически приемлемую соль. В предпочтительном варианте осуществления изобретения стадию расщепления проводятN-бензоил-D-фенилглицином. В ещ одном предпочтительном варианте осуществления изобретения очистку (S)-3-(N-метил-Nбензиламино)-1-(2-тиенил)-1-пропанола формулы IV предпочтительно в форме его оксалатной соли выполняют фракционной кристаллизацией. В дополнительном предпочтительном варианте осуществления изобретения очистку (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V предпочтительно в форме его малеатной соли проводят также фракционной кристаллизацией. В ещ одном предпочтительном варианте осуществления изобретения (S)-(+)-N-метил-3-(1 нафталинилокси)-3-(2-тиенил)пропанамин формулы I выделяют в форме его малеатной соли, затем либо выделяют его в свободное основание в растворе, не получая в чистом виде, подвергают основание взаимодействию с безводным хлористым водородом и получают активное вещество дулоксетинHCl либо, не выделяя малеатную соль в свободное основание, подвергают е взаимодействию с конц. водной хлористо-водородной кислотой и получают активное вещество дулоксетинHCl. Кроме того, настоящее изобретение относится к (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанолу формулы IV и его кислотно-аддитивным солям, предпочтительно его оксалатной соли. Ещ одним объектом настоящего изобретения является способ получения (S)-3-(N-метил-Nбензиламино)-1-(2-тиенил)-1-пропанола формулы IV включающий расщепление рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы II где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси.-7 015828 В предпочтительном варианте осуществления изобретения стадию расщепления рацемата проводятN-бензоил-D-фенилглицином. В ещ одном предпочтительном варианте осуществления изобретения очистку (S)-3-(N-метил-Nбензиламино)-1-(2-тиенил)-1-пропанола формулы IV предпочтительно в форме его оксалатной соли проводят фракционной кристаллизацией. Настоящее изобретение, кроме того, относится к (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамину формулы V и его фармацевтически приемлемым солям, предпочтительно его малеатной соли. Кроме того, настоящее изобретение относится к способу получения (S)-N-метил-Н-бензил-3-(1 нафталинилокси)-3-(2-тиенил)пропанамина формулы V, включающему где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси, и взаимодействие полученного выше (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы с 1-фторнафталином. В предпочтительном варианте осуществления вышеописанного способа расщепление проводятN-бензоил-D-фенилглицином. В ещ одном предпочтительном варианте осуществления способа очистку (S)-3-(N-метил-Nбензиламино)-1-(2-тиенил)-1-пропанола формулы IV предпочтительно в форме его оксалатной соли осуществляют фракционной кристаллизацией. В дополнительном предпочтительном варианте осуществления способа очистку (S)-N-метил-Nбензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V предпочтительно в форме его малеатной соли осуществляют фракционной кристаллизацией. В изобретении используют следующие аналитические методы. Для идентификации соединений по изобретению использованы спектры 1 Н ЯМР, полученные на спектрометре "VarianUNITY INOVA 500". Величины химических сдвигов определены в м.д. (миллионных долях) относительно тетраметилсилана в качестве внутреннего стандарта. Оптическую чистоту оптически активных промежуточных соединений определяют методами хиральной ВЭЖХ. 1. Чистоту (S)-3-(N-метил-N-бензиламиноамино)-1-(2-тиенил)-1-пропанола формулы IV определяют методом ВЭЖХ следующим образом: колонка: CHIRALPAK IA (Chiral Technologies Europe) 2504,6 мм, размер частиц 5 мкм; температура: 25 С; элюент: н-гексан:хлороформ:диэтиламин=92:8:0,1;-8 015828 скорость потока: 1 мл/мин; детектирование: УФ-детектор;(длина волны) = 254 нм; приблизительные времена удерживания для (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола - 19,9 мин,для (R)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола - 23,1 мин. 2. Чистоту (S)-N-метил-N-бензил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V определяют методом ВЭЖХ следующим образом: колонка: CHIRALPAK IB (Chiral Technologies Europe) 2504,6 мм, размер частиц 5 мкм; температура: 25 С; элюент: н-гексан:2-пропанол=99,5:0,5 (+ 0,1% диэтиламина); скорость потока: 1 мл/мин; детектирование: УФ-детектор;(длина волны) = 255 нм; приблизительные времена удерживания для (S)-N-метил-N-бензил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина - 7,7 мин,для (R)-N-метил-N-бензил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина - 11,4 мин; температура плавления малеатной соли (S)-N-метил-N-бензил-N-3-(1-нафталинилокси)-3-(2 тиенил)пропанамина: 145-147 С. 3. Чистоту (S)-N-метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы I определяют методом ВЭЖХ следующим образом: колонка: CHIRALPAK IA (Chiral Technologies Europe) 2504,6 мм, размер частиц 5 мкм; температура: 35 С; элюент: н-гексан:2-пропанол:этанол=95:4,5:0,5 (+ 0,3% диэтиламина); скорость потока: 1 мл/мин; детектирование: УФ-детектор;(длина волны) = 254 нм; приблизительные времена удерживания для (S)-N-метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина - 11,2 мин,для (R)-N-метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина - 14,0 мин. Изобретение иллюстрируется следующими неограничивающими примерами. Пример 1. Получение (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV. Метод расщепления А. 3-(N-Метил-N-бензиламино)-1-(2-тиенил)-1-пропанол (60 г, 0,23 моль) растворяют в 300 см 3 ацетона и раствор нагревают до температуры кипения. К полученному раствору основания прибавляют смесь 29,3 г (0,115 моль) N-бензоил-D-фенилглицина и 6,5 см 3 (0,115 моль) уксусной кислоты, растворнную в 600 см 3 горячего ацетона. Полученную суспензию кипятят в течение 30 мин, затем после охлаждения до температуры 20-25 С смесь дополнительно перемешивают в течение 1 ч, фильтруют и осадок промывают ацетоном. Масса сухого вещества: 50 г (84%). Полученную на предшествующей стадии соль суспендируют в смеси 700 см 3 н-гексана и 700 см 3 воды. К образовавшейся суспензии прибавляют 20 см 3 раствора аммиака (25%), затем смесь перемешивают и получают два гомогенных слоя. Слои разделяют и водный слой экстрагируют н-гексаном(2300 см 3). Объединнные органические экстракты промывают водой (1300 см 3), затем сушат над сульфатом натрия и упаривают. Масса сухого вещества полученного масла составляет 25 г (83%), оптическая чистота 65-70% ее. Повторением способа ращепления с использованием 0,8 экв. расщепляющего агента получают 18,1 г указанного в заголовке соединения с оптической чистотой 80% ее. Доводят рН объединнных водных слоев до 2, прибавляя раствор хлористо-водородной кислоты(2 М), и полученный кристаллический N-бензоил-D-фенилглицин отфильтровывают, нейтрализуют, промывают деионизированной водой, сушат и получают 22 г N-бензоил-D-фенилглицина (в стереомерной соли содержится 90% N-бензоил-D-фенилглицина). Метод расщепления В. К раствору 2,6 г (0,01 моль) 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола в 13 см 3 ацетона прибавляют 1,3 см 3 воды, 0,1 см 3 конц. раствора серной кислоты и 1,53 г (0,006 моль) N-бензоил-Dфенилглицина. Реакционную смесь нагревают до температуры кипения и после полного растворения охлаждают до температуры 20-25 С. Суспензию кристаллов перемешивают в течение 20 ч, затем кристаллы отфильтровывают и промывают ацетоном (25 см 3), получая 2,2 г (85%) указанного в заголовке соединения (75-80% ее).-9 015828 Очистка в форме оксалатной соли. К раствору 17,8 г (0,068 моль) (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола (80% ее) в 180 см 3 метанола прибавляют безводную щавелевую кислоту (6,1 г, 0,068 моль) и реакционную смесь перемешивают при температуре 20-25 С в течение 1 ч. Полученную кристаллическую оксалатную соль(около 6 г) отфильтровывают, фильтрат упаривают до объма около 30 см 3, затем прибавляют 90 см 3 трет-бутилметилового эфира и получают очищенную оксалатную соль (S)-3-(N-метил-N-бензиламино)-1(2-тиенил)-1-пропанола в форме кристаллов. Полученное кристаллическое вещество отфильтровывают,промывают трет-бутилметиловым эфиром и сушат. Выход 16,6 г (69%). Из оксалатной соли выделяют свободное основание по методике, описанной в способе А. Оптическая чистота полученного (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола составляет 98% ее. 1H ЯМР (500 МГц, ДМСО-d6, 25 С)2,00-2,15 (м, 2 Н), 2,57 (с, 3 Н), 2,92-3,11 (м, 2 Н), 4,06-4,24 (м,2 Н), 4,88 (дд, J=7,6; 4,9 Гц, 1H), 6,93-6,99 (м, 2 Н), 7,37-7,50 (м, 6 Н) м.д. Рацемизация R-3-(N-метил-N-бензиламино)-1-(2-тионил)-1-пропанола из обогащнного им маточного раствора. Около 1 дм 3 ацетонового маточного раствора, полученного на стадии расщепления, упаривают в вакууме и остаток растворяют в 300 см 3 1 М серной кислоты. Нерастворимый расщепляющий агентN-бензоил-D-фенилглицин осаждают в форме кристаллов, затем кристаллы отфильтровывают и фильтрат кипятят в колбе объмом 500 см 3 с обратным холодильником при 50 С и при перемешивании. Ход реакции контролируют хиральной ВЭЖХ. Для завершения процесса рацемизации требуется около 1,5-2 ч. После завершения рацемизации реакционную смесь охлаждают до температуры 20-25 С и прибавляют 100 см 3 н-гексана, затем по каплям прибавляют 500 см 3 10% раствора карбоната калия при перемешивании. Слои разделяют и водный слой экстрагируют н-гексаном (2100 см 3). Объединнные органические слои сушат над сульфатом натрия, сульфат натрия отфильтровывают и фильтрат упаривают в вакууме. Полученный рацемический 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол (34 г) можно очищать в форме его оксалатной соли. Таким образом, 34 г рацемического 3-(N-метил-N-бензиламино)1-(2-тиенил)-1-пропанола растворяют в 150 см 3 трет-бутилметилового эфира и прибавляют 15 см 3 метанола. Затем смесь охлаждают до температуры 0-5 С. К охлажднной смеси медленно прибавляют раствор 11,7 г безводной щавелевой кислоты и 30 см 3 метанола при перемешивании, при этом смесь охлаждают ледяной водой. Выпавшую в осадок оксалатную соль отфильтровывают через стеклянный фильтр,промывают трет-бутилметиловым эфиром (220 см 3), затем сушат в открытом сосуде при комнатной температуре и получают белый кристаллический порошок. Масса: 45 г Пример 2. Получение (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина. В колбу объмом 250 см 3, снабжнную магнитной мешалкой и термометром, загружают 10,4 г(0,04 моль) (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола, 15 см 3 ДМСО, 45 мл тетрагидрофурана, 15,7 г (0,28 моль, 7 экв.) порошкообразного гидроксида калия и 5,3 см 3 (0,048 моль, 1,2 экв.) 1-фторнафталина и смесь перемешивают при температуре 40-45 С в течение 9-12 ч. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции смесь охлаждают до температуры 20-25 С и выливают в 150 см 3 смеси вода-лд, затем экстрагируют н-гексаном (350 см 3). Объединнные органические фазы промывают водой (350 мл), сушат над Na2SO4, упаривают в вакууме, получая неочищенное соединение, указанное в заголовке. Масса: 17,0 г, оптическая чистота 95% ее (по данным хиральной ВЭЖХ). Получение малеата (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина. Неочищенное основание (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина(17,0 г) растворяют в 150 мл этилацетата и прибавляют 4,7 г (0,04 моль) малеиновой кислоты. Выпавшие в осадок кристаллы перемешивают в течение 1 ч, затем отфильтровывают и промывают этилацетатом. Масса кристаллической малеатной соли: 16,2 г (0,032 моль) (85%), оптическая чистота 99% ее (по данным метода хиральной ВЭЖХ). Перекристаллизация соли (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина. Малеатную соль (S)-N-метил-N-бензил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина (16,2 г,0,032 моль), полученную на предыдущей стадии, растворяют в 90 мл горячего этанола и реакционную смесь охлаждают до температуры 0 С в течение примерно 30 мин, выпавшие в осадок кристаллы отфильтровывают и промывают холодным этанолом. Масса сухого вещества: 14,5 г (90%). 1- 10015828 Выделение свободного основания (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина из его малеатной соли. Малеат(14,5 г,0,029 моль) суспендируют в смеси 200 см 3 дихлорметана и 250 см 3 раствора хлорида калия (10%) и смесь перемешивают до полного растворения малеатной соли. Слои разделяют, водный слой экстрагируют дихлорметаном (220 см 3) и объединнные органические слои промывают водой (230 см 3). Дихлорметановый раствор сушат над сульфатом натрия и упаривают, получая указанное в заголовке соединение в маслообразном виде. Выход 11,0 г (0,028 моль), оптическая чистота 99,5% ее (по данным хиральной ВЭЖХ). Пример 3. Получение малеата (S)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина (малеата дулоксетина). При комнатной температуре в колбу объмом 250 см 3, снабжнную магнитной мешалкой и термометром, загружают 13,6 г (0,035 моль) (S)-N-метил-N-бензил-(3-(1-нафталинилокси)-3-(2 тиенил)пропанамина, 150 см 3 дихлометана и 7,4 см 3 (0,042 моль) диизопропилэтиламина, затем прибавляют по каплям 4,5 см 3 (0,042 моль) 1-хлорэтилхлорформиата. Реакционную смесь перемешивают при температуре 25 С в течение примерно 2 ч, при этом ход реакции контролируют тонкослойной хроматографией. После завершения реакции смесь выливают в 150 см 3 раствора лимонной кислоты (5%) и разделяют слои. Водный слой экстрагируют 100 см 3 дихлорметана, затем объединнные органические слои промывают 100 см 3 воды до полного удаления кислоты. Органический слой сушат над Na2SO4, сушат в вакууме и получают 16,0 г неочищенного карбамата (VI). В колбе объмом 250 см 3, снабжнной магнитной мешалкой, термометром и прибором для барботирования газа, растворяют неочищенный карбамат (VI) в 80 см 3 метанола и перемешивают до тех пор,пока происходит образование газа. Ход реакции контролируют тонкослойной хроматографией. После завершения реакции к смеси прибавляют 80 см 3 воды и полученный раствор экстрагируют 250 см 3 н-гексана. Водно-метанольный слой подщелачивают 30 см 3 раствора карбоната калия (10%), затем смесь экстрагируют этилацетатом (2200 см 3). Объединнные органические слои промывают примерно 100 мл воды, сушат над Na2SO4 и упаривают в вакууме до объма около 80 см 3. Затем к смеси при перемешивании прибавляют 3,3 г (0,0366 моль) малеиновой кислоты. Полученную малеатную соль перемешивают в течение примерно 1 ч, отфильтровывают и промывают этилацетатом (220 см 3), затем сушат в вакууме и получают 10,9 г кристаллического малеата дулоксетина. Выход 74%. Свободное основание дулоксетина может быть получено из его малеатной соли по методике, описанной в примере 2. Пример 4. Получение хлорида (S)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина (гидрохлорида дулоксетина). Образование соли под действием конц. водного раствора хлористо-водородной кислоты в органическом растворителе В колбу объмом 250 см 3, снабжнную магнитной мешалкой, загружают 4,0 г (0,0097 моль) малеата(S)-N-метил-N-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина, 100 см 3 раствора карбоната калия (10%) и 100 см 3 н-гексана. Гетерогенную смесь перемешивают при комнатной температуре до полного растворения малеатной соли, затем слои разделяют и водный слой экстрагируют н-гексаном (250 см 3). Объединнные органические слои промывают водой (150 см 3) до нейтральной реакции. Органический слой сушат над 20 г сульфата натрия, затем сушат в вакууме. Остаток 2,9 г (0,0097 моль) основания дулоксетина растворяют в 29 см 3 ацетона, полученный раствор основания охлаждают ледяной водой до температуры 0-5 С и по каплям прибавляют 0,8 см 3 (0,0097 моль) хлористо-водородной кислоты (37%). Затем в смесь вносят затравку кристаллов и перемешивают при температуре 0-5 С в течение примерно 1-2 ч. Выпавшие в осадок кристаллы отфильтровывают и промывают ацетоном (25 см 3), затем сушат на воздухе и получают 2,65 г кристаллического гидрохлорида дулоксетина (82%). Образование соли под действием безводного хлористого водорода в органическом растворителе. Основание дулоксетина (1,0 г, 0,0033 моль) растворяют в 8 см 3 этилацетата и при комнатной температуре прибавляют по каплям 1,0 см 3 этилацетата, содержащего 0,0033 моль сухого хлористого водорода. После начала кристаллизации суспензию перемешивают в течение 15 мин, фильтруют, промывают этилацетатом (21 см 3) и получают 0,71 г кристаллического гидрохлорида дулоксетина. Выход 64,5%. Получение гидрохлоридной соли из малеатной соли. Малеат дулоксетина (0,82 г, 0,002 моль) суспендируют в 8 см 3 ацетона и охлаждают до температуры 0-5 С. К охлажднной суспензии прибавляют по каплям 0,17 см 3 (0,002 моль) конц. раствора хлористо-водородной кислоты. После растворения малеатной соли выпадает в осадок гидрохлоридная соль дулоксетина. Суспензию кристаллов перемешивают при температуре 0-5 С в течение 1-2 ч, затем кристаллы отфильтровывают, промывают ацетоном (22 см 3), сушат на воздухе и получают 0,42 г (64%) кристаллического гидрохлорида дулоксетина.S)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей, который включает следующие стадии: расщепление рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы II где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; с образованием (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV Взаимодействие полученного соединения формулы IV с 1-фторнафталином с образованием (S)-Nметил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V взаимодействие полученного соединения формулы V с 1-хлорэтилхлорформиатом с образованием дулоксетина формулы I. 2. Способ по п.1, отличающийся тем, что расщепление проводят N-бензоил-D-фенилглицином. 3. Способ по п.1 или 2, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV очищают фракционной кристаллизацией. 4. Способ по п.3, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в(S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV. 5. Способ по п.1 или 2, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин формулы V очищают фракционной кристаллизацией. 6. Способ по п.5, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин формулы V осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V. 7. Способ по п.1 или 2, отличающийся тем, что дулоксетин S)-(+)-N-метил-3-(1-нафталинилокси)3-(2-тиенил)пропанамин) формулы I очищают фракционной кристаллизацией.- 12015828 8. Способ по п.7, отличающийся тем, что дулоксетин S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин) формулы I осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в дулоксетин S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин) формулы I. 9. Способ по п.1 или 2, отличающийся тем, что S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин) формулы I осаждают в форме его малеатной соли, которую затем в растворе выделяют в свободное основание и, не получая в чистом виде, подвергают его взаимодействию с безводным хлористым водородом и получают активное вещество гидрохлорид дулоксетина. 10. Способ по п.1 или 2, отличающийся тем, что (S)-(+)-N-метил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин формулы I выделяют в форме его малеатной соли, затем, не выделяя в свободное основание, малеатную соль подвергают взаимодействию с концентрированной водной хлористоводородной кислотой в органическом растворителе и получают активное вещество гидрохлорид дулоксетина. 11. Способ получения (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IV где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; и получают (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV и, если требуется,полученное соединение формулы IV осаждают в форме его кислотно-аддитивной соли. 12. Способ по п.11, отличающийся тем, что расщепление осуществляют N-бензоил-Dфенилглицином. 13. Способ по п.11 или 12, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV очищают фракционной кристаллизацией. 14. Способ по п.13, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV. 15. Способ получения (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина формулы V где R1, R2 и R3 представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещнную одной или несколькими C1-C4-алкильными группами; трифторметил, C1-C4-алкил, C1-C4-алкокси, -C(=O)-NH2, C1-C4-алкоксикарбонил, трифторметокси или гидрокси; и полученный (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV подвергают взаимодействию с 1-фторнафталином и получают (S)-N-метил-N-бензил-3-(1 нафталинилокси)-3-(2-тиенил)пропанамин формулы V и, если требуется, полученное соединение формулы V осаждают в форме его кислотно-аддитивной соли. 16. Способ по п.15, отличающийся тем, что расщепление осуществляют N-бензоил-Dфенилглицином. 17. Способ по п.15 или 16, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV очищают фракционной кристаллизацией. 18. Способ по п.17, отличающийся тем, что (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1 пропанол формулы IV осаждают в форме его оксалатной соли, затем полученную оксалатную соль превращают в (S)-3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанол формулы IV. 19. Способ по п.15 или 16, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3(2-тиенил)пропанамин формулы V очищают фракционной кристаллизацией. 20. Способ по п.19, отличающийся тем, что (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2 тиенил)пропанамин формулы V осаждают в форме его малеатной соли, затем полученную малеатную соль превращают в (S)-N-метил-N-бензил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамин формулы V.
МПК / Метки
МПК: C07D 333/20
Метки: новые, ключевые, получения, дулоксетина, применения, способ, интермедиаты
Код ссылки
<a href="https://eas.patents.su/15-15828-sposob-polucheniya-duloksetina-i-novye-klyuchevye-intermediaty-dlya-ego-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения дулоксетина и новые ключевые интермедиаты для его применения</a>
Способ получения (s)-n-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина гидрохлорида (дулоксетина)

Номер патента: 14559
Опубликовано: 30.12.2010
Авторы: Плацек Лукас, Цинибулк Йосеф, Ярраг Камаль, Ридван Лудек, Затопкова Моника
МПК: C07D 333/20
Метки: гидрохлорида, s)-n-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина, способ, дулоксетина, получения
Формула / Реферат:
1. Способ получения (S)-N-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина формулы Iи его фармацевтически приемлемых солей, включающий:а) взаимодействие (RS)-N,N-диметил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина формулы IIIс оптически активной D-винной кислотой или кислой солью, являющейся производным D-винной кислоты, с образованием смеси диастереоизомерных солей N,N-диметил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина и D-винной кислоты (2:1),б)...
Способ получения (s)-n-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина гидрохлорида (дулоксетина)

Номер патента: 11768
Опубликовано: 30.06.2009
Авторы: Груби Петр, Кухар Мирослав, Плацек Лукас, Ридван Лудек
МПК: C07D 333/20, A61K 31/38
Метки: s)-n-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина, способ, получения, дулоксетина, гидрохлорида
Формула / Реферат:
1. Способ получения (S)-N-метил-3-(1-нафтилокси)-3-(2-тиенил)пропиламина формулы I или его фармацевтически приемлемой соли, отличающийся тем, что (RS)-N,N-диметил-3-(1-нафтилокси)-3-(2-тиенил)пропиламин формулы III подвергают реакции с оптически активной кислотой, после чего осуществляют кристаллизацию этого диастереоизомера, что дает при реакции с неорганическим или органическим основанием...
Новые бициклические аминопиразиноновые соединения, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 3209
Опубликовано: 27.02.2003
Авторы: Валлез Мари-Одиль, Де Нантей Гийом, Глоанек Филипп, Рюпэн Ален, Верберен Тони
МПК: A61P 7/02, C07D 471/04, A61K 31/4985...
Метки: способ, композиции, фармацевтические, аминопиразиноновые, получения, новые, содержащие, соединения, бициклические
Формула / Реферат:
1. Соединение формулы (I) где R1 обозначает атом водорода или линейную или разветвленную (C1-C6)алкильную группу (необязательно замещенную одним или более одинаковыми или различными заместителями, выбранными из арильной, гетероарильной, циклоалкильной, гетероциклоалкильной, карбокси, линейной или разветвленной (C1-C6)алкоксикарбонильной группы, карбамоильной группы или гидроксигруппы), циклоалкильную группу, гетероциклоалкильную группу или...
Новые соединения аминокислоты, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 6389
Опубликовано: 29.12.2005
Авторы: Де-Нантёй Гийом, Глоане Филипп, Рюпэн Ален, Верберен Тони
МПК: A61K 31/196, A61P 7/04, C07C 257/18...
Метки: получения, новые, соединения, аминокислоты, фармацевтические, композиции, содержащие, способ
Формула / Реферат:
1. Соединение формулы (I) в которой R1 представляет собой арильную группу, гетероарильную группу или линейную или разветвлённую (C1-C6)алкильную группу, необязательно замещённую одной или более одинаковыми или различными группами, выбранными из арила и гетероарила, или R1 представляет собой группу формулы -(CO)-CR6R7NR8R9, в которой R6 представляет собой атом водорода или группу, выбранную из арила, гетероарила, гетероциклоалкила,...
Промышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино) фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона и ключевые промежуточные соединения данного способа

Номер патента: 15478
Опубликовано: 31.08.2011
Авторы: Молнар Чаба, Боди Йожеф, Хорват Золтан, Туба Золтан, Араньи Антал, Балог Габор, Чёргей Янош, Терди Ласло, Санта Чаба, Махо Шандор, Виски Дьёрдь, Селеш Янош
МПК: C07J 21/00, C07J 41/00, C07J 51/00...
Метки: соединения, промышленный, способ, фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона, синтеза, ключевые, промежуточные, данного, способа, 17-ацетокси-11b-[4-(диметиламино
Формула / Реферат:
1. Промышленный способ синтеза 17-ацетокси-11b-[4-(диметиламино)фенил]-21-метокси-19-норпрегн-4,9-диен-3,20-диона формулы (I)из 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)отличающийся тем, что включает стадии:i) образование эпоксида по двойной связи в положении 5(10) взаимодействием 3,3-[1,2-этандиил-бис-(окси)]оэстр-5(10),9(11)-диен-17-она формулы (II)с перекисью водорода;ii) присоединение полученного in situ...
Предыдущий патент: Ингибиторы вируса гепатита с
Следующий патент: Механизм автоматического регулирования зазора дискового тормоза
Случайный патент: Экономящие топливо способ и система изоляции двигателя внутреннего сгорания