Циклопропилконденсированные индолбензазепиновые ингибиторы белка ns5b вируса гепатита с
Номер патента: 15286
Опубликовано: 30.06.2011
Авторы: Джентлс Роберт Г., Бендер Джон А., Хевавасам Пиасена, Динг Мин









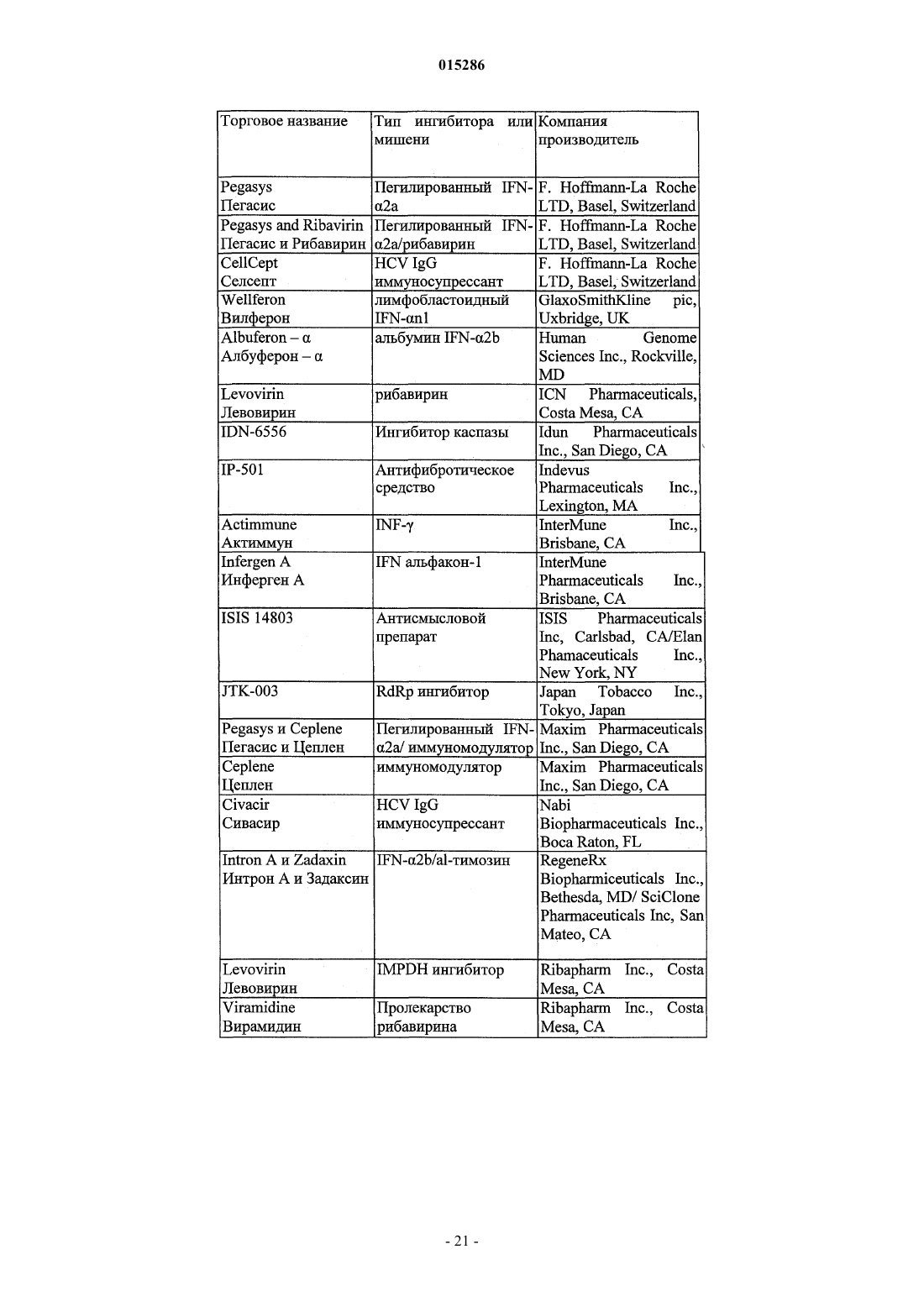







Формула / Реферат
1. Соединение формулы I

где R1представляет собой CO2R5или CONR6R7;
R2 представляет собой ![]() или
или ![]()
R3 представляет собой водород, галоген, (С1-С6)алкил, (С2-С6)алкенил, гидрокси, бензилокси, (С1-С6)алкокси или галоген(С1-С6)алкокси;
R4 представляет собой (С3-С7)циклоалкил;
R5 представляет собой водород или (С1-С6)алкил;
R6 представляет собой водород, (С1-С6)алкил, (С1-С6)алкилSO2, (С3-С7)циклоалкилSO2, галоген(С1-С6)алкилSO2, (R9)2NSO2или (R10)SO2;
R7 представляет собой водород или (С1-С6)алкил;
R8 представляет собой водород, (С1-С6)алкил, (С3-С7)циклоалкил, (С3-С7)циклоалкил(С1-С6)алкил, (С1-С6)алкилкарбонил, (С3-С7)циклоалкилкарбонил, галоген(С1-С6)алкилкарбонил, (С1-С6)алкоксикарбонил, (С1-С6)алкилSO2, (С3-С7)циклоалкилSO2, галоген(С1-С6)алкилSO2, аминокарбонил, ((С1-С6)алкиламино)карбонил, (ди(С1-С6)алкиламино)карбонил, бензил, бензилоксикарбонил или пиридинил;
R9 представляет собой водород, (С1-С6)алкил или (С3-С7)циклоалкил и
R10 представляет собой азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперидинил или гомоморфолинил и замещен 0-3 (С1-С6)алкилами;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где
R3 представляет собой водород, галоген, (С1-С6)алкил, (С2-С6)алкенил, гидрокси, бензилокси или (С1-С6)алкокси;
R8 представляет собой водород, (С1-С6)алкил, (С3-С7)циклоалкил, (С3-С7)циклоалкил(С1-С6)алкил, (С1-С6)алкилкарбонил, (С1-С6)алкоксикарбонил, бензил, бензилоксикарбонил или пиридинил;
R9 представляет собой водород или (С1-С6)алкил и
R10 представляет собой азетидинил, пирролидинил, пиперидинил, пиперазинил, N-(С1-С6алкил)пиперазинил, морфолинил, тиоморфолинил, гомопиперидинил или гомоморфолинил.
3. Соединение по п.1, где
R1 представляет собой CONR6R7;
R6 представляет собой (С1-С6)алкилSO2, (С3-С7)циклоалкилSO2, галоген(С1-С6)алкилSO2, (R9)2NSO2 или (R10)SO2 и
R7 представляет собой водород.
4. Соединение по п.1, где R3 представляет собой водород.
5. Соединение по п.1, где R3 представляет собой метокси.
6. Соединение по п.1, где R4 представляет собой циклогексил.
7. Соединение по п.1, где R6 представляет собой (С1-С6)алкилSO2, (R)2NSO2или (R10)SO2.
8. Соединение по п.1 со следующей стереохимией

9. Соединение по п.1 со следующей стереохимией

10. Соединение по п.1, выбранное из группы, состоящей из






или его фармацевтически приемлемая соль.
11. Соединение по п.1

или его фармацевтически приемлемая соль.
12. Соединение по п.11, где соль представляет собой гидрохлорид.
13. Соединение по п.1

или его фармацевтически приемлемая соль.
14. Композиция, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
15. Композиция по п.14, содержащая по крайней мере одно дополнительное соединение, имеющее терапевтическую эффективность для HCV и выбранное из группы, состоящей из интерферонов, циклоспоринов, интерлейкинов, HCV ингибиторов металлопротеазы, HCV ингибиторов серинпротеазы, HCV ингибиторов полимеразы, HCV ингибиторов хеликазы, HCV NS4B ингибиторов протеина, HCV ингибиторов входа, HCV ингибиторов сборки, HCV ингибиторов выхода, HCV NS5A ингибиторов протеина, HCV NS5B ингибиторов протеина и HCV ингибиторов репликона.
16. Композиция по п.14, где соединение по п.1 представляет собой

или его фармацевтически приемлемая соль.
17. Композиция по п.15, где соединение по п.1 представляет собой

или его фармацевтически приемлемая соль.
18. Способ лечения инфекционного гепатита С, включающий введение терапевтически эффективного количества соединения по п.1 пациенту.
19. Способ по п.18, включающий введение по крайней мере одного дополнительного соединения, имеющего терапевтическую эффективность для HCV, выбранного из группы, состоящей из интерферонов, циклоспоринов, интерлейкинов, HCV ингибиторов металлопротеазы, HCV ингибиторов серинпротеазы, HCV ингибиторов полимеразы, HCV ингибиторов хеликазы, HCV NS4B ингибиторов протеина, HCV ингибиторов входа, HCV ингибиторов сборки, HCV ингибиторов выхода, HCV NS5A ингибиторов протеина, HCV NS5B ингибиторов протеина и HCV ингибиторов репликона.
20. Способ по п.18, где соединение по п.1 представляет собой

или его фармацевтически приемлемая соль.
21. Способ по п.19, где соединение по п.1 представляет собой

или его фармацевтически приемлемая соль.
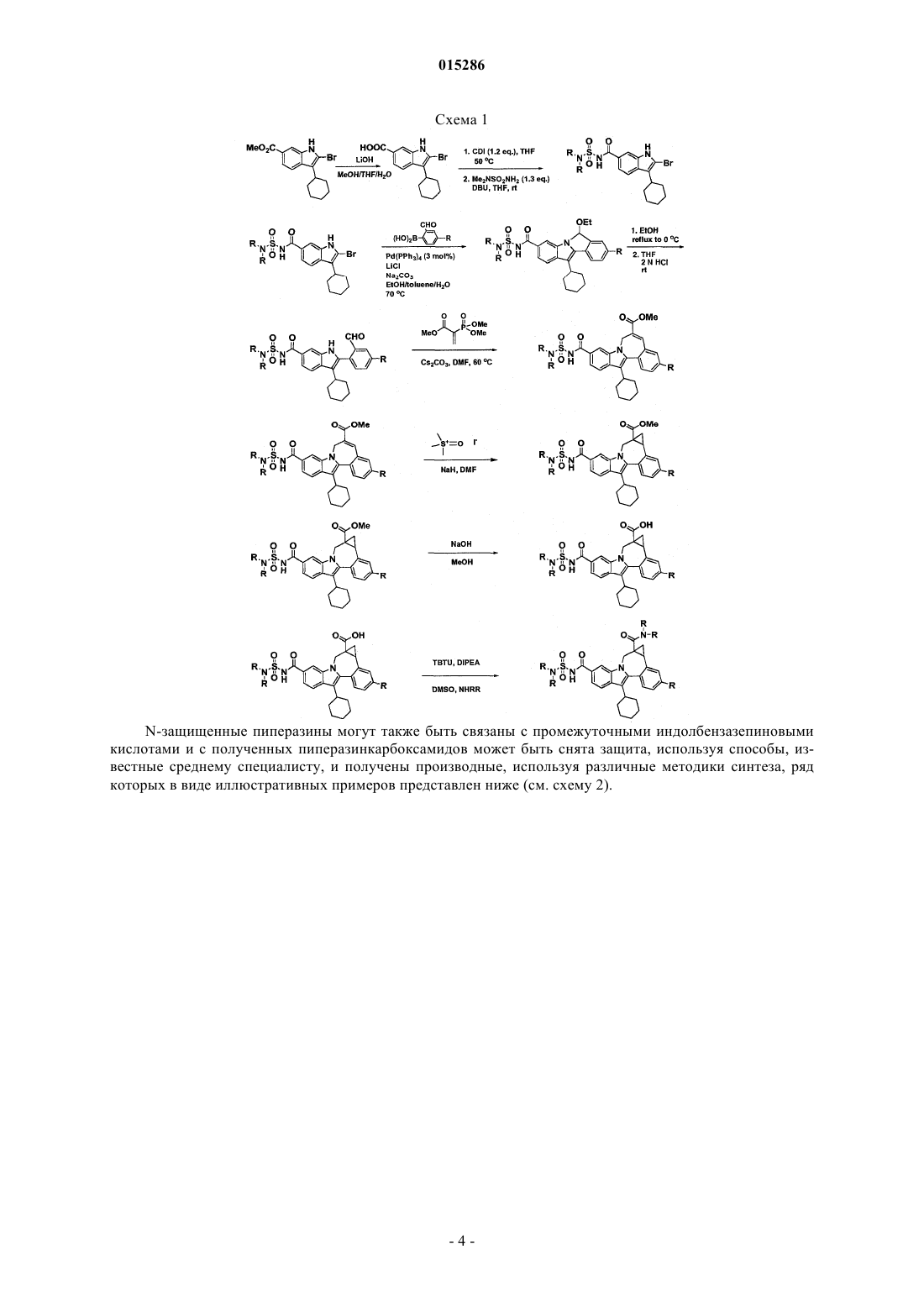
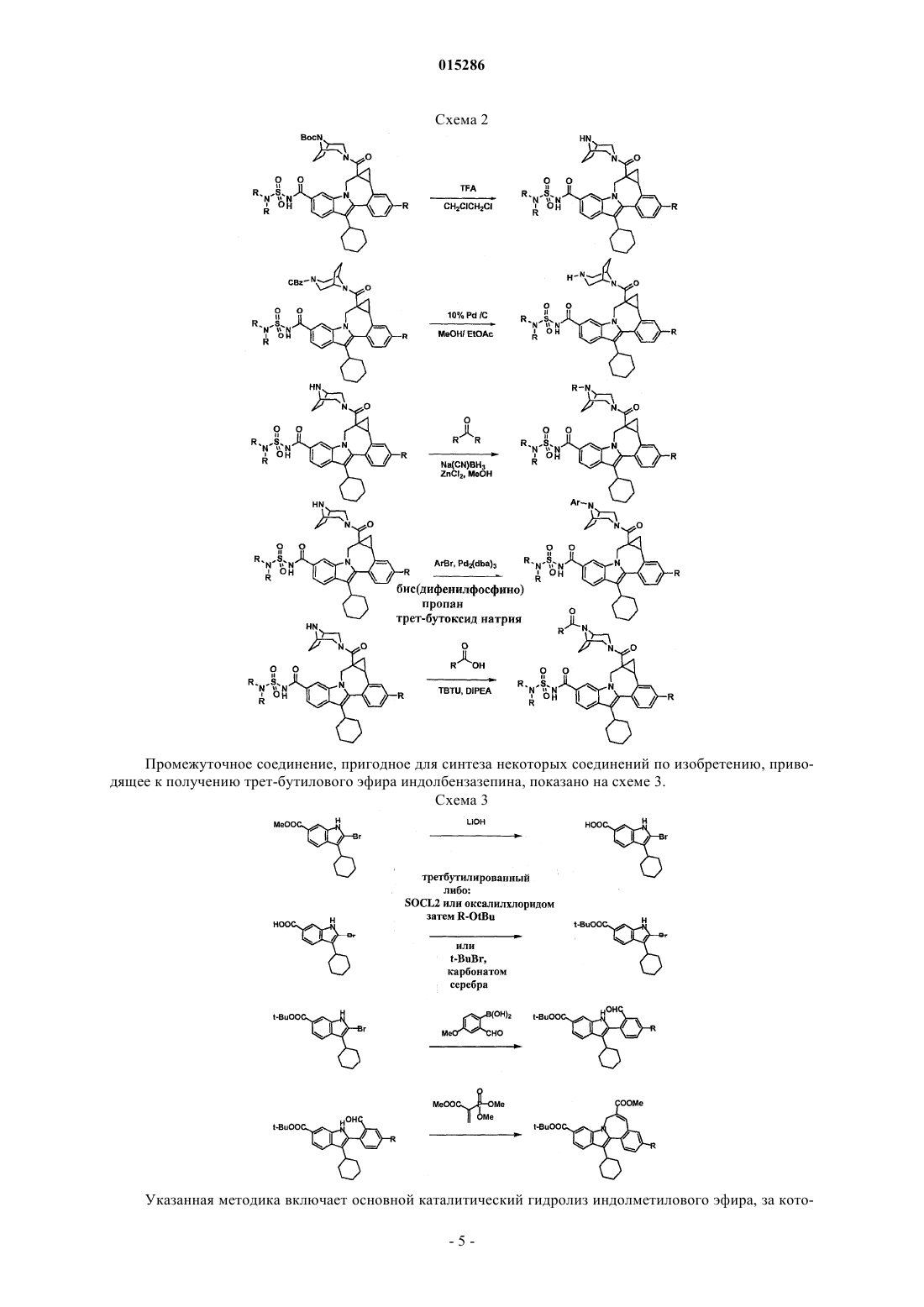
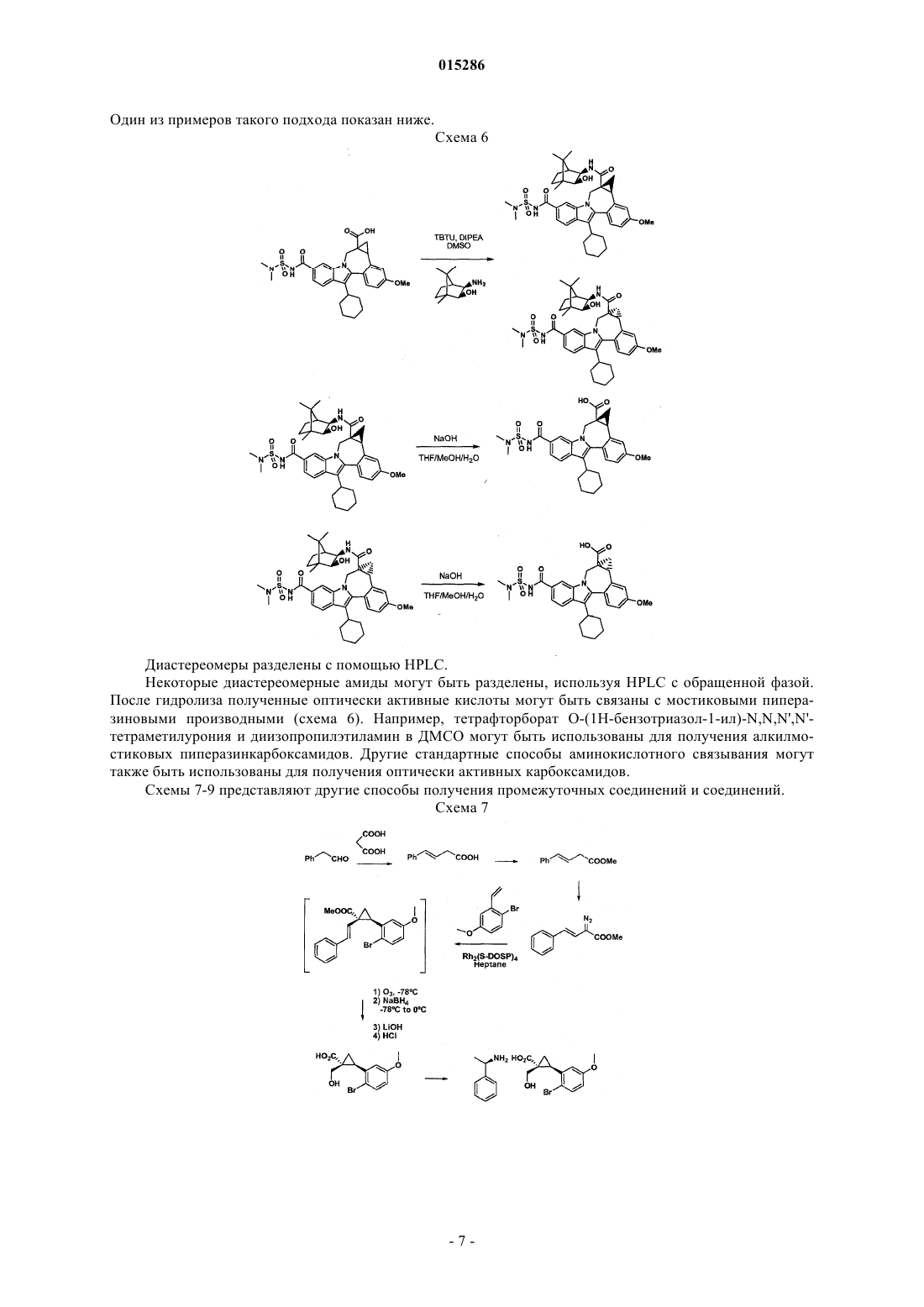
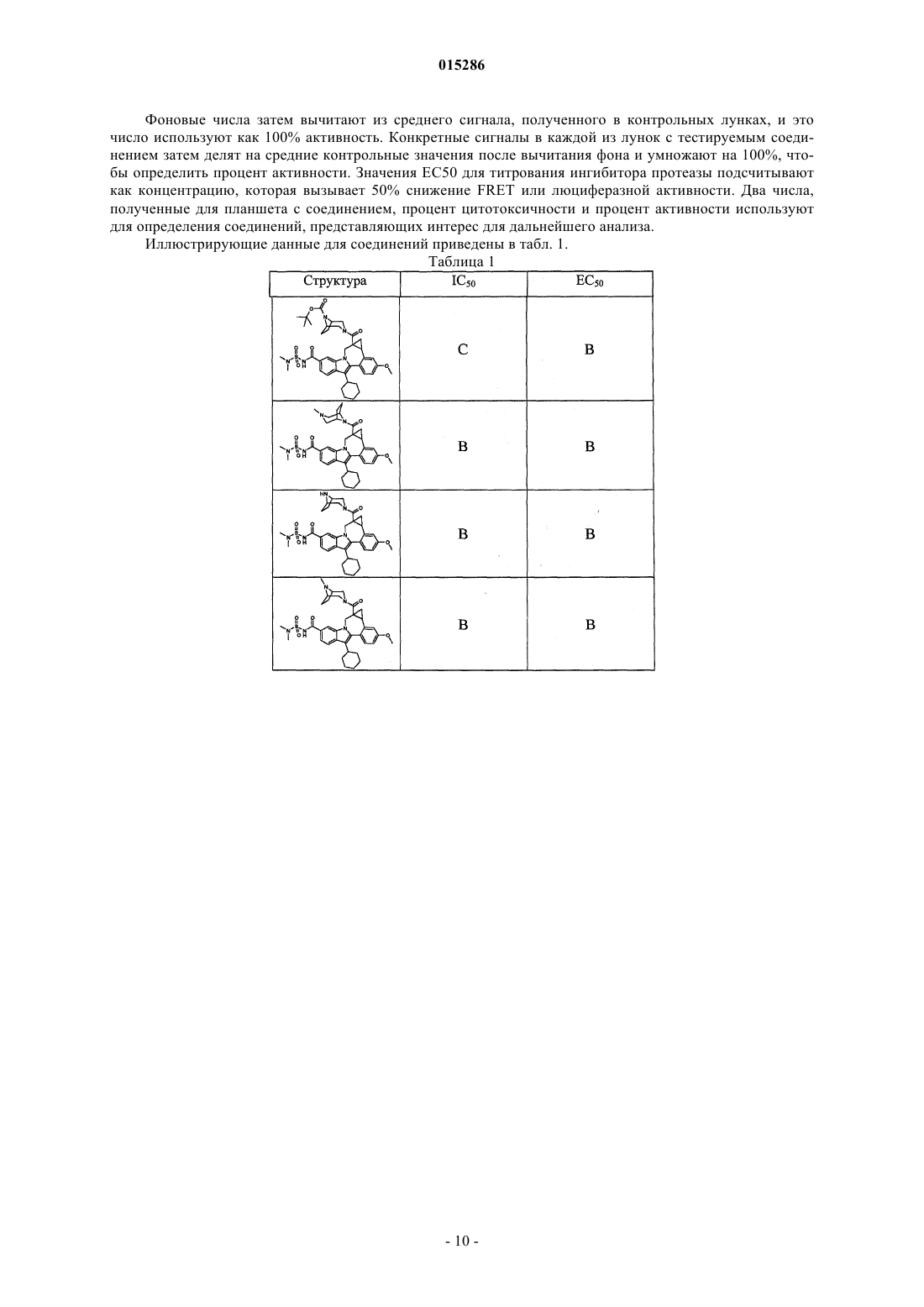
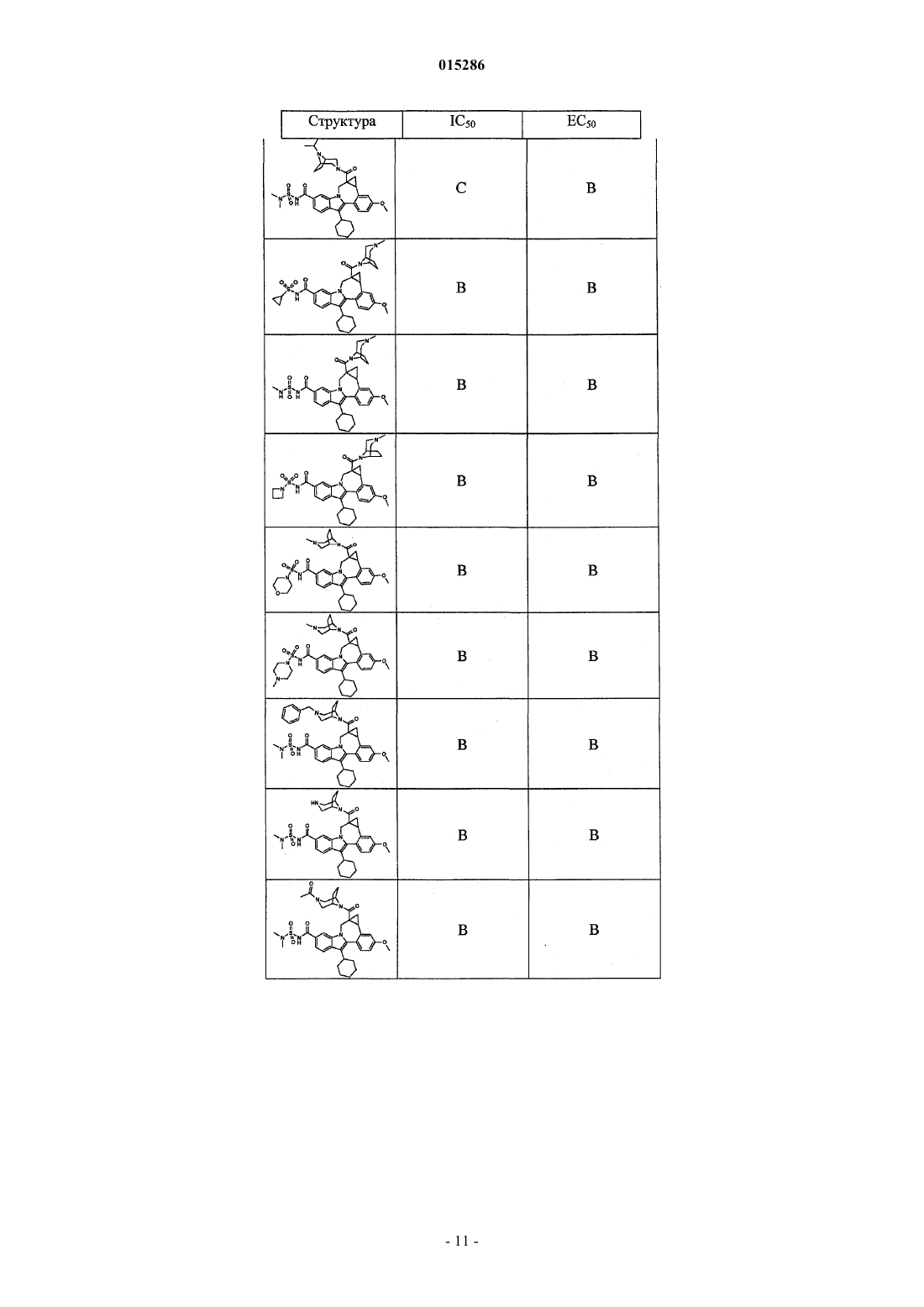
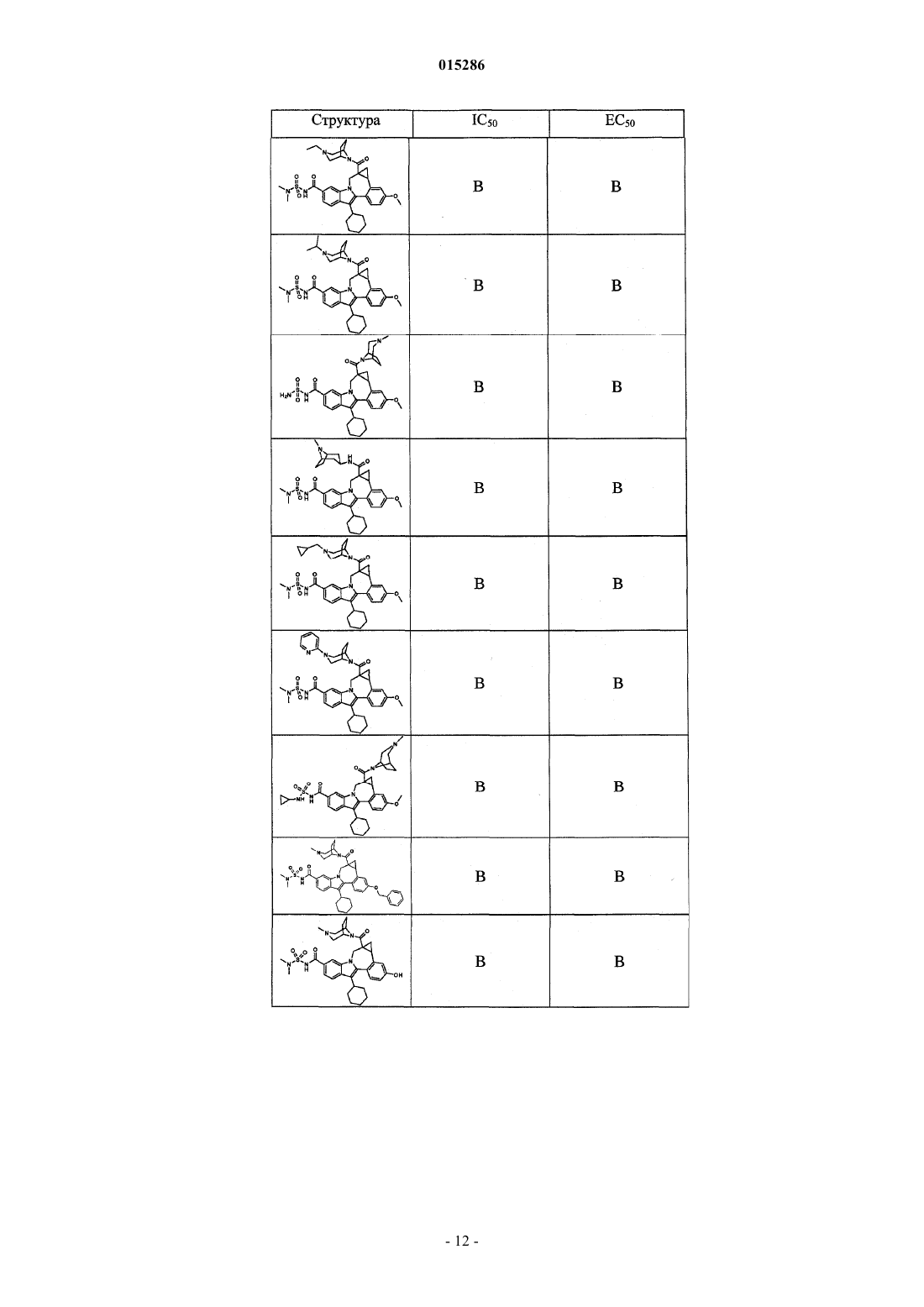
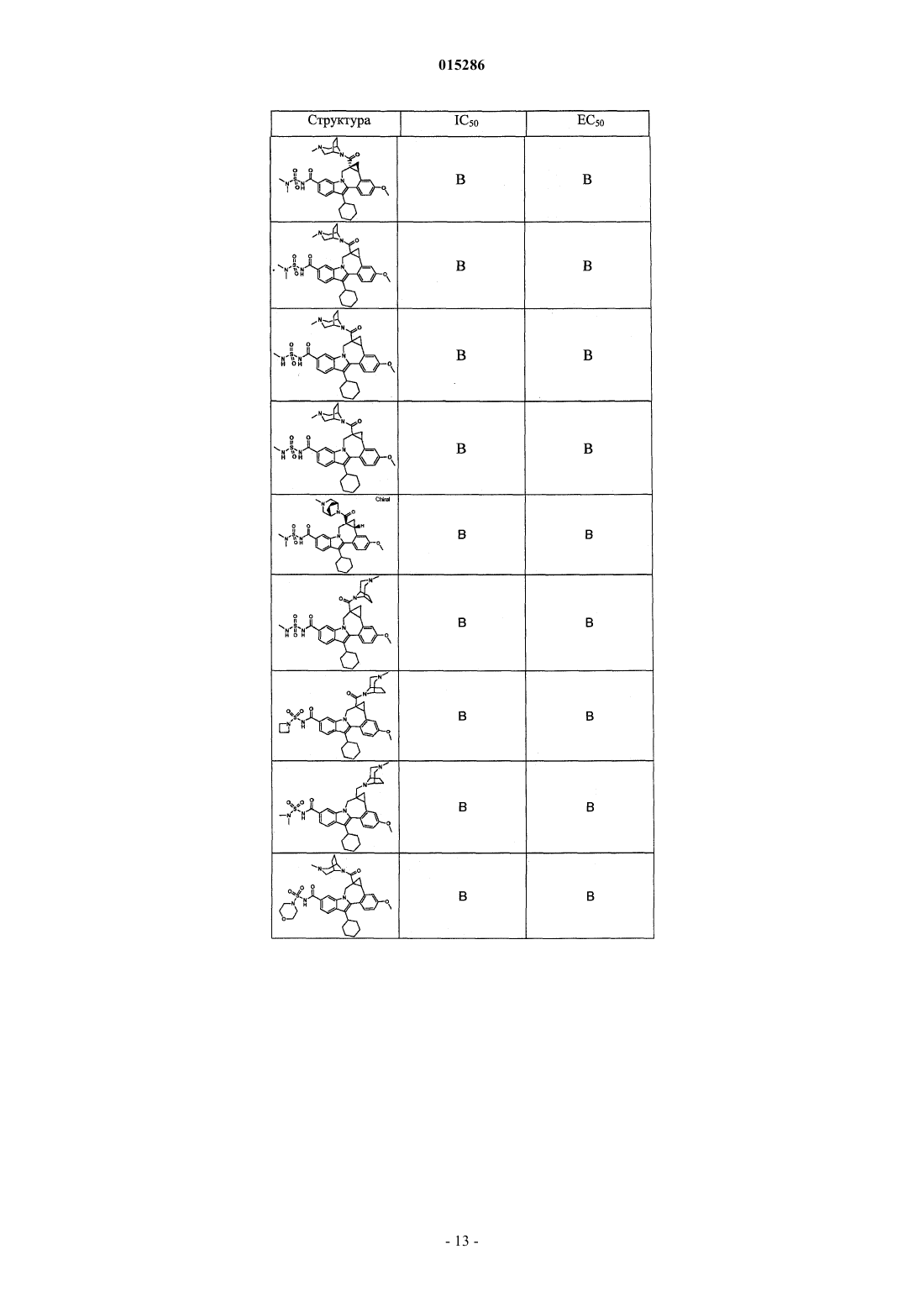
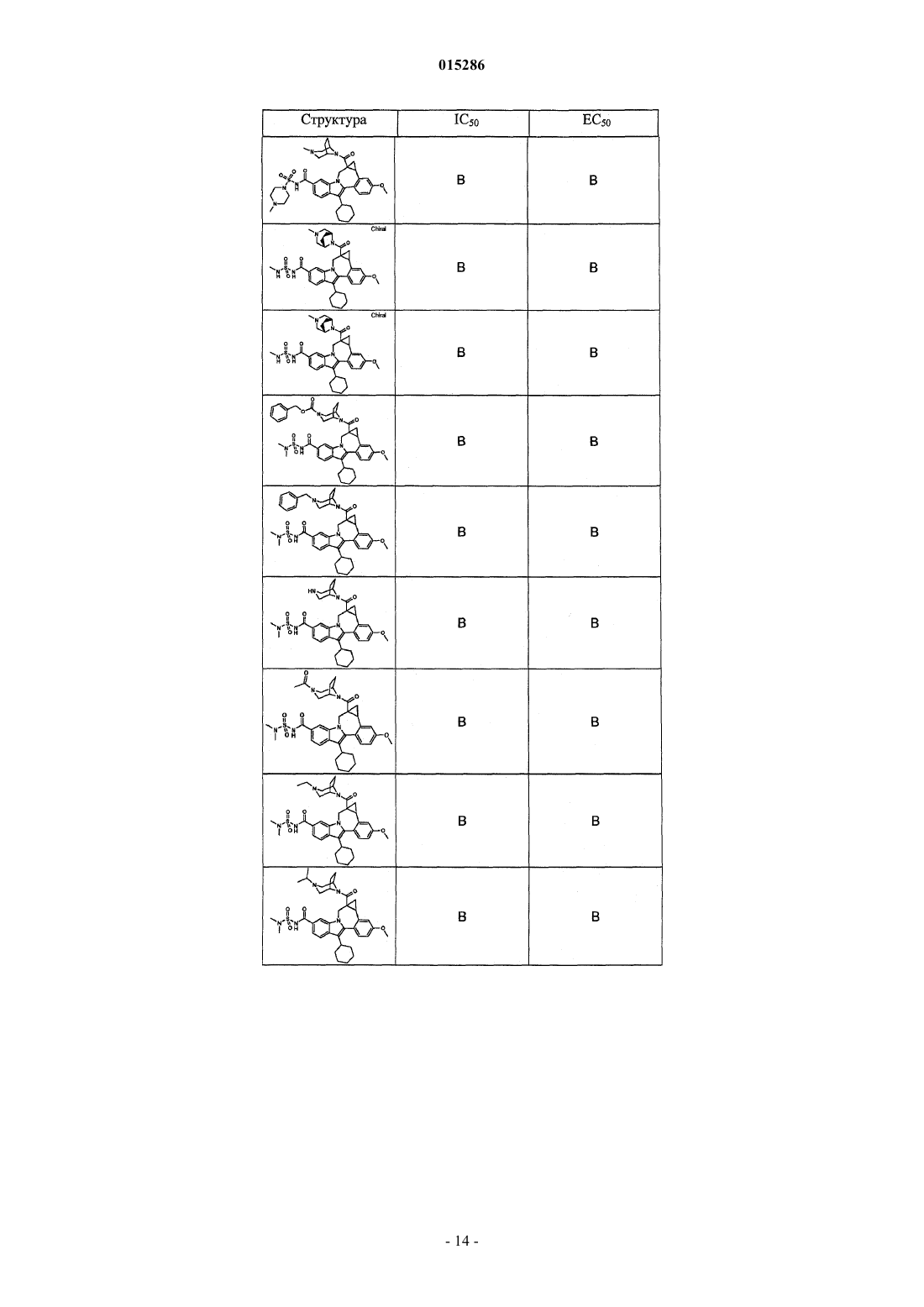
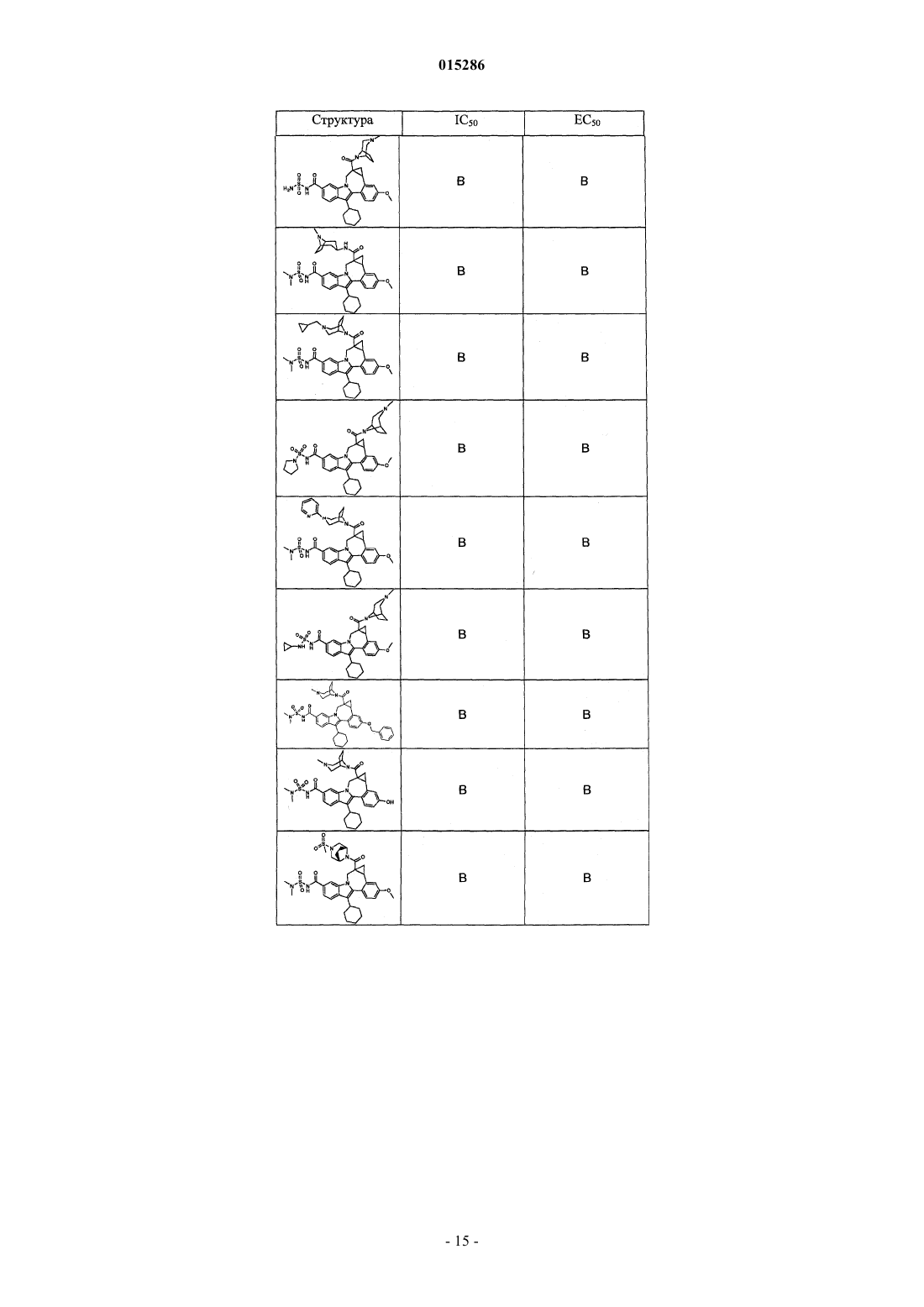
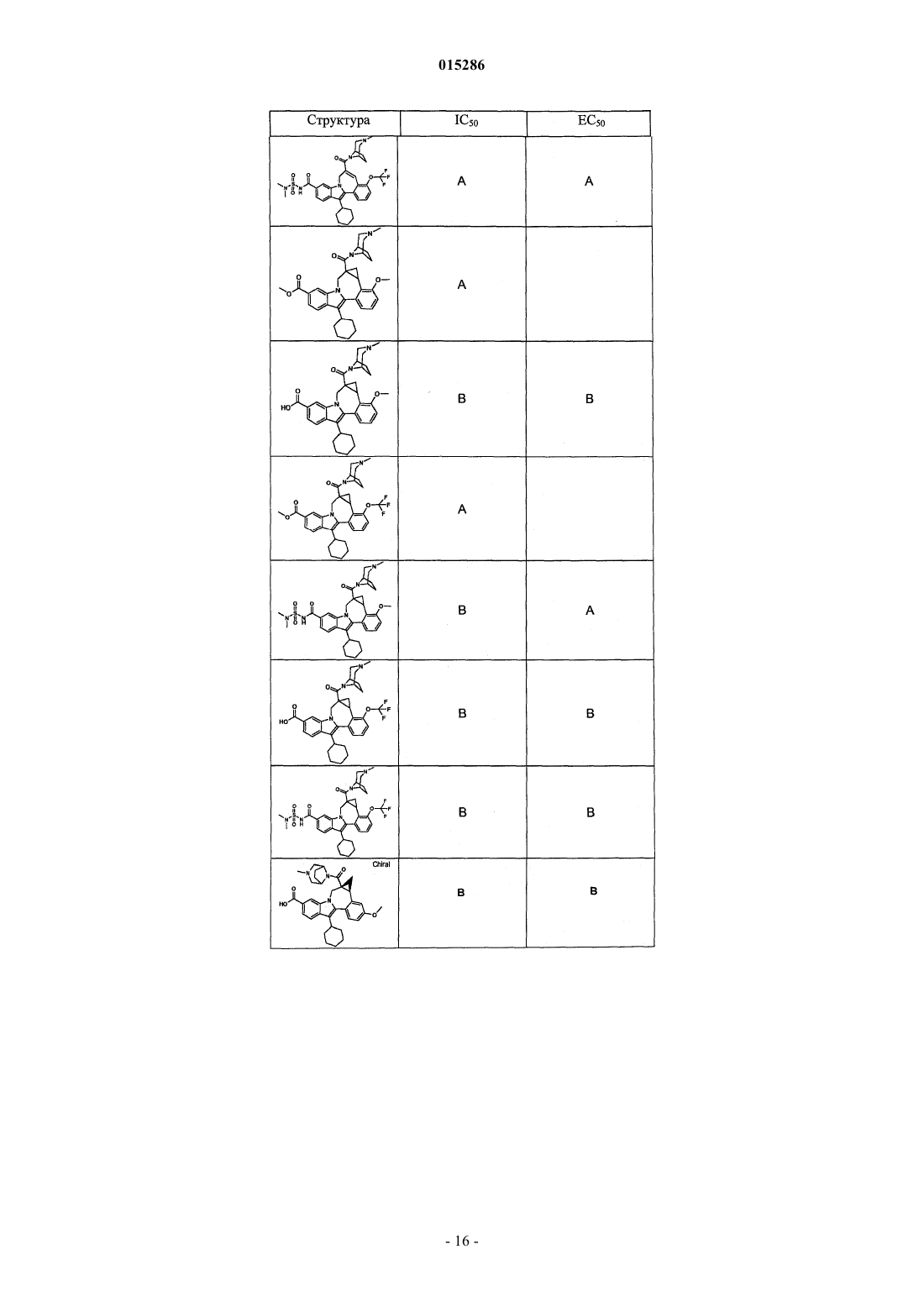
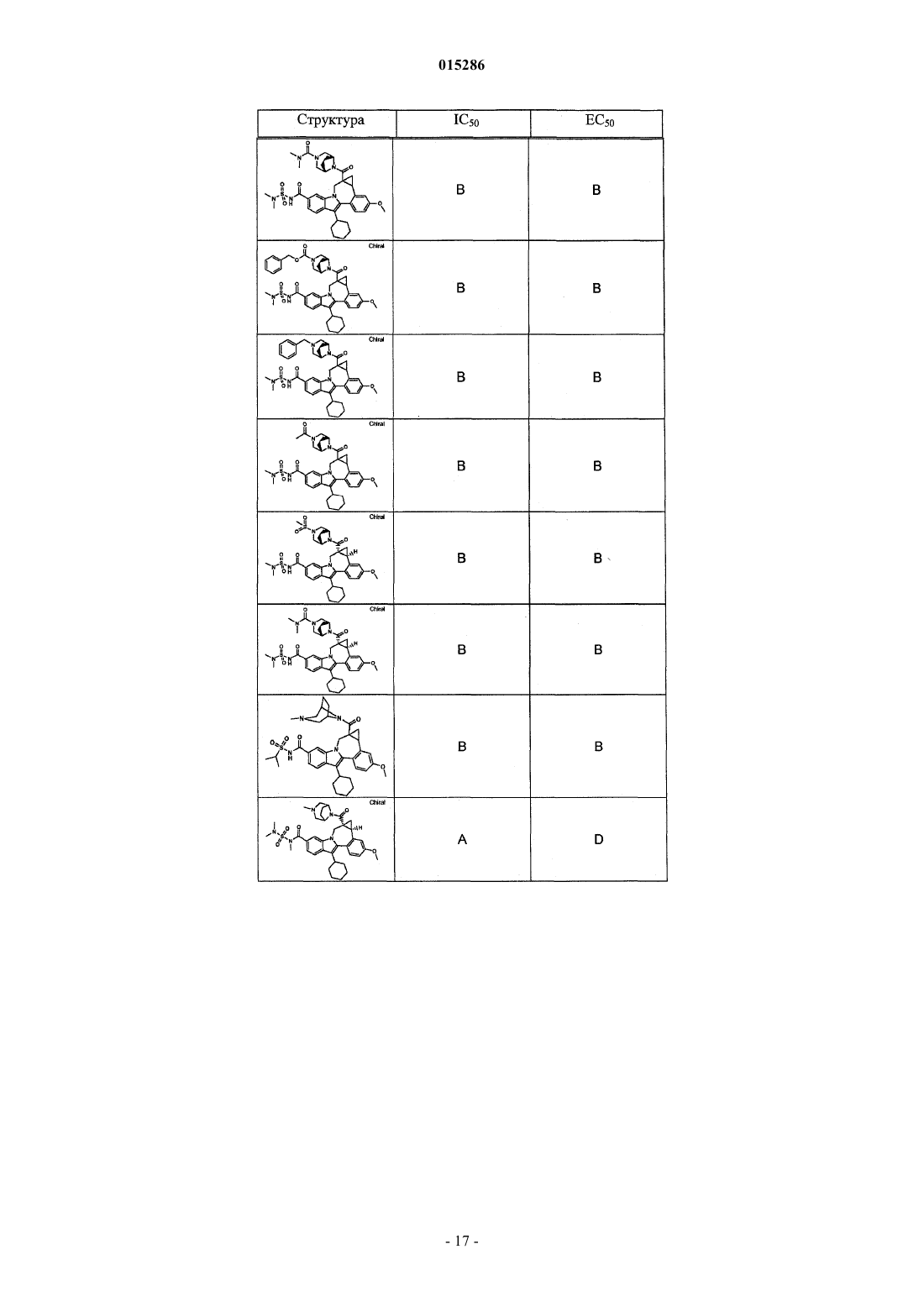
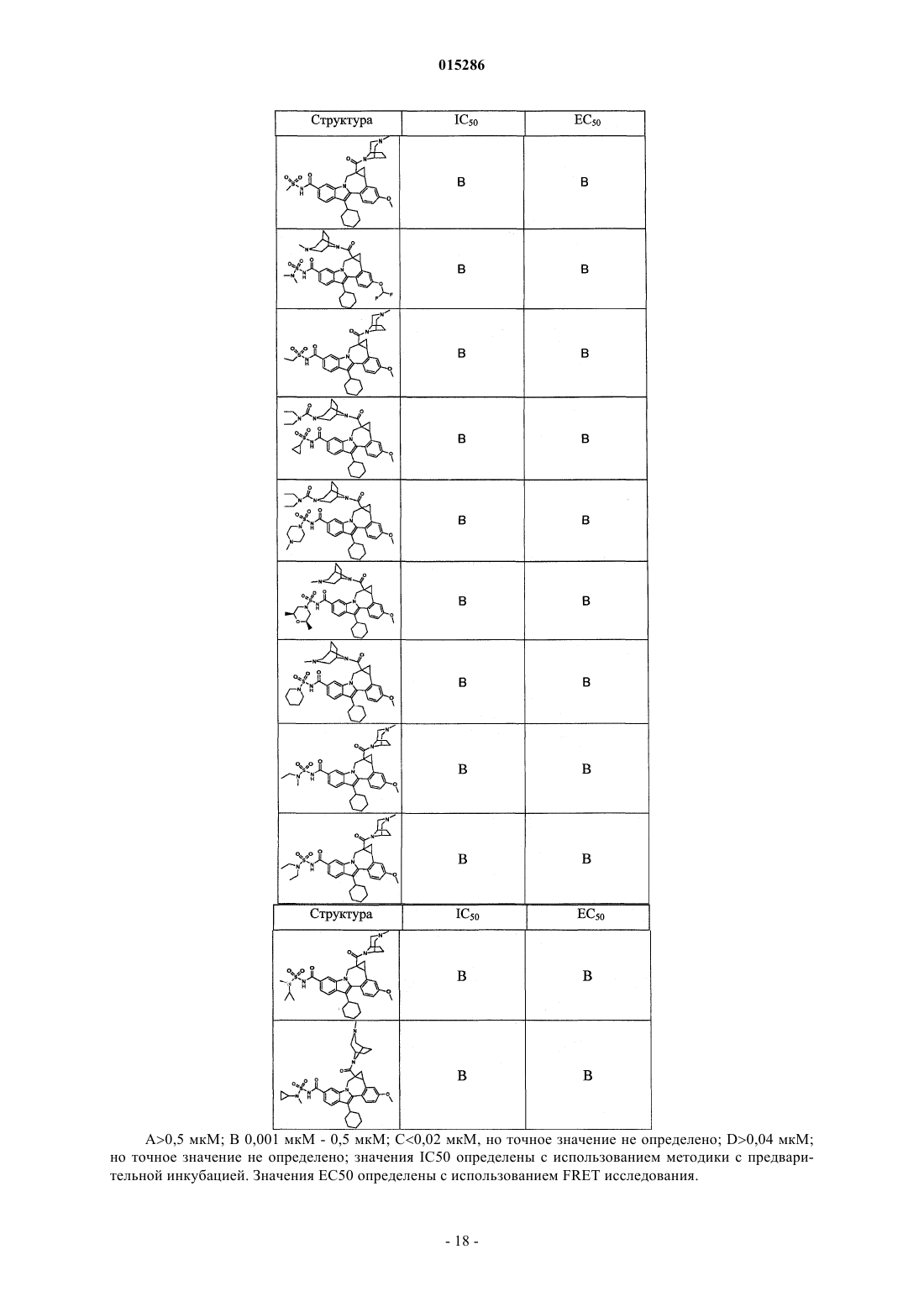
Текст
ЦИКЛОПРОПИЛКОНДЕНСИРОВАННЫЕ ИНДОЛБЕНЗАЗЕПИНОВЫЕ ИНГИБИТОРЫ БЕЛКА NS5B ВИРУСА ГЕПАТИТА С Изобретение охватывает соединения формулы (I) также как композиции и способы использования соединений. Соединения имеют активность против инфекционного заболевания гепатита С (HCV) и являются пригодными при лечении указанных инфекций. 015286 Уровень техники Вирус гепатита С (HCV) является важным патогеном человека, инфицировавшим предположительно 170 млн человек по всему миру - грубо в пять раз больше, чем инфицированных вирусом иммунодефицита человека 1 типа. У значительного числа инфицированных HCV индивидуумов развивается серьезное прогрессирующее заболевание печени, включая цирроз и печеночно-клеточный рак (Lauer, G. M.;HCV является положительно закрученным РНК вирусом. Основываясь на сравнении вычисленной последовательности аминокислот и значительном сходстве в 5'-нетранслируемой области, HCV классифицируется как отдельный род в семействе Flaviviridae. Все представители семейства Flaviviridae имеют оболочечные вирионы, которые содержат положительно закрученный РНК геном, кодирующий все известные вирус-специфические белки с помощью трансляции единичной непрерывной открытой рамки считывания. Существенная гетерогенность найдена в нуклеотидной и кодированной аминокислотной последовательности по всему геному HCV. По меньшей мере шесть основных генотипов и более чем 50 подтипов были описаны. Основные генотипы HCV отличаются по распространенности в мире и клиническое значение генетической гетерогенности HCV остается неясным, несмотря на множество исследований возможного действия генотипов на патогенез и терапию. Геном одноцепочечной РНК HCV приблизительно 9500 нуклеотидов длиной и имеет одну открытую рамку считывания (ORF), кодирующую один большой полипротеин из примерно 3000 аминокислот. В инфицированных клетках этот полипротеин расщепляется по многим сайтам клеточными и вирусными протеазами до получения структурных и неструктурных (NS) белков. В случае HCV поколение зрелых неструктурных белков (NS2, NS3, NS4A, NS4B, NS5A и NS5B) подвергается воздействию двух вирусных протеаз. Первая, как предполагают, является металлопротеазой и расщепляет NS2-NS3 связь; вторая является сериновой протеазой, содержащей на N-конце NS3 (также обозначаемая как NS3 протеаза), и опосредует все последующие расщепления ниже по каскаду NS3, как в цис-положении, в сайте расщепленияNS3-NS4A, и в транс-положении, для остальных сайтов NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. NS4A белок, как представляется, выполняет множество функций, действуя как кофактор для NS3 протеазы, и возможно способствует расположению в мембране NS3 и других компонентов вирусной репликазы. Образование комплекса NS3 белка и NS4A представляется необходимым для этапов процессинга, повышая эффективность протеолиза во всех сайтах. Белок NS3 также проявляет активность нуклеозидной трифосфатазы и РНК хеликазы. NS5B (также обозначаемая как HCV полимераза) является РНК-зависимой РНК полимеразой, которая вовлечена в репликацию HCV. Белок HCV NS5B описан в "Structural AnalysisVirology 2002, 3482-3492; and Defrancesco and Rice, Clinics in Liver Disease 2003,7, 211-242). В настоящее время наиболее эффективная терапия HCV включает комбинацию альфа-интерферона и рибавирина, которые приводят к устойчивому эффекту у 40% пациентов (Poynard, Т. et al. Lancet 1998,352, 1426-1432). Последние клинические результаты демонстрируют, что пегилированный альфаинтерферон предпочтителен по сравнению с немодифицированным альфа-интерфероном в виде монотерапии (Zeuzem, S. et al. N. Engl. J. Med. 2000, 343, 1666-1672). Тем не менее, даже при экспериментальных терапевтических режимах, включающих комбинации пегилированного альфа-интерферона и рибавирина, существенная часть пациентов не отмечают устойчивого снижения вирусной нагрузки. Таким образом, существует очевидная и важная необходимость развивать эффективные лекарственные средства для лечения HCV инфекции. Описание изобретения Одним из объектов изобретения является соединение формулы IR5 представляет собой водород или (С 1-С 6)алкил;R7 представляет собой водород или (С 1-С 6)алкил;R10 представляет собой азетидинил, пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперидинил или гомоморфолинил и замещен 0-3 (С 1-С 6)алкиламил; или его фармацевтически приемлемая соль. Другим вариантом изобретения является соединение формулы I, гдеR9 представляет собой водород или (С 1-С 6)алкил; иR10 представляет собой азетидинил, пирролидинил, пиперидинил, пиперазинил, N-(С 1 С 6 алкил)пиперазинил, морфолинил, тиоморфолинил, гомопиперидинил или гомоморфолинил. Предпочтительным является соединение формулы I, гдеR7 представляет собой водород. Другим предпочтительным соединением является соединение формулы I, где R3 представляет собой водород. Другим предпочтительным соединением является соединение формулы I, где R3 представляет собой метокси. Другим предпочтительным соединением является соединение формулы I, где R4 представляет собой циклогексил. Другим предпочтительным соединением является соединение формулы I, где R6 представляет собой 9(R )2NSO2,(R10)SO2. Другим предпочтительным соединением является соединение формулы I, где R6 представляет собой(диметиламино)SO2. Другим предпочтительным соединением является соединение формулы I, где R6 представляет собой алкилSO2. Другим предпочтительным соединением является соединение формулы I, где R6 представляет собой изопропилSO2. Другим объектом изобретения является соединение формулы I в соответствии со следующей стереохимией. Другим объектом изобретения является соединение формулы I в соответствии со следующей стереохимией. Любое значение любого переменного, включая R1, R2, R3, R4, R5, R6, R7, R8, R9 или R10, может быть использовано независимо от значения любого другого переменного радикала. Если иное не указано, данные термины имеют следующие значения. Термин "алкил" означает алкильную группу с прямой или разветвленной цепью, состоящую из от 1 до 6 атомов углерода. Термин"алкенил" означает алкильную группу с прямой или разветвленной цепью, состоящую из от 2 до 6 атомов углерода с по крайней мере одной двойной связью. Термин "циклоалкил" означает моноциклическую кольцевую систему, содержащую от 3 до 7 атомов углерода. Термины "гидроксиалкил," "алкокси" и другие термины с замещенным алкильным остатком включают изомеры с прямой или разветвленной цепью, содержащие от 1 до 6 атомов углерода для алкильного остатка. Термины "галогеналкил" и "гало-2 015286 геналкокси" включают все галогенированные изомеры от моногалогензамещенного алкила до пергалогензамещенного алкила. Термин "арил" включает карбоциклические и гетероциклические ароматические заместители. Побочные и мультипобочные термины предназначены для понимания средним специалистом связывания зависимостей. Например, термин, такой как R)алкил), означает алкильный заместитель в дальнейшем замещенный заместителем R. Изобретение включает все фармацевтически приемлемые формы солей соединений. Фармацевтически приемлемыми солями являются те, в которых противоположенные ионы не вносят значительный вклад в физиологическую активность или токсичность соединений и по существу функционируют как фармакологические эквиваленты. Указанные соли могут быть получены в соответствии с общими органическими методиками, используя коммерчески доступные реагенты. Некоторые анионные формы солей включают ацетат, ацистрат, безилат, бромид, камзилат, хлорид, цитрат, фумарат, глюкоуронат, гидробромид, гидрохлорид, гидроиодид, иодид, лактат, малеат, мезилат, нитрат, памоат, фосфат, сукцинат,сульфат, тартрат, тозилати ксинофоат. Некоторые катионоактивные формы солей включают аммоний,алюминий, бензатин, висмут, кальций, холин, диэтиламин, диэтаноламин, литий, магний, меглумин, 4 фенилциклогексиламин, пиперазин, калий, натрий, трометамин и цинк. Некоторые соединения по изобретению имеют асимметричные атомы углерода (смотри, например,структуры, представленные ниже). Изобретение включает все стереоизомерные формы, включая энантиомеры и диастереомеры, также как смеси стереоизомеров, такие как рацематы. Некоторые стереоизомеры могут быть получены, используя способы, известные среднему специалисту. Стереоизомерные смеси соединений и связанные промежуточные соединения могут быть разделены на индивидуальные изомеры в соответствии с общими способами, известными среднему специалисту. Использование промежуточных или отдельных данных в описании молекулярных структур в нижеследующих схемах и таблицах предназначены только, чтобы указать на относительность стереохимии и не должены интерпретироваться как допущение абсолютных стереохимических значений. Соединения могут быть получены с помощью способов, известных среднему специалисту, включая те, которые описаны ниже. Некоторые реагенты и промежуточные соединения являются известными среднему специалисту. Другие реагенты и промежуточные соединения может быть получены с помощью способов, известных среднему специалисту, используя легко доступные продукты. Радикалы (например,нумерованные "R" заместителей) использованы, чтобы описать синтез соединений, предназначеный только для иллюстрации получения, и не должны быть смешаны с радикалами, приведенными в формуле изобретения или в других разделах описания. Сокращения, использованные в схемах, в целом следуют традициям, используемым средним специалистом. Метил-2-бром-3-циклогексил-1H-индол-6-карбоксилат может быть гидролизован до 2-бром-3 циклогексил-1 Н-индол-6-карбоновой кислоты (см. схему 1). Данное соединение может быть конденсировано с различными сульфонилмочевинами, используя, например, 1,1-карбонилдиимидазол в комбинации с 1,8-диазабицикло[5.4.0]ундец-7-еном в безводном ТГФ. Полученные ацилсульфонамиды могут быть подвергнуты известным реакциям связывания с разнообразными 2-формилбороновыми кислотами или эфирами, используя, например, условия связывание по Сузуки, чтобы обеспечить циклические гемиаминальные промежуточные соединения из представленного типа. Эти соединения могут быть превращены в индолбензазепиновые производные путем обработки метил-2-(диметоксифосфорил)акрилатом в присутствии карбоната цезия в ДМФА через механизм реакций по Михаэлю и Хорнеру Эммонсу. Родственные конденсированные циклопропилэфирные производные могут быть произведены с помощью способов, известных среднему специалисту, включая обработку индолбензазепиновых эфиров иодидом триметилсульфоксония в сильно основных условиях в ДМСО. Оставшийся остаток алифатического эфира в полученных конденсированных циклопропанах может быть гидролизован и целевые кислоты могут быть конденсированы с различными алкилмостиковыми пиперазинами. Например, тетрафторборат O-(1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония и диизопропилэтиламин в ДМСО может дать алкилмостиковые пиперазинкарбоксамиды.N-защищенные пиперазины могут также быть связаны с промежуточными индолбензазепиновыми кислотами и с полученных пиперазинкарбоксамидов может быть снята защита, используя способы, известные среднему специалисту, и получены производные, используя различные методики синтеза, ряд которых в виде иллюстративных примеров представлен ниже (см. схему 2). Промежуточное соединение, пригодное для синтеза некоторых соединений по изобретению, приводящее к получению трет-бутилового эфира индолбензазепина, показано на схеме 3. Схема 3 Указанная методика включает основной каталитический гидролиз индолметилового эфира, за кото-5 015286 рым следует его реакция или с тионилхлоридом и третичным бутоксидом калия или алкилирование карбонатом серебра и третичными бутилбромидами. Полученное соединение может быть превращено, используя химические аналоги, которые представлены ранее, чтобы обеспечить смешанные индолбензазепиновые эфиры, показанные выше. Указанные промежуточные соединения являются пригодными в альтернативной методике, которая может быть применена для получения ацилсульфамида и ацилсульфонамид алкилмостиковых пиперазинов, как показано на Схеме 4. Циклопропанирование промежуточного трет-бутилового эфира индолбензазепина и последующее отщепление группы трет-бутилового эфира может генерировать кислоту, которая может быть связана с разнообразными сульфонамидами и сульфонилмочевинами. Последующий гидролиз дает родственную алифатическую кислоту, которая может быть связана с разнообразными алкилмостиковыми пиперазинами. Например, тетрафторборат O-(1H-бензотриазол-1-ил)-N,N,N',N'тетраметилурония и диизопропилэтиламин в ДМСО могут дать алкилмостиковые пиперазинкарбоксамиды. Схема 4 Некоторые примеры представлены как стереоизомерные смеси. Изобретение охватывает все стереоизомеры соединений. Способы фракционирования стереоизомерных смесей хорошо известны среднему специалисту и включают, но без ограничения; препаративную хиральную сверхкритическую жидкостную хроматографию (SFC) и хиральную высокоэффективную жидкостную хроматографию (HPLC). Пример использования такого подхода показан на схеме 5. Схема 5 Дополнительный способ осуществления таких разделений включает получение смеси диастереомеров, которая может быть разделена, используя различные способы, известные среднему специалисту.-6 015286 Один из примеров такого подхода показан ниже. Схема 6 Диастереомеры разделены с помощью HPLC. Некоторые диастереомерные амиды могут быть разделены, используя HPLC с обращенной фазой. После гидролиза полученные оптически активные кислоты могут быть связаны с мостиковыми пиперазиновыми производными (схема 6). Например, тетрафторборат O-(1 Н-бензотриазол-1-ил)-N,N,N',N'тетраметилурония и диизопропилэтиламин в ДМСО могут быть использованы для получения алкилмостиковых пиперазинкарбоксамидов. Другие стандартные способы аминокислотного связывания могут также быть использованы для получения оптически активных карбоксамидов. Схемы 7-9 представляют другие способы получения промежуточных соединений и соединений. Схема 7 Биологические методы Соединения демонстрируют активность против HCV NS5B, что определено в нижеследующих исследованиях HCV RdRp. Клонирование, экспрессия и очистка HCVNS5B RdRp. кДНК, кодирующая NS5B белок HCV, генотип 1b, клонирована в рЕТ 21 векторе экспрессии. Белок экспрессирован с укорочением на 18 аминокислот с С-конца для повышения растворимости. Е. coli компетентная линия клеток BL21(DE3) используется для экспрессии белка. Культуры выращивают при 37 С в течение примерно 4 ч до того, как культуры достигают оптической плотности 2,0 при 600 нм. Культуры охлаждают до 20 С и индуцируют 1 мМIPTG. Свежий ампициллин добавляют до конечной концентрации 50 мкг/мл и клетки выращивают в течение ночи при 20 С. Осадки клеток (3L) лизируют для очистки, чтобы получить 15-24 мг очищенного NS5B. Литический буфер состоит из 20 мМ Tris-HCl, pH 7,4, 500 мМ NaCl, 0,5% тритон Х-100, 1 мМ DTT, 1 мМ EDTA,20% глицерин, 0,5 мг/мл лизоцима, 10 мМ MgCl2, 15 мкг/мл дезоксирибонуклеазы I и таблетки ингибитора протеаз Complete TM (Roche). После добавления литического буфера замороженные осадки клеток ресуспендируют, используя гомогенизатор тканей. Для уменьшения вязкости образца, аликвоты лизата разрушают ультразвуком на льду, используя микронаконечник, прикрепленный к ультразвуковому аппарату Branson. Разрушенный ультразвуком лизат центрифугируют при 100000 g в течение 1 ч при 4 С и фильтруют через 0,2 мкм фильтр (Corning). Белок очищают, используя три последовательных этапа хроматографии: гепарин сефароза CL-6B,поли-U сефароза 4 В и Hitrap SP сефароза (Pharmacia). Хроматографические буферы были идентичны литическому буферу, но не содержали лизоцима, дезоксирибонуклеазы I, MgCl2 или ингибитор протеаз и концентрация NaCl в буфере была откорректирована в соответствии с потребностью загрузить белок в колонку. Каждая колонка была элюирована с градиентом NaCl, который варьировал по длине от 5 до 50 объемов колонки в зависимости от вида колонки. После заключительного этапа хроматографии итоговая чистота фермента составляет более 90%, основываясь на SDS-PAGE анализе. Фермент аликвотируют и-8 015286 хранят при -80 С. Стандартное исследование фермента HCVNS5B RdRp. Исследования HCV RdRp генотипа 1b проводят в конечном объеме 60 мкл в 96-луночных планшетах (Costar 3912). Буфер исследования состоит из 20 мМ Hepes, рН 7,5, 2,5 мМ KCl, 2,5 мМ MgCl2, 1 мМ DTT, 1,6 U ингибитора РНКазы (PromegaN2515),0,1 мг/мл BSA (Promega R3961) и 2% глицерина. Все соединения последовательно разводят (3-кратно) в ДМСО и разводят далее в воде так, что конечная концентрация ДМСО в исследовании составляет 2%. Фермент HCV RdRp генотипа lb используют в конечной концентрации 28 нМ. Шаблон поли-А используют при 6 нМ и биотинилированный праймер олиго-dT12 используют при конечной концентрации 180 нМ. Шаблон приобретают (Amersham 27-4110). Биотинилированный праймер изготовливает Sigma Genosys. 3H-UTP используют при 0,6 мкКи (0,29 мкМ общей UTP). Реакции запускают путем добавления фермента, инкубируют при 30 С в течение 60 мин и останавливают путем добавления 25 мкл 50 мМPackard Top Count NXT после 1 ч инкубации при комнатной температуре. Исследование модифицированного фермента HCVNS5B RdRp. Исследование модифицированного фермента выполняют преимущественно, как это описано для стандартного исследования фермента за исключением следующего: биотинилированный олиго dT12 праймер предварительно захватывается на покрытые стрептавидином SPA гранулы путем смешивания праймера и гранул в буфере исследования и инкубирования при комнатной температуре в течение одного часа. Несвязанный праймер удаляют после центрифугирования. Связанные с праймером гранулы ресуспендируют в 20 мМ HEPES буфере, рН 7,5 и используют в исследовании в конечной концентрации 20 нМ праймера и 0,67 мкг/мкл гранул. Порядок добавления реактивов в исследовании: фермент (14 нМ) добавляют к разбавленному соединению, после чего добавляют смесь матрицы (0,2 нМ), 3H-UTP (0,6 мкКи, 0,29 мкМ) и связанные с праймером гранулы, чтобы инициировать реакцию; приведенные концентрации являются конечными. Реакции проводят в течение 4 ч при 30 С. Значения IC50 для соединений определяют, используя семь разных [I]. Значения IC50 подсчитывают исходя из ингибирования, используя формулу у = A+B-A)/(1+C/x)D. Подготовка исследования FRET Для выполнения скринингового HCV FRET исследования 96-луночные планшеты с клеточными культурами были использованы. Пептид FRET (Anaspec, Inc.) (Taliani et al. Anal. Biochem. 1996, 240, 6067) содержит флуоресцентный донор, EDANS, рядом с одним концом пептида и акцептор DABCYL рядом с другим концом. Флуоресценцию пептида гасят с помощью межмолекулярного резонансного переноса энергии (RET) между донором и акцептором, и по мере того как NS3 протеаза расщепляет пептид,продукты высвобождаются из RET гашения и флуоресценция донора становится заметной. Реактив исследования получают следующим образом: 5 Х литический реактив люциферазы клеточной культуры отPromega (E153A) разбавляют до 1X dH2O, NaCl добавляют до конечной концентрации 150 мМ, FRET пептид разбавляют до конечной концентрации 20 мкМ из 2 мМ исходного раствора. Для приготовления планшетов клетки с HCV репликоном с или без репортерного гена люциферазыRenilla трипсинизируют и помещают в каждую лунку 96-луночного планшета с титрованными тестируемыми соединениями, добавленными в колонки с 3 по 12; колонки 1 и 2, содержащие контрольное соединение (ингибитор HCV протеазы), и нижний ряд, содержащий клетки без соединения. Планшеты затем помещают в СО 2 инкубатор при 37 С. Исследования После добавления тестируемых соединений, описанных выше (приготовление исследования FRET),в различное время планшеты удаляют и раствор Alamar синий (Trek Diagnostics, 00-100) добавляют в каждую лунку в качестве меры клеточной токсичности. После считывания в приборе Cytoflour 4000 (РЕBiosystems) планшеты отмывают PBS и затем используют для FRET исследования путем добавления 30 мкл реактива исследования FRET пептида, описанного выше (приготовление исследования FRET) на лунку. Планшет затем помещают в прибор Cytoflour 4000, который устанавливают на 340 возбуждение/490 эмиссия, автоматический режим на 20 циклов и планшет считывают в кинетическом режиме. Обычно, отношение сигнал/шум при использовании анализа конечных точек после считывания составляет по меньшей мере три. Альтернативно, после считывания Alamar синего планшеты отмывают PBS, 50 мкл DMEM (с высоким содержанием глюкозы) без фенолового красного добавляют и планшеты затем используют для исследования люциферазы, применяя систему исследования люциферазы Promega DualGlo Luciferase Assay System. Анализ соединения проводят путем количественного определения относительного ингибированияHCV репликона и значений относительной цитотоксичности. Для расчета уровней цитотоксичности среднее значение флуоресцентных сигналов Alamar синего из контрольных лунок принимают за 100% нетоксичное. Отдельные сигналы в каждой из лунок с тестируемым соединением затем делят на средний контрольный сигнал и умножают на 100% для определения процента цитотоксичности. Для подсчета значений ингибирования HCV репликона берут средний фоновый уровень, который получается из двух лунок, содержащих наибольшее количество ингибитора HCV протеазы в конце периода исследования. Эти числа были схожими с теми, что получены в интактных клетках Huh-7.-9 015286 Фоновые числа затем вычитают из среднего сигнала, полученного в контрольных лунках, и это число используют как 100% активность. Конкретные сигналы в каждой из лунок с тестируемым соединением затем делят на средние контрольные значения после вычитания фона и умножают на 100%, чтобы определить процент активности. Значения ЕС 50 для титрования ингибитора протеазы подсчитывают как концентрацию, которая вызывает 50% снижение FRET или люциферазной активности. Два числа,полученные для планшета с соединением, процент цитотоксичности и процент активности используют для определения соединений, представляющих интерес для дальнейшего анализа. Иллюстрирующие данные для соединений приведены в табл. 1. Таблица 1 А 0,5 мкМ; В 0,001 мкМ - 0,5 мкМ; С 0,02 мкМ, но точное значение не определено; D0,04 мкМ; но точное значение не определено; значения IC50 определены с использованием методики с предварительной инкубацией. Значения ЕС 50 определены с использованием FRET исследования.- 18015286 Фармацевтические композиции и способы лечения Соединения демонстрируют активность в отношении HCV NS5B и могут быть полезными в лечении HCV и HCV инфекции. Следовательно, другой аспект изобретения является композицией, содержащей соединение или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Другим аспектом изобретения является композиция, дополнительно содержащая соединение, обладающее анти-HCV активностью. Другим аспектом изобретения является композиция, где соединение, обладающее анти-HCV активностью, является интерфероном. В другом аспекте изобретения интерферон выбран из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. Другим аспектом изобретения является композиция, где соединение, обладающее анти-HCV активностью, является циклоспорином. В другом аспекте изобретения циклоспорин является циклоспорином А. Другим аспектом изобретения является композиция, где соединение, обладающее анти-HCV активностью, выбрано из группы, состоящей из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое усиливает развитие ответа Т-хелперов 1 типа, интерферирующей РНК, антисмысловой РНК, имиквимода, рибавирина, ингибитора инозина 5'-монофосфат дегидрогеназы, амантадина и римантадина. Другим аспектом изобретения является композиция, где соединение, обладающее анти-HCV активностью, эффективно ингибирует функцию мишени, выбранной из следующих: HCV металлопротеаза,HCV сериновая протеаза, HCV полимераза, HCV хеликаза, HCV NS4B белок, вход HCV, сборка HCV,выход HCV, HCV NS5A белок, IMPDH и нуклеозидный аналог, для лечения HCV инфекции. Другим аспектом изобретения является композиция, содержащая соединение или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель, интерферон и рибавирин. Другим аспектом изобретения является способ ингибирования функции HCV репликона, включающий контактирование HCV репликона с соединением или его фармацевтически приемлемой солью. Другим аспектом изобретения является способ ингибирования функции HCV NS5B белка, включающий контактирование HCV NS5B белка с соединением или его фармацевтически приемлемой солью. Другим аспектом изобретения является способ лечения HCV инфекции у пациента, включающий введение этому пациенту терапевтически эффективного количества соединения или его фармацевтически приемлемой соли. В другом воплощении это соединение эффективно ингибирует функцию HCV репликона. В другом воплощении это соединение эффективно ингибирует функцию HCV NS5B белка. Другим аспектом изобретения является способ лечения HCV инфекции у пациента, включающий введение этому пациенту терапевтически эффективного количества соединения или его фармацевтически приемлемой соли в сочетании (перед, после или одновременно) с другим соединением, обладающим анти-HCV активностью. Другим аспектом изобретения является способ, в котором другое соединение, обладающее антиHCV активностью, является интерфероном. Другим аспектом изобретения является способ, в котором интерферона выбран из интерферона альфа-2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа-2 А и лимфобластоидного интерферона тау. Другим аспектом изобретения является способ, в котором другое соединение, обладающее антиHCV активностью, является циклоспорином. Другим аспектом изобретения является способ, в котором циклоспорин является циклоспорином А. Другим аспектом изобретения является способ, в котором другое соединение, обладающее антиHCV активностью, является выбранным из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое усиливает развитие ответа Т-хелперов 1 типа, интерферирующей РНК, антисмысловой РНК, имиквимода, рибавирина, ингибитора инозина 5'-монофосфат дегидрогеназы, амантадина и римантадина. Другим аспектом изобретения является способ, в котором другое соединение, обладающее антиHCV активностью, эффективно ингибирует функцию мишени, выбранной из следующих: HCV металлопротеаза, HCV сериновая протеаза, HCV полимераза, HCV хеликаза, HCV NS4B белок, вход HCV, сборка HCV, выход HCV, HCV NS5A белок, IMPDH и нуклеозидный аналог, для лечения HCV инфекции. Другим аспектом изобретения является способ, в котором другое соединение, обладающее антиHCV активностью, эффективно ингибирует функцию мишени в жизненном цикле HCV, другую нежели белок HCV NS5B. Термин "терапевтически эффективный" означает количество агента, требуемого для обеспечения убедительной пользы для пациента в понимании врачей, лечащих гепатит и HCV инфекцию. Термин "пациент" означает человека, инфицированного вирусом HCV и подходящего для терапии в понимании врачей, лечащих гепатит и HCV инфекцию. Термины "лечение," "терапия," "режим," "HCV инфекция" и сходные термины используются в значениях, принятых врачами, которые лечат гепатит и HCV инфекцию. Соединения по настоящему изобретению, как правило, вводятся в виде фармацевтических компо- 19015286 зиций, содержащих терапевтически эффективное количество соединения или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя, и могут содержать традиционные наполнители. Терапевтически эффективным является количество, которое необходимо для обеспечения существенной пользы для пациента. Фармацевтически приемлемыми носителями являются такие традиционно известные носители, обладающие приемлемыми профилями безопасности. Композиции охватывают все обычные твердые и жидкие формы, включая капсулы, таблетки, пастилки и порошки, так же как и жидкие суспензии, сиропы, эликсиры и растворы. Композиции получают, используя обычные методики приготовления и традиционные наполнители (такие как связывающие и смачивающие агенты), и носители (такие как вода и спирты) обычно применяются в композициях. Твердые композиции, как правило, готовят в дозированных формах и композиции, содержащие от около 1 до 1000 мг активного ингредиента на дозу, являются предпочтительными. Некоторыми примерами доз являются 1, 10, 100, 250, 500 и 1000 мг. Обычно другие агенты будут представлены в единице дозирования в диапазоне, сходном с агентами того класса, который применяется клинически. Обычно это 0,25-1000 мг/единицу. Жидкие композиции обычно представлены в дозированных лекарственных формах. Как правило,жидкая композиция будет в диапазоне дозированных лекарственных форм 1-100 мг/мл. Некоторыми примерами дозировок являются 1, 10, 25, 50 и 100 мг/мл. Как правило, другие агенты будут представлены в единице дозирования в диапазоне, сходном с агентами того класса, который применяется клинически. Обычно это составляет 1-100 мг/мл. Настоящее изобретение охватывает все традиционные способы введения; пероральный и парентеральный способы являются предпочтительными. Обычно, режим дозирования будет сходным с другими агентами, применяемыми клинически. Как правило, суточная доза составляет 1-100 мг/кг массы тела ежедневно. Обычно, больше соединения требуется при пероральном приеме и меньше при парентеральном. Конкретный режим дозирования, тем не менее, будет определен лечащим врачом с использованием обоснованного медицинского суждения. Настоящее изобретение также охватывает способы, в которых соединение вводится в комбинированной терапии. Это означает, что соединение может быть использовано в сочетании, но отдельно от других агентов, применяемых в лечении гепатита и HCV инфекции. В этих комбинированных способах соединение обычно вводят в суточной дозе 1-100 мг/кг массы тела ежедневно в сочетании с другими агентами. Другие агенты обычно будут вводиться в количествах, используемых терапевтически. Конкретный режим дозирования, тем не менее, будет определен лечащим врачом с использованием обоснованного медицинского суждения. Некоторые примеры соединений, подходящих для композиций и способов, перечислены в табл. 2. Таблица 2 Описание конкретных воплощений Если иное не указано, данные аналитической LCMS на следующие промежуточные соединения и примеры получают, используя следующие колонки и условия. Время выдержки: градиент времени + 1 мин; Начальная концентрация: 0% В, если иное не отмечено; Элюент А: 5% CH3CN/95% Н 2 О с 10 мМ NH4OAc (для колонок A, D и Е); 10% МеОН/90% H2O с 0,1% TFA (для колонок В и С); Элюент В: 95% CH3CN/5% H2O с 10 мМ NH4OAc (для колонок A, D и Е); 90% МеОН/10% H2O с 0,1% TFA (для колонок В и С); Колонка A: Phenomenex 10 мк 4,650 мм С 18; Колонка В: Phenomenex 1H-Индол-6-карбоновой кислоты 2-бром-3-циклогексгилметиловый эфир. Свежеперекристаллизованный трибромид ниридиния (перекристаллизация из горячего АсОН (5 мл на 1 г), промывают холодным АсОН и сушат в высоком вакууме над KOH) добавляют порционно (в течение более 10 мин) к перемешиваемому раствору метил-3-циклогексил-1 Н-индол-6-карбоксилата (60 г, 233 ммоль) (получают, используя методики, описанные в WO 2004/065367) в смеси CHCl3/ТГФ (1:1, 1.25 л) при температуре 2 С. Реакционный раствор перемешивают при температуре 0-5 С в течение 2,5 ч и промывают насыщенным водным раствором NaHSO3 (1 л), 1N HCl (1 л) и рассолом (1 л). Органический слой сушат (MgSO4) и концентрируют. Полученное масло красного цвета разбавляют Et2O и концентрируют. Полученное твердое вещество розового цвета растворяют в Et2O (200 мл), обрабатывают гексанами (300 мл) и частично концентрируют. Твердые вещества собирают фильтрованием и промывают гексанами. Маточный раствор концентрируют досуха и методику повторяют. Твердые вещества объединяют, чтобы получить 1Hиндол-6-карбоновой кислоты, 2-бром-3-циклогексилметиловый эфир (64 г, 190 ммоль, 82%) в виде пушистого твердого вещества розового цвета, которое используют без дополнительной очистки. 2-Бром-3-циклогексил-1H-индол-6-карбоновая кислота. Раствор метил-2-бром-3-циклогексил-1Hиндол-6-карбоксилат (20 г, 60 ммоль) и LiOH (3,8 г, 160 ммоль) в смеси МеОН/ТГФ/Н 2 О (1:1:1, 300 мл) нагревают при температуре 90 С в течение 2 ч. Реакционную смесь охлаждают на бане со смесью лед/Н 2 О, нейтрализуют 1 М раствором HCl (160 мл), разбавляют H2O (250 мл) и перемешивают в течение часа при комнатной температуре. Осадки собирают с помощью фильтровальной промывки H2O и сушат, чтобы получить 2-бром-3-циклогексил-1Hиндол-6-карбоновую кислоту (количественный выход), который используют без дополнительной очистки. Альтернативная методика, которая может быть использована, чтобы обеспечить 2-бром-3 циклогексил-1H-индол-6-карбоновую кислоту, описана ниже: Раствор метил-2-бром-3-циклогексил-1H-индол-6-карбоксилата (117 г, 349 ммоль) и LiOH.H2O(26.4 г, 629 ммоль) в смеси МеОН/ТГФ/H2O (1:1:1, 1.8 л) нагревают при температуре кипения с обратным холодильником в течение 3-х ч. Реакционную смесь охлаждают на бане со смесью лед/H2O до температуры 2 С, нейтрализуют 1 М раствором HCl (650 мл) (добавляют с такой скоростью, чтобы температура не превышала 5 С), разбавляют H2O (1 л) и перемешивают, пока происходит нагревание до температуры окружающей среды. Осадки собирают с помощью фильтровальной промывки H2O и сушат, чтобы получить моно ТГФ сольват 2-бром-3-циклогексил-1 Н-индол-6-карбоновой кислоты (135,5 г, 345 ммоль,99%) в виде твердого вещества желтого цвета, который используют без дополнительной очистки. 1 Н ЯМР (300 МГц, CDCl3)11,01 (уширенный с, 1H), 8,77 (с, 1H), 8,07 (д, J = 1,5 Гц, 1H), 7,82 (дд, J = 1,5, 8,8 Гц, 1H), 7,72 (д, J = 8,8 Гц, 1H), 3,84-3,74 (м, 4 Н), 2,89 (м, 1H), 1,98-1,72 (м, НН), 1,50-1,24 (м, 3 Н). 13 С ЯМР (75 МГц, CDCl3)172,7, 135,5, 130,7, 122,3, 120,9(2), 118,8, 113,3, 111,1, 67,9(2), 37,0,32,2(2), 27,0(2), 26,1, 25,5(2). LCMS: м/е 320 (М-Н)-; время задержки 2,21 мин, колонка А, 4 мин градиент. Промежуточное соединение 3 2-Бром-3-циклогексил-N-[(диметиламино)сульфонил]-1 Н-индол-6-карбоксамид. 1,1'-Карбонилдиимидазол (1,17 г, 7,2 ммоль) добавляют к перемешиваемому раствору 2-бром-3 циклогексил-1H-индол-6-карбоновой кислоты (2,03 г, 6,3 ммоль) в ТГФ (6 мл) при температуре 22 С. Выделение CO2 происходит немедленно и тогда этот успокоившийся раствор нагревают при температуре 50 С в течение часа и затем охлаждают до температуры 22 С. Добавляют N,N-диметилсульфамид (0,94 г,7,56 ммоль) с последующим добавлением по каплям раствора DBU (1,34 г, 8,8 ммоль) в ТГФ (4 мл). Перемешивание продолжают в течение 24 ч. Смесь распределяют между этилацетатом и разбавленной HCl. Этилацетатный слой промывают водой, а затем рассолом и сушат над Na2SO4. Экстракт концентрируют досуха, чтобы предоставить указанный в заголовке продукт в виде рыхлой пены бледно-желтого цвета,(2.0 г, 74%, 90% чистота, вычислена из ЯМР). 1 Н ЯМР (300 МГц, ДМСО-D6)част. на млн. 1,28-1,49 (м, 3 Н), 1,59-2,04 (м, 7 Н), 2,74-2,82 (м, 1 Н),2,88 (с, 6 Н), 7,57 (дд, J = 8,42, 1,46 Гц, 1 Н), 7,74 (д, J = 8,78 Гц, 1 Н), 7,91 (с, 1 Н), 11,71 (с, 1 Н), 12,08 (с,1 Н). Альтернативной способ получения 2-бром-3-циклогексил-N-[(диметиламино)сульфонила]-1Hиндол-6-карбоксамида описан ниже. В 4-горлую круглодонную колбу объемом объемом 1 л, оборудованную механической мешалкой,термореле, N2 вводом и холодильником, в атмосфере N2, добавляют 2-бром-3-циклогексил-1H-индол-6 карбоновую кислоту (102,0 г, 0,259 моль) и сухой ТГФ (300 мл). После перемешивания в течение 10 мин добавляют порционно CDI (50,3 г, 0,31 моль). Реакционную смесь затем нагревают до температуры 50 С в течение 2 ч. После охлаждения до температуры 30 С добавляют одной порцией N,Nдиметиламиносульфонамид (41,7 г, 0,336 моль) с последующим добавлением DBU (54,1 мл, 0,362 моль)- 24015286 по каплям в течение более часа. Реакционную смесь затем перемешивают при комнатной температуре в течение 20 ч. Растворитель удаляют в вакууме и остаток распределяют между EtOAc и 1N HCl (1 : 1,2 л). Органический слой отделяют и водный слой экстрагируют EtOAc (500 мл). Объединенные органические слои промывают рассолом (1,5 л) и сушат над MgSO4. Раствор фильтруют и концентрируют в вакууме для получения сырого продукта (111,0 г). Сырой продукт суспендируют в EtOAc (400 мл) при температуре 60 С. К суспензии медленно добавляют гептан (2 л). Полученную суспензию перемешивают и охлаждают до температуры 0 С. Затем ее фильтруют. Фильтровальную лепешку промывают небольшим количеством гептана и в атмосфере лабораторного вакуума сушат в течение 2 дней. Продукт собирают в виде твердого вещества белого цвета (92,0 г, 83%). 1 Н ЯМР (MeOD, 300 МГц)7,89 (с, Н), 7,77 (д, J = 8,4 Гц, 1H), 7,55 (дд, J = 8,4 и 1,8 Гц, 1H), 3,01 (с,6 Н), 2,73-2,95 (м, 1 Н), 1,81-2,05 (м, 8 Н), 1,39-1,50 (м, 2 Н); м/е 429 (М+Н)+. Промежуточное соединение 4 3-Циклогексил-N-[(диметиламино)сульфонил]-2-(2-формил-4-метоксифенил)-1 Н-индол-6-карбоксамид. Смесь 2-бром-3-циклогексил-N-[(диметиламино)сульфонил]-1H-индол-6-карбоксамида (4,28 г, 0,01 моль), 4-метокси-2-формилфенилбороновой кислоты (2,7 г, 0,015 моль), 2-дициклогексилфосфино-2',6'диметоксибифенила (41 мг, 0,0001 моль), ацетат палладия (11,2 мг) и хорошо измельченный карбонат калия (4,24 г, 0,02 моль) в толуоле (30 мл) перемешивают при температуре кипения с обратным холодильником и в атмосфере азота в течение 30 мин, в течение которых LC/MS анализ показывает завершение реакции. Реакционную смесь затем разбавляют этилацетатом и водой и затем подкисляют избытком разбавленной HCl. Этилацетатный слой затем собирают и промывают разбавленной HCl, водой и рассолом. После этого органический раствор сушат (сульфат магния), фильтруют и концентрируют для получения смолы. Смолу разбавляют гексанами (250 мл) и этилацетатом (25 мл) и полученную смесь перемешивают в течение 20 ч при температуре 22 С, в течение которых продукт превращается в гранулированное твердое вещество ярко-желтого цвета (4,8 г), которое используют непосредственно без дополнительной очистки. Альтернативная методика получения 3-циклогексил-N-[(диметиламино)сульфонил]-2-(2-формил-4 метоксифенила)-1H-индол-6-карбоксамида обеспечена ниже: К перемешиваемому раствору 2-бром-3-циклогексил-N-[(диметиламино)сульфонил]индол-6 карбоксамида (54,0 г, 126 ммоль), 4-метокси-2-формилфенилбороновой кислоты (29,5 г, 164 ммоль) иLiCl (13,3 г, 315 ммоль) в смеси EtOH/толуол (1:1, 1 л) добавляют раствор Na2CO3 (40,1 г, 379 ммоль) в воде (380 мл). Реакционную смесь перемешивают в течение 10 мин и затем добавляют Pd(PPh3)4 (11,3 г,10,0 ммоль). Реакционный раствор продувают азотом и нагревают при температуре 70 С (внутреннее контролирование) в течение ночи и затем охлаждают до комнатной температуры. Реакционную смесь разбавляют EtOAc (1 л) и EtOH (100 мл), промывают осторожно 1N водным раствором HCl (1 л) и рассолом (500 мл), сушат (MgSO4), фильтруют и концентрируют. Оставшиеся твердые вещества перемешивают с Et2O (600 мл) в течение часа и собирают фильтрованием, чтобы получить 3-циклогексил-N[(диметиламино)сульфонил]-2-(2-формил-4-метоксифенил)-1H-индол-6-карбоксамид (52,8 г, 109 ммоль,87%) в виде порошка желтого цвета, который используют без дополнительной очистки. 1 Н ЯМР (300 МГц, d6-ДМСО)11,66 (с, 1H), 8,17 (с, 1H), 7,75 (д, J = 8,4 Гц, 1H), 7,74 (д, J = 8,4 Гц,1H), 7,59 (дд, J = 1,4, 8,4 Гц, 1H), 7,23-7,16 (м, 2 Н), 7,08 (дд, J=2,6, 8,4 Гц, 1H), 6,54 (д, J = 8,8 Гц, 1H), 3,86 11-Циклогексил-N-[(диметиламино)сулъфонил]-6-этокси-8-метокси-6 Н-изоиндоло[2,1-а]индол-3 карбоксамид. В 4-горлую круглодонную колбу объемом 5 л, оборудованную термореле, холодильником,N2 вводом и механической мешалкой, загружают толуол (900 мл), EtOH (900 мл), 2-бром-3-циклогексилN-(N,N-диметилсульфамоил)-1H-индол-6-карбоксамид(90 г,0,21 моль),2-формил-4- 25015286 метоксифенилбороновую кислоту (49,2 г, 0,273 моль) и LiCl (22,1 г, 0,525 моль). Через полученный раствор пробулькивают N2 в течение 15 мин. Добавляют раствор Na2CO3 (66,8 г, 0,63 моль) в Н 2 О (675 мл) и через реакционную смесь пробулькивают N2 в течение еще 10 мин. Добавляют Pd(PPh3)4 (7,0 г, 6,3 ммоль) и реакционную смесь нагревают до температуры 70 С в течение 20 ч. После охлаждения до температуры 35 С добавляют медленно 1N раствор HCl (1,5 л). Полученную смесь переносят в делительную воронку объемом 6 л и экстрагируют EtOAc (21,5 л). Объединенные органические экстракты промывают рассолом (2 л), сушат над MgSO4, фильтруют и концентрируют в вакууме для получения твердого вещества желтого цвета, которое растирают в порошок с 20% EtOAc в гексане (450 мл, от 50 до 0 С) для получения 3-циклогексил-N-(N,N-диметилсульфамоил)-2-(2-формил-4-метоксифенил)-1H-индол-6 карбоксамида (65,9 г) в виде твердого вещества желтого цвета. HPLC чистота, 98%. Маточный раствор от растирания концентрируют в вакууме. Остаток нагревают при температуре кипения с обратным холодильником с EtOH (50 мл) в течение 3 ч. Раствор затем охлаждают до температуры 0 С. Осадки фильтруют и промывают охлажденным ТВМЕ (5 С) (20 мл). Фильтровальную лепешку в атмосфере лабораторного вакуума сушат для получения в дальнейшем количественно указанного в заголовке соединения в виде твердого вещества белого цвета (16,0 г). HPLC чистота, 99%. 1 Н ЯМР (CDCl3, 300 МГц)8,75 (с, 1H), 7,96 (с, 1H), 7,73 (д, J = 8,4 Гц, 1H), 7,67 (д, J = 8,4 Гц, 1H),7,45 (дд, J = 8,4 и 1,4 Гц, 1H), 7,09 (д, J = 2,2 Гц, 1H), 6,98 (дд, J = 8,4 и 2,2 Гц, 1H), 6,50 (с, 1H), 3,86 (с,3 Н), 3,05 (с, 6 Н), 2,92-3,13 (м, 3 Н), 1,85-1,93 (м, 7 Н), 1,40-1,42 (м, 3 Н), 1,05 (t, J= 7,1 Гц, 3 Н). м/е 512 (М + Н)+. Промежуточное соединение 7 13-Циклогексил-10-(диметиламино)сульфонил]амино]карбонил]-3-метокси-7 Н-индоло[2,1-а][2]бензазепин-6-карбоновой кислоты метиловый эфир. Смесь 3-циклогексил-N-(N,N-диметилсульфамоил)-2-(2 формил-4-метоксифенил)-1 Н-индол-6-карбоксамида (4,8 г, 0,01 моль), метил-2-(диметоксифосфорил)акрилата (9,7 г, 0,02 моль) и карбоната цезия (7,1 г, 0,02 моль) в ДМФА (28 мл) перемешивают в течение 20 ч при температуре масляной бани 55 С. Смесь выливают в воду со льдом и подкисляют разбавленной HCl для выпадения в осадок сырого продукта. Твердое вещество собирают, сушат и флешхроматографируют на SO2 (110 г), используя раствор этилацетата и хлористого метилена (1:10), содержащего 2% уксусную кислоту. Гомогенные фракции объединяют и упаривают, чтобы получить указанное в заголовке соединение в виде твердого вещества бледно-желтого цвета (3,9 г, 71% выход). MS: 552(М=Н+). Альтернативная методика получения 13-циклогексил-10-(диметиламино)сульфонил]амино]карбонил]-3-метокси-7 Н-индоло[2,1-а][2]бензазепин-6-карбоновой кислоты метилового эфира обеспечена ниже. Раствор 11-циклогексил-N-[(диметиламино)сульфонил]-6-гидрокси-8-метокси-6 Н-изоиндоло[2,1 а]индол-3-карбоксамида(63,0 г,130 ммоль),метил-2(диметоксифосфорил)акрилата (60 г, 261 ммоль), карбоната цезия (106 г, 326 ммоль) в ДМФА (400 мл) нагревают при температуре 60 С в течение 4,5 ч. Вводят дополнительное количество метил-2(диметоксифосфорил)акрилата (15 г, 65 ммоль) и карбонат цезия (21,2 г, 65 ммоль) и реакционную смесь нагревают при температуре 60 С в течение ночи и затем охлаждают до комнатной температуры. Перемешиваемую реакционную смесь разбавляют Н 2 О (1 л), медленно нейтрализуют 1N водным растворомHCl (800 мл), перемешивают в течение 3-х ч и затем осадки собирают фильтрованием. Твердые вещества растирают в порошок с Et2O (800 мл) и сушат, чтобы получить 13-циклогексил-10(диметиламино)сульфонил]амино]карбонил]-3-метоксиметил-7 Н-индоло[2,1-а][2]бензазепин-6-карбоновой кислоты метиловый эфир (70,2 г, 127 ммоль, 98%) в виде твердого вещества желтого цвета, который используют без дополнительной очистки. 1 Н ЯМР (300 МГц, CDCl3)8,67 (с, 1H), 8,09 (с, 1H), 7,86 (д, J = 8,4 Гц, 1H), 7,80 (с, 1H), 7,50 (д, J = 8,4 Гц, 1H), 7,42 (д, J = 8,8 Гц, 1H), 7,08 (дд, J = 2,6, 8,8 Гц, 1H), 6,98 (д, J = 2,6 Гц, 1H), 5,75-5,51 (м, 1H),4,29-4,01 (м, 1H), 3,89 (с, 3 Н), 3,82 (с, 3 Н), 3,05 (с, 6 Н), 2,87-2,73 (м, 1H), 2,11-1,12 (м, 10 Н). LCMS: м/е 550 (М-Н)-, время задержки 3,21 мин, колонка А, 4 мин градиент.(+/-)-8-Циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты метиловый эфир. ДМСО (5 мл) добавляют к смеси иодида триметилсульфоксония (199 мг, 0,906 ммоль) и NaH (38 мг в 60% масло дисперсия, 0,953 ммоль) в круглодонной колбе. Реакционную смесь перемешивают при комнатной температуре в течение 0,5 ч. Затем добавляют 13-циклогексил-10-(диметиламино)сульфонил]амино]карбонил]-3-(метокси)-7 Н-индоло[2,1-а][2]бензазепин-6-карбоновой кислоты метиловый эфир (125 мг, 0,227 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 3 ч и затем при температуре 50 С в течение еще 3 ч. Затем реакцию гасят водой и подкисляют 1N раствором HCl. Сырой продукт затем высаживают в виде твердого вещества светло-желтого цвета, которое собирают фильтрованием и сушат на воздухе (106 мг, 83% выход). 6 мг указанного продукта затем очищают с помощью препаративной HPLC, чтобы получить указанное в заголовке соединение в виде твердого вещества светло-желтого цвета (1,8 мг). MS м/е 566 (МН+), Время задержки: 3,850 мин. 1 Н ЯМР (500 МГц, MeOD)част. на млн. 0,28 (м, 0,36 Н), 1,19-2,20 (м, 11,64 Н), 2,70-3,02 (м, 2 Н),3,03 (с, 2,16 Н), 3,05 (с, 3,84 Н), 3,49 (д, J = 15,26 Гц, 0,64 Н), 3,54 (с, 1,92 Н), 3,83 (с, 1,08 Н), 3,91 (с, 3 Н),4,08 (д, J = 15,26 Гц, 0,36 Н), 5,29 (д, J = 15,26 Гц, 0,36 Н), 5,50 (д, J = 14,95 Гц, 0,64 Н), 6,98-7,06 (м, 1 Н),7,16 (д, J = 2,44 Гц, 0,36 Н), 7,23 (д, J = 2,44 Гц, 0,64 Н), 7,30 (д, J = 8,55 Гц, 0,64 Н), 7,34 (д, J = 8,55 Гц,0,36 Н), 7,56 (дд, J = 8,55, 1,53 Гц, 0,64 Н), 7,63 (дд, J = 8,55, 1,53 Гц, 0,36 Н), 7,88 (д, J = 8,55 Гц, 0,64 Н),7,91 (д, J = 8,55 Гц, 0,36 Н), 8,12 (с, 0,36 Н), 8,33 (д, J = 1,53 Гц, 0,64 Н). Альтернативная методика получения указанных в заголовке соединений обеспечена ниже. В высушенную на пламени 4-горлую круглодонную колбу объемом 1 л, оборудованную механической мешалкой, N2 вводом и термометром, в атмосфере N2, загружают гидрид натрия (95%) (3,09 г, 129,2 ммоль) и сухой ДМФА (200 мл). При энергичном перемешивании порционно загружают иодид триметилсульфоксония (32,5 г, 147,3 ммоль), в течение которых температура повышается до 30 С. После перемешивания в течение 30 мин быстро добавляют раствор 13-циклогексил-10(диметиламино)сульфонил]амино]карбонил]-3-(метокси)-7 Н-индоло[2,1-а][2]бензазепин-6-карбоновой кислоты метилового эфира (33,8 г, 61,3 ммоль) в сухом ДМФА (70 мл). Реакционную смесь перемешивают при температуре ниже 30 С в течение 30 мин и затем порционно выливают в охлажденный льдом раствор 1N HCl (130 мл) в Н 2 О (2 л). Затем полученную суспензию механически перемешивают в течение часа, осадки фильтруют и фильтровальную лепешку промывают Н 2 О (100 мл). Фильтровальную лепешку распределяют между EtOAc и 0,5N HCl (1:1, 4 л). Органическую фазу отделяют, промывают Н 2 ОEtOAc (150 мл) и раствор фильтруют через плотный слой из силикагеля (300 г в гексане) и промывают 50% EtOAc в гексане (5 л). Фильтрат концентрируют в вакууме для получения слегка твердого вещества желтого цвета, которое растирают в порошок с 10% EtOAc в ТВМЕ (220 мл) в интервале температур от 50 до 0 С для получения (+/-)-8-циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12bдигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты метилового эфира в виде твердого вещества белого цвета (26,1 г, 75% выход). HPLC чистота, 100%. 1 Н ЯМР (ДМСО-d6, 300 МГц)11,61 (с, 1H), 8,47 (с, 0,5 Н), 8,25 (с, 0,5 Н), 7,81-7,88 (м, 1H), 7,577,63 (м, 1H), 7,23-7,29 (м, 2 Н), 7,01-7,07 (м, 1H), 5,43 (д, J = 15,0 Гц, 0,5 Н), 5,22 (д, J = 15 Гц, 0,5 Н), 4,04(-)-8-Циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты метиловый эфир. Образец (+/-)-8-циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11 метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты метилового эфира раство- 27015286 ряют в смеси EtOH/CH3CN 1/1 + 0,5% DEA при концентрации 50 мг/мл (добавление DEA обеспечивает сохранение соединения в растворе в течение процесса введения). Указанный раствор затем вводят наThar SFC-350 препаративную SFC при условиях, представленных ниже. Препаративные условия на Thar SFC-350: Колонка: Chiralcel OJ-H 525 см; мобильная фаза : 25% МеОН/ CH3CN (1/1) в СО 2; давление (бар): 100; скорость потока (мл/ минуту): 240; концентрация раствора (мг/мл): 50; вводимое количество (мл): 4,5-5; Продолжительность цикла (минуты/ин.): 6,5-7; Температура (С): 45; производительность (г/час): 2; Значение длины волны (нм): 254. Из 371,4 г рацемического исходного продукта получают общее количество 177,3 г (-) изомера желаемого вторичного элюирования, содержащего 1 Мэкв. диэтиламина. Указанный продукт очищают, используя следующую методику. Смесь (24,7 г) растворяют в дихлорметане (800 мл) промывают последовательно; 0,5N HCl (1400 мл, 1240 мл), Н 2 О (2240 мл) и рассолом (2240 мл). Органический слой затем сушат (безводный Na2SO4, фильтруют и упаривают для получения 22,33 г (-)-8-циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11 метокси-(циклопроп[d]индоло[2,1-а] [2]бензазепид-1 а(2 Н)-карбоновой кислоты метилового эфира в виде твердого вещества желтого цвета (92% восстановление). HPLC199% (Время задержки 2,38 мин);LC/MS (ES+) 566,51 (М+Н, 100); []D25C- 194,64 (с 1,03, МеОН). Вычислено для C30H35N3O6S0,33H2O: С, 63,04; Н, 6,29; N, 7,35; S, 5,61; H2O, 1,04. Найдено: С, 63,07; Н, 6,01; N, 7,24; S, 5,58; H2O, 1,03. ЯМР показывает отсутствие Et2NH. ЕЕ указанного продукта определяют равным 99%, используя следующую аналитическую HPLC методику. Аналитические условия определения ее на Thar аналитической SFC. Аналитическая колонка:(-)-8-Циклогексгсил-5-(диметиламино)сульфонил]амино]карбонгил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновая кислота. К раствору (-)-8-циклогексил-5(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2] бензазепин-1 а(2 Н)-карбоновой кислоты метилового эфира (22,33 г, 39,5 ммоль) в МеОН (300 мл) медленно добавляют 1N NaOH (120 мл) в течение более 20 мин, пока значение температуры реакционной смеси станет равной 30 С. Смесь перемешивают при комнатной температуре в атмосфере N2 в течение 18 ч. HPLC показывает завершение реакции. В реакционный раствор добавляют 1N HCl (130 мл). После завершения добавления значение рН реакционной смеси станет равным приблизительно 2. Метанол в реакционной смеси упаривают. К смеси добавляют воду (300 мл), которую затем экстрагируют CH2Cl2(1600 мл, 1200 мл). Объединенные экстракты промывают H2O (2300 мл), рассолом (2300 мл),сушат (Na2SO4) и упаривают для получения 20,82 г (96% выход) указанного в заголовке соединения в виде твердого вещества желтого цвета. HPLC условия колонки: Phenomenoex Synergi Polar-RP 4 ум 4,650 мм; УФ: 220 нм; градиент времени: 4 мин; скорость потока: 4 мл/мин, 75-100% В; растворитель А: 10% МеОН/90% H2O с 0,2% H3PO4, растворитель В: 90% МеОН/10% H2O с 0,2% H3PO4. HPLC99%(+/-)-8-Циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновая кислота. К раствору (+/-)-8-циклогексил-5(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты метилового эфира (100 мг, 0,177 ммоль) в смеси ТГФ/метанол (2,0- 28015286 мл/2,0 мл) добавляют 2N раствор NaOH (1,0 мл). Реакционную смесь нагревают при температуре 90 С в условиях микроволнового облучения в течение 5 мин. Затем ее концентрируют, подкисляют 1N раствором HCl и экстрагируют этилацетатом (220 мл). Органические слои объединяют, сушат (MgSO4),фильтруют и концентрируют. Остаток очищают с помощью препаративной HPLC, чтобы получить желаемый продукт в виде твердого вещества светло-желтого цвета (59 мг, 60% выход). MS м/е 552(МН+),Время задержки: 3,850 мин. 1 Н ЯМР (300 МГц, MeOD)част. на млн. 0,25 (м, 0,38 Н), 1,14-2,22 (м, 11,62 Н), 2,69-2,98 (м, 2 Н),3,02 (с, 2,28 Н), 3,02 (с, 3,72 Н), 3,41 (д, J = 15,00 Гц, 0,62 Н), 3,88 (с, 3 Н), 4,01 (д, J = 15,00 Гц, 0,38 Н),5,26 (д, J = 15,00 Гц, 0,38 Н), 5,45 (д, J = 14,64 Гц, 0,62 Н), 6,94-7,02 (м, 1 Н), 7,13 (д, J = 2,56 Гц, 0,38 Н),7,21 (д, J = 2,20 Гц, 0,62 Н), 7,26 (д, J = 8,42 Гц, 0,62 Н), 7,30 (д, J = 8,78 Гц, 0,38 Н), 7,53 (дд, J = 8,42, 1,46 Гц, 0,62 Н), 7,61 (дд, J = 8,60, 1,65 Гц, 0,38 H), 7,85 (д, J = 8,42 Гц, 0,62 Н), 7,89 (д, J = 8,42 Гц, 0,38 Н),8,10 (с, 0,38 Н), 8,28(д, J = 1,46 Гц, 0,62 H). Промежуточное соединение 12(1aR)-[неполный]-8-Циклогексил-N5-[(диметиламино)сульфонил]-1,12b-дигидро-N1a-[(2R,3S)-3 гидрокси-4,7,7-триметилбицикло[2.2.1]гепт-2-ил]-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин 1 а,5(2 Н)-дикарбоксамид. TBTU (437 мг, 1,36 ммоль) и DIPEA (0,95 мл, 5,436 ммоль) добавляют к раствору 8-циклогексил-5-(диметиламино)сульфонил]амино]карбонил]-1,12b-дигидро-11-метокси-(+/-)циклопроп[d]индоло[2,1-а][2]бензазепин-1 а(2 Н)-карбоновой кислоты (500 мг, 0,906 ммоль) в ДМСО (20.0 мл). Реакционную смесь перемешивают при комнатной температуре в течение 15 мин. Затем добавляют(2S,3R)-3-амино-1,7,7-триметилбицикло[2.2.1]гептан-2-ол (280 мг, 1,36 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Затем ее гасят водой и подкисляют 1N раствором HCl. Отделенное твердое вещество коричневого цвета собирают фильтрованием. Указанный продукт затем фракционируют с помощью препаративной HPLC в следующих условиях. Колонка: Waters Sunfire 19 мм 100 мм; Растворитель А: 10% CH3CN-90% H2O-0,1% TFA; Растворитель В: 90% CH3CN-10% Н 2 О-0,1% TFA; Программа: Начало с 65% растворителя В, исходное время удержания в течение 5 мин,затем постепенное увеличение до 90% растворителя В в течение 30 мин со скоростью потока 25 мл/минуту. Загрузка: 50-60 мг/цикл.(1aR)-[неполный]-8-Циклогексил-N5-[(диметиламино)сульфонил]-1,12b-дигидро-N1a-[(2R,3S)-3 гидрокси-4,7,7-триметилбицикло[2.2.1]гепт-2-ил]-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин 1 а,5(2 Н)-дикарбоксамид элюируют перед (1aS)-[неполный]-8-циклогексил-N5-[(диметиламино)сульфонил]-1,12b-дигидро-N1a-[(2R,3S)-3-гидрокси-4,7,7-триметилбицикло[2.2.1]гепт-2-ил]-11-метоксициклопроп[d]индоло[2,1-а][2]бензазепин-1 а,5(2 Н)-дикарбоксамидом при HPLC условиях, описанных ниже. Продукт получают в виде твердого вещества светло-желтого цвета, 230 мг, 36% выход). MS м/е 703
МПК / Метки
МПК: A61K 31/55, A61P 31/12, C07D 519/00
Метки: ингибиторы, вируса, гепатита, белка, циклопропилконденсированные, индолбензазепиновые
Код ссылки
<a href="https://eas.patents.su/30-15286-ciklopropilkondensirovannye-indolbenzazepinovye-ingibitory-belka-ns5b-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Циклопропилконденсированные индолбензазепиновые ингибиторы белка ns5b вируса гепатита с</a>
Ингибиторы ns3 протеазы вируса гепатита с

Номер патента: 13331
Опубликовано: 30.04.2010
Авторы: Ливертон Найджел Дж., Людмерер Стивен У., Радд Майкл Т., Холловэй М.Катарин, Олсен Дэвид Б., Вакка Джозеф П., Макинтайр Чарльз Дж., Макколи Джон А.
МПК: A61K 38/06, A61P 31/14, C07K 5/08...
Метки: протеазы, гепатита, вируса, ингибиторы
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где р и q оба равны 1;R1 является CONR10SO2R6;R2 является C1-С6алкилом или С2-С6алкенилом, где указанный алкил или алкенил необязательно замещен 1-3 атомами галогена;R3 является C1-С8алкилом или C3-С8циклоалкилом;R5 является Н;R6 является C3-С6циклоалкилом;Y является С(=O);Z является О;М является С1-С12алкиленом или С2-С12алкениленом икаждый R10 независимо является Н или...
Трипептидные ингибиторы вируса гепатита с

Номер патента: 3906
Опубликовано: 30.10.2003
Авторы: Верник Доминик М., Гиро Элиза, Ллина-Брюне Монсе, Ранкур Жан, Пупар Марк-Андре, Байли Мюррей Д., Симоно Брюно, Гудреу Натали, Тсантризо Иула С., Камерон Даль, Альмос Тедди, Фоше Анн-Мари
МПК: A61K 38/06, C07C 229/48, A61P 31/12...
Метки: гепатита, трипептидные, вируса, ингибиторы
Формула / Реферат:
1. Соединение формулы (I), включая его рацематы, диастереоизомеры и оптические изомеры в которой B обозначает H, C6- или C10арил, C7-C16аралкил; Het или (низш.)алкил-Het, все необязательно замещенные C1-C6алкилом; C1-C6алкоксигруппу; C1-C6алканоил; гидроксигруппу; гидроксиалкил; галоген; галоалкил; нитрогруппу; цианогруппу; цианалкил; аминогруппу, необязательно замещенную C1-C6алкилом; амидогруппу; или (низш.)алкиламид; или B обозначает...
Пептидные ингибиторы вируса гепатита с

Номер патента: 4765
Опубликовано: 26.08.2004
Авторы: Камерон Даль, Пупар Марк-Андре, Ранкур Жан, Ллина-Брюне Монсе, Гудреу Натали, Гиро Элиза, Байли Мюррей Д., Тсантризо Иула С.
МПК: A61K 38/55, C07K 14/81, A61P 31/14...
Метки: гепатита, вируса, ингибиторы, пептидные
Формула / Реферат:
1. Рацематы, диастериоизомеры и оптические изомеры соединения формулы (I) , в которой a равно 0 или 1, b обозначает 0 или 1, Y обозначает H или C1-C6алкил, B обозначает H, ацильное производное формулы R7-C(O)- или сульфонил формулы R7-SO2, где R7 обозначает C1-C10алкил, необязательно замещенный карбоксилом, C1-C6аланоилокси- или C1-C6алкоксигруппой, C6- или C10арил или C7-C16аралкил, необязательно замещенный C1-C6алкилом, гидрокси- или...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 13475
Опубликовано: 30.04.2010
Авторы: Айеса Алварес Сусана, Самуэльссон Бенгт Бертил, Классон Бьерн Олоф, Нильссон Карл Магнус, Йенссон Карл Эрик Даниель, Де Кок Херман Аугустинус, Росенквист Оса Анника Кристина, Симмен Кеннет Алан, Валльберг Ханс Кристиан
МПК: A61K 31/538, A61P 31/14, C07D 401/12...
Метки: ингибиторы, вируса, макроциклические, гепатита
Формула / Реферат:
1. Соединение формулыего N-оксид, соль или стереоизомер,где каждая пунктирная линия (представленная как представляет собой необязательную двойную связь;X представляет собой N, СН и, когда X содержит двойную связь, он представляет собой С;R1 представляет собой -OR6, -NH-SO2R7;R2 представляет собой водород и, когда X представляет собой С или СН, R2 также может представлять собой C1-6-алкил;R3 представляет собой водород, C1-6-алкил,...
Макроциклические ингибиторы вируса гепатита с

Номер патента: 14188
Опубликовано: 29.10.2010
Авторы: Нильссон Карл Магнус, Самуэльссон Бенгт Бертил, Классон Бьерн Олоф, Линдквист Карин Карлотта, Де Кок Херман Аугустинус, Канберг Пиа Сесилия, Симмен Кеннет Алан, Бельфраге Анна Карин Гертруд Линнеа, Росенквист Оса Анника Кристина, Ху Лили, Рабуассон Пьер Жан-Мари Бернар, Валльберг Ханс Кристиан, Линдстрем Матс Стефан, Вехлинг Хорст Юрген, Сальберг Свен Кристер
МПК: A61P 31/12, C07D 239/72, A61K 31/517...
Метки: ингибиторы, макроциклические, гепатита, вируса
Формула / Реферат:
1. Соединение формулы (I)и его N-оксиды, соли и стереоизомеры,где А представляет собой OR1, NHS(=O)pR2; гдеR1 представляет собой водород, C1-C6-алкил;R2 представляет собой C3-C7-циклоалкил, причем указанный C3-C7-циклоалкил необязательно замещен 1-3 заместителями, независимо выбранными из C1-C6-алкила;р независимо равно 2;n равно 3, 4, 5 или 6;---- означает необязательную двойную связь;L представляет собой N или CRz;Rz представляет собой Н;Rq...
Предыдущий патент: Реактор для переработки твердого топлива
Случайный патент: Способ и устройство для приготовления радиофармацевтических препаратов для инъекций