Производные пиразолизохинолинмочевины в качестве ингибиторов p38 киназы
Номер патента: 15123
Опубликовано: 30.06.2011
Авторы: Чжун Боюй, Гарсиа-Паредес Кристина, Мэйдер Мэри Маргарет, Лопес Де Уралде-Гармениа Беатрис, Де Дьес Альфонсо, Ших Чуан, Побанс Марк Эндрю







Формула / Реферат
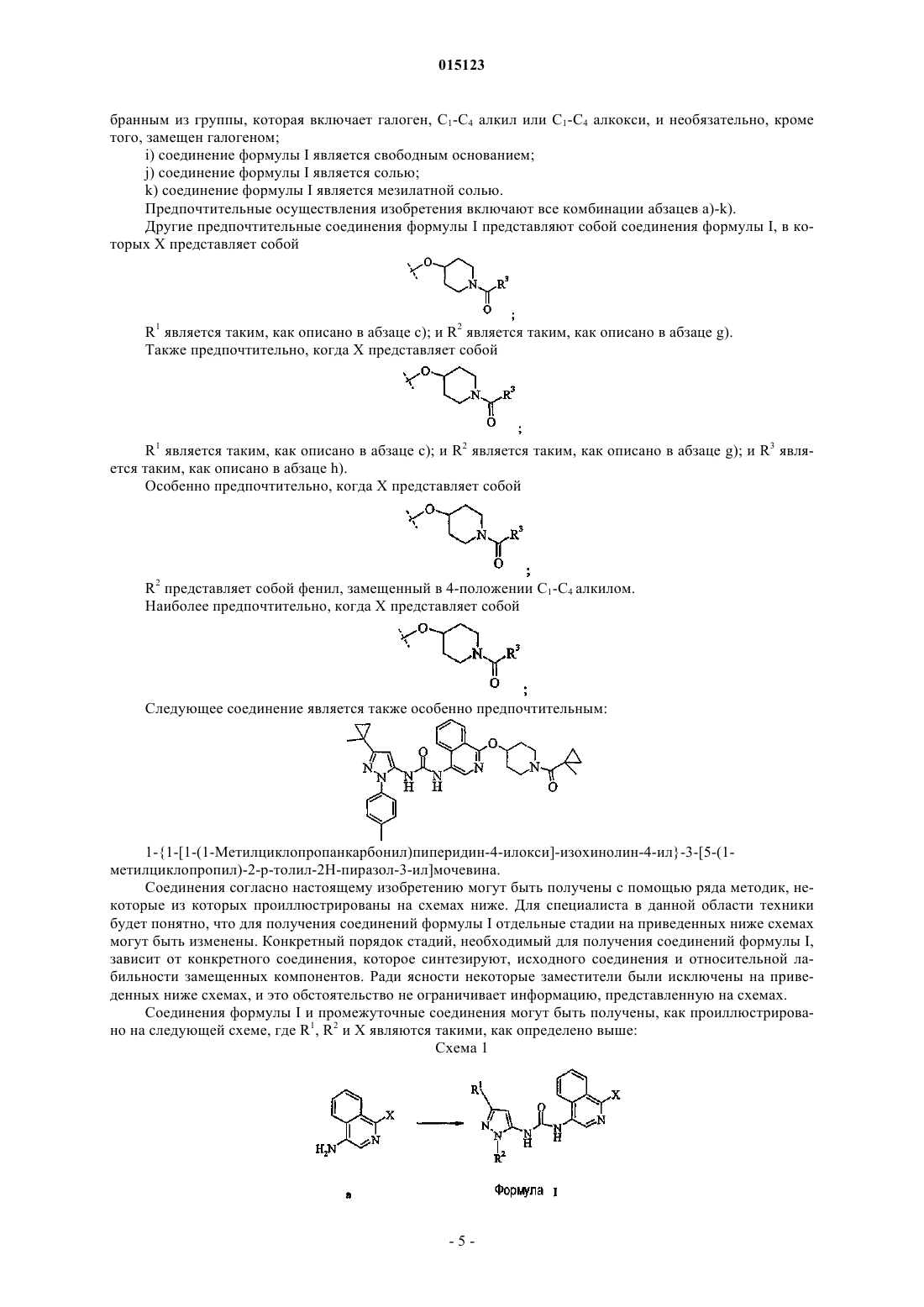
1. Соединение формулы I

где X выбран из группы, включающей

R1 представляет собой C1-C4 алкил, C3-C4 циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, включающей C1-C4 алкокси, метил и трифторметил; или C1-C4 алкилгалоген;
R2 представляет собой фенил, необязательно замещенный C1-C4 алкилом, или пиридинил, необязательно замещенный C1-C4 алкилом;
R3 представляет собой амино, C1-C4алкил, C1-C4алкокси, C1-C4алкилгалоген, C3-C4циклоалкил, необязательно замещенный заместителем, выбранным из метила, трифторметила или галогена; фенил или тиенил, каждый необязательно замещенный первым заместителем, выбранным из группы, включающей галоген, C1-C4 алкил и C1-C4алкокси, и необязательно, дополнительно замещенный вторым заместителем, выбранным из галогена;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где X представляет собой

3. Соединение по п.1 или 2, где R2 представляет собой 4-толил.
4. Соединение по любому из пп.1-3, где R1 представляет собой 1-метил-1-циклопропил, 2-фтор-1,1-диметилэтил или 2-фтор-1-фторметил-1-метилэтил.
5. Соединение по любому из пп.1-4, где R3 представляет собой 1-метил-1-циклопропил.
6. Соединение формулы I

1-{1-[1-(1-Метилциклопропанкарбонил)пиперидин-4-илокси]изохинолин-4-ил}-3-[5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-ил]мочевина или его фармацевтически приемлемая соль.
7. Фармацевтическая композиция, содержащая соединение по любому из пп.1-6 в комбинации с фармацевтически приемлемым наполнителем, носителем или разбавителем.
8. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для лечения рака.
9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака.
Текст
Изобретение предлагает ингибиторы киназы формулы I где R1, R2 и X являются такими, как описано в данной заявке, или их фармацевтически приемлемую соль. Де Дьес Альфонсо (US), Гарсиа-Паредес Кристина, Лопес Де Уралде-Гармениа Беатрис (ES), Мэйдер Мэри Маргарет, Побанс Марк Эндрю, Ших Чуань, Чжун Боюй(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)p38 киназа представляет собой активированную митогеном протеин (MAP) киназу, которая принадлежит к суперсемейству серин/треонин киназ. Эта киназа активируется внеклеточными факторами, такими как тепло, УФ-свет и осмотическое давление, а также воспалительными раздражителями, такими как липополисахарид. Будучи активированной, p38 киназа фосфорилирует внутриклеточные протеиновые субстраты, которые регулируют биосинтез провоспалительных цитокинов фактора некроза опухоли(TNF) и интерлейкина-1 (IL-1). Эти цитокины вовлечены в патологии многих хронических воспалительных расстройств (Lee, et al., Ann, N.Y. Acad. Sci., 696, 149-170 (1993); Muller-Ladner, Curr, Opin.Rheumatol. 8, 210-220 (1996, сердечно-сосудистых расстройств и расстройств центральной нервной системы (Salituro, et al., Current Medicinal Chemistry, 6, 807-823 (1999 и аутоиммунных расстройств (Pargellis, et al., Nature Structural Biology, 9(4), 268-272 (2002. Кроме того, фосфорилированная форма активированной митогеном протеинкиназы - протеин киназы 2 (или pMAPKAPK2) также является киназой вp38 MAPK пути и непосредственно может быть активирована p38 MAPK. Исследования MAPKAPK2 на нокаутированных мышах показывают снижение продуцирования цитокинов, предполагая, чтоMAPKAPK2 может быть ключевым регулятором воспалительной реакции и также может быть потенциальной мишенью для противовоспалительной терапии (WO 2005120509). Многие соединения мочевины (например, в WO 9923091, WO 01012188, WO 04004720, WO 04037789, WO 99/32111, US 2004/0058961, EP 1609789, WO 03072569 и WO 0043384) были идентифицированы как ингибиторы p38 киназы или ингибиторы цитокинов. Ингибиторы p38 киназы или ингибиторы цитокинов могут быть дорогостоящими для производства и могут иметь проблемы, связанные с биодоступностью и абсорбцией, которые ограничивают эффекты in vivo и терапевтическое применение. Таким образом, существует потребность в новых лекарственных средствах на основе малых молекул, которые подавляют цитокины, то есть соединений, которые способны ингибировать p38 киназу с большей эффективностью и большей биодоступностью. Настоящее изобретение предлагает новые ингибиторы p38 киназы, полезные для лечения состояний, которые являются результатом чрезмерного продуцирования цитокинов. Краткое описание изобретения Настоящее изобретение предлагает соединения формулы IR1 представляет собой C1-C4 алкил, C3-C4 циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, которая включает C1-C4 алкокси, метил и трифторметил; или C1C4 алкилгалоген;R3 представляет собой амино, C1-C4 алкил, C1-C4 алкокси, C1-C4 алкилгалоген, C3-C4 циклоалкил,необязательно замещенный заместителем, выбранным из метила, трифторметила или галогена; фенил или тиенил, каждый необязательно замещен первым заместителем, выбранным из группы, которая включает галоген, C1-C4 алкил и C1-C4 алкокси, и необязательно, кроме того, замещен вторым заместителем,выбранным из галогена; или их фармацевтически приемлемую соль. Настоящее изобретение также предлагает способ ингибирования p38 киназы у млекопитающего,включающий введение млекопитающему, который нуждается в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также предлагает способ подавления продуцирования фактора некроза опухолей(TNF) у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также предлагает способ подавления продуцирования интерлейкина-1 (IL1) у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении,эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение, кроме того, предлагает способ лечения состояний, которые являются ре-1 015123 зультатом чрезмерного продуцирования цитокинов, у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении, соединения формулы I или его фармацевтически приемлемой соли в количестве, которое подавляет цитокины. Настоящее изобретение также предлагает способ ингибирования роста чувствительного новообразования у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении, соединения формулы I или его фармацевтически приемлемой соли в количестве, которое ингибирует p38. Настоящее изобретение также предлагает способ ингибирования метастаза у млекопитающего,включающий введение млекопитающему, который нуждается в таком лечении, соединения формулы I или его фармацевтически приемлемой соли в количестве, которое ингибирует p38. Настоящее изобретение также предлагает способ лечения ревматоидного артрита у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении, соединения формулыI или его фармацевтически приемлемой соли в количестве, которое ингибирует p38. Настоящее изобретение также предлагает фармацевтическую композицию, которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с фармацевтически приемлемым наполнителем, носителем или разбавителем. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для ингибирования p38 киназы. Кроме того, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую соль для применения при ингибировании p38 киназы у млекопитающих. Кроме того, настоящее изобретение предлагает фармацевтическую композицию, подходящую для ингибирования p38 киназы, которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для подавления продуцирования фактора некроза опухолей(TNF ). Дополнительно, настоящее изобретение предлагает соединение формулыI или его фармацевтически приемлемую соль для применения при подавлении продуцирования фактора некроза опухолей(TNF) у млекопитающих. Настоящее изобретение предлагает также фармацевтическую композицию, подходящую для подавления продуцирования фактора некроза опухолей(TNF),которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для подавления продуцирования интерлейкина-1 (IL-1). Дополнительно, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую соль для применения при подавлении продуцирования интерлейкина-1(IL-1) у млекопитающего. К тому же, настоящее изобретение предлагает фармацевтическую композицию, подходящую для подавления продуцирования интерлейкина-1 (IL-1), которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения состояний, которые являются результатом чрезмерного продуцирования цитокинов. Дополнительно, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую с,оль для применения при лечении состояний, которые являются результатом чрезмерного продуцирования цитокинов у млекопитающих. К тому же, настоящее изобретение предлагает фармацевтическую композицию, подходящую для лечения состояний, которые являются результатом чрезмерного продуцирования цитокинов, которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для ингибирования роста чувствительного новообразования. Дополнительно, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую соль для применения при ингибирования роста чувствительного новообразования у млекопитающих. К тому же, настоящее изобретение предлагает фармацевтическую композицию, подходящую для ингибирования роста чувствительного новообразования, которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для ингибирования метастаза. Дополнительно, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую соль для применения при ингибировании метастаза у млекопитающих. К тому же, настоящее изобретение предлагает фармацевтическую композицию, подходящую для ингибирования метастаза, которая со-2 015123 держит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Настоящее изобретение также предлагает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения ревматоидного артрита. Дополнительно, настоящее изобретение предлагает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении ревматоидного артрита у млекопитающих. К тому же,настоящее изобретение предлагает фармацевтическую композицию, подходящую для лечения ревматоидного артрита, которая содержит соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или больше фармацевтически приемлемыми наполнителями, носителями или разбавителями. Подробное описание изобретения Термин "p38 киназа" обозначает изоформы p38 и/или p38 киназы. Термин "подавление продуцирования TNF (IL-1, цитокинов)" обозначает снижение избыточных уровней TNF, IL-1 или другого цитокина in vivo у млекопитающего до нормальных или субнормальных уровней. Такой эффект может быть достигнут с помощью ингибирования высвобождения in vivoTNF, IL-1 или другого цитокина всеми клетками, включая макрофаги, с помощью понижающего регулирования, на геномном уровне, избыточных уровней TNF, IL-1 или другого цитокина in vivo у млекопитающего до нормальных или суб-нормальных уровней с помощью ингибирования синтеза TNF, IL1 или другого цитокина как пост-трансляционного процесса или с помощью понижающего регулирования TNF, IL-1 или другого цитокина на трансляционном уровне. Специалисту в данной области будет понятно, что соединения согласно настоящему изобретению способны образовывать кислотно-аддитивные соли. Во всех случаях фармацевтически приемлемые соли всех соединений включены в их название. Соединения согласно настоящему изобретению представляют собой амины и соответственно будут реагировать с любой из многочисленных неорганических и органических кислот с образованием фармацевтически приемлемых кислотно-аддитивных солей. Термин "фармацевтически приемлемая соль", как используется в данной заявке, относится к солям соединений формулы I, которые не являются существенно токсичными для живых организмов. Типичные фармацевтически приемлемые соли включают соли, полученные с помощью реакции соединений согласно настоящему изобретению с фармацевтически приемлемыми органическими или неорганическими кислотами. Такие соли включают фармацевтически приемлемые соли, приведенные в Journal of Pharmaceutical Science, 66,2-19 (1977), который известен специалисту в данной области техники. Наиболее предпочтительными являются мезилатные соли соединений формулы I. Соединения формулы I являются ингибиторами p38 киназы. Таким образом, настоящее изобретение также предлагает способ ингибирования p38 киназы у млекопитающего, включающий введение млекопитающему, который нуждается в таком лечении, соединения формулы I в количестве, которое ингибирует p38 киназу. Предпочтительно млекопитающим, которое лечат введением соединений формулы I,является человек. Как ингибиторы p38 киназы соединения согласно настоящему изобретению полезны для подавления продуцирования провоспалительных цитокинов фактора некроза опухоли(TNF) и интерлейкина 1 (IL-1), и, таким образом, для лечения расстройств, которые являются результатом избыточного продуцирования цитокинов. Таким образом, считают, что настоящие соединения будут полезными при печении воспалительных расстройств, включая экзему, атопический дерматит, ревматоидный артрит, остеоартрит, воспалительное заболевание кишечника и синдром токсического шока. Также считают, что соединения согласно настоящему изобретению будут полезными при лечении сердечно-сосудистых расстройств, таких как острый инфаркт миокарда, хроническая сердечная недостаточность, атеросклероз,вирусный миокардит, отторжение сердечного аллотрансплантата и сердечная дисфункция, связанная с сепсисом. К тому же, также считают, что соединения согласно настоящему изобретению будут полезными при лечении расстройств центральной нервной системы, таких как менингококковый менингит, болезнь Альцгеймера, болезнь Паркинсона и рассеянный склероз - WO 99/32111, WO 9923091, WO 04004720, WO 03072569. Увеличение массы твердых опухолей обусловлено пролиферацией злокачественных клеток и клеток стромы, включая эндотелиальные клетки. Для увеличения опухоли до размера, больше 2-3 мм в диаметре, она должна сформировать сосудистую сеть, этот процесс известен под названием ангиогенез. Было сообщено, что подавление вызванного опухолью ангиогенеза под воздействием ангиостатина и эндостатина приводит к противоопухолевому эффекту (O'Reilly, et al., Cell, 88, 277-285 (1997. Было показано, что селективный ингибитор p38 киназы SB22025 ингибирует ангиогенез (J.R. Jackson, et al., J. Pharmacol. Exp. Therapeutics. 284, 687 (1998. Поскольку ангиогенез является решающим фактором при увеличении массы большинства твердых опухолей, разработка новых ингибиторов p38 киназы для ингибирования этого процесса является перспективным подходом к противоопухолевой терапии. Этот подход к противоопухолевой терапии может быть лишен токсичных побочных эффектов или развития резистентности к лекарственным средствам, имеющего место при традиционной химиотерапии (Judah Folkman,-3 015123Endogenous Inhibitors of Angiogenesis, The Harvey Lectures, Series 92, pages 65-82, Wiley-Liss Inc. (1998. Как ингибиторы p38 киназы соединения согласно настоящему изобретению, таким образом, также полезны при ингибировании роста чувствительных новообразований. Schultz, R. M. Potential of p38 MAPkinase inhibitors in the treatment of cancer. B: E. Jucker (ed.), Progress in Drug Research, 60, 59-92, (2003). Чувствительное новообразование определено как новообразование, выживание, рост или метастаз которого зависят от p38 киназы. Чувствительные новообразования включают опухоли головного мозга, мочеполового тракта, лимфатической системы, желудка, гортани и легких (патент US 5717100). Предпочтительно, термин "чувствительные новообразования", как используется в данной заявке, включает раки у людей, включая немелкоклеточный рак легких (A. Greenberg, et al., Am. J. Respir. Cell Mol. Biol., 26, 558(2002, карциному молочной железы (J. Chen, et al., J, Biol. Chem., 276, 47901 (2001); B. Salh, et al., Int. J.al., Proc. Am. Assoc. Cancer Res., 43, 9 (2002, колоректальные карциномы (S. Xiong, et al., Cancer Res.,61, 1727 (2001 и злокачественную меланому (C. Denkert, et al. , Clin. Exp. Metastasis, 19, 79 (2002. Также было сообщено, что ингибирование ангиогенеза путем подавлением TNF является полезным при ингибировании или профилактике метастаза (патент US 6414150; патент US 6335336). К тому же подавление TNF предписано при лечении и профилактике кахексии, изнурительного синдрома, от которого страдает приблизительно половина всех больных раком (T. Yoneda, et al., J. Clin. Invest., 87, 977(1991. К тому же ингибирование p38 киназы может быть эффективно при лечении некоторых вирусных состояний, таких как грипп (K. Kujime, et al., J. Immunology., 164, 3222-3228 (2000, риновирус (S.Griego, et al., J. Immunology, 165, 5211-5220 (2000 и ВИЧ (L. Shapiro, et al., Proc. Natl. Acad. Sci. USA,95, 7422-7426, (1998. Как используется в данной заявке, термин "C1-C4 алкил" относится к линейной или разветвленной,моновалентной, насыщенной алифатической цепи, содержащей от одного до четырех атомов углерода, и включает, но не ограничивается этим, метил, этил, пропил, изопропил, бутил, изобутил и трет-бутил. Как используется в данной заявке, термин "C1-C4 алкокси" относится к прямой или разветвленной алкильной цепи, содержащей от одного до четырех атомов углерода, присоединенных к атому кислорода. Типичные C1-C4 алкоксигруппы включают метокси, этокси, пропокси, изопропокси, бутокси, третбутокси и подобные. Термин "C1-C4 алкокси" включает в свое определение термин "C1-C3 алкокси". Как используется в данной заявке, термин "галоген" относится к атому хлора, брома, йода или фтора, если не указано другое. Как используется в данной заявке, термин "C1-C4 алкилгалоген" относится к C1-C4 алкилу, замещенному до пяти атомами галогена. Типичные C1-C4 алкилгалогенгруппы включают метилгалоген, трифторметил, этилгалоген, бис-фторметилэтил, пропилгалоген, изопропилгалоген, бутилгалоген, третбутилгалоген и подобные. Термин "C1-C4 алкилгалоген" включает в свое определение термин "C1-C3 алкилгалоген". Как используется в данной заявке, термин "C3-C4 циклоалкил" обозначает неароматическое кольцо,содержащее атомы углерода и водорода, и включает циклопропил и циклобутил. Как используется в данной заявке, термин "1-метил-1-циклопропил" относится к следующему остатку: Определенные классы соединений формулы I представляют собой предпочтительные ингибиторыp38 киназы. В следующих абзацах описаны такие предпочтительные классы: а) X представляет собойh) R3 представляет собой C3-C4 циклоалкил, необязательно замещенный C1-C4 алкилом; или тиенил,необязательно замещенный метилом; или фенил, необязательно замещенный первым заместителем, вы-4 015123 бранным из группы, которая включает галоген, C1-C4 алкил или C1-C4 алкокси, и необязательно, кроме того, замещен галогеном;k) соединение формулы I является мезилатной солью. Предпочтительные осуществления изобретения включают все комбинации абзацев a)-k). Другие предпочтительные соединения формулы I представляют собой соединения формулы I, в которых X представляет собой Следующее соединение является также особенно предпочтительным: 1-1-[1-(1-Метилциклопропанкарбонил)пиперидин-4-илокси]-изохинолин-4-ил-3-[5-(1 метилциклопропил)-2-p-толил-2H-пиразол-3-ил]мочевина. Соединения согласно настоящему изобретению могут быть получены с помощью ряда методик, некоторые из которых проиллюстрированы на схемах ниже. Для специалиста в данной области техники будет понятно, что для получения соединений формулы I отдельные стадии на приведенных ниже схемах могут быть изменены. Конкретный порядок стадий, необходимый для получения соединений формулы I,зависит от конкретного соединения, которое синтезируют, исходного соединения и относительной лабильности замещенных компонентов. Ради ясности некоторые заместители были исключены на приведенных ниже схемах, и это обстоятельство не ограничивает информацию, представленную на схемах. Соединения формулы I и промежуточные соединения могут быть получены, как проиллюстрировано на следующей схеме, где R1, R2 и X являются такими, как определено выше: Схема 1-5 015123 Амин (a) подвергают реакции с подходящим изоцианатом или карбаматом, таким как пиразолил 2,2,2-трихлорэтилкарбамат, с получением соединений формулы I. Например, нагревают раствор амина (1 эквивалент), трихлорэтилкарбамата (1 эквивалент) и подходящего основания, такого как диизопропилэтиламин (2 эквивалента) или карбонат калия, в подходящем растворителе, таком как ацетонитрил или диметилсульфоксид (DMSO). Желаемое соединение затем может быть выделено и, если необходимо и желательно, очищено с использованием способов, хорошо известных из уровня техники, таких как хроматография, с получением соединения формулы I. Необходимые амины получают, как проиллюстрировано ниже на схеме 2, где X является таким, как определено выше: Схема 2 Нитрокомпонент (b) восстанавливают в стандартных условиях восстановления, например, водородом в присутствии катализатора на основе палладия, в подходящем растворителе, таком как низшие алканолы или этилацетат, с получением соответствующего амина (a). Такие стадии восстановления хорошо известны и определены в данной области техники. См. Larock, R., "Comprehensive Organic Transformations," 412, VCH Publishing, Inc., New York, 1989. Необходимые нитросоединения получают, как проиллюстрировано на схеме 3 ниже, где R3 является таким, как описано выше, и X' представляет собой C(O)R3 или подходящую защитную группу PG: Схема 3 1-хлор-4-нитроизохинолин (e) в подходящем органическом растворителе, таком как THF, подвергают реакции с N-защищенным (PG) гидроксипиперидином и гидридом натрия с получением соответствующего замещенного пиперидина b(i). Если необходимо или желательно, может быть использована подходящая амино-защитная группа "Pg", такая как трет-бутоксикарбонильный (BOC) фрагмент. Способы введения этих групп хорошо известны специалисту в данной области техники. Специалист в данной области техники примет во внимание, что азот-защитные группы могут быть удалены на любой подходящей стадии синтеза соединений согласно настоящему изобретению. Способы удаления аминозащитных групп хорошо известны из уровня техники; (см., например, T.W. Greene, "Protective Groups inOrganic Synthesis", John Wiley and Sons, New York, N.Y., 1999). Альтернативно, 1-хлор-4-нитроизохинолин (e) подвергают реакции с N-BOC защищенным пиперазином в карбонате калия в полярном растворителе, таком как ацетонитрил, с получением соответствующего замещенного пиперазина b(ii). Необходимые пиразолилкарбаматы могут быть получены, как описано на следующей схеме, где R1 2 и R являются такими, как определено выше: Схема 4-6 015123 3-аминопиразолы (m) получают, используя условия, хорошо известные из уровня техники; Larock,R., "Comprehensive Organic Transformations," 79, VCH Publishing, Inc., New York, 1989. Например, цианокетон (j) и подходящий гидразин или соль гидразина (k) в подходящем органическом растворителе,таком как этанол, подвергают реакции при повышенной температуре, и полученный продукт может быть очищен с использованием стандартных способов, таких как хрома.тография на колонке с силикагелем. 2,2,2-Трихлорэтилхлорформиат (n) подвергают реакции с подходяще замещенным 3-аминопиразолом (m) и основанием, например карбонатом натрия, в подходящем растворителе, например, THF, с получением соответствующего 2,2,2-трихлорэтилкарбамата (o). Специалист в данной области техники примет во внимание, что соответствующий карбамат может быть получен с помощью реакции 3 аминопиразола с другими активными карбонатами. Соединения формулы I(i) могут быть получены, как продемонстрировано на схеме 5 ниже, где R1,2 3 Из соединения формулы (f) удаляют защитную группу в условиях, хорошо известных из уровня техники. Например, когда защитная группа представляет собой трет-бутоксикарбонил, соединение формулы (f) растворяют в подходящем органическом растворителе, таком как дихлорметан, или смеси растворителей и обрабатывают кислотой, такой как соляная кислота в диоксане или трифторуксусная кислота. Снятием защитной группы с N-защищенной-пиперидин-замещенной мочевины (f) получают замещенный пиперидин (g), который подвергают реакции с замещенной карбоновой кислотой в стандартных условиях сочетания для органических кислот и органических аминов в присутствии агента сочетания, такого как гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида (EDCI), каталитического количества 4-диметиламинопиридина (DMAP) и 1-гидроксибензотриазола (HOBt), с получением соединения формулы I (i). Специалист в данной области техники примет во внимание, что примеры формулы I(i) могут быть получены, исходя из других защищенных пиперидинов, включая различные N-защитные группы, такие как формил, для которых могут быть необходимы другие методики снятия защитных групп, с получением промежуточных соединений (g). Специалист в данной области техники также примет во внимание, что не все заместители в соединениях формулы 1 будут толерантными в определенных условиях реакции, которые используются при синтезе соединений. Эти заместители могут быть введены на подходящей стадии синтеза или могут быть защищены, а затем защитная группа, при необходимости или желании, может быть удалена. Специалист в данной области техники примет во внимание, что защитные группы могут быть удалены на любой подходящей стадии синтеза соединений согласно настоящему изобретению. Способы введения и удаления азот- и кислородзащитных групп хорошо известны из уровня техники; смотрите, например, Greeneand Wuts, Protective Groups in Organic Synthesis, John Wiley and Sons, New York, (1999). К тому же, специалист в данной области техники примет во внимание, что во многих случаях порядок, в котором вводят компоненты, не является критическим. Конкретный порядок стадий, необходимых для получения соединений формулы I, зависит от конкретного соединения, которое синтезируют, исходного соединения и относительной лабильности замещенных компонентов. Сокращения, обозначения и условия, которые используются в примерах и исследованиях, имеют следующие значения: AcOH = уксусная кислота, DCC = дициклогексилкарбодиимид, DIEA = N,Nдиизопропилэтиламин, DMSO = диметилсульфоксид, DMF = N,N-диметилформамид, ч = час(ы), HOBt = 1-гидроксибензотриазол, LDA = диизопропиламид лития, EDCl = гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, EtOAc = этилацетат, EtOH = этанол, MeOH = метанол, NaBH(OAc)3 триацетоксиборгидрид натрия, TBAF = фторид тетрабутиламмония, Tf2O = ангидрид трифторметансульфоновой кислоты, THF = тетрагидрофуран. Пример получения 1. Метиловый эфир 1-трифторметилциклопропанкарбоновой кислоты К раствору 1-трифторметилциклопропан-1-карбоновой кислоты (3,65 г, 23,7 ммоль) в метанолегексанах (2,5 мл-22,5 мл) добавляют 2 M раствор диазометана в гексанах (14,2 мл, 28,45 ммоль). Растворитель удаляют при пониженном давлении и остаток перегоняют, получая желтое масло (2,93 г, выход Раствор 2M LDA в THF (29,1 мл, 58,3 ммоль) добавляют к охлажденному сухим льдом-ацетоном раствору метилового эфира 1-метоксициклопропанкарбоновой кислоты (WO 2005/014577) (3,45 г, 26,5 ммоль) и ацетонитрила (2,17 г мл, 53,0 ммоль) в THF (30 мл). Реакционную смесь перемешивают при-78C в течение 1 ч и затем при 22C в течение 0,5 ч. Выпаривают растворитель, получая коричневое твердое вещество. Фильтруют и промывают гексанами. Добавляют 2 N соляную кислоту и три раза экстрагируют диэтиловым эфиром (50 мл каждого). Объединенные органические фазы сушат над сульфатом натрия. Растворитель удаляют, получая красное масло (3,55 г, выход 96%, ES+(m/z) 140,1 [M+H]). Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше.(100 г) в MeOH (1 л, градиентный растворитель ВЭЖХ) и перемешивают при комнатной температуре в течение приблизительно 70 ч. Растворитель удаляют, и остаток разделяют между EtOAc (1 л) и H2O (100 мл). Водный слой повторно экстрагируют EtOAc, и объединенные органические фракции сушат надMgSO4. Фильтруют и концентрируют при пониженном давлении. Неочищенная смесь может быть использована без дополнительной очистки. 1H ЯМР (CDCl3, 300 МГц):м.д. 3,9 (д, 2H, J=11,1 Гц), 3,76 (с,3H), 3,71 (д, 2H, J=11,1 Гц), 2,8 (ушир.с, 2H), 1,1 (с, 3H). Следующее соединение получают, используя методику, по существу аналогичную методике, описанной выше. Пример получения 7. Бензиловый эфир 3-гидрокси-2,2-диметилпропионовой кислоты. Гидроксид калия (486,7 ммоль, 32,1 г) добавляют к раствору 2,3-дигидрокси-2-метилпропионовой кислоты (423,2 ммоль, 50 г) в 300 мл DMF. Перемешивают в течение 1 ч при 100C. Затем добавляют бензилбромид (58 4,04 ммоль, 69,46 мл) и перемешивают в течение ночи. Смесь охлаждают и разбавляют этилацетатом. Органический слой промывают водой. Водный слой несколько раз промывают этилацетатом. Органические слои объединяют и сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении. 1H ЯМР (CDCl3, 300 МГц):м.д.: 7,36-7,32 (м, 5H), 5,1 (с, 2H), 3,5 (с, 2H), 1,21 (с,6H). Пример получения 8. 5-Пентафторэтил-2-p-толил-2H-пиразол-3-иламин-8 015123 ммоль) в этаноле (20 мл) нагревают до 95C в герметичной колбе в течение 15 ч. После охлаждения до комнатной температуры растворитель удаляют при пониженном давлении, получая коричневый остаток. Остаток подвергают хроматографии на силикагеле, элюируя этилацетатом и гексанами, получая указанное в заглавии соединение (3,24 г, выход 52%, ES+(m/z) 292,1 [M+H]). Следующее соединение получают, используя методику, по существу, аналогичную методике, описанной выше:AgF (4,5 эквивалентов, 4,56 г) добавляют к раствору этилового эфира 1-фторциклобутанкарбоновой кислоты (1,6 г, 8 ммоль) в ацетонитриле 22 мл, содержащему 274 мл воды. Смесь нагревают при 80C в герметичной пробирке в течение 20 ч с энергичным перемешиванием. Смесь оставляют охлаждаться и фильтруют через целит. Растворитель удаляют при пониженном давлении, получая этиловый эфир 1-фторметилциклопропанкарбоновой кислоты в виде масла (0,81 г, выход 61%). ES+(m/z) 147,1 [М+1].i-Pr2NH (1,7 мл, 2,2 эквивалента, 12,1 ммоль) и n-BuLi (1,6 М в гексанах, 7,5 мл, 2,2 эквивалента,12,1 ммоль) в 12 мл THF перемешивают при -78C в течение 30 мин под N2, затем к раствору LDA добавляют раствор этилового эфира 1-фторметилциклопропанкарбоновой кислоты (0,81 г, 5,5 ммоль) в 7 мл THF. Перемешивают и смесь оставляют нагреваться от -78C до комнатной температуры, затем перемешивают при комнатной температуре в течение 5 ч. Добавляют 10 мл насыщенного водного раствораNH4Cl. Добавляют AcOEt, органический слой отделяют, промывают насыщенным водным раствором хлорида натрия, сушат над Na2SO4 и растворитель удаляют, получая коричневое масло (0,42 г, выход 54%). Соединение растворяют в 10 мл EtOH и добавляют р-толилгидразин (0,47 г, 1 эквивалент, 3 ммоль). Смесь нагревают в герметичной пробирке при 90C в течение ночи. Затем смесь оставляют охлаждаться и растворитель удаляют, получая остаток. Очищают с помощью хроматографии Смесь 3-(1-метоксициклопропил)-3-оксопропионитрила (пример получения 2, 3,55 г, 25,5 ммоль) и гидрохлорида-р-толилгидразина (12,15 г, 76,5 ммоль) в этаноле (50 мл) нагревают при 90C в герметичной пробирке в течение 18 ч. Затем растворитель удаляют, остаток подвергают хроматографии на силикагеле, элюируя 0-5% метанола в дихлорметане, получая указанное в заглавии соединение в виде желтого твердого вещества (3,29 г, выход 53%, ES+(m/z) 244,2 [M+H]). Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше: К охлажденному смесью льда с солью раствору 5-(1-метоксициклопропил)-2-p-толил-2H-пиразол-3 иламина (пример получения 11, 2,43 г, 10 ммоль) и пиридина (0,9 мл, 11 ммоль) в THF (30 мл) по каплям добавляют раствор 2,2,2-трихлорэтилхлорформиата (2,12 г, 10 ммоль) в THF (10 мл). Перемешивают при-15C в течение 0,5 ч, затем при 22C в течение 1 ч. Реакционную смесь разделяют между дихлорметаном (50 мл) и насыщенным водным бикарбонатом натрия (50 мл). Водную фазу отделяют и дважды экстрагируют дихлорметаном (25 мл каждого). Объединенные органические фазы сушат над сульфатом натрия и концентрируют. Остаток подвергают хроматографии на силикагеле, элюируя гексанами и этилацетатом, получая белое твердое вещество (3,73 г, выход 89%, ES+(m/z) 418,1 [M+H]). Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше. Пример получения 17. 2,2,2-Трихлорэтиловый эфир 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)карбаминовой кислоты. К раствору 5-трет-бутил-2-p-толил-2H-пиразол-3-иламина (Regan et al. J. Med. Chem. 2002, 45, 29943008, 400 г, 1,74 моль) в THF (8 л) добавляют насыщенный раствор карбоната натрия (2,4 л) и смесь охлаждают до 0C. Затем по каплям добавляют 2,2,2-трихлорэтилхлорформиат (406,77 г, 1,92 моль) и смесь перемешивают при 0C в течение 2 ч. Реакционную смесь экстрагируют этилацетатом (36,5 л), сушат над безводным сульфатом магния и растворитель выпаривают. Твердое вещество растворяют в минимальном количестве этилацетата и добавляют избыток гексанов для осаждения твердого вещества. Твердое вещество собирают фильтрованием и сушат, получая указанное в заглавии соединение в виде не совсем белого твердого вещества (586 г, выход 83%). (ES+): m/z 406,1 (М+Н). Пример получения 18. 2,2,2-Трихлорэтиловый эфир [5-(1-метилциклопропил)-2-p-толил-2Hпиразол-3-ил]-карбаминовой кислоты. Ацетонитрил (87 мл, 1,6 моль) добавляют к суспензии гидрида натрия (66 г, 1,6 моль) в THF (700 мл) при комнатной температуре и перемешивают в течение 10 мин. Затем добавляют метиловый эфир 1 метилциклопропанкарбоновой кислоты (90,0 г, 0,78 моль) и белую суспензию кипятят с обратным холо- 10015123 дильником в течение 3 ч. Смесь охлаждают, затем добавляют метанол (200 мл) и смесь выливают в воду(500 мл). Фазы разделяют и pH водной фазы доводят до pH 3-4 1,0 N HCl. Водную фазу экстрагируют диэтиловым эфиром (2350 мл), органические слои объединяют и промывают водным хлоридом натрия. Сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 3-(1 метилциклопропил)-3-оксопропионитрил в виде желтого масла. 1H ЯМР (CDCl3): 3,59 (с, 2H), 1,37 (с,3H), 1,30 (кв., J=4 Гц, 2H), 0,86 (кв., J=4 Гц, 2H). Альтернативно в 5 л трехгорлую круглодонную колбу, снабженную верхней мешалкой, термопарой, обратным холодильником и капельной воронкой, помещают трет-бутоксид калия в THF (3,00 л, 3,00 моль). Этиловый эфир 1-метилциклопропанкарбоновой кислоты (264,00 г, 2,06 моль) смешивают с ацетонитрилом (123,00 г, 3,00 моль), затем эту смесь добавляют через капельную воронку в течение 0,5 ч к раствору бутоксида. Полученную смесь нагревают до кипения с обратным холодильником. Кипятят с обратным холодильником в течение 2 ч, затем охлаждают до 40, добавляя метанол (96,00 г, 3,00 мл). Смесь перемешивают в течение 10 мин, затем помещают в 12 л делительную воронку, которая содержит энергично перемешанную смесь воды (3,96 л, 219,81 моль) и MTBE (3,96 л, 33,32 моль). Слои разделяют и водный слой экстрагируют MTBE (3,96 л, 33,32 моль). pH водного слоя корректируют от 12,5 до 3,5,используя 5N HCl (610,00 мл, 3,05 моль). Водный слой экстрагируют MTBE (2x1,32 л, 11,11). Органические слои объединяют, сушат над сульфатом натрия (62,00 г, 436,49 моль) и фильтруют, получая 3-(1 метилциклопропил)-3-оксопропионитрил. Раствор 3-(1-метилциклопропил)-3-оксо-пропионитрила (60 г, 487,2 ммоль) и гидрохлорида ртолилгидразина (78 г, 478,2 ммоль) в этаноле (975 мл) нагревают до кипения с обратным холодильником в течение 4 ч. Растворитель выпаривают и оставшееся твердое вещество растворяют в воде. pH раствора увеличивают до pH 8, используя 1,0 N раствор гидроксида натрия. Осадок фильтруют, получая 5-(1 метилциклопропил)-2-p-толил-2H-пиразол-3-иламин в виде белого твердого вещества. 1H ЯМР (DMSO): 7,39 (д, J=8 Гц, 2H), 7,22 (д, J=8 Гц, 2H), 5,12 (с, 2H), 2,31 (с, 3H), 1,30 (с, 3H), 0,80 (кв., J=4 Гц, 2H), 0,61(кв., J=4 Гц, 2H). 2,2,2-трихлорэтилхлорформиат (3,0 мл, 23 ммоль) по каплям добавляют к раствору 5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-иламина (4,75 г, 21 ммоль) в тетрагидрофуране (105 мл) и насыщенном водном карбонате натрия (32 мл) при 0C. Перемешивают при этой температуре в течение 2 ч. Смесь выливают в воду и фазы разделяют. Водную фазу экстрагируют этилацетатом. Обработка A. Органические слои объединяют и промывают водным хлоридом натрия, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая желтое твердое вещество. Твердое вещество растворяют в минимальном количестве этилацетата и добавляют гексаны до помутнения при перемешивании. Указанное в заглавии соединение кристаллизуют и фильтруют в виде белого твердого вещества. 1H ЯМР (DMSO): 9,89 (ушир. с, 1H), 7,31 (кв., J=8 Гц, 2H), 7,23 (кв., J=8 Гц,2H), 6,12 (с, 1H), 4,82 (с, 2H), 2,31 (с, 3H), 1,37 (с, 3H), 0,89 (кв., J=4 Гц, 2H), 0,71 (кв., J=4 Гц, 2H). Обработка B. Растворитель этилацетат заменяют на изопропиловый спирт (91,56 моль). Суспензию перемешивают при 0C в течение 2 ч, фильтруют, промывают охлажденным изопропиловым спиртом(13,08 моль) и сушат при 40C при пониженном давлении в течение ночи, получая указанное в заглавии соединение, в виде белого кристаллического твердого вещества. Пример получения 19. 2,2,2-трихлорэтиловый эфир [5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]карбаминовой кислоты.Tf2O (80 мл) по каплям добавляют к охлажденному раствору (-78C) метилового эфира 3-гидрокси 2-гидроксиметил-2-метилпропионовой кислоты (пример получения 5, 32,5 г) в дихлорметане (400 мл) и 2,6-лутидине (80 мл). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение приблизительно 2 ч. Разбавляют дихлорметаном (400 мл) и промывают HCl (3% водный раствор). Органический слой сушат над MgSO4, фильтруют и концентрируют. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексаны/этилацетат 5%, получая метиловый эфир 2 метил-2,3-бис-трифторметансульфонилокси-пропионовой кислоты в виде бесцветного масла. 1H ЯМРTBAF 1 M (132 ммоль, 132 мл) добавляют к раствору метилового эфира 2-метил-2,3-бистрифторметансульфонилокси-пропионовой кислоты (65,9 ммоль, 26,3 г) в 500 мл безводного THF, охлажденного до 0C. Перемешивают в течение ночи. Концентрируют при пониженном давлении и добавляют дихлорметан. Органический слой промывают насыщенным водным хлоридом натрия. Органические слои объединяют и сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая метиловый эфир 3-фтор-2-фторметил-2-метилпропионовой кислоты. 1H ЯМР (CDCl3, 300 МГц):м.д.: 4,7-4,4 (м, 4H), 3,5 (с, 3H). 0,98 (т, 3H, J=l,7 Гц).LDA 2,0 M (62,0 ммоль, 31 мл) добавляют к раствору метилового эфира 3-фтор-2-фторметил-2 метилпропионовой кислоты (28,2 ммоль, 4,3 г) в 100 мл безводного THF, охлажденного до -78C, затем добавляют безводный ацетонитрил (56,4 ммоль, 2,9 мл). Перемешивают в течение 2 ч при -78C и затем- 11015123 раствор оставляют нагреваться до комнатной температуры в течение ночи. Концентрируют при пониженном давлении и добавляют дихлорметан. Органический слой промывают насыщенным водным хлоридом натрия и водной 10% HCl. Органические слои объединяют и сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая остаток. Гидрохлорид p-толилгидразина (15,5 ммоль, 2,5 г) и остаток, полученный выше (15,5 ммоль, 2,5 г),в 31 мл этанола перемешивают при 90C в течение ночи. Растворитель выпаривают, и остаток растворяют в воде. Добавляют 10% раствор гидроксида натрия и экстрагируют в этилацетате. Органические слои объединяют и сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении,получая 5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-иламин. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексаны/этилацетат в качестве элюента (от 15 до 50%). ЖХМС ES+ (m/z) 266 [M+H]). 2,2,2-трихлорэтилхлорформиат (8,1 ммоль, 1,1 мл) и водный раствор карбоната натрия (4,8 мл) добавляют к раствору 5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-иламина (7,3 ммоль,1,9 г) в 37 мл THF. Перемешивают в течение 24 ч. Раствор выливают в воду и экстрагируют в этилацетате. Органические слои объединяют и промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая 2,2,2 трихлорэтиловый эфир [5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]-карбаминовой кислоты. ЖХМС ES+ (m/z) 440 [M+H]. Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше. Серную кислоту (900 мл) охлаждают до 5C, используя баню лед/ацетон, затем добавляют 1 аминоизохинолин (208,8 г, 1448 ммоль) в течение 45 мин, поддерживая внутреннюю температуру смеси 20C. Темную смесь охлаждают до 0C под азотом с механическим перемешиванием, затем обрабатывают KNO3 (149,4 г, 1477 ммоль) порциями в течение 45 мин, поддерживая температуру реакционной смеси 10C. Смесь перемешивают в течение 2 ч, одновременно нагревая до 15C. Смесь выливают в воду/лед (3 кг), затем разбавляют дополнительной водой (6 л). Суспензию перемешивают в течение 45 мин, затем фильтруют. Осадок промывают водой (61,5 л). Золотистое вещество частично сушат на воздухе в течение ночи, затем помещают в вакуумную печку (50-55C, приблизительно 10 Торр, отвод азота, 24 ч), получая 1-амино-4-нитроизохинолин-H2SO4 (256,8 г, выход 62%). (ES+):m/z 190(MH). Суспензию 1-амино-4-нитроизохинолина-H2SO4 (120 г, 418 ммоль) в водной HCl (6 N, 2 л) механически перемешивают под азотом при 35C. Добавляют раствор NaNO2 (72,1 г, 1044 ммоль,) в воде (300 мл) в течение 1 ч, одновременно нагревая суспензию до 50C. Смесь нагревают в течение дополнительных 30 мин, затем оставляют охлаждаться до комнатной температуры и перемешивают в течение ночи. Смесь нагревают до 50C, затем добавляют раствор NaNO2 (36 г в 150 мл воды) в течение 30 мин. Смесь перемешивают в течение 2 ч, затем нагревают до 60C, оставляют медленно охлаждаться до комнатной температуры на 3 ч. Полученную суспензию фильтруют и осадок промывают водой (3600 мл). Частично сушат на воздухе (180 г). Влажное твердое вещество суспендируют в i-PrOH (1 л) и EtOH (1 л) при кипении с обратным холодильником, добавляют THF (350 мл), чтобы добиться полного растворения. К- 12015123 раствору добавляют воду (400 мл) и охлаждают до 10C в течение 3 ч. Фильтруют и промывают EtOH(2300 мл), затем диэтиловым эфиром (3150 мл). Сушат на воздухе в течение ночи, получая 1-гидрокси 4-нитроизохинолин (62,62 г, выход 79%) в виде светло-коричневого твердого вещества. (ES+): m/z 191(MH). Суспензию 1-гидрокси-4-нитроизохинолина (62,2 г, 327 ммоль) в POCl3 (180 мл) механически перемешивают под азотом и нагревают до 100-105C в течение 1 ч, получая гомогенный темно-коричневый раствор. Холодильник заменяют на насадку для молекулярной перегонки, и избыток POCl3 удаляют при пониженном давлении (приблизительно 10-30 Торр), что приводит к температуре реактора 55C. К этой смеси добавляют 1,2-дихлорэтан (150 мл), и смесь нагревают до 70C, получая гомогенный раствор. Раствор охлаждают до 15C, затем добавляют i-PrOH (450 мл) в течение 5 мин, происходит выделение тепла до 33C. Суспензию перемешивают в течение 3,5 ч при 15C, затем фильтруют и промывают i-PrOH(3100 мл). Полученное твердое вещество сушат на воздухе в течение ночи, получая 1-хлор-4 нитроизохинолин (52,08, выход 76%) в виде желто-коричневого твердого вещества. (ES+): m/z 209/211NaH (60% в минеральном масле, непромытый; 6,23 г, 156 ммоль) частями добавляют к раствору 1 хлор-4-нитроизохинолина (26,0 г, 125 ммоль) и 1-трет-бутоксикарбонил-4-гидроксипиперидина (27,6 г,137 ммоль) в THF (350 мл), перемешанному при комнатной температуре под потоком азота. Темнокрасноватую смесь перемешивают при 40C в течение 1,5 ч, затем при 55C в течение 1,5 ч. Добавляют дополнительный NaH (1,3 г), и полученную смесь перемешивают при 55C в течение 2 ч, затем охлаждают до комнатной температуры в течение ночи. Добавляют гексаны (75 мл), затем медленно добавляют воду (600 мл). pH реакционной смеси корректируют до pH 7 водной HCl (1 N), затем слои разделяют. Водный слой отбрасывают, и к органическому слою добавляют гексаны (200 мл). Смесь частично концентрируют при пониженном давлении до объема 50-100 мл. Добавляют диэтиловый эфир (150 мл) и гексаны (250 мл), затем полученную суспензию фильтруют. Осадок промывают смесью диэтиловый эфир/гексаны (1:1) и сушат на воздухе, получая трет-бутиловый эфир 4-(4-нитроизохинолин-1-илокси)пиперидин-1-карбоновой кислоты (33,84 г, выход 73%) в виде желто-коричневого твердого вещества.(ES+): m/z 374 (MH). К суспензии трет-бутилового эфира 4-(4-нитроизохинолин-1-илокси)-пиперидин-1-карбоновой кислоты (33,5 г, 89,7 ммоль) в THF (300 мл) и этаноле (300 мл) добавляют 10% Pd/C (1,80 г, 1,69 ммоль) в виде суспензии в этаноле (25 мл) . Смесь помещают на встряхивающий аппарат Парра в атмосферу водорода (25-40 фунт/дюйм 2) при комнатной температуре на 8 ч. Смесь фильтруют через прокладку из диатомовой земли и промывают этанолом, пока фильтрат не станет бесцветным. Фильтрат концентрируют при пониженном давлении, получая темное масло. Остаток подвергают хроматографии на силикагеле,элюируя 1%, затем 2,5% смесью MeOH/дихлорметан, получая указанное в заглавии соединение (30,3 г,выход 98%) в виде оранжевого стеклоподобного вещества. (ES+): m/z 344 (MH). Пример получения 23. [4-(4-Аминоизохинолин-1-илокси)пиперидин-1-ил]-(1-метилциклопропил) метанон Изохинолин (500,00 г, 3,75 моль) и этилацетат (7,60 л, 77,62 моль) помещают в 22 л трехгорлую круглодонную колбу, снабженную верхней мешалкой, термопарой, трубкой входа/выхода азота и капельной воронкой, на водяной бане. Перемешивают до растворения. При комнатной температуре по каплям добавляют надуксусную кислоту (1,25 л, 5,94 моль) в течение 0,5 ч. Перемешивают при комнатной температуре в течение ночи. Реакционную колбу охлаждают на водяной бане со льдом, затем реакционную смесь гасят, добавляя по каплям диметилсульфид (525,00 мл, 7,14 моль) в течение 45 мин. Перемешивают в течение ночи, одновременно нагревая до комнатной температуры. Реакционную смесь тестируют на пероксид. Две партии реакционной смеси объединяют. Реакционную смесь переносят в 50 л делительную воронку и добавляют воду (2,00 л, 111,02 моль) и дихлорметан (12,00 л, 187,21 моль). Частями добавляют карбонат натрия (2,07 кг, 19,53 моль), затем слои разделяют. Водный слой экстрагируют дихлорметаном(34 л), органические слои объединяют и сушат над сульфатом натрия; фильтруют и растворитель удаляют при пониженном давлении, получая неочищенное темно-красное масло/жидкость. К неочищенному темно-красному маслу/жидкости добавляют этилацетат (8,00 л, 81,76 моль), затем перемешивают при пониженном давлении, пока не удалятся 6,8 л этилацетата. Осажденное твердое вещество фильтруют, промывают холодным этилацетатом (750,00 мл, 7,66 моль), затем промывают гептаном (800,00 мл, 5,46 моль). Твердое вещество сушат, получая изохинолин-2-оксид, 714,20 г (64%) в виде мелкозернистого пескоподобного твердого вещества. В качестве альтернативной обработки перед промыванием гептаном из фильтрата удаляют 1 л рас- 13015123 творителя. Фильтрат выдерживают в течение ночи. Твердое вещество фильтруют и промывают холодным этилацетатом (500,00 мл, 5,11 моль), затем промывают гептаном (500,00 мл, 3,41 моль). Твердое вещество сушат, получая изохинолин-2-оксид, 11.0,10 г (10%) в виде мелкозернистого пескоподобного твердого вещества - партия 2 (общий выход 74%). Изохинолин 2-оксид (4,82 моль; 699,1 г) и ангидрид уксусной кислоты (73,95 моль; 6,99 л) помещают в 22 л колбу с трубкой входа N2, с насадкой для перегонки и термопарой. Реакционную смесь нагревают до слабого кипения с обратным холодильником и летучие вещества удаляют с помощью перегонки. Нагревание продолжают и дистиллят собирают в течение 2 ч (удаляется приблизительно половина общего объема реакционной смеси). Реакционную смесь охлаждают до 50C. По каплям добавляют метанол (2 л), и реакционную смесь оставляют нагреваться до 70C. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Изохинолин-1-ол кристаллизуют из раствора, выделяют с помощью фильтрования и сушат при пониженном давлении при 50C. Выход = 246,7 г (35%). Изохинолин-1-ол (2,35 моль; 341,0 г) и 1705 мл смеси 1:4 воды и уксусной кислоты помещают в 5 л колбу. Реакционную смесь нагревают до 60C. По каплям добавляют раствор азотной кислоты (7,04 моль; 443,0 мл) в 1705 мл смеси 1:4 воды и уксусной кислоты в течение 3 ч. При добавлении азотной кислоты температуру реакционной смеси поддерживают между 68 и 70C. Через 3 ч реакционную смесь охлаждают до комнатной температуры. К реакционной смеси добавляют воду (5564 мл), затем фильтруют. Осадок промывают водой (1 л) и сушат в вакууме при 50C. Выход = 196,8 г (44%). 1-гидрокси-4-нитроизохинолин (880,3 ммоль; 167,4 г) и POCl3 (4,95 моль; 460 мл) помещают в 2 л колбу с трубкой входа N2, холодильником и термопарой. Полученную суспензию нагревают до 100C в течение приблизительно 1 ч. Реакционную смесь концентрируют досуха, используя ротационный испаритель. Остаток суспендируют в 1,2-дихлорэтане (402 мл) и охлаждают до 15C. По каплям добавляютi-PrOH (1015 мл), поддерживая температуру резервуара ниже 30C. Реакционную смесь перемешивают в течение 2 ч при 20C и в течение 1 ч при 10C. Реакционную смесь фильтруют, и осадок промывают изопропиловым спиртом (100 мл). Сушат при пониженном давлении при 50C. Выход = 118,5 г (65%). Этиловый эфир 1-метилциклопропанкарбоновой кислоты (405,0 г, 3,16 моль), 5N гидроксид натрия(1,0 л, 5,00 моль) и метанол (400,0 мл, 9,88 моль) помещают в 3 л трехгорлую круглодонную колбу. Реакционную смесь нагревают при температуре от 50C до 60C в течение 5 ч, затем оставляют охлаждаться до температуры окружающей среды в течение ночи. Органический растворитель удаляют и основной раствор экстрагируют МТВЕ (2500 мл). pH водного раствора корректируют до pH 1, используя 600 мл концентрированной HCl и экстрагируют МТВЕ (4500 мл). Органические слои объединяют, промывают насыщенным водным хлоридом натрия, сушат над сульфатом магния, фильтруют и растворитель удаляют при пониженном давлении. Остаток оставляют затвердевать, затем добавляют 100 мл гептана и полученную суспензию перемешивают при 0-5C. Суспензию фильтруют, затем полученное твердое вещество сушат при пониженном давлении. Фильтрат концентрируют и охлаждают на ледяной бане, получая дополнительное белое твердое вещество. Две партии вещества объединяют, восстанавливая 265 г (84%) 1-метилциклопропанкарбоновой кислоты в виде белого твердого вещества. 1-метилциклопропанкарбоновую кислоту (260,0 г, 2,60 моль), 2-бутанон (2,5 л, 27,92 моль) и Nметилморфолин (325,0 мл, 2,95 моль) помещают в 5 л трехгорлую круглодонную колбу, снабженную верхней мешалкой. Смесь перемешивают при 0C и частями добавляют 2-хлор-4,6-диметокси-[1,3,5] триазин (510,0 г, 2,86 моль) в течение 30 мин. Перемешивание продолжают при 0C в течение 15 мин,затем при температуре окружающей среды в течение 2 ч. Полученный гидрохлорид N-метилморфолина фильтруют и промывают 2200 мл 2-бутанона. Фильтрат концентрируют и остаток растворяют в 1500 млTHF, получая раствор A. Карбонат калия (550,0 г, 3,94 моль) в 2 л воды помещают в 5 л трехгорлую круглодонную колбу. Добавляют 4-гидроксипиперидин (275,0 г, 2,66 моль), получая раствор B. Раствор B охлаждают на водяной бане со льдом и по каплям добавляют раствор A. Охлаждающую баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Органический растворитель удаляют при пониженном давлении. Оставшийся основной водный раствор экстрагируют 62 л дихлорметана. Органические слои объединяют, промывают смесью 1,5 л насыщенного водного хлорида натрия и 200 мл концентрированной HCl. Кислотный водный раствор экстрагируют 31 л дихлорметана. Органические слои объединяют и сушат над 500 г сульфата магния и 100 г карбоната калия в течение ночи. Сушильные реагенты отфильтровывают и растворитель удаляют, пока не останется приблизительно 500 мл растворителя. Добавляют 1 л гептана и растворитель удаляют, пока происходит кристаллизация. Твердое вещество фильтруют, сильно промывая гептаном и сушат при пониженном давлении, получая (4-гидроксипиперидин-1-ил)-(1-метилциклопропил)метанон, 330 г (69%) в виде белого кристаллического твердого вещества. Гидрид натрия (38,40 г, 960,09 моль) и THF (1,54 л, 18,92 моль) помещают в 5 л колбу Мортона,снабженную верхней мешалкой, капельной воронкой и термопарой. Перемешивают в течение нескольких минут, затем добавляют (4-гидроксипиперидин-1-ил)-(1-метилциклопропил)метанон (149,00 г,- 14015123 813,09 моль). Перемешивают в течение 0,5 ч Добавляют 1-хлор-4-нитроизохинолин (154,00 г, 738,24 моль) в THF (1,54 л, 18,92 моль) в течение 1 ч. Реакционную смесь перемешивают в течение 2 ч при комнатной температуре, затем при 40C в течение 6 ч, затем охлаждают до комнатной температуры в течение ночи. К реакционной смеси по каплям добавляют воду (500 мл, 27,78 моль). Реакционную смесь гасят, выливая в энергично перемешиваемую смесь воды (2,5 л, 138,89 моль) и МТВЕ (3,08 л, 25,92 моль). Слои разделяют, органический слой промывают водой (3,08 л, 170,97 моль) и сушат над сульфатом натрия (142,00 г, 999,70 моль). Сушильный реагент отфильтровывают, получая фильтрат 1. Описанную выше процедуру повторяют, получая фильтрат 2. Фильтраты 1 и 2 объединяют. Растворитель удаляют при пониженном давлении (150 мм) и собирают дистиллят с температурой выпаривания 20-28C, пока не останется приблизительно 1,5 л. В перегонную колбу добавляют изопропиловый спирт (4,62 л, 60,43 моль) и перегонку продолжают при пониженном давлении (120 мм), собирая,дистиллят с температурой выпаривания 35-44C, пока не останется приблизительно 1 л. Оставшуюся суспензию охлаждают на льду в течение ночи. Твердое вещество фильтруют и промывают холодным изопропиловым спиртом (300,00 мл, 3,92 моль), затем 3100 мл гептана (300,00 мл, 2,05 моль). Твердое вещество сушат при 40C при пониженном давлении в течение ночи, получая (1-метилциклопропил)-[4(4-нитроизохинолин-1-илокси)пиперидин-1-ил]метанон, 368 г (72% - вместе) в виде оранжевого твердого вещества. В автоклав на три галлона помещают (1-метилциклопропил)-[4-(4-нитроизохинолин-1-илокси)пиперидин-1-ил]метанон (368,0 г, 1,04 моль), THF (4,42 л, 54,32 моль) и 5% Pd на угле (сухой, 40,50 г,19,03 моль). Автоклав герметизируют и вводят водород до 50 фунт/дюйм 2. Смесь перемешивают при 1000 об/мин при комнатной температуре под 50 фунт/дюйм 2 водорода в течение 4,5 ч. Фильтруют и промывают дополнительным THF (2,0 л, 24,58 моль). Фильтрат обрабатывают углем (20-40 меш, 42,00 г) и нагревают до 40C в течение 1 ч. Добавляют дополнительный уголь (60 меш, 47,00 г) и нагревание продолжают в течение 1 ч. Уголь фильтруют через микроволокнистую бумагу и слой Hyflo Super Cel и промывают минимальным количеством THF. Растворитель удаляют, получая [4-(4-аминоизохинолин-1 илокси)-пиперидин-1-ил]-(1-метилциклопропил)-метанон, 348 г (103%) в виде темно-оранжево-красного масла/пены. 1H ЯМР (500 МГц, CDCl3):0,59 (т, J=6,0 Гц, 2H), 0,96 (т, J=6,0 Гц, 2H), 1,34 (с, 3H), 1,881,94 (м, 2H), 2,06-2,10 (м, 2H), 3,61-3,70 (м, 2H), 3,93-4,00 (м, 2H), 5,48-5,50 (м, 1H), 7,52 (с, 1H), 7,58 (т,J=7,0 Гц, 1H), 7,73 (т, J=7,0 Гц, 1H), 7,81 (д, J=7,0 Гц, 1H), 8,27 (д, J=8,5 Гц, 1H). Пример получения 24. [4-(4-Аминоизохинолин-1-илокси)пиперидин-1-ил]-(2-фторфенил)метанон. К холодному раствору трет-бутилового эфира 4-(4-нитроизохинолин-1-илокси)пиперидин-1 карбоновой кислоты (9, 68 г, 25,9 ммоль, 0C) в DCM (100 мл) добавляют TFA (80 мл). Перемешивают при 22C в течение 2 ч, затем растворитель удаляют при пониженном давлении. Остаток растворяют вDCM и обрабатывают 1 N гидроксидом натрия до pH 14. Водную фазу отделяют и дважды экстрагируют DCM. Органические фазы объединяют и сушат безводным сульфатом натрия. Растворитель удаляют,получая (4-нитроизохинолин-1-илокси)пиперидин (7,06 г, выход 99%, ES+(m/z) 274,3 [M+H]). Реакционную смесь 4-(4-нитроизохинолин-1-илокси)пиперидина (4,0 г, 14,6 ммоль), 2 фторбензойной кислоты (2,45 г, 17,5 ммоль), DCC (3,6 г, 17,5 ммоль) и HOBt (2,37 г, 17,5 ммоль) в THF(100 мл) перемешивают при 22C в течение ночи. Фильтруют, затем концентрируют. Остаток подвергают хроматографии на силикагеле, элюируя гексанами и этилацетатом, получая (2-фторфенил)-[4-(4 нитроизохинолин-1-илокси)пиперидин-1-ил]-метанон в виде желтого твердого вещества (6,82 г, выход 92%, ES+(m/z) 396,3 [M+H]). Суспензию (2-фторфенил)-[4-(4-нитроизохинолин-1-илокси)пиперидин-1-ил]-метанона (5,85 г, 14,3 ммоль) и палладия на угле (10%, 2,9 г) в метаноле (250 мл) перемешивают под водородом в течение ночи. Катализатор удаляют фильтрованием. Фильтрат концентрируют, получая бледно-желтое твердое вещество (4,7 г, выход 87%, ES+(m/z) 366,3 [M+H]). Следующее соединение получают, используя методику, по существу, аналогичную методике, описанной выше. Суспензию 1-хлор-4-нитроизохинолина (2,5 г, 12 ммоль) в ацетонитриле (100 мл) обрабатывают твердым пиперазином (5,2 г, 60 ммоль). Полученную желтую смесь нагревают в течение ночи при 60C. После охлаждения до температуры окружающей среды реакционную смесь разделяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Добавляют MeOH, дихлорметан и этилацетат, и всю смесь фильтруют, получая блестящее желтое твердое вещество. (2,15 г, 69%; ЖХМС ES+ (m/z) 259 [M+H]). Пример получения 27. Циклопропил-[4-(4-нитроизохинолин-1-ил)пиперазин-1-ил]метанон Раствор или суспензию 4-нитро-1-пиперазин-1-ил-изохинолина (пример получения 26, 517 мг, 2 ммоль), циклопропанкарбоновой кислоты (258 мг, 3 ммоль) и каталитического DMAP (24 мг, 0,2 ммоль) в дихлорметане (20 мл) обрабатывают EDCI (575 мг, 3 ммоль). Полученную смесь встряхивают при температуре окружающей среды в течение ночи, затем промывают насыщенным водным раствором бикарбоната натрия. Органический слой сушат над сульфатом натрия и концентрируют при пониженном давлении. Остаток подвергают хроматографии на силикагеле, используя градиент 0-2% 2 M аммиак-метанол в дихлорметане и градиент 0-70% этилацетата в гексанах, получая желтое твердое вещество после двух очисток. (513 мг, выход 79%, ЖХМС ES+ (m/z) 327 [M+H]). Следующие соединения получают, используя методику, по существу, аналогичную методике, описанной выше. Суспензию циклопропил-[4-(4-нитроизохинолин-1-ил)пиперазин-1-ил]метанона (пример получения 27; 513 мг, 1,57 ммоль) и 5% палладия на угле (91 мг) в этилацетате (50 мл) в встряхивающем аппарате Парра при температуре окружающей среды подвергают действию атмосферы водорода при 60 фунт/дюйм 2. Через 8 ч реакционную смесь фильтруют и концентрируют при пониженном давлении, получая коричневую пену. (Количественный выход; ЖХМС ES+(m/z) 297 [M+H]). Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше. К раствору трет-бутилового эфира пиперазин-1-карбоновой кислоты (10 г, 53,7 ммоль) в дихлорметане (300 мл) добавляют триэтиламин (15,1 мл, 107,4 ммоль) и 2-фторбензоилхлорид (6,4 мл, 53,7 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. Затем добавляют воду (200 мл) и органический слой отделяют, сушат над сульфатом натрия, фильтруют и выпаривают растворитель при пониженном давлении, получая 17,3 г трет-бутилового эфира 4-(2-фторбензоил)-пиперазин-1 карбоновой кислоты. ES+ (m/z) 309 [M+H]. К раствору трет-бутилового эфира 4-(2-фторбензоил)пиперазин-1-карбоновой кислоты (16,3 г, 53 ммоль) в дихлорметане (100 мл) добавляют раствор 4M HCl в 1,4-диоксане (40 мл, 159 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Et2O добавляют к полученной белой суспензии и растворители выпаривают при пониженном давлении, получая 12,6 г гидрохлорида (2 фторфенил)пиперазин-1-ил-метанона. ES+ (m/z) 209 [M+H]).(2-фторфенил)пиперазин-1-ил-метанон (3,16 г, 12,9 ммоль) и K2CO3 (8,9 г, 64,5 ммоль) добавляют к 1-хлор-4-нитроизохинолину (2,85 г, 13,7 ммоль) в ацетонитриле (100 мл) и перемешивают в течение 24 ч. Нерастворимое твердое вещество фильтруют и осадок промывают AcOEt. Растворитель выпаривают при пониженном давлении, получая остаток. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексан:AcOEt 20-90%, получая 3,72 г (2-фторфенил)-[4-(4-нитроизохинолин-1-ил)пиперазин-1 ил]метанона в виде твердого вещества. ЖХМС ES+ (m/z) 381,2 [M+H].Na2S2O4 (6,87 г, 39,4 ммоль) добавляют к (2-фторфенил)-[4-(4-нитроизохинолин-1-ил)пиперазин-1 ил]метанону (3 г, 7,89 ммоль) в 170 мл смеси 1:1 THF:H2O, затем добавляют NH4OH 32% (15 мл) и перемешивают в течение 90 мин. Разбавляют водой и экстрагируют AcOEt несколько раз. Органические фазы объединяют и промывают насыщенным водным хлоридом натрия, сушат над Na2SO4 и выпаривают при пониженном давлении, получая 1,8 г указанного в заглавии соединения в виде твердого вещества. ЖХМС ES+(m/z) 351,2 [M+H]. Пример получения 36. Дигидрохлорид 1-[5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-ил]-3[1-(пиперидин-4-илокси)изохинолин-4-ил]мочевина 2,2,2-трихлорэтиловый эфир [5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-ил]карбаминовой кислоты (пример получения 18, 3,20 г, 7,94 ммоль и 2,0 эквивалента) и трет-бутиловый эфир 4-(4 аминоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты (пример получения 22, 1,37 г, 3,97 ммоль,1,0 эквивалент) растворяют в 7 мл безводного DMSO, добавляют диизопропилэтиламин (DIPEA, 1,36 мл,7,94 ммоль, 2,0 эквивалента) и нагревают в герметичной пробирке с перемешиванием в течение 20 ч при 80C. Раствор выливают в смесь 1:1 об./об. дихлорметана и ледяной воды. Водную фазу экстрагируют дихлорметаном (250 мл), и объединенные органические слои промывают водой (250 мл) и водным хлоридом натрия (100 мл). Органический раствор сушат над сульфатом натрия и выпаривают при пони- 17015123 женном давлении. Остаток подвергают хроматографии на силикагеле, элюируя с градиентом от 15% до 80% EtOAc в гексанах, получая 1,40 г трет-бутилового эфира 4-(4-3-[5-(1-метилциклопропил)-2-pтолил-2H-пиразол-3-ил]уреидоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты в виде чистого пенистого кремового твердого вещества, выход 59%. ES+(m/z) 597,4 [M+H]. 4M HCl в диоксане (2,4 мл, 9,36 ммоль) добавляют к трет-бутиловому эфиру 4-(4-3-[5-(1 метилциклопропил)-2-p-толил-2H-пиразол-3-ил]уреидоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты (1,4 г, 2,34 ммоль) в дихлорметане (20 мл) при комнатной температуре и перемешивают в течение ночи. Растворитель выпаривают при пониженном давлении, получая указанное в заглавии соединение (1,3 г) в виде белого твердого вещества (количественный). ES+(m/z): 497,4 (M+H). Пример получения 37. Гидрохлорид 1-[5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол 3-ил]-3-[1-(пиперидин-4-илокси)изохинолин-4-ил]мочевины. 2,2,2-трихлорэтиловый эфир [5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]карбаминовой кислоты (пример получения 27, 2,7 ммоль, 1,2 г) и DIPEA (2,9 ммоль, 0,5 мл) добавляют к раствору трет-бутилового эфира 4-(4-аминоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты(пример получения 22, 2,9 ммоль, 1,0 г) в 4 мл DMSO и перемешивают при 85C в течение ночи. Охлаждают, добавляют воду и экстрагируют дихлорметаном. Органические слои объединяют и промывают насыщенным водным хлоридом натрия. Сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая остаток. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексаны/этилацетат с градиентом (от 15 до 70%), получая трет-бутиловый эфир 4-(4-3-[5-(2 фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]уреидоизохинолин-1-илокси)пиперидин-1 карбоновой кислоты. ЖХМС ES+ (m/z) 635 [M+H]. трет-Бутиловый эфир 4-(4-3-[5-(2-фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]уреидоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты (1,1 ммоль, 0,6 г) перемешивают и растворяют в 5 мл дихлорметана и 4,0 M соляной кислоте в диоксане (5,3 ммоль, 1,3 мл) при комнатной температуре в течение ночи. Концентрируют при пониженном давлении. Полученное белое твердое вещество растирают в порошок с диэтиловым эфиром. ЖХМС ES+ (m/z) 535 [M+H]. Следующие соединения получают, используя методику, по существу, аналогичную методике, описанной выше. Газообразный азот барботируют через раствор 2,2,2-трихлорэтилового эфира 1-(5-трет-бутил-2-pтолил-2H-пиразол-3-ил)карбаминовой кислоты (пример получения 17, 589 мг, 1,456 ммоль) и третбутилового эфира 4-(4-аминоизохинолин-1-илокси)пиперидин-1-карбоновой кислоты (пример получения 22, 500 мг, 1,456 ммоль) в DMSO (5 мл) в течение 5 мин. Затем добавляют N,N-диизопропилэтиламин(507 мкл, 2,912 ммоль). Смесь перемешивают при 60C в течение 6 ч, затем оставляют охлаждаться до- 18015123 комнатной температуры в течение ночи. Реакционную смесь разделяют между 200 мл CH2Cl2 и 100 мл дистиллированной воды. Промывают водой (1100 мл), затем водные слои объединяют и экстрагируютCH2Cl2 (350 мл). Органические слои объединяют и промывают насыщенным водным хлоридом натрия(2100 мл). Объединенные органические фазы сушат над сульфатом натрия и концентрируют. Остаток подвергают хроматографии на силикагеле, элюируя гексанами и этилацетатом, получая трет-бутиловый эфир 4-4-[3-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)уреидо]изохинолин-1-илоксипиперидин-1-карбоновой кислоты в виде коричневого твердого вещества. ЖХМС ES+ (m/z) 599,5 [M+H]. К охлажденному раствору трет-бутилового эфира 4-4-[3-(5-трет-бутил-2-p-толил-2H-пиразол-3 ил)уреидо]изохинолин-1-илоксипиперидин-1-карбоновой кислоты (820 мг, 1,37 ммоль) в дихлорметане(50 мл) добавляют трифторуксусную кислоту (15 мл). Реакционную смесь перемешивают при 22C в течение 1,5 ч. Затем растворитель удаляют, остаток обрабатывают 1 N гидроксидом натрия, насыщеннымNaCl, (50 мл) и пять раз экстрагируют дихлорметаном (50 мл каждого). Объединенные органические фазы сушат над сульфатом натрия. Смесь фильтруют и остаток подвергают хроматографии на силикагеле,элюируя дихлорметаном и метанолом, получая белое твердое вещество (.515 мг, выход 75%, ЖХМСES+(m/z) 499,5 [M+H]). Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше. Таблица 3(0,2 64 мл) в 4 мл DMSO нагревают при 60C в течение 5 ч. Полученную смесь охлаждают до температуры окружающей среды и добавляют воду. Осадок фильтруют, промывают водой, затем пентаном и сушат в вакуумной печи при 60C. Остаток подвергают хроматографии на силикагеле, элюируя с градиентом 2M аммиак-метанол в дихлорметане (от 0 до 2%), получая 206 мг продукта (чистота 93% с помощью ЖХМС). Полученный продукт растворяют в небольшом количестве дихлорметана и MeOH, обрабатывают 1 эквивалентом 2 M метансульфоновой кислоты в дихлорметане и концентрируют в потоке азота. Полученное твердое вещество растирают в порошок с несколькими миллилитрами смеси DMSO/MeOH/вода и фильтруют, получая 145 мг свободного основания (ЖХМС ES+ (m/z) 550 [M+H]). Следующее соединение получают после дополнительной очистки с помощью обратной фазы на колонке MC C18 Xterra 3075 мм 5 мкм, используя градиент: 10 мМ водный бикарбонат аммония в ацетонитриле, применяя методику, по существу аналогичную методике, описанной выше. Пример 3. 1-1-[4-(2,6-Дифторбензоил)пиперазин-1-ил]изохинолин-4-ил-3-[5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-ил]мочевина. Раствор [4-(4-аминоизохинолин-1-ил)пиперазин-1-ил]-(2,6-дифторфенил)метанона (пример получения 32, приблизительно 0,88 ммоль), 2,2,2-трихлорэтилового эфира [5-(1-метилциклопропил)-2-р-толил 2H-пиразол-3-ил]карбаминовой кислоты (пример получения 18, 3 62 мг, 0,9 ммоль) и диизопропилэтиламина (0,314 мл) в 5 мл DMSO нагревают при 60C в течение ночи. Полученную смесь охлаждают до температуры окружающей среды и разделяют между водой и этилацетатом, используя насыщенный водный хлорид натрия, чтобы способствовать разделению фаз. Водный слой экстрагируют этилацетатом, и объединенные органические слои сушат над сульфатом натрия. Концентрируют при пониженном давлении. Остаток подвергают хроматографии на силикагеле, элюируя с градиентом 2M аммиак-метанол в дихлорметане (от 0 до 2%). Растворяют в небольшом количестве дихлорметана и MeOH, обрабатывают 1 эквивалентом 2M метансульфоновой кислоты в дихлорметане и концентрируют под потоком азота. Дополнительной очисткой с помощью обратной фазы на колонке MC C18 Xterra 3075 мм 5 мкм, используя градиент: 10 мМ водный бикарбонат аммония в ацетонитриле, получают 73 мг свободного основания. ЖХМС ES+ (m/z) 622 [M+H]). Свободное основание следующего соединения получают, используя методику, по существу аналогичную методике, описанной выше. Затем растворяют 0,13 г в 1 мл DCM и добавляют 1 N раствор метансульфоновой кислоты в DCM (1 эквивалент). Растворитель выпаривают при пониженном давлении,получая 0,15 г соединения, описанного ниже. Пример 5. 1-(5-трет-Бутил-2-p-толил-2H-пиразол-3-ил)-3-1-[1-(1-метилциклопропанкарбонил) пиперидин-4-илокси]изохинолин-4-илмочевина. Газообразный азот барботируют через раствор 2,2,2-трихлорэтилового эфира 1- (5-трет-бутил-2-pтолил-2 Н-пиразол-3-ил)-карбаминовой кислоты (пример получения 17, 203 мг, 0,5 ммоль) и [4-(4 аминоизохинолин-1-илокси)пиперидин-1-ил]-(1-метилциклопропил) метанона (163 мг, 0,5 ммоль) вDMSO (2 мл) в течение 5 мин. Затем добавляют N,N-диизопропилэтиламин (200 мкл, 1,1 ммоль). После перемешивания при 60C в течение ночи реакционную смесь разделяют между дихлорметаном (15 мл) и насыщенным бикарбонатом натрия (50 мл). Водную фазу отделяют и дважды экстрагируют дихлорметаном (15 мл каждого). Объединенные органические фазы сушат над сульфатом натрия и концентрируют. Остаток очищают с помощью хроматографии на силикагеле, элюируя гексанами и этилацетатом, получая белое твердое вещество (226 мг, выход 78%, ES+ (m/z) 581,3 [M+H]). Свободные основания следующих соединений получают, используя методику, по существу аналогичную методике, описанной выше. Затем растворяют в 1 мл DCM и добавляют раствор 1 N метансульфоновой кислоты в DCM (1 эквивалент). Растворитель выпаривают при пониженном давлении, получая соединения, описанные ниже. Пример 11. Мезилат 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)-3-1-[4-(2,2-диметилпропионил) пиперазин-1-ил]изохинолин-4-илмочевины. Перемешивают 1-[4-(4-аминоизохинолин-1-ил)-пиперазин-1-ил]-2,2-диметилпропан-1-он (пример получения 3, 0,9 ммоль, 0,3 г), растворенный в 18 мл ацетонитрила. Добавляют карбонат калия (0,99 ммоль, 0,1 г) и 2,2,2-трихлорэтиловый эфир 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)-карбаминовой кислоты (пример получения 17, 0,9 ммоль, 0,4 г) и перемешивают в течение ночи при комнатной температуре. Затем добавляют 1 мл DMF и перемешивают при 60C в течение 24 ч. Охлаждают, добавляют воду и экстрагируют дихлорметаном. Органические слои объединяют и промывают насыщенным водным хлоридом натрия, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении, получая остаток. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексаны/этилацетат с градиентом (от 5 до 20%). К полученному маслу добавляют дихлорметан и образовавшийся осадок фильтруют, получая 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)-3-1-[4-(2,2-диметилпропионил)пиперазин-1-ил]изохинолин-4-илмочевину. ES+(m/z) 568 [M+H]). 1-(5-трет-Бутил-2-p-толил-2H-пиразол-3-ил)-3-1-[4-(2,2-диметилпропионил)-пиперазин-1-ил]изохинолин-4-илмочевину перемешивают при комнатной температуре. Добавляют 1 N раствор метансульфоновой кислоты в дихлорметане, получая указанное в заглавии соединение. ЖХМС ES+ (m/z) 568N2. Добавляют дихлорметан (5 мл), затем добавляют DIPEA (0,22 мл, 1,28 ммоль) и перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают, получая остаток. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексан:AcOEt 50-100%, получая 1-1-[1-(1 метилциклопропанкарбонил)пиперидин-4-илокси]изохинолин-4-ил-3-[5-(1-метилциклопропил)-2-pтолил-2H-пиразол-3-ил]мочевину в виде свободного основания. ES+(m/z) 579,3 (M+H). 0,3 г 1-1-[1-(1-метилциклопропанкарбонил)пиперидин-4-илокси]изохинолин-4-ил-3-[5-(1-метилциклопропил)-2-p-толил-2H-пиразол-3-ил]мочевины растворяют в дихлорметане 1 мл и добавляют 321 мкл 1 N раствора метансульфоновой кислоты в дихлорметане. Растворитель выпаривают при пониженном давлении, получая 0,357 г указанного в заглавии соединения в виде белого твердого вещества (количественный). ES+(m/z) 579,3 (M+H). Следующие соединения получают, используя способ, по существу аналогичный способам примеров получения, описанных выше. Газообразный азот барботируют через раствор 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)-3-[1(пиперидин-4-илокси)изохинолин-4-ил]мочевины (пример получения 39, 100 мг, 0,2 ммоль) и фенилового эфира карбаминовой кислоты (32,9 мг, 0,24 ммоль) в DMSO (1 мл) в течение 5 мин. Затем добавляютN,N-диизопропилэтиламин (200 мкл, 1,1 ммоль). После перемешивания при 85C в течение ночи реакционную смесь разделяют между этилацетатом (15 мл) и насыщенным бикарбонатом натрия (50 мл). Водную фазу отделяют и дважды экстрагируют этилацетатом (15 мл каждого). Объединенные органические фазы сушат над сульфатом натрия и концентрируют. Остаток подвергают хроматографии на силикагеле,элюируя гексанами и этилацетатом, получая белое твердое вещество (40 мг, выход 37%, ES+ (m/z) 542,3CH2Cl2 (100 мл). Промывают 1 N NaOH (220 мл), водные слои объединяют и экстрагируют CH2Cl2(250 мл), затем объединенные органические слои промывают насыщенным водным хлоридом натрия(150 мл). Объединенные органические фазы сушат над сульфатом натрия, смесь фильтруют и остаток подвергают хроматографии на силикагеле, элюируя дихлорметаном и метанолом, получая белое твердое вещество (109,8 мг, выход 80%, ES+ (m/z) 621,5 [M+H]). 0,107 г 1-(5-трет-бутил-2-p-толил-2H-пиразол-3-ил)-3-3-хлор-1-(2-фторбензоил)пиперидин-4-илокси]изохинолин-4-ил)мочевины растворяют в дихлорметане (2 мл) и метаноле (2 мл) и добавляют метансульфоновую кислоту (16,56 мг, 0,1723 ммоль). Растворитель выпаривают при пониженном давлении,получая указанное в заглавии соединение в виде белого твердого вещества (119,7 мг, 97%, ES+ (m/z) 621,5 [M+H-MsOH]. Следующие соединения получают, используя методику, по существу аналогичную методике, описанной выше.(0,6 ммоль, 0,07 мл) и триэтиламин (1,7 ммоль, 0,2 мл) в 5 мл дихлорметана перемешивают при комнатной температуре в течение ночи. Добавляют немного воды и экстрагируют в дихлорметане. Органический слой промывают насыщенным водным хлоридом натрия; сушат над безводным сульфатом натрия и- 25015123 концентрируют при пониженном давлении. Остаток подвергают хроматографии на силикагеле, элюируя смесью гексаны/этилацетат в качестве элюента (30%-70%). ЖХМС ES+ (m/z) 675 [M+H]. Следующие соединения получают, используя методику, по существу, аналогичную методике, описанной выше. Пример 45. Мезилат 1-1-[1-(2,6-дифторбензоил)пиперидин-4-илокси]изохинолин-4-ил-3-[5-(2 фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]мочевины. 0,13 г (0,19 ммоль) 1-1-[1-(2,6-дифторбензоил)пиперидин-4-илокси]-изохинолин-4-ил-3-[5-(2 фтор-1-фторметил-1-метилэтил)-2-p-толил-2H-пиразол-3-ил]мочевины растворяют в дихлорметане (2 мл) и добавляют 0,19 мл 1N раствора метансульфоновой кислоты в дихлорметане. Растворитель выпаривают при пониженном давлении, получая 0,09 г указанного в заглавии соединения в виде белого твердого вещества. ES+ (m/z) 675 (M+H). Следующие соединения получают, используя методику, по существу, аналогичную методике, описанной выше.[4-(4-Аминоизохинолин-1-илокси)пиперидин-1-ил]-(1-метилциклопропил)метанон (пример полу- 26015123 чения 23, 331,00 г, 1,02 моль), THF (3,97 л, 48,79 моль), 2,2,2-трихлорэтиловый эфир [5-(1 метилциклопропил)-2-p-толил-2H-пиразол-3-ил]карбаминовой кислоты (пример получения 18, 410,00 г,1,02 моль) и DIPE (159,00 г, 1,22 моль) помещают в 12 л колбу Мортона, снабженную верхней мешалкой,термопарой и обратным холодильником. Смесь кипятят с обратным холодильником в течение 22 ч. Реакционную смесь охлаждают до 50C и обрабатывают углем (Darco G-60; 33,00 г, 2,75 моль). Суспензию перемешивают в течение 30 мин при 43-50C, затем фильтруют через микроволокнистую бумагу иHyflo Super Cel (27,00 г). Фильтрат затравливают и оставляют охлаждаться при температуре окружающей среды во время осаждения. Полученную суспензию помещают на лед и перемешивают в течение ночи. Твердые вещества фильтруют, промывают THF (300,00 мл, 3,69 моль) и сушат при пониженном давлении при 40C. Остаток подвергают хроматографии на силикагеле, элюируя смесью 1:3 ацетон/DCM, затем 30% ацетона в DCM, получая 401 г (68%) 1-1-[1-(1-метилциклопропанкарбонил)пиперидин-4-илокси]изохинолин-4-ил-3-[5-(1-метилциклопропил)-2-р-толил-2H-пиразол-3-ил)мочевины в виде белого твердого вещества. 1H ЯМР (300 МГц, DMSO-d6):0,56 (т, J=6,0 Гц, 2H), 0,73 (т,J=6,0 Гц, 2H), 0,84 (т, J=6,0 Гц, 2H), 0,92 (т, J=6,0 Гц, 2H), 1,27 (с, 3H), 1,41 (с, 3H), 1,73-1,81 (м, 2H), 1,992,08 (м, 2H), 2,41 (с, 3H), 3,52-3,65 (м, 2H), 3,83-3,96 (м, 2H), 5,47-5,58 (м, 1H), 6,22 (с, 1H), 7,37 (д, J=8,l Гц, 2H), 7,44 (д, J=8,l Гц, 2H), 7,65-7,71 (м, 1H), 7,78-7,83 (м, 2H), 8,12 (с, 1H), 8,27 (д, J=8,4 Гц, 1H), 8,58(с, 1H), 8,69 (с, 1H). Свободное основание (400,00 г, 6,91 моль) и дихлорметан (3,00 л, 46,80 моль) помещают в 5 л колбу, снабженную верхней мешалкой. По каплям добавляют метансульфоновую кислоту (72,00 г, 7,49 моль) при комнатной температуре. Реакционную смесь перемешивают в течение 1 ч. Фильтруют и растворитель удаляют при пониженном давлении. Добавляют диэтиловый эфир (4 л) и перемешивают в течение ночи. Фильтруют и сушат при пониженном давлении, получая указанное в заглавии соединение,467,8 г. 1H ЯМР (DMSO-d6):0,52 (т, J=2,5 Гц, 2H), 0,70 (т, J=2,5 Гц, 2H), 0,80 (т, J=2,5 Гц, 2H), 0,90 (т,J=2,5 Гц, 2H), 1,24 (с, 3H), 1,38 (с, 3H), 1,76 (ушир., 2H), 2,02 (ушир., 2H), 2,38 (с, 3H), 2,40 (с, 3H), 3,55 5-(1-фторметилциклопропил)-2-p-толил-2H-пиразол-3-иламин (160,00 мг; 652,26 мкмоль), растворенный в 1 мл DCM, медленно добавляют к раствору 1,1'-карбонилдиимидазола (158,65 мг, 978,39 мкмоль) в DCM. Смесь перемешивают при 40C в течение 7 ч. Затем добавляют раствор [4-(4-аминоизохинолин-1-илокси)-пиперидин-1-ил]-(1-метилциклопропил)метанона (пример получения 23, 212,25 мг, 652,26 мкмоль, 1,00 эквивалент) в 1 мл DCM. Смесь перемешивают в течение ночи. Растворитель выпаривают под N2. Сначала очищают с помощью картриджа HLB (6 г), используя NH4CO3/MeOH (от 100:0 до 0:100) и затем в конце с помощью ВЭЖХ (колонка Kromasil C18 (10021,2 мм, 5 мкм (HiChrom). Мобильной фазой является вода (растворитель A) и ацетонитрил (растворитель B), оба содержат 0,05% трифторуксусной кислоты (TFA). Градиентный режим: 2 мин при 40% B, от 40 до 60% B через 6 мин., в конце, 2 мин при 95% B. Скорость потока: 25 мл/мин), получая 1-[5-(1-фторметилциклопропил)2-p-толил-2H-пиразол-3-ил]-3-1-[1-(1-метилциклопропанкарбонил)пиперидин-4-илокси]изохинолин-4 илмочевину в виде твердого вещества (19 мг, выход 5%). ES+ (m/z): 597 [М+1]. 1M метансульфоновую кислоту (30,17 мкмоль; 30,17 мкл) добавляют к раствору 1-[5-(1 фторметилциклопропил)-2-p-толил-2 Н-пиразол-3-ил]-3-1-[1-(1-метилциклопропанкарбонил)пиперидин 4-илокси]изохинолин-4-илмочевины (18,00 мг, 30,17 мкмоль) в 0,5 мл дихлорметана. Смесь перемешивают при комнатной температуре в течение 1 ч. Растворитель выпаривают под азотом, получая указанное в заглавии соединение в виде твердого вещества (20 мг, выход 96%). ES+(m/z): 597,4 [M+1]. Ингибирование p38 киназы Приготовление стандартных растворов. Буферный раствор киназы получают с помощью смешивания 2,5 мл 1 M Tris-HCl (pH 7,5), 0,1 мл 1M дитиотреитола. 1,0 мл 1 М хлорида магния и 300 мкл 1% Тритона X-100 и разбавления до 100 мл водой. 84 мл этого буферного раствора киназы смешивают с 16 мл DMSO, получая 16% раствор DMSO. 200 мкМ раствор АТФ получают с помощью добавления 102,6 мкл 10 мМ водного АТФ, 25 мкл 33PАТФ и 163,5 мкл 4 мМ водного раствора пептида 661-681 эпидермального фактора роста (Biomol,кат.P-121) в 5 мл буферного раствора киназы. Раствор фермента p38 киназы получают с помощью растворения 9,5 мкл концентрированного рас- 27015123 твора фермента (250 нг p38 фермента/мкл буферного раствора киназы) в 1536 мкл буферного раствора киназы. Подготовка проб 80 мкМ раствор каждого тестируемого соединения и контрольного соединения получают с помощью растворения 2 мкл 10 мМ исходного раствора соответствующих соединений в диметилсульфоксиде в 248 мкл 16% раствора DMSO в 96-луночном микротитровальном планшете Costar. Планшет помещают на автоматизированный манипулятор для жидкостей Tecan Genesis для последовательных разведений 1:3. Исследование 10 мкл последовательно разведенного соединения помещают в 96-луночный исследовательский планшет автоматизированного манипулятора для жидкостей Beckman Multimek. Добавляют 20 мкл 200 мкМ раствора АТФ с помощью 8-канального манипулятора для жидкостей Titertek Multidrop. 10 мкл раствора фермента p38 киназы помещают в исследовательский планшет, используя Multimek. Смесь оставляют для прохождения реакции на 40 мин при 30C и затем реакцию останавливают путем добавления 60 мкл свежеприготовленного 5% раствора ледяной AcOH с помощью Multidrop. 80 мкл этого раствора переносят в планшет "MAPH", используя Multimek. Планшеты выдерживают в течение 30 мин при комнатной температуре, после чего промывают/отсасывают на экстракторе Titertek MAP свежеприготовленным 0,5% раствором ледяной AcOH (1300 мкл, 2200 мкл). Планшеты промокают фильтровальной бумагой и добавляют 100 мкл сцинтилляционной жидкости MicroScint-20 (Packard Bioscience) с помощью Multidrop. Планшеты выдерживают в течение 30 мин и радиоактивность подсчитывают на сцинтилляционном счетчике PE/Wallac Microbeta Trilux для изотопа 33P. Соединение, описанное в примере 12, изначально исследовали при 10 концентрациях (20 мкМ - 1 нМ, используя последовательные разведения 1:3). Соединения со значениями IC50 меньше 25 нМ повторно исследовали при начальной концентрации от 2 мкМ до 0,1 нМ (последовательные разведения 1:3). Значения IC50 рассчитывали (программное обеспечение IDBS Activity Base) для каждого соединения, используя нелинейную регрессию. Соединение, описанное в примере 12, исследовали, по существу,как описано выше, и было найдено, что оно ингибирует фермент p38 киназы при IC50 34 нМ. Ингибирование р-МАРКАРК 2 in vitro Клетки RAW 264.7 (линия моноцитов/макрофагов АТСС мышей) высевают с плотностью 50000 клеток/лунка в 96-луночные планшеты со средой RPMI-1640, дополненной 10% эмбриональной бычьей сывороткой (FBS), и оставляют осаждаться и прилипать ко дну лунки на 48 ч. После достижения слияния клетки обрабатывают в течение 2 ч 10 последовательными разведениями разных соединений. Всегда включают контрольное соединение. Через 2 ч добавляют анизомицин (100 мкг/мл) и клетки инкубируют в течение 30 мин при 37C в атмосфере 5% CO2. Затем клетки фиксируют и обрабатывают пероксидом водорода, чтобы удалить эндогенную пероксидазу. В заключение, планшеты блокируют FBS. промывают и проводят исследование ELISA, используя антифосфо- MAPKAPK2 антитело (Thr 334, Cell Signalling, кат. 3041) и ahP-конъюгированное вторичное антитело. Эту реакцию определяют, используя FEMTO(Pierce), который представляет собой усиленный хемолюминесцентный субстрат ahP, который вызывает быструю кинетическую светоотдачу и высокую сигнальную интенсивность. Соединение, описанное в примере 12, исследовали, по существу, как описано выше, и было найдено, что оно ингибирует продуцирование фермента рМАРКАРК 2 при IC50 7 нМ. Соединения, описанные в примерах, исследовали, по существу, как описано выше, и было найдено,что они имеют значения IC50, которые меньше чем или равняются 200 нМ. Следующие соединения были исследованы, по существу, как описано выше, и было найдено, что они имеют такую активность: Ингибирование TNF in vitro Мышиные брюшинные макрофаги 1 мл тиогликолевого бульона (5,0 г экстракта дрожжей, 15,0 г казитона или триптиказы, 5,0 г глюкозы, 2,5 г хлорида натрия, 0,75 г L-цистина, 0,5 тиогликолята натрия, 1,0 мг резазурина и 0,75 г агара в 1,0 л дистиллированной воды) впрыскивают в брюшинную впадину самок мышей Balb/C. На 4-й или 5-й день после инъекции мышей умерщвляют и затем впрыскивают внутрибрюшинно 4 мл среды RPMI-1640(Bio Whittaker) и брюшинные макрофаги отбирают с помощью шприца. Продуцирование цитокинов Мышиные брюшинные макрофаги подсчитывают с помощью гемоцитометра и их концентрацию доводят до 5105 клеток/лунка в 96-луночных планшетах в среде RPMI-1640 с помощью 10% эмбрио- 28015123 нальной бычьей сыворотки. 200 мкл/лунка высеивают в 96-луночные планшеты и клетки оставляют осаждаться и прилипать ко дну лунки, как минимум, на 3 ч. Исследуемое соединение или стандартный ингибитор p38 киназы предварительно обрабатывают, используя серии 8 концентраций, в течение 1 ч при 37C (20 мкл/лунка). Клетки обрабатывают смесью 50 нг/мл липополисахарида (LPS) и 10 Ед/мл интерферона- в течение 18 ч при 37C (20 мкл/лунка) . Кондиционированную среду собирают и исследуют на продуцирование TNF, используя методику выявления Luminex. Исследование TNF с выявлением Luminex (набор Bio-Rad Bio-Plex-кат. 171-G12221) Лиофилизированный предварительно смешанный стандарт TNF (1 стандартная ампула/два 96 луночных планшета) восстанавливают в 50 мкл стерильной воды (500000 пг/мл). Образцы взбалтывают в течение 5 с, инкубируют на льду в течение 30 мин и взбалтывают в течение 5 с перед использованием. Набор с двенадцатью пробирками объемом 1,5 мл нумеруют от 1 до 12 и затем в соответствующие пробирки добавляют указанные ниже клеточные среды (стандартные концентрации являются такими,как указано ниже: 50000; 25000; 12500; 6250; 3125; 1562,5; 781,3; 390,6; 195,3; 97,7; 48,8; и 24,4 пг/мл). Предварительно смешанные гранулы, связанные с цитокином, энергично взбалтывают (25X) в течение 30 с. Гранулы, связанные с цитокином, разбавляют до IX концентрации, используя IX буфер исследования Bio-Plex. Для каждого планшета 240 мкл предварительно смешанных гранул добавляют к 5760 мкл буфера исследования Bio-Plex. 96-луночный фильтровальный планшет Millipore блокируют добавлением 100 мкл/лунка блокирующего буфера. Блокирующий буфер фильтруют через лунки, используя систему фильтрования Millipore и затем вытирают тканью насухо. Проводят два промывания на фильтровальном планшете, используя 100 мкл/лунка буфера исследования Bio-Plex, и вытирают тканью насухо. IX гранул, связанных с антицитокином, взбалтывают в течение 15 с и добавляют в каждую лунку по 50 мкл. Фильтруют и вытирают тканью насухо. Проводят два промывания на планшетах, используя 100 мкл/лунка буфера промывания Bio-Plex. Снова фильтруют и вытирают тканью насухо. 50 мкл образца или стандарта добавляют в каждую лунку. Пробы инкубируют в течение 60 с при комнатной температуре на встряхивающем аппарате, защищенном от света, при положении регулятора 6 и затем в течение 30 мин при положении регулятора 3, и затем помещают на ночь в холодильник. Проводят 3 промывания буфером промывания Bio-Plex. Фильтруют и вытирают тканью насухо. Приготавливают антитело для выявления цитокина (за 10 мин до использования) для каждого планшета, и 60 мкл предварительно смешанного исходного раствора антитела для выявления цитокина добавляют к 5940 мкл разбавителя для выявления антитела Bio-Plex. Добавляют 50 мкл антитела для выявления цитокина и инкубируют в течение 60 с при комнатной температуре на встряхивающем аппарате, защищенном от света, при положении регулятора 6 и затем в течение 30 мин при положении регулятора 3. Проводят 3 промывания буфером промывания Bio-Plex. Фильтруют и вытирают тканью насухо. Приготавливают стрептавидин-PE (за 10 мин до использования) для каждого планшета и добавляют 60 мкл к 5940 мкл буфера исследования Bio-Plex. В каждую лунку добавляют 50 мкл стрептавидина-PE и инкубируют в течение 60 с при комнатной температуре на встряхивающем аппарате, защищенном от света, при положении регулятора 6 и затем в течение 10 мин при положении регулятора 3. Проводят 3 промывания буфером промывания Bio-Plex. Фильтруют. Гранулы повторно суспендируют в 100 мкл/лунка буфера исследования Bio-Plex. Стандарты и пробы считывают на устройстве Luminex. Считанные значения интенсивности затем переводят в единицы пикограмм/миллилитр, используя 12-точечную стандартную кривую, полученную по данным двух параллельных исследований с применением способа четырехпараметро'вой логистической регрессии (Bio-PlexManager 2.0, Bio-Rad) и рассчитывают значения IC50. Соединение, описанное в примере 12, исследовали, по существу, как описано выше, и выявили подавление TNF in vitro при IC50 меньше, чем 9 нМ. Ингибирование TNF in vivo Соединения вводят перорально (30, 10, 3 и 1 мг/кг) самкам мышей Balb/C (6 мышей/доза). Через 1 ч после введения соединения в 4 дозах (перорально в объеме 0,1 мл/мышь; основа: 1% NaCMC/0,25%Tween-80 в воде) мышам делают внутрибрюшинную инъекцию LPS с дозой 400 мкг/кг. Через 1,5 ч после стимулирования LPS мышей анестезируют изофлураном и обескровливают с помощью сердечной пункции. TNF-уровни в плазме определяют, используя набор ELISA от систем RD и определяют зависимое от дозы ED50. Соединение, описанное в примере 12, исследовали, по существу, как описано выше, и выявили подавление TNF in vivo при TMED50 1,0 мг/кг. Действующая минимальная доза (TMED) 50 представляет собой дозу, при которой достигается ингибирование, большее чем или равное 50%, и которое является статистически отличимым от контроля/плацебо. Влияние перорального введения Соединения исследуют для определения влияния перорального введения на самцах крыс Fischer 344. Животные голодают в течение ночи и им вводят исследуемые соединения в виде суспензий в карбоксиметилцеллюлозе натрия (1% масс./об.), которая содержит Tween 80 (0,25% об./об.) и антивспениватель (0,1% масс./об.). Дозы суспензий получают с концентрацией 1 мг/мл и вводят в дозе 1 мл/кг с по- 29
МПК / Метки
МПК: C07D 403/14, A61P 25/00, A61P 35/00, A61P 29/00, C07D 403/12, A61K 31/4725
Метки: ингибиторов, киназы, пиразолизохинолинмочевины, производные, качестве
Код ссылки
<a href="https://eas.patents.su/30-15123-proizvodnye-pirazolizohinolinmocheviny-v-kachestve-ingibitorov-p38-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразолизохинолинмочевины в качестве ингибиторов p38 киназы</a>
Производные хиназолина в качестве ингибиторов киназы

Номер патента: 5809
Опубликовано: 30.06.2005
Авторы: Иде Синити, Панди Анджали, Мацуно Кендзи, Скарборо Роберт М., Ода Содзи, Цукуда Еидзи, Номото Юдзи, Итимура Митио, Ирие Дзунко
МПК: C07D 403/12
Метки: киназы, ингибиторов, качестве, производные, хиназолина
Формула / Реферат:
1. Азотсодержащее гетероциклическое соединение формулы в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи; R2 и R4 каждый представляют собой -O-CH3 или -O(-CH2)n-R3; причем, когда R2 представляет собой -O-CH3, R4 представляет...
Производные триазолопиримидина в качестве ингибиторов киназы гликогенсинтазы-3

Номер патента: 11300
Опубликовано: 27.02.2009
Авторы: Эмбрехтс Вернер Констант Йохан, Лав Кристофер Джон, Коиманс Людвиг Поль, Вандермасен Неле, Фрейн Эдди Жан Эдгар, Бейнстерс Петер Якобус Йоханнес Антониус, Виллемс Марк
МПК: C07D 487/00
Метки: киназы, триазолопиримидина, гликогенсинтазы-3, ингибиторов, производные, качестве
Формула / Реферат:
1. Производные триазолопиримидина формулы и его фармацевтически приемлемая аддитивная соль, где кольцо A представляет собой фенил; R1 представляет собой водород или C1-6алкил; X1 представляет собой прямую связь или -(CH2)n3-, причем n3 означает целое число, равное 1, 2 или 3; R2 представляет собой C3-7циклоалкил; фенил; 6-членный моноциклический гетероцикл, содержащий N, который может быть замещен C1-6-алкоксикарбонилом; или радикал формулы ...
Замещённые пирролопиразольные производные в качестве ингибиторов киназы

Номер патента: 10596
Опубликовано: 30.10.2008
Авторы: Певарелло Паоло, Амичи Раффаэлла, Вульпетти Анна, Руссель Патрик, Фанчелли Даниеле, Нези Марчелла, Орзини Паоло, Орци Фабрицио, Браска Мария Габриелла
МПК: A61P 17/00, A61K 31/4162, A61P 25/28...
Метки: производные, пирролопиразольные, киназы, качестве, ингибиторов, замещённые
Формула / Реферат:
1. Способ лечения заболеваний, связанных с пролиферацией клеток, которые вызваны и/или связаны с измененной киназной активностью, зависящей от клеточного цикла, путем введения нуждающемуся в лечении млекопитающему эффективного количества соединения, представленного формулами (Ia) и (Ib) в которых R означает группу -CORa, -CONHRa или -CONRaRb, где Ra и Rb, каждый независимо, означают водород или необязательно замещенную группу, выбранную из...
Производные 1н-тиено [2,3-c] пиразола, предназначенные для использования в качестве ингибиторов киназы

Номер патента: 11032
Опубликовано: 30.12.2008
Авторы: Вианелло Паола, Фанчелли Даниеле, Виольо Серджо, Бинди Симона, Тезеи Даниа, Варази Марио
МПК: A61P 17/06, A61K 31/4162, A61P 19/02...
Метки: пиразола, производные, киназы, 1н-тиено, 2,3-c, ингибиторов, предназначенные, использования, качестве
Формула / Реферат:
1. Способ лечения клеточных пролиферативных расстройств, вызванных и/или связанных с измененной активностью протеинкиназы, включающий введение нуждающемуся в нем млекопитающему эффективного количества производного 1Н-тиено[2,3-c]пиразола формулы (I) где R представляет собой фенил или 5- или 6-членную гетероарильную группу, содержащую 1 или 2 гетероатома, выбранных из N, S или О, где R необязательно замещен в любом из его свободных положений 1-6...
Производные триазолопиридинсульфанила в качестве ингибиторов p38 мар киназы

Номер патента: 12119
Опубликовано: 28.08.2009
Авторы: Льютуайт Рассел Эндрю, Матиас Джон Пол, Миллан Дэвид Саймон, Филлипс Кристофер
МПК: A61K 31/4745, C07D 471/04, A61P 29/00...
Метки: киназы, триазолопиридинсульфанила, мар, производные, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль и/или сольват (включая гидрат), где R1 представляет CH3, SCH3, SCH2CH3, CH2CH3, Н или CH2SCH3; R1a представляет CH3 или CH2CH3; R2 представляет пиридил, тетрагидронафтил или арил, причем указанные пиридил, тетрагидронафтил и арил, каждый, необязательно были замещены одним или более из заместителей, независимо выбранных из группы, состоящей из галогена, -CN, -CO2H, OH, CONR5R6,...
Предыдущий патент: Фармацевтическая композиция и ее применение для лечения тромбоза
Следующий патент: Применение соединения, содержащего кремний (варианты)
Случайный патент: Устройство для смешивания и передачи лекарственного препарата