Производные пиридо-пиримидина, их получение, их применение в терапии
Номер патента: 11406
Опубликовано: 27.02.2009
Авторы: Жегам Самир, Бурри Бернар, Казелла Пьер, Перро Пьер







Формула / Реферат
1. Соединение, отвечающее формуле (I)

в которой R1 выбран из группы, состоящей из (C1-C6)алкила, (C3-С7)циклоалкила, CH2COR4, фенила или фенила, замещенного гидроксилом, и/или галогеном, и/или (C1-C6)алкилом;
R4 обозначает гидроксильную группу, (С1-С4)алкоксигруппу, аминогруппу, (C1-C4)алкиламиногруппу, ди(С1-С4)алкиламиногруппу;
Ar1 обозначает радикал, выбранный из

R5 обозначает цианогруппу, гидрокси(С1-С4)алкил, (C1-С6)алкокси(C1-C6)алкил или группу (CH2)nNR7R8, CO2R7, CONHNR7R8, CONR7R8, CONR8OR9, (CH2)nNR7COR8, (CH2)nNR7COOR8;
R6 обозначает атом водорода, (С1-С4)алкил или одно из значений R5;
или, R5 и R6, такие как определенные перед этим, связаны вместе с образованием цикла, от 4- до
7-членного, содержащего от 0 до 2 гетероатомов, выбранных из N и О, причем указанный цикл, состоящий 4-7 звеньев, может быть замещен одним или несколькими заместителями, независимо выбранными из следующих групп: галоген, (C1-C4)алкил, галогенированный (C1-С4)алкил, гидрокси(C1-C4)алкил, (C1-C4)алкокси(C1-C4)алкил, (СН2)mNR7R8, трет-бутоксикарбонил;
R7 и R8 обозначают, каждый независимо один от другого, заместитель, выбранный из следующих: Н, (С1-С4)алкил, (C1-С4)алкил-OH, (С3-С7)циклоалкил, (С3-С7)циклоалкил-NH2, (C1-С4)алкил-(С3-С7)циклоалкил, C(=NH)NH2, SO2(C1-C6)алкил, SO2-фенил, R8 может также обозначать трет-бутоксикарбонильную группу или бензилоксикарбонильную группу;
или R7 и R8 вместе с атомом азота, с которым они связаны, образуют радикал азетидинил, пирролидинил, пиперидинил, пиперазинил или морфолинил, причем указанный радикал является незамещенным или замещенным, один или несколько раз, (C1-С6)алкилом, (C1-C4)алкил-ОН, COO(C1-C6)алкилом, F;
R9 обозначает атом водорода или (C1-C4)алкил;
Ar2 обозначает фенильную группу, незамещенную или замещенную от 1 до 5 раз одинаковыми или разными заместителями, выбранными из атома галогена, (С1-С4)алкильной группы, трифторметильной группы или (С1-С4)алкоксигруппы;
n обозначает 1, 2 или 3;
m обозначает 0, 1, 2 или 3.
2. Соединение формулы (I) по п. 1, отличающееся тем, что
R1 обозначает трет-бутил, этил или фенил;
и/или Ar1 обозначает радикал, выбранный из

и/или R5 обозначает группу (CH2)nNR7R8, CONHNR7R8, CONR7R8, гидрокси(С1-С4)алкил или (СН2)nNR7COR8;
и/или R6 обозначает атом водорода, метил или группу (CH2)nNR7R8 или гидроксиметил;
и/или Ar2 обозначает арильную группу, замещенную 1-2 заместителями, независимо выбранными из галогена, (С1-С4)алкила, (С1-С4)алкоксигруппы;
n, R7 и R8 такие, как определенные перед этим для соединения формулы (I);
в форме основания или соли присоединения с кислотой, а также в форме гидрата или сольвата.
3. Соединение по любому из предыдущих пунктов, отличающееся тем, что R5 выбран из (CH2)nNR7R8, CONR7R8 и (СН2)nNR7COR8.
4. Соединение по любому из предыдущих пунктов, отличающееся тем, что оно находится в форме:
нехиральной, или
рацемической, или
обогащенной одним стереоизомером, или
обогащенной одним энантиомером;
и тем, что оно может находиться в форме сольвата, или гидрата, или соли.
5. Промежуточный продукт для получения соединения по любому из пп.1-4, отличающийся тем, что он отвечает следующей общей формуле:

в которой R1 и Ar2 такие, как определено в одном из пп.1-4.
6. Промежуточный продукт для получения соединения по любому из пп.1-5, отличающийся тем, что он отвечает следующей общей формуле:

в которой R1 и Ar2 такие, как определено в одном из пп.1-6.
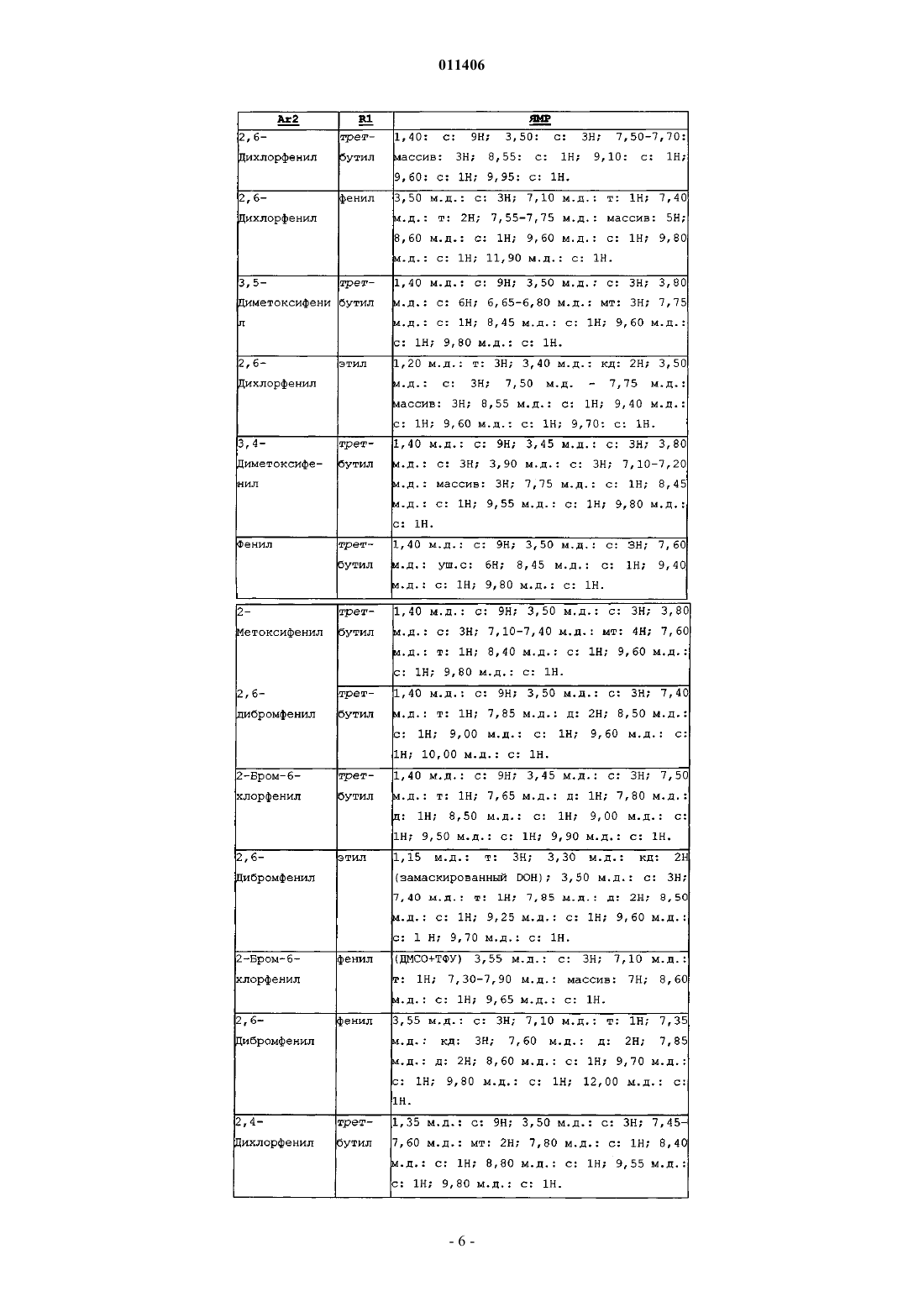
7. Промежуточный продукт по п.5 или 6, отличающийся тем, что заместитель Ar2 выбран из следующих: фенил, 2-метоксифенил, 2,6-дихлорфенил, 3,5-диметоксифенил, 3,4-диметоксифенил, 2,6-дибромфенил, 2-бром-6-хлорфенил, 2,4-дихлорфенил и 3,5-дихлорфенил.
8. Промежуточный продукт по п.5 или 6, отличающийся тем, что заместитель R1 выбран из следующих: этил, трет-бутил и фенил.
9. Способ получения соединения формулы (I) по любому из пп.1-3, отличающийся тем, что вводят в реакцию с соединением формулы

в которой R1 и Ar2 такие, как определены для (I), амин формулы Ar'1NH2 (III), в которой Ar'1 представляет собой Ar1, такой как определен для (I), или предшественника Ar1, представляющего собой группу а), b), с), d) или е), такую как определена для (I), в которой заместители R5 и/или R6 являются такими, как определены для (I); в случае необходимости превращают группу Ar'1 соединения, полученного таким образом, в группу Ar1.
10. Способ получения соединения формулы (I) по любому из пп.1-3, отличающийся тем, что вводят в реакцию:
i) соединение формулы

в которой R10 обозначает удаляемую группу, такую как (а) галоген, в частности Cl или Br, или (b) алкил-S(О)m-, в которой m=0, 1 или 2; в которой R11 обозначает NHC(=R12)-NH-R1, с R12=O или S; и
ii) амин формулы Ar'1NH2 (III), в которой Ar'1 представляет собой Ar1, такой как определен для (I), или предшественника Ar1, представляющего собой группу а), b), с), d) или е), такую как определена для (I), в которой заместители R5 и/или R6 являются такими, как определены для (I); в случае необходимости превращают группу Ar'1 соединения, полученного таким образом, в группу Ar1;
в котором:
(a) когда R10 представляет собой галоген или алкил-S(O)m- с m=2, реакцию осуществляют в растворителе;
(b) когда R10 представляет собой алкил-S(O)m- с m=0 или 1, реакцию осуществляют с Ar'1NH2 (III) в расплавленном состоянии при 200шС;
в случае необходимости, аминогруппы, присутствующие в группе Ar'1 соединения (III), предварительно превращают в солевые или защищают.
11. Способ по п.10, отличающийся тем, что когда R10 представляет собой галоген или алкил-S(O)m- с m=2, реакцию осуществляют в растворителе, предпочтительно полярном:
(i) например, тетрагидрофуране, диметилсульфоксиде или этаноле, в случае необходимости, в присутствии следового количества кислоты, такой как соляная кислота; или
(ii) в диметилсульфоксиде в присутствии сильного основания, такого как t-BuOK;
при температуре, находящейся в интервале от комнатной температуры до температуры кипения растворителя.
12. Лекарственное средство, отличающееся тем, что оно содержит соединение формулы (I) по любому из пп.1-2, или его соль присоединения с фармацевтически приемлемой кислотой, или гидрат, или сольват соединения формулы (I).
13. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение формулы (I) по любому из пп.1-3, или фармацевтически приемлемую соль, гидрат или сольват упомянутого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
14. Применение соединения формулы (I) по любому из пп.1-3 для получения лекарственного средства, предназначенного для лечения и профилактики заболеваний, вызываемых или обостряемых пролиферацией клеток.
15. Применение по п.14 для профилактики и лечения лейкозов, первичных и метастатических солидных опухолей, карцином и раков.
16. Фармацевтическая композиция по п.13, отличающаяся тем, что она дополнительно содержит одно (или несколько) другое(их) цитостатическое(их) действующее(их) начало(начал).
Текст
011406 Объектом настоящего изобретения являются производные пиридо[2,3-d]пиримидина, их получение и их применение в терапии. Соединения, являющиеся производными пиридо[2,3-d]пиримидина, описаны в патентных заявкахWO 01/55147 и WO 03/000011 и в патентах ЕР-В-790997 и US 5733913. Эти соединения являются потенциально пригодными для лечения нарушений пролиферации клеток. Согласно первому аспекту объектом настоящего изобретения являются соединения, отвечающие формуле (I)R6 обозначает атом водорода, (С 1-С 4)алкил или одно из значений R5; или R5 и R6, такие как определенные перед этим, связаны вместе с образованием цикла, от 4- до 7 членного, содержащего от 0 до 2 гетероатомов, выбранных из N и О, причем указанный цикл, состоящий из 4-7 звеньев, может быть замещен одним или несколькими заместителями, независимо выбранными из следующих групп: галоген, (C1-C4)алкил, галогенированный (С 1-С 4)алкил, гидрокси(С 1-С 4)алкил, (C1 С 4)алкокси(С 1-С 4)алкил, (CH2)mNR7R8, трет-бутоксикарбонил;R7 и R8 обозначают, каждый независимо один от другого, заместитель, выбранный из следующих: Н, (С 1-С 4)алкил, (C1-С 4)алкил-ОН, (С 3-С 7)циклоалкил, (С 3-С 7)циклоалкил-NH2, (C1-С 4)алкил-(C3 С 7)циклоалкил, C(=NH)NH2, SO2(C1-C6)алкил, SO2-фенил, R8 может также обозначать третбутоксикарбонильную группу или бензилоксикарбонильную группу; или R7 и R8 вместе с атомом азота, с которым они связаны, образуют радикал азетидинил, пирролидинил, пиперидинил, пиперазинил или морфолинил, причем указанный радикал является незамещенным или замещенным, один или несколько раз, (C1-C6)алкилом, (С 1-С 4)алкил-ОН, COO(C1-C6)алкилом, F;R9 обозначает атом водорода или (С 1-С 4)алкил;Ar2 обозначает фенильную группу, незамещенную или замещенную от 1 до 5 раз одинаковыми или разными заместителями, выбранными из атома галогена, (С 1-С 4)алкильной группы, трифторметильной группы или (С 1-С 4)алкоксигруппы;m обозначает 0, 1, 2 или 3. Соединения формулы (I) могут содержать один или несколько асимметрических атомов углерода. Следовательно, они могут существовать в форме энантиомеров или диастереоизомеров. Упомянутые энантиомеры, диастереоизомеры, а также их смеси, включая рацемические смеси, являются частью изобретения. Соединения формулы (I) могут существовать в форме оснований или солей присоединения с кислотами. Когда соединения формулы (I) содержат свободные кислотные группы, например карбоксильную,сульфоновую, фосфоновую, упомянутые группы могут быть превращены в соль при помощи оснований с получением солей присоединения. Такие соли присоединения составляют часть изобретения. Предпочтительно, соли присоединения с кислотами или основаниями получают, соответственно, с фармацевтически приемлемыми кислотами или основаниями, но соли с другими кислотами или основаниями, используемыми, например, для очистки или выделения соединений формулы (I), также являются частью изобретения. Равным образом, соединение формулы (I) может существовать в форме гидратов или сольватов, а именно, в форме ассоциатов или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты или сольваты также являются частью изобретения. В рамках настоящего изобретения подразумевают атом галогена: фтор, хлор, бром или йод; алкильная группа: насыщенная алифатическая группа, прямая или разветвленная. В качестве примера можно назвать следующие группы: метил, н-пропил, н-бутил, н-пентил, н-гексил, н-гептил, 1-1 011406 метилэтил, 1-метилпропил, 2-метилпропил, 1,1-диметил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,1 диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-метилпентил, 2-метилпентил, 3-метилпентил,4-метилпентил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,2-диметилбутил, 2,3 диметилбутил, 3,3-диметилбутил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этил-1-метилпропил,1-этил-2-метилпропил, 1-этилбутил, 2-этилбутил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4 метилгексил, 5-метилгексил, 1,1-диметилпентил, 1,2-диметилпентил, 1,3-диметилпентил, 1,4 диметилпентил, 2,2-диметилпентил, 2,3-диметилпентил, 2,4-диметилпентил, 3,3-диметилпентил, 3,4 диметилпентил, 4,4-диметилпентил, 1,1,2-триметилбутил, 1,1,3-триметилбутил, 1,2,2-триметилбутил,1,2,3-триметилбутил, 1,3,3-триметилбутил, 2,2,3-триметилбутил, 2,3,3-триметилбутил, 1,1,2,2 тетраметилпропил, 1-этилпентил, 2-этилпентил, 3-этилпентил, 1-этил-1-метилбутил, 1-этил-2 метилбутил, 1-этил-3-метилбутил, 2-этил-1-метилбутил, 2-этил-2-метилбутил, 2-этил-3-метилбутил, 1 пропилбутил, 1-(1-метилэтил)бутил, 1-(1-метилэтил)-2-метилпропил; циклоалкильная группа: циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, бицикло[2.2.1]гептил, циклооктил, бицикло[2.2.2]октил, бицикло[3.2.1]октил, адамантил. Из соединений формулы (I), предметов изобретения, можно назвать предпочтительные соединения,которые определяются следующим образом:(CH2)nNR7COR8; и/или R6 обозначает атом водорода, метил или группу (CH2)nNR7R8 или гидроксиметил; и/или Ar2 обозначает арильную группу, замещенную 1-2 заместителями, независимо выбранными из галогена, (С 1-С 4)алкила, (С 1-С 4)алкоксигруппы;n, m, R7 и R8 такие, как определенные перед этим для соединения формулы (I); в форме основания или соли присоединения кислоты, а также в форме гидрата или сольвата. Продукты согласно изобретению, предпочтительно, будут иметь заместитель R5, выбранный из(CH2)nNR7R8, CONR7R8 и (CH2)nNR7COR8. Продукт согласно изобретению может находиться в нехиральной, или рацемической, или обогащенной одним стереоизомером, или обогащенной одним энантиомером форме; может находиться в сольватированной или гидратированной форме и может быть превращен в соль. Согласно второму аспекту изобретение касается промежуточных продуктов синтеза, используемых при получении продуктов согласно его первому аспекту, причем указанные промежуточные продукты отвечают следующей общей формуле: в которой R1 и Ar2 такие, как определенные перед этим. Согласно третьему аспекту изобретение касается получения промежуточных продуктов согласно его первому и его второму аспектам, отвечающих следующей общей формуле: в которой R1 и Ar2 такие, как определенные перед этим. Согласно четвертому аспекту изобретение касается получения промежуточных продуктов согласно его первому, второму и третьему аспектам, отвечающих следующей общей формуле: в которой Ar2 такой, как определенный перед этим. Промежуточные продукты синтеза согласно второму, третьему и четвертому аспектам изобретения-2 011406 содержат заместитель Ar2, выбранный из следующих: фенил, 2-метоксифенил, 2,6-дихлорфенил, 3,5 диметоксифенил, 3,4-диметоксифенил, 2,6-дибромфенил, 2-бром-6-хлорфенил, 2,4-дихлорфенил и 3,5 дихлорфенил. Промежуточные продукты синтеза согласно второму и третьему аспектам изобретения содержат заместитель Ar1, выбранный из следующих: этил, трет-бутил и фенил. В соответствии с изобретением соединения формулы (I) можно получить согласно способу, отличающемуся тем, что вводят в реакцию:ii) амин формулы Ar'1NH2 (III), в которой Ar'1 представляет собой Ar1, такой как определенный для(I), или предшественника Ar1; в случае необходимости превращают группу Ar'1 соединения, полученного таким образом, в группу Ar1. Когда R10 представляет собой галоген или алкил-S(O)m- с m=2, реакцию осуществляют в растворителе, предпочтительно полярном:(i) например, тетрагидрофуране, диметилсульфоксиде или этаноле, в известных случаях, в присутствии следового количества кислоты, такой как соляная кислота; или(ii) в диметилсульфоксиде в присутствии сильного основания, такого как t-BuOK; при температуре, находящейся в интервале от комнатной температуры до температуры кипения растворителя. Когда R10 представляет собой алкил-S(O)m- с m=1, можно осуществить реакцию с Ar'1NH2 (III) в расплаве, предпочтительно, при температуре, близкой к 200 С, без катализатора. В случае необходимости, аминогруппы, присутствующие в группе Ar'1 соединения (III), предварительно превращают в солевые или защищают. Под предшественником Ar1 подразумевают группу а), b), с), d) или е), такую как определенную выше для (I), в которой заместители R5 и/или R6 являются такими, как определенные выше для (I), или являются предшественниками R5 и/или R6. Соединения формулы (II) получают, следуя методике, описанной в европейском патенте 790997 и американском патенте US 5733913, как представлено на схеме 1, следующей ниже. Схема 1 м-ПХБК: мета-перхлорбензойная кислота. Амины формулы (III) известны или получены известными способами, исходя из соответствующих нитропроизводных Ar'1NO2 (IV), восстановлением либо (i) в кислой среде в присутствии металла, такого как железо или цинк в порошкообразном состоянии, либо (ii) водородом в присутствии катализатора,такого как Pd/C. Соединения формулы (IV) известны или получены известными способами. Так, 5-нитро-1,3-бенздиоксолы, монозамещенные в положении 2 группой R5=метоксикарбонил, могут быть получены действием метилдихлорацетата на 4-нитрокатехин(4-нитробензол-1,2-диол). 5-Нитро-1,3-бенздиоксолы, гем-дизамещенные в положении 2, могут быть получены, согласноPharmazie, 2003, 58 (1), 13-17, действием этилдиброммалоната на 4-нитрокатехин(4-нитробензол-1,2 диол). 7-Нитро-2,3-дигидро-1,4-бенздиоксины, замещенные в положении 2 или 3 группами R5 и R6, могут-3 011406 быть получены согласно методу, описанному в международной заявке на патент WO 01/021577, исходя из 4-нитрокатехина или при помощи известных химических превращений. 1,3-Дигидро-2-бензофуран-5-амин описан в J. Med. Chem., 1978, 21, 965-978; 4H-1,3-бенздиоксин-6 амин описан в J. Org. Chem., 1994, 59 (4), 754-757; 4H-1,3-бенздиоксин-7-амин описан в Chimie Therapeutique, 1972, 7,443-449. Действуют известными методами, такими как описанные в мартовском номере Advanced OrganicChemistry, 5th Edition, 2005, ISBN 0471585890, для того, чтобы превратить группу R5 и/или R6 соединений формулы (IV), соответственно, в заместители R5 и/или R6, желательные для соединений формулы (I). Равным образом, можно превратить группы R5 и/или R6 соединений формулы (I) для того, чтобы получить новые соединения формулы (I), несущие желаемые заместители R5 и/или R6. Так, группа R5=CO2Me позволяет получить соединения формулы (IV) или (I), в которых R5 представляет собой группу CO2H, CN, CH2OH, CONR7R8, CONHNR7R8, CONR8OR9, CH2NR7R8, методами,известными специалисту. Исходя из соединения формулы (IV) или (I), содержащего группу R5=(CH2)nOH с n=1, 2 или 3, можно получить соединение формулы (IV) или (I), в котором R5=мезилоксиметил, действием мезилхлорида,затем соединение формулы (IV) или (I), в котором R5=-CH2NR7R8, действием HNR7R8, при этом R7 и R8 такие, как определенные для соединений формулы (I). Соединения согласно изобретению получают в рацемической форме; потом можно получить оптически чистые изомеры, используя методы разделения, известные специалисту, такие как кристаллизация в результате образования солей с хиральными агентами. Можно также получить соединения согласно изобретению в оптически чистой форме, используя методы асимметрического или стереоспецифического синтеза, или хроматографические методики с применением хиральной фазы. Кроме того, продукты согласно изобретению могут быть разделены через образование диастереоизомеров, их разделение, затем расщепление фармакологически применимого диастереоизомера с образованием его энантиомерно чистого продукта. Ферментативные методики также могут быть применены. Могут быть использованы известные дополнительные методики разделения. Они включают в себя методики, опубликованные в книгеEnantiomers, Racemates and Resolutions, John Wiley and Sons, New York (1981). Начиная с получения промежуточных продуктов синтеза, соединения согласно изобретению могут быть также получены в форме, обогащенной одним стереоизомером. Так, разделение энантиомеров аминов формулы (III) или нитрованных предшественников (IV) может быть осуществлено одним из указанных методов. Следующие примеры описывают получение некоторых промежуточных продуктов и соединений согласно изобретению. Приведенные примеры не являются ограничивающими и служат только для иллюстрации настоящего изобретения. В примерах используют следующие сокращения: Тпл: температура плавленияBOP: гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (БОФ) ТГФ (THF): тетрагидрофуран ТК (ТА): температура комнатная ДХМ (DCM): дихлорметанZ: бензилоксикарбонил Спектры ядерного магнитного резонанса (ЯМР) протонов зарегистрированы при 200 МГц или при 250 МГц в DMSO-d6, если не указано противоположное. Сигнал DMSO-d6 находится при 2,5 м.д. и служит стандартом сравнения. При интерпретации спектров используют следующие сокращения: с - синглет, d - дублет, t - триплет, m - массив, mt - мультиплет, se - уширенный синглет, dd - дублет дублетов,qd - квартет, qt - квинтет. Получение соединения формулы (II). Синтез 1.N-(трет-Бутил)-N'-[6-(2,6-дихлорфенил)-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7 ил]карбамид. 1.1. 4-Амино-2-(метилтио)пиримидин-5-этилкарбоксилат. К суспензии 50,7 г 4-хлор-2-(метилтио)пиримидин-5-этилкарбоксилата в 400 мл EtOH за 20 мин добавляют, поддерживая температуру при 20 С, 140 мл 20%-ного раствора NH4OH. После 20 ч перемешивания при комнатной температуре реакционную среду концентрируют в вакууме почти досуха, затем остаток извлекают в 350 мл воды, перемешивают 20 мин, фильтруют, промывают 360 мл водой, затем сушат в вакууме в присутствии P2O5. Получают белое твердое вещество, Тпл=134-135 С, m=39,9 г. 1.2. [4-Амино-2-(метилтио)пиримидин-5-ил]метанол.-4 011406 К 39,68 г сложного эфира, полученного на предыдущей стадии, растворенного в 1 л ТГФ, за 45 мин добавляют 210 мл раствора 1 М LiAlH4 в ТГФ, поддерживая температуру меньше 30 С. Перемешивают еще 1 ч, затем понижают температуру до 5 С и последовательно, по каплям, добавляют 9 мл воды, 6,5 мл 5 н. гидроксида натрия, затем 32 мл воды. После 10 мин перемешивания твердое вещество отфильтровывают, затем промывают ТГФ. Фильтрат концентрируют в вакууме досуха, затем остаток вновь растворяют в 600 мл толуола при кипении, быстро фильтруют в горячем состоянии, чтобы удалить небольшое количество нерастворимого вещества, и дают фильтрату остыть в течение ночи. Полученные белые кристаллы отфильтровывают, промывают небольшим количеством толуола, затем простого эфира и сушат,Тпл=124-127 С, m=23,9 г. 1.3. 4-Амино-2-(метилтио)пиримидин-5-карбальдегид. К суспензии 23,8 г спирта, полученного на предыдущей стадии, в 1600 мл хлороформа за 2 мин добавляют 79,5 г активного MnO2 и перемешивают в течение 1 ночи при комнатной температуре; твердое вещество отфильтровывают, промывают 375 мл CHCl3 и фильтрат концентрируют в вакууме досуха; белый твердый остаток извлекают простым эфиром, фильтруют, сушат, Тпл=184-186 С, m=21,05 г. 1.4. 6-(2,6-Дихлорфенил)-2-(метилтио)пиридо[2,3-d]пиримидин-7-амин. К 21 г альдегида, полученного на предыдущей стадии, растворенного в 240 мл ДМФ (DMF) и охлажденного до 5 С, за 5 мин добавляют 5,47 г 60%-ного NaH, затем за 20 мин, небольшими порциями,29,05 г 2,6-дихлорфенилацетонитрила. Перемешивание продолжают в течение 30 мин при 5 С, затем в течение ночи при комнатной температуре. Реакционную среду охлаждают до 5 С и добавляют 65 мл насыщенного раствора NH4Cl, затем 500 мл смеси вода/лед; образуется красный осадок, который отфильтровывают, промывают 2 раза водой, максимально обезвоживают, промывают простым эфиром, 100 мл хлороформа, затем вновь простым эфиром; после сушки получают бежевое твердое вещество, Тпл=250253 С, m=29,92 г. Фазы простого эфира и хлороформа, образовавшиеся при промывке, концентрируют досуха, извлекают небольшим количеством хлороформа, к которому добавляют простой эфир: получают вторую порцию 3,15 г, Мобщая=33,07 г. 1.5. N-(трет-Бутил)-N'-[6-(2,6-дихлорфенил)-2-(метилтио)пиридо[2,3-d]пиримидин-7-ил]карбамид. К раствору 29,9 г амина, полученного выше, в 300 мл ДМФ за 10 мин добавляют, поддерживая температуру меньше 25 С, 4 г 60%-ного NaH; перемешивают еще 20 мин, затем за 20 мин добавляют 12,2 мл трет-бутилизоцианата, потом перемешивают в течение ночи. Реакционную среду медленно выливают на 800 мл смеси вода/лед + 100 мл 6N HCl; образовавшийся осадок отфильтровывают, промывают простым эфиром и сушат. Получают бежевое твердое вещество, Тпл=195-196C (разл.), m=26,5 г. 1.6. N-(трет-Бутил)-N'-[6-(2,6-дихлорфенил)-2-(метилсульфонил)пиридо[2,3-d]пиримидин-7-ил]карбамид. К раствору 21,95 г карбамида, полученного выше, в 300 мл хлороформа за 25 мин добавляют, поддерживая температуру меньше 25 С, 27 г метаперхлорбензойной кислоты. Образуется осадок. Через 2 ч реакционную среду разбавляют 1 л дихлорметана и добавляют Na2SO4, затем 14 г Са(ОН)2; после 30 мин перемешивания твердое вещество отфильтровывают промывают дихлорметаном, затем фильтрат концентрируют досуха; остаток растирают в 80 мл простого эфира при нагревании; дают остыть, затем белое твердое вещество отфильтровывают, промывают простым эфиром и сушат, Тпл=138-140 С, m=20,5 г. Тем же самым образом, что для соединения, описанного в синтезе 1, можно получить следующие соединения общей формулы (II):-6 011406 Получение соединений формулы (III). Используемые номера синтезов соответствуют номерам соединений в табл. 1 и 2, следующих ниже. Когда они содержат асимметрический атом углерода, данные соединения получены в форме рацемической смеси, если не указано противоположное. Синтез 2. 2.1. 5-Нитро-1,3-бенздиоксол-2-метилкарбоксилат. К суспензии 17,6 г 60%-ного NaH в 300 мл ДМФ добавляют за 1 ч 31,0 г 4-нитрокатехина, охлаждая для того, чтобы поддерживать температуру меньше 30 С. Перемешивают еще 15 мин, затем за 1 ч добавляют 104 мл метилдихлорацетата, затем перемешивают 4 ч при 90 С. Реакционную среду выливают на 2 л смеси лед/вода, затем 4 раза экстрагируют 400 мл AcOEt. Объединенные органические фазы промывают 1 раз насыщенным раствором NaCl, затем сушат и концентрируют в вакууме (выпаривание ДМФ). Остаток извлекают в смесь AcOEt/H2O и доводят до pH 8,6 при помощи Na2CO3; органическую фазу декантируют, промывают насыщенным NaHCO3, Н 2 О, 5%-ным KHSO4/K2SO4, H2O, насыщенным NaCl,затем сушат и упаривают в вакууме; получают полутвердый остаток, который извлекают, затем растирают в гептане с получением твердого вещества, m=27,7 г, Тпл=90-92 С. 2.2. 5-Амино-1,3-бенздиоксол-2-метилкарбоксилат. К 900 мл сложного эфира, полученного на предыдущей стадии, растворенного в 30 мл ТГФ, добавляют 3,92 порошкообразного цинка и, после охлаждения до -5 С, за 30 мин добавляют 4 мл уксусной кислоты, разбавленной 4 мл ТГФ, затем позволяют температуре снова подняться. Через 1,5 ч фильтруют,промывают твердое вещество небольшим количеством ТГФ и метанола. Фильтрат разбавляют AcOEt и промывают H2O, насыщенным NaHCO3, H2O, насыщенным NaCl; после сушки и концентрирования в вакууме получают желтый воск, идентифицируемый методом ЯМР, m=800 мг. Синтез 3. 3.1. 5-Нитро-1,3-бенздиоксол-2-карбоксамид. К 1,12 г сложного метилового эфира, полученного в синтезе 2.1, приливают 20 мл раствора 2 М аммиака в метаноле. Через 25 мин концентрируют в вакууме, извлекают твердый остаток в Et2O, фильтруют и сушат, m=0,99 г, Тпл=202-207C. 3.2. 5-Амино-1,3-бенздиоксол-2-карбоксамид. К 0,98 г амида, полученного в синтезе 3.1, в 35 мл ТГФ добавляют 4,57 г порошкообразного цинка; после охлаждения до -5 С, за 30 мин добавляют 5 мл уксусной кислоты, разбавленной 5 мл ТГФ. По окончании добавления позволяют температуре снова подняться. Через 1 ч твердое вещество отфильтровывают, промывают небольшим количеством ТГФ, метанола, AcOEt. Фильтрат разбавляют AcOEt, добавляют туда воду и доводят до pH 6 насыщенным NaHCO3. Образовавшийся осадок удаляют фильтрованием, фильтрат декантируют, затем органическую фазу промывают насыщенным NaHCO3, H2O, насыщенным NaCl, сушат и упаривают, получают воск, который затвердевает при охлаждении, m=0,63 г. Синтез 4. 4.1. 5-Нитро-(1,3-бенздиоксол-2-ил)метанол. К 5,02 г сложного метилового эфира, полученного в синтезе 2.1, растворенным в 25 мл ТГФ, при-5 С за 1 ч 15 мин добавляют 22,3 мл раствора 1 М LiAlH4 в ТГФ; через 20 мин после окончания добавления, по каплям, добавляют 20 мл AcOEt, затем 9 мл 1 н. NaOH; образовавшийся осадок выделяют фильтрованием, промывают AcOEt, фильтрат разбавляют AcOEt и промывают H2O, 5%-ным KHSO4/K2SO4,H2O, насыщенным NaCl; после сушки и концентрирования в вакууме получают воск, который кристаллизуется, m=2,74 г, Тпл=80-82 С. На следующей стадии нитропроизводное, полученное в синтезе 4.1, восстанавливают согласно методикам, описанным выше, с получением амина формулы (III) синтеза 4.2. Синтез 5. 5.1. (5-Нитро-1,3-бенздиоксол-2-ил)метилсульфонат. К 4,12 г спирта, полученного на стадии 4.1, растворенного в 30 мл CH2Cl2, при 5 С добавляют 3 мл триэтиламина, затем за 15 мин 1,85 г мезилхлорида. Через 15 мин убирают ледяную баню. Через 55 мин реакционную среду разбавляют CH2Cl2 и водой; органическую фазу декантируют, промывают H2O, насыщенным NaCl, сушат, упаривают. После растирания в гептане получают твердое вещество каштанового цвета, m=5,20 г, Тпл=112-115 С. 5.2. [(5-Нитро-1,3-бенздиоксол-2-ил)метил]диэтиламин. К 2,91 г мезилата, полученного на предыдущей стадии, в 18 мл ДМФ добавляют 2,19 г диэтиламина и нагревают до 80 С. Через 15 ч добавляют 0,73 г диэтиламина, затем через 8 ч еще 0,73 г. Через 48 ч, в общей сложности, реакционную среду разбавляют AcOEt, промывают водой, затем насыщенным NaCl; после сушки AcOEt упаривают, затем остаток извлекают в 40 мл Et2O+10 мл AcOEt и экстрагируют 2 раза 60 мл 0,25 н. HCl; кислые фазы смешивают, приводят в контакт с AcOEt и доводят до pH 9 при помощи 10 н. NaOH; органическую фазу декантируют, промывают водой, затем насыщенным NaCl, затем сушат и упаривают. Получают масло, m=1,55 г. 5.3. [(5-Амино-1,3-бенздиоксол-2-ил)метил]диэтиламин. К 1,93 г нитросоединения, полученного на предыдущей стадии, растворенного в 70 мл ТГФ, добав-7 011406 ляют 7,45 г порошкообразного цинка, затем при -5 С за 25 мин добавляют 7,6 мл AcOEt, затем продолжают перемешивание при температуре в интервале от 0 до 5 С. Через 1,5 ч твердое вещество отфильтровывают, промывают ТГФ и небольшим количеством метанола; фильтрат разбавляют AcOEt+H2O и доводят до pH 9 при помощи 10 н. NaOH; образовавшийся осадок удаляют фильтрованием; фильтрат декантируют; органическую фазу промывают водой, насыщенным NaCl, сушат, упаривают, получают черное масло, m=1,75 г. Синтез 6. 6.1. 1-Метил-4-5-нитро-1,3-бенздиоксол-2-ил)метил)пиперазин. Реакция согласно методике 5.2, выделенный продукт в форме дихлоргидрата. 6.2. 2-Метилпиперазин-1-ил)метил)-1,3-бенздиоксол-5-амин. Реакция по методике 5.3. Синтез 7. 7.1. 5-Нитро-1,3-бенздиоксол-2-карбоновая кислота. К 12 г сложного эфира, полученного в синтезе 2.1, в 12 мл метанола за 30 мин добавляют 1,5 мл 5 н. гидроксида натрия. Через 35 мин после окончания добавления реакционную среду разбавляют AcOEt и H2O и доводят до pH 2 при помощи 2 н. HCl; органическую фазу декантируют, промывают водой и насыщенным NaCl,затем сушат, затем упаривают, получают 1,25 г масла. 7.2. N,N-Диметил-5-нитро-1,3-бенздиоксол-2-карбоксамид. К 0,84 г кислоты, полученной на предыдущей стадии, растворенной в 15 мл дихлорметана, добавляют 0,36 г хлоргидрата диметиламина, 0,77 мл ДИПЭА (DIPEA), затем 0,91 г ДЦКДИ (DCCl). После 3 ч перемешивания при комнатной температуре реакционную среду фильтруют, затем фильтрат разбавляютCH2Cl2, который последовательно промывают насыщенным раствором NHCO3, H2O, 5%-нымKHSO4/K2SO4, H2O, насыщенным NaCl, после сушки растворитель выпаривают, затем остаток хроматографируют на диоксиде кремния смесью дихлорметан/метанол 99/1. Получают 0,5 г твердого продукта с Тпл=109 С. Синтез 9. 9.1. 5-Нитро-1,3-бенздиоксол-2-карбонитрил. К 45 мл ДМФ, охлажденного до 5 С, медленно добавляют 3,7 мл POCl3. После 30 мин перемешивания при 5 С добавляют за 1 раз 1,67 г амида, полученного на стадии 3.1. После 3 ч перемешивания при комнатной температуре реакционную смесь выливают на 250 мл смеси вода/лед. Образовавшийся осадок фильтруют, промывают водой, затем сушат, m=1,32 г, Тпл=105-110 С. 9.2. 5-Амино-1,3-бенздиоксол-2-карбонитрил. Восстановление NO2 группы продукта, полученного на стадии 9.1, в NH2, осуществляют согласно методике, описанной перед этим, применяя смесь Zn/AcOH. Синтез 11. 11.1. 5-Нитро-1,3-бенздиоксол-2,2-диэтилкарбоксилат. Данное соединение получают согласно методике, описанной в Pharmazi 2003 58 (1) 13-17. 11.2. 5-Нитро-1,3-бенздиоксол-2,2-дикарбоксамид. К 14 мл раствора 2 М аммиака в метаноле за 1 раз добавляют 1,24 г сложного диэфира, полученного на предыдущей стадии. После 30 мин перемешивания реакционную среду концентрируют досуха, затем извлекают остаток в простой эфир, фильтруют и сушат, m=1,01 г, Тпл=231-233 С. Синтез 12. 12.1. (5-Нитро-1,3-бенздиоксол-2,2-диил)диметанол. К 1,87 г сложного диэфира, полученного на стадии 11.1, в 60 мл ТГФ при комнатной температуре за 1 ч добавляют 1,50 г NaBH4; через 25 мин после окончания добавления разбавляют 250 мл AcOEt, затем 5 мл метанола, затем 40 мл воды по каплям. Органическую фазу декантируют, промывают водой, 5%ным раствором KHSO4/K2SO4, затем H2O, насыщенным раствором NaCl. После сушки и выпаривания остаток хроматографируют на диоксиде кремния смесью хлороформ/метанол (98/2), получают 0,64 г масла, которое схватывается в массе, Тпл=111-113 С. Синтез 13. 13.1. 3-(2-Метил-5-нитро-1,3-бенздиоксол-2-ил)этилпропаноат. К суспензии 15,51 г 4-нитрокатехина в 22,10 г этилацетоацетата добавляют при 70 С за 15 мин 22,4 г фосфорного ангидрида. Через 1 ч 45 мин реакционную смесь охлаждают, затем экстрагируют 4150 мл теплым толуолом. Толуольные фазы объединяют, промывают H2O, 1 н. NaOH, H2O, 5%-нымKHSO4/K2SO4, H2O, насыщенным раствором NaCl. После сушки и выпаривания продукт очищают хроматографией на диоксиде кремния, элюируя хлороформом, получают 2,44 г твердого вещества, Тпл=7678 С. 13.2. 3-(2-Метил-5-нитро-1,3-бенздиоксол-2-ил)пропан-1-ол. К 2,33 г сложного эфира, полученного выше, растворенного в 40 мл ТГФ, добавляют при -5 С за 45 мин 8 мл 1 М LiAlH4 в ТГФ. Через 35 мин по каплям добавляют 8 мл этилацетата, затем 1 мл воды,-8 011406 затем 1 мл 1 н. гидроксида натрия. Твердое вещество выделяют фильтрованием; фильтрат разбавляютAcOEt, промывают H2O, 5%-ным KHSO4/K2SO4, H2O, насыщенным раствором NaCl; после сушки органическую фазу концентрируют в вакууме; получают масло m=1,90 г. 13.3. 3-(2-Метил-5-нитро-1,3-бенздиоксол-2-ил)пропилметансульфонат. К 1,89 г спирта, полученного перед этим на стадии 13.2, в 40 мл дихлорметана при 5 С добавляют 1,01 г триэтиламина, затем 1,14 г мезилхлорида за 20 мин. По окончании добавления ледяную баню убирают и продолжают перемешивание 1 ч; реакционную среду разбавляют CH2Cl2 и промывают H2O, затем насыщенным раствором NaCl; после сушки растворитель выпаривают; получают 2,46 г воска, который при охлаждении затвердевает. 13.4. N,N-Диэтил-3-(2-Метил-5-нитро-1,3-бенздиоксол-2-ил)пропан-1-амин. К 1,21 г мезилата в 20 мл ДМФ добавляют 0,87 г диэтиламина, затем нагревают до 80 С. Через 8,5 ч добавляют 0,44 г диэтиламина и продолжают нагрев в течение 14 ч. Реакционную среду концентрируют в вакууме, остаток вновь растворяют в AcOEt и доводят до pH 9,5 при помощи 1 н. NaOH, органическую фазу декантируют, промывают H2O, насыщенным раствором NaCl, затем сушат, после выпаривания получают неочищенный продукт, который вновь растворяют в 20 мл AcOEt плюс 20 мл Et2O, затем 2 раза экстрагируют 50 мл 0,5 н. HCl; 2 водные фазы смешивают, приводят в контакт с AcOEt и доводят до pH 9 при помощи 10 н. NaOH; органическую фазу декантируют, повторно промывают H2O, затем насыщенным раствором NaCl, сушат, упаривают, получают 0,66 г масла. 13.5. Заместитель NO2 продукта, полученного на стадии 13.4, восстанавливают в NH2 способом, описанным перед этим, используя смесь Zn/AcOH для получения восстановленного продукта 13.5. 13.6. К 2,00 г продукта, полученного на стадии 13.3, в 20 мл ДМФ добавляют 0,85 г азида натрия и перемешивают 5 дней при комнатной температуре, затем экстрагируют простым эфиром и промывают органическую фазу водой, затем насыщенным раствором NaCl. Получают масло, m=1,50 г. ЯМР соответствует. 13.7. Смесь 1,49 г продукта, полученного на стадии 13.6, 1,71 г трифенилфосфина и 0,12 г воды в 25 мл ТГФ перемешивают в течение 24 ч. Затем реакционную среду экстрагируют AcOEt и промывают водой. Полученный неочищенный продукт растворяют в смеси AcOEt/Et2O и экстрагируют водным раствором 1 н. HCl. Кислую водную фазу приводят в контакт с AcOEt и доводят до pH 9 при помощи 10 н. NaOH. Органическую фазу выделяют, промывают водой, затем насыщенным раствором NaCl. Органическую фазу концентрируют при пониженном давлении с получением 1,10 г масла. 13.8. В 10 мл дихлорметана (ДХМ) (DCM) растворяют 1,09 г амина, полученного на стадии 13.7, затем добавляют 0,20 г триэтиламина, затем 1,18 г Boc2O. Через 5 ч реакционную среду разбавляют ДХМ, затем промывают, последовательно, 5%-ным раствором KHSO4/K2SO4, водой, насыщенным растворомNaCl. После сушки и выпаривания при пониженном давлении органическую фазу выделяют, получают 1,36 г масла. 13.9 (предшественник примера 55). Продукт, полученный на стадии 13.8, в количестве 1,35 г обрабатывают Zn/AcOH, как описано на стадии 2.2, чтобы восстановить группу NO2 в NH2. Получают 1,13 г воска. Синтез 14. 2-(2-Метоксиметил)-5-нитро-1,3-бенздиоксол. В 25 мл ТГФ растворяют 1,45 г гидроксиметила, полученного на стадии 4.1; после охлаждения до 5 С небольшими порциями добавляют 353 мг 60%-ного NaH; через 30 мин добавляют 0,92 мл метилиодида, затем перемешивают в течение одной ночи при комнатной температуре; добавляют 1 мл метилиодида и продолжают перемешивание в течение 5 ч; к реакционной среде добавляют 30 мл насыщенного-9 011406 раствора NH4Cl, затем воду и этилацетат; органическую фазу декантируют, повторно промывают H2O,затем насыщенным NaCl, сушат и упаривают. Неочищенный продукт хроматографируют на диоксиде кремния, элюируя смесью CHCl3/гептан 9/1. Получают 595 мг масла, которое идентифицируют методом ЯМР. Синтез 16. 16.1. трет-Бутил-2-5-нитро-1,3-бенздиоксол-2-ил)карбонил)гидразинкарбоксилат. Смесь 900 мг сложного эфира (стадия 2.1) и 2,114 г трет-бутилкарбазата в 40 мл метанола нагревают при 60 С в течение 60 ч; добавляют 600 мг трет-бутилкарбазата и греют еще 3 ч. Метанол выпаривают, остаток извлекают этилацетатом и промывают водой, 0,2 н. соляной кислотой, насыщенным раствором бикарбоната натрия, водой и насыщенным раствором NaCl. Получают воск, который схватывается в массе, m=1,27 г. 16.2. трет-Бутил-2-5-амино-1,3-бенздиоксол-2-ил)карбонил)гидразинкарбоксилат. К 1,30 г продукта, такого как полученный на предыдущей стадии, растворенного в 25 мл ТГФ, добавляют 3,92 г порошкообразного цинка, затем при -5 С за 30 мин 4,8 г уксусной кислоты; убирают ледяную баню и продолжают перемешивание 2 ч; твердое вещество отфильтровывают, промывают небольшим количеством ТГФ, затем AcOEt; добавляют к фильтрату воду и доводят до pH 6,5 15%-ным раствором Na2CO3; AcOEt декантируют, промывают насыщенным NaHCO3, H2O, насыщенным NaCl, сушат и упаривают. Получают ожидаемое соединение в форме черного масла, m=1,12 г. Синтез 21. 21.1. К 2,14 г мезилата, полученного на стадии 5,1, растворенного в 17 мл ДМФ, добавляют 1,51 г азида натрия и нагревают при 70 С в течение 3 ч. Реакционную среду экстрагируют AcOEt, который промывают водой, затем насыщенным раствором NaCl. Получают масло. m=1,71 г. 21.2. К 1,7 г продукта, полученного на стадии 21.1, растворенного в 20 мл AcOEt, небольшими порциями добавляют 3,41 г трифенилфосфина, потом через 10 мин 2,34 мл воды и нагревают до 60 С. Через 1 ч реакционную среду упаривают досуха, затем извлекают в Et2O. Нерастворимые вещества удаляют, затем добавляют избыток насыщенного раствора HCl в простом эфире. Образовавшееся твердое вещество отфильтровывают, промывают простым эфиром, затем сушат с получением ожидаемого продукта в форме хлоргидрата. Соответствующий амин получают высвобождением из хлоргидрата. 21.3. Продукт, полученный на стадии 21.2, может быть разделен на два его энантиомера. К 2 г амина, полученного на стадии 21.2, растворенного в 70 мл воды и 7 мл диоксана при 70 С, добавляют 1,97 г (S) (+) кислоты. Затем реакционной среде дают медленно вернуться к 30 С при перемешивании магнитной мешалкой. Полученный осадок отфильтровывают, растворяют в 40 мл воды и 4 мл диоксана при 70 С, затем дают медленно вернуться к 30 С при перемешивании. Твердое вещество, которое образуется при охлаждении, отфильтровывают и сушат, m=0,49 г. Полученный таким образом продукт извлекают 20 мл воды и 100 мл AcOEt, затем доводят до pH 9,5 добавлением 1 н. NaOH. Смесь декантируют, органическую фазу выделяют, промывают несколько раз водой, затем насыщенным раствором NaHCO3, водой и, наконец, насыщенным раствором NaCl. Органическую фазу собирают, сушат и растворитель выпаривают при пониженном давлении. Получают 0,27 г воска, который постепенно затвердевает. []D=+94,7 при 25 С; С=0,5 (MeOH); оптическая чистота: хиральная ВЭЖХ: 96/4 (вращательная способность энантиомера, очищенного до 100%=102). 21.4. К продукту, полученному на стадии 21.2, в 30 мл ДХМ добавляют 1,49 мл триэтиламина, затем небольшими порциями 2,53 г Boc2O. Через 1 ч реакционную среду промывают 5%-ным KHSO4/K2SO4, H2O,насыщенным раствором NaCl. После сушки органическую фазу концентрируют досуха, затем остаток растирают в гептане; получают 2 г твердого вещества. 21.5. К 480 мг 60%-ного NaH в 20 мл ТГФ при 5 С за 30 мин добавляют 1,79 г продукта, полученного на стадии 21.4, растворенного в 15 мл ТГФ. Смесь перемешивают 45 мин при комнатной температуре, затем за 10 мин добавляют 1,2 мл иодметана. Через 3 ч реакционную среду приливают к 60 мл насыщенного водного раствора лимонной кислоты, затем экстрагируют этилацетатом. Органическую фазу выделяют,промывают водой, затем насыщенным раствором NaCl, сушат и упаривают досуха. Полученный неочищенный продукт очищают импульсной хроматографией с градиентом дихлорметана в циклогексане. Получают белое твердое вещество: m=1,4 г. 21.6. Продукт, полученный на стадии 21.5, восстанавливают согласно обычной методике при помощи Смесь 11,65 г 4-N-Z-пиперидона, 6,35 г метилортоформиата и 40 мг паратолуолсульфокислоты нагревают в колбе Кляйзена (Claisen) при 60 С в течение 1 ч, затем при 70 С в течение 1 ч, не мешая перегонке метилформиата. Остаток разбавляют AcOEt+H2O и добавляют несколько капель 1 н. NaOH, чтобы довести до pH 7. Органическую фазу декантируют, выделяют, промывают водой, затем насыщенным раствором NaCl, сушат, упаривают. Получают 14 г неокрашенного масла. 22.2. Смесь 16,09 г кеталя, полученного на стадии 22.1, 10,80 г 4-нитрокатехина и 60 мг паратолуолсульфокислоты нагревают в 120 мл толуола с медленной перегонкой толуола. Через 4 ч 30 мин реакционную среду разбавляют толуолом, охлаждают; фильтрованием отделяют нерастворимое вещество, промывают фильтрат 1 н. NaOH, H2O, 5%-ным раствором KHSO4/K2SO4, H2O, насыщенным раствором NaCl. После сушки и выпаривания неочищенный продукт хроматографируют на диоксиде кремния, элюируя смесью К 0,60 г продукта, полученного выше, в 5 мл трифторуксусной кислоты добавляют 0,5 мл тиоанизола. Через 3 ч реакционную среду концентрируют в вакууме, остаток извлекают CH2Cl2 с водой и доводят до pH 0 при помощи 1 н. NaOH. После декантации органическую фазу вновь промывают водой, затем насыщенным раствором NaCl. Органическую фазу сушат и упаривают. Неочищенный продукт собирают и хроматографируют на диоксиде кремния, элюируя смесью CHCl3/MeOH/NH4OH 95/5/0,1; получают 100 мг ожидаемого твердого продукта. 22.4. В течение 1 ч 95 мг амина, полученного на стадии 22,3, обрабатывают 98 мг Boc2O и 20 мг триэтиламина в 3 мл дихлорметана. После реакции и обычных обработок получают белое твердое вещество,m=130 мг. 22.5. Группу NO2 восстанавливают в амин при помощи Zn/AcOH согласно уже описанной методике. Синтез 23. 23.1. К 985 мг продукта стадии 4.1, растворенного в 25 мл ДХМ, добавляют 0,46 мл пиридина. Затем, при 5 С за 20 мин добавляют 0,84 мл ангидрида трифторуксусной кислоты, растворенного в 3 мл ДХМ. Через 1 ч при 5 С реакционную среду промывают смесью воды со льдом, затем насыщенным растворомNaCl. Органическую фазу сушат и концентрируют в вакууме. Получают 1,42 г твердого вещества. 23.2. К 1,15 г продукта, полученного на стадии 23.1, растворенного в 10 мл ДХМ+0,5 мл ДМФ, добавляют 75 мг диэтаноламина. После перемешивания в течение ночи, реакционную среду разбавляют 100 мл ДХМ, промывают водой, промывают насыщенным раствором NaCl, сушат и концентрируют при пониженном давлении. Остаток очищают импульсной хроматографией с градиентом метанола от 0 до 15% в хлороформе. Получают 730 мг твердого вещества. 23.3. Группу NO2 восстанавливают в амин при помощи Zn/AcOH, как описано перед этим. Исходя из 720 мг продукта, полученного на стадии 23.2, получают 400 мг ожидаемого продукта в форме смолоподобного вещества. Продукт получают согласно методике, описанной в Org. Lett., 2001, 3(9), 1399-1402. Синтез 24.2. К 2,73 г продукта, полученного на стадии 24.1, в 30 мл ДХМ при 5 С за 2 мин добавляют 5 мл 69%ной азотной кислоты. Через 2,5 ч перемешивания реакционную среду разбавляют Et2O; органическую фазу промывают 2 раза H2O, 2 раза охлажденным 7%-ным раствором Na2CO3, 1 раз водой, 1 раз 5%-ным раствором KHSO4/K2SO4, 1 раз водой, затем 1 раз насыщенным раствором NaCl. После сушки и выпаривания получают белое твердое вещество, m=3,20 г. Синтез 24.3. При -5 С в течение 1 ч 3,63 г продукта, полученного по методике, описанной выше, в 80 мл ТГФ обрабатывают 532 мг LiAlH4. После обычной обработки выделяют 2,60 г продукта в форме густого масла. Синтез 24.4. Спирт, полученный на стадии 24.3, в количестве 2,59 г обрабатывают мезилхлоридом по методике,описанной в синтезе 13.3, с получением 3,52 г мезилата. Идентификация методом ЯМР. Синтез 24.5. Продукт, полученный на стадии 24.4, в количестве 3,51 г обрабатывают 1,97 г азида натрия по методике, описанной в синтезе 13.6. Получают 2,60 г ожидаемого продукта. Синтез 24.6 Продукт, полученный на стадии 24.5, в количестве 2,59 г обрабатывают 4,90 г трифенилфосфина и 2 мл воды по методике, описанной в синтезе 13.7. Получают 2 г ожидаемого продукта в форме масла. Синтез 24.7. Продукт, полученный на стадии 24.6, в количестве 1,99 г обрабатывают BOC2O по методике, описанной в синтезе 13.8. Получают 2,41 г твердого вещества. Синтез 24.8. Продукт, полученный на стадии 24.7, обрабатывают Zn/AcOH согласно обычному способу восстановления нитрогруппы в аминогруппу. На основе 0,93 г исходного продукта получают 0,84 г ожидаемого продукта в форме воска. Соединения формулы (III) и промежуточные продукты формулы (IV), являющиеся производными бенздиоксола, охарактеризованы в таблице, следующей ниже. Превращение соединения формулы (IV) в соединение формулы (III) было осуществлено согласно синтезу 5.3 для следующих соединений: 7.2, 9.1, 11.1, 12.1, 13.4 и 14.1.- 12011406 Таблица 1 Синтезы соединений формулы (III) Синтез 18. 18.1. Хлоргидрат 7-нитро-2,3-дигидро-1,4-бенздиоксин-2-ил)метил)диэтиламина. К 1,50 г (7-нитро-2,3-дигидро-1,4-бенздиоксин-2-ил)метансульфоната, описанного в международной заявке на патент WO 01/021577, в 30 мл ДМФ добавляют 800 мкл диэтиламина и нагревают до 80 С,- 15011406 затем с интервалом в 12 часов 3 раза добавляют по 800 мкл диэтиламина. Через 48 ч реакционную среду упаривают досуха; остаток извлекают 100 мл AcOEt плюс 5 мл 4 н. NaOH; органическую фазу декантируют, промывают водой, затем насыщенным NaCl; после сушки и выпаривания AcOEt остаток извлекают 20 мл AcOEt плюс 50 мл 0,5 н. HCl, перемешивают и затем декантируют; водную фазу упаривают досуха и остаток растирают в простом эфире, фильтруют и сушат, m=0,95 г, Тпл=192-194 С. 18.2. 7-Амино-2,3-дигидро-1,4-бенздиоксин-2-ил)метил)диэтиламин. К суспензии 0,94 г нитропроизводного, полученного на предыдущей стадии, в 40 мл ТГФ добавляют 3,05 г порошкообразного цинка. После охлаждения до 0 С, за 20 мин добавляют 3,1 мл уксусной кислоты; через 10 мин ледяную баню убирают и продолжают перемешивание в течение 2 ч. Твердое вещество отфильтровывают, промывают ТГФ, затем небольшим количеством метанола. Фильтрат упаривают досуха, извлекают этилацетатом, добавляют воду и доводят до pH 8 при помощи 10 н. NaOH; образовавшийся осадок выделяют фильтрованием, промывают AcOEt; фильтрат декантируют, промывают насыщенным NaCl, сушат и упаривают; получают масло каштанового цвета, m=0,51 г. Соединения формулы (III) и промежуточные продукты формулы (IV), являющиеся производными бенздиоксина, получены в форме рацемической смеси и охарактеризованы в табл. 2, следующей ниже. Таблица 2 Синтез соединений формулы (III) Номера соединений, приведенных в примерах, соответствуют номерам, присвоенным в табл. 3, следующей ниже, которая иллюстрирует химические структуры и физические свойства некоторых соединений согласно изобретению. Когда они содержат асимметрический атом углерода, данные соединения получены в форме рацемической смеси. Пример 1. Соединение 2. 5-7-трет-Бутиламино)карбонил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-2 ил)амино-1,3-бенздиоксол-2-метилкарбоксилат. Смесь 0,97 г соединения синтеза 2.2 и 1,40 г соединения синтеза 1 в 15 мл ТГФ кипятят в колбе с обратным холодильником. Через 6 ч реакционную среду концентрируют в вакууме. Продукт очищают хроматографией на диоксиде кремния, элюируя смесью AcOEt/толуол (2/3), затем повторной хроматографией, элюируя смесью CHCl3/MeOH 98/2, получают 0,45 г желтого твердого вещества, идентифицированного методом масс-спектрометрии, МН+=583.- 16011406 Пример 2. Соединение 4. 5-7-трет-Бутиламино)карбонил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-2 ил)амино-1,3-бенздиоксол-2-карбоновая кислота. К 0,25 г сложного эфира, полученного в примере, приведенном выше, в 10 мл метанола добавляют 0,3 мл 2 н. гидроксида натрия. Через 1 ч 10 мин перемешивания при комнатной температуре реакционную среду разбавляют AcOEt+H2O и доводят до pH 4 при помощи 1 н. HCl. Органическую фазу декантируют, промывают H2O, затем насыщенным NaCl, сушат и упаривают. Твердый желтый остаток извлекают простым эфиром, растирают, фильтруют и сушат. Получают 205 мг продукта, идентифицированного методом масс-спектрометрии, МН+=569. Пример 3. Соединение 3. 5-7-трет-Бутиламино)карбонил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-2 ил)амино-1,3-бенздиоксол-2-карбоксамид. Смесь 0,62 г соединения синтеза 3.2 и 1,40 г сульфопроизводного синтеза 1 в 20 мл ТГФ кипятят в колбе с обратным холодильником. Через 3 ч реакционную среду концентрируют в вакууме, затем остаток хроматографируют на диоксиде кремния, элюируя смесью CHCl3/MeOH 97/3 об./об. Получают 315 мг желтого твердого вещества, идентифицированного методом масс-спектрометрии, МН+=568. Пример 4. Соединение 9.N-(трет-Бутил)-N'-6-(2,6-дихлорфенил)-2-2-гидроксиметил)-1,3-бенздиоксол-5 ил)амино)пиридо[2,3-d]пиримидин-7-ил)карбамид. К смеси 350 мг соединения синтеза 4.2 и 937 мг соединения синтеза 1 в 15 мл EtOH добавляют 43 мкл концентрированной HCl и греют при 55 С в течение 6 ч. Реакционную среду концентрируют в вакууме, затем остаток хроматографируют на диоксиде кремния, получают 490 мг желтого твердого вещества, идентифицированного методом масс-спектрометрии, МН+=555. Пример 5. Соединение 6. 5-7-трет-Бутиламино)карбонил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-2 ил)амино-1,3-бенздиоксол-2-карбонитрил. К 1,00 г соединения синтеза 1 и 450 мг соединения синтеза 9.2 в 15 мл EtOH добавляют 50 мкл концентрированной HCl и несильно нагревают с обратным холодильником. Через 1,5 ч реакционную среду концентрируют в вакууме. Остаток хроматографируют на диоксиде кремния, элюируя смесьюCHCl3/AcOEt (90/10; об./об.). Получают 465 мг желтого твердого вещества, идентифицированного методом масс-спектрометрии, МН+=550. Пример 6. Соединение 12.N-2-Аминометил)-1,3-бенздиоксол-5-ил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-7 ил)-N'-(трет-бутил)карбамид. К 530 мг нитрильного производного предыдущего примера, растворенного в 25 мл ТГФ, при -10 С за 30 мин добавляют 1,9 мл раствора 1 М LiAlH4 в ТГФ. Через 15 мин после окончания добавления приливают 1,5 мл AcOEt, затем 10 мл насыщенного раствора NH4Cl, затем позволяют температуре подняться. После разбавления AcOEt, промывают водой, затем насыщенным NaCl. После сушки органическую фазу концентрируют в вакууме и остаток хроматографируют на диоксиде кремния, элюируя смесьюCHCl3/MeOH (95/5 об./об.). Получают 165 мг желтого твердого вещества, идентифицированного методом масс-спектрометрии, МН+=554. Пример 7. Соединение 19. трет-Бутилкарбоксилат 2-5-7-трет-бутиламино)карбонил)амино)-6-(2,6-дихлорфенил)пиридо[2,3-d]пиримидин-2-ил)амино-1,3-бенздиоксол-2-ил)карбонил)гидразина. К 0,645 г продукта, полученного в синтезе 1, и 0,63 г продукта, полученного в синтезе 16.2, в 25 млEtOH добавляют 0,025 мл концентрированной HCl и перемешивают 5 ч при 70 С. Реакционную среду упаривают и остаток извлекают CHCl3, который промывают водой, насыщенным раствором NaHCO3,водой, насыщенным NaCl, затем сушат и упаривают в вакууме; остаток хроматографируют на диоксиде кремния. Получают 450 мг желтого твердого вещества, идентифицированного методом массспектрометрии, МН+= 683. Пример 8. Соединение 20.N-(трет-Бутил)-N'-6-(2,6-дихлорфенил)-2-2-гидразинкарбонил)-1,3-бенздиоксол-5 ил)амино)пиридо[2,3-d]пиримидин-7-ил)карбамид. Соединение предыдущего примера в количестве 360 мг перемешивают 45 мин в смеси 4 мл CH2Cl2 и 14 мл ТФУ (TFA); реакционную среду упаривают в вакууме; остаток извлекают CHCl3, который промывают H2O, 15%-ным водным раствором Na2CO3, H2O, насыщенным раствором NaCl; после сушкиCHCl3 выпаривают в вакууме, затем остаток растирают в простом эфире, отфильтровывают. Получают 225 мг желтого твердого вещества, МН+=583. Пример 9. Соединение 34. При комнатной температуре 431 мг соединения 33 перемешивают 30 мин в 5 мл ДХМ и 5 мл ТФУ. После выпаривания остаток извлекают смесью хлороформ/вода и доводят до pH 9 добавлением 15%-ного водного раствора Na2CO3. Органическую фазу декантируют, промывают водой, затем насы- 17011406 щенным раствором NaCl, сушат и концентрируют при пониженном давлении. Собирают 116 мг твердого вещества. [М+Н]+=568. Пример 10. Соединение 35. Стадия 1. Стадию осуществляют согласно методике, описанной в J. Organometall. Chem., 1996, 507, 1-21. Стадия 2. Стадию осуществляют согласно J. Med. Chem. 1988, 31, 84-91. Стадия 3. К 2,91 г продукта, полученного на предыдущей стадии, в 40 мл метанола добавляют при 8 С за 35 мин 800 мг боргидрида натрия. Через 50 мин реакционную среду выливают в 150 мл вода/лед плюс 400 мл этилацетата; после 5 мин перемешивания проводят декантацию и промывают органическую фазу 5%-ным раствором KHSO4/K2SO4, водой, насыщенным раствором NaCl; после сушки и выпаривания органической фазы выделяют твердое вещество коричневого цвета; m=2,25 г. Стадия 4. К 2,24 г предыдущего спирта в 60 мл ДХМ последовательно добавляют 1,43 г триэтиламина, затем за 25 мин 1,67 г метансульфонилхлорида. Через 45 мин реакционную среду разбавляют 100 мл ДХМ; промывают 2 раза смесью воды со льдом и 1 раз насыщенным раствором NaCl, органическую фазу сушат и концентрируют в вакууме. Получают твердое вещество каштанового цвета, m=3,01 г. Стадия 5. Смесь 3,0 г продукта, полученного на стадии 4, и 1,82 г азида натрия нагревают в течение 7,5 ч в 20 мл ДМФ при 65 С. Затем реакционную среду выливают в 75 мл смеси воды со льдом и 300 мл простого эфира. Органическую фазу выделяют, промывают несколько раз водой, затем насыщенным растворомNaCl. Затем органическую фазу сушат и концентрируют в вакууме с получением твердого вещества каштанового цвета, m=2,20 г. Стадия 6. К 2,19 г продукта, полученного на стадии 5, растворенного в 50 мл этилацетата, за 15 мин добавляют 4,33 г трифенилфосфина, затем через 10 мин 1,8 мл воды за 2 мин. Реакционную среду перемешивают в течение 1 ч 40 мин при 60 С, затем разбавляют 150 мл этилацетата. Полученный таким образом раствор промывают 2 раза водой, 1 раз насыщенным раствором NaCl, сушат, упаривают. Остаток после выпаривания растворяют в смеси 50 мл этилацетата и 50 мл диэтилового эфира и экстрагируют 2 раза 50 мл 1 н. HCl. Кислые водные фазы объединяют и экстрагируют смесью 25 мл этилацетата и 25 мл диэтилового эфира, затем приводят в контакт с 300 мл этилацетата и доводят pH до 9 при помощи 10 н. NaOH. После декантации органическую фазу промывают водой, насыщенным раствором NaHCO3, водой, затем насыщенным раствором NaCl. Остаточную органическую фазу сушат, затем концентрируют в вакууме. Получают масло, m=1,40 г. К 1,39 г амина, полученного на стадии 6, в 35 мл ДХМ при 5 С в течение 10 мин добавляют 0,35 г триэтиламина, затем 1,75 г Boc2O. После перемешивания в течение 1 ночи при комнатной температуре реакционную среду разбавляют 150 мл ДХМ, промывают водой, 5%-ным раствором KHSO4/K2SO4, водой, затем насыщенным раствором NaCl. После сушки и выпаривания ДХМ полученное твердое вещество растворяют в минимальном количестве диэтилового эфира затем добавляют гептан до полного осаждения. Получают твердое вещество: m=1,90 г. ЯМР: 1,30 м.д. : с : 9 Н; 3,40-3,55 м.д. : мт : 2 Н; 6,55 м.д. : т : 1 Н; 7,00 м.д. : т : 1 Н; 7,15-7,30 м.д. : мт : 2 Н; 7,55 м.д. : д : 1 Н. Стадия 8. К 0,60 г продукта, полученного на стадии 7, в 25 мл ТГФ добавляют 1,96 г порошкообразного цинка, затем при -3 С в течение 25 мин 2 мл АсОН. По окончании добавления реакционной среде дают возвратиться к комнатной температуре. Через 1 ч 15 мин реакционную среду фильтруют, фильтрат разбавляют 150 мл этилацетата и 30 мл воды, pH доводят до 9 добавлением 15%-ного раствора Na2CO3. После декантации органическую фазу выделяют, промывают насыщенным раствором NaHCO3, водой, затем насыщенным раствором NaCl. Органическую фазу выделяют, сушат, затем концентрируют при пониженном давлении. Получают 0,53 г вязкого желтого масла. ЯМР: 1,35 м.д. : с : 9 Н; 3,30 м.д. : мт : 2 Н; 4,80 м.д. : с : 2 Н; 6,05 м.д. : т : 1 Н; 6,10-6,25 м.д. : мт : 2 Н; 6,50 м.д. : т : 1 Н; 7,10 м.д. : т : 1 Н. Стадия 9. Связывание, для получения соединения 35, в стандартных условиях, таких как описанные перед этим. Пример 11. Соединение 45. К 1 г соединения 12, растворенного в 15 мл метанола и 2 мл ДМФ, добавляют 1,035 мл ДИПЭА, затем 0,675 г формамидинилсульфокислоты (H2N-C(=NH)-SO3H). После перемешивания в течение ночи при 25 С к реакционной среде добавляют воду, образовавшийся осадок отфильтровывают, промывают водой, затем сушат при пониженном давлении. Неочищенный продукт подвергают импульсной хроматографии на силикагеле с градиентом MeOH, от 10 до 50%, в ДХМ. Получают 170 мг твердого вещества,превращенного в соль в соотношении моль/моль H2SO4. MC (MS): МН+=596. Пример 12. Соединение 39. Синтез 39.1. К 555 мг соединения 9 (пример 4) в 10 мл ДХМ при 5 С добавляют 0,11 мл пиридина, затем за 15 мин 0,20 мл ангидрида трифторуксусной кислоты, разведенных в 2 мл ДХМ. Через 45 мин реакционную среду разбавляют 50 мл ДХМ, промывают последовательно смесью воды со льдом, водой, 5%-ным раствором KHSO4/K2SO4, водой, затем насыщенным раствором NaCl. Органическую фазу потом сушат и концентрируют при пониженном давлении. Получают 593 мг твердого вещества. ЯМР 1 Н: 1,40 м.д. : с : 9 Н; 4,65 м.д. : уш.с : 2 Н; 6,50 м.д. : т : 1 Н; 6,90 м.д. : д : 1 Н; 7,40 м.д. : уш.д : 1 Н; 7,50-7,70 м.д. : массив : 3H; 8,00 м.д. : уш.с : 1 Н; 8,05 м.д. : с : 1 Н; 8,20 м.д. : уш.с : 1 Н; 9,00 м.д. : с : 1 Н; 10,15 м.д. : с : 1 Н; 10,70 м.д. : с : 1 Н. Синтез 39.2. К 579 мг соединения полученного на стадии 39.1, в 10 мл ДХМ добавляют 0,17 мл морфолина. Реакционную среду перемешивают 3 ч 30 мин при комнатной температуре, затем систему упаривают при пониженном давлении. Остаток очищают импульсной хроматографией на силикагеле с градиентомAcOEt, от 0 до 20%, в ДХМ. Собирают 300 мг желтого порошка. МС: МН+=624. Пример 13. Соединения 40-43. Соединения с 40 по 43 получают тем же самым образом, что соединение 39, описанное в примере 12, используя соединение, полученное на стадии 39.1 и заменяя морфолин стадии 39.2, соответственно,трет-бутиламином, 1-БОК-пиперазином (БОК (для текста) - Boc (для химических формул и схем реакции), циклопропиламином, цис-2,6-диметилпиперидином. Пример 14. Соединение 44. Соединение 44 получают удалением защитной группы из соединения 41, полученного в примере 13, способом, описанным перед этим, использующим ТФУ.- 19011406 Пример 15. Соединения 46-48. Соединение 12 вводят в реакцию с кислотой в активированной форме, например, ангидрида, хлорида кислоты, кислота + связывающий агент, например, ДЦКДИ, БОФ. Так, соединения с 46 по 48 получают в результате реакции между соединением 12 и, соответственно, уксусным ангидридом, бензоилхлоридом и циклопропанкарбонилхлоридом. Пример 16. Соединение 49. Охлаждают до 5 С 444 мг соединения 12 в 12 мл ацетонитрила. Добавляют 0,14 мл триэтиламина,затем 0,075 мл метансульфонилхлорида. Через 40 мин перемешивания при 25 С реакционную среду извлекают AcOEt. Органическую фазу промывают водой, промывают насыщенным раствором NaCl, сушат и упаривают при пониженном давлении. Остаток очищают импульсной хроматографией на силикагеле с градиентом от 0 до 5% метанола в хлороформе. Получают 220 мг твердого вещества. МС: МН+=632. Пример 17. Соединение 50. К 284 мг соединения 4 (пример 2) в 5 мл ДМФ добавляют 40 мг трет-бутиламина, 71 мг диизопропиламина и 176 мг тетрафторбората О-(1 Н-бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (ТБТУ)(TBTU). Реакционную среду перемешивают 1 ч при 25 С затем разбавляют AcOEt. Органическую фазу последовательно промывают водой, насыщенным раствором NaHCO3, водой, затем насыщенным раствором NaCl. Органическую фазу сушат, упаривают при пониженном давлении и остаток очищают импульсной хроматографией на силикагеле с градиентом от 0 до 5% метанола в ДХМ. Получают 196 мг желтого твердого вещества. МС: МН+=624. Пример 18. Соединения 51-54. Соединения с 51 по 54 получают тем же самым образом, что соединение 50, исходя из соединения 4 и заменяя трет-бутиламин, соответственно, на циклопропиламин, пирролидин, N-изопропилметиламин и метиламин. Пример 19. Соединение 62. В 25 мл абсолютного спирта, содержащего 0,02 мл концентрированной HCl, при 70 С в течение 3 ч перемешивают 819 мг соединения, описанного в синтезе 1, и 0,314 мг 6-амино-2,3 дигидробензо[b]фурана, который может быть получен согласно Eur. J. Med. Chem. Chimica Therapeutics,1977, Vol. 12, 231-235. После охлаждения осадок отфильтровывают, промывают теплым MeOH, затемEt2O. Получают 637 мг желтого твердого вещества, плавящегося при 197 С. МС: МН+=523. Пример 20. Соединение 69. Стадия 1. К 3 г продукта, полученного на стадии 1.4, в 25 мл ДМФ за 10 минут добавляют 432 мг 60%-ногоNaH. Через 30 мин при перемешивании добавляют 1,40 г этилизоцианатацетата за 10 мин, затем реакционную среду оставляют на 3,5 ч при перемешивании при комнатной температуре. Реакционную среду экстрагируют AcOEt, последовательно промывают водой, 5%-ным раствором KHSO4/K2SO4, водой и насыщенным раствором NaCl. Органическую фазу сушат, концентрируют в вакууме и полученный неочищенный продукт очищают хроматографией на силикагеле, элюент: CHCl3/AcOEt 85/15 об./об. с получением 1,52 г ожидаемого продукта. Стадия 2. Продукт, полученный на стадии 1, окисляют метахлорпербензойной кислотой (МХПБК) (МСРВА) согласно способу, описанному в синтезе 1.6. Получают 900 мг ожидаемого продукта в форме бежевого твердого вещества. При 65 С в течение 6 ч в присутствии 15 мл EtOH и 0,06 мл концентрированной HCl нагревают 798 мг продукта стадии 2 и 295 мг 1,3-дигидро-2-бензофуран-5-амина. Реакционную среду разбавляютCHCl3 и водой, затем доводят водную фазу до pH 9 добавлением насыщенного раствора NaHCO3. После декантации органическую фазу выделяют, промывают водой, затем насыщенным раствором NaCl, сушат и концентрируют при пониженном давлении. Продукт перекристаллизовывают из AcOEt. Получают 0,67 г желтого твердого вещества. Стадия 4. В течение 5 ч 0,66 г сложного эфира, полученного на стадии 3, в 25 мл этанола и 2 мл ДМФ обрабатывают 1,5 мл 2 н. NaOH. Реакционную среду разбавляют CHCl3, затем доводят до pH 4 добавлением 1 н.NaCl, сушат и концентрируют при пониженном давлении с получением 0,61 г желтого порошка. Стадия 5. В течение 1 ч 15 мин в 2,5 мл ДМФ перемешивают 105 мг продукта, полученного на стадии 4, 16 мг трет-бутиламина, 28 мг ДИПЭА (диизопропилэтиламин) и 70 мг ТБТУ. Затем реакционную среду экстрагируют CHCl3, органическую фазу последовательно промывают водой, 5%-ным растворомKHSO4/K2SO4, насыщенным раствором NaCl, затем сушат и упаривают при пониженном давлении. Неочищенный продукт очищают хроматографией на диоксиде кремния, элюируя смесью CHCl3/MeOH, 94/6 об./об. Получают 90 мг ожидаемого продукта в форме желтого твердого вещества. МС: МН+=580. Пример 21. Соединения 31 и 32. Соединение 12, полученное в примере 6, может являться объектом для разделения его энантиомеров хроматографией на хиральной неподвижной фазе следующим образом:- 21011406 Исходя из 10 г рацемической смеси, после разделения получают 4,147 г правовращающего оптического изомера (соединение 31) и 4,077 г левовращающего оптического изомера (соединение 32). Табл. 3 и 4, следующие ниже, иллюстрируют химическое строение и физические свойства некоторых примеров согласно изобретению. В упомянутых таблицах Me, Et, iPr и tBu обозначают, соответственно, метильную, этильную, изопропильную и трет-бутильную группы, и Boc обозначает третбутоксикарбонильную группу. Если не указано противоположное, продукты, содержащие асимметрический атом углерода, получены в форме рацемической смеси. Таблица 3
МПК / Метки
МПК: A61K 31/519, C07D 417/04, A61P 35/02, A61P 35/00
Метки: производные, получение, пиридо-пиримидина, применение, терапии
Код ссылки
<a href="https://eas.patents.su/30-11406-proizvodnye-pirido-pirimidina-ih-poluchenie-ih-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиридо-пиримидина, их получение, их применение в терапии</a>
Производные 4-арилморфолин-3-она, их получение и их применение в терапии

Номер патента: 11035
Опубликовано: 30.12.2008
Авторы: Эмон -Альт Ксавье, Проиетто Винченцо
МПК: A61K 31/537, A61P 25/00, A61P 1/00...
Метки: терапии, производные, применение, 4-арилморфолин-3-она, получение
Формула / Реферат:
1. Соединение формулы (I) в которой Ar обозначает фенил, дважды замещенный атомом галогена; R1 обозначает фенил, не замещенный или замещенный один или два раза атомом галогена; R2 обозначает пиридил; фенил, не замещенный или замещенный один или два раза одним или двумя заместителями, независимо выбранными из атома галогена, (C1-C4)алкила, (C1-C4)алкокси, трифторметильной группы, трифторметоксигруппы; кроме того, R2 может обозначать...
Производные пиримидина для использования в качестве лигандов ваниллоидных рецепторов и их применение для терапии боли

Номер патента: 10265
Опубликовано: 30.06.2008
Авторы: Доерти Элизабет М., Бэлан Ченера, Норман Марк Г., Вонг Ху-Линг, Кэтон Джоди, Фэлси Джеймс Р., Гор Виджей Кешев
МПК: A61K 31/506, C07D 401/12, A61P 29/00...
Метки: ваниллоидных, использования, рецепторов, боли, качестве, терапии, лигандов, производные, пиримидина, применение
Формула / Реферат:
1. Соединение, обладающее структурой или его любая фармацевтически приемлемая соль, где R2 является независимо частично замещённым или незамещённым 8-, 9-, 10- или 11-членным бициклическим кольцом, содержащим 1, 2, 3 или 4 атома N, О и S, где атомы углерода кольца замещены 0, 1 или 2 оксогруппами и кольцо замещено 0, 1, 2 или 3 заместителями, выбранными из ряда групп: С1-8алкил-, С1-4галогеналкил-, галоген-, циано-, нитро-, -C(=O)Rb,...
Производные тетрагидроизохинолилсульфонамидов, их получение и применение в терапии

Номер патента: 10234
Опубликовано: 30.06.2008
Авторы: Диас Мартин Хуан Антонио, Химинес Баргуэно Мария Долорес
МПК: A61K 31/4725, A61K 31/472, A61K 31/496...
Метки: получение, тетрагидроизохинолилсульфонамидов, терапии, производные, применение
Формула / Реферат:
1. Соединение формулы I где n может принимать значения от 1 до 6; -(С)n- представляет собой C1-6-алкилиден, при необходимости замещенный заместителями в числе от 1 до 4, выбранными из атома галогена, гидрокси, нитро, циано, амино, C1-3-моноалкиламино, C2-6-диалкиламино или C1-3-алкокси; R1 представляет собой атом водорода; C1-6-алкил; R2 представляет собой атом водорода; C1-6-алкил или C3-6-циклоалкил, при необходимости замещенные заместителями...
Производные 5-(пиридин-3-ил)-1-азабицикло[3.2.1]октана, их получение и применение в терапии

Номер патента: 7793
Опубликовано: 27.02.2007
Авторы: Галли Фредерик, Леклерк Одиль, Лочид Алистер, Неделек Ален
МПК: A61K 31/439, A61P 25/00, C07D 209/00...
Метки: применение, терапии, 5-(пиридин-3-ил)-1-азабицикло[3.2.1]октана, получение, производные
Формула / Реферат:
1. Соединение в форме чистого энантиомера или в форме смеси энантиомеров, соответствующее общей формуле (I) где R представляет атом галогена или фенильную группу, замещенную одной или двумя группами, выбранными из атома галогена или группы (С1-C6)алкил, (C1-C6)алкокси, нитро, трифторметил, трифторметокси, гидрокси, ацетил или метилендиокси, или группу пиперидинил, или морфолин-4-ил, или пирролидин-1-ил, или азетидин-1-ил, или азепин-1-ил, или...
Получение арилалкилкарбаматных производных и их применение в терапии

Номер патента: 8801
Опубликовано: 31.08.2007
Авторы: Раве Антуан, Хурнер Кристан, Альмарио Гарсия Антонио, Абуабделла Ахмед
МПК: A61K 31/33, C07C 269/00, A61K 31/27...
Метки: получение, терапии, применение, производных, арилалкилкарбаматных
Формула / Реферат:
1. Соединение формулы (I) в которой n представляет собой целое число в интервале от 1 до 7; А выбран из одной или более чем одной группы X, Y и/или Z; X представляет собой С1-2-алкиленовую группу, возможно замещенную одной или более чем одной С1-12-алкильной, С3-7-циклоалкильной или С3-7-циклоалкил-С1-6-алкиленовой группой; Y представляет собой либо С2-алкениленовую группу, возможно замещенную одной или более чем одной С1-12-алкильной,...
Предыдущий патент: Способ получения газообразных диазоалканов
Случайный патент: Соль производного нафтиридинкарбоновой кислоты