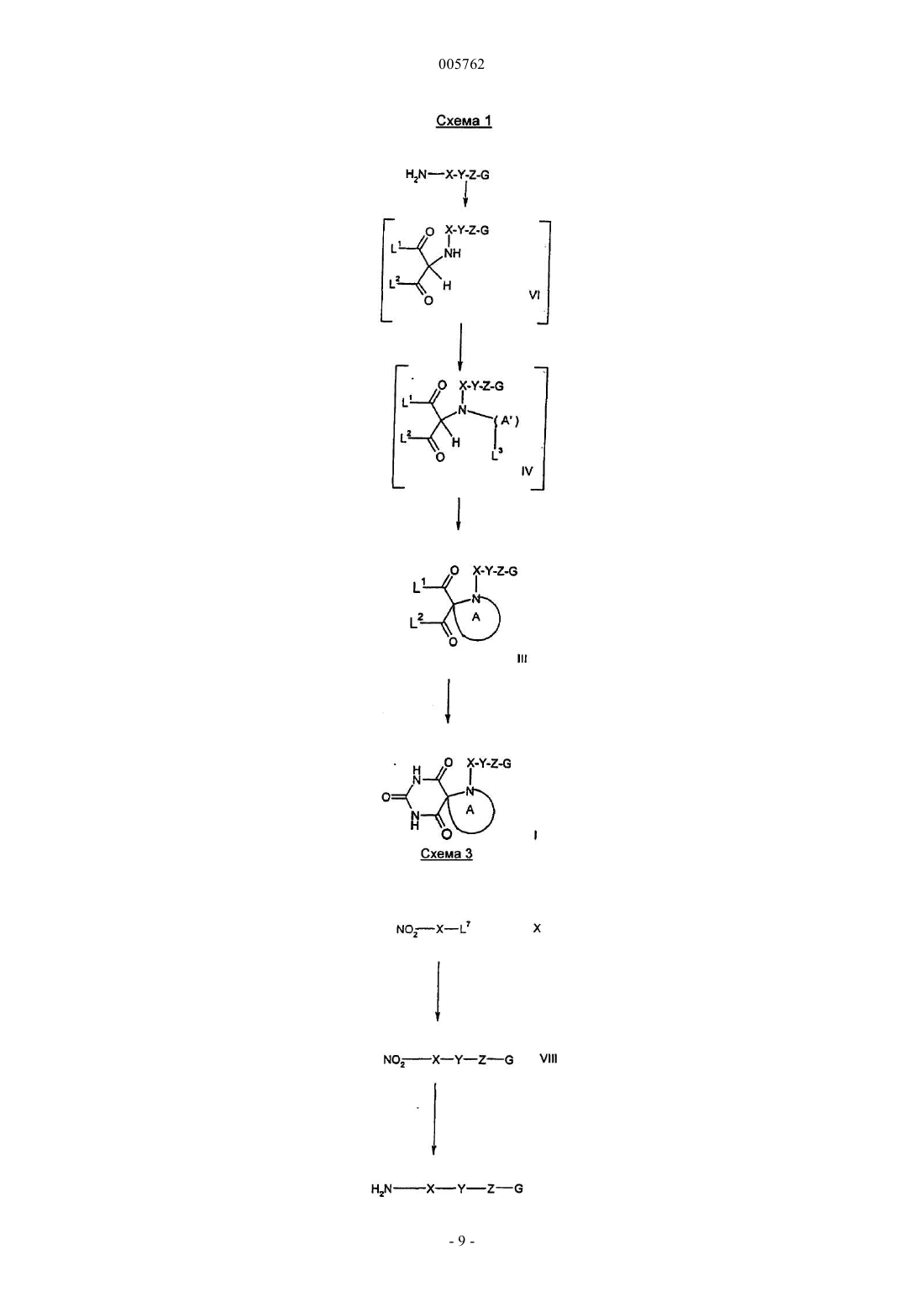
Спиро-пиримидин-2,4,6-трионовые ингибиторы металлопротеиназы
Номер патента: 5762
Опубликовано: 30.06.2005
Авторы: Ноу Марк Карл, Уайтс Мартин Джеймс, Бронк Брайан Скотт


























Формула / Реферат
1. Соединение формулы

где указанное A выбрано из группы, состоящей из

где каждый из R1, R2, R3, R4, R5 и R6 независимо представляет собой водород;
X представляет собой пиридинил;
Y представляет собой кислород;
Z представляет собой фенил;
G представляет собой R15-(CR16R17)p-, где G представляет собой заместитель по любому кольцевому атому углерода Z, способному к образованию дополнительной связи, и ориентирован в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y;
p представляет собой целое число от 0 до 1;
R15 независимо выбран из группы, состоящей из галогено, -CN, R18- и R19-(C=O)-(NR21)-;
каждый из R16 и R17 независимо представляет собой водород;
R18, R19 и R21 независимо выбраны из группы, состоящей из водорода, (C1-C4)алкила, (C3-C8)циклоалкила, оксадиазолила и пиразолила;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где указанный G представляет собой R15-(CR16R17)p-, где p равно 0.
3. Соединение по п.1, где указанный G представляет собой R15-(CR16R17)p-, где p представляет собой целое число 1.
4. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из
1-[6-(4-фторфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона;
1-[6-(4-фторфенокси)пиридин-3-ил]-1,8,10-триазаспиро[5,5]ундекан-2,7,9,11-тетраона;
4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензонитрила;
1-[6-(4-[1,3,4]оксадиазол-2-илфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона;
1-[6-(4-этилфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона;
N-{4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензил}ацетамида;
N-{4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензил}пропионамида;
N-{4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензил}бутирамида;
пентановой кислоты 4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензиламида;
циклобутанкарбоновой кислоты 4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензиламида;
1-[6-(4-бромфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона;
1-[6-(4-пиразол-1-илметилфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона
и их фармацевтически приемлемых солей.
5. Фармацевтическая композиция для лечения состояния, выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств, иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств, кожных расстройств и рака, у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель.
6. Способ лечения состояния, выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств, иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств, кожных расстройств и рака, у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1, эффективное при лечении такого состояния.
7. Фармацевтическая композиция для лечения состояния, которое можно лечить путем ингибирования матриксных металлопротеиназ, у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель.
Текст
005762 Настоящее изобретение относится к 5-спиропиримидин-2,4,6-трионовым ингибиторам металлопротеиназы и к фармацевтическим композициям, и способам лечения воспаления, рака и других расстройств. Соединения по настоящему изобретению представляют собой ингибиторы цинковых металлоэндопептидаз, особенно тех, которые принадлежат к классу матриксных металлопротеиназ (также называемых ММР или матриксин). ММР подсемейство ферментов в настоящее время содержит семнадцать членов (ММР-1, ММР-2,ММР-3, ММР-7, ММР-8, ММР-9, ММР-10, ММР-11, ММР-12, ММР-13, ММР-14, ММР-15, ММР-16,ММР-17, ММР-18, ММР-19, ММР-20). ММР наиболее хорошо известны по их роли в регуляции обмена внеклеточных матриксных белков и как таковые играют важную роль в нормальных физиологических процессах, таких как репродукция, развитие и дифференцировка. Кроме того, ММР экспрессируются во многих патологических ситуациях, в которых имеет место аномальный обмен соединительной ткани. Например, было продемонстрировано, что ММР-13, фермент с сильной активностью при деградации коллагена типа II (основной коллаген в хряще), сверхэкспрессируется в остеоартритном хряще (Mitchell,et al., J. Clin. invest., 97,761 (1996. Другие ММР (ММР-2, ММР-3, ММР-8, ММР-9, ММР-12) также сверхэкспрессируется в остеоартритном хряще, и ожидается, что ингибирование некоторых или всех из этих ММР замедляет или блокирует ускоренную потерю хряща, типичную для суставных болезней, таких как остеоартрит или ревматоидный артрит. Известно, что различные комбинации ММР экспрессируются в различных патологических ситуациях. Как таковые, ингибиторы со специфическими селективностями для конкретных ММР могут быть предпочтительны для конкретных болезней. Ингибиторы матриксных металлопротеиназ хорошо известны в литературе. Ингибиторы ММР,представляющие собой гидроксамовую кислоту, приведены в публикации европейского патента 606046,опубликованного 13 июля 1994. На некоторые пиримидин-2,4,6-трионовые ингибиторы ММР ссылаются в РСТ публикации WO 98/58925, опубликованной 30 декабря 1998. РСТ публикация WO 00/47565, опубликованная 17 августа 2000, относится к некоторым арилзамещенным пиримидин-2,4,6-трионовым ингибиторам ММР. Heпредварительная заявка на патент США 09/635156, поданная 9 Августа 2000 (которая испрашивает приоритет к предварительной заявке на патент США 60/148547, поданной 12 Августа 1999) относится к гетероарилзамещенным пиримидин-2,4,6-трионовым ингибиторам ММР. Предварительная заявка на патент США, озаглавленная "Пиримидин-2,4,6-трионовые ингибиторы металлопротеиназ", поданная 26 октября 2000, относится к некоторым пиримидин-2,4,6-трионам. Барбитуровые кислоты и способы их получения хорошо известны в данной области техники, смотри, например, Goodman andGilman's, The Phamacoloqical Basis of Therapeutics." 345-382 (Eighth Edition, McGraw Hill, 1990). Каждая из вышеуказанных публикаций и заявок включена здесь путем ссылки целиком. Сущность изобретения Настоящее изобретение относится к соединениям формулы где каждый из R1, R2, R3, R4, R5 и R6 независимо представляет собой водород;G представляет собой заместитель по любому кольцевому атому углерода Z, способному к образованию дополнительной связи, и ориентирован в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y; р представляет собой целое число от 0 до 1;R15 независимо выбран из группы, состоящей из галогено, -CN, R18- и R19-(C=O)-(NR21)-; каждый из R16 и R17 независимо представляет собой водород;R18, R19 и R21 независимо выбраны из группы, состоящей из водорода, (С 1-С 4)алкила, (С 3-С 8)циклоалкила,оксадиазолила и пиразолила; или его фармацевтически приемлемая соль. Настоящее изобретение также относится к фармацевтически приемлемым солям присоединения кислот соединений формулы I. Кислоты, которые используют для получения фармацевтически приемлемых солей присоединения кислот вышеупомянутых основных соединений по настоящему изобретению,представляют собой те, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат,битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат (то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат. Настоящее изобретение также относится к солям присоединения основания формулы I. Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых солей основания тех соединений формулы I, которые являются кислыми по своей природе,представляют собой те, которые образуют нетоксичные соли основания с такими соединениями. Такие нетоксичные соли основания включают в себя те, которые получены от таких фармакологически приемлемых катионов, как катионы щелочных металлов (например калия и натрия) и катионы щелочноземельных металлов (например кальция и магния), аммония или соли присоединения водорастворимых аминов, таких как N-метилглюкамин (меглумин), и низшего алканоламммония, и другие соли основания фармацевтически приемлемых органических аминов, но не ограничены ими. Пунктирные линии, которые используются в каждом из гетероциклических колец "А" формул (а) и(b), относятся к возможным двойным связям. Точные положения возможных двойных связей для каждого из гетероциклических колец "А" формул (а) и (b) такие, как определено в описании. Везде, где пунктирная линия распространяется на два атома углерода, специалисту в данной области будет понятно, что два углерода являются тетравалентными, и что дополнительный заместитель(и) (то есть любой из R1, R2,R3, R4, R5 или R6) может отсутствовать. Термин "алкил", как используется здесь, если не указано иначе, включает в себя насыщенные моновалентные углеводородные радикалы, имеющие прямые, разветвленные группировки или их комбинации. Алкильные группы, где бы они не встречались, могут быть возможно замещены подходящим заместителем. Термин "циклоалкил", как используется здесь, если не указано иначе, включает в себя моно или бициклическое карбоциклическое кольцо (например, циклопропил, циклобутил, циклопентил, циклогексил,циклогептил, циклооктил, циклононил, циклопентенил, циклогексенил, бицикло[2,2,1]гептанил, бицикло[3,2,1]октанил и бицикло[5,2,0]нонанил и так далее); возможно содержащее 1 или 2 двойных связи и возможно замещенное подходящими заместителями в количестве от одного до трех, как определено ниже, такими как фтор, хлор, трифторметил, (С 1-С 4)алкокси, (С 6-С 10)арилокси, трифторметокси, дифторметокси или (С 1-С 4)алкил, более предпочтительно фтор, хлор, метил, этил и метокси. Термин "алкокси", как используется здесь, включает в себя О-алкильные группы, где "алкил" такой,как определено выше. Термин "галогено", как используется здесь, если не указано иначе, включает в себя фтор, хлор,бром или иод, предпочтительно фтор или хлор. Термин "арил", как используется здесь, если не указано иначе, включает в себя органический радикал, полученный от ароматического углеводорода путем удаления одного или более чем одного водорода, такой как фенил или нафтил, возможно замещенный подходящими заместителями в количестве от 1 до 3, такими как фторо, хлоро, циано, нитро, трифторметил, (С 1-С 6)алкокси, (С 6-С 10)арилокси, (С 3 С 8)циклоалкилокси, трифторметокси, дифторметокси или (С 1-С 6)алкил. Термин "гетероарил", как используется здесь, если не указано иначе, включает в себя органический радикал, полученный от ароматического гетероциклического соединения путем удаления одного или более чем одного водорода, такой как бензимидазолил, бензофуранил, бензофуразанил, 2 Н-1 бензопиранил, бензотиадиазин, бензотиазинил, бензотиазолил, бензотиофенил, бензоксазолил, хроманил,циннолинил, фуразанил, фуропиридинил, фурил, имидазолил, индазолил, индолинил, индолизинил, индолил, 3 Н-индолил, изоиндолил, изохинолинил, изотиазолил, изоксазолил, нафтиридинил, оксадиазолил,оксазолил, фталазинил, птеридинил, пуринил, пиразинил, пиридазинил, пиридинил, пиримидинил, пиразолил, пирролил, хиназолинил, хинолинил, хиноксалинил, татразолил, тиазолил, тиадиазолил, тиенил,триазинил и триазолил, причем указанный (С 1-С 10)гетероарил возможно замещен по любому из кольцевых атомов углерода, способных к образованию дополнительной связи, одним или двумя заместителями,независимо выбранными из F, Cl, Br, CN, ОН, (С 1-С 4)алкила, (С 1-С 4)перфторалкила, (С 1-С 4)перфторалкокси, (С 1-С 4)алкокси и (С 3-С 8)циклоалкилокси. Вышеупомянутые группы могут быть Сприсоединенные или N-присоединенные, где это возможно. Например, пирролил может представлять собой пиррол-1-ил (N-присоединенный) или пиррол-3-ил (С-присоединенный). Фраза "подходящий заместитель" предназначена для обозначения химически и фармацевтически приемлемой функциональной группы, то есть группировки, которая не устраняет ингибирующую актив-2 005762 ность соединений по изобретению. Такие подходящие заместители могут быть обычным образом выбраны специалистом в данной области. Иллюстративные примеры подходящих заместителей включают в себя галогено группы, перфторалкильные группы, перфторалкоксигруппы, алкильные группы, гидроксигруппы, оксо группы, меркаптогруппы, алкилтиогруппы, алкоксигруппы, арильные или гетероарильные группы, арилокси или гетероарилоксигруппы, аралкильные или гетероаралкильные группы, аралкокси или гетероаралкоксигруппы, карбоксигруппы, аминогруппы, алкил- и диалкиламиногруппы, карбамоильные группы, алкилкарбонильные группы, алкоксикарбонилыные группы, алкиламинокарбонильные группы, диалкиламинокарбонильные группы, арилкарбонильные группы, арилоксикарбонильные группы, алкилсульфонильные группы, арилсульфонильные группы и тому подобное, но не ограничены ими. Фраза "в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y", как используется здесь, если не указано иначе, предназначена для обозначения химически и фармацевтически приемлемой ориентации связи, соединяющей группу Z с G (Z-G связь), по отношению к связи, соединяющей группу Y с Z (Y-Z связь). Такая относительная ориентация может быть мета, когда Z-G связь находится в 1,3 положении по отношению к Y-Z связи. Другая относительная ориентация может быть пара, когда Z-G связь находится в 1,4 положении по отношению к Y-Z связи. Некоторые соединения формулы I содержат хиральные центры и, следовательно, существуют в различных энантиомерных формах. Данное изобретение относится ко всем оптическим изомерам, энантиомерам, диастереомерам и стереоизомерам соединений формулы I и их смесям. Соединения по изобретению также существуют в различных таутомерных формах. Данное изобретение относится ко всем таутомерам формулы I. Специалистам в данной области хорошо известно, что пиримидин-2,4,6-трионовые ядра существуют в виде смеси таутомеров в растворе. Различные соотношения таутомеров в твердой и жидкой форме зависят от различных заместителей на молекуле, а также от конкретной методики кристаллизации, используемой для выделения соединения. В одном воплощении изобретения указанный G представляет собойR15-(CR16R17)p-, где р равно 0. В еще одном воплощении изобретения указанный G представляет собой R15-(CR16R17)p-, где р представляет собой целое число 1. В другом воплощении изобретения гетероциклическое кольцо "А" имеет формулу (а). В другом воплощении изобретения гетероциклическое кольцо "А" имеет формулу (b). В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 0; R15 выбран из группы, состоящей из галогено, -CN и R18, где указанный R18 выбран из группы, состоящей из водорода, (С 1-С 4)алкила, (С 3-С 8)циклоалкила, оксадиазолила и пиразолила. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно целому числу от 0 до 1; R15 выбран из группы, состоящей из галогено, -CN, R18- и R19-(C=O)-(NR21)-; каждый из R16 иR17 независимо представляет собой водород; R18 выбран из группы, состоящей из водорода, (С 1 С 4)алкила, (С 3-С 8)циклоалкила, оксадиазолила и пиразолила. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1;R15 выбран из группы, состоящей из галогено, -CN и R18; каждый из R16 и R17 независимо представляет собой водород; причем указанный R18 выбран из группы, состоящей из водорода, (С 1-С 4)алкила, (С 3 С 8)циклоалкила, оксадиазолила и пиразолила. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1;R15 выбран из группы, состоящей из R18; каждый из R16 и R17 независимо представляет собой водород; причем указанный R18 выбран из группы, состоящей из водорода и (С 1-С 4)алкила. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1;R15 представляет собой R19-(C=O)-(NR21)-; каждый из R16 и R17 независимо представляет собой водород; каждый из R19 и R21 независимо выбран из группы, состоящей из водорода, (С 1-С 4)алкила, (С 3 С 8)циклоалкила, оксадиазолила и пиразолила. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 0, a G ориентирован в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 0, a G ориентирован в положении мета по отношению к точке присоединения Z кольца к Y. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1, a G ориентирован в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y. В другом воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1, a G ориентирован в положении мета по отношению к точке присоединения Z кольца к Y. В другом предпочтительном воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно О, R15 выбран из группы, состоящей из галогено, -CN, оксадиазолила и пиразолила. Более предпочтительно R15 представляет собой бромо, фторо, -CN или оксадиазолил, предпочтительно[1,3,4]оксадиазол-2-ил. В другом предпочтительном воплощении изобретения G представляет собой R15-(CR16R17)p-, где р-3 005762 равно 0 или 1, R15 представляет собой R18, каждый из R16 и R17 представляет собой водород, a R18 независимо представляет собой водород или (С 1-С 4)алкил, предпочтительно метил. В другом предпочтительном воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 0 или 1, причем G ориентирован в положении пара по отношению к точке присоединения Z кольца к Y. В другом предпочтительном воплощении изобретения G представляет собой R15-(CR16R1)p-, где р равно 1, R15 представляет собой R19-(C=O)-(NR21)-, каждый из R16 или R17 независимо представляет собой водород, R19 представляет собой (С 1-С 4)алкил, более предпочтительно метил, этил или бутил, либо (С 3 С 8)циклоалкил, более предпочтительно циклобутил, a R21 представляет собой водород. В другом предпочтительном воплощении изобретения G представляет собой R15-(CR16R17)p-, где р равно 1, R15 представляет собой 2-пиразолил; и каждый из R16 и R17 независимо представляет собой водород. Другие предпочтительные соединения по изобретению включают в себя соединения формулы I, где гетероциклическое кольцо "А" имеет формулу (а) или (b), как определено выше; X представляет собой пиридинил, наиболее предпочтительно, когда пиридинил вместе с кольцом "А" и группой Y-Z-G имеет формулу где Y представляет собой кислород. Другие предпочтительные соединения по изобретению включают в себя соединения формулы I, где гетероциклическое кольцо "А" имеет формулу (а) или (b), как определено выше; X представляет собой пиридинил, наиболее предпочтительно, когда пиридинил вместе с кольцом "А" и группой Y-Z-G имеет формулу (а") или (b"), как определено выше; Y представляет собой кислород; Z представляет собой фенил; G представляет собой R15-(CR16R17)p-, где р равно 1; R15 представляет собой 2-пиразолил; каждый изR16 и R17 независимо представляет собой водород; и где G ориентирован в положении пара по отношению к точке присоединения Z кольца к Y. Наиболее предпочтительные соединения по изобретению включают в себя соединения формулы I,где гетероциклическое кольцо "А" имеет формулу (а) или (b), как определено выше; X представляет собой пиридинил, наиболее предпочтительно, когда пиридинил вместе с кольцом "А" и группой Y-Z-G имеет формулу (а") или (b"), как определено выше; Y представляет собой кислород; Z представляет фенил; G представляет собой R15-(CR16R17)P-, где р равно О, R15 выбран из группы, состоящей из -CN, галогено и оксадиазолила; и где G ориентирован в положении пара по отношению к точке присоединения Z кольца K Y. Другие наиболее предпочтительные соединения по изобретению включают в себя соединения формулы I, где гетероциклическое кольцо "А" имеет формулу (а) или (b), как определено выше; X представляет собой пиридинил, наиболее предпочтительно, когда пиридинил вместе с кольцом "А" и группой YZ-G имеет формулу (а") или (b"), как определено выше; Y представляет собой кислород; Z представляет собой фенил; G представляет собой R15-(CR16R17)P-, где р равно 1, R15 представляет собой R19-(C=O)(NR21)-; каждый из R16 и R17 независимо представляет собой водород; R19 выбран из группы, состоящей из (С 1-С 4)алкила и (С 3-С 8)циклоалкила, такого как метил, этил, пропил, бутил или циклобутил; R21 выбран из группы, состоящей из водорода или (С 1-С 4)алкила; и где G ориентирован в положении пара в положении к точке присоединения Z кольца к Y. Другие соединения по изобретению выбраны из группы, состоящей из: 1-[6-(4-Фторфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; 1-[6-(4-Фторфенокси)-пиридин-3-ил]-1,8,10-триазаспиро[5,5]ундекан-2,7,9,11-тетраона; 4-[5-(2,6,8,10-Тетраоксо-1,7,9-триазаспиро[4,5]дец-1 -ил)-пиридин-2-илокси]-бензонитрила; 1-[6-(4-[1,3,4]Оксадиазол-2-ил-фенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10 тетраона; 1-[6-(4-Этилфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,51 декан-2,6,8,10-тетраона;-4 005762 Пентановой кислоты 4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)-пиридин-2-илокси]бензиламида; Циклобутанкарбоновой кислоты 4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)-пиридин 2-илокси]-бензиламида; 1-[6-(4-Бромфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; 1-[6-(4-пиразол-1-илметилфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; и их фармацевтически приемлемых солей. Настоящее изобретение также относится к фармацевтической композиции для лечения состояния,выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств,иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств, кожных расстройств и рака, у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к фармацевтической композиции для лечения состояния,которое можно лечить путем ингибирования матриксных металлопротеиназ, у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу лечения состояния, выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств, иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечнососудистых заболеваний, болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств,кожных расстройств и рака, у млекопитающего, включая человека, при котором указанному млекопитающему вводят количество соединения по п.1, эффективное при лечении такого состояния. Авторы настоящего изобретения также обнаружили, что возможно идентифицировать ингибиторы формулы I с характерной металлопротеазной активностью (предпочтительно ММР-13 ингибиторная активность). Одна группа предпочтительных ингибиторов формулы I, идентифицированных изобретателям, включает в себя те, которые селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1. Соединения по изобретению также проявляют селективность по сравнению с родственной группой ферментов, известных как репролизины, такие как ТАСЕ (TNF- (фактор некроза опухоли )конвертирующий фермент) и аггреканаза. Еще одна группа предпочтительных ингибиторов формулы I,идентифицированных изобретателями, включает в себя те, которые селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1 и ММР-14. Еще одна группа предпочтительных ингибиторов формулы I, идентифицированных изобретателями, включает в себя те, которые селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1 и 12. Еще одна группа предпочтительных ингибиторов формулы I, идентифицированных изобретателями, включает в себя те, которые селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1, 12 и 14. Еще одна группа предпочтительных ингибиторов формулы I, идентифицированных изобретателями, включает в себя те, которые селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1, 2, 3, 7, 9 и 14. Наиболее предпочтительные соединения по изобретению селективно ингибируют ММР-13 преимущественно по сравнению с ММР-1, 2, 3, 7, 9,12 и 14 и репролизинами млекопитающих. Термин "лечение", как он используется здесь, относится к реверсированию, облегчению, подавлению развития или предупреждению расстройства или состояния, к которым такой термин применим,либо одного или более чем одного симптома такого расстройства или состояния. Термин "лечение", как он используется здесь, относится к акту лечения, как "лечение" определено непосредственно выше."Расстройства соединительной ткани", как используется здесь, относится к таким расстройствам,как дегенеративная потеря хряща после травматического повреждения сустава, остеоартрит, остеопороз,болезнь Педжета, расшатывание искуственных суставных протезов, периодонтальное заболевание и гингивит."Деструкция суставного хряща", как используется здесь, относится к расстройствам соединительной ткани, приводящим к деструкции суставного хряща, предпочтительно повреждению сустава, реактивному артриту, острому пирофосфатному артриту (псевдоподагре), псориатическому артриту или болезни Стилла, более предпочтительно остеоартриту."Иммунологические/аллергические расстройства", как используется здесь, относится к таким расстройствам, как токсичность органного трансплантата, аллергические реакции, аллергическая контактная гиперчувствительность, аутоиммунные расстройства, такие как те расстройства, которые связаны с гранулематозным воспалением/реструктурированием ткани (таким как астма), иммуносупрессия и саркоид."Инфекционные заболевания", включая те, которые опосредованы вирусами, бактериями, грибами или микобактериальной инфекцией, как используется здесь, относятся к таким расстройствам, как септический артрит, СПИД, лихорадка, прионные заболевания, миастения гравис, малярия, сепсис, гемодинамический шок и септический шок."Респираторные заболевания", как используется здесь, относятся к таким расстройствам, как хроническое обструктивное легочное заболевание (включая эмфизему), острый респираторный дистресс синдром, астма, гипероксическое альвеолярное повреждение и идиопатический легочный фиброз и другие фиброзные болезни легких."Сердечно-сосудистые заболевания", как используется здесь, относятся к таким расстройствам, как атеросклероз, включая разрыв атеросклеротических бляшек, аневризма аорты, включая абдоминальную аневризму аорты и аневризму аорты головного мозга, застойная сердечная недостаточность, инфаркт миокарда и церебральный инфаркт, удар, церебральная ишемия, коагуляция и острофазный ответ, дилатация левого желудочка, пост-ишемическое реперфузионное повреждение, ангиофибромы, гемангиомы и рестеноз."Болезни глаз", как используется здесь, относятся к таким расстройствам, как аберрантный ангиогенез, глазной ангиогенез, глазное воспаление, кератоконус, синдром Шегрена, миопия, глазные опухоли, отторжение трансплантата роговицы, повреждение роговицы, неоваскулярная глаукома, изъязвление роговицы, рубцевание роговицы, дегенерация желтого пятна (включая связанную с возрастом дегенерацию желтого пятна (ARMD), включая как влажную, так и сухую форму), пролиферативная витреоретинопатия и ретролентальная фиброплазия."Расстройства центральной нервной системы (ЦНС)", как используется здесь, относятся к таким расстройствам, как травма головы, повреждение спинного мозга, воспалительные заболевания центральной нервной системы, нейродегенеративные расстройства (острое и хроническое), болезнь Альцгеймера,демиелирующие заболевания нервной системы, болезнь Хантингтона, болезнь Паркинсона, периферическая навропатия, боль, церебральная амилоидная ангиопатия, ноотропное усиление или усиление познания, боковой амиотрофический склероз, рассеянный склероз, мигрень, депрессия и анорексия."Заболевания печени/почек", как используется здесь, относятся к таким расстройствам, как нефротические синдромы, такие как гломерулонефрит и гломерулярная болезнь почек, протеинурия, цирроз печени и интерстициальный нефрит."Расстройства репродуктивного здоровья", как используется здесь, относятся к таким расстройствам, как эндометриоз, контрацепция (мужская/женская), дисменорея, нефункциональное маточное кровотечение, преждевременный разрыв плодного пузыря и выкидыш."Желудочные расстройства", как используется здесь, относятся к таким расстройствам, как анастомоз ободочной кишки и язвы желудка."Кожные расстройства", как используется здесь, относятся к таким расстройствам, как старение кожи, пролежни, псориаз, экзема, дерматит, радиационное повреждение, изъязвление ткани, декубитальные язвы (пролежни), врожденный буллезный эпидермолиз, аномальное заживление ран (местные или пероральные препараты), ожоги и склерит."Рак", как используется здесь, относится к таким расстройствам, как твердоопухолевый рак, включая рак ободочной кишки, рак молочной железы, рак легкого и рак простаты, инвазия опухоли, рост опухоли, опухолевый метастаз, рак ротовой полости и глотки (губы, языка, рта, глотки), пищевода, желудка,тонкого кишечника, толстого кишечника, прямой кишки, печени и желчных протоков, поджелудочной железы, гортани, легкого, кости, соединительной ткани, кожи, шейки матки, эндометрия, яичника, яичка,мочевого пузыря, почки и других уринарных тканей, глаза, головного мозга и центральной нервной системы, щитовидной железы и других эндокринных желез, болезнь Ходжкина, неходжкинские лимфомы,множественная миелома и гематопоэтические злокачественные образования, включая лейкемии и лимфомы, включая лимфоцитическую, гранулоцитическую и моноцитическую. Настоящее изобретение также включает в себя меченные изотопом соединения, которые идентичны таковым, приведенным в формуле I, но при этом один или более чем один атом замещены атомом,имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно встречаемого в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2 Н, 3 Н, 13 С, 14 С, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl, соответственно. Соединения по настоящему изобретению, их пролекарства и фармацевтически приемлемые соли указанных соединений или указанных пролекарств, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, входят в объем настоящего изобретения. Некоторые меченные изотопом соединения по настоящему изобретению,например такие, в которые включены такие радиоактивные изотопы, как 3 Н и 14 С, полезны в анализах распределения лекарства и/или субстрата в ткани. Тритированный, то есть 3 Н, и углерод-14, то есть 14 С,изотопы особо предпочтительны благодаря легкости их получения и обнаружения. Далее, замещение-6 005762 более тяжелыми изотопами, такими как дейтерий, то есть 2 Н, может дать некоторые терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например увеличенный период полураспада in vivo либо необходимость в пониженной дозировке, и, следовательно, могут быть предпочтительны в некоторых случаях. Меченные изотопом соединения формулы I по настоящему изобретению и их пролекарства могут обычно быть приготовлены путем осуществления процедур, описанных в схемах и/или примерах и получениях ниже, путем замещения легкодоступным меченным изотопом реагентом не меченного изотопом реагента. Данное изобретение также включает в себя фармацевтические композиции, содержащие пролекарства соединений формулы I. Данное изобретение также включает в себя способы лечения или предупреждения расстройств, которые можно лечить или предупреждать путем ингибирования матриксных металлопротеиназ или ингибирования репролизина млекопитающих, при которых вводят пролекарства соединений формулы I. Соединения формулы I, имеющие свободные амино, амидо, гидрокси, сульфонамидные или карбоксильные группы, могут быть превращены в пролекарства. Пролекарства включают в себя соединения, где аминокислотный остаток или полипептидная цепь из двух или более (то есть двух,трех или четырех) аминокислотных остатков ковалентно соединены через пептидные связи со свободными амидо, амино, гидрокси группами или группами карбоксильной кислоты соединений формулы I. Аминокислотные остатки включают в себя 20 встречающихся в природе аминокислот, обычно обозначаемых трехбуквенными символами, а также включают в себя 4-гидроксипролин, гидроксилизин, демозин, изодемозин, 3-метилгистидин, норвалин, бета-аланин, гамма-аминомасляную кислоту, цитруллин,гомоцистеин, гомосерин, орнитин и метионина сульфон. Пролекарства также включают в себя соединения, где карбонаты, карбаматы, амиды и алкильные эфиры ковалентно связаны с вышеуказанными заместителями формулы I через карбонил-углеродную боковую цепь пролекарства. Пролекарства также включают в себя димеры соединений формулы I. Специалисту в данной области будет понятно, что соединения по изобретению полезны при лечении широкого спектра заболеваний. Специалисту в данной области также будет понятно, что при использовании соединений по настоящему изобретению при лечении конкретного заболевания, соединения по изобретению можно комбинировать с различными существующими терапевтическими агентами, используемыми для такого заболевания. Для лечения ревматоидного артрита, соединения по изобретению можно комбинировать с такими агентами, как ингибиторы TNF-, такие как анти-TNF моноклональные антитела (такие как инфликсимаб, D2E7 и CDP-870) и TNF рецепторные иммуноглобулиновые молекулы (такие как этанерцепт), ICE(интерлейкин-1-конвертирующий фермент) ингибиторы, МЕКК 1 ингибиторы, СОХ-2 ингибиторы, такие как целекоксиб, рофекоксиб, валдекоксиб и эторикоксиб; низкие дозы метотрексата, лефунимида,стероидов, глюкозаминов, хондрозаминов/сульфатов, габапентина, А-агонистов, ингибиторы образования и высвобождения IL-1, рецепторные антагонисты IL-1, такие как Kineret, антагонисты CCR-1, гидроксихлорохин, d-пенициламин, ауранофин либо парентеральное или пероральное золото. Соединения по изобретению могут также применяться в комбинации с существующими терапевтическими агентами для лечения остеоартрита. Подходящие агенты для использования в комбинации включают в себя стандартные нестероидные противовоспалительные агенты (здесь и далее NSAID's),такие как пироксикам, диклофенак, пропионовые кислоты, такие как напроксен, флубипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, апазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин, ингибиторы СОХ-2, такие как целекоксиб, валдекоксиб, паракоксиб, эторикоксиб и рофекоксиб; анальгетики, стероиды, глюкозамины,хондрозамины/сульфаты, габапентин, А-агонисты, ингибиторы образования и высвобождения IL-1, антагонисты CCR-1, ингибиторы LTD-4, LTB-4 и 5-LO, ингибиторы киназы р 38 и внутрисуставные терапевтические средства, такие как кортикостероиды и гиалуроновые кислоты, такие как гиалган и синвиск. Соединения по настоящему изобретению могут также быть использованы в комбинации с противораковыми агентами, такими как эндостатин и ангиостатин, или цитотоксическими лекарствами, такими как адриамицин, дауномицин, цис-платина, этопосид, паклитаксель, доцетаксель, и алкалоидами, такими как винкристин, и антиметаболитами, такими как метотрексат. Соединения по настоящему изобретению могут также быть использованы в комбинации с сердечно-сосудистыми агентами, такими как блокаторы кальциевых каналов (такие как амлодипин и нифедипин), агенты, понижающие уровень липидов, такие как статины (такие как ловастатин, аторвастатин,правастатин и симвастатин), такими как адренэргические средства, как, например, доксазоцин и теразоцин; фибраты, бета-блокаторы, ингибиторы Асе (такие как каптоприл, лизиноприл, фозиноприл, эналаприл и квинаприл), ангиотензин-2 рецепторные антагонисты, такие как лосартан и ирбесартан; нитраты,препараты ССВ, диуретики, такие как препараты наперстянки и ингибиторы аггрегации тромбоцитов. Соединения по настоящему изобретению могут также быть использованы в комбинации со средствами для предотвращения разрыва бляшек, такими как статины, зитромакс, NSAIDs, включая аспирин, гепарин, урарфарин, абциксимаб, ТРА и ингибиторы тромбоцитов. Соединения по настоящему изобретению могут также быть использованы в комбинации с агентами для лечения удара, такими как антагонистыNIF, NHEI's и CCRIR. Соединения по настоящему изобретению могут также быть использованы в комбинации с ЦНС агентами, такими как антидепрессанты (таким как сертралин), антипаркинсонические лекарства, такие как депренил, карбадопа, L-дофа, агонисты дофаминовых рецепторов, такие какропинирол, перголид и прамипексол; ингибиторы МАОВ, такие как селеджилин и разаджилин, ингибиторы катехол-Ометилтрансферазы, такие как толкапон, ингибиторы А-2, ингибиторы обратного захвата дофамина,NMDA антагонисты, никотиновые агонисты, ингибиторы NK-1, дофаминовые агонисты и ингибиторы нейрональной синтазы оксида азота и лекарства против болезни Альцгеймера, такие как донепезил, такрин, ингибиторы СОХ-2, пропентофиллин или метрифонат. Соединения по настоящему изобретению могут также быть использованы в комбинации с агентами против остеопороза, такими как ролоксифен, дролоксифен, лазофоксифен или фозомакс, и иммунносупрессантными агентами, такими как FK-506 и рапамицин. Соединения по настоящему изобретению могут также быть использованы в комбинации с агентами для лечения респираторных заболеваний, такими как ингибиторы PDE-IV, стероиды, такие как флутиказон, триамцинолон, будесонид и беклометазон; антихолинэргические средства, такие как ипратропиум,симпатомиметики, такие как салметерол, альбутерол и Ксопенекс, противоотечные средства, такие как фексофенадин, лоратадин и цетиризин; лейкотриеновые антагонисты, такие как зафирлукаст и мотелукаст; и стабилизаторы тучных клеток, такие как зилеутон. Соединения по настоящему изобретению могут также быть использованы в комбинации с агентами для лечения кожных расстройств, такими как третиноин, изотретиноин, стероиды, такие как кортизон и мометазон, антибиотики, такие как тетрациклин, противогрибковые средства, такие как клотримазол,миконазол и флуконазол, и ингибиторы PDE-IV. Соединения по настоящему изобретению могут также быть использованы в комбинации с агентами для лечения диабета, такими как инсулин, включая человеческий или гуманизированный инсулин и ингалируемый инсулин, ингибиторы альдозоредуктазы, ингибиторы сорбитолдегидрогеназы, противодиабетические агенты, такие как бигуаниды, такие как метформин, глитазоны, ингибиторы глюкозидазы,такие как акарбоза, сульфонилмочевины, такие как глимепирид и глипизид; и тиазолидиндионы, такие как пиоглитазон, розиглизатон и троглиазон. Предпочтительные комбинации полезны для лечения побочных эффектов диабета, таких как ретинопатия, нефропатия и нейропатия, предпочтительно ретинопатия. Подробное описание изобретения Следующие реакционные Схемы иллюстрируют получение соединений по настоящему изобретению. Если не указано иначе, каждый из X, Y, Z, G, R1, R2, R3, R4, R5, R6, R15, R16, R17, R18, R19 и R21 в реакционных схемах и обсуждении, которое следует, определен как выше.-9 005762 Схема 1 относится к получению соединений формулы I. Ссылаясь на схему 1, соединения формулы I, где гетероциклическое кольцо "А" имеет формулы а-b (то есть соединения формул Ia-Ib соответственно) могут быть получены путем взаимодействия соединений формул IIIa-IIIb соответственно где L1 и L2 представляют собой уходящие группы, такие как алкокси, предпочтительно метокси, этокси или бензилокси, более предпочтительно метокси или этокси, с мочевиной формулы II (то есть, H2N(CO)-NH2) в присутствии подходящего основания в полярном растворителе. Подходящие основания включают в себя алкоксидные основания, такие как метоксид натрия, этоксид натрия или mpemбутоксид калия, предпочтительно этоксид натрия. Подходящие растворители включают в себя тетрагидрофуран, диметилформамид или спирты (такие как этанол), предпочтительно тетрагидрофуран или диметилформамид. Вышеуказанную реакцию проводят при температуре от примерно 20 до примерно 90 С,предпочтительно от примерно 50 до примерно 80 С, в течение периода времени от примерно 5 мин до примерно 8 ч. Соединения формул IIIa-IIIb, соответственно, могут быть получены путем взаимодействия соединения формул IVa-IVb соответственно где L1 и L2 представляют собой уходящие группы, такие как алкокси, предпочтительно метокси, этокси или бензилокси, более предпочтительно метокси или этокси, и где L3 представляет собой подходящую уходящую группу, такую как галогено, пара-толилсульфонилокси (OTs) или метилсульфонилокси (OMs),предпочтительно галогено, такую как бромо или иодо, с подходящим основанием в полярном растворителе. Подходящие основания включают в себя третичные амины, такие как триэтиламин. Другие подходящие основания включают в себя сильно щелочную макросетчатую смолу или смолу гелевого типа,такую как смола Amberlyst 400 (гидроксидная форма). Подходящие растворители включают в себя спиртовые растворители, предпочтительно этанол. Вышеуказанную реакцию можно проводить при температуре от примерно -10 С до примерно 50 С, предпочтительно примерно 20 С, в течение периода от примерно 6 до примерно 36 ч. Соединения формул IVa-IVb, соответственно, могут быть получены путем взаимодействия соединения формулы VI с соединением общей формулы L3-(A')-L4 (V) (то есть с соединениями формул Va-Vb соответственно) где каждый из L3 и L4 представляет собой подходящую уходящую группу, такую как галогено, паратолилсульфонилокси (OTs) или метилсульфонилокси (OMs). Предпочтительно L3 представляет собой галогено, такую как бромо, хлоро или иодо. Предпочтительно L4 представляет собой хлоро или фторо. Возможно, вышеупомянутую реакцию можно проводить в присутствии третичного аминного основания,такого как N,N-диметиланилин или пиридин, в присутствии подходящего растворителя, такого как углеводородный растворитель (бензол или толуол), тетрагидрофурана или метиленхлорида. Вышеупомянутую реакцию проводят при температуре от примерно 20 до примерно 90 С, предпочтительно от пример- 10005762 но 50 до примерно 80 С, в течение периода времени от примерно 30 мин до примерно 6 ч. Предпочтительно, вышеупомянутую реакцию проводят в ароматическом углеводородном растворителе, таком как бензол или толуол, в отсутствии вышеупомянутого основания. Соединение формулы VI может быть получено путем взаимодействия соединения формулы H2N-XY-Z-G с соединением формулы VII где L1 и L2 представляют собой уходящие группы, такие как метокси, этокси или бензилокси, предпочтительно этокси; a L5 представляет собой уходящую группу, такую как галогено, пара-толилсульфонилокси(OTs) или метилсульфонилокси (OMs), предпочтительно галогено, наиболее предпочтительно хлоро или бромо. Вышеупомянутую реакцию можно осуществлять либо в чистом виде, либо в присутствии подходящего растворителя, предпочтительно в чистом виде, в присутствии подходящего основания. Подходящие растворители включают в себя тетрагидрофуран или диметилформамид. Подходящие основания включают в себя слабое третичное аминное основание, предпочтительно третичные анилиновые основания, наиболее предпочтительно N,N-диметиланилин. Предпочтительно, вышеупомянутую реакцию проводят при температуре от примерно 23 до примерно 100 С, предпочтительно от примерно 50 до примерно 90 С, в течение периода времени от примерно 30 мин до примерно 24 ч. Соединения формулы H2N-X-Y-Z-G коммерчески доступны или могут быть приготовлены способами, хорошо известными специалистам в данной области. Альтернативно, соединения формулы H2N-XY-Z-G могут быть получены, как описано на схеме 3. Соединение формулы VII может быть приготовлено способами, хорошо известными в данной области техники, такими как те, которые описаны в РСТ публикации патента WO 98/58925 или изложены вThe Organic Chemistry of Drug Synthesis. D. Lednicer and L. A. Mitscher, Volume 1, pages 167 to 277, и приведенных там ссылках. Каждая из вышеуказанных публикаций и заявок включена здесь путем ссылки во всей ее полноте. Соединения формулы II коммерчески доступны или могут быть приготовлены способами, хорошо известными специалистам в данной области. Схема 3 относится к получению соединений формулы H2N-X-Y-Z-G, которые являются промежуточными соединениями, полезными в получении соединений формулы I, в схеме 1. Ссылаясь на схему 3,соединения формулы H2N-X-Y-Z-G могут быть получены путем взаимодействия соединения формулыVIII с восстанавливающим агентом, таким как хлорид олова (II), в присутствии подходящей кислоты,такой как соляная кислота, в полярном протонном растворителе. Подходящие растворители включают в себя спиртовой растворитель, воду или их смеси, предпочтительно смесь этанола и воды. Вышеуказанную реакцию можно проводить при температуре от примерно 40 до примерно 100 С, в течение периода от примерно 1 до примерно 12 ч. Альтернативно, соединения формулы H2N-X-Y-Z-G могут быть получены путем взаимодействия соединения формулы VIII с водородным газом, при давлении между атмосферным давлением и 50 psi(фунт/кв. дюйм, 344,74 КПа), в присутствии катализатора и полярного растворителя. Подходящие катализаторы включают в себя палладиевый или платиновый катализатор, предпочтительно катализатор Адамса (то есть оксид платины) или палладий, адсорбированный на угле. Подходящие растворители включают в себя спиртовой растворитель, предпочтительно метанол. Вышеуказанную реакцию можно проводить при температуре от примерно 20 до примерно 50 С, предпочтительно примерно 23 С, в течение периода от примерно 30 мин до примерно 6 ч. Соединение формулы VIII, где Y представляет собой кислород, может быть получено путем взаимодействия соединения формулы X, где группа L7 представляет собой фторо или хлоро, с соединением формулы:(IX) где Y представляет собой кислород, в присутствии основания в полярном апротонном растворителе. Подходящие основания включают в себя основание гидрид щелочного металла, предпочтительно гидрид натрия. Подходящие растворители включают в себя диметилформамид, тетрагидрофуран или 1,2 диметоксиэтан; предпочтительно диметилформамид. Вышеуказанную реакцию можно проводить при температуре от примерно 40 до примерно 140 С, предпочтительно от примерно 80 до примерно 120 С, в течение от примерно 1 до примерно 24 ч. Альтернативно, вышеуказанное соединение формулы VIII, где Y представляет собой кислород, может быть получено в присутствии основания гидроксида щелочного металла, предпочтительно гидроксида калия, возможно в присутствии межфазного катализатора, такого как четвертичная соль аммония или фосфония, предпочтительно тетрабутиламмония бромид, в ароматическом углеводородном растворителе. Предпочтительно растворитель представляет собой бензол или толуол. Вышеуказанную реакцию можно проводить при температуре от примерно 0 до примерно 120 С, предпочтительно примерно при- 11005762 23 С, в течение от примерно 1 до примерно 12 ч. Альтернативно, вышеуказанное соединение формулы VIII, где Y представляет собой кислород, может быть получено в так называемых условиях "сочетания Ульмана". В таких условиях, вышеуказанное соединение формулы VIII может быть получено путем взаимодействия соединения формулы X, где группа L7 представляет собой бромо или хлоро, с соединением формулы(IX) где Y представляет собой кислород, в присутствии основания и катализатора в полярном апротонном растворителе. Подходящие основания включают в себя карбонат щелочного металла или гидроксидное основание, предпочтительно карбонат калия. Подходящие катализаторы включают в себя катализатор меди (0), предпочтительно тонко измельченную порошкообразную медную бронзу. Подходящие растворители включают в себя диметилформамид или 1-метил-2-пирролидинон. Вышеуказанную реакцию можно проводить при температуре от примерно 80 до примерно 140 С, в течение от примерно 6 до примерно 24 ч. Соединения формул X и IX (то есть, соединения формул G-Z-Y-H, G-Z-W-Н или G-Z-Y-L9) являются либо коммерчески доступными, либо хорошо известны и могут быть получены способами, известными специалистам в данной области. Соединения формулы I, которые являются основными по своей природе, способны к образованию широкого разнообразия различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемы для введения животным, часто желательно на практике первоначально выделить соединение формулы I из реакционной смеси в виде фармацевтически неприемлемой соли, а затем просто превратить последнюю обратно в соединение свободного основания путем обработки щелочным реагентом, и последовательно превратить свободное основание в фармацевтически приемлемую соль присоединения кислоты. Соли присоединения кислоты основных соединений по настоящему изобретению легко получают путем обработки основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. При осторожном выпаривании растворителя получают желаемую твердую соль. Кислоты, которые используют для получения фармацевтически приемлемых солей присоединения кислоты основных соединений по настоящему изобретению, представляют собой те, которые образуют нетоксичные соли присоединения кислоты, то есть соли, содержащие фармакологически приемлемые анионы, такие как соли гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, лактат, цитрат или кислый цитрат, тартрат или битартрат, сукцинат, малеат,фумарат, глюконат, сахарат, бензоат, метансульфонат и памоат (то есть 1,1'-метилен-бис-(2-гидрокси-3 нафтоат. Те соединения формулы I, которые являются также кислыми по своей природе, способны к образованию солей оснований с различными фармакологически приемлемыми катионами. Примеры таких солей включают в себя соли щелочных металлов или щелочноземельных металлов и, в частности, соли натрия и калия. Эти соли получают с помощью общепринятых методик. Химические основания, которые используют в качестве реагентов для получения фармацевтически приемлемых солей оснований по настоящему изобретению, представляют собой те, которые образуют нетоксичные соли оснований с описанными здесь кислыми соединениями формулы I. Эти нетоксичные соли оснований включают в себя те,которые получены от таких фармакологически приемлемых катионов, как натрий, калий, кальций и магний, и так далее. Эти соли можно легко получать путем обработки соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, а затем упаривания полученного раствора до сухости, предпочтительно при пониженном давлении. Альтернативно, эти соли могут также быть получены путем смешивания растворов низших спиртов кислых соединений и желаемого алкоксида щелочного металла вместе, а затем упаривания полученного раствора досуха и тем же самым способом, как ранее. В любом случае, предпочтительно используют стехиометрические количества реагентов для того, чтобы гарантировать завершение реакции и максимальные выходы продукта. Биологические анализы Способность соединений формулы I или их фармацевтически приемлемых солей (которые здесь и далее также называют соединениями по настоящему изобретению) ингибировать металлопротеиназы или репролизины млекопитающих и, следовательно, демонстрировать их эффективность для лечения заболеваний, характеризующихся металлопротеиназной активностью, показана следующими in vitro и invivo тест-анализами. ММР анализы Селективные ингибиторы ММР-13 могут быть идентифицированы путем скрининга ингибиторов по настоящему изобретению с помощью ММР флуоресцентных анализов, описанных ниже, и отбора тех агентов, у которых ИК 50 для соотношения ММР-13/ММР-Х ингибирования составляет 100 или более и эффективностью менее чем 100 нМ, где ММР-Х относится к одному или более чем одному другому- 12005762 Неселективные ингибиторы коллагеназы, которые используются здесь, если не указано иначе, относятся к агентам, которые проявляют менее чем 100-кратную селективность для ингибирования активности ММР-13 фермента по сравнению с активностью ММР-Х фермента или эффективность более чем 100 нМ, как определено с помощью результатов ИК 50 из ММР-13/ММР-Х флуоресцентных анализов,описанных ниже. Способность ингибиторов коллагеназы ингибировать коллагеназную активность хорошо известна в данной области техники. Степень ингибирования конкретной ММР для некоторых соединений хорошо подтверждена документально в данной области техники, и специалисты в данной области техники должны знать, как нормализовать результаты анализа, отличного от тех анализов, о которых сообщается здесь. Следующие анализы могут быть использованы для идентификации ингибиторов матриксных металлопротеиназ. Ингибирование человеческой коллагеназы (ММР-1) Человеческую рекомбинантную коллагеназу активируют трипсином. Количество трипсина оптимизировано для каждой партии коллагеназы-1, но в типичной реакции используют следующее соотношение: 5 мкг трипсина на 100 мкг коллагеназы. Трипсин и коллагеназу инкубируют при комнатной температуре в течение 10 мин, затем добавляют пятикратный избыток (50 мг/10 мг трипсина) ингибитора трипсина из соевых бобов. Исходные растворы (10 мМ) ингибиторов готовят в диметилсульфоксиде, а затем разбавляют с использованием следующей схемы: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ Двадцать пять микролитров каждой концентрации затем добавляют в трехкратной повторности в соответствующие лунки 96-луночного микротитрационного планшета. Конечная концентрация ингибитора будет составлять 1:4 разведение после добавления фермента и субстрата. Позитивные контроли(фермент, нет ингибитора) расположены в лунках D7-D12, а негативные контроли (нет фермента, нет ингибиторов) расположены в лунках D1-D6. Коллагеназу-1 разбавляют до 240 нг/мл, и 25 мкл затем добавляют в соответствующие лунки микротитрационного планшета. Конечная концентрация коллагеназы в анализе составляет 60 нг/мл. Субстрат (DNP-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2) готовят в качестве 5 мМ исходного раствора в диметилсульфоксиде, а затем разбавляют до 20 мкМ в буфере для анализа. Анализ инициируют добавлением 50 мкл субстрата на лунку микротитрационного планшета с получением конечной концентрации 10 мкМ. Показания флуоресценции (360 нМ возбуждение, 460 нМ испускание) снимают в момент времени 0, а затем с 20 минутными интервалами. Анализ проводят при комнатной температуре с обычным временем анализа 3 ч. Флуоресценцию против времени затем наносят на график как для пустых образцов, так и для содержащих коллагеназу (данные от трехкратных определений усреднены). Точку времени, которая дает хороший сигнал (по меньшей мере, пятикратный по сравнению с пустым образцом) и которая находится на линейной части кривой (обычно около 120 мин) выбирают для определения величин ИК 50. Время ноль используют в качестве контроля для каждого соединения для каждой концентрации, и эти величины вычитают из 120-минутных данных. Данные наносят на график в виде концентрации ингибитора против % контроля (флуоресценция ингибитора, деленная на флуоресценцию коллагеназы отдельно х 100). Величины ИК 50 определяют из концентрации ингибитора, которая дает сигнал, который составляет 50% от контроля. Если сообщают, что величины ИК 50, чем 0,03 мкМ, тогда ингибиторы используют в анализе в концентрациях 0,3 мкМ, 0,03 мкМ и 0,003 мкМ. Ингибирование желатиназы (ММР-2) Человеческую рекомбинантную 72 кДа желатиназу (ММР-2, желатиназа А) активируют в течение 16-18 часов 1 мМ пара-аминофенил-ацетатом ртути (из свежеприготовленного 100 мМ исходного раствора в 0,2 н NaOH) при 4 С, аккуратно качая на шейкере. 10 мМ диметилсульфоксидные исходные растворы ингибиторов разбавляют серийно в буфере для анализа (50 мМ Трис, рН 7,5, 200 мМ NaCl, 5 мМ СаСl2, 20 мкМ ZnCl2 и 0,02% BRIJ-35 (об./об., с использованием следующей схемы: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ Дальнейшие разбавления готовят, как необходимо, следуя этой самой схеме. Минимум четыре концентрации ингибитора для каждого соединения осуществляют в каждом анализе. 25 мкл каждой концентрации затем добавляют в трехкратной повторности в лунки черного 96-луночного U-донного микротитрационного планшета. Когда конечный объем анализа составляет 100 мкл, конечные концентрации ингибитора являются результатом дальнейшего 1:4 разведения (то есть 30 мкМ 3 мкМ 0,3 мкМ 0,03 мкМ, и так далее). Пустую пробу (нет фермента, нет ингибитора) и позитивный ферментативный контроль (с ферментом, без ингибитора) также готовят в трехкратной повторности. Активированный фермент разбавляют до 100 нг/мл в буфере для анализа, 25 мкл на лунку добавля- 13005762 ют в соответствующие лунки микропланшета. Конечная концентрация фермента в анализе составляет 25 нг/мл (0,34 нМ). Диметилсульфоксидный исходный раствор субстрата (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2) с концентрацией 5 мМ разбавляют в буфере для анализа до 20 мкМ. Анализ инициируют добавлением 50 мкл разбавленного субстрата, с получением конечной концентрации в анализе 10 мкМ субстрата. Показания флуоресценции (320 возбуждение, 390 испускание) снимают непосредственно в момент времени 0,и последующие показания снимают каждые пятнадцать минут при комнатной температуре с помощью устройства для чтения планшетов PerSeptive Biosystems CytoFluor Multi-Well Reader с приростом 90 единиц. Среднюю величину флуоресценции фермента и пустой пробы наносят на график против времени. Раннюю точку времени на линейной части этой кривой выбирают для определений ИК 50. Точку времени ноль для каждого соединения при каждом разбавлении вычитают из последней точки времени, и данные затем выражают как процент ферментативного контроля (флуоресценция ингибитора, деленная на флуоресценцию позитивного ферментативного контроля х 100). Данные наносят на график как концентрацию ингибитора против процента ферментативного контроля. ИК 50 определяют как концентрацию ингибитора, которая дает сигнал, который составляет 50% от позитивного ферментативного контроля. Ингибирование активности стромелизина (ММР-3) Человеческий рекомбинантный стромелизин (ММР-3, стромелизин-1) активируют в течение 20-22 ч 2 мМ пара-аминофенилацетатом ртути (из свежеприготовленного 100 мМ исходного раствора в 0,2 нNaOH) при 37 С. 10 мМ диметилсульфоксидные исходные растворы ингибиторов разбавляют серийно в буфере для анализа (50 мМ Трис, рН 7,5, 150 мМ NaCl, 10 мМ СаСl2 и 0,05% BRIJ-35 (об./об., с использованием следующей схемы: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ Дальнейшие разбавления готовят, как необходимо, следуя этой самой схеме. Минимум четыре концентрации ингибитора для каждого соединения осуществляют в каждом анализе. 25 мкл каждой концентрации затем добавляют в трехкратной повторности в лунки черного 96-луночного U-донного микротитрационного планшета. Когда конечный объем анализа составляет 100 мкл, конечные концентрации ингибитора являются результатом дальнейшего 1:4 разведения (то есть 30 мкМ 3 мкМ 0,3 мкМ 0,03 мкМ, и так далее). Пустую пробу (нет фермента, нет ингибитора) и позитивный ферментативный контроль (с ферментом, без ингибитора) также готовят в трехкратной повторности. Активированный фермент разбавляют до 200 нг/мл в буфере для анализа, 25 мкл на лунку добавляют в соответствующие лунки микропланшета. Конечная концентрация фермента в анализе составляет 50 нг/мл (0,875 нМ). Диметилсульфоксидный исходный раствор субстрата (Mca-Arg-Pro-Lys-Pro-Val-Glu-Nva-Trp-ArgLys(Dnp)-NH2) с концентрацией 10 мМ разбавляют в буфере для анализа до 6 мкМ. Анализ инициируют добавлением 50 мкл разбавленного субстрата, с получением конечной концентрации в анализе 3 мкМ субстрата. Показания флуоресценции (320 возбуждение, 390 испускание) непосредственно снимают в момент времени 0, и последующие показания снимают каждые пятнадцать минут при комнатной температуре с помощью устройства для чтения планшетов PerSeptive Biosystems CytoFluor Multi-Well Reader с приростом 90 единиц. Среднюю величину флуоресценции фермента и пустой пробы наносят на график против времени. Раннюю точку времени на линейной части этой кривой выбирают для определений ИК 50. Точку времени ноль для каждого соединения при каждом разбавлении вычитают из последней точки времени, и данные затем выражают как процент ферментативного контроля (флуоресценция ингибитора, деленная на флуоресценцию позитивного ферментативного контроля х 100). Данные наносят на график как концентрацию ингибитора против процента ферментативного контроля. ИК 50 определяют как концентрацию ингибитора, которая дает сигнал, который составляет 50% от позитивного ферментативного контроля. Ингибирование человеческой 92 кДа желатиназы (ММР-9) Ингибирование активности 92 кДа желатиназы (ММР-9) анализируют с использованием Mca-ProLeu-Gly-Leu-Dpa-Ala-Arg-NH2 субстрата (10 мкМ) в условиях, подобных тем, которые описаны выше для ингибирования человеческой коллагеназы (ММР-1). Человеческую рекомбинантную 92 кДа желатиназу (ММР-9, желатиназа В) активируют в течение 2 часов 1 мМ пара-аминофенил-ацетатом ртути (из свежеприготовленного 100 мМ исходного раствора в 0,2 Н NaOH) при 37 С. 10 мМ диметилсульфоксидные исходные растворы ингибиторов разбавляют серийно в буфере для анализа (50 мМ Трис, рН 7,5, 200 мМ NaCl, 5 мМ СаСl2, 20 мкМ ZnCl2, 0,02% BRIJ-35 (об./об., с использованием следующей схемы: 10 мМ 120 мкМ 12 мкМ 1,2 мкМ 0,12 мкМ Дальнейшие разбавления готовят, как необходимо, следуя этой самой схеме. Минимум четыре концентрации ингибитора для каждого соединения осуществляют в каждом анализе. 25 мкл каждой концен- 14005762 трации затем добавляют в трехкратной повторности в лунки черного 96-луночного U-донного микротитрационного планшета. Когда конечный объем анализа составляет 100 мкл, конечные концентрации ингибитора являются результатом дальнейшего 1:4 разведения (то есть 30 мкМ 3 мкМ 0,3 мкМ 0,03 мкМ, и так далее). Пустую пробу (нет фермента, нет ингибитора) и позитивный ферментативный контроль (с ферментом, без ингибитора) также готовят в трехкратной повторности. Активированный фермент разбавляют до 100 нг/мл в буфере для анализа, 25 мкл на лунку добавляют в соответствующие лунки микропланшета. Конечная концентрация фермента в анализе составляет 25 нг/мл (0,27 нМ). Диметилсульфоксидный исходный раствор субстрата (Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2) в концентрации 5 мМ разбавляют в буфере для анализа до 20 мкМ. Анализ инициируют добавлением 50 мкл разбавленного субстрата, с получением конечной концентрации в анализе 10 мкМ субстрата. Показания флуоресценции (320 возбуждение, 390 испускание) снимают непосредственно в момент времени 0,и последующие показания снимают каждые пятнадцать минут при комнатной температуре с помощью устройства для чтения планшетов PerSeptive Biosystems CytoFluor Multi-Well Reader с приростом 90 единиц. Среднюю величину флуоресценции фермента и пустой пробы наносят на график против времени. Раннюю точку времени на линейной части этой кривой выбирают для определений ИК 50. Точку времени ноль для каждого соединения при каждом разбавлении вычитают из последней точки времени, и данные затем выражают как процент ферментативного контроля (флуоресценция ингибитора, деленная на флуоресценцию позитивного ферментативного контроля х 100). Данные наносят на график как концентрацию ингибитора против процента ферментативного контроля. ИК 50 определяют как концентрацию ингибитора, которая дает сигнал, который составляет 50% от позитивного ферментативного контроля. Ингибирование ММР-13 Человеческую рекомбинантную ММР-13 активизируют 2 мМ АРМА (пара-аминофенил-ацетатом ртути) в течение 1,5 часов при 37 С и разбавляют до 400 мг/мл в буфере для анализа (50 мМ Трис, рН 7,5, 200 мМ хлорид натрия, 5 мМ хлорид кальция, 20 мкМ хлорид цинка, 0,02% brij). Двадцать пять микролитров разбавленного фермента добавляют на лунку 96-луночного микротитрационного планшета. Фермент затем разбавляют в соотношении 1:4 в анализе путем добавления ингибитора и субстрата, с получением конечной концентрации в анализе 100 мг/мл. 10 мМ исходные растворы ингибиторов готовят в диметилсульфоксиде, а затем разбавляют в буфере для анализа как для схемы разбавления ингибиторов для ингибирования человеческой коллагеназы(ММР-1): Двадцать пять микролитров каждой концентрации добавляют в трехкратной повторности в микротитрационный планшет. Конечные концентрации в анализе составляют 30 мкМ, 3 мкМ, 0,3 мкМ и 0,03 мкМ. Субстрат (Dnp-Pro-Cha-Gly-Cys(Me)-His-Ala-Lys(NMA)-NH2) готовят как для ингибирования человеческой коллагеназы (ММР-1), и 50 мкл добавляют в каждую лунку с получением конечной концентрации в анализе 10 мкМ. Показания флуоресценции (360 нМ возбуждение, 460 нМ испускание) снимают в момент времени 0 и каждые 5 мин в течение 1 ч. Позитивные контроли состоят из фермента и субстрата при отсутствии ингибитора, а пустые пробы состоят только из субстрата. ИК 50 определяют как для ингибирования человеческой коллагеназы (ММР-1). Если про величины ИК 50 сообщают, что они меньше, чем 0,03 мкМ, тогда ингибиторы используют в анализе в конечных концентрациях 0,3 мкМ, 0,03 мкМ, 0,003 мкМ и 0,0003 мкМ. ММР-13 анализ коллагеновой пленки Крысиный коллаген типа I метили радиоактивной меткой с помощью 14 С уксусного ангидрида (Т.Е.Cawston and A.J. Barrett, Anal. Biochem 99, 340-345 (1979 и использовали для получения 96 луночных планшетов, содержащих коллагеновые пленки с радиоактивной меткой (Barbara Johnson-Wint, Anal. Biochem 104, 175-181 (1980. Когда раствор, содержащий коллагеназу, добавляют в лунку, фермент расщепляет нерастворимый коллаген, который раскручивается и, таким образом, солюбилизируется. Активность коллагеназы прямо пропорциональна количеству солюбилизированного коллагена, определенному с помощью пропорции радиоактивности, высобожденной в супернатант, как измеряют в стандартном сцинтилляционном счетчике. Ингибиторами коллагеназы являются, следовательно, соединения, которые уменьшают радиоактивные импульсы, высвобожденные по отношению к контролям без ингибитора. Одно конкретное воплощение этого анализа описано более детально ниже. Для определения селективности соединений для ММР-13 против ММР-1, с использованием коллагена в качестве субстрата, используют следующую процедуру. Рекомбинантную человеческую проММР 13 или проММР-1 активируют в соответствии с процедурами, приведенными выше. Активированную ММР-13 или ММР-1 разбавляют до 0,6 мкг/мл буфером (50 мМ Трис, рН 7,5, 150 мМ NaCl, 10 мМ CaCl2, 1 мкМ ZnCl2, 0,05% Brij-35, 0,02% азид натрия). Готовят исходные растворы тестируемого соединения (10 мМ) в диметилсульфоксиде. Разбавления тестируемых соединений в Трис-буфере, выше, готовят до 0,2; 2,0; 20; 200; 2000 и 20000 нМ. 100 мкл разведения соответствующего лекарства и 100 мкл разбавленного фермента пипетируют в- 15005762 лунки 96 луночного планшета, содержащего коллагеновые пленки, меченные 14 С-коллагеном. Конечная концентрация фермента составляет 0,3 мкг/мл, тогда как конечная концентрация лекарства составляет 0,1, 1,0, 10, 100, 1000 нМ. Каждую концентрацию лекарства и контроля анализируют в трехкратной повторности. Трехкратные контроли также осуществляют для условий, в которых отсутствует фермент, и для фермента в отсутствии какого-либо соединения. Планшеты инкубируют при 37 С в течение периода времени, так что около 30-50% доступного коллагена солюбилизировано, определяют путем подсчета дополнительных контрольных лунок в различные моменты времени. В большинстве случаев требуется около 9 ч инкубации. Когда анализ достаточно развился, супернатант из каждой лунки удаляют и подсчитывают в сцинтилляционнном счетчике. Фоновые импульсы (определенные путем подсчетов импульсов в лунках без фермента) вычитают из каждого образца, и % высвобождения вычисляют по отношению к лункам только с ферментом и без ингибитора. Трехкратные повторности для каждой точки усредняют, и данные наносят на график как процент высвобождения против концентрации лекарства. ИК 50 определяют из точки, в которой получают 50% ингибирования высвобождения коллагена, меченного радиоактивной меткой. Для определения идентичности активных коллагеназ в кондиционированной среде хряща, анализы проводили с использованием коллагена в качестве субстрата, кондиционированной среды хряща, содержащей коллагеназную активность и ингибиторы варьирующей селективности. Кондиционированную среду хряща собирали в течение времени, в которое происходила деградация коллагена, и, таким образом, она является репрезентативной для коллагеназ, ответственных за распад коллагена. Анализы проводили как указано выше, за исключением того, что вместо использования рекомбинантной ММР-13 или рекомбинантной ММР-1, кондиционированная среда хряща была источником фермента.IL-1 индуцированная деградация коллагена хряща из носового хряща быка В этом анализе используют экспланты носового хряща быка, которые обычно используют для тестирования эффективности различных соединений на ингибирование либо IL-1 индуцированной деградации протеогликанов, либо IL-1 индуцированной деградации коллагена. Носовой хрящ быка представляет собой ткань, которая очень сходна с суставным хрящом, то есть хондроциты, окруженные матриксом,который представляет собой преимущественно коллаген типа II и аггрекан. Эту ткань используют потому, что она (1) очень сходна с суставным хрящом, (2) легко доступна, (3) относительно гомогенна и (4) деградирует с предсказуемой кинетикой после стимуляции IL-1. Два варианта этого анализа были использованы для анализа соединений. Оба варианта дают похожие данные. Эти два варианта описаны ниже: Вариант 1. Три пробки носового хряща быка (приблизительно 2 мм в диаметре х 1,5 мм в длину) помещают в каждую лунку 24 луночного планшета для культуры тканей. Один мл среды без сыворотки добавляют затем в каждую лунку. Соединения готовят в виде 10 мМ исходных растворов в DMSO (диметилсульфоксид), а затем разбавляют подходящим образом в среде без сыворотки до конечных концентраций,например 50, 500 и 5000 нМ. Каждую концентрацию анализируют в трехкратной повторности. Человеческий рекомбинантный IL-1(5 нг/мл) (IL-1) добавляют в трехкратные контрольные лунки и в каждую лунку, содержащую лекарство. Также готовят трехкратные контрольные лунки, в которые не добавляют ни лекарства, ни IL-1. Среду удаляют, и свежую среду, содержащую IL-1 и подходящие концентрации лекарства, добавляют на день 6, 12, 18 и 24, либо каждые 3-4 дня, если необходимо. Среду,удаленную в каждый момент времени хранят при -20 С для дальнейшего анализа. Когда хрящ в лунках только с IL-1 почти полностью резорбируется (примерно 21 день), эксперимент завершают. Среду удаляют и хранят. Аликвоты (100 мкл) из каждой лунки в каждый момент времени объединяют, подвергают расщеплению с помощью папаина, и затем анализируют на содержание гидроксипролина. Фоновый гидроксипролин (среднее лунок с отсутствием IL-1 и отсутствием лекарства) вычитают из каждой точки данных, и среднее вычисляют для каждой трехкратной повторности. Данные затем выражают как процент средней величины одного IL-1 и наносят на график. ИК 50 определяют из этого графика. Вариант 2. Экспериментальные установки являются такими же, как приведено выше в варианте 1, до дня 12. На 12 день кондиционированную среду из каждой лунки удаляют и замораживают. Затем один мл забуференного фосфатом солевого раствора (ФСБ - фосфатно-солевой буфер), содержащего 0,5 мкг/мл трипсина добавляют в каждую лунку и инкубацию продолжают в течение дополнительных 48 ч при 37 С. После 48 ч инкубации в трипсине раствор ФСБ удаляют. Аликвоты (50 мкл) раствора ФСБ/трипсин и предыдущих двух точек времени (день 6 и 12) объединяют, гидролизуют и определяют содержание гидроксипролина. Фоновый гидроксипролин (среднее лунок с отсутствием IL-1 и отсутствием лекарства) вычитают из каждой точки данных, и среднее вычисляют для каждой трехкратной повторности. Данные затем выражают как процент средней величины одного IL-1 и наносят на график. ИК 50 определяют из этого графика. В этом варианте время эксперимента значительно укорачивается. Добавление трипсина в течение 48 ч после 12 дней стимуляции IL-1 вероятно высвобождает любой коллаген типа II, который был поврежден коллагеназной активностью, но еще не высвободился из хрящевого матрикса. В отсутст- 16005762 вии стимуляции IL-1 обработка трипсином дает только низкие фоновые уровни деградации коллагена в хрящевых эксплантах. Ингибирование продукции TNF Способность или неспособность соединений или их фармацевтически приемлемых солей ингибировать продукцию TNF показана с помощью следующего in vitro анализа: Анализ человеческих моноцитов Человеческие мононуклеарные клетки выделяли из антикоагулированной человеческой крови, используя одностадийную методику разделения Ficoll-hypaque. (2) Мононуклеарные клетки промывали три раза в сбалансированном солевом растворе Хенка (HBSS) с двухвалентными катионами и ресуспендировали до плотности 2 х 106/мл в HBSS, содержащем 1% BSA (бычий сывороточный альбумин). Различные подсчеты, определенные с использованием анализатора Abbott Cell Dyn 3500, указывали на то, что моноциты варьируют от 17 до 24% всех клеток в этих препаратах. Аликвоты по 180 мкл клеточной суспензии помещали в плоскодонные 96 луночные планшеты (Costar). Добавления соединений и ЛПС (липополисахаридов) (100 нг/мл конечная концентрация) дало конечный объем 200 мкл. Все условия осуществляли в трехкратной повторности. После четырех часов инкубации при 37 С в увлажненном СО 2 инкубаторе, планшеты удаляли и центрифугировали (10 мин при приблизительно 250 х д), и супернатанты удаляли и анализировали на TNF, используя RD набор дляELISA (твердофазного иммуноферментного анализа). Аггреканазный анализ Первичные свиные хондроциты из суставного хряща выделяли путем последовательного расщепления трипсином и коллагеназой, за чем следовало расщепление коллагеназой в течение ночи, и помещали в концентрации 2x105 клеток на лунку в 48 луночные планшеты с 5 мкКю/мл 35S (1000 Кю/ммоль) серы в планшеты, покрытые коллагеном типа I. Клеткам давали включить метку в их протеогликановый матрикс (примерно 1 неделя) при 37 С, в атмосфере 5% СО 2. За ночь перед инициацией анализа монослои хондроцитов промывали два раза в DMEM (модифицированная по способу Дульбекко среда Игла)/1% PSF/G, а затем оставляли инкубироваться в свежейDMEM/1% ФСБ в течение ночи. На следующее утро хондроциты промывали один раз в DMEM/1%PSF/G. Конечной промывке давали осесть на планшетах в инкубаторе во время приготовления разведений. Среды и разведения можно сделать, как описано в таблице ниже. Контрольная средаDMEM + IL-1 (5 нг/мл) Исходные растворы всех соединений готовят в концентрации 10 мМ в концентрации DMSO. Готовят 100 мкМ исходный раствор каждого соединения в DMEM в 96 луночном планшете. Хранят в холодильнике в течение ночи. На следующий день осуществляют серийные разведения в DMEM с IL-1 до 5 мкМ, 500 нМ и 50 нМ. Отсасывают конечные промывки из лунок, и добавляют 50 мкл соединения из вышеуказанных разбавлений к 450 мкл IL-1 среды в соответствующие лунки 48 луночных планшетов. Конечные концентрации соединений равны 500 нМ, 50 нМ и 5 нМ. Все образцы выполняют в трехкратной повторности с отдельными контрольным и IL-1 образцами на каждом планшете. Планшеты метят, и только внутренние 24 лунки планшета используют. На одном из планшетов несколько колонок обозначают как IL-1 (нет лекарства) и Контроль (нет IL-1, нет лекарства). Эти контрольные колонки периодически подсчитывают для наблюдения высвобождения 35S-протеогликана. Контрольную и IL-1 среду добавляют в лунки (450 мкл), за чем следует соединение (50 мкл), так чтобы инициировать анализ. Планшеты инкубируют при 37 С, с 5% СО 2 атмосферой. При 40-50 % высвобождении (когда СРМ из IL-1 среды составляет 4-5 раз контрольной среды), как оценивают с помощью подсчета (импульсов) в жидкой фазе (LSC) образцов среды, анализ завершают (912 ч). Среду удаляют из всех лунок и помещают в сцинтилляционные пробирки. Добавляют сцинтиллят,и получают радиоактивные импульсы (LSC). Для солюбилизации клеточных слоев 500 мкл папаинового буфера для расщепления (0,2 М Трис, рН 7, 0, 5 мМ EDTA (этилендиаминтетрауксусная кислота), 5 мМDTT (дитиотреитол) и 1 мг/мл папаина) добавляют в каждую лунку. Планшеты с расщепляющим раствором инкубируют при 60 С в течение ночи. Клеточный слой удаляют с планшетов на следующий день и помещают в сцинтилляционные пробирки. Затем добавляют сцинтиллят, и подсчитывают образцы (LSC).- 17005762 Определяют процент высвобожденных импульсов от общего количества, присутствующего в каждой лунке. Средние от трехкратных повторностей делают с помощью контрольного фона, вычтенного из каждой лунки. Процент ингибирования соединения основан на IL-1 образцах в качестве 0% ингибирования (100% от общего количества импульсов). Соединения по настоящему изобретению, которые были протестированы, по меньшей мере в одном из вышеуказанных анализов, все имели ИК 50 менее, чем 100 мкМ, предпочтительно менее чем 100 нМ. Некоторые предпочтительные группы соединений проявляют различную селективность в отношении различных MMP's или ADAMs. Одна группа предпочтительных соединений проявляет селективную активность в отношении ММР-13 по сравнению с ММР-1. Еще одна предпочтительная группа соединений проявляет селективную активность в отношении ММР-13 по сравнению с ММР-1, ММР-3 и ММР-7. Еще одна предпочтительная группа соединений проявляет селективную активность в отношении ММР-13 по сравнению с ММР-1, ММР-3, ММР-7 и ММР-17. Еще одна предпочтительная группа соединений проявляет селективную активность в отношении ММР-13 по сравнению с ММР-1, ММР-2, ММР-3, ММР-7,ММР-9 и ММР-14. Еще одна предпочтительная группа соединений проявляет селективную активность в отношении ММР-13 по сравнению с ММР-12 и ММР-14. Для введения млекопитающим, включая людей, для ингибирования матриксных металлопротеиназ,разнообразие общепринятых путей может быть использовано, включая пероральный, парентеральный(например внутривенный, внутримышечный или подкожный), трансбуккальный, анальный или местный. В общем, соединения по изобретению (здесь и далее также известные как активные соединения) следует вводить в дозировках примерно 0,1 и 25 мг/кг массы тела субъекта, которого лечат, в сутки, предпочтительно от примерно 0,3 до 5 мг/кг. Предпочтительно активное соединение следует вводить перорально или парентерально. Однако, некоторые вариации в дозировке будут необходимым образом встречаться, в зависимости от состояния субъекта, которого лечат. Лицо, ответственное за введение, должно, в любом случае, определять соответствующую дозу для конкретного субъекта. Соединения по настоящему изобретению можно вводить в широком разнообразии различных лекарственных форм, в общем, терапевтически эффективные соединения по настоящему изобретению присутствуют в таких лекарственных формах на концентрационных уровнях, варьирующих от примерно 5,0 до примерно 70% по массе. Для перорального введения таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальция фосфат и глицин, могут быть использованы вместе с различными дезинтегрантами, такими как крахмал (и предпочтительно кукурузный,картофельный или крахмал тапиока), альгиновая кислота и некоторые сложные силикаты, вместе со связующими веществами гранулирования, наподобие поливинилпирролидона, сахарозы, желатина и акации. Дополнительно, смазывающие агенты, такие как стеарат магния, лаурил сульфат натрия и тальк, часто очень полезны для целей таблетирования. Твердые композиции подобного типа могут также быть использованы как наполнители в желатиновых капсулах, предпочтительные материалы в этой связи также включают в себя лактозу или молочный сахар, а также полиэтиленгликоли высокой молекулярной массы. Когда водные суспензии и/или эликсиры желательны для перорального введения, активный ингредиент может быть объединен с различными подслащивающими агентами или корригентами, красящим веществом или красителями, и, если желательно, также с эмульгирующим и/или суспендирующим агентами, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные подобные их комбинации. В случае животных,они преимущественно содержатся в пище животных или питьевой воде в концентрации 5-5000 млн'1,предпочтительно 25-500 млн-1. Для парентерального введения (внутримышечное, интраперитониальное, подкожное и внутривенное применение) обычно готовят стерильный инъецируемый раствор активного ингредиента. Растворы терапевтического соединения по настоящему изобретению либо в масле кунжута или арахиса, либо в водном пропиленгликоле, могут быть использованы. Водные растворы должны быть подходящим образом отрегулированы и забуферены, предпочтительно при рН более, чем 8, если необходимо, и жидкий разбавитель сначала сделан изотоничным. Эти водные растворы подходят для целей внутривенной инъекции. Масляные растворы подходят для целей внутрисуставной, внутримышечной и подкожной инъекции. Приготовление всех этих растворов в стерильных условиях легко осуществимо с помощью стандартных фармацевтических методик, хорошо известных специалистам. В случае животных, соединения можно вводить внутримышечно или подкожно в уровнях дозировки от примерно 0,1 до 50 мг/кг/сутки,преимущественно от 0,2 до 10 мг/кг/сутки в однократной дозе или вплоть до 3 разделенных доз. Активные соединения по настоящему изобретению могут также быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например содержащие общепринятые основы для суппозитория, такие как масло кокоса или другие глицериды. Для интраназального введения или введения путем ингаляции активные соединения по настоящему изобретению удобным образом доставляют в форме раствора или суспензии из контейнера с нагнетаемым аэрозолем, который сжимает или нагнетает пациент, или в виде представления распыляемого аэрозоля из герметичного контейнера или распылителя, с использованием подходящего пропеллента, напри- 18005762 мер дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящего газа. В случае герметичного аэрозоля стандартную дозу можно определить путем обеспечения клапана для доставки отмеренного количества. Герметичный контейнер или распылитель может содержать раствор или суспензию активного соединения. Капсулы и картриджи (сделанные, например,из желатина) для использования в ингаляторе или инсуффляторе могут быть приготовлены таким образом, чтобы содержать порошкообразную смесь соединения по изобретению и подходящую порошкообразную основу, такую как лактоза или крахмал. Для местного глазного введения, может быть использовано прямое нанесение на поврежденный глаз в форме такого препарата, как глазные капли, аэрозоль, гели или мази, или может быть включено в коллаген (такой как поли-2-гидроксиэтилметакрилат и его сополимеры), или в виде гидрофильной полимерной защиты. Материалы также могут быть нанесены в виде контактных линз или посредством локального резервуара или в виде субконъюнктивального препарата. Для интраорбитального введения обычно готовят стерильный инъецируемый раствор активного ингредиента. Могут быть использованы растворы терапевтического соединения по настоящему изобретению в водном растворе или суспензии (размер частиц менее, чем 10 микрон). Водные растворы должны быть подходящим образом отрегулированы и забуферены, предпочтительно при рН между 5 и 8, если необходимо, и жидкий разбавитель сначала сделан изотоничным. Небольшие количества полимеров могут быть добавлены для увеличения вязкости или для замедленного высвобождения (такие как целлюлозные полимеры, декстран, полиэтиленгликоль или альгиновая кислота). Эти растворы подходят для целей интраорбитальной инъекции. Приготовление всех этих растворов в стерильных условиях легко осуществимо с помощью стандартных фармацевтических методик, хорошо известных специалистам в данной области. В случае животных, соединения можно вводить интраорбитально в уровнях дозировки от примерно 0,1 до 50 мг/кг/сутки, преимущественно от 0,2 до 10 мг/кг/сутки в однократной дозе или вплоть до 3 разделенных доз. Что касается других путей введения и соответствующих лекарственных форм, описанных здесь, лекарственные формы, предназначенные для перорального введения, также подходящим образом готовят,чтобы обеспечить регулируемое, замедленное и/или задержанное высвобождение активного ингредиента. Типично, они будут включать в себя пероральные таблетки, капсулы и мультипартикуляты с задержанным высвобождением, а также таблетки и капсулы, покрытые энтеросолюбильной оболочкой, которые предупреждают высвобождение и адсорбцию активного ингредиента в желудке пациента и облегчают доставку по кишечнику дистальнее желудка, то есть в кишечнике. Другие типичные пероральные лекарственные формы будут включать в себя пероральные таблетки, капсулы и мультипартикуляты с замедленным высвобождением, которые обеспечивают системную доставку активного ингредиента регулируемым способом в течение пролонгированного периода времени, например 24-часового периода. Когда быстрая доставка активного ингредиента необходима или желательна, пероральная лекарственная форма с регулируемым высвобождением может быть приготовлена в форме быстрорастворимой таблетки, которая будет предпочтительно также включать в себя высокорастворимые солевые формы активного ингредиента. Следующие примеры иллюстрируют получение соединений по настоящему изобретению. Точки плавления не откорректированы. Данные ЯМР представлены в частях на миллиони относятся к сигналу синхронизации дейтерия из образца растворителя (дейтериохлороформ, если не указано иначе). Коммерческие реагенты используют без дополнительной очистки. Хроматография относится к колоночной хроматографии, осуществляемой с использованием 32-63 мм силикагеля и выполняемой в условиях давления азота (флэш-хроматография). Комнатная или температура окружающей среды относится к 2025 С. Все неводные реакции осуществляют в атмосфере азота для удобства и чтобы максимизировать выходы. Концентрирование при пониженном давлении или в вакууме означает, что использовался роторный испаритель. Общие экспериментальные данные Общий пример: Соединения формулы I могут быть получены путем взаимодействия соответствующего соединения формулы III с мочевиной формулы II (т.е., H2N(CO)-NH2) в присутствии подходящего основания, такого как алкоксидное основание, предпочтительно этоксид натрия, в полярном растворителе, таком как спиртовой растворитель, предпочтительно этанол, при температуре от 20 С до точки кипения растворителя,предпочтительно 80 С, в течение от 15 мин до 3 ч. Общее получение: Соединение формулы III может быть получено путем взаимодействия соответствующего соединения формулы IV с подходящим основанием, таким как третичное аминовое основание или основание, связанное с полимером, предпочтительно смолой Amberlyst-400 (гидроксидная форма), в полярном растворителе, таком как спиртовой растворитель, предпочтительно этанол, при температуре от примерно 0 до примерно 50 С,предпочтительно примерно 20 С, в течение периода от примерно 6 до примерно 36 ч. Соединение формулы IV может быть получено путем взаимодействия соответствующего соединения формулы VI с соединением формулы V, которое имеет общую формулу L3-(A')-L4 или L3-(А'), в ап- 19005762 ротонном растворителе, предпочтительно ароматическом углеводородном растворителе, таком как бензол или толуол, при температуре от примерно 40 С и до точки кипения растворителя, предпочтительно примерно 80 С, в течение периода от примерно 1 до примерно 6 ч. Соединение формулы VI может быть получено путем взаимодействия соответствующего соединения формулы NH2-X-Y-Z-G с соединением формулы VII, которое представляет собой 2 галогеномалонатный эфир, предпочтительно 2-бромдиэтиловый эфир малоновой кислоты, в присутствии подходящего основания, такого как третичное аминовое основание, предпочтительно N,Nдиметиланилин, при температуре от примерно 20 до примерно 100 С, предпочтительно примерно 80 С, в течение периода от примерно 4 до примерно 48 ч. Пример 1. 1-[6-(4-Бромфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраон: Металлический натрий (29 мг, 1,26 ммоль) добавляли к 1,3 мл этанола и перемешивали до гомогенности. Добавляли диэтиловый эфир 1-[6-(4-бромфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (0,20 г, 0,42 ммоль), затем мочевину (75 мг, 1,26 ммоль) и данную смесь перемешивали в течение 5 мин при 80 С. Смесь охлаждали до температуры окружающей среды, подкисляли 1 М соляной кислотой и экстрагировали 3 раза этилацетатом. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии на силикагеле (3:1, гексан : этилацетат) с получением 28 мг 1-[6-(4-бромфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона в виде бесцветного твердого вещества. ВЭЖХ: время удерж.: 2,201 мин; МС (APCI(химическая ионизация при атмосферном давлении), m/z): 436 [М-Н]-; 438 [М+Н]+. Получение 1: 2-(4-Бромфенокси)-5-нитропиридин: 4-Бромфенол (5,5 г, 32 ммоль) добавляли к 42 мл 50% мас./мас. водного гидроксида натрия. После перемешивания в течение 30 мин добавляли 44 мл толуола, затем следует 2-хлор-5-нитропиридин (5,0 г,32 ммоль) и тетрабутиламмония бромид (10 г, 32 ммоль). После перемешивания в течение 1,5 ч при 23 С данную смесь разбавляли 200 мл воды, нейтрализовали 12 М водной соляной кислотой, и данную смесь трижды экстрагировали эфиром. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали в вакууме с получением 6 г 2-(4-бромфенокси)-5-нитропиридина. 1 Н ЯМР (CDCl3, 500 МГц): 9,05 (d, 1H, J = 3,5 Гц), 8,51 (dd, 1H, J = 3,5; 9,5 Гц), 7,58 (d, 2H, J = 9,0 Гц), 7,08 (m, 3 Н) млн-1. МС(APCI, m/z): 295 [М+Н]+. 6-(4-Бромфенокси)-пиридин-3-иламин: Смесь 2-(4-бромфенокси)-5-нитропиридина (6,0 г, 22,7 ммоль), 200 мл метанола и 50 мг РtO2 встряхивали при 50 psi (344,74 КПа) Н 2 в течение 1 ч при 23 С. Смесь фильтровали через подложку из целита, и фильтрат концентрировали в вакууме с получением 6 г 6-(4-бромфенокси)-пиридин-3-иламина. 1 Н ЯМР (CD3OD, 500 МГц): 7,65 (d, 1H, J = 3,5 Гц), 7,48 (d, 2H, J = 8,5 Гц), 7,25 (dd, 1H, J = 3,5; 9,0 Гц),6,91 (d, 2H, J = 9,0 Гц), 6,80 (d, 1H, J= 9,0 Гц) млн-1. МС (APCI, m/z): 265 [М+Н]+. Получение 2: Диэтиловый эфир 1-[6-(4-бромфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты: Смесь 6-(4-бромфенокси)-пиридин-3-иламина (4,5 г, 16,9 ммоль), 2-бромдиметилмалоната (4,1 г, 17 ммоль) и N,N-диметиланилина (2,1 г, 17 ммоль) перемешивали при 80 С в течение 24 ч. Данную смесь охлаждали до 23 С, разбавляли 50 мл бензола и обрабатывали 7 мл 2-бромпропионилхлорида. После перемешивания при кипячении с обратным холодильником в течение 3 ч данную смесь охлаждали до 23 С,концентрировали в вакууме и разбавляли 750 мл этанола. Добавляли смолу Amberlyst-400 (гидроксидная форма) (75 г) и данную смесь перемешивали в течение 24 ч при 23 С. Данную смесь фильтровали, и смолу промывали 50 мл метанола. Фильтрат концентрировали в вакууме и остаток очищали с помощью хроматографии на силикагеле (2:1, гексан:этилацетат) с получением 6 г диэтиловго эфира 1-[6-(4-бромфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты. 1 Н ЯМР (CDCl3, 500 МГц): 8,06 (d, 1H,- 20005762[М+Н]+. Следующие соединения были получены в соответствии со способами, аналогичными таковому примеру 1 с заменой, где необходимо, правильного пиридина и диэфира: Таблица 1 Следуя процедуре для образования пиримидинтриона, приведенной в примере 1, реакция диэтилового эфира 1-[6-(4-цианофенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (58 мг, 0,14 ммоль) с мочевиной (0,030 г, 0,5 ммоль) в 0,5 мл 1 М этоксида натрия в этаноле давала 14,3 мг 4-[5(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)-пиридин-2-илокси]-бензонитрила в виде бесцветного твердого вещества. 1 Н ЯМР (CD3OD, 500 МГц): 8,06 (d, 1H, J = 3,5 Гц), 7,78 (m, 3H), 7,31 (d, 2H, J = 8,5 Гц), 7,13 (d, 1H, J = 9,0 Гц), 2,75 (m, 2 Н), 2,68 (m, 2 Н) млн-1. МС (APCI, m/z): 390 [М-Н]-; 392 [М+Н]+. Получение 1: Диэтиловый эфир 1-[6-(4-цианофенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты: Смесь диэтилового эфира 1-[6-(4-бромфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (0,28 г, 0,53 ммоль), цианида цинка (0,037 г, 0,32 ммоль), тетракистрифенилфосфина палладия (0)(0,024 г, 0,021 ммоль) и 0,66 мл диметилформамида нагревали до 80 С в течение 24 ч. Добавляли дополнительные 37 мг цианида цинка и 24 мг тетракистрифенилфосфина палладия (0), и данную смесь перемешивали при 80 С в течение дополнительных 48 ч. После охлаждения до комнатной температуры данную смесь разбавляли толуолом и промывали 2 М гидроксидом аммония (дважды), солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка с помощью радиальной хроматографии (этилацетат-гексаны, затем метанол) дала 58 мг диэтилового эфира 1-[6-(4-цианофенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. 1 Н ЯМР (CDCl3, 500 МГц): 8,07 (d, 1H, J = 3,5 Гц), 7,80 (dd, 1H, J = 2,5; 9,0 Гц), 7,69 (d, 2H, J = 9,0 Гц),7,25 (d, 2H, J = 9,0 Гц), 7,02 (d, 1 Н, J = 9,0 Гц), 4,21 (q, 4 Н, J = 7,5 Гц), 2,74 (m, 2 Н), 2,66 (m, 2 Н), 1,19 (t,6 Н, J = 7,0 Гц) млн-1. МС (APCI, m/z): 424 [М+Н]+. Пример 5. 1-[6-(4-[1,3,4]Оксадиазол-2-ил-фенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан 2,6,8,10-тетраон: Следуя процедуре для образования пиримидинтриона, приведенной в примере 1, реакция диэтилового эфира 1-[6-(4-[1,3,4]оксадиазол-2-ил-фенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (200 мг, 0,44 ммоль) с мочевиной (0,080 г, 1,3 ммоль) в 1,3 мл 1 М этоксида натрия в этаноле дала 25 мг 1-[6-(4-[1,3,4]оксадиазол-2-ил-фенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10 тетраона в виде бесцветного твердого вещества. 1 Н ЯМР (CD3OD, 500 МГц): 9,02 (s, 1H), 8,14 (d, 2H, J = 8,0 Гц), 8,06 (d, 1H, J = 2,0 Гц), 7,78 (dd, 1H, J = 2,5; 9,0 Гц), 7,35 (d, 2 Н, J = 9,0 Гц), 7,12 (d, 1H, J = 9,0 Гц),2,74 (m, 2 Н), 2,66 (m, 2 Н) млн-1. МС (APCI, m/z): 435 [М+Н]+. Получение 1: Диэтиловый эфир 1-[6-(4-карбоксифенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты: Смесь диэтилового эфира 1-[6-(4-формилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (0,70 г, 1,64 ммоль), карбоната натрия (0,26 г, 1,64 ммоль) и 16,4 мл смеси трет-бутилового спирта и воды в соотношении 1:1 обрабатывали перманганатом калия (0,26 г, 1,64 ммоль). После пермешивания в течение 2 ч при комнатной температуре данную смесь гасили сульфитом натрия, подкисляли 1 М соляной кислотой и трижды экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением диэтилового эфира 1-[6(4-карбокси-фенокси)-пиридин-3-ил]-5-оксо-пирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа (0,5 г). 1 Н ЯМР (CDCl3, 500 МГц): 8.14 (d, 2H, J = 8,5 Гц), 8,10 (d, 1H, J = 3,0 Гц), 7,80 (dd, 1 Н, J = 2,5; 8,5 Гц), 7,24 (d, 2H, J = 8,0 Гц), 7,02 (d, 1H, J = 9,0 Гц), 4,21 (q, 4H, J = 7,0 Гц), 2,74 (m, 2 Н), 2,66 (m,2 Н), 1,21 (t, 6 Н, J = 7,5 Гц) млн-1. МС (APCI, m/z): 443 [М+Н]+. Получение 2: Диэтиловый эфир 1-[6-(4-гидразинокарбонилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2 дикарбоновой кислоты: Смесь полученного диэтилового эфира 1-[6-(4-карбоксифенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (0,4 г, 0,97 ммоль), 1-гидроксибензотриазола гидрата (0,176 г, 1,3 ммоль), 1,2-дихлорэтана (0,25 г, 1,3 ммоль) и 6 мл метиленхлорида перемешивали при комнатной температуре в течение 20 мин. Смесь обрабатывали bос-гидразидом (0,17 г, 1,3 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь разбавляли этилацетатом, промывали 1 М соляной кислотой, раствором бикарбоната натрия, солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток растворяли в 5 мл 1:1 об./об. смеси метиленхлорида и трифторуксусной кислоты, перемешивали в течение 1 ч при температуре окружающей среды и концентрировали в вакууме. Остаток растворяли в этилацетате, промывали 1 М гидроксидом натрия, солевым раствором,сушили над сульфатом натрия, фильтровали и концентрировали с получением диэтилового эфира 1-[6(4-гидразинокарбонилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (0,20 г) в виде бесцветного сиропа. ВЭЖХ: 2,770 мин. Получение 3: Диэтиловый эфир 1-[6-(4-[1,3,4]оксадиазол-2-ил-фенокси)-пиридин-3-ил]-5-оксопирролидин-2,2 дикарбоновой кислоты Смесь полученного диэтилового эфира 1-[6-(4-гидразинокарбонилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (0,20 г, 0,44 ммоль), триметилортоформиата (0,1 мл, 0,91 ммоль) и 1 мл ксилола кипятили с обратным холодильником в течение 24 ч. Данную смесь концентрировали в вакууме с получением диэтилового эфира 1-[6-(4-[1,3,4]оксадиазол-2-ил-фенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (0,2 г) в виде бесцветного сиропа. МС (APCI, m/z): 467,2 [М+Н]+. Следуя процедуре для образования пиримидинтриона, приведенной в примере 1, реакция диэтилового эфира 1-[6-(4-этилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты (200 мг,0,41 ммоль) с мочевиной (0,088 г, 1,4 ммоль) в 1,4 мл 1 М этоксида натрия в этаноле дала 25 мг 1-[6-(4[1,3,4]оксадиазол-2-ил-фенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона в виде бесцветного твердого вещества. 1 Н ЯМР (CDCl3, 500 МГц): 8,73 (bs, 2 Н), 7,97 (d, 1H, J = 2,0 Гц), 7,78 (d,1H, J = 8,5 Гц), 7,22 (d, 2H, J = 8,5 Гц), 7,05 (d, 2H, J = 9,0 Гц), 6,91 (d, 1H, J = 9,0 Гц), 2,81 (q, 2H, J = 7,5 Гц), 2,74 (m, 2 Н), 2,64 (m, 2 Н), 1,26 (t, 3 Н, J = 8,0 Гц) млн-1. Получение 1: Диэтиловый эфир 1-[6-(4-этилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2 дикарбоновой кислоты: Смесь диэтилового эфира 1-[6-(4-винилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (0,20 г), 50 мг 10% палладия на угле и 20 мл этилацетата встряхивали при 50 psi (344,74 КПа) газообразного водорода в течение 2 ч. Данную смесь фильтровали и концентрировали в вакууме с получением 0,20 г диэтилового эфира 1-[6-(4-этилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. 1 Н ЯМР (CDCl3, 500 МГц): 8,08 (d, 1H, J = 2,5 Гц), 7,73 (dd, 1H, J = 2,5; 8,5 Гц), 7,24 (d, 2H, J = 7,5 Гц), 7,05 (d, 2H, J = 8,0 Гц), 6,89 (d, 1H, J = 9,0 Гц), 4,21 (q, 4H, J = 7,0 Гц),2.74 (m, 2 Н), 2.65 (m, 4 Н), 1,27 (t, 3 Н, J = 8,0 Гц), 1,20 (t, 6H, J = 7,5 Гц) млн-1. Пример 7. N-4-[5-(2,6,8,10-Тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)-пиридин-2-илокси]-бензилацетамид: Смесь диэтилового эфира 1-6-[4-(трет-бутоксикарбониламинометил)-фенокси]-пиридин-3-илпирролидин-2,2-дикарбоновой кислоты (0,52 ммоль) и 2 мл смеси 1:1 об./об. раствора трифторуксусной кислоты в метиленхлориде перемешивали в течение 1 ч при температуре окружающей среды, а затем концентрировали в вакууме. Остаток растворяли в 2,6 мл метиленхлорида и обрабатывали ММР-смолой(связанное с полимером основание N-метилморфолинового типа, 0,86 г, 1,75 ммоль) и обрабатывали ацетилхлоридом (0,055 г, 0,7 ммоль). После встряхивания в течение 24 ч, смесь фильтровали, и смолу промывали метиленхлоридом. Объединенные фильтраты концентрировали в вакууме, растворяли в 1,5 мл 1 М этоксида натрия в этаноле и обрабатывали 94 мг мочевины. После перемешивания в течение 10 мин при 80 С, смеси обрабатывали 2 г полистиролсвязанного полимера сульфоновой кислоты, фильтровали и смолу промывали 10 мл 2 М аммиака в метаноле. Объединенные фильтраты концентрировали в вакууме и очищали с помощью хроматографии с обращенной фазой (элюент ацетонитрил-вода-трифторуксусная кислота), за которой следовала радиальная хроматография (10% метанол-метиленхлорид), с получениемN-4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)-пиридин-2-илокси]-бензил-ацетамида в виде бесцветного твердого вещества. ВЭЖХ: время удерж.: 2,201 мин; МС (APCI, m/z): 436 [М-Н]-; 438[М+Н]+. Получение 1: Диэтиловый эфир 1-[6-(4-винилфенокси)-пиридин-3-ил]-пирролидин-2,2 дикарбоновой кислоты: Смесь диэтилового эфира 1-[6-(4-бромфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (5,8 г, 12,2 ммоль), винилтрибутилолова (3,9 мл, 12,3 ммоль), тетракистрифенилфосфина палладия(0) (0,60 г, 0,52 ммоль) и 24 мл толуола нагревали при температуре дефлегмации в течение 1 ч. После охлаждения до комнатной температуры данную смесь концентрировали в вакууме и очищали с помощью хроматографии на силикагеле (Flash (флэш) 40, 20% - 50% этилацетат-гексаны), с получением 4,8 г диэтилового эфира 1-[6(4-винилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. Получение 2: Диэтиловый эфир 1-[6-(4-формилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты Смесь диэтилового эфира 1-[6-(4-винилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты(189 мл) перемешивали в течение 6 ч при температуре окружающей среды. Смесь гасили сульфитом натрия,разбавляли водой и трижды экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением диэтилового эфира 1-[6-(4 формилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа (4,6 г). Получение 3: Диэтиловый эфир 1-6-[4-(трет-бутоксикарбониламинометил)-фенокси]-пиридин-3-ил)-пирролидин-2,2 дикарбоновой кислоты Смесь диэтилового эфира 1-[6-(4-формилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты(0,1 г, 0,24 ммоль), трет-бутоксикарбониламида (0,083 г, 0,71 ммоль), триэтилсилана (0,11 мл, 0,083 г, 0,71 ммоль) и ацетонитрила (1 мл) обрабатывали трифторуксусной кислотой (0,035 мл, 0,46 ммоль) и перемешивали в течение 48 ч при температуре окружающей среды. Данную смесь разбавляли этилацетатом, промывали насыщенным раствором бикарбоната натрия, солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением диэтилового эфира 1-6-[4-(трет-бутоксикарбониламинометил)фенокси]-пиридин-3-ил-пирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. Следующие соединения были получены в соответствии со способами, аналогичными способу по примеру 7, с представлением, где необходимо, правильного пиридина и диэфира: Таблица 2 1-[6-(4-Пиразол-1-илметилфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6, Следуя процедуре для образования пиримидинтриона, приведенной в примере 1, реакция диэтилового эфира 5-оксо-1-[6-(4-пиразол-1-илметилфенокси)-пиридин-3-ил]-пирролидин-2,2-дикарбоновой кислоты (0,2 г, 0,4 ммоль) с мочевиной (0,074 г, 1,2 ммоль) в 1,2 мл 1 М этоксида натрия в этаноле дала 6 мг 1-[6-(4-пиразол-1-илметилфенокси)-пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона в виде бесцветного твердого вещества. 1 Н ЯМР (CD3OD, 500 МГц): 7,99 (d, 1H, J = 2,5 Гц), 7,72 (m, 2H), 7,53 (d,1H, J = 2,5 Гц), 7,29 (d, 2H, J = 8,5 Гц), 7,10 (d, 2H, J = 8,5 Гц), 6,97 (d, 1H, J = 8,5 Гц), 6,35 (t, 1 Н, J = 2,0 Гц), 5,38 (s, 2 Н), 2,75 (m, 2 Н), 2,65 (m, 2 Н) млн-1. МС (APCI, m/z): 447,2 [М+Н]+. Получение 1: Диэтиловый эфир 1-[6-(4-гидроксиметилфенокси)-пиридин-3-ил]-5-оксопирролидин 2,2-дикарбоновой кислоты: Диэтиловый эфир 1-[6-(4-гидроксиметилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты: к раствору диэтилового эфира 1-[6-(4-формилфенокси)-пиридин-3-ил]-пирролидин-2,2 дикарбоновой кислоты (1,0 г, 2,3 ммоль) в 30 мл этанола добавляли боргидрид натрия (0,090 г, 2,3 ммоль) при 0 С. После перемешивания в течение 3 ч данную смесь концентрировали в вакууме, разбавляли этилацетатом и водой, и водный слой осторожно подкисляли 1 М соляной кислотой, затем нейтрализовали насыщенным водным бикарбонатом натрия. Смесь экстрагировали трижды этилацетатом и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением 0,80 г (80%) диэтилового эфира 1-[6-(4-гидроксиметилфенокси)-пиридин-3-ил]-5 оксопирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. 1 Н ЯМР (CDCl3, 400 МГц): 8,04(APCI, m/z): 429,1 [М+Н]+. Получение 2: Диэтиловый эфир 1-[6-(4-бромметилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты: К раствору диэтилового эфира 1-[6-(4-гидроксиметилфенокси)-пиридин-3-ил]-5-оксопирролидин 2,2-дикарбоновой кислоты (0,80 г, 1,9 ммоль) в 9,4 мл метиленхлорида добавляли триэтиламин (0,46 мл,0,33 г, 3,3 мл). После охлаждения до -40 С смесь обрабатывали метансульфонилхлоридом (0,20 мл, 0,30 г, 2,61 ммоль). После перемешивания в течение 1 ч добавляли дополнительные 0,10 мл метансульфонилхлорида и 0,4 мл триэтиламина, и перемешивание продолжали в течение 1 ч. Раствор безводного бромида лития (1,6 г, 19 ммоль, высушенный в пламени в вакууме перед применением) в тетрагидрофуране (20 мл) добавляли через канюлю, и смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Смесь разбавляли этилацетатом, и органическую фазу промывали водой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток фильтровали через подложку из силикагеля,элюируя смесью 1:1 этилацетат:гексаны, с получением 0,65 г диэтилового эфира 1-[6-(4-бромметилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2-дикарбоновой кислоты в виде бесцветного сиропа. 1 Н ЯМР (CDCl3, 500 МГц): 8,07 (d, 1H, J = 3,0 Гц), 7,76 (dd, 1H, J = 2,5; 8,5 Гц), 7,44 (d, 2 Н, J = 8,5 Гц), 7,12(d, 2 Н, J = 8,0 Гц), 6,95 (d, 1 Н, J = 9,0 Гц), 4,53 (s, 2 Н), 4,22 (q, 4H, J = 7,0 Гц), 2,75 (m, 2H), 2,65 (m, 2H),1.20 (t, 6H, J = 7,0 Гц) млн-1. Получение 3: Диэтиловый эфир 5-оксо-1-[6-(4-пиразол-1-илметилфенокси)-пиридин-3-ил]-пирролидин-2,2 дикарбоновой кислоты: К раствору диэтилового эфира 1-[6-(4-бромметилфенокси)-пиридин-3-ил]-5-оксопирролидин-2,2 дикарбоновой кислоты (0,2 г, 0,4 ммоль) в 0,8 мл диметилформамида добавляли пиразол (0,056 г, 0,82 ммоль) и карбонат калия (0,11 г, 0,82 ммоль). После перемешивания в течение 24 ч при 50 С смесь разбавляли водой, трижды экстрагировали этилацетатом, и объединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта в- 25005762 виде бесцветного сиропа, который использовали непосредственно на следующей стадии. МС (APCI,m/z): 479,2 [М+Н]+. Хотя данное изобретение описано и проиллюстрировано со ссылкой на его некоторые конкретные воплощения, специалистам в данной области будет понятно, что различные адаптации, изменения, модификации, замены, удаления или добавления процедур и протоколов могут быть сделаны без отступления от объема изобретения. Например, эффективные дозировки, отличные от конкретных дозировок,которые приведены здесь выше, могут быть применимы вследствие вариаций в чувствительности млекопитающего, которое лечат, для любого из показаний, с помощью соединений по изобретению, указанных выше. Подобным образом, конкретные наблюдаемые фармакологические реакции могут варьировать в соответствии с и в зависимости от конкретных выбранных активных соединений, или от того, присутствует ли фармацевтические носители по настоящему изобретению, а также используемый тип препарата и способ введения, и такие ожидаемые вариации или различия в результатах рассматриваются в соответствии с задачами и практикой настоящего изобретения. Следовательно, предполагается, что изобретение будет определено объемом формулы, которая следует, и что эта формула должна быть интерпретирована так широко, как это допустимо. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где каждый из R1, R2, R3, R4, R5 и R6 независимо представляет собой водород;G представляет собой R15-(CR16R17)p-, где G представляет собой заместитель по любому кольцевому атому углерода Z, способному к образованию дополнительной связи, и ориентирован в положении, отличном от альфа, по отношению к точке присоединения Z кольца к Y; р представляет собой целое число от 0 до 1;R15 независимо выбран из группы, состоящей из галогено, -CN, R18- и R19-(C=O)-(NR21)-; каждый из R16 и R17 независимо представляет собой водород;R18, R19 и R21 независимо выбраны из группы, состоящей из водорода, (С 1-С 4)алкила, (С 3-С 8)циклоалкила, оксадиазолила и пиразолила; или его фармацевтически приемлемая соль. 2. Соединение по п.1, где указанный G представляет собой R15-(CR16R17)p-, где р равно 0. 3. Соединение по п.1, где указанный G представляет собой R15-(CR16R17)p-, где р представляет собой целое число 1. 4. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из 1-[6-(4-фторфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; 1-[6-(4-фторфенокси)пиридин-3-ил]-1,8,10-триазаспиро[5,5]ундекан-2,7,9,11-тетраона; 4-[5-(2,6,8,10-тетраоксо-1,7,9-триазаспиро[4,5]дец-1-ил)пиридин-2-илокси]бензонитрила; 1-[6-(4-[1,3,4]оксадиазол-2-илфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; 1-[6-(4-этилфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона;- 26005762 1-[6-(4-бромфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона; 1-[6-(4-пиразол-1-илметилфенокси)пиридин-3-ил]-1,7,9-триазаспиро[4,5]декан-2,6,8,10-тетраона и их фармацевтически приемлемых солей. 5. Фармацевтическая композиция для лечения состояния, выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств, иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний,болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств, кожных расстройств и рака,у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель. 6. Способ лечения состояния, выбранного из группы, состоящей из расстройств соединительной ткани, воспалительных расстройств, иммунологических/аллергических расстройств, инфекционных заболеваний, респираторных заболеваний, сердечно-сосудистых заболеваний, болезней глаз, болезней обмена веществ, расстройств центральной нервной системы, заболеваний печени/почек, расстройств репродуктивного здоровья, желудочных расстройств, кожных расстройств и рака, у млекопитающего,включая человека, при котором указанному млекопитающему вводят количество соединения по п.1, эффективное при лечении такого состояния. 7. Фармацевтическая композиция для лечения состояния, которое можно лечить путем ингибирования матриксных металлопротеиназ, у млекопитающего, включая человека, содержащая количество соединения по п.1, эффективное при таком лечении, и фармацевтически приемлемый носитель.
МПК / Метки
МПК: C07D 471/10, A61K 31/527, A61P 29/00, C07D 487/10
Метки: металлопротеиназы, ингибиторы, спиро-пиримидин-2,4,6-трионовые
Код ссылки
<a href="https://eas.patents.su/28-5762-spiro-pirimidin-246-trionovye-ingibitory-metalloproteinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Спиро-пиримидин-2,4,6-трионовые ингибиторы металлопротеиназы</a>
Пиримидин-2, 4, 6-трионовые ингибиторы металлопротеиназ

Номер патента: 4680
Опубликовано: 24.06.2004
Автор: Блэгг Джулиан
МПК: C07D 239/60, A61P 43/00, A61K 31/513...
Метки: металлопротеиназ, ингибиторы, пиримидин-2, 6-трионовые
Формула / Реферат:
1. Соединение формулы где R1 представляет собой (C1-C8)алкил или (C3-C8)циклоалкил; R2 и R3 представляют собой водород; X выбран из группы, состоящей из кислорода и серы; Y выбран из группы, состоящей из связи, кислорода и серы; Ar1 представляет собой (C6-C10)арил и Z представляет собой бензил или (C6-C10)арил, возможно замещенный по любому кольцевому атому углерода, способному образовывать дополнительную связь, одним заместителем, выбранными...
Ингибиторы металлопротеиназы, фармацевтические композиции, их содержащие, и их фармацевтические применения

Номер патента: 3294
Опубликовано: 24.04.2003
Авторы: Мелник Майкл Дж., Бендер Стивен Л., Жук Скотт Е., Дагнино Раймонд Джр., Дизон Майкл Е.
МПК: A61K 31/54, A61K 31/495, A61K 31/535...
Метки: фармацевтические, применения, металлопротеиназы, композиции, содержащие, ингибиторы
Формула / Реферат:
1. Соединение с формулой 1 где Z является O или S; Ar является моноциклической арильной или гетероарильной группой, выбранной из фенильной группы, или пяти- или шестичленной азот- или кислородсодержащей гетероарильной группы, незамещенной или замещенной одним или более заместителями, выбранными из галогрупп и цианогруппы; X выбран из S, S=O, O, N-R3 и N+(O-)-R4, где R3 является атомом водорода или C1-C6-алкильной группой, где указанная...
Гетероарильные производные сукцинамидов и их применение в качестве ингибиторов металлопротеиназы

Номер патента: 3127
Опубликовано: 27.02.2003
Авторы: Дил Юдит Г., Чен Джиан Джеффри, Билледо Рональд Дж., Бендер Стивен Л., Чонг Уэсли К.М., Абрео Мелвин А., Кастелано Арлиндо Л.
МПК: A61K 31/40, C07D 207/327, A61P 35/00...
Метки: применение, гетероарильные, ингибиторов, производные, металлопротеиназы, сукцинамидов, качестве
Формула / Реферат:
1. Соединение с формулой где X является одинарной связью или группой -C(O)CH2-; Y является -CH(OH)-; R1 является H, фенилом, возможно замещенным C3-алкилом, цианогруппой, фтором или пиридинилом; бифенилом, возможно замещенным цианогруппой или карбамоилом; или пиридинилом; R2 является H, алкильной группой, арильной группой, гетероарильной группой, циклоалкильной группой, гетероциклоалкильной группой или C(O)R10, где R10 является H, алкильной...
Гидрогалогениды 1′[4-[1-(4-фторфенил)-1н-индол-3-ил]-1-бутил]-спиро[изобензофуран-1(3н),4'- пиперидина]

Номер патента: 2092
Опубликовано: 24.12.2001
Авторы: Мерк Нильс, Лопес Де Диего Эйди, Нильсен Оле
МПК: A61K 31/452, C07D 491/107, A61P 25/14...
Метки: пиперидина, гидрогалогениды, 1'[4-[1-(4-фторфенил)-1н-индол-3-ил]-1-бутил]-спиро[изобензофуран-1(3н),4
Формула / Реферат:
1. Гидрогалогенид 1'-[4-[1-(4-фторфенил)-1H-индол-3-ил]-1-бутил]-спиро[изобензофуран-1(3Н),4'-пиперидина] и его гидраты и/или сольваты. 2. Гидрогалогенид по п.1, который является гидрохлоридом 1'-[4-[1-(4-фторфенил)-1H-индол-3-ил]-1-бутил]-спиро[изобензофуран-1(3Н),4'-пиперидина], его гидрат и/или сольват. 3. Фармацевтическая композиция, содержащая гидрогалогенид по п.1 вместе, по меньшей мере, с одним фармацевтически приемлемым носителем или...
Производные 2-пиридинил-6,7,8,9-тетрагидропиримидо[1,2-а]пиримидин-4-она и 7-пиридинил-2,3-дигидроимидазо[1,2-а]пиримидин-5(1н)она

Номер патента: 5297
Опубликовано: 30.12.2004
Авторы: Саади Мурад, Галле Тьерри, Маргери Северен, Альмарио Гарсия Антонио, Неделек Ален, Ли Адриен Так, Локхид Алистер, Йеш Филипп
МПК: A61P 25/00, C07D 487/04, A61K 31/519...
Метки: 2-пиридинил-6,7,8,9-тетрагидропиримидо[1,2-а]пиримидин-4-она, производные, 7-пиридинил-2,3-дигидроимидазо[1,2-а]пиримидин-5(1н)она
Формула / Реферат:
1. Производное пиримидона, представленное формулой (I), или его соль, или их сольват либо их гидрат где X представляет собой атомы водорода, атом серы, атом кислорода или C1-2алкильную группу и атом водорода; Y является связью, этениленовой группой, этиниленовой группой, 1,2-циклопропиленом, атомом кислорода, атомом серы, сульфонильной группой, сульфинильной группой, карбонильной группой, атомом азота, возможно замещенным C1-6алкильной группой,...
Предыдущий патент: 4-аминозамещенные-2-замещенные-1,2,3,4-тетрагидрохинолины в качестве ингибиторов сетр (хэпб)
Следующий патент: Синергетические гербицидные смеси
Случайный патент: 7- аминоалкилиденил-гетероциклические хинолоны и нафтиридоны