Азольные нуклеозиды и их применение в качестве ингибиторов вариальных рнк- и днк-полимераз
Номер патента: 16830
Опубликовано: 30.07.2012
Авторы: Артербёрн Джефри Б., Паркер Вильям Б., Джонсон Коллин Б.

















Формула / Реферат
1. Соединение, представленное формулами

где А = N;
В = С или N;
X = Н, NH2;
Z = H;
Е = (C1-C6)алкил(OH)C(=O)NH2;
каждый из W, Y, R отдельно выбирают из группы, включающей Н; С1-С6алкил, алкинил, фенил, галоген; О, ОН, Оалкил, галогеналкил, NH-фенил;
при условии, что по меньшей мере один из W, Y и R отличается от Н и NH2, и при этом W и Y могут вместе представлять собой О; и
каждый D отдельно представляет собой ОН,
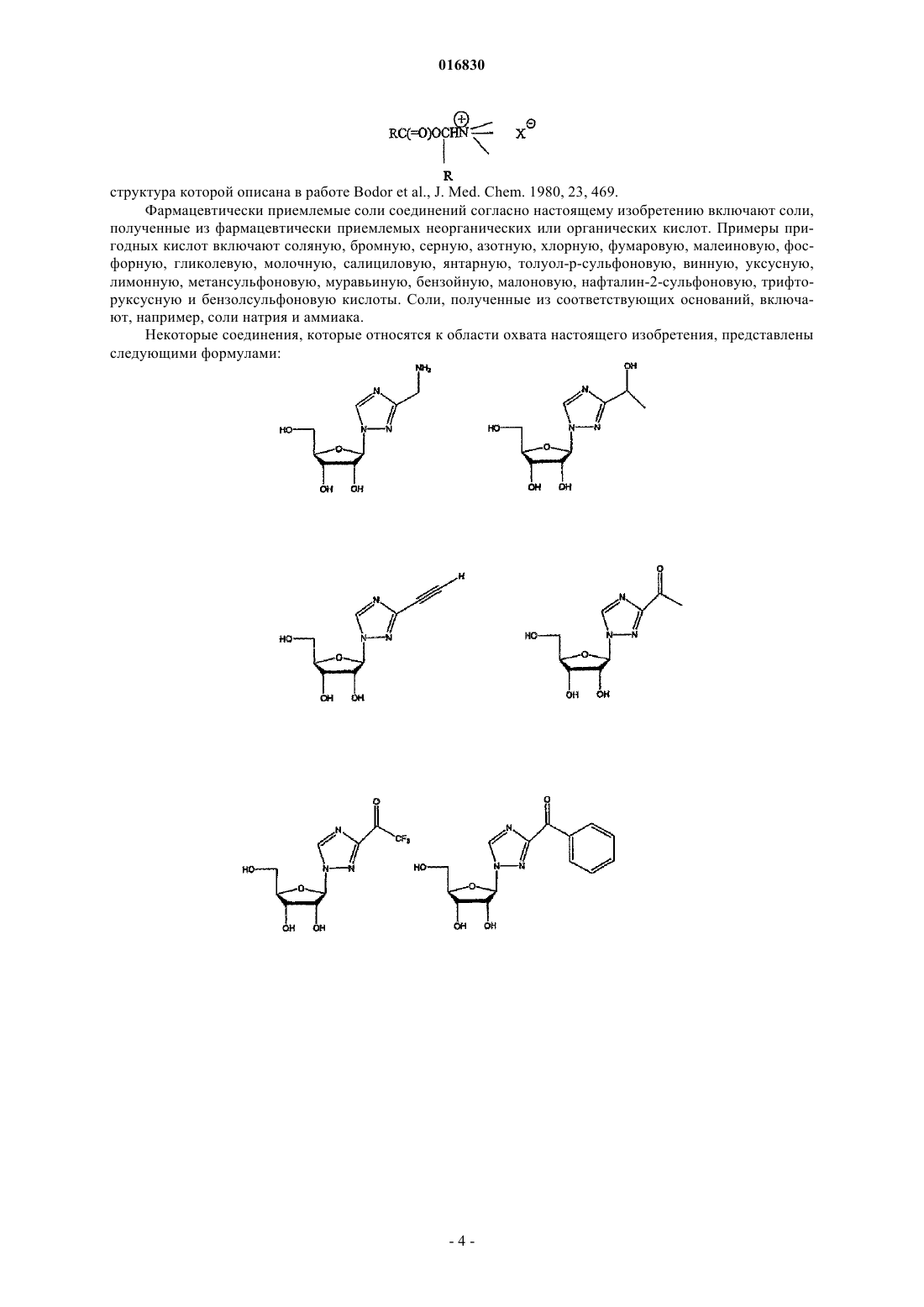
а также его фармацевтически приемлемые соли и смеси.
2. Соединение по п.1, отличающееся тем, что представляет собой 5-амино-4-N-3-фторфенилкарбоксамид-1-β-D-рибофуранозил-1Н-имидазол.
3. Соединение по п.1, отличающееся тем, что представляет собой 3-этинил-1-(β-D-рибофуранозил)-[1,2,4]-триазол.
4. Соединение по п.1, отличающееся тем, что представляет собой 1-(1-β-D-рибофуранозил [1,2,4]триазол-3-ил)фенилметанол.
5. Соединение по п.1, отличающееся тем, что представляет собой 1-(1-β-D-рибофуранозил [1,2,4]триазол-3-ил)фенилметанон.
6. Соединение по п.1, отличающееся тем, что представляет собой 3-(1,1-дифторэтил)-1-β-D-рибофуранозил[1,2,4]триазол.
7. Соединение по п.1, отличающееся тем, что представляет собой 1-(1-β-D-рибофуранозил[1,2,4]триазол-3-ил)-2,2,2-трифторэтанол.
8. Фармацевтический состав, содержащий соединение по п.1 и фармацевтически приемлемый носитель.
9. Способ ингибирования у пациента вирусной РНК-полимеразы путем введения пациенту по меньшей мере одного соединения по п.1.
10. Применение соединения по пп.1-7 для ингибирования РНК и ДНК вирусных инфекций.
11. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой грипп.
12. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус Хантаан.
13. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус конго-крымской геморрагической лихорадки.
14. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус семейства Bunyaviridae.
15. Применение по любому из пп.10-14, дополнительно включающее использование по меньшей мере одного терапевтического агента, выбранного из группы, включающей интерферон (INF), интерферон α-2а, интерферон α-2b, консенсусный интерферон (CIFN), рибавирин, амантадин, ремантадин, интерлейкин-12, урсодеоксихолевую кислоту (UDCA) и глицирризин.
16. Применение по п.10, отличающееся тем, что указанная вирусная инфекция содержит по меньшей мере один элемент из группы, включающей инфекцию вируса гриппа, вируса Хантаан, вируса конго-крымской геморрагической лихорадки, вируса гепатита В, вируса гепатита С, вируса коксаки А, вируса коксаки В, ЕСНО-вирусов, риновирусную инфекцию, вирусную инфекцию ортопоксвируса (оспы), вирусную инфекцию Эбола, вирусную инфекцию полиомиелита и вирусную инфекцию Западного Нила.
Текст
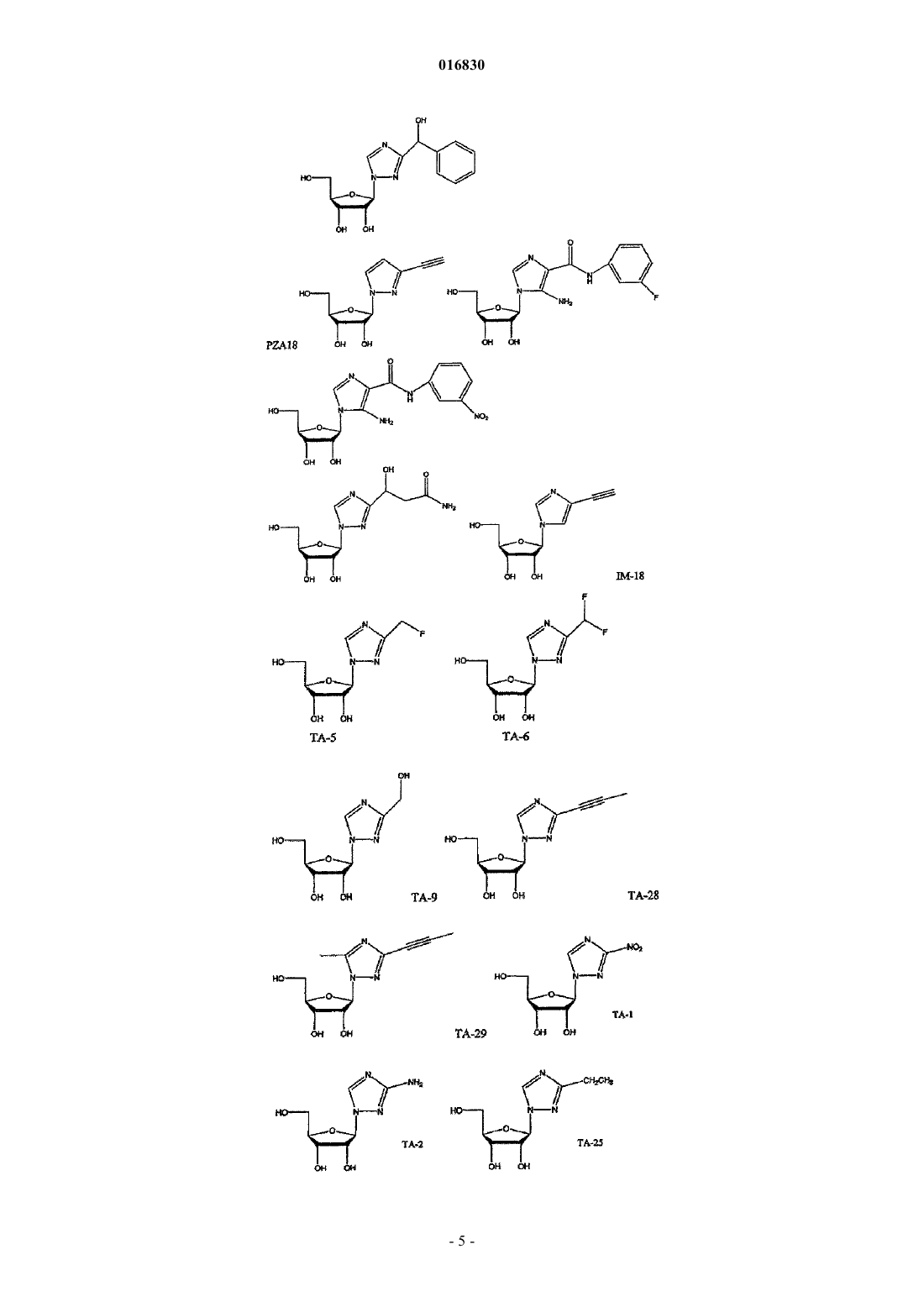
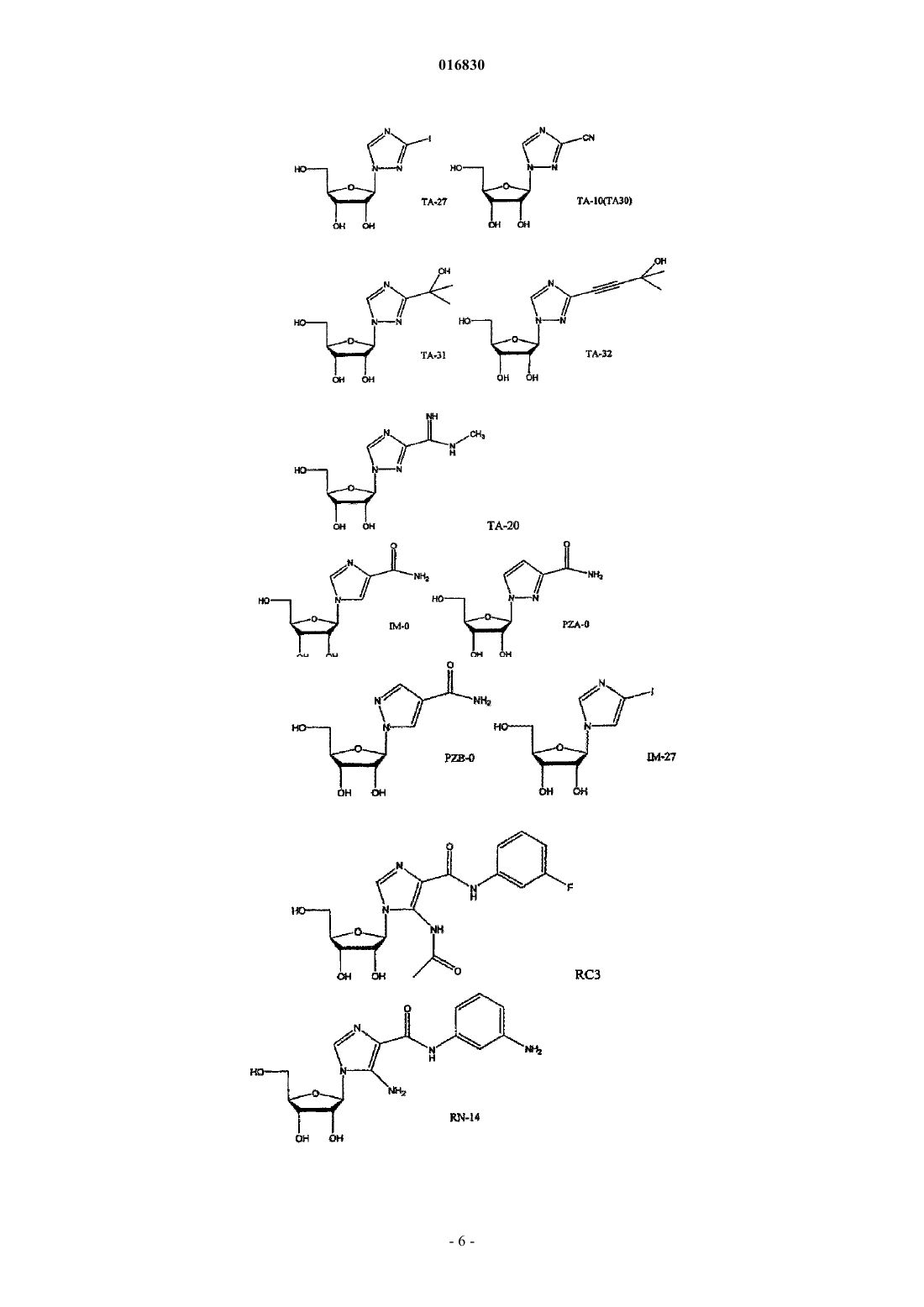
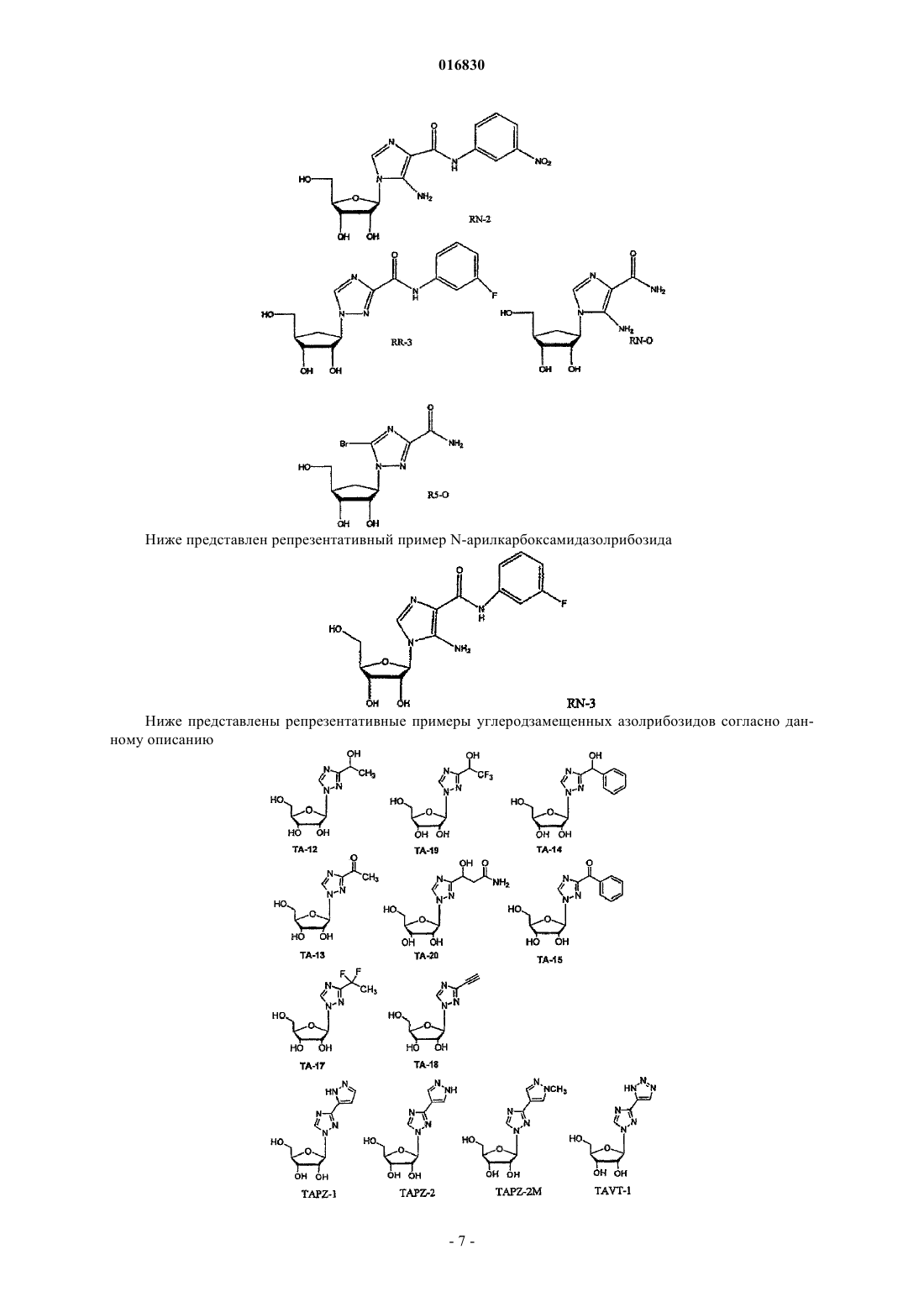
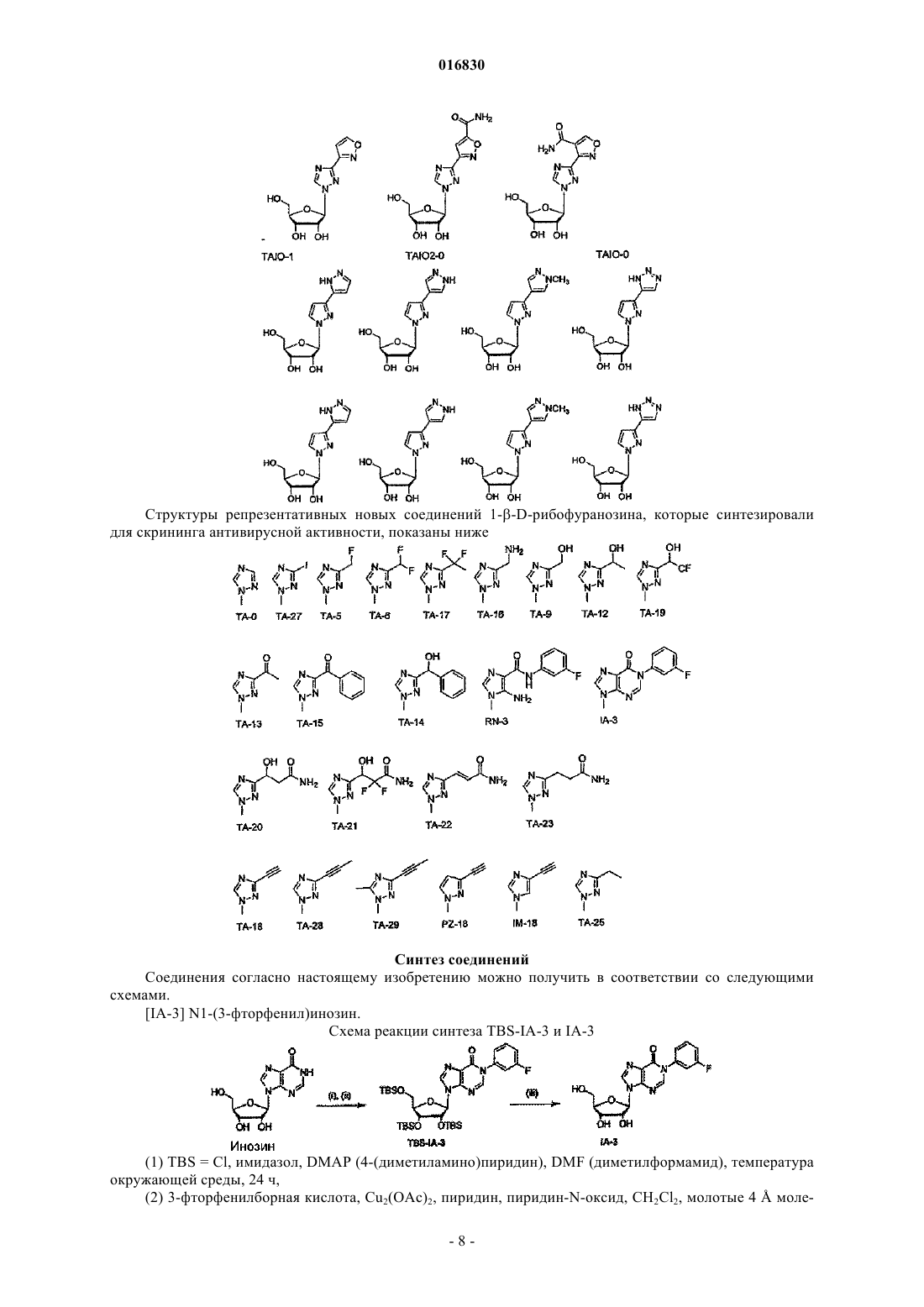
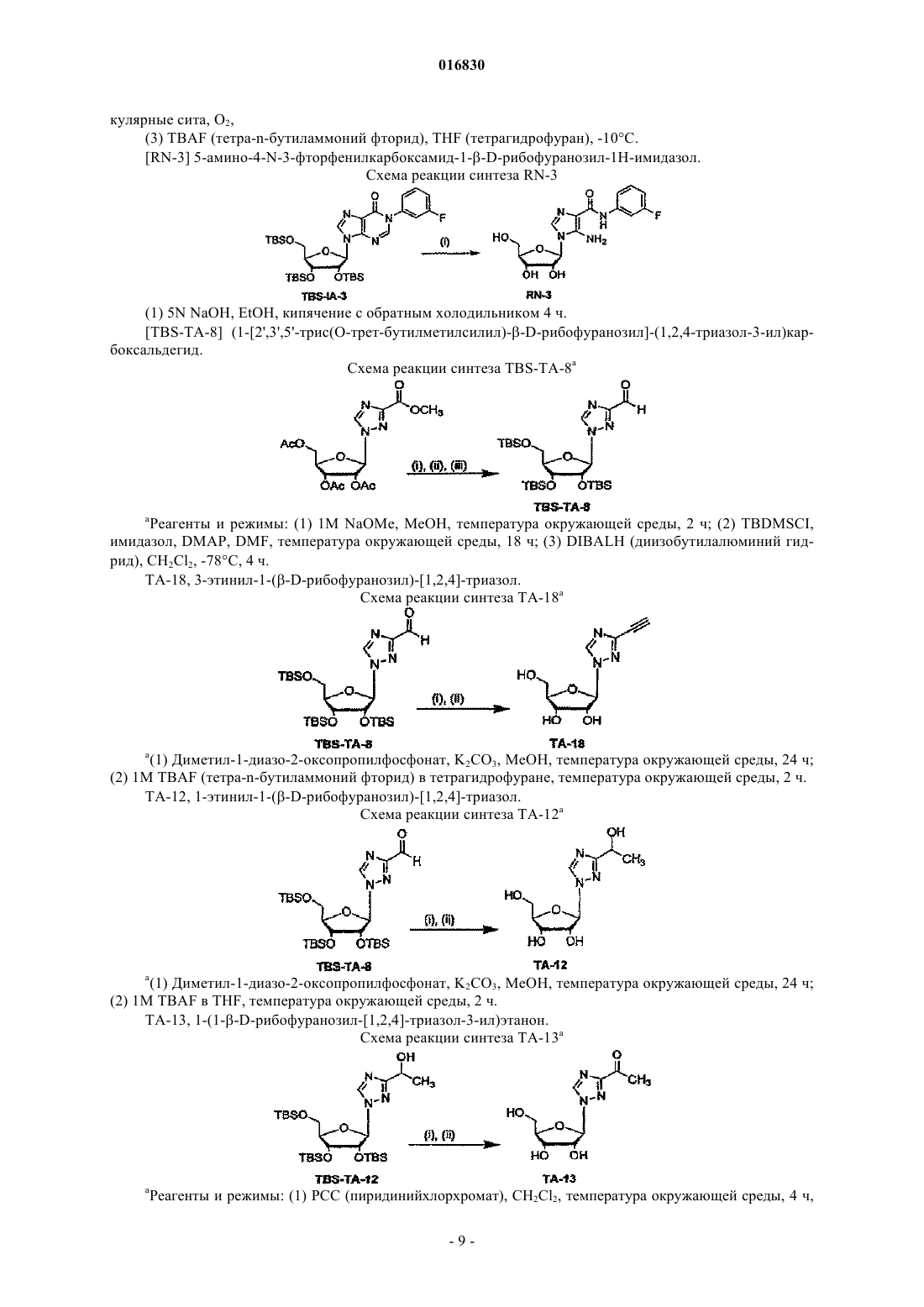
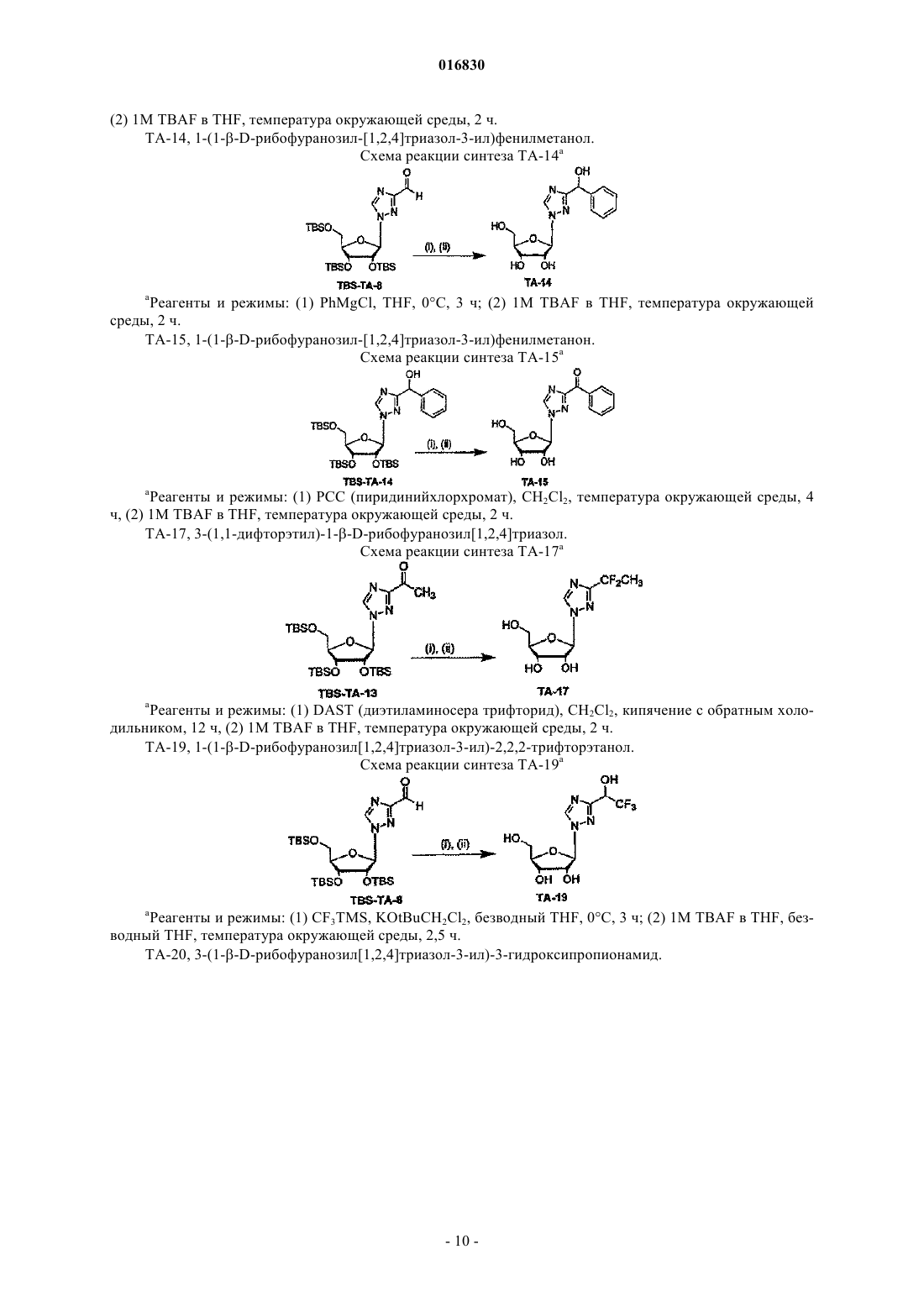
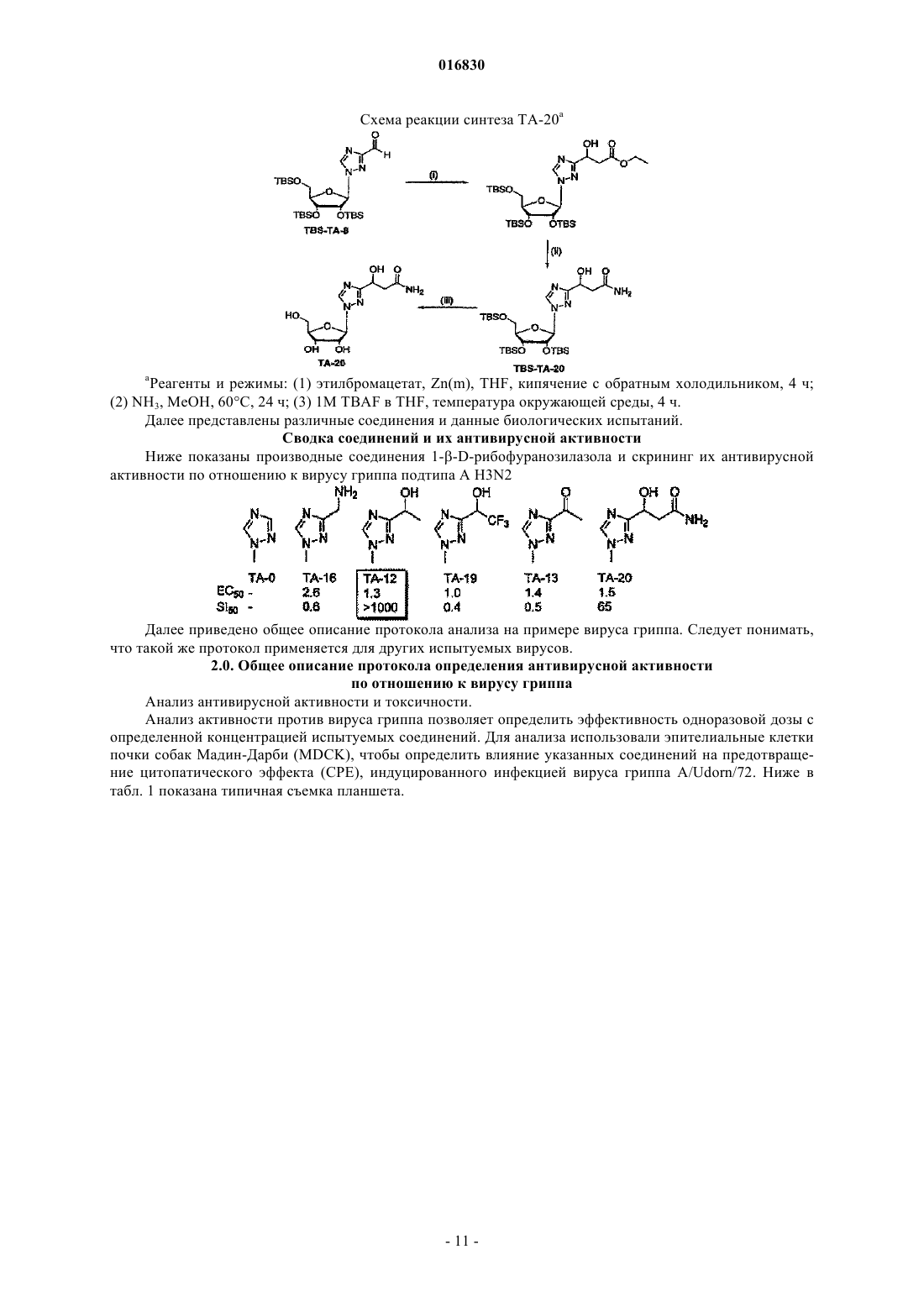
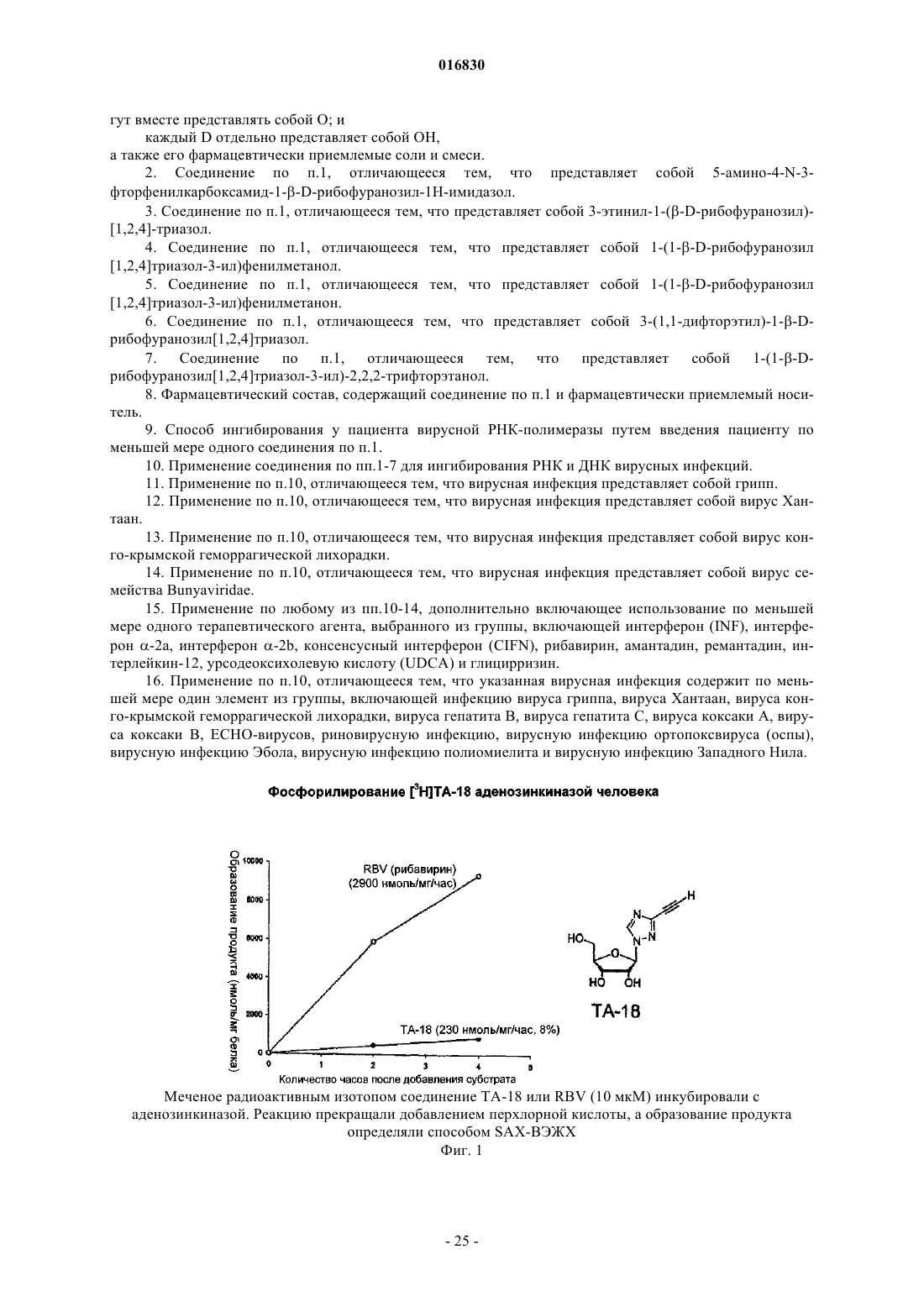
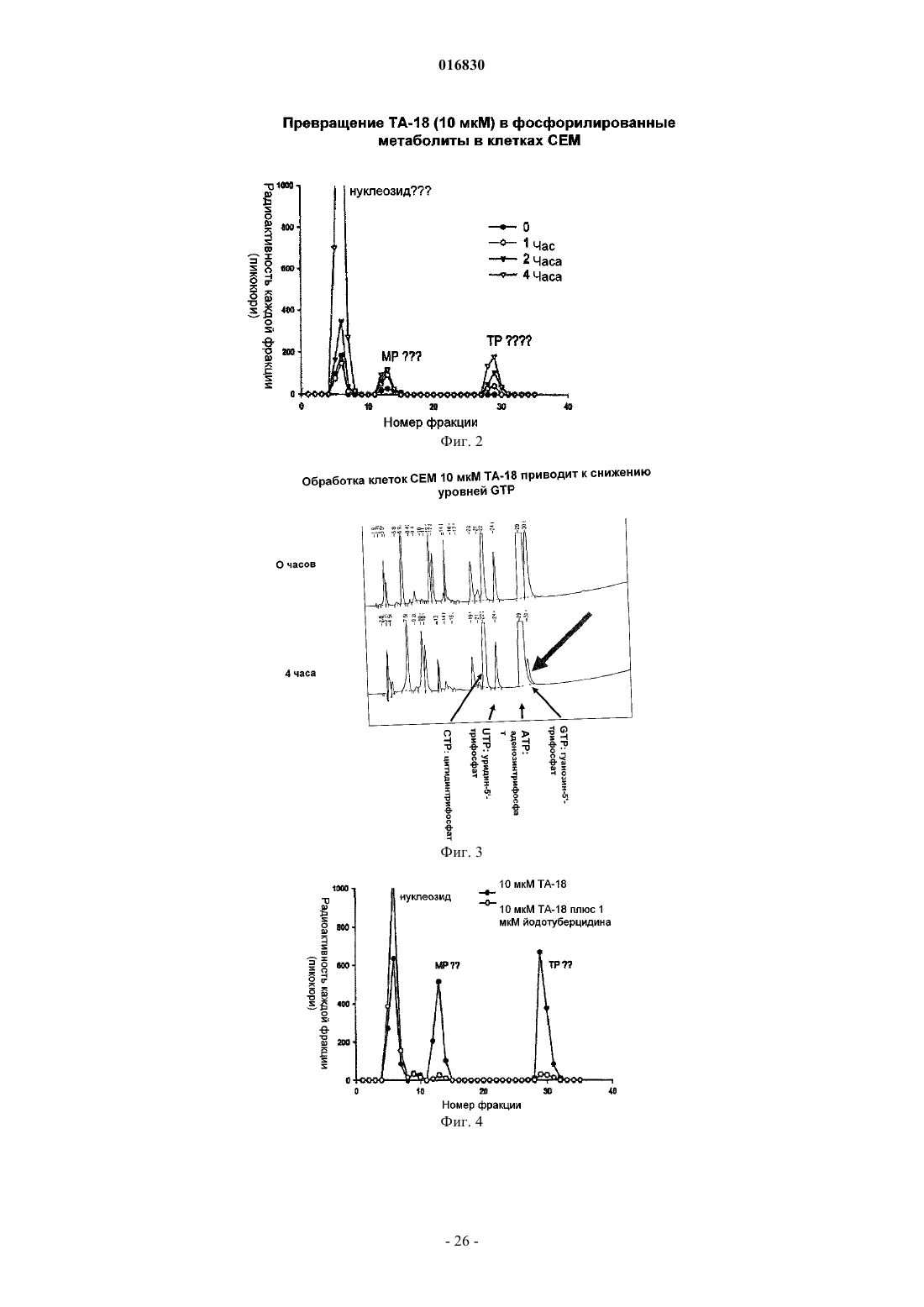
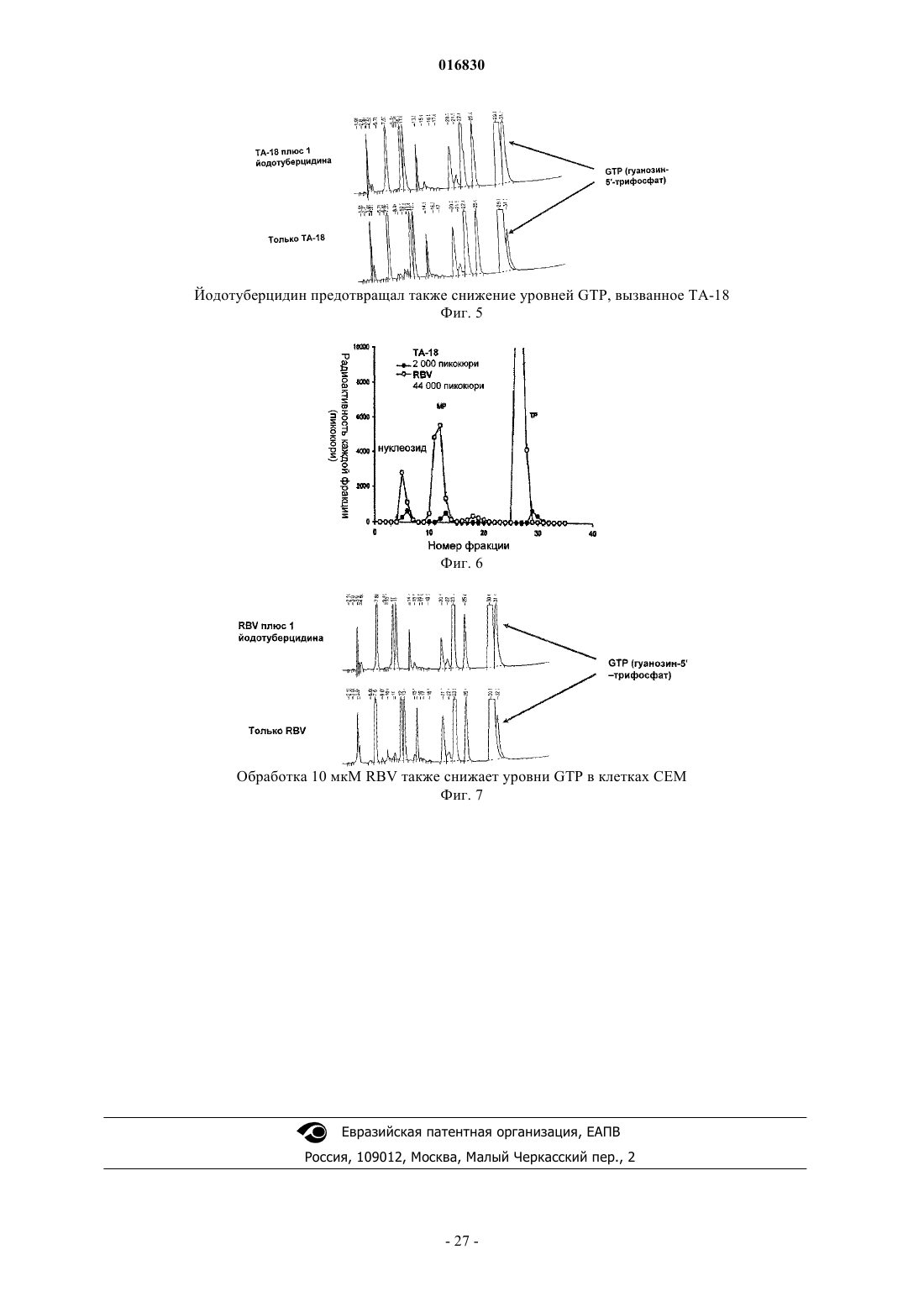
АЗОЛЬНЫЕ НУКЛЕОЗИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАРИАЛЬНЫХ РНК- И ДНК-ПОЛИМЕРАЗ Изобретение обеспечивает азольные нуклеозиды, представленные формулами (I) и (II), где А = С или N; В = С или N; X = Н; С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил; гетероцикло, галоген, в частности F, Cl, Br и I; ОН, NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло), Z = Н, С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероцикло,галоген, в частности F, Cl, Br и I; ОН, NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло); Е = (CH2)nONHR1, n - целое число от 0 до 6, более типично 0-3; R1 = арил или гетероцикло; каждый из W, Y, R отдельно выбирают из группы, включающей Н; С 1-С 6 алкил, циклоалкил,алкенил, циклоалкенил, алкинил, арил, гетероцикло, галоген, в частности F, Cl, Br и I; О, ОН,Оалкил, Оарил, NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло), при условии, что по меньшей мере один из W, Y и R отличается от Н и NH2, и при этом как W, так и Y могут вместе = О, и каждый D отдельно представляется собой ОН, Оалкил, Оарил, Fl и Н,а также их фармацевтически приемлемые соли, пролекарства и смеси. Соединения согласно настоящему изобретению являются полезными в качестве ингибиторов вирусных РНК- и ДНКполимераз, включая, в частности, но без ограничения, полимеразы вируса гриппа, вируса Хантаан,вируса конго-крымской геморрагической лихорадки, гепатита В, гепатита С, полиомиелита, вируса коксаки А и В, риновируса, ЕСНО-вирусов, ортопоксвируса (оспы), ВИЧ, вируса Эбола и вируса Западного Нила, и в особенности - ортопоксвируса, ВИЧ и гепатита В. 016830 Область применения Настоящее изобретение относится к азолу и в особенности - к диазинам, в частности к пиразолу и к имидазолу, триазиновым и пуриновым соединениям, которые являются полезными в качестве ингибиторов вирусных РНК- и ДНК-полимераз, включая, в частности, но без ограничения, полимеразы вируса гриппа, вируса Хантаан (HTNV), вируса конго-крымской геморрагической лихорадки (CCHFV), вируса лихорадки долины Рифт (RVFV), гепатита В, гепатита С, полиомиелита, вируса коксаки А и В, риновируса, ЕСНО-вирусов (энтерических цитопатических "сиротских" вирусов человека), ортопоксвируса (оспы), ВИЧ, вируса Эбола и вируса Западного Нила, и в особенности вируса гриппа, и вирусов семействаBunyaviridae, в частности вируса Хантаан, вируса конго-крымской геморрагической лихорадки и вируса лихорадки долины Рифт. Настоящее изобретение относится также к фармацевтическим составам, которые содержат вышеуказанные соединения, и к способам применения этих соединений для ингибирования вирусных РНК- и ДНК-полимераз и лечения пациентов, страдающих заболеваниями, которые вызывают различные РНК- и ДНК-вирусы, и различными раковыми заболеваниями. Кроме того, настоящее изобретение относится к способу получения соединений согласно изобретению. Предпосылки изобретения Вирусные заболевания являются одной из основных причин смертности и экономических потерь в мире. Из разнообразных вирусных болезней инфекции гриппа, ВИЧ, вируса гепатита В и вируса гепатита С являются наиболее важными и ответственными за большое количество смертельных случаев. Существует несколько препаратов для лечения ВИЧ, лишь немного - для лечения вируса гепатита В, в то время как для лечения вируса гепатита С хорошие препараты отсутствуют. Гепатит С представляет собой вирусное заболевание печени, которое вызывает инфекция вируса гепатита С (Hepatitis С Virus, HCV). Во всем мире носителями хронической инфекции HCV являются примерно 170 млн человек, из которых около 2.7 млн проживает в Соединенных Штатах Америки. HCV является основной причиной цирроза,распространенной причиной гепатоцеллюлярного рака и основной причиной трансплантации печени в Соединенных Штатах. В настоящее время единственными апробированными способами лечения HCV являются монотерапия -интерфероном и комбинационная терапия -интерфероном-рибавирином. В этой связи желательным является нахождение ингибиторов вирусных РНК- и ДНК-полимераз. Краткое описание изобретения Настоящее изобретение относится, в частности, к соединениям, представленным формуламиZ = Н; С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероцикло, галоген, в частности F, Cl, Br и I; ОН, NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло); Е = (CH2)nONHR1; n - целое число от 0 до 6, более типично 0-3;R1 = арил или гетероцикло; каждый из W, Y, R отдельно выбирают из группы, включающей Н; С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероцикло, галоген, в частности F, Cl, Br и I; О, ОН, Оалкил, Оарил,NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло); при условии, что по меньшей мере один из W, Y и R отличается от Н и NH2, и при этом как W, так иY могут вместе = О; и каждый D отдельно представляется собой ОН, Оалкил, Оарил, Fl и Н,а также к их фармацевтически приемлемой соли, пролекарству и смесям. Другой аспект настоящего изобретения относится к фармацевтическому составу, содержащему по меньшей мере одно из вышеописанных соединений. Следующий аспект настоящего изобретения относится к способу ингибирования вирусной РНКполимеразы у пациента путем введения данному пациенту по меньшей мере одного из вышеописанных-1 016830 соединений в количестве, эффективном для ингибирования вирусной РНК-полимеразы. Еще один аспект настоящего изобретения относится к способу лечения пациента, страдающего вирусной РНК-инфекцией, который включает введение указанному пациенту эффективной дозы по меньшей мере одного из вышеописанных соединений. Другие задачи и достоинства настоящего изобретения станут очевидными для специалистов в данной области техники из следующего подробного описания, в котором предпочтительные варианты реализации показаны и описаны только в целях иллюстрации оптимальных условий. При этом следует понимать, что изобретение может иметь различные другие варианты реализации, и некоторые детали изобретения могут быть модифицированы в различных очевидных аспектах без отклонения от сути изобретения. В соответствии с этим данное описание следует рассматривать как иллюстративное, но не в качестве ограничительного. Краткое описание фигур Фиг. 1 - график, показывающий, что ТА-18 представляет собой субстрат для человеческой аденозинкиназы; фиг. 2 - график, показывающий, что ТА-18 превращается в фосфорилированные метаболиты в клетках СЕМ человека; фиг. 3 - графики, показывающие, что обработка ТА-18 приводит к снижению уровней GTP (гуанозин-5'-трифосфата); фиг. 4 - график, показывающий, что ингибирование активности аденозинкиназы йодотуберцидином ингибирует метаболизм ТА-18 в клетках человека; фиг. 5 - график, показывающий, что ингибирование активности аденозинкиназы йодотуберцидином также препятствует снижению уровней GTP, которое вызывается ТА-18; фиг. 6 - график, показывающий, что из ТА-18 образуется гораздо меньше внутриклеточных метаболитов, чем из рибавирина; фиг. 7 - график, показывающий, что обработка рибавирином также приводит к снижению уровнейGTP в клетках человека. Оптимальный и другие варианты реализации Настоящее изобретение относится, в частности, к соединениям, представленным следующими формулами:Z = Н; С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероцикло, галоген, в частности F, Cl, Br и I; ОН, NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло); Е = (CH2)nONHR1; n - целое число от 0 до 6, более типично 0-3;R1 = арил или гетероцикло; каждый из W, Y, R отдельно выбирают из группы, включающей Н; С 1-С 6 алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероцикло, галоген, в частности F, Cl, Br и I; О, ОН, Оалкил, Оарил,NH2, NH-(С 1-С 6 алкил, циклоалкил, арил или гетероцикло); при условии, что по меньшей мере один из W, Y и R отличается от Н и NH2, и при этом как W, так иY могут вместе = О; и каждый D отдельно представляется собой ОН, Оалкил, Оарил, Fl и Н,а также к их фармацевтически приемлемой соли, пролекарству и смесям. Стереохимия заместителей в этих соединениях может представлять собой вариант (R) или (S) в замещенных позициях. Смеси различных стереоизомеров также включаются в область распространения изобретения. Далее приведены определения терминов, используемых в описании настоящего изобретения. Эти определения относятся к соответствующим терминам, употребляемым в описании отдельно или применительно к части более крупной группы, если в конкретных случаях не указаны иные ограничения. Термин "алкил" относится к не содержащим замещений прямым или разветвленным углеводородным-2 016830 группам, типично содержащим от 1 до 6 атомов углерода и более типично - от 1 до 3 атомов углерода. Примеры пригодных алкильных групп включают метил, этил и пропил. Примеры разветвленных алкильных групп включают изопропил и t-бутил. Примерами пригодных аклкоксигрупп являются метокси, этокси и пропокси. Циклоалкильные группы обычно содержат 3-6 атомов углерода и включают циклопропил, циклобутил, циклопентил и циклогексил. Примерами галоидных групп являются Cl, F, Br и I. Алкенильные группы обычно содержат 2-6 атомов углерода и включают этенил, пропенил и бутенил. Циклоалкенильные группы обычно содержат 3-6 атомов углерода и включают циклопропенил, циклобутенил, циклопентенил и циклогексенил. Алкинильные группы обычно содержат 2-6 атомов углерода и включают ацетиленил и пропинил. Термин "арил" относится к моноциклическим или многоядерным ароматическим углеводородным группам, которые обычно содержат в ядре от 6 до 14 атомов углерода и включают, в частности, фенил, 2 нафтил, 1-нафтил, 4-бифенил, 3-бифенил, 2-бифенил и дифенильные группы, все из которых могут содержать замещения. Термин "гетероцикло" относится к насыщенным или ненасыщенным одно- или многоядерным группам. Примерами многоядерных ароматических (ненасыщенных) гетероциклических групп являются 2 хинолинил, 3-хинолинил, 5-хинолинил, 6-хинолинил, 7-хинолинил, 1-изохинолинил, 3-изохинолинил, 6 изохинолинил, 7-изохинолинил, 3-циннолил, 6-циннолил, 7-циннолил, 2-хиназолинил, 4-хиназолинил, 6 хиназолинил, 7-хиназолинил, 2-хиноксалинил, 5-хиноксалинил, 6-хиноксалинил, 1-фталазонил, 6 фталазинил, 1-5-нафтаридин-2-ил, 1,5-нафтаридин-3-ил, 1,6-нафтаридин-3-ил, 1,6-нафтаридин-7-ил, 1,7 нафтаридин-3-ил, 1,7-нафтаридин-6-ил, 1,8-нафтаридин-3-ил, 2,6-нафтаридин-6-ил, 2,7-нафтаридин-3-ил,индолил, 1 Н-индазолил, пуринил и птеридинил. Примерами одноядерных гетероциклических групп являются пирролил, пиранил, оксазолил, тиазолил, тиофенил, фуранил, имидазолил, пиразолил, пиридинил, пиразинил, пиримидинил, 4-пиримидинил,3-пиримидинил и 2-пиримидинил, пиридазинил, изотиазолили и изоксазолил. Примеры насыщенных гетероциклических групп включают пирролидинил, имидазолидинил, пиразолидинил, пипериденил, пиперазинил и морфолинил. Гетероциклические группы содержат N, О и/или S и обычно имеют от 5 до 10 атомов в ядре(ядрах),а также обычно содержат в ядре 1, 2 или 3 гетероатома (например, N, О и S). Если требуется, вышеуказанные алкильные, циклоалкильные, алкенильные, циклоалкенильные, арильные и гетероциклические группы могут содержать замещения. В случае замещений такие группы типично содержат галоидные и/или алкильные заместители и/или (CH2)nONH2, где n представляет собой целое число от 0 до 6, более типично 0-3. Следует понимать, что соединения согласно настоящему изобретению включают все оптические изомеры и стереоизомеры с различными возможными атомами молекулы. Соединения согласно настоящему изобретению могут образовывать пролекарства с гидроксильными или аминными функциональными группами с использованием алкокси, аминокислотных и других групп в качестве элементов, образующих пролекарства. Так, например, гидроксиметильные группы можно превращать в -ОСН 2 Р(О)(ОН)2 и пролекарства фосфонатов. Кислородный атом гидрометила можно превращать в СН 2, а затем - в СН 2 Р(О)(ОН)2 и пролекарства. Пролекарственные формы соединений, содержащих различные азотные функциональные группы(амино, гидроксиамино, амид и т.п.), могут включать следующие типы производных, где группа R может отдельно представлять собой водород, замещенную или незамещенную алкильную, арильную, алкенильную, алкинильную, гетероциклическую, алкиларильную, аралкильную, аралкенильную, аралкинильную,циклоалкильную или циклоалкенильную группы, как определено выше.(а) карбоксамиды, -NHC(O)R,(б) карбаматы, -NHC(O)OR,(в) (ацилокси)алкилкарбаматы, -NHC(O)OROC(O)R,(г) энамины, -NHCR(=CHCO2R) или -NHCR(=CHCONR2),(д) основания Шиффа, -N=CR2,(е) основания Манниха (из карбоксимидных соединений), RCONHCH2NR2. Получение таких пролекарственных производных обсуждается в различных литературных источниках (см., например, Alexander et al., J. Med. Chem, 1988, 31, 318; Aligas-Martin et al., PCT WO pp/41531,p. 30). Азотная функциональная группа, которая подвергается превращению при получении этих производных, представляет собой один или несколько атомов азота соединения согласно изобретению. Пролекарственные формы карбоксилсодержащих соединений согласно изобретению включают сложные эфиры (-CO2R), где группа R соответствует спирту, выделение которого в организме в результате ферментных или гидролитических процессов происходит на фармацевтически приемлемых уровнях. Другое пролекарство, которое получают из карбоновой кислотной формы согласно изобретению, может представлять собой соль четырехзамещенного основания структура которой описана в работе Bodor et al., J. Med. Chem. 1980, 23, 469. Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают соли,полученные из фармацевтически приемлемых неорганических или органических кислот. Примеры пригодных кислот включают соляную, бромную, серную, азотную, хлорную, фумаровую, малеиновую, фосфорную, гликолевую, молочную, салициловую, янтарную, толуол-р-сульфоновую, винную, уксусную,лимонную, метансульфоновую, муравьиную, бензойную, малоновую, нафталин-2-сульфоновую, трифторуксусную и бензолсульфоновую кислоты. Соли, полученные из соответствующих оснований, включают, например, соли натрия и аммиака. Некоторые соединения, которые относятся к области охвата настоящего изобретения, представлены следующими формулами: Ниже представлен репрезентативный пример N-арилкарбоксамидазолрибозида Ниже представлены репрезентативные примеры углеродзамещенных азолрибозидов согласно данному описанию Структуры репрезентативных новых соединений 1D-рибофуранозина, которые синтезировали для скрининга антивирусной активности, показаны ниже Синтез соединений Соединения согласно настоящему изобретению можно получить в соответствии со следующими схемами.[IA-3] N1-(3-фторфенил)инозин. Схема реакции синтеза TBS-IA-3 и IA-3[RN-3] 5-амино-4-N-3-фторфенилкарбоксамид-1D-рибофуранозил-1 Н-имидазол. Схема реакции синтеза RN-3[TBS-TA-8] (1-[2',3',5'-трис(О-трет-бутилметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил)карбоксальдегид. Схема реакции синтеза TBS-TA-8a Реагенты и режимы: (1) 1 М NaOMe, MeOH, температура окружающей среды, 2 ч; (2) TBDMSCI,имидазол, DMAP, DMF, температура окружающей среды, 18 ч; (3) DIBALH (диизобутилалюминий гидрид), CH2Cl2, -78C, 4 ч. ТА-18, 3-этинил-1-(-D-рибофуранозил)-[1,2,4]-триазол. Схема реакции синтеза ТА-18a(2) 1 М TBAF (тетра-n-бутиламмоний фторид) в тетрагидрофуране, температура окружающей среды, 2 ч. ТА-12, 1-этинил-1-(-D-рибофуранозил)-[1,2,4]-триазол. Схема реакции синтеза ТА-12a(2) 1 М TBAF в THF, температура окружающей среды, 2 ч. ТА-13, 1-(1D-рибофуранозил-[1,2,4]-триазол-3-ил)этанон. Схема реакции синтеза ТА-13a(2) 1 М TBAF в THF, температура окружающей среды, 2 ч. ТА-14, 1-(1D-рибофуранозил-[1,2,4]триазол-3-ил)фенилметанол. Схема реакции синтеза ТА-14a Реагенты и режимы: (1) PhMgCl, THF, 0C, 3 ч; (2) 1 М TBAF в THF, температура окружающей среды, 2 ч. ТА-15, 1-(1D-рибофуранозил-[1,2,4]триазол-3-ил)фенилметанон. Схема реакции синтеза ТА-15 а Реагенты и режимы: (1) РСС (пиридинийхлорхромат), CH2Cl2, температура окружающей среды, 4 ч, (2) 1 М TBAF в THF, температура окружающей среды, 2 ч. ТА-17, 3-(1,1-дифторэтил)-1D-рибофуранозил[1,2,4]триазол. Схема реакции синтеза ТА-17 а Реагенты и режимы: (1) DAST (диэтиламиносера трифторид), CH2Cl2, кипячение с обратным холодильником, 12 ч, (2) 1 М TBAF в THF, температура окружающей среды, 2 ч. ТА-19, 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)-2,2,2-трифторэтанол. Схема реакции синтеза ТА-19 а- 10016830 Схема реакции синтеза TA-20a(2) NH3, МеОН, 60C, 24 ч; (3) 1 М TBAF в THF, температура окружающей среды, 4 ч. Далее представлены различные соединения и данные биологических испытаний. Сводка соединений и их антивирусной активности Ниже показаны производные соединения 1D-рибофуранозилазола и скрининг их антивирусной активности по отношению к вирусу гриппа подтипа А H3N2 Далее приведено общее описание протокола анализа на примере вируса гриппа. Следует понимать,что такой же протокол применяется для других испытуемых вирусов. 2.0. Общее описание протокола определения антивирусной активности по отношению к вирусу гриппа Анализ антивирусной активности и токсичности. Анализ активности против вируса гриппа позволяет определить эффективность одноразовой дозы с определенной концентрацией испытуемых соединений. Для анализа использовали эпителиальные клетки почки собак Мадин-Дарби (MDCK), чтобы определить влияние указанных соединений на предотвращение цитопатического эффекта (СРЕ), индуцированного инфекцией вируса гриппа A/Udorn/72. Ниже в табл. 1 показана типичная съемка планшета. СС = контрольные клетки, CD = лунки с положительными контрольными соединениями,VC = контрольные вирусы. Цифры указывают номера отдельных соединений в лунках. В качестве положительного контрольного соединения в каждый цикл включали рибавирин. Субконфлюэнтные культуры клеток MDCK поместили в планшеты с 384 лунками для анализа антивирусной активности (СРЕ). Через 24 ч к клеткам добавили лекарственные препараты. В назначенное время в лунки для определения СРЕ добавили также 100 доз тканевой культуры (100 TCID50s), инфицированной вирусом A/Udorn/72. Через 72 ч определили жизнеспособность клеток при помощи набора CellTiter-Glo(Promega). Эффективными считали соединения, которые ингибировали вирус-индуцированный СРЕ более чем на 50%. Анализ жизнеспособности клеток с использованием набора CellTiter-Glo. Определение СРЕ, индуцированного вирусом гриппа, основано на количественном анализе АТР(аденозинтрифосфат), который является показателем метаболически активных клеток. Для анализа использовали серийно выпускаемый люминесцентный набор для определения жизнеспособности клетокCellTiter-Glo (Promega, Мэдисон, Висконсин). Этот метод является надежным для определения цитотоксичности и пролиферации клеток в культуре. Процедура включает добавление одного реагента (реагент CellTiter-Glo) непосредственно в предварительно культивированные в среде субконфлюэнтные клетки. Это индуцирует лизис клеток и генерацию биолюминесентного сигнала (время полужизни более 5 ч в зависимости от типа клеток), который пропорционален содержанию присутствующего АТР (являющегося биомаркером жизнеспособности). 3.0. Материалы и методики 3.1. Материалы. КлеткиA/Udorn/72; H3N2; пассаж 2; 140 СТ 05 Реагент конечной точки CellTiter-Glo-Promega Субстрат каталожныйG755B Буфер - каталожныйG756B Контрольный препарат рибавирин - МР Biomedicals, каталожный 196066. 3.2. Методики. В первый день клетки MDCK вырастили до конфлюэнтности 90%, затем трипсинизировали, регенерировали, центрифугировали и дважды промыли в забуференном фосфатом физиологическом растворе, чтобы удалить остаточную серу. Затем клетки разбавили в бессывороточной среде Игла, модифицированной по способу Дульбекко, поместили аликвотами в планшеты с 384 лунками (20 мкл/лунка) и выдержали для соединения с планшетом в течение ночи при 37C. На второй день произвели визуальный контроль морфологии клеток на небольшой случайной выборке планшетов. В отдельные лунки планшетов добавили испытуемые соединения (5 мкл) до конечной концентрации 10 мкМ и концентрации диметилсульфоксида (DMSO) 0,5%. Затем планшеты снова выдерживали при температуре 37 С в течение 72 ч. На пятый день клетки исследовали с помощьюCellTiter-Glo люминесцентным методом. Дальнейшие детали, касающиеся протокола анализа, можно найти в работе Noah et al. Клеточный люминесцентный анализ является эффективным для высокопроизводительного скрининга противогриппозных антивирусных соединений, см. работу Antiviral Research (2006), doi:10.1-16/j. antiviral. 2006.07.006 (копия размещена на сайте www.sciencedirect.com), все содержание которой включено в данное описание в качестве ссылки. На основании предварительных исследований аденозинкиназы и Т-18 можно сделать следующие заключения. 1. Определили активность субстрата с аденозинкиназой и рядом синтезированных аналогов (см. далее табл. 2). 2. Используя меченый радиоизотопом ТА-18, подтвердили, что он является субстратом для аденозинкиназы человека (см. фиг. 1). Расхождение активности между результатами, показанными в таблице на следующей странице, и результатами с соединением, меченым радиоизотопом, вероятно, вызваны применением различных концентраций соединений в экспериментах (100 мкМ использовали для результатов, показанных в таблице, и 10 мкМ - для всех остальных экспериментов). 3. ТА-18 превратили в фосфорилированные метаболиты в клетках человека (см. фиг. 2). 4. Обработка Т-18 привела к снижению уровней GTP (см. далее табл. 3 и фиг. 3). 5. Ингибирование активности аденозинкиназы йодотуберцидином (см. фиг. 4) привело к ингибированию метаболизма Т-18 в клетках человека. Это показывает, что аденозинкиназа является главным ферментом, участвующим в метаболизме Т-18 в этой клеточной линии. 6. Ингибирование активности аденозинкиназы йодотуберцидином (см. фиг. 5) также предотвратило снижение уровней GTP, вызванное Т-18. Это показывает, что метаболит ТА-18 является ответственным за уменьшение уровней GTP, которое наблюдали в клетках, обработанных ТА-18. 7. Обработка рибавирином также уменьшала уровни GTP в клетках человека (см. фиг. 7). Поскольку внутриклеточных метаболитов из ТА-18 было гораздо меньше, чем из рибавирина (см. фиг. 6), этот результат показывает, что метаболиты ТА-18 оказывают более сильное влияние на уменьшение уровнейGTP, чем метаболиты рибавирина. 8. Эти предварительные результаты позволяют предположить, что антивирусный механизм дейст- 13016830 вия ТА-18 связан с уменьшением внутриклеточных уровней GTP, возможно, вследствие ингибирования активности IMP (инозинмонофосфат) дегидрогеназы. Таблица 2 Активность аденозинкиназы с некоторыми нуклеозидными аналогами Аденозинкиназу человека инкубировали со 100 мкМ каждого соединения и АТР. После инкубации в течение желаемого времени при 37C реакцию останавливали и при помощи ВЭЖХ определяли превращение соединения в соответствующий 5'-монофосфат. При испытании этих соединений мы не отметили разницы в уровне ингибирования указанных трех вирусов и гриппа. Следующие результаты показали важное обстоятельство, касающееся антивирусной активности испытуемых соединений согласно данному изобретению. Так, например, антивирусный скрининг при проведении испытаний с вирусом Хантаан (HTNV), вирусом конго-крымской геморрагической лихорадки (CCHFV), вирусом лихорадки долины Рифт (RVFV) и вирусом гриппа показал селективность соединений согласно данному изобретению в пределах семейства Bunyaviridae. В частности,соединение 18-0 показало антивирусную активность по отношению к HTNV и вирусу гриппа. Соединение IA-3 показало антивирусную активность по отношению к HTNV, a IM-18-антивирусную активность по отношению к вирусу гриппа. Соединение PZA-O показало антивирусную активность по отношению к вирусу гриппа. Соединение RC-3 показало антивирусную активность по отношению к HTNV и вирусу гриппа, a RN-3 - антивирусную активность по отношению к HTNV. Соединение ТА-1 показало антивирусную активность по отношению к CCHFV, TA12 - антивирусную активность по отношению к HTNV,ТА-14 и 16 - антивирусную активность по отношению к HTNV, ТА-18 - антивирусную активность по отношению к HTNV, вирусу гриппа и CCHFV, a ТА-23 - антивирусную активность по отношению кRVFV. Предпочтительными являются соединения серии Т. Далее приведены неограничительные примеры, которые дополнительно иллюстрируют настоящее изобретение. Пример 1 2',3',5'-трис-(О-трет-бутилдиметилсилил)инозин (TBS-1). Инозин (5,36 г, 20 ммоль) защитили TBSCI (18,1 г, 120 ммоль) и имидазолом (10.9 г, 160 ммоль) в сухом DMF (100 мл) при температуре окружающей среды в течение 48 ч. После концентрирования в вакууме разбавили CH2Cl2 до 200 и промыли порциями по 100 мл воды (4 промывки), насыщенным NH4Cl (3 промывки) и насыщенным NaCl с последующей рекристаллизацией в EtOAc, получив белый кристаллический осадок (10,9 г, 17,8 ммоль, 90%). Инфракрасная Фурье-спетроскопия (тефлоновая плата, см-1) 1706; 1 Н ЯМР (400 МГц, CDCl3-d)13.30 (1 Н, 1s), 8.31 (1 Н, 1s), 5.98 (1 Н, d, J=4.8 Гц), 4.46 (1 Н, m), 4.26N1-(3-фторфенил)-2',3'5'-трис-(О-трет-бутилдиметилсилил)инозин (TBS-IA-3). В сосуд Шленка, высушенный в печи, добавили TBS-IA (2,4 г, 4,0 ммоль), 3-фторфенилборную кислоту (1,1 г, 8,0 моль), безводную соль Cu(ОАс)2 (800,0 мг, 4,4 ммоль), пиридин-N-оксид (800 мг, 4,0 ммоль), молотые молекулярные сита 4(1 г) и якорь магнитной мешалки. Сосуд герметично закрыли резиновой мембраной, откачали и продули кислородом. Затем добавили сухой пиридин (647 мкл, 8,0 ммоль) и осушенный при помощи молекулярного сита CH2Cl2 (20 мкл) и интенсивно перемешали реакционную смесь при температуре окружающей среды в течение 24 ч. Затем реакцию погасили насыщенным NH4OH в МеОН (0,5 мл в 5 мл соответственно) с последующим разбавлением гексаном до 500 мл. Органический состав промыли порциями по 250 мл: воды, насыщенного NH4Cl, 1 М NaCl и насыщенного NaCl. Затем осушили органический состав над Na2SO4 и сконцентрировали в вакууме. Все соединения очистили способом флешхроматографии среднего давления (Isco CombiFlash GRADUATE) с CH2Cl2/МеОН в качестве элюента,получив аморфный белый осадок (F. W. = 705,1, 1,93 г, 2,74 ммоль, 67%). Инфракрасная Фурьеспетроскопия (тефлоновая плата, см-1) 1706; 1 Н ЯМР (400 МГц, CDCl3-d)8.20 (1 Н, 1s), 7.99 (1 Н, 1s), 7.45 (1 Н, m), 7.16-7.13 (3H, m), 5.99 (1 Н, d,J=4.8 Гц), 4.46 (1 Н, m), 4.29 (1 Н, m), 4.11 (1 Н, m), 3.97 (1 Н, m), 3.77 (1 Н, m), 0.93-0.80 (27 Н, mult. s), 0.120.16 (18 Н, mult. s); 13 С ЯМР (400 МГц, CDCl3-d)162.6 (J=248.1 Гц), 156.0, 147.1, 146.4, 138.4 (J=9.5 Гц), 130.7 (J=9.0 Гц), 124.7, 123.0, 116.3 (J=20.0 Гц), 115.2 (J=23.9 Гц), 88.1, 85.4, 76.7, 71.6, 62.3,TBS - не зарегистрировано;N1-(3-фторфенил)инозин (IA-3). В круглодонную колбу добавили TBS3-IA-3 (1,06 г, 1,5 ммоль), сухой THF (25 мл) и якорь магнитной мешалки, а затем начали перемешивание при -10 С. Далее добавили 5,0 мл 1 М раствора тетратбутиламмонийфторида/THF и через 1,5 ч (завершение реакции согласно анализу методом тонкослойной хроматографии) и загрузили раствор непосредственно в гравитационную колонку диаметром 5 см с силикагелем (350 мл силикагеля 60 , 70-230 меш) с ацетоном в качестве элюента для удаления основной массы тетрабутиламмониевых солей. Затем осадок очистили способом флеш-хроматографии среднего давления (Isco CombiFlash GRADUATE) с толуолом/EtOH в качестве элюента, получив аморфный белый осадок (F. W. = 362,3, 469 мг, 1,29 ммоль, 86%). Инфракрасная Фурье-спетроскопия (KBr, см-1) 3394, 2931, 1699, 1601, 1578, 1546, 1489, 1226; 1 Н ЯМР (CD3OD, 400 МГц)8.39 (1 Н, 1s), 8.30 (1 Н, 1s), 7.57 (1 Н, m), 7.35-7.26 (3H, m), 6.04 (1 Н, d,J=5.9 Гц), 4.63 (1 Н, m), 4.33 (1 Н, m), 4.13 (1 Н, m), 3.86 (1 Н, m), 3.75 (1 Н, т); 13 С ЯМР (CD3OD, 400 МГц)164.1 (J=245.4 Гц), 157 9, 149.2, 148.7, 141.5, 139.9 (J=10.2 Гц), 132.1- 15016830 г, 2 ммоль) добавили в круглодонную колбу, растворили в абсолютном EtOH (30 мл) и довели до кипения при перемешивании. Затем в раствор добавили 5N NaOH (10 мл) и кипятили с обратным холодильником в течение 4 ч. Колбу сняли с нагревателя, охладили до температуры окружающей среды и нейтрализовали (рН 7) 6 Н HCl. Водную смесь экстрагировали 3 порциями EtOAc, которые затем осушили над Сложный метиловый эфир (1-[2',3'5'-трис-(О-трет-бутилдиметилсилил)D-рибофуранозил]-(1,2,4 триазол-3-ил)карбоновой кислоты. В раствор метил-1-(-D-рибофуранозил)-1,2,4-триазол-3 карбоксилата (5,1345 г, 19,8 ммоль), имидазола (10,78 г, 158,3 ммоль) и DMAP (50 мг) в сухом DMF (50 мл) добавили трет-бутилдиметилсилилхлорид (11,74 г, 77,9 ммоль). Реакционную смесь перемешали при температуре окружающей среды в течение ночи. После этого анализ способом тонкослойной хроматографии (5% МеОН/CH2Cl2, Rf=62) показал полное превращение исходного материала в единственный продукт. Белую суспензию перелили в двухслойный состав из воды (100 мл) и DCM (дихлорметан) (100 мл). Органический слой отделили, а водную фазу повторно экстрагировали DCM (350 мл). Объединенные органические экстракты осушили (безводным Na2SO4), профильтровали и выпарили при пониженном давлении, получив белый осадок, который рекристаллизовали из гексана, и получили желаемый продукт в виде белого порошка (F. W. = 602,00, 10,07 г, 84%). 1 Н ЯМР (200 МГц, CDCl3)8.57 (s, 1H), 5.84 (d, 1 Н, J1',2'=4.9 Гц, Н-1'), 4.45 (m, 1 Н, Н-2'), 4.22 (m,1 Н, Н-3'), 4.17-4.09 (m, 1 Н, Н-4'), 3.99 (s, 3H), 3.98-3.90 (dd, 1H, J5'a,5'b=11.9 и J5'a,4'=3.7 Гц, Н-5 а), 3.80-3.73(1-[2',3',5'-трис-(О-трет-бутилдиметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил)карбоксальдегид [TBS-TA-8]. В раствор сложного метилового эфира (1-[2',3',5'-трис-(О-трет-бутилдиметилсилил)-D-рибофуранозил]-(1,2,4-триазол-3-ил)карбоновой кислоты (4,2140 г, 7,0 ммоль) в сухом CH2Cl2 (15 мл) при -78 С медленно добавили DIBAL-H (17,5 мл, 1 М раствор в CH2Cl2), чтобы поддерживать внутреннюю температуру ниже -65 С. Реакционную смесь перемешали в течение 4 ч при -78 С, затем погасили путем медленного добавления холодного (-78 С) МеОН (7 мл) при поддержании внутренней температуры ниже -65 С. Образовавшуюся белую эмульсию выдержали для нагревания до температуры окружающей среды при перемешивании в течение 2 ч. Затем реакционную смесь разбавили CH2Cl2 (25 мл) и промыли 0,5 М NaOH (25 мл). Водную смесь экстрагировали CH2Cl2 (3). Объединенный органический раствор промыли солевым раствором, осушили над безводным Na2SO4 и сконцентрировали при пониженном давлении, получив сырой продукт в форме светло-желтого масла, которое очистили на колонке с силикагелем (5% МеОН/CH2Cl2), и получили очищенный продукт в виде бесцветного масла. После его сушки при пониженном давлении в течение 5 дней получили белый осадок (F. W. = 571,97, 3,1668 г,78%). 1 Н ЯМР (200 МГц, CDCl3)10.01 (s, 1H), 8.57 (s, 1H), 5.82 (d, 1H, J1',2'=4.2 Гц, Н-1'), 4.48 (m, 1 Н, Н 2'), 4.25 (m, 1 Н, Н-3'), 4.18-4.09 (m, 1 Н, Н-4'), 3.95-3.88 (dd, 1H, J5'a,5'b=11.9 и J5'a,4'=3.7 Гц, Н-5 а), 3.79-3.72 3-Этинил-1-(2',3',5'-трис-(О-трет-бутилдиметилсилил)D-рибофуранозил)-1,2,4-триазол [TBS-TA18]. В раствор (1-[2',3',5'-трис-(О-трет-бутилдиметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил) карбоксальдегида [TBS-TA-8] (572 мг, 1 ммоль) и диметил-1-диазо-2-оксопропилфосфоната (249 мг, 1,3 ммоль) в безводном метаноле (5 мл) добавили безводный K2CO3 (208 мг, 2,1 ммоль). Образовавшийся светло-желтый раствор перемешали в течение 24 ч. Смесь погасили водой (10 мл) и экстрагировали Et2O(420 мл). Объединенные экстракты промыли NaHCO3(водный) (насыщенный, 10 мл) и солевым раствором(насыщенный, 10 мл), а затем осушили над Na2SO4. После удаления растворителя в вакууме получили неочищенный продукт, который очистили способом флеш-хроматографии (5-20% EtOAC/гексан), получив белый осадок. После его рекристаллизации из гексана образовался желаемый продукт в форме белого порошка (F. W. = 567,98, 435 мг, 76%). 1 Н ЯМР (200 МГц, CDCl3)8.72 (s, 1 Н), 5.69 (d, 1 Н, J1',2'=4.03 Гц, Н-1'), 4.458 (m, 1 Н, Н-2'), 4.235 (m,1 Н, Н-3'), 4.11 (m, 1 Н, Н-4'), 3.95-3.88 (dd, 1H, J5'a,5'b=11.9 и J5'a,4'=4.03 Гц, Н-5 а), 3.79-3.72 (dd, 1H,J5'a,5'b=11.35 и J5'b,4'=2.9 Гц, Н-5b), 3.06 (s, 1H), 0.95-0.78 (mult. s, 27H), 0.14-0.09 (mult. s, 18H),жидкостная хроматография с масс-спектроскопией (химическая ионизация при атмосферном давлении): рассчитали для C27H53N3O4Si3[М+1]+ 568.34 m/z, получили в результате эксперимента: 568.28 m/z. ВЭЖХ 100% CH3CN, температура окружающей среды, 6,62 мин. Пример 9 3-Этинил-1-(-D-рибофуранозил)-[1,2,4]триазол [ТА-18]. В перемешанный раствор 3-этинил-1(2',3',5'-трис-(О-трет-бутилдиметилсилил)D-рибофуранозил)-1,2,4-триазола [TBS-TA-18] (125 мг, 0,22 ммоль) в безводном THF (3 мл) добавили 1 М TBAF в THF (0,8 мл, 0,8 ммоль). Смесь перемешали при температуре окружающей среды в течение 2 ч до окончания реакции согласно анализу методом тонкослойной хроматографии (50%-ацетон/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флеш-хроматографии (50%-ацетон/CH2Cl2), получив белый осадок, который рекристаллизовали из 5% MeOH/CH2Cl2, и получили желаемый продукт в виде белого кристаллического порошка (F. W. = 225,20, 41 мг, 82%). 1 Н ЯМР (200 МГц, CD3OD)8.72 (s, 1H), 5.84 (d, 1H, J1',2'=3.5 Гц, Н-1'), 4.43 (m, 1 Н, Н-2'), 4.29 (m,1 Н, Н-3'), 4.09 (m, 1 Н, Н-4'), 3.83-3.79 (dd, 1H, J5'a,5'b=12.3 и J5'a,4'=3.3 Гц, Н-5 а), 3.73 (s, 1H), 3.70-3.65 (dd,1H, J5'b,5'a=12.9 и J5'b,4'=4.7 Гц, Н-5b). 13 С ЯМР (CD3OD, 400 МГц,)148.4, 145.6, 93.9, 86.9, 80.3, 75.0, 76.5, 71.6, 62.8. Жидкостная хроматография с масс-спектроскопией (ионизация электрораспылением): рассчитали для C9H11N3O4 [M+1]+ 226.08 m/z, получили в результате эксперимента: 225.23 m/z. Пример 10[TBSTA-12]. В раствор (1-[2',3',5'-трис(О-трет-бутилметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил) карбоксальдегида [TBS-TA-8] (1,1420 г, 2 ммоль) в атмосфере аргона в THF (50 мл) при 0 С по каплям добавили CH3MgCl (1,35 мл, 3 М раствор в THF). Реакционную смесь перемешали и контролировали протекание реакции при помощи тонкослойной хроматографии (5% MeOH/CH2Cl2, Rf=0.3). Полное исчезно- 17016830 вение исходного материала наблюдали через 3 ч. Затем реакционную смесь погасили насыщеннымNH4Cl(водный) (20 мл) и экстрагировали простым диэтиловым эфиром (325 мл). Объединенные органические экстракты осушили (безводным Na2SO4), профильтровали и выпарили при пониженном давлении,получив бесцветное масло, которое очистили в колонке с силикагелем (5% МеОН/CH2Cl2), и получили продукт в виде бесцветного масла (F. W. = 588,02, 1,0216 г, 87%). Пример 11THF (0,8 мл, 0,8 ммоль). Смесь перемешали при температуре окружающей среды в течение 2 ч до окончания реакции согласно анализу методом тонкослойной хроматографии (5% MeOH/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флешхроматографии (50%-ацетон/CH2Cl2), получив бесцветное масло (F. W. = 245,10, 130 мг, 79%). 1 Н ЯМР (200 МГц, CD3OD)8.63 (s, 1H), 5.82 (d, 1H, J1',2'=3.91 Гц, Н-1'), 4.89 (q, 1H, J=6 64 Гц), 4.45 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)этанон [ТА-13]. В суспензию 1-(2',3',5'-трис-(О-третбутилдиметилсилил)D-рибофуранозил[1,2,4]триазол-3-ил)этанола [TBS-TA-12] (1,764 г, 3 ммоль) и молотых молекулярных сит (0,3 г) в атмосфере аргона в CH2Cl2 (15 мл) добавили РСС (0,970 г, 4,5 ммоль) и перемешали при температуре окружающей среды, контролируя протекание реакции при помощи тонкослойной хроматографии (5% MeOH/CH2Cl2, Rf=0,7). Полное исчезновение исходного материала наблюдали через 4 ч. Затем реакционную смесь профильтровали через фторсил и сконцентрировали при пониженном давлении. Полученный осадок разделили между водой и простым диэтиловым эфиром и экстрагировали простым диэтиловым эфиром (325 мл). Объединенные органические экстракты осушили (безводным Na2SO4), профильтровали при пониженном давлении, а продукт выделили способом флеш-хроматографии (1% MeOH/CH2Cl2) в форме белого осадка (F. W. = 243,22, 65 мг, 76%). Затем этот продукт (207 мг, 0,35 ммоль) растворили в безводном THF (3 мл) и добавили 1 М TBAF в THF (1 мл, 1 ммоль). Смесь перемешали при температуре окружающей среды в течение 2 ч до завершения реакции согласно анализу методом тонкослойной хроматографии (5% MeOH/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флеш-хроматографии(50% ацетон/CH2Cl2), получив желаемый продукт в виде белого осадка (F. W. = 243,22, 65 мг, 76%). 1 Н ЯМР (200 МГц, CD3OD)8.84 (s, 1 Н), 5.94 (d, 1 Н, J1',2'=3.30 Гц, Н-1'), 4.49 (m, 1 Н, Н-2'), 4.35 (m,1 Н, Н-3'), 4.13 (m, 1 Н, Н-4'), 3.88-3.81 (dd, 1H, J5'a,5'b=12.1 и J5'a,4'=3.3 Гц, Н-5 а), 3.74-3.66 (dd, 1H,J5'b,5'a=12.10 и J5b,4'=4.4 Гц, Н-5b), 2.61 (s, 3H). Жидкостная хроматография с масс-спектроскопией (ионизация электрораспылением): рассчитали для C9H13N3O5 [М+1]+ 244,09 m/z, получили в результате эксперимента: 244.25 m/z. 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанол [ТА-14]. В раствор (1-[2',3',5'-трис-(Отрет-бутилметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил)карбоксальдегида [TBS-TA-8] (320 г,0,56 ммоль) в THF (2 мл) в атмосфере аргона при 0 С по каплям добавили PhMgCl (0,56 мл, 2 М раствор вTHF). Реакционную смесь перемешали, контролируя протекание реакции при помощи тонкослойной хроматографии (5% МеОН/CH2Cl2, Rf=0,33). Полное исчезновение исходного материала наблюдали через 2 ч. Затем реакционную смесь погасили насыщенным MH4Cl(водный) (20 мл) и экстрагировали простым диэтиловым эфиром (325 мл). Объединенные органические экстракты высушили (безводным Na2SO4),профильтровали и выпарили при пониженном давлении, получив бесцветный неочищенный продукт в виде масла, которое очистили способом флеш-хроматографии (5% МеОН/CH2Cl2), и получили 1-(2',3',5'трис-(О-трет-бутилдиметилсилил)-1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанол [TBS-TA-14] в виде бесцветного масла (F. W. = 650,08, 269 мг, 74%). В перемешанный раствор TBS-TA-14 (195 мг, 0,3 ммоль) в безводном THF (3 мл) добавили 1 МTBAF в THF (1 мл, 1 ммоль). Смесь перемешали при температуре окружающей среды в течение 2 ч до окончания реакции согласно анализу методом тонкослойной хроматографии (5% MeOH/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флеш-хроматографии (50%-ацетон/CH2Cl2), получив продукт в виде бесцветного масла (F. W. = 307,30,68 мг, 74%). 1 Н ЯМР (400 МГц, CD3OD, сложная смесь диаст.)8.62 (s, 1H), 7.45-7.23 (m, 5H), 5.82 (d, 1H,J1',2'=3.71 Гц, Н-1'), 4.45 (m, 1 Н), 4.31 (m, 1 Н), 4.01 (m, 1 Н), 3.82-3.59 (m, 2 Н), 2.31 (s, 1 Н). Жидкостная хроматография с масс-спектроскопией (химическая ионизация при атмосферном давлении): рассчитали для C14H17N3O5 [М+1]+ 308.12 m/z, получили в результате эксперимента: 308.24 m/z. Пример 14 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанон [ТА-15]. В суспензию 1-(2',3',5'-трис-(Отрет-бутилдиметилсилил)-1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанола [TBS-TA-14] [TBS14] (0,749 г, 1,15 ммоль) и молотых молекулярных сит (0,2 г) в атмосфере аргона в CH2Cl2 (5 мл) добавили РСС (0,373 г, 1,73 ммоль) и перемешали при температуре окружающей среды, контролируя протекание реакции при помощи тонкослойной хроматографии (5% MeOH/CH2Cl2, Rf=0,75). Полное исчезновение исходного материала наблюдали через 4 ч. Затем реакционную смесь профильтровали через фторсил и сконцентрировали при пониженном давлении. Полученный осадок разделили между водой и простым диэтиловым эфиром и экстрагировали простым диэтиловым эфиром (325 мл). Объединенные органические экстракты осушили (безводным Na2SO4), профильтровали и выпарили при пониженном давлении,получив неочищенный продукт в виде белого осадка (F. W. = 305,29, 0,5021 г, 67%). Затем этот продукт(0,198 г, 0,3 ммоль) растворили в безводном THF (3 мл) и добавили 1 М TBAF в THF (1 мл, 1 ммоль). Смесь перемешали при температуре окружающей среды в течение 2 ч до завершения реакции согласно анализу методом тонкослойной хроматографии (5% MeOH/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флеш-хроматографии (ацетон),получив желаемый продукт в виде белого осадка (F. W. = 305,29, 90 мг, 98%). 1 Н ЯМР (200 МГц, D2O)8.82 (s, 1H), 8.10 (m, 2 Н), 7.72 (m, 1 Н), 7.56 (m, 1 Н), 6.08 (d, 1 Н, J1',2'=3.30 Гц, Н-1'), 4.62 (m, 1 Н, Н-2'), 4.44 (m, 1 Н, Н-3'), 4.19 (m, 1 Н, Н-4'), 3.88-3.80 (dd, 1H, J5'a,5'b=12.82 и J5'a,4'=3.3 Гц, Н-5 а), 3.74-3.65 (dd, 1H, J5'b,5'a=12.82 и J5b,4'=5.1 Гц, Н-5b). Жидкостная хроматография с масс-спектроскопией (ионизация электрораспылением): рассчитали для C9H13N3O5 [M+1]+ 306.11 m/z, получили в результате эксперимента: 306.29 m/z.CH2Cl2 (5 мл) добавили DAST (20 мкл, 0,16 ммоль) и кипятили с обратным холодильником, контролируя протекание реакции при помощи тонкослойной хроматографии (5% MeOH/CH2Cl2, Rf=0,7). Через 12 ч реакционную смесь погасили приливаемой по каплям Н 2 О (25 мл), добавили CH2Cl2 (25 мл), органический слой отделили и промыли насыщенным NaHCO3 и Н 2 О (325 мл). Затем органический слой осушили (безводным Na2SO4), профильтровали, выпарили при пониженном давлении и выделили продукт TBSTA-17 способом флеш-хроматографии (5% MeOH/CH2Cl2) в виде белого осадка (F. W. = 608, 36,4 мг,42%). Раствор 1 М TBAF в THF (0,2 мл, 1 ммоль) добавили в раствор TBS-TA-17 (36,4 мг, 0,06 ммоль) в безводном THF (3 мл). Смесь перемешали при температуре окружающей среды в течение 2 ч до окончания реакции согласно анализу способом тонкослойной хроматографии (5% MeOH/CH2Cl2) и погасили МеОН (2 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флешхроматографии (50%-ацетон/CH2Cl2), получив желаемый продукт в виде белого осадка (F. W. = 265,21,12 мг, 75%). 1 Н ЯМР (400 МГц, CD3OD)8.79 (s, 1 Н), 5.88 (d, 1H, J1',2'=3.52 Гц, Н-1'), 4.46 (m, 1H, Н-2'), 4.32 (m,1 Н, Н-3'), 4.10 (m, 1 Н, Н-4'), 3.88-3.81 (dd, 1H, J5'a,5'b=12.1 и J5a,4'=3.5 Гц, Н-5 а), 3.74-3.66 (dd, 1H, J5'b,5'a=12.1 и J5'b,4'=4.7 Гц, H-5b), (t, 3H, J=18.5 Гц). Пример 16(100 мг, 0,17 ммоль) в сухом THF (2 мл) при 0 С добавили триметил(трифторметил)силан (33 мкл, 0,21 ммоль) и катализатор KO'Bu (1 мг). Реакционную смесь перемешали при указанной температуре в атмосфере аргона в течение 4,5 ч. Затем реакционную смесь выпарили при температуре окружающей среды,маслянистый осадок растворили в простом диэтиловом эфире (4 мл) и промыли водой (2 мл), осушили над безводным Na2SO4 и выпарили растворитель. Неочищенный продукт очистили в колонке с силикагелем (градиент мобильной фазы этилацетата в гексане от 10 до 20%), получив чистый продукт TBS-TA-19 в виде бесцветного масла (94 мг, 86%). Инфракрасная Фурье-спетроскопия (NaCl, см-1) 2955, 2931, 1473,1258, 1172, 1136, 837, 779. 1 Н ЯМР (200 МГц, CDCl3)8.35 (s, 1H), 5.75 (d, 1H, J=4.9 Гц), 5.16 (q, 1H, J=6.6 Гц), 4.49-4.58 (m,1H), 4.21-4.56 (m, 1H), 4.06-4.13 (m, 1H), 3.82-3.91 (m, 1H), 3.67-3.76 (m, 1H), 0.92 (s, 18H), 0.83 (s, 9H),0.14 (s, 3H), 0.13 (s, 6H), 0.09 (s, 6H), 0.01 (s, 3H). 13 С ЯМР (100 МГц, CDCl3)158.69, 144.34, 123.48 (q, J=282 Гц), 91.91, 86.17, 76.10, 71.90, 67.63 (q,J=34 Гц), 62.47, 25.97 (3C), 25.78 (3C), 25.61 (3C), 18.43, 18.01, 17.89, -4.51, -4.69 (2 С), -5.40, -5.51 (2 С). В перемешанный раствор TBS-TA-19 (434 мг, 0,68 ммоль) в безводном THF (3 мл) добавили 1 МTBAF в THF (1,4 мл, 1,4 ммоль). Смесь перемешали при температуре окружающей среды в течение 2,5 ч и погасили МеОН (1 мл). Растворитель удалили при пониженном давлении и выделили продукт способом флеш-хроматографии (30% ацетон, 70% гексан), получив желаемый продукт в форме масла (95 мг,47%). Инфракрасная Фурье-спетроскопия (NaCl, см-1) 3350, 1660, 1524, 1270, 1183, 1134, 867. 1 Н ЯМР (200 МГц, CDCl3)8.66 (s, 1H), 5.86 (d, 1 Н, J=3.3 Гц), 5.24 (q, 1H, J=7.0 Гц), 4.50 (m, 1H),4.37 (m, 1H), 4.07 (m, 1H), 3.77 (dd, 1H, J1=11.9 Гц, J1=3.3 Гц,), 3.72 (dd, 1H, J1=11.9 Гц, J1=3.3 Гц,). 13 С ЯМР (100 МГц, CDCl3)160.11, 145.55, 125.10 (q, J=282 Гц), 93.29, 86.94, 76.41, 71.59, 68.08 (q,J=33 Гц), 62.75. 3-(1D-рибофуранозил[1,2,4]триазол-3-ил)-3-гидроксипропионамид [ТА-20]. Металлический цинк промыли EtOH, ацетоном, простым диэтиловым эфиром и высушили. Затем Zn (130 мг, 2,1 ммоль) добавили в раствор (1-[2',3',5'-трис(О-трет-бутилметилсилил)D-рибофуранозил]-(1,2,4-триазол-3-ил)карбоксальдегида [TBS-TA-8] (572 мг, 1,0 ммоль), этилбром ацетата (0,35 мл, 3,1 ммоль) в THF (10 мл). Реакционную смесь кипятили с обратным холодильником в течение 3,5 ч. Затем раствор разбавили CH2Cl2(25 мл), промыли 3 порциями воды, осушили над Na2SO4 и сконцентрировали при пониженном давлении. Образовавшееся масло очистили в колонке с силикагелем (10-20% EtOAc/гексан), получив чистый продукт - сложный этиловый эфир 3-(2',3',5'-О-трис-(трет-бутилдиметилсилил)D-рибофуранозил)-[1,2,4]триазол-3-ил)-3-оксипропионовой кислоты (F. W. = 660,08, 455 мг, 69%). Этот материал использовали на следующей операции. В напорной трубке МеОН (15 мл) насытили газообразным NH3 и в раствор добавили сложный этиловый эфир 3-(2',3',5'-О-трис-(трет-бутилдиметилсилил)D-рибофуранозил)-[1,2,4]-триазол-3-ил)-3 оксипропионовой кислоты (306 мг, 0,46 ммоль). Реакционную смесь нагрели при 60 С в течение 24 ч. Образовавшийся раствор сконцентрировали при пониженном давлении. Неочищенный продукт очистили способом флеш-хроматографииEtOAc/гексан),получив 3-(2',3',5'-О-трис-(третбутилдиметилсилил)D-рибофуранозил)-[1,2,4]-триазол-3-ил)-3-гидроксипропионамид (F. W. = 631,04,240 мг, 88%). Этот материал использовали на следующей операции. 3-(2',3',5'-O-трис-(трет-бутилдиметилсилил)D-рибофуранозил)-[1,2,4]-триазол-3-ил)-3-гидроксипропионамид (150 мг, 0,24 ммоль) соединили с 1 М раствором тетрабутиламмония фторида (0,8 мл, 0,8 ммоль) в сухом THF (4 мл) и перемешали при температуре окружающей среды в течение 4 ч. Реакционную смесь погасили 5 мл МеОН, затем сконцентрировали при пониженном давлении. Неочищенный продукт очистили способом флеш-хроматографии (50% EtOAc/толуол); (F. W. = 288,26, 24 мг, 35%). 1 Н ЯМР (200 МГц, CD3OD)8.64 (s, 1 Н),5.83 (d, 1H, J=3.7 Гц, Н-1'),5.16 (m, 1 Н), m 4.45 (m, 1 Н,Н-2'),4.33 (m, 1 Н, Н-3'),4.09 (m, 1 Н, Н-4'),3.77-3.84 (dd, 1 Н, J5'a,4'=2.9, J5'a,5'b=12.1, Н-5'а),3.63-3.71(dd, 1 Н, J5'b,4'=4.8, J5'b,5'a=12.8, Н-5b')2.75-2.81 (m, 2 Н). Лекарственные формы Соединения согласно настоящему изобретению можно вводить любыми традиционными способами, доступными для применения, в сочетании с фармацевтическими препаратами в качестве отдельных терапевтических агентов или в комбинации терапевтических агентов. Их можно вводить отдельно, однако, обычно их вводят с фармацевтическим носителем, который выбирают в соответствии с предполагаемым способом введения и стандартной фармацевтической практикой. Соединения можно также вводить в сочетании с другими терапевтическими агентами, в частности с интерфероном (INF), интерфероном 2 а, интерфероном -2b, консенсусным интерфероном (CIFN), рибавирином, амантадином, ремантадином, интерлейкином-12, урсодеоксихолевой кислотой (UDCA) и глицирризином. Описанные здесь фармацевтически приемлемые носители, например транспортирующие агенты,адъюванты, наполнители или разбавители, хорошо известны специалистам в данной области техники. Обычно фармацевтически приемлемый носитель является химически инертным по отношению к активным соединениям и не имеет вредных побочных эффектов или токсичности при условиях применения. Фармацевтически приемлемые носители могут включать полимеры и полимерные матрицы. Соединения согласно настоящему изобретению можно вводить любыми традиционными средствами, доступными для применения в сочетании с фармацевтическими препаратами в качестве отдельных терапевтических агентов или в комбинации терапевтических агентов. Вводимые дозы, разумеется, изменяются в зависимости от таких известных факторов, как фармакодинамические характеристики конкретного агента и его режима и способа введения, возраст, состояние здоровья и вес реципиента, природа и степень тяжести симптомов, вид параллельно проводимого лечения, частота лечения и желаемый эффект. Ежедневная доза активного ингредиента может составлять примерно от 0,001 до 1000 мг на 1 кг массы тела, при этом предпочтительная доза составляет от 0,1 до примерно 30 мг/кг. Лекарственные формы (составы, пригодные для введения) содержат примерно от 1 до примерно 500 мг активного ингредиента на единицу формы. В таких фармацевтических составах доля активного ингредиента обычно лежит в пределах примерно 5-95 мас.% относительно общей массы состава. Активный ингредиент можно вводить перорально в твердых лекарственных формах, в частности в- 21016830 капсулах, таблетках и порошках, или в жидких лекарственных формах, в частности в эликсирах, сиропах и суспензиях. Его можно вводить также парентерально в стерильных жидких лекарственных формах. Кроме того, активный ингредиент можно вводить интраназально (капли для носа) или путем ингаляции аэрозоля с лекарственным порошком. Потенциально возможны также другие лекарственные формы, в частности пластырь или притирание для трансдермального введения. Лекарственные формы, пригодные для перорального введения, могут включать: (а) жидкие растворы, в частности эффективная доза соединения, растворенного в разбавителях, например в воде, физиологическом растворе или апельсиновом соке, (б) капсулы, саше, таблетки, пастилки и лепешки, все из которых содержат предварительно установленную дозу активного ингредиента в форме твердого вещества или гранул, (в) порошки, (г) суспензии в соответствующей жидкости и (д) пригодные эмульсии. Жидкие лекарственные формы могут включать разбавители, в частности воду, и спирты, например этанол, бензиловый спирт, пропиленгликоль, глицерин и полиэтиленовые спирты с добавлением или без добавления фармацевтически приемлемого поверхностно-активного агента, суспендирующего агента или эмульгирующего агента. Капсульные формы могут представлять собой обычные твердые или мягкие желатиновые оболочки, содержащие, например, поверхностно-активные агенты, смазывающие вещества и инертные наполнители, в частности, лактозу, сахарозу, фосфат кальция и кукурузный крахмал. Таблеточные формы могут содержать один или несколько следующих компонентов: лактозу, сахарозу, манит, кукурузный крахмал, картофельный крахмал, альгиновую кислоту, микрокристаллическую целлюлозу, гуммиарабик, желатин, гуаровую камедь, коллоидный диоксид кремния, кроскармеллозу натрия, тальк,стеарат магния, стеарат кальция, стеарат цинка, стеариновую кислоту и другие наполнители, красители,разбавители, буферирующие агенты, разлагающие агенты, увлажняющие агенты, консерванты, вкусовые добавки и фармакологически совместимые носители. Формы пастилок могут содержать активный ингредиент во вкусовом веществе, обычно - в сахарозе и в гуммиарабике или трагаканте, а также пастилки,содержащие активный ингредиент в инертной основе, в частности желатин, или глицерин, или сахарозу и гуммиарабик, эмульсии и гели, содержащие кроме активного ингредиента носители, которые известны специалистам в данной области техники. Соединения согласно настоящему изобретению отдельно или в сочетании с другими пригодными компонентами можно использовать в аэрозольных лекарственных формах для введения посредством ингаляции. Такие аэрозольные лекарственные формы можно помещать в контейнеры с допустимым газомвытеснителем, в частности с дихлордифторметаном, пропаном и азотом. Их можно также получать в форме фармацевтических препаратов, не подлежащих сжатию под давлением, в частности в распылителе или в аэрозольном ингаляторе. Лекарственные формы, пригодные для парентерального введения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы,бактериостаты и растворы, которые придают составу изотоничность по отношению к крови предполагаемого реципиента, а также водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты, солюбилизаторы, загустители, стабилизаторы и консерванты. Соединение можно вводить в физиологически растворимом разбавителе в фармацевтическом носителе, в частности в стерильной жидкости или смеси жидкостей, включая воду, физиологический раствор, водную декстрозу и соответствующие сахарные растворы, спирт, в частности этанол, изопропанол или гексадециловый спирт,гликоли, в частности пропиленгликоль или полиэтиленгликоль, например поли(этиленгликоль) 400, глицеринкетали, в частности 2,2-диметил-1,3-диоксолан-4-метанол, простые эфиры, масло, жирную кислоту,сложный эфир или глицерид жирной кислоты или ацетилированный глицерид жирной кислоты с добавлением или без добавления фармацевтически приемлемого поверхностно-активного агента, в частности мыла или детергента, суспендирующего агента, в частности пектина, карбомеров, метилцеллюлозы, гидроксипропилметилцеллюлозы или карбоксиметилцеллюлозы, или эмульгирующих агентов и других фармацевтических адъювантов. Масла, которые можно использовать в составах для парентерального введения, включают вазелин,животные, растительные или синтетические масла. Конкретные примеры масел включают арахисовое,соевое, кунжутное, хлопковое, кукурузное, оливковое, вазелиновое и минеральное масла. Жирные кислоты, пригодные для применения в составах для парентерального введения, включают олеиновую кислоту, стеариновую кислоту и изостеариновую кислоту. Примерами пригодных сложных эфиров жирных кислот являются этилолеат и изопропилмиристат. Пригодные мыла для применения в парентеральных составах включают соли жирных кислот и щелочных металлов, аммония и триэтаноламина, а пригодные детергенты включают (а) катионные детергенты, например галоиды диметилдиалкиламмония и галоиды алкилпиридина, (б) анионные детергенты, например алкильные, арильные и олефиновые сульфонаты,алкильные, олефиновые эфирные и моноглицеридные сульфаты и сульфосукцинаты, (в) неионные детергенты, в частности жирные аминные оксиды, алканоламиды жирных кислот, и сополимеры полиоксиэтилена и полипропилена, (г) амфотерные детергенты, например алкильные -аминопропионаты, четвертичные аммониевые соли 2-алкилимидозолина и (д) их смеси. Составы для парентерального введения содержат в растворе примерно от 0,5 до примерно 25 мас.% активного ингредиента. В таких лекарственных формах можно использовать пригодные консерванты и- 22016830 буферы. Для минимизации или предотвращения раздражения в месте инъекции такие составы могут содержать одно или более неионных поверхностно-активных веществ, имеющих гидрофильнолипофильный баланс (HLB) примерно от 12 до примерно 17. Содержание поверхностно-активного вещества в таких составах лежит в пределах примерно от 5 до примерно 15 мас.%. Пригодные поверхностноактивные вещества включают сложные эфиры полиэтиленсорбитана и жирных кислот, в частности сорбитанолеат, и высокомолекулярные аддукты этиленоксида с гидрофобным основанием, которые получают путем конденсации пропиленоксида с пропиленгликолем. Фармацевтически приемлемые наполнители также хорошо известны специалистам в данной области техники. Выбор наполнителя частично определяется конкретным соединением, а также конкретным способом, который используют для введения состава. В соответствии с этим существует широкий выбор пригодных лекарственных форм фармацевтического состава согласно настоящему изобретению. Следующие способы и наполнители описаны в качестве примеров, которые ни в какой степени не ограничивают изобретение. Фармацевтически приемлемые наполнители предпочтительно не оказывают влияния на действие активных ингредиентов и не вызывают вредных побочных эффектов. Пригодные носители и наполнители включают растворители, в частности воду, спирт и пропиленгликоль, твердые абсорбенты и разбавители, поверхностно-активные агенты, суспендирующий агент, связующие для таблетирования,смазывающие вещества, вкусовые вещества и красители. Лекарственные формы можно поставлять в герметичных контейнерах с однократной или многократной дозой, в частности в ампулах и флаконах, и можно хранить в сублимированном (лиофилизированном) состоянии, при этом непосредственно перед употреблением требуется только добавить стерильный жидкий наполнитель, например воду для инъекций. Растворы и суспензии для незамедлительного введения инъекций можно приготовить из стерильных порошков, гранул и таблеток. Требования к эффективным фармацевтическим носителям, которые используют в составах для инъекций, хорошо известны специалистам в данной области техники. См. Pharmaceutics and Pharmacy Practice, J. В. LippincottCo., Philadelphia, PA, Banker and Chalmers, Eds., 238-250 (1982) и ASHP Handbook on Injectable Drugs,Toissel, 4th ed., 622-630 (1986). Лекарственные формы, пригодные для топического введения, включают таблетки для рассасывания, содержащие активный ингредиент во вкусовом веществе, обычно - в сахарозе и гуммиарабике или трагаканте, пастилки, содержащие активный ингредиент в инертной основе, в частности в желатине и глицерине или в сахарозе и гуммиарабике, и жидкости для полоскания рта, содержащие активный ингредиент в пригодном жидком носителе, а также кремы, эмульсии и гели, содержащие кроме активного ингредиента носители, известные специалистам в данной области техники. Кроме того, лекарственные формы, пригодные для ректального введения, можно получить в виде суппозиториев путем смешивания с различными основами, в частности с эмульгирующими основами или водорастворимыми основами. Составы, пригодные для вагинального введения, можно получить в форме пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих кроме активного ингредиента носители, известные специалистам в данной области техники. Пригодные фармацевтические носители описаны в стандартном справочнике для данной областиRemington's Pharmaceutical Sciences, Mack Publishing Company. Величина дозы, вводимой животному, в том числе человеку, в контексте настоящего изобретения должна быть достаточной для того, чтобы вызвать терапевтическую реакцию у животного в течение разумного периода времени. Для специалиста в данной области техники очевидно, что величина дозы будет зависеть от различных факторов, включая состояние животного, массу тела животного, а также тяжесть и стадию патологического состояния, подлежащего лечению. Пригодной является доза, которая обеспечивает концентрацию активного агента в организме пациента, заведомо вызывающую желаемую реакцию. Предпочтительная доза приводит к максимальному ингибированию патологического состояния, подвергаемого лечению, без неуправляемых побочных эффектов. Величина дозы также определяется способом, временным режимом и частотой введения, а также наличием, природой и степенью тяжести вредных побочных эффектов, которые могут сопровождать введение соединения и желаемый физиологический эффект. Применяемые фармацевтические лекарственные формы для введения соединений согласно настоящему изобретению можно иллюстрировать следующим образом. Капсулы с твердой оболочкой. Большое количество сборных капсул получают путем заполнения стандартных двухэлементных твердых желатиновых капсул, каждая из которых содержит 100 мг порошкообразного активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния. Мягкие желатиновые капсулы. Смесь активного ингредиента в съедобном масле, в частности в соевом масле, хлопковом масле или оливковом масле, приготавливают и впрыскивают при помощи насоса вытесняющего действия в расплавленный желатин для получения мягких желатиновых капсул, содержащих 100 мг активного ингредиента. Капсулы промывают и сушат. Активный ингредиент можно растворить в смеси полиэтиленгли- 23016830 коля, глицерина и сорбита для получения лекарственной смеси, смешивающейся с водой. Таблетки. Большое количество таблеток получают традиционными способами таким образом, чтобы лекарственная форма содержала 100 мг активного ингредиента, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Для повышения вкусовой привлекательности, улучшения внешнего вида и стабильности или замедления поглощения можно наносить соответствующие водные и неводные покрытия. Таблетки/капсулы с незамедлительным высвобождением. Они представляют собой твердые лекарственные формы для перорального приема, изготовленные традиционными и новыми способами. Эти лекарственные формы принимают перорально, не запивая водой, для незамедлительного растворения и доставки активного компонента с целью медикаментозного лечения. Активный ингредиент смешивают в жидкости, содержащей такие ингредиенты, как сахар, желатин, пектин и подсластители. Эти жидкости отверждают, получая твердые таблетки или каплеты при помощи сублимации и твердофазной экстракции. Лекарственные соединения можно прессовать с вязкоупругими и термоупругими сахарами и полимерами или бурно выделяющими газ компонентами для получения пористых матриц, предназначенных для незамедлительного высвобождения при отсутствии воды. Кроме того, соединения согласно настоящему изобретению можно вводить в форме капель в нос или мерной дозы назального или трансбуккального ингалятора. Препарат доставляется из назального раствора в форме мелкодисперсного тумана или из порошка в форме аэрозоля. Приведенное выше описание иллюстрирует и описывает настоящее изобретение. При этом в описании показаны и описаны только предпочтительные варианты реализации, однако, как указано выше, следует понимать изобретение можно использовать в других различных комбинациях, модификациях и условиях, при этом в него можно вносить изменения и модификации в рамках описанной здесь концепции в соответствии с вышеуказанными положениями и/или опытом или знаниями специалистов в данной области техники. Термин "содержащий" (и его грамматические варианты), используемый в данном описании, употребляется в инклюзивном смысле "имеющий" или "включающий", а не в исключительном смысле "состоящий только из". Употребляемые термины следует понимать как включающие единственное и множественное число. Предполагается также, что описанные выше варианты реализации поясняют наилучшие режимы,известные из практики, и позволяют другим специалистам в данной области техники использовать изобретение в тех или иных вариантах с различными модификациями, которые требуются для конкретного случая применения или использования. В соответствии с этим имеется в виду, что описание не ограничивает изобретение изложенной здесь формой. Кроме того, предполагается, что прилагаемая формула изобретения включает альтернативные варианты реализации. Все публикации, патенты и патентные заявки, которые цитируются в данном описании, включены в него в качестве ссылки таким же образом, как, если бы было указано, что каждая отдельная публикация,патент или патентная заявка включается в качестве ссылки. В случае расхождений превалирующим является настоящее описание изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, представленное формуламиZ = H; Е = (C1-C6)алкил(OH)C(=O)NH2; каждый из W, Y, R отдельно выбирают из группы, включающей Н; С 1-С 6 алкил, алкинил, фенил, галоген; О, ОН, Оалкил, галогеналкил, NH-фенил; при условии, что по меньшей мере один из W, Y и R отличается от Н и NH2, и при этом W и Y мо- 24016830 гут вместе представлять собой О; и каждый D отдельно представляет собой ОН,а также его фармацевтически приемлемые соли и смеси. 2. Соединение по п.1, отличающееся тем, что представляет собой 5-амино-4-N-3 фторфенилкарбоксамид-1D-рибофуранозил-1 Н-имидазол. 3. Соединение по п.1, отличающееся тем, что представляет собой 3-этинил-1-(-D-рибофуранозил)[1,2,4]-триазол. 4. Соединение по п.1, отличающееся тем, что представляет собой 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанол. 5. Соединение по п.1, отличающееся тем, что представляет собой 1-(1D-рибофуранозил[1,2,4]триазол-3-ил)фенилметанон. 6. Соединение по п.1, отличающееся тем, что представляет собой 3-(1,1-дифторэтил)-1Dрибофуранозил[1,2,4]триазол. 7. Соединение по п.1, отличающееся тем, что представляет собой 1-(1Dрибофуранозил[1,2,4]триазол-3-ил)-2,2,2-трифторэтанол. 8. Фармацевтический состав, содержащий соединение по п.1 и фармацевтически приемлемый носитель. 9. Способ ингибирования у пациента вирусной РНК-полимеразы путем введения пациенту по меньшей мере одного соединения по п.1. 10. Применение соединения по пп.1-7 для ингибирования РНК и ДНК вирусных инфекций. 11. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой грипп. 12. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус Хантаан. 13. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус конго-крымской геморрагической лихорадки. 14. Применение по п.10, отличающееся тем, что вирусная инфекция представляет собой вирус семейства Bunyaviridae. 15. Применение по любому из пп.10-14, дополнительно включающее использование по меньшей мере одного терапевтического агента, выбранного из группы, включающей интерферон (INF), интерферон -2 а, интерферон -2b, консенсусный интерферон (CIFN), рибавирин, амантадин, ремантадин, интерлейкин-12, урсодеоксихолевую кислоту (UDCA) и глицирризин. 16. Применение по п.10, отличающееся тем, что указанная вирусная инфекция содержит по меньшей мере один элемент из группы, включающей инфекцию вируса гриппа, вируса Хантаан, вируса конго-крымской геморрагической лихорадки, вируса гепатита В, вируса гепатита С, вируса коксаки А, вируса коксаки В, ЕСНО-вирусов, риновирусную инфекцию, вирусную инфекцию ортопоксвируса (оспы),вирусную инфекцию Эбола, вирусную инфекцию полиомиелита и вирусную инфекцию Западного Нила. Меченое радиоактивным изотопом соединение ТА-18 или RBV (10 мкМ) инкубировали с аденозинкиназой. Реакцию прекращали добавлением перхлорной кислоты, а образование продукта определяли способом SAX-ВЭЖХ Фиг. 1 Йодотуберцидин предотвращал также снижение уровней GTP, вызванное ТА-18 Фиг. 5 Обработка 10 мкМ RBV также снижает уровни GTP в клетках СЕМ Фиг. 7
МПК / Метки
МПК: C07H 19/056, C07H 19/044, C07H 5/04, A01N 43/04, A61K 31/70, C07H 5/06
Метки: рнк, применение, днк-полимераз, вариальных, азольные, качестве, ингибиторов, нуклеозиды
Код ссылки
<a href="https://eas.patents.su/28-16830-azolnye-nukleozidy-i-ih-primenenie-v-kachestve-ingibitorov-varialnyh-rnk-i-dnk-polimeraz.html" rel="bookmark" title="База патентов Евразийского Союза">Азольные нуклеозиды и их применение в качестве ингибиторов вариальных рнк- и днк-полимераз</a>
Азольные соединения для применения в качестве fxr модуляторов

Номер патента: 16475
Опубликовано: 30.05.2012
Авторы: Варшавски Алан М., Ахехас-Чичарро Франсиско Хавьер, Маннинен Питер Рудольф, Генин Майкл Джеймс, Буэно Мелендо Ана Белен
МПК: C07D 261/08, A61K 31/42, A61P 3/06...
Метки: применения, соединения, модуляторов, качестве, азольные
Формула / Реферат:
1. Соединение формулы Iгде q равен 1 или 2 при условии, что, если X представляет собой С, q равен 1;U представляет собой О, N или С при условии, что если U представляет собой О или N, то R3a отсутствует; и при условии, что, если U представляет собой N или С, UN-связь представляет собой двойную связь; и при условии, что, если W представляет собой С, WN-связь представляет собой двойную связь;W представляет собой С или N;X представляет собой С или...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Вилл Дэвид Вильям, Бодари Сара Кэтрин, Пейман Ануширван, Кнолле Йохен, Катбертсон Роберт Эндрю, Карниато Дени, Гурвест Жан-Франсуа, Шойнеманн Карлхайнц, Гадек Томас, Макдауэлл Роберт
МПК: C07D 239/42, A61K 31/505, A61P 19/10...
Метки: фармацевтическая, адгезии, ткани, качестве, ингибиторов, рассасывания, получения, производные, клеток,способ, композиция, сульфонамидные, костной, применение
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Новые производные бензимидазолов и бензотиазолов, способ их получения, их применение в качестве лекарственных средств, фармацевтические композиции и новое применение, в частности, в качестве ингибиторов cmet

Номер патента: 14315
Опубликовано: 29.10.2010
Авторы: Клерк Франсуа, Немесек Консепсьон
МПК: C07D 235/32, C07D 277/82, C07D 235/30...
Метки: новое, бензимидазолов, производные, фармацевтические, лекарственных, применение, способ, ингибиторов, частности, композиции, качестве, бензотиазолов, средств, получения, новые
Формула / Реферат:
1. Соединения формулы (I)в которой А обозначает NH или S;R1 и R2, одинаковые или разные, выбирают из атомов водорода, радикала NH2, атомов галогена и алкильных радикалов, необязательно замещенных одним или несколькими атомами галогена,и R3 обозначает атом водорода или выбран из значений R1 и R2, причем по меньшей мере один из R1, R2 и R3 не является водородом,R обозначает циклоалкил или алкил, необязательно замещенный фенилом, гетероарилом,...
Производные тетрагидронафталина и их применение в качестве ингибиторов воспаления

Номер патента: 15834
Опубликовано: 30.12.2011
Авторы: Шэке Хайке, Бергер Маркус, Шмеес Норберт, Ревинкель Хартмут, Бэурле Штефан
МПК: A61K 31/47, A61K 31/502, A61K 31/517...
Метки: ингибиторов, производные, тетрагидронафталина, воспаления, применение, качестве
Формула / Реферат:
1. Соединения общей формулы (Ia)гдеI) остатки R1, R2и R3 независимо друг от друга выбраны из -ОН, О-СН3, Cl, F, Н;II) остаток R4 выбран из 2-метилхинолин-5-ила, 2-метилхиназолин-5-ила, 2-этилхиназолин-5-ила, 7-фтор-2-метилхиназолин-5-ила, 8-фтор-2-метилхиназолин-5-ила, 7,8-дифтор-2-метилхиназолин-5-ила, хинолин-2(1Н)-он-5-ила, 7-фторхинолин-2(1Н)-он-5-ила, 8-фторхинолин-2(1Н)-он-5-ила, изохромен-1-он-5-ила, 2-метилфталазин-1-он-5-ила,...
N-аминоацетилпирролидин-2-карбонитрилы и их применение в качестве ингибиторов ddp-iv

Номер патента: 7410
Опубликовано: 27.10.2006
Авторы: Варга Мартон, Бови Филипп, Урбан-Сабо Каталин, Араньи Петер, Батори Шандор, Боронкаи Эва, Канаи Карой, Шушан Эдит, Балаж Ласло, Сабо Тибор, Надь Лайош Т., Бата Имре, Капуи Зольтан
МПК: C07D 211/58, C07D 207/16, C07D 401/04...
Метки: ddp-iv, применение, качестве, ингибиторов, n-аминоацетилпирролидин-2-карбонитрилы
Формула / Реферат:
1. Соединения общей формулы (I) где R1 означает азотсодержащий ароматический фрагмент, состоящий из одного или двух ароматических колец, предпочтительно кольцо, такое как пиридил, пиридазинил, пиримидинил, пиразинил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, оксадиазолил, хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, хиноксалинил, бензимидазолил, индазолил, бензотиазолил, бензизотиазолил,...
Предыдущий патент: Производные n-(аминогетероарил)-1h-индол-2-карбоксамидов, их получение и их применение в терапии
Следующий патент: Дозирующий ингалятор
Случайный патент: Применение фармацевтической композиции, содержащей бривудин, для лечения герпесвирусных инфекций