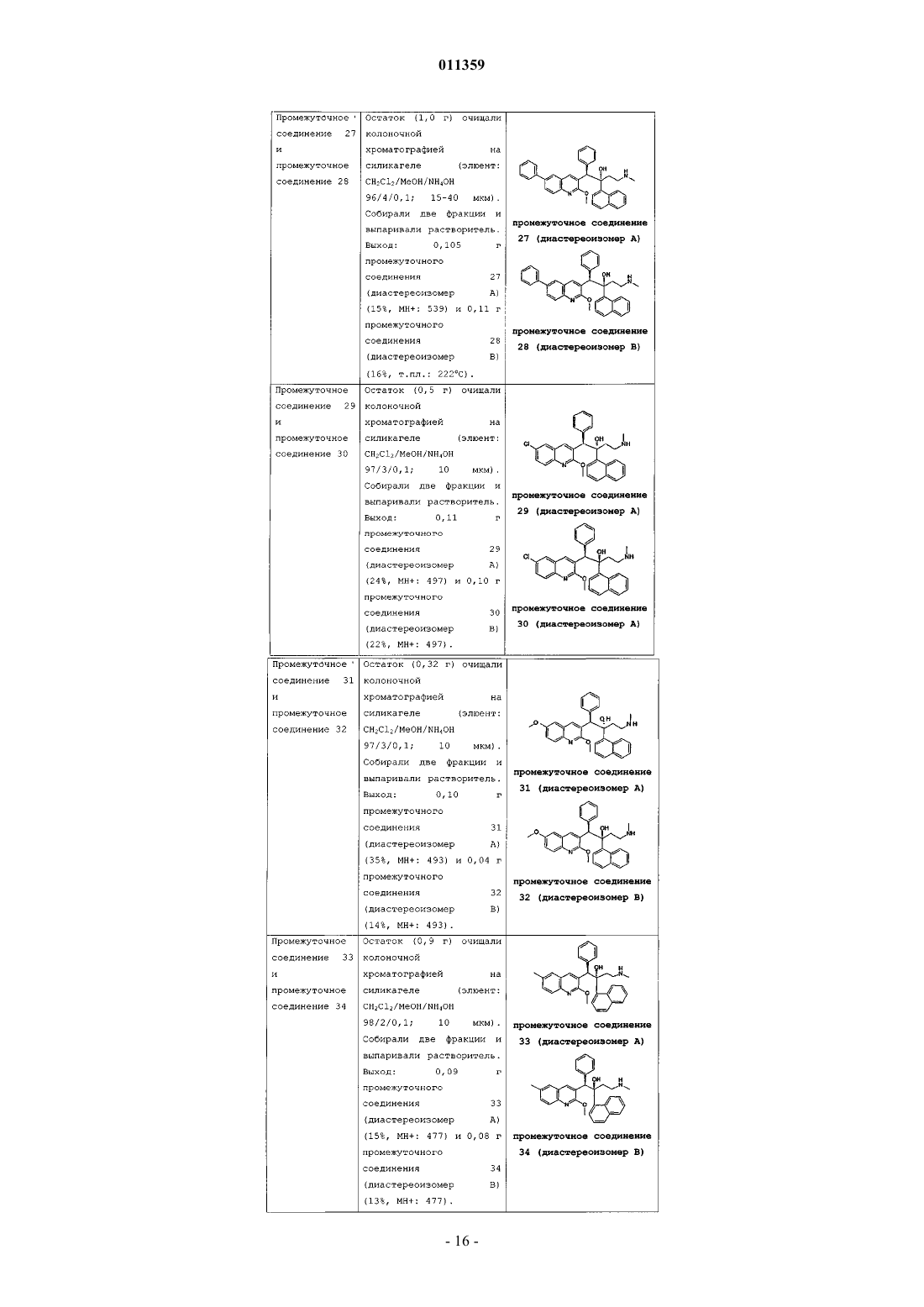
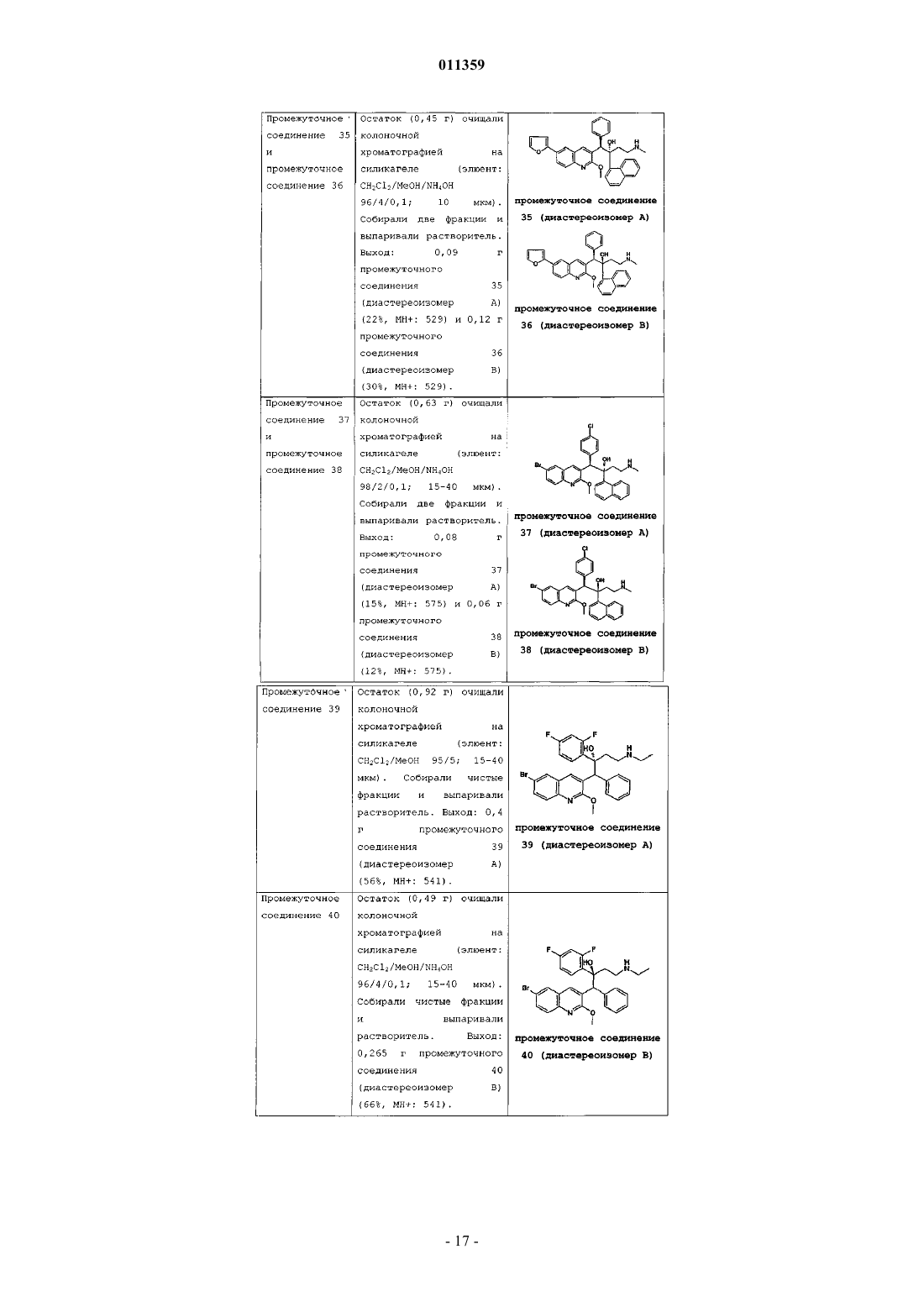
Замещённые хинолины и их применение в качестве микобактериальных ингибиторов
Номер патента: 11359
Опубликовано: 27.02.2009
Авторы: Гийемон Жером Эмиль Жорж, Паскье Элизабет Тереза Жанна












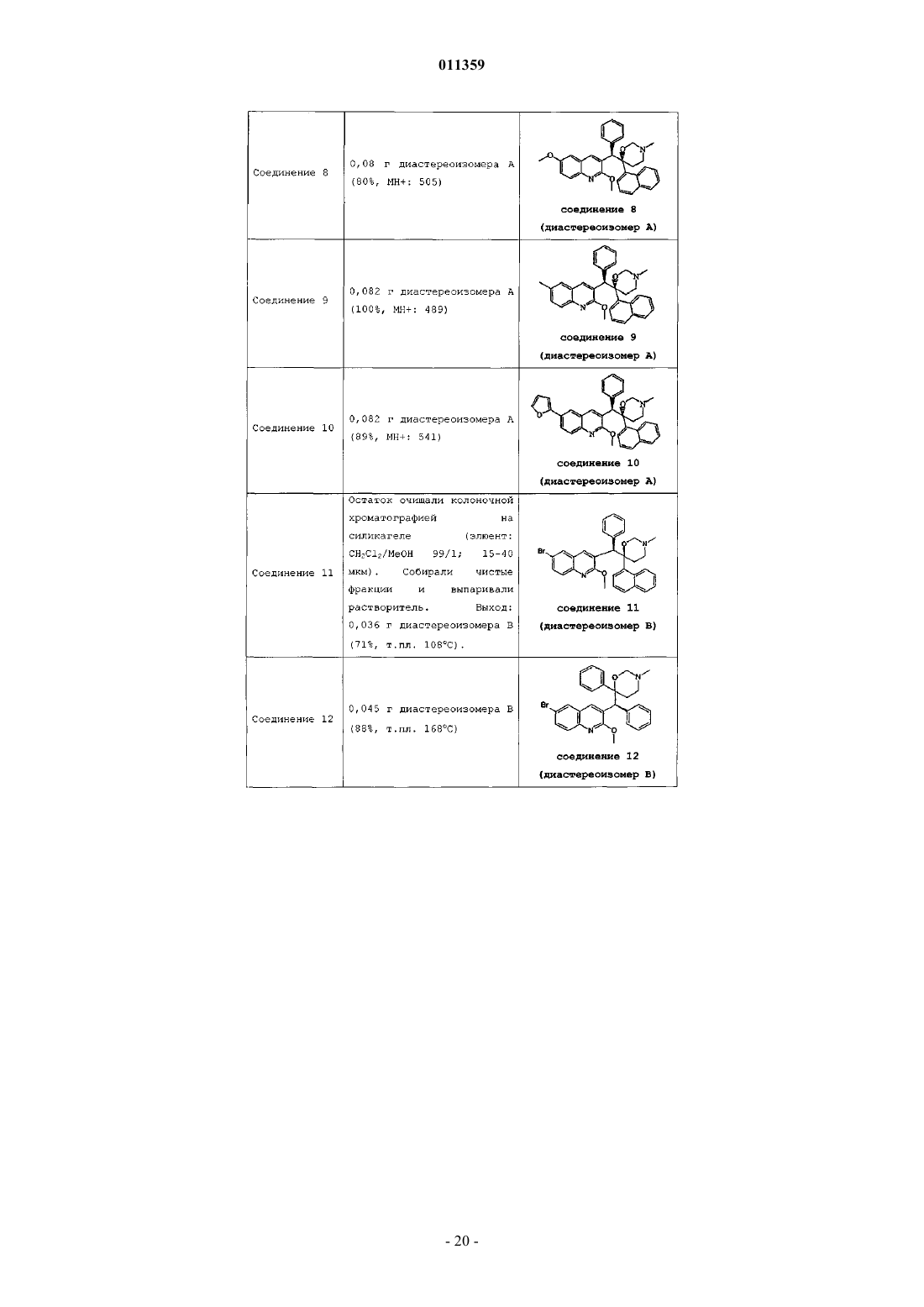
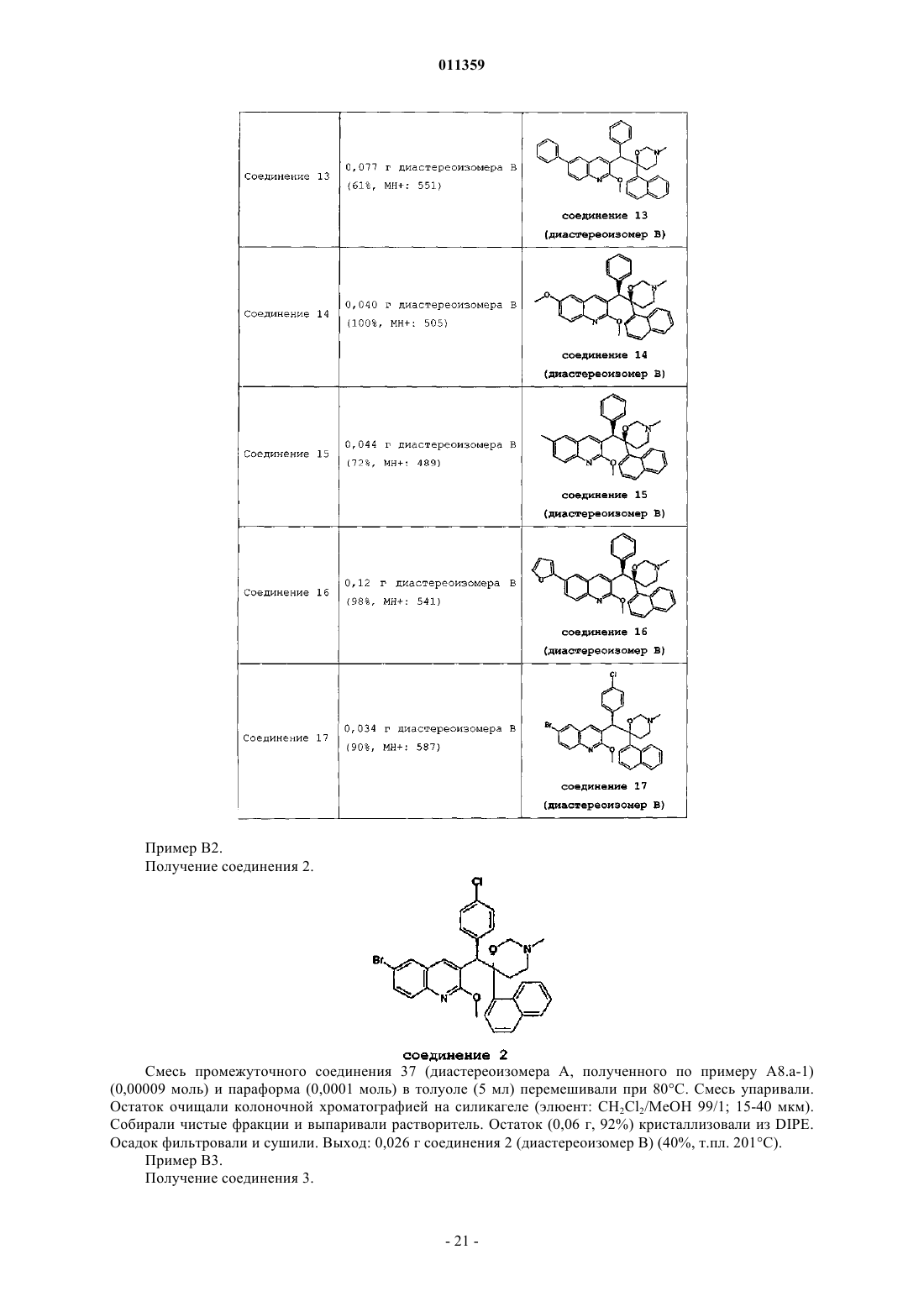




Формула / Реферат
1. Соединение формулы

его фармацевтически приемлемые соли присоединения кислоты или основания, его четвертичные амины, его стерохимически изомерные формы, его таутомерные формы и его N-оксидные формы, в котором
R1 представляет собой водород, галоген, Ar, Het, C1-6алкил, C1-6алкилокси;
р представляет собой целое число, равное 1 или 2;
R2 представляет собой C1-6алкилокси;
R3 представляет собой Ar или Het;
R4 представляет собой C1-6алкил;
R5 представляет собой водород или галоген;
r представляет собой целое число, равное 1 или 2;
R6 представляет собой водород;
Z представляет собой СН2 или С(=O);
Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1, 2 или 3 галогеновыми заместителями;
Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей фуранил и тиенил;
галоген представляет собой заместитель, выбранный из группы, включающей фтор, хлор, бром и йод.
2. Соединение по п.1, в котором Z представляет собой СН2.
3. Соединение по п.1 или 2, в котором
R1 представляет собой водород, галоген, Ar, Het, C1-6алкил и C1-6алкилокси;
р представляет собой целое число, равное 1;
R2 представляет собой C1-6алкилокси;
R3 представляет собой Ar или Het;
R4 представляет собой C1-6алкил;
R5 представляет собой водород, галоген;
r представляет собой целое число, равное 1;
R6 представляет собой водород;
Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1 или 2 галогеновыми заместителями;
Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей фуранил и тиенил.
4. Соединение по любому из предшествующих пунктов, которое представляет собой соединение формулы (Ia) и в котором
R1 представляет собой водород, галоген, Ar, Het, C1-6алкил или C1-6алкилокси;
р=1;
R2 представляет собой C1-6алкилокси;
R3 представляет собой нафтил, фенил или Het, где каждый нафтил и фенил необязательно замещен 1 или 2 заместителями, выбранными из группы, включающей галоген;
R4 представляет собой C1-6алкил;
R5 представляет собой водород или галоген;
r равен 1; a
R6 представляет собой водород.
5. Соединение по любому из пп.1, 3 или 4, которое представляет собой соединение формулы (Ia), где
R1 представляет собой водород, галоген, C1-6алкил или фуранил;
R2 представляет собой C1-6алкилокси;
R3 представляет собой нафтил, фенил или Het, где каждый нафтил и фенил необязательно замещен галогеном;
R4 представляет собой C1-6алкил;
R5 представляет собой водород или галоген;
R6 представляет собой водород;
Z представляет собой СН2 или С(=O).
6. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения, по любому из пп.1-5.
7. Применение соединения по любому из пп.1-5 или композиции по п.6 для получения лекарственного средства для лечения микобактериальных заболеваний.
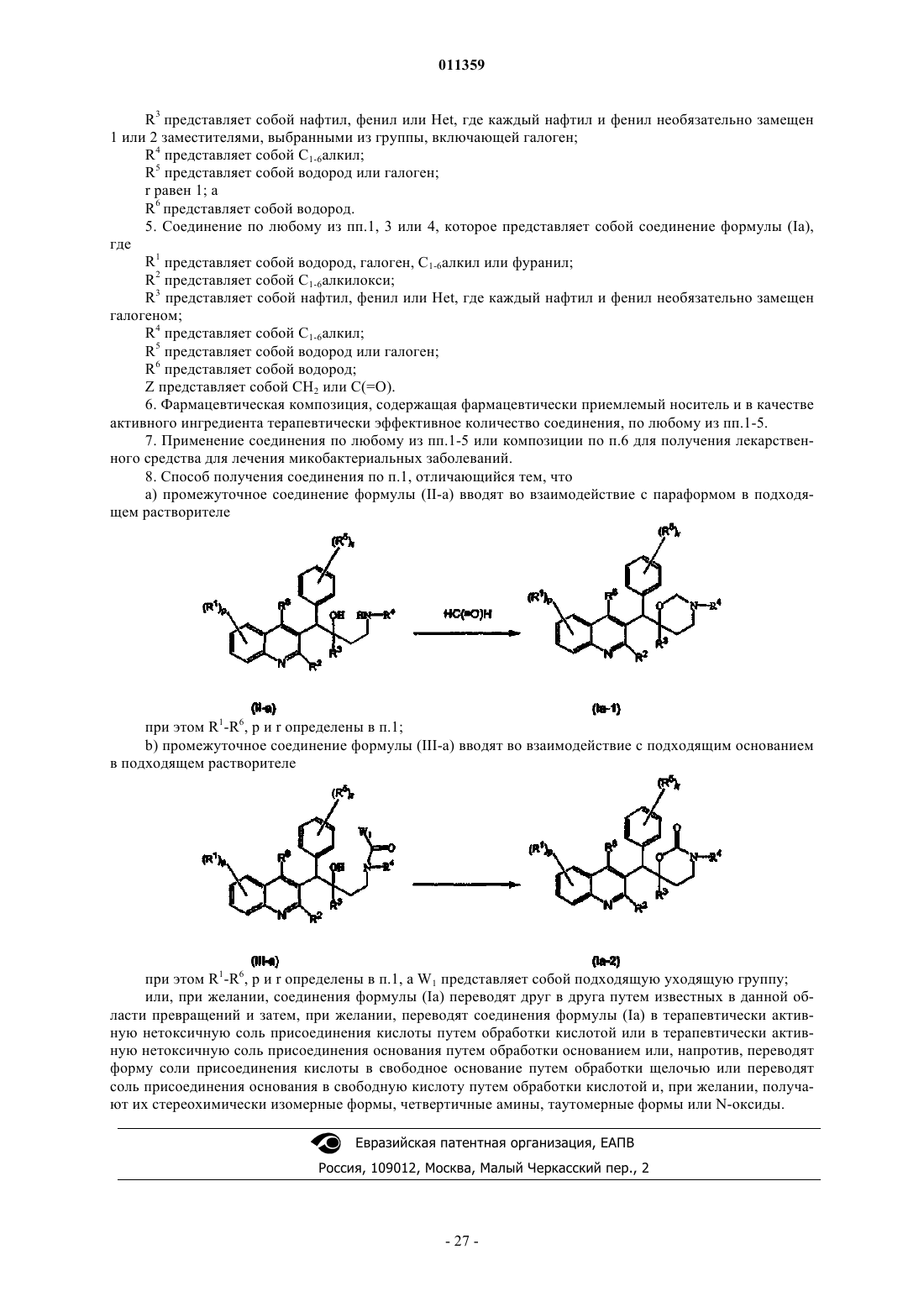
8. Способ получения соединения по п.1, отличающийся тем, что
а) промежуточное соединение формулы (II-а) вводят во взаимодействие с параформом в подходящем растворителе

при этом R1-R6, p и r определены в п.1;
b) промежуточное соединение формулы (III-a) вводят во взаимодействие с подходящим основанием в подходящем растворителе

при этом R1-R6, p и r определены в п.1, a W1 представляет собой подходящую уходящую группу;
или, при желании, соединения формулы (Ia) переводят друг в друга путем известных в данной области превращений и затем, при желании, переводят соединения формулы (Ia) в терапевтически активную нетоксичную соль присоединения кислоты путем обработки кислотой или в терапевтически активную нетоксичную соль присоединения основания путем обработки основанием или, напротив, переводят форму соли присоединения кислоты в свободное основание путем обработки щелочью или переводят соль присоединения основания в свободную кислоту путем обработки кислотой и, при желании, получают их стереохимически изомерные формы, четвертичные амины, таутомерные формы или N-оксиды.
Текст
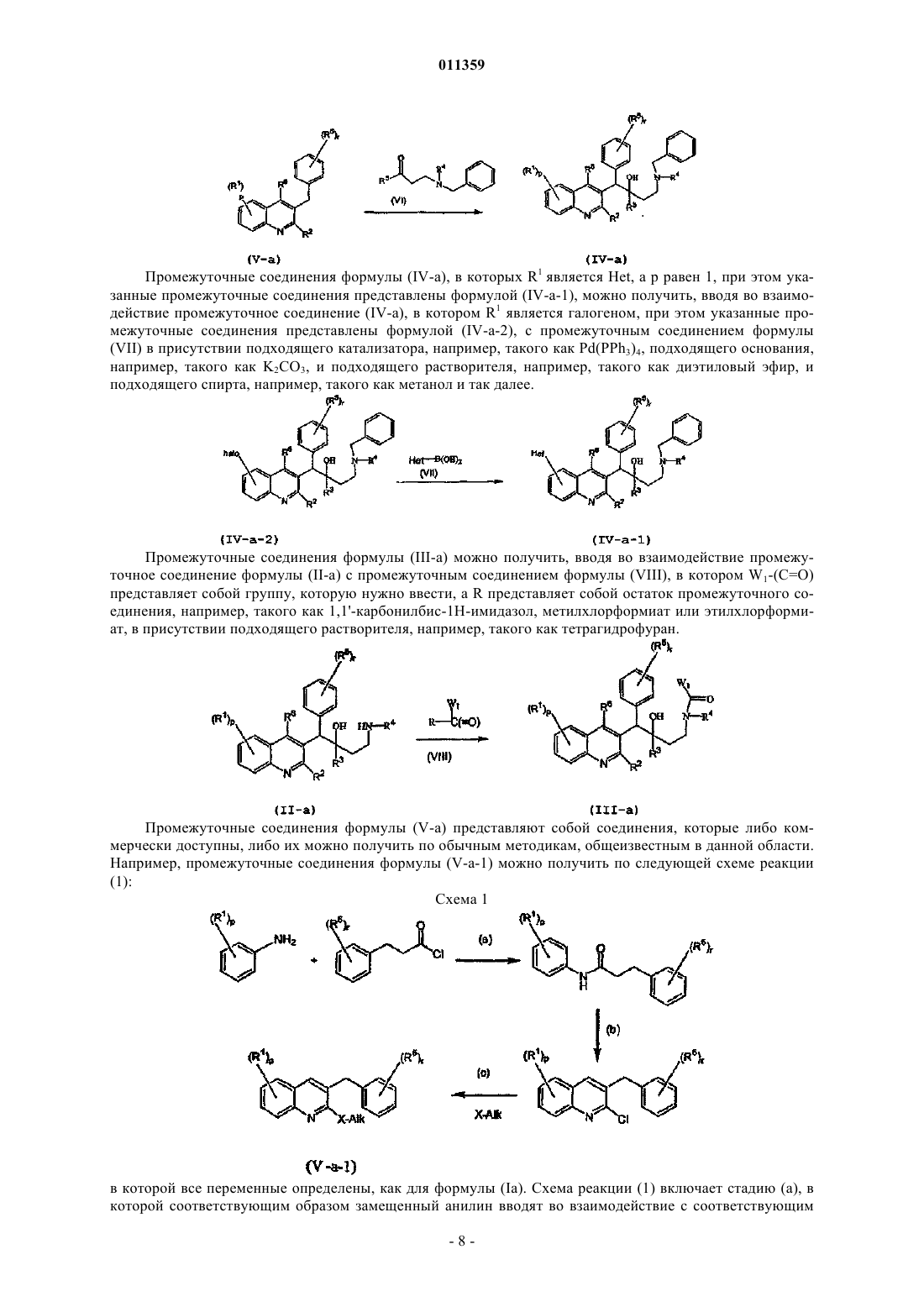
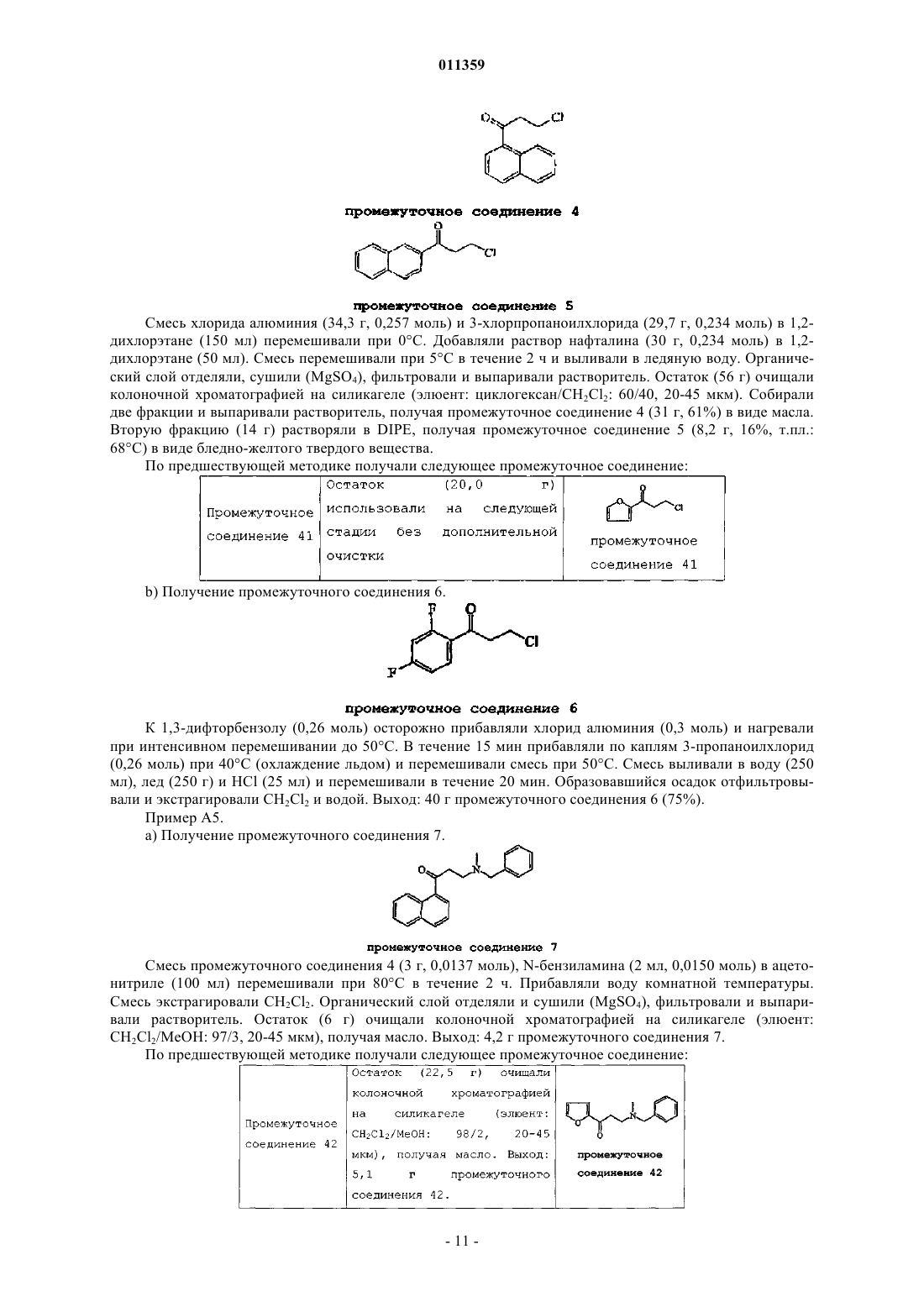
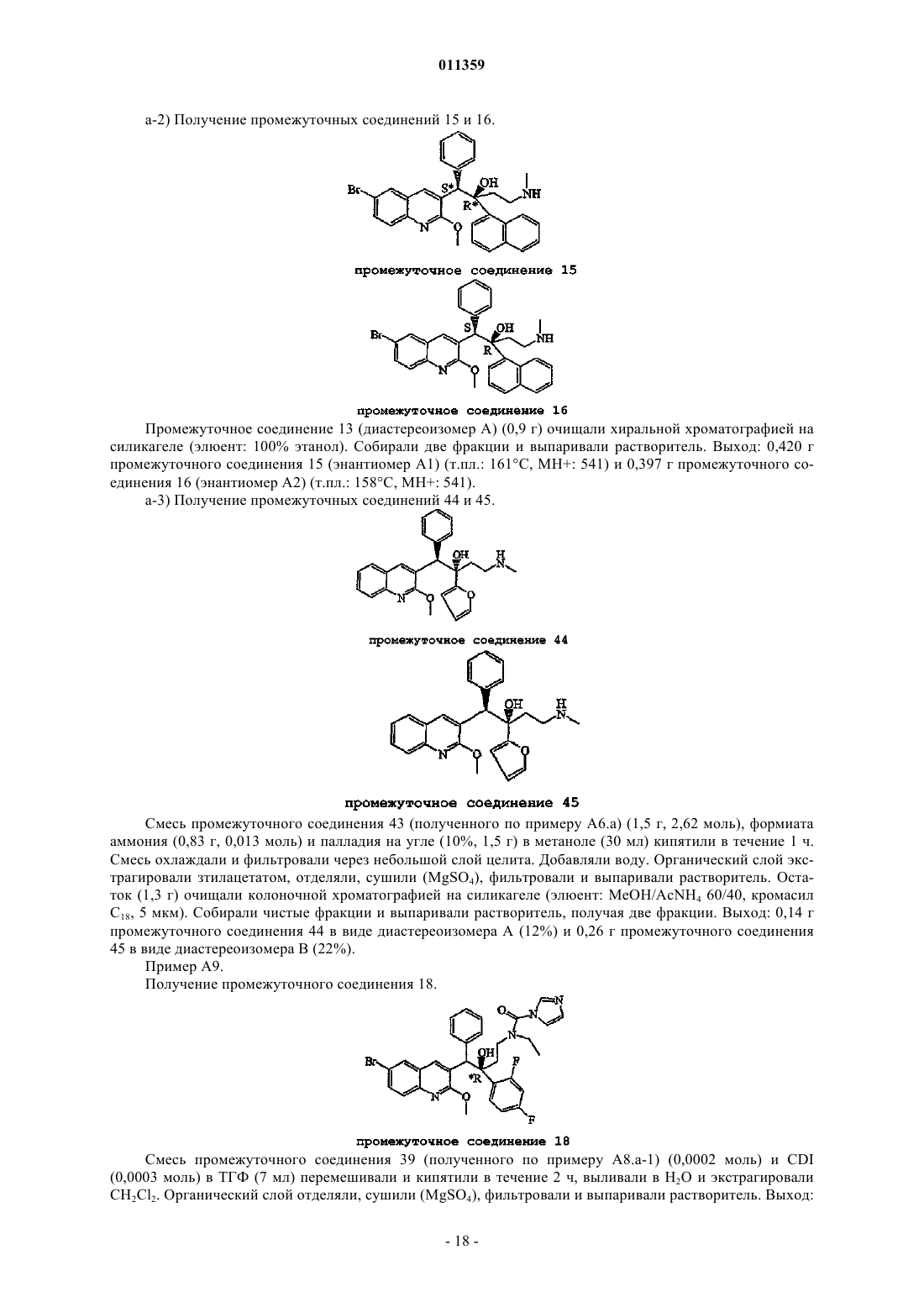
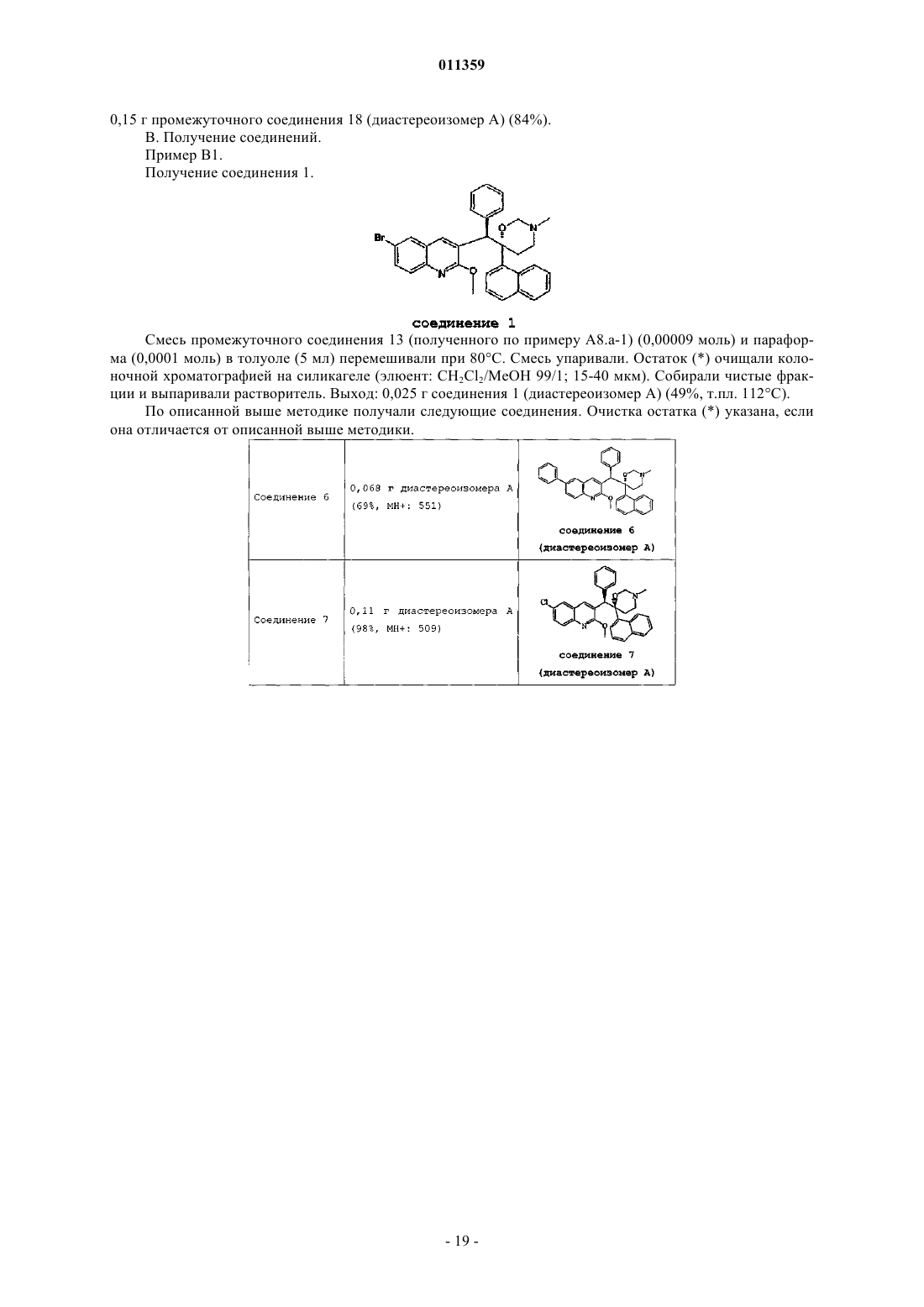
011359 Настоящее изобретение относится к новым замещенным производным хинолинов, применимым для лечения микобактериальных заболеваний, в частности заболеваний, вызванных такими патогенными микобактериями, как Mycobacterium, (М.) tuberculosis, М. bovis, M. avium и М. marinum. Уровень техникиMycobacterium tuberculosis является возбудителем туберкулеза (ТБ), серьезной и потенциально смертельной инфекции, распространенной по всему миру. По оценкам Всемирной Организации Здравоохранения более 8 миллионов человек заболевают ТБ каждый год, а 2 миллиона человек умирают от туберкулеза ежегодно. В течение последнего десятилетия количество случаев заболевания ТВ по всему миру выросло на 20%, причем наибольший груз приходится на наиболее развитые государства. Если подобная тенденция продолжится, в последующие 20 лет число случаев заболевания ТБ вырастет на 41%. Через пятьдесят лет после введения эффективной химиотерапии ТБ остается после СПИД в числе лидирующих инфекций, вызывающих смертность среди взрослого населения в мире. Усложняет эпидемию ТБ возрастающий поток штаммов, устойчивых к множеству лекарственных препаратов, и смертельный симбиоз с ВИЧ. Люди, обладающие положительной ВИЧ-реакцией, и инфицированные ТБ, распространят активный ТБ с вероятностью в 30 раз большей, чем люди, обладающие отрицательной ВИЧреакцией, а ТБ является причиной смерти одного из каждых трех человек-носителей ВИЧ/СПИД по всему миру. Все известные подходы к лечению туберкулеза включают сочетание многочисленных агентов. Например, режим, рекомендованный системой здравоохранения США, включает сочетание изониазида,рифампицина и пиразинамида в течение двух месяцев, затем только изониазид и рифампицин еще четыре месяца. Пациенты, инфицированные ВИЧ, продолжают прием этих лекарственных препаратов еще в течение семи месяцев. Для пациентов, инфицированных штаммами М. tuberculosis, устойчивыми к многочисленным лекарственным препаратам, к комбинационной терапии добавляют такие агенты, как этамбутол, стрептомицин, канамицин, амикацин, капреомицин, этионамид, циклосерин, ципрофлоксацин и офлоксацин. Не существует отдельного агента, который был бы эффективен для клинического лечения туберкулеза, а также никакого сочетания агентов, которые давали бы возможность терапии продолжительностью менее шести месяцев. Для медицины крайне необходимы новые лекарственные средства, которые улучшат настоящее лечение, сделав возможным режимы, способствующие его соблюдению пациентом и поставщиком. Менее продолжительные короткие режимы и режимы, требующие меньшего контроля, представляют собой наилучший путь для достижения этого. Наибольшая польза от лечения достигается в течение первых двух месяцев во время интенсивной или бактерицидной фазы, когда одновременно дают четыре лекарственных препарата; бактериальная нагрузка значительно уменьшается, и пациенты становятся незаразными. Для фазы продолжительностью от 4 до 6 месяцев или стерилизационной необходимо вывести из организма оставшиеся микробы и свести к минимуму опасность рецидива. Было бы крайне полезно эффективное стерилизующее лекарственное средство, сокращающее продолжительность лечения до 2 месяцев. Кроме того, необходимы лекарственные препараты, которые способствуют соблюдению режима, требуя менее интенсивного контроля. Очевидно, наибольшую пользу принесло бы соединение, уменьшающее как общую продолжительность лечения, так и частоту введения лекарственных препаратов. Усложняет эпидемию ТБ возрастающий процент штаммов, устойчивых к большому числу лекарственных препаратов или MDR-ТБ. Вплоть до четырех процентов от всех случаев по всему миру считаются MDR-ТБ - штаммы, устойчивые к наиболее эффективным лекарственным средствам четырехпрепаратного стандарта, изониазида и рифампина. При отсутствии лечения MDR-ТБ смертелен и не может быть адекватно вылечен при помощи обычной терапии, так что для режима необходимо до 2 лет применения лекарственных средств второй линии. Эти лекарственные средства часто токсичны, дорогостоящи и эффективны в самой малой степени. В отсутствие эффективной терапии пациенты, инфицированные MDR-ТБ, продолжают распространять данное заболевание, приводя к новым инфекциям со штаммами MDR-ТБ. В медицине существует крайняя необходимость в новом лекарственном препарате с новым механизмом действия, который, возможно, проявит активность в отношении MDR штаммов. Использованный ранее или в дальнейшем термин лекарственная устойчивость является термином, понятным специалисту в микробиологии. Лекарственно устойчивая Micobacterium представляет собой Micobacterium, которая больше не является чувствительной по меньшей мере к одному из ранее эффективных лекарственных средств, которая обрела способность противостоять антибиотическому действию по меньшей мере одного из ранее эффективных лекарственных средств. Лекарственно устойчивый штамм может передавать эту способность своему потомству. Причиной указанной устойчивости могут являться случайные генетические мутации в бактериальной клетке, изменяющие ее чувствительность к одному лекарственному средству или к различным лекарственным средствам.MDR туберкулез представляет собой особый вид лекарственно устойчивого туберкулеза из-за бактерии, устойчивой, по меньшей мере, к изониазиду и рифампицину (при наличии или в отсутствие устойчивости к другим лекарственным средствам), которые в настоящее время являются двумя наиболее мощными противо-ТБ лекарственными средствами. Цель настоящего изобретения заключается в предоставлении новых соединений, в частности, за-1 011359 мещенных производных хинолина, обладающих способностью ингибировать рост микобактерий, включая лекарственно устойчивые или устойчивые к большому числу лекарственных средств микобактерий и, таким образом, применимых для лечения микобактериальных заболеваний, в частности микобактериальных заболеваний, вызванных патогенными микобактериями, такими как Micobacterium tuberculosis,М. bovis, M. avium, M. smegmatis и М. marinum. Замещенные хинолины уже были описаны в патенте США 5965572 для лечения инфекций, устойчивых к антибиотикам, и в WO 00/34265 для ингибирования роста бактериальных микроорганизмов. Ни в одной из этих публикаций замещенные производные хинолина не были описаны в соответствии с настоящим изобретением. Сущность изобретения Настоящее изобретение относится к новым замещенным производным хинолина формулы (Ia) и их фармацевтически активным солям присоединения кислоты или основания, их четвертичным аминам,их стереохимически изомерным формам, их таутомерным формам и их N-оксидным формам, в которыхR1 представляет собой водород, галоген, Ar, Het, C1-6 алкил или C1-6 алкилокси; р представляет собой целое число, равное 1 или 2;R5 представляет собой водород или галоген;r представляет собой целое число, равное 1 или 2;Z представляет собой СН 2 или С(=O);Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1, 2 или 3 галогеновыми заместителями;Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей фуранил и тиенил; галоген представляет собой заместитель, выбранный из группы, включающей фтор, хлор, бром и йод. Также изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения по изобретению. Кроме этого, предлагается применение соединения по изобретению или композиции на его основе для получения лекарственного средства для лечения микобактериальных заболеваний, а также способ получения соединений по изобретению. Подробное описание В рамках данной заявки алкил представляет собой линейный или разветвленный насыщенный углеводородный радикал, содержащий от 1 до 6 атомов углерода, или циклический насыщенный углеводородный радикал, содержащий от 3 до 6 атомов углерода, или циклический насыщенный углеводородный радикал, содержащий от 3 до 6 атомов углерода, связанный с линейным или разветвленным насыщенным углеводородным радикалом, содержащим от 1 до 6 атомов углерода, где каждый атом углерода может быть необязательно замещен галогеном, гидрокси, алкилокси или оксо. Предпочтительно алкил представляет собой метил, этил или циклогексилметил. В рамках данной заявки Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1, 2 или 3 галоидными заместителями. В рамках данной заявки Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей тиенил или фуранил, наиболее предпочтительно Het представляет собой фуранил. В рамках данной заявки галоген является заместителем, выбранным из группы, включающей фтор,хлор, бром и йод. Предпочтительно галоген представляет собой бром, фтор или хлор. Где бы это ни использовалось в дальнейшем, подразумевается, что термин соединения формулы(Ia) включает также формы их N-оксидов, их соли, их четвертичные амины, их таутомерные формы и их стереохимически изомерные формы. Особый интерес представляют те из соединений формулы (Ia),которые являются стереохимически чистыми. Интересный вариант осуществления настоящего изобретения относится к тем соединениям форму-2 011359 лы (Ia), их фармацевтически приемлемым солям присоединения кислоты или основания, их стереохимически изомерным формам, их таутомерным формам и формам их N-оксидов, в которыхR1 представляет собой водород, галоген, Ar, Het, C1-6 алкил, C1-6 алкилокси; р представляет собой целое число, равное 1;r представляет собой целое число, равное 1; иR6 представляет собой водород. Интересной группой соединений являются такие соединения формулы (Ia), их фармацевтически приемлемые соли присоединения кислоты или основания, их четвертичные амины, их стереохимически изомерные формы, их таутомерные формы и формы их N-оксидов, в которых R1 представляет собой водород, галоген, Ar, Het, C1-6 алкил или C1-6 алкилокси; р равен 1; R2 представляет собой C1-6 алкилокси; R3 представляет собой нафтил, фенил или Het, где нафтил или фенил необязательно замещен 1 или 2 галогеновыми заместителями; R4 представляет собой C1-6 алкил; R5 представляет собой водород или галоген; r равен 1, a R6 представляет собой водород. Интересной группой соединений являются такие соединения формулы (Ia), в которых R1 представляет собой водород, галоген, например бром; C1-6 алкил, например метил; или Het, например фуранил; R2 представляет собой C1-6 алкилокси, например метилокси; R3 представляет собой нафтил, фенил или Het,где фенил или нафтил необязательно замещен галогеном, например фенил, необязательно замещенный галогеном, нафтил или фуранил; R4 представляет собой C1-6 алкил, например метил или этил; R5 представляет собой водород; R6 представляет собой водород; a Z является СН 2 или С(=O). По определению фармацевтически приемлемые соли присоединения кислоты включают формы терапевтически активных нетоксичных солей присоединения кислот, которые способны образовывать соединения формулы (Ia). Указанные соли присоединения кислот можно получить обработкой формы основания соединений формулы (Ia) соответствующими кислотами, например неорганическим кислотами,например галогеноводородными кислотами, в частности хлористо-водородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой и фосфорной кислотой; органическими кислотами, например уксусной кислотой, гидроксиуксусной кислотой, пропановой кислотой, молочной кислотой, пировиноградной кислотой, щавелевой кислотой, малоновой кислотой, янтарной кислотой, малеиновой кислотой, фумаровой кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой, цикламовой кислотой, салициловой кислотой, п-аминосалициловой кислотой и памовой кислотой. Соединения формулы (Ia), содержащие кислые протоны, можно также перевести в формы их терапевтически активных нетоксичных солей присоединения оснований при обработке соответствующими органическими и неорганическими основаниями. Соответствующие формы солей оснований включают,например, аммониевые соли, соли щелочных и щелочно-земельных металлов, в частности соли лития,натрия, калия, магния и кальция, соли органических оснований, например соли бензатина, N-метил-Dглюкамина, гибрамина, и соли аминокислот, например аргинина и лизина. Напротив, формы указанных солей присоединения кислот или оснований можно перевести в свободные формы обработкой соответствующим основанием или кислотой. Использованный в рамках данной заявки термин соль присоединения включает также сольваты, которые способны образовывать соединения формулы (Ia), а также их соли. Подобными сольватами являются, например, гидраты и алкилоголяты. Использованный ранее термин четвертичный амин определяет четвертичные амины, которые способны образовывать соединения формулы (Ia) при реакции основного атома азота соединения формулы (Ia) и соответствующим кватернизующим агентом, например необязательно замещенным алкилгалогенидом, арилгалогенидом или арилалкилгалогенидом, например метилйодидом или бензилйодидом. Можно использовать другие реагенты с хорошими уходящими группами, такие как алкилтрифторметансульфонаты, алкилметансульфонаты и алкил-п-толуолсульфонаты. Четвертичный амин имеет положительно заряженный атом азота. Фармацевтически приемлемые противоионы включают хлор, бром, йод,трифторацетат и ацетат. Выбранный противоион можно ввести при помощи ионообменных смол. Использованный здесь термин стереохимически изомерные формы определяет все возможные изомерные формы, которыми могут обладать соединения формулы (Ia). Если не упомянуто или не указано иначе, химическое обозначение соединений означает смесь всех возможных стереохимически изомерных форм, при этом указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители двухвалентных циклических (частично) насыщенных радикалов могут иметь либо цис-, либо транс-конфигурацию. Понятно, что стереохимически изомерные формы соединений формулы (Ia) вклю-3 011359 чены в рамки данного изобретения. В соответствии с правилами CAS-номенклатуры, если в молекуле имеются два стереогенных центра известной абсолютной конфигурации, признак R или S (на основании правила последовательности Канна-Ингольда-Прелога) приписывают низшему хиральному центру, центру отсчета. Конфигурацию второго стереогенного центра указывают с использованием относительных признаков [R,R] или[R,S], где R всегда определяют как центр отсчета, a [R,R] обозначает центры с одинаковой хиральностью и [R,S] обозначает центры с различной хиральностью. Например, если низший хиральный центр в молекуле имеет S конфигурацию, а второй центр является R, стерео-признак был бы обозначен как S-[R,S]. При использованиииположение самого старшего заместителя у асимметрического атома углерода в циклической системе с самым низшим номером цикла произвольно всегда находится вположении средней плоскости, определенной циклической системой. Положение у другого асимметрического атома углерода в циклической системе относительно положения самого старшего заместителя у базового атома обозначают , если он находится по ту же сторону средней плоскости, определенной циклической системой, или , если он находится по другую сторону от средней плоскости, определенной циклической системой. Соединения формулы (Ia) и некоторые промежуточные соединения без исключений имеют по меньшей мере два стереогенных центра в своей структуре, что может приводить по меньшей мере к 4 стереохимически различным структурам. Соединения формулы (Ia), получаемые описанными ниже способами, можно синтезировать в виде рацемических смесей энантиомеров, которые можно отделить друг от друга при помощи известных в данной области методов разделения. Рацемические соединения формулы (Ia) можно перевести в соответствующую диастереомерную соль реакцией с подходящей хиральной кислотой. Указанные формы диастереомерных солей можно затем разделить, например, селективной или дробной кристаллизацией и выделить из них энантиомеры действием щелочи. Другой способ разделения энантиомерных форм соединений формулы (Ia) включает жидкостную хроматографию на хиральной неподвижной фазе. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии, что реакция протекает стереоспецифично. Предпочтительно, если требуется определенный стереоизомер, указанное соединение синтезируют стереоспецифичными методами получения. В подобных методах предпочтительно будут использоваться энантиомерно чистые исходные вещества. Подразумевается, что таутомерные формы соединений формулы (Ia), включают такие соединения формулы (Ia) в которых, например, енольная группа превращается в кетогруппу (кетоенольная таутомерия). Подразумевается, что формы N-оксидов соединений формулы (Ia) включают такие соединения формулы (Ia), в которых один или несколько атомов азота окислены до так называемого N-оксида, в частности, такие N-оксиды, в которых атом азота аминного радикала окислен. Также описаны производные соединений (называемые обычно пролекарствами) фармакологически активных соединений в соответствии с изобретением, которые разлагаются in vivo, образуя соединения в соответствии с изобретением. Пролекарства обычно (но не всегда) менее эффективны по отношению к целевому рецептору, чем соединения, до которых они разлагаются. В частности, пролекарства применимы в тех случаях, когда требуемое соединение имеет химические или физические свойства, делающие их введение сложным или неэффективным. Например, требуемое соединение может быть крайне малорастворимо, оно может плохо переноситься через эпителий слизистой оболочки или оно может иметь нежелательно короткий период полупревращения в плазме. Дальнейшее обсуждение по поводу пролекарств можно найти у Stella, V.J. et al., "Prodrugs", Drug Delivery Systems, 1985, pp. 112-176 и Drugs,1985, 29, pp. 455-473. Формы пролекарств фармакологически активных соединений в соответствии с данным изобретением, как правило, будут представлять собой соединения формулы (Ia), их фармацевтически приемлемые соли присоединения кислоты или основания, их стерохимически изомерные формы, их таутомерные формы и формы их N-оксидов, содержащие кислотную группу, которую подвергают этерификации или амидированию. В число подобных этерифицированных кислотных групп входят группы формулы-COORx, где Rx представляет собой C1-6 алкил, фенил, бензил или одну из следующих групп: Амидированные группы включают группы формулы -CONRyRz, где Ry представляет собой Н, C1-6 алкил, фенил или бензил, a Rz представляет собой -ОН, Н, C1-6 алкил, фенил или бензил. Из соединений в соответствии с настоящим изобретением, содержащих аминогруппу, можно получить производные действием кетона или альдегида, такого как формальдегид, с образованием основания Манниха. Это основание будет гидролизоваться в водном растворе по кинетике первого порядка.-4 011359 Неожиданно, было обнаружено, что соединения в соответствии с настоящим изобретением подходят для лечения микобактериальных заболеваний, в частности микобактериальных заболеваний, вызванных патогенными микобактериями, включая лекарственно устойчивые и устойчивые к действию многочисленных лекарственных средств, такими как Mycobacterium tuberculosis, M. bovis, М. avium, M. smegmatis и М. marinum. Таким образом, настоящее изобретение относится также к соединениям формулы или (Ia), или (Ib), их фармацевтически приемлемым солям присоединения кислоты или основания, их стерохимически изомерным формам, их таутомерным формам и формам их N-оксидов, для использования в качестве лекарственного средства. Кроме того, изобретение относится к композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения в соответствии с настоящим изобретением. Из соединений в соответствии с настоящим изобретением могут быть получены различные фармацевтические препаративные формы в целях введения. В качестве подходящих композиций можно привести все композиции, которые обычно используются для системно вводимых лекарственных препаратов. Для получения фармацевтических композиций по данному изобретению эффективное количество конкретного соединения, необязательно в форме соли присоединения в качестве активного ингредиента тщательно смешивают с фармацевтически приемлемым носителем,при этом носитель может принимать самые различные формы в зависимости от формы препарата, желательного для введения. Подобные фармацевтические композиции желательны в виде дозированной лекарственной формы, подходящей, в частности, для перорального введения или в виде парентеральной инъекции. Например, при получении композиции в виде пероральной дозированной формы, можно использовать любые обычные фармацевтические среды, например, такие как вода, гликоли, масла, спирты и так далее, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы, или твердые носители, такие как крахмалы, сахара, каолин, разбавители, смазывающие вещества, связующие вещества, разрыхляющие агенты и так далее в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения, таблетки и капсулы представляют собой наиболее предпочтительные пероральные разовые лекарственные формы, в которых обычно используют твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по крайней мере, большей частью, хотя в него могут входить другие ингредиенты, например, для улучшения растворимости. Например, можно приготовить растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Кроме того, можно приготовить растворы для инъекций, в которых можно использовать подходящие жидкие носители, суспендирующие агенты и так далее. Сюда также входят твердые препараты, предназначенные для перевода в жидкие препараты незадолго до использования. В зависимости от способа введения фармацевтическая композиция предпочтительно будет содержать от 0,05 до 99 мас.%, более предпочтительно от 0,1 до 70 мас.% активного ингредиента формулы (Ia) и от 1 до 99,95 мас.%, более предпочтительно от 30 до 99,9 мас.% фармацевтически приемлемого носителя, при этом все процентные соотношения даны из расчета на общую композицию. Кроме того, фармацевтическая композиция может содержать различные прочие ингредиенты, известные в данной области, например смазывающее средство, стабилизирующий агент, буферирующий агент, эмульгатор, регулятор вязкости, поверхностно-активное вещество, консервант, ароматизатор или окрашивающий агент. Особенно предпочтительно получать препараты из упомянутых выше фармацевтических композиций в виде единичных лекарственных форм для простоты введения и равномерности дозировки. Использованная здесь единичная лекарственная форма относится к физически дискретным единицам, пригодным для единичных доз, при этом каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное таким образом, чтобы получить требуемый терапевтический эффект, в сочетании с желательным фармацевтическим носителем. Примерами подобных единичных лекарственных форм являются таблетки (включая таблетки с насечкой или с покрытием), капсулы, пилюли, пакетики с порошком, пластинки, суппозитории, растворы для инъекций или суспензии и так далее и их отдельные множества. Вне сомнения, дневная доза соединения в соответствии с изобретением будет изменяться в зависимости от используемого соединения, способа введения, необходимого лечения и указанного микобактериального заболевания. Однако в общем, удовлетворительные результаты будут получены в том случае,когда соединение в соответствии с изобретением вводят в виде дневной дозы, не превышающей 1 г, например, в интервале от 10 до 50 мг/кг массы тела. Кроме того, настоящее изобретение относится к применению соединения формулы (Ia), его фармацевтически приемлемой соли присоединения кислоты или основания, его стерохимически изомерных форм, его таутомерных форм и форм его N-оксидов, а также любой из упомянутых выше фармацевтических композиций для получения лекарственного средства в целях предупреждения или лечения микобактериальных заболеваний. Соответственно, в другом аспекте описан способ лечения пациента, страдающего или подвергающегося риску микобактериального заболевания, включающий введение данному пациенту фармацевти-5 011359 ческой композиции в соответствии с изобретением. Соединения настоящего изобретения могут также быть соединены с одним или более антимикобактериальными агентами. Другие микобактериальные агенты, которые можно соединить с соединениями формулы (Ia), представляют собой, например, рифампицин (=рифампин), изониазид, пиразинамид, амикацин, этионамид,моксифлоксацин, этамбутол, стрептомицин, парааминосалициловая кислота, циклосерин, капреомицин,канамицин, тиоацетазон, РА-824, хинолоны/фторхинолоны, например, такие как офлоксацин, ципрофлоксацин, спарфлоксацин, макролиды, например, такие как кларитромицин, клофазимин, амоксициллин с клавулановой кислотой, рифамицины, рифабутин, рифапентин. Предпочтительно настоящие соединения формулы (Ia) соединяют с рифапентином и моксифлоксацином. Общие способы плучения В целом, соединения в соответствии с данным изобретением могут быть получены в результате последовательности стадий, каждая из которых известна специалисту в данной области. Соединения формулы (Ia), в которых Z является СН 2, которые представлены формулой (Ia-1), можно получить, вводя во взаимодействие промежуточное соединение формулы (II-а) с параформом в подходящем растворителе, например, в таком как толуол. Соединения формулы (Ia), в которых Z является С(=O), которые представлены формулой (Ia-2),можно получить, вводя во взаимодействие промежуточное соединение формулы (III-а), где W1 является подходящей уходящей группой, например, такой как имидазол, алкилоксигруппы, например метилокси,с подходящим основанием, например, таким как гидрид натрия, тритиобутилат калия, в подходящем растворителе, например, таком как тетрагидрофуран, диэтиловый эфир, диоксан. В приведенных выше реакциях полученное соединение формулы (Ia) можно выделить и, при необходимости, очистить при помощи общеизвестных в данной области методов, например, таких как экстракция, кристаллизация, перегонка, растирание и хроматография. В том случае, если соединение формулы (Ia) выкристаллизовывается, его можно выделить фильтрованием. В ином случае, кристаллизацию можно вызвать добавлением соответствующего растворителя, например, такого как вода, ацетонитрил,любой спирт, например, такой как метанол, этанол, и сочетания указанных растворителей. Альтернативно, реакционную смесь можно также упарить досуха, а затем очистить остаток хроматографией (например, ВЭЖХ с обращенной фазой, флэш-хроматографией и так далее). Реакционную смесь можно также очистить хроматографией без предварительного выпаривания растворителя. Соединение формулы (Ia) можно выделить также выпариванием растворителя с последующей перекристаллизацией из соответствующего растворителя, например, такого как вода, ацетонитрил, любой спирт, например, такой как метанол, и сочетания указанных растворителей. Специалист в данной области поймет, какой способ следует выбрать, какой растворитель наиболее подойдет для использования или же поиск наиболее подходящего способа выделения относится к обычной экспериментальной работе. Кроме того, соединения формулы (Ia) можно получить, превращая соединения формулы (Ia) друг в друга в соответствии с известными в данной области реакциями превращения групп. Соединения формулы (Ia) можно перевести в форму соответствующих N-оксидов по известным в данной области методикам превращения трехвалентного атома азота в форму его N-оксида. В общем,указанную реакцию N-окисления можно осуществить при взаимодействии исходного соединения формулы (Ia) с соответствующей органической или неорганической перекисью. Соответствующие неоргани-6 011359 ческие перекиси включают, например, перекись водорода, перекиси щелочных или щелочно-земельных металлов, например перекись натрия, перекись калия; соответствующие органические перекиси могут включать пероксикислоты, например, такие как надбензойная кислота или галогензамещенная надбензойная кислота, например 3-хлорнадбензойная кислота, пероксоалкановые кислоты, например пероксоуксусная кислота, алкилгидроперекиси, например гидроперекись третбутила. Подходящими растворителями являются, например, вода, низшие спирты, например этанол и так далее, углеводороды, например толуол, кетоны, например 2-бутанон, галогензамещенные углеводороды, например хлористый метилен, и смеси подобных растворителей. Соединения формулы (Ia), в которых R4 представляет собой C1-6 алкил, можно перевести в подходящий четвертичный амин реакцией с подходящим кватернизующим агентом, например, таким как необязательно замещенный алкилгалогенид, например ICH3, в присутствии подходящего растворителя,например ацетона. Некоторые соединения формулы (I) и некоторые промежуточные соединения настоящего изобретения могут состоять из смеси стереохимически изомерных форм. Чистые стереохимически изомерные формы указанных соединений и указанных промежуточных соединений можно получить при использовании известных в данной области методов. Например, диастереоизомеры можно разделить физическими методами, такими как дробная кристаллизация, или хроматографическими методами, например противопоточным распределением, жидкостной хроматографией и тому подобными методами. Энантиомеры можно выделить из рацемических смесей, переведя сначала указанные рацемические смеси в смеси диастереомерных солей или соединений, при помощи подходящих разделяющих агентов, например, таких как хиральные кислоты, затем физически разделяя указанные смеси диастереомерных солей или соединений, например, при помощи дробной кристаллизации или хроматографических методов, например жидкостной хроматографией и тому подобными методами, и, наконец, переводя указанные разделенные диастереомерные соли или соединения в соответствующие энантиомеры. Чистые стереохимически изомерные формы можно также получить из чистых стереохимически изомерных форм соответствующих промежуточных соединений и исходных веществ при условии, что промежуточные реакции протекают стереоспецифично. Альтернативный способ разделения энантиомерных форм соединений формулы (I) и промежуточных соединений включает жидкостную хроматографию, в частности жидкостную хроматографию с использованием хиральной неподвижной фазы. Необходимо понять, что в приведенных выше или последующих получениях продукты реакции могут быть выделены из реакционной среды и, при необходимости, очищены далее при помощи в общеизвестных в данной области методов, например, таких, как экстракция, кристаллизация, перегонка, растирание и хроматография. Некоторые промежуточные соединения и исходные вещества являются известными соединениями и могут быть коммерчески доступными или могут быть получены по известным в данной области методикам. Промежуточные соединения формулы (II-а) можно получить, вводя во взаимодействие промежуточное соединение формулы (IV-a) с подходящим агентом для снятия защиты, например, таким как 1 хлорэтилхлорформиат, в подходящем растворителе, например, таком как 1,2-дихлорэтан, и подходящим спиртом, например, таким как метанол и так далее. Промежуточные соединения формулы (II-а) можно также получить, вводя во взаимодействие промежуточное соединение формулы (IV-a) с формиатом аммония в присутствии палладия на угле и в присутствии подходящего растворителя, например, такого как спирт, например метанол. Промежуточные соединения формулы (IV-a), в которых R1 является галогеном, могут терять указанный галоидный заместитель при превращении их в промежуточные соединения формулы (II-а). Промежуточные соединения формулы (IV-a) можно получить, вводя во взаимодействие промежуточное соединение (V-a) с промежуточным соединением формулы (VI) в присутствии подходящего восстанавливающего агента, например, такого как н-BuLi, в присутствии подходящего основания, например,такого как N,N-диизопропиламин, и в присутствии подходящего растворителя, например, такого как тетрагидрофуран. Промежуточные соединения формулы (IV-a), в которых R1 является Het, a p равен 1, при этом указанные промежуточные соединения представлены формулой (IV-a-1), можно получить, вводя во взаимодействие промежуточное соединение (IV-a), в котором R1 является галогеном, при этом указанные промежуточные соединения представлены формулой (IV-a-2), с промежуточным соединением формулы(VII) в присутствии подходящего катализатора, например, такого как Pd(PPh3)4, подходящего основания,например, такого как K2CO3, и подходящего растворителя, например, такого как диэтиловый эфир, и подходящего спирта, например, такого как метанол и так далее. Промежуточные соединения формулы (III-a) можно получить, вводя во взаимодействие промежуточное соединение формулы (II-a) с промежуточным соединением формулы (VIII), в котором W1-(C=O) представляет собой группу, которую нужно ввести, a R представляет собой остаток промежуточного соединения, например, такого как 1,1'-карбонилбис-1 Н-имидазол, метилхлорформиат или этилхлорформиат, в присутствии подходящего растворителя, например, такого как тетрагидрофуран. Промежуточные соединения формулы (V-a) представляют собой соединения, которые либо коммерчески доступны, либо их можно получить по обычным методикам, общеизвестным в данной области. Например, промежуточные соединения формулы (V-a-1) можно получить по следующей схеме реакции в которой все переменные определены, как для формулы (Ia). Схема реакции (1) включает стадию (а), в которой соответствующим образом замещенный анилин вводят во взаимодействие с соответствующим-8 011359 ацилхлоридом, таким как 3-фенилпропионилхлорид, 3-фторбензолпропионилхлорид или пхлорбензолпропионилхлорид, в присутствии подходящего основания, такого как триэтиламин, и подходящего нереакционноспособного растворителя, такого как хлористый метилен или дихлорэтан. Данную реакцию удобно проводить при температуре в интервале от комнатной температуры до температуры кипения. На следующей стадии (b) аддукт, полученный на стадии (а), вводят во взаимодействие с хлорокисью фосфора (POCl3) в присутствии подходящего растворителя, например, такого как N,Nдиметилформамид (формилирование Вильсмайера-Хаака с последующей циклизацией). Данную реакцию удобно проводить при температуре в интервале от комнатной температуры до температуры кипения. На следующей стадии (с) вводят определенную R2-группу, где R2 представляет собой алкилокси или алкилтио-радикал, вводя во взаимодействие промежуточное соединение, полученное на стадии (b), и соединение -X-Alk, в котором X=S или О, a Alk представляет собой алкильную группу, определенную в формуле (Ia), например, такое как метилат натрия, в присутствии подходящего растворителя, например,такого как спирт, например метанол. Промежуточные соединения формулы (V-a-2) можно получить по следующей схеме реакции (2), в которой на первой стадии (а) замещенный индол-2,3-дион вводят во взаимодействие с замещенным 3 фенилпропионовым альдегидом в присутствии подходящего основания, такого как гидроксид натрия(реакции Пфитцингера), после чего соединение карбоновой кислоты на следующей стадии (b) декарбоксилируют при высокой температуре в присутствии подходящего нереакционноспособного растворителя,такого как дифениловый эфир. Схема 2 Очевидно, что в упомянутых выше и в последующих реакциях продукты реакции можно выделить из реакционной среды и, при необходимости, очистить далее в соответствии с методами, общеизвестными в данной области, например, такими как экстракция, кристаллизация и хроматография. Кроме того,очевидно, что продукты реакции, существующие более чем в одной энантиомерной форме, можно выделить из их смеси известными методами, в частности препаративной хроматографией, такой как ВЭЖХ. Как правило, соединения формулы (Ia) можно разделить на их изомерные формы. Промежуточные соединения формулы (VI) представляют собой соединения, которые либо коммерчески доступны, либо их можно получить по обычным методикам, общеизвестным в данной области. Например, промежуточные соединения формулы (VI) можно получить по следующей схеме реакции (3): Схема 3 Схема реакции (3) включает стадию (а), в которой R3, например, соответствующим образом замещенный фенил, нафтил или Het, вводят во взаимодействие по реакции Фриделя-Крафтса с соответствующим ацилхлоридом, таким как 3-хлорпропионилхлорид или 4-хлорбутирилхлорид, в присутствии подходящей кислоты Льюиса, такой как AlCl3, FeCl3, SnCl4, TiCl4 или ZnCl2 и, необязательно, в подходящем нереакционноспособном растворителе, таком как хлористый метилен или дихлорэтан. Реакцию удобно проводить при температуре в интервале от комнатной температуры до температуры кипения. На следующей стадии (b) вводят аминогруппу (-NR4(CH2-C6H5 при взаимодействии промежуточного соединения, полученного на стадии (а), с первичным или вторичным амином в присутствии подходящего растворителя, например, такого как ацетонитрил, и, необязательно, подходящего основания, например,такого как K2CO3. Следующие примеры иллюстрируют изобретение, не ограничивая его при этом. Экспериментальная часть Для некоторых соединений абсолютную конфигурацию стереогенного атома(атомов) углерода экспериментально не определяли. В этих случаях стереохимически изомерную форму, выделенную первой,обозначали А, а вторую - В, независимо от действительной стереохимической конфигурации. Однако-9 011359 специалист в данной области может однозначно охарактеризовать указанные изомерные формы А и В при помощи известных в данной области методов, например, таких как дифракция рентгеновских лучей. Способ выделения подробно описан ниже. Для некоторых промежуточных соединений и некоторых конечных соединений стереохимические конфигурации указаны в структурах. Данные конфигурации являются относительными конфигурациями,указывающими, что рассматриваемые группы расположены в одной и той же или в противоположных плоскостях молекулыR означает, что хиральный центр имеет абсолютную конфигурацию R или S.S означает, что хиральный центр имеет абсолютную конфигурацию R или S. В дальнейшем, термин т.пл. (М.Р.) означает температуру плавления, ТГФ означает тетрагидрофуран, EtOAc означает этилацетат, МеОН означает метанол, DME означает диметиловый эфир,DIPE означает диизопропиловый эфир, ДМФА означает N,N-диметилформамид, Et3N означает триэтиламин, Pd(PPh3)4 означает тетракис(трифенилфосфин)палладий, CDI означает 1,1'карбонилбис-1 Н-имидазол. А. Получение промежуточных соединений. Пример А 1. Получение промежуточного соединения 1. К раствору 4-бромбензоламина (0,407 моль) в Et3N (70 мл) и CH2Cl2 (700 мл) прибавляли по каплям бензолпропаноилхлорид (0,488 моль) при комнатной температуре и перемешивали смесь при комнатной температуре в течение ночи. Смесь выливали в воду и концентрированный NH4OH и экстрагировалиCH2Cl2. Органический слой сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток кристаллизовали из диэтилового эфира. Остаток (119,67 г) растворяли в CH2Cl2 и промывали HCl 1N. Органический слой сушили (MgSO4), фильтровали и выпаривали растворитель. Выход: 107,67 г промежуточного соединения 1. Пример А 2. Получение промежуточного соединения 2. Реакцию проводили дважды. К ДМФА (0,525 моль) по каплям прибавляли POCl3 (1,225 моль) при 10 С. Затем при комнатной температуре прибавляли промежуточное соединение 1 (0,175 моль). Смесь перемешивали в течение ночи при 80 С, выливали на лед и экстрагировали CH2Cl2. Органический слой сушили (MgSO4), фильтровали и выпаривали растворитель, получая 77,62 г (67%) промежуточного соединения 2. Продукт использовали без дополнительной очистки. Пример A3. Получение промежуточного соединения 3. Смесь промежуточного соединения 2 (0,233 моль) в CH3ONa (30%) в МеОН (222,32 мл) и МеОН(776 мл) перемешивали и кипятили в течение ночи, затем выливали на лед и экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/циклогексан 20/80, а. затем 100/0, 20-45 мкм). Чистые фракции собирали и выпаривали растворитель. Выход: 25 г (33%) промежуточного соединения 3 (т.пл.: 84 С). Пример А 4. а) Получение промежуточных соединений 4 и 5. Смесь хлорида алюминия (34,3 г, 0,257 моль) и 3-хлорпропаноилхлорида (29,7 г, 0,234 моль) в 1,2 дихлорэтане (150 мл) перемешивали при 0 С. Добавляли раствор нафталина (30 г, 0,234 моль) в 1,2 дихлорэтане (50 мл). Смесь перемешивали при 5 С в течение 2 ч и выливали в ледяную воду. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (56 г) очищали колоночной хроматографией на силикагеле (элюент: циклогексан/CH2Cl2: 60/40, 20-45 мкм). Собирали две фракции и выпаривали растворитель, получая промежуточное соединение 4 (31 г, 61%) в виде масла. Вторую фракцию (14 г) растворяли в DIPE, получая промежуточное соединение 5 (8,2 г, 16%, т.пл.: 68 С) в виде бледно-желтого твердого вещества. По предшествующей методике получали следующее промежуточное соединение:b) Получение промежуточного соединения 6. К 1,3-дифторбензолу (0,26 моль) осторожно прибавляли хлорид алюминия (0,3 моль) и нагревали при интенсивном перемешивании до 50 С. В течение 15 мин прибавляли по каплям 3-пропаноилхлорид(0,26 моль) при 40 С (охлаждение льдом) и перемешивали смесь при 50 С. Смесь выливали в воду (250 мл), лед (250 г) и HCl (25 мл) и перемешивали в течение 20 мин. Образовавшийся осадок отфильтровывали и экстрагировали CH2Cl2 и водой. Выход: 40 г промежуточного соединения 6 (75%). Пример А 5. а) Получение промежуточного соединения 7. Смесь промежуточного соединения 4 (3 г, 0,0137 моль), N-бензиламина (2 мл, 0,0150 моль) в ацетонитриле (100 мл) перемешивали при 80 С в течение 2 ч. Прибавляли воду комнатной температуры. Смесь экстрагировали CH2Cl2. Органический слой отделяли и сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (6 г) очищали колоночной хроматографией на силикагеле (элюент:CH2Cl2/МеОН: 97/3, 20-45 мкм), получая масло. Выход: 4,2 г промежуточного соединения 7. По предшествующей методике получали следующее промежуточное соединение:b) Получение промежуточного соединения 8. Смесь промежуточного соединения 6 (0,015 моль), N-этилбензолметанамина (0,016 моль) и K2CO3(0,016 моль) в ацетонитриле (30 мл) перемешивали при 70 С в течение 2 ч, выливали в воду и экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Выход: 4 г промежуточного соединения 8 (88%). Пример A6. а) Получение промежуточного соединения 9. К раствору диизопропиламина (0,0075 моль) в ТГФ (50 мл) прибавляли н-бутиллитий (0,0075 моль) при -20 С. Смесь охлаждали до -70 С. Прибавляли промежуточное соединение 3 (0,0062 моль). Смесь перемешивали при -70 С в течение 1 ч и 30 мин. Прибавляли промежуточное соединение 7 (0,0075 моль). Смесь перемешивали в течение 1 ч и 30 мин. Добавляли воду. Смесь экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (3 г) очищали колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc: 90/10, 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Выход: 1,5 г смеси двух диастереоизомеров(38%), то есть промежуточное соединение 9. По предшествующей методике получали следующее промежуточное соединение:b) Получение промежуточного соединения 10. К раствору диизопропиламина (0,0075 моль) в ТГФ (50 мл) прибавляли н-бутиллитий (0,0075 моль) при -20 С. Смесь охлаждали до -70 С. Прибавляли промежуточное соединение 3 (0,0061 моль). Смесь перемешивали при -70 С в течение 1 ч и 30 мин. Прибавляли 4-[метил(фенилметил)амино]-1-фенил-1 бутанон (0,0073 моль). Смесь перемешивали в течение 1 ч и 30 мин. Добавляли воду. Смесь экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (4,9 г) очищали колоночной хроматографией на силикагеле (элюент: 100% CH2Cl2; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель, получая 1,4 3 г промежуточного соединения 10(40%, смесь диастереизомеров: 60/40). По предшествующей методике получали следующие промежуточные соединения: с) Получение промежуточных соединений 11 и 12. К раствору диизопропиламина (0,0075 моль) в ТГФ (50 мл) прибавляли н-бутиллитий (0,0075 моль) при -20 С. Смесь охлаждали до -70 С. Прибавляли промежуточное соединение 3 (0,00824 моль). Смесь перемешивали при -70 С в течение 1 ч и 30 мин. Прибавляли промежуточное соединение 8 (0,0099 моль). Смесь перемешивали в течение 1 ч и 30 мин. Добавляли воду. Смесь экстрагировали CH2Cl2. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (5,4 г) очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/циклогенсан 60/40; 15-40 мкм). Собирали две фракции и выпаривали растворитель. Выход: 0,95 г промежуточного соединения 11 в виде диастереоизомера А (15%, т.пл.: 171 С) и 0,83 г промежуточного соединения 12 в виде диастереоизомера В (13%, МН+: 631). Пример А 7. Получение промежуточного соединения 17.(0,158 ммоль), DME (30 мл), МеОН (10 мл) и K2CO3 (1,6 мл) нагревали при микроволновом облучении(300 Вт, 68 С) в течение 10 мин. Смесь охлаждали, выливали в воду и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (1,4 г) очищали колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 90/10; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Выход: 0,47 г промежуточного соединения 17 в виде смеси(60/40) (41%). Пример А 8. а-1) Получение промежуточных соединений 13 и 14. К смеси промежуточного соединения 9 (0,0023 моль) в 1,2-дихлорэтане (30 мл) прибавляли при комнатной температуре 1-хлорэтилхлорформиат (15 мл). Смесь перемешивали при 80 С в течение 1 ч. Растворитель выпаривали. Добавляли МеОН (15 мл). Смесь перемешивали и кипятили в течение 30 мин. Растворитель выпаривали. Остаток(1,49 г) очищали колоночной хроматографией на силикагеле(элюент: CH2Cl2/MeOH/NH4OH 97/3/0,1; 15-40 мкм). Собирали две фракции и выпаривали растворитель. Первый остаток (0,23 г) кристаллизовали из DIPE. Осадок отфильтровывали и сушили, получая 0,168 г(13%) промежуточного соединения 13 (диастереоизомер А) (т.пл.: 204 С). Второй остаток (0,32 г) кристаллизовали из DIPE. Осадок отфильтровывали и сушили. Выход: 0,298 г (23%) промежуточного соединения 14 (диастереоизомер В) (т.пл.: 225 С). По описанной выше методике получали следующие промежуточные соединения. Очистка полученного остаткауказана для каждого промежуточного соединения отдельно.- 17011359 а-2) Получение промежуточных соединений 15 и 16. Промежуточное соединение 13 (диастереоизомер А) (0,9 г) очищали хиральной хроматографией на силикагеле (элюент: 100% этанол). Собирали две фракции и выпаривали растворитель. Выход: 0,420 г промежуточного соединения 15 (энантиомер А 1) (т.пл.: 161 С, МН+: 541) и 0,397 г промежуточного соединения 16 (энантиомер А 2) (т.пл.: 158 С, МН+: 541). а-3) Получение промежуточных соединений 44 и 45. Смесь промежуточного соединения 43 (полученного по примеру A6.а) (1,5 г, 2,62 моль), формиата аммония (0,83 г, 0,013 моль) и палладия на угле (10%, 1,5 г) в метаноле (30 мл) кипятили в течение 1 ч. Смесь охлаждали и фильтровали через небольшой слой целита. Добавляли воду. Органический слой экстрагировали зтилацетатом, отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (1,3 г) очищали колоночной хроматографией на силикагеле (элюент: MeOH/AcNH4 60/40, кромасилC18, 5 мкм). Собирали чистые фракции и выпаривали растворитель, получая две фракции. Выход: 0,14 г промежуточного соединения 44 в виде диастереоизомера А (12%) и 0,26 г промежуточного соединения 45 в виде диастереоизомера В (22%). Пример А 9. Получение промежуточного соединения 18. Смесь промежуточного соединения 39 (полученного по примеру А 8.а-1) (0,0002 моль) и CDI Смесь промежуточного соединения 13 (полученного по примеру А 8.а-1) (0,00009 моль) и параформа (0,0001 моль) в толуоле (5 мл) перемешивали при 80 С. Смесь упаривали. Остатокочищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/МеОН 99/1; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Выход: 0,025 г соединения 1 (диастереоизомер А) (49%, т.пл. 112 С). По описанной выше методике получали следующие соединения. Очистка остаткауказана, если она отличается от описанной выше методики. Смесь промежуточного соединения 37 (диастереоизомера А, полученного по примеру А 8.а-1)(0,00009 моль) и параформа (0,0001 моль) в толуоле (5 мл) перемешивали при 80 С. Смесь упаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/МеОН 99/1; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Остаток (0,06 г, 92%) кристаллизовали из DIPE. Осадок фильтровали и сушили. Выход: 0,026 г соединения 2 (диастереоизомер В) (40%, т.пл. 201 С). Пример B3. Получение соединения 3. Смесь промежуточного соединения 30 (диастереоизомера В, полученного по примеру А 8.а-1)(0,00009 моль) и параформа (0,0001 моль) в толуоле (5 мл) перемешивали при 80 С. Смесь упаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/МеОН 99/1; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Остаток (0,11 г, 100%) кристаллизовали из диэтилового эфира. Осадок фильтровали к сушили. Выход: 0,033 г соединения 3 (диастереоизомер В) (33%,т.пл. 189 С). Пример В 4. Получение соединения 4. Смесь соединения 7 (диастереоизомера А, полученного по примеру В 1) (0,1 ммоль) и йодистого метила (0,1 ммоль) в ацетоне (2 мл) перемешивали при комнатной температуре в течение 2,5 ч. Осадок фильтровали, промывали ацетоном и сушили. Выход: 0,031 г соединения 4 в виде гидройодида (диастереоизомер А) (48%, т.пл.: 211 С). По описанной выше методике получали следующее соединение. К смеси промежуточного соединения 18 (полученного по примеру А 9) (0,0001 моль) в ТГФ (10 мл) прибавляли гидрид натрия (0,011 г) при 0 С в токе N2. Смесь перемешивали при 0 С в течение 30 мин,выливали в H2O и экстрагировали EtOAc. Органический слой отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: циклогексан/EtOAc 80/20; 15-40 мкм). Собирали чистые фракции и выпаривали растворитель. Остаток (0,09 г,67%) кристаллизовали из DIPE. Осадок фильтровали и сушили. Выход: 0,034 г соединения 5 (диастереоизомер А) (т.пл.: 192 С). По описанной выше методике получали следующее соединение. Смесь промежуточного соединения 44 (диастереоизомера А, полученного по примеру А 8.а-3) (0,12 г, 0,298 ммоль) и параформа (0,358 моль) в толуоле (5 мл) перемешивали при 80 С в течение 2 ч. Добавляли воду при комнатной температуре и экстрагировали органический слой этилацетатом, отделяли, сушили (MgSO4), фильтровали и выпаривали растворитель. Остаток (0,13 г) очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH 99/1/0,1; 10 мкм). Собирали чистые фракции и выпаривали растворитель. Выход: 0,075 г соединения 20 (диастереоизомер А) (61%, МН+: 415). По описанной выше методике, но без колоночной хроматографии, получали следующее соединение. С. Аналитические методы. Массу соединений определяли при помощи LCMS (жидкостная хроматография с массспектрометрией). Использовали три описанных ниже метода. Данные сведены в приведенной далее табл. 1.LCMS-метод 1. Анализ методом LCMS (ионизация электроспреем в положительном режиме, режим сканирования от 100 до 900 атомных единиц массы (amu на колонке Kromasil C18 (Interchim, Montlucon, FR; 5 мкм,4,6150 мм) при скорости потока 1 мл/мин. Использовали две подвижные фазы (подвижная фаза А: 30%ный 6,5 мМ ацетат аммония+40%-ный ацетонитрил+30%-ная муравьиная кислота (2 мл/л); подвижная фаза В: 100%-ный ацетонитрил) для получения условия градиента от 100% А в течение 1 мин до 100% В в течение 4 мин, от 100% В в течение 5 мин до 100% А в течение 3 мин и повторного установления равновесия при 100% А в течение 2 мин.(импульсном) режиме сканирования от 100 до 1000 атомных единиц массы (amu на колонке KromasilC18 (Interchim, Montlucon, FR; 3,5 мкм, 4,6100 мм) при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: 35%-ный 6,5 мМ ацетат аммония+30%-ный ацетонитрил+35%-ная муравьиная кислота (2 мл/л); подвижная фаза В: 100%-ный ацетонитрил) для получения условия градиента от 100% А в течение 1 мин до 100% В в течение 4 мин, от 100% В при скорости потока 1,2 мл/мин в течение 4 мин до 100% А при 0,8 мл/мин в течение 3 мин и повторного установления равновесия при 100% А в течение 1,5 мин.(импульсном) режиме сканирования от 100 до 1000 атомных единиц массы (amu на колонке Sunfire C18(Waters, Millford USA; 3,5 мкм, 4,6100 мм) при скорости потока 0,8 мл/мин. Использовали две подвиж- 23011359 ные фазы (подвижная фаза А: 35%-ный 6,5 мМ ацетат аммония+30%-ный ацетонитрил+35%-ная муравьиная кислота (2 мл/л); подвижная фаза В: 100%-ный ацетонитрил) для получения условия градиента от 100% А в течение 1 мин до 100% В в течение 4 мин, от 100% В при скорости потока 1,2 мл/мин в течение 4 мин до 100% А при 0,8 мл/мин в течение 3 мин и повторного установления равновесия при 100% А в течение 1,5 мин.(импульсном) режиме сканирования от 100 до 1000 атомных единиц массы (amu на колонке Sunfire C18(Waters, Millford USA; 3,5 мкм, 4,6100 мм) при скорости потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: 25%-ный 6,5 мМ ацетат аммония+50%-ный ацетонитрил+25%-ная муравьиная кислота (2 мл/л); подвижная фаза В: 100%-ный ацетонитрил) для получения условия градиента от 100% А в течение 1 мин до 100% В в течение 4 мин, от 100% В при скорости потока 1,2 мл/мин в течение 4 мин до 100% А при 0,8 мл/мин в течение 3 мин и повторного установления равновесия при 100% А в течение 1,5 мин. Таблица 1 Исходный пик LCMSD.1. In vitro способ тестирования соединений против М. tuberculosis.- 24011359 Плоскодонные стерильные 96-луночные пластиковые микротитровочные планшеты заполняли 100 мкл бульонной средой Middlebrook (1). После этого добавляли основные растворы (10 конечная тестовая концентрация) соединений в объемах 25 мкл к ряду парных лунок в колонке 2 с тем, чтобы оценить их влияние на бактериальный рост. Последовательные пятикратные разбавления проводили непосредственно в микротитровочных планшетах с колонки 2 по 11 при помощи выполненного по заказу робототехнического комплекса (Zymark Corp., Hopkinton, MA). Наконечники пипеток меняли после каждых трех разбавлений, чтобы свести к минимуму ошибки пипеток с высокогидрофобными соединениями. Необработанные контрольные образцы, содержащие (колонка 1) и не содержащие (колонка 12) прививочный материал, входили в каждый микротитровочный планшет. К рядам с А по Н, за исключением колонки 12, добавляли приблизительно 5000 CFU на лунку Mycobacterium tuberculosis (штамм H37RV) в объеме 100 мкл в бульонной среде Middlebrook (1). Тот же объем бульонной среды без прививочного материала добавляли в колонку 12 в ряд с А по Н. Культуры инкубировали при 37 С в течение 7 дней в увлажненной атмосфере (инкубатор с открытым воздушным клапаном и непрерывной вентиляцией). За день до окончания инкубации, через 6 дней после инокуляции во все лунки добавляли Resazurin (1:5) в объеме 20 мкл и инкубировали планшеты еще в течение 24 ч при 37 С. На 7 день проводили количественную оценку бактериального роста методом флуоресценции. Флуоресценцию измеряли на управляемом компьютером флуориметре (Spectramax Gemini EM, Molecular Devices) при длине волны возбуждения 530 нм и длине волны эмиссии 590 нм. Процентный рост ингибирования, достигнутый соединениями, рассчитывали стандартными методами и рассчитывали данные MIC (представляющие величины IC90, выраженные в мкг/мл).D.2. Способ тестирования соединений на антибактериальную активность против штамма М. Smegmatis ATCC607 in vitro. Плоскодонные стерильные 96-луночные пластиковые микротитровочные планшеты заполняли 180 мкл стерильной деионизованной водой с добавкой 0,25% BSA. После этого добавляли основные растворы (7,8 конечная тестовая концентрация) соединений в объеме 45 мкл к ряду парных лунок в колонке 2 с тем, чтобы оценить их влияние на бактериальный рост. Последовательные пятикратные разбавления(45 мкл в 180 мкл) проводили непосредственно в микротитровочных планшетах с колонки 2 по 11 при помощи выполненного по заказу робототехнического комплекса (Zymark Corp., Hopkinton, MA). Наконечники пипеток меняли после каждых трех разбавлений, чтобы свести к минимуму ошибки пипеток с высокогидрофобными соединениями. Необработанные контрольные образцы, содержащие (колонка 1) и не содержащие (колонка 12) прививочный материал, входили в каждый микротитровочный планшет. В ряды с А по Н. за исключением колонки 12, добавляли приблизительно 250 CFU на лунку бактериального прививочного материала, в объеме 100 мкл в 2,8 бульонной среды Mueller-Hinton. Тот же объем бульонной среды без прививочного материала добавляли в колонку 12 в ряд с А по Н. Культуры инкубировали при 37 С в течение 48 ч в увлажненной атмосфере, содержащей 5% CO2 (инкубатор с открытым воздушным клапаном и непрерывной вентиляцией). В конце инкубации, через два дня после инокуляции проводили количественную оценку бактериального роста методом флуоресценции. Поэтому во все лунки добавляли Alamar Blue (10) в объеме 20 мкл и инкубировали планшеты еще 2 ч при 50 С. Флуоресценцию измеряли на управляемом компьютером флуориметре (Cytofluor, Biosearch) при длине волны возбуждения 530 нм и длине волны эмиссии 590 нм (увеличение 30). Процентный рост ингибирования, достигнутый соединениями, рассчитывали стандартными методами. pIC50 определяли как 50% ингибирующую концентрацию бактериального роста. Результаты представлены в табл. 2. Таблица 2 Результаты in vitro-скрининга соединений в соответствии с данным изобретением для М. smegmatis (pIC50) его фармацевтически приемлемые соли присоединения кислоты или основания, его четвертичные амины, его стерохимически изомерные формы, его таутомерные формы и его N-оксидные формы, в которомR1 представляет собой водород, галоген, Ar, Het, C1-6 алкил, C1-6 алкилокси; р представляет собой целое число, равное 1 или 2;R5 представляет собой водород или галоген;r представляет собой целое число, равное 1 или 2;Z представляет собой СН 2 или С(=O);Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1, 2 или 3 галогеновыми заместителями;Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей фуранил и тиенил; галоген представляет собой заместитель, выбранный из группы, включающей фтор, хлор, бром и йод. 2. Соединение по п.1, в котором Z представляет собой СН 2. 3. Соединение по п.1 или 2, в которомR1 представляет собой водород, галоген, Ar, Het, C1-6 алкил и C1-6 алкилокси; р представляет собой целое число, равное 1;r представляет собой целое число, равное 1;Ar представляет собой гомоцикл, выбранный из группы, включающей фенил и нафтил, каждый из которых необязательно замещен 1 или 2 галогеновыми заместителями;Het представляет собой моноциклический гетероцикл, выбранный из группы, включающей фуранил и тиенил. 4. Соединение по любому из предшествующих пунктов, которое представляет собой соединение формулы (Ia) и в которомR3 представляет собой нафтил, фенил или Het, где каждый нафтил и фенил необязательно замещен 1 или 2 заместителями, выбранными из группы, включающей галоген;R5 представляет собой водород или галоген;R6 представляет собой водород. 5. Соединение по любому из пп.1, 3 или 4, которое представляет собой соединение формулы (Ia),гдеR3 представляет собой нафтил, фенил или Het, где каждый нафтил и фенил необязательно замещен галогеном;R5 представляет собой водород или галоген;Z представляет собой СН 2 или С(=O). 6. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения, по любому из пп.1-5. 7. Применение соединения по любому из пп.1-5 или композиции по п.6 для получения лекарственного средства для лечения микобактериальных заболеваний. 8. Способ получения соединения по п.1, отличающийся тем, что а) промежуточное соединение формулы (II-а) вводят во взаимодействие с параформом в подходящем растворителеb) промежуточное соединение формулы (III-a) вводят во взаимодействие с подходящим основанием в подходящем растворителе при этом R1-R6, p и r определены в п.1, a W1 представляет собой подходящую уходящую группу; или, при желании, соединения формулы (Ia) переводят друг в друга путем известных в данной области превращений и затем, при желании, переводят соединения формулы (Ia) в терапевтически активную нетоксичную соль присоединения кислоты путем обработки кислотой или в терапевтически активную нетоксичную соль присоединения основания путем обработки основанием или, напротив, переводят форму соли присоединения кислоты в свободное основание путем обработки щелочью или переводят соль присоединения основания в свободную кислоту путем обработки кислотой и, при желании, получают их стереохимически изомерные формы, четвертичные амины, таутомерные формы или N-оксиды. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: A61K 31/5355, C07D 413/06, C07D 413/14, A61P 31/10
Метки: микобактериальных, хинолины, замещённые, ингибиторов, качестве, применение
Код ссылки
<a href="https://eas.patents.su/28-11359-zameshhyonnye-hinoliny-i-ih-primenenie-v-kachestve-mikobakterialnyh-ingibitorov.html" rel="bookmark" title="База патентов Евразийского Союза">Замещённые хинолины и их применение в качестве микобактериальных ингибиторов</a>
Производные хинолина и их применение в качестве микобактериальных ингибиторов

Номер патента: 8937
Опубликовано: 26.10.2007
Авторы: Чока Имре Кристиан Франсис, Ван Гестел Йозеф Франс Элизабета, Андрис Конрад Йозеф Лодевийк Марсель, Оддс Френк Кристофер, Вене Марк Гастон, Вернье Даниэль Ф.Ж., Гийемон Жером Эмиль Жорж, Декран Лоранс Франсуаз Бернадетт
МПК: A61K 31/47, C07D 215/22, A61P 31/06...
Метки: производные, хинолина, микобактериальных, применение, качестве, ингибиторов
Формула / Реферат:
1. Соединение общей формулы (Iа) или общей формулы (Ib) их фармацевтически приемлемые аддитивные соли с кислотами или основаниями, стереохимически изомерные формы, таутомерные формы и N-оксиды, где R1 означает водород, галоген, галогеналкил, циано, гидрокси, Ar, Het, алкил, алкилокси, алкилтио, алкилоксиалкил, алкилтиоалкил, Ar-алкил или ди(Аr)алкил; р означает целое число, равное 0, 1, 2, 3 или 4; R2 означает водород, гидрокси, тио,...
Замещённые бензазолы и их применение в качестве ингибиторов киназы raf

Номер патента: 7987
Опубликовано: 27.02.2007
Авторы: Пун Дэниел Дж., Субраманиан Шарадха, Фэнтл Уэнди, Рамуртхи Савитхри, Сунг Леонард, Ренхауэ Пол А., Эмайри Пэйман, Левайн Берри Хаскелл
МПК: A61K 31/41, C07D 401/12, C07D 401/14...
Метки: качестве, ингибиторов, замещённые, применение, бензазолы, киназы
Формула / Реферат:
1. Соединение формулы (I) где X1 и Х2 независимо выбраны из =N-, -NR4-, -О- или -S- при условии, что если X1 представляет собой -NR4-, -О- или -S-, тогда Х2 представляет собой =N-, или если Х2 представляет -NR4-, -О- или -S-, тогда X1 представляет собой =N-, и как X1, так и Х2 не являются =N-; Y представляет О или S; Y представляет О или S; А1 представляет замещенный или незамещенный С1-С12алкил, С3-С8циклоалкил, С3-С8гетероциклоалкил,...
Замещённые производные пиррола и их применение в качестве ингибиторов hmg-cоa

Номер патента: 9646
Опубликовано: 28.02.2008
Авторы: Саттигери Джитендра, Чугх Анита, Кумар Ятендра, Раманатан Викрам Кришна, Салман Мохаммад, Ариян Рам Чандер
МПК: A61K 31/40, C07D 207/34, A61K 31/366...
Метки: hmg-cоa, ингибиторов, производные, применение, замещённые, качестве, пиррола
Формула / Реферат:
1. Соединения, имеющие структурную формулу I их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, полиморфы, таутомеры, или N-оксиды, где или R1 может быть С1-С6алкил, С3-С6 циклоалкил или необязательно замещенный фенил (где до трех заместителей независимо выбраны из галоидов, C1-С6алкила, циано или С1-С3перфторалкила); R2 может быть необязательно замещенный фенил (где до трех заместителей независимо выбраны из циано,...
4-(n-фениламино)хиназолины/-хинолины в качестве ингибиторов тирозинкиназы

Номер патента: 9300
Опубликовано: 28.12.2007
Авторы: Солка Флавио, Юнг Биргит, Химмельсбах Франк
МПК: C07D 239/94, A61P 35/00, A61K 31/517...
Метки: ингибиторов, тирозинкиназы, качестве
Формула / Реферат:
1. Бициклические гетероциклы общей формулы в которой Ra обозначает атом водорода, Rb обозначает фенил, в котором фенильное ядро замещено остатками R1-R3, где R1 и R2 могут иметь идентичные или различные значения и обозначают атом водорода, фтора, хлора, брома или иода, C1-4алкильную группу, C1-4алкоксигруппу, С2-3алкинильную группу, арилметоксигруппу, гетероарилоксигруппу, гетероарилметоксигруппу и R3 обозначает атом водорода, Rc обозначает...
4-замещённые бензимидазолы и их применение в качестве ингибиторов желудочной секреции

Номер патента: 8779
Опубликовано: 31.08.2007
Авторы: Кромер Вольфганг, Пальмер Андреас, Грундлер Герхард, Постиус Штефан, Циммерманн Петер Ян, Брем Кристоф, Зенн-Билфингер Йёрг, Бур Вильм, Зимон Вольфганг-Александер, Хиеза Витториа
МПК: A61K 31/422, A61K 31/4245, A61K 31/4184...
Метки: качестве, бензимидазолы, желудочной, 4-замещённые, применение, ингибиторов, секреции
Формула / Реферат:
1. Соединение формулы 1 в котором R1 представляет собой водород, C1-4-алкил, С3-7-циклоалкил, С3-7-циклоалкил-C1-4-алкил, C1-4-алкокси, C1-4-алкокси-C1-4-алкил, C1-4-алкоксикарбонил, С2-4-алкенил, С2-4-алкинил, фтор-C1-4-алкил, гидрокси-C1-4-алкил, моно- или ди-C1-4-алкиламино или С1-4-алкилкарбонилокси-С1-4-алкил, R2 представляет собой водород, C1-4-алкил, арил, С3-7-циклоалкил, С3-7-циклоалкил-С1-4-алкил, C1-4-алкоксикарбонил, моно- или...
Предыдущий патент: Композиция минерального нефтетоплива, содержащая смесь присадок, способ ее получения и ее применение
Следующий патент: 5,6-диалкил-7-аминотриазолопиримидины, способ их получения и их применение для борьбы с патогенными грибами, а также содержащие их средства
Случайный патент: Инсектициды на основе неоникотиноидов и защитных веществ