Производные тиенопиридона в качестве активаторов амр-активированной протеинкиназы (амрк)
Номер патента: 20773
Опубликовано: 30.01.2015
Авторы: Краво Даньель, Шарон Кристине, Лепифре Франк, Аллаку-Бозек Софи

























Формула / Реферат
1. Соединение формулы (I)

в которой R1 представляет собой Н или Hal, R2 представляет собой фенил, который может быть замещен CN, или представляет собой пиридин, B1 представляет собой 2,5-пиридинилен, 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН, B2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил, 1,3-бензодиоксан-5-ил или фенил, необязательно замещенные одной или двумя группами, выбранными из ОН, метоксигруппы, гидроксиметила, Hal, метила, трифторметила, CN, метилсульфонила, метоксикарбонила и СООН, Hal представляет собой F, Cl, Br или I, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях.
2. Соединение по п.1, в котором В2 представляет собой фенил, необязательно замещенный одной или двумя группами, выбранными из метила, Hal, метоксигруппы, ОН, СООН, метилсульфонила и метоксикарбонила, или В2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил или 1,3-бензодиоксан-5-ил, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях.
3. Соединение по п.1 или 2, в котором В1 представляет 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях.
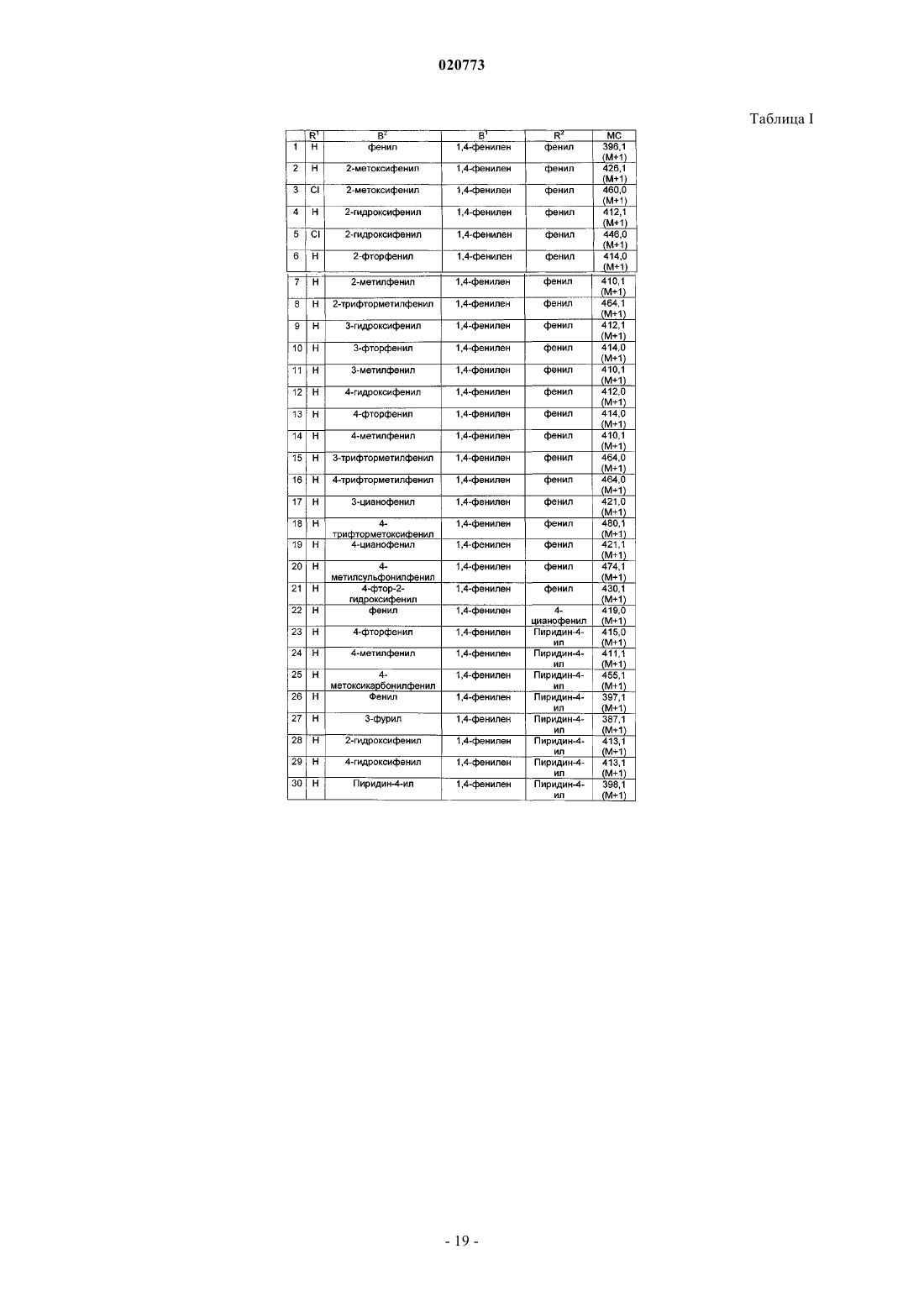
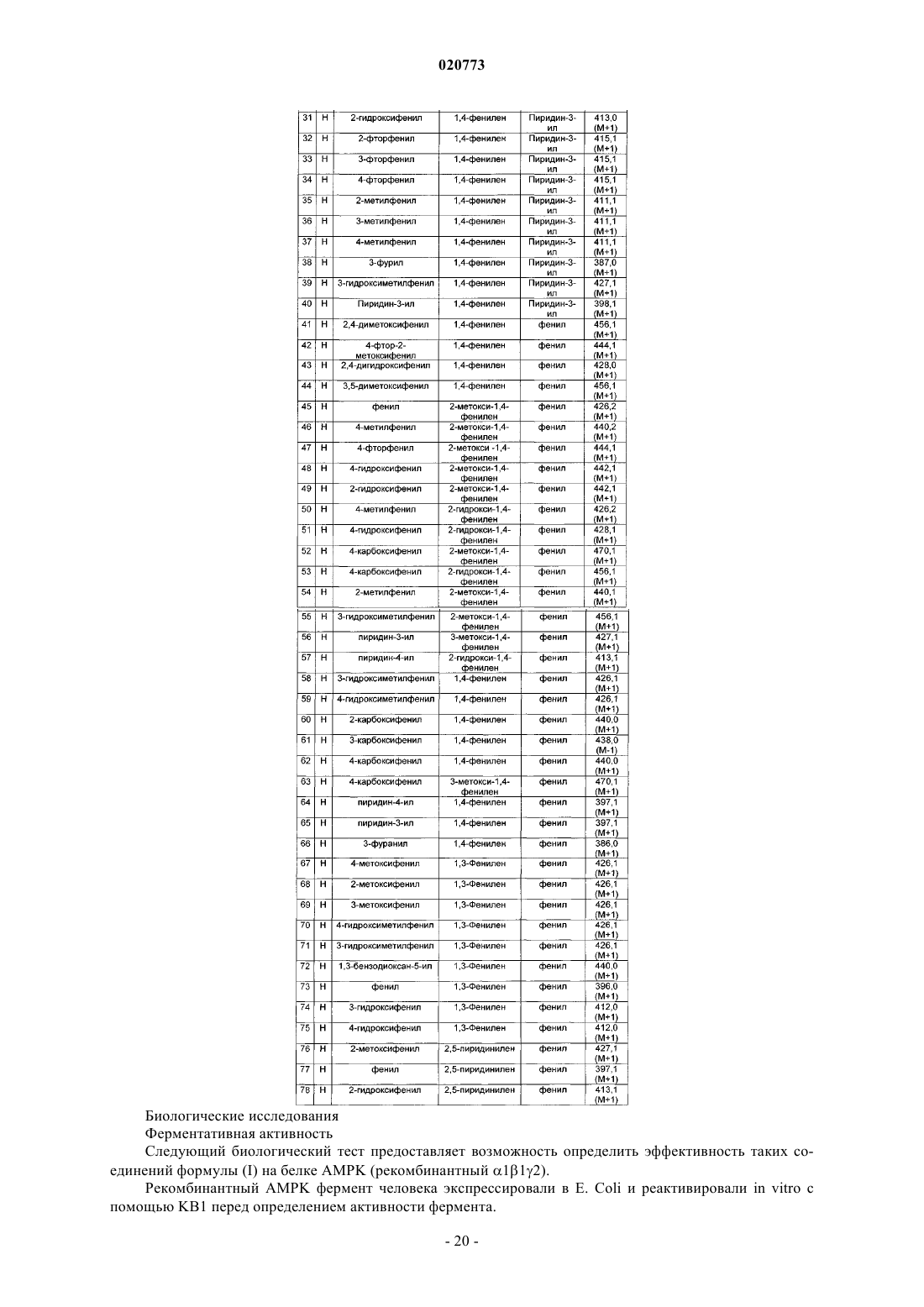
4. Соединение по п.1, выбранное из группы, включающей





5. Соединение по п.1, выбранное из группы, включающей



6. Промежуточное соединение формулы (II) для приготовления соединения формулы (I)

где R1, R2, В1 имеют значения, указанные в п.1, и X представляет собой атом галогена, и его соли.
7. Способ получения соединений формулы (I) по любому из пп.1-5 и их фармацевтически пригодных сольватов, солей и стереоизомеров, который характеризуется тем, что соединение формулы (II)

где R1, R2 и В1 имеют значения, указанные в п.1, и X представляет собой Cl, Br или I, подвергают взаимодействию в реакции Сузуки с производным бороновой кислоты В2-В(ОН)2, где В2 имеет значения, указанные в п.1, и/или соединение формулы I в виде основания или кислоты превращают в одну из его солей.
8. Лекарственные средства, содержащие по меньшей мере одно соединение формулы (I) по любому из пп.1-5 и/или его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях, и необязательно, наполнители и/или вспомогательные вещества.
9. Применение соединений по любому из пп.1-5 и их фармацевтически пригодных солей, сольватов и стереоизомеров, включая их смеси во всех соотношениях, для приготовления лекарственного средства для лечения метаболического синдрома, диабета, ожирения, злокачественного новообразования, воспаления, сердечно-сосудистых заболеваний.
10. Применение соединений по любому из пп.1-5 для приготовления лекарственного средства, повышающего поглощение глюкозы в мышечных клетках.
Текст
ПРОИЗВОДНЫЕ ТИЕНОПИРИДОНА В КАЧЕСТВЕ АКТИВАТОРОВ АМРАКТИВИРОВАННОЙ ПРОТЕИНКИНАЗЫ (АМРK) формула (II) Настоящее изобретение относится к соединениям формулы (I), где R1, R2 и В 1, В 2 имеют указанные далее значения, а также к содержащим их лекарственным средствам и к их применению для лечения и/или предотвращения заболеваний и нарушений, модулируемых агонистами AMP. Изобретение также относится к новому промежуточному соединению формулы (II) и к способу получения соединений формулы (I).(71)(73) Заявитель и патентовладелец: МЕРК ПАТЕНТ ГМБХ (DE) Область техники, к которой относится изобретение Изобретение относится к производным тиенопиридона, которые являются активаторами AMPKактивированной протеинкиназы (AMPK) формулы (I) формула (I) Изобретение также относится к получению и применению этих тиенопиридонов для лечения нарушений, таких как диабет, метаболический синдром, ожирение, злокачественное новообразование, воспаление. Предпосылки создания изобретения Объектом изобретения является выявление новых соединений, обладающих ценными свойствами, в особенности тех, которые могут применяться для приготовления лекарственных средств. Настоящее изобретение относится к соединениям, которые пригодны для лечения и/или предотвращения заболеваний, таких как диабет, метаболический синдром, ожирение, злокачественное новообразование, воспаление. Также обеспечиваются способы лечения заболеваний и нарушений, которые можно лечить путем активации AMPK, включающие введение эффективного количества соединения согласно настоящему изобретению. Следовательно, настоящее изобретение относится к соединениям в соответствии с изобретением в качестве лекарственных средств и/или активных компонентов лекарственных средств для лечения и/или профилактики указанных заболеваний и к применению соединений в соответствии с изобретением для приготовления лекарственного средства для лечения и/или профилактики указанных заболеваний, а также к способу лечения указанных заболеваний, который включает введение одного или нескольких соединений в соответствии с изобретением пациенту, который нуждается в таком введении. Неожиданно нами было обнаружено, что производные тиенопиридона активируют AMPK; следовательно, эти соединения особенно пригодны для предотвращения и лечения диабета, метаболического синдрома, ожирения, злокачественного новообразования, воспаления. Было обнаружено, что соединения в соответствии с изобретением и их соли обладают чрезвычайно ценными фармакологическими свойствами, а также обладают хорошей переносимостью. В частности, они проявляют активирующие действия на AMPK. Хозяин или пациент может принадлежать к любому виду млекопитающих, например, такому как приматы, в особенности человек; грызуны, включая мышей, крыс и хомячков; кролики; лошади, коровы,собаки, коты и т.д. Животные модели представляют интерес для экспериментальных исследований, поскольку они обеспечивают модель для лечения заболевания человека.AMPK хорошо известна в качестве чувствительного элемента и регулятора гомеостаза клеточной энергии (Hardie, D.G. и Hawley, S.A., "AMP-activated protein kinase: the energy charge hypothesis revisited",Bioassays, 23, 1112, (2001), Kemp, В.Е. и др., "AMP-activated protein kinase, super metabolic regulator", Biochem. Soc. Transactions, 31, 162 (2003. Аллостерическая активация этой киназы в результате повышения уровней AMP происходит при состояниях истощения клеточной энергии. Последующее фосфорилирование серина/треонина целевых ферментов приводит к адаптации клеточного метаболизма к сниженному энергетическому состоянию. Результирующим эффектом изменений, вызванных активацией AMPK, является ингибирование процессов потребления АТР и активация путей образования АТР, следовательно,регенерация запасов АТР. Примерами субстратов AMPK являются ацетил-СоА-карбоксилаза (АСС) иenzymes of fatty acid and cholesterol biosynthesis", FEBS Letters, 223, 217 (1987. Фосфорилирование и,следовательно, ингибирование АСС приводит к снижению синтеза жирных кислот (АТР-потребление) и в то же время к повышению окисления жирных кислот (АТР-образование). Фосфорилирование и последующее ингибирование HMG-CoA редуктазы приводит к снижению синтеза холестерина. Другими субстратами AMPK является липаза, чувствительная к действию гормонов (Garton, A.J. и др., "Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism", Eur. J. Biochem., 179, 249, (1989, глицерин-3-фосфат ацилтрансфераза (Muoio D.M. и др., "AMPactivated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target", Biochem. J., 338, 783 (1999, малонилСоА декарбоксилаза (Saha А.K. и др., "Activation of malonyl-CoA decarboxylase in rat skeletal muscle byAMPK также вовлечен в регуляцию обмена веществ в печени. Повышенная выработка глюкозы печенью является основной причиной гипергликемии натощак у T2D (Saltiel и др., "New perspectives intothe molecular pathogenesis и treatment of type 2 diadetes", cell 10, 517-529 (2001. Глюконеогенез в печени регулируется различными ферментами, такими как фосфоенолпируват карбоксикиназа (PEPCK) и глю-1 020773 козо-6-фосфатаза (G6Pase). Активация AMPK подавляет транскрипцию этих генов в клетках гепатомыthe 2 key gluconeogenic genes PEPCK and glucose-6-phosphatase", Diabetes, 49, 896-903 (2000. Активация AMPK также понижающе регулирует глюконеогенез, действуя на экспрессию некоторых других генов. Эти эффекты также могут быть обусловлены ее способностью понижающе регулировать ключевые транскрипционные факторы, такие как SREBP-1c (Zhou G. и др.,"Role of AMP-activatednovel target of AMP-activated protein kinase". Diabetes, 50, 1515 (2001 или путем прямого фосфорилирования транскрипционных кофакторов, таких как р 300 (Yang W. и др., "Regulation of transcription by AMPactivated protein kinase; Phosphorylation of р 300 blocks its interaction with nuclear receptors". J. Biol. Chem. 276, 38341 (2001 и TORC2.AMPK рассматривают в качестве перспективного кандидата для поглощения глюкозы, индуцированного сокращениями скелетных мышц, поскольку она действует одновременно с повышением AMP и уменьшением энергетических запасов креатин-фосфата (Hutber и др. "Electrical stimulation inactivatesmuscle acetyl-CoA carboxylase and increases AMP-activated protein kinase". Am. J. Physiol. Endocrinol. Metab. 272, E262-E66 (1997. Кроме того, AICAR-индуцированная активация AMPK повышает поглощение глюкозы (Merrill и др. "AICA Riboside increases AMP-activated protein kinase, fatty acid oxidation and glucose uptake in rat muscle". Am. J. Physiol. Endocrinol. Metab. 273, E1107-E1112 (1997 совместно с глюкоза-транспортером 4 (GLUT4) слиянием в плазматической мембране (Kurth-Kraczek "5'-AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle, Diabetes, 48, 2, 1667-1671 (1999. Сверхэкспрессия мертвой субъединицы киназы в скелетных мышцах отменяет AICAR, но частично нарушает поглощение глюкозы, стимулированное сокращением (Mu J. и др. "A role for AMP-activated protein kinase in contraction and hypoxia-regulated glucose transport in skeletal muscle", Mol. Cell. 7, 1085-1094(2001. Эти данные свидетельствуют о том, что дополнительные пути опосредуют поглощение глюкозы,индуцированное сокращением, в то время как AMPK опосредует влияния AICAR на поглощение глюкозы. Несмотря на всестороннее изучение вышерасположенных стимулов, которые активируют AMPK,отсутствуют исследования относительно нижерасположенного(ых) субстрата(ов)AMPKопосредованного поглощения глюкозы. В более современных исследованиях было показано, что Akt субстрат с молекулярным весом 160 кДа (AS160) является важным субстратом, нижерасположенным по отношению к Akt, который вовлечен в стимулированное инсулином поглощение глюкозы. Дополнительно к инсулину сокращение и активация AMPK с помощью AICAR связана с повышенным фосфорилированием AS160 в скелетных мышцах грызунов. Фосфорилирование AS160 нарушено или отменено в скелетных мышцах мышей AMPK а 2 "выключенной", g3 "выключенной" и а 2-"мертвой" киназы в ответ на лечение с помощью AICAR (Treeback и др. AMPK-mediated AS 160 phosphorylation in skeletal muscle isdependent on AMPK catalytic and regulatory subunits, Diabetes (2006. Это согласуется с данными о нарушении AICAR-стимулированного поглощения глюкозы в скелетных мышцах этих мышей (Jorgensen S.B. и др. Knockout of the a2 but not a1 5'-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4carboxamide-1b-4 ribofuranoside but not contraction-induced glucose uptake in skeletal muscle, J. Biol. Chem. 279, 1070-1079 (2004. Следовательно, предполагают, что AS160 является нижерасположенной мишенью AMPK при опосредовании поглощения глюкозы в скелетных мышцах. Совместно все эти метаболические действия предоставляют подтверждение того, что AMPK подавляет глюконеогенез в печени и продукцию липидов, в то время как снижает отложение липидов посредством усиления окисления липидов, улучшая таким образом профиль глюкозы и липидов в T2D. Недавно было обнаружено, что AMPK задействована в регуляции обмена энергии не только в клетке, но и на уровне всего организма. Было показано, что гормон лептин, имеющий происхождение из адипоцитов,приводит к стимуляции AMPK и, следовательно, к повышению окисления жирных кислот в скелетных мышцах (Minokoshi Y. и др., "Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase", Nature, 415, 339 (2002. Также было показано, что адипонектин, другой гормон, которые имеет происхождение из адипоцитов, вызывающий улучшенный метаболизм углеводов и липидов, стимулирует AMPK печени и скелетных мышц (Yamauchi Т. и др., "Adiponectin stimulates glucose utilization and fattyinhibition and AMP-activated protein kinase activation", PNAS, 99, 16309 (2002. Активация AMPK с учетом этих данных, возможно, является независимой от повышенных уровней AMP в клетках, но более вероятно, происходит в результате фосфорилирования одной или нескольких еще не идентифицированных вышерасположенных киназ. Исходя из знаний относительно вышеописанных условий активации AMPK, чрезвычайно благоприятные действия ожидаются от активации AMPK в условиях in vivo. В печени, снижение экспрессии ферментов глюконеогенеза будет уменьшать выработку глюкозы в печени и улучшать общий гомеостаз глюкозы, и оба процесса: непосредственное ингибирование и/или уменьшение экспрессии ключевых ферментов обмена липидов - будут повышать поглощение глюкозы и окисление жирных кислот с итоговым улучшением гомеостаза глюкозы и, вследствие уменьшения накопления триглицеридов внутри миоцитов, улучшать действие инсулина. В завершение, повышение потребления энергии будет приводить к снижению веса тела. Комбинация этих действий при метаболическом синдроме, как ожидают,будет значительно уменьшать риск приобретенных сердечнососудистых заболеваний. Некоторые исследования у грызунов подтверждают эту гипотезу (Bergeron R. и др., "Effect of 5aminoimidazole-4-carboxamide-1(beta)-D-ribofuranoside infusion on in vivo glucose metabolism in lean andof the insulin resistance syndrome", Diabetes, 51, 2199 (2002. До недавнего времени большинство исследований в условиях in vivo проводились с использованием AMPK-активатора AICAR, предшественника проницаемости в клетки ZMP. ZMP действует в качестве внутриклеточного имитатора AMP и при его накоплении в достаточно высоких концентрациях способен стимулировать активность AMPK (Cortonprotein kinase in intact cells", Eur. J. Biochem., 229, 558 (1995. Тем не менее, ZMP также действует в качестве имитатора AMP при регуляции других ферментов и поэтому не является специфическим активатором AMPK (Musi N. и Goodyear L.J. "Targeting the AMP-activated protein kinase for the treatment of type 2diabetes", Current Drug Targets-immune, Endocrine and Metabolic Disorders, 2, 119 (2002. В некоторых исследованиях в условиях in vivo были показаны положительные действия как острого, так и хронического введения AICAR на модели ожирения и диабета типа 2 у грызунов (Bergeron R. и др., "Effect of 5aminoimidazole-4-carboxamide-1b-D-ribofuranoside infusion on in vivo glucose metabolism in lean and obese(fa/fa), страдающим ожирением, приводит к уменьшению концентрации в плазме триглицеридов и свободных жирных кислот, повышению ЛВП холестерина и нормализации метаболизма глюкозы, что оценивают с помощью перорального теста - проба на толерантность к глюкозе (Minokoshi Y. и др., "Leptinstimulates fatty-acid oxidation by activating AMP-activated protein kinase", Nature, 415, 339 (2002. У обоих мышей ob/ob и db/db введение AICAR в течение 8 дней уменьшает концентрацию глюкозы в крови на 35% (Halseth A.E. и др., "Acute and chronic treatment of ob/ob and db/db mice with AICAR decreases bloodglucose concentrations", Biochem. Biophys. Res. Comm., 294, 798 (2002. Дополнительно к AICAR было обнаружено, что диабетическое средство метформин может активировать AMPK в условиях in vivo в высоких концентрациях (Zhou G. и др., "Role of AMP-activated protein kinase in mechanism of metforminactivity in skeletal muscle of subjects with type 2 diabetes", Diabetes, 51, 2074 (2002, несмотря на то что необходимо установить, в какой степени его противодиабетическое действие зависит от этой активации. Как в случае с лептином и адипонектином, стимулирующее действие метформина происходит косвенно путем активации вышерасположенной киназы (Zhou G. и др., "Role of AMP-activated protein kinase inmechanism of metformin action", J. Clin. Invest., 108, 1167 (2001. Недавно был описан низкомолекулярный активатор AMPK. Этот прямой AMPK активатор, названный А-769662, представитель тиенопиридонового семейства, in vivo индуцирует снижение уровня глюкозы и триглицеридов в плазме (Cool. В. и др., "Identification and characterization of a small moleculeAMPK activator that treats key components of type 2 diabetes and the metabolic syndrome", cell Metab., 3, 403416 (2006. Дополнительно к фармакологическому вмешательству, в последнее время были разработаны некоторые модели трансгенных мышей и стали доступными начальные результаты. При экспрессии доминантно-негативной AMPK в скелетных мышцах трансгенных мышей было показано, что AICAR влияет на стимуляцию транспорта глюкозы зависимо от активации AMPK (Mu J. и др., "Role for AMP-activatedprotein kinase in contraction and hypoxia-regulated glucose transport in skeletal muscle", Molecular Cell, 7,1085 (2001 и, следовательно, вероятно, не вызывается неспецифическими действиями ZMP. Сходные исследования в других тканях помогли дополнительно прояснить последствия активации AMPK. Полагают, что фармакологическая активация AMPK будет оказывать благоприятное влияние при метаболическом синдроме, улучшая метаболизм глюкозы и липидов и уменьшая вес тела. Для постановки диагноза метаболический синдром для пациента следует принимать во внимание следующие пять критериев: по-3 020773 вышенное кровяное давление около 130/85 мм рт.ст., концентрация глюкозы в крови натощак около 110 мг/дл, абдоминальное ожирение около 40 дюймов (мужчины) или 35 дюймов (женщины) окружности талии и изменения концентрации липидов в плазме крови, как определено путем повышения триглицеридов около 150 мг/дл или снижения ЛВП холестерина ниже 40 мг/дл (мужчины) или 50 мг/дл (женщины). Следовательно, комбинированные действия, которых можно достичь путем активации AMPK у пациента, квалифицированного как имеющий метаболический синдром, будут являться чрезвычайно интересными для этой цели. Было показано, что стимуляция AMPK стимулирует экспрессию разобщающего белка 3 (UCP3) скелетных мышц (Zhou M. и др., "UCP-3 expression in skeletal muscle: effects of exercise, hypoxia, andAMP-activated protein kinase", Am. J. Physiol. Endocrinol. Metab., 279, E622 (2000 и, следовательно, может являться путем предотвращения повреждений, вызываемых реакционно-способными формами кислорода. Было показано, что эндотелиальная NO-синтаза (eNOS) активируется путем AMPKопосредованного фосфорилирования (Chen Z.-P. и др., "AMP-activated protein kinase phosphorylation ofendothelial NO synthase", FEBS Letters, 443, 285 (1999, следовательно, активация AMPK может использоваться для улучшения локальных кровеносных систем.AMPK принимает участие в регулировании пути mTOR. mTOR представляет собой серин/треонин киназу и является ключевым регулятором синтеза белка. Для ингибирования роста клеток и защиты клеток от апоптоза, индицированного глюкозным истощением, AMPK фосфорилирует TSC2 на Thr-1227 иSer-1345, повышая активность TSC1 и TSC-2 комплекса для ингибирования m-TOR. Дополнительно,AMPK ингибирует действие mTOR путем фосфорилирования на Thr-2446. Таким образом, AMPK опосредованно и прямо ингибирует активность mTOR, ограничивая синтез белка. AMPK также может являться терапевтической мишенью для многих злокачественных новообразований, при которых имеет место конститутивная активация пути передачи сигналов PI3K-Akt. При обработке различных раковых клеточных линий с помощью AICAR наблюдается ослабление пролиферации клеток как в условиях invitro, так и in vivo (Giri R.,"5-Aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase (AMPK", J. Biol. Chem. (2005. Два отчета связывают лечение метформином со снижением риска злокачественного новообразования у пациентов с диабетом (Evans J.M. "Metformin and reduced risk of cancer in diabetic patients", BMJ, 330, 1304-1305(2005. Показано, что активация AMPK с помощью AICAR уменьшает экспрессию липогенных ферментовFAS и АСС, приводя к супрессии пролиферации в клетках рака предстательной железы. Многие раковые клетки проявляют существенно повышенную скорость синтеза жирных кислот de novo, которая коррелирует с высокими уровнями FAS. Ингибирование FAS подавляет пролиферацию раковых клеток и индуцирует гибель клеток. Таким образом, активация AMPK и ингибирование FAS активности является очевидной мишенью для фармакологической терапии злокачественных новообразований. В некоторых публикациях было показано, что AICAR в качестве активатора AMPK оказывает действие на воспалительные заболевания. Было установлено, что AICAR ослабляет продукцию провоспалительных цитокинов и медиаторов (S. Giri и др. J. Neuroscience 2004, 24:479-487), AICAR на моделях у крыс и in vitro ослабляет ЕАЕ прогрессию путем ограничения инфильтрации лейкоцитов через гематоэнцефалический барьер(ВВВ) (N. Nath. и др. J. of Immunology 2005, 175:566-574; R. Prasad и др. J. Neurosci Res. 2006, 84:614-625) и в последнее время было показано, что средства, активирующие AMPK, действуют в качестве противовоспалительных средств и могут обладать терапевтическим потенциалом при болезни Краббе/"судорожной" болезни (врожденное неврологическое заболевание) (S.Giri и др. J. Neurochem. 2008,Mar 19). Уровень техники В US 5602144 описаны производные тиенопиридона для лечения ишемии головного мозга или шизофрении. В US 7119205 описаны производные тиенопиридонов для лечения, пригодные для лечения диабета,ожирения в качестве AMPK активаторов. В WO 2007019914 описаны производные тиенопиридона для лечения, пригодные для лечения диабета, ожирения в качестве AMPK активаторов. Сущность изобретения Изобретение относится к соединениям формулы (I) формула (I) в которой R1 представляет собой Н или Hal,R2 представляет собой фенил, который может быть замещен CN, или представляет собой пиридин,B1 представляет собой 2,5-пиридинилен, 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН,-4 020773B2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил, 1,3-бензодиоксан-5-ил или фенил,необязательно замещенные одной или двумя группами, выбранными из ОН, метоксигруппы, гидроксиметила, Hal, метила, трифторметила, CN, метилсульфонила, метоксикарбонила и СООН,Hal представляет собой F, Cl, Br или I,и их фармацевтически пригодным солям, сольватам и стереоизомерам, включая их смеси во всех соотношениях. Некоторыми предпочтительными соединениями формулы (I) являются следующие соединения: Выбранными из этих предпочтительных соединений являются соединения Выбранными из этих предпочтительных соединений являются соединения Изобретение относится к соединениям формулы (I) и их фармацевтически пригодных сольватов,солей и стереоизомеров и к способу получения соединений формулы (I), который характеризуется тем,что соединение формулы (II) формула (II) где R1, R2, В 1 имеют значения, указанные выше, и X представляет собой атом галогена, Cl, Br, I,предпочтительно атом Cl, Br,подвергают реакции с производным бороновой кислоты В 2-В(ОН)2, где В 2 имеет значения, указанные выше, в условиях реакции Сузуки (Journal of organometallic chemistry, 1999, 576(1-2), 147-168/Applied Homogeneous Catalysis with Organometallic Compounds (2-oe изд.) (2002), 1, 591-598),и/или соединение формулы I в виде основания или кислоты превращают в одну из его солей. Соединения формулы (II) синтезируют путем реакции циклизации из соединений формулы (III) в щелочных условиях Соединение формулы (IV), соединение формулы (V) являются исходными веществами химического способа формула (IV) где R1, R2, В 1 имеют значения, указанные выше, ALK представляет собой C1-С 6 алкил и Y представляет собой ОН или галоген, предпочтительно Cl или Br. 2-Аминотиофеновое исходное соединение (IV) является коммерчески доступным (chemos Gmbh,Fluorochem, Acros, Interchim) или легко может быть приготовлено специалистом в данной области техники с помощью реакции Gewald, описанной в (Journal Heterocycle Chemistry, том 36, с. 333, 1999). В соединениях формулы IV Y предпочтительно представляет собой Cl, Br, I или свободную или реакционно-способную модифицированную ОН группу, такую как, например, активированный сложный эфир, имидазолид или алкилсульфонилокси, содержащий 1-6 атомов углерода (предпочтительно метилсульфонилокси или трифторметилсульфонилокси) или арилсульфонилокси, содержащий 6-10 атомов углерода (предпочтительно фенил- или п-толилсульфонилокси), используя связующее средство, такое как карбодиимидазол (CDI), дициклогексилкарбодиимин (DCC), предпочтительно DCC. Если связующее средство представляет собой карбодиимин, то предпочтительное производное описано в указанной интернет-ссылке (http://chemicalland21.com/lifescience/phar/HBTU.htm). Соединение формулы (I) приготавливают с помощью трехстадийного химического способа а) стадия 1 Аминотиазол (V) взаимодействует с соединениями IV если Y представляет собой атом галогена, предпочтительно Cl, то его подвергают реакции в инертном растворителе, таком как тетрагидрофуран, диоксан, предпочтительно диоксан при температуре от нуля до 100 градусов в течение 5 мин-24 ч, получая соединения формулы (III); если Y представляет собой ОН, то его подвергают реакции в непротонном растворителе, таком как тетрагидрофуран, диоксан, предпочтительно тетрагидрофуран с конденсирующим средством, таким как карбодиимидазол, диклогексилкарбодиимин (DCC), предпочтительно DCC при температуре от нуля до температуры флегмы растворителя в течение 15 мин-24 ч, предпочтительно при температуре от комнатной температуры до температуры флегмы растворителя в течение ночи. Подходящими инертными растворителями являются, например, углеводороды, такие как гексан,петролейный эфир, бензол, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен,1,2-дихлорэтан, четыреххлористый углерод, хлороформ или дихлорметан; спирты, такие как метанол,этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (ТГФ) или диоксан; гликолевые простые эфиры, такие как этиленгликольмонометиловый или моноэтиловый эфир, этиленгликольдиметиловый эфир (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (ДМСО); сероуглерод; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей. После этого соединение формулы (III) циклизируют в щелочных условиях, синтезируя соединение формулы (II) путем реакции циклизации, используя основание, такое как, например, гексаметилдисилилазан, соль калия или натрия, например, натрий или калий тертиоамилат, этилат натрия предпочтительно с применением гексаметилдисилазида в инертном растворителе, предпочтительно в тетрагидрофуране,диоксане, толуоле при температуре от 20 до 150 С, предпочтительно при комнатной температуре в течение от 30 мин до 24 ч и более предпочтительно от 30 мин до 1 ч. Подходящими инертными растворителями являются, например, углеводороды, такие как гексан,петролейный эфир, бензол, толуол или ксилол; хлорированные углеводороды, такие как трихлорэтилен,1,2-дихлорэтан, четыреххлористый углерод, хлороформ или дихлорметан; спирты, такие как метанол,этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (ТГФ) или диоксан; гликолевые простые эфиры, такие как этиленгликольмонометиловый или моноэтиловый эфир, этиленгликольдиметиловый эфир (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид или диметилформамид (ДМФА); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (ДМСО); сероуглерод; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей. с) стадия 3 Соединение формулы (I) приготавливают путем реакции Сузуки, используя производное бороновой кислоты, такое как, например, арил бороновую кислоту в присутствии основания, предпочтительно карбонатную соль, более предпочтительно карбонат цезия в присутствии палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий в инертной атмосфере в смеси растворителей, таких как полярный апротонный растворитель/инертный растворитель/протонный растворитель/вода. Комбинация растворителей предпочтительно представляет собой диметилформамид/толуол/этанол/воду при соотношении 10/1/6/3 или 25/2,6/15/7,5 при температуре от 20 градусов до температуры флегмы растворителя в течение 1-48 ч, предпочтительно от 6 до 24 ч. Трехстадийный химический процесс обобщен на схеме 1 Соединения формулы (I) могут существовать в форме рацематов и таутомеров, энантиомеров, диастереоизомеров, эпимеров и органических или минеральных солей, а также в виде кристаллических форм, включая их полиморфные формы и полиморфные формы соединений формулы (I). Настоящее изобретение также относится к отдельным стереоизомерам и/или диастереоизомерам как таковым или в виде их смесей во всех соотношениях. Изобретение также относится к стереоизомерам (включая Е, Z изомеры) и гидратам и сольватам этих соединений. Под сольватами соединений подразумевают аддукты молекул инертного растворителя на соединениях, которые образуются благодаря их силе взаимного притяжения. Сольваты представляют собой, например, моно- или дигидраты или алкоголяты. Под фармацевтически пригодными производными понимают соли, а также так называемые пролекарства соединений. Под производными пролекарств понимают соединения, которые являются модифицированными,например, алкильной или ацильной группами, сахарами или олигопептидами и которые быстро расщепляются в организме с образованием активных соединений. Это понятие также включает производные биоразлагаемых полимеров соединений в соответствии с изобретением, как описано (например, в Int. J. Pharm. 115, 61-67 (1995. Термин "пролекарство", как используется в настоящей заявке, относится к любому соединению, ко- 10020773 торое при введении в биологическую систему образует "лекарственную" субстанцию (биологически активное соединение) в результате самопроизвольной(ых) химической(их) реакции(й), и/или метаболической(их) химической(их) реакции(й). Выражение "эффективное количество" обозначает количество лекарственного средства или фармацевтического активного компонента, которое вызывает в ткани, системе, животном или человеке биологическую или медицинскую ответную реакцию, которую предполагает или желает получить, например,исследователь или лечащий врач. Дополнительно, выражение "терапевтически эффективное количество" обозначает то количество,которое имеет следующие последствия по сравнению с соответствующим субъектом, который не получал этого количества: улучшение лечения, излечение, предотвращение или элиминацию заболевания,синдрома, состояния, жалобы, расстройства или предотвращения побочных действий или также уменьшения прогрессирования заболевания, состояния, расстройства или побочных действий или также уменьшение прогрессирования заболевания, состояния или расстройства. Выражение "терапевтически эффективное количество" также охватывает количества, которые эффективны для повышения нормальной физиологической функции. Изобретение также относится к смесям соединений формулы I в соответствии с изобретением, например, смесям двух диастереомеров, например, в соотношении 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 или 1:1000. Особенно предпочтительными являются смеси стереоизомерных соединений. Для всех радикалов, которые встречаются более одного раза, их значения являются независимыми друг от друга. Предпочтительно R1 представляет собой фенил. Предпочтительно В 2 представляет собой фенил, необязательно замещенный одной или двумя группами, выбранными из метила, Hal, метоксигруппы, ОН, СООН, метилсульфонила и метоксикарбонила,или В 2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил или 1,3-бензодиоксан-5-ил. Предпочтительно В 1 представляет 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН. Таким образом, изобретение относится, в частности, к соединениям формулы I, в которых по меньшей мере один из указанных радикалов имеет одно из предпочтительных значений, указанных выше. Соединения согласно настоящему изобретению могут быть получены с помощью различных методов, хорошо известных специалисту в данной области техники, включая, но не ограничиваясь только теми, которые перечислены ниже, или посредством модификаций этих методов путем применения стандартных техник, хорошо известных специалисту в области органического синтеза. Все процессы, раскрытые в связи с настоящим изобретением, могут быть практически осуществлены в любом объеме,включая миллиграмм, грамм, несколько грамм, килограмм, несколько килограмм или коммерческом промышленном объеме. Следует принять во внимание, что соединения согласно настоящему изобретению могут содержать один или несколько ассиметрично замещенных атомов углерода и могут быть выделены в оптически активных или рацемических формах. Таким образом, все хиральные, диастереомерные, рацемические формы и все геометрические изомерные формулы структуры охватываются, если специфично не указана конкретная стереохимия или изомерная форма. В данной области техники хорошо известно, каким образом получают такие оптически активные формы. Например, смеси стереоизомеров могут быть разделены с помощью стандартных техник, включая, но не ограничиваясь только ими, разделение рацемических форм, с нормальной фазой, с обращенной фазой, и хиральной хроматографией, образование предпочтительной соли, перекристаллизацию и др., или путем хирального синтеза либо из активных исходных вещества или путем высвобождения хирально синтезированных целевых центров. В реакциях, описанных в настоящей заявке ниже, может быть необходимым защищать реакционноспособные функциональные группы, например, гидрокси, амино, имино, тио или карбокси группы, если они являются желательными в конечном продукте, для избегания их нежелательного участия в реакциях. Можно использовать общепринятые защитные группы в соответствии со стандартной практикой (см.,например, T.W. Greene и P.G.M. Wuts в Protective Groups in Organic Chemistry, John Wiley и Sons, 1991;J.F.W. McOmie в Protective Groups in Organic Chemistry, Plenum Press, 1973). Некоторые реакции можно осуществлять в присутствии основания. Отсутствуют конкретные ограничения относительно природы основания, используемого в этих реакциях, и в настоящем изобретении можно равным образом использовать любое основание, которое общепринято используется в реакциях такого типа, при условии, что не оказывается нежелательного воздействия на другие части молекулы. Примерами подходящих оснований являются гидроксид натрия, карбонат калия, тертиобутилат калия,тертиоамилат натрия, триэтиламин, гексаметилдисилазил калия, гидриды щелочных металлов, такие как гидрид натрия и гидрид калия; алкиллитиевые соединения, такие как метиллитий и бутиллитий; и алкоголяты щелочных металлов, такие как метилат натрия и этилат натрия. Обычно реакции осуществляют в подходящем растворителе. Можно использовать различные растворители, при условии, что не оказывается неблагоприятного воздействия на реакцию или на задействованные реагенты. Примеры подходящих растворителей включают углеводороды, которые могут пред- 11020773 ставлять собой ароматические, алифатические или циклоалифатические углеводороды, такие как гексан,циклогексан, бензол, толуол и ксилол; амиды, такие как диметилформамид; спирты, такие как этанол и метанол, и простые эфиры, такие как диэтиловый эфир, диоксан и тетрагидрофуран. Реакции можно осуществлять в широком температурном интервале. В целом, нами было обнаружено, что подходящим является осуществление реакции при температуре от 0 до 150 С (более предпочтительно от приблизительно комнатной температуры до 100 С). Время, необходимое для осуществления реакции, также может изменяться в широком диапазоне, в зависимости от многих факторов, в особенности от температуры реакции и природы реагентов. Однако, при условии, что реакцию осуществляют в предпочтительных условиях, описанных выше, предпочтительным является период от 3 до 20 ч. Таким образом приготовленные соединения могут быть восстановлены из реакционной смеси с помощью общепринятых методов. Например, соединения могут быть восстановлены путем отгонки растворителя из реакционной смеси или, при необходимости, после отгонки растворителя из реакционной смеси, промывания остатка в воде с последующей экстракцией с несмешиваемым с водой органическим растворителем и отгонки растворителя из экстракта. Дополнительно, продукт, при необходимости, можно подвергать дальнейшей очистке с помощью различных хорошо известных техник, таких как перекристаллизация, повторное осаждение или различные хроматографические техники, в основном колоночная хроматография или препаративная тонкослойная хроматография. Фармацевтические соли и другие формы Соединения, раскрытые в изобретении, могут использоваться в своей заключительной, несолевой форме. С другой стороны, настоящее изобретение также относится к применению таких соединений в форме их фармацевтически приемлемых солей, которые могут быть получены с помощью разнообразных органических и неорганических кислот и оснований в соответствии со способами, хорошо известными в данной области техники. Фармацевтически приемлемые формы солей соединений формулы I готовят, главным образом, при использовании традиционных способов. В случае, если соединение формулы I содержит группу карбоновой кислоты, то его приемлемая соль может быть образована с помощью реакции соединения с приемлемым основанием для получения соответствующей соли присоединения основания. Примерами таких оснований являются гидроксиды щелочных металлов, включая гидроксид калия, гидроксид натрия и гидроксид лития; гидроксиды щелочно-земельных металлов, такие как гидроксид бария и гидроксид кальция; алкоксиды щелочных металлов, например, этанолят калия и пропанолят натрия; а также различные органические основания, такие как пиперидин, диэтаноламин и Nметилглутамин. Сюда также включены соли алюминия соединений формулы I. Для некоторых соединений формулы I соли присоединения кислоты могут быть образованы путем обработки указанных соединений фармацевтически приемлемыми органическими и неорганическими кислотами, например, гидрогалогенидами, такими как гидрохлорид, гидробромид или гидройодид; другими минеральными кислотами, и их соответствующими солями, такими как сульфат, нитрат или фосфат и др.; и алкил- и моноарилсульфонатами, такими как этансульфонат, толуолсульфонат и бензолсульфонат и др. органическими кислотами, их соответствующими солями, такими как ацетат, трифторацетат, тартрат, малеат, сукцинат,цитрат, бензоат, салицилат, аскорбат и др. Таким образом, фармацевтически приемлемые соли присоединения кислоты соединений формулы I включают следующие соли, но не ограничиваясь только ими: ацетат, адипат, альгинат, аргинат, аспартат,бензоат, бензолсульфонат (безилат), бисульфат, бисульфит, бромид, бутират, камфорат, камфорсульфонат, каприлат, хлорид, хлорбензоат, цитрат, циклопентанпропионат, диглюконат, дигидрофосфат, динитробензоат, додецилсульфат, этансульфонат, фумарат, галактерат (из слизевой кислоты), галактуронат,глюкогептаноат, глюконат, глутамат, глицерофосфат, гемисукцинат, гемисульфат, гептаноат, гексаноат,гиппурат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, йодид, изетионат, изобутират, лактат, лактобионат, малат, малеат, малонат, манделат, метафосфат, метансульфонат, метилбензоат, моногидрофосфат, 2-нафталинсульфонат, никотинат, нитрат, оксалат, олеат, пальмоат, пектинат, персульфат, фенилацетат, 3-фенилпропионат, фосфат, фосфонат, фталат. Кроме того, основные соли соединений в соответствии с изобретением включают, но не ограничиваясь только ими, соли алюминия, аммония, кальция, меди, железа(III), железа(II), лития, магния, марганца(III), марганца(II), калия, натрия и цинка. Предпочтительными среди перечисленных выше солей являются аммонийные; соли щелочных металлов натрия и калия; и соли щелочно-земельных металлов кальция и магния. Соли соединений формулы I, которые имеют происхождение от фармацевтически приемлемых органических нетоксических оснований, включают, но не ограничиваясь только ими, соли первичных, вторичных и третичных аминов, замещенных аминов, также включая природные замещенные амины, циклические амины и основные ионообменные смолы, например, аргинин, бетаин, кофеин,хлорпрокаин, холин, N,N'-дибензилэтилендиамин (бензатин), дициклогексиламин, диэтаноламин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, Nэтилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лидокаин, лизин, меглумин, N-метил-D-глюкамин, морфолин, пиперазин, пиперидин, полиаминные смолы, прокаин, пурины,теобромин, триэтаноламин, триэтиламин, триметиламин, трипропиламин и трис-(гидроксиметил) мети- 12020773 ламин (трометамин). Соединения в соответствии с настоящим изобретением, которые включают основные азотсодержащие группы, могут быть кватернизированы с помощью таких агентов, как (С 1-С 4)алкилгалогениды, например, метил-, этил-, изопропил- и трет-бутилхлориды, бромиды и йодиды; ди(С 1-С 4)алкилсульфаты,например, диметил-, диэтил- и диамилсульфаты; (С 10-С 18)алкилгалогениды, например, децил-, додецил-,лаурил-, миристил- и стеарилхлориды, бромиды и йодиды; и арил(С 1-С 4)алкилгалогениды, например,бензилхлорид и фенетилбромид. Указанные соли позволяют получать как растворимые в воде, так и растворимые в масле соединения в соответствии с изобретением. Предпочтительные фармацевтические соли, указанные выше, включают, но не ограничиваясь только ими, ацетат, трифторацетат, безилат, цитрат, фумарат, глюконат, гемисукцинат, гиппурат, гидрохлорид, гидробромид, изотионат, манделат, меглумин, нитрат, олеат, фосфонат, пивалат, фосфат натрия,стеарат, сульфат, сульфосалицилат, тартрат, тиомалат, тозилат и трометамин. Кислотно-аддитивные соли основных соединений формулы I получают путем приведения в контакт формы свободных оснований с достаточным количеством желаемой кислоты для получения соли традиционным способом. Свободное основание можно регенерировать путем приведения в контакт формы соли с основанием и выделения свободного основания традиционным способом. Формы свободного основания в некоторой степени отличаются от своих соответствующих форм солей своими определенными физическими свойствами, такими как растворимость в полярных растворителях, однако во всем остальном соли являются эквивалентными своим соответствующим формам свободных оснований для целей настоящего изобретения. Как было указано, фармацевтически приемлемые соли присоединения основания соединений формулы I образуются с металлами или аминами, такими как щелочные металлы и щелочно-земельные металлы или органические амины. Предпочтительные металлы представляют собой натрий, калий, магний и кальций. Предпочтительные органические амины представляют собой N,N'-дибензилэтилендиамин,хлорпрокаин, холин, диэтаноламин, этилендиамин, N-метил-D-глюкамин и прокаин. Соли присоединения основания кислых соединений в соответствии с изобретением получают путем приведения в контакт формы свободной кислоты с достаточным количеством желаемого основания для получения соли традиционным способом. Форма свободной кислоты может быть регенерирована путем приведения в контакт формы соли с кислотой и выделения формы свободной кислоты известным способом. Формы свободной кислоты в некоторой степени отличаются от своих соответствующих форм солей определенными физическими свойствами, такими как растворимость в полярных растворителях, однако во всем остальном соли являются эквивалентными своим соответствующим формам свободных кислот для целей настоящего изобретения. Если соединение в соответствии с изобретением включает более чем одну группу, которая способна к образованию фармацевтически приемлемых солей этого типа, то изобретение также охватывает составные соли. Примеры типичных составных форм солей включают, но не ограничиваясь только ими,битартрат, диацетат, дифумарат, димеглумин, дифосфат, динатрий и тригидрохлорид. В свете описанного выше можно увидеть, что выражение "фармацевтически приемлемая соль" в контексте данной заявки предназначено для обозначения активного компонента, который включает соединение формулы I в форме своей соли, особенно в том случае, если указанная форма соли обеспечивает указанному активному компоненту улучшенные фармакокинетические свойства по сравнению со свободной формой указанного активного компонента или другой солью указанного активного компонента,которые использовались ранее. Фармацевтически приемлемая форма соли активного компонента может также изначально обеспечивать желаемое фармакокинетическое свойство указанному активному компоненту, которым он ранее не обладал, а также может даже положительно влиять на фармакодинамику указанного активного компонента в отношении его терапевтической активности в организме. Соединения формулы I в соответствии с изобретением могут быть хиральными благодаря их молекулярной структуре и, соответственно, могут встречаться в различных энантиомерных формах. Поэтому они могут существовать в рацемической или в оптически активной форме. Так как фармацевтическая активность рацематов или стереоизомеров соединений в соответствии с изобретением может отличаться, то может являться желательным использование энантиомеров. В этих случаях, конечный продукт или даже промежуточные продукты могут быть разделены на энантиомерные соединения химическими или физическими способами, известными специалисту в данной области техники, или даже использоваться как таковые в синтезе. В случае рацемических аминов, диастереомеры выделяют из смеси реакцией с оптически активным разделяющим агентом. Примерами подходящих разделяющих агентов являются оптически активные кислоты,такие как R и S формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты, миндальной кислоты, яблочной кислоты, молочной кислоты, подходящих N-защищенных аминокислот (например, Nбензоилпролина или N-бензолсульфонилпролина), или различные оптически активные камфорсульфоновые кислоты. Также предпочтительным является хроматографическое разделение энантиомеров с помощью оптически активного разделяющего агента (например, динитробензоилфенилглицина, триацетата целлюлозы или других производных углеводородов или хирально модифицированных полимеров метакрилата, иммобилизо- 13020773 ванных на силикагеле). Подходящими для этой цели элюентами являются водные или спиртовые смеси растворителей, такие как, например, гексан/изопропанол/ацетонитрил, например, в отношении 82:15:3. Для хирального разделения рацематов можно использовать следующие кислоты и амины. В качестве примеров можно использовать следующие хиральные кислоты: (+)-D-ди-О-бензоилвинная кислота, (-)-L-ди-О-бензоилвинная кислота, (-)-L-ди-О,О'-п-толуил-L-винная кислота, (+)-D-ди-О,О'-п-толуилL-винная кислота, (R)-(+)-яблочная кислота, (S)-(-)-яблочная кислота, (+)-камфорная кислота, (-)-камфорная кислота, R-(-)1,1'-бинафталин-2,2'-диил гидрогенофосфиновая, (+)-камфановая кислота, (-)-камфановая кислота, (S)-(+)-2-фенилпропионовая кислота, (R)-(+)-2-фенилпропионовая кислота, О-(-)-миндальная кислота, L(+)-миндальная кислота, D-винная кислота, L-винная кислота, или любую их смесь. В качестве примеров можно использовать следующие хиральные амины: хинин, бруцин, (S)-1-(бензилоксиметил)пропиламин(III),(-)-эфедрин, (4S,5R)-(+)-1,2,2,3,4-тетраметил-5-фенил-1,3-оксазолидин, (R)-1-фенил-2-п-толилэтиламин, (S)фенилглицинол, (-)-N-метилэфедрин, (+)-(2S,3R)-4-диметиламино-3-метил-1,2-дифенил-2-бутанол, (S)фенилглицинол, (S)метилбензиламин или любую их смесь. Изобретение также относится к применению соединений и/или их физиологически приемлемых солей для приготовления лекарственного средства (фармацевтической композиции), в частности, при помощи нехимических методов. Они могут быть превращены в подходящую дозированную форму совместно с по меньшей мере одним твердым, жидким и/или полужидким наполнителем или вспомогательным веществом и, при необходимости, в комбинации с одним или несколькими другими активными компонентами. Изобретение, кроме того, относится к лекарственным средствам, содержащим по меньшей мере одно соединение в соответствии с изобретением и/или его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях, и, необязательно, наполнители и/или вспомогательные вещества. Лекарственные препараты могут вводиться в виде дозированных единиц, которые содержат заранее установленное количество активного компонента на дозированную единицу. Такая единица может включать, например, от 0,5 мг до 1 г, предпочтительно от 1 до 700 мг, более предпочтительно от 5 до 100 мг, соединения в соответствии с изобретением, в зависимости от заболевания, подвергаемого лечению,способа введения, а также возраста, веса тела и состояния пациента, или фармацевтические композиции могут вводиться в виде дозированных единиц, которые содержат заранее установленное количество активного компонента на дозированную единицу. Предпочтительными дозированными единицами лекарственных препаратов являются те, которые содержат суточную дозу или часть суточной дозы, как указано выше, или соответствующую порцию их активного компонента. Лекарственные средства этого типа также могут быть получены способом, который хорошо известен в области фармацевтики. Лекарственные препараты могут адаптироваться для введения при помощи любого подходящего способа, например, путем перорального (включая буккальное или подъязычное), ректального, назального, местного (включая буккальное, подъязычное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или внутрикожное) введения. Такие препараты могут быть приготовлены с помощью любого способа, известного в области фармацевтики, например,путем объединения активного компонента с наполнителем(ями) или вспомогательным(ыми) веществом(ами). Лекарственные препараты, адаптированные для перорального введения, могут вводиться в виде отдельных единиц, таких как, например, капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; пищевых пен или пенистых пищевых продуктов; или жидких эмульсий масло-в-воде или жидких эмульсий вода-в-масле. Так, например, в случае перорального введения в виде таблетки или капсулы активный компонент может быть объединен с пероральным, нетоксичным и фармацевтически приемлемым инертным наполнителем, таким как, например, этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до подходящего небольшого размера и смешивания его с фармацевтическим наполнителем,измельченным аналогичным способом, таким как, например, пищевой углеводород, такой как, например,крахмал или маннит. Также можно добавлять ароматизатор, консервант, диспергирующее вещество и краситель. Капсулы получают путем приготовления порошковой смеси, как описано выше, и заполняют ею желатиновые капсулы определенной формы. Перед заполнением капсул к порошковой смеси можно добавлять скользящие и смазывающие вещества, такие как, например, высокодисперсная кремниевая кислота, тальк, стеарат магния, стеарат кальция или полиэтиленгликоль в твердой форме. Для улучшения доступности лекарственного средства, заключенного в капсулу, также можно добавлять дезинтегрирующее вещество или солюбилизатор, такой как, например, агар-агар, карбонат кальция или карбонат натрия. Дополнительно, если это является желательным или необходимым, в смесь также можно добавлять подходящие связующие, смазывающие вещества, дезинтеграторы, а также красители. Подходящими связующими являются крахмал, желатин, природные сахара, такие как, например, глюкоза или бета-лактоза,подсластители, приготовленные из кукурузы, естественных и синтетических резин, такие как, например,- 14020773 аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и т.п. Смазывающие вещества, которые могут применяться в таких дозированных формах,включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Дезинтеграторы включают, но не ограничиваясь только ими, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. Лекарственные средства в виде таблеток получают, например, путем приготовления порошковой смеси, гранулирования или сухого прессования смеси, добавления смазывающего вещества и дезинтегратора и прессования полученной смеси в таблетки. Порошковую смесь готовят путем смешивания соединения, измельченного подходящим образом, с разбавителем или основанием,как описано выше, и необязательно со связующим, таким как, например, карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как, например, парафин,усилителем поглощения, таким как, например, четвертичная соль, и/или абсорбентом, таким как, например, бентонит, каолин или дикальцийфосфат. Порошковую смесь можно гранулировать путем смачивания со связующим, таким как, например, сироп, крахмальная паста, слизь акации или растворы целлюлозы или полимерных веществ и прессования ее через сито. В качестве альтернативы грануляции, порошковую смесь можно пропускать через таблетировочную машину, получая куски неправильной формы,которые распадаются, образуя гранулы. Гранулы можно замасливать путем добавления стеариновой кислоты, стеарата, талька или минерального масла для предотвращения слипания в таблетировочной литейной форме. После этого смазанную смесь спрессовывают, получая таблетки. Соединения в соответствии с изобретением также можно объединять с сыпучим инертным наполнителем и затем подвергать прямому прессованию, получая таблетки без осуществления стадий грануляции или сухого прессования. Таблетки также можно покрывать прозрачным или светонепроницаемым защитным слоем, состоящим из шеллакового запечатывающего слоя, слоя сахара или полимерного вещества и глянцевого слоя воска. К этим покрытиям также можно добавлять красители для возможности различения между разными дозируемыми единицами. Жидкости для перорального введения, такие как, например, раствор, сиропы и эликсиры, могут быть приготовлены в виде дозируемых единиц таким образом, чтобы они содержали заранее установленное количество соединений. Сиропы могут быть получены путем растворения соединения в водном растворе с подходящим ароматизатором, тогда как эликсиры готовят с применением нетоксичного спиртового наполнителя. Суспензии могут быть приготовлены путем диспергирования соединения в нетоксичном наполнителе. Также можно добавлять солюбилизаторы и эмульсификаторы, такие как, например,этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматические добавки, такие как, например, масло мяты перечной, или натуральные заменители сахара или сахарин, или другие искусственные заменители сахара и т.п. Лекарственные препараты для перорального введения в виде дозированных единиц могут быть инкапсулированы в микрокапсулы, если это является желательным. Также лекарственный препарат может быть приготовлен таким образом, чтобы пролонгировать или замедлить высвобождение, например, путем применения покрытий или заделывания требуемого вещества в полимеры, воск и т.п. Соединения в соответствии с изобретением и их соли, сольваты и физиологически функциональные производные также могут вводиться в виде липосомных систем доставки, таких как, например, небольшие однослойные пузырьки, большие однослойные пузырьки и многослойные пузырьки. Липосомы могут быть образованы с помощью различных фосфолипидов, таких как, например, холестерин, стеариламин или фосфатидилхолины. Соединения в соответствии с изобретением и их соли, сольваты и физиологически функциональные производные также могут доставляться с помощью моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения также могут быть соединены с растворимыми полимерами в качестве нацеливающих носителей лекарственных средств. Такими полимерами могут являться поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксид полилизина, замещенный пальмитоиловыми радикалами. Кроме того, соединения можно связывать с биоразлагаемыми полимерами, которые пригодны для обеспечения контролируемого высвобождения лекарственного средства, например, полимолочной кислотой, поли-эпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами,полиацеталями, полидигидроксипиранами, полицианоакрилатами и перекрестно-сшитыми или амфипатическими блок-сополимерами гидрогелей. Лекарственные препараты, адаптированные для трансдермального введения, могут вводиться в виде независимых пластырей для удлиненного, тесного контакта с эпидермисом реципиента. Таким образом, например, активный компонент может доставляться из пластыря путем ионофореза, как в общем описано (см. Pharmaceutical Research, 3(6), 318 (1986. Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Для лечения глаз или других наружных тканей, например рта и кожи, предпочтительно применяются лекарственные препараты в виде местной мази или крема. При приготовлении лекарственного препа- 15020773 рата в виде мази активный компонент может применяться с парафиновым или смешивающимся с водой мазевым основанием. Альтернативно, для получения крема активный компонент может быть приготовлен с основой для крема типа масло-в-воде или основой вода-в-масле. Лекарственные препараты, адаптированные для местного введения в глаза, включают глазные капли, в которых активный компонент растворен или суспендирован в подходящем носителе, предпочтительно в водном растворителе. Лекарственные препараты, адаптированные для местного введения в полость рта, включают лепешки, пастилки и жидкости для полоскания рта. Лекарственные препараты, адаптированные для ректального введения, могут вводиться в виде суппозиториев или клизм. Лекарственные препараты, адаптированные для интраназального введения, в которых носитель представляет собой твердое вещество, включают крупный порошок, имеющий размер частичек, например, в интервале 20-500 мкм, который вводится путем вдыхания, то есть путем быстрого вдоха через нос из контейнера, содержащего порошок, который придерживают возле носа. Подходящие лекарственные препараты для введения в виде интраназального аэрозоля или носовых капель с жидкостью в качестве носителя включают растворы активного вещества в воде или в масле. Лекарственные препараты, адаптированные для введения путем ингаляции, включают тонкоизмельченные частички в виде пыли или тумана, которые могут быть получены с помощью различных диспергирующих устройств под давлением с аэрозолями, распылителями или инсуффляторами. Лекарственные препараты, адаптированные для вагинального введения, могут вводиться в виде пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей. Лекарственные препараты, адаптированные для парентерального введения, включают водные или неводные стерильные растворы для инъекций, содержащие антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, с помощью которых лекарственное средство поддерживается изотоническим по отношению к крови реципиента, подвергаемого лечению; и водные или неводные стерильные суспензии, которые могут содержать суспензионную среду и загустители. Лекарственные препараты могут вводиться с помощью емкостей для однократного или многократного введения, например,запечатанных ампул и флаконов, и храниться в лиофилизированном состоянии, при этом непосредственно перед введением необходимо только добавить стерильную жидкость-носитель, например, воду для инъекций. Растворы и суспензии для инъекций, приготовленные согласно рецептуре, могут быть приготовлены из стерильных порошков, гранул и таблеток. Также является очевидным, что дополнительно к предпочтительным вышеописанным составляющим лекарственные препараты также могут содержать другие вещества, которые используются в данной области для конкретных типов лекарственных средств; например, лекарственные препараты, пригодные для перорального введения, могут содержать ароматизаторы. Терапевтически эффективное количество соединения в соответствии с изобретением зависит от многих факторов, включая, например, возраст и вес человека или животного, определенное болезненное состояние, которое необходимо лечить, и его тяжесть, природу лекарственного средства и способ введения, и в конечном счете оно может быть определено лечащим врачом или ветеринаром. Тем не менее,эффективное количество соединения в соответствии с изобретением, как правило, находится в интервале от 0,1 до 100 мг/кг веса тела реципиента (млекопитающего) в сутки и предпочтительно обычно находится в интервале от 1 до 10 мг/кг веса тела в сутки. Следовательно, действующее суточное количество для взрослого млекопитающего весом 70 кг обычно может составлять от 70 до 700 мг, причем это количество может вводиться в виде отдельной дозы один раз в день или обычно в виде циклов частичных доз (таких как, например, два, три, четыре, пять или шесть раз) в день, таким образом, что общая суточная доза является аналогичной. Эффективное количество его соли или сольвата или физиологически функционального производного может быть определено в виде доли эффективного количества соединения в соответствии с изобретением per se. Также предполагается, что подобные дозы пригодны для лечения других состояний, указанных в настоящей заявке. Примеры Следующие примеры иллюстрируют изобретение, но никоим образом его не ограничивают. Используемые исходные вещества являются известными продуктами или продуктами, приготовленными в соответствии с известными методиками. Проценты выражены на основе веса, если специально не указано иначе. Соединения характеризовали в особенности с помощью следующих аналитических методик. ЯМР спектры измеряли с помощью ЯМР-спектрофотометра Bruker Avance DPX 300 МГц. Массы определяли с помощью ВЭЖХ, соединенной с масс-детектором Agilent серии 1100. Точки плавления (t пл.) измеряли на блоке Stuart Scientific. Промежуточное соединение 1. 3-(4-Бромфенил)-4-гидрокси-5-фенил-6,7-дигидро-тиено[2,3-b]пиридин-6-он Стадия 1. К раствору этил 2-амино-4-(4-бромфенил)тиофен-3-карбоксилата (12 г, 36,8 ммоль) в ди- 16020773 оксане (100 мл) по каплям добавляли раствор фенилацетил хлорида (5,84 мл) в диоксане (100 мл). Через 15 мин при комнатной температуре реакционную смесь нагревали до 70 С в течение 1 ч. Растворитель удаляли при пониженном давлении и желтоватое оставшееся твердое вещество ресуспендировали в небольшом количестве этилацетата. Добавляли петролейный эфир и восстанавливали не совсем белое твердое вещество (14,3 г). 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 10,98 (bs, 1H), 7,53-7,20 (m, 9H), 6,93 (s, 1H), 4,01 (q,2H), 3,92 (s, 2H), 0,91 (t, 3 Н). Стадия 2. К раствору вышеуказанного соединения (14,3 г) в тетрагидрофуране (630 мл) добавляли гексаметилдисилазан калия (256 мл, 0,5 М в толуоле). Через 30 мин при комнатной температуре реакционную смесь закаливали с помощью раствора соляной кислоты (4 М) и экстрагировали этилацетатом. Органическую фазу высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении. Оставшееся желтоватое масло ресуспендировали в небольшом количестве этилацетата. Добавляли петролейный эфир и восстанавливали образованное не совсем более твердое вещество (11 г). 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 7,55-7,40 (dd, 4H), 7,35-7,25 (m, 5 Н), 7,06 (s, 1H). Промежуточное соединение 2. 3-(4-Бромфенил)-4-гидрокси-5-(пиридин-3-ил)-6,7-дигидро-тиено[2,3-b]пиридин-6-он Стадия 1. К раствору этил 2-амино-4-(4-бромфенил)тиофен-3-карбоксилата (10 г, 30,7 ммоль), гидрохлорида 3-пиридил уксусной кислоты (12,8 г) в тетрагидрофуране (200 мл) добавляли дициклогексилкарбодиимид (15 г). Раствор нагревали в колбе с обратным холодильником в течение ночи, затем фильтровали. Органическую фазу ресуспендировали в этилацетате и промывали раствором бикарбоната натрия. Органический раствор высушивали над сульфатом натрия и растворитель удаляли при пониженном давлении. Полученное неочищенное твердое вещество промывали смесью петролейный эфир/небольшое количество этилацетата. Восстанавливали не совсем белое твердое вещество (9,8 г). 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 11,10 (bs, 1H), 8,59 (s, 1H), 8,53 (d, 1 Н), 7,82 (d, 1H),7,54 (d, 2H), 7,41 (dd, 1H), 7,27 (d, 2H), 6,99 (s, 1H), 4,06 (q, 2H), 4,04 (s, 2H), 0,96 (t, 3 Н). Стадия 2. К вышеописанному соединению (5 г, 11,2 ммоль) в тетрагидрофуране (250 мл) по каплям добавляли гексаметилдисилазан калия (90 мл, 0,5 М раствор в толуоле). Через 1 ч реакционную смесь концентрировали насухо и ресуспендировали в смеси этилацетат/вода. Водную фазу частично концентрировали до осаждения твердого вещества. Его фильтровали (3,46 г) и промывали этилацетатом; МС: 399,0 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 8,83 (s, 1H), 8,03 (m, 2H), 7,41 (m, 5 Н), 7,09 (m, 1 Н),6,59 (s, 1 Н). Пример 1. 2-Хлор-4-гидрокси-3-(2'-метоксибифенил-4-ил)-5-фенил-6,7-дигидро-тиено[2,3-b]пиридин-6-он Стадия 1. Раствор промежуточного соединения 1 со стадии 1 (2 г, 4,50 ммоль), 2-метоксифенил бороновой кислоты (1,37 г), карбоната цезия (4,40 г) и тетракис(трифенилфосфин) палладия (468 мг) в атмосфере аргона в смеси толуол (55 мл)/этанол (65 мл)/вода (32 мл) нагревали в течение ночи при 80 С. Раствор фильтровали через слой целита и ресуспендировали в этилацетате. Органический раствор промывали раствором соляной кислоты (4 М), после этого высушивали над сульфатом натрия. Растворитель удаляли при пониженном давлении и полученное неочищенное твердое вещество (1,57 г) промывали смесью петролейный эфир/небольшое количество этилацетата. 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 11,01 (bs, 1H), 7,67-7,03 (m, 13H), 6,99 (s, 1 Н), 4,08 (q,2H), 3,96 (s, 2H), 3,78 (s, 3 Н), 0,96 (t, 3 Н). Стадия 2. Раствор вышеуказанного соединения (1,54 г, 3,27 ммоль), N-хлорсукцинимида (0,48 г) в хлороформе (20 мл) нагревали в течение ночи при 50 С. Растворитель упаривали и неочищенное вещество ресуспендировали в этилацетате. Этот раствор промывали водой, после этого высушивали над сульфатом натрия. Растворитель удаляли при пониженном давлении и оставшееся масло кристаллизовали со смесью изопропиловый эфир/небольшое количество этилацетата. Образованное твердое вещество (1,18 г) фильтровали. 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 11,51 (bs, 1H), 7,50-7,13 (m, 13H), 3,97 (s, 2H), 3,95 (q,2H), 3,76 (s, 3 Н), 0,79 (t, 3 Н). Стадия 3. К раствору гексаметилдисилазана калия (18,6 мл, 0,5 М в толуоле) по каплям добавляли вышеописанное соединение (1,18 г, 2,32 ммоль, в 45 мл тетрагидрофурана). Через 1 ч растворители удаляли при пониженном давлении и к неочищенному твердому веществу добавляли раствор соляной кислоты (4 н.). После перемешивания в течение нескольких минут твердое вещество фильтровали (1 г) и промывали смесью петролейный эфир/небольшое количество этилацетата; МС: 460,0 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 7,55-7,06 (m, 13H), 3,80 (s, 3 Н). Пример 2. 4-Гидрокси-3-(2'-гидроксибифенил-4-ил)-5-фенил-6,7-дигидро-тиено[2,3-b]пиридин-6-он Раствор промежуточного соединения 1 (0,4 г, 1 ммоль), 2-гидроксифенил бороновой кислоты (277 мг), карбоната цезия (981 мг) и тетракис(трифенилфосфин) палладия (100 мг) в атмосфере аргона в смеси диметилформамида (10 мл)/толуола (1 мл)/этанола (6 мл)/воды (3 мл) нагревали в течение ночи при 80 С. Раствор фильтровали через слой целита и концентрировали при пониженном давлении. Добавляли ацетонитрил. Осажденное твердое вещество (244 мг) фильтровали и промывали водой, раствором соляной кислоты (4 М), ацетонитрилом и петролейным эфиром; МС: 412,1 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 7,52-7,45 (m, 4 Н), 7,37-7,23 (m, 6 Н), 7,17-7,11 (m, 1 Н),7,04 (s, 1 Н), 6,96-6,84 (m, 2 Н). Пример 3. 4-Гидрокси-3-(2'-гидроксибифенил-4-ил)-5-(пиридин-3-ил)-6,7-дигидро-тиено[2,3-b]пиридин-6-он Раствор промежуточного соединения 2 (1 г, 2,50 ммоль), 2-гидроксифенил бороновой кислоты (0,69 г),карбоната цезия (2,45 г) и тетракис(трифенилфосфин) палладия (260 мг) в атмосфере аргона в смеси диметилформамид (25 мл)/толуол (2,6 мл)/этанол (15 мл)/вода (7,5 мл) нагревали в течение 6 ч при 80 С. Раствор фильтровали через слой целита и концентрировали при пониженном давлении. Добавляли ацетонитрил. Осажденное твердое вещество (629 мг) фильтровали и промывали водой, ацетоном, этилацетатом и петролейным эфиром; МС: 413 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 9,66 (bs, 1H), 8,90 (m, 1H), 8,79-8,77 (m, 1 Н), 8,56-8,53(m, 1 Н), 8,06-8,01 (m, 1 Н), 7,54-7,47 (m, 4 Н), 7,26-7,09 (m, 3 Н), 7,00-6,83 (m, 2 Н). Пример 4. 4-Гидрокси-3-(3'-гидроксиметилбифенил-4-ил)-5-(пиридин-3-ил)-6,7-дигидро-тиено[2,3-b]пиридин 6-он Раствор промежуточного соединения 2 (1 г, 2,50 ммоль), 3-гидроксиметилфенил бороновой кислоты (0,76 г), карбоната цезия (2,45 г) и тетракис(трифенилфосфин) палладия (260 мг) в атмосфере аргона в смеси диметилформамид (25 мл)/толуол (2,6 мл)/этанол (15 мл)/вода (7,5 мл) нагревали в течение ночи при 80 С. Раствор фильтровали через слой целита и концентрировали при пониженном давлении. Добавляли ацетонитрил и раствор соляной кислоты (5 М). Осажденное твердое вещество (527 мг) фильтровали и промывали водой, этилацетатом и петролейным эфиром; МС: 427,1 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 8,89-8,01 (m, 4 Н), 7,65-7,29 (m, 8 Н), 7,12 (s, 1H), 4,56(s, 2H). Пример 5. 4-Гидрокси-3-[2-(2-метоксифенил)пиридин-5-ил]-5-фенил-6,7-дигидро-тиено[2,3-b]пиридин-6-он Стадия 1. Раствор 5-ацетил-2-хлор-пиридина (4,45 г, 28,6 ммоль), 2-метоксифенил бороновой кислоты (8,96 г), карбоната цезия (27,9 г) и тетракис(трифенилфосфин) палладия (2,97 г) в атмосфере аргона в смеси толуола (10 мл)/этанола (12 мл)/воды (6 мл) нагревали в течение ночи при 80 С. Раствор фильтровали через слой целита и концентрировали при пониженном давлении. Неочищенное вещество очищали через силикагель (дихлорметан, затем диизопропиловый эфир); МС: 228,1 (М+1). Стадия 2. Раствор вышеуказанного соединения (5,73 г, 25 ммоль), серы (6,47 г), этил цианоацетата(3,28 мл), морфолина (4,4 мл) и абсолютного этанола (100 мл) нагревали до 50 С в течение ночи. Реакционную смесь фильтровали и растворитель упаривали. Оставшееся масло ресуспендировали в смеси вода/этилацетат. Водную фазу экстрагировали этилацетатом и цельную органическую фазу промывали соляным раствором, высушивали над сульфатом натрия и концентрировали при пониженном давлении. Неочищенное масло очищали через силикагель (петролейный эфир 80/этилацетат 20), получая желательное соединение (2,38 г); МС: 355,1 (М+1). Стадия 3. К вышеописанному соединению (2,38 г) в диоксане (15 мл) добавляли фенилацеил хлорид(1,15 мл в 5 мл диоксана) по каплям. Через 2 ч реакционную смесь упаривали насухо. К оставшемуся маслу добавляли смесь воды со льдом и диизопропиловый эфир. Осажденное твердое вещество (2,4 г) фильтровали и промывали водой и диизопропиловым эфиром. К этому твердому веществу в тетрагидрофуране (40 мл) добавляли гексаметилдисилазан калия (40 л, 0,5 М в толуоле) и реакционную смесь нагревали при 70 С в течение ночи. Растворители удаляли при пониженном давлении. К оставшемуся маслу добавляли воду и уксусную кислоту (до достижения рН 4). Осажденное твердое вещество (1,77 г) фильтровали и промывали водой; МС: 427,1 (М+1); 1 Н ЯМР (ДМСО-d6, 300 МГц)[част. на млн] 12,09 (bs, 1H), 9,65 (bs, 1H), 8,67 (s, 1 Н), 7,85-7,71 (m,3H), 7,37 (m, 9H), 3,78 (s, 3 Н). Следующие соединения могут быть получены аналогично. Биологические исследования Ферментативная активность Следующий биологический тест предоставляет возможность определить эффективность таких соединений формулы (I) на белке AMPK (рекомбинантный 112). Рекомбинантный AMPK фермент человека экспрессировали в Е. Coli и реактивировали in vitro с помощью KB1 перед определением активности фермента. Активность AMPK фермента исследовали, используя технологию A Delfia. Активность AMPK фермента определяли в микротитровальных планшетах (50 мМ Hepes буфера,рН 7,4 с 125 мкМ АТР соответственно) в присутствии синтетического пептидного субстрата (AMARAASAAALARRR, "AMARA" пептид) и активаторов в серийных разведениях. Реакции запускали путем добавления AMPK (50-100 нг). После смешивания планшеты инкубировали в течение 30 мин при комнатной температуре. Активность фермента анализировали с помощью антифосфосеринового антитела для изменения количества фосфата, инкорпорированного в AMARAA.: номер молекулы Активность Соотношение между % контроля (спонтанной активности) соединения формулы (I) при 30 мкМ и % контроля (спонтанной активности) AMP (природный субстрат) при 30 мкМ. Соединения формулы (I) в табл. II рассматриваются как прямой активатор AMPK, если соотношение равно 90% или выше. Таблица II Поглощение глюкозы в мышцах Скелетные мышцы являются основным участком стимулированного инсулином использования глюкозы и резистентность к инсулину в этой целевой ткани в течение продолжительного времени рассматривалась в качестве фактора, способствующего патогенезу диабета 2 типа (T2D). Следовательно,альтернативные пути, которые стимулируют поглощение скелетными мышцами глюкозы независимо от передачи сигналов посредством инсулина, потенциально могут улучшать гликемический контроль у субъектов с T2D. Несмотря на попытки стимулировать поглощение глюкозы в скелетных мышцах независимо от инсулинового пути, молекулярные механизмы, лежащие в основе этого процесса, остаются плохо изученными. Полученные данные, указывающие на то, что транспорт глюкозы может быть повышен в изолированной скелетной мышце в ответ на сокращение in vitro, свидетельствуют о том, что пути передачи сигналов,активируемые недостаточностью энергетических запасов внутри клетки, частично подконтрольны индуцированному сокращением поглощению глюкозы. AMPK рассматривается как перспективный кандидат для индуцированного сокращением скелетных мышц поглощения глюкозы, поскольку он активируется параллельно с повышением AMP и уменьшением энергетических запасов креатин-фосфата (Hubter C.A., Am. J. Physiol. Endocrinol. Metab. 272:E262-E266; 1997). Кроме того, AICAR-индуцированная активация AMPK повышает поглощение глюкозы (Merrill G.F. и др., Am. J. Physiol. Endocrinol. Metab. 273:E1107-E1112; 1997). Поглощение глюкозы в Н-2Kb клетках (in vitro клеточный тест) Следующий клеточный тест предоставляет возможность определения влияния активаторов AMPK,- 21020773 таких как соединения формулы (I), на поглощение глюкозы на модели мышечных клеток. Н-2Kb клетки, имеющие происхождение из гетерозиготных H-2Kb tsA58 трансгенных мышей, выращивали в планшетах на 24 лунки, покрытых матригелем, и культивировали при 33 С в течение 4 дней в пермиссивных условиях, как было описано ранее Fryer и др. (Diabetes 49 (12): 1978, 2000). Для дифференциации в мышцы клетки переключали на условия не-пермиссивой культуры (37 С при отсутствии интерферон-). Через 3 дня клетки инкубировали в течение 4 ч в DMEM 1 г/л глюкозы культуральной среде, содержащей различные концентрации тестируемых молекул. После этого определяли поглощение глюкозы путем инкубирования клеток в течение 10 мин с радиоактивно меченой 2-дезокси-D-[1,23 Н] глюкозой. Поглощение глюкозы останавливали путем быстрого промывания планшет 2 раза с ледянымNaCl 0,9%. После этого клетки солюбилизировали в 0,1 н. NaOH в течение 30 мин. Радиоактивность определяли путем жидкостно-сцинтилляционного измерения активности.: номер молекулы Активность (табл. III): концентрация соединения (I) для поглощения глюкозы, равного или выше поглощения глюкозы, индуцированного инсулином (170 нМ)концентрация соединения (I) 10 мкмоль/лконцентрация соединения (I) 10 мкмоль/л Таблица III Соединения согласно изобретению способны повышать поглощение глюкозы в мышечной клеточной линии, а именно Н-2Kb, независимо от инсулина. Эти данные, полученные в результате осуществления ферментативного теста с последующим клеточным тестом, свидетельствуют о том, что производные тиенопиридона, как определено в формуле (I), являются прямыми активаторам AMPK и эти соединения способны повышать поглощение глюкозы предпочтительно в мышечных клетках. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) в которой R1 представляет собой Н или Hal,R2 представляет собой фенил, который может быть замещен CN, или представляет собой пиридин,B1 представляет собой 2,5-пиридинилен, 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН,B2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил, 1,3-бензодиоксан-5-ил или фенил, необязательно замещенные одной или двумя группами, выбранными из ОН, метоксигруппы, гидроксиметила, Hal, метила, трифторметила, CN, метилсульфонила, метоксикарбонила и СООН,Hal представляет собой F, Cl, Br или I, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях. 2. Соединение по п.1, в котором В 2 представляет собой фенил, необязательно замещенный одной или двумя группами, выбранными из метила, Hal, метоксигруппы, ОН, СООН, метилсульфонила и метоксикарбонила, или В 2 представляет собой фуранил, пиридин-4-ил, пиридин-3-ил или 1,3-бензодиоксан 5-ил, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях. 3. Соединение по п.1 или 2, в котором В 1 представляет 1,3-фенилен или 1,4-фенилен, которые могут быть замещены метоксигруппой или ОН, и его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях. 4. Соединение по п.1, выбранное из группы, включающей 6. Промежуточное соединение формулы (II) для приготовления соединения формулы (I)X представляет собой атом галогена, и его соли. 7. Способ получения соединений формулы (I) по любому из пп.1-5 и их фармацевтически пригодных сольватов, солей и стереоизомеров, который характеризуется тем, что соединение формулы (II)X представляет собой Cl, Br или I,подвергают взаимодействию в реакции Сузуки с производным бороновой кислоты В 2-В(ОН)2, где 2 В имеет значения, указанные в п.1, и/или соединение формулы I в виде основания или кислоты превращают в одну из его солей. 8. Лекарственные средства, содержащие по меньшей мере одно соединение формулы (I) по любому из пп.1-5 и/или его фармацевтически пригодные соли, сольваты и стереоизомеры, включая их смеси во всех соотношениях, и необязательно, наполнители и/или вспомогательные вещества. 9. Применение соединений по любому из пп.1-5 и их фармацевтически пригодных солей, сольватов и стереоизомеров, включая их смеси во всех соотношениях, для приготовления лекарственного средства для лечения метаболического синдрома, диабета, ожирения, злокачественного новообразования, воспаления, сердечно-сосудистых заболеваний. 10. Применение соединений по любому из пп.1-5 для приготовления лекарственного средства, повышающего поглощение глюкозы в мышечных клетках.
МПК / Метки
МПК: C07D 333/38, A61P 35/00, C07D 495/04, A61P 3/10, A61P 29/00, A61P 3/04
Метки: производные, качестве, активаторов, амрк, амр-активированной, тиенопиридона, протеинкиназы
Код ссылки
<a href="https://eas.patents.su/27-20773-proizvodnye-tienopiridona-v-kachestve-aktivatorov-amr-aktivirovannojj-proteinkinazy-amrk.html" rel="bookmark" title="База патентов Евразийского Союза">Производные тиенопиридона в качестве активаторов амр-активированной протеинкиназы (амрк)</a>
Производные тиенопиридона в качестве активаторов амр-активированной протеинкиназы (амрк)

Номер патента: 17756
Опубликовано: 28.02.2013
Авторы: Аллаку-Бозек Софи, Шарон Кристине, Лепифре Франк, Краво Даньель
МПК: A61P 29/00, A61K 31/4365, A61P 3/00...
Метки: амр-активированной, амрк, протеинкиназы, качестве, тиенопиридона, производные, активаторов
Формула / Реферат:
1. Соединение формулы (I)в которой R1 представляет собой Hal;R2 представляет собой фенил, который незамещен или моно-, ди- или тризамещен A, Hal, ОА, ОН, СООН и/или бензилокси, или представляет собой нафтил, который незамещен или моно- или дизамещен ОА и/или ОН, или представляет собой пиридил или фуранил, каждый из которых незамещен или монозамещен ОА;R3 представляет собой фенил, который незамещен или моно-, ди- или тризамещен CN, А, ОА, ОН,...
Производные индола и их применение в качестве активаторов глюкокиназы

Номер патента: 19640
Опубликовано: 30.05.2014
Авторы: Ясума Цунео, Такакура Нобуюки
МПК: A61K 31/427, C07D 417/14, A61P 3/10...
Метки: применение, активаторов, производные, качестве, глюкокиназы, индола
Формула / Реферат:
1. Соединение формулы (I)где цикл А обозначает бензол, замещенный -W1-R1 и -W2-R2 и необязательно замещенный 1-3 атомами галогена;цикл В обозначает тиазолин, тиазол или тиадиазол, каждый из которых необязательно замещен 1-3 заместителями, выбранными из:(1) C1-6алкильной группы, необязательно замещенной 1-3 заместителями, выбранными из:(a) гидроксильной группы,(b) карбамоильной группы, необязательно моно- или дизамещенной заместителем(ями),...
Производные n-(пиразол-3-ил)бензамида в качестве активаторов глюкокиназы

Номер патента: 17114
Опубликовано: 28.09.2012
Авторы: Бургдорф Ларс Торе, Шарон Кристин, Глайтц Иоганнес, Байер Норберт, Карниато Денис, Эмде Ульрих
МПК: A61P 3/04, A61K 31/4155, A61P 3/10...
Метки: активаторов, глюкокиназы, производные, n-(пиразол-3-ил)бензамида, качестве
Формула / Реферат:
1. Соединения, выбранные из группыи их фармацевтически пригодные соли и стереоизомеры, включая их смеси во всех соотношениях.2. Лекарственное средство, обладающее активностью активаторов глюкокиназы, содержащее по меньшей мере одно соединение в соответствии с п.1 и/или его фармацевтически пригодные соли и стереоизомеры, включая их смеси во всех соотношениях, и необязательно наполнители и/или вспомогательные...
Производные 2-пиридин-2-ил-пиразол-3(2h)-она, их получение и применение в терапии в качестве активаторов hif

Номер патента: 19591
Опубликовано: 30.04.2014
Авторы: Манетт Жеральдин, Фоссе Валери, Иллиано Стефан, Альтенбюрже Жан-Мишель
МПК: A61K 31/4545, A61K 31/4439, A61K 31/444...
Метки: терапии, получение, качестве, производные, 2-пиридин-2-ил-пиразол-3(2h)-она, применение, активаторов
Формула / Реферат:
1. Соединение, соответствующее следующей формуле (I)где R представляет собой -SO2-NR3R4 или -SO2R4, где R3, R4 и R5 имеют значения, определенные далее;R1 представляет собой гетероциклоалкил, не содержащий атом азота, -W-(С3-С6)циклоалкил, -W-арил, -W-гетероарил, -W-гетероциклоалкил, -W-COOR5 или -W-CONR5R6;причем арил, гетероарил и гетероциклоалкил по меньшей мере у одного атома углерода при необходимости имеют по меньшей мере один заместитель,...
Замещенные амидные производные в качестве ингибиторов протеинкиназы

Номер патента: 13231
Опубликовано: 30.04.2010
Авторы: Домингуез Селия, Боезио Алессандро, Потейшман Майкл, Беллон Стивен, Сигмунд Аарон К., Феллоус Ингрид М., Боер Дэвид, Букер Шон, Янг Кевин, Ким Тэ-Сеонг, Арманж Жан-Кристоф, Ли Мэтью, Джермейн Джули, Д`анжело Ноел, Кси Нинг, Лиу Лонгбин, Грасеффа Рассел, Норман Марк Х., Ла Дэниэл, Ровето Филип, Чокитте Дебора, Хйраи Сатоко, Д`амико Дерин К
МПК: C07D 401/14, C07D 401/12, A61K 31/435...
Метки: производные, качестве, замещенные, протеинкиназы, амидные, ингибиторов
Формула / Реферат:
1. Соединение формулы Iего энантиомеры, диастереомеры, соли, сольваты и N-оксиды,где R представляет собойТ выбран из фенила, 5-6-членного гетероарила или 5-6-членного гетероциклила;Z выбран из N или CR7;Z1 выбран из N или CR7;W представляет собой замещенный или незамещенный фенил, замещенный или незамещенный бензоморфолинил, замещенный или незамещенный 6-членный гетероарил, содержащий атом азота; C3-7 циклоалкил, C1-6-алкил и C1-6-алкинил;X...
Предыдущий патент: 6-замещенные пиримидины, ингибирующие вич
Следующий патент: Центральная буферная сцепка для движущегося в колее транспортного средства
Случайный патент: Трипептиды мар и iтр или их соли, белковые гидролизаты и смеси, содержащие указанные трипептиды или их соли, для снижения кровяного давления