Производные биариловых эфиров, полезные в качестве ингибиторов обратного захвата моноамина





Формула / Реферат
1. Соединение формулы

где n и m независимо выбраны из единицы и двух;
R1 и R2 независимо выбраны из водорода и (C1-C4)алкила;
R3 и R4 независимо выбраны из водорода и (C1-C4)алкила;
каждый X независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, O и S, где каждый X может быть дополнительно замещен водородом, галогено, (C1-C4)алкилом, оксо, амино и группировкой NR5(C=O)(C1-C4)алкил, где R5 представляет собой водород;
каждый Y независимо выбран из водорода и галогено и
каждый Z независимо выбран из водорода и галогено;
или его фармацевтически приемлемая соль.
2. Соединение или соль по п.1, где X выбран из фурана, тиофена, пиридина, 1,2,3-триазола и 1-пирролидин-2-она и где X может быть дополнительно замещен.
3. Соединение или соль по п.1, где один или оба R3 и R4 представляют собой водород.
4. Соединение по п.1, выбранное из группы, состоящей из
[4-(3,4-дихлорфенокси)бифенил-3-илметил]метиламина;
[2-(3,4-дихлорфенокси)-5-тиофен-3-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-4-тиофен-3-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-4-фуран-2-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-фуран-2-илбензил]метиламина;
N-[4'-(3,4-дихлорфенокси)-3'-метиламинометилбифенил-3-ил]ацетамида;
[2-(3,4-дихлорфенокси)-5-тиофен-2-илбензил]метиламина;
[4-(3,4-дихлорфенокси)-4'-фторбифенил-3-илметил]метиламина;
[2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-1-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-2-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-пиридин-2-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-пиридин-3-илбензил]метиламина;
1-[4-(3,4-дихлорфенокси)-3-метиламинометилфенил]-1H-пиразол-3-иламина;
[2-(3,4-дихлорфенокси)-5-пиридин-4-илбензил]метиламина;
[3-(3,4-дихлорфенокси)бифенил-4-илметил]метиламина;
[4-(3,4-дихлорфенокси)-4'-метилбифенил-3-илметил]метиламина;
[2-(3,4-дихлорфенокси)-4-тиофен-2-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-тиазол-2-илбензил]метиламина;
[2-(3,4-дихлорфенокси)-5-фуран-3-илбензил]метиламина;
{1-[2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-1-илфенил]этил}метиламина;
{1-[2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-2-илфенил]этил}метиламина;
{1-[2-(3,4-дихлорфенокси)-5-тиазол-2-илфенил]этил}метиламина;
{1-[2-(3,4-дихлорфенокси)-4-[1,2,4]триазол-1-илфенил]этил}метиламина;
[2-(3,4-дихлорфенокси)-5-(5-метилтиофен-2-ил)бензил]метиламина;
[2-(3,4-дихлорфенокси)-5-[1,2,4]триазол-4-илбензил]метиламина;
1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пирролидин-2-она;
1-[4-(3,4-дихлорфенокси)-3-(1-метиламиноэтил)фенил]пирролидин-2-она;
1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пиперидин-2-она
и их фармацевтически приемлемых солей.
5. Фармацевтическая композиция, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
6. Соединение формулы

где n и m независимо выбраны из единицы и двух;
X выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, O и S, где X может быть дополнительно замещен водородом, галогено, (C1-C4)алкилом, (C1-C4)алкокси, оксо и группировкой NR5(C=O)(C1-C4)алкил, где R5 представляет собой водород;
каждый Y независимо выбран из водорода и галогено;
каждый Z независимо выбран из водорода и галогено;
Q представляет собой -C(=O)R3 и
R3 представляет собой H или C1-C4алкил.
Текст
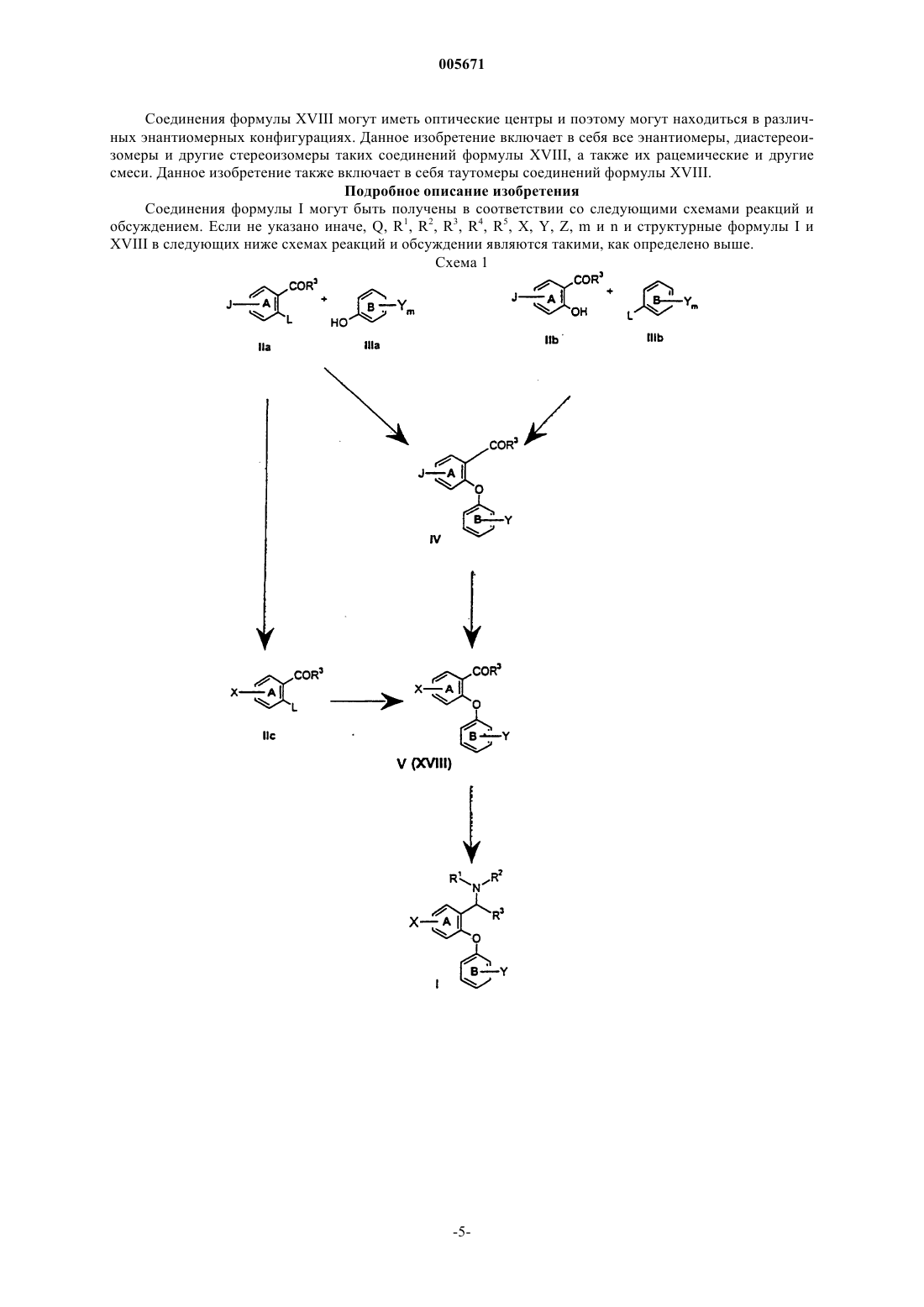
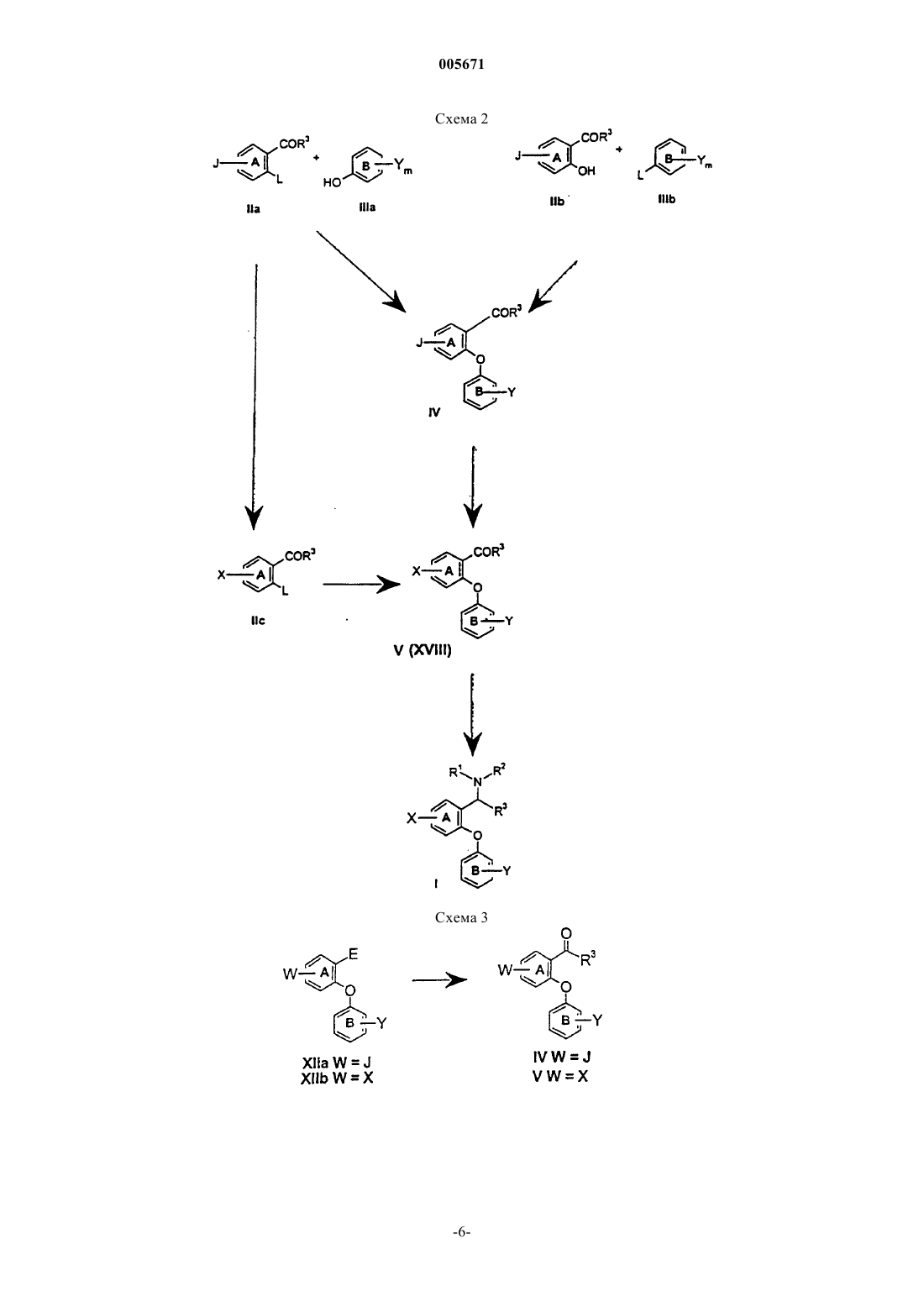
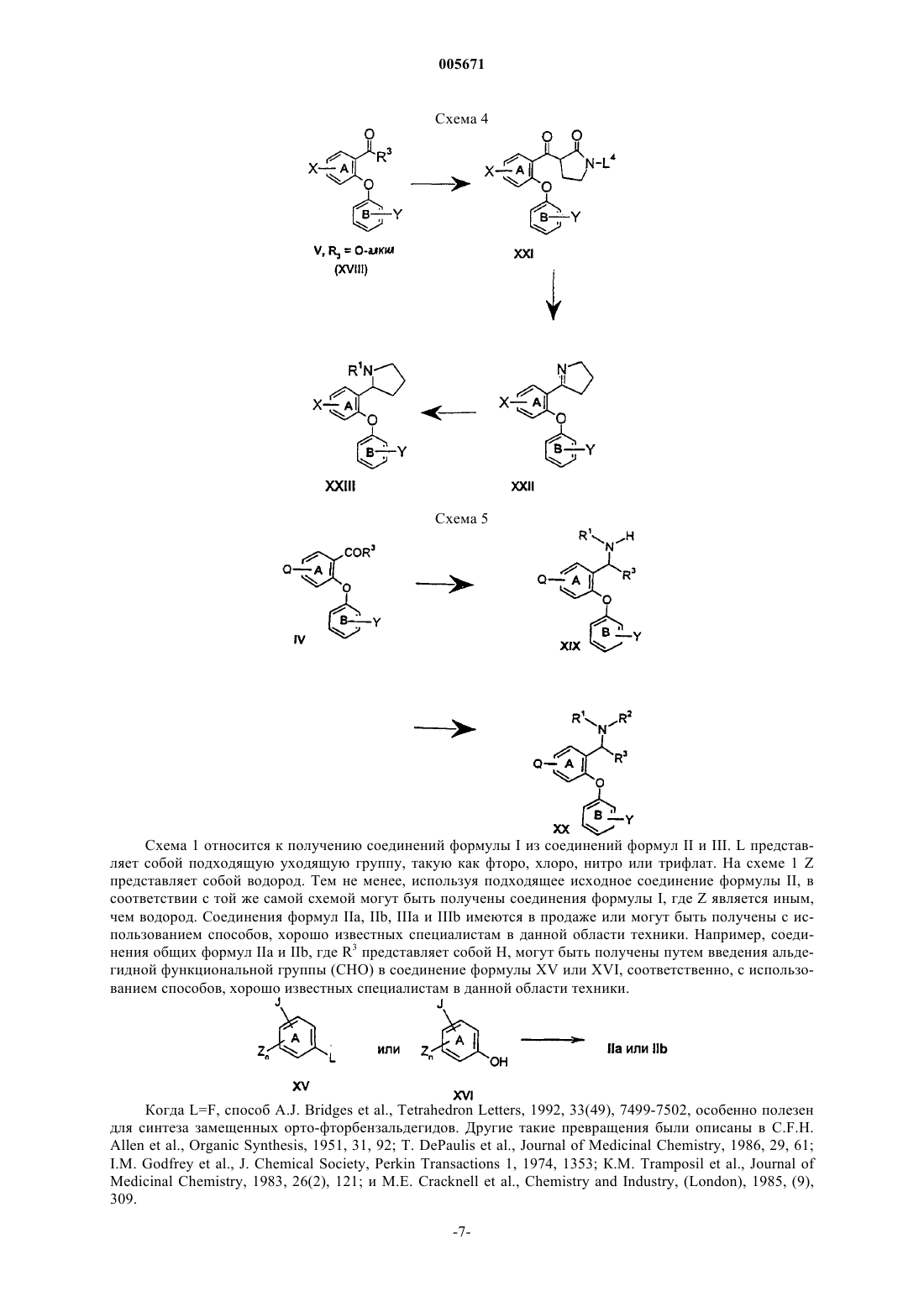
005671 Предшествующий уровень техники Селективные ингибиторы обратного захвата серотонина (СИОЗС) в настоящее время обеспечивают эффективность при лечении большого депрессивного расстройства (БДР) и обычно рассматриваются психиатрами и врачами первичной медико-санитарной помощи как эффективные, хорошо переносимые и легко вводимые. Тем не менее, они ассоциируются с нежелательными свойствами, такими как сообщения о сексуальной дисфункции, замедленное начало действия и уровень невосприимчивости, оцениваемый как 30% (см. М.J. Gitlin, Journal of Clinical Psychiatry, 1994, 55, 406-413 и R.T. Segraves, Journal ofClinical Psychiatry, 1992, 10(2), 4-10). Преклинические и клинические данные указывают на то, что сексуальная дисфункция, ассоциированная с лечением при помощи СИОЗС, может быть уменьшена путем использования ингибиторов обратного захвата дофамина (ИОЗД), таких как бупропион (см. А.К. Ashton,Journal of Clinical Psychiatry, 1998, 59(3), 112-115). Кроме того, комбинация ингибиторов обратного захвата серотонина (ИОЗС) и ИОЗД может ускорить начало действия, а также принести облегчение не поддающимся лечению пациентам, возможно посредством синергического механизма (см. R.D. Marshallet al., Journal of Psychopharmacology, 1995, 9(3), 284-286), и оказаться полезной при лечении злоупотребления веществами и болезни гиперактивного дефицита внимания (БГДВ), в соответствии с Barrickman etal., Journal of the American Academy of Child and Adolescent Psychology, 1995, 34 (5), 649 и Shekim et al.,Journal of Nervous and Mental Disease, 1989, 177 (5), 296. Данное изобретение относится к новым производным биариловых эфиров, которые проявляют активность ингибиторов обратного захвата моноамина (например, дофамина, серотонина), к фармацевтическим композициям, содержащим такие соединения, и к способам применения таких соединений для лечения расстройств центральной нервной системы (ЦНС) и других расстройств. Патент США 4018830, выданный 19 апреля 1997 года, относится к фенилтиоаралкиламинам и 2 фенилтиобензиламинам, которые являются активными в качестве антиаритмических средств.WO 97/17325, дата международной публикации 15 мая 1997 г., относится к производным N,Nдиметил-2-(арилтио)бензиламина, которые селективно влияют на транспорт серотонина в центральной нервной системе и полезны в качестве антидепрессантов. Патент США 5190965, выданный 2 марта 1993 г., и патент США 5430063, выданный 4 июля 1995 г.,относятся к феноксифенильным производным, которые полезны при лечении депрессии. Патент США 4161529, выданный 17 июля 1979 г., относится к пирролидиновым производным, которые обладают антихолестеримической и гиполипемической активностью. Предварительная заявка на патент США 60/121313, поданная 23 февраля 1999 г., относится к биариловым эфирам, которые обладают активностью при ингибировании обратного захвата как серотонина, так и дофамина. Краткое изложение сущности изобретения Настоящее изобретение относится к соединениям формулы где n и m независимо выбраны из единицы и двух;R1 и R2 независимо выбраны из водорода и (С 1-С 4)алкила;R3 и R4 независимо выбраны из водорода и (С 1-С 4)алкила; каждый X независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, где каждый Х может быть дополнительно замещен водородом,галогено, (C1-С 4)алкилом, оксо, амино и группировкой NR5(C=O)(C1-C4)aлкил, где R5 представляет собой водород; каждый Y независимо выбран из водорода и галогено; и каждый Z независимо выбран из водорода и галогено; или его фармацевтически приемлемая соль. Еще в одном воплощении данное изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, как описано выше, где Х выбран из фурана, тиофена, пиридина, 1,2,3 триазола и 1-пирролидин-2-она, и где Х может быть дополнительно замещен. Еще в одном воплощении данное изобретение относится к соединениям формулы I или их фармацевтически приемлемым солям, как описано выше, где один или оба R3 и R4 представляют собой водород.-1 005671 К предпочтительным соединениям формулы I относятся соединения, выбранные из группы, состоящей из[2-(3,4-дихлорфенокси)-5-[1,2,4]триазол-4-илбензил]метиламина; 1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пирролидин-2-она; 1-[4-(3,4-дихлорфенокси)-3-(1-метиламиноэтил)фенил]пирролидин-2-она; 1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пиперидин-2-она и их фармацевтически приемлемых солей. В соответствии с еще одним аспектом изобретения предложена фармацевтическая композиция, содержащая эффективное количество соединения формулы I или его фармацевтически приемлемой соли,как они описаны выше, и фармацевтически приемлемый носитель. В соответствии еще с одним аспектом изобретения предложено соединение формулыn и m независимо выбраны из единицы и двух; Х выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, где Х может быть дополнительно замещен водородом, галогено, (С 1-С 4)алкилом, (С 1 С 4)алкокси, оксо и группировкой NR5(C=O)(C1-C4)aлкил, где R5 представляет собой водород; каждый Y независимо выбран из водорода и галогено; каждый Z независимо выбран из водорода и галогено;Q представляет собой -C(=O)R3, и R3 представляет собой Н или С 1-С 4 алкил. Соединения формулы XVIII являются полезными в качестве промежуточных соединений для получения соединений формулы I. Данное изобретение также относится к фармацевтически приемлемым солям присоединения кислоты соединений формулы I. Примеры фармацевтически приемлемых солей присоединения кислоты соединений формулы I представляют собой соли соляной килоты, пара-толуолсульфоновой кислоты, фумаровой кислоты, лимонной кислоты, янтарной кислоты, салициловой кислоты, щавелевой кислоты,бромоводородной кислоты, фосфорной кислоты, метансульфоновой кислоты, винной кислоты, малеиновой кислоты, ди-пара-толуоилвинной кислоты, уксусной кислоты, серной кислоты, йодоводородной кислоты и миндальной кислоты. Если не указано иначе, используемый здесь термин "галогено" включает в себя фторо, хлоро, бромо и йодо.-2 005671 Если не указано иначе, используемый здесь термин "алкил" может быть линейным, разветвленным или циклическим и может включать в себя линейные и циклические группировки, а также разветвленные и циклические группировки. Если не указано иначе, используемый здесь термин "гетероцикл" относится, во-первых, к ароматическим группам, содержащим один или более чем один гетероатом (О, S или N), предпочтительно от одного до четырех гетероатомов. Мультициклическая группа, содержащая один или более чем один гетероатом, где по меньшей мере одно кольцо группы является ароматическим, также, если не указано иначе,представляет собой "гетероарильную" группу для целей настоящего изобретения. Примеры таких гетероциклических групп представляют собой пиридинил, пиридазинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, хинолил, изохинолил, тетразолил, фурил, тиофенил, изоксазолил, тиазолил,оксазолил, изотиазолил, пирролил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил,индолицинил, фталазинил, триазинил, изоиндолил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотриазолил, бензотиазолил, бензизотиазолил, бензоксазолил, бензизоксазолил, бензимидазолил, хиназолинил, хиноксалинил, нафтиридинил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил, тетрагидроизохинолил, бензофурил, фуропиридинил, пирролопиримидинил и азаиндолил. Термин "гетероцикл" также относится к неароматическим циклическим группам, содержащим один или более чем один гетероатом, предпочтительно от одного до четырех гетероатомов, каждый из которых выбран из О, S и N. "Гетероцикл", если не указано иначе, включает гетеробициклические группы. Гетеробицикл относится к неароматическим двухкольцевым циклическим группам, где указанные кольца разделяют один или два атома, и где по меньшей мере одно из колец содержит гетероатом (О, S или N). Гетеробициклические группы для целей настоящего изобретения, и если не указано иначе, включают в себя спирогруппы и конденсированные кольцевые группы. В первом воплощении каждое кольцо в гетеробицикле содержит до четырех гетероатомов (то есть от нуля до четырех гетероатомов, при условии, что по меньшей мере одно кольцо содержит по меньшей мере один гетероатом). Примеры таких гетероциклических групп включают в себя азиридинил, азетидинил, пирролидинил, пиперидинил, азепинил, пиперазинил, 1,2,3,6-тетрагидропиридинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, оксазолидинил, морфолино, тиоморфолино, тиазолидинил, тиоксанил, пирролинил, индолинил, 2 Н-пиранил, 4 Н-пиранил, диоксанил, 1,3-диоксоланил,пиразолинил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3,1,0]гексанил, 3-азабикло[4,1,0]гептанил, хинолизинил, хинуклидинил, 1,4 диоксаспиро[4,5]децил, 1,4-диоксаспиро[4,4]нонил, 1,4-диоксаспиро[4,3]октил и 1,4-диоксаспиро[4,2]гептил, но не ограничиваются ими. Вышеупомянутые гетероциклические группы могут быть С-присоединенными или Nприсоединенными, там, где такое присоединение является возможным. Например, группа, полученная из пиррола, может представлять собой пиррол-1-ил (N-присоединенный) или пиррол-3-ил (Сприсоединенный). Термины, относящиеся к группам, также охватывают все возможные таутомеры. Термины "лечение", "лечить" и тому подобное относятся к реверсированию, ослаблению или ингибированию прогрессирования заболевания или состояния, к которому применяется такой термин, или одного или более чем одного симптома такого заболевания или состояния. Используемые здесь эти термины также охватывают в зависимости от состояния пациента предотвращение заболевания или состояния, включая предотвращение начала заболевания или состояния или симптомов, ассоциированных с заболеванием или состоянием, и включая снижение тяжести заболевания или состояния или симптомов,ассоциированных с ними, перед тем, как указанное заболевание или состояние поражает пациента. Такое предотвращение или снижение перед тем, как заболевание поражает пациента, относится к введению соединения по данному изобретению субъекту, который в момент введения не страдает этим заболеванием или состоянием. "Предотвращение" также охватывает предотвращение рецидива заболевания или состояния или симптомов, ассоциированных с ними. Используемый здесь термин "млекопитающее" относится к любому представителю класса "Млекопитающие", включая людей, собак и кошек, но не ограничен ими. Когда здесь ссылаютсяна расстройство или состояние, которое можно лечить путем ингибирования обратного захвата серотонина, дофамина или норэпинефрина, это означает, что расстройство или состояние имеет в качестве способствующего фактора по меньшей мере один тип нейропередачи, опосредованный серотонином, дофамином или норэпинефрином. Расстройство или состояние может иметь в качестве способствующего фактора один, два или все три вышеупомянутые типа нейропередачи. Кроме того, фактор или факторы, отличающиеся от путей нейропередачи, опосредованных серотонином, дофаином или норэпинефрином, могут также способствовать этому расстройству или состоянию. Расстройства или состояния, которым способствуют пути нейропередачи, опосредованные серотонином,дофамином или норэпинефрином, могут быть установлены специалистом в данной области техники, и включают в себя, например, привыкание к веществам и злоупотребление ими, депрессию и фобию, но не ограничиваются ими.-3 005671 Соединения формулы I могут иметь оптические центры и поэтому могут находиться в различных энантиомерных конфигурациях. Данное изобретение включает в себя все энантиомеры, диастереоизомеры и другие стереоизомеры таких соединений формулы I, а также их рацемические и другие смеси. Данное изобретение также включает в себя таутомеры соединений формулы I. Объектом изобретения также являются соединения, меченные изотопами, которые идентичны соединениям, соответствующим формуле I, но за исключением того, что один или более чем один атом заменен атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по данному изобретению, включают в себя изотопы водорода, углерода, азота, кислорода,фосфора, фтора, йода и хлора, такие как 3H, 11 С, 14 С, 18F,123I и 125I. Соединения по настоящему изобретению и фармацевтически приемлемые соли указанных соединений, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов, находятся в объеме данного изобретения. Соединения по настоящему изобретению, меченные изотопами, например соединения, в которые включены радиоактивные изотопы, такие как 3H и 14 С, являются полезными в анализах распределения в тканях лекарственного средства и/или субстрата. Изотопы тритий, то есть 3H, и углерод-14, то есть 14 С, особенно предпочтительны по причине их легкого получения и обнаруживаемости. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может представить некоторые терапевтические преимущества, являющиеся результатом большей метаболической устойчивости, например увеличенным периодом полураспада in vivo или уменьшенными потребностями в дозировке и, следовательно, могут быть предпочтительны в некоторых случаях. Соединения формулы I по данному изобретению, меченные изотопами, обычно могут быть получены путем осуществления процедур, раскрытых на схемах и/или в следующих ниже примерах, путем замещения реактива, не меченного изотопами, легко доступным реактивом, меченным изотопом. Использованный здесь термин "химическая зависимость" обозначает ненормальную потребность или желание лекарственного средства или привыкание к лекарственному средству. Такие лекарственные средства обычно вводят страдающему индивидууму посредством любого из множества способов введения, включая пероральное, парентеральное, назальное введение или введение путем ингаляции. Примеры химических зависимостей, которые можно лечить при помощи способов по настоящему изобретению,представляют собой зависимости от спирта, никотина, кокаина, героина, фенобарбитала и бензодиазепинов (например Valium (товарный знак. Используемый здесь термин "лечение химической зависимости" обозначает уменьшение или ослабление такой зависимости. Соединения формулы I можно использовать в комбинации с антагонистом NK-1-рецепторов или антагонистом 5 НТ 1D-рецепторов. Антагонист NK-1-рецепторов, как изложено выше, представляет собой вещество, которое способно к антагонизации NK-1-рецепторов, посредством этого ингибируя реакции, опосредованные тахикининами, такие как реакции, опосредованные веществом Р. Различные антагонисты NK-1-рецепторов известны из области техники,и любой такой антагонист NK-1-рецепторов может быть использован по настоящему изобретению, как описано выше, в комбинации с соединением формулы I. Антагонисты NK-1-рецепторов описаны, например, в патенте США 5716965 (выданном 10 февраля 1998 г.); патенте США 5852038 (выданном 22 декабря 1998 г.); WO 90/05729 (дата международной публикации 31 мая 1990 г.); патенте США 5807867 (выданном 15 сентября 1998 г.); патенте США 5886009 (выданном 23 марта 1999 г.); патенте США 5939433 (выданном 17 августа 1999 г.); патенте США 5773450 (выданном 30 июня 1998 г.); патенте США 5744480 (выданном 28 апреля 1998 г.); патенте США 5232929 (выданном 3 августа 1993 г.); патенте США 5332817 (выданном 26 июля 1994 г.); патенте США 5122525 (выданном 16 июня 1992 г.); патенте США 5843966 (выданном 1 декабря 1998 г.); патенте США 5703240 (выданном 30 декабря 1997 г.); патенте США 5719147 (выданном 17 февраля 1998 г.) и патенте США 5637699 (выданном 10 июня 1997 г.). Каждый из вышеупомянутых патентов США и вышеуказанная опубликованная международная заявка согласно РСТ включены здесь путем ссылки. Соединения, описанные в указанных ссылках, обладающие антагонистической активностью в отношении NK-1-рецепторов, могут быть использованы по настоящему изобретению. Тем не менее, другие антагонисты NK-1-рецепторов могут также быть использованы по данному изобретению. Антагонист 5 НТ 1D-рецепторов, как здесь изложено, представляет собой вещество, которое является антагонистом подтипа 5 НТ 1D серотонинового рецептора. Любое такое вещество может быть использовано по настоящему изобретению, как описано выше, в комбинации с соединением формулы I. Вещества,обладающие антагонистической активностью в отношении 5 НТ 1D-рецепторов, могут быть определены специалистом в данной области техники. Например антагонисты 5 НТ 1D-рецепторов описаны в WO 98/14433 (дата международной публикации 9 апреля 1998 г.); WO 97/36867 (дата международной публикации 9 октября 1997 г.); WO 94/21619 (дата международной публикации 29 сентября 1994 г.); патенте США 5510350 (выданном 23 апреля 1996 г.); патенте США 5358948 (выданном 25 октября 1994 г.); и GB 2276162 А (опубликованном 21 сентября 1994 г.). Эти антагонисты 5 НТ 1D-рецепторов, а также другие,могут быть использованы по настоящему изобретению. Вышеупомянутые опубликованные заявки на патент и патенты включены здесь путем ссылки.-4 005671 Соединения формулы XVIII могут иметь оптические центры и поэтому могут находиться в различных энантиомерных конфигурациях. Данное изобретение включает в себя все энантиомеры, диастереоизомеры и другие стереоизомеры таких соединений формулы XVIII, а также их рацемические и другие смеси. Данное изобретение также включает в себя таутомеры соединений формулы XVIII. Подробное описание изобретения Соединения формулы I могут быть получены в соответствии со следующими схемами реакций и обсуждением. Если не указано иначе, Q, R1, R2, R3, R4, R5, X, Y, Z, m и n и структурные формулы I иXVIII в следующих ниже схемах реакций и обсуждении являются такими, как определено выше. Схема 1 Схема 1 относится к получению соединений формулы I из соединений формул II и III. L представляет собой подходящую уходящую группу, такую как фторо, хлоро, нитро или трифлат. На схеме 1 Z представляет собой водород. Тем не менее, используя подходящее исходное соединение формулы II, в соответствии с той же самой схемой могут быть получены соединения формулы I, где Z является иным,чем водород. Соединения формул IIа, IIb, IIIa и IIIb имеются в продаже или могут быть получены с использованием способов, хорошо известных специалистам в данной области техники. Например, соединения общих формул IIа и IIb, где R3 представляет собой Н, могут быть получены путем введения альдегидной функциональной группы (СНО) в соединение формулы XV или XVI, соответственно, с использованием способов, хорошо известных специалистам в данной области техники. Когда L=F, способ A.J. Bridges et al., Tetrahedron Letters, 1992, 33(49), 7499-7502, особенно полезен для синтеза замещенных орто-фторбензальдегидов. Другие такие превращения были описаны в С.F.H.-7 005671 Ссылаясь на схему 1, соединение (то есть альдегид или кетон) формулы IIа подвергают взаимодействию с соединением (то есть фенолом) формулы IIIa в присутствии основания с образованием соответствующего соединения формулы IV. Эту реакцию обычно осуществляют при температуре от приблизительно 0 до приблизительно 150 С, в течение от приблизительно 1 ч до приблизительно 3 дней, предпочтительно при приблизительно 90-95 С в течение приблизительно 18 ч, в полярном растворителе, таком как диметилсульфоксид (ДМСО), N,N-диметилформамид (ДМФ), N,N-диметилацетамид (ДМА) или Nметил-2-пирролидинон (ММП), предпочтительно ДМФ. Подходящие основания включают в себя безводный карбонат натрия (Nа 2 СО 3), карбонат калия (К 2 СО 3), гидроксид натрия (NaOH), гидроксид калия(КОН) и амины, такие как пирролидин, триэтиламин и пиридин, причем безводный К 2 СО 3 является предпочтительным. Подробности осуществления этого способа могут быть найдены в G.W. Yeager et al.,Synthesis, 1995, 28-30; J.R. Dimmock et al., Journal of Medicinal Chemistry, 1996, 39(20), 3984-3997. В качестве альтернативы, фенолы формулы IIb и соединения формулы IIIb могут быть превращены в альдегиды или кетоны общих формул IV в соответствии со способами, описанными К. Tomisawa et al., Chemical andPharmaceutical Bulletin, 1984, 32(8), 3066-3074. Затем соединение формулы IV, где J представляет собой уходящую группу, например бром, йод,трифлат, фторсульфонат или метансульфонат, может быть превращено в соединение формулы V путем взаимодействия с соединением общей формулы X-G, где G определен как реакционноспособная уходящая группа, такая как В(ОН)2, Sn[(С 1-С 6)алкил], Zn(Hal) и тому подобное, обычно в присутствии каталитического количества катализатора, например, среди прочих, тетракис(трифенилфосфин)палладия(0),тетракис(трифенилфосфин)никеля(0) или дихлорбис(трифенилфосфин)палладия (II), и в присутствии основания, такого как карбонат натрия, карбонат калия или триэтиламин. Эти реакции могут быть проведены во множестве органических растворителей (например бензоле, диметоксиэтане) или в смесях,таких как водный N,N-диметилформамид или водный этанол, при температурах, находящихся в диапазоне от приблизительно 0 до приблизительно 100 С. Хорошая общая ссылка на этот способ может быть найдена в обзоре Stephen Stanforth, Tetrahedron, 1998, 54, 263-303. Другие специфические ссылки включают в себя М.J. Sharp et al., Synthetic Communications, 1981, 11(7), 513; R.В. Miller et al., Tetrahedron Letters, 1989, 30(3), 297; W.J. Thompson et al., Journal of Organic Chemistry, 1984, 49(26), 5237. Соединения общей формулы X-G во многих случаях имеются в продаже или могут быть получены специалистом в области органического синтеза (например, см. способы в М.J. Sharp and V. Snieckus, Tetrahedron Letters,1985, 26(49), 5997-6000; G.W. Kabalka et al., Journal of Organometallic Chemistry, 1983, 259, 269-274). В качестве альтернативы, промежуточное соединение формулы IIа может быть превращено в соединение формулы IIc, где Х является таким, как определено выше, с использованием способов, описанных выше для превращения соединений формулы IV в V. Эти промежуточные соединения формулы IIc затем можно подвергать взаимодействию с соединением общей формулы IIIа с получением простых эфиров общей формулы V с использованием способов, описанных выше для превращения соединений формулы IIа в IV. Дополнительно, соединения формул IIа или IV, где J представляет собой функциональную группу,как CN, могут быть превращены в соединения формулы IIс или V, где Х представляет собой группировку, такую как и где R10 независимо выбран из водорода, (С 1-С 6)алкила, арил-(С 1-С 6)алкила или арила, возможно замещенного водородом, галогено, (С 1-С 6)алкилом, (С 1-С 6)алкоксилом или (С 1-С 6)SOr, где r равно нулю,единице или двум. Способы для этого превращения хорошо описаны в химической литературе; например, путем использования азида натрия и хлорида лития в 2-метоксиэтаноле в соответствии со способом,описанным J. Sauer et al., Tetrahedron, 1960, 11, 241. В этих условиях может быть необходимо сначала ввести защиту в группу COR3 соединения IIа или IV для эффективного превращения группы J в соответствующую группу Х соединений IIc или V (промежуточные соединения формулы XVIII), соответственно. Существует множество доступных защитных групп, которые могут быть использованы в этом способе, включая ацетали и кетали, которые описаны и на которые ссылаются Р.G.M. Wuts и Т.W. Green вProtective Groups in Organic Synthesis, 2nd ed., John Wiley and Sons, New York, 1991, стр. 175-223. Выбор подходящей защитной группы может быть осуществлен, исходя из присутствия других реакционноспособных групп в молекуле. Затем соединения формулы V (R3=Н или (С 1-С 4)алкил) могут быть превращены в соединения формулы I путем подвергания их воздействию условий гидроаминирования. Например, соединение формулыV можно подвергать взаимодействию с соединением формулы HNR1R2 с образованием промежуточного соединения формулы XVII: которое может быть выделено или превращено непосредственно на той же самой реакционной стадии в соединение формулы I. Это превращение, или осуществляемое in situ, или начинающееся с выделенного соединения формулы XVII, может быть осуществлено с использованием одного или более чем одного способа, известного специалистам в данной области техники. Например, соединение формулы V и подходящее соединение формулы HNR1R2 можно объединять в присутствии дегидратирующего реактива, такого как тетрахлорид титана (IV) или изопропоксид титана(IV), в реакционно-инертном растворителе, таком как бензол, толуол, этанол или похожий растворитель,до тех пор, пока взаимодействие не посчитают завершившимся, в соответствии со способом S. Bhattarcharyya (Journal of Organic Chemistry, 1995, 60(15), 4928-4929). В качестве альтернативы, соединение формулы V и соединение формулы HNR1R2 можно объединять в инертном растворителе, таком как бензол или толуол, в присутствии или отсутствии акцептора воды, такого как молекулярные сита, и можно нагревать для удаления воды, образовавшейся в процессе образования промежуточного соединения формулы XVII. О степени завершения превращения соединений формулы IV в вышеуказанное промежуточное(ые) соединение(ия) формулы XVII можно судить, используя один или более чем один известный аналитический способ, включая 1H-ЯМР спектроскопию. В некоторых случаях может быть возможным или желательным выделить промежуточное(ые) соединение(ия) формулы XVII, или их дополнительно можно подвергать взаимодействию с восстановителем, избирательным для восстановления промежуточного соединения до желаемых соединений формулыI. Такие восстановители широко известны специалистам в данной области техники и включают в себя,например, борогидрид натрия (NaBH4), цианоборогидрид натрия (NаВН 3 СN) и триацетоксиборогидрид натрия (NаВН(ОАс)3), как описано A.F. Abdel-Magid et al. в Tetrahedron Letters, 1990, 31, 5595. Это восстановление обычно осуществляют в полярном растворителе, таком как метанол, этанол, изопропанол или похожий растворитель, и температурах от приблизительно 0 до приблизительно 100 С, предпочтительно при комнатной температуре. В способе, описанном Bhattarcharyya, промежуточное соединение формулы XVII образуется в этанольном растворителе, и его без выделения восстанавливают до продукта формулы I с использованием NaBH4. В качестве альтернативы альдегидным или кетоновым промежуточным соединениям формул IV иV, специалист в данной области техники может также получить соединения формулы VII (то есть нитрилы), начиная с соединений формул IIIa и VI, как проиллюстрировано на схеме 2, используя способ образования дифенилового эфира, описанный на схеме 1. Эти соединения могут затем служить в качестве промежуточных соединений для синтезов желаемых соединений формулы I. Способы получения соединений формулы VI, используемые в этом способе, могут быть адаптированы из способов, имеющихся в литературе. (См., например, D.С. Remy et al., Journal of Medicinal Chemistry, 1975, 18(2), 142-148; E.A.Schmittling et al., Journal of Organic Chemistry, 1993, 58(12), 3229-3230). Превращение нитрилов формулы VII, полученных таким образом, в желаемые продукты формулы I,может быть достигнуто несколькими путями, как изображено на схеме 2. Например, нитрильная группаVII может быть гидролизована в кислотных условиях с использованием способов, хорошо известных специалистам в данной области техники, с получением производного карбоновой кислоты формулы VIIIOrganic Chemistry, 1951, 16, 1275). Это производное карбоновой кислоты может затем быть превращено в соединение формулы V (R3=ОН) с использованием способов, ранее описанных на схеме 1 для превращения IV в V; затем соединение V (R3=ОН) может быть превращено в соединение V (R3=NR1R2) и затем в соединения формулы I, как описано ниже. В качестве альтернативы, соединение VIII может быть превращено в карбоксамидное производное формулы IX с использованием одного или более чем одного стандартного способа, который раскрыт в химической литературе, например путем взаимодействия галоидангидрида, полученного из соединения формулы VIII, с амином общей формулы HNR1R2 (см. R.Е. Kent et al., Organic Synthesis, Coll. Vol. III,1955, 490; и R.M. Herbst et al., Organic Synthesis, Coll. Vol. II, 1943, 11 для обсуждений реакции ШоттенБаумана). Эти карбоксамиды формулы IX затем могут быть превращены в соответствующие карбоксамиды формулы V (R3=NR1R2) путем замены заместителя J подходящей группой Х с использованием условий, похожих на условия, описанные для превращения IV в V на схеме 1. Карбоксамиды формул V, полученные таким образом, могут затем быть восстановлены до конечных продуктов формул I с использованием подходящего способа восстановления. В зависимости от при-9 005671 сутствия заместителей X, Y и Z в карбоксамидах V, это восстановление может быть осуществлено с использованием одного или более чем одного из различных реактивов, включающих в себя алюмохлорид лития (например, J. Lehmann et al., Archiv. Pharm. (Weinheim, Ger.), 1982, 315 (11), 967; N.S. NarasimhanBrown et al., Journal of the American Chemical Society, 1970, 92, 1637 и 1973, 38, 912; N.M. Moon et al.,Journal of Organic Chemistry, 1973, 38, 2786; Н.С. Brown and V. Verma, Journal of Organic Chemistry, 1974,39, 1631), тексилборан/диэтиланилин (A. Pelter et al., Tetrahedron Letters, 1978, 4715), трихлорид фосфора/толуол с последующим этанольным борогидридом натрия (A. Rahman et al., Tetrahedron Letters, 1976,219) или гидрид алюминия (Н.С. Brown et al., Journal of the American Chemical Society, 1966, 88, 1464;A.F. Burchat et al., Journal of Organic Chemistry, 1996, 61(21), 7627-7630). Получающиеся в результате карбоксамиды формулы IX, где R1 и R2 представляют собой водород,могут также быть получены непосредственно из соответствующих нитрилов формулы VII с использованием способов специфического гидролиза, используя например пероксид водорода или сильные водные соли щелочных металлов. (См. ChemistryIndustry, 1961, 1987; С.R. Noller, Organic Synthesis, Coll. Vol.II, 1943, 586; и J.H. Hall and M. Gisler, Journal of Organic Chemistry, 1976, 41, 3769). Затем карбоксамидные производные формулы IX могут быть превращены в карбоксамидные соединения формулы V(R3=NR1R2) с использованием способа, только что описанного для превращения VIII в V. Наконец, нитрилы формулы X, полученные из нитрилов формулы VII, аналогично превращению соединений формул IV в V, могут быть восстановлены до желаемых соединений общей формулы I, гдеR1 и R2 оба представляют собой водород, путем использования одного из множества восстановителей,раскрытых в химической литературе, которые являются избирательными для этого превращения (включая каталитическое гидрирование с использованием газа водорода и оксида платины(II), как описано J.A.Secrist, III и M.V. Logue в Journal of Organic chemistry, 1972, 37, 335; гидрата гидразина и никеля Ренея в этаноле, как описано W.W. Zajac, Jr. et al., в Journal of Organic Chemistry, 1971, 36, 3539; и трифторацетоксиборогидрида натрия в ТГФ (тетрагидрофуране), как описано N. Umino et al., в Tetrahedron Letters,1976, 2875). Такие восстановители могут также включать в себя алюмогидрид лития в нереакционноспособном растворителе, таком как диэтиловый эфир или тетрагидрофуран (см., например, А.С. Соре et al.,Organic Synthesis, Coll. Vol. IV, 1963, 339, для использования алюмогидрида лития в растворителе диэтиловом эфире или ТГФ). Нитрилы формулы VII могут также быть превращены в соответствующие альдегиды общей формулы IV, где R3 представляет собой водород, с использованием реактивов и условий, которые являются специфическими для этого превращения, такие как триэтоксиалюмогидрид лития в растворителе, таком как ТГФ или диэтиловый эфир, как описано H.С. Brown и С.Р. Garg в Journal of the American ChemicalSociety, 1964, 86, 1085 и J. Malek and M. Cerny в Synthesis, 1972, 217. В дополнение к способам, описанным выше на схемах 1 и 2 для получения промежуточных соединений альдегидов и кетонов формулы I, существуют другие способы, которые могут дать возможность получить соединения формулы I. Например, в способе, изображенном на схеме 3, соединение формулыXIIa,b, где Е представляет собой атом водорода, можно подвергать взаимодействию в условиях ацилирования по Фриделю-Крафтсу (например AlCl3/CH2Cl2/R3COCl) с получением кетонов формулы IV или V,где R3 представляет собой С 1-С 4 алкил (С.F.H. Allen, Organic Synthesis, Coll. Vol. II, 3, 1943). В качестве альтернативы, ангидрид кислоты, то есть (R3 СО)2 О можно подвергать взаимодействию в похожих условиях (О. Grummitt et al., Organic Synthesis, Coll. Vol. III, 109, 1955) с получением промежуточных соединений формулы IV или V. Когда желательно получить соединения формулы IV или V, где R3 представляет собой водород, указанное соединение может быть получено из соединений формулы XIIa,b путем ацилирования по Вильсмайеру-Хааку с использованием способов, описанных Е. Campaigne and W.L.IV, 98, 1955. Расположение ацильной группы (COR3), введенной таким образом, может быть определено по природе и расположению присутствующих заместителей J, Х и/или Y, а также по условиям, используемым в реакции. В тех случаях, когда желательно получить соединения формулы IV (R3=H) из XIIa (E=H), введение альдегидной функциональной группы (СНО) может также быть достигнуто с использованием условий, описанных выше для получения промежуточных соединений IIа и IIb на схеме 1. Например, получение соединений формулы IV, где R3=H (то есть альдегидов) может быть осуществлено с использованием одного или более чем одного из известных способов для формилирования ароматических колец,включая взаимодействие дихлорметилметилового эфира и тетрахлорида титана (IV) в метиленхлориде в соответствии со способом, описанным М.L. Mancini et al., Synthetic Communications, 1989, 2001-2007, илиH. Chikashita et al., Journal of Organic Chemistry, 1991, 56, 1692. Для получения соединений общей формулы I, где R2 и R3, взятые вместе с атомом азота, к которому присоединен R2, и атомом углерода, к которому присоединен R3, образуют кольцо, содержащее азот, может быть использована модификация способа, описанного L.S. Bleicher et al. (Journal of OrganicChemistry, 1998, 63, 1109), как изображено на схеме 4. Так, сложный эфир общей формулы V (R3=Oалкил) (промежуточное соединение формулы XVIII), полученный путем этерификации соответствующей-10 005671 карбоновой кислоты формулы V (R3=ОН) (также формулы XVIII), подвергают взаимодействию с циклическим лактамом общей формулы XXV где L4 представляет собой реакционно-лабильную группу, такую как -СН=СН 2, в присутствии сильного основания, такого как метоксид натрия, с получением дикето-промежуточного соединения общей формулы XXI. Это промежуточное соединение затем может быть превращено в соответствующий циклический имин формулы XXII в присутствии сильной кислоты, такой как соляная кислота, обычно в условиях дефлегмации. Затем соединения формулы XXII могут быть восстановлены с образованием циклических аминов формулы XXIII (где R1=Н) с использованием, например, борогидрида натрия в метаноле, как описано ранее. Такие соединения формулы XXIII могут дополнительно быть превращены в соединения формулы XXIII (где R1 является таким, как определено для соединений формулы I), как обсуждалось ранее. Для получения соединений общей формулы I, где группа Х представляет собой лактам, присоединенный к фенильному или нафтильному кольцу А через атом N лактама, предпочтительным является способ, изображенный на схеме 5. В этом способе альдегид или кетон общей формулы IV (R3=Н или С 1 С 4 алкил, соответственно), где Q представляет собой NO2, превращают в амин общей формулы XIX, гдеR1 является таким, как определено ранее, в соответствии со способом, описанным на схеме 1. Это промежуточное соединение XIX затем превращают в соединение общей формулы XX, где R2 представляет собой защитную группу, предпочтительно трет-бутоксикарбонильную группу (тpeт-БОК), которая устойчива к условиях гидрирования, но может быть легко удалена на более поздней стадии последовательности синтеза; предложения относительно таких групп могут быть найдены у Wuts and Green ранее, на стр. 309. Это последнее промежуточное соединение XX, где Q представляет собой NO2, может затем быть обработано в условиях восстановления с образованием аналогичного промежуточного соединения формулы XX, где Q представляет собой NH2, в то же время, оставляя группу тpeт-БОК нетронутой. Такие условия восстановления для этого превращения известны специалистам в данной области техники и включают в себя использование газа водорода и катализатора, предпочтительно палладия на углероде, в реакционно-инертном растворителе, таком как низший спирт (например, метанол, этанол), сложный эфир (например, этилацетат) или простой эфир (например, тетрагидрофуран, 1,4-диоксан) и в присутствии или отсутствии небольшого количества кислоты, предпочтительно небольшого количества уксусной кислоты. Группа NH2 получающихся в результате соединений формулы XX может затем быть превращена в циклические амиды (лактамы), где R2 остается тpeт-БОК, путем их взаимодействия с омегахлоралканоилхлоридом или -бромидом или омега-бромалканоилхлоридом или -бромидом в нейтральном растворителе, таком как ТГФ, и в присутствии акцептора кислоты, такого как Nа 2 СО 3, К 2 СО 3, СS2 СО 3 и тому подобное, и нагревания смеси при температуре кипения растворителя. В результате этого происходит замыкание кольца и образуется циклический амид (то есть лактам). Наконец, защитная группа может быть удалена с получением соединений общей формулы I, где Х представляет собой лактам, и R2 представляет собой Н; в случае защитной группы трет-БОК, смесь этилацетата, насыщенного газом HCl, эффективна при таком удалении. Фармацевтически приемлемые соли соединения формулы I могут быть получены обычным образом путем обработки раствора или суспензии соответствующих свободного основания или кислоты одним химическим эквивалентом фармацевтически приемлемых кислоты или основания. Обычные способы концентрирования или кристаллизации могут быть использованы для выделения солей. В качестве иллюстрации подходящих кислот выступают уксусная, молочная, янтарная, малеиновая, винная, лимонная, глюконовая, аскорбиновая, бензойная, коричная, фумаровая, серная, фосфорная,соляная, бромоводородная, йодоводородная, сульфаминовая, сульфоновые кислоты, такие как метансульфоновая, бензолсульфоновая, пара-толуолсульфоновая и родственные кислоты. В качестве иллюстрации оснований выступают натрий, калий и кальций. Соединение по данному изобретению может быть введено само по себе или в комбинации с фармацевтически приемлемыми носителями в виде разовой или множественных доз. Подходящие фармацевтические носители включают в себя инертные твердые разбавители или наполнители, стерильные водные растворы и различные органические растворители. Фармацевтические композиции, образованные путем объединения соединения формулы 1 или его фармацевтически приемлемой соли, могут затем быть легко введены в виде множества лекарственных форм, таких как таблетки, порошки, лепешки, сиропы, инъекционные растворы и тому подобное. Эти фармацевтические композиции могут, если желательно, содержать дополнительные ингредиенты, такие как корригенты, связывающие вещества, эксципиенты и тому подобное. Таким образом, для целей перорального введения могут использоваться таблетки, содержащие различные эксципиенты, такие как цитрат натрия, карбонат кальция и фосфат кальция, наряду с различ-11 005671 ными разрыхлителями, такими как крахмал, метилцеллюлоза, альгиновая кислота и некоторые комплексные силикаты, вместе со связывающими агентами, такими как поливинилпирролидон, сахароза,желатин и аравийская камедь. Дополнительно, смазывающие вещества, такие как стеарат магния, лаурилсульфат натрия и тальк, часто полезны для целей таблетирования. Твердые композиции похожего типа могут также использоваться в качестве наполнителей в твердых и мягких наполненных желатиновых капсулах. Предпочтительные для этого вещества включают в себя лактозу, или молочный сахар, и полиэтиленгликоли с высокой молекулярной массой. Когда для перорального введения желательны водные суспензии или эликсиры, необходимый активный ингредиент в них можно объединить с различными подсластителями или корригентами, окрашивающими веществами или красителями и, если желательно, эмульгаторами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин и их комбинации. Для парентерального введения могут использоваться растворы, содержащие соединение по данному изобретению или его фармацевтически приемлемую соль в кунжутном или арахисовом масле, водном пропиленгликоле или в стерильном водном растворе. Такие водные растворы должны быть в случае необходимости подходящим образом забуферены, и жидкий разбавитель сначала делают изотоническим,используя достаточное количество физиологического раствора или глюкозы. Эти специфические водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. Используемые стерильные водные среды легко доступны путем использования стандартных способов, известных специалистам в данной области техники. Соединение формулы I или его фармацевтически приемлемую соль можно вводить перорально,трансдермально (например, путем использования пластыря), парентерально (например, внутривенно или ректально) или местно. Как правило, суточная дозировка для лечения расстройства или состояния в соответствии со способами, описанными выше, обычно будет находиться в диапазоне от приблизительно 0,01 до приблизительно 10,0 мг/кг массы тела пациента, подлежащего лечению. В качестве примера, соединение формулы I или его фармацевтически приемлемая соль могут быть введены для лечения, например, депрессии взрослому человеку со средней массой тела (приблизительно 70 кг) в дозе, находящейся в диапазоне от приблизительно 0,7 мг до приблизительно 700 мг в сутки, предпочтительно от приблизительно 1 до приблизительно 500 мг в сутки, в виде разовой или дробных (то есть множественных) доз. Изменения, основанные на вышеупомянутых диапазонах дозировок, могут быть сделаны врачом,обладающим обычной квалификацией, принимая во внимание такие факторы, как масса, возраст и состояние лица, подлежащего лечению, тяжесть заболевания и конкретный выбранный путь введения. Активность in vitro соединений по настоящему изобретению в отдельных сайтах обратного захвата моноамина может быть определена с использованием синаптосом крыс или клеток НЕК-293, трансфицированных переносчиком серотонина, дофамина или норэпинефрина человека, в соответствии со следующим способом, адаптированным из способа, описанного S. Snyder et al., (Molecular Pharmacology,1971, 7, 66-80), D.T. Wong et al., (Biochemical Pharmacology, 1973, 22, 311-322), H.F. Bradford (Journal ofNeurochemistry, 1969, 16, 675-684) и D.J.К. Balfour (European Journal of Pharmacology, 1973, 23, 19-26). Синаптосомы. Самцов крыс Sprague Dawley декапитируют и головной мозг быстро вынимают. Кору головного мозга, гиппокамп и полосатое тело вырезают и помещают в ледяной сахарозный буфер, 1 г в 20 мл буфера (буфер готовят, используя 320 мМ сахарозу, содержащую 1 мг/мл глюкозы, 0,1 мМ этилендиаминтетрауксусную кислоту (ЭДТА), доведенную до рН 7,4 основанием трис(гидроксиметил)аминометаном (TRIS. Ткани гомогенизируют в стеклянной гомогенизационной пробирке пестиком из Teflon при 350 об./мин,используя гомогенизатор Potters. Гомогенат центрифугируют при 1000 х g в течение 10 мин при 4 С. Получающуюся в результате надосадочную жидкость повторно центрифугируют при 17000 х g в течение 20 мин при 4 С. Конечный осадок ресуспендируют в подходящем объеме сахарозного буфера, что позволяет получить менее чем 10% выход. Приготовление клеток: клетки НЕК-293, трансфицированные переносчиком серотонина (5-ГТ), норэпинефрина (НЭ) или дофамина (ДА) человека, выращивают на среде Игла, модифицированной по способу Дульбекко (СИМД) (Life Technologies Inc., 9800 Medical Center Dr., Gaithersburg, MD, номер по каталогу 11995-065), дополненной 10% диализованной фетальной бычьей сывороткой (ФБС) (от Life Technologies, номер по каталогу 26300-053), 2 мМ L-глутамином и 250 мкг/мл G418 для переносчика 5-ГТ и НЭ или 2 мкг/мл пуромицина для переносчика ДА для давления отбора. Клетки растут в трехгорлых колбах Gibco, их собирают с помощью забуференного фосфатом физиологического раствора (Life Technologies, номер по каталогу 14190-136) и разбавляют подходящим количеством для того, чтобы выход составлял менее 10%. Анализ захвата нейромедиатора: анализы захвата проводят в стеклянных пробирках, содержащих 50 мкл растворителя, ингибитора или 10 мкМ сертралина, дезипрамина или номифензина для анализа неспецифического захвата 5-ГТ, НЭ или ДА, соответственно. В каждой пробирке содержится 400 мкл[3 Н]5-ГТ (конечная концентрация 5 нМ), [3 Н]НЭ (конечная концентрация 10 нМ) или [3 Н]ДА (конечная концентрация 5 нМ), приготовленные в модифицированном растворе Кребса, содержащем 100 мкМ паргилина и глюкозу (1 мг/мл). Пробирки помещают на лед и в каждую пробирку добавляют 50 мкл синаптосом или клеток. Пробирки затем инкубируют при 37 С в течение 7 мин (5-ГТ, ДА) или 10 мин (НЭ).-12 005671 Инкубацию останавливают путем фильтрации (фильтры GB/F), используя коллектор клеток Brandel на 96 лунок, фильтры промывают модифицированным буфером Кребса и подсчитывают, используя сцинтилляционный счетчик или Wallac Model 1214 или Wallac Beta Plate Model 1205. Определение активности ингибирования обратного захвата серотонина in vivo и силы действия соединений по настоящему изобретению может быть осуществлено путем измерения способности соединения блокировать утечку серотонина из передней части коры головного мозга крыс, вызванную (+/-)пара-хлорамфетамином (ПХА), в соответствии со способом, адаптированным из R.W. Fuller, H.D. Snoddyand M.L. Cohen в Neuropharmacology 23: 539-544 (1984). Как правило, самцов белых крыс Sprague-Dawley массой 160-230 г каждый распределяют или в контрольную (получают носитель), или тестируемую группу. Когда тестируемое соединение вводят подкожно (пк) в заданной дозе, его вводят вместе с 5 мг/кг пара-хлорамфетамина (ПХА). Через 3 ч после введения дозы животных умерщвляют путем декапитации и переднюю часть коры головного мозга удаляют, заключают в парафильм и замораживают в сухом льду (-78 С). Когда крысам вводят дозы перорально (по), крысам не дают есть в течение ночи перед экспериментом и затем обрабатывают тестируемым соединением в заданной дозе за 30 мин перед введением ПХА (5 мг/кг, пк). Через 3 ч животных умерщвляют и ткани удаляют, как изложено выше. Для определения уровней серотонина (5-ГТ) замороженные ткани гомогенизируют с помощью ультразвукового дезинтегратора Branson в 0,5 мл подвижной фазы в центрифужных пробирках Эппендорфа. Образцы затем центрифугируют при 11000 об./мин в течение двадцати минут в роторе Sorval SHMT в центрифуге Sorval RC5C. Полученную таким образом надосадочную жидкость отбирают пипеткой в пробирки для высокоэффективной жидкостной хроматографии (ВЭЖХ) и уровни 5-ГТ измеряют на приборе ВЭЖХ с электрохимическим детектором ВЭЖХ-ЭХ. Объяснение результатов следующее. В каждом эксперименте имеется группа животных, обработанных носителем, и группа животных, обработанных лишь ПХА. Среднее значение 5-ГТ у животных,обработанных ПХА, вычитают из среднего значения 5-ГТ у животных, обработанных носителем. Эта величина представляет собой сигнал или "окно" ответа. Определяют среднее значение 5-ГТ каждой тестируемой группы, из него вычитают среднее значение группы ПХА, и полученная величина, деленная на"окно", представляет собой процент защиты от воздействия ПХА для этой дозы. Для представления ID50 математически проводится линия через процентные величины защиты и рассчитывается 50-процентный уровень. Все указанные соединения формулы I в следующих примерах анализировали in vitro в отношении ингибирования обратного захвата серотонина, дофамина и норэпинефрина, и все они имели значенияIC50 приблизительно менее чем 250 нМ или равные ей в отношении ингибирования обратного захвата серотонина, приблизительно менее чем 1000 нМ или равные ей в отношении ингибирования обратного захвата дофамина и приблизительно менее чем 1000 нМ или равные ей в отношении ингибирования обратного захвата норэпинефрина. Примеры Получение 1. 5-Бром-2-(3,4-дихлорфенокси)бензальдегид. В атмосфере N2 в круглодонную колбу на 1 л, оснащенную дефлегматором и магнитной мешалкой помещали 51,1 г (370 ммоль) К 2 СО 3 и 20,1 г (123 ммоль) 3,4-дихлорфенола (Aldrich Chem. Co.,Milwaukee, WI) в 500 мл безводного N,N-диметилформамида (ДМФ). После перемешивания смеси в течение 30 мин добавляли 25 г (123 ммоль) 5-бром-2-фторбензальдегида (Aldrich) в 150 мл ДМФ и смесь нагревали до 90-100 С в течение ночи. После того, как реакционную смесь оставляли охлаждаться до комнатной температуры, ее концентрировали при пониженном давлении на роторном испарителе, и получающийся в результате маслянистый остаток затем разбавляли водой и EtOAc. Водный слой затем экстрагировали дополнительным количеством EtOAc и органические слои объединяли, промывали Н 2O и насыщенным NaCl и сушили над Na2SO4. Удаление растворителя в вакууме позволяло получить светложелтое масло, которое дополнительно сушили в вакууме в течение ночи с получением указанного в заголовке продукта в виде бледно-желтого твердого вещества, 40,2 г; т.пл. 129-132 С. 1(m, 1H), 6,94 (m, 1H). Масс-спектр (ГХМС, m/z): 346 (m+2), 344 (m+). Получение 2. 2-(3,4-Дихлорфенокси)-5-фенилбензальдегид. В атмосфере N2 в круглодонную колбу на 50 мл, оснащенную магнитной мешалкой, помещали следующие реактивы в следующем порядке: 15 мл толуола, 500 мг (1,4 ммоль) 5-бром-2-(3,4-13 005671 дихлорфенокси)бензальдегида (из получения 1), 341 мг (2,8 ммоль) фенилборной кислоты (Aldrich Chem.Co.), 1,5 мл этанола и 774 мг (5,6 ммоль) Nа 2 СО 3 в 3 мл воды. К ним добавляли 45 мг (0,04 ммоль) тетракис(трифенилфосфин)палладия(0) (Aldrich Chem. Co.) и смесь дегазировали с использованием N2. Реакционную смесь затем нагревали до температуры дефлегмации в течение 4 ч, по истечении которых тонкослойная хроматография (ТСХ) с использованием смеси 1:1 СН 2 Сl2:гексан на пластинках, покрытых силикагелем, показала отсутствие исходного альдегида. После охлаждения эту смесь разбавляли 100 мл ЕtOАс, дважды промывали водой, дважды 2 н. NaOH, дважды водой, и наконец, насыщенным воднымNaCl. После сушки над МgSO4 растворитель удаляли в вакууме с получением маслянистого остатка, 690 мг. Его подвергали хроматографии на силикагеле, элюируя смесью СН 2 Сl2:гексан (1:1) с получением указанного в заголовке продукта в виде масла, 462 мг. 1(m, 1H), 7,20 (m, 1H), 7,00 (dd, 1H), 6,96 (m, 1H). Масс-спектр (ГХМС, m/z): 344 (m+2), 342 (m+). Таким же образом были получены 4- или 5-замещенные 2-(3,4-дихлорфенокси)бензальдегиды: Получение 20. 5-(2-Пиридил)бензальдегид. В атмосфере N2 в высушенную пламенем круглодонную колбу на 25 мл, оборудованную магнитной мешалкой, помещали 200 мг (0,58 ммоль) 5-бром-2-(3,4-дихлорфенокси)бензальдегида, 162 мг (0,64 ммоль) бис(пинаколато)диборана (Frontier Scientific Co.), 170 мг (1,7 ммоль) ацетата калия и 13 мг (0,018 ммоль) дихлорметанового аддукта дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия(II)(РdСl2(дффф (Strem Chemicals) в 5 мл безводного ДМФ. Смесь дегазировали с использованием N2 в течение 5 мин и затем нагревали при 80 С в течение 2,5 ч. К ней добавляли 110 мкл (1,2 ммоль) 2 бромпиридина с последующими 13 мг РdCl2(дффф) и 0,7 мл 2 н. водного Nа 2 СО 3. Смесь снова нагревали до 80 С в атмосфере N2 в течение 10,5 ч, затем оставляли охлаждаться до комнатной температуры в течение ночи. Смесь затем распределяли между ЕtOАс и H2O, органический слой промывали водой, рассолом и сушили над Na2CO3, затем концентрировали в вакууме до масла, 359 мг. Хроматография на силикагеле с элюцией градиентной системой СНСl3 (100-97%) и СН 3 ОН (0-3%) позволяла получить указанный в заголовке продукт в виде светло-коричневого масла, 44 мг. Масс спектр (ГХМС, m/z): 346 (m+2), 344 (m+). Получение 21. 5-Циано-2-(3,4-дихлорфенокси)бензальдегид. В атмосфере N2 в высушенной пламенем трехгорлой круглодонной колбе, оснащенной дефлегматором и магнитной мешалкой, при комнатной температуре при дегазировании с использованием N2 в течение 5 мин перемешивали смесь 5-бром-2-(3,4-дихлорфенокси)бензальдегида (3,0 г, 8,7 ммоль), цианида цинка(II) (1,5 г, 13 ммоль) и тетракис(трифенилфосфин)палладия(0) (1,5 г, 1,3 ммоль) в безводном ДМФ(145 мл). После нагревания при приблизительно 80 С в течение 90 мин о завершении реакции судили по тонкослойной хроматографии (1:1 СН 2 Сl2:гексаны) и реакционную смесь оставляли охлаждаться до комнатной температуры. Реакционную смесь затем разбавляли водой и этилацетатом и перемешивали в течение дополнительных 10 мин. Водный слой отделяли, дважды экстрагировали ЕtOАс и объединяли с исходным органическим слоем, и промывали водным раствором сегнетовой соли (тетрагидрат тартрата калия-натрия) с последующим водным NaCl. Органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме до масла. Это масло подвергали флэш-хроматографии на колонке с силикагелем 5 х 15 см (230-400 меш), элюируя смесью СН 2 Сl2:гексаны (1:1) с получением указанного в заголовке продукта в виде белого твердого вещества, 1,5 г (60%), т.пл. 122-126 С. Масс-спектр (ГХ/МС, m/z): 291 (m+), 262. 1H-ЯМР (CDCl3):10,47 (s, 1 Н), 8,22 (d, 1H), 7,75 (dd, 1 Н), 7,53 (d, 1H), 7,25 (m, 1H), 6,98 (dd, 1H),6,92 (d, 1H). Таким же образом был получен 4-циано-2-(3,4-дихлорфенокси)бензальдегид из соответствующего 4-бром-2-(3,4-дихлорфенокси)бензальдегида в виде прозрачного масла, 16%. Масс-спектр (ГХ/МС, m/z): 291 (m+). 1 Н-ЯМР (CDCl3):10,45 (s, 1H), 8,02 (d, 1H), 7,55 (m, 2H), 7,23 (m, 1H), 7,14 (m, 1H), 6,96 (dd, 1H).-17 005671 Пример 1. 2-(3,4-Дихлорфенокси)-5-Фенил-N-метилбензиламин. В круглодонную колбу, оснащенную магнитной мешалкой и входом для N2, помещали 1,34 мл (2,68 ммоль) метиламина (2,0 М раствор в метаноле, Aldrich Chemical Co.) в 8,0 мл этанола при перемешивании до тех пор, пока раствор не становился прозрачным. При комнатной температуре через шприц добавляли 0,8 мл (2,68 ммоль) изопропоксида титана (IV) с последующими 0,460 г (1,34 ммоль) 2-(3,4 дихлорфенокси)-5-фенилбензальдегида в 15 мл ЕtOН, которые затем перемешивали в течение ночи. К полученному в результате раствору добавляли 0,076 г (2,01 ммоль) борогидрида натрия и перемешивание продолжали в течение дополнительных 24 ч. Реакционную смесь затем гасили приблизительно 3 мл 6 н. HCl и 10 мл воды, рН доводили до 10,0 насыщенным водным Nа 2 СО 3 и перемешивали в течение дополнительных 2 ч перед экстракцией EtOAc. Слой ЕtOАс объединяли с дополнительными экстрактами водного слоя, и объединенные органические слои промывали насыщенным водным NaCl, сушили с использованием Na2SO4 и концентрировали в вакууме до масла, 0,47 г. Масс-спектр (химическая ионизация при атмосферном давлении, m/z): 357 (m+). 1(m, 1H), 6,82 (d, 1H), 4,37 (m, 2H), 2,30 (m, 3H). Это масло, растворенное в безводном ЕtOАс, обрабатывали 1,3 мл 1 н. HCl в Et2O и затем перемешивали при комнатной температуре, полученное в результате твердое вещество (0,276 мг) фильтровали и промывали Et2O и сушили в вакууме, т.пл. 170-173 С. Элементарный анализ для C16H14Cl2F3NOHCl1/4H2O: рассчитано: С, 60,17, Н, 4,67, N, 3,51. Обнаружено: С 60,17, Н, 4,36, N, 3,42. Таким же образом были получены следующие соединения формулы I: Пример 15. 5-Бром-2-(3,4-дихлорфенокси)-N-метилбензиламин. В атмосфере N2 раствор метиламина (2,9 мл, 5,8 ммоль, 2,0 М раствор в СН 3 ОН) в 20 мл этанола обрабатывали изопропоксидом титана (IV) (1,7 мл, 5,8 ммоль) при комнатной температуре. Через 5 мин добавляли суспензию 5-бром-2-(3,4-дихлорфенокси)бензальдегида (1,0 г, 2,9 ммоль, указаное в заголовке соединение из получения 1) в 20 мл этанола и перемешивали в течение 16 ч при комнатной температуре. Затем добавляли борогидрид натрия (0,165 г, 4,4 ммоль) и перемешивание продолжали в течение дополнительных 24 ч, по истечении которых реакцию гасили путем добавления 5 мл 6 н. HCl и 5 мл воды, перемешивали в течение 30 мин и подщелачивали путем добавления насыщенного водного Na2CO3. Полученную в результате смесь экстрагировали ЕtOАс и органические экстракты очищали путем фильтрации через диатомовую землю (д.з.), промывали насыщенным NaCl, сушили над Na2CO3 и концентрировали в вакууме до получения прозрачного масла, 0,987 мг. Пример 16. Дигидрохлорид 1-[4-(3,4-дихлорфенокси)-3-метиламинометилфенил]-1 Н-пиразол-3 иламина. В атмосфере N2 в высушенную пламенем круглодонную колбу на 15 мл, оснащенную магнитной мешалкой, помещали 318 мг (0,88 ммоль) 5-бром-2-(3,4-дихлорфенокси)-N-метилбензиламина (указанное в заголовке соединение из примера 15), 1,50 г (18 ммоль) 3-аминопиразола, 56 мг (0,88 ммоль) медного порошка и 122 мг (0,88 ммоль) карбоната калия. Эту смесь нагревали до 130 С в течение, в общей сложности, 1 ч, охлаждали и перемешивали при комнатной температуре в течение ночи. Дегтеобразный остаток распределяли между ЕtOАс и разбавленным водным раствором ЭДТА (этилендиаминтетрауксусной кислоты), органический слой промывали водой и насыщенным водным NaCl и затем сушили надNа 2SO4. После фильтрации растворитель удаляли в вакууме с получением масла в количестве 287 мг,которое элюировали на силикагеле градиентной системой NН 4OН:СН 3 ОН:СНСl3 (от 2:2:96 до 2:10:88). Фракции продукта концентрировали до масла (110 мг), которое растворяли в 25 мл ЕtOАс и обрабатывали 0,6 мл 1 н. HCl в Et2O. Твердое вещество, которое осаждалось, фильтровали, промывали небольшим количеством Et2O и сушили в вакууме с получением 60 мг указанного в заголовке продукта, т.пл. 225233 С. 1(d, 1H), 7,10 (dd, 1H), 6,33 (s, 1H), 4,17 (s, 2H), 2,56 (s, 3 Н). Масс-спектр (химическая ионизация при атмосферном давлении, m/z): 363 (m+), 365. Элементарный анализ: рассчитано для С 17 Н 16 Сl2N4O2 НСl1/3 Н 2 О: С, 46,18, Н, 4,26, N, 12,67. Обнаружено: С, 46,37, Н, 4,30, N, 12,30.-19 005671 Пример 17. Гидрохлорид [2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-1-ил-бензил]метиламина и гидрохлорид [2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-2-ил-бензил]метиламина. Смесь 390 мг (1,08 ммоль) 5-бром-2-(3,4-дихлорфенокси)-N-метилбензиламина, 1,8 г (26 ммоль) 1,2,3-триазола, 69 мг (1,08 ммоль) медного порошка и 149 мг (1,08 ммоль) карбоната калия нагревали в атмосфере N2 при 160 С в течение ночи, затем оставляли охлаждаться до комнатной температуры. Смесь распределяли между ЕtOАс и разбавленным водным раствором ЭДТА, органический слой отделяли,промывали водой, насыщенным водным NaCl и сушили над Na2SO4. Концентрирование в вакууме позволяло получить 1,25 г масла, которое подвергали хроматографии на силикагеле, элюируя градиентной системой, начиная с СНСl3 и заканчивая смесью 2:10:88 триэтиламин:СН 3 ОН:СНСl3. Были выделены два основных новых продукта. Первый продукт с Rf=0,54 (2:10:98 - NН 4OН:СН 3 ОН:СНСl3) превращали в соль гидрохлорид [2-(3,4 дихлорфенокси)-5-[1,2,3]триазол-2-илбензил]метиламина, 52 мг, т.пл. 235-238 С. 1H-ЯМР (ДМСО-d6, 400 МГц, соль гидрохлорид):9,14 (bs, 2H), 8,32 (d, 1H), 8,13 (s, 2H), 8,01 (dd,1 Н), 7,70 (d, 1 Н), 7,50 (d, 1 Н), 7,17 (dd, 1H), 7,10 (d, 1 Н), 4,25 (t, 2H), 2,59 (t, 3H). Масс-спектр (химическая ионизация при атмосферном давлении, m/z): 349 (m+), 351. Элементарный анализ: рассчитано для C16H14Cl2N4OHCl: С, 49,83, Н, 3,92, N, 14,53. Обнаружено: С, 49,81, Н, 3,69, N, 14,41. Второй продукт с Rf=0,25 превращали в гидрохлорид [2-(3,4-дихлорфенокси)-5-[1,2,3]триазол-1 илбензил]метиламина, т.пл. 180-185 С. 1H-ЯМР (ДМСО-d6, 400 МГц, соль гидрохлорид):9,26 (bs, 2H), 8,78 (d, 1H), 8,31 (d, 1H), 7,98 (d,1H), 7,89 (dd, 1H), 7,70 (d, 1H), 7,50 (d, 1H), 7,18 (dd, 1H), 7,14 (d, 1H), 4,24 (s, 2H), 2,60 (s, 3H). Масс-спектр (химическая ионизация при атмосферном давлении, m/z): 349 (m+), 351. Элементарный анализ: рассчитано для С 16 Н 14 Сl2N4OНСl0,75 Н 2 О: С, 47,86, Н, 4,17 N, 15,03. Обнаружено: С, 47,90, Н, 3,72, N, 15,26. Пример 18. Гидрохлорид 1-[4-(3,4-дихлорфенокси)-3-(1-метиламиноэтил)фенил]пирролидин-2-она. А. 5-Нитро-2-(3,4-дихлорфенокси)ацетофенон. В атмосфере N2 объединяли смесь 2-фтор-5-нитроацетофенона (1,24 г, 6,77 ммоль, полученного в соответствии со способом, обнаруженным в J. Med. Chem., 1991, 28(3), 673-683), 3,4-дихлорфенола (1,15 г, 7,1 ммоль), К 2 СО 3 (2,8 г, 20,3 ммоль) и 15 мл ДМФ и перемешивали при комнатной температуре в течение 1 ч. В этот момент времени ТСХ (40% ЕtOАс:60% гексаны) показала, что реакция завершилась. Реакционную смесь гасили 50 мл воды и экстрагировали ЕtOАс. Органические экстракты несколько раз промывали водой и водным NaCl и сушили над Na2SO4. После фильтрации растворитель удаляли в вакууме с получением 2,14 г желтого твердого вещества, которое очищали посредством флэшхроматографии, элюируя 10% ЕtOАс в гексанах. Продукт (2,02 г (92% представлял собой белое твердое вещество, т.пл. 118-126 С. Масс-спектр (М+): 325, 327. Б. 1-[2-(3-Дихлорфенокси)-5-нитрофенил]этилметиламин. Смесь ацетофенона, полученного на стадии А (2,0 г, 6,1 ммоль), и 2,0 М метиламина в метаноле (6,1 мл, 12,2 ммоль) в 25 мл этанола перемешивали в течение ночи при 25 С. Добавляли изопропоксид титана(IV) (3,6 мл, 12,2 ммоль) и смесь перемешивали в течение дополнительных 24 ч. Затем добавляли борогидрид натрия (0,346 г, 9,4 ммоль) и перемешивание продолжали в течение дополнительных 24 ч, по истечении которых ТСХ (тонкослойная хроматография) (10% метанол:хлороформ) показала, что реакция завершилась. Реакционную смесь гасили путем добавления 5 мл 6 н. HCl, перемешивания в течение 20 мин и затем добавления водного NаНСО 3 до тех пор, пока рН не стал щелочным. Эту смесь экстрагировали ЕtOАс и объединенные экстракты промывали Н 2 О, сушили над Na2SO4, фильтровали и концентрировали до 1,7 г бесцветного масла. Это масло подвергали флэш-хроматографии с использованием 2% МеОН в СНСl3 и очищенный продукт выделяли в виде масла, 1,37 г. В. тpeт-Бутиловый эфир 1-[2-(3,4-дихлорфенокси)-5-нитрофенил]этилметилкарбаминовой кислоты. Раствор амина, полученного на стадии Б (1,36 г, 4 ммоль), в 20 мл CH2Cl2 перемешивали с ди-третбутилдикарбонатом (БОК ангидрид, 0,96 г, 4,4 ммоль) и триэтиламином (1,2 мл, 8,6 момль) при комнатной температуре в течение ночи. Удаление растворителя в вакууме позволило получить желтое масло,2,04 г, которое очищали с использованием флэш-хроматографии (15% ЕtOАс:гексаны) с получением 1,6 г (94%) желаемого промежуточного нитросоединения в виде бледно-желтого масла. Г. трет-Бутиловый эфир 1-[5-амино-2-(3,4-дихлорфенокси)фенил]этилметилкарбаминовой кислоты. Нитросоединение, полученное на стадии В (0,839 г), в 20 мл этанола обрабатывали 120 мг 10% Pd на углероде в атмосфере N2 и затем гидрировали на шейкере Парра при давлении 50 фунт/кв.дюйм(344,75 кПа) в течение 25 мин. Реакционную смесь затем фильтровали, например, через д.з., фильтровальный осадок промывали CH2Cl2. Объединенные фильтраты концентрировали в вакууме с получением 1,3 г бесцветного масла, которое подвергали флэш-хроматографии, элюируя 40% смесью ЕtOАс:гексаны.-20 005671 Концентрирование фракций элюанта позволило получить 0,62 г указанного в заголовке промежуточного амино-соединения данной стадии в виде пены. Д. трет-Бутиловый эфир 1-[2-(3,4-дихлорфенокси)-5-(2-оксо-пирролидин-1-ил)фенил]этилметилкарбаминовой кислоты. Указанное в заголовке соединение стадии Г выше (0,615 г, 1,5 ммоль) в 20 мл безводного ТГФ объединяли с карбонатом цезия (1,0 г, 3,1 ммоль) и перемешивали в атмосфере N2 при комнатной температуре, добавляя с помощью шприца 4-хлорбутирилхлорид (0,17 мл, 1,5 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 24 ч, охлаждали до комнатной температуры и распределяли между ЕtOАс и водой. Органический слой сушили над Na2SO4, концентрировали в вакууме с получением 740 мг твердого вещества. Это твердое вещество подвергали флэш-хроматографии, элюируя 40% ЕtOАс в гексанах с получением двух основных фракций. Менее полярную фракцию, представляющую собой 250 мг бесцветного масла, идентифицировали как нециклическое промежуточное соединение, основываясь на его 1H-ЯМР спектре. Более полярную фракцию, представляющую собой 558 мг белого твердого вещества, идентифицировали как пактам, защищенный ВОС. 1E. Гидрохлорид 1-[4-(3,4-дихлорфенокси)-3-(1-метиламиноэтил)фенил]пирролидин-2-она. Указанную более полярную фракцию из предыдущей стадии Д растворяли в 20 мл EtOAc, охлаждали на льду и ацетоновой бане и насыщали газом НСl приблизительно в течение 5 мин, затем оставляли нагреваться до комнатной температуры в течение ночи. Растворитель затем удаляли в вакууме и остаток растирали с Et2O с образованием белых твердых веществ, которые фильтровали и сушили в вакууме с получением 429 мг указанной соли гидрохлорида в виде белого твердого вещества, т.пл. 195-200 С. Элементарный анализ: рассчитано для C19H20Cl2N2O2HCl: С, 54,89; Н, 5,09; N, 6,74. Обнаружено: С, 54,86; Н, 5,40; N, 6,94. Получение 22. [2-(3,4-Дихлорфенокси)-5-нитробензил]метиламин. Указанное в заголовке соединение получали в виде масла таким же образом, как указанное в заголовке соединение из примера 18, стадия Б. Масс-спектр (М+): 326, 328. 1H-ЯМР (CDCl3, ) 8,36 (d, 1H), 8,08 (dd, 1H), 7,46 (d, 1H), 7,15 (d, 1H), 6,90 (dd, 1 Н), 6,85 (d, 1 Н),3,87 (s, 2H), 2,48 (s, 3 Н). Получение 23. тpeт-Бутиловый эфир [2-(3,4-дихлорфенокси)-5-нитробензил]метилкарбаминовой кислоты. Указанное в заголовке соединение получали в виде белого твердого вещества таким же образом,как указанное в заголовке соединение из примера 18, стадия В. Т.пл. 102-108 С. Получение 24. трет-Бутиловый эфир [5-амино-2-(3,4-дихлорфенокси)бензил]метилкарбаминовой кислоты. Указанное в заголовке соединение получали в виде бесцветного масла таким же образом, как указанное в заголовке соединение из примера 18, стадия Г. Получения 25 и 26. Следующие промежуточные соединения получали таким же образом, как указанное в заголовке соединение из примера 18, cтадия Д. трет-Бутиловый эфир [2-(3,4-дихлорфенокси)-5-(2-оксопиперидин-1-ил)бензил]метилкарбаминовой кислоты. Бесцветное масло, 1,82 г (76%). тpeт-Бутиловый эфир [2-(3,4-дихлорфенокси)-5-(2-оксопирролидин-1-ил)бензил]метилкарбаминовой кислоты. Бесцветное масло, 0,867 г (98%). 1H-ЯМР (CDCl3, ) 7,65 (dd, 1 Н), 7,41 (d, 1 Н), 7,33 (d, 1H), 6,96 (d, 1H), 6,92 (d, 1H), 6,75 (dd, 1H),4,39 (bs, 2H), 3,83 (t, 2 Н), 2,82 (d, 3H), 2,60 (t, 2H), 2,16 (m, 2H), 1,43 (s,9H). Следующее соединение получали таким же образом, как соединение со стадии Е из примера 18. Пример 19. 1-[4-(3,4-Дихлорфенокси)-3-(метиламинометил)фенил]пирролидин-2-он. Т.пл. 166-170 С. Элементарный анализ: рассчитано для C18H18Cl2N2O2HCl: С, 53,82; Н, 4,77; N, 6,97. Обнаружено: С,54,03; Н, 4,80; N, 6,88. Пример 20. Гидрохлорид 1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пиперидин-2-она. Т.пл. 191-196 С. 1 где n и m независимо выбраны из единицы и двух;R1 и R2 независимо выбраны из водорода и (С 1-С 4)алкила;R3 и R4 независимо выбраны из водорода и (С 1-С 4)алкила; каждый Х независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, где каждый Х может быть дополнительно замещен водородом,галогено, (C1-С 4)алкилом, оксо, амино и группировкой NR5(С=O)(С 1-C4)алкил, где R5 представляет собой водород; каждый Y независимо выбран из водорода и галогено и каждый Z независимо выбран из водорода и галогено; или его фармацевтически приемлемая соль. 2. Соединение или соль по п.1, где Х выбран из фурана, тиофена, пиридина, 1,2,3-триазола и 1 пирролидин-2-она и где Х может быть дополнительно замещен. 3. Соединение или соль по п.1, где один или оба R3 и R4 представляют собой водород. 4. Соединение по п.1, выбранное из группы, состоящей из[2-(3,4-дихлорфенокси)-5-[1,2,4]триазол-4-илбензил]метиламина; 1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пирролидин-2-она; 1-[4-(3,4-дихлорфенокси)-3-(1-метиламиноэтил)фенил]пирролидин-2-она; 1-[4-(3,4-дихлорфенокси)-3-(метиламинометил)фенил]пиперидин-2-она и их фармацевтически приемлемых солей. 5. Фармацевтическая композиция, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. где n и m независимо выбраны из единицы и двух; Х выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, О и S, где Х может быть дополнительно замещен водородом, галогено, (С 1-С 4)алкилом, (С 1 С 4)алкокси, оксо и группировкой NR5(C=O)(C1-C4)aлкил, где R5 представляет собой водород; каждый Y независимо выбран из водорода и галогено; каждый Z независимо выбран из водорода и галогено;
МПК / Метки
МПК: C07D 333/20, A61P 25/24, A61K 31/13, C07C 217/58
Метки: биариловых, полезные, эфиров, моноамина, качестве, производные, ингибиторов, обратного, захвата
Код ссылки
<a href="https://eas.patents.su/24-5671-proizvodnye-biarilovyh-efirov-poleznye-v-kachestve-ingibitorov-obratnogo-zahvata-monoamina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные биариловых эфиров, полезные в качестве ингибиторов обратного захвата моноамина</a>
Производные бензо[3,4] циклобута [1,2-c] пиррола и их применение в качестве ингибиторов обратного захвата серотонина

Номер патента: 3302
Опубликовано: 24.04.2003
Авторы: Декейн Анн, Гумен Бертран, Дессанж Эме, Риве Жан-Мишель, Миллан Марк, Пеглион Жан-Луи
МПК: C07D 209/58, A61P 25/24, A61K 31/403...
Метки: обратного, циклобута, серотонина, бензо[3,4, пиррола, производные, захвата, 1,2-c, применение, ингибиторов, качестве
Формула / Реферат:
1. Соединение формулы (I), имеющее цис-соединение колец в которой R1, R2 и R3, которые могут быть одинаковыми или различными, каждый независимо от других, представляет группу, выбранную из атома водорода, атома галогена, линейной или разветвленной (C1-C6)алкоксигруппы, линейной или разветвленной (C1-C6)тригалогеналкильной группы, или два из R1, R2 и R3 в смежных положениях представляют (C1-C2)алкилендиоксигруппу; R4 представляет группу,...
Производные 6-фенилпиридил-2-амина, полезные в качестве ингибиторов nos

Номер патента: 2907
Опубликовано: 31.10.2002
Автор: Лоу Джон Адамс III
МПК: C07D 213/73, A61P 11/06, A61K 31/4418...
Метки: ингибиторов, производные, 6-фенилпиридил-2-амина, качестве, полезные
Формула / Реферат:
1. Соединение формулы где R1 и R2 независимо выбраны из водорода, гидрокси, метила и метокси; и G является группой формулы где n равно нулю или единице; Y представляет собой NR3R4, (С1-С6)алкил или аралкил, где арильная группировка указанного аралкила является фенилом или нафтилом, а алкильная группировка является нормальной или разветвленной и содержит от 1 до 6 атомов углерода и где указанный (С1-С6)алкил и арильная группировка указанного...
Никотинамидные бензоконденсированные гетероциклические производные, полезные в качестве селективных ингибиторов pde4-изозимов

Номер патента: 4885
Опубликовано: 26.08.2004
Авторы: Чэмбер Роберт Джеймс, Марфэт Энтони
МПК: A61K 31/665, C07D 413/12, C07D 417/12...
Метки: полезные, качестве, селективных, производные, pde4-изозимов, бензоконденсированные, гетероциклические, ингибиторов, никотинамидные
Формула / Реферат:
1. Соединение формулы (1.0.0) где m равен 1; n равен 1; RA и RB представляют собой -H, -CF3 или -(C1-C6)алкил, замещенный -F, -Cl, -CF3, -CN, -NH2 или -C(=O)NH2 в количестве 0 или 1, или оба RA и RB, взятые вместе, представляют собой спиро-(C3-C6)циклоалкил, замещенный -F, -Cl, -CF3 или -CN в количестве 0 или 1; один из RC и RD представляет собой -H, а другой представляет собой -H, -(C1-C4)алкил или фенил, каждый замещенный -F, -Cl или -CN в...
Применение селективных ингибиторов обратного захвата серотонина быстрого запуска для лечения половой дисфункции

Номер патента: 4101
Опубликовано: 25.12.2003
Автор: Тор Карл Брюс
МПК: A61P 15/00, A61K 31/138
Метки: дисфункции, захвата, ингибиторов, запуска, быстрого, обратного, селективных, серотонина, лечения, применение, половой
Формула / Реферат:
1. Применение селективного ингибитора обратного захвата серотонина быстрого запуска или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения или регуляции половой дисфункции у млекопитающего, где лекарственное средство адаптировано к введению в случае необходимости. 2. Применение по п.1, где селективный ингибитор обратного захвата серотонина быстрого запуска является краткодействующим. 3. Применение по п.1 или...
4,4-дизамещенные 3,4-дигидро-2(1н)-хиназолиноны, полезные в качестве ингибиторов обратной транскриптазы вич

Номер патента: 1991
Опубликовано: 22.10.2001
Авторы: Корбетт Джеффри В., Ко Соо Сунг
МПК: A61P 31/18, A61K 31/517, C07D 239/80...
Метки: 4,4-дизамещенные, полезные, вич, обратной, 3,4-дигидро-2(1н)-хиназолиноны, транскриптазы, ингибиторов, качестве
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомер, или фармацевтически приемлемая соль, где R1 обозначает C1-3-алкил, замещенный 1-7 атомами галогена; R2 выбран из C1-5-алкила, замещенного 1-2 R4, С2-5-алкенила, замещенного 1-2 R4, и С2-5-алкинила, замещенного одним R4; R3 каждый независимо выбран из C1-4-алкила, ОН, C1-4-алкокси, F, Cl, Br, I, NR5R5a, NO2, CN, C(O)R6, NHC(O)R7 и NHC(О)NR5R5a; альтернативно, если присутствуют два заместителя R3...
Предыдущий патент: Конденсатор с электрическим двойным слоем
Следующий патент: Хемосенсибилизатор
Случайный патент: Устройство и способ записи и считывания оптической информации (варианты)