Производные аминотиазола и их применение в качестве лигандов рецепторов крф
Номер патента: 5159
Опубликовано: 30.12.2004
Авторы: Галли Даниэль, Роже Пьер, Прадине Антуан, Фонтен Эвелин, Жеслен Мишель







Формула / Реферат
1. Соединения, в рацемической форме или в форме чистого энантиомера, формулы

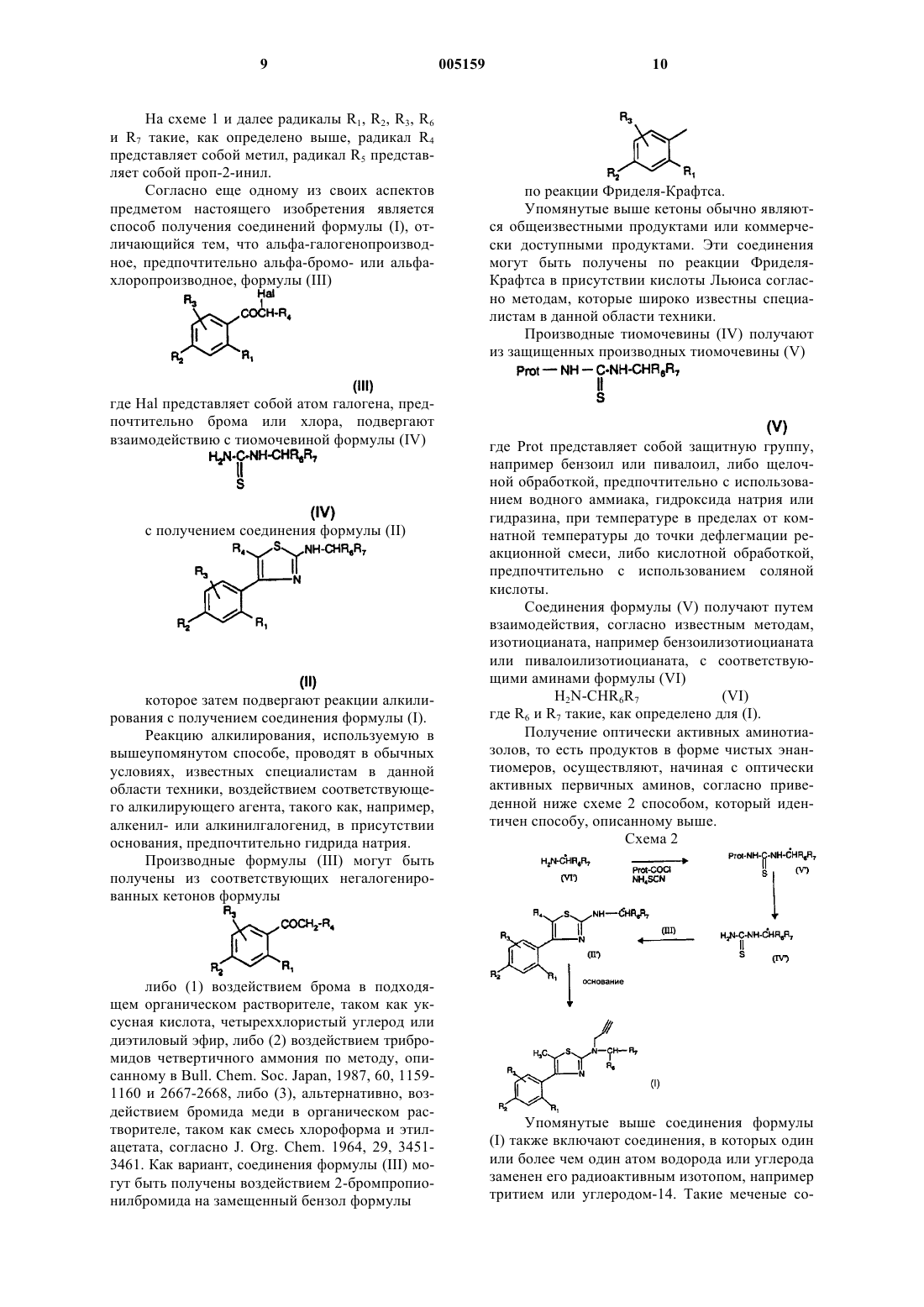
где R1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; гидрокси(C1-C5)алкил; (C1-C5)алкил; аралкил, в котором арильная часть представляет собой (C6-C8), а алкильная часть представляет собой (C1-C4); (C1-C5)алкокси; трифторметильную группу; нитрогруппу; нитрильную группу; группу -SR, где R представляет собой водород, (C1-C5)алкил или аралкил, в котором арильная часть представляет собой (C6-C8), а алкильная часть представляет собой (C1-C4); группу -S-CO-R, где R представляет собой (C1-C5)алкильный или аралкильный радикал, в котором арильная часть представляет собой (C6-C8), а алкильная часть представляет собой (C1-C4); группу -COORa, где Ra представляет собой водород или (C1-C5)алкил; группу -CONRaRb с Ra и Rb такими, как определено выше для Ra; группу -NRaRb с Ra и Rb такими, как определено выше для Ra; группу -CONRcRd или -NRcRd, где Rс и Rd составляют с атомом азота, к которому они присоединены, 5-7-членный гетероцикл; или группу -NHCO-NRaRb с Ra и Rb такими, как определено выше для Ra;
R3 представляет собой водород или является таким, как определено выше для R1 и R2;
или, альтернативно, R2 составляет с R3, когда последний является заместителем фенила в положении 5, группу -X-CH2-X-, где X независимо представляет собой CH2 или атом кислорода или серы;
R6 представляет собой (C1-C6)алкил; (C1-C6)алкокси(C1-C3)алкил; (C3-C5)циклоалкил; (C3-C6)циклоалкил(C1-C6)алкил; (C1-C6)алкилтио(C1-C3)алкил; (C1-C6)алкилсульфокси(C1-C3)алкил; (C1-C6)алкилсульфодиокси(C1-C3)алкил;
R7 представляет собой фенил, который не замещен, моно-, ди- или тризамещен в положении 3, 4 или 5 галогеном, (C1-C5)алкилом, группой -O-CH2-O- по двум соседним атомам углерода фенила, группой -CF3, -NO2 или -CN, группой -COOR8 или -CONR8R9, или группой -CH2OR8, где R8 и R9 представляют собой (C1-C3)алкил, OR10, где R10 представляет собой (C1-C5)алкил; или, альтернативно, R7 представляет собой пиридильную, тиофеновую, пиразолильную, имидазолильную, (C3-C5)циклоалкильную или (C3-C6)циклоалкил(C1-C6)алкильную группу;
их соли присоединения, их гидраты и/или их сольваты.
2. Соединения по п.1, отличающиеся тем, что
R1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; (C1-C5)алкил; (C1-C5)алкокси;
R3 представляет собой водород или является таким, как определено выше для R1 и R2;
R6 представляет собой (C1-C6)алкил; (C1-C6)алкокси(C1-C3)алкил; (C3-C5)циклоалкил; (C3-C6)циклоалкил(C1-C6)алкил;
R7 представляет собой фенил, который не замещен или моно- или дизамещен в положении 3 или 4 галогеном, (C1-C5)алкильной группой, группой -CH2OR8, где R8 представляет собой (C1-C3)алкил, или группой -O-CH2-O- в положении 3, 4; или, альтернативно, R7 представляет собой (C3-C5)циклоалкильную группу.
3. Соединения по п.1, отличающиеся тем, что R3 находится в положении 5 фенила.
4. Соединения по п.1, выбранные из
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1R)-(1-(3-фтор-4-метилфенил)-2-метоксиэтил)]проп-2-иниламина гидрохлорида (пример 31);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-фенилбутил)]проп-2-иниламина гидрохлорида (пример 33);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-фенилэтил)]проп-2-иниламина гидрохлорида (пример 34);
[4-(2-хлор-4-метоксифенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-фенилэтил)]проп-2-иниламина гидрохлорида (пример 35);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(4-фторфенил)этил)]проп-2-иниламина гидрохлорида (пример 36);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-фенилпентил)]проп-2-иниламина гидрохлорида (пример 37);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1R)-(2-метокси-1-(4-метоксиметилфенил)этил)]проп-2-иниламина гидрохлорида (пример 40);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(4-метоксиметилфенил)пентил)]проп-2-иниламина гидрохлорида (пример 42);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(4-фторфенил)пентил)]проп-2-иниламина гидрохлорида (пример 45);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)(циклопропилфенилметил)]проп-2-иниламина гидрохлорида (пример 47);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(3-фтор-4-метилфенил)пентил)]проп-2-иниламина гидрохлорида (пример 49);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(3-фтор-4-метилфенил)этил)]проп-2-иниламина гидрохлорида (пример 50);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(4-фторфенил)бутил)]проп-2-иниламина гидрохлорида (пример 51);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(3-фтор-4-метоксиметилфенил)бутил)]проп-2-иниламина гидрохлорида (пример 52);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(4-хлорфенил)этил)]проп-2-иниламина гидрохлорида (пример 53);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(4-метилфенил)этил)]проп-2-иниламина гидрохлорида (пример 54);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклобутил-1-(4-фторфенил)этил)]проп-2-иниламина гидрохлорида (пример 55);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(4-бромфенил)этил)]проп-2-иниламина гидрохлорида (пример 56);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(3,4-метилендиоксифенил)этил)]проп-2-иниламина гидрохлорида (пример 57);
[4-(2-хлор-4-метоксифенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(3-фтор-4-метилфенил)этил)]проп-2-иниламина гидрохлорида (пример 58);
[4-(2,4-диметокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(3-фтор-4-метилфенил)этил)]проп-2-иниламина гидрохлорида (пример 59);
[4-(4-метокси-2,5-диметилфенил)-5-метилтиазол-2-ил][(1S)-(2-циклопропил-1-(3-фтор-4-метилфенил)этил)]проп-2-иниламина гидрохлорида (пример 60);
[4-(2-хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил][(1S)-(1-(3,4-метилендиоксифенил)бутил)]проп-2-иниламина гидрохлорида (пример 61),
а также их соответствующих оснований, других солей присоединения и сольватов и/или гидратов.
5. Способ получения соединений формулы (I) по п.1, отличающийся тем, что эти соединения получают путем алкилирования соединений формулы

где R4 представляет собой метил, а R1, R2, R3, R6 и R7 такие, как определено для (I).
6. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение по п.1 в качестве активного начала.
7. Применение соединения по п.1 для приготовления лекарственных продуктов, предназначенных для предупреждения и/или лечения кортикотропин-рилизинг-фактор (КРФ)-зависимых состояний.
8. Применение соединения по п.1 для приготовления лекарственного продукта, предназначенного для превентивного или куративного лечения патологии, в которую вовлечен КРФ, выбранной из связанных со стрессом состояний, болезни Кушинга, нервно-психиатрических расстройств, таких как депрессивные, тревожные, панические, обсессивно-компульсивные расстройства, расстройства настроения, посттравматический стресс, расстройства поведения, агрессивность, анорексия, булимия, гипергликемия, преждевременные роды, беременность с повышенным риском, замедленное развитие, нарушения сна, эпилепсия и все типы депрессии; болезни Альцгеймера, болезни Паркинсона, хореи Гентингтона; бокового амиотрофического склероза; сосудистых, сердечных и церебральных расстройств; расстройств сексуальной деятельности и расстройств фертильности; иммунодепрессии, иммуносупрессии, воспалительных процессов, множественных инфекций, ревматоидного артрита, остеоартрита, увеита, псориаза и диабета; раковых заболеваний; желудочно-кишечных функциональных расстройств и являющихся их результатом воспалений, раздраженного и воспаленного кишечника, диареи; расстройств восприятия боли, фибромиалгий, которые могут быть связаны или не связаны с нарушениями сна, утомлением или мигренью; симптомов, ассоциированных с алкогольной зависимостью и отменой лекарственных средств.
Текст
1 Настоящее изобретение относится к новым разветвленным производным аминотиазола, к способу их получения и к содержащим их фармацевтическим композициям. Эти новые производные тиазола обладают антагонистической активностью в отношении кортикотропин-рилизинг-фактора (КРФ) и, таким образом, могут быть активными началами для фармацевтических композиций. Кортикотропин-рилизинг-фактор(КРФ) представляет собой пептид, последовательность которого, состоящая из 41 аминокислоты, была охарактеризована Вэйлом и др. в 1981 году (W.Vale et al., Science, 1981, 213, 1394-1397). КРФ является основным эндогенным фактором, вовлеченным в регулирование системы гипоталамусгипофиз-надпочечники (высвобождение адренокортикотропного гормона: АКТГ) и ее патологий, а также в депрессивные синдромы, являющиеся их результатом. Кроме того, КРФ вызывает секрецию -эндорфина, -липотропина и кортикостерона. Таким образом, КРФ является физиологическим регулятором секреции адренокортикотропного гормона (АКТГ) и вообще пептидов, получаемых из проопиомеланокортина(ПОМК). Помимо того, что он локализован в гипоталамусе, КРФ также широко распространен в центральной нервной системе, а также во вненейрональных тканях, таких как надпочечники и яички. Присутствие КРФ было продемонстрировано также в ходе воспалительных процессов. Многочисленные эксперименты на животных показали, что центральное введение КРФ вызывает различные анксиогенные эффекты,такие как изменение поведения вообще, например неофобия, снижение сексуальной восприимчивости, уменьшение потребления пищи и сокращение медленного сна у крыс. Интрацеребровентрикулярная инъекция КРФ также увеличивает возбуждение норадренергических нейронов голубого пятна, которое у животных часто связывают с состоянием тревоги. Центральное или периферическое введение КРФ или сходных пептидов (например, урокортина или саувагина) индуцирует у крыс, в дополнение к центральным эффектам, таким как повышение умственных способностей и эмоциональной реактивности по отношению к окружающей среде, изменения опорожнения желудка, кислотной секреции, прохождения через кишечник и фекальной экскреции, а также эффекты напряжения. КРФ также вовлечен в сложную регуляцию воспалительных ответов, во-первых,играя провоспалительную роль в конкретных животных моделях, и, во-вторых, как ингибитор эффектов, индуцируемых при увеличении проницаемости сосудов вследствие воспаления. Использование пептидного антагониста,альфа-спирального КРФ(9-41) (-КРФ), или специфических антител (Rivier J. et al., Science,1984, 224, 889-891) подтверждает роль этого 2 пептида во всех этих эффектах. Эти эксперименты также подтвердили важную роль КРФ у человека в интеграции комплексных ответных реакций, наблюдаемых при физиологических,психологических или иммунологических стрессах, как в нейроэндокринологических, так и в висцеральных, а также в поведенческих выражениях (Morley J.E. et al., Endocrine Review, 1987, 8,3, 256-287; Smith M.A. et. al., Horm. Res., 1989, 31,66-71). Кроме того, клинические данные свидетельствуют в пользу эффективного участия КРФ во многих расстройствах, являющихся результатом состояния стресса (Gulley L.R. et al., J. Clin.Psychiatry, 1993, 54, 1 (suppl.), 16-19), например создание КРФ-теста (внутривенное (в.в.) введение) на человеке дало возможность продемонстрировать изменение АКТГ-ответа у пациентов в состоянии депрессии (Breier A. et al.,Am. J. Psychiatry, 1987, 144, 1419-1425); обнаружение гиперсекреции эндогенного КРФ при некоторых видах патологий, например повышенного уровня КРФ в цефалопозвоночной жидкости у не подвергнутых лечению лекарственными средствами пациентов, которые находятся в подавленном состоянии или страдают деменцией, такой как болезнь Альцгеймера (Nemeroff C.B. et al., Science, 1984, 226, 4680,1342-1343; Regul. Pept., 1989, 25, 123-130), или пониженной плотности рецепторов КРФ в коре головного мозга жертв суицида (Nemeroff C.B.et al., Arch. Gen. Psychiatry, 1988, 45, 577-579); нарушение функционирования КРФ-зависимых нейронов предполагается также при тяжелых патологиях, таких как болезнь Альцгеймера, болезнь Паркинсона, хорея Гентингтона и боковой амиотрофический склероз (De Souza Е.В., Hospital Practice, 1988, 23, 59). Центральное введение КРФ у многих видов животных вызывает поведенческие эффекты,подобные эффектам у человека в стрессовых состояниях. Эти эффекты, если они повторяются через какое-то время, могут приводить к различным патологиям, таким как усталость, гипертензия, сердечные расстройства или расстройства напряжения, изменение опорожнения желудка или фекальной экскреции (колит, раздраженный кишечник), изменение секреции кислоты, гипергликемия, задержка развития, анорексия, неофобия, мигрени, репродуктивные нарушения, иммуносупрессия (воспалительные процессы, множественные инфекции и раковые болезни) и различные нейропсихиатрические расстройства (депрессия, нервно-психическая анорексия и тревога). Интрацеребровентрикулярная инъекция упомянутого пептидного антагониста, -КРФ,предотвращает эффекты, получаемые либо в результате введения экзогенного КРФ, либо в результате применения вызывающих стресс агентов (эфира, лишения свободы, неприятного звука, электрического шока, симптомов этанольной абстиненции или хирургического вмешательства), которые сами способны индуциро 3 вать увеличение уровня эндогенного КРФ. Эти результаты подтверждены исследованием многих антагонистических пептидных молекул, которые структурно подобны КРФ и которые имеют продолжительную длительность действия по сравнению с -КРФ (Rivier J. et al., J. Med. Chem.,1993, 36, 2851-2859; Menzaghi F. et al., J. Pharmacol.et al., J. Med. Chem., 1993, 36, 2860-2867). Такие КРФ-антагонистические пептидные соединения описаны, например, в патентах США 5109111, 5132111 и 5245009 и в патентных заявках WO 92/22576 и WO 96/19499. Кроме того, предварительные исследования показали, что трициклические антидепрессанты могут модулировать уровень КРФ, а также количество рецепторов КРФ в мозге (Grigoriadis D.E.et al., Neuropsychopharmacology, 1989, 2, 53-60). Аналогичным образом, бенздиазепиновые анксиолитические агенты способны реверсировать эффект КРФ (Britton K.T. et al., Psychopharmacology,1988, 94, 306), хотя механизм действия этих веществ объяснен не полностью. Эти результаты подкрепляют, если необходимо, растущую потребность в непептидных молекулах-антагонистах для рецепторов КРФ. Также важно указать на три возможных последствия состояний хронического стресса, а именно: иммунодепрессия, расстройства фертильности и развитие диабета. КРФ вызывает такие эффекты, взаимодействуя со специфическими мембранными рецепторами, которые обнаружены и охарактеризованы в гипофизе и мозге многих видов (мышей,крыс и человека), равно как и в сердце, скелетной мышце (крыс и мышей) и в миометрии и плаценте во время беременности. Уже известно много производных 2-аминотиазола. В ЕР 462264 описаны производные 2-аминотиазола, в которых третичный амин в положении 2 имеет два заместителя, каждый из которых содержит по меньшей мере один гетероатом, включая аминопроизводное. Эти соединения являются антагонистами фактора активации тромбоцитов (PAF-acether) и находят применение при лечении астмы, определенных аллергических или воспалительных состояний,сердечно-сосудистых заболеваний, гипертензии и различных почечных патологий или, альтернативно, в качестве контрацептивных агентов. В GB 2022285 описаны соединения с регуляторным действием на иммунный ответ и с противовоспалительными свойствами. Они являются производными тиазола, замещенными в положении 2 вторичными аминогруппами. Некоторые 2-ациламинотиазольные производные описаны в ЕР 432040. Эти соединения являются антагонистами холецистокинина и гастрина. Известны также 2-амино-4,5-дифенилтиазольные производные с противовоспалительными свойствами (заявка Японии JP-0175475). 4 Известны также 2-амино-4-(4-гидроксифенил)тиазольные производные, которые полезны в качестве синтетических промежуточных соединений для получения 2,2-диарилхроменотиазольных производных (заявка ЕР 205069). В J. Chem. Soc. Perkin, Trans. 1, 1984, 2,147-153 и в J. Chem. Soc. Perkin, Trans. 1, 1983,2, 341-347 описаны также 2-(N-метил-N-бензиламино)тиазольные производные. В WO 94/01423 описаны производные 2 аминотиазола. Эти соединения используют в качестве инсектицидов; они не замещены в положении 5 гетероцикла. В WO 96/16650 описаны также соединения, получаемые из 2-аминотиазола. Эти соединения используют в качестве антибиотиков. В заявке ЕР 283390 среди других производных тиазола описаны 2-(N-алкил-N-пиридилалкиламино)тиазольные производные, в которых амин в положении 2 замещен неразветвленным пиридилалкильным радикалом. Эти соединения, в частности, оказывают стимулирующее действие на холинергическую передачу в центральной нервной системе. Их можно использовать в качестве агонистов мускариновых рецепторов, и они могут найти применение при лечении нарушений памяти и сенильных деменций. Производные 2-аминотиазола, в которых амин в положении 2 является третичным амином, несущим разветвленный алкильный или аралкильный заместитель, описаны в ЕР 576350 и в ЕР 659747 как обладающие аффинностью к рецепторам КРФ. Ни одно из этих соединений не несет замещенного фенила в качестве заместителя третичного амина в положении 2 тиазольного ядра. В патенте США 5063245 описаны антагонисты КРФ, которые в концентрации примерно 1 мкмоль in vitro обеспечивают замещение связи КРФ с его специфическими рецепторами. Впоследствии были опубликованы многочисленные заявки, относящиеся к непептидным молекулам,например WO 94/13643, WO 94/13644, WO 94/13661, WO 94/13676, WO 94/13677, WO 94/10333, WO 95/00640, WO 95/10506, WO 95/13372, WO 95/33727, WO 95/33750, WO 95/34563, ЕР 691128 или ЕР 729758. Теперь согласно настоящему изобретению обнаружено, что некоторые разветвленные производные аминотиазола, которые являются предметом настоящего изобретения, обладают превосходной аффинностью к рецепторам КРФ. К тому же, благодаря своей структуре эти молекулы хорошо диспергируются и/или растворяются в растворителях или растворах, обычно используемых в терапевтических целях, что стимулирует их фармакологическое действие и облегчает приготовление пероральных и парентеральных фармацевтических форм. Удивительным и неожиданным оказалось то,что соединения по данному изобретению in vivo 5 более активны, чем соединения подобной структуры, ввиду, в частности, более сильного ингибирования ответной реакции, индуцированной КРФ в системе гипоталамус-гипофиз-надпочечники. Одним из объектов настоящего изобретения являются соединения, в рацемической форме или в форме чистого энантиомера, формулы где R1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; гидрокси(С 1-С 5)алкил; (С 1 С 5)алкил; аралкил, в котором арильная часть представляет собой (С 6-С 8), а алкильная часть представляет собой (С 1-С 4); (С 1-С 5)алкокси; трифторметильную группу; нитрогруппу; нитрильную группу; группу -SR, где R представляет собой водород, (С 1-С 5)алкил или аралкил, в котором арильная часть представляет собой (С 6 С 8), а алкильная часть представляет собой (С 1 С 4); группу -S-CO-R, где R представляет собой(С 1-С 5)алкильный или аралкильный радикал, в котором арильная часть представляет собой (С 6 С 8), а алкильная часть представляет собой (С 1 С 4); группу -COORa, где Ra представляет собой водород или (С 1-С 5)алкил; группу -CONRaRb сRa и Rb такими, как определено выше для Ra; группу -NRaRb с Ra и Rb такими, как определено выше для Ra; группу -CONRcRd или -NRcRd,где Rс и Rd составляют с атомом азота, к которому они присоединены, 5-7-членный гетероцикл; или группу -NHCO-NRaRb с Ra и Rb такими, как определено выше для Ra;R3 представляет собой водород или является таким, как определено выше для R1 и R2; или, альтернативно, R2 составляет с R3, когда последний является заместителем фенила в положении 5, группу -X-CH2-X-, где Х независимо представляет собой CH2 или атом кислорода или серы;-O-СН 2-O- по двум соседним атомам углерода фенила, группой -СF3, -NO2 или -CN, группой 6 дазолильную, (С 3-С 5)циклоалкильную или (С 3 С 6)циклоалкил(С 1-С 6)алкильную группу; их соли присоединения, их гидраты и/или их сольваты. В настоящем описании изобретения алкильные группы и алкоксигруппы являются линейными или разветвленными. Термин атом галогена означает атом фтора, хлора, брома или йода. Гетероциклы в определении R7, возможно,могут быть замещены такими же заместителями, как и заместители на фениле. Согласно другим своим аспектам данное изобретение относится к соединениям, в рацемической форме или в форме чистого энантиомера, формулы (I), гдеR1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; (С 1-С 5)алкил; (С 1-С 5)алкокси;R3 представляет собой водород или является таким, как определено выше для R1 и R2;R7 представляет собой фенил, который не замещен или моно- или дизамещен в положении 3 или 4 галогеном, (С 1-С 5)алкильной группой,группой -CH2OR8, где R8 представляет собой(С 1-С 3)алкил, или группой -O-СН 2-O- в положении 3, 4; или, альтернативно, R7 представляет собой (С 3-С 5)циклоалкильную группу; их солям присоединения, их гидратам и/или их сольватам. Данное изобретение также относится к соединениям формулы (I), в рацемической форме или в форме чистого энантиомера, где R3 находится в положении 5 фенила, а также их солям присоединения, их гидратам и/или их сольватам. Согласно другим своим аспектам данное изобретение относится к соединениям, выбранным из(пример 61),а также их соответствующих оснований,других солей присоединения и сольватов и/или гидратов. Соединения по изобретению в свободной форме обычно обладают слабоосновными свойствами. Однако в зависимости от природы заместителей некоторые из них могут проявлять кислотные свойства. Соли соединений формулы (I) с фармацевтически приемлемыми кислотами или основаниями (когда это возможно) являются предпочтительными солями, однако, соли, которые могут обеспечить возможность выделения соединений формулы (I), в особенности возможность очистки или получения чистых изомеров, также входят в объем изобретения. Среди фармацевтически приемлемых кислот для получения солей присоединения к соединениям формулы (I) можно упомянуть соляную кислоту, бромоводородную кислоту, фосфорную кислоту, фумаровую кислоту, лимонную кислоту, щавелевую кислоту, серную кислоту, аскорбиновую кислоту, винную кислоту,малеиновую кислоту, миндальную кислоту, метансульфоновую кислоту, лактобионовую кислоту, глюконовую кислоту, глюкаровую кислоту, янтарную кислоту, сульфоновую кислоту и гидроксипропансульфоновую кислоту. Среди фармацевтически приемлемых оснований для получения солей присоединения к соединениям формулы (I), если эти соединения обладают кислотными свойствами, можно упомянуть гидроксид натрия, гидроксид калия и гидроксид аммония. Соединения по изобретению и промежуточные соединения, которые пригодны для их получения, получают способами, которые общеизвестны специалистам в данной области техники, в частности согласно ЕР 576350 и ЕР 659747. Представленная ниже реакционная схема иллюстрирует один способ синтеза соединений формулы (I). Схема 1 9 На схеме 1 и далее радикалы R1, R2, R3, R6 и R7 такие, как определено выше, радикал R4 представляет собой метил, радикал R5 представляет собой проп-2-инил. Согласно еще одному из своих аспектов предметом настоящего изобретения является способ получения соединений формулы (I), отличающийся тем, что альфа-галогенопроизводное, предпочтительно альфа-бромо- или альфахлоропроизводное, формулы (III) где Hal представляет собой атом галогена, предпочтительно брома или хлора, подвергают взаимодействию с тиомочевиной формулы (IV) с получением соединения формулы (II) которое затем подвергают реакции алкилирования с получением соединения формулы (I). Реакцию алкилирования, используемую в вышеупомянутом способе, проводят в обычных условиях, известных специалистам в данной области техники, воздействием соответствующего алкилирующего агента, такого как, например,алкенил- или алкинилгалогенид, в присутствии основания, предпочтительно гидрида натрия. Производные формулы (III) могут быть получены из соответствующих негалогенированных кетонов формулы либо (1) воздействием брома в подходящем органическом растворителе, таком как уксусная кислота, четыреххлористый углерод или диэтиловый эфир, либо (2) воздействием трибромидов четвертичного аммония по методу, описанному в Bull. Chem. Soc. Japan, 1987, 60, 11591160 и 2667-2668, либо (3), альтернативно, воздействием бромида меди в органическом растворителе, таком как смесь хлороформа и этилацетата, согласно J. Оrg. Chem. 1964, 29, 34513461. Как вариант, соединения формулы (III) могут быть получены воздействием 2-бромпропионилбромида на замещенный бензол формулы по реакции Фриделя-Крафтса. Упомянутые выше кетоны обычно являются общеизвестными продуктами или коммерчески доступными продуктами. Эти соединения могут быть получены по реакции ФриделяКрафтса в присутствии кислоты Льюиса согласно методам, которые широко известны специалистам в данной области техники. Производные тиомочевины (IV) получают из защищенных производных тиомочевины (V) где Prot представляет собой защитную группу,например бензоил или пивалоил, либо щелочной обработкой, предпочтительно с использованием водного аммиака, гидроксида натрия или гидразина, при температуре в пределах от комнатной температуры до точки дефлегмации реакционной смеси, либо кислотной обработкой,предпочтительно с использованием соляной кислоты. Соединения формулы (V) получают путем взаимодействия, согласно известным методам,изотиоцианата, например бензоилизотиоцианата или пивалоилизотиоцианата, с соответствующими аминами формулы (VI)(VI) где R6 и R7 такие, как определено для (I). Получение оптически активных аминотиазолов, то есть продуктов в форме чистых энантиомеров, осуществляют, начиная с оптически активных первичных аминов, согласно приведенной ниже схеме 2 способом, который идентичен способу, описанному выше. Схема 2 Упомянутые выше соединения формулы(I) также включают соединения, в которых один или более чем один атом водорода или углерода заменен его радиоактивным изотопом, например тритием или углеродом-14. Такие меченые со 11 единения полезны для научных исследований,для изучения метаболизма или фармакокинетики, или, альтернативно, для использования в биохимических анализах в качестве рецепторных лигандов. Соединения по настоящему изобретению были подвергнуты биохимическим и фармакологическим исследованиям. Они демонстрируют чрезвычайно полезные фармакологические свойства. Соединения по данному изобретению в концентрациях менее 10 мкМ замещают связывание КРФ или родственных йодинированных пептидов (уротензина, саувагина), например 125I-тирозин-КРФ, с рецепторами, имеющимися на мозговых мембранах или на клетках в культуре, согласно методу, описанному Е.В. DeSouza (J. Neurosci., 1987, 7, 1, 88-100). Антагонистическая активность соединений по данному изобретению продемонстрирована по их способности ингибировать некоторые виды активности, связанные с КРФ. В частности,соединения формулы (I) способны ингибировать секрецию адренокортикотропного гормона(АКТГ), индуцированную КРФ. Исследование по секреции АКТГ, индуцированной КРФ, проводили in vivo на находящихся в сознании крысах согласно адаптированному для данного случая методу, описанному в С. Rivier et al.,Endocrinology, 1982,110 (1), 272-278. КРФ является нейропептидом, который контролирует активность системы гипоталамусгипофиз-надпочечники. Этот фактор ответственен за связанные со стрессом поведенческие и эндокринные ответные реакции. Конкретно, было продемонстрировано, что КРФ может модулировать поведение, а также определенные функции автономной нервной системы (G.F. Koob, F.E. Bloom, Fed. Рrос.,1985, 44, 259; M.R. Brown, L.A. Fisher, Fed.Proc., 1985, 44, 243). Более конкретно, КРФ индуцирует секрецию кортикотропина (АКТГ), эндорфина и других пептидов, получаемых из проопиомеланокортина (A. Tazi et al., Regul.J. Physiol., 1987, G 582, 253). Таким образом, соединения по данному изобретению могут быть полезными в регуляции секреции этих эндогенных веществ. Они находят применение, главным образом, в качестве активных начал лекарственных продуктов для снижения ответной реакции на стресс (поведение, эмоциональные состояния, желудочнокишечные и сердечно-сосудистые расстройства,расстройства иммунной системы) и, более широко, при патологиях, в которые вовлечен КРФ,например при психиатрических расстройствах,состоянии тревоги, депрессии, нервно-психической анорексии, эпилепсии, расстройствах сексуальной деятельности и расстройствах фертильности, болезни Альцгеймера и тому подобных. 12 Соединения по данному изобретению очень стабильны и поэтому особенно подходят для формирования активного начала готовых лекарственных средств. Данное изобретение распространяется также на фармацевтические композиции, содержащие в качестве активного начала соединение формулы (I) или одну из его фармацевтически приемлемых солей, возможно, в комбинации с одним или более чем одним инертным и подходящим эксципиентом. В каждой лекарственной единице активное начало формулы (I) присутствует в количествах,которые соответствуют предусмотренным суточным дозам. Каждую лекарственную единицу выверяют в соответствии с дозировкой и типом предусмотренного введения, например таблетки, гелевые капсулы и тому подобное, саше,ампулы, сиропы и тому подобное, капли, трансдермальные или трансмукозальные пластыри,так, чтобы такая лекарственная единица содержала от 0,5 до 800 мг активного начала, предпочтительно от 0,5 до 200 мг. Соединения по данному изобретению также могут быть использованы в комбинации с другим активным началом, которое полезно для требуемого лечения, таким как, например, анксиолитические агенты, антидепрессанты или анорексигенные агенты. Соединения формулы (I) относительно нетоксичны; их токсичность совместима с их использованием в качестве лекарственных продуктов для лечения вышеупомянутых расстройств и заболеваний. Соединения формулы (I) могут быть введены в состав фармацевтических композиций для введения млекопитающим, включая человека, для лечения вышеупомянутых заболеваний. Полученные таким образом фармацевтические композиции преимущественно находятся в разных формах, таких как, например, растворы для инъекций и питья, таблетки с сахарным покрытием, таблетки или гелевые капсулы. Фармацевтические композиции, содержащие в качестве активного начала по меньшей мере одно соединение формулы (I) или одну из его солей,полезны, в частности, для превентивного или куративного лечения связанных со стрессом состояний и, более широко, при лечении любой патологии, в которую вовлечен КРФ, такой как,например, болезнь Кушинга, нервно-психиатрические расстройства, такие как депрессивные,тревожные, панические, обсессивно-компульсивные расстройства, расстройства настроения,посттравматический стресс, расстройства поведения, агрессивность, анорексия, булимия, гипергликемия, преждевременные роды, беременность с повышенным риском, замедленное развитие, нарушения сна, эпилепсия и все типы депрессии; болезнь Альцгеймера, болезнь Паркинсона, хорея Гентингтона; боковой амиотрофический склероз; сосудистые, сердечные и 13 церебральные расстройства; расстройства сексуальной деятельности и расстройства фертильности; иммунодепрессия, иммуносупрессия,воспалительные процессы, множественные инфекции, ревматоидный артрит, остеоартрит,увеит, псориаз и диабет; раковые заболевания; желудочно-кишечные функциональные расстройства и являющиеся их результатом воспаления (раздраженный и воспаленный кишечник,диарея); расстройства восприятия боли, фибромиалгии, которые могут быть связаны или могут не быть связаны с нарушениями сна, утомлением или мигренью; симптомы, ассоциированные с (алкогольной) зависимостью и отменой лекарственных средств. Дозировка может варьировать в широких пределах в зависимости от возраста, массы и состояния здоровья пациента, природы и серьезности осложнения, а также пути введения. Эта дозировка включает в себя ежедневное введение одной или более чем одной дозы приблизительно от 0,5 до 800 мг, предпочтительно приблизительно от 0,5 до 200 мг. Активное начало в фармацевтических композициях по настоящему изобретению для перорального, сублингвального, подкожного,внутримышечного, внутривенного, трансдермального, трансмукозального, местного или ректального введения может быть введено животным и людям в стандартных формах введения в виде смеси с традиционными фармацевтическими вспомогательными веществами. Соответствующие стандартные формы введения включают пероральные формы, такие как таблетки, гелевые капсулы, порошки, гранулы и пероральные растворы или суспензии, формы для сублингвального и трансбуккального введения, формы для подкожного, внутримышечного,внутривенного, интраназального или внутриглазного введения и формы для ректального введения. Когда твердую композицию готовят в форме таблеток, основное активное начало смешивают с фармацевтическим носителем,таким как желатин, крахмал, лактоза, стеарат магния, тальк, аравийская камедь или тому подобное. Таблетки могут быть покрыты сахарозой или другими подходящими материалами или, альтернативно, они могут быть обработаны так, чтобы они обладали замедленным или пролонгированным действием, и так, чтобы они непрерывно высвобождали предопределенное количество активного начала. Препарат в виде гелевых капсул получают путем смешения активного начала с разбавителем и вливания полученной смеси в мягкие или твердые гелевые капсулы. Препарат в форме сиропа или эликсира может содержать активное начало вместе с подсластителем, предпочтительно бескалорийным подсластителем, метилпарабеном и пропилпа 005159 14 рабеном в качестве антисептика, а также корригентом и подходящим красителем. Диспергируемые в воде порошки или гранулы могут содержать активное начало в смеси с диспергирующими агентами, или увлажняющими агентами, или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или корригентами. Для ректального введения используют суппозитории, которые изготавливают с использованием связующих, которые плавятся при ректальной температуре, например масла какао или полиэтиленгликолей. Для парентерального, интраназального или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные инъецируемые растворы, которые содержат фармакологически совместимые диспергирующие агенты и/или увлажняющие агенты, например пропиленгликоль или бутиленгликоль. Для трансмукозального введения препарат активного начала может быть приготовлен в присутствии промотора, такого как соль желчной кислоты, гидрофильного полимера, такого как, например, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза, этилцеллюлоза, карбоксиметилцеллюлоза, декстран, поливинилпирролидон, пектины,крахмалы, желатин, казеин, акриловые кислоты,акриловые эфиры и их сополимеры, виниловые полимеры или сополимеры, виниловые спирты,алкоксиполимеры, полиэтиленоксидные полимеры, полиэфиры или их смеси. Активное начало может быть приготовлено также в форме микрокапсул, возможно, с одним или более чем одним вспомогательным веществом или добавкой. Активное начало может быть представлено также в форме комплекса с циклодекстрином,например -, - или -циклодекстрином, 2-гидроксипропилциклодекстрином или метил-циклодекстрином. Следующие ниже примеры иллюстрируют данное изобретение, не ограничивая его. Способы синтеза различных промежуточных соединений для получения соединений по данному изобретению описаны в подготовительных примерах. Все эти промежуточные соединения получают способами, которые общеизвестны для специалистов в данной области техники. Точки плавления измерены методом MicroKofler и выражены в градусах Цельсия. Спектры протонного ядерного магнитного резонанса (1 Н ЯМР) сняты в CDCl3, за исключением особо упомянутых случаев, при 200 МГц или при 300 МГц. Химические сдвиги выражены в миллионных долях (м.д.), а константы взаимодействия выражены в Герцах. 15 Энантиомерные избытки (ее) оценивали по хроматограммам, полученным либо посредством ВЭЖХ с хиральной фазой, либо посредством хиральной хроматографии со сверхкритической подвижной фазой (SFC). Оптические вращения оптически активных продуктов охарактеризованы их []tD (концентрации (с) анализируемых растворов выражены в граммах на 100 мл). Использованы следующие сокращения:q=квартет. Результаты элементного анализа соединений по изобретению соответствуют теоретическим результатам. Соединения по изобретению, охарактеризованные в табл. 3 и 5, демонстрируют также спектры ЯМР, которые соответствуют их структуре. Получение -бромкетонов формулы (III) 2-Бром-1-(2-хлор-4-метокси-5-метилфенил) пропан-1-он, соединение III.1. Раствор 46 г (280 ммоль) 4-хлор-2-метокситолуола в 150 мл дихлорметана перемешивают при 0 С и добавляют 29,4 г (280 ммоль) 2 бромпропионилбромида. К этой смеси порциями добавляют 39,2 г (294 ммоль) трихлорида алюминия. Эту смесь перемешивают, давая возможность температуре постепенно подняться до комнатной температуры. После перемешивания в течение 4 ч реакционную смесь медленно выливают на лед. К этой перемешиваемой смеси добавляют 50 мл 1 н. соляной кислоты и 1 л воды, после чего осуществляют экстракцию 1,2 л трет-бутилметилового эфира. Органическую фазу промывают водой, насыщенным водным раствором гидрокарбоната натрия, водой, а затем насыщенным раствором хлорида натрия. Высушивают над безводным сульфатом натрия,а затем упаривают досуха. Неочищенный остаток очищают хроматографией на силикагелеH ЯМР: 7,44 (s, Аr, 1 Н); 6,86 (s, Аr, 1 Н); 5,41 (q, J=5,35 Гц, СН, 1 Н); 3,90 (s, ОСН 3, 3 Н); 2,23 (s, СН 3, 3 Н); 1,91 (d, J=5,35 Гц, СН 3, 3 Н). Этим же способом синтезируют следующие соединения: 2-бром-1-(2-хлор-4-метоксифенил)пропан 1-он, cоединение III.2; 2-бром-1-(2,4-дихлор-5-метилфенил)пропан 1-он, cоединение III.3; 2-бром-1-(2,4-диметокси-5-метилфенил) пропан-1-он, cоединение III.4; 2-бром-1-(4-метокси-2,5-диметилфенил) пропан-1-он, cоединение III.5. Получение рацемических аминов формулы (VI) Первый способ. а) 2-Амино-2-(4-фторфенил)этанол, соединение I.1. 60 мл (60 ммоль) 1 М раствора алюмогидрида лития в тетрагидрофуране перемешивают 16 при температуре дефлегмации, затем добавляют порциями 5 г (29 ммоль) 4-фтор-DLфенилглицина (Fluka). После перемешивания при температуре дефлегмации в течение 6 ч реакционную смесь перемешивают при 0 С, а затем медленно добавляют 2,5 мл воды, 2,5 мл водного 15% раствора гидроксида натрия и затем 7,5 мл воды. Полученную суспензию фильтруют через целит. Фильтрат концентрируют и переносят в 300 мл дихлорметана. Раствор промывают насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия, а затем упаривают досуха. Получают 3,3 г маслянистого желтого продукта. Выход=73%. МС (масс-спектрометрия) (MH+=156). 1(m, Ar, 2H); 4,08 (m, CH, 1H); 3,45-3,86 (m,CH2O, 2H); 2,03 (s, NН 2 и ОН, 3 Н). б) 1-(4-Фторфенил)-2-метоксиэтиламин,cоединение VI.1. 0,94 г (23 ммоль) гидрида калия, полученного промывкой 2,2 г масляной суспензии пентаном, суспендируют в 18 мл тетрагидрофурана и перемешивают при 10 С. Медленно добавляют раствор 3,3 г (21 ммоль) cоединения I.1 в 43 мл тетрагидрофурана. После перемешивания в течение 16 ч при комнатной температуре добавляют раствор 1,3 мл (20,8 ммоль) йодметана в 25 мл тетрагидрофурана за 1 ч 30 мин. Реакционную смесь перемешивают в течение 3 ч при комнатной температуре, а затем вливают в 300 мл охлажденной во льду воды, содержащей соль. Смесь экстрагируют 500 мл тpeт-бутилметилового эфира. Органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия, после чего упаривают досуха. Получают 3,2 г маслянистого амина. Выход=88%. 1VI.2, получают таким же способом. Второй способ. а) Синтез замещенных фенилкетонов, cоединений 3. Методика А. 1-(3-Фтор-4-метилфенил)-2-метоксиэтан-1 он, cоединение 3.1. Для получения магниевого реагента 14 г(583 ммоль; 1 экв.) магниевых стружек оставляют перемешиваться в присутствии дробленого стекла в атмосфере аргона в течение ночи. Их покрывают 400 мл диэтилового эфира с последующим добавлением йода на кончике шпателя. Медленно, так чтобы поддерживалось слабое кипение, добавляют 110 г (582 ммоль) 4-бром-2 фтортолуола, растворенного в 700 мл диэтилового эфира, и реакционную смесь затем нагревают при температуре дефлегмации в течение 3 ч. Добавляют 39 мл метоксиацетонитрила (610 17 ммоль; 1,1 экв.) и смесь оставляют реагировать в течение 2 ч. После завершения реакции реакционную смесь вливают в 1,5 кг льда, а затем при перемешивании добавляют 300 мл концентрированной соляной кислоты. Эту смесь экстрагируют диэтиловым эфиром, высушивают над сульфатом натрия и упаривают. Получают 77 г соединения 3.1, которое прямо без очистки используют на второй стадии. Следующие соединения получают таким же способом: 1-(4-хлор-3-фторфенил)-2-метоксиэтан-1 он, cоединение 3.2; 1-(4-хлорфенил)-2-метоксиэтан-1-он, cоединение 3.3; 3-циклопропил-1-(4-фторфенил)пропан-1 он, cоединение 3.4. Методика Б. 2-Метокси-1-(4-метоксиметилфенил)этан 1-он, cоединение 3.5. Раствор 62 г (308 ммоль) 1-бром-4-метоксиметилфенила в 600 мл тетрагидрофурана перемешивают при -70 С и медленно добавляют 200 мл (320 ммоль) 1,6 М раствора бутиллития. Реакционную смесь перемешивают в течение 30 мин при -70 С, затем медленно добавляют раствор 50 г (380 ммоль) 2-N-диметокси-N-метилацетамида. Реакционную смесь перемешивают,давая возможность температуре постепенно подняться до комнатной. После перемешивания в течение 4 ч ее охлаждают до 0 С и медленно добавляют насыщенный водный раствор хлорида аммония. Смесь экстрагируют этилацетатом и органическую фазу промывают водой и затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и затем упаривают досуха. Полученный остаток очищают хроматографией на силикагеле (растворитель: смесь циклогексан/этилацетат 9/1, а затем 3/1). Получают 32 г кетона. Выход=53%. 1cоединения 3.1, разбавленного 30 мл этанола. По окончании добавления добавляют 60 г предварительно измельченных гранул гидроксида натрия (1,5 моль; 5 экв.) и температуру поддерживают ниже 30 С. Реакционную смесь оставляют стоять при комнатной температуре в течение ночи, а затем при 0 С для нейтрализации концентрированной соляной кислотой (рН 7). Затем эту смесь экстрагируют этилацетатом и органическую фазу промывают водой и насыщенным раствором хлорида натрия. Органическую фазу высушивают над сульфатом натрия и 18 упаривают. Полученное таким образом масло хроматографируют на силикагеле, используя в качестве элюента смесь этилацетат/циклогексан 1/9 (об./об.). Получают 26 г (Z)-изомера и 9 г(Е)-изомера, то есть выход (Z) составляет 45%, а выход (Е) составляет 16%. Методика Б. 47 г гидроксиламина гидрохлорида (676 ммоль; 1,6 экв.) смешивают с 275 мл пиридина. При 0 С добавляют 77 г (423 ммоль) cоединения 3.1. Реакционную смесь оставляют стоять при комнатной температуре в течение 5 ч. По окончании реакции пиридин выпаривают, а остаток затем экстрагируют дихлорметаном. Органическую фазу промывают водой, затем насыщенным раствором хлорида натрия. Органическую фазу высушивают над сульфатом натрия и упаривают. Полученное таким образом масло хроматографируют на силикагеле, используя в качестве элюента смесь этилацетат/циклогексан 1/9 (об./об.), с получением 42,5 г соединения (Z) и 14 г соединения (Е), то есть выход Z составляет 51%, а выход Е составляет 17%. 1(ОСН 3, s, 3H); 2,22 (СН 3-Ph, s, 3H). По одной из двух вышеописанных методик получают следующие соединения: 1-(4-хлор-3-фторфенил)-2-метоксиэтан-1 она оксим, cоединение 4.2; 1-(4-хлорфенил)-2-метоксиэтан-1-она оксим,cоединение 4.3; 1-фенилбутан-1-она оксим, cоединение 4.4; 1-(4-метоксиметилфенил)-2-метоксиэтан 1-она оксим, cоединение 4.5; 1-(4-метоксиметилфенил)бутан-1-она оксим,cоединение 4.6; дициклобутилкетона оксим, cоединение 4.7; 1-фенилпентан-1-она оксим, cоединение 4.8. в) Синтез аминов, cоединений VI. 1-(3-Фтор-4-метилфенил)-2-метоксиэтиламин, cоединение VI.3. Раствор 1 г cоединения 4.1 (5 ммоль), растворенного в 15 мл тетрагидрофурана, медленно при 0 С добавляют к 10 мл 1 М раствора алюмогидрида лития в тетрагидрофуране (10 ммоль; 8 экв.). Реакционной смеси дают возможность нагреться до комнатной температуры, а затем оставляют реагировать в течение 2 ч и кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до 0 С, чтобы добавить 10 мл воды. Водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы экстрагируют 2 н. раствором соляной кислоты. Полученную кислотную водную фазу перемешивают при 0 С и добавляют 35% раствор гидроксида натрия. Полученный щелочной раствор экстрагируют дихлорметаном. Органическую фазу промывают насыщенным раство 19 ром хлорида натрия, а затем высушивают над сульфатом натрия и упаривают досуха. После фильтрования на диоксиде кремния с использованием смеси дихлорметан/метанол 95/5(об./об.) в качестве элюента получают 0,6 г соединения VI.3. Выход=65%. 1H ЯМР: 6,90-7,20 (Аr, m, 3 Н); 4,14 (CH-N,dd, J=4 и 8,5, 1 Н); 3,47 (-СН 2-O, dd, J=4 и 9, 1 Н); 3,37 (ОСН 3, s, 3 Н); 3,32 (-CH2-O, dd, 1H, J=8,5 и 9); 2,24 (СН 3-Ph, d, J=1,8, 3 Н); 1,68 (-NH2, s, 2H). Следующие соединения получают таким же способом: 1-(4-хлор-3-фторфенил)-2-метоксиэтиламин,cоединение VI.4; дициклобутилметиламин, cоединение VI.5. Третий способ. а) Синтез O-алкилоксимов, cоединений 6. Методика А. 1-Фенилбутан-1-она O-метилоксим, cоединение 6.1. 18 г (0,45 моль) 55% гидрида натрия в масле в течение 1 ч при 0 С порциями добавляют к 66 г (0,40 моль) 1-фенилбутан-1-она оксима (cоединение 4.4) в смеси 400 мл диметилформамида и тетрагидрофурана (1:1). После добавления 31 мл (0,5 моль) метилйодида реакционная смесь постепенно становится очень густой. После добавления 50 мл этанола, а затем воды реакционную смесь экстрагируют этилацетатом 4 х 250 мл. Органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом натрия, а затем упаривают под вакуумом. Получают 75 г бледно-желтого масла в виде смеси геометрических изомеров(7% (Z) и 93% (Е. Выход=94% (Z+Е). Эти два изомера могут быть разделены хроматографией на диоксиде кремния с элюированием смесью циклогексан/этилацетат. 1(m, Аr, 3 Н); 3.95 [s, ОСН 3, (Е)]; 3,82 [s, ОСН 3,(Z)]; 2,71 [m, OCH2, (Е)]; 2,50 [m, CH2, (Z)]; 1,411,64 (m, CH2, 2H); 0,84-1,03 (m, СН 3, 3 Н). Следующие алкилированные оксимы получают таким же способом: 1-фенилпентан-1-она O-метилоксим, cоединение 6.2; 1-(4-хлорфенил)-2-метоксиэтан-1-она O-бензилоксим, cоединение 6.3; 2-метокси-1-(4-метоксиметилфенил)этан-1 она O-метилоксим, cоединение 6.4; 1-(4-Метоксиметилфенил)бутан-1-она O-метилоксим, cоединение 6.5. Методика Б. Циклобутил-4-фторфенилкетона O-бензилоксим, cоединение 6.6. Раствор 15 г (84 ммоль) циклобутил-4 фторфенилкетона в 80 мл этанола перемешивают при комнатной температуре и добавляют 20,2 г (126 ммоль) O-бензилгидроксиламина гидрохлорида. Затем к этой смеси порциями добавляют 8,4 г (210 ммоль) гидроксида натрия,и смесь перемешивают в течение 4 ч при ком 005159 20 натной температуре. К этой смеси добавляют воду, а затем экстрагируют этилацетатом. Органическую фазу промывают водой до нейтральной реакции, затем насыщенным водным раствором хлорида натрия. Ее высушивают над сульфатом натрия, после чего упаривают досуха. Получают 28,4 г смеси изомеров (58% Е,42% Z). 1[m, CH циклобутил, (E)]; 3,36-3,52 [m, CH циклобутил, (Z)]; 1,58-2,30 (m, CH2 циклобутил, 6 Н]). Следующее соединение получают таким же способом: 3-циклопропил-1-(4-фторфенил)пропан-1 она O-бензилоксим, соединение 6.7. б) Синтез аминов, cоединений VI. 1-Фенилбутиламин, cоединение VI.6. Раствор 14,2 г (0,085 моль) 1-фенилбутан 1-она O-метилоксима (cоединение 6.1) в 85 мл тетрагидрофурана по каплям в атмосфере аргона добавляют к 85 мл 1 М раствора алюмогидрида лития в тетрагидрофуране. По окончании добавления реакционную смесь кипятят с обратным холодильником в течение 1 ч 30 мин. Оставляют на ночь при комнатной температуре,после чего добавляют 3,5 мл H2O, а затем добавляют 3,5 мл 15% гидроксида натрия и 10,5 млH2O. Осадок отфильтровывают и промывают диэтиловым эфиром. Тетрагидрофуран/диэтилэфирный фильтрат промывают водой, а затем трижды экстрагируют 1 н. раствором соляной кислоты. Кислотные водные фазы объединяют,а затем подщелачивают при 0 С 35% гидроксидом натрия. После экстракций дихлорметаном,промывок водой, высушивания над сульфатом натрия и последующего упаривания под вакуумом получают 9,3 г масла. Выход=73%. 1(m, CH, 1 Н); 1,73 (s, NH2, 2 Н), 1,60-1,70 (m, CH2,2 Н); 1,15-1,36 (m, CH2, 2H); 0,95-0,98 (m, СН 3, 3 Н). Следующие амины получают таким же способом: 1-фенилпентиламин, cоединение VI.7; 1-(4-хлорфенил)-2-метоксиэтиламин, cоединение VI.8; 2-метокси-1-(4-метоксиметилфенил)этиламин, cоединение VI.9; 1-(4-метоксиметилфенил)бутиламин, cоединение VI.10; циклобутил-(4-фторфенил)метиламин, cоединение VI.11; 3-циклопропил-1-(4-фторфенил)пропиламин,cоединение VI.12. Четвертый способ. 1. (4-Фторфенил)пентиламин, cоединениеVI.13. Раствор 1,21 г (10 ммоль) 4-фторбензнитрила в 10 мл тетрагидрофурана перемешивают при 0 С и по каплям добавляют 10 мл (10 ммоль) 1 М раствора боран-тетрагидрофурана. Смесь перемешивают в течение 1 ч 30 мин при ком 21 натной температуре, а затем медленно с перемешиванием переносят в 18,9 мл 1,6 М раствора бутиллития в гексане, который предварительно был охлажден до -78 С. Реакционную смесь перемешивают в течение 2 ч при -78 С, а затем гидролизуют при этой температуре 10 мл 2 н. соляной кислоты. Органическую фазу экстрагируют 2 н. соляной кислотой. Полученную кислотную водную фазу нейтрализуют при 0 С,медленно добавляя 35% гидроксид натрия, а затем экстрагируют этилацетатом. Органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и после этого упаривают досуха. Получают 0,95 г маслянистого амина. Выход=53%. 1(m, Аr, 2H); 4,13 (t, CH, 1H); 1,59-1,75 (m, CH2,2H); 1,49 (s, NH2, 2H), 1,24-1,33 (m, CH2-CH2,4 Н); 0,85 (t, СН 3, 3 Н). Следующее соединение получают таким же способом: 1-(3-фтор-4-метилфенил)пентиламин, cоединение VI.14. Пятый способ. 1-(4-Фторфенил)бутиламин, cоединение VI.15. Один кристалл йода добавляют к суспензии 2,4 г (100 ммоль) магния в 30 мл диэтилового эфира, после чего добавляют 17,4 г (100 ммоль) 4-бромфторбензола (разбавленного 70 мл диэтилового эфира), чтобы вызвать легкую дефлегмацию. Реакционную смесь кипятят с обратным холодильником в течение 1 ч, затем охлаждают до комнатной температуры и добавляют 5,75 г(85 ммоль) бутиронитрила, разбавленного 30 мл диэтилового эфира. Реакционную смесь кипятят с обратным холодильником в течение 2 ч, затем охлаждают и фильтруют через стекловату. Фильтрат перемешивают при комнатной температуре и медленно добавляют 100 мл (100 ммоль) 1 М раствора алюмогидрида лития в тетрагидрофуране. Реакционную смесь кипятят с обратным холодильником в течение 18 ч, а затем охлаждают до 0 С, после чего последовательно добавляют 3,8 мл воды, 3,8 мл 15% гидроксида натрия и 11,4 мл воды. Смесь фильтруют через целит и фильтрат упаривают досуха. Полученный остаток фильтруют через диоксид кремния,элюируя смесью дихлорметан/метанол 98/2N-[1-(4-Фторфенил)-2-метоксиэтил]тиомочевина, соединение IV.1. 6,5 мл (56,6 ммоль) бензоилхлорида добавляют при 0 С к перемешиваемому раствору 4,5 г(58 ммоль) изотиоцианата аммония в 115 мл ацетона. Через 30 мин медленно добавляют 8,6 г(56 ммоль) соединения VI.1, растворенного в 100 мл ацетона. Реакционную смесь перемешивают в течение 2 ч при комнатной температуре,а затем концентрируют при пониженном давлении. Суспензию переносят в 200 мл тpeтбутилметилового эфира и 200 мл воды. Органическую фазу промывают водой, затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и после этого упаривают досуха. Остаток после упаривания растворяют в 180 мл этанола и к полученному раствору добавляют 5,85 мл (116 ммоль) гидразина моногидрата. После перемешивания в течение 16 ч при комнатной температуре, так как реакция не проходит до конца, добавляют следующие 1,7 мл гидразина. После перемешивания в течение 24 ч при комнатной температуре реакционную смесь упаривают. Остаток после упаривания растворяют в 500 мл этилацетата и органическую фазу промывают водой, затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и упаривают досуха. Остаток после упаривания хроматографируют на колонке из диоксида кремния,элюируя смесью циклогексан/этилацетат 1/1H ЯМР (ДМСО-d6): 8,10 (d, NH, 1H); 7,287,32 (m, Аr, 2 Н); 7,08-7,17 (m, Ar и NH2, 4 Н); 5,45 (m, СН, 1H); 3,54-3,62 (m, CH-CH2, 2H); 3,24 (s, ОСН 3, 3 Н). Таким же способом получают приведенные в табл. 1 тиомочевины. Таблица 1[4-(2-Хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил]-[1-(4-метоксиметилфенил)бутил] амин, соединение II.1. 1,92 г (6 ммоль) 2-бром-1-(2-хлор-4-метокси 5-метилфенил)пропан-1-она (cоединение III.1) и 1,5 мл триэтиламина добавляют к 1,4 г (5,54 ммоль) 1-(4-метоксиметилфенил)бутилтиомочевины (cоединение IV.7) в 60 мл этанола. Реакционную смесь перемешивают при 85 С в течение 3 ч, а затем концентрируют при пониженном давлении. Остаток переносят в 100 мл дихлорметана и 50 мл воды. Органическую фазу промывают насыщенным водным раствором хлорида натрия, высушивают над сульфатом натрия, а затем упаривают досуха под вакуумом. Неочищенный экстракт очищают хроматографией на колонке из силикагеля, элюируя смесью циклогексан/этилацетат 9/1 (об./об.). Получают 2,35 г аминотиазола. Выход=96%. МС (МН+)=445. 1(s, OCH2, 2H); 4,17-4,33 (m, CH, 1H); 3,81 (s,ОСН 3, 3 Н); 3,39 (s, ОСН 3, 3 Н); 2.14 (s, СН 3, 3 Н); 2,05 (s, СН 3, 3 Н); 1,63-1,88 (m, CH2, 2H); 1,231,48 (m, CH2, 2H); 0,90 (t, СН 3, 3 Н). Описанные в табл. 2 продукты получают таким же способом. Таблица 2 Получение N-замещенных тиазолов Пример 1. [4-(2-Хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил]-[1-(4-фторфенил)2-метоксиэтил)проп-2-иниламин. 50 мг 60% гидрида натрия в масле добавляют с перемешиванием и при 0 С к 500 мг (1,2 ммоль) соединения II.2 в 6 мл безводного диметилформамида. Реакционную смесь перемешивают в течение 20 мин при 0 С, после чего добавляют 0,22 мл (2 ммоль) 80% раствора пропаргилбромида в толуоле. Реакционную смесь перемешивают в течение 1 ч при 10 С, затем добавляют 0,5 мл этанола и затем 10 мл воды. Смесь дважды экстрагируют 50 мл этилацетата. Органическую фазу промывают водой, а затем насыщенным водным раствором хлорида натрия, высушивают над безводным сульфатом натрия и после этого упаривают досуха. Неочищенный остаток хроматографируют на колонке из силикагеля [элюент: смесь циклогексан/этилацетат 9/1 (об./об.)]. Получают 400 мг чистого ожидаемого соединения. Выход=73%; гидрохлорида полугидрат: т.пл.=94 С. Описанные в табл. 3 продукты получают таким же способом. Получение аминов в форме энантиомера,соединение VI' Первый способ. а) (R)-2-Амино-2-(4-фторфенил)этанол, cоединение 1'.1. 240 мл (240 ммоль) 1 М раствора алюмогидрида лития в тетрагидрофуране перемешивают при температуре дефлегмации, затем порциями добавляют 20 г (118 ммоль) (R)-(4 фторфенил)глицина. После перемешивания при температуре дефлегмации в течение 6 ч 30 мин реакционную смесь перемешивают при 0 С,затем медленно добавляют 9,5 мл воды, 9,5 мл 15% раствора гидроксида натрия, а затем 28,5 мл воды. Полученную суспензию фильтруют через целит. Фильтрат концентрируют и переносят в 1 л дихлорметана. Раствор промывают насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия, после чего упаривают досуха. Кристаллизация из изопропилового эфира дает 13,22 г (85,2 ммоль) кристаллического продукта. Выход=72%; т.пл.=95 С; МСH ЯМР: (ДМСО-d6): 7,30-7,41 (m, Аr, АН); 7,01-7,13 (m, Аr, 2 Н); 4,73 (s, ОН, 1 Н); 3,84 (m, CH,1 Н); 3,35-3,45 (m, CH2O, 2H); 1,82 (s, NH2, 2H). б) (R)-1-(4-Фторфенил)-2-метоксиэтиламин,cоединение VI'.1 3,64 г (91 ммоль) гидрида калия, полученного промывкой 8,1 г масляной суспензии пентаном, суспендируют в 70 мл тетрагидрофурана и перемешивают при 10 С. Медленно добавляют раствор 13,22 г (85 ммоль) cоединения 1'.1 в 175 мл тетрагидрофурана. После перемешивания в течение 16 ч при комнатной температуре за 2 ч добавляют раствор 5,2 мл (83,5 ммоль) йодметана в 105 мл тетрагидрофурана. Реакционную смесь перемешивают в течение 3 ч при комнатной температуре, а затем вливают в 1 л охлажденной во льду воды, содержащей соль. Смесь экстрагируют 1 л тpeт-бутилметилового эфира. Органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и по 27 сле этого упаривают досуха. Получают 11,87 г(m, Аr, 2H); 4,16 (m, CH, 1H); 3,45 (dd, CH2, 1H); 3,36 (s, ОСН 3, 3 Н); 3,29 (d, CH2, 1H); 1,66 (s,NH2, 2H). Следующее соединение получают таким же способом, исходя из (R)-фенилглицина:(1790 ммоль) в диэтиловом эфире перемешивают при 0 С и разбавляют 300 мл ТГФ, после чего порциями добавляют 50 г (298 ммоль) Lвалинметилового эфира гидрохлорида, поддерживая при этом температуру ниже 10 С. После перемешивания в течение 3 ч при комнатной температуре реакционную смесь медленно вливают в охлажденный во льду раствор хлорида аммония. К этой смеси добавляют 500 мл диэтилового эфира и 500 мл этилацетата, затем перемешивают в течение ночи при комнатной температуре. После разделения фаз расслоением водную фазу повторно экстрагируют 1 л ТВМЕ(трет-бутилметилового эфира). Объединенные органические фазы перемешивают при 0 С и медленно подкисляют приблизительно 40 мл 35% соляной кислоты в воде. Образовавшийся в результате этого осадок гидрохлорида отфильтровывают и промывают ТВМЕ. Далее смесь переносят в 1 л дихлорметана и 1 л воды и подщелачивают при 0 С приблизительно 50 мл 35% каустической соды. После разделения фаз расслаиванием водную фазу повторно экстрагируют 1 л дихлорметана. Объединенные органические фазы промывают водой, а затем рассолом, высушивают над сульфатом натрия и концентрируют. После кристаллизации из изопропилового эфира получают 61 г cоединения 2'.1 (выход=87%).(-СН 3, 2d, J=7, 6 Н). Следующий продукт получают аналогичным способом, исходя из D-валинметилового эфира гидрохлорида:(R)-2-aмино-3-метил-1,1-дифенилбутан-1 ол, cоединение 2'.2. Эти соединения используют как хиральные вспомогательные вещества в энантиоселективном восстановлении O-бензилоксимов 6'. б) Синтез замещенных фенилкетонов, cоединений 3'. Методика А. 2-Циклопропил-1-(3-фтор-4-метилфенил) этан-1-он, cоединение 3'.1. 50 мл диэтилового эфира и один кристалл йода добавляют к 10,2 г (418 ммоль) магниевых 28 стружек и эту смесь перемешивают при комнатной температуре. За период времени 3 ч добавляют раствор 75,35 г (398 ммоль) 4-бром-2 фтортолуола в 370 мл диэтилового эфира, чтобы поддерживать умеренную дефлегмацию. Затем реакционную смесь кипятят с обратным холодильником в течение 1 ч 30 мин, после чего охлаждают и фильтруют через стекловату. Полученный раствор медленно добавляют к раствору 32,3 г (398 ммоль) циклопропилацетонитрила в 230 мл диэтилового эфира, перемешиваемого при 0 С. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Затем ее перемешивают при 0 С и медленно добавляют 200 мл 2 н. соляной кислоты. После отделения эфирной фазы кислотную водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают водой, а затем насыщенным раствором хлорида натрия,высушивают над безводным сульфатом натрия и после этого упаривают досуха. Неочищенный экстракт очищают хроматофафией на силикагеле (растворитель для элюирования: смесь циклогексан/этилацетат 20/1). Получают 53,3 г кетона 3'-1 (выход=70%). 1(m, 2H, СН 2, циклопропил). Следующие кетоны синтезируют аналогичным способом: 1-(4-этилфенил)-2-метоксиэтан-1-он, cоединение 3'.2; 2-циклопропил-1-(4-метилфенил)этан-1-он,cоединение 3'.3; 2-циклобутил-1-(4-фторфенил)этан-1-он, cоединение 3'.4. Методика Б. Способ, описанный для cоединения 3.5O-Бензилоксимы получают O-бензилированием соответствующих оксимов согласно описанному ниже способу (исходные оксимы получают из кетонов одним из двух описанных ранее для cоединения 4.1 способов синтеза). 29 1-(3-Фтор-4-метилфенил)-2-метоксиэтан-1 она O-бензилоксим, Z-изомер, cоединение 6'.1. Раствор 42,5 г (217 ммоль) 1-(3-фтор-4 метилфенил)-2-метоксиэтан-1-она оксима Z (соединение 4.1) в 100 мл диметилформамида перемешивают при 0C и порциями добавляют 15,6 г (325 ммоль; 1,5 экв.) гидрида натрия (50% в масле). Реакционную смесь перемешивают в течение 15 мин, затем медленно добавляют раствор, содержащий 30 мл (280 ммоль; 1,3 экв.) бензилбромида в 100 мл диметилформамида. Реакционную смесь перемешивают в течение 2 ч при комнатной температуре, после чего охлаждают до 0 С и добавляют 5 мл этанола, а затем 50 мл воды. Полученную смесь экстрагируют этилацетатом. Органическую фазу промывают водой, а затем насыщенным раствором хлорида натрия. Затем ее высушивают над сульфатом натрия и упаривают досуха. Полученное таким образом масло очищают хроматографией на силикагеле (элюент: смесь циклогексан/дихлорметан 7/3 (об./об Получают 39 г cоединения 6'.1 (Z); выход=63%. 1H ЯМР: 7,10-7,50 (Аr, m, 8 Н); 5,22 (-OCH2-Ph, s, 2 Н); 4,58 (-СН 2-O, s, 2H); 3,28 (ОСН 3,s, 3 Н); 2,26 (СН 3-Ph, d, J=1,8, 3 Н). Следующие соединения получают таким же способом: 1-(4-хлор-3-фторфенил)-2-метоксиэтан-1 она O-бензилоксим (Z), cоединение 6'.2; 1-(4-хлорфенил)-2-метоксиэтан-1-она O-бензилоксим (Z), cоединение 6'.3; 2-метокси-1-(3,4-метилендиоксифенил)этан 1-она O-бензилоксим (Z), cоединение 6'.4; 1-(4-этилфенил)-2-метоксиэтан-1-она O-бензилоксим (Z), cоединение 6'.5; 2-метокси-1-(4-метоксиметилфенил)этан 1-она O-бензилоксим (Z), cоединение 6'.6; 1-фенилбутан-1-она O-бензилоксим (Е), cоединение 6'.7; 1-(4-метоксиметилфенил)бутан-1-она O-бензилоксим (Е), cоединение 6'.8; 1-(4-метоксиметилфенил)пентан-1-она Oбензилоксим (Е), cоединение 6'.9; 2-циклопропил-1-фенилэтан-1-она O-бензилоксим (Е), cоединение 6'.10; 2-циклопропил-1-(4-фторфенил)этан-1-она(R)-1-(3-Фтор-4-метилфенил)-2-метоксиэтиламин, cоединение VI'.3. Раствор 86,5 г cоединения 2'.1 (330 ммоль) в 600 мл тетрагидрофурана перемешивают при температуре ниже 30 С, затем медленно добавляют 670 мл 1 М боран-тетрагидрофуранового раствора (670 ммоль). Температуре дают возможность подняться до комнатной температуры в течение 2 ч. Реакционную среду затем перемешивают при 0 С и добавляют 39 г (132 ммоль) соединения 6'.1, предварительно растворенного в 100 мл тетрагидрофурана. После перемешивания в течение 20 ч при комнатной температуре реакционную смесь охлаждают до 0 С и добавляют 1 л 2 н. соляной кислоты. Эту смесь оставляют перемешиваться в течение 16 ч. Смесь подщелачивают при 0 С добавлением 35% гидроксида натрия, затем экстрагируют этилацетатом. Этот экстракт промывают водой и насыщенным водным раствором хлорида натрия,затем высушивают над сульфатом натрия и упаривают досуха. Полученный остаток хроматографируют на колонке из силикагеля (элюент: смесь дихлорметан/метанол 95/5 (об./об Получают 17 г cоединения VI'. 3; выход=79%. 1(ВЭЖХ или хиральной SFC) этих аминов или соответствующих тиомочевин IV. Следующие соединения получают таким же способом:(S)-1-(3,4-метилендиоксифенил)бутиламин,cоединение VI'.26 (ее=84%). Третий способ. Для увеличения энантиомерного избытка указанные выше амины можно обработать органическими кислотами в форме чистых энантиомеров (например, N-ацетил-L-лейцином) и перекристаллизовать.(S)-1-(Фенил)бутиламин, cоединение VI'.9. Солеобразование с использованием N-ацетил-L-лейцина. Раствор 10,4 г (60 ммоль) N-ацетил-Lлейцина в 70 мл безводного метанола перемешивают при 60 С, затем по каплям добавляют раствор 9,0 г (60 ммоль) (S)-(1-фенил)бутиламина, cоединения VI'.9 (ее=97,1%), в 30 мл безводного метанола. В конце добавления метанольный раствор доводят до точки кипения(полного растворения) и оставляют стоять в течение ночи. После фильтрования и промывки 20 мл холодного безводного метанола выделяют 7,7 г кристаллов, которые растворяют в минимальном количестве воды. После подщелачивания 1 н. гидроксидом натрия и экстракции дихлорметаном органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над сульфатом натрия и упаривают под вакуумом. Получают 3,4 г амина в виде масла.N-[(R)-1-(4-Фторфенил)-2-метоксиэтил] тиомочевина, cоединение IV'.1. 4,23 мл (36,6 ммоль) бензоилхлорида добавляют при 0 С к перемешиваемому раствору 2,83 г(37,2 ммоль) аммонийизотиоцианата в 75 мл ацетона. Через 30 мин медленно добавляют 6 г(35,5 ммоль) cоединения VI',1, растворенного в 75 мл ацетона. Реакционную смесь перемешивают в течение 2 ч при комнатной температуре, а затем концентрируют при пониженном давлении. Суспензию переносят в 200 мл трет-бутилметилового эфира и 200 мл воды. Органическую фазу промывают водой, затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и после этого упаривают досуха. Остаток после упаривания растворяют в 180 мл этанола и к полученному раствору добавляют 3,75 мл (75 ммоль) гидразина моногидрата. После перемешивания в течение 24 ч при комнатной температуре реакционную смесь упаривают. Остаток после упаривания растворяют в 200 мл этилацетата и органическую фазу промывают водой, затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и упаривают досуха. Остаток после упаривания хроматографируют на колонке из силикагеля, элюируя смесью циклогексан/этилацетат 1/1 (об./об.). Получают 5 г (23 ммоль) твердого белого продукта; выход=63%; т.пл.=119 С. 1[]D19=+32,0 (с=0,87, СН 2 Сl2). Сверхкритическая хиральная хроматография: ее=100%. Следующие продукты получают таким же способом.N-[(5)-2-Циклопропил-1-фенилэтил]тиомочевина, cоединение IV'.10. Исходя из смеси, содержащей S энантиомер в качестве основного продукта (ее=95,8%), и после разделения хроматографией на фазе ChiracelOJ с элюированием смесью изогексан/этанол 97/3, получают чистый энантиомер (ее=100%). Т.пл.=84 С; []D19=+59,3 (с=1,06 CH2Cl2).F=140 С; []D20=+40,3 (с=1,18 CH2Cl2); ее=100%. б) Получение оптически активных тиомочевин (ее 99%) из рацемических тиомочевин с помощью хроматографии.N-[(S)-1-Фенилпентил]тиомочевина, cоединение IV'.27. Исходя из рацемической N-(1-фенилпентил)тиомочевины, и после разделения хроматографией на фазе Chiracel OJ с элюированием смесью изогексан/этанол 95/5 получают S энантиомер с энантиомерной чистотой 99,8%. Т.пл.=147 С; []D19=+46,0 (с=1,00 CH2Cl2). Получение NН-аминотиазолов в форме энантиомеров, соединения II'[4-(2-Хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил]-[(1R)-1-(4-фторфенил)-2-метоксиэтил]амин, соединение II'.1. 4,23 г (14,5 ммоль) 2-бром-1-(2'-хлор-4'метокси-5'-метилфенил)пропан-1-она (соединение III.1) и 4,2 мл (30 ммоль) триэтиламина добавляют к 3,28 г (14,3 ммоль) тиомочевины (cоединения IV'.1), растворенной в 70 мл этанола. Реакционную смесь перемешивают при 90 С в течение 3 ч и концентрируют при пониженном давлении. Остаток переносят в 200 мл дихлорметана и 100 мл воды. Органическую фазу промывают насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и упаривают досуха. Неочищенный экстракт очищают хроматографией на колонке из силикагеля, элюируя смесью циклогексан/этилацетат 4/1 (об./об.). Получают 5,27 г (12,5 ммоль)(m, CH, 1H); 3,81 (s, ОСН 3, 3H); 3,46-3,62 (m,OCH2, 2H); 3,35 (s, ОСН 3, 3H); 2,14 (s, СН 3, 3H); 2,04 (s, СН 3, 3H). Следующие промежуточные соединения получают таким же способом. Таблица 4 37 Получение N-замещенных аминотиазолов в форме энантиомера Пример 25. [4-(2-Хлор-4-метокси-5-метилфенил)-5-метилтиазол-2-ил]-[(1R)-1-(4-фторфенил)-2-метоксиэтил]проп-2-иниламин. Раствор 2,5 г (5,9 ммоль) cоединения II'.1 в 30 мл диметилформамида перемешивают при 0 С и добавляют 260 мг (6,5 ммоль) гидрида натрия, 60% в масле. Реакционную смесь перемешивают в течение 20 мин при 0 С, затем добавляют 0,83 мл (7,5 ммоль) 80% раствора пропаргилбромида в толуоле. Реакционную смесь перемешивают в течение 1 ч при 10 С с последующим добавлением при 0 С 2 мл этанола, а затем 50 мл воды. Смесь экстрагируют 200 мл этилацетата. Органическую фазу промывают водой, затем насыщенным раствором хлорида натрия, высушивают над безводным сульфатом натрия и после этого упаривают досуха. Неочищенный остаток хроматографируют на колонке из силикагеля, элюируя смесью циклогексан/этилацетат 9/1 (об./об.). Получают 2,14 г вязкого чистого продукта; выход=80%; MS ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединения, в рацемической форме или в форме чистого энантиомера, формулы где R1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; гидрокси(С 1-С 5)алкил; (С 1 С 5)алкил; аралкил, в котором арильная часть представляет собой (С 6-С 8), а алкильная часть представляет собой (С 1-С 4); (С 1-С 5)алкокси; трифторметильную группу; нитрогруппу; нитрильную группу; группу -SR, где R представляет собой водород, (С 1-С 5)апкил или аралкил, в котором арильная часть представляет собой (С 6-С 8),а алкильная часть представляет собой (С 1-С 4); группу -S-CO-R, где R представляет собой (С 1 С 5)алкильный или аралкильный радикал, в котором арильная часть представляет собой (С 6-С 8),а алкильная часть представляет собой (С 1-С 4); группу -COORa, где Ra представляет собой водород или (С 1-С 5)алкил; группу -CONRaRb с Ra и Rb такими, как определено выше для Ra; группу -NRaRb с Ra и Rb такими, как определено выше для Ra; группу -CONRcRd или -NRcRd,где Rс и Rd составляют с атомом азота, к которому они присоединены, 5-7-членный гетероцикл; или группу -NHCO-NRaRb с Ra и Rb такими, как определено выше для Ra;R3 представляет собой водород или является таким, как определено выше для R1 и R2; или, альтернативно, R2 составляет с R3, когда последний является заместителем фенила в положении 5, группу -Х-СН 2-Х-, где Х независимо представляет собой СН 2 или атом кислорода или серы;-O-СН 2-O- по двум соседним атомам углерода фенила, группой -СF3, -NO2 или -CN, группойR1 и R2, которые могут быть одинаковыми или разными, каждый независимо представляет собой атом галогена; (С 1-С 5)алкил; (С 1-С 5)алкокси;R3 представляет собой водород или является таким, как определено выше для R1 и R2;R7 представляет собой фенил, который не замещен или моно- или дизамещен в положении 3 или 4 галогеном, (С 1-С 5)алкильной группой,группой -CH2OR8, где R8 представляет собой(пример 61),а также их соответствующих оснований,других солей присоединения и сольватов и/или гидратов. 5. Способ получения соединений формулы(I) по п.1, отличающийся тем, что эти соединения получают путем алкилирования соединений формулы где R4 представляет собой метил, a R1, R2, R3, R6 и R7 такие, как определено для (I). 6. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение по п.1 в качестве активного начала. 7. Применение соединения по п.1 для приготовления лекарственных продуктов, предназначенных для предупреждения и/или лечения кортикотропин-рилизинг-фактор (КРФ)-зависимых состояний. 8. Применение соединения по п.1 для приготовления лекарственного продукта, предназначенного для превентивного или куративного лечения патологии, в которую вовлечен КРФ,выбранной из связанных со стрессом состояний,болезни Кушинга, нервно-психиатрических расстройств, таких как депрессивные, тревожные,панические, обсессивно-компульсивные расстройства, расстройства настроения, посттравматический стресс, расстройства поведения,агрессивность, анорексия, булимия, гипергликемия, преждевременные роды, беременность с повышенным риском, замедленное развитие,нарушения сна, эпилепсия и все типы депрессии; болезни Альцгеймера, болезни Паркинсона,хореи Гентингтона; бокового амиотрофического склероза; сосудистых, сердечных и церебральных расстройств; расстройств сексуальной деятельности и расстройств фертильности; иммунодепрессии, иммуносупрессии, воспалительных процессов, множественных инфекций, ревматоидного артрита, остеоартрита, увеита, псориаза и диабета; раковых заболеваний; желудочно-кишечных функциональных расстройств и являющихся их результатом воспалений, раздраженного и воспаленного кишечника, диареи; расстройств восприятия боли, фибромиалгий,которые могут быть связаны или не связаны с нарушениями сна, утомлением или мигренью; симптомов, ассоциированных с алкогольной зависимостью и отменой лекарственных средств.
МПК / Метки
МПК: C07D 277/42, C07D 417/12, A61K 31/426, A61P 25/00
Метки: лигандов, аминотиазола, производные, рецепторов, качестве, крф, применение
Код ссылки
<a href="https://eas.patents.su/22-5159-proizvodnye-aminotiazola-i-ih-primenenie-v-kachestve-ligandov-receptorov-krf.html" rel="bookmark" title="База патентов Евразийского Союза">Производные аминотиазола и их применение в качестве лигандов рецепторов крф</a>
Производные индолин-2-она, их получение и их применение в качестве лигандов рецепторов окситоцина

Номер патента: 5093
Опубликовано: 28.10.2004
Авторы: Фулон Луак, Серрадейль-Ле Галь Клодин, Валетт Жерар, Гарсия Жорж
МПК: C07D 209/34, A61P 5/10, A61K 31/404...
Метки: применение, качестве, лигандов, окситоцина, получение, индолин-2-она, рецепторов, производные
Формула / Реферат:
1. Соединения в форме чистого энантиомера или смеси энантиомеров формулы в которой R0 представляет собой группу, выбранную из 1) в которой Z1 представляет собой атом хлора, брома, йода или фтора или (C1-C4)алкильную, (C1-C4)алкокси или трифторметильную группу; Z2 представляет собой атом водорода, хлора, брома, йода или фтора или (C1-C4)алкильную, (C3-C5)циклоалкильную, (C1-C4)алкокси, (C3-C5)циклоалкокси или полифтор(C1-C4)алкильную...
Новые производные 1,3-дигидро-2н-индол-2-она и их применение в качестве лигандов рецепторов аргинин-вазопрессина v1b и v1a

Номер патента: 4628
Опубликовано: 24.06.2004
Авторы: Серрадель-Ле Галь Клодин, Ру Ришар, Тоннерр Бернар, Ваньон Жан
МПК: A61K 31/404, A61P 43/00, C07D 403/04...
Метки: производные, лигандов, рецепторов, качестве, новые, применение, 1,3-дигидро-2н-индол-2-она, аргинин-вазопрессина
Формула / Реферат:
1. Соединение формулы где R1 представляет собой атом галогена; (C1-C4)алкил; (C1-C4)алкокси; трифторметильный радикал; трифторметокси радикал; R2 представляет собой атом водорода; атом галогена; (C1-C4)алкил; (C1-C4)алкокси; трифторметильный радикал; или R2 находится в положении -6-индол-2-онового кольца и R1 и R2 вместе представляют собой двухвалентный триметиленовый радикал; R3 представляет собой атом галогена; гидроксил; (C1-C2)алкил;...
Производные триазолопиридазина в качестве лигандов для гамк рецепторов

Номер патента: 2755
Опубликовано: 29.08.2002
Авторы: Мэйдин Эндрю, Расселл Майкл Джеффри Нейл, Карлинг Вилльям Роберт, Харрисон Тимоти, Гвиблин Александер Ричард, Стрит Лесли Джозеф, Мур Кевин Вилльям, Коллинз Ян Джеймс, Кастро Пинейро Хосе Луис, Скотт Гейл
МПК: A61P 25/08, A61K 31/5025, C07D 487/04...
Метки: рецепторов, триазолопиридазина, качестве, гамк, производные, лигандов
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль где Z представляет собой трифторметил, 2-метилпропил, 2,2-диметилпропил, 3-метилбутил, 1-фторбут-3-енил, циклобутил, 1-метилциклобутил, 1-фторциклобутил, 3-фторциклобутил, 3,3-дифторциклобутил, 3-гидроксициклобутил, 3-бензилоксициклобутил, 3-оксоциклобутил, 1-метилциклогексил, 4,4-дифтор-1-метилциклогексил, циклопентилметил, 4-фторциклогекс-3-енил, 3-фторфенил, тетрагидрофур-2-ил,...
Производные 3-азабицикло[3.1.0]гексана в качестве лигандов опиатных рецепторов

Номер патента: 4672
Опубликовано: 24.06.2004
Авторы: Ланн Грэм, Петтман Алан Джон, Гибсон Стивен Пол, Банкс Бернард Джозеф, Крук Роберт Джеймс
МПК: A61P 17/02, C07D 209/52, A61K 31/40...
Метки: производные, лигандов, 3-азабицикло[3.1.0]гексана, качестве, рецепторов, опиатных
Формула / Реферат:
1. Вещество, которое представляет собой соединение формулы I где кольцо "Ar" представляет собой фенил или 6-членное гетероарильное кольцо; R1, взятый в отдельности, представляет собой H, галоген, NH2, NY2WY1, Het1 или OE, Y2 представляет собой H, W представляет собой SO2 или C(O)O, Y1 представляет собой C1-10алкил (возможно замещенный одним или более чем одним заместителем, независимо выбранным из галогена, OH, C1-4алкокси, CONH2,...
Замещенные производные триазоло-пиридазина в качестве лигандов рецепторов гамк

Номер патента: 2436
Опубликовано: 25.04.2002
Авторы: Гиблин Александр Ричард, Кастро Пинейро Хосе Луис, Карлинг Вилльям Роберт, Стрит Лесли Джозеф, Мур Кевин Вилльям, Мэдин Эндрю, Бротон Говард Барфф, Расселл Майкл Джеффри
МПК: C07D 487/00, A61K 31/5025, A61P 25/00...
Метки: лигандов, качестве, замещенные, триазоло-пиридазина, рецепторов, производные, гамк
Формула / Реферат:
1. Соединение формулы I, его соль или пролекарство где Y означает водород или C1-6алкил; и Z означает C1-6алкил, С3-7циклоалкил, С4-7циклоалкенил, арил, С3-7гетероциклоалкил, гетероарил или ди(C1-6)алкиламино, причем любая из групп может быть необязательно замещена; или Y и Z, взятые вместе с двумя расположенными между ними атомами углерода, образуют кольцо, выбранное из С5-9циклоалкенила, С6-10бициклоалкенила, тетрагидропиридинила, пиридинила...
Предыдущий патент: Применение ингибиторов cyp2d6 в комбинированных способах лечения
Следующий патент: Новые соединения октагидро-2h-пиридо[1,2-α]пиразина, способ их получения и фармацевтические препараты их содержащие
Случайный патент: Способ получения массы для производства профилированных строительных элементов и масса для производства профилированных строительных элементов