Пиразольные соединения














Формула / Реферат
1. Соединение формулы

где X представляет собой следующий фрагмент:

или фармацевтически приемлемая соль указанного соединения.
2. Соединение или фармацевтически приемлемая соль по п.1, которое представляет собой

3. Соединение по п.2, которое представляет собой

4. Способ лечения диабета у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-3.
5. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 в терапии заболеваний и расстройств, связанных с гипергликемией.
6. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета.
7. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета 1 типа.
8. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета 2 типа.
9. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета.
10. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета 1 типа.
11. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета 2 типа.
12. Фармацевтическая композиция для лечения диабета, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-3 в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами.
Текст
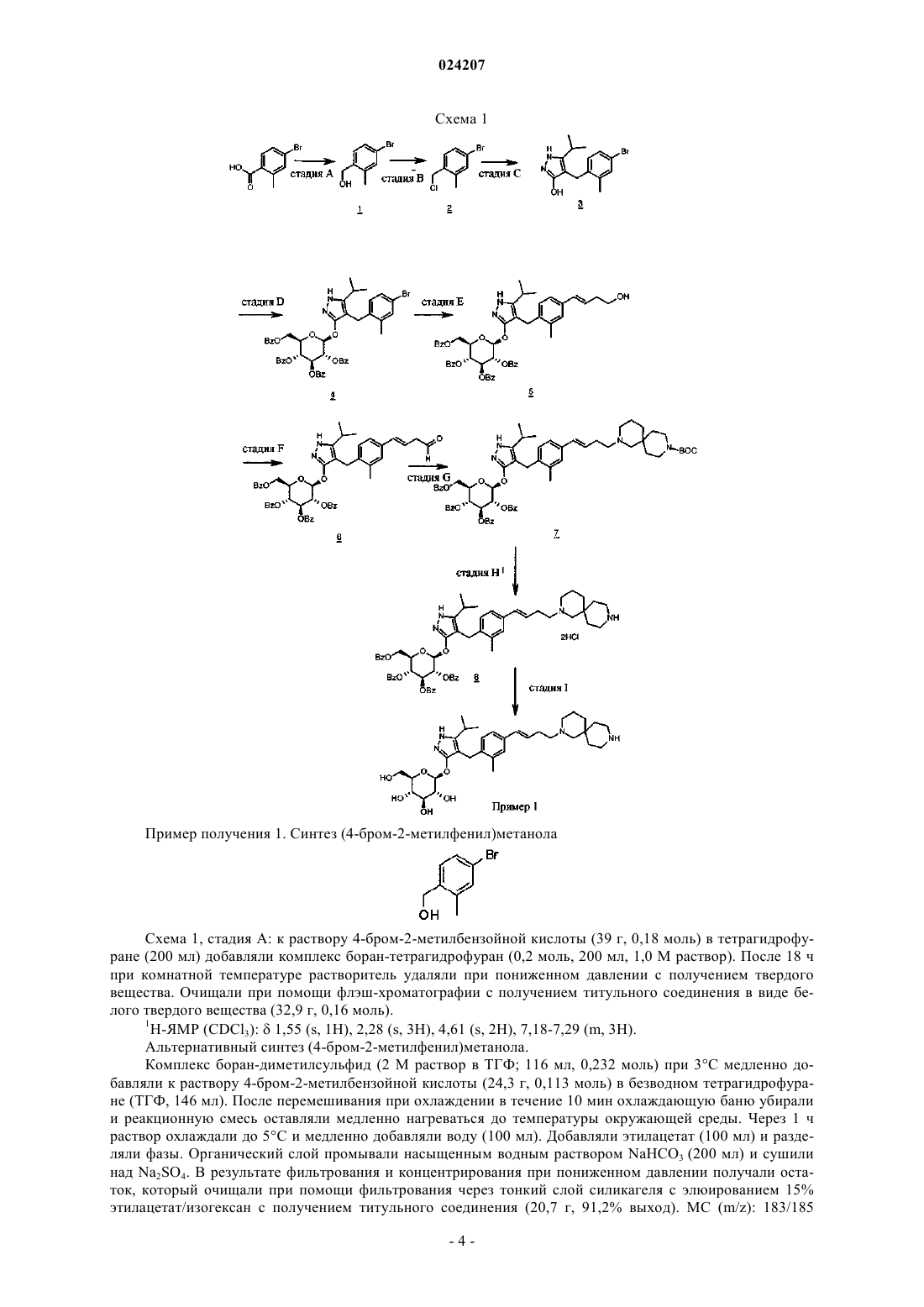
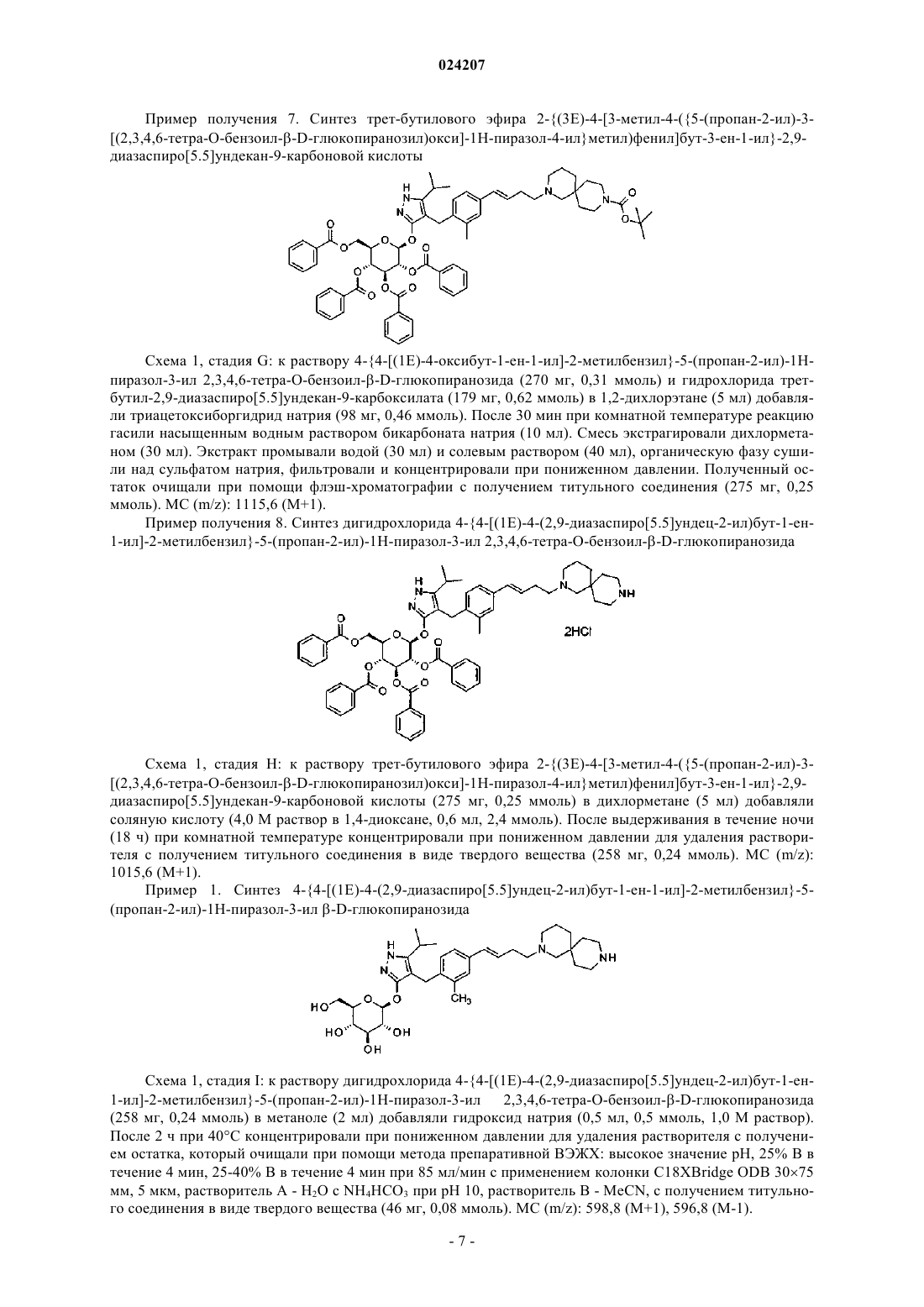
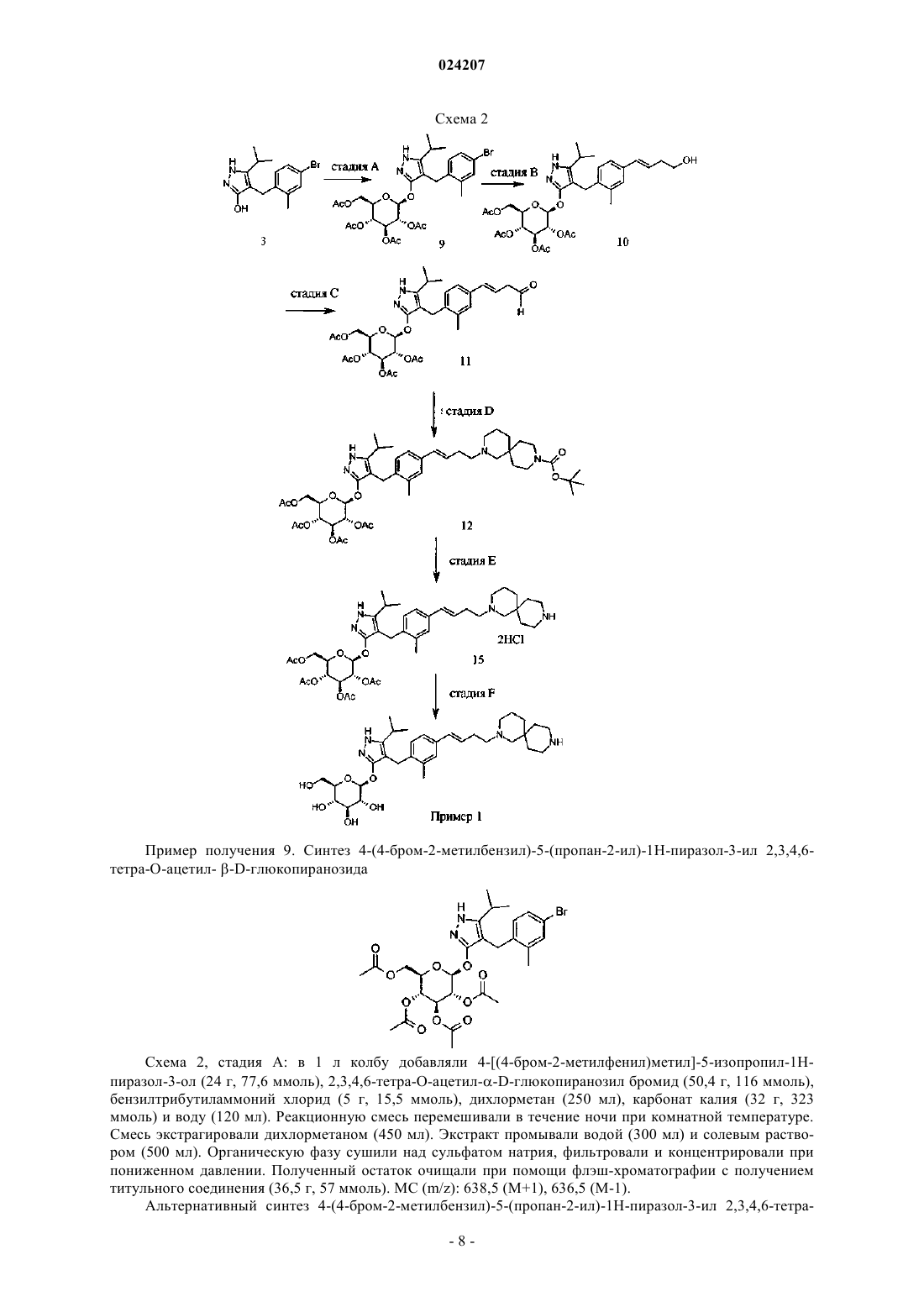
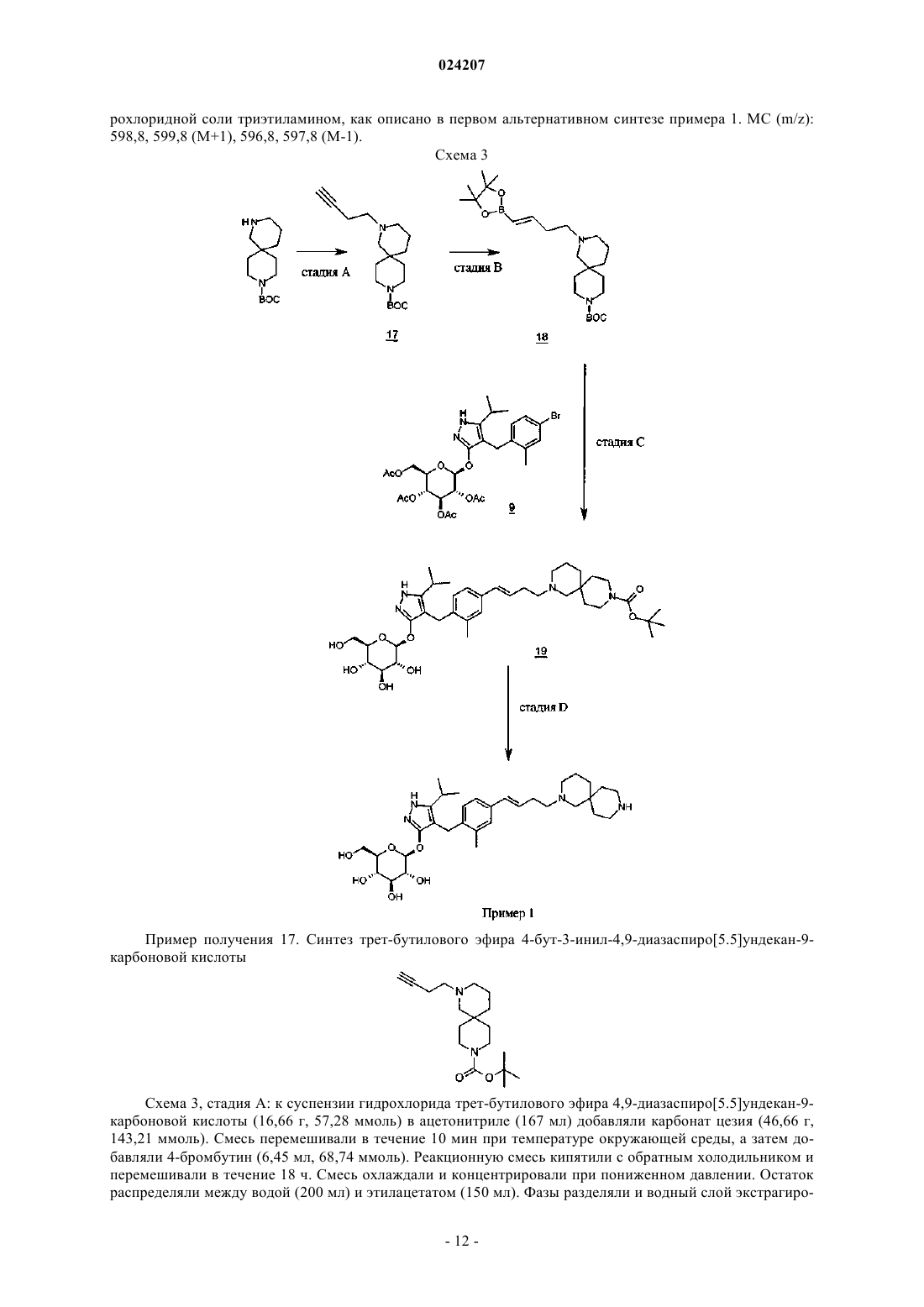
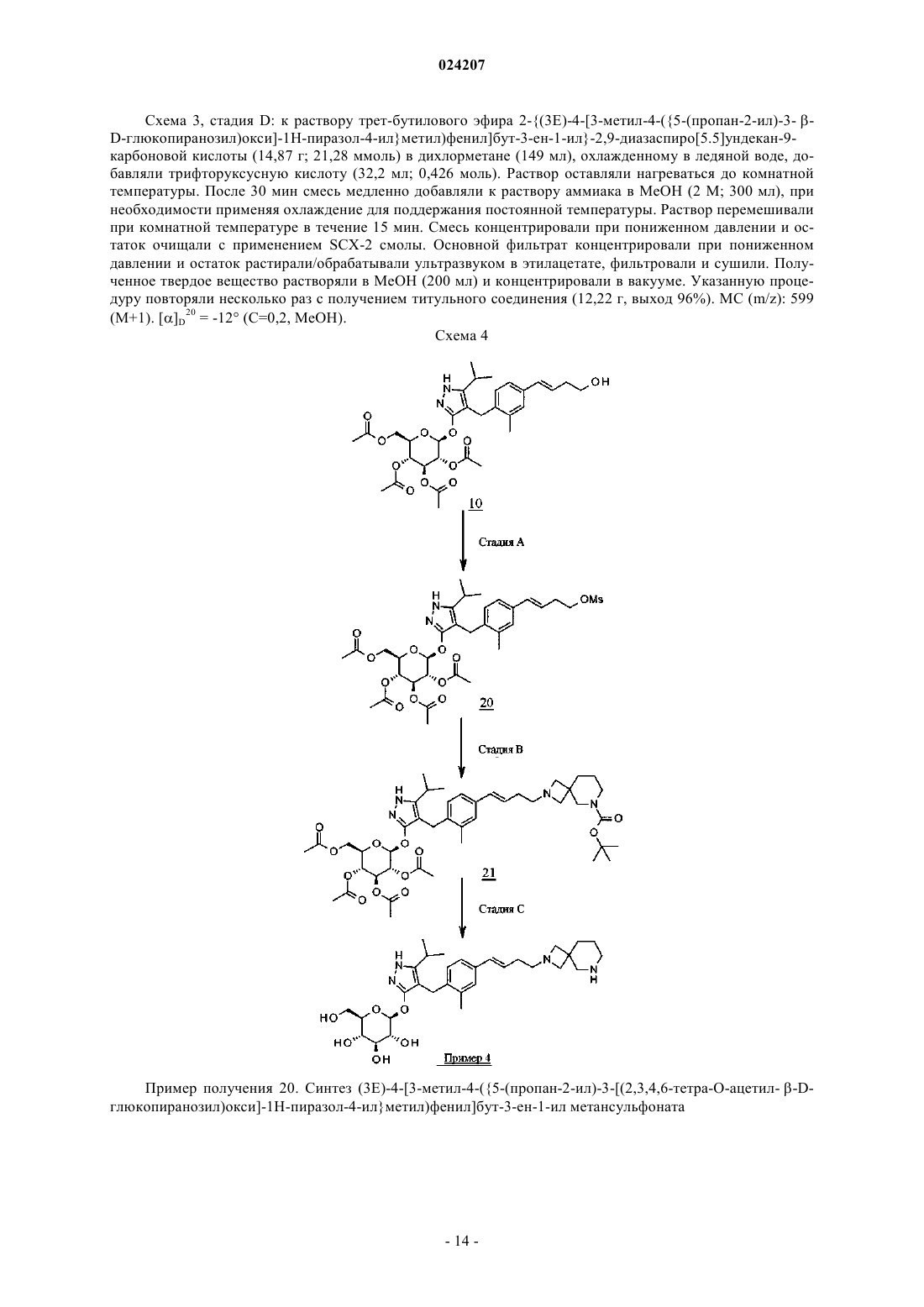
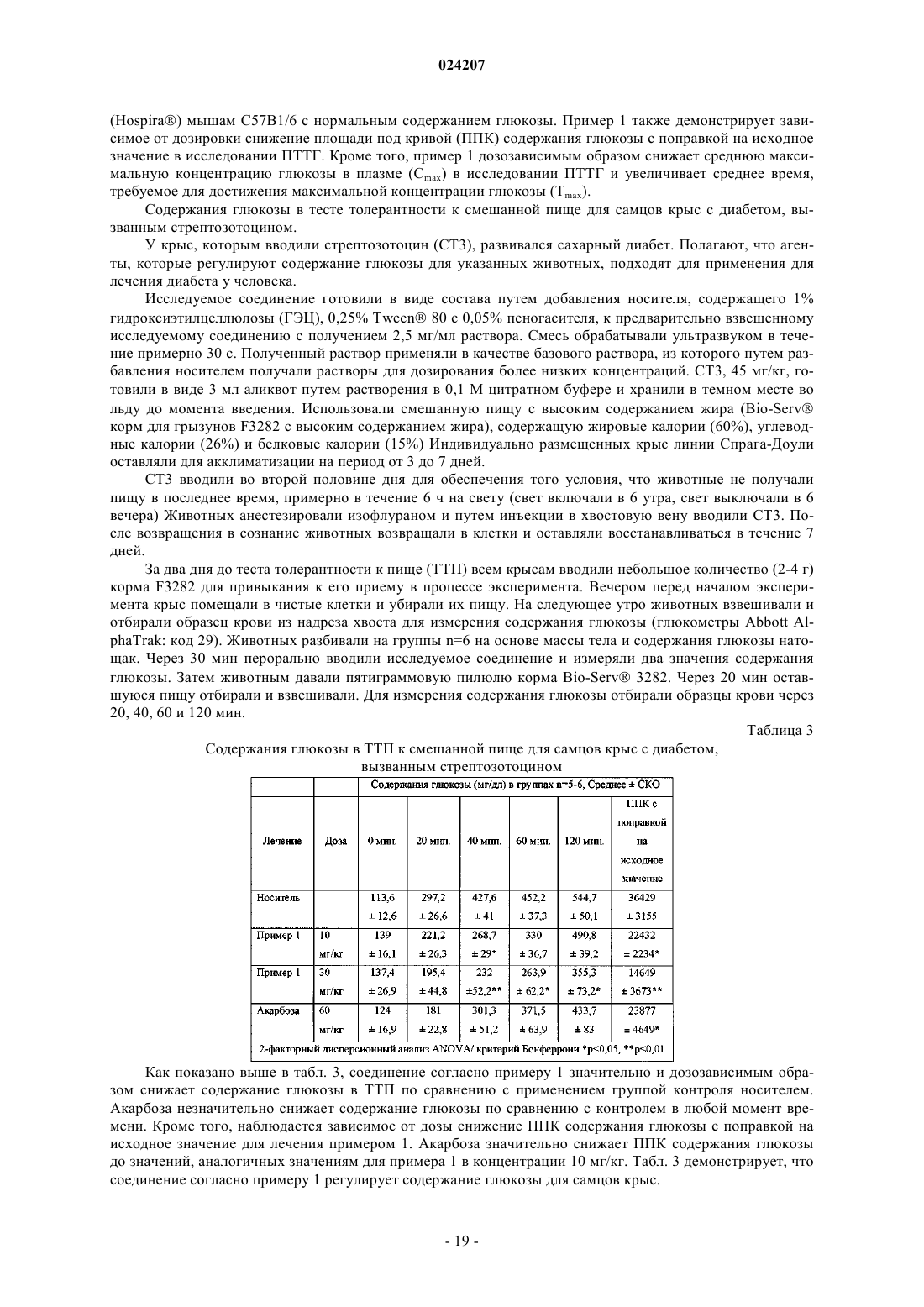
В настоящем изобретении предложено соединение формулы II где X представляет собой следующий фрагмент: или фармацевтически приемлемая соль указанного соединения.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к новым пиразольным соединениям, к фармацевтическим композициям, содержащим такие соединения, к способам применения таких соединений для лечения физиологических расстройств и к промежуточным соединениям и способам, подходящим для применения для синтеза таких соединений. Настоящее изобретение относится к области лечения диабета и других заболеваний и расстройств,связанных с гипергликемией. Диабет представляет собой группу заболеваний, характеризующихся высоким содержанием глюкозы в крови. Ему подвержены примерно 25 млн человек в Соединенных Штатах,и он является седьмой по значимости причиной смерти в США согласно национальному информационному бюллетеню по вопросам диабета 2011 (Министерство здравоохранения и социальных служб США,центры контроля и профилактики заболеваний). Натрийзависимые котранспортеры глюкозы (SGLT) представляют собой переносчики, которые отвечают за поглощение углеводов, таких как глюкоза. В частности, SGLT1 отвечает за перенос глюкозы через мембрану клеток щеточной каемки тонкого кишечника. Ингибирование SGLT1 может приводить к снижению абсорбции глюкозы в тонком кишечнике, что обеспечивает подходящий для применения способ лечения диабета. В патенте США 7655632 описаны конкретные производные пиразола, обладающие ингибирующей активностью в отношении SGLT1 человека, которые также описаны в качестве подходящих для применения для предотвращения или лечения заболевания, связанного с гипергликемией, такого как диабет. Кроме того, в публикации международной заявки на патент WO 2011/039338 описаны конкретные производные пиразола, обладающие ингибирующей активностью в отношении SGLT1/SGLT2, которые также описаны в качестве подходящих для применения для лечения заболеваний костей, таких как остеопороз. Существует потребность в альтернативных лекарственных средствах и лечении диабета. В настоящем изобретении предложены некоторые новые ингибиторы SGLT1, которые могут быть подходящими для лечения диабета. Соответственно, в настоящем изобретении предложено соединение формулы II где X представляет собой следующий фрагмент: или фармацевтически приемлемая соль указанного соединения. В настоящем изобретении также предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения. В настоящем изобретении также предложен способ лечения диабета у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формул I или II или фармацевтически приемлемой соли указанного соединения. В настоящем изобретении также предложен способ лечения диабета 1 типа у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формул I или II или фармацевтически приемлемой соли указанного соединения. Кроме того, в настоящем изобретении предложен способ лечения диабета 2 типа у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного коли-1 024207 чества соединения формул I или II или фармацевтически приемлемой соли указанного соединения. В настоящем изобретении также предложен способ лечения нарушения толерантности к глюкозе (НТГ),нарушения гликемии натощак (НГН) или метаболического синдрома у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формул I или II или фармацевтически приемлемой соли указанного соединения. Кроме того, в настоящем изобретении предложено соединение формул I или II или фармацевтически приемлемая соль указанного соединения для применения в терапии, в частности для лечения диабета. Кроме того, в настоящем изобретении предложено соединение формул I или II или фармацевтически приемлемая соль указанного соединения для применения для лечения диабета 1 типа. Кроме того, в настоящем изобретении предложено соединение формул I или II или фармацевтически приемлемая соль указанного соединения для применения для лечения диабета 2 типа. В настоящем изобретении также предложено применение соединения формул I или II или фармацевтически приемлемой соли указанного соединения для получения лекарственного средства для лечения диабета. Кроме того, в настоящем изобретении предложено применение соединения формул I или II или фармацевтически приемлемой соли указанного соединения для получения лекарственного средства для лечения диабета 1 типа. В настоящем изобретении также предложено применение соединения формул I илиII или фармацевтически приемлемой соли указанного соединения для получения лекарственного средства для лечения диабета 2 типа. В настоящем изобретении также предложено применение соединения формул I или II или фармацевтически приемлемой соли указанного соединения для получения лекарственного средства для лечения НТГ,НГН или метаболического синдрома. В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формул I или II или фармацевтически приемлемую соль указанного соединения в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами. В конкретном варианте реализации композиция дополнительно содержит один или более других терапевтических агентов. Настоящее изобретение также охватывает новые промежуточные соединения и способы синтеза соединения формул I или II. Термины "лечение" или "лечить", применяемые в настоящей заявке, включают предотвращение, ограничение, замедление, прекращение или обращение развития или тяжести имеющегося симптома или расстройства. Термин "пациент", применяемый в настоящей заявке, относится к млекопитающему, такому как мышь, морская свинка, крыса, собака или человек. Понятно, что предпочтительным пациентом является человек. Термин "эффективное количество", применяемый в настоящей заявке, относится к количеству или дозе соединения согласно настоящему изобретению или фармацевтически приемлемой соли указанного соединения, которое при введении пациенту в виде одной или множества доз обеспечивает целевой эффект у пациента, подвергающегося диагностике или лечению. Эффективное количество может быть легко определено лечащим врачом, как специалистом в данной области, при помощи известных способов и изучения результатов, полученных в аналогичных условиях. При определении эффективного количества для пациента лечащий врач рассматривает ряд факторов, включая, но, не ограничиваясь ими, вид млекопитающего; его размер, возраст и общее состояние здоровья; конкретное заболевание или расстройство; степень, поражение или тяжесть заболевания или расстройства; ответную реакцию у конкретного пациента; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого препарата; выбранный режим дозировки; применение сопутствующего лечения и другие соответствующие условия. Соединения формул I и II обычно являются эффективными в широком диапазоне доз. Например,дневная доза обычно находится в диапазоне от примерно 0,01 до примерно 30 мг/кг массы тела. В некоторых случаях более подходящими могут быть дозы меньше нижнего предела вышеуказанного диапазона, в то время как в других случаях могут быть применены более высокие дозировки без проявления вредных побочных эффектов, и, следовательно, описанный выше диапазон доз не ограничивает объем настоящего изобретения никоим образом. Соединения согласно настоящему изобретению предпочтительно готовят в виде фармацевтических композиций, вводимых при помощи любого из способов, обеспечивающих биодоступность соединения. Наиболее предпочтительно указанные композиции предназначены для перорального введения. Указанные фармацевтические композиции и способы их получения хорошо известны в данной области техники.(См., например, Remington: The Science and Practice of Pharmacy (D.B. Troy, Editor, 21st Edition., Lippincott, WilliamsWilkins, 2006). В другом аспекте настоящего изобретения соединения вводят в комбинации с одним или более терапевтическими агентами, такими как противодиабетические агенты. Введение в комбинации включает одновременное или последовательное введение. Кроме того, одновременное введение комбинации может проводиться в виде одной комбинированной дозы или отдельных доз каждого терапевтического агента. Примеры противодиабетических агентов включают метформин; ДПП-IV ингибитор (ингибитор дипептидилпептидазы IV), такой как ситаглиптин или линаглиптин; сульфонилмочевину, такую как глимепи-2 024207 рид; тиазолидиндион, такой как пиоглитазон; базальный инсулин, такой как гларгин; быстродействующий инсулин, такой как HUMALOG или NOVOLOG; агонист ГПП-1 (глюкагоноподобный пептид-1),такой как эксенатид или лираглутид; ингибитор SGLT2, такой как дапаглифлозин или эмпаглифлозин; антагонист глюкагонового рецептора, такой как LY2409021; и т.п. Соединения формул I и II получают, как показано ниже в примерах и схемах. Реагенты и исходные вещества легко доступны специалисту в данной области техники. Если не указано иное, все заместители являются такими, как определено ранее. Понятно, что указанные схемы, примеры получения и примеры не ограничивают объем настоящего изобретения никоим образом. Примеры методов разделения включают способы селективной кристаллизации или хиральную хроматографию. (См., например, J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley andSons, Inc., 1981, и E.L. Eliel and S.H. Wilen, "Stereochemistry of Organic Compounds", Wiley-Interscience,1994). Специалисту в данной области техники должно быть понятно, что разделение и выделение при помощи хроматографии, хиральной хроматографии или селективной кристаллизации индивидуальных диастереомеров или геометрических изомеров соединения формулы I или II или индивидуальных диастереомеров или геометрических изомеров промежуточных соединений, позволяющих получить соединение формулы I или II, может быть проведено в любой подходящий момент в процессе синтеза. Согласно настоящей заявкеотносится к сдвигу относительно тетраметилсилана в долях на миллион; "мин" относится к минуте или минутам; "ТГФ" относится к тетрагидрофурану; "MeOH" относится к метанолу или метиловому спирту; "ВЭЖХ" относится к высокоэффективной жидкостной хроматографии; термин "Ac" относится к ацетильному заместителю следующей структуры: Термин "Bz" относится к бензоильному заместителю следующей структуры: Термин "Boc" относится к трет-бутилоксикарбонильной защитной группе. Фармацевтически приемлемые соли и общепринятые способы их получения хорошо известны в данной области. См., например, Gould, PL., "Salt selection for basic drugs", International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin et al. "Salt Selection and Optimization Procedures for Pharmaceutical NewChemical Entities", Organic Process Research and Development, 4: 427-435 (2000); и S.M. Berge, et al.,"Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Специалисту в области синтеза понятно, что соединения формулы I и II, такие как амины, представляют собой органические основания и могут быть легко превращены и выделены в виде фармацевтически приемлемых солей,таких как тартрат или HCl соли, с применением способов и условий, хорошо известных специалисту в данной области техники. Схема 1, стадия A: к раствору 4-бром-2-метилбензойной кислоты (39 г, 0,18 моль) в тетрагидрофуране (200 мл) добавляли комплекс боран-тетрагидрофуран (0,2 моль, 200 мл, 1,0 М раствор). После 18 ч при комнатной температуре растворитель удаляли при пониженном давлении с получением твердого вещества. Очищали при помощи флэш-хроматографии с получением титульного соединения в виде белого твердого вещества (32,9 г, 0,16 моль). 1H-ЯМР (CDCl3):1,55 (s, 1H), 2,28 (s, 3H), 4,61 (s, 2H), 7,18-7,29 (m, 3H). Альтернативный синтез (4-бром-2-метилфенил)метанола. Комплекс боран-диметилсульфид (2 М раствор в ТГФ; 116 мл, 0,232 моль) при 3C медленно добавляли к раствору 4-бром-2-метилбензойной кислоты (24,3 г, 0,113 моль) в безводном тетрагидрофуране (ТГФ, 146 мл). После перемешивания при охлаждении в течение 10 мин охлаждающую баню убирали и реакционную смесь оставляли медленно нагреваться до температуры окружающей среды. Через 1 ч раствор охлаждали до 5C и медленно добавляли воду (100 мл). Добавляли этилацетат (100 мл) и разделяли фазы. Органический слой промывали насыщенным водным раствором NaHCO3 (200 мл) и сушили над Na2SO4. В результате фильтрования и концентрирования при пониженном давлении получали остаток, который очищали при помощи фильтрования через тонкий слой силикагеля с элюированием 15% этилацетат/изогексан с получением титульного соединения (20,7 г, 91,2% выход). MC (m/z): 183/185(200 мл) и диметилформамиде (0,025 моль, 2,0 мл) при 0C добавляли тионилхлорид (14,31 мл, 0,2 моль). После 1 ч при комнатной температуре смесь выливали в ледяную воду (100 г), экстрагировали дихлорметаном (300 мл), экстракт промывали 5% вод. раствором бикарбоната натрия (30 мл) и солевым раствором(200 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного титульного соединения в виде белого твердого вещества (35,0 г, 0,16 моль). Вещество применяли на следующей стадии реакции без дополнительной очистки. 1H-ЯМР (CDCl3):2,38 (s, 3H),4,52 (s, 2H), 7,13-7,35 (m, 3H). Альтернативный синтез 4-бром-1-хлорметил-2-метилбензола. Метансульфонилхлорид (6,83 мл, 88,3 ммоль) медленно добавляли к раствору (4-бром-2 метилфенил)метанола (16,14 г, 80,27 ммоль) и триэтиламина (16,78 мл; 120,4 ммоль) в дихлорметане(80,7 мл), охлажденному в смеси лед/вода. Смесь оставляли медленно нагреваться до температуры окружающей среды и перемешивали в течение 16 ч. Добавляли еще метансульфонилхлорида (1,24 мл; 16,1 ммоль) и смесь перемешивали при температуре окружающей среды в течение 2 ч. Добавляли воду (80 мл) и фазы разделяли. Органический слой промывали соляной кислотой (1 н.; 80 мл), затем насыщенным водным раствором гидрокарбоната натрия (80 мл), затем водой (80 мл) и сушили над Na2SO4. В результате фильтрования и концентрирования при пониженном давлении получали остаток, который очищали при помощи флэш-хроматографии (с элюированием гексаном) с получением титульного соединения Схема 1, стадия C: к раствору метилового эфира 4-метил-3-оксовалериановой кислоты (27,1 мл,0,19 моль) в тетрагидрофуране при 0C добавляли гидрид натрия (8,29 г, 0,21 моль, 60% дисперсия в масле). После 30 мин при комнатной температуре добавляли раствор 4-бром-1-хлорметил-2 метилбензола (35,0 г, 0,16 моль) в тетрагидрофуране (50 мл). Полученную смесь грели при 70C в течение ночи (18 ч). Реакцию гасили добавлением 1,0 М раствора HCl (20 мл). Экстрагировали этилацетатом(200 мл), экстракт промывали водой (200 мл) и солевым раствором (200 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток растворяли в толуоле (200 мл) и добавляли моногидрат гидразина (23,3 мл, 0,48 моль). Смесь грели при 120C в течение 2 ч с применением прибора Дина-Старка для удаления воды. Смесь охлаждали и растворитель удаляли при пониженном давлении, остаток растворяли в дихлорметане (50 мл) и метаноле (50 мл). Указанный раствор медленно переносили в стакан с водой (250 мл). Полученный продукт, выпавший в осадок, собирали при помощи вакуумного фильтрования. Сушили под вакуумом в сушильном шкафу при 40C в течение ночи с получением титульного соединения в виде твердого вещества (48,0 г, 0,16 моль). MC (m/z): 311,0(M+1), 309,0 (M-1). Альтернативный синтез 4-[(4-бром-2-метилфенил)метил]-5-изопропил-1H-пиразол-3-ола. Готовили раствор 4-бром-1-хлорметил-2-метилбензола (13,16 г, 59,95 ммоль) в ацетонитриле (65,8 мл). Добавляли карбонат калия (24,86 г, 179,9 ммоль), йодид калия (11,94 г, 71,94 ммоль) и метиловый эфир 4-метил-3-оксовалериановой кислоты (8,96 мл; 62,95 ммоль). Полученную смесь перемешивали при температуре окружающей среды в течение 20 ч. Добавляли соляную кислоту (2 н.) до получения pH 3. Раствор экстрагировали этилацетатом (100 мл), органическую фазу промывали солевым раствором (100 мл) и сушили над Na2SO4. Смесь фильтровали и концентрировали при пониженном давлении. Остаток растворяли в толуоле (65,8 мл) и добавляли моногидрат гидразина (13,7 мл, 0,180 моль). Полученную смесь кипятили с обратным холодильником и удаляли воду при помощи прибора Дина-Старка. Через 3 ч смесь охлаждали до 90C и добавляли еще моногидрат гидразина (13,7 мл; 0,180 моль), смесь кипятили с обратным холодильником в течение 1 ч. Смесь охлаждали и концентрировали при пониженном давлении. Полученное твердое вещество растирали с водой (200 мл), фильтровали и сушили в вакуумном сушильном шкафу над Р 2 О 5 при 60C. Твердое вещество растирали в изогексане (200 мл) и фильтровали с получением титульного соединения (14,3 г; 77,1% выход). MC (m/z): 309/311 (M+1). Пример получения 4. Синтез 4-(4-бром-2-метилбензил)-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6 тетра-O-бензоилD-глюкопиранозида(100 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Экстрагировали дихлорметаном (500 мл). Экстракт промывали водой (300 мл) и солевым раствором (500 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали при помощи флэш-хроматографии с получением титульного соединения (37 г, 64 ммоль). MC (m/z): 889,2 (M+1), 887,2 (M-1). Пример получения 5. Синтез 4-4-[(1E)-4-гидроксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)1H-пиразол-3-ил 2,3,4,6-тетра-O-бензоилD-глюкопиранозида Схема 1, стадия E: к раствору 4-(4-бром-2-метилбензил)-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6 тетра-O-бензоилD-глюкопиранозида (3 г, 3,4 ммоль) в ацетонитриле (30 мл) и триэтиламине (20 мл) добавляли 3-бутен-1-ол (0,58 мл, 6,8 ммоль). Раствор дегазировали азотом в течение 10 мин. Добавляли три-o-толилфосфин (205 мг, 0,67 ммоль) и ацетат палладия (76 мг, 0,34 ммоль). Кипятили с обратным холодильником при 90C в течение 2 ч. Охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления растворителя. Остаток очищали при помощи флэш-хроматографии с получением титульного соединения (2,1 г, 2,4 ммоль). MC (m/z): 878,4 (M+1). Пример получения 6. Синтез 4-4-[(1E)-4-оксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1Hпиразол-3-ил 2,3,4,6-тетра-О-бензоилD-глюкопиранозида(134 мг, 0,96 ммоль). После 15 мин при комнатной температуре реакцию гасили насыщенным водным раствором тиосульфата натрия (10 мл). Смесь экстрагировали дихлорметаном (30 мл). Экстракт промывали водой (30 мл) и солевым раствором (40 мл). Органическую фазу сушили над сульфатом натрия,фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии с получением титульного соединения (270 мг, 0,31 ммоль). MC (m/z): 876,5 (M+1),874,5 (M-1). Схема 1, стадия G: к раствору 4-4-[(1E)-4-оксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1Hпиразол-3-ил 2,3,4,6-тетра-O-бензоилD-глюкопиранозида (270 мг, 0,31 ммоль) и гидрохлорида третбутил-2,9-диазаспиро[5.5]ундекан-9-карбоксилата (179 мг, 0,62 ммоль) в 1,2-дихлорэтане (5 мл) добавляли триацетоксиборгидрид натрия (98 мг, 0,46 ммоль). После 30 мин при комнатной температуре реакцию гасили насыщенным водным раствором бикарбоната натрия (10 мл). Смесь экстрагировали дихлорметаном (30 мл). Экстракт промывали водой (30 мл) и солевым раствором (40 мл), органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии с получением титульного соединения (275 мг, 0,25 ммоль). MC (m/z): 1115,6 (M+1). Пример получения 8. Синтез дигидрохлорида 4-4-[(1E)-4-(2,9-диазаспиро[5.5]ундец-2-ил)бут-1-ен 1-ил]-2-метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6-тетра-O-бензоилD-глюкопиранозида Схема 1, стадия H: к раствору трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-O-бензоилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,9 диазаспиро[5.5]ундекан-9-карбоновой кислоты (275 мг, 0,25 ммоль) в дихлорметане (5 мл) добавляли соляную кислоту (4,0 М раствор в 1,4-диоксане, 0,6 мл, 2,4 ммоль). После выдерживания в течение ночи(18 ч) при комнатной температуре концентрировали при пониженном давлении для удаления растворителя с получением титульного соединения в виде твердого вещества (258 мг, 0,24 ммоль). MC (m/z): 1015,6 (M+1). Пример 1. Синтез 4-4-[(1E)-4-(2,9-диазаспиро[5.5]ундец-2-ил)бут-1-ен-1-ил]-2-метилбензил-5(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида(258 мг, 0,24 ммоль) в метаноле (2 мл) добавляли гидроксид натрия (0,5 мл, 0,5 ммоль, 1,0 М раствор). После 2 ч при 40C концентрировали при пониженном давлении для удаления растворителя с получением остатка, который очищали при помощи метода препаративной ВЭЖХ: высокое значение pH, 25% В в течение 4 мин, 25-40% В в течение 4 мин при 85 мл/мин с применением колонки C18XBridge ODB 3075 мм, 5 мкм, растворитель A - H2O с NH4HCO3 при pH 10, растворитель B - MeCN, с получением титульного соединения в виде твердого вещества (46 мг, 0,08 ммоль). MC (m/z): 598,8 (M+1), 596,8 (M-1). Схема 2, стадия A: в 1 л колбу добавляли 4-[(4-бром-2-метилфенил)метил]-5-изопропил-1Hпиразол-3-ол (24 г, 77,6 ммоль), 2,3,4,6-тетра-O-ацетилD-глюкопиранозил бромид (50,4 г, 116 ммоль),бензилтрибутиламмоний хлорид (5 г, 15,5 ммоль), дихлорметан (250 мл), карбонат калия (32 г, 323 ммоль) и воду (120 мл). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь экстрагировали дихлорметаном (450 мл). Экстракт промывали водой (300 мл) и солевым раствором (500 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии с получением титульного соединения (36,5 г, 57 ммоль). MC (m/z): 638,5 (M+1), 636,5 (M-1). Альтернативный синтез 4-(4-бром-2-метилбензил)-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6-тетра-8 024207(4,94 г, 15,52 ммоль), карбонат калия (32,18 г, 232,9 ммоль), дихлорметан (250 мл) и воду (120 мл) и смесь перемешивали при температуре окружающей среды в течение 18 ч. Смесь распределяли между дихлорметаном (250 мл) и водой (250 мл). Органическую фазу промывали солевым раствором (250 мл),сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии (с элюированием с градиентом от 10% этилацетата в дихлорметане до 70% этилацетата в дихлорметане) с получением титульного соединения (36,5 г, 74% выход). MC (m/z): 639/641 (M+1). Пример получения 10. Синтез 4-4-[(1E)-4-гидроксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2 ил)-1H-пиразол-3-ил 2,3,4,6-тетра-O-ацетилD-глюкопиранозида Схема 2, стадия B: к раствору 4-(4-бром-2-метилбензил)-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6 тетра-O-ацетилD-глюкопиранозида (15 г, 23,5 ммоль) в ацетонитриле (200 мл) и триэтиламине (50 мл) добавляли 3-бутен-1-ол (6,1 мл, 70 ммоль). Раствор дегазировали азотом в течение 10 мин. Добавляли три-o-толилфосфин (1,43 г, 4,7 ммоль) и ацетат палладия (526 мг, 2,35 ммоль). После кипячения с обратным холодильником при 90C в течение 2 ч охлаждали и концентрировали при пониженном давлении для удаления растворителя. Полученный остаток очищали при помощи флэш-хроматографии с получением титульного соединения (7,5 г, 11,9 ммоль). MC (m/z): 631,2 (M+1), 629,2 (M-1). Пример получения 11. Синтез 4-4-[(1E)-4-оксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1Hпиразол-3-ил 2,3,4,6-тетра-O-ацетилD-глюкопиранозида(2 г, 23,8 ммоль) в дихлорметане (50 мл) при 0C добавляли 3,3,3-триацетокси-3-йодфталид (2,1 г, 4,76 ммоль). После 15 мин при комнатной температуре реакцию гасили насыщенным водным раствором тиосульфата натрия (10 мл). Смесь экстрагировали дихлорметаном (30 мл), экстракт промывали водой (30 мл) и солевым раствором (40 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэшхроматографии с получением титульного соединения (0,95 г, 1,51 ммоль). MC (m/z): 628,8 (M+1), 626,8 ли триацетоксиборгидрид натрия (303 мг, 1,4 ммоль). После 30 мин при комнатной температуре реакцию гасили насыщенным водным раствором бикарбоната натрия (15 мл). Смесь экстрагировали дихлорметаном (60 мл). Экстракт промывали водой (30 мл) и солевым раствором (60 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии с получением титульного соединения (500 мг, 0,58 ммоль). MC (m/z): 866,8, 867,8 (M+1), 864,8, 865,8 (M-1). Пример получения 13. Синтез трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,8 диазаспиро[4.5]декан-8-карбоновой кислоты Титульное соединение получали, по существу, при помощи способа согласно примеру получения 12. MC (m/z): 852,8, 853,6 (M+1). 850,8, 851,6 (M-1). Пример получения 14. Синтез трет-бутилового эфира 9-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4.6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-3,9 диазаспиро[5.5]ундекан-3-карбоновой кислоты Титульное соединение получали, по существу, при помощи способа согласно примеру получения 12. MC (m/z): 866,8, 867,6 (M+1), 864,8, 865,6 (M-1). Пример получения 15. Синтез дигидрохлорида 4-4-[(1E)-4-(2,9-диазаспиро[5.5]ундец-2-ил)бут-1 ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6-тетра-O-ацетилD-глюкопиранозида Схема 2, стадия E: к раствору трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-O-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,9 диазаспиро[5.5]ундекан-9-карбоновой кислоты (500 мг, 0,58 ммоль) в дихлорметане (20 мл) добавляли соляную кислоту (4,0 М раствор в 1,4-диоксане, 1,5 мл, 5,8 ммоль). После 2 ч при комнатной температуре смесь концентрировали при пониженном давлении для удаления растворителя с получением титульного соединения в виде твердого вещества (480 мг, 0,57 ммоль). Титульное соединение получали по существу при помощи способа согласно примеру получения 15.MC (m/z): 752,8, 753,8 (M+1), 750,8 (M-1). Первый альтернативный синтез примера 1. Первый альтернативный синтез 4-4-[(1E)-4-(2,9 диазаспиро[5.5]ундец-2-ил)бут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Схема 2, стадия F: к дигидрохлориду 4-4-[(1E)-4-(2,9-диазаспиро[5.5]ундец-2-ил)бут-1-ен-1-ил]-2 метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил 2,3,4,6-тетра-O-ацетилD-глюкопиранозида (480 мг,0,24 ммоль) добавляли метанол (5 мл), триэтиламин (3 мл) и воду (3 мл). После выдерживания в течение 18 ч (в течение ночи) при комнатной температуре смесь концентрировали досуха при пониженном давлении. Полученный остаток очищали при помощи препаративной ВЭЖХ: высокое значение pH, 25% В в течение 4 мин, 25-40% В в течение 4 мин при 85 мл/мин с применением колонки C18XBridge ODB 3075 мм, 5 мкм, растворитель A - H2O с NH4HCO3 при pH 10, растворитель B - MeCN, с получением титульного соединения в виде твердого вещества (50 мг, 0,08 ммоль). MC (m/z): 598,8 (M+1), 596,8 (M-1). 1 Титульное соединение получали, по существу, при помощи способа из первого альтернативного синтеза примера 1. MC (m/z): 584,7 (M+1), 582,8 (M-1). Пример 3. Синтез 4-4-[(1 Е)-4-(3,9-диазаспиро[5.5]ундец-3-ил)бут-1-ен-1-ил]-2-метилбензил-5(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Титульное соединение получали, по существу, путем обработки соединения согласно примеру получения 14 с применением HCl, как описано в примере получения 15, а затем обработки полученной гид- 11024207 рохлоридной соли триэтиламином, как описано в первом альтернативном синтезе примера 1. MC (m/z): 598,8, 599,8 (M+1), 596,8, 597,8 (M-1). Схема 3 Схема 3, стадия A: к суспензии гидрохлорида трет-бутилового эфира 4,9-диазаспиро[5.5]ундекан-9 карбоновой кислоты (16,66 г, 57,28 ммоль) в ацетонитриле (167 мл) добавляли карбонат цезия (46,66 г,143,21 ммоль). Смесь перемешивали в течение 10 мин при температуре окружающей среды, а затем добавляли 4-бромбутин (6,45 мл, 68,74 ммоль). Реакционную смесь кипятили с обратным холодильником и перемешивали в течение 18 ч. Смесь охлаждали и концентрировали при пониженном давлении. Остаток распределяли между водой (200 мл) и этилацетатом (150 мл). Фазы разделяли и водный слой экстрагиро- 12024207 вали этилацетатом (100 мл). Объединенные органические слои промывали водой (200 мл), затем солевым раствором (150 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (17,2 г, 98% выход). 1H-ЯМР (300,11 МГц, CDCl3):3,43-3,31 (m,4H). 2,53-2,48 (m, 2H), 2,37-2,29 (m, 4H), 2,20 (s, 2H), 1,94 (t, J=2,6 Гц, 1H), 1,44 (s, 17H). Пример получения 18. Синтез трет-бутилового эфира 4-[(E)-4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)бут-3-енил]-4,9-диазаспиро[5.5]ундекан-9-карбоновой кислоты Схема 3, стадия B: к трет-бутиловому эфиру 4-бут-3-инил-4,9-диазаспиро[5.5]ундекан-9 карбоновой кислоты (17,21 г, 56,16 ммоль) добавляли триэтиламин (5,62 ммоль; 0,783 мл), 4,4,5,5 тетраметил-1,3,2-диоксаборолан (8,56 мл, 59,0 ммоль) и цирконоцен хлорид (1,45 г, 5,62 ммоль). Полученную смесь грели при 65C в течение 3,5 ч. Смесь охлаждали и растворяли в дихлорметане (150 мл). Полученный раствор пропускали через 4 см слой силикагеля, элюируя дихлорметаном (2200 мл). Фильтрат концентрировали при пониженном давлении с получением титульного соединения (21,2 г, 87% выход). 1H-ЯМР (300,11 МГц, CDCl3):6,65-6,55 (m, 1H), 5,49-5,43 (m, 1H), 3,42-3,29 (m, 4H), 2,40-2,27(16,3 г, 37,5 ммоль) и карбоната калия (12,97 г, 93,82 ммоль) в тетрагидрофуране (200 мл) и воде (40 мл) дегазировали в течение 15 мин путем продувания газообразным азотом. Добавляли Pd(OAc)2 (140 мг, 625 мкмоль) и 2-дициклогексилфосфино-2',4',6'-три-изопропил-1,1'-бифенил (0,596 г, 1,25 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 16 ч. Раствор охлаждали до температуры окружающей среды и добавляли метанол (200 мл). После 30 мин растворитель удаляли при пониженном давлении. Смесь распределяли между этилацетатом (500 мл) и солевым раствором (500 мл) с добавлением водного раствора MgSO4 (1 M; 500 мл) для облегчения разделения фаз. Слои разделяли и органический слой сушили над MgSO4 и фильтровали через 10 см слой силикагеля, элюируя этилацетатом (1,5 л). Фильтрат отбрасывали и слой силикагеля промывали 5% раствором MeOH в ТГФ (2 л). Метанольный фильтрат концентрировали при пониженном давлении с получением титульного соединения (20,1 г,92%).MC (m/z):699(M+l). Второй альтернативный синтез примера 1. Второй альтернативный синтез 4-4-[(1E)-4-(2,9 диазаспиро[5.5]ундец-2-ил)бут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Схема 3, стадия D: к раствору трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3- D-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,9-диазаспиро[5.5]ундекан-9 карбоновой кислоты (14,87 г; 21,28 ммоль) в дихлорметане (149 мл), охлажденному в ледяной воде, добавляли трифторуксусную кислоту (32,2 мл; 0,426 моль). Раствор оставляли нагреваться до комнатной температуры. После 30 мин смесь медленно добавляли к раствору аммиака в MeOH (2 М; 300 мл), при необходимости применяя охлаждение для поддержания постоянной температуры. Раствор перемешивали при комнатной температуре в течение 15 мин. Смесь концентрировали при пониженном давлении и остаток очищали с применением SCX-2 смолы. Основной фильтрат концентрировали при пониженном давлении и остаток растирали/обрабатывали ультразвуком в этилацетате, фильтровали и сушили. Полученное твердое вещество растворяли в MeOH (200 мл) и концентрировали в вакууме. Указанную процедуру повторяли несколько раз с получением титульного соединения (12,22 г, выход 96%). MC (m/z): 599 Схема 4, стадия A: к раствору 4-4-[(1 Е)-4-гидроксибут-1-ен-1-ил]-2-метилбензил-5-(пропан-2-ил)1H-пиразол-3-ил 2,3,4,6-тетра-O-ацетилD-глюкопиранозида (3,7, 5,87 ммоль) в дихлорметане (15 мл) и триэтиламине (4 мл, 29 ммоль) при 0C добавляли метансульфонилхлорид (0,54 мл, 7 ммоль). После кипячения с обратным холодильником при комнатной температуре в течение 30 мин смесь концентрировали при пониженном давлении для удаления растворителя. Очищали остаток при помощи флэшхроматографии с получением титульного соединения (2,9 г, 4,1 ммоль). MC (m/z): 708,5 (M+1), 706,5 (M-1). Пример получения 21. Синтез трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,6 диазаспиро[3.5]нонан-6-карбоновой кислоты. Схема 4, стадия B: к раствору (3E)-4-[3-метил-4-(5-(пропан-2-ил)-3-[(2,3,4,6-тетра-O-ацетилDглюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил метансульфоната (200 мг, 0,28 ммоль) и трет-бутилового эфира 2,6-диазаспиро[3.5]нонан-6-карбоновой кислоты (77 мг, 0,34 ммоль) в ацетонитриле (3 мл) добавляли диизопропилэтиламин (0,2 мл, 1,1 ммоль). Смесь грели при 80C в течение ночи. Концентрировали при пониженном давлении и очищали остаток при помощи флэшхроматографии с получением титульного соединения (127 мг, 0,15 ммоль). MC (m/z): 838,8, 839,6 (M+1),836,8, 837,6 (M-1). Пример получения 22. Синтез трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,7 диазаспиро[3.5]нонан-7-карбоновой кислоты Титульное соединение получали, по существу, при помощи способа согласно примеру получения 21. MC (m/z): 838,8, 839,6 (M+1), 836,8, 837,6 (M-1). Пример получения 23. Синтез трет-бутилового эфира 7-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,7 диазаспиро[3.5]нонан-2-карбоновой кислоты Титульное соединение получали, по существу, при помощи способа согласно примеру получения 21. MC (m/z): 838,8, 839,6 (M+1), 836,8, 837,6 (M-1). Пример получения 24. Синтез трет-бутилового эфира 1-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3- 15024207 Титульное соединение получали, по существу, при помощи способа согласно примеру получения 21. MC (m/z): 852,8, 853,6 (M+1), 850,8, 852,8 (M-1). Пример получения 25. Синтез трет-бутилового эфира 8-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-О-ацетилD-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,8 диазаспиро[4.5]декан-2-карбоновой кислоты Титульное соединение получали, по существу, при помощи способа согласно примеру получения 21. MC (m/z): 852,8, 853,6 (M+1), 850,8, 851,6 (M-1). Пример 4. Синтез 4-4-[(1 Е)-4-(2,6-диазаспиро[3.5]нон-2-ил)бут-1-ен-1-ил]-2-метилбензил-5(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Схема 4, стадия C: к раствору трет-бутилового эфира 2-(3E)-4-[3-метил-4-(5-(пропан-2-ил)-3[(2,3,4,6-тетра-O-ацетил- -D-глюкопиранозил)окси]-1H-пиразол-4-илметил)фенил]бут-3-ен-1-ил-2,6 диазаспиро[3.5]нонан-6-карбоновой кислоты в дихлорметане (2 мл) добавляли 4,0 М раствор HCl/1,4 диоксан (1,5 мл, 1,5 ммоль) и перемешивали при КТ в течение 4,0 ч. Смесь концентрировали при пониженном давлении с получением пенообразного твердого вещества. Твердое вещество в течение ночи обрабатывали 2,0 М раствором аммиака в MeOH (2 мл). После 18 ч при комнатной температуре смесь концентрировали при пониженном давлении для удаления растворителя. Полученный остаток очищали при помощи препаративной ВЭЖХ: высокое значение pH, 19% В в течение 3 мин, 19-34% В в течение 5 мин при 85 мл/мин с применением колонки C18XBridge ODB 3075 мм, 5 мкм, растворитель A - Н 2 О сNH4HCO3 при pH 10, растворитель B - MeCN, с получением титульного соединения в виде твердого вещества (47 мг, 0,08 ммоль). MC (m/z): 570,8, 571.8 (M+1), 568,7, 569,8 (M-1). Пример 5. Синтез 4-4-[(1 Е)-4-(2,7-диазаспиро[3.5]нон-2-ил)бут-1-ен-1-ил]-2-метилбензил-5(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Титульное соединение получали, по существу, при помощи способа согласно примеру 4. MC (m/z): 570,8, 571,8 (M+1), 568,7, 569,8 (M-1). Пример 6. Синтез трифторацетата 4-4-[(1 Е)-4-(2,7-диазаспиро[3.5]нон-7-ил)бут-1-ен-1-ил]-2 метилбензил-5-(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида (1:2) Титульное соединение получали, по существу, при помощи способа согласно примеру 4 с очисткой конечного соединения при помощи метода препаративной ВЭЖХ при низком значении pH (низкое значение pH, 16% В в течение 3 мин, 16-33% В в течение 5 мин при 85 мл/мин с применением колонки Титульное соединение получали, по существу, при помощи способа согласно примеру 4. MC (m/z): 584,7, 585,8 (M+1), 582,8, 583,8 (M-1). Пример 8. Синтез 4-4-[(1E)-4-(2,8-диазаспиро[4.5]дец-8-ил)бут-1-ен-1-ил]-2-метилбензил-5(пропан-2-ил)-1H-пиразол-3-ил -D-глюкопиранозида Титульное соединение получали, по существу, при помощи способа согласно примеру 4. MC (m/z): 584,7, 585,8 (M+1), 582,8, 583,8 (M-1). Исследования натрийзависимого транспортера глюкозы 1 (SGLT1) и SGLT2. кДНК, кодирующую SGLT1 человека (slc5al, NM000343), SGLT2 человека (slc5a2, NM003041) иSGLT1 мыши (slc5al, NM019810.4) приобретали в Openbiosystems, Invitrogen и Openbiosystems соответственно. кДНК клонировали в pcDNA3.1+ для экспрессии у млекопитающих и устойчиво трансфицировали в клетки яичника китайского хомячка (CHO)-K1 при помощи стандартных процедур трансфицирования млекопитающих. Субклон, экспрессирующий SGLT, каждой из сверхэкспрессирующих клеточных линий выбирали на основе устойчивости к неомицину (Geneticin, Invitrogen) и активности в исследовании усваивания 14Cметил-D-глюкопиранозида (14C-AMG) (см. ниже). Клетки, устойчиво экспрессирующие SGLT, выдерживали с применением стандартных способов выращивания клеточных культур. Активность SGLT измеряли как зависящий от натрия захват 14C-AMG в указанных выше клеточных линиях, как описано ниже. 100 мкл питательной среды, содержащей 30000 клеток, высеивали в каждую лунку 96-луночного планшета BioCoat с покрытием из поли-D-лизина (Becton Dickson) и культивировали при 37C в течение ночи. Питательную среду аспирировали и клетки дважды промывали 200 мкл рабочего буферного раствора (140 мМ NaCl, 2 мМ KCl, 1 мМ CaCl2, MgCl2 и 14 мМ N-2-гидроэтилпиперазинN'-2-этансульфокислоты (Hepes), pH 7,5). Избыток буфера удаляли при помощи бумажных полотенец. В каждую лунку добавляли 35 мкл рабочего буферного раствора. В каждую лунку вносили 5 мкл 10% раствора диметилсульфоксида (ДМСО) в рабочем буферном растворе, содержащем различные концентрации исследуемого соединения или не содержащем соединения в случае контроля. Реакцию инициировали путем добавления 10 мкл 14C-AMG в рабочий буферный раствор с получением конечной концентрации 4 мкМ. Планшет инкубировали при 37C в течение 125 мин. Реакцию останавливали путем аспирации рабочего буферного раствора, а затем трижды промывали 200 мкл ледяного рабочего буферного раствора. Дня обеспечения полного удаления рабочего буферного раствора применяли ручную аспирацию. В каждую лунку добавляли 10 мкл 0,1 н. раствора NaOH, а затем добавляли 100 мкл сцинтилляционной смеси Supermix (PerkinElmer). После смешивания подсчитывали сцинтилляционный сигнал на планшете при помощи MicroBeta (PerkinElmer). Для определения концентрации ингибитора при полумаксимальном ингибировании (IC50) строили кривую зависимости доза-ответ по десяти точкам для дозы путем под- 17024207 становки данных в четырехпараметровой эмпирической модели с применением программного обеспечения ActivityBase (ID Business Solution). Соединения из представленных в настоящей заявке примеров 1-8,которые исследовали, по существу, как описано выше, имели значения IC50 для SGLT1 менее чем примерно 500 нМ. Таблица 1 Активность in vitro примера 1 в отношении SGLT1 и SGLT2 В частности, данные из табл. 1 демонстрируют, что соединение согласно примеру 1 ингибируетSGLT1 человека и мыши in vitro и обладает более высокой активностью в случае SGLT1 человека и мыши по сравнению с SGLT2 человека in vitro. Эффекты, понижающие уровень глюкозы в пероральном тесте толерантности к глюкозе (ПТТГ). Исследуемое соединение готовили в виде состава путем добавления носителя, содержащего 1% гидроксиэтилцеллюлозы, 0,25% Tween 80 с 0,05% пеногасителя, к предварительно взвешенному исследуемому соединению с получением 1 мг/мл раствора. Смесь обрабатывали ультразвуком в течение примерно 30 с. Полученный раствор применяли в качестве базового раствора, из которого путем разбавления носителем получали растворы для дозирования более низких концентраций. Индивидуально размещенных мышей С 57 В 1/6 не кормили в течение ночи путем прекращения доступа к пище с позднего вечера перед днем исследования. На следующее утро мышей взвешивали и отбирали один образец крови натощак из надреза хвоста для измерения содержания глюкозы на глюкометре(Roche AccuChek). Исследуемые группы (n=5) формировали на основе содержания глюкозы в крови натощак, и они содержали преимущественно животных с содержанием глюкозы в диапазоне 80-100 мг/дл. После формирования групп первым мышам перорально при помощи зонда вводили 10 мл/кг препарата исследуемого соединения и начинали отсчет времени. Каждому следующему животному вводили дозу через полторы минуты после предыдущего. Через 3 ч после начала лечения первым соединением отбирали базовый образец крови для измерения содержания глюкозы (из надреза хвоста первого животного). Сразу после этого животному вводили пероральную дозу 50% декстрозы (Hospira) в концентрации 3 г/кг. С интервалом ровно полторы минуты из хвостовой вены других животных отбирали образцы крови для определения содержания глюкозы таким образом,что отбор крови для каждого из животных проводили через 20, 40, 60 и 120 мин после введения дозы декстрозы. Таблица 2 Эффекты, понижающие глюкозу в ПТТГ Как показано выше в табл. 2, соединение согласно примеру 1 обеспечивает зависимое от дозировки снижение амплитуды колебаний содержания глюкозы после перорального введения 50% декстрозы(Hospira) мышам С 57 В 1/6 с нормальным содержанием глюкозы. Пример 1 также демонстрирует зависимое от дозировки снижение площади под кривой (ППК) содержания глюкозы с поправкой на исходное значение в исследовании ПТТГ. Кроме того, пример 1 дозозависимым образом снижает среднюю максимальную концентрацию глюкозы в плазме (Cmax) в исследовании ПТТГ и увеличивает среднее время,требуемое для достижения максимальной концентрации глюкозы (Tmax). Содержания глюкозы в тесте толерантности к смешанной пище для самцов крыс с диабетом, вызванным стрептозотоцином. У крыс, которым вводили стрептозотоцин (СТ 3), развивался сахарный диабет. Полагают, что агенты, которые регулируют содержание глюкозы для указанных животных, подходят для применения для лечения диабета у человека. Исследуемое соединение готовили в виде состава путем добавления носителя, содержащего 1% гидроксиэтилцеллюлозы (ГЭЦ), 0,25% Tween 80 с 0,05% пеногасителя, к предварительно взвешенному исследуемому соединению с получением 2,5 мг/мл раствора. Смесь обрабатывали ультразвуком в течение примерно 30 с. Полученный раствор применяли в качестве базового раствора, из которого путем разбавления носителем получали растворы для дозирования более низких концентраций. СТ 3, 45 мг/кг, готовили в виде 3 мл аликвот путем растворения в 0,1 М цитратном буфере и хранили в темном месте во льду до момента введения. Использовали смешанную пищу с высоким содержанием жира (Bio-Serv корм для грызунов F3282 с высоким содержанием жира), содержащую жировые калории (60%), углеводные калории (26%) и белковые калории (15%) Индивидуально размещенных крыс линии Спрага-Доули оставляли для акклиматизации на период от 3 до 7 дней. СТ 3 вводили во второй половине дня для обеспечения того условия, что животные не получали пищу в последнее время, примерно в течение 6 ч на свету (свет включали в 6 утра, свет выключали в 6 вечера) Животных анестезировали изофлураном и путем инъекции в хвостовую вену вводили СТ 3. После возвращения в сознание животных возвращали в клетки и оставляли восстанавливаться в течение 7 дней. За два дня до теста толерантности к пище (ТТП) всем крысам вводили небольшое количество (2-4 г) корма F3282 для привыкания к его приему в процессе эксперимента. Вечером перед началом эксперимента крыс помещали в чистые клетки и убирали их пищу. На следующее утро животных взвешивали и отбирали образец крови из надреза хвоста для измерения содержания глюкозы (глюкометры Abbott AlphaTrak: код 29). Животных разбивали на группы n=6 на основе массы тела и содержания глюкозы натощак. Через 30 мин перорально вводили исследуемое соединение и измеряли два значения содержания глюкозы. Затем животным давали пятиграммовую пилюлю корма Bio-Serv 3282. Через 20 мин оставшуюся пищу отбирали и взвешивали. Для измерения содержания глюкозы отбирали образцы крови через 20, 40, 60 и 120 мин. Таблица 3 Содержания глюкозы в ТТП к смешанной пище для самцов крыс с диабетом,вызванным стрептозотоцином Как показано выше в табл. 3, соединение согласно примеру 1 значительно и дозозависимым образом снижает содержание глюкозы в ТТП по сравнению с применением группой контроля носителем. Акарбоза незначительно снижает содержание глюкозы по сравнению с контролем в любой момент времени. Кроме того, наблюдается зависимое от дозы снижение ППК содержания глюкозы с поправкой на исходное значение для лечения примером 1. Акарбоза значительно снижает ППК содержания глюкозы до значений, аналогичных значениям для примера 1 в концентрации 10 мг/кг. Табл. 3 демонстрирует, что соединение согласно примеру 1 регулирует содержание глюкозы для самцов крыс. где X представляет собой следующий фрагмент: или фармацевтически приемлемая соль указанного соединения. 2. Соединение или фармацевтически приемлемая соль по п.1, которое представляет собой 4. Способ лечения диабета у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1-3. 5. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 в терапии заболеваний и расстройств, связанных с гипергликемией. 6. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета. 7. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета 1 типа. 8. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для лечения диабета 2 типа. 9. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета. 10. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета 1 типа. 11. Применение соединения или его фармацевтически приемлемой соли по любому из пп.1-3 для получения лекарственного средства для лечения диабета 2 типа. 12. Фармацевтическая композиция для лечения диабета, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп.1-3 в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами.
МПК / Метки
МПК: C07D 471/10, C07D 487/10, A61P 3/10, A61K 31/438, A61K 31/4155
Метки: пиразольные, соединения
Код ссылки
<a href="https://eas.patents.su/21-24207-pirazolnye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Пиразольные соединения</a>
Пиразольные соединения для борьбы с беспозвоночными вредителями

Номер патента: 20661
Опубликовано: 30.12.2014
Авторы: Зёргель Зебастиан, Калбертсон Дебора Л., Дешмукх Прашант, Кайзер Флориан, Ле-Везуэ Ронан, Анспо Дуглас Д., Фон Дейн Вольфганг, Гросс Штеффен, Олоуми-Садегхи Хассан, Кёрбер Карстен, Польман Маттиас, Дикхаут Йоахим
МПК: C07D 403/12, A01N 43/56
Метки: вредителями, борьбы, беспозвоночными, соединения, пиразольные
Формула / Реферат:
1. Пиразольные соединения формулы (I.A) и их соли и N-оксидыгде А означает пиразольный радикал формул А1, А2 или A3# означает место присоединения к оставшейся части формулы (I.A);R41, R42, R43 и R51 независимо друг от друга выбирают из водорода, галогена, CN, NO2, C1-C10-алкила, C2-C10-алкенила и C2-C10-алкинила, где три упомянутых последними радикала могут быть незамещенными, могут быть частично или полностью галогенированными или могут нести...
Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения

Номер патента: 12786
Опубликовано: 30.12.2009
Авторы: Каццолла Никола, Драгоне Патриция, Руссо Винченцо, Колетта Изабелла, Поленцани Лоренцо, Ализи Мария Алессандра, Фурлотти Гвидо, Мангано Джорджина
МПК: C07D 209/88, A61P 35/00, A61K 31/403...
Метки: способ, фармацевтическая, композиция, получения, содержащая, 3-аминокарбазола, указанные, соединения
Формула / Реферат:
1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы и их фармацевтически приемлемые соли. 2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Лидли Роберт Дж., Юзан Андре, Кюродо Алан Х., Перроне Марк Х., Данвидди Кристофер Т.
МПК: A61K 31/715, A61P 7/02
Метки: сопутствующих, применение, тромбообразованию, эти, соединения, заболеваний, содержащий, профилактики, способ, тромбоцитов, содержащая, фармкомпозиция, агрегации, анти-ха, антагониста, набор, лечения, активностью
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Йеом Зи-Хо, Ким Хие Дзин, Йим Хиеон Дзоо, Кох Дзонг Сунг, Ким Геун Тае, Коо Ки Донг, Хур Гвонг-Чеунг, Ли Чанг-Сеок, Ким Киоунг-Хее, Бу Сеонг Чеол, Ким Мин-Дзунг, Ким Сунг Хо, Хонг Санг Йонг, Квон Ох Хван, Лим Донгчул, Хан Хее Оон, Ким Сунгсуб, Йео Донг-Дзун, Ким Дзи Янг
МПК: A61P 3/10, A61K 31/452, A61K 31/444...
Метки: соединения-ингибиторы, ингредиента, также, композиции, содержащие, соединения, дипептидилпептидазы-iv, фармацевтические, указанные, качестве, активного, способы, получения
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Соединения, ингибирующие ферменты гистондеацетилазы, и фармацевтическая композиция, содержащая данные соединения

Номер патента: 22964
Опубликовано: 31.03.2016
Авторы: Гхош Шомир, Балоглу Эркан, Шмидт Дарби, Лобера Мерседес
МПК: A61K 31/497
Метки: соединения, ингибирующие, гистондеацетилазы, содержащая, фармацевтическая, композиция, ферменты, данные
Формула / Реферат:
1. Соединение формулы (I-а)где R1 представляет собой галоген(C1-C2)алкил, где указанный галоген(C1-C2)алкил содержит по меньшей мере 2 атома фтора;А представляет собой необязательно замещенный (С3-С6)циклоалкил, фенил или 5-6-членный гетероарил, где указанный циклоалкил, фенил или гетероарил необязательно замещен 1-3 группами, независимо выбранными из (C1-C4)алкила, галогена, циано и (C1-C4)алкокси;Z представляет собой -C(=O)NRX-; -NRXC(=O)-,...
Предыдущий патент: Всасывающая головка для подводного горного инструмента
Следующий патент: Тонкая крончатая крышка
Случайный патент: Мост закрытого типа