Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения
Номер патента: 12786
Опубликовано: 30.12.2009
Авторы: Мангано Джорджина, Поленцани Лоренцо, Каццолла Никола, Драгоне Патриция, Руссо Винченцо, Ализи Мария Алессандра, Колетта Изабелла, Фурлотти Гвидо







Формула / Реферат
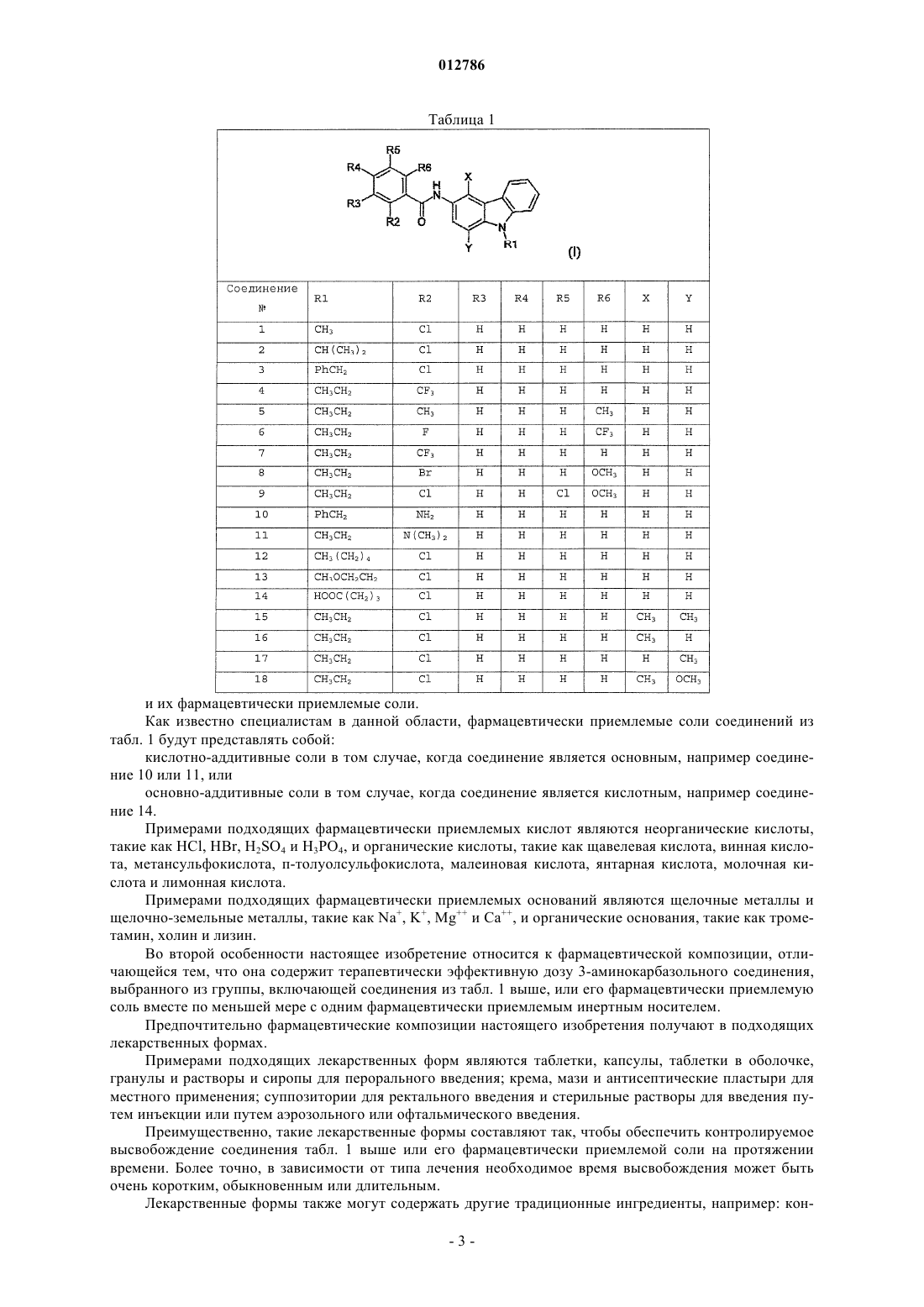
1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы

и их фармацевтически приемлемые соли.
2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным носителем.
3. Способ лечения или предупреждения воспалительных процессов, опухолей, болезни Альцгеймера и атеросклероза у млекопитающих, включающий введение терапевтически эффективного количества соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемые соли, субъекту, который нуждается в этом.
4. Способ получения 3-аминокарбазола из таблицы по п.1, отличающийся тем, что он включает следующие стадии:
а) реагирование амина формулы (II)

где R1, X и Y имеют значения, указанные в таблице,
с соединением формулы (III)

где R2, R3, R4, R5 и R6 имеют значения, указанные в упомянутой таблице, и Z выбирают из группы, включающей Cl, Br, ОН, OR и OC(O)R, где R представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода,
получая соединение 3-аминокарбазола формулы (I)

где R1, R2, R3, R4, R5, R6, X и Y имеют значения, указанные в упомянутой таблице, и
b) необязательно, образование фармацевтически приемлемой соли соединения формулы (I), полученного таким образом.
Текст
012786 Область техники, к которой относится изобретение Настоящее изобретение относится к соединению 3-аминокарбазола, к фармацевтической композиции, содержащей указанное соединение, и к способу его получения. Более конкретно, настоящее изобретение относится к соединению 3-аминокарбазола, которое полезно в лечении или в предупреждении нарушений, связанных с продуцированием простагландина Е 2(PGE2), например воспалительных процессов, опухолей, болезни Альцгеймера и атеросклероза. Известный уровень техники Ценность простагландинов Е 2 (PGE2) обусловлена той ролью, которую они играют в качестве биорегуляторов, вместе с другими простаноидами, продуцируемыми метаболически из арахидоновой кислоты, и в качестве медиаторов воспалительных процессов. Простаноиды представляют собой класс соединений, включающий простагландины, тромбоксаны и простациклины. Простаноиды являются медиаторами липидов, которые действуют в качестве локальных гормонов на клетках, расположенных рядом с центром их высвобождения. Простаноиды в основном образуются из арахидоновой кислоты путем ферментативного окисления, активированного циклооксигеназой. Циклооксигеназы (простагландин-G/Н-синтазы) катализируют последовательное образование PGG2 и PGH2 из арахидоновой кислоты. PGH2 затем превращается посредством ферментов со специфической активностью в различные простаноиды. Таким образом образуются простагландины D2 (PGD2), простагландины Е 2 (PGE2), простагландины F2 (PGF2), простагландины I2 (PGI2) и тромбоксаны А 2 (ТХА 2). С выбросом семенной жидкости простаноиды не накапливаются. После воздействия различных раздражителей (воспалительных, иммунологических, гормональных, ультрафиолетового света, опухолевых агентов и также механического встряхивания) простаноиды синтезируются и высвобождаются в межклеточное пространство, из которого они поступают в плазму крови, в мочу и другие биологические жидкости. Простаноиды играют важную роль в защитных механизмах функционирования органов и всего организма в целом. Это продемонстрировано цитопротекторной функцией в желудочно-кишечном тракте,посредством регуляции функции почек и капиллярного кровообращения, посредством регуляции агрегации тромбоцитов и свертывания крови, вовлечения в дифференцировку иммунных клеток и в заживление раны, костного метаболизма и овуляции. В частности, следует подчеркнуть вазопротекторное действие простаноидов PGI2, которые необходимы для поддержания тонуса сосудов и для предупреждения тромбоэмболии и атеросклероза на эндотелиальном уровне, и противовоспалительное и антипролиферативное действие простаноидов PGD2, метаболит которых, 15d-PGJ2, способен оказывать противовоспалительные воздействия посредством активирования ядерных рецепторов PPAR (рецептор-гамма, активированный пролифератором пероксисомы)(Inoue et al., 2 000). Простаноиды, таким образом, являются биорегуляторами, но также важными медиаторами воспаления и других патологий. В частности, простаноиды PGE2 имеются в избытке в центрах воспаления и ответственны за различные патологические особенности острого и хронического воспаления, например отек, образование эритематозных дерматитов, болезненное течение воспалительного процесса, воспаление суставов и повышенную температуру. По сути, простаноиды PGE2 представляют собой сильнодействующие противовоспалительные и алгогенные средства. Антитела анти-РСЕ 2 обладают противовоспалительной активностью, и животные, испытывающие недостаток в рецепторах PGE2, показывают сниженную реакцию на воспалительные раздражители (Portanova et al., 1996, Ueno et al., 2001) и отсутствующую пиретическую реакцию на пирогенные раздражители (Ushikubi et al., 1998). Нестероидные противовоспалительные лекарственные средства (NSAIDs) и лекарственные средства, селективные в отношении СОХ-2, применяемые в настоящее время, уменьшают симптомы, связанные с воспалением, посредством неселективного ингибирования образования эйкозаноидов (PGE2, PGD2,PGF2, PGI2 и ТХА 2) вследствие их ингибирующего действия на циклооксигеназы 1 и 2 (FitzGerald andPatrono, 2001). В частности, лекарственные средства, селективные в отношении СОХ-2, имеющиеся в настоящее время на рынке сбыта лекарственных средств, имеют пониженную желудочно-кишечную токсичность по сравнению с традиционно применяемыми нестероидными противовоспалительными средствами(NSAIDs). Однако указанные лекарственные средства, селективные в отношении СОХ-2, снижают продуцирование сосудистого простациклина (PGI2, который образуется преимущественно из СОХ-2), изменяя обычное равновесие между протромботическими и антитромботическими эйкозаноидами в пользу протромботических (ТХА 2, который образуется преимущественно из СОХ-1), и приводят к повышенному риску тромботических сердечно-сосудистых явлений (S. Malhotra, MD, DM; N. Shafiq, MD; P. Pandhi,MD Medscape General Medicine 6(1), 2004; D.Mukherjee and E.J. Topol Cardiovascular risk and COX-2 inhibitors, Arthritis Res. Ther. 2003, 5:8-11-2002). Различные 3-аминокарбазольные соединения были изучены на предмет их способности селективно связываться с рецептором Y5 человека и модулировать его активность. Такая способность делает их по-1 012786 лезными в лечении нарушений аппетита и обмена веществ, например ожирения, нейрогенной булимии,нервно-психической анорексии, нарушений сна, морфинной наркомании и приступов эпилепсии (WO 01/07409 Al, WO 02/051806, WO 02/096902 и US 6399631). Краткое изложение сущности изобретения Неожиданно было обнаружено, что некоторые 3-аминокарбазольные соединения способны селективно ингибировать продуцирование простагландина Е 2(PGE2). Такие соединения способны снижать продуцирование PGE2 и, следовательно, являются активными во всех патологических состояниях, в которых PGE2 действует в качестве медиатора (например, воспалительные процессы, болевые состояния, повышенная температура, опухоли, болезнь Альцгеймера и атеросклероз). Типичными примерами таких воспалительных процессов являются отек, эритематозный дерматит,воспаление суставов, ревматоидный артрит и артроз. Типичными примерами таких опухолей являются карцинома и аденокарцинома кишечника и легких. Соединения настоящего изобретения селективно ингибируют синтез PGE2. Такая селективность имеет преимущество ингибирования сильнодействующего медиатора воспаления, болевого состояния и повышенной температуры, при оставлении неизменным продуцирование других простаноидов, вырабатываемых одновременно по пути активации арахидоновой кислоты, таких как PGF2a, TXA2, PGI2 и PGD2. Все защитные механизмы функционирования органов и всего организма в целом, которые являются типичными в действии других простаноидов, следовательно, остаются неизмененными. Подобно традиционно применяемым нестероидным противовоспалительным средствам соединения настоящего изобретения обладают противовоспалительными, жаропонижающими и аналгетическими свойствами, и, следовательно, являются активными в патологиях, таких как воспаление, болевое состояние, повышенная температура, ревматоидный артрит и артроз. Кроме того, так как вовлечение PGE2 в опухоли, болезнь Альцгеймера и атеросклероз известно в литературе, соединения настоящего изобретения также имеют применения в предупреждении и лечении этих патологий. Преимущественно такие соединения, однако, показывают несколько побочных эффектов по сравнению с NSAIDs и с лекарственными средствами, селективными в отношении СОХ-2, заключающихся в том, что, посредством ингибирования циклооксигеназ, они не делают различий между простаноидами. В частности, такие производные полезны как в лечении, так и в предупреждении воспалительных процессов. В частности, соединения настоящего изобретения показывают пониженную желудочно-кишечную,почечную и сосудистую токсичность. Подробное описание изобретения В первой особенности настоящее изобретение относится к соединению 3-аминокарбазола, отличающемуся тем, что его выбирают из группы, включающей соединения из табл. 1 ниже. и их фармацевтически приемлемые соли. Как известно специалистам в данной области, фармацевтически приемлемые соли соединений из табл. 1 будут представлять собой: кислотно-аддитивные соли в том случае, когда соединение является основным, например соединение 10 или 11, или основно-аддитивные соли в том случае, когда соединение является кислотным, например соединение 14. Примерами подходящих фармацевтически приемлемых кислот являются неорганические кислоты,такие как HCl, HBr, H2SO4 и H3PO4, и органические кислоты, такие как щавелевая кислота, винная кислота, метансульфокислота, п-толуолсульфокислота, малеиновая кислота, янтарная кислота, молочная кислота и лимонная кислота. Примерами подходящих фармацевтически приемлемых оснований являются щелочные металлы и щелочно-земельные металлы, такие как Na+, K+, Mg и Са, и органические основания, такие как трометамин, холин и лизин. Во второй особенности настоящее изобретение относится к фармацевтической композиции, отличающейся тем, что она содержит терапевтически эффективную дозу 3-аминокарбазольного соединения,выбранного из группы, включающей соединения из табл. 1 выше, или его фармацевтически приемлемую соль вместе по меньшей мере с одним фармацевтически приемлемым инертным носителем. Предпочтительно фармацевтические композиции настоящего изобретения получают в подходящих лекарственных формах. Примерами подходящих лекарственных форм являются таблетки, капсулы, таблетки в оболочке,гранулы и растворы и сиропы для перорального введения; крема, мази и антисептические пластыри для местного применения; суппозитории для ректального введения и стерильные растворы для введения путем инъекции или путем аэрозольного или офтальмического введения. Преимущественно, такие лекарственные формы составляют так, чтобы обеспечить контролируемое высвобождение соединения табл. 1 выше или его фармацевтически приемлемой соли на протяжении времени. Более точно, в зависимости от типа лечения необходимое время высвобождения может быть очень коротким, обыкновенным или длительным. Лекарственные формы также могут содержать другие традиционные ингредиенты, например: кон-3 012786 серванты, стабилизаторы, поверхностно-активные вещества, буферные растворы, соли для регулирования осмотического давления, эмульгаторы, подслащивающие вещества, красители, ароматизирующие добавки и т.п. Более того, в том случае, когда требуются особые виды лечения, фармацевтическая композиция настоящего изобретения также может содержать другие фармакологически активные ингредиенты, одновременное введение которых является полезным. Количество соединения настоящего изобретения в фармацевтической композиции может варьироваться в пределах широкого диапазона в функциональной зависимости от известных факторов, например, от типа заболевания, которое должно быть вылечено, от серьезности заболевания, от веса тела больного, от лекарственной формы, от выбранного пути введения, от количества введений за сутки и от эффективности выбранного соединения. Однако оптимальное количество может быть легко и в обычном порядке определено специалистом в данной области. Обычно количество соединения настоящего изобретения в фармацевтической композиции будет таким, чтобы гарантировать уровень введения от 0,0001 до 100 мг/кг/день и даже более предпочтительно от 0,01 до 10 мг/кг/день. Лекарственные формы фармацевтической композиции настоящего изобретения могут быть изготовлены в соответствии с методиками, которые хорошо известны химикам-фармацевтам, включая смешение, гранулирование, таблетирование, растворение, стерилизацию и т.п. В третьей особенности настоящее изобретение относится к способу лечения и предупреждения воспалительных процессов, опухолей, болезни Альцгеймера и атеросклероза у млекопитающих, включающему введение терапевтически эффективного количества 3-аминокарбазольного соединения, выбранного из группы, включающей соединения из табл. 1 выше или их фармацевтически приемлемую соль, субъекту, который нуждается в этом. Предпочтительно воспалительный процесс выбирают из группы, включающей отек, эритематозный дерматит, воспаление суставов, ревматоидный артрит и артроз, и опухоль выбирают из группы, включающей карциному и аденокарциному кишечника или легких. 3-Аминокарбазолы из табл. 1 выше могут быть получены в соответствии с известными способами,например путем реагирования кислоты или ее реакционноспособного производного с выбранным амином (патентная заявка WO 02/096902 A1, WO 02/051806, J. Med. Chem. 2002, vol.45, pp.3509-3523). Типичными примерами реакционноспособных производных являются ацилгалогениды, ангидриды или сложные эфиры. В четвертой особенности настоящее изобретение, следовательно, относится к способу получения 3 аминокарбазола из табл. 1 выше, отличающемуся тем, что он включает следующие стадии: а) реагирование амина формулы (II) где R2, R3, R4, R5 и R6 имеют значения, указанные в табл. 1, и Z выбирают из группы, включающей Cl, Br, ОН, OR и OC(O)R, где R представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода,получая соединение 3-аминокарбазола формулы (I)b) необязательно, образование фармацевтически приемлемой соли соединения формулы (I), полученного таким образом. Обычно стадию (а) проводят в присутствии подходящего разбавителя при температуре в пределах диапазона между 0 и 140 С, в течение времени в пределах диапазона между 0,5 и 24 ч. Предпочтительно температура реакции находится в пределах диапазона между 15 и 40 С. Преимущественно время реакции колеблется в диапазоне от 2 до 18 ч.-4 012786 Предпочтительно разбавитель является апротонным, полярным или аполярным. Еще более предпочтительно он представляет собой полярный апротонный разбавитель. Примерами подходящих полярных не содержащих протоны разбавителей являются диметилформамид и дихлорметан. В варианте осуществления, в котором Z представляет собой Cl или Br, реакцию преимущественно проводят в присутствии подходящего органического или неорганического акцептора кислоты. Примерами подходящих органических акцепторов являются диизопропилэтилендиамин, триэтиленамин, пиридин и диметиламинопиридин. Примерами подходящих неорганических акцепторов являются карбонаты или бикарбонаты щелочных металлов. В варианте осуществления, в котором Z представляет собой ОН, реакцию предпочтительно проводят в присутствии подходящего связующего агента, например дициклогексилкарбодиимида (также нанесенного на полистирольный полимер) или карбонилдиимидазола. Примеры, которые следуют далее, приведены для того, чтобы более подробно проиллюстрировать изобретение, однако не ограничивая его. Пример 1. Получение соединения 1 (R1=CH3, R2=Cl, R3=R4=R5=R6=X=Y=H).a) 9-Метил-9H-карбазол. К раствору карбазола (5 г; 0,030 моль) в диметилформамиде (50 мл) добавляли порциями гидрид натрия (60% суспензия в минеральном масле, 1,15 г; 0,030 моль), и полученную суспензию перемешивали при комнатной температуре в течение 0,5 ч. Затем по каплям добавляли йодометан (1,43 мл; 0,030 моль). Суспензию, таким образом полученную, перемешивали в течение 16 ч и затем осторожно добавляли H2O до тех пор, пока не завершилось выпадение осадка. Осадок собирали посредством фильтрации под вакуумом и сушили в вакуум-сушильном шкафу. Таким образом, получали 5,3 г желаемого продукта и использовали без дополнительной очистки. 1b) 9-Метил-3-нитро-9H-карбазол. Продукт, полученный так, как описано для стадии а) выше, (3,9 г; 0,022 моль) растворяли в ледяной уксусной кислоте (70 мл). Затем в течение 30 мин по каплям добавляли смесь, содержащую дымящую азотную кислоту (1,05 мл) в ледяной уксусной кислоте (3,11 мл). Полученный таким образом раствор перемешивали в течение следующих 30 мин. Затем реакционную смесь выливали в H2O и лед, перемешивали в течение 15 мин и фильтровали. Осадок, полученный таким образом, (4 г) очищали с помощью флэш-хроматографии при элюировании смесью гексан/этилацетат 85/15. Таким образом, получали желаемый продукт (2,2 г). 1c) 9-Метил-9H-карбазол-3-амин. Продукт, полученный так, как описано в стадии b) выше, (1,94 г; 0,009 моль) растворяли в безводном этаноле (20 мл). Затем добавляли дигидрат-хлорид олова (8,16 г; 0,043 моль). Полученную таким образом смесь кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и затем удаляли растворитель при пониженном давлении. Остаток смывали водой. РН доводили до 7,5 путем добавления насыщенного раствора бикарбоната натрия, и смесь перемещали в делительную воронку. Органическую фазу экстрагировали этилацетатом (2100 мл). Органические экстракты объединяли и промывали водой, насыщенной NaCl (2100 мл). Органическую фазу отделяли и сушили над Na2SO4. Органическую фазу отделяли и сушили над Na2SO4. Затем удаляли растворитель путем выпаривания при пониженном давлении, и остаток, полученный таким образом, (1,5 г) использовали без дополнительной очистки. 1d) 2-Хлор-N-(9-метил-9H-карбазол-3-ил)бензамид. Продукт, полученный так, как описано для стадии с) выше, (1,5 г; 0,008 моль) растворяли в дихлорметане (20 мл). Затем к раствору добавляли диизопропилэтилендиамин (1,19 г; 0,009 моль) и 2 хлорбензоилхлорид (1,16 мл; 0,009 моль). Полученную таким образом смесь перемешивали в течение 3 ч. Реакционную смесь перемещали в делительную воронку и промывали посредством 1N раствора HCl(50 мл), посредством 1N раствора NaOH (50 мл) и посредством Н 2O (50 мл). Органическую фазу отделяли и сушили над Na2SO4. Затем удаляли растворитель путем выпаривания при пониженном давлении, и остаток, полученный таким образом, (3 г) выкристаллизовывали из смеси гексан/этилацетат 1/2. Таким образом получали 2-хлор-N-(9-метил-9H-карбазол-3-ил)бензамид (1,2 г). Температура плавления: 206 С. Элементный анализ для C20H15ClN2Oa) 9-(1-Метилэтил)-9H-карбазол. Карбазол (5 г; 0,03 моль) подвергали реакции с изопропилбромидом (5,61 мл; 0,06 моль), действуя аналогично способу, описанному в примере 1 а). Твердый продукт, получаемый после фильтрации, (4 г) очищали посредством флэш-хроматографии, при элюировании смесью гексан/этилацетат 97/3, что дает 3,3 г желаемого продукта. 1b) 9-(1-Метилэтил)-3-нитро-9H-карбазол. Продукт, полученный так, как описано для стадии а) выше, (3,3 г; 0,016 моль) подвергали реакции,действуя аналогично способу, описанному в примере 1b). Твердый продукт, получаемый после фильтрации, (3,7 г) очищали посредством флэш-хроматографии, при элюировании смесью гексан/этилацетат 95/5, получая 1,8 г желаемого продукта. 1c) 9-(1-Метилэтил)-9H-карбазол-3-амин. Продукт, полученный так, как описано для стадии b) выше, (1,7 г; 0,007 моль) подвергали реакции,действуя аналогично способу, описанному в примере 1 с). Полученный таким образом продукт (1,3 г) использовали без дополнительной очистки. 1d) 2-Хлор-N-[9-(1-метилэтил)-9H-карбазол-3-ил]бензамид. Продукт, полученный так, как описано для стадии с) выше, (1,2 г; 0,005 моль) подвергали реакции,действуя аналогично способу, описанному в примере Id). Продукт, полученный таким образом, (2,2 г) выкристаллизовывали дважды из смеси гексан/этилацетат 1/9. Таким образом получали 2-хлор-N-[9-(1 метилэтил)-9H-карбазол-3-ил]бензамид (1,1 г). Температура плавления: 195 С. Элементный анализ для C22H19ClN2Ob) 3-Нитро-9-(фенилметил)-9 Н-карбазол. Продукт, полученный так, как описано для стадии а) выше, (6,7 г; 0,026 моль) подвергали реакции,действуя аналогично способу, описанному в примере 1b). Твердый продукт, получаемый после фильтрации, (7 г) очищали посредством флэш-хроматографии при элюировании смесью гексан/этилацетат 95/5,получая 2,5 г желаемого продукта. 1c) 9-(Фенилметил)-9H-карбазол-3-амин. Продукт, полученный так, как описано для стадии b) выше, (1 г; 0,003 моль) подвергали реакции,действуя аналогично способу, описанному в примере 1 с). Полученный таким образом продукт (1 г) использовали без дополнительной очистки. 1d) 2-Хлор-N-[9-(фенилметил)-9H-карбазол-3-ил]бензамид. Продукт, полученный так, как описано для стадии с) выше, (0,9 г; 0,003 моль) подвергали реакции,действуя аналогично способу, описанному в примере 1d). Продукт, таким образом полученный, (1,5 г)-6 012786 выкристаллизовывали из этилацетата. Таким образом получали 2-хлор-N-[9-(фенилметил)-9 Н-карбазол-3-ил]бензамид (550 мг). 1a) N-(9-Этил-9 Н-карбазол-3-ил)-2-(трифторметил)бензамид. К раствору 3-амино-9-этилкарбазола (4 г; 0,019 моль) в дихлорметане (30 мл) добавляли триэтиламин (2,9 мл; 0,021 моль) и 2-(трифторметил)бензоилхлорид (3,1 мл; 0,021 моль). Полученную таким образом смесь перемешивали в течение 16 ч. Реакционную смесь перемещали в делительную воронку и промывают посредством H2O (250 мл). Органическую фазу отделяли и сушили над Na2SO4, удаляли растворитель путем выпаривания при пониженном давлении, и остаток, полученный таким образом, (4 г) выкристаллизовывали из этанола. Таким образом получали N-(9-этил-9H-карбазол-3-ил)-2-(трифторметил)бензамид (3,5 г). Температура плавления: 206-207 С. Элементный анализ для C22H17F3N2O(3,6 мл; 0,05 моль). Смесь, полученную таким образом, кипятили с обратным холодильником в течение 2 ч. Растворитель и избыток тионилхлорида затем выпаривавали при пониженном давлении. Остаток, полученный таким образом, смывали толуолом (50 мл) три раза и выпаривали при пониженном давлении. Полученный таким образом продукт (3,6 г) использовали без дополнительной очистки.b) N-(9-Этил-9 Н-карбазол-3-ил)-2,6-диметилбензамид. Продукт, полученный так, как описано для стадии а) выше, (3,3 г; 0,020 моль) подвергали реакции,действуя аналогично способу, описанному в примере 4. Полученный продукт (2,4 г) выкристаллизовывали из изопропанола. Таким образом получали N-(9-этил-9H-карбазол-3-ил)-2,6-диметилбензамид (1,4 г). Температура плавления: 192-193 С. Элементный анализ для C23H22N2OH ЯМР (300 МГц, DMSO-d6)м.д. 1,31 (т, J=7,1 Гц, 3H), 2,34 (с, 6 Н), 4,43 (кв., J=7,1 Гц, 2 Н), 7,087,27 (м, 4 Н), 7,41-7,49 (м, 1 Н), 7,54-7,62 (м, 2 Н), 7,68 (дд, J=8,8, 1,8 Гц, 1 Н), 8,10 (д, J=7,6 Гц, 1 Н), 8,59 (д,J=1,8 Гц, 1 Н), 10,53 (шир.с, 1 Н). Примеры 6-9. Получение соединений 6-9. Суспензию, содержащую 9-этил-3-аминокарбазол (0,1 г; 0,48 ммоль) и соответствующую бензойную кислоту (0,70 ммоль), в которой R2, R3, R4, R5 и R6 имеют значения, указанные в табл. 1, в дихлорметане (4 мл) и диметилформамиде (0,8 мл), перемешивали в течение 10 мин. К полученной таким образом реакционной смеси, в инертной атмосфере, добавляли полистиролдивинилбензолкарбодиимидный полимер (ПС-карбодиимид) (0,73 г; 0,9 ммоль). Затем суспензию перемешивали в течение 16 ч. Реакционную смесь фильтровали, и полученный твердый продукт промывали дихлорметаном. Затем раствор обрабатывали полимером Amberlyst 15 (200 мг; 0,8 ммоль) и полимеромAmberlyst A21 (250 мг; 1 ммоль) и перемешивали в течение 2 ч. Полимеры отделяли путем фильтрации и промывали дихлорметаном (25 мл), и органический растворитель выпаривали при пониженном давлении, получая желаемый продукт.a) 2-[(трет-Бутоксикарбонил)амино]бензойная кислота. Смесь антраниловой кислоты (6,8 г; 0,050 моль), ди-трет-бутилдикарбоната (16,4 мл; 0,071 моль),0,5N NaOH (100 мл), диоксана (50 мл) и ацетонитрила (10 мл), поддерживали при перемешивании при комнатной температуре в течение 16 ч. Растворитель удаляли путем выпаривания при пониженном давлении, и остаток обрабатывали смесью 100 г льда и 100 мл 10% водного раствора лимонной кислоты. Раствор экстрагировали этилацетатом (3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (50 мл) и затем сушили над Na2SO4. После фильтрации растворитель удаляли путем выпаривания при пониженном давлении. Полученный таким образом остаток выкристаллизовывали из смеси гексан:этилацетат = 3:1, получая продукт,указанный в заглавии, (4,9 г). 1b) 2-Амино-N-(9-бензил-9H-карбазол-3-ил)бензамид гидрохлорид. 4-Диметиламинопиридин (0,31 г, 0,0025 моль) и 9-(фенилметил)-9 Н-карбазол-3-амин, полученный так, как описано в примере 3 с), (0,626 г, 0,0023 моль) добавляли в раствор продукта, полученного на предыдущей стадии а), (0,496 г, 0,0021 моль) в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение 5 мин, добавляли N,N-дициклогексилкарбодиимид (0,475 г, 0,0023 моль) и оставляли перемешиваться при комнатной температуре в течение 2 дней. Впоследствии смесь разбавляли дихлорметаном (15 мл) и фильтровали через слой силикагеля. Раствор промывали 10%-ным водным раствором лимонной кислоты (310 мл), водой (210 мл) и насыщенным раствором NaCl (10 мл) в указанном порядке. Органическую фазу сушили над Na2SO4 и растворитель удаляли при пониженном давлении. Полученный таким образом продукт (1,3 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 7:3, и затем выкристаллизовывали из смеси гексан:этилацетат = 95:5. Полученный таким образом продукт (350 мг, 0,712 ммоль) растворяли в этилацетате (50 мл) и обрабатывали посредством 3 М раствора HCl в этаноле (26 мл). Растворитель удаляли путем выпаривания при пониженном давлении, и остаток выкристаллизовывали из смеси безводный этанол:этилацетат = 1:1. Таким образом получали 2-амино-N-(9-бензил-9H-карбазол-3-ил)бензамид гидрохлорид (0,2 г). 1 Н ЯМР (300 МГц, DMSO-d6)м.д. 5,66 (с, 2 Н), 7,01 (т, J=7,51 Гц, 1 Н), 7,08-7,50 (м, 12 Н), 7,62 (д,J=8,59 Гц, 2 Н), 7,70 (дд, J=8,82, 1,80 Гц, 1 Н), 7,84 (дд, J=1,16, 0,99 Гц, 1 Н), 8,10 (д, J=7,60 Гц, 1 Н), 8,56 (д,J=1,65 Гц, 1 Н), 10,34 (шир.с, 1 Н). Пример 11. Получение соединения 11 (R1=CH3CH2, R2=N(CH3)2, R3=R4=R5=R6=X=Y=H). а) 2-Диметиламино-N-(9-этил-9H-карбазол-3-ил)бензамид гидрохлорид. 3-Амино-9-этилкарбазол (0,484 г, 0,0023 моль) подвергали реакции с N-диметилантраниловой кислотой (0,347 г, 0,0021 моль), действуя аналогично способу примера 10b. Таким образом полученный продукт (0,9 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 8:2. Впоследствии, его растворяли в этаноле, обрабатывали 3 М раствором HCl в этаноле (2,0 мл) и оставляли при комнатной температуре на 3 ч. Затем растворитель удаляли путем выпаривания при пониженном давлении, и остаток выкристаллизовывали из смеси изопропиловый спирт:изопропиловый эфир =1:3. Таким образом получали 2-диметиламино-N-(9-этил-9H-карбазол-3-ил)бензамид гидрохлорид (0,25 г). 1a) 9-Пентил-9H-карбазол. Карбазол (5 г, 0,030 моль) подвергали реакции с 1-бромпентаном (7,5 мл, 0,06 моль), действуя аналогично способу примера 1 а). Твердый продукт, полученный после холодной фильтрации, (8 г) очищали посредством фильтрации через слой силикагеля при элюировании смесью гексан:этилацетат = 8:2. Таким образом получали желаемый продукт (6 г). 1b) 3-Нитро-9-пентил-9H-карбазол. Продукт, полученный так, как описано в предыдущей стадии а), (1 г, 0,0042 моль) подвергали реакции, действуя аналогично способу примера 1b). Полученный таким образом продукт (0,9 г) очищали посредством флэш-хроматографии при элюи-8 012786 ровании смесью гексан:этилацетат = 10:1, и получающийся в результате продукт (0,54 г) использовали без дополнительной очистки. 1c) 9-Пентил-9H-карбазол-3-амин. Продукт, полученный так, как описано в предыдущей стадии b), (2,6 г, 0,0092 моль) подвергали реакции, действуя аналогично способу примера 1 с). Полученный таким образом продукт (2 г) очищали посредством кристаллизации из гексана, получая желаемый продукт (1,3 г). Газовая хроматография/масс-спектрометрия GC/MS (m/z): 252 (молекулярный ион), 195 (основной пик).d) 2-Хлор-N-(9-пентил-9H-карбазол-3-ил) бензамид. Продукт, полученный так, как описано в предыдущей стадии с), (0,6 г, 0,0024 моль) подвергали реакции, действуя аналогично способу примера 1d). Полученный таким образом продукт (0,9 г) очищали посредством кристаллизации из смеси гексан:этилацетат = 4:1. Таким образом получали 2-хлор-N-(9 пентил-9H-карбазол-3-ил)бензамид (0,3 г). 1(т, J=6,97 Гц, 2 Н), 7,19 (тд, J=7,90, 7,00, 0,80 Гц, 1 Н), 7,41-7,71 (м, 8 Н), 8,09 (д, J=7,49 Гц, 1 Н), 8,56 (д,J=1,74 Гц, 1 Н), 10,47 (с, 1 Н). Пример 13. Получение соединения 13 (R1=CH3OCH2CH2, R2=Cl, R3=R4=R5=R6=X=Y=H). а) 9-(2-Метоксиэтил)-9H-карбазол. Карбонат цезия (19,5 г, 0,06 моль) добавляли в раствор карбазола (5 г, 0,0 30 моль) в диметилформамиде (100 мл), и смесь оставляли перемешиваться на 1 ч при комнатной температуре. Добавляли 2 бромэтилметиловый эфир (5,6 мл, 0,06 моль) и смесь оставляли перемешиваться при 90 С в течение 5 ч. Впоследствии реакционную смесь выливали в воду (300 мл) и перемешивали при комнатной температуре в течение 2 ч. Твердое вещество (4,8 г) отфильтровывали и использовали без какой-либо дополнительной очистки. 1b) 3-Нитро-9-(2-метоксиэтил)-9H-карбазол. Продукт, полученный так, как описано в предыдущей стадии а), (6 г, 0,027 моль) подвергали реакции, действуя аналогично способу примера 1b). Полученный таким образом продукт (7,2 г) очищали посредством кристаллизации из толуола, получая желаемый продукт (4,5 г). 1c) 9-(2-Метоксиэтил)-9 Н-карбазол-3-амин. Продукт, полученный так, как описано в предыдущей стадии b), (3 г, 0,011 моль) подвергали реакции, действуя аналогично способу примера 1 с). Полученный таким образом продукт (2,7 г) выкристаллизовывали из смеси гексан:этилацетат = 2:1, получая желаемый продукт (1,1 г). 1d) 2-Хлор-N-[9-(2-метоксиэтил)-9H-карбазол-3-ил]бензамид. Продукт, полученный так, как описано в предыдущей стадии с), (0,58 г, 0,0024 моль) подвергали реакции с 2-хлорбензоилхлоридом (0,36 мл, 0,0028 моль), действуя аналогично способу примера 1d). Полученный таким образом продукт (0,9 г) выкристаллизовывали из смеси гексан:этилацетат = 2:1. Таким образом получали 2-хлор-N-[9-(2-метоксиэтил)-9H-карбазол-3-ил]бензамид (0,5 г). 1a) 4-(9H-Карбазол-9-ил)бутаннитрил. Карбазол (10 г, 0,060 моль) подвергали реакции с 4-бромбутиронитрилом (11,9 мл, 0,12 моль), действуя аналогично способу примера 1 а). Твердый продукт, получаемый после холодной фильтрации, (18 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 8:2. Таким образом получали желаемый продукт (10 г). 1b) 4-(3-Нитро-9H-карбазол-9-ил)бутаннитрил. Продукт, полученный так, как описано в предыдущей стадии а), (5 г, 0,0213 моль) подвергали реакции, действуя аналогично способу примера 1b). Полученный таким образом продукт (5,2 г) выкристаллизовывали из 95 этанола, получая желаемый продукт (4,8 г). Газовая хроматография/масс-спектрометрияc) 4-(3-Амино-9 Н-карбазол-9-ил)бутаннитрил. Продукт, полученный так, как описано в предыдущей стадии b), (2,3 г, 0,0082 моль) подвергали реакции, действуя аналогично способу примера 1 с). Полученный таким образом продукт (2,3 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 4:6. Таким образом получали желаемый продукт (1,4 г). 1d) 4-(3-Амино-9H-карбазол-9-ил)бутановая кислота. Продукт, полученный так, как описано в предыдущей стадии с), (0,7 г, 2,81 ммоль) подвергали реакции в воде (5 мл) с 95% серной кислотой (2,8 мл, 50 ммоль) при кипячении с обратным холодильником в течение одной ночи. Затем реакционную смесь охлаждали, выливали на лед (50 г) и твердый продукт собирали фильтрацией. Таким образом получали желаемый продукт (0,83 г), который использовали в следующей реакции без какой-либо дополнительной очистки.e) 4-[3-(2-Хлорбензамидо)]-9H-карбазол-9-илбутановая кислота. Продукт, полученный так, как описано в предыдущей стадии d), (0,3 г, 1,19 ммоль) подвергали реакции с 2-хлорбензоилхлоридом (0,171 мл, 1,35 ммоль), действуя аналогично способу примера 1d). Реакционную смесь промывали посредством 1N HCl (36 мл). Органические фазы отделяли. Удаляли растворитель из объединенных органических фаз путем выпаривания при пониженном давлении. Таким образом получали 4-[3-(2-хлорбензамидо)]-9H-карбазол-9-илбутановую кислоту (0,15 г). 1a) 6-Бром-9-этил-1,4-диметил-3-нитро-9H-карбазол. 6-Бром-1,4-диметил-3-нитро-9H-карбазол, полученный так, как описано в Chem. Pharm. Bull 35(1),425-428 (1987), (4,8 г, 0,015 моль) подвергали реакции с йодоэтаном, действуя аналогично способу примера 1 а). Твердый продукт, полученный после фильтрации, (5,1 г) использовали без какой-либо дополнительной очистки. 1b) 6-Бром-9-этил-1,4-диметил-3-амино-9H-карбазол. Продукт, полученный так, как описано в предыдущей стадии а), (5 г, 0,014 моль) подвергали реакции, действуя аналогично способу примера 1 с). Твердый продукт, получаемый после фильтрации, (5,1 г) использовали без какой-либо дополнительной очистки. Газовая хроматография/масс-спектрометрияc) 9-Этил-1,4-диметил-3-амино-9H-карбазол. Раствор продукта, полученного на предыдущей стадии b), (2 г, 0,006 моль) в тетрагидрофуране(THF) (25 мл) добавляли в суспензию LiAlH4 (0,95 г, 0,025 моль) в безводном тетрагидрофуране (25 мл). Суспензию кипятили с обратным холодильником в течение 24 ч. Впоследствии суспензию оставляли остывать до комнатной температуры и добавляли смесь H2O и THF 1:1 и NaOH (1,9 г). THF удаляли путем выпаривания при пониженном давлении и водный остаток помещали в делительную воронку, и экстрагировали этилацетатом (250 мл). Органическую фазу отделяли и сушили над Na2SO4. Растворитель удаляли путем выпаривания при пониженном давлении и полученный таким образом остаток (1,3 г) использовали без какой-либо дополнительной очистки. Газовая хроматография/масс-спектрометрия GC/MSd) 2-Хлор-N-(9-этил-1,4-диметил-9H-карбазол-3-ил)бензамид. Продукт, полученный так, как описано в предыдущей стадии с), (1,2 г, 0,005 моль) подвергали реакции, действуя аналогично способу примера 1d). Полученный таким образом продукт (2 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 8:2 и затем выкристаллизовывали из смеси хлороформ:гексан = 1:1. Таким образом получали 2-хлор-N-(9-этил-1,4-диметил 9 Н-карбазол-3-ил)бензамид (0,2 г). 1H ЯМР (300 МГц, DMSO-d6)м.д. 1,32 (т, J=7,00 Гц, 3H), 2,72 (с, 3H), 2,79 (с, 3H), 4,65 (кв., J=6,94 Гц, 2 Н), 7,17 (с, 1 Н), 7,22 (тд, J=8,00, 7,00, 0,80 Гц, 1 Н), 7,42-7,70 (м, 6 Н), 8,21 (д, J=7,93 Гц, 1 Н), 10,03 (с,- 10012786 1 Н). Пример 16. Получение соединения 16 (R1=CH3CH2, R2=Cl, X=CH3, R3=R4=R5=R6=Y=H). а) 9-Этил-9H-карбазол. Карбазол (4 г, 0,024 моль) подвергали реакции с йодоэтаном, действуя аналогично способу примера 1 а). Твердый продукт, получаемый после фильтрации, (4,4 г) использовали без какой-либо дополнительной очистки. 1b) 9-Этил-3-нитро-9H-карбазол. Продукт, полученный так, как описано в предыдущей стадии а), (4 г, 0,0014 моль) подвергали реакции, действуя аналогично способу примера 1b). Полученный таким образом продукт (6 г) очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 8:2. Таким образом получали желаемый продукт (2,8 г). 1c) 9-Этил-4-метил-3-нитро-9 Н-карбазол. Продукт, полученный так, как описано в предыдущей стадии b), (2,8 г, 0,0012 моль) растворяли в безводном тетрагидрофуране (THF) (приблизительно 100 мл). Раствор помещали в инертную атмосферу при -15 С. Добавляли 3 М раствор CH3MgCl в THF (5,7 мл, 0,012 моль), и реакционную смесь оставляли перемешиваться в течение 1 ч при -15 С. Впоследствии добавляли 2,3-дихлор-5,6-дициано-1,4 бензохинон) (4,5 г, 0,0020 моль), и реакционной смеси давали нагреться до комнатной температуры, и поддерживали при перемешивании в течение 48 ч. Реакционную смесь разбавляли дихлорметаном (100 мл), и промывали водой (260 мл). Органическую фазу отделяли и сушили над Na2SO4. Затем удаляли растворитель путем выпаривания при пониженном давлении, и полученный таким образом остаток очищали посредством флэш-хроматографии при элюировании смесью гексан:этилацетат = 8:2. Чистые фракции объединяли, получая желаемый продукт (0,6 г). 1d) 9-Этил-4-метил-9 Н-карбазол-3-амин. Продукт, полученный так, как описано в предыдущей стадии с), (0,4 г, 0,0016 моль) подвергали реакции, действуя аналогично способу примера 1 с). Полученный таким образом продукт (0,5 г) очищали посредством флэш-хроматографии при элюировании трихлорметаном (CHCl3). Таким образом получали желаемый продукт (0,3 г). 1 Н ЯМР (300 МГц, DMSO-d6)м.д. 1,24 (т, J=7,02 Гц, 3H), 2,57 (с, 3H), 4,32 (кв., J=7,02 Гц, 2 Н),4,55 (с, 2 Н), 6,91 (д, J=8,48 Гц, 1 Н), 7,09 (ддд, J=8,18, 7,00, 1,00 Гц, 1 Н), 7,18 (д, J=8,48 Гц, 1 Н), 7,35 (ддд,J=8,00, 7,20, 1,20 Гц, 1 Н), 7,48 (д, J=8, 18 Гц, 1 Н), 8,15 (д, J=7,89 Гц, 1 Н). е) 2-Хлор-N-(9-этил-4-метил-9H-карбазол-3-ил)бензамид. Продукт, полученный так, как описано в предыдущей стадии d), (0,3 г, 0,0013 моль) подвергали реакции, действуя аналогично способу примера 1d). Полученный таким образом продукт (0,5 г) выкристаллизовывали 2 раза из смеси гексан:этилацетат = 1:2. Таким образом получали 2-хлор-N-(9-этил-4-метил 9 Н-карбазол-3-ил)бензамид (0,23 г). 1H ЯМР (300 МГц, DMSO-d6)м.д. 1,31 (т, J=7,10 Гц, 3H), 2,77 (с, 3H), 4,47 (кв., J=7,05 Гц, 2 Н),7,23 (ддд, J=8,00, 7,00, 1,00 Гц, 1 Н), 7,39-7,72 (м, 8 Н), 8,23 (д, J=7,93 Гц, 1 Н), 10,09 (с, 1 Н). Примеры 17 и 18. Получение соединений 17 и 18. Соединения 17 и 18 могут быть получены, действуя аналогично способу, описанному в примерах 116 выше. Пример 19. Получение сравнительного соединения А (R1=CH3CH2, R2=OH, R3=R4=R5=R6=a) N-(9-Этил-9H-карбазол-3-ил)-2-метоксибензамид. 3-Амино-9-этилкарбазол (5,6 г; 0,027 моль) подвергали реакции с 2-метоксибензоилхлоридом (4,35 мл; 0,029 моль), действуя аналогично способу, описанному в примере 4. Остаток, полученный таким образом, (3,5 г) выкристаллизовывали из изопропанола, получая желаемый продукт (2,5 г). 1b) N-(9-Этил-9 Н-карбазол-3-ил)-2-гидроксибензамид. К раствору, содержащему трибромид бора (1 М в дихлорметане, 0,2 мл; 0,001 моль) в дихлорметане- 11012786 моль). Полученную таким образом смесь перемешивали в течение 16 ч. Реакционную смесь перемещали в делительную воронку и промывали посредством H2O (250 мл). Органическую фазу отделяли и сушили над Na2SO4. Растворитель удаляли путем выпаривания при пониженном давлении, и полученный таким образом остаток (0,4 г) выкристаллизовывали из этанола. Таким образом получали N-(9-этил-9Hкарбазол-3-ил)-2-гидроксибензамид (0,25 г). 1(шир.с, 1 Н). Пример 20. Получение сравнительного соединения В (R1=CH3CH2, R2=SCH3, R3=R4=R5=R6=X=Y=H). Сравнительное соединение В получали аналогично способу, описанному в отношении соединений 6-9, начиная с 9-этил-3-аминокарбазола и 2-метилмеркаптобензойной кислоты. Жидкостная хроматография/масс-спектрометрия LC/MS (М+Н)+ =361,2. Пример 21. Испытание активности in vitro. Данное испытание позволяет оценить ингибирующую способность в отношении продуцирования простагландинов PGE2 и селективность относительно продуцирования простагландинов PGF2. Использовали клеточную линию А 549, аденокарциному легкого человека, которая особенно чувствительна к стимуляции провоспалительными циокинами, например IL-1, и, реагируя на такую стимуляцию, является особенно активной в продуцировании и высвобождении двух простаноидов: PGE2 и PGF2a (Thoren S.Jakobsson P-J, 2000). Клетки стимулировали посредством IL-1 (1 нг/мл) и одновременно обрабатывали испытываемым соединением в течение 18 ч в соответствующей культуральной среде (DMEM - Dulbecco's Modified Eagle's Medium), дополненной 5% фетальной телячьей сывороткой и L-глутамином (4 мМ конечная концентрация), в инкубаторе при 37 С и при концентрации CO2 5%. После инкубирования количества PGE2 и PGF2, продуцируемые и высвобождаемые в надосадочную жидкость, оценивали при использовании набора реактивов для иммуноферментного анализа (EIA kit = Enzyme Immunoassay kit)(произведенных и продаваемых компанией Cayman Chemicals, Ann Arbor, MI, USA). Используемые сравнительные соединения представляют собой индометацин (Sigma-Aldrich), нестероидное противовоспалительное средство, которое ингибирует в равной мере как PGE2, так и PGF2a, и также соединения формулы(I), где R1=CH3CH2, R2=OH, R3=R4=R5=R6=X=Y=H (сравнительное соединение А) и R1=CH3CH2,R2=SCH3, R3=R4=R5=R6=X=Y=H (сравнительное соединение В). Результаты, приведенные в процентах ингибирования продуцирования PGE2 и PGF2a при концентрации 10 мкМ, показаны в табл. 2. Таблица 2 Не активно = не активно при концентрации, используемой в эксперименте. В целях иллюстрации, табл. 3 показывает значения pIC50 некоторых соединений изобретения, гдеpIC50 представляет собой отрицательный логарифм значения IC50, которое, в свою очередь, представляет собой концентрацию соединения, при которой ингибируется 50% продуцирования PGE2 или PGF2 относительно клеток, которые были подвергнуты стимуляции, но не были обработаны аналогичным соединением. Пример 22. Испытание активности in vivo. Это испытание позволяет оценить активность соединений изобретения в испытании ноцицептивной реакции воспалительного происхождения. Испытываемые соединения оценивали в экспериментальной модели, которая вызывает вытягивание мышей, индуцируемое уксусной кислотой (Stock J.L. et al., J. Clin. Inv. 2001, 107:325-331). Для испытания использовали самок мышей CD-1, имеющих массу 25-30 г. Животных обрабатывали перорально соединением (30 мг/кг), суспендированным в метилцеллюлозе (МТС). Контрольных животных обрабатывали перорально только носителем (МТС). По истечении одного часа после обработки животным вводили внутрибрюшинный инъекционный раствор уксусной кислоты (0,7% в/в в физиологическом растворе, 16 мкл/г массы тела) для того, чтобы вызвать боль воспалительного происхождения и проверить влияние обработки на ноцицептивную реакцию. Сразу после введения уксусной кислоты и в течение последующих 20 мин измеряли число вытягиваний, которое представляет собой параметр для оценки ноцицептивной реакции. Животные, обработанные соединениями изобретения, показали значительное снижение вытягивания в течение 20 мин после введения уксусной кислоты, по сравнению с животными, обработанными только МТС. Результаты, полученные с соединением 4, показаны на чертеже. Пример 23. Испытание на первичных эндотелиальных клетках человека (HUVEC). Это испытание позволяет оценить способность соединений изобретения ингибировать продуцирование PGI2. Отсутствие ингибирующей активности на этом простаноиде может гарантировать сохранение вазопротекторного действия простаноидов PGI2 и обеспечить полезную фармакологическую информацию, касающуюся отсутствия неблагоприятных побочных эффектов на эндотелии. Действие тестовых соединений оценивали в клетках HUVEC при базальных условиях и в условиях стимуляции (J. Immunol. 1989, June 1; 142(11): 3993-9. Результаты приведены в процентах ингибирования относительно контрольной ферментативной активности. Индометацин используют в качестве эталонного соединения. Соединения изобретения не показали никакого значительного ингибирования секреции PGI2. Результаты показаны в табл. 4. Таблица 4 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы и их фармацевтически приемлемые соли. 2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным носителем. 3. Способ лечения или предупреждения воспалительных процессов, опухолей, болезни Альцгеймера и атеросклероза у млекопитающих, включающий введение терапевтически эффективного количества соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемые соли, субъекту, который нуждается в этом. 4. Способ получения 3-аминокарбазола из таблицы по п.1, отличающийся тем, что он включает следующие стадии: а) реагирование амина формулы (II) где R2, R3, R4, R5 и R6 имеют значения, указанные в упомянутой таблице, и Z выбирают из группы, включающей Cl, Br, ОН, OR и OC(O)R, где R представляет собой линейный или разветвленный алкил, содержащий от 1 до 6 атомов углерода,получая соединение 3-аминокарбазола формулы (I)b) необязательно, образование фармацевтически приемлемой соли соединения формулы (I), полученного таким образом.
МПК / Метки
МПК: A61P 35/00, C07D 209/88, A61K 31/403
Метки: 3-аминокарбазола, композиция, фармацевтическая, соединения, содержащая, способ, указанные, получения
Код ссылки
<a href="https://eas.patents.su/16-12786-soedineniya-3-aminokarbazola-farmacevticheskaya-kompoziciya-soderzhashhaya-ukazannye-soedineniya-i-sposob-ih-polucheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения</a>
Спироимидазолиновые соединения, способ их получения и фармацевтическая композиция, содержащая их

Номер патента: 2979
Опубликовано: 26.12.2002
Авторы: Брокко Морисетт, Корди Алекс, Ньюмен-Танкреди Адриан, Миллан Марк
МПК: C07D 235/02, A61P 25/24, A61K 31/4184...
Метки: соединения, содержащая, способ, спироимидазолиновые, композиция, получения, фармацевтическая
Формула / Реферат:
1. Спироимидазолиновые соединения формулы (I) в которой А представляет бензольное кольцо, незамещенное или замещенное 1-4 одинаковыми или различными группами, выбранными из линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, гидрокси, полигалоген-(С1-С6)алкила, в котором алкильный фрагмент является линейным или разветвленным, и галогена, В представляет имидазолиновое кольцо, как представлено в формулах (Iа)...
Новые замещенные димерные соединения, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 3300
Опубликовано: 24.04.2003
Авторы: Декамп-Франсуа Кароль, Лезьер Даниель, Гийоме Жераль, Вио Мари-Клод, Ренар Пьер, Иус Саид, Делягранж Филипп, Лефулон Франсуа, Беннежан Каролин
МПК: A61K 31/353, C07D 403/12, C07C 233/72...
Метки: новые, фармацевтическая, соединения, композиция, содержащая, димерные, замещенные, способ, получения
Формула / Реферат:
1. Соединения формулы (I) A-G1-Cy-G2-Cy-G3-B (I), где A представляет собой группы формулы -NHC(Q)R1 или -NHC(Q)NHR2, где Q обозначает атом кислорода или серы, R1 обозначает линейную или разветвленную (C1-C6)алкильную группу, замещенную или незамещенную атомом галогена, линейную или разветвленную (C2-C6)алкенильную группу, (C3-C8)циклоалькильную группу, арильную или гетероарильную группу, R2 обозначает линейную или разветвленную...
Бета- карболиновые соединения, способ их получения и фармацевтическая композиция, их содержащая

Номер патента: 4140
Опубликовано: 26.02.2004
Авторы: Гольдстейн Соло, Пуассонне Гийом, Декейн Анн, Миллан Марк, Парментье Жан-Жилль, Бутэн Жан, Брион Жан-Даниель
МПК: A61K 31/437, A61P 25/00, C07D 471/04...
Метки: соединения, карболиновые, способ, фармацевтическая, beta, композиция, получения, содержащая
Формула / Реферат:
1. Соединения формулы (I) в которой --- представляет простую или двойную связь, способную необязательно сообщать ароматический характер кольцу, несущему ее, R1 представляет группу, выбранную из водорода, прямого или разветвленного (C1-C6)алкила, -R6-арила, -R6-циклоалкила, -R6-гетероцикла, где группы R6 представляют прямую или разветвленную (C1-C6)алкиленовую группу, -CO2R7, где R7 представляет прямую или разветвленную (C1-C6)алкильную группу,...
Новые производные арилглицинамида, способ их получения и фармацевтическая композиция , содержащая эти соединения

Номер патента: 2201
Опубликовано: 28.02.2002
Авторы: Шромм Курт, Юнг Биргит, Эссер Франц, Шнорренберг Герд, Шпек Георг, Доллингер Хорст
МПК: A61P 9/00, A61K 31/395, C07D 295/14...
Метки: фармацевтическая, композиция, эти, новые, соединения, производные, получения, содержащая, арилглицинамида, способ
Формула / Реферат:
1. Производные арилглицинамида общей формулы (I) или их фармацевтически приемлемые соли, где Ar означает незамещенный или 1-5-кратнозамещенный фенил или незамещенный или 1-2-кратнозамещенный нафтил, причем заместители фенила и нафтила независимо друг от друга являются галогеном (фтором, хлором, бромом, йодом), алкилом с 1-4 атомами углерода, O-(С1-С4)алкилом, трифторметилом, трифторметокси или NR12R13, где R12 и R13 независимо друг от друга...
Фармацевтическая композиция, содержащая фенофибрат, и способ ее получения

Номер патента: 4294
Опубликовано: 26.02.2004
Авторы: Криер Брюно, Шеневьер Филипп, Сюпли Паскаль
МПК: A61K 31/216
Метки: композиция, фенофибрат, фармацевтическая, содержащая, получения, способ
Формула / Реферат:
1. Фармацевтическая композиция, содержащая микронизированный фенофибрат, поверхностно-активное вещество и связующее производное целлюлозы в качестве адъюванта солюбилизации, отличающаяся тем, что она содержит количество фенофибрата выше или равное 60 мас.%. 2. Композиция по п.1, отличающаяся тем, что связующим производным целлюлозы, являющимся адъювантом солюбилизации, является гидроксипропилметилцеллюлоза. 3. Композиция по п.2, отличающаяся...
Предыдущий патент: Спирокетальзамещённые циклические кетоенолы
Следующий патент: Способ получения эсциталопрама
Случайный патент: Буровой раствор на водной основе