Тетразамещенные пиридазины в качестве антагонистов пути hedgehog
Номер патента: 17386
Опубликовано: 28.12.2012
Авторы: Клэй Джулия Мари, Коэн Джеффри Дэниел, Лобб Карен Линн, Бастиан Джоли Энн, Сэлл Дэниел Джон, Томпсон Майкл Ли, Хипскинд Филип Артур

















Формула / Реферат
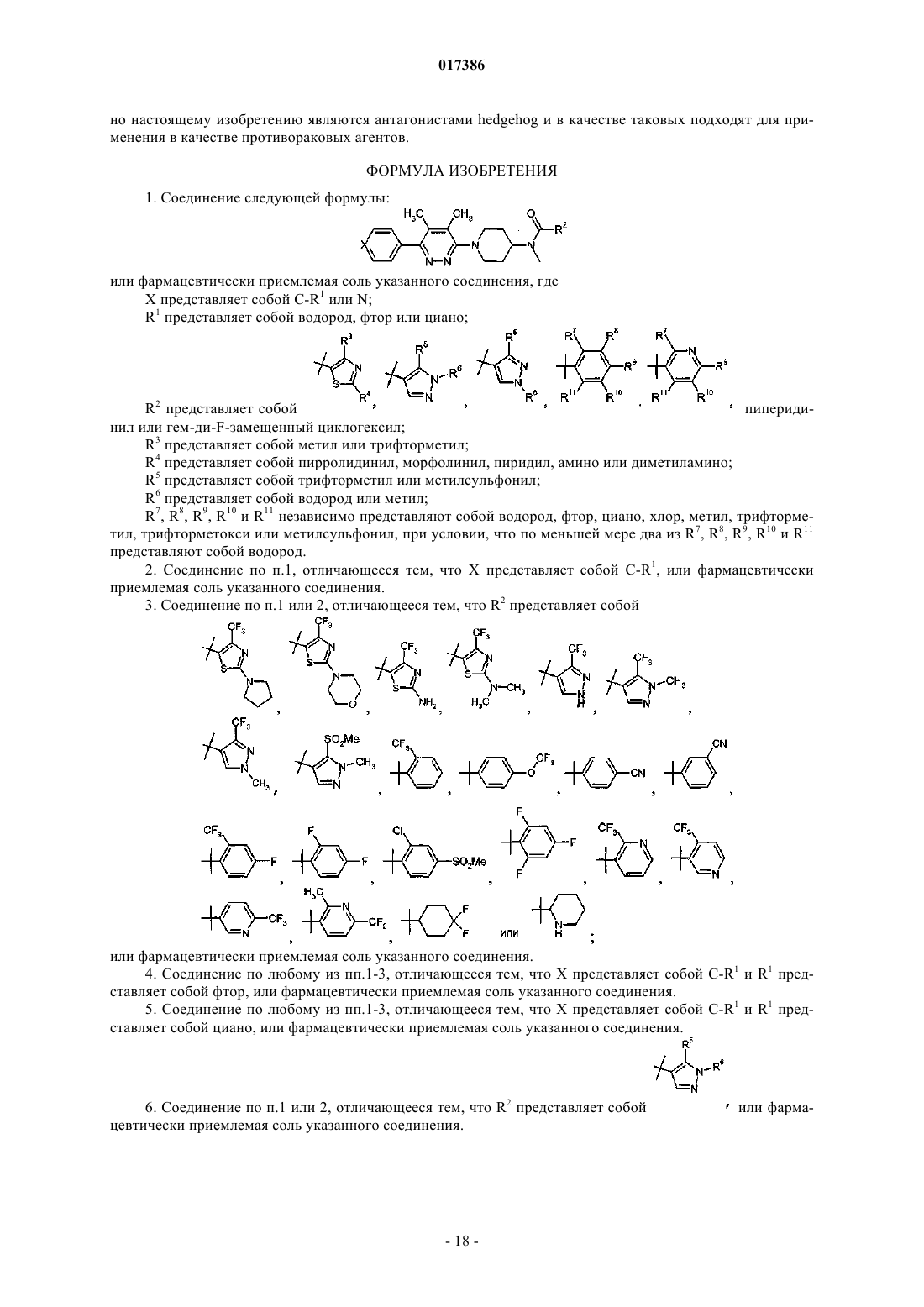
1. Соединение следующей формулы:

или фармацевтически приемлемая соль указанного соединения, где
X представляет собой C-R1 или N;
R1 представляет собой водород, фтор или циано;
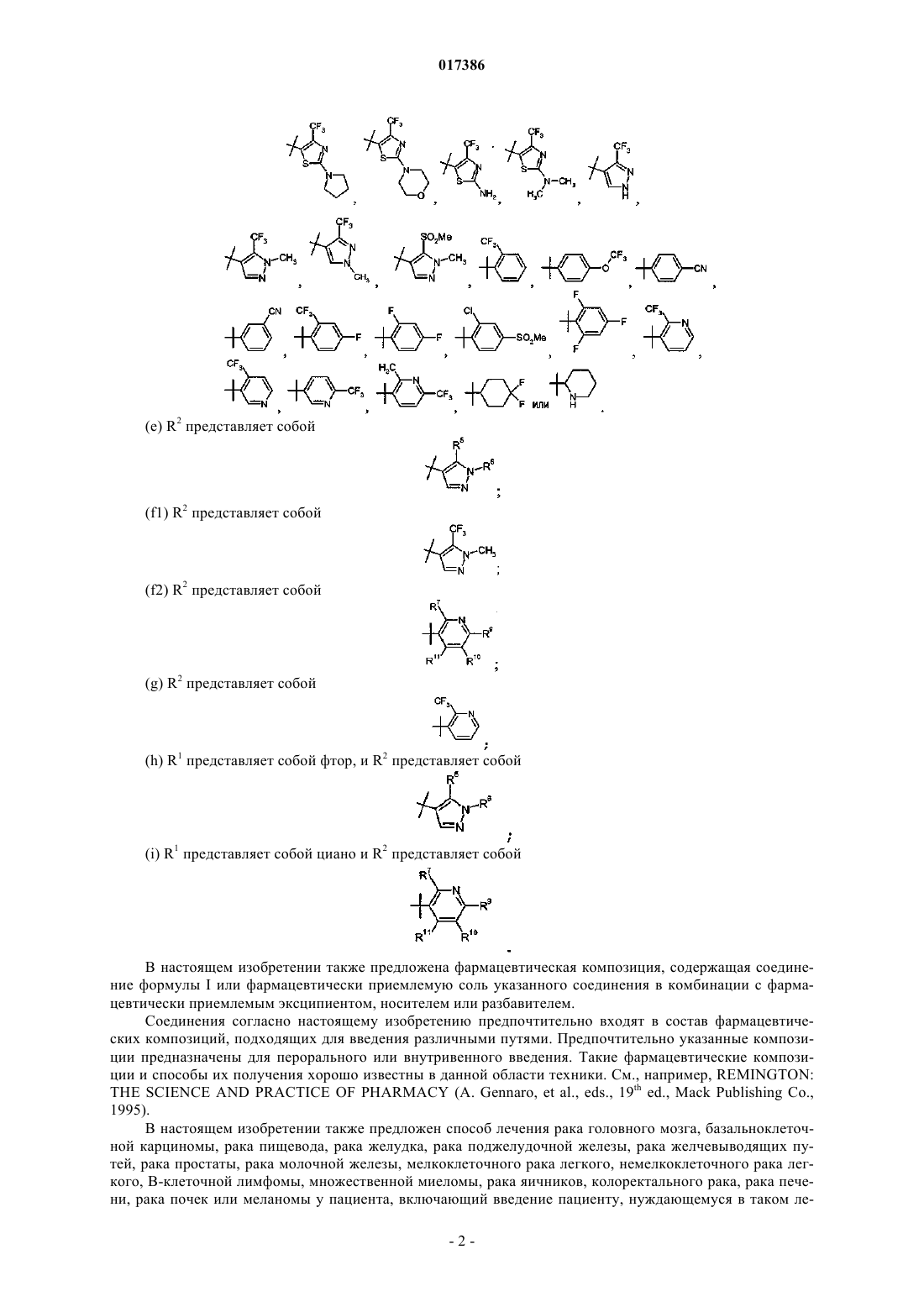
R2 представляет собой
пиперидинил или гем-ди-F-замещенный циклогексил;
R3 представляет собой метил или трифторметил;
R4 представляет собой пирролидинил, морфолинил, пиридил, амино или диметиламино;
R5 представляет собой трифторметил или метилсульфонил;
R6 представляет собой водород или метил;
R7, R8, R9, R10 и R11 независимо представляют собой водород, фтор, циано, хлор, метил, трифторметил, трифторметокси или метилсульфонил, при условии, что по меньшей мере два из R7, R8, R9, R10 и R11 представляют собой водород.
2. Соединение по п.1, отличающееся тем, что X представляет собой C-R1, или фармацевтически приемлемая соль указанного соединения.
3. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой


или фармацевтически приемлемая соль указанного соединения.
4. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой C-R1 и R1 представляет собой фтор, или фармацевтически приемлемая соль указанного соединения.
5. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой C-R1 и R1 представляет собой циано, или фармацевтически приемлемая соль указанного соединения.
6. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой
или фармацевтически приемлемая соль указанного соединения.
7. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой
или фармацевтически приемлемая соль указанного соединения.
8. Соединение по п.6, отличающееся тем, что R5 представляет собой трифторметил и R6 представляет собой метил, или фармацевтически приемлемая соль указанного соединения.
9. Соединение по п.7, отличающееся тем, что R7 представляет собой трифторметил и R9, R10 и R11 представляют собой водород, или фармацевтически приемлемая соль указанного соединения.
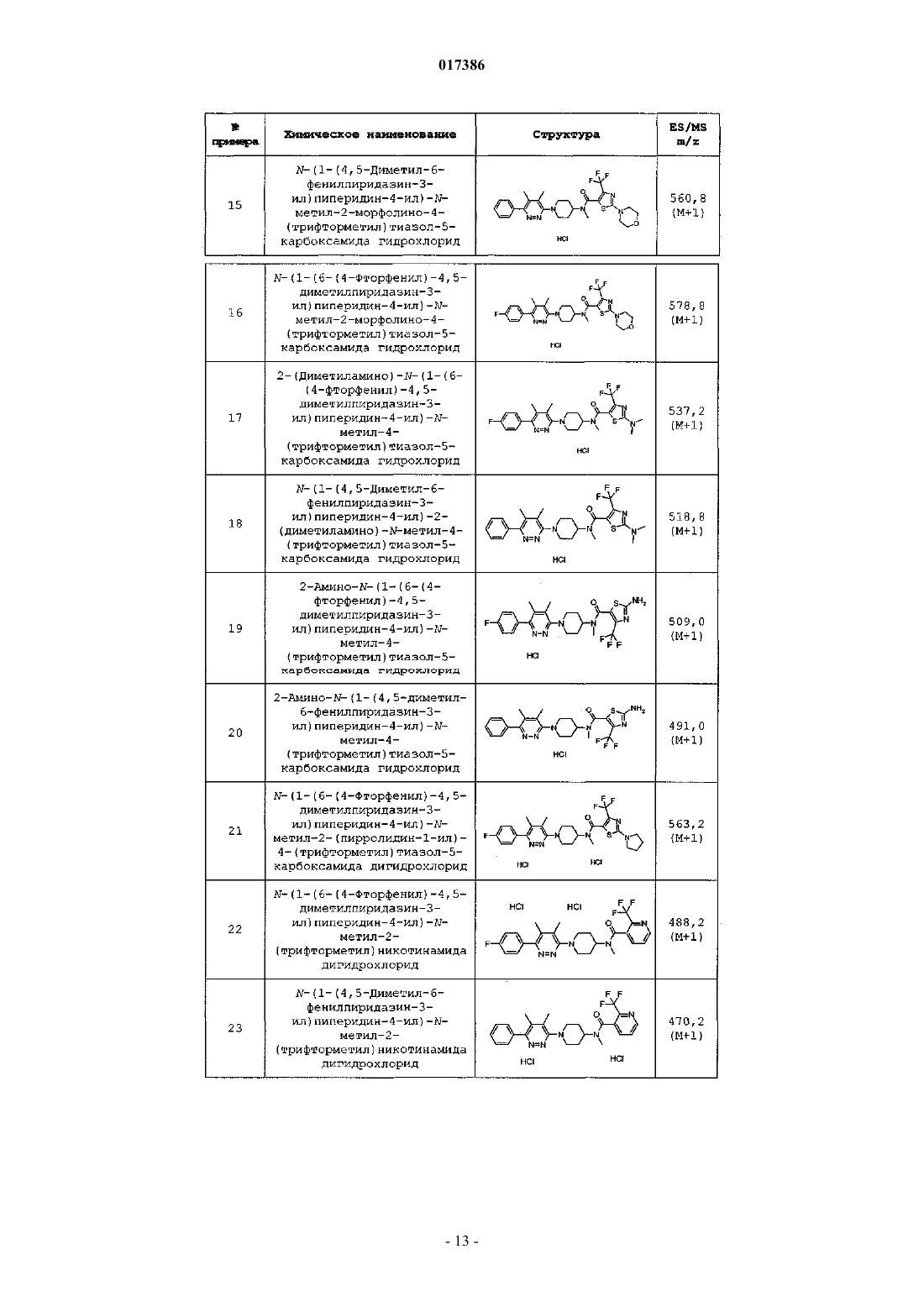
10. Соединение по п.1, представляющее собой N-(1-(6-(4-цианофенил)-4,5-диметилпиридазин-3-ил)пиперидин-4-ил)-N-метил-2-(трифторметил)никотинамид.
11. Соединение по п.1, представляющее собой N-(1-(6-(4-цианофенил)-4,5-диметилпиридазин-3-ил)пиперидин-4-ил)-N-метил-2-(трифторметил)никотинамида дигидрохлорид.
12. Фармацевтическая композиция, содержащая соединение по любому из пп.1-11 или фармацевтически приемлемую соль указанного соединения в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
13. Применение соединения по любому из пп.1-11 или фармацевтически приемлемой соли указанного соединения в качестве лекарственного средства.
14. Применение соединения по любому из пп.1-11 или фармацевтически приемлемой соли указанного соединения для лечения рака.
15. Применение по п.14, отличающееся тем, что указанный рак выбран из группы, состоящей из рака головного мозга, базальноклеточной карциномы, рака пищевода, рака желудка, рака поджелудочной железы, рака желчевыводящих путей, рака простаты, рака молочной железы, мелкоклеточного рака легкого, немелкоклеточного рака легкого, В-клеточной лимфомы, множественной миеломы, рака яичников, колоректального рака, рака печени, рака почек и меланомы.
Текст
В настоящем изобретении предложены новые тетразамещенные пиридазины в качестве антагонистов пути hedgehog, подходящие для лечения рака.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 017386 Настоящее изобретение относится к антагонистам пути Hedgehog и, в частности, к новым тетразамещенным пиридазинам и применению указанных соединений в терапии. Сигнальный путь Hedgehog(Hh) играет важную роль в образовании эмбриональной структуры и обеспечении жизнедеятельности зрелой ткани за счет регулирования дифференцировки и пролиферации клеток. Семейство белковHedgehog (Hh), включающее Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) и Desert Hedgehog (Dhh), представляет собой секретируемые гликопротеины, подвергающиеся посттрансляционным модификациям,включая автокаталитическое отщепление и присоединение холестерина к аминоконцевому пептиду с образованием фрагмента, обладающего активностью в отношении передачи сигналов. Hh связывается с состоящим из 12 доменов трансмембранным белком Ptch (Ptch1 и Ptch2), тем самым способствуя опосредуемому Ptch подавлению Smoothened (Smo). Активация Smo запускает ряд внутриклеточных процессов,завершающихся стабилизацией транскрипционных факторов Gli (Gli1, Gli2 и Gli3) и экспрессией Gliзависимых генов, отвечающих за пролиферацию клеток, выживаемость клеток, ангиогенез и инвазию. Сигнальный путь Hh в последнее время привлекает значительный интерес, обусловленный обнаружением того факта, что нарушенная активация сигнального пути Shh приводит к образованию различных опухолей, например вызывает рак поджелудочной железы, медуллобластому, базальноклеточную карциному, мелкоклеточный рак легкого и рак простаты. В данной области техники сообщалось о нескольких антагонистах Hh, таких как стероидное алкалоидное соединение IP-609, аминопролин CUR61414 и 2,4 дизамещенный тиазол JK18. В WO 2005033288 предложены некоторые 1,4-дизамещенные фталазины,которые, как утверждают, являются антагонистами hedgehog. Аналогично, в документе WO 2008110611 предложены некоторые 1,4-дизамещенные фталазины, связанные с диагностикой и лечением патологий,связанных с путем hedgehog. По-прежнему существует потребность в мощных ингибиторах пути hedgehog, в частности ингибиторах, обладающих желаемыми фармакодинамическими, фармакокинетическими и токсикологическими характеристиками. В настоящем изобретении предложены новые тетразамещенные пиридазины, являющиеся мощными антагонистами указанного пути. Согласно настоящему изобретению предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения, гдеR2 представляет собой или гем-дифторзамещенный циклогексил;R3 представляет собой метил или трифторметил;R5 представляет собой трифторметил или метилсульфонил;R6 представляет собой водород или метил;R7, R8, R9, R10 и R11 независимо представляют собой водород, фтор, циано, хлор, метил, трифторметил, трифторметокси или метилсульфонил, при условии, что по меньшей мере два из R7, R8, R9, R10 и R11 представляют собой водород. Специалисту в данной области техники понятно, что соединения согласно настоящему изобретению содержат фрагмент, представляющий собой третичный амин, и способны взаимодействовать с рядом неорганических и органических кислот, образуя фармацевтически приемлемые соли присоединения кислот. Такие фармацевтически приемлемые соли присоединения кислот и общая методика получения указанных солей известны в данной области техники. См., например, P. Stahl, et al., HANDBOOK OFBerge, et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, Vol. 6,1, January 1977. Конкретные варианты реализации настоящего изобретения включают соединения формулы I или фармацевтически приемлемую соль указанных соединений, где:(i) R1 представляет собой циано и R2 представляет собой В настоящем изобретении также предложена фармацевтическая композиция, содержащая соединение формулы I или фармацевтически приемлемую соль указанного соединения в комбинации с фармацевтически приемлемым эксципиентом, носителем или разбавителем. Соединения согласно настоящему изобретению предпочтительно входят в состав фармацевтических композиций, подходящих для введения различными путями. Предпочтительно указанные композиции предназначены для перорального или внутривенного введения. Такие фармацевтические композиции и способы их получения хорошо известны в данной области техники. См., например, REMINGTON:THE SCIENCE AND PRACTICE OF PHARMACY (A. Gennaro, et al., eds., 19th ed., Mack Publishing Co.,1995). В настоящем изобретении также предложен способ лечения рака головного мозга, базальноклеточной карциномы, рака пищевода, рака желудка, рака поджелудочной железы, рака желчевыводящих путей, рака простаты, рака молочной железы, мелкоклеточного рака легкого, немелкоклеточного рака легкого, В-клеточной лимфомы, множественной миеломы, рака яичников, колоректального рака, рака печени, рака почек или меланомы у пациента, включающий введение пациенту, нуждающемуся в таком ле-2 017386 чении, эффективного количества соединения формулы I или фармацевтически приемлемой соли указанного соединения. Следует понимать, что фактически вводимое количество соединения будет определять лечащий врач с учетом соответствующих обстоятельств, включая состояние, лечение которого проводят, выбранный путь введения, конкретное вводимое соединение или соединения, возраст, массу тела и ответ конкретного пациента, а также степень тяжести симптомов заболевания у пациента. Суточные дозировки обычно составляют примерно от 0,1 примерно до 5 мг/кг массы тела. В некоторых случаях более чем достаточными могут быть уровни дозировок ниже нижней границы вышеуказанного диапазона, тогда как в других случаях можно применять еще более высокие дозы. Следовательно, вышеуказанный диапазон дозировок никоим образом не ограничивает объем настоящего изобретения. Согласно настоящему изобретению также предложено соединение формулы I или фармацевтически приемлемая соль указанного соединения для применения в качестве лекарственного средства. Кроме того, согласно настоящему изобретению предложено применение соединения формулы I или фармацевтически приемлемой соли указанного соединения для получения лекарственного средства для лечения рака. В частности, указанный рак выбран из группы, состоящей из рака головного мозга, базальноклеточной карциномы, рака пищевода, рака желудка, рака поджелудочной железы, рака желчевыводящих путей, рака простаты, рака молочной железы, мелкоклеточного рака легкого, немелкоклеточного рака легкого, В-клеточной лимфомы, множественной миеломы, рака яичников, колоректального рака,рака печени, рака почек и меланомы. Далее, в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение формулы I или фармацевтически приемлемую соль указанного соединения в качестве активного агента, для лечения рака головного мозга, базальноклеточной карциномы, рака пищевода, рака желудка,рака поджелудочной железы, рака желчевыводящих путей, рака простаты, рака молочной железы, мелкоклеточного рака легкого, немелкоклеточного рака легкого, В-клеточной лимфомы, множественной миеломы, рака яичников, колоректального рака, рака печени, рака почек или меланомы. Соединения формулы I или соли указанных соединений можно получить при помощи множества методик, известных в данной области техники, а также описанных на схемах и в синтезах и примерах,приведенных ниже. Для получения соединений формулы I или солей указанных соединений можно различными способами комбинировать конкретные стадии синтеза в каждом из описанных путей, или использовать их в сочетании со стадиями из других схем. Если не указано иное, заместители являются такими, как определено ранее. Реагенты и исходные вещества в общем случае легко доступны среднему специалисту в данной области техники. Другие реагенты и исходные вещества можно получить при помощи стандартных методик органической и гетероциклической химии, методик, аналогичных синтезу известных похожих по структуре соединений, и методик, описанных в синтезах и примерах, приведенных далее, включая любые новые методики. В настоящем описании следующие термины имеют указанные значения: "boc" или "t-boc" относится к трет-бутоксикарбонилу; "ВОР" относится к гексафторфосфату бензотриазол-1-илокси-трис(диметиламино)фосфония; "ДМАА" относится к N,N-диметилацетамиду; "ДМФА" относится к N,Nдиметилформамиду; "ДМСО" относится к диметилсульфоксиду; "EDCI" относится к гидрохлориду 1-(3 диметиламинопропил)-3-этилкарбодиимида; "Et2O" относится к диэтиловому эфиру; "EtOAc" относится к этилацетату; "iPrOH" относится к изопропанолу; "МеОН" относится к метанолу; "ТФУК" относится к трифторуксусной кислоте; "SCX" относится к сильному катиониту; "РуВОР" относится к гексафторфосфату бензотриазол-1-илокситрипирролидинофосфония; и "IC50" относится к концентрации агента, обеспечивающей 50% от максимально возможного для данного агента ингибиторного ответа. Схема 1 Соединение формулы I можно получить в соответствии с реакциями, приведенными на схеме 1. На схеме 1, стадия 1, 3,6-дихлор-4,5-диметилпиридазин (1) взаимодействует с третбутилметил(пиперидин-4-ил)карбаматом (2) в реакции ароматического нуклеофильного замещения(SNAr) в полярном апротонном растворителе, таком как ДМФА, ДМАА или ДМСО, в присутствии орга-3 017386 нического основания, такого как триэтиламин или диизопропилэтиламин, и/или неорганического основания, такого как карбонат калия, при нагревании до 100-140 С с получением трет-бутил-1-(6-хлор-4,5 диметилпиридазин-3-ил)пиперидин-4-ил(метил)карбамата (3). На стадии 2 оставшийся хлорид в диметилпиридазине может взаимодействовать с арилбороновой кислотой (4) в реакции перекрестного сочетания Сузуки-Мияура, с образованием соответствующего 4,5-диметил-6-замещенного арилпиридазин-3 замещенного пиперидина (5). Специалисту в данной области техники понятно, что существует множество условий, способствующих протеканию указанных реакций перекрестного сочетания. При проведении реакции применяют подходящий растворитель, такой как диоксан или диоксан/вода, и проводят реакцию в присутствии основания, такого как карбонат цезия или фторид цезия, и палладиевого катализатора,такого как хлорид (1,1'-бис-(дифенилфосфино)ферроцен)палладия(II) или (SP-4-1)-бис-[бис-(1,1 диметилэтил)(4-метоксифенил)фосфин-кР]дихлорпалладий (полученный в соответствии с J. Org. Chem. 2007, 72, 5104-5112) в инертной атмосфере при температуре примерно 80-160 С, с получением соединения формулы (5). Удалить защиту с аминной группы можно при помощи обычных способов удаления защиты. Способы введения и удаления защитных групп при атоме азота хорошо известны в данной области техники (см., например, Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John Wileyand Sons, New York, (1999. Например, удаление защитной группы boc из амина формулы (5) можно проводить в кислой среде, например, при помощи хлористого водорода или трифторуксусной кислоты, с получением соединения формулы (6). Ацилирование амина на стадии 4 можно осуществить при помощи замещенного хлорангидрида (7) в инертном растворителе, таком как дихлорметан, или, альтернативно,соединение формулы (6) можно ацилировать при помощи замещенной карбоновой кислоты и подходящего агента для реакции сочетания, такого как РуВОР, пентафторфенилдифенилфосфинат, ВОР илиEDCI, и подходящего основания, такого как триэтиламин или диизопропилэтиламин, в подходящем растворителе, таком как ДМФА и/или ДМСО или дихлорметан, с получением нейтральных соединений формулы I. Соединения формулы I можно превратить в соль, такую как соль HCl, при помощи способов,известных специалистам в данной области техники, таких как воздействие HCl в Et2O, или HCl может быть получена in situ путем добавления по каплям ацетилхлорида к раствору в спиртовом растворителе,таком как метанол, при 0-20 С. Схема 2 Целевые карбоновые кислоты (7) (Y=OH, стадия 4, схема 1) можно получить, как показано на схеме 2. Первичный амин в положении 2 тиазола (8) замещают хлоридом по реакции Зандмейера с использованием хлорида меди и изопентилнитрита в подходящем растворителе, таком как ацетонитрил, как показано на стадии 1, с образованием 2-хлор-4,5-замещенного тиазола (9). Затем указанный хлорид заменяют на целевой амин (10) на стадии 2 в полярном апротонном растворителе, таком как ДМСО, с получением соответствующего аминотиазола (11). Гидролиз сложного эфира на стадии 3 с применением подходящего основания, такого как водный раствор гидроксида натрия или водный раствор гидроксида лития, в подходящем растворителе, таком как МеОН или диоксан, позволяет получить целевую карбоновую кислоту (12). Схема 3 В еще одном примере получения целевой карбоновой кислоты (7) (Y=OH, стадия 4, схема 1), представленном на схеме 3, в пиразол (13) вводят подходящую защитную группу, такую как 4 метоксибензил, как показано на стадии 1, при помощи неорганического основания, такого как карбонат калия, в растворителе, таком как ацетон, с образованием защищенного пиразола (14). Затем сложный эфир подвергают гидролизу с применением подходящего основания, как показано на стадии 3, с образованием соединения формулы (15). После ацилирования на стадии 4, схема 1, может быть завершено удаление защитной группы в кислой среде, такой как ТФУК, с получением соединения формулы 1. На схеме 4 показан еще один пример получения карбоновой кислоты (7) (Y=OH, стадия 4, схема 1). Изопентилнитрит можно добавлять по каплям к раствору аминопиразола (16) и диметилдисульфида в инертном растворителе, таком как хлороформ, с обеспечением превращения первичного амина в трифторметильную группу с образованием этил-1-метил-5-(метилтио)-1H-пиразол-4-карбоксилата (17), как показано на стадии 1. Трифторметильную группу соединения (17) можно окислить до метилсульфона при помощи окислителя, такого как пероксид водорода, в подходящем растворителе, таком как уксусная кислота, с образованием соединения формулы (18), стадия 2. Гидролиз сложного эфира, как описано ранее, приводит к образованию карбоновой кислоты, как показано на стадии 3, соединение (7). Следующие синтезы и примеры приведены с целью более подробной иллюстрации настоящего изобретения и представляют собой типичные способы синтеза соединений формулы (I). Названия соединений согласно настоящему изобретению в общем случае получены при помощи ChemDraw Ultra 10.0. Синтез 1. трет-Бутил-1-(6-хлор-4,5-диметилпиридазин-3-ил)пиперидин-4-ил(метил)карбамат(11,0 г,62,1 ммоль),третбутилметил(пиперидин-4-ил)карбамата (23,3 г, 109 ммоль) и порошкообразного K2CO3 (17,2 г, 124 ммоль) в ДМСО (310 мл) при 120 С в течение 2 дней. Реакционную смесь охлаждали, разбавляли Н 2 О и отфильтровывали твердое вещество. Полученное твердое вещество промывали Н 2 О и высушивали в вакууме при 45 С. Твердое вещество растворяли в CH2Cl2 и пропускали полученный раствор через слой силикагеля, элюируя CH2Cl2. Органический слой концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде желтого твердого вещества (14,3 г, 65%).Org. Chem. 2007, 72, 5104-5112) (29 мг, 0,042 ммоль) в смеси 1,4-диоксана (30 мл) и H2O (10 мл) в атмосфере N2 при 90 С в течение ночи. Реакционную смесь распределяли между Н 2 О и CH2Cl2. Разделяли слои и экстрагировали водный слой CH2Cl2. Органические слои объединяли, высушивали над Na2SO4,фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 2% 2 М NH3/МеОН в CH2Cl2), получая указанное в заголовке соединение в виде белой пены (1,05 г, 60%).ES/MS m/z 415,2 (М+1). Альтернативная методика. Дегазированную при помощи N2 смесь трет-бутил-1-(6-хлор-4,5-диметилпиридазин-3 ил)пиперидин-4-ил(метил)карбамата (3,01 г, 8,48 ммоль), 4-фторфенилбороновой кислоты (1,23 г,8,80 ммоль) и CsF (4,08 г, 26,8 ммоль) в 1,4-диоксане (80 мл) обрабатывали хлоридом (1,1'-бис(дифенилфосфино)ферроцен)палладия(II) (1,10 г, 1,35 ммоль). Полученную смесь нагревали в атмосфереN2 при 95 С в течение ночи. Реакционную смесь распределяли между H2O и EtOAc. Разделяли слои и промывали органический слой раствором соли. Органический слой высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэшхроматографии на силикагеле (градиент от 20 до 80% EtOAc в гексане), получая указанное в заголовке соединение (3,05 г, 87%).(29 мг, 0,042 ммоль) в смеси 1,4-диоксана (30 мл) и Н 2 О (10 мл) в атмосфере N2 при 90 С в течение ночи. Реакционную смесь распределяли между EtOAc и Н 2 О с растворенным NaHCO3. Разделяли слои и экстрагировали водный слой с помощью EtOAc. Органические слои объединяли, высушивали над Na2SO4,фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 2% 2 M NH3/МеОН в CH2Cl2), получая указанное в заголовке соединение в виде желтого твердого вещества (1,68 г, 94%).ES/MS m/z 422,2 (М+1). Замещенные фенилпиридазины, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в синтезе 3, с использованием соответствующей арилбороновой кислоты. В синтезе 5 неочищенную реакционную смесь непосредственно фильтровали через слой силикагеля,элюируя 5% M NH3/МеОН в CH2Cl2. Элюат концентрировали и очищали, не подвергая воздействию воды. Раствор трет-бутил-1-(6-(4-фторфенил)-4,5-диметилпиридазин-3-ил)пиперидин-4-ил(метил)карбамата (1/04 г, 2,51 ммоль) в 1,4-диоксане (10 мл) обрабатывали 4 M HCl в 1,4-диоксане (15,0 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток растворяли в МеОН и помещали на колонку SCX (Varian, 10 г). Промывали колонку МеОН и CH2Cl2 и элюировали продукт смесью 1:1 CH2Cl2 и 2 М NH3/MeOH. Концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде не совсем белого твердого вещества (784 мг, 99%). Раствор трет-бутил-1-(6-(4-цианофенил)-4,5-диметилпиридазин-3-ил)пиперидин-4-ил(метил)карбамата (1,68 г, 3,99 ммоль) в 1,4-диоксане (20 мл) обрабатывали 4 М HCl в 1,4-диоксане (20 мл). Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток растворяли в МеОН и помещали на колонкуSCX (Varian, 20 г). Промывали колонку МеОН и CH2Cl2 и элюировали продукт смесью 1:1 CH2Cl2 и 2 МNH3/МеОН. Концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде желтого твердого вещества (1,28 г, количественно).N-метиламинопиперидин с удаленной защитной группой, представленный в таблице ниже, получали, по существу, в соответствии с методикой, описанной в синтезе 7, с использованием соответствующего пиперидина с защитной группой boc.(1,19 г, 2,99 ммоль) добавляли CH2Cl2 (20 мл) и трифторуксусную кислоту (20 мл). Перемешивали при комнатной температуре в течение 3 дней. Концентрировали при пониженном давлении, получая остаток. Распределяли полученный остаток между CH2Cl2 и 1 н. NaOH. Разделяли слои и дважды экстрагировали водный слой CH2Cl2. Органические экстракты объединяли, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение (790 мг, 89%). К смеси CuCl2 (671 мг, 4,99 ммоль) и изопентилнитрита (732 мг, 6,24 ммоль) в ацетонитриле (10 мл) при 0 С медленно добавляли этил-2-амино-4-(трифторметил)тиазол-5-карбоксилат (1,0 г, 4,16 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 ч. Нагревали при 50 С в течение 1 ч. Удаляли большую часть растворителей и выливали в смесь льда и концентрированной HCl. Экстрагировали CH2Cl2. Органический слой промывали раствором соли, сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэшхроматографии на силикагеле (20:1 гексан: EtOAc), получая указанное в заголовке соединение (762 мг,71%). 1 К раствору морфолина (4,03 г, 46,2 ммоль) в ДМСО (10 мл) добавляли этил 2-хлор-4(трифторметил)тиазол-5-карбоксилат (4,00 г, 15,4 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи. Добавляли CH2Cl2 и промывали полученную смесь Н 2 О. Органическую фазу высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (20:5:1 гексан: EtOAc: 2 М К смеси 1 н. NaOH (20 мл) в МеОН (20 мл) добавляли этил-2-морфолино-4-(трифторметил)тиазол-5 карбоксилат (4,58 г, 14,8 ммоль) и нагревали реакционную смесь при 50 С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и к полученному остатку добавляли Н 2 О. Подкисляли полученную смесь до рН 4 и отфильтровывали твердое вещество. Промывали твердое вещество Н 2 О и высушивали, получая указанное в заголовке соединение (4,13 г, 99%).ES/MS m/z 283,0 (М+1). Аминотиазольные кислоты, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в синтезе 15, с использованием соответствующего сложного эфира. Раствор 1-(6-(4-фторфенил)-4,5-диметилпиридазин-3-ил)-N-метилпиперидин-4-амина (100 мг,0,318 ммоль) в CH2Cl2 (3,2 мл) последовательно обрабатывали (S)-1-(трет-бутоксикарбонил)пиперидин 2-карбоновой кислотой (109 мг, 0,477 ммоль), триэтиламином (0,067 мл, 0,477 ммоль) и EDCI (92 мг,0,477 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 2 дней. Выливали реакционную смесь в Н 2 О, содержащую NaHCO3. Разделяли слои и экстрагировали водный слой CH2Cl2. Органические слои объединяли, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 2% 2 М NH3/MeOH в CH2Cl2), получая указанное в заголовке соединение в виде белого твердого вещества (82 мг, 49%). К раствору этил-3-(трифторметил)-1H-пиразол-4-карбоксилата (500 мг, 2,40 ммоль) в ацетоне (8 мл) добавляли K2CO3 (503 мг, 3,60 ммоль), при комнатной температуре в атмосфере N2. К полученной смеси добавляли по каплям 1-бромметил-4-метоксибензол (0,51 мл, 3,6 ммоль) и перемешивали полученную смесь в течение ночи в атмосфере N2. Реакцию гасили Н 2 О и дважды экстрагировали EtOAc. Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 15% EtOAc в гексане), получая указанное в заголовке соединение (789 мг, количественно). Раствор LiOH (122 мг, 5,03 ммоль) в Н 2 О (3 мл) добавляли при перемешивании к раствору этил-1(4-метоксибензил)-3-(трифторметил)-1H-пиразол-4-карбоксилата (550 мг, 1,68 ммоль) в 1,4-диоксане(10 мл). Перемешивали в течение ночи при комнатной температуре. Подкисляли до рН 5 при помощи 1 н.HCl и экстрагировали CH2Cl2 и 220% iPrOH в CHCl3. Объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая 140 мг указанного в заголовке соединения в виде белого твердого вещества. Водный слой подкисляли до рН 2-3 при помощи 1 н. HCl и дважды экстрагировали 20% iPrOH в CHCl3. Объединенные органические слои высушивали над MgSO4, фильтровали и добавляли полученный раствор к первоначально полученным 140 мг белого твердого вещества. Концентрировали, получая указанное в заголовке соединение в виде белого твердого вещества (426 мг, 85%). К раствору 1-(6-(4-фторфенил)-4,5-диметилпиридазин-3-ил)-N-метилпиперидин-4-амина (157 мг,0,50 ммоль) и 1-(4-метоксибензил)-3-(трифторметил)-1 Н-пиразол-4-карбоновой кислоты (165 мг,0,55 ммоль) в безводном ДМФА (2,5 мл) добавляли при перемешивании РуВОР (343 мг, 0,65 ммоль) и триэтиламин (0,21 мл, 1,50 ммоль). Полученную смесь перемешивали при комнатной температуре в атмосфере N2 в течение ночи. Концентрировали, добавляли МеОН, отфильтровывали твердое вещество и концентрировали фильтрат. Полученный остаток разбавляли МеОН и переносили на колонку SCX(Thermo Scientific, 10 г). Промывали МеОН, затем элюировали продукт 2 М NH3/MeOH. Концентрировали и очищали при помощи флэш-хроматографии на силикагеле с использованием градиента от 0 до 10%(10% 2 М NH3/MeOH в EtOAc) в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (170 мг, 43%). К раствору этил-1-метил-5-амино-1H-пиразол-4-карбоксилата (1,69 г, 10,0 ммоль) и диметилсульфида (1,79 мл, 20,0 ммоль) в CHCl3, помещенному в 3-горлую колбу, снабженную термометром и холо-9 017386 дильником, добавляли при перемешивании изопентилнитрит (0,5 мл, 3,75 ммоль), при 5 С в атмосфере азота. Оставляли реакционную смесь нагреться до 20 С на водяной бане и добавляли по каплям дополнительную порцию изопентилнитрита (1,5 мл, 11,3 ммоль). Через 15 мин убирали из-под реакционного сосуда 20 С водяную баню (из-за экзотермического эффекта температура повышалась до 50 С приблизительно в течение 1 мин). Перемешивали при комнатной температуре в атмосфере N2 в течение ночи. Промывали H2O и разделяли слои. Водный слой экстрагировали CHCl3, органические слои объединяли,высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 35% EtOAc в гексане),получая указанное в заголовке соединение (1,94 г, 97%). Смешивали этил-1-метил-5-(метилтио)-1H-пиразол-4-карбоксилат (1,68 г, 8,39 ммоль), ледяную уксусную кислоту (11 мл) и пероксид водорода (5,10 мл, 50,3 ммоль) и нагревали полученную смесь при 100 С в течение 1,5 ч. Оставляли реакционную смесь остывать до комнатной температуры и перемешивали в течение ночи. Добавляли лед и дважды экстрагировали CH2Cl2. Органические слои объединяли,высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 50% EtOAc в гексане),получая указанное в заголовке соединение (1,95 г, 100%). Раствор LiOH (22 мг, 0,90 ммоль) в Н 2 О (1 мл) добавляли при комнатной температуре и интенсивном перемешивании к раствору этил 1-метил-5-(метилсульфонил)-1H-пиразол-4-карбоксилата (175 мг,0,75 ммоль) в 1,4-диоксане (3 мл). Добавляли к реакционной смеси дополнительную порцию LiOH (5 мг,0,21 ммоль) и перемешивали в течение ночи. Подкисляли до рН 2 при помощи 1 н. HCl и дважды экстрагировали CH2Cl2. Органические слои объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении, получая указанное в заголовке соединение в виде белого твердого вещества (98 мг, 64%). Раствор 4-(4,5-диметил-6-(4-(метиламино)пиперидин-1-ил)пиридазин-3-ил)бензонитрила (90 мг,0,28 ммоль) и триэтиламина (0,12 мл, 0,84 ммоль) в CH2Cl2 (2,8 мл) обрабатывали 4 цианобензоилхлоридом (56 мг, 0,34 ммоль). Перемешивали реакционную смесь при комнатной температуре в течение ночи. Промывали Н 2 О и разделяли слои. Очищали органический слой непосредственно при помощи флэш-хроматографии на силикагеле (градиент от 0 до 2% 2 М NH3/MeOH в CH2Cl2). Концентрировали, получая белое твердое вещество. Полученное вещество растворяли в МеОН и добавляли 1,1 экв. метанольного раствора HCl (полученного при добавлении по каплям ацетилхлорида в МеОН). Концентрировали полученную смесь в токе N2 и высушивали полученный остаток в вакуум-сушильном шкафу при 45 С, получая указанное в заголовке соединение в виде желтого твердого вещества (130 мг,95%).ES/MS m/z 451,2 (М+1). Пиперидиниламиды, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в примере 1, с использованием хлорангидрида соответствующей кислоты, время реакции варьировали от 6 ч до 3 дней. В примерах 7, 8 и 13 для получения солей применяли избыток 1 МHCl в Et2O. В примере 10 для получения соли применяли 3 эквивалента предварительно приготовленного метанольного раствора HCl.(102 мг, 0,32 ммоль), 2-(трифторметил)никотиновую кислоту (70 мг, 0,38 ммоль) и триэтиламин (0,13 мл,0,96 ммоль) в ДМФА (10 мл). К полученной смеси добавляли РуВОР (2 00 мг, 0,38 ммоль) и перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли CH2Cl2 и промывали раствором соли. Высушивали органическую фазу над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле(20:5:1 гексан:EtOAc:2 М NH3/MeOH). Добавляли к раствору свободного основания в CH2Cl2/MeOH избыток 1 М HCl в Et2O и выпаривали растворители в токе N2, получая указанное в заголовке соединениеES/MS m/z 495,2 (M+1). Альтернативная методика проведения реакции сочетания. Смешивали 4-(4,5-диметил-6-(4-(метиламино)пиперидин-1-ил)пиридазин-3-ил)бензонитрил (300 мг, 0,93 ммоль), 2-(трифторметил)никотиновую кислоту (210 мг, 1,12 ммоль) и диизопропилэтиламин(0,79 мл, 4,51 ммоль) в смеси 4:1 ДМФА и ДМСО (20 мл). Полученную смесь кратковременно нагревали при 60 С для растворения твердых веществ, а затем охлаждали до 0 С. Добавляли по каплям раствор перфторфенилдифенилфосфината (750 мг, 1,96 ммоль) в смеси 4:1 ДМФА и ДМСО (1 мл). Полученную смесь нагревали при 60 С в течение ночи. Распределяли реакционную смесь между водным растворомNaHCO3 и CH2Cl2. Органический слой промывали раствором соли, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэшхроматографии на силикагеле (20:5:1 гексан:EtOAc:2 М NH3/МеОН), получая указанное в заголовке соединение в форме свободного основания (346 мг, 75%).ES/MS m/z 495,2 (М+1). Соль HCl получали, как описано выше. Пиперидиниламиды, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в примере 14, с использованием соответствующих диметилпиридазинов и карбоновых кислот. В примере 28, применяли 1,1 экв. 1 М HCl в МеОН (предварительно полученного прикапыванием ацетилхлорида в МеОН), затем концентрировали полученный раствор, получая соль моногидрохлорид. В примере 28 следовали альтернативной методике из примера 14. Раствор 4-(4,5-диметил-6-(4-(метиламино)пиперидин-1-ил)пиридазин-3-ил)бензонитрила (102 мг,0,32 ммоль), 4-(трифторметил) никотиновой кислоты (81 мг, 0,42 ммоль) и триэтиламина (0,07 мл,0,5 ммоль) в CH2Cl2 (4 мл) обрабатывали EDCI (99 мг, 0,52 ммоль) и перемешивали в течение 3 дней. Выливали реакционную смесь в Н 2 О и экстрагировали EtOAc. Органический слой промывали H2O, высушивали над Na2SO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле(градиент от 0 до 10% МеОН в CH2Cl2). Полученное свободное основание растворяли в МеОН (2 мл) и добавляли 1 М HCl в Et2O (0,5 мл). Концентрировали, получая указанное в заголовке соединение (82 мг,49%).ES/MS m/z 495,2 (М+1). Пиперидиниламиды, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в примере 29, с использованием соответствующей карбоновой кислоты. В примерах 31-33 перемешивание проводили в течение ночи. Для получения солей HCl в примерах 31-33 растворяли соответствующее свободное основание в МеОН и добавляли 1,1 эквивалента метанольного раствора HCl(предварительно полученного прикапыванием ацетилхлорида в МеОН), затем концентрировали.(S)-трет-бутил-2-1-(6-(4-фторфенил)-4,5-диметилпиридазин-3-ил)пиперидин-4 ил)(метил)карбамоил)пиперидин-1-карбоксилата (80 мг, 0,152 ммоль) в CH2Cl2 (2 мл) добавляли 4 М HCl в 1,4-диоксане (1,00 мл, 4,00 ммоль). Полученную смесь перемешивали в течение 4 ч при комнатной температуре. Концентрировали при пониженном давлении и высушивали полученный остаток в вакуумсушильном шкафу при 45 С, получая указанное в заголовке соединение в виде бледно-желтой пены(1,08 г, 3,44 ммоль), триэтиламин (1,44 мл, 10,3 ммоль) и РуВОР (2,68 г, 5,15 ммоль). Перемешивали при комнатной температуре в течение 3 ч. Концентрировали реакционную смесь и очищали полученный остаток при помощи флэш-хроматографии на силикагеле с использованием градиента 0-100% (5:1 EtOAc: 2 М NH3/MeOH) в гексане. Выделенный продукт растворяли в CH2Cl2 (10 мл) и добавляли 2 М HCl вES/MS (m/z) 491,2 (М+1). Амиды, представленные в таблице ниже, получали, по существу, в соответствии с методикой, описанной в примере 34, с использованием соответствующей карбоновой кислоты. В примере 36 очистку проводили при помощи колонки SCX (элюируя 2 M NH3/MeOH) с последующей флэш-хроматографией.N-(1-(6-(4-фторфенил)-4,5-диметилпиридазин-3-ил)пиперидин-4-ил)-1-(4-метоксибензил)-Nметил-3-(трифторметил)-1H-пиразол-4-карбоксамиду (99 мг, 0,12 ммоль) добавляли трифторуксусную кислоту (10 мл) и кипятили с обратным холодильником в атмосфере N2 в течение ночи. Концентрировали при пониженном давлении. Полученный остаток растворяли в 20% iPrOH в CHCl3 и промывали насыщенным водным раствором Na2CO3. Водный слой экстрагировали 2 0% iPrOH в CHCl3. Органические слои объединяли, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали при помощи флэш-хроматографии на силикагеле (градиент от 0 до 10% 2 М NH3/MeOH в CH2Cl2). Очищенное свободное основание растворяли в CH2Cl2 (2 мл) и добавляли по каплям 1 М HCl в Et2O (0,5 мл). Перемешивали в течение 30 мин. Концентрировали и высушивали в вакуум-сушильном шкафу при 50 С в течение ночи, получая указанное в заголовке соединение (55 мг,60%).ES/MS m/z 477,0 (М+1). Биология. Было показано, что Hedgehog как фактор выживаемости связан со следующими видами рака: базальноклеточная карцинома; рак верхней части желудочно-кишечного тракта (пищевода, желудка, поджелудочной железы и желчевыводящих путей); рак простаты; рак молочной железы; мелкоклеточный рак легкого; немелкоклеточный рак легкого; В-клеточная лимфома; множественная миелома; рак желудка; рак яичников; колоректальный рак; рак печени; меланома; рак почек; и рак головного мозга. Было доказано, что элементы сигнального пути hedgehog являются потенциальными мишенями лекарственных средств для лечения различных видов рака. Линия клеток Daoy, полученная из опухоли медуллобластомы (АТСС, НТВ-186), чувствительна к лигандам Hh. При воздействии на указанные клетки экзогенно внесенной Shh-кондиционированной среды происходит активация сигнального пути Hh, приводящая к увеличенной экспрессии Gli1. Циклопамин - алкалоид, выделенный из полевого вьюнкаVeratrum californicum - является слабым антагонистом hedgehog и, как показано, подавляет экспрессиюGli1 в ответ на стимуляцию Shh. Последние наблюдения позволяют предположить, что циклопамин ингибирует рост клеток медуллобластомы в культуре и аллотрансплантатов. С использованием модельной системы на клетках Daoy можно идентифицировать мощные ингибиторы сигнального пути hedgehog. Поскольку соединения согласно настоящему изобретению представляют собой антагонисты hedgehog,они подходят для лечения указанных выше типов опухолей. Определение биологической активности IC50. Следующий протокол и результаты исследования дополнительно демонстрируют применимость и эффективность соединений и способов согласно настоящему изобретению. Функциональные исследования подтверждают, что соединения согласно настоящему изобретению проявляют способность ингибировать сигнальный путь Shh. Все лиганды, растворители и реактивы, применяемые в следующем исследовании, легко доступны из коммерческих источников или легко могут быть получены специалистом в данной области техники. Биологическую активность определяли при помощи функционального исследования на клетках нейронального рака Daoy и измеряли уровни рибонуклеиновой кислоты Gli1 при помощи системы ана- 16017386 лиза рДНК (разветвленной дезоксирибонуклеиновой кислоты) (Panomics, Inc., Fremont, CA). Gli первоначально открыт в линии клеток глиобластомы и кодирует белок "цинковый палец" (zinc finger protein),активируемый сигнальным путем Shh. Максимальный отклик получен при включении транскрипцииGli1 в клетках Daoy с кондиционированной средой (клетки мезонефроса человека, HEK-293, стабильно экспрессирующие рекомбинантный Shh) в течение 24 ч и последующем измерении количества стимулированного транскрипта Gli1. Минимальный отклик представляет собой количество транскрипта Gli1,ингибированного контрольным соединением в клетках Daoy, которое было стимулировано кондиционированной средой (клетки мезонефроса человека, HEK-293, стабильно экспрессирующие рекомбинантныйShh) в течение 24 ч. Функциональное исследование для измерения ингибирования Gli1 в клетках Daoy. В системе анализа рДНК используют технологию ДНК с разветвленной цепью, чтобы сделать возможной амплификацию рибонуклеиновой кислоты-мишени (транскрипта). В указанной технологии применяют три типа синтетических гибридных коротких специфических к Gli1 кДНК-зондов, которые определяют специфичность транскрипта-мишени [усилители захвата (СЕ), усилители метки (LE) и блокаторы(BL)], которые гибридизируются в виде комплекса с транскриптами-мишенями для усиления сигнала гибридизации. Добавление на стадии амплификации хемолюминогенного субстрата позволяет проводить детектирование при помощи люминесценции. Линия клеток Daoy, полученная из American Type Culture collection (ATCC), представляет собой чувствительную к Shh линию клеток нейрональной опухоли человека, и была получена в 1985 из опухоли десмопластической медуллобластомы мозжечка, физиологически значимой линии опухолевых клеток. Эндогенные уровни транскриптов Gli1 в клетках Daoy низкие, но их можно стимулировать за счет использования кондиционированной среды, взятой из клеток человека стабильной сверхэкспрессирующихShh (линия клеток HEK-293 стабильно трансфицированная hShh). Клетки Daoy выращивали до монослоя в сосудах для культуры клеток Т 225 в среде для выращивания клеток Daoy, содержащей минимально обогащенную среду (MEM) плюс 10% фетальной телячьей сыворотки (FBS) с 0,1 нМ неосновных аминокислот и 1 мМ пирувата натрия. Клетки удаляли из сосудов Т 225 при помощи трипсина с этилендиаминтетрауксусной кислотой (ЭДТА), центрифугировали, вновь суспендировали в среде, а затем подсчитывали. Затем клетки Daoy высевали по 50000 клеток на ячейку в среде для выращивания в прозрачные 96 луночные планшеты для культуры тканей Costar и оставляли для инкубирования в течение ночи при 37 С в атмосфере 5% диоксида углерода (СО 2). Клетки промывали один раз фосфатно-солевым буферным раствором (PBS) с последующим добавлением 100 мкл кондиционированной Shh среды (Shh-CM) для стимуляции уровней экспрессирования Gli1. Shh-CM разбавляли для получения максимальной стимуляции контрольной средой для выращивания - 0,1% FBS/DMEM (среда Игла, модифицированная по Дульбекко). На клетки Daoy, обработанные Shh-CM, затем воздействовали различными концентрациями ингибиторов hedgehog в диапазоне приблизительно от 1 мкМ до 0,1 нМ. Исследуемые соединения оставляли для инкубирования в течение 24 ч при 37 С в атмосфере 5% СО 2. Измерение транскрипта Gli1 проводили при помощи теста Quantigene 2.0 Gli1 согласно описанию производителя (Panomics, Inc.). Готовили разбавленную смесь буфера для лизирования (DLM), содержащего Протеиназу К. Через 24 ч инкубирования с соединением клетки однократно промывали PBS и добавляли к клеткам 180 мкл DLM. Планшеты с клетками, содержащие буфер для лизирования, запечатывали и выдерживали при 55 С от 30 до 45 мин. Полученные лизаты клеток затем растирали 5 раз. Рабочий набор зондов, содержащий зонды Gli1, получали, разбавляя зонды в DLM согласно указаниям производителя, и затем добавляли 20 мкл рабочего набора зондов к планшетам для теста рДНК вместе с 80 мкл лизатов Daoy. Планшеты запечатывали и инкубировали в течение ночи при 55 С. Затем планшеты с рДНК обрабатывали согласно указаниям производителя. Проводили количественное измерение сигнала,считывая планшеты на считывающем устройстве Perkin Elmer Envision reader, детектирующем люминесценцию. Сигнал люминесценции прямо пропорционален количеству транскрипта-мишени, присутствующего в образце. Данные о люминесцентном сигнале, полученные в функциональном исследовании, применяли для расчета IC50 при исследовании in vitro. Данные вычисляли на основании максимальных контрольных значений (клетки Daoy, обработанные Shh-CM) и минимального контрольного значения (клетки Daoy,обработанные Shh-CM и ингибирующей концентрацией контрольного соединения, 1 мкМ N-(3-(1 Нбензо[d]имидазол-2-ил)-4-хлорфенил)-3,5-диметоксибензамида). Для получения значений IC50 использовали четырехпараметрический логистический подбор кривой при помощи программного обеспеченияand NIH Chemical Genomics Center). Согласно описанному протоколу для соединений согласно настоящему изобретению, приведенных в качестве примеров в настоящем изобретении, было показано, что IC5015 нМ. Например, для соединения из примера 14 IC50 составляет приблизительно 1,27 нМ со стандартной ошибкой 0,114 (n=4), а для соединения из примера 34 IC50 составляет приблизительно 1,22 нМ со стандартной ошибкой 0,293 (n=3) в описанном выше исследовании. Полученные результаты подтверждают тот факт, что соединения соглас- 17017386 но настоящему изобретению являются антагонистами hedgehog и в качестве таковых подходят для применения в качестве противораковых агентов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение следующей формулы: или фармацевтически приемлемая соль указанного соединения, где пиперидиR2 представляет собой нил или гем-ди-F-замещенный циклогексил;R3 представляет собой метил или трифторметил;R5 представляет собой трифторметил или метилсульфонил;R6 представляет собой водород или метил;R7, R8, R9, R10 и R11 независимо представляют собой водород, фтор, циано, хлор, метил, трифторметил, трифторметокси или метилсульфонил, при условии, что по меньшей мере два из R7, R8, R9, R10 и R11 представляют собой водород. 2. Соединение по п.1, отличающееся тем, что X представляет собой C-R1, или фармацевтически приемлемая соль указанного соединения. 3. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой или фармацевтически приемлемая соль указанного соединения. 4. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой C-R1 и R1 представляет собой фтор, или фармацевтически приемлемая соль указанного соединения. 5. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой C-R1 и R1 представляет собой циано, или фармацевтически приемлемая соль указанного соединения. 6. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой цевтически приемлемая соль указанного соединения. 7. Соединение по п.1 или 2, отличающееся тем, что R2 представляет собой или фармацевтически приемлемая соль указанного соединения. 8. Соединение по п.6, отличающееся тем, что R5 представляет собой трифторметил и R6 представляет собой метил, или фармацевтически приемлемая соль указанного соединения. 9. Соединение по п.7, отличающееся тем, что R7 представляет собой трифторметил и R9, R10 и R11 представляют собой водород, или фармацевтически приемлемая соль указанного соединения. 10. Соединение по п.1, представляющее собой N-(1-(6-(4-цианофенил)-4,5-диметилпиридазин-3 ил)пиперидин-4-ил)-N-метил-2-(трифторметил)никотинамид. 11. Соединение по п.1, представляющее собой N-(1-(6-(4-цианофенил)-4,5-диметилпиридазин-3 ил)пиперидин-4-ил)-N-метил-2-(трифторметил)никотинамида дигидрохлорид. 12. Фармацевтическая композиция, содержащая соединение по любому из пп.1-11 или фармацевтически приемлемую соль указанного соединения в комбинации с фармацевтически приемлемым носителем, разбавителем или эксципиентом. 13. Применение соединения по любому из пп.1-11 или фармацевтически приемлемой соли указанного соединения в качестве лекарственного средства. 14. Применение соединения по любому из пп.1-11 или фармацевтически приемлемой соли указанного соединения для лечения рака. 15. Применение по п.14, отличающееся тем, что указанный рак выбран из группы, состоящей из рака головного мозга, базальноклеточной карциномы, рака пищевода, рака желудка, рака поджелудочной железы, рака желчевыводящих путей, рака простаты, рака молочной железы, мелкоклеточного рака легкого, немелкоклеточного рака легкого, В-клеточной лимфомы, множественной миеломы, рака яичников,колоректального рака, рака печени, рака почек и меланомы.
МПК / Метки
МПК: C07D 401/14, A61K 31/501, C07D 417/14, C07D 401/04
Метки: тетразамещенные, пиридазины, пути, hedgehog, качестве, антагонистов
Код ссылки
<a href="https://eas.patents.su/20-17386-tetrazameshhennye-piridaziny-v-kachestve-antagonistov-puti-hedgehog.html" rel="bookmark" title="База патентов Евразийского Союза">Тетразамещенные пиридазины в качестве антагонистов пути hedgehog</a>
Бензил- и пиридинилпроизводные в качестве модуляторов пути передачи сигнала hedgehog

Номер патента: 16564
Опубликовано: 30.05.2012
Авторы: Пойкерт Штефан, Льямас Луис, Миллер-Мослин Карен, Макюан Майкл А., Юсуфф Наим, Дай Миао, Перес Лоренс Блас, Ли Джон, Хэ Фэн, Карки Раджеш, Келлехер Джозеф III, Джейн Риши Кумар
МПК: A61K 31/4545, C07D 237/28, A61K 31/502...
Метки: пиридинилпроизводные, передачи, бензил, сигнала, hedgehog, пути, качестве, модуляторов
Формула / Реферат:
1. Соединение формулы Iи его фармацевтически приемлемые соли, гдеR1 обозначает фенил или het, которые могут быть незамещенными или замещенными;R2 обозначает het, в котором по меньшей мере одним гетероатомом является N и который может быть незамещенным или замещенным;L обозначает C1-С6-алкил;X обозначает N;Y обозначает связь, СН2;R3 обозначает арил или het, который является замещенным;Z обозначает Н, C1-С6-алкил, оксогруппу, C(O)OR6; иm равно 0,...
Соединения 2-(2-галоген-4-аминофенил)пиридиновых ингибиторов передачи сигналов белком hedgehog (варианты), способ их получения, композиция и способы лечения рака и ингибирований ангиогенеза и сигнального пути hedgehog в клетках на их основе

Номер патента: 17262
Опубликовано: 30.11.2012
Авторы: Гунцнер Джанет, Рейнолдс Марк, Сатерлин Даниэл, Малески Кимберли, Сэвидж Скотт, Лалонд Ребекка, Ван Шумэй, Дайна Майкл, Бао Лян, Кастанедо Джорджетт, Стэнли Марк
МПК: C07D 213/40, C07D 213/56, C07D 213/38...
Метки: сигналов, композиция, передачи, клетках, ингибирований, получения, hedgehog, 2-(2-галоген-4-аминофенил)пиридиновых, ангиогенеза, способы, пути, лечения, рака, белком, способ, сигнального, основе, варианты, соединения, ингибиторов
Формула / Реферат:
1. Соединения 2-(2-галоген-4-аминофенил)пиридиновых ингибиторов передачи сигналов белком hedgehog, охватываемые общей структурной формулойгде R1 выбран из группы, содержащей алкил, карбоцикл или гетероцикл, каждый из которых может содержать заместители, выбранные из группы, включающей гидроксил, галоген, амино, карбоксил, амидино, гуанидино, карбонил, нитро, циано, ацил, алкил, галоалкил, сульфонил, сульфинил, алкокси, алкилтио, карбамоил,...
Производные пиридилкарбамоилиндолинов в качестве антагонистов 5-нт2с-рецепторов

Номер патента: 1780
Опубликовано: 27.08.2001
Авторы: Бромидж Стивен Марк, Форбес Ян Томсон
МПК: C07D 401/14, A61P 25/28, A61K 31/4439...
Метки: производные, 5-нт2с-рецепторов, качестве, антагонистов, пиридилкарбамоилиндолинов
Формула / Реферат:
1. Соединение формулы (I) или его соль где Х обозначает СН или N; R1 обозначает водород или C1-6-алкил; R2 и R3 обозначают независимо C1-6-алкил или трифторметил. 2. Соединение по п.1, в котором Х обозначает СН. 3. Соединение по п.1 или 2, в котором R1 обозначает метил. 4. Соединение по любому из пп.1-3, в котором R2 обозначает СF3. 5. Соединение по любому из пп.1-4, в котором R3 обозначает C1-6-алкил. 6. Соединение по любому из пп.1-5, в...
Производные циклопентена в качестве антагонистов рецептора мотилина

Номер патента: 3252
Опубликовано: 27.02.2003
Авторы: Чен Роберт Х., Мур Джон Б.Мл., Ксианг Мин, Биверз Мэри Пэт
МПК: A61K 31/5375, C07C 233/41, A61P 1/00...
Метки: циклопентена, качестве, рецептора, антагонистов, производные, мотилина
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет H, C1-5-алкил, замещенный C1-5-алкил (где заместителями являются один или несколько галогенов), амино-C1-5-алкил, C1-5-алкиламино-C1-5-алкил, ди-C1-5-алкиламино-C1-5-алкил, RaRbN-C1-5-алкил (где Ra и Rb независимо выбраны из H и C1-5-алкила или вместе образуют морфолин, пиперазин, пиперидин или N-замещенный пиперидин, где N-заместитель представляет C1-5-алкил или фенил-C1-5-алкил),...
Замещенные производные азабициклогексана в качестве антагонистов мускаринового рецептора

Номер патента: 9059
Опубликовано: 26.10.2007
Авторы: Гупта Джанг Бахадур, Силамкоти Арундутт Висванатхам, Мехта Анита
МПК: A61P 11/00, A61K 31/403, A61P 1/00...
Метки: замещенные, качестве, производные, мускаринового, антагонистов, азабициклогексана, рецептора
Формула / Реферат:
1. Соединения структурной формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереомеры, N-оксиды, полиморфные формы, где Ar представляет собой арильное или гетероарильное кольцо, имеющее 1-2 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота, арильное или гетероарильное кольца могут быть незамещенными или замещенными от одного до трех заместителями, независимо...