Соединения в качестве ингибиторов вируса гепатита с
Номер патента: 9295
Опубликовано: 28.12.2007
Авторы: Гудро Натали, Ранкур Жан, Бейли Мюррей Д., Бордело Жозе, Гори Вида, Гуле Сильвия, Форжион Паскаль, Гиро Элиза, Халмос Тедди, Бхардвай Пунит, Ллина-Брюне Монтсе


























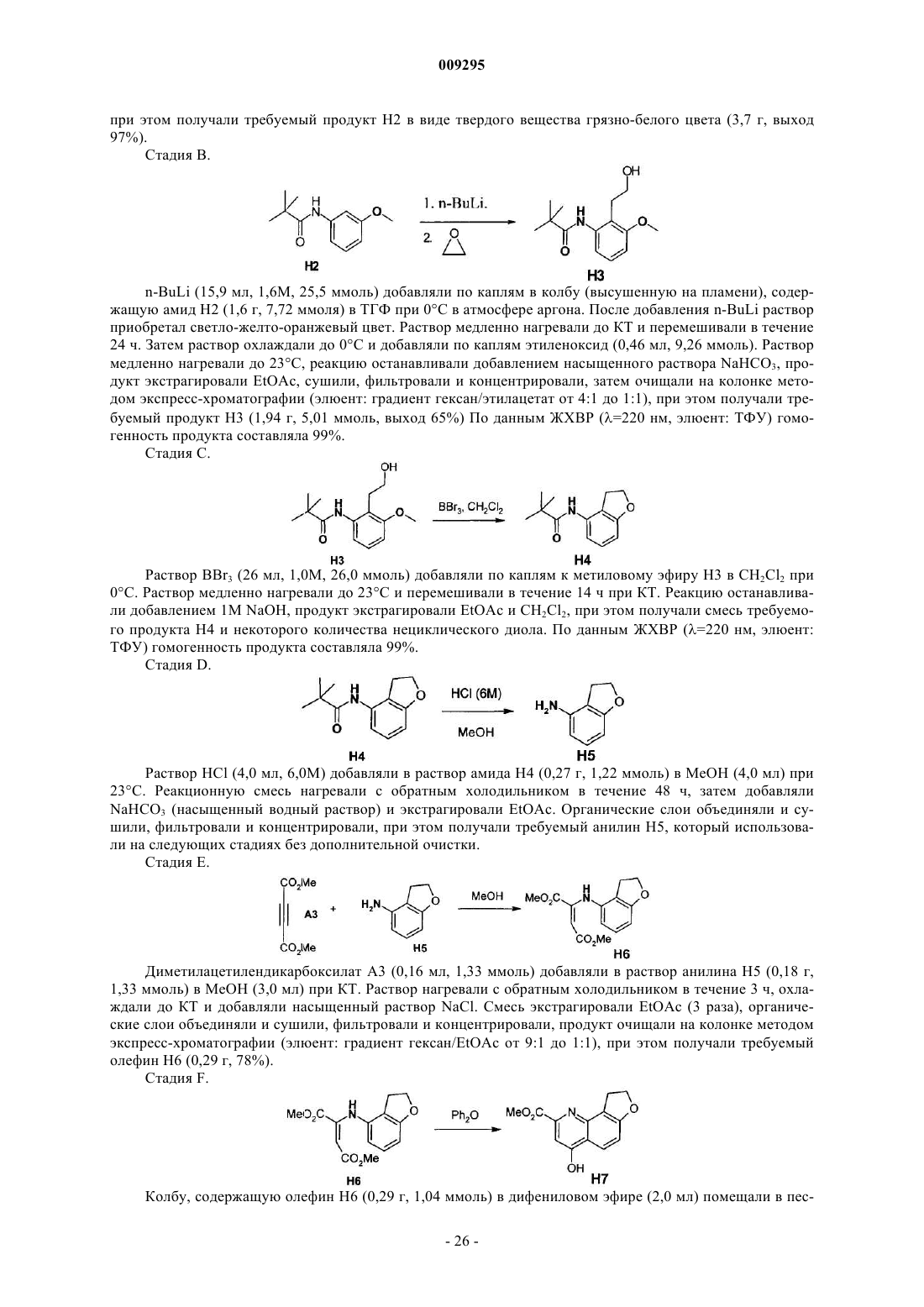



Формула / Реферат
1. Рацемат, диастереоизомер или оптический изомер соединения формулы (I)

где В означает (С1-С10)алкил или (С3-С7)циклоалкил,
а) где указанный алкил и циклоалкил может быть моно-, ди- или тризамещенным группой (С1-С3)алкил и
б) где указанный алкил может быть моно- или дизамещенным группой О-(С1-С4)алкил и
в) где указанный алкил может быть моно-, ди- или тризамещен атомами галогена и
г) где каждая из указанных циклоалкильных групп означает 4-, 5-, 6- или 7-членное кольцо, необязательно содержащее одну (в случае 4-, 5-, 6- или 7-членных колец) или две (в случае 5-, 6- или 7-членных колец) -СН2-групп, соединенных друг с другом не напрямую и замещенных группой -О- таким образом, что атом кислорода соединен с группой X по крайней мере через два атома углерода;
X означает О или NH;
R3 означает (С2-С8)алкил, который может быть моно-, ди- или тризамещенным группой (С1-С4)алкил;
L0 означает Н, галоген, (С1-С4)алкил, -ОН, -О-(С1-С4)алкил, -NH2, -NH(C1-С4)алкил или -N((С1-С4)алкил)2;
L1 означает галоген, циано, (С1-С4)алкил, -О-(С1-С4)алкил, -S-(C1-C4)алкил, -SO-(С1-С4)алкил или
-SO2-(С1-С4)алкил, где каждая из указанных алкильных групп необязательно замещена от 1 до 3 атомов галогена, и
L2 означает галоген, (С1-С4)алкил- или -О-(С1-С4)алкил, и L1 или L2 (но не оба одновременно) могут означать Н; или L0 и L1 могут быть связаны ковалентно и вместе с двумя атомами углерода, к которым они присоединены, образуют 5- или 6-членное карбоциклическое кольцо, в котором одна или две -СН2-группы связаны друг с другом не напрямую, и независимо разделены -О-;
R2 означает (С1-С8)алкил, -NR22COR20, -NR22COOR20 или -NR22R21, где R20 выбирают из группы, включающей (С1-С8)алкил и (С1-С4)алкил(С3-С7)циклоалкил;
R21 означает Н или выбран из группы, включающей (С1-С8)алкил, (С3-С7)циклоалкил и (С1-С4)алкил(С3-С7)циклоалкил,
R22 выбирают из группы, включающей водород и метил,
R1 означает этил или винил;
RC означает гидрокси или NHSO2RS, где RS означает (С3-С7)циклоалкил; или RS означает
-N(RN2)RN1), где RN1 и RN2 независимо выбирают из группы, включающей (С1-С6)алкил;
или его фармацевтически приемлемая соль или сложный эфир.
2. Соединение по п.1, где B означает (С1-С10)алкил или (С3-С7)циклоалкил,
а) где указанный циклоалкил может быть моно-, ди- или тризамещен группой (С1-С3)алкил, и
б) где указанный алкил может быть моно- или дизамещен О-(С1-4)алкилом; и
в) где все указанные алкильные группы могут быть моно-, ди- или тризамещены атомами галогена, и
г) где все указанные циклоалкильные группы означают 4-, 5-, 6- или 7-членное кольцо, в котором необязательно одна (в случае 4-, 5-, 6- или 7-членного кольца) или две (в случае 5-, 6- или 7-членного кольца) -СН2-группы не связаны напрямую друг с другом и замещены группой -О- таким образом, что атом О соединен с группой X по крайней мере через два атома углерода;
X означает О или NH;
R3 означает (С2-С8)алкил, который может быть моно-, ди- или тризамещен группой (С1-С4)алкил;
L0 означает Н, -ОН, -О-(С1-С4)алкил, -NH2, -NH(C1-C4)алкил или -N((C1-С4)алкил)2;
L1 означает галоген, (С1-С4)алкил, -О-(С1-С4)алкил или -S-(С1-С4)алкил (в окисленном состоянии, такой как SO или SO2), и
L2 означает галоген, (С1-С4)алкил- или -О-(С1-С4)алкил, и
L1 или L2 (но не оба одновременно) могут означать Н; или
L0 и L1 могут быть связаны ковалентно и вместе с двумя атомами углерода, к которым они присоединены, образуют 5- или 6-членное карбоциклическое кольцо, в котором одна или две -СН2-группы связаны друг с другом не напрямую, и независимо разделены -О-;
R2 означает (С1-С8)алкил, -NR22COR20, -NR22COOR20 или -NR22R21, где
R20 выбирают из группы, включающей (С1-С8)алкил и (С1-С4)алкил(С3-С7)циклоалкил;
R21 означает Н или выбран из группы, включающей (С1-С8)алкил, (С3-С7)циклоалкил и (С1-С4)алкил(С3-С7)циклоалкил,
R22 выбирают из группы, включающей водород и метил,
R1 означает этил или винил;
RC означает гидрокси или NHSO2RS, где RS означает (С3-С7)циклоалкил; или RS может быть дополнительно выбран из -N((С1-С6)алкил)2;
или его фармацевтически приемлемая соль или эфир.
3. Соединение по одному или более предыдущих пунктов, где B выбирают из группы, включающей (С2-С8)алкил- и (С3-С7)циклоалкил,
а) где указанные алкил и циклоалкил могут быть моно-, ди- или тризамещены группой (С1-С3)алкил, и
б) где указанный алкил может быть моно- или дизамещен -O-(C1-С4)алкилом, и
в) где каждая из указанных алкильных групп может быть моно-, ди- или тризамещена атомами фтора или монозамещена атомами хлора или брома, и
г) где каждая из указанных циклоалкильных групп означает 5-, 6- или 7-членное кольцо, в котором одна или две -СН2-группы не связаны напрямую друг с другом и заменены на группу -О- таким образом, что атом О соединен с группой X по крайней мере через два атома углерода.
4. Соединение по п.3, где B выбирают из группы, включающей этил, н-пропил, трет-бутил, 2-метилпропил, 1,2-диметилпропил, 1,2,2-триметилпропил, 2-фторэтил, 3-фторпропил, 3,3,3-трифторпропил, циклопропил, циклобутил, циклопентил, циклогексил, 1-метилциклопентил и 1-метилциклогексил, и группу, которую выбирают из следующих формул:

5. Соединение по п.4, где B выбирают из группы, включающей этил, н-пропил, трет-бутил, циклопентил, 1-метилциклопентил, 2-фторэтил или 3-фторпропил.
6. Соединение по одному или более предыдущих пунктов, где X означает О.
7. Соединение по одному или более предыдущих пунктов, где X означает NH.
8. Соединение по одному или более предыдущих пунктов, где R3 означает (С2-С6)алкил, который необязательно замещен 1-3 заместителями, выбранными из (C1-С4)алкила.
9. Соединение по п.8, где R3 представляет собой 1,1-диметилэтил.
10. Соединение по одному или более предыдущих пунктов, где L0 выбирают из группы, включающей Н, галоген, СН3, -ОН, -ОСН3, -ОС2Н5, -ОС3Н7, -OCH(CH3)2, -NHCH3, -NHC2H5, -NHC3H7,
-NHCH(CH3)2, -N(CH3)2, -N(CH3)C2H5, -N(CH3)C3H7 и -N(CH3)CH(CH3)2.
11. Соединение по п.10, где L0 выбирают из группы, включающей Н, -ОН, -ОСН3 галоген и
-N(CH3)2.
12. Соединение по п.11, где L0 выбирают из группы, включающей Н, -ОН или -ОСН3.
13. Соединение по одному или более предыдущих пунктов, где L1 выбирают из группы, включающей галоген, -СН3, -C2H5, -ОСН3, -OC2H5, -ОС3Н7, -ОСН(СН3)2, CF3, -SMe, -SOMe и SO2Me, и L2 выбирают из группы, включающей галоген, -СН3, -С2Н5, -ОСН3, -ОС2Н5, -ОС3Н7, -ОСН(СН3)2, где L1 или L2 может означать Н.
14. Соединение по п.13, где или L1 означает -СН3, -F, -Cl, -Br, -ОМе, -SMe или -SO2Me; или L2 означает -СН3, -F, -Cl, -Br или -ОМе; а другой из заместителей L1 и L2 означает Н.
15. Соединение по п.14, где L1 означает СН3, -F, -Cl, -Br, -ОМе, -SMe или -SO2Me, a L2 означает Н.
16. Соединение по одному или более предыдущих пунктов, где L0 выбирают из группы, включающей Н, -ОН и -ОСН3; и/или L1 означает -СН3, -F, -Cl, -Br, -ОМе, -SMe или -SO2Me; или L2 означает -СН3, -F, -Cl, -Br или -ОМе; а другой из заместителей L1 и L2 означает Н.
17. Соединение по п.16, где L0 выбирают из группы, включающей Н, -ОН и -ОСН3; L1 означает
-СН3, -F, -Cl, -Br, -OMe, -SMe или -SO2Me, a L2 означает Н.
18. Соединение по п.17, где L0 выбран из Н или -ОСН3, L1 означает -СН3, -Cl или -Br; a L2 означает Н.
19. Соединение по одному или более предыдущих пунктов, где L0 и L1 ковалентно связаны и вместе с хинолиновым остатком, к которому они присоединены, образуют циклическую систему, которую выбирают из следующих групп:

где L2 имеет значения, как определено в п.1.
20. Соединение по п.19, где L2 означает Н или метил.
21. Соединение по одному или более предыдущих пунктов, где R2 означает (С1-С8)алкил,
-NHCOR20, -NHCOOR20 или -NHR21, где
R20 выбирают из группы, включающей (С1-С8)алкил и (C1-С3)алкил(С3-С7)циклоалкил, и
R21 означает Н или выбран из группы, включающей (С1-С8)алкил, (С3-С7)циклоалкил и (С1-С3)алкил(С3-С7)циклоалкил.
22. Соединение по п.21, где R2 означает -NHCOR20, -NHCOOR20 или -NHR21.
23. Соединение по п.21, где R20 выбирают из группы, включающей метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил и циклогексилметил,
R21 выбирают из группы, включающей метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил и циклогексилметил.
24. Соединение по п.23, где R20 выбирают из группы, включающей метил, этил, н-пропил, изопропил и циклопентилметил,
R21 выбирают из группы, включающей метил, этил, н-пропил, изопропил, циклопентил и циклопентилметил.
25. Соединение по одному или более предыдущих пунктов, где R1 означает винил.
26. Соединение по одному или более предыдущих пунктов, где RC означает гидрокси, NHSO2-циклопропил, NHSO2-циклобутил или NHSO2-циклопентил.
27. Соединение по п.26, где RC означает гидрокси.
28. Соединение по п.26, где RC означает NHSO2-циклопропил.
29. Соединение по одному или более предыдущих пунктов, где RC означает NHSO2N(RN2)RN1), где RN1 и RN2 независимо выбирают из (С1-С4)алкила.
30. Соединение по п.1, где B означает (С2-С8)алкил или (С3-С7)циклоалкил,
а) где указанный алкил и циклоалкил могут быть моно-, ди- или тризамещены группой (С1-С3)алкил; и
б) где указанный алкил может быть моно- или дизамещенным группой О-(С1-С4)алкил; и
в) где каждая из указанных алкильных групп может быть моно-, ди- или тризамещена атомами фтора или монозамещена атомами хлора или брома; и
г) где каждая из указанных циклоалкильных групп означает 4-, 5-, 6- или 7-членное кольцо, необязательно содержащее одну (в случае 4-, 5-, 6- или 7-членных колец) или две (в случае 5-, 6- или 7-членных колец) -СН2-групп, соединенных друг с другом не напрямую и разделенных группой -О- таким образом, что атом кислорода соединен с группой X по крайней мере через два атома углерода;
X означает О или NH;
R3 означает (С2-С6)алкил, который может быть моно-, ди- или тризамещенными группой (С1-С4)алкил;
L0 означает Н, -ОН, -ОСН3, галоген или -N(CH3)2;
L1 означает галоген, -СН3, -С2Н5, -ОСН3, -ОС2Н5, -ОС3Н7, -ОСН(СН3)2, -CF3, -SMe, -SOMe и SO2Me;
L2 означает означает галоген, -СН3, -C2H5, -ОСН3, -ОС2Н5, -ОС3Н7 и -ОСН(СН3)2, где L1 или L2 могут означать Н;
R2 означает (С1-С8)алкил, -NHCOR20, -NHCOOR20 или -NHR21, где
R20 выбирают из группы, включающей (С1-С8)алкил и (С1-С3)алкил(С3-С7)циклоалкил;
R21 означает Н или выбран из группы, включающей (С1-С8)алкил, (С3-С7)циклоалкил и (С1-С3)алкил(С3-С7)циклоалкил,
R1 означает этил или винил;
RC означает гидрокси, NHSO2циклопропил, NHSO2циклобутил или NHSO2циклопентил.
31. Соединение по п.30, где B выбирают из группы, включающей этил, н-пропил, трет-бутил, 2-метилпропил, 1,2-диметилпропил, 1,2,2-триметилпропил, 2-фторэтил, 3-фторпропил, 3,3,3-трифторпропил, циклопропил, циклобутил, циклопентил, циклогексил, 1-метилциклопентил и 1-метилциклогексил и группу, которую выбирают из следующих формул:

R3 выбирают из группы, включающей 1,1-диметилэтил, L0 означает Н, -ОН или -ОСН3; L1 означает СН3, -F, -Cl, -Br, -OMe, -SMe или -SO2Me; a L2 означает Н;
R2 означает -NHCOR20, -NHCOOR20 или -NHR21, где R20 выбирают из группы, включающей метил, этил, н-пропил, изопрояшы и циклометилпентил;
R21 выбирают из группы, включающей метил, этил, н-пропил, изопропил, циклопентил и циклометилпентил,
R1 означает винил, a RC означает гидрокси или NHSO2-циклопропил.
32. Соединение по п.31, где B выбирают из группы, включающей этил, н-пропил, трет-бутил, циклопентил, 1-метилциклопентил, 2-фторэтил и 3-фторпропил, R3 представляет собой 1,1-диметилэтил, L0 означает Н или -ОСН3;
L1 означает СН3, -Cl или -Br, L2 означает Н, a RC означает гидрокси.
33. Соединение по п.1 формулы

где B, L0, L1 и R2 имеют значения, показанные ниже в таблице


















34. Соединение по п.1, формулы

где B, L0, L1 и R2 имеют значения, показанные ниже в таблице


35. Соединение по п.1 формулы

где B, W1, W2 и R2 имеют значения, показанные ниже в таблице


36. Соединение по п.1 формулы

где B, L0, L2 и R2 имеют значения, показанные ниже в таблице



37. Соединение по п.1 формулы

где B, L0, L2 и R2 имеют значения, показанные ниже в таблице


38. Соединение по п.1 формулы

где B, L0, L1, L2 и R2 имеют значения, показанные ниже в таблице:



39. Соединение по п.1 формулы

где B, RQ и RS имеют значения, показанные ниже в таблице


40. Фармацевтическая композиция, включающая эффективное количество соединения формулы I по одному или более из пп.1-39, оказывающее противовирусное действие на вирус гепатита C, или его фармацевтически приемлемой соли или эфира в смеси по крайней мере с одним фармацевтически приемлемым носителем или вспомогательным агентом.
41. Фармацевтическая композиции по п.40, которая, кроме того, включает терапевтически эффективное количество по крайней мере одного другого противовирусного агента.
42. Фармацевтическая композиция по п.41, в которой упомянутым противовирусным агентом является рибавирин.
43. Фармацевтическая композиция по п.41, в которой упомянутый противовирусный агент выбирают из группы, включающей другой агент против ВГС, ингибитор ВИЧ, ингибитор ВГА и ингибитор ВГВ.
44. Фармацевтическая композиция по п.43, в которой упомянутый агент против ВГС выбирают из группы, включающей иммуномодуляторы, другие ингибиторы протеазы NS3 ВГС, ингибиторы полимеразы ВГС и ингибиторы другой мишени жизненного цикла ВГС.
45. Фармацевтическая композиция по п.44, в которой упомянутый иммуномодулятор выбирают из группы, включающей a-интерферон и ПЭГилированный a-интерферон.
46. Фармацевтическая композиция по п.44, в которой упомянутый ингибитор другой мишени жизненного цикла ВГС выбирают из группы, включающей ингибиторы геликазы, протеазы NS2/3 и внутреннего входного участка рибосомы (IRES).
47. Способ лечения или профилактики инфекции вируса гепатита C (ВГС) у млекопитающего, включающий введение млекопитающему эффективного в отношении ВГС количества соединения формулы I по одному или более из пп.1-39 или его фармацевтически приемлемых соли или сложного эфира.
48. Способ лечения или профилактики инфекции вируса гепатита C (ВГС) у млекопитающего, включающий введение млекопитающему эффективного в отношении ВГС количества соединения формулы I по одному или более из пп.1-39 или его фармацевтически приемлемых соли или сложного эфира в комбинации по крайней мере с одним другим противовирусным агентом.
49. Способ по п.48, в котором упомянутым противовирусным агентом является рибавирин.
50. Способ по п.48, в котором упомянутый другой противовирусный агент выбирают из группы, включающей другой агент против ВГС, ингибитор ВИЧ, ингибитор ВГА и ингибитор ВГВ.
51. Способ по п.50, в котором упомянутый другой агент против ВГС выбирают из группы, включающей иммуномодуляторы, другие ингибиторы протеазы NS3 ВГС, ингибиторы полимеразы ВГС и ингибиторы другой мишени жизненного цикла ВГС.
52. Способ по п.51, в котором упомянутый иммуномодулятор выбирают из группы, включающей a-интерферон и ПЭГилированный a-интерферон.
53. Способ по п.51, в котором упомянутый ингибитор другой мишени жизненного цикла ВГС выбирают из группы, включающей ингибиторы геликазы, протеазы NS2/3 и внутреннего входного участка рибосомы (IRES).
54. Применение соединения формулы I, включая их фармацевтически приемлемые соль или сложный эфир, по одному или более из пп.1-39, для получения лекарственного средства, предназначенного для лечения или профилактики инфекции вируса гепатита C у млекопитающего.
55. Способ ингибирования репликации вируса гепатита C при контактировании вируса с ингибирующим протеазу NS3 вируса гепатита количеством соединения формулы I по одному или более из пп.1-39 или его фармацевтически приемлемых соли или сложного эфшЁр.
56. Набор, включающий упаковку, в которой содержится композиция, предназначенная для эффективного лечения инфекции ВГС или для ингибирования протеазы NS3 ВГС, причем упаковка включает этикетку, на которой указано, что композицию можно использовать для лечения инфекции вируса гепатита C, и упомянутая композиция включает соединение формулы I по одному или более из пп.1-39 или его фармацевтически приемлемые соль или сложный эфир.
Текст
009295 Область изобретения Настоящее изобретение относится к соединениям, способам их синтеза и композициям и к способам лечения инфекции вируса гепатита C. Прежде всего настоящее изобретение относится к новым аналогам пептидов, фармацевтическим композициям, содержащим такие аналоги, и к способам применения таких аналогов для лечения инфекции вируса гепатита C человека (ВГС). Предпосылки создания изобретения Вирус гепатита C (ВГС) является основным этиологическим фактором, вызывающим широко распространенные во всем мире гепатит после переливания крови и домашний гепатит (в отличии от гепатита A и гепатита B). Известно, что во всем мире около 200 млн человек инфицированы этим вирусом. Высокий процент носителей этой инфекции страдают от хронической инфекции, а у большинства развивается хроническое заболевание печени, так называемый хронический гепатит C. Эти больные в свою очередь относятся к группе высокого риска тяжелых заболеваний печени, таких как цирроз печени, гепатоцеллюлярный рак и терминальное заболевание печени, которые приводят к летальному исходу. Механизмы, по которым развивается персистенция вируса и высокая частота хронических заболеваний печени до конца не изучены. В настоящее время не известно, каким образом ВГС взаимодействует с иммунной системой организма хозяина и происходит инвазия вируса. Кроме того, до сих пор не изучена роль клеточной и гуморальной иммунной системы при защите от инфекции ВГС и связанных с ней заболеваний. Установлено, что иммуноглобулины можно использовать для профилактики вирусного гепатита, связанного с переливанием крови, однако, в настоящее время иммуноглобулины не рекомендованы Центром по контролю заболеваемости для лечения указанных заболеваний. Отсутствие эффективной защитной реакции иммунной системы препятствует созданию вакцины или проведению соответствующих мероприятий вторичной профилактики, и таким образом в настоящее время существует острая необходимость в разработке новых противовирусных средств. В последнее время проводятся различные клинические испытания для выявления фармацевтических агентов, обладающих эффективностью при лечении инфекции ВГС у пациентов, пораженных хроническим гепатитом C. Были исследованы -интерферон в отдельности и в комбинации с другими противовирусными агентами. Установлено, что значительное число добровольцев не чувствительны к такому лечению, а у пациентов, на которых указанные агенты оказывали благоприятное действие, наблюдался рецидив заболевания после прекращения лечения. До настоящего времени единственным испытанным в клинике средством, оказывающим благоприятное действие при лечении пациентов, страдающих от хронического гепатита C, является интерферон. Однако при лечении интерфероном наблюдается низкая степень пролонгированной ответной реакции и тяжелые побочные действия (например, ретинопатия, тироидит, острый панкреатит, депрессия), что снижает качество жизни пациентов, проходящих курс лечения. Недавно было установлено, что интерферон действует в комбинации с рибавирином на пациентов, не чувствительных к лечению одним интерфероном, но при такой комбинационной терапии не снижаются побочные действия интерферона. ПЭГилированные формы интерферонов, такие как PEG-Intron и Pegasys частично снижают указанные отрицательные побочные действия, однако, проблема выбора противовирусных лекарственных средств для перорального введения при лечении ВГС все еще остается актуальной. Следовательно, в настоящее время существует необходимость в разработке эффективных противовирусных агентов для лечения ВГС, которые помогут исключить существующие ограничения при медикаментозном лечении. ВГС является РНК-вирусом семейства Flaviviridae и включает положительную цепь РНК с оболочкой. Одноцепочечная вирусная РНК генома ВГС включает приблизительно 9500 нуклеотидов и единственную открытую рамку считывания (ORF), кодирующую один белок-предшественник, содержащий приблизительно 3000 аминокислотных остатков. В инфицированных клетках этот белок расщепляется по многочисленным связям клеточными и вирусными протеазами с образованием структурных и неструктурных (NS) белков. В случае ВГС зрелые неструктурные белки (NS2, NS3, NS4A, NS4B, NS5A и NS5B) образуются при действии двух вирусных протеаз. Первая протеаза, в настоящее время мало изученная,расщепляет связь NS2-NS3 (названная протеазой NS2/3), вторая протеаза является сериновой протеазой,расположенной в N-концевом фрагменте NS3 (протеаза NS3), и опосредует расщепление NS3 на всех последующих стадиях, как в цис-конформации участков расщепления NS3-NS4A, так и в трансконформации остальных участков расщепления NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. NS4A является многофункциональным белком, то есть действует в качестве кофактора протеазы NS3 и возможно способствует локализации NS3 и других компонентов вирусной репликазы в мембране. Предполагается, что образование комплекса протеазы NS3 с белком NS4A необходимо для процессинга, причем комплекс повышает протеолитическую эффективность расщепления всех участков расщепления. Белок NS3 проявляет также активность нуклеозидтрифосфатазы и РНК геликазы. Белок NS5B является РНК-зависимой РНК-полимеразой, которая включена в процесс репликации ВГС. Основным подходом при создании противовирусных агентов является инактивация кодируемых вирусом ферментов, играющих важную роль при репликации вируса. Сравнительно недавно установлено, что протеаза NS3 проявляет дополнительное действие, а имен-1 009295 но блокирует опосредованную интерфероном противовирусную активность клеток в инфицированных клетках (Foy и др., Science, 17 апреля (2003. В связи с этим можно предположить, что протеазаNS3/NS4A представляет собой двойную терапевтическую мишень, при ингибировании которой можно блокировать вирусную репликацию и поддерживать ответную реакцию на интерферон в инфицированных ВГС клетках. В заявке WO 00/09543 описаны соединения формулы где R2 предпочтительно означает незамещенный или моно- или дизамещенный хинолинильный остаток,определенный в данном контексте, причем указанные соединения являются ингибиторами протеазы NS3 ВГС, то есть основного фермента, ответственного за репликацию ВГС. В настоящем изобретении предлагаются производные трипептида, характеризующиеся повышенной активностью в отношении протеазы NS3 ВГС. Более того, соединения проявляют высокую активность в клеточных культурах. Один предпочтительный объект настоящего изобретения относится к соединениям по настоящему изобретению, которые специфически ингибируют протеазу NS3 и практически не ингибируют другие вирусные сериновые протеазы, такие как эластаза из лейкоцитов человека (HLE) или цистеиновые протеазы, такие как катепсин В из печени человека (cat В). По сравнению с соединениями, описанными в заявке WO 00/09543, соединения по настоящему изобретению обладают наожиданными преимуществами. В основном они характеризуются одним или более следующими преимуществами: более низкие значения IC50 по данным определения активности протеазы NS3-NS4A,более низкие значения ЕС 50 по данным анализа репликации РНК ВГС с использованием клеток,более высокая растворимость и/или более высокий уровень в плазме после перорального введения крысам. Краткое описание изобретения В объем настоящего изобретения включены рацемат, диастереоизомер или оптический изомер соединения формулы (I) где В означает (С 1-С 10)алкил, (С 3-С 7)циклоалкил или (С 1-С 4)алкил(С 3-С 7)циклоалкил,а) где указанный алкил, циклоалкил и алкилциклоалкил являются моно-, ди- или тризамещенным группой (С 1-С 3)алкил и б) где указанный алкил, циклоалкил и алкилциклоалкил являются моно- или дизамещенным группой, которую выбирают из групп гидрокси и О-(С 1-С 4)алкил и в) где указанный алкил, циклоалкил и алкилциклоалкил являются моно-, ди- или тризамещенным атомами водорода и г) где каждая из указанных циклоалкильных групп означает 4-, 5-, 6- или 7-членное кольцо, необязательно содержащее одну (в случае 4-, 5-, 6- или 7-членных колец) или две (в случае 5-, 6- или 7 членных колец) -СН 2-групп, соединенных друг с другом не напрямую и разделенных группой -О- таким образом, что атом кислорода соединен с группой X по крайней мере через два атома углерода;R3 означает (С 2-С 8)алкил, (С 3-С 7)циклоалкил или (С 1-С 3)алкил(С 3-С 7)циклоалкил, где указанный алкил, циклоалкил и алкилциклоалкил являются моно-, ди- или тризамещенными группой (С 1-С 4)алкил;L1, L2, каждый независимо, означает галоген, циано, (С 1-С 4)алкил, -O-(C1-С 4)алкил, -S-(С 1-С 4)алкил,-SO-(С 1-С 4)алкил или -SO2-(С 1-С 4)алкил, где каждая из указанных алкильных групп необязательно замещена от 1 до 3 атомов галогена и L1 или L2 (но не оба одновременно) означают Н; илиL0 и L2 связаны ковалентно и вместе с двумя атомами углерода, к которым они присоединены, образуют 5- или 6-членное карбоциклическое кольцо, в котором одна или две -СН 2-группы связаны друг с другом не напрямую, и независимо разделены группами -О- или NRa, где Ra означает Н или (С 1-4)алкил и где указанное карбо- или гетероциклическое кольцо необязательно является моно- или дизамещенным группой (С 1-С 4)алкил;R2 означает R20, -NR22COR20, -NR22COOR20, -NR22R21 и -NR22CONR21R23, где R20 выбирают из группы, включающей (С 1-С 8)алкил, (С 3-С 7)циклоалкил или (С 1-С 4)алкил(С 3-С 7)циклоалкил, причем указанный циклоалкил и алкилциклоалкил являются моно-, ди- или тризамещенными группой (С 1-С 3)алкил;R21 означает Н или R20, как определено выше,R22 и R23 независимо выбирают из группы, включающей водород и метил,R1 означает этил или винил;RC означает гидрокси или NHSO2RS, где RS означает (С 1-С 6)алкил, (С 3-С 7)циклоалкил, (С 1 С 6)алкил(С 3-С 7)циклоалкил, фенил, нафтил, пиридинил, (C1-С 4)алкилфенил, (С 1-С 4)алкилнафтил или (С 1 С 4)алкилпиридинил, каждый из которых необязательно является моно-, ди- или тризамещенным группами, которые выбирают из группы, включающей галоген, гидрокси, циано, (С 1-С 4)алкил, О-(С 1-С 6)алкил,-CO-NH2, -CO-NH(С 1-С 4)алкил, -CO-NC1-C4)алкил)2, -NH2, -NH(C1-C4)алкил и -NC1-C4)алкил)2, причем (C1-C4)алкил и О-(С 1-С 6)алкил необязательно замещены от 1 до 3 атомов галогена и каждый из которых необязательно монозамещен нитрогруппой; илиRS означает -N(RN2)RN1, где RN1 и RN2 независимо выбирают из группы, включающей Н, (С 1 С 6)алкил, (С 3-С 7)циклоалкил, (С 1-С 6)алкил(С 3-С 7)циклоалкил, арил и (С 1-6)алкиларил; причем указанные(С 1-С 6)алкил, (С 3-С 7)циклоалкил, (С 1-С 6)алкил(С 3-С 7)циклоалкил, арил и (С 1-С 6)алкиларил необязательно замещены одним или более заместителями, которые независимо выбирают из группы, включающей галоген, (С 1-С 6)алкил, гидрокси, циано, О-(С 1-С 6)алкил, -NH2, -NH(С 1-С 4)алкил 1, -NС 1-С 4)алкил)2, -CONH2, -СО-NH(С 1-С 4)алкил, -СО-NС 1-4)алкил)2, -СООН и -COO(C1-C6)алкил; илиRN2 и RN1 соединены и вместе с атомом азота, к которому они присоединены, образуют 3-7-членный моноциклический насыщенный или ненасыщенный гетероцикл или 9-10-членный бициклический, насыщенный или ненасыщенный гетероцикл, каждый из которых необязательно содержит от 1 до 3 дополнительных гетероатомов, которые независимо выбирают из N, S и О и каждый из которых необязательно замещен одним или более заместителями, которые независимо выбирают из группы, включающей галоген, (С 1-С 6)алкил, гидрокси, циано, О-(С 1-С 6)алкил, -NH2, -NH(С 1-С 4)алкил, -NС 1-С 4)алкил)2, -CO-NH2,-СО-NH(С 1-С 4)алкил, -СО-NС 1-С 4)алкил)2, -СООН и -СОО(С 1-С 6)алкил; или его фармацевтически приемлемых солей или сложных эфиров. В объем настоящего изобретения включена фармацевтическая композиция, включающая эффективное количество соединения формулы I, оказывающее противовирусное действие на вирус гепатита C,или его фармацевтически приемлемой соли или эфира в смеси по крайней мере с одним фармацевтически приемлемым носителем или вспомогательным агентом. Другой объект настоящего изобретения относится к фармацевтической композиции, которая, кроме того, включает терапевтически эффективное количество по крайней мере одного другого противовирусного агента. Еще один важный объект настоящего изобретения относится к способу лечения или профилактики инфекции вируса гепатита C у млекопитающего, причем способ включает введение млекопитающему эффективного количества соединения формулы I в отношении вируса гепатита C, или его фармацевтически приемлемой соли или эфира, или композиции, описанной выше, отдельно или в комбинации по крайней мере с одним другим противовирусным агентом, который вводят отдельно или в комбинации с указанным соединением. Настоящее изобретение включает также применение соединения формулы I или его фармацевтически приемлемых соли или эфира, как описано выше, для получения лекарственного средства, предназначенного для лечения или профилактики инфекции вируса гепатита C у млекопитающих. Подробное описание предпочтительных вариантов осуществления настоящего изобретения Определения. В настоящем описании используются следующие определения, если не указано иное. Символы (R) или (S), использованные в названии соединений, означают абсолютную конфигурацию заместителя или асимметрического центра соединения формулы I, при этом эти символы относятся к соединению в целом, а не к заместителю или асимметическому центру соединения в отдельности. Символы P1, P2 и Р 3, использованные в данном контексте, означают номера аминокислотных остатков, начиная с C-концевого фрагмента пептидных аналогов по направлению к N-концевому фрагменту (т.е. P1 означает аминокислотный остаток 1 от C-концевого фрагмента, P2 означает аминокислотный остаток 2 от C-концевого фрагмента и т.п.) (см. статью Berger А. и Schechter I., Transactions of the Royal а именно (1R,2S) 1-амино-2-этенилциклопропанкарбоновую кислоту. Термин (С 1-Cn)алкил, использованный в данном контексте отдельно или в комбинации с другим заместителем, означает ациклический, прямой или разветвленный алкильный заместитель, содержащий от 1 до n атомов углерода. Термин (С 1-С 6)алкил включает, без ограничения перечисленным, метил, этил, н-пропил, н-бутил,1-метилэтил (изопропил), 1-метилпропил, 2-метилпропил, 1,1-диметилэтил (трет-бутил), пентил и гексил. Сокращения Me и Pr означают метильную и н-пропильную группы соответственно. Термин (С 3-С 7)циклоалкил, использованный в данном контексте отдельно или в комбинации с другим заместителем, означает циклоалкильный заместитель, содержащий от 3 до 7 атомов углерода, и включает, без ограничения перечисленным, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термин (С 1-Cn)алкил(С 3-С 7)циклоалкил, использованный в данном контексте, означает алкиленовый радикал, содержащий от 1 до n атомов углерода, к которому напрямую присоединен циклоалкильный радикал, содержащий от 3 до 7 атомов углерода, и включает, без ограничения перечисленным, циклопропилметил, циклобутилметил, циклопентилметил, 1-циклопентилэтил, 2-циклопентилэтил, циклогексилметил, 1-циклогексилэтил, 2-циклогексилэтил и циклогептилпропил. Термин арил или С 6 или С 10 арил, использованный в данном контексте взаимозаменяемо, отдельно или в комбинации с другим заместителем, означает ароматическую моноциклическую группу,содержащую 6 атомов углерода, или ароматическую бициклическую группу, содержащую 10 атомов углерода. Термин арил включает, без ограничения перечисленным, фенил, 1-нафтил или 2-нафтил. Термин (С 1-Cn)алкиларил, использованный в данном контексте, означает алкильный радикал, содержащий от 1 до п атомов углерода, к которому присоединен арил. Примеры радикалов (С 1 С 3)алкиларил включают, без ограничения перечисленным, бензил(фенилметил), 1-фенилэтил, 2-фенилэтил и фенилпропил. Термин О-(С 1-Cn)алкил или (С 1-Cn)алкокси, использованный в данном контексте отдельно или в комбинации с другим заместителем, означает радикал -O-(С 1-Cn)алкил, где алкил имеет значения, определенные выше, содержит от 1 до n атомов углерода и включает метокси, этокси, пропокси, 1 метилэтокси, бутокси и 1,1-диметилэтокси. Последний радикал обычно называется трет-бутокси. Термин галоген, использованный в данном контексте, означает заместитель, который выбирают из группы, включающей фтор, хлор, бром или иод. Термин фармацевтически приемлемый сложный эфир, использованный в данном контексте отдельно или в комбинации с другим заместителем, означает сложные эфиры соединения формулы I, в которых любые карбоксильные функциональные группы, предпочтительно концевые карбоксильные группы, заменены на алкоксикабонильную группу где остаток R в составе эфира выбирают из группы, включающей алкил (включая, без ограничения перечисленным, метил, этил, н-пропил, трет-бутил, н-бутил), алкоксиалкил (включая, без ограничения перечисленным, метоксиметил), алкоксиацил (включая, без ограничения перечисленным, ацетоксиметил),алкиларил (включая, без ограничения перечисленным, бензил), арилоксиалкил (включая, без ограничения нижеперечисленным, феноксиметил), арил (включая, без ограничения нижеперечисленным, фенил),необязательно замещенные группами галоген, (С 1-С 4)алкил или (С 1-С 4)алкокси. Другие пригодные эфиры пролекарств описаны в книге Design of prodrugs, Bundgaard, H. Ed. Elsevier (1985). Такие фармацевтически приемлемые эфиры обычно гидролизуются in vivo при введении их млекопитающему и превращаются в кислотную форму соединения формулы I. При описании упомянутых выше эфиров, если не указано иное, все алкильные группы преимущественно содержат от 1 до 16 атомов углерода, прежде всего от 1 до 6 атомов углерода. Все арильные группы в таких эфирах преимущественно включают фенильную группу. Прежде всего эфиры включают С 1-С 16 алкиловый эфир, незамещенный бензиловый эфир или бензиловый эфир, замещенный по крайней мере одним атомом галогена, одной группой C1-С 6 алкил, С 1 С 6 алкокси, нитро или трифторметил. Термин фармацевтически приемлемая соль означает соль соединения формулы I, которая является пригодной при использовании в медицине при контактировании с тканями человека и животных и не оказывает отрицательного действия, т.е. не обладает токсичностью, не вызывает раздражения и аллергической реакции и т.п., соответствует достаточно высокому соотношению выгода/риск, причем указанная соль образует раствор или дисперсию в воде или масле и оказывает предназначенное действие. Термин включает фармацевтически приемлемые кислотно-аддитивные соли и фармацевтически приемлемые-4 009295 основно-аддитивные соли. Список пригодных солей приведен в литературе, например, в статье S.M.Birge и др., J. Pharm. Sci., 66, cc. 1-19 (1977). Термин фармацевтически приемлемые кислотно-аддитивные соли означает соли, которые сохраняют биологическую эффективность и свойства свободных оснований и которые не оказывают отрицательного действия в биологическом или ином отношении, причем указанные соли образуются при взаимодействии с неорганическими кислотами, такими как хлористо-водородная кислота, бромистоводородная кислота, серная кислота, сульфаминовая кислота, азотная кислота, фосфорная кислота и т.п.,и с органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, адипиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота,бутановая кислота, камфорная кислота, камфорсульфоновая кислота, коричная кислота, лимонная кислота, диглюконовая кислота, этансульфоновая кислота, глутаминовая кислота, гликолевая кислота, глицерофосфорная кислота, гемисерная кислота, гексановая кислота, муравьиная кислота, фумаровая кислота,2-гидроксиэтансульфоновая кислота (изетионовая кислота), молочная кислота, гидроксималеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, мезитиленсульфоновая кислота, метансульфоновая кислота, нафталинсульфоновая кислота, никотиновая кислота, 2-нафталинсульфоновая кислота, щавелевая кислота, памоевая кислота, пектиновая кислота, фенилуксусная кислота, 3 фенилпропионовая кислота, пиваловая кислота, пропионовая кислота, пировиноградная кислота, салициловая кислота, стеариновая кислота, янтарная кислота, сульфаниловая кислота, винная кислота, паратолуолсульфоновая кислота, ундекановая кислота и т.п. Термин фармацевтически приемлемые основно-аддитивные соли означает соли, которые сохраняют биологическую эффективность и свойства свободных кислот и которые не оказывают отрицательного действия в биологическом или ином отношении, причем указанные соли образуются при взаимодействии с неорганическими основаниями, такими как аммиак или гидроксид, карбонат или бикарбонат аммония, или катион металла, такого как натрий, калий, литий, кальций, магний, железо, цинк, медь,марганец, алюминий и т.п. Прежде всего предпочтительными являются соли аммония, калия, натрия,кальция и магния. Соли фармацевтически приемлемых органических нетоксичных оснований включают соли первичных, вторичных и третичных аминов, соединения четвертичного амина, замещенные амины,которые включают природные замещенные амины, циклические амины и основные ионообменные смолы, такие как метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, изопропиламин, трипропиламин, трибутиламин, этаноламин, диэтаноламин, 2-диметиламиноэтанол, 2 диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, гидрабамин, холин, бетаин,этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперазин, пиперидин, Nэтилпиперидин, соединения тетраметиламмония, соединения тетраэтиламмония, пиридин, N,Nдиметиланилин, N-метилпиперидин, N-метилморфолин, дициклогексиламин, дибензиламин, N,Nдибензилфенетиламин, 1-эфенамин, N,N'-дибензилэтилендиамин, полиаминовые смолы и т.п. Прежде всего предпочтительные органические нетоксичные основания включают изопропиламин, диэтиламин,этаноламин, триметиламин, дициклогексиламин, холин и кофеин. Термин млекопитающее, использованный в данном контексте, означает человека, а также других млекопитающих, чувствительных к инфекции вируса гепатита С (ВГС), включая домашних животных,таких как коровы, свиньи, лошади, собаки и кошки, а также недомашних животных. Термин противовирусный агент, использованный в данном контексте, означает агент (химический или биологический), который эффективно ингибирует формирование и/или репликацию вируса в организме млекопитающего. Такие агенты включают агенты, которые блокируют механизмы в организме хозяина или механизмы вируса, необходимые для формирования и/или репликации вируса в организме млекопитающего. Такие агенты выбирают из группы, включающей другой противовирусный агент в отношении ВГС, ингибитор ВИЧ, ВГА и ВГВ. Противовирусные агенты включают, например, рибавирин, амантадин, VX-497 (меримеподиб, фирмы Vertex Pharmaceuticals), VX-498 (фирмы Vertex Pharmaceuticals), левовирин, вирамидин, цеплен (максамин), XTL-001 и XTL-002 (фирмы XTL Biopharmaceuticals). Термин другой противовирусный агент, использованный в данном контексте, означает агенты,которые проявляют эффективность за счет снижения интенсивности или профилактики симптомов заболевания, связанного с развитием вируса ВГС. Такие агентывыбирают из группы, включающей иммуномодуляторы, ингибиторы протеазы NS3 ВГС, ингибиторы полимеразы ВГС или ингибиторы другой мишени жизненного цикла ВГС. Термин иммуномодуляторы, использованный в данном контексте, означает такие агенты (химические или биологические), которые проявляют эффективность за счет увеличения или потенциирования ответной реакции иммунной системы в организме млекопитающего. Иммуномодуляторы включают, например, интерфероны класса I (такие как -, -, - , - и -интерфероны, консенсус-интерфероны и асиалоинтерфероны), интерфероны класса II (такие как -интерфероны) и их ПЭГилированные формы. Термин ингибитор протеазы NS3 ВГС, использованный в данном контексте, означает агент (химический или биологический), который проявляет эффективность при ингибировании функции протеазыNS3 ВГС в организме млекопитающего. Ингибиторы протеазы NS3 ВГС включают, например, соединения, описанные в заявках WO 99/07733, WO 99/07734, WO 00/09558, WO 00/09543, WO 00/59929, WO 03/064416, WO 03/064455, WO 03/064456, WO 02/060926, WO 03/053349, WO 03/099316 или WO 03/099274, и препарат фирмы Vertex (сокращенное обозначение VX-950), находящийся на стадии предварительных испытаний. Термин ингибитор полимеразы ВГС, использованный в данном контексте, означает агент (химический или биологический), который проявляет эффективность при ингибировании функции полимеразы ВГС в организме млекопитающего. Такие ингибиторы включают, например, ингибиторы полимеразыNS5B ВГС. Ингибиторы полимеразы ВГС включают ненуклеотидные соединения, например, соединения, описанные в следующих документах: заявка на выдачу патента США 60/441674, поданная 22 января 2003 г, включенная в настоящее описание в виде ссылки (Boehringer Ingelheim),заявка на выдачу патента США 60/441871, поданная 22 января 2003 г, включенная в настоящее описание в виде ссылки (Boehringer Ingelheim),заявки WO 04/005286 (Gilead), WO 04/002977 (Pharmacia), WO 04/002944 (Pharmacia), WO 04/002940 (Pharmacia), WO 03/101993 (Neogenesis), WO 03/099824 (Wyeth), WO 03/099275 (Wyeth), WO 03/099801 (GSK), WO 03/097646 (GSK), WO 03/095441 (Pfizer), WO 03/090674 (Viropharma), WO 03/084953 (BC Biopharm), WO 03/082265 (Fujisawa), WO 03/082848 (Pfizer), WO 03/062211 (Merck), WO 03/059356 (GSK), EP 1321463 (Shire), WO 03/040112 (Rigel), WO 03/037893 (GSK), WO 03/037894 (GSK),WO 03/037262 (GSK), WO 03/037895 (GSK), WO 03/026587 (BMS), WO 03/002518 (Dong Wha), WO 03/000254 (Japan Tobacco), WO 02/100846 A1 (Shire), WO 02/100851 A2 (Shire), WO 02/098424 A1 (GSK),WO 02/079187 (Dong Wha), WO 03/02/20497 (Shionogi), WO 02/06246 (Merck), WO 01/47883 (Japan Tobacco), WO 01/85172 A1 (GSK), WO 01/85720 (GSK), WO 01/77091 (Tularik), WO 00/18231 (Viropharma),WO 00/13708 (Viropharma), WO 01/10573 (Viropharma) WO 00/06529 (Merck), EP 1256628 A2 (Agouron),WO 02/04425 (Boehringer Ingelheim), WO 03/007945 (Boehringer Ingelheim), WO 03/010140 (BoehringerIngelheim) и WO 03/010141 (Boehringer Ingelheim). Более того, другие ингибиторы полимеразы ВГС включают также аналоги нуклеозидов, например, соединения, описанные в заявках WO 04/007512(Hoffmann-La Roche), WO 02/069903 (Biocryst Pharmaceuticals Inc.), WO 02/057287 (Merck/Isis), WO 02/057425 (Merck/Isis), WO 01/90121 (Idenix), WO 01/60315 (Shire) и WO 01/32153 (Shire). Примеры ингибиторов полимеразы ВГС включают JTK-002, JTK-003 и JTK-109 (Japan Tobacco). Термин ингибитор другой мишени жизненного цикла ВГС, использованный в данном контексте,означает агент (химический или биологический), который проявляет эффективность при ингибировании формирования и/или репликации вируса ВГС в организме млекопитающего, по другому механизму в отличие от ингибирования функции протеазы NS3 ВГС. Такие агенты включают агенты, которые блокируют механизмы в организме хозяина или механизмы вируса ВГС, необходимые для формирования и/или репликации вируса в организме млекопитающего. Ингибиторы другой мишени жизненного цикла ВГС включают, например, агенты, ингибирующие мишень, которую выбирают из группы, включающей геликазу, протеазу NS2/3 и внутренний входной участок рибосом (IRES). Примеры ингибиторов другой мишени жизненного цикла вируса ВГС включают ISIS-14803 (ISIS Pharmaceuticals). Термин ингибитор ВИЧ, использованный в данном контексте, означает агент (химический или биологический), который проявляет эффективность при ингибировании формирования и/или репликации вируса ВИЧ в организме млекопитающего. Такие агенты включают агенты, которые блокируют механизмы в организме хозяина или механизмы вируса, необходимые для формирования и/или репликации вируса ВИЧ в организме млекопитающего. Ингибиторы ВИЧ включают, например, нуклеозидные ингибиторы, ненуклеозидные ингибиторы, ингибиторы протеазы, ингибиторы слияния, ингибиторы интегразы. Термин ингибитор ВГА, использованный в данном контексте, означает агент (химический или биологический), который проявляет эффективность при ингибировании формирования и/или репликации вируса ВГА в организме млекопитающего. Такие агенты включают агенты, которые блокируют механизмы в организме хозяина или механизмы вируса, необходимые для формирования и/или репликации вируса ВГА в организме млекопитающего. Ингибиторы ВГА включают вакцины против вируса гепатита А, например, Havrix (фирмы GlaxoSmithKline), VAQTA (фирмы Merck) и Avaxim (фирмы Aventis Pasteur). Термин ингибитор ВГВ, использованный в данном контексте, означает агент (химический или биологический), который проявляет эффективность при ингибировании формирования и/или репликации вируса ВГВ в организме млекопитающего. Такие агенты включают агенты, которые блокируют механизмы в организме хозяина или механизмы вируса, необходимые для формирования и/или репликации-6 009295 вируса ВГВ в организме млекопитающего. Ингибиторы ВГВ включают, например, агенты, которые ингибируют ДНК полимеразу вируса ВГВ, или вакцины против ВГВ. Примеры ингибиторов ВГВ включают ламивудин (Epivir-HBV), адефовир дипивоксил, энтекавир, FTC (Coviracil), DAPD (DXG), LFMAU (Clevudine), AM365 (Amrad), Ldt (Telbivudine), моновал-LdC (Valtorcitabine), ACH-126443 (LFd4C Achillion), MCC478 (фирмы Eli Lilly), расивир (RCV), нуклеозиды фтор-L и -D, робустафлавон, ICN 2001-3 (ICN), бам 205 (Novelos), XTL-001 (XTL), иминосахара (Nonyl-DNJ Synergy), HepBzyme и иммуномодуляторы, такие как интерферон альфа 2b, HE2000 (Hollis-Eden), терадигм (Epimmune), EHT899HBV DNA (фирмы Jefferon Center), антиген HBV (фирмы OraGen), BayHep В (фирмы Bayer), NabiHB (фирмы Nabi) и Anti-hepatitis В (фирмы Cangene); а также вакцины HBV, такие как: Engerix В, Recombivax HB, GenHevac В, Hepacare, Bio-Hep В, TwinRix, Comvax, Hexavac. Термин интерферон класса I, использованный в данном контексте, означает интерферон, который выбирают из группы интерферонов, которые связываются с рецептором типа I. Такие интерфероны включают природные и синтетические интерфероны класса I. Примеры интерферонов класса I включают-, , -, - и -интерфероны, консенсус-интерфероны, асиало-интерфероны и их ПЭГилированные формы. Термин интерферон класса II, использованный в данном контексте, означает интерферон, который выбирают из группы интерферонов, которые связываются с рецептором типа II. Примеры интерферонов класса II включают -интерфероны. Наиболее предпочтительные примеры некоторых из указанных агентов перечислены ниже: противовирусные агенты: рибавирин или амантадин,иммуномодуляторы: интерфероны класса I, интерфероны класса II или их ПЭГилированные формы,ингибиторы полимеразы ВГС: нуклеозидные аналоги и ненуклеозидные соединения,ингибитор другой мишени жизненного цикла ВГС, который ингибирует мишень, которую выбирают из группы, включающей геликазу NS3, протеазу NS2/3 или внутренний входной участок рибосомы(IRES),ингибиторы ВИЧ: нуклеозидные ингибиторы, ненуклеозидные ингибиторы, ингибиторы протеаз,ингибиторы слияния или ингибиторы интегразы или ингибиторы ВГВ: агенты, ингибирующие ДНК полимеразу, или вакцины против ВГВ. Как описано выше, комбинационная терапия означает введение соединения формулы (I) или его фармацевтически приемлемой соли одновременно по крайней мере с одним дополнительным агентом,который выбирают из группы, включающей противовирусный агент, иммуномодулятор, ингибитор протеазы NS3 ВГС, ингибитор полимеразы ВГС, ингибитор другой мишени жизненного цикла ВГС, ингибитор ВИЧ, ингибитор ВГА и ингибитор ВГВ. Примеры таких агентов представлены в разделе Определения выше. Такие дополнительные агенты можно смешивать с соединениями по настоящему изобретению и получать единую фармацевтическую лекарственную форму. В другом варианте такие дополнительные агенты вводят пациенту отдельно как часть многокомпонентной лекарственой формы, например, при использовании набора лекарственных форм. Такие дополнительные агенты вводят пациенту перед, одновременно или после введения соединения формулы (I) или его фармацевтически приемлемой соли. Термин лечение, использованный в данном контексте, означает введение соединения или композиции по настоящему изобретению для снижения интенсивности или устранения симптомов гепатита C и/или снижения содержания вируса в организме пациента. Термин профилактика, использованный в данном контексте, означает введение соединения или композиции по настоящему изобретению после инфицирования субъекта вирусом, но до появления симптомов заболевания и/или до обнаружения вируса в крови для предотвращения появления симптомов заболевания. Связь с заместителем R изображена как линия, выходящая из центра кольца, такая как, например, или которая означает, что заместитель R может быть присоединен в любое свободное положение в кольцо,которое в другом варианте будет замещено водородом, если не указано иное. Символилииспользуют взаимозаменяемо при изображении связи, с помощью которой остаток присоединен к молекуле.-7 009295 Предпочтительные варианты осуществления настоящего изобретения В данном разделе подробно описаны предпочтительные варианты осуществления настоящего изобретения, группы и заместители соединений по настоящему изобретению.B предпочтительно выбирают из группы, включающей (С 2-С 8)алкил, (С 3-С 7)циклоалкил и (С 1 С 3)алкил(С 3-С 7)циклоалкил,а) где указанные алкил, циклоалкил и алкилциклоалкил являются моно-, ди- или тризамещенными группой (С 1-С 3)алкил, и б) где указанные алкил, циклоалкил и алкилциклоалкил являются моно- или дизамещенными заместителями, которые выбирают из группы, включающей гидрокси и О-(С 1-С 4)алкил, и в) где каждая из указанных алкильных групп является моно-, ди- или тризамещенной атомами фтора или монозамещенной атомами хлора или брома, и г) где каждая из указанных циклоалкильных групп означает 5-, 6- или 7-членное кольцо, в котором одна или две -СН 2-группы не связаны напрямую друг с другом, замещены группой -О- таким образом,что атом О соединен с группой X по крайней мере через два атома углерода. Более предпочтительно B выбирают из группы, включающей этил, н-пропил, изопропил, н-бутил,1-метилпропил, 2-метилпропил, трет-бутил, циклопропил, циклобутил, циклопентил и циклогексил,а) где каждая из указанных групп необязательно замещена 1-3 заместителями, которые выбирают из группы, включающей метил и этил, и б) где каждая из указанных групп необязательно моно- или дизамещена заместителями, которые выбирают из группы, включающей гидрокси, метокси и этокси, и в) где каждая из указанных алкильных групп является моно-, ди- или тризамещенной атомами фтора или монозамещеной атомами хлора или брома, и г) где каждая из указанных циклоалкильных групп означает 5-, 6- или 7-членное кольцо, в котором одна или две -СН 2-группы не связаны напрямую друг с другом и замещены группой -О- таким образом,что атом О соединен с группой X по крайней мере через два атома углерода. Еще более предпочтительно B выбирают из группы, включающей этил, 1-метилэтил, 1,1 диметилэтил, пропил, 1-метилпропил, 2-метилпропил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2 диметилпропил, 1,1,2-триметилпропил, 1,2,2-триметилпропил, 1-этилпропил, 1-этил-2-метилпропил, 1(1-метилэтил)-2-метилпропил, 1-этил-2,2-диметилпропил, бутил, 1-метилбутил, 2-метилбутил, 3 метилбутил, 1,2-диметилбутил, 1,1-диметилбутил, 1,3-диметилбутил, 2,2-диметилбутил, 2,3 диметилбутил, 3,3-диметилбутил, 1,2,2-триметилбутил, 1,2,3-триметилбутил, 2,2,3-триметилбутил, 2,3,3 триметилбутил и 2,2,3-триметилбутил, причем данные алкильные группы замещены атомами хлора, или брома, или 1, 2 или 3 атомами фтора. Примеры предпочтительных фторированных алкильных групп включают, без ограничения перечисленным, 2-фторэтил, 3-фторпропил и 3,3,3-трифторпропил. Еще более предпочтительно B означает циклопропил, циклобутил, циклопентил или циклогексил или заместитель B выбирают из следующих формул, где одна или две СН 2-группы в составе циклоалкильной группы заменены на кислород: Из приведенного выше списка циклоалкильная и алкилциклоалкильная группы, необязательно включающие 1 или 2 атома кислорода, необязательно замещены 1, 2 или 3 метильными группами. Прежде всего предпочтительными являются циклоалкильные группы, необязательно включающие 1 или 2 атома кислорода, где атом углерода в -положении замещен метильной группой. Другие примеры предпочтительных замещенных циклических групп включают Еще более предпочтительно B выбирают из группы, включающей этил, н-пропил, трет-бутил, циклопентил, 1-метилциклопентил, 2-фторэтил или 3-фторпропил. В одном варианте осуществления настоящего изобретения X означает O. В другом варианте осуществления настоящего изобретения X означает NH.R3 предпочтительно означает (С 2-С 6)алкил, (С 3-С 7)циклоалкил или (C1-С 3)алкил(С 3-С 7)циклоалкил,каждый из которых необязательно замещен 1-3 заместителями (С 1-С 4)алкил.R3 более предпочтительно выбирают из группы, включающей этил, пропил, бутил, циклопропил,циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил или циклогексилметил, каждый из которых необязательно замещен 1 или 2 заместителями, которые выбирают из группы, включающей метил, этил и пропил. Еще более предпочтительно R3 выбирают из группы, включающей 1-метилэтил, 1,1-диметилэтил, 1 метилпропил, 2-метилпропил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, циклопентил,циклогексил, 1-метилциклопентил, 1-метилциклогексил, циклопентилметил, циклогексилметил, (1 метилциклопентил)метил и (1-метилциклогексил)метил. Наиболее предпочтительно R3 выбирают из группы, включающей 1,1-диметилэтил, циклопентил,циклогексил и 1-метилциклогексил. Еще более предпочтительно R3 выбирают из группы, включающей 1,1-диметилэтил и циклогексил.L0 предпочтительно выбирают из группы, включающей Н, галоген, СН 3, -ОН, -ОСН 3, -ОС 2 Н 5,-ОС 3 Н 7, -ОСН(СН 3)2, -NHCH3, -NHC2H5, -NHC3H7, -NHCH(CH3)2, -N(CH3)2, -N(CH3)C2H5, -N(CH3)C3H7 иL0 более предпочтительно выбирают из группы, включающей Н, -ОН, -ОСН 3; галоген и -N(CH3)2. Еще более предпочтительно L0 выбирают из группы, включающей Н, -ОН или -ОСН 3. Наиболее предпочтительно L0 выбирают из группы, включающей Н или -ОСН 3.L1 и L2 независимо друг от друга предпочтительно выбирают из группы, включающей галоген,-СН 3, -С 2 Н 5, -ОСН 3, -ОС 2 Н 5, -ОС 3 Н 7, -OCH(CH3)2, CF3, -SMe, -SOMe, и SO2Me, причем L1 или L2 может означать Н. Более предпочтительно один из заместителей L1 и L2 означает -СН 3, -F, -Cl, -Br, -OMe, -Sme или-SO2Me, а другой из заместителей L1 и L2 означает Н. Наиболее предпочтительно L означает СН 3, -F, -Cl, -Br, -OMe, -SMe, или -SO2Me, a L2 означает Н. Таким образом, предпочтительно L0 выбирают из группы, включающей Н, -ОН и -ОСН 3; один из заместителей L1 и L2 означает СН 3, -F, -Cl, -Br, -OMe, -SMe или -SO2Me, а другой из заместителей L1 и L2 означает Н. Более предпочтительно L0 выбирают из группы, включающей Н, -ОН и -ОСН 3; L1 означает -СН 3, -F,-Cl, -Br, -OMe, -SMe, или -SO2Me, a L2 означает Н. Наиболее предпочтительно L0 означает Н или -ОСН 3, L1 означает -СН 3, -Cl или -Br; a L2 означает Н. Если L0 и L1 ковалентно связаны и вместе с хинолиновым остатком, к которому они присоединены,образуют циклическую систему, причем указанную циклическую систему предпочтительно выбирают из следующих групп: где Ха, Xb независимо выбирают из группы, включающей СН 2, О и NRa; наиболее предпочтительно О;L2 определен выше, предпочтительно означает Н или метил, прежде всего Н. Если L0 и L2 ковалентно связаны и вместе с хинолиновым остатком, к которому они присоединены,образуют циклическую систему, то указанную циклическую систему предпочтительно выбирают из следующих групп: где Ха, Xb независимо выбирают из группы, включающей СН 2, О и NRa; наиболее предпочтительно О;L1 определен выше, предпочтительно означает Н или метил, прежде всего Н. Более предпочтительно L0 и L1 ковалентно связаны и вместе с хинолиновым остатком, к которому они присоединены, образуют циклическую систему, которую выбирают из следующих групп: где каждый заместитель R незачисимо означает (С 1-С 4)алкил, a L2 определен выше, предпочтительно означает Н или метил, прежде всего Н. Наиболее предпочтительно L0 и L1 ковалентно связаны и вместе с хинолиновым остатком, к которому они присоединены, образуют циклическую систему, которую выбирают из следующих групп:R20 выбирают из группы, включающей (С 1-С 8)алкил, (С 3-С 7)циклоалкил, (С 1-С 3)алкил-(С 3 С 7)циклоалкил, причем указанные циклоалкил и алкилциклоалкил моно-, ди- или тризамещены группойR20 выбирают из группы, включающей метил, этил, н-пропил, изопропил, н-бутил, 1-метилпропил,2-метилпропил, трет-бутил, 2,2-диметилпропил, 1,1-диметилпропил, 1,2-диметилпропил, 1,2,2 триметилпропил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил и циклогексилметил, при этом каждая из указанных циклоалкильных и алкилциклоалкильных групп необязательно замещена 1-3 заместителями, которые выбирают из группы,включающей метил и этил, прежде всего метил, иR23 означает Н или метил, наиболее предпочтительно Н. Предпочтительно R выбирают из группы, включающей а) амино, N-метиламино, N-этиламино, N-пропиламино, N-(1-метилэтил)амино, N-(1,1-диметилэтил)амино, N-(2-метилпропил)амино, N-(1-метилпропил)амино, N-(2,2-диметилпропил)амино, N-(1,2 диметилпропил)амино, N-(1,1-диметилпропил)амино, N-циклопропиламино, N-циклобутиламино, Nциклопентиламино, N-циклогексиламино, N-(циклопропилметил)амино, N-(циклобутилметил)амино, N(циклопентилметил)амино и N-(циклогексилметил)амино; б) метилкарбониламино, этилкарбониламино, 1-метилэтилкарбониламино, пропилкарбониламино,2-метилпропилкарбониламино, 1-метилпропилкарбониламино, 2,2-диметилпропилкарбониламино, 1,2 диметилпропилкарбониламино, 1,1-диметилпропилкарбониламино, циклопропилкарбониламино, цикло- 10009295 бутилкарбониламино, циклопентилкарбониламино, циклогексилкарбониламино, циклопропилметилкарбониламино, циклобутилметилкарбониламино, циклопентилметилкарбониламино, циклогексилметилкарбониламино и в) метоксикарбониламино, этоксикарбониламино, 1-метилэтоксикарбониламино, пропоксикарбониламино, трет-бутоксикарбониламино, циклопропилоксикарбониламино, циклобутилоксикарбониламино, циклопентилоксикарбониламино, циклогексилоксикарбониламино, циклопропилметоксикарбониламино, циклобутилметоксикарбониламино, циклопентилметоксикарбониламино и циклогексилметоксикарбониламино,причем все указанные циклоалкильные или алкилциклоалкильные группы моно- или дизамещены метильной группой. Наиболее предпочтительно R2 означает -NHCOR20, -NHCOOR20 или -NHR21, где R20 и R21 определены выше. Предпочтительно R20 и R21 независимо выбирают из группы, включающей метил, этил, н-пропил,изопропил, н-бутил, 1-метилпропил, 2-метилпропил, трет-бутил, 2,2-диметилпропил, 1,1-диметилпропил,1,2-диметилпропил, 1,2,2-триметилпропил, циклопропил, циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил и циклогексилметил, при этом каждая из указанных циклоалкильных или алкилциклоалкильных групп необязательно моно- или дизамещена заместителями, которые выбирают из группы, включающей метил и этил. Более предпочтительно R20 и R21 независимо выбирают из группы, включающей метил, этил, нпропил, изопропил, 2,2-диметилпропил, циклопентил и циклопентилметил. Заместитель R1 и карбонильная группа в составе остатка Р 1 характеризуются син-ориентацией. Таким образом, если R означает этил, то асимметричные атомы углерода в циклопропильной группе характеризуются конфигурацией R,R, как показано ниже: если R1 означает винил, то асимметричные атомы углерода в циклопропильной группе характеризуются конфигурацией R,S, как показано ниже:RC предпочтительно выбирают из группы, включающей гидрокси или NHSO2RS, где RS означает метил, этил, н-пропил, изопропил, н-бутил, 1-метилпропил, 2-метилпропил, трет-бутил, циклопропил,циклобутил, циклопентил, циклогексил, циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил, фенил, нафтил, пиридинил, фенилметил(бензил), нафтилметил или пиридинилметил,а) причем каждый заместитель необязательно моно-, ди- или тризамещен заместителями, которые выбирают из группы, включающей фтор и метил, и б) причем каждый заместитель необязательно моно- или дизамещен заместителями, которые выбирают из группы, включающей гидрокси, трифторметил, метокси и трифторметокси, и в) причем каждый заместитель необязательно монозамещен заместителями, которые выбирают из группы, включающей хлор, бром, циано, нитро, -CO-NH2, -CO-NHCH3, -CO-N(CH3)2, -NH2, -NH(CH3) и-N(CH3)2. В другом варианте предпочтительно RC означает NHSO2RS, где RS означает -N(RN2)RN1, где RN1 иR независимо выбирают из группы, включающей Н, (C1-С 4)алкил, (С 3-С 7)циклоалкил, (С 1-С 3)алкил(С 3 С 7)циклоалкил, фенил и (С 1-С 3)алкилфенил; причем указанные (С 1-С 4)алкил, (С 3-С 7)циклоалкил, (С 1 С 3)алкил(С 3-С 7)циклоалкил, фенил и (С 1-С 3)алкилфенил необязательно замещены 1, 2 или 3 заместителями, которые независимо выбирают из группы, включающей галоген, (С 1-С 6)алкил, гидрокси, циано, О(С 1-С 6)алкил, -NH2, -NH(C1-C4)алкил, -NС 1-С 4)алкил)2, -CO-NH2, -CO-NH(C1-C4)алкил, -СО-NС 1 С 4)алкил)2, -СООН и -СОО(С 1-С 6)алкил, илиRN2 и RN1 вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членный моноциклический гетероцикл, который является насыщенным или ненасыщенным, необязательно содержащим от 1 до 3 дополнительных гетероатомов, которые независимо выбирают из N, S и О, и необязательно замещены 1, 2 или 3 заместителями, которые независимо выбирают из группы, включающей галоген,(С 1-С 6)алкил, гидрокси, циано, О-(С 1-С 6)алкил, -NH2, -NH(С 1-С 4)алкил, -NС 1-С 4)алкил)2, -CO-NH2, -СОNH(С 1-С 4)алкил, -СО-NС 1-С 4)алкил)2, -СООН и -СОО(С 1-С 6)алкил. Группа RC предпочтительно означает гидрокси, NHSO2-метил, NHSO2-этил, NHSO2-(1-метил)этил,NHSO2-пропил, NHSO2-циклопропил, NHSO2-CH2-циклопропил, NHSO2-циклобутил, NHSO2-цикло- 11009295 пентил или NHSO2-фенил. Более предпочтительно RC означает гидрокси или NHSO2-циклопропил. В наиболее предпочтительном варианте осуществления настоящего изобретения группа RC означает гидрокси. В другом наиболее предпочтительном варианте группа RC означает NHSO2-циклопропил. В самом предпочтительном варианте группа RC означает NHSO2N(CH3)2. В объем настоящего изобретения включены также соединения формулы (I), гдеB означает (С 1-С 10)алкил, (С 3-С 7)циклоалкил или (С 1-С 4)алкил(С 3-С 7)циклоалкил,а) причем указанные циклоалкил и алкилциклоалкил моно-, ди- или тризамещены группой (С 1 С 3)алкил, и б) причем указанные алкил, циклоалкил и алкилциклоалкил моно- или дизамещены заместителями,которые выбирают из группы, включающей гидрокси и О-(С 1-4)алкил, и в) причем все указанные алкильные группы моно-, ди- или тризамещены атомами галогена, и г) причем все указанные циклоалкильные группы означают 4-, 5-, 6- или 7-членное кольцо, в котором необязательно одна (в случае 4-, 5-, 6- или 7-членного кольца) или две (в случае 5-, 6-, 7-членного кольца) -СН 2-группы не связаны напрямую друг с другом, и замещены группой -О- таким образом, что атом О соединен с группой X по крайней мере через два атома углерода;R3 означает (С 2-С 8)алкил, (С 3-С 7)циклоалкил или (С 1-С 3)алкил(С 3-С 7)циклоалкил, где указанные циклоалкильные группы моно-, ди- или тризамещены группой (С 1-С 4)алкил;L1 , L2 каждый независимо означает галоген, (С 1-С 4)алкил, -О-(С 1-С 4)алкил или -S-(С 1-С 4)алкил (в любом окисленном состоянии, таком как SO или SO2); a L1 или L2 (но не оба одновременно) означают Н; илиL0 и L2 ковалентно соединены и вместе с двумя атомами углерода, к которым они присоединены,образуют 5- или 6-членное карбоциклическое кольцо, в котором одна или две -СН 2-группы напрямую не связаны друг с другом и каждая независимо заменена на группы -О- или NRa, где Ra означает Н или (С 1 С 4)алкил, причем указанное карбо- или гетероциклическое кольцо необязательно моно- или дизамещено группой (С 1-С 4)алкил;R2 означает R20, -NR22COR20, -NR22COOR20, -NR22R21 и -NR22CONR21R23, где R20 выбирают из группы, включающей (С 1-С 8)алкил, (С 3-С 7)циклоалкил и (С 1-С 4)алкил(С 3-С 7)циклоалкил, причем указанные циклоалкильная и алкилциклоалкильная группы моно-, ди- или тризамещены группой (С 1-С 3)алкил,R21 означает Н или имеет одно из значений группы R20, как определено выше,R22 и R23 независимо выбирают из группы, включающей Н и метил,R1 означает этил или винил;RC означает гидрокси или NHSO2RS, где RS означает (С 1-С 6)алкил, (С 3-С 7)циклоалкил, (С 1 С 6)алкил(С 3-С 7)циклоалкил, фенил, нафтил, пиридинил, (С 1-С 4)алкилфенил, (С 1-С 4)алкилнафтил или (С 1 С 4)алкилпиридинил; причем все указанные заместители необязательно моно-, ди- или тризамещены заместителями, которые выбирают из группы, включающей галоген, гидрокси, циано, (С 1-С 4)алкил, О-(С 1 С 6)алкил, -CO-NH2, -СО-МН(С 1-С 4)алкил, -СО-NС 1-С 4)алкил)2, -NH2, -NH(С 1-С 4)алкил и -NС 1 С 4)алкил)2, и каждый из которых необязательно монозамещен нитрогруппой, или RS выбирают из группы, включающей -NH(С 1-С 6)алкил, NС 1-С 6)алкил)2, -Het, или или их фармацевтически приемлемые соли или эфиры. ПредпочтительноB означает (С 2-С 8)алкил, (С 3-С 7)циклоалкил или (С 1-С 3)алкил(С 3-С 7)циклоалкил,а) где указанные алкил, циклоалкил и алкилциклоалкил моно, ди- или тризамещены группой (С 1 С 3)алкил, и б) где указанные алкил, циклоалкил и алкилциклоалкил моно- или дизамещены заместителями, которые выбирают из группы, включающей гидрокси и О-(С 1-С 4)алкил, и в) где каждая из указанных алкильных групп моно-, ди- или тризамещена атомами фтора или монозамещена атомами хлора или брома, и г) где каждая из указанных циклоалкильных групп означает 5-, 6- или 7-членное кольцо, в котором одна или две -СН 2-группы не связаны напрямую друг с другом, и заменены на группу -О- таким образом,что атом О соединен с группой X по крайней мере через два атома углерода;R3 означает (С 2-С 6)алкил или (С 3-С 7)циклоалкил, каждый из которых необязательно замещен 1-3R20 выбирают из группы, включающей (С 1-С 8)алкил, (С 3-С 7)циклоалкил, (С 1-С 3)алкил(С 3 С 7)циклоалкил, где каждая из указанных циклоалкильных и алкилциклоалкильных групп моно-, ди- или тризамещена группой (С 1-С 3)алкил, иR23 означает Н или метил,R1 означает этил или винил; аNHSO2 фенил. Более предпочтительно B выбирают из группы, включающей этил, н-пропил, трет-бутил, 2 метилпропил,1,2-диметилпропил,1,2,2-триметилпропил,2-фторэтил,3-фторпропил,3,3,3 трифторпропил, циклопропил, циклобутил, циклопентил, циклогексил, 1-метилциклопентил и 1 метилциклогексил, группу, которую выбирают из следующих формул:R2 означает -NHCOR20, -NHCOOR20 или -NHR21, где R20 и R21 независимо выбирают из группы,включающей метил, этил, н-пропил, изопропил, 2,2-диметилпропил, циклопентил и циклометилпентил,R1 означает винил, a RC означает гидрокси или NHSO2-циклопропил. Наиболее предпочтительно B выбирают из группы, включающей этил, н-пропил, трет-бутил, циклопентил, 1-метилциклопентил, 2-фторэтил, 3-фторпропил, R выбирают из групп 1,1-диметилэтил и циклогексил, L0 означает Н или -ОСН 3; L1 означает СН 3, -Cl или -Br, L2 означает Н, a RC означает гидрокси. Примеры предпочтительных вариантов осуществления настоящего изобретения включают отдельные соединения, перечисленные в табл. 1, 2, 3, 4, 5 и 6. В другом варианте фармацевтическая композиция по настоящему изобретению дополнительно включает по крайней мере один другой противовирусный агент в отношении ВГС. Примеры противовирусных агентов в отношении ВГС включают - (альфа), - (бета), - (дельта), - (гамма), - (омега) или- (тау) интерфероны, ПЭГилированный -интерферон, рибавирин или амантадин. В одном варианте фармацевтическая композиция по настоящему изобретению дополнительно включает по крайней мере один другой ингибитор протеазы NS3 ВГС. В другом варианте фармацевтическая композиция по настоящему изобретению дополнительно включает по крайней мере один ингибитор полимеразы ВГС. В еще одном варианте фармацевтическая композиция по настоящему изобретению дополнительно включает по крайней мере один ингибитор других мишеней жизненного цикла ВГС, которые включют,без ограничения перечисленным, геликазу, протеазу NS2/3 или внутренний входной участок рибосомы(IRES). Фармацевтическую композицию по настоящему изобретению вводят пероральным, парентеральным способом или с использованием имплантированного резервуара. Предпочтительным является пероральное введение или введение с использованием инъекций. Фармацевтическая композиция по настоящему изобретению содержит любые стандартные нетоксичные фармацевтически приемлемые носители,адьюванты или наполнители. В некоторых случаях pH состава доводят с использованием фармацевтически приемлемых кислот, оснований или буферных растворов с целью повышения устойчивости соединения или формы, в которой его вводят. Термин парентеральное введение, использованный в данном контексте, включает подкожное,внутрикожное, внутривенное, внутримышечное, интраартикулярное, внутрисуставное, внутриподложечное, подоболочечное введение и введение в очаг патологического изменения или вливание. Фармацевтические композиции получают в виде стерильных растворов для инъекций, например, в виде стерильных водных растворов или масляных суспензий для инъекций. Такие суспензии получают- 13009295 по стандартным методикам, известным в данной области техники, с использованием пригодных диспергирующих или смачивающих агентов (таких как, например, Твин-80) и суспендирующих агентов. Фармацевтическую композицию по настоящему изобретению вводят перорально в любой пероральной пригодной лекарственной форме, включая, без ограничения перечисленным, капсулы, таблетки,водные суспензии и растворы. В случае таблеток для перорального введения используют стандартные носители, включая лактозу и кукурузный крахмал. Используют также замасливатели, такие как стеарат магния. В случае капсул для перорального введения используемые разбавители включают лактозу и сухой кукурузный крахмал. При пероральном введении водных суспензий активный компонент смешивают с эмульгирующими и суспендирующими агентами. При необходимости добавляют соответствующие подсластители, и/или ароматизаторы, и/или красители. Другие пригодные наполнители или носители для указанных выше составов композиций описаны в известных фармацевтических изданиях, например, в книге "Remington's Pharmaceutical Sciences", TheScience and Practice of Pharmacy, 19 Ed. Mack Publishing Company, Easton, Penn., (1995). При монотерапии для профилактики и лечения заболеваний, вызванных ВГС, используют дозы в интервале от приблизительно 0,01 до приблизительно 100 мг/кг массы тела в сутки, предпочтительно от приблизительно 0,1 до приблизительно 50 мг/кг массы тела в сутки ингибитора протеазы, описанного в данном контексте. Прежде всего, фармацевтическую композицию по настоящему изобретению вводят от приблизительно 1 до приблизительно 5 раз в сутки или, в другом варианте, в виде непрерывного вливания. Такое введение используют при лечении хронических или острых заболеваний. Количество активного компонента, который смешивают с материалами носителя с образованием единой лекарственной формы, зависит от состояния организма хозяина, который нуждается в лечении и, прежде всего, от конкретного способа введения. Стандартная лекарственная форма содержит от приблизительно 5 до приблизительно 95% (мас./мас.) активного соединения. Такие препараты предпочтительно содержат от приблизительно 20 до приблизительно 80% активного соединения. Специалистам в данной области техники представляется очевидным, что при необходимости можно использовать более низкие или более высокие дозы по сравнению с указанными выше. Дозировка и курс лечения для каждого конкретного пациента зависят от ряда факторов, включая активность используемого соединения, возраст, массу тела, общее состояние здоровья, пол, режим питания, время введения, скорость выведения, комбинацию лекарственных средств, тяжесть и развитие инфекции, чувствительность пациента к инфекции и квалификацию лечащего врача. В основном, лечение начинают с небольших доз,значительно меньших по сравнению с оптимальной дозой пептида. Затем дозу повышают с небольшим приростом до достижения оптимального эффекта в конкретной ситуации. В основном, соединения наиболее предпочтительно вводят в концентрации, при которой наблюдается эффективное противовирусное действие без любых отрицательных или опасных побочных явлений. Если композиция по настоящему изобретению включает комбинацию соединения формулы I и одного или более дополнительных терапевтических или профилактических агентов, то доза как соединения, так и дополнительного агента находится в интервале от приблизительно 10 до 100% и более предпочтительно от приблизительно 10 до 80% от дозы, которую обычно вводят при монотерапии. Если указанные соединения, включая их фармацевтически приемлемые соли и эфиры, включены в композицию в смеси с фармацевтически приемлемым носителем, то полученную композицию можно вводить in vivo млекопитающим, таким как человек, для ингибирования протеазы NS3 ВГС или лечения или профилактики вирусной инфекции ВГС. Такое лечение проводят также с использованием соединения по настоящему изобретению в комбинации с другим противовирусным агентом. Предпочтительные другие противовирусные агенты описаны в разделе Определения и в разделе предпочтительных фармацевтических композиций по настоящему изобретению и включают, без ограничения перечисленным,-, -, -, -, - или -интерфероны, рибавирин, амантадин, другие ингибиторы протеазы NS3 ВГС, ингибиторы полимеразы ВГС, ингибиторы других мишеней жизненного цикла ВГС, включая, без ограничения перечисленным, геликазу, протеазу NS2/3 или внутренний входной участок рибосомы (IRES), или их комбинации. Дополнительные агенты смешивают с соединениями по настоящему изобретению и получают единую лекарственную форму. В другом варианте такие дополнительные агенты вводят млекопитающим в отдельности в составе многокомпонентной лекарственной формы. Соответственно в другом варианте осуществления настоящего изобретения предлагается способ ингибирования активности протеазы NS3 ВГС у млекопитающих при введения соединения формулы I,включая его фармацевтически приемлемую соль или эфир. В предпочтительном варианте данный способ используют для снижения активности протеазы NS3 ВГС в организме инфицированного млекопитающего. Как описано выше, комбинационная терапия включает совместное введение соединения формулы(I) или его фармацевтически приемлемой соли или эфира в смеси по крайней мере с одним дополнительным противовирусным агентом. Предпочтительные противовирусные агенты описаны в данном контексте и примеры таких агентов перечислены в разделе Определения. Такие дополнительные агенты смешивают с соединениями по настоящему изобретению и получают единую лекарственую форму. В другом варианте такие дополнительные агенты вводят пациенту в отдельности в виде части многокомпо- 14009295 нентной лекарственной формы, например, при использовании набора лекарственных форм. Такие дополнительные агенты вводят пациенту перед, одновременно или после введения соединения формулы (I) или его фармацевтически приемлемой соли или эфира. Соединение формулы (I) или его фармацевтически приемлемая соль или эфир, описанные в данном контексте, используют также в качестве лабораторного реагента. Более того, соединение по настоящему изобретению, включая его фармацевтически приемлемые соль или эфир, используют также для обработки или предотвращения от вирусного загрязнения различных материалов и, следовательно, для снижения риска вирусной инфекции лабораторного или медицинского персонала или пациентов, контактирующих с такими материалами (например, кровь, ткани, хирургические инструменты и материалы, лабораторные инструменты и материалы, а также устройства и материалы для отбора крови). Соединение формулы (I), включая его фармацевтически приемлемую соль или эфир, описанные в данном контексте, используют также в качестве реагента для исследований. Соединение формулы (I),включая его фармацевтически приемлемую соль или эфир, описанные в данном контексте, используют также в качестве положительного контроля для подтверждения результатов анализа с использованием заменителей клеток или анализа вирусной репликации in vitro или in vivo. Методы. Некоторые способы синтеза соединений формулы I с использованием различных производных хинолина описаны в патенте WO 00/09558. Промежуточное производное дипептида 15 (схема 2) и 2 карбометокси-4-гидрокси-7-метоксихинолин 9 (схема 1) синтезировали по общим методикам, описанным в патенте WO 00/09543. Соединение формулы I, где RC означает NHSO2RS, как определено выше, получали при конденсации соответствующей кислоты формулы I (например, R означает гидрокси) с соответствующим сульфонамидом формулы RSSO2NH2 в присутствии конденсирующего агента в стандартных условиях. Несмотря на то, что можно использовать различные стандартные конденсирующие агенты, наиболее пригодными являются TBTU и HATU. Сульфонамиды являются коммерческими продуктами или их получают по стандартным методикам. На показанных ниже схемах представлено два стандартных способа с использованием стандартных методик для получения соединений формулы 1, где Синтез дипептида 3 проводили при конденсации остатка Р 1 с соответственно защищенным трансгидроксипролином в стандартных условиях. Стереохимическую конформацию гидроксильной группы изменяли в условиях реакции Мицунобу в присутствии паранитробензойной кислоты. При конденсации дипептида с остатком Р 3 (полученного с использованием стандартных методик и описанного в разделе Примеры) получали трипептид 6. Введение хинолинового остатка по гидроксильной группе трипептида 7 с одновременным стереохимическим обращением можно проводить в условиях реакции Мицунобу или при превращении свободной гидроксильной группы в эффективную уходящую группу (такую как парабромбензолсульфогруппа, Brs) и замене ее на гидроксильное производное хинолина 9. При синтезе 2-(2-амино-4-тиазолил)производных используют хинолин, содержащий 2-карбометоксигруппу, как показано в соединении 9. Превращение- 15009295 карбоксилатной группы в производное аминотиазола проводили по стандартной методике, которая описана в заявках WO 00/09543 и WO 00/09598. И наконец, C-концевой эфир гидролизовали в основноводных условиях. Схема 2 На схеме 2 показана другая последовательность реакций для получения соединений формулы I. В данном случае хинолиновый остаток вводили в дипептид аналогично тому, как показано на схеме 1. Наконец, остаток Р 3 конденсировали с дипептидом 21 в стандартных условиях. Превращение полученного трипептида в конечное соединение проводили, как показано на схеме 1. Синтез фрагментов Р 1. Фрагменты Р 1 соединений формулы (I) получали по методикам, описанным в заявках WO 00/59929,поданной 12 октября 2000 г., или WO 00/09543, поданной 24 февраля 2000 г. Прежде всего методики описаны на стр. 33-35, пример 1, заявки WO 00/59929 и на стр. 56-69, примеры 9-20 заявки WO 00/09543,в которых описано получение фрагментов 1-аминоциклопропанкарбоновой кислоты Р 1. Синтез фрагментов Р 2. Соединения формулы 9 синтезировали из коммерческих материалов по методикам, описанным в заявках WO 00/59929, WO 00/09543, WO 00/09558 и патенте USA 6323180 В 1. Примеры синтеза различных производных 2-карбометокси-4-гидроксихинолина описаны ниже. Синтез производных 2-карбометокси-4-гидроксихинолина проводили с использованием соответствующих анилинов по методике, описанной в работе Unangst Р.С, Connor D.T., J. Heterocyc. Chem. 29, 5,1097-1100 (1992). Методика показана ниже на схеме 3. Схема 3 Соответственно замещенные анилины в положениях 2, 3 и/или 4 конденсировали с диметилацетилендикарбоксилатом и полученный енамин нагревали при высокой температуре, при которой происходит циклизация. Соответствующие анилины являются коммерческими продуктами или их получают по стандартным методикам. Например, нитропроизводное является коммерческим продуктом, который затем превращают в соответствующий амин в присутствии восстановителя. Если карбоновая кислота также является коммерческим продуктом, то ее превращают в соответсвующий амин в условиях перегруппировки Курциуса. Более подробно настоящее изобретение описано в следующих примерах, которые не ограничивают его объем, как определено в пунктах формулы изобретения. Другие специальные методы синтеза или разделения соединений по настоящему изобретению, описаны в заявках WO 00/09543, WO 00/09558 иWO 00/59929, а также в одновременно поданной заявке 09/368670, которые включены в настоящее изобретение в виде ссылок. Примеры Если не указано иное, процентная концентрация растворов означает отношение массы к объему,объемная концентрация растворов означает отношение объема к объему. Спектры ядерного магнитного резонанса (ЯМР) получены на спектрометре фирмы Bruker 400 МГц, величина химического сдвига (5) выражена в част./млн. Экспресс-хроматографию проводили на силикагеле (SiO2) по методу Стилла (W.C.Still и др., J. Org. Chem., 43, 2923, (1978. Сокращения, использованные в примерах: ВОС или Boc означает трет-бутилоксикарбонил, DBU означает 1,8-диазабицикло[5.4.0]ундец-7-ен,ДХМ означает дихлорметан, DIAD означает диизопропилазодикарбоксилат, DIPEA означает диизопропилэтиламин, ДМФА означает N,N-диметилформамид, DMAP означает 4-(диметиламино)пиридин,ДМСО означает диметилсульфоксид, EtOAc означает этилацетат, HATU означает гексафторфосфат О-(7 азабензотриазол-1-ил)-1,1,3,3-тетраметилурония, ЖХВР означает жидкостную хроматографию высокого разрешения, МС означает масс-спектр (получен методом ионизации лазерной десорбции в матрице с времяпролетным масс-анализатором (MALDI-TOF), FAB означает бомбардировку быстрыми атомами),Me означает метил, MeOH означает метанол, Ph означает фенил, КТ означает комнатную температуру(от 18 до 22 С), трет-бутил означает 1,1-диметилэтил, Tbg означает трет-бутилглицин (трет-лейцин),TBTU означает тетрафторборат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, ТЭА означает триэтиламин, ТФУ означает трифторуксусную кислоту, а ТГФ означает тетрагидрофуран. Пример 1. Получение производных P3. Получение карбамата 4 а. ТГФ (350 мл) добавляли в колбу, содержащую циклопентиловый, 2,5-диоксопирролидин-1-иловый диэфир карбоновой кислоты (9,00 г, 39,6 ммоль) и трет-бутилглицин (6,24 г, 47,5 ммоль), при этом получали суспензию. Затем при интенсивном перемешивании добавляли дистиллированную воду (100 мл),при этом в смеси оставалось небольшое количество твердого вещества. Затем добавляли триэтиламин(16,6 мл, 119 ммоль), при этом получали гомогенный раствор, который перемешивали при КТ. Через 2,5 ч ТГФ упаривали, а водный остаток разбавляли водой (100 мл). Реакционную смесь подщелачивали доpH 10 добавлением 1 н. NaOH (25 мл). Полученный раствор промывали EtOAc (2 раза по 200 мл), водную фазу подкисляли до pH2 добавлением 1 н. HCl (приблизительно 70 мл). Мутный раствор экстрагировали EtOAc (200+150 мл). Экстракт сушили (MgSO4) и упаривали, при этом получали карбамат 4 а в виде твердого вещества белого цвета (8,68 г). Получение мочевины 5 а. Раствор гидрохлорида бензилового эфира трет-бутилглицина (2,55 г, 9,89 ммоль) в ТГФ (20 мл) и пиридине (2,0 мл, 24,73 ммоль) охлаждали до 0 С. Затем к холодному раствору по каплям добавляли фениловый эфир хлормуравьиной кислоты (1,30 мл, 10,19 ммоль). Полученную суспензию перемешивали в течение 5 мин при 0 С, затем при КТ в течение 1,5 ч. Реакционную смесь разбавляли EtOAc, промывали 10% лимонной кислотой (2 раза), водой (2 раза), насыщенным раствором NaHCO3 (2 раза), водой (2 раза) и солевым раствором (1 раз), сушили (MgSO4), фильтровали и упаривали, при этом получали неочищенный продукт в виде почти бесцветного масла (3,73 г, 100%, приблизительно 9,89 ммоль). Неочищенный продукт (1,01 г, 2,97 ммоль) растворяли в ДМСО (6,5 мл) и добавляли по каплям циклопентиламин. Реакционную смесь перемешивали при КТ в течение 45 мин, затем разбавляли EtOAc. Органическую фазу промывали 10% лимонной кислотой (2 раза), водой (2 раза), насыщенным раствором NaHCO3 (2 раза),водой (2 раза) и солевым раствором (1 раз), сушили (MgSO4), фильтровали и упаривали, при этом получали неочищенное Tbg-OBn-производное циклопентилмочевины в виде почти бесцветного масла. Неочищенный продукт очищали методом экспресс-хроматографии на колонке с силикагелем, менее полярные примеси удаляли смесью гексан/EtOAc (9:1), очищенный продукт элюировали смесью гексан/EtOAc(7:3), при этом получали вязкое бесцветное масло (936 мг, 95%). Защитные бензиловые группы в полученном продукте (936 мг, 2,82 ммоль) удаляли при гидрировании водородом, подаваемым из баллона,при КТ в абсолютном этаноле (15 мл) при перемешивании в присутствии 10% Pd/C (93,6 мг) в течение 5,5 ч. Реакционную смесь фильтровали через фильтр с диаметром пор 0,45 мкм и упаривали досуха, при этом получали мочевину 5 а в виде твердого вещества белого цвета (669 мг, 98%). Н ЯМР (400 МГц, ДМСО-d6):12,39 (s, 1 Н), 6,09 (d, J=7,4 Гц, 1 Н), 5,93 (d, J=9,4 Гц, 1 Н), 3,90 (d,J=9,4 Гц, 1 Н), 3,87-3,77 (m, 1 Н), 1,84-1,72 (m, 2 Н), 1,63-1,42 (m, 4 Н), 1,30-1,19 (m, 2 Н), 0,89 (s, 9H). МС (ES, электроспрей): 241,0 (М-Н)- 243,0, (М+Н)+. По данным обращенно-фазовой ЖХВР (элюент: 0,06% ТФУ, CH3CN/H2O) гомогенность продукта составляла 99%. Пример 2. Получение производных тиомочевины 8. Получение тиомочевины 8 а. К раствору трет-бутилизотиоцианата (5,0 мл, 39,4 ммоля) в ДХМ (200 мл) добавляли циклопентиламин (4,67 мл, 47,3 ммоль), DIPEA и реакционную смесь перемешивали при КТ в течение 2 ч. Смесь разбавляли EtOAc, промывали 10% водным раствором лимонной кислоты (2 раза), насыщенным раствором NaHCO3 (2 раза), Н 2 О (2 раза) и солевым раствором (1 раз). Органический слой сушили над безводным MgSO4, фильтровали и упаривали, при этом получали N-трет-бутил-N'-циклопентилтиомочевину в виде твердого вещества белого цвета (3,70 г, выход 47%). N-трет-Бутил-N'-циклопентилтиомочевину(3,70 г) растворяли в конц. HCl (46 мл). Раствор темно-желтого цвета нагревали при слабом кипении растворителя с обратным холодильником. Через 40 мин реакционную смесь охлаждали до КТ, затем охлаждали на ледяной бане и подщелачивали до pH 9,5 добавлением твердого NaHCO3 и насыщенного водного раствора. Продукт экстрагировали EtOAc (3 раза). Слои EtOAc объединяли, промывали Н 2 О (2 раза) и солевым раствором (1 раз). Органический слой сушили (MgSO4), фильтровали и концентрировали, при этом получали твердое вещество бежевого цвета (2,46 г, неочищенный продукт). Неочищенный продукт растирали в смеси гексан/EtOAc (95:5) и фильтровали, при этом получали N-циклопентилтиомочевину 8 а в виде твердого вещества белого цвета (2,38 г, выход 90%). 1(8,88 г, 66 ммоль). Смесь нагревали с обратным холодильником в течение 14 ч, при этом получали раствор желтого цвета. Смесь упаривали досуха, остаток распределяли между EtOAc и насыщенным раствором NaHCO3. Органическую фазу желтого цвета сушили над MgSO4, фильтровали и концентрировали, при этом получали твердое вещество желтого цвета. Твердое вещество растворяли в минимальном количестве EtOAc и растирали в гексане, при этом получали соединение 8b в виде твердого вещества белого цвета (8,52 г, 75%). МС (ES): 173 (М-Н)-, 175 (М+Н)+. По данным обращенно-фазовой ЖХВР (элюент: 0,06% ТФУ,CH3CN/H2O) гомогенность продукта составляла 99%. Получение тиомочевины 8 с. Тиомочевину 8 с получали по методике, описанной выше, но при замене трет-бутилацетилхлорида на коммерческий циклопентилацетилхлорид. Получение производных 2-карбометокси-4-гидроксихинолина. Пример 3 А. Получение 2-карбометокси-4-гидрокси-7-метокси-8-метилхинолина (А 5). Стадия А.(подается из баллона) при атмосферном давлении и комнатной температуре в течение 19 ч. Реакционную смесь фильтровали через слой целита, промывали и упаривали досуха, при этом получали 2-метил-3- 18009295 метоксианилин A2 в виде масла розовато-лилового цвета (4,1 г, 29,81 ммоль, выход 98%). МС: 137 (МН)+. По данным обращенно-фазовой ЖХВР (=220 нм, элюент: 0,06% ТФУ, CH3CN/H2O) гомогенность продукта составляла 99%. Стадия B. Диметилацетилендикарбоксилат A3 (3,6 мл, 29,28 ммоль) добавляли по каплям к раствору 2-метил 3-метоксианилина A2 (3,95 г, 28,79 ммоль) в МеОН (100 мл, экзотермическая реакция). Смесь нагревали при слабом кипении растворителя с обратным холодильником в течение 5 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке с силикагелем методом экспрессхроматографии (элюент: гексан/EtOAc, 95:5), фракции, содержащие чистый продукт, упаривали, при этом получали соединение A4 (6,5 г, 23,27 ммоль, выход 81%). По данным обращенно-фазовой ЖХВР (=220 нм, элюент: 0,06% ТФУ, CH3CN/H2O) гомогенность продукта составляла 95%. Стадия C. Диэфир A4 (6,5 г, 23,27 ммоль) растворяли в дифениловом эфире (12 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до температуры 350-400 С. После того как температура реакционной смеси достигала 240 С (при 230-240 С наблюдалось выделение MeOH) смесь нагревали еще в течение 6 мин (реакцию останавливали при температуре 262 С), баню удаляли, а смесь охлаждали до комнатной температуры. Твердое вещество, полученное после охлаждения, разбавляли эфиром,фильтровали и сушили, при этом получали твердое вещество желто-коричневого цвета (3,48 г, неочищенный продукт). Неочищенный продукт очищали хроматографией на колонке с силикагелем. Примеси удаляли смесью гексан/EtOAc (5:5), продукт элюировали смесью гексан/EtOAc (2:8), а затем 100%EtOAc, после упаривания получали соединение A5 в виде твердого вещества бледно-желтого цвета (2,1 г,выход 37%). МС: 246 (М+Н)+, 248,1 (М-Н)-. По данным обращенно-фазовой ЖХВР (=220 нм, элюент: 0,06% ТФУ, CH3CN/H2O) гомогенность продукта составляла 99%. Пример 3B. Получение 2-карбометокси-8-бром-4-гидрокси-7-метоксихинолина (B6). Стадия A. 2-Амино-3-нитрофенол B1 (5 г, 32,4 ммоль) растворяли в Н 2 О (29,5 мл) и 1,4-диоксане (14,7 мл). Смесь нагревали с обратным холодильником и добавляли по каплям в течение 20 мин бромистоводородную кислоту (48%, 16,7 мл, 147 ммоль). Затем смесь нагревали с обратным холодильником в течение еще 15 мин. Реакционную смесь охлаждали до 0 С (ледяная баня), затем добавляли в течение 30 мин нитрит натрия (2,23 г, 32,3 ммоль) в Н 2 О (20 мл) и перемешивали в течение еще 15 мин при 0 С, затем смесь переносили в капельную воронку с рубашкой (0 С) и добавляли при 0 С по каплям в перемешиваемую смесь Cu(I)Br (5,34 г, 37,2 ммоль) в Н 2 О (29,5 мл) и HBr (48%, 16,7 мл, 147 ммоль). Реакционную смесь перемешивали в течение 15 мин при 0 С, нагревали до 60 С и перемешивали в течение еще 15- 19009295 мин, охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь переливали в делительную воронку и экстрагировали эфиром (3 раза по 150 мл). Органические слои объединяли, промывали солевым раствором (1 раз), сушили (Na2SO4), фильтровали и концентрировали, при этом получали неочищенный продукт (7,99 г) в виде масла красно-коричневого цвета. Неочищенный продукт очищали на колонке методом экспресс-хроматографии (ультрачистый силикагель 1:25, 230-400 меш, 4060 мм, 60, элюент: CH2Cl2), при этом получали чистый 2-бром-3-нитрофенол B2 (45%, 3,16 г) в виде твердого вещества оранжево-коричневого цвета. МС: 217,8 (МН)-. По данным ЖХВР (-220 нм, элюент: ТФУ) гомогенность продукта составляла 97%. Стадия B. Нитрофенол B2 (3,1 г, 14,2 ммоль) растворяли в ДМФА (20 мл) и к раствору добавляли измельченный карбонат цезия (5,58 г, 17,1 ммоль) и MeI (2,6 мл, 42,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. ДМФА упаривали, остаток переносили в эфир (1 раз, 200 мл), промывали водой (1 раз, 200 мл), солевым раствором (4 раза по 100 мл), сушили (MgSO4), фильтровали и упаривали,при этом получали неочищенный 2-бром-3-нитроанизол B3 (94%, 3,1 г) в виде твердого вещества оранжевого цвета. МС: 234 (М+2 Н)+, по данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 98%. Стадия C. 2-Бром-3-нитроанизол B3 (1,00 г, 4,31 ммоль) растворяли в ледяной уксусной кислоте (11,0 мл) и этаноле (11,0 мл). К полученному раствору добавляли порошкообразное железо (0,98 г, 17,5 ммоль). Смесь перемешивали при нагревании с обратным холодильником в течение 3,5 ч. Реакционную смесь разбавляли водой (35 мл), нейтрализовали добавлением твердого Na2CO3, продукт экстрагировалиCH2Cl2 (3 раза по 50 мл). Экстракты сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали неочищенный 2-бром-3-метоксианилин B4 (91%, 0,79 г) в виде масла бледно-желтого цвета. МС: 201,8 (МН)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 95%. Стадия D. К раствору 2-бром-3-метоксианилина B4 (0,79 г, 3,9 ммоль) в МеОН (7,6 мл) добавляли по каплям диметилацетилендикарбоксилат A3 (0,53 мл, 4,3 ммоль) при 0 С (осторожно, экзотермическая реакция). Раствор нагревали с обратным холодильником в течение ночи. MeOH упаривали, а неочищенный продукт сушили в высоком вакууме, при этом получали смолу красного цвета. Продукт очищали на колонке методом экспресс-хроматографии (ультрачистый силикагель 1:30, 230-400 меш, 40-60 мм, 60, элюент: гексан/EtOAc, 4:1), при этом получали аддукт B5 (86%, 1,16 г) в виде твердого вещества бледно-желтого цвета. МС: 344,0 (МН)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 72%. Стадия E. В песчаную баню, предварительно нагретую до температуры приблизительно 440 С (температура бани), помещали диэфир В 5 (1,1 г, 3,16 ммоль) в дифениловом эфире (3,6 мл). Реакционную смесь перемешивали при температуре от 230 до 245 С (температура реакционной смеси, при приблизительно 215 С наблюдалось выделение MeOH) в течение 7 мин, а затем охлаждали до комнатной температуры. При охлаждении раствора продукт кристаллизовался из реакционной смеси. Полученное твердое вещество коричневого цвета фильтровали, промывали эфиром и сушили в высоком вакууме, при этом получали неочищенный бромхинолин В 6 (74%, 0,74 г) в виде твердого вещества коричневого цвета. По данным ЯМР полученный продукт содержал смесь таутомеров в соотношении приблизительно 1:1. ЯМР (ДМСО, 400 МГц, смесь таутомеров, 1:1), МС: 311,9 (МН)+. По данным ЖХВР (=220 нм,элюент: ТФУ) гомогенность продукта составляла 96%. Пример 3 С. Получение 2-карбометокси-8-хлор-4-гидрокси-7-метоксихинолина (С 6). Стадия A. 2-Амино-3-нитрофенол В 1 (5 г, 32,4 ммоль) растворяли в конц. HCl (75 мл) и 1,4-диоксане (14,7 мл). Смесь нагревали при 70 С до практически полного растворения твердых веществ. Реакционную смесь охлаждали до 0 С (ледяная баня) и к раствору коричневого цвета добавляли нитрит натрия (2,23 г,32,3 ммоль) в Н 2 О (5,4 мл) в течение 3 ч при температуре не выше 10 С, затем смесь перемешивали при 0 С в течение еще 15 мин. Полученное промежуточное производное диазония выливали в растворCu(I)Cl (3,8 г, 38,9 ммоль) в Н 2 О (18,5 мл) и конц. HCl (18,5 мл) при 0 С. Реакционную смесь перемешивали в течение 15 мин при 0 С, нагревали до 60 С и перемешивали в течение 15 мин. Затем реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь переносили в делительную воронку и экстрагировали эфиром (3 раза по 150 мл). Органические слои объединяли, промывали солевым раствором (1 раз), сушили (Na2SO4), фильтровали и концентрировали, при этом получали неочищенный продукт (5,83 г) в виде масла красно-коричневого цвета. Неочищенный продукт очищали на колонке методом экспресс-хроматографии (ультрачистый силикагель 1:25, 230-400 меш, 40-60 мм, 60 , элюент: гексан/EtOAc, 3:1), при этом получали чистый 2-хлор-3-нитрофенол C2(48%, 2,7 г) в виде твердого вещества оранжевого цвета. МС: 171,8 (МН)-. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 96%. Методика описана в работе Sandmeyer Reaction, J. Med. Chem, 25 (4), 446-451, (1982). Стадия B. Нитрофенол C2 (1,3 г, 7,49 ммоль) растворяли в ДМФА (10 мл) и к полученному раствору добавляли измельченный карбонат цезия (2,92 г, 8,96 ммоль), а затем MeI (1,4 мл, 22,5 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. ДМФА упаривали в вакууме, остаток переносили в эфир (150 мл), промывали водой (150 мл), солевым раствором (4 раза по 100 мл), затем сушили (MgSO4). Органическую фазу фильтровали и упаривали, при этом получали неочищенный 2-хлор-3-нитроанизол С 3 (98%, 1,38 г) в виде твердого вещества оранжевого цвета. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 93%. Стадия C. 2-Хлор-3-нитроанизол C3 (1,38 г, 7,36 ммоль) растворяли в смеси ледяная уксусная кислота (19 мл)/этанол (19 мл). К полученному раствору добавляли порошкообразное железо (1,64 г, 29,4 ммоль). Смесь перемешивали при нагревании с обратным холодильником в течение 3,5 ч. Реакционную смесь разбавляли водой (70 мл), нейтрализовали добавлением твердого Na2CO3 и продукт экстрагировалиCH2Cl2 (3 раза по 150 мл). Экстракты объединяли и промывали насыщенным солевым раствором, затем сушили (Na2SO4), фильтровали и концентрировали в вакууме, при этом получали неочищенный 2-хлор 3-метоксианилин C4 (100%, 1,2 г) в виде масла желтого цвета. Полученный продукт использовали на следующих стадиях без дополнительной очистки. МС: 157,9 (МН)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 86%. Стадия D. К раствору 2-хлор-3-метоксианилина С 4 (1,2 г, 7,61 ммоль) в MeOH (15 мл) добавляли по каплям диметилацетилендикарбоксилат A3 (1,0 мл, 8,4 ммоль) при 0 С (осторожно, экзотермическая реакция). Раствор нагревали с обратным холодильником в течение ночи. MeOH упаривали, неочищенный продукт сушили в высоком вакууме, при этом получали смолу красного цвета, которую очищали на колонке методом экспресс-хроматографии (ультрачистый силикагель 1:30, 230-400 меш, 40-60 мм, 60 , элюент: гексан/EtOAc, 4:1), при этом получали аддукт C5 (74%, 1,68 г) в виде твердого вещества желтого цвета. МС: 300 (МН)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 90%. Стадия E. В песчаную баню, предварительно нагретую до температуры приблизительно 440 С (температура бани), помещали диэфир С 5 (1,68 г, 5,6 ммоль) в дифениловом эфире (6,3 мл). Реакционную смесь пере- 21009295 мешивали при температуре от 230 до 245 С (температура реакционной смеси, при приблизительно 215 С наблюдалось выделение MeOH) в течение 7 мин, затем охлаждали до комнатной температуры. При охлаждении раствора продукт кристаллизовался из реакционной смеси. Полученное твердое вещество коричневатого цвета фильтровали, промывали эфиром и сушили в высоком вакууме, при этом получали хинолин C6 (83%, 1,25 г) в виде твердого вещества бежевого цвета. По данным ЯМР полученный продукт содержал смесь таутомеров (кето/фенольные формы, соотношение таутомеров приблизительно 1:1). МС: 267,9 (МН)+. По данным ЖХВР (-220 нм, элюент: ТФУ) гомогенность продукта составляла 92%. Пример 3D. Получение 2-карбометокси-8-фтор-4-гидрокси-7-метоксихинолина (D5). Стадия A. Раствор 2-фтор-3-метоксибензойной кислоты D1 (1,68 г, 9,87 ммоль) и DIPEA (2,07 мл, 11,85 ммоль, 1,2 экв.) в смеси толуола (8 мл) и трет-BuOH (8 мл) перемешивали с активированными молекулярными ситами (4 ) в течение 1 ч, затем добавляли дифенилфосфорилазид (DPPA, 2,55 мл, 11,85 ммоль) и полученную смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь фильтровали, фильтрат концентрировали в вакууме, остаток переносили в EtOAc (50 мл), промывали Н 2 О (2 раза по 30 мл) и солевым раствором (1 раз, 30 мл). Органическую фазу сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Неочищенный продукт D2 (2,38 г, 96%) использовали на следующей стадии без дополнительной очистки. По данным МС наблюдается отщепление Boc группы, МС: 141,9 М+Н)-Вос)+, 139,9 М-Н)-Вос)-. Стадия В. Соединение D2 (2,28 г, 9,45 ммоль) обрабатывали 4 н. раствором HCl в диоксане (Aldrich, 10 мл, 40 ммолей), по данным ЖХВР через 60 мин наблюдалось полное потребление исходного материала. Реакционную смесь концентрировали в вакууме, остаток растворяли в EtOAc и промывали водой, насыщенным раствором NaHCO3 (водный раствор) и насыщенным солевым раствором. Органическую фазу сушили (MgSO4), фильтровали и концентрировали, при этом получали 1,18 г (88%) соединения D3 в виде масла коричневого цвета, которое использовали на следующей стадии без дополнительной очистки. МС: 141,9 (М+Н)+, 139,9 (М-Н)-. Стадия С. Анилин D3 (1,18 г, 8,36 ммоль) смешивали с диметилацетилендикарбоксилатом A3 (1,45 мл, 10,0 ммоль) в метаноле (25 мл). Реакционную смесь нагревали с обратным холодильником в течение 2 ч, затем упаривали досуха. Неочищенный продукт очищали методом экспресс-хроматографии (элюент: гексан/EtOAc, 9:1), при этом получали аддукт Михаэля D4 в виде масла желтого цвета (1,27 г, 54%). Стадия D. Аддукт Михаэля D4 растворяли в теплом дифениловом эфире (6 мл) и помещали в песочную баню,предварительно нагретую до 350 С. Температуру реакционной смеси поддерживали на уровне 245 С в течение приблизительно 5 мин, при этом получали раствор коричневого цвета. После охлаждения до КТ из раствора выпадал осадок в виде требуемого 4-гидроксихинолина. Твердое вещество коричневого цвета фильтровали и промывали несколько раз диэтиловым эфиром. После высушивания получали хинолинD5 в виде твердого вещества коричневого цвета (0,51 г, 45%). МС: 252 (М+Н)+, 249,9 (М-Н)-. Смесь таутомеров (1:1). 1 Н ЯМР (ДМСО-d6, 400 МГц): 12,04 (s, 1H), 11,02 (s, 1H), 8,0 (d, 1H), 7,88 (d, 1H), 7,65 (m, 1 Н), 7,39 Стадия А. Амид Е 1 (5,0 г, 30,63 ммоля) растворяли в смеси уксусной кислоты (5 мл) и серной кислоты (10 мл),охлаждали до 0 С. Затем добавляли по каплям смесь азотной кислоты (70%, 3 мл) и серной кислоты (2 мл), раствор нагревали до КТ и перемешивали в течение 1 ч. Затем реакционную смесь выливали в ледяную крошку, холодный раствор фильтровали (после полного таяния льда), при этом получали требуемое соединение Е 2 (5,8 г, 91%), которое использовали на следующей стадии без дополнительной очистки. МС (ES+): 209,0, МС (ES-): 206,9. Методика описана в работе Giumanini A.G., Verardo G., Polana M., J. Prak. Chem., 181, (1988). Стадия В. Соединение E2 (5,8 г, 27,86 ммоль) обрабатывали 6 М раствором HCl (5 мл) в MeOH (10 мл) и нагревали с обратным холодильником в течение 48 ч, при этом получали требуемый продукт Е 3 (4,6 г,99%). По данным обращенно-фазовой ЖХВР наблюдалось полное потребление исходного материала(время удерживания (ву) (Е 2)=2,6 мин, ву (Е 3)=3,9 мин). Смесь концентрировали и использовали на следующей стадии без дополнительной очистки. Стадия С. Серную кислоту (18 мл) добавляли в раствор анилина Е 3 (4,20 г, 25,27 ммоль) в воде (36 мл) при 0 С, затем добавляли нитрит натрия (2,3 г, 33,33 ммоль в 6 мл воды). В отдельном сосуде готовили раствор серной кислоты (1,5 мл) в воде (14 мл). Полученный раствор нагревали с обратным холодильником,затем при постоянном кипении по каплям добавляли смесь исходных реагентов. После завершения добавления реагентов полученную смесь кипятили в течение еще 5 мин, затем выливали в смесь лед/карбонат натрия и охлаждали на ледяной бане. Продукт экстрагировали водным EtOAc и концентрировали, при этом получали соединение Е 4 в виде вязкой жидкости темно-коричневого цвета (2,00 г,47%), которую использовали на следующей стадии без дополнительной очистки. МС (ES-): 210,9. Стадия D.MeI (1,42 мл, 22,74 ммоль) добавляли при КТ в раствор фенола Е 4 (1,9 г, 11,37 ммоль) и карбоната калия (2 г) в ДМФА (25 мл). Смесь нагревали при 50 С в течение 2 ч, затем охлаждали до КТ, добавлялиEtOAc и промывали водой (3 раза), затем водный слой экстрагировали EtOAc. Органические слои объединяли и сушили, фильтровали и концентрировали, при этом получали требуемый метиловый эфир Е 5(s, 3 Н). Стадия Е. К раствору нитропроизводного Е 5 (2,0 г, 11,04 ммоль) в EtOH добавляли 10% Pd/C (200 мг) и переносили в аппарат Парра. Реакцию проводили в атмосфере Н 2 под давлением 40 фунтов/кв. дюйм в течение 2 ч. Раствор фильтровали через слой диоксида кремния/целит, промывали MeOH и концентрировали,при этом получали требуемый анилин Е 6 (1,5 г, 90%), который использовали без дополнительной очистки. Стадия F. Анилин Е 6 (1,9 г, 12,65 ммоль) добавляли в раствор диметилацетилендикарбоксилата A3 (2,32 мл,18,85 ммоль) в метаноле (3 мл). Реакционную смесь нагревали с обратным холодильником в течение 2 ч,затем упаривали досуха. Неочищенный продукт очищали методом экспресс-хроматографии (элюент: гексан/EtOAc, 9:1), при этом получали соединение Е 7 (аддукт Михаэля) в виде масла желтого цвета (2,8 г,- 23009295 76%).H ЯМР (CDCl3, 400 МГц) 9,48 (ушир. s, 1 Н), 6,89 (d, J=7,9 Гц, 1 Н), 6,47 (d, J=7,9 Гц, 1 Н), 5,35 (s,1H), 3,74 (s, 3H), 3,70 (s, 3H), 3,65 (s, 3H), 2,27 (s, 3H), 2,24 (s, 3 Н). Стадия G. Соединение Е 7 (аддукт Михаэля) растворяли в теплом дифениловом эфире (10 мл) и помещали в песочную баню, предварительно нагретую до 350 С. Температуру реакционной смеси поддерживали на уровне 245 С в течение приблизительно 5 мин, при этом получали раствор коричневого цвета. После охлаждения до КТ из раствора выпадал осадок в виде требуемого 4-гидроксихинолина. Твердое вещество коричневого цвета фильтровали и промывали несколько раз диэтиловым эфиром. После высушивания получали хинолин Е 8 в виде твердого вещества желто-коричневого цвета (1,10 г, 88%). 1 Н ЯМР (CHCl3, 400 МГц): 8,80 (ушир. s, 1H), 8,06 (s, 1H), 7,26 (s, 1H), 6,93 (s, 1H), 4,04 (s, 3 Н), 3,80 В суспензию 2-метил-5-нитроанизола F1 (1,54 г, 9,21 ммоль) в абсолютном этаноле (15 мл) добавляли 10% Pd/C (249 мг) в качестве катализатора. Суспензию гидрировали водородом, подаваемым из баллона, при атмосферном давлении и комнатной температуре в течение 6,5 ч. Реакционную смесь фильтровали через фильтр Millex с диаметром пор 0,45 мкм и упаривали досуха, при этом получали 4 метил-мета-анизидин F2 (1,22 г, 8,89 ммоль, выход 97%). Стадия В. Диметилацетилендикарбоксилат A3 (1,1 мл, 8,95 ммоль) добавляли по каплям в раствор 4-метилмета-анизидина F2 (1,22 г, 8,89 ммоль) в МеОН (20 мл). Осторожно, экзотермическая реакция. Смесь нагревали при слабом кипении растворителя с обратным холодильником в течение 4 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке с силикагелем методом экспрессхроматографии (элюент: гексан/EtOAc, 92,5:7,5), фракции, содержащие чистый продукт, упаривали, при этом получали аддукт, диэфир F3 (1,8 г, 6,44 ммоль, выход 73%). Стадия С. Диэфир F3 (1,8 г, 6,44 ммоль) растворяли в дифениловом эфире (5 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до 350-400 С. После того, как температура реакционной смеси достигала 240 С, смесь нагревали еще в течение 5 мин, затем баню удаляли, а смесь охлаждали до комнатной температуры в течение ночи. Твердое вещество, полученное после охлаждения, смешивали с эфиром, фильтровали и сушили, при этом получали неочищенный продукт (0,97 г), содержащий региоизомеры в приблизительно равном соотношении, в виде твердого вещества коричневого цвета. Неочищенный продукт растирали в МеОН и EtOAc, фильтровали и сушили, при этом получали требуемый региоизомер метилхинолина (продукт F4) в виде твердого вещества желтого цвета (245 мг, выход 15%). По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 90%. Пример 3G. Получение 2-карбометокси-4-гидрокси[1,3]диоксоло[4,5-h]хинолина (G5). Стадия А.- 24009295 К раствору 2,3-метилендиоксибензойной кислоты G1 (коммерческий препарат, 485 мг, 2,92 ммоль) в 1,4-диоксане (8,0 мл) и трет-бутаноле (2,5 мл), при кипячении с обратным холодильником, добавляли ТЭА (430 мкл, 3,08 ммоль) и дифенилфосфорил азид (DPPA, 630 мкл, 2,92 ммоль) и нагревали с обратным холодильником в течение 10 ч. Смесь упаривали, разбавляли хлороформом, промывали 5% лимонной кислотой (3 раза), водой, насыщенным раствором бикарбоната натрия и солевым раствором, сушили(MgSO4), фильтровали и упаривали, при этом получали неочищенный продукт, который очищали на колонке методом экспресс-хроматографии (силикагель, элюент: гексан/EtOAc, 75:25), при этом получали чистое Вос-аминопроизводное G2 (257 мг, 37%). Стадия В.Boc-Производное G2 (257 мг, 1,08 ммоль) растворяли в 4 М HCl/диоксан (5,0 мл) и перемешивали при комнатной температуре в течение 1 ч. Растворитель упаривали, а остаток разбавляли насыщенным раствором бикарбоната натрия (несколько мл) и 1 М NaOH (1 мл), экстрагировали EtOAc (2 раза), сушили(MgSO4), фильтровали и упаривали досуха, при этом получали неочищенный 2,3-метилендиоксианилин Диметилацетилендикарбоксилат A3 (130 мкл, 1,06 ммоль) добавляли по каплям в раствор неочищенного 2,3-метилендиоксианилина G3 (148 мг, 1,08 ммоль) в МеОН (2,5 мл). Осторожно, экзотермическая реакция. Смесь нагревали при слабом кипении с обратным холодильником в течение 3 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке с силикагелем методом экспресс-хроматографии (элюент: гексан/EtOAc, 9:1), фракции, содержащие чистый продукт упаривали, при этом получали диэфир G4 (185 мг, 0,662 ммоль, выход 61%). Стадия D. Диэфир G4 (180 мг, 0,645 ммоль) растворяли в дифениловом эфире (2,5 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до 350-400 С. После того как температура реакционной смеси достигала 250 С (при 220-230 С наблюдалось выделение MeOH), смесь нагревали еще в течение 6 мин (реакцию останавливали при температуре 262 С), затем баню удаляли, а смесь охлаждали до комнатной температуры. Твердое вещество, полученное после охлаждения, смешивали с эфиром,фильтровали и сушили, при этом получали неочищенный диоксихинолин G5 (125 мг, 78%). Полученное соединение использовали на следующих стадиях без дополнительной очистки. МС: 246 (М+Н)+, 248,1 (М-Н)-. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 88%. Пример 3 Н. Получение 2-карбометокси-4-гидрокси-8,9-дигидрофуро[2,3-h]хинолина. Стадия А. Триэтиламин (5,0 мл, 35,6 ммоль) добавляли в колбу, содержащую амин H1 (2,0 мл, 17,8 ммоль) и дихлорметан (100 мл) в атмосфере азота. Смесь охлаждали на ледяной бане, затем по каплям добавляли триметилацетилхлорид (3,3 мл, 26,7 ммоль). Реакционную смесь медленно нагревали до КТ и перемешивали в течение 14 ч при КТ. Реакцию останавливали добавлением насыщенного раствора NaHCO3 и экстрагировали EtOAc. Органические слои объединяли и сушили, фильтровали и концентрировали, продукт очищали на колонке методом экспресс-хроматографии (элюент: градиент гексан/EtOAc от 4:1 до 1:1),- 25009295 при этом получали требуемый продукт Н 2 в виде твердого вещества грязно-белого цвета (3,7 г, выход 97%). Стадия В.n-BuLi (15,9 мл, 1,6 М, 25,5 ммоль) добавляли по каплям в колбу (высушенную на пламени), содержащую амид Н 2 (1,6 г, 7,72 ммоля) в ТГФ при 0 С в атмосфере аргона. После добавления n-BuLi раствор приобретал светло-желто-оранжевый цвет. Раствор медленно нагревали до КТ и перемешивали в течение 24 ч. Затем раствор охлаждали до 0 С и добавляли по каплям этиленоксид (0,46 мл, 9,26 ммоль). Раствор медленно нагревали до 23 С, реакцию останавливали добавлением насыщенного раствора NaHCO3, продукт экстрагировали EtOAc, сушили, фильтровали и концентрировали, затем очищали на колонке методом экспресс-хроматографии (элюент: градиент гексан/этилацетат от 4:1 до 1:1), при этом получали требуемый продукт Н 3 (1,94 г, 5,01 ммоль, выход 65%) По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 99%. Стадия С. Раствор BBr3 (26 мл, 1,0 М, 26,0 ммоль) добавляли по каплям к метиловому эфиру H3 в CH2Cl2 при 0 С. Раствор медленно нагревали до 23 С и перемешивали в течение 14 ч при КТ. Реакцию останавливали добавлением 1 М NaOH, продукт экстрагировали EtOAc и CH2Cl2, при этом получали смесь требуемого продукта Н 4 и некоторого количества нециклического диола. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 99%. Стадия D. Раствор HCl (4,0 мл, 6,0 М) добавляли в раствор амида Н 4 (0,27 г, 1,22 ммоль) в МеОН (4,0 мл) при 23 С. Реакционную смесь нагревали с обратным холодильником в течение 48 ч, затем добавлялиNaHCO3 (насыщенный водный раствор) и экстрагировали EtOAc. Органические слои объединяли и сушили, фильтровали и концентрировали, при этом получали требуемый анилин Н 5, который использовали на следующих стадиях без дополнительной очистки. Стадия Е. Диметилацетилендикарбоксилат A3 (0,16 мл, 1,33 ммоль) добавляли в раствор анилина Н 5 (0,18 г,1,33 ммоль) в MeOH (3,0 мл) при КТ. Раствор нагревали с обратным холодильником в течение 3 ч, охлаждали до КТ и добавляли насыщенный раствор NaCl. Смесь экстрагировали EtOAc (3 раза), органические слои объединяли и сушили, фильтровали и концентрировали, продукт очищали на колонке методом экспресс-хроматографии (элюент: градиент гексан/EtOAc от 9:1 до 1:1), при этом получали требуемый олефин Н 6 (0,29 г, 78%). Стадия F. Колбу, содержащую олефин Н 6 (0,29 г, 1,04 ммоль) в дифениловом эфире (2,0 мл) помещали в пес- 26009295 чаную баню, предварительно нагретую до 350 С. После того как температура реакционной смеси достигала 225 С, колбу нагревали в течение еще 6-7 мин, при этом температура реакционной смеси достигала 240 С. Затем баню удаляли, а реакционную смесь медленно охлаждали до КТ, при этом получали продукт в виде осадка. Затем добавляли диэтиловый эфир, раствор фильтровали и промывали диэтиловым эфиром, при этом получали продукт Н 7 (0,20 г, 77%) в виде твердого вещества светло-коричневого цвета. МС: 246,0 (МН)+. Пример 3J. Получение 2-карбометокси-4-гидрокси-8-метилтиохинолина (J3). Стадия А. Диметилацетилендикарбоксилат A3 (5,21 мл, 35,91 ммоль) добавляли по каплям в раствор 2 метилмеркаптоанилина J1 (5,0 г, 35,91 ммоль) в MeOH (100 мл). Осторожно, экзотермическая реакция. Смесь нагревали при слабом кипении растворителя с обратным холодильником в течение 2 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке методом экспрессхроматографии (элюент: гексан/EtOAc, 90:10), фракции, содержащие чистый продукт, упаривали, при этом получали диэфир J2 (10,53 г, 37,43 ммоль, выход 99%). По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 85%. Стадия В. Диэфир J2 (10,53 г, 37,43 ммоль) растворяли в дифениловом эфире (35 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до 350-400 С. После того, как температура реакционной смеси достигала 245 С смесь нагревали в течение еще 6 мин, затем баню удаляли, а смесь охлаждали до комнатной температуры. Полученный осадок суспендировали в эфире, фильтровали и промывали эфиром, при этом получали хинолин C8-SMe, J3 (6,15 г, 66%). МС: 250 (М+Н)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 99%. Пример 3K. Получение 7-трет-бутилокси-2-карбометокси-4-гидрокси-8-метилхинолина (K6). Стадия 1.(5,36 мл, 28,73 ммоль) и каталитическое количество эфирата трифторида бора (143,8 мкл, 1,14 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 ч, при этом по данным аналитической ЖХВР реакция не завершена. Затем добавляли дополнительное количество третбутилтрихлорацетимида (1,4 мл, 7,51 ммоль), при этом не наблюдалось выпадение осадка. Реакция завершалась после перемешивания при комнатной температуре в течение 5 ч. Затем добавляли твердый бикарбонат натрия и смесь фильтровали, промывали дихлорметаном и упаривали досуха, при этом получали твердое вещество белого цвета. Полученное твердое вещество растирали в дихлорметане, твердое вещество белого цвета (трихлорацетимид) удаляли фильтрованием. Фильтрат концентрировали, продукт очищали на колонке методом экспресс-хроматографии (элюент: гексан/EtOAc, 9:1), при этом получали чистый 2-метил-3-трет-бутоксинитробензол K3 (1,17 г, 78%). По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 96%. В раствор 2-метил-3-трет-бутоксинитробензола K3 (1,31 г, 6,26 ммоль) в абсолютном этаноле (30 мл) добавляли 10% Pd/C (130 мг) в качестве катализатора. Раствор гидрировали в атмосфере водорода,подаваемого из баллона, при атмосферном давлении и комнатной температуре в течение 63 ч. Реакционную смесь фильтровали через слой целита, промывали абсолютным EtOH и упаривали досуха, при этом получали 2-метил-3-трет-бутоксианилин K4 (1,11 г, 6,14 ммоль, выход 98%). МС: 180 (М+Н)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 96%. Стадия 3. Диметилацетилендикарбоксилат A3 (749 мкл, 5,97 ммоль) добавляли по каплям в раствор 2-метил 3-трет-бутоксианилина K4 (1,07, 5,97 ммоль) в МеОН (14 мл). Смесь нагревали при слабом кипении растворителя с обратным холодильником в течение 2 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке методом экспресс-хроматографии (элюент: гексан/EtOAc, 95:5),фракции, содержащие чистый продукт, упаривали, при этом получали диэфир (1,13 г, 3,52 ммоль, выход 59%). МС: 320,0 (М-Н)-, 322,1 (М+Н)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 92%. Стадия 4. Диэфир K5 (1,13 г, 3,52 ммоль) растворяли в дифениловом эфире (3,0 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до 400-440 С. После того как температура реакционной смеси достигала 230 С (при 220 С наблюдалось выделение MeOH), смесь нагревали в течение 6 мин (реакцию останавливали при температуре 242 С), затем баню удаляли, а смесь охлаждали до комнатной температуры. После охлаждения выделение осадка не наблюдалось, продукт выделяли из неочищенной смеси экспресс-хроматографией, дифениловый эфир элюировали смесью гексан/EtOAc, 8:2, а продукт элюировали смесью гексан/EtOAc, 4:6, при этом получали хинолин С 7-О-трет-Bu,С 8-Ме, K6(838 мг, 82%) в виде твердого вещества бежевого цвета. МС: 288,0 (М-Н)-, 290,0 (М+Н)+. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 99%. Полученный хинолин K6 использовали для синтеза соединений 1032 и 1033 представленных в табл. 1, а также для получения соединений 1034, 1035, 1057 и 1058, также представленных в табл. 1. С 7-третБутилэфирную группу превращали в гидроксильную группу при обработке конечного соединения 50% ТФУ в дихлорметане в течение 30 мин при 0 С, затем в течение 30 мин при КТ, затем продукт упаривали досуха, разбавляли водой и лифиолизовали. Пример 3L. Получение 2-карбометокси-4-гидрокси-8-метоксихинолина (L3). Стадия А. Диметилацетилендикарбоксилат A3 (5,5 мл, 44,74 ммоль) добавляли по каплям в раствор ортоанизидина L1 (5,0 мл, 44,33 ммоль) в МеОН (100 мл). Осторожно, экзотермическая реакция. Смесь нагревали при слабом кипении с обратным холодильником в течение 5 ч, охлаждали и концентрировали в вакууме. Неочищенный продукт очищали на колонке методом экспресс-хроматографии (элюент: гради- 28009295 ент гексан/EtOAc от 95:5 до 90:10), фракции, содержащие чистый продукт, упаривали, при этом получали диэфир L2 (10 г, 37,70 ммоль, выход 85%). По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 82%. Стадия В. Диэфир L2 (10 г, 37,70 ммоль) растворяли в дифениловом эфире (15 мл) и реакционную смесь помещали в песчаную баню, предварительно нагретую до 350-400 С. После того, как температура реакционной смеси достигала 240 С смесь нагревали в течение 6 мин, затем баню удаляли, а смесь охлаждали до комнатной температуры. После охлаждения выделение осадка не наблюдалось, продукт выделяли из неочищенной смеси на колонке методом экспресс-хроматографией. Примеси элюировали в градиенте гексан/EtOAc от 6:4 до 5:5, затем продукт элюировали смесью гексан/EtOAc, 2:8, при этом получали хинолин С 8-ОМе, L3 (4,56 г, 52%). МС: 231,9 (М-Н)-. По данным ЖХВР (=220 нм, элюент: ТФУ) гомогенность продукта составляла 99%. Пример 4. Получение дипептидов. Получение дипептида 1. Смесь Вос-гидроксипролина Р 2 (50,0 г, 216 ммоль), метилового эфира винил-АССА Р 1 (42,25 г, 238 ммоль, 1,1 экв.), TBTU (76,36 г, 238 ммоль, 1,1 экв.) и DIPEA (113 мл, 649 ммоль, 3 экв.) в ДМФА (800 мл) перемешивали при КТ в атмосфере азота. Через 3,5 ч растворитель упаривали и остаток экстрагировали EtOAc. Экстракт промывали соляной кислотой (10%), насыщенным раствором бикарбоната натрия и солевым раствором. Затем органическую фазу сушили над сульфатом магния, фильтровали и упаривали, при этом получали масло. Продукт сушили в высоком вакууме в течение ночи, при этом получали дипептид 1 в виде пены желтого цвета (72,0 г, 94%, по данным ЖХВР чистота продукта 95%). Получение дипептида 3. Дипептид 1 (72,0 г, 203 ммоль), трифенилфосфин (63,94 г, 243,8 ммоль, 1,2 экв.) и 4 нитробензойную кислоту (41,08 г, 245,8 ммоль, 1,2 экв.) растворяли в безводном ТГФ (1,4 л). Раствор охлаждали до 0 С при перемешивании в атмосфере азота. Затем по каплям добавляли диэтилазодикарбоксилат (38,4 мл, 244 ммоль, 1,2 экв.) в течение 45 мин и реакционную смесь нагревали до КТ. Через 4 ч растворитель упаривали. Остаток разделили на четыре части. Каждую часть очищали хроматографией(тонкодисперсный силикагель, 10-40 мкм (меш), диаметр колонки 12 см, высота колонки 16 см, элюент: градиент гексан/EtOAc от 2:1 до 1:1, затем до чистого EtOAc). Затем растворитель упаривали, остаток сушили в высоком вакууме при 70 С в течение 1 ч, при этом получали эфир Вос-дипептида 2 (108,1 г,количественный выход) в виде аморфного твердого вещества белого цвета. К эфиру Вос-дипептида 2(108 г, 243 ммоль) добавляли 4 н. раствор хлористого водорода в диоксане, при этом получали бесцветный раствор. Раствор перемешивали при КТ в течение 1 ч. Растворитель упаривали и остаток сушили в высоком вакууме в течение 3 ч, при этом получали соединение 3 в виде гидрохлорида (твердое аморфное вещество), которое использовали без дополнительной очистки. Пример 5. Получение трипептидов. Получение трипептида 6 а. Карбамат 4b (6,15 г, 22,5 ммоль) и TBTU (7,72 г, 24,7 ммоль) суспендировали в ДХМ и суспензию интенсивно перемешивали. Затем при КТ добавляли DIPEA (3,92 мл, 22,5 ммоль) и через 10 мин получали практически гомогенный раствор. Затем в реакционную смесь добавляли раствор дипептида 3 (10,39 г, 23,6 ммоль) в безводном ДХМ (100 мл), содержащий DIPEA (4,11 мл, 23,62 ммоль). Полученный раствор желтого цвета перемешивали в течение 14 ч. Затем растворитель упаривали, при этом получали сироп желтого цвета, который экстрагировали EtOAc (300 + 150 мл) и промывали 0,05 н. HCl (2 раза по 200 мл), насыщенным раствором Na2CO3 (300 мл) и солевым раствором (150 мл). Экстракты объединяли,сушили над MgSO4 и упаривали, при этом получали трипептид 6 а в виде пены светло-желтого цвета Трипептид 6 а (15,68 г) растворяли в ТГФ (200 мл) и добавляли воду (30 мл). Полученный раствор охлаждали до 0 С и добавляли раствор гидроксида лития (моногидрат, 1,18 г, 28,12 ммоль) в течение 3 мин при интенсивном перемешивании. Смесь инкубировали в течение 3 ч при 0 С, затем избыток основания нейтрализовали добавлением 1 н. HCl (рН приблизительно 6) и ТГФ упаривали, при этом получали продукт в виде водной суспензии (смола желтого цвета). Смесь экстрагировали EtOAc (2 раза по 200 мл) и промывали насыщенным раствором NaHCO3 (2 раза по 300 мл). Экстракты объединяли, сушили надMgSO4 и упаривали, при этом получали пену бледно-желтого цвета. Продукт очищали экспрессхроматографией (силикагель, элюент EtOAc), при этом получали соединение 7 а в виде твердого аморфного вещества белого цвета (9,77 г, 91%). Получение трипептида 6b. Циклопентилмочевину-Tbg 5 (2,21 г, 9,10 ммоль) и TBTU (3,12 г, 10,0 ммоль) растворяли/суспендировали в безводном дихлорметане (40 мл) и добавляли DIPEA (1 экв.). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение приблизительно 10 мин, при этом получали практически гомогенный раствор. К реакционной смеси добавляли раствор дипептида Р 1 Р 2 (4,20 г, 9,56 ммоль) в безводном дихлорметане (35 мл, 1 экв. DIPEA) и полученную смесь желтого цвета перемешивали в течение 14 ч, затем подщелачивали добавлением DIPEA (приблизительно 1,5 мл). Растворитель упаривали, при этом получали сироп желтого цвета, который экстрагировали этилацетатом(150 + 50 мл) и промывали 0,1 н. HCl (150 мл), водой (100 мл, эмульсию разрушали солевым раствором),насыщенным раствором Na2CO3 (150 мл) и солевым раствором (100 мл). Экстракты объединяли, сушили над MgSO4 и упаривали, при этом получали соединение 6b в виде твердого вещества бледно-желтого цвета (6,21 г, по данным ЖХВР чистота продукта 95%). Получение трипептида 7b.
МПК / Метки
МПК: A61P 31/00, C07K 5/08, C07D 417/04
Метки: гепатита, ингибиторов, соединения, вируса, качестве
Код ссылки
<a href="https://eas.patents.su/30-9295-soedineniya-v-kachestve-ingibitorov-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения в качестве ингибиторов вируса гепатита с</a>
Гетероциклические трипептиды в качестве ингибиторов вируса гепатита с

Номер патента: 7739
Опубликовано: 29.12.2006
Авторы: Бейли Марри Д., Ллина-Брюне Монсе, Гиро Элиза
МПК: C07D 417/14, A61K 31/47, A61P 31/00...
Метки: гетероциклические, трипептиды, гепатита, вируса, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) в котором R1 обозначает гидрокси или NHSO2R1A, где R1A обозначает C1-С8алкил, С3-С7циклоалкил или С1-С6алкил-С3-С7циклоалкил, которые все необязательно замещены 1-3 заместителями, выбранными из ряда, включающего галоген, циано, нитро, OC1-С6алкил, амидо, амино или фенил, или R1A обозначает C6- или С10арил, необязательно замещенный 1-3 заместителями, выбранными из ряда, включающего галоген, циано, нитро, С1-С6алкил,...
Производные &beta-d-нуклеозида в качестве лекарственного средства для лечения инфекции вируса гепатита c у хозяина

Номер патента: 7178
Опубликовано: 25.08.2006
Авторы: Лаколла Пауло, Соммадосси Жан-Пьер
МПК: A61K 31/7068, A61P 31/14, A61K 31/7076...
Метки: лекарственного, средства, инфекции, хозяина, производные, beta-d-нуклеозида, качестве, гепатита, вируса, лечения
Формула / Реферат:
1. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или пролекарства, необязательно, в фармацевтически приемлемом носителе или разбавителе при получении лекарственного средства для лечения инфекции вируса гепатита С у хозяина. 2. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или...
Способ и промежуточные соединения для получения ненуклеозидных ингибиторов ревертазы вируса вич-1

Номер патента: 8149
Опубликовано: 27.04.2007
Авторы: Ким Дзиюн, Эрикссон Магнус С., Бусакка Карл А.
МПК: C07D 243/00, C07D 471/14, C07D 221/00...
Метки: получения, ненуклеозидных, способ, вируса, ревертазы, промежуточные, соединения, ингибиторов, вич-1
Формула / Реферат:
1. Способ получения соединения формулы I в которой R2 выбран из группы, включающей Н, F, Cl, C1-C4алкил, C3-C4диклоалкил и CF3, R4 обозначает Н или Me, R5 обозначает Н, Me или Et, при условии, что оба R4 и R5 одновременно не обозначают Me и, если R4 обозначает Me, то R5 не может обозначать Et, R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбран из группы, включающей заключающийся в том, что (а) исходное соединение...
Производные пиримидина в качестве ингибиторов репликации вируса иммунодефицита человека

Номер патента: 2973
Опубликовано: 26.12.2002
Авторы: Де Корт Барт, Койманс Люсьен Мария Хенрикус, Андрис Кунрад Йозеф Лодевейк Марсель, Янссен Поль Адриан Жан, Янссен Марсель Огюст Констант, Лудовики Дональд Уилльям, Ван Акен Кун Жанн Альфонс, Де Жонж Марк Рене, Хо Чих Юнг, Кукла Майкл Джозеф, Хэрес Ян
МПК: C07D 239/50, A61K 31/505, A61P 31/18...
Метки: пиримидина, ингибиторов, качестве, вируса, человека, иммунодефицита, репликации, производные
Формула / Реферат:
1. Применение соединения формулы его N-оксида, фармацевтически приемлемой соли присоединения или стереохимически изомерной формы, где А является СН или N; n равен 0, 1, 2, 3 или 4; Q является водородом или -NR1R2; R1 и R2, каждый независимо, выбран из водорода, гидрокси, C1-12aлкила, С1-12алкилокси, С1-12алкилкарбонила, С1-12алкилоксикарбонила, где каждая из указанных С1-12алкильных групп необязательно и каждая независимо может быть замещена...
Производные бензимидазола и имидазопиридина в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 4746
Опубликовано: 26.08.2004
Авторы: Вене Марк Гастон, Янссенс Франс Эдуард, Лякрамп Жан Фернан Арман, Гийемон Жером Эмиль Жорж, Андрис Кунрад Йозеф Лодевейк Марсель
МПК: C07D 401/14, A61P 11/00, A61K 31/4709...
Метки: вируса, имидазопиридина, бензимидазола, производные, качестве, ингибиторов, репликации, респираторно-синцитиального
Формула / Реферат:
1. Соединение формулы его аддитивная соль или стереохимически изомерная форма, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- (a-2); -CH=N-CH=CH- (a-3); -CH=CH-N=CH- (a-4) или -CH=CH-CH=N- (a-5); где каждый атом водорода в...