Производные нуклеозидов в качестве ингибиторов рнк-зависимой рнк вирусной полимеразы
Номер патента: 7491
Опубликовано: 27.10.2006
Авторы: Маккосс Малкольм, Сонг Кванлай, Бхат Неелима, Кук Филлип Дэн, Олсен Дэвид Б., Элдруп Энн Б., Прхавк Мариджа, Пракаш Тхазха П., Кэрролл Стивен С., Бхат Балкришен





























Формула / Реферат
1. Соединение структурной формулы II

или его фармацевтически приемлемая соль;
где R1 представляет метил, фторметил, гидроксиметил, дифторметил, трифторметил или аминометил;
R2 представляет гидрокси, фтор или метокси;
R3 представляет водород, фтор, гидрокси, амино или метокси;
R5 представляет водород или Р3О9Н4;
R8 представляет водород или амино;
R9 представляет водород, циано, метил, галоген или CONH2 и
R10 и R11, каждый независимо, представляет водород, фтор, гидрокси или амино;
при условии, что, если R1 представляет b -метил, R2 и R3 представляют a -гидрокси, R10 представляет амино и R5, R8 и R11 представляют водород, тогда R9 не представляет циано или CONH2.
2. Соединение по п.1, выбранное из группы, состоящей из
4-амино-7-(2-С-метил-b -D-арабинофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
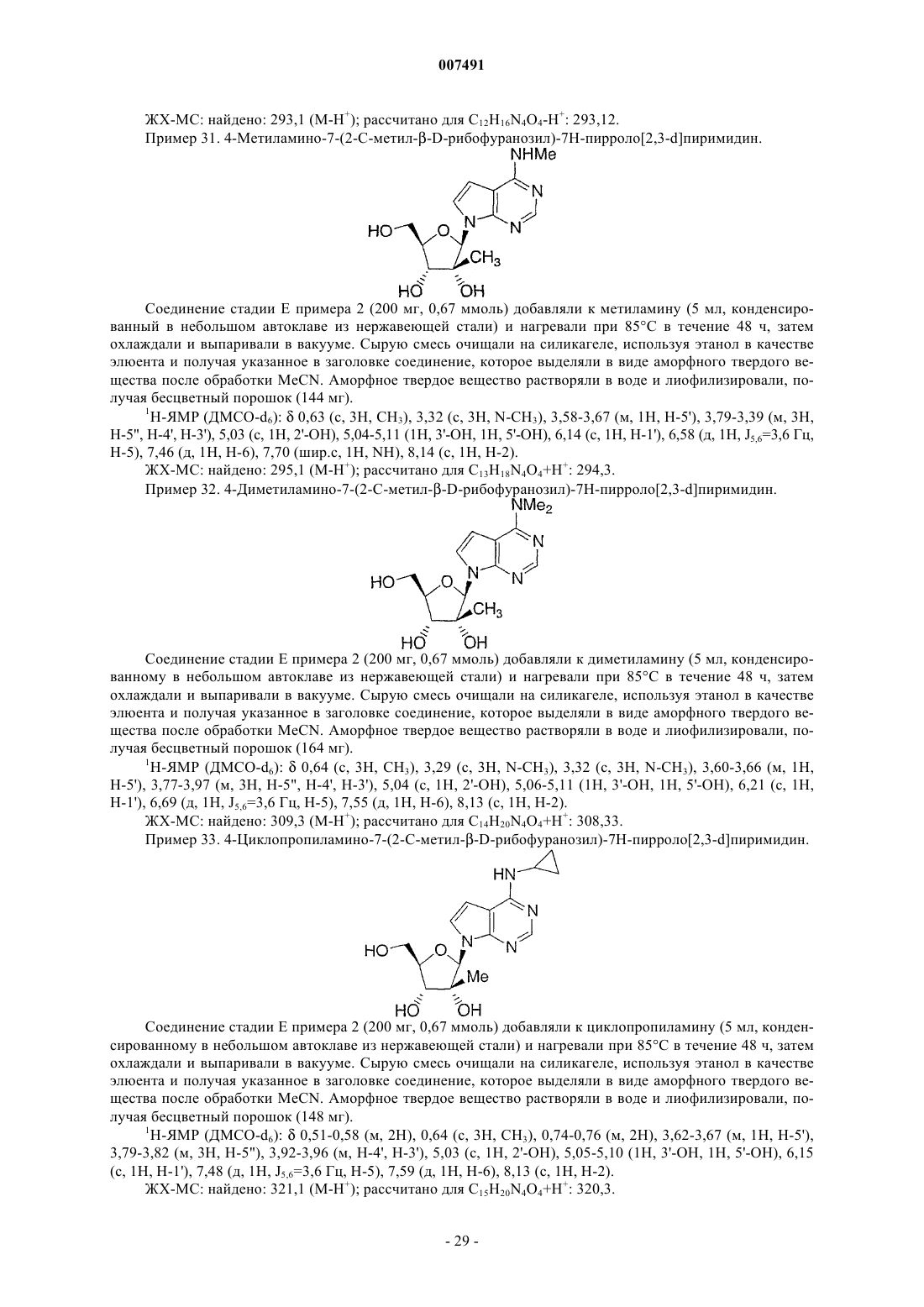
4-метиламино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-диметиламино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-циклопропиламино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-винил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-гидроксиметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-фторметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-метил-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидин-5-карбоновой кислоты,
4-амино-5-бром-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-хлор-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-фтор-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
2,4-диамино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
2-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
2-амино-4-циклопропиламино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
2-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидин-4(3Н)-она,
4-амино-7-(2-С-этил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С,2-O-диметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидин-4(3Н)-она,
2-амино-5-метил-7-(2-С,2-O-диметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидин-4(3Н)-она,
4-амино-7-(3-дезокси-2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(3-дезокси-2-С-метил-b -D-арабинофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-2-фтор-7-(2-С-метил-b -D-рибофуранозил) -7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(3-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(3-С-метил-b -D-ксилофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2,4-ди-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина и
4-амино-7-(3-дезокси-3-фтор-2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
и соответствующих им 5'-трифосфатов;
или их фармацевтически приемлемых солей.
3. Соединения по п.2, выбранные из группы, состоящей из
4-амино-7-(2-С-метил-b -D-арабинофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-7-(2-С-фторметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-метил-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-бром-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-хлор-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
4-амино-5-фтор-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина и
4-амино-7-(2-С,2-O-диметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидина,
и соответствующих им 5'-трифосфатов;
или их фармацевтически приемлемых солей.
4. Соединение по п.3, которое является 4-амино-7-(2-С-метил-b -D-арабинофуранозил)-7Н-пирроло[2,3-d]пиримидином или его фармацевтически приемлемой солью.
5. Соединение по п.3, которое является 4-амино-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидином или его фармацевтически приемлемой солью.
6. Соединение по п.3, которое является 4-амино-7-(2-С-фторметил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидином или его фармацевтически приемлемой солью.
7. Соединение по п.3, которое является 4-амино-5-хлор-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидином или его фармацевтически приемлемой солью.
8. Соединение по п.3, которое является 4-амино-5-бром-7-(2-С-метил-b -D-рибофуранозил)-7Н-пирроло[2,3-d]пиримидином или его фармацевтически приемлемой солью.
9. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель.
10. Фармацевтическая композиция по п.9, предназначенная для ингибирования РНК-зависимой РНК вирусной полимеразы, ингибирования РНК-зависимой РНК репликации и/или для лечения РНК-зависимой РНК вирусной инфекции.
11. Фармацевтическая композиция по п.10, где указанная РНК-зависимая РНК вирусная полимераза является HCV NS5B полимеразой, указанная РНК-зависимая РНК вирусная репликация является HCV репликацией и указанная РНК-зависимая РНК вирусная инфекция является HCV инфекцией.
12. Способ ингибирования РНК-зависимой РНК вирусной полимеразы и/или ингибирования РНК-зависимой РНК вирусной инфекции, включающий введение нуждающемуся в таком ингибировании млекопитающему эффективного количества соединения по п.1.
13. Способ по п.12, где указанная РНК-зависимая РНК вирусная полимераза является HCV NS5B полимеразой и указанная РНК-зависимая РНК вирусная репликация является HCV вирусной репликацией.
14. Способ лечения РНК-зависимой РНК вирусной инфекции, включающий введение нуждающемуся в таком лечении млекопитающему эффективного количества соединения по п.1.
15. Способ по п.14, где указанная РНК-зависимая РНК вирусная инфекция является HCV инфекцией.
16. Способ по п.15, включающий введение соединения по п.1 в комбинации с терапевтически эффективным количеством другого агента, активного против HCV.
17. Способ по п.16, где указанный агент, активный против HCV, является рибавирином, левовирином, тимозином альфа-1; ингибитором NS3 серинпротеазы; ингибитором инозинмонофосфатдегидрогеназы; интерфероном-a или пэгилированным интерфероном-a , одним или в комбинации с рибавирином или левовирином.
18. Способ по п.17, где указанным агентом, активным против HCV, является интерферон-a или пэгилированный интерферон-a , один или в комбинации с рибавирином.
19. Применение соединения по п.1 для ингибирования РНК-зависимой РНК вирусной полимеразы или ингибирования РНК-зависимой РНК вирусной репликации у млекопитающих.
20. Применение соединения по п.1 для лечения РНК-зависимой РНК вирусной инфекции у млекопитающих.
21. Применение по п.20, где указанной РНК-зависимой РНК вирусной инфекцией является инфекция гепатита С.
22. Применение соединения по п.1 для изготовления лекарства для ингибирования РНК-зависимой РНК вирусной полимеразы или для ингибирования РНК-зависимой РНК вирусной репликации у млекопитающих.
23. Применение соединения по п.1 для изготовления лекарства для ингибирования РНК-зависимой РНК вирусной инфекции у млекопитающих.
24. Применение по п.23, где указанной РНК-зависимой РНК вирусной инфекцией является инфекция гепатита С.
Текст
007491 Область изобретения Настоящее изобретение относится к нуклеозидным соединениям и некоторым их производным, к их получению и к их использованию в качестве ингибиторов РНК-зависимой РНК вирусной полимеразы. Соединения настоящего изобретения являются ингибиторами РНК-зависимой РНК вирусной репликации и могут быть использованы для лечения РНК-зависимой РНК вирусной инфекции. Они особенно полезны в качестве ингибиторов (HCV) NS5B полимеразы вируса гепатита С, в качестве ингибиторов репликации HCV и для лечения инфекции гепатита С. Предпосылки изобретения Инфекция вируса гепатита С (HCV) является основной проблемой для здоровья, которая приводит к хроническим заболеваниям печени, таким как цирроз и гепатоклеточная карцинома, у значительного числа инфицированных индивидуумов, число которых оценивается как 2-15% населения всего мира. Только в Соединенных Штатах диагностировано 4,5 миллиона инфицированных людей, по данным Центра Здравоохранения США. В соответствии с данными Всемирной Организации Здравоохранения во всем мире инфицировано более 200 миллионов человек, причем, по меньшей мере, от 3 до 4 миллионов людей инфицируется ежегодно. После инфицирования около 20% людей избавляются от вируса, но остальные являются носителями вируса HCV до конца своей жизни. У 10-20% хронически инфицированных индивидуумов, в конечном счете, развивается разрушающий печень цирроз или рак печени. Вирусное заболевание переносится парентерально через зараженную кровь и продукты, включающие кровь,загрязненные иглы или половым путем и непосредственно от инфицированных матерей или беременных матерей их потомству. Существующие в настоящее время способы лечения HCV инфекции, которые ограничиваются иммунотерапией с использованием одного рекомбинантного интерферона- или в комбинации с нуклеозидным аналогом рибавирина, дают ограниченные положительные клинические результаты. Более того, для HCV не существует признанной вакцины. Соответственно, существует острая необходимость в усовершенствованных терапевтических агентах, которые эффективно боролись бы с хронической HCV инфекцией. Существующее состояние дел в отношении лечения HCV инфекции приведено в обзорах, приводятся ссылки на следующие публикации: В. Dymock, et al., "Novel approaches to theDrugs, 6: 13-42 (2001); и С. Crabb, "Hard-Won Advances Spark Excitement about Hepatitis C", Science: 506507 (2001); содержание которых включено в качестве ссылки. Были предприняты различные подходы к лечению HCV, которые включают ингибирование вирусной серинпротеиназы (NS3 протеаза), геликазы и РНК-зависимой РНК полимеразы (NS5B) и разработку вакцины.HCV вирион является оболочковым РНК вирусом с позитивной цепью, с единственной олигонуклеотидной геномной последовательностью примерно из 9600 оснований, которая кодирует полипротеин,состоящий из примерно 3010 аминокислот. Белковые продукты HCV гена состоят из структурных белков С, Е 1 и Е 2 и неструктурных белков NS2, NS3, NS4A и NS4B и NS5A и NS5B. Считают, что неструктурные белки (NS) обеспечивают каталитический механизм вирусной репликации. NS3 протеаза выделяетNS5B, РНК-зависимую РНК полимеразу из полипротеиновой цепи. HCV NS5B полимераза необходима для синтеза двухцепочечной РНК из одноцепочечной вирусной РНК, которая служит матрицей в цикле репликации HCV. Поэтому NS5B полимеразу считают существенным компонентом в HCV комплексе репликации [см. K. Ishi, et al., "Expression of Hepatitis С Virus NS5B Protein: Characterization of Its RNAand Kinetic Analyses of NS5B RNA-Dependent RNA Polymerase of the Hepatitis С Virus", Virology, 249: 108118 (1998)]. Ингибирование HCV NS5B полимеразы предотвращает образование двухцепочечной HCV РНК и поэтому является привлекательным подходом к разработке HCV-специфических противовирусных способов лечения. В настоящее время было обнаружено, что нуклеозидные соединения настоящего изобретения и некоторые их производные являются эффективными ингибиторами РНК-зависимой РНК вирусной репликации, и в частности HCV репликации. 5'-Трифосфатные производные этих нуклеозидных соединений являются ингибиторами РНК-зависимой РНК вирусной полимеразы, и в частности HCV NS5B полимеразы. Рассматриваемые нуклеозидные соединения являются их производными и могут быть полезны при лечении РНК-зависимой РНК вирусной инфекции, и в частности HCV инфекции. Поэтому задачей настоящего изобретения является создание нуклеозидных соединений и некоторых их производных, которые могли бы быть полезны в качестве ингибиторов РНК-зависимой РНК вирусной полимеразы, и в частности в качестве ингибиторов HCV NS5B полимеразы. Другой задачей настоящего изобретения является создание нуклеозидных соединений и некоторых их производных, которые могли бы быть полезны в качестве ингибиторов репликации РНК-зависимой-1 007491 РНК вируса, и в частности в качестве ингибиторов репликации вируса гепатита С. Другой задачей настоящего изобретения является создание нуклеозидных соединений и некоторых их производных, которые были бы полезны при лечении РНК-зависимой РНК вирусной инфекции, и в частности при лечении HCV инфекции. Другой задачей настоящего изобретения является создание фармацевтических композиций, включающих нуклеозидные соединения вместе с фармацевтически приемлемым носителем. Другой задачей настоящего изобретения является создание фармацевтических композиций, включающих нуклеозидные соединения и их производные, для использования в качестве ингибиторов РНКзависимой РНК вирусной полимеразы, и в частности в качестве ингибиторов HCV NS5B полимеразы. Другой задачей настоящего изобретения является создание фармацевтических композиций, включающих нуклеозидные соединения и их производные, для использования в качестве ингибиторов РНКзависимой РНК вирусной репликации, и в частности в качестве ингибиторов HCV репликации. Другой задачей настоящего изобретения является создание фармацевтических композиций, включающих нуклеозидные соединения и их производные, для использования при лечении РНК-зависимой РНК вирусной инфекции, и в частности при лечении HCV инфекции. Другой задачей настоящего изобретения является создание фармацевтических композиций, включающих нуклеозидные соединения и их производные в комбинации с другими агентами, активными против РНК-зависимого РНК вируса, и в частности против HCV. Другой задачей настоящего изобретения является создание способов ингибирования РНК-зависимой РНК вирусной полимеразы, и в частности ингибирования HCV NS5B полимеразы. Другой задачей настоящего изобретения является создание способов ингибирования РНКзависимой РНК вирусной репликации, и в частности ингибирования HCV репликации. Другой задачей настоящего изобретения является создание способов лечения РНК-зависимой РНК вирусной инфекции, и в частности лечения HCV инфекции. Другой задачей настоящего изобретения является создание способов лечения РНК-зависимой РНК вирусной инфекции в комбинации с другими агентами, активными против РНК-зависимой РНК вируса, и в частности лечения HCV инфекции в комбинации с другими агентами, активными против HCV. Другой задачей настоящего изобретения является создание нуклеозидных соединений и некоторых их производных и их фармацевтических композиций для использования в качестве лекарства для ингибирования РНК-зависимой РНК вирусной репликации и/или для лечения РНК-зависимой РНК вирусной инфекции, и в частности для ингибирования HCV репликации и/или лечения HCV инфекции. Другой задачей настоящего изобретения является создание для использования нуклеозидных соединений и некоторых их производных и их фармацевтических композиций для получения лекарства для ингибирования РНК-зависимой РНК вирусной репликации и/или лечения РНК-зависимой РНК вирусной инфекции, и в частности для ингибирования HCV репликации и/или лечения HCV инфекции. Эти и другие задачи станут очевидны из последующего подробного описания. Краткое содержание изобретения Настоящее изобретение относится к соединениям структурной формулы II указанной стереохимической конфигурации: или его фармацевтически приемлемым солям; где R1 представляет метил, фторметил, гидроксиметил, дифторметил, трифторметил или аминометил;R5 представляет водород или Р 3 О 9 Н 4;R8 представляет водород или амино;R10 и R11, каждый независимо представляет водород, фтор, гидрокси или амино; при условии, что, если R1 представляет -метил, R2 и R3 представляют -гидрокси, R10 представляет амино и R5, R8 и R11 представляют водород, тогда R9 не представляет циано или CONH2. Предпочтительно данное соединение выбрано из группы, состоящей из: 4-амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина,-2 007491 4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-метиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-диметиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-винилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-гидроксиметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-5-карбоновой кислоты,4-амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,2,4-диамино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,2-амино-4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,2-амино-7-(2-С-метилD-рибофуранозил) -7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-она,4-амино-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-она,2-амино-5-метил-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-она,4-амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(3-дезокси-2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-2-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(3-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(3-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2,4-ди-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина и 4-амино-7-(3-дезокси-3-фтор-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,и соответствующих им 5'-трифосфатов; или их фармацевтически приемлемых солей. Кроме того, указанное соединение может быть выбрано из группы, состоящей из: 4-амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина и 4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,и соответствующих им 5'-трифосфатов; или их фармацевтически приемлемых солей. Настоящее изобретение также включает фармацевтическую композицию, содержащую соединение по п.1 и фармацевтически приемлемый носитель. Предпочтительно данная фармацевтическая композиция предназначена для ингибирования РНКзависимой РНК вирусной полимеразы, ингибирования РНК-зависимой РНК репликации и/или для лечения РНК-зависимой РНК вирусной инфекции. При этом указанная РНК-зависимая РНК вирусная полимераза может являться HCV NS5B полимеразой, указанная РНК-зависимая РНК вирусная репликация может являться HCV репликацией, а указанная РНК-зависимая РНК вирусная инфекция может являтьсяHCV инфекцией. Кроме того, изобретение относится к способу ингибирования РНК-зависимой РНК вирусной полимеразы и/или ингибирования РНК-зависимой РНК вирусной инфекции, включающему введение нуждающемуся в таком ингибировании млекопитающему эффективного количества соединения структурной формулы II, указанной выше. Предпочтительно указанная РНК-зависимая РНК вирусная полимераза является HCV NS5B полимеразой и указанная РНК-зависимая РНК вирусная репликация является HCV вирусной репликацией. Данное изобретение также относится к способу лечения РНК-зависимой РНК вирусной инфекции,включающему введение нуждающемуся в таком лечении млекопитающему эффективного количества соединения структурной формулы II, указанной выше. Предпочтительно указанная РНК-зависимая РНК вирусная инфекция является HCV инфекцией, а соединение структурной формулы II вводят в комбинации с терапевтически эффективным количеством-3 007491 другого агента, активного против HCV. При этом агент, активный против HCV, является рибавирином, левовирином, тимозином альфа-1; ингибитором NS3 серинпротеазы; ингибитором инозинмонофосфатдегидрогеназы; интерфероном- или пэгилированным интерфероном-, одним или в комбинации с рибавирином или левовирином. Следующим объектом изобретения является применение соединения структурной формулы II для ингибирования РНК-зависимой РНК вирусной полимеразы или ингибирования РНК-зависимой РНК вирусной репликации у млекопитающих. Предпочтительно соединение структурной формулы II применяется для лечения РНК-зависимой РНК вирусной инфекции у млекопитающих, при этом указанной РНК-зависимой РНК вирусной инфекцией является инфекция гепатита С. Настоящее изобретение относится также к применению соединения структурной формулы II для изготовления лекарства для ингибирования РНК-зависимой РНК вирусной полимеразы или для ингибирования РНК-зависимой РНК вирусной репликации у млекопитающих. В предпочтительном варианте изобретение относится к применению соединения структурной формулы II для изготовления лекарства для ингибирования РНК-зависимой РНК вирусной инфекции у млекопитающих, где указанной РНК-зависимой РНК вирусной инфекцией является инфекция гепатита С. Подробное описание изобретения Настоящее изобретение относится к соединениям структурной формулы I указанной стереохимической конфигурации: или его фармацевтически приемлемым солям; гдеR1 представляет С 2-4-алкенил, С 2-4-алкинил или C1-4-алкил, где алкил является незамещенным или замещен гидрокси, амино, C1-4-алкокси, C1-4-алкилтио или 1-3 атомами фтора;R2 представляет водород, фтор, гидрокси, меркапто, С 1-4-алкокси или C1-4-алкил; или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют 3-6-членную насыщенную моноциклическую кольцевую систему, необязательно содержащую гетероатом, выбранный из О, S и NC0-4-алкила;R3 и R4, каждый независимо выбирают из группы, состоящей из водорода, циано, азидо, галогена,гидрокси, меркапто, амино, C1-4-алкокси, С 2-4-алкенила, С 2-4-алкинила и C1-4-алкила, где алкил является незамещенным или замещен гидрокси, амино, C1-4-алкокси, C1-4-алкилтио или 1-3 атомами фтора;R6 и R7, каждый независимо представляет водород, метил, гидроксиметил или фторметил;R8 представляет водород, C1-4-алкил, С 2-4-алкинил, галоген, циано, карбокси, C1-4-алкилоксикарбонил, азидо, амино, С 1-4-алкиламино, ди(C1-4-алкил)амино, гидрокси, С 1-6-алкокси, C1-6-алкилтио, C1-6-алкилсульфонил, (C1-4-алкил)0-2-аминометил или С 4-6-циклогетероалкил, незамещенный или замещенный одной-двумя группами, независимо выбранными из галогена, гидрокси, амино, C1-4-алкила и C1-4-алкокси;R9 представляет водород, циано, нитро, C1-3-алкил, NHCONH2, CONR12R12, CSNR12R12, COOR12,C(=NH)NH2, гидрокси, C1-3-алкокси, амино, C1-4-алкиламино, ди(C1-4-алкил)амино, галоген, (1,3-оксазол 2-ил), (1,3-тиазол-2-ил) или (имидазол-2-ил); где алкил является незамещенным или замещен однойтремя группами, независимо выбранными из галогена, амино, гидрокси, карбокси и C1-3-алкокси;R10 и R11, каждый независимо представляет водород, гидрокси, галоген, C1-4-алкокси, амино, C1-4-алкиламино, ди(С 1-4-алкил)амино, С 3-6-циклоалкиламино, ди(С 3-6-циклоалкил)амино или С 4-6-циклогетероалкил, незамещенный или замещенный одной-двумя группами, независимо выбранными из галогена,гидрокси, амино, С 1-4-алкила и C1-4-алкокси; каждый R12 независимо представляет водород или C1-6-алкил; иR13 и R14, каждый независимо представляет гидрокси, ОСН 2 СН 2SС(=O)С 1-4-алкил, ОСН 2O(С=O)OC1-4 алкил, NHCHMeCO2Me, ОСН(C1-4-алкил)О(С=O)C1-4-алкил, при условии, что, если R1 представляет -метил и R4 представляет водород или R4 представляет метил и R1 представляет водород, R2 и R3 представляют -гидрокси, R10 представляет амино и R5, R6, R7,R8 и R11 представляют водород, тогда R9 не представляет циано или CONH2.-4 007491 Соединения формулы I можно использовать в качестве ингибиторов РНК-зависимой РНК вирусной полимеразы. Они также являются ингибиторами РНК-зависимой РНК вирусной репликации, и их можно использовать для лечения РНК-зависимой РНК вирусной инфекции. В одном из вариантов соединения структурной формулы I являются соединениями структурной формулы II или их фармацевтически приемлемой солью; гдеR1 представляет C1-3-алкил, где алкил необязательно замещен гидрокси, амино, C1-3-алкокси, C1-3-алкилтио или 1-3 атомами фтора;R10 и R11, каждый независимо представляет водород, галоген, гидрокси, амино, C1-4-алкиламино,ди(C1-4-алкил)амино или С 3-6-циклоалкиламино; при условии, что, если R1 представляет -метил, R2 и R3 представляют -гидрокси, R10 представляет амино и R5, R8 и R11 представляют водород, тогда R9 не представляет циано или CONH2. Во втором варианте соединения структурной формулы I представляют соединения структурной формулы II, гдеR5 представляет водород или Р 3O9 Н 4;R8 представляет водород или амино;R10 и R11, каждый независимо представляет водород, фтор, гидрокси или амино; при условии, что, если R1 представляет -метил, R2 и R3 представляют -гидрокси, R10 представляет амино и R5, R8 и R11 представляют водород, тогда R9 не представляет циано или CONH2. Иллюстративными, но не лимитирующими примерами соединений настоящего изобретения структурной формулы I, которые можно использовать в качестве ингибиторов РНК-зависимой РНК вирусной полимеразы, являются следующие: 4-амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-метиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-диметиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С-винилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С-гидроксиметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-5-карбоновая кислота,4-амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,2, 4-диамино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,2-амино-4-циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,2-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он,4-амино-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,-5 007491 7- (2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он,2-амино-5-метил-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он,4-амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(3-дезокси-2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-2-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(3-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(3-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидин,4-амино-7-(2,4-ди-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин и 4-амино-7-(3-дезокси-3-фтор-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин и соответствующие 5'-трифосфаты; или их фармацевтически приемлемые соли. Дополнительными иллюстрациями настоящего изобретения являются соединения, выбранные из группы, состоящей из: 4-амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-бром-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина,4-амино-5-фтор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина и 4-амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина и соответствующих 5'-трифосфатов; или их фармацевтически приемлемых солей. В одном воплощении настоящего изобретения нуклеозидные соединения можно использовать в качестве ингибиторов позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной полимеразы,ингибиторов позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной репликации и/или для лечения позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной инфекции. В классе этого воплощения позитивно-смысловой одноцепочечный РНК-зависимый РНК вирус является Flaviviridae вирусом или Picornaviridae вирусом. В подклассе этого класса Picornaviridae вирус является риновирусом, полиовирусом или вирусом гепатита А. Во втором подклассе этого класса Flaviviridae вирус выбирают из группы, состоящей из вируса гепатита С, вируса желтой лихорадки, вируса Денге, вируса Западного Нила, вируса японского энцефалита, вируса Банзи и вируса коровьей вирусной диареи (BVDV). В подгруппе этого подкласса Flaviviridae вирус является вирусом гепатита С. Другой аспект настоящего изобретения относится к способу ингибирования РНК-зависимой РНК вирусной полимеразы, к способу ингибирования РНК-зависимой РНК вирусной репликации и/или к способу лечения РНК-зависимой РНК вирусной инфекции у нуждающихся в этом млекопитающих, включающий введение млекопитающему терапевтически эффективного количества соединения структурной формулы I. В одном воплощении этого аспекта настоящего изобретения РНК-зависимая РНК вирусная полимераза является позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной полимеразой. В классе этого воплощения позитивно-смысловая одноцепочечная РНК-зависимая РНК вирусная полимераза является Flaviviridae вирусной полимеразой или Picornaviridae вирусной полимеразой. В подклассе этого класса Picornaviridae вирусная полимераза является риновирусной полимеразой, полиовирусной полимеразой или полимеразой вируса гепатита А. Во втором подклассе этого класса Flaviviridae вирусную полимеразу выбирают из группы, состоящей из полимеразы вируса гепатита С, полимеразы вируса желтой лихорадки, полимеразы вируса Денге, полимеразы вируса Западного Нила, полимеразы вируса японского энцефалита, полимеразы вируса Банзи и полимеразы вируса коровьей вирусной диареи (BVDV). В подгруппе этого подкласса Flaviviridae вирусная полимераза является полимеразой вируса гепатита С. Во втором воплощении этого аспекта настоящего изобретения РНК-зависимая РНК вирусная репликация является позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной репликацией. В классе этого воплощения позитивно-смысловая одноцепочечная РНК-зависимая РНК вирусная репликация является Flaviviridae вирусной репликацией или Picornaviridae вирусной репликацией. В подклассе этого класса Picornaviridae вирусная репликация является риновирусной репликацией, полиовирусной репликацией или репликацией вируса гепатита А. Во втором подклассе этого класса Flaviviridae вирусную репликацию выбирают из группы, состоящей из репликации вируса гепатита С, репликации вируса желтой лихорадки, репликации вируса Денге, репликации вируса Западного Нила, репликации вируса японского энцефалита, репликации вируса Банзи и репликации вируса коровьей вирусной диареи. В подгруппе этого подкласса Flaviviridae вирусная репликация является репликацией вируса гепатита С. В третьем воплощении этого аспекта настоящего изобретения РНК-зависимая РНК вирусная инфекция является позитивно-смысловой одноцепочечной РНК-зависимой РНК вирусной инфекцией. В классе этого воплощения позитивно-смысловая одноцепочечная РНК-зависимая РНК вирусная инфекция является Flaviviridae вирусной инфекцией или Picornaviridae вирусной инфекцией. В подклассе этого-6 007491 класса Picornaviridae вирусная инфекция является риновирусной инфекцией, полиовирусной инфекцией или инфекцией вируса гепатита А. Во втором подклассе этого класса Flaviviridae вирусную инфекцию выбирают из группы, состоящей из инфекции вируса гепатита С, инфекции вируса желтой лихорадки,инфекции вируса Денге, инфекции вируса Западного Нила, инфекции вируса японского энцефалита, инфекции вируса Банзи и инфекции вируса коровьей вирусной диареи. В подгруппе этого подклассаFlaviviridae вирусная инфекция является инфекцией вируса гепатита С. В тексте рассматриваемой заявки следующие термины имеют указанные далее значения. Указанные выше алкильные группы включают алкильные группы указанной длины в линейной или разветвленной конфигурации. Примерами таких алкильных групп являются метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, гексил, изогексил и т.п. Термин "алкенил" означает неразветвленную или разветвленную алкеновую цепь, содержащую от 2 до 6 атомов углерода или любое число в этом интервале (например, этенил, пропенил, бутенил, пентенил и т.д.). Термин "алкинил" означает неразветвленную или разветвленную алкиновую цепь, содержащую от 2 до 6 атомов углерода или любое число в этом интервале (например, этинил, пропинил, бутинил, пентинил и т.д.). Термин "циклоалкил" означает циклические алкановые кольца, содержащие от 3 до 8 атомов углерода или любое число в этом интервале (т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил). Термин "циклогетероалкил" включает неароматические гетероциклы, содержащие 1 или 2 гетероатома, выбранные из атомов азота, кислорода и серы. Примеры 4-6-членных циклогетероалкилов включают азетидинил, пирролидинил, пиперидинил, морфолинил, тиаморфолинил, имидазолидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиофенил, пиперазинил и т.п. Термин "алкокси" относится к неразветвленной или разветвленной цепи алкоксидов, содержащей указанное количество атомов углерода (например, С 1-4-алкокси) или любое число в указанном интервале(т.е. метокси (МеО-), этокси, изопропокси и т.д.). Термин "алкилтио" относится к неразветвленной или разветвленной цепи алкилсульфидов, содержащей указанное число атомов углерода (например, C1-4-алкилтио) или любое число в указанном интервале (т.е. метилтио (MeS-), этилтио, изопропилтио и т.д.). Термин "алкиламино" относится к неразветвленным или разветвленным алкиламинам, содержащим указанное число атомов углерода (например, C1-4-алкиламино) или любое число в указанном интервале(т.е. метиламино, этиламино, изопропиламино, трет-бутиламино и т.д.). Термин "алкилсульфонил" относится к неразветвленным или разветвленным алкилсульфонам, содержащим указанное число атомов углерода (например, C1-6-алкилсульфонил) или любое число в указанном интервале (т.е. метилсульфонил (МеSО 2-), этилсульфонил, изопропилсульфонил и т.д.). Термин "алкилоксикарбонил" относится к сложным эфирам с неразветвленной или разветвленной цепью производного карбоновой кислоты настоящего изобретения, содержащего указанное число атомов углерода (например, C1-4-алкилоксикарбонил) или любое число в указанном интервале (т.е. метилоксикарбонил (МеОСО-), этилоксикарбонил или бутилоксикарбонил). Термин "арил" включает как фенил, нафтил, так и пиридил. Арильная группа необязательно замещена одной-тремя группами, независимо выбранными из C1-4-алкила, галогена, циано, нитро, трифторметила, С 1-4-алкокси и С 1-4-алкилтио. Термин "галоген" включает атомы галогенов: фтора, хлора, брома и йода. Термин "замещенный" включает множественную степень замещения указанными заместителями. Когда раскрыты или заявлены множество замещающих групп, замещенное соединение может быть независимо замещено одним или более из раскрытых или заявленных групп заместителей, по одному или несколько. Термин "5'-трифосфат" относится к производному 5'-гидроксильной группы сложного эфира трифосфорной кислоты группы нуклеозидных соединений настоящего изобретения, имеющих следующую общую структурную формулу III: где R1-R11 имеют указанные выше значения. Предполагается, что соединения настоящего изобретения включают фармацевтически приемлемые соли трифосфатного сложного эфира, а также фармацевтически приемлемые соли производных 5'-монофосфатных и 5'-дифосфатных сложных эфиров структурных формул IV и V, соответственно: Термин "5'-(S-ацил-2-тиоэтил)фосфат" или "SATE" относится к производным сложных моно- или диэфиров производных 5'-монофосфатнуклеозидов настоящего изобретения структурных формул VI иVII, соответственно, а также к фармацевтически приемлемым солям сложных моноэфиров: Термин "композиция" в выражении "фармацевтическая композиция" включает активный ингредиент (ингредиенты) и инертный ингредиент (ингредиенты), который составляет носитель, а также любой продукт, который образован прямо или косвенно в результате комбинирования, комплексообразования или агрегации любых двух или более ингредиентов, или в результате распада одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции настоящего изобретения включают любые композиции,полученные в результате смешивания соединения настоящего изобретения и фармацевтически приемлемого носителя. Термины "прием" и "введение" соединения следует понимать как обеспечение соединением настоящего изобретения или пролекарственной формой соединения нуждающегося в этом индивидуума. Другой аспект настоящего изобретения относится к способу ингибирования HCV NS5B полимеразы, ингибирования HCV репликации или лечения HCV инфекции соединением настоящего изобретения в комбинации с одним или более агентов, которые можно использовать для лечения HCV инфекции. Такие агенты, активные против HCV, включают (но этим не ограничиваются) рибавирин, левовирин, вирамидин, тимозин альфа-1, интерферон-, пэгилированный интерферон- (пэгинтерферон-), комбинацию интерферона- и рибавирина, комбинацию пэгинтерферона- и рибавирина, комбинацию интерферонаи левовирина и комбинацию пэгинтерферона- и левовирина. Интерферон- включает (но этим не ограничивается) рекомбинантный интерферон-2 а (такой, как Roferon интерферон, доступный от HoffmannLaRoche, Nutley, NJ), пэгилированный интерферон-2 а (Pegasys), интерферон-2b (такой, как Intron-A интерферон, доступный от Schering Corp., Kenilworth, NJ), пэгилированный интерферон-2b (PegIntron),рекомбинантный консенсусный интерферон (такой, как интерферон alphacon-1) и очищенный продукт интерферона-. Рекомбинантный консенсусный интерферон Amgen имеет торговую марку Infergen. Левовирин представляет собой L-энантиомер рибавирина, который демонстрирует иммуномодуляторную активность, аналогичную рибавирину. Вирамидин представляет собой аналог рибавирина, раскры-8 007491 тый в WO 01/60379 (правоприемник ICN Pharmaceuticals). В соответствии со способом настоящего изобретения отдельные компоненты комбинации можно вводить отдельно в различные моменты времени на протяжении курса лечения или одновременно в разделенной или единой комбинированной форме. Поэтому настоящее изобретение следует понимать как включающее все такие схемы одновременного или чередующегося лечения, и термин "введение" следует интерпретировать соответствующим образом. Следует учитывать, что объем комбинаций соединений настоящего изобретения с другими агентами,которые можно использовать для HCV инфекции, включает, в принципе, любую комбинацию с любой фармацевтической композицией для лечения HCV инфекции. Когда соединение настоящего изобретения или его фармацевтически приемлемую соль используют в комбинации со вторым терапевтическим агентом, активным против HCV, доза каждого из соединений может быть одинаковой или отличающейся от дозы в том случае, когда это соединение используют отдельно. Для лечения HCV инфекции соединения настоящего изобретения можно также вводить в комбинации с агентом, который является ингибитором HCV NS3 серинпротеазы. HCV NS3 серинпротеаза является основным вирусным ферментом, и было показано, что она является прекрасной мишенью для ингибирования HCV репликации. Ингибиторы HCV NS3 протеазы, как на субстрате, так и без субстрата, раскрыты в WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442,WO 00/09543, WO 00/59929 и GB-2337262. HCV NS3 протеаза, как мишень для разработки ингибиторовHCV репликации и для лечения HCV инфекции, обсуждена в B.W. Dymock, "Emerging therapies for hepatitis С virus infection", Emerging Drugs, 6: 13-42 (2001). Рибавирин, левовирин и вирамидин могут проявлять свое анти-HCV действие, модулируя внутриклеточный объем гуаниннуклеотидов за счет ингибирования внутриклеточного фермента инозинмонофосфатдегидрогеназы (IMPDH). IMPDH является ферментом, ограничивающим скорость в биосинтетической схеме биосинтеза гуаниннуклеотида de novo. Рибавирин легко фосфорилируется внутриклеточно,и монофосфатное производное является ингибитором IMPDH. Таким образом, ингибирование IMPDH представляет собой еще одну подходящую мишень для обнаружения ингибиторов HCV репликации. Поэтому соединения настоящего изобретения можно также вводить в комбинации с ингибитором IMPDH,таким как VX-497, который раскрыт в WO 97/41211 и WO 01/00622 (правопреемник Vertex); еще одним(Suppl.): 165 (1993)]. Для лечения HCV инфекции соединения настоящего изобретения можно также вводить в комбинации с противовирусным агентом амантадином (1-аминоадамантан) (исчерпывающее описание этого агента см.: J. Kirschbaum, Anal. Profiles Drug Subs. 12: 1-36 (1983. Термин "фармацевтически приемлемый" подразумевает, что носитель, разбавитель или эксципиент должен быть совместим с другими ингредиентами композиции и не должен оказывать вредного воздействия на реципиента. В объем настоящего изобретения включены также фармацевтические композиции, включающие нуклеозидные соединения и их производные вместе с фармацевтически приемлемым носителем. Другим примером настоящего изобретения является фармацевтическая композиция, полученная в результате комбинации любого из описанных выше соединений и фармацевтически приемлемого носителя. Другой иллюстрацией изобретения является способ получения фармацевтической композиции, включающий комбинирование любого из описанных выше соединений и фармацевтически приемлемого носителя. В объем настоящего изобретения включены также фармацевтические композиции, пригодные для ингибирования РНК-зависимой РНК вирусной полимеразы, в частности HCV NS5B полимеразы, содержащие эффективное количество соединения настоящего изобретения и фармацевтически приемлемый носитель. Фармацевтические композиции, пригодные для лечения РНК-зависимой РНК вирусной инфекции, в частности HCV инфекции, также включены в объем настоящего изобретения, так же, как и способ ингибирования РНК-зависимой РНК вирусной полимеразы, в частности HCV NS5B полимеразы, и способ ингибирования РНК-зависимой вирусной репликации, и в частности HCV репликации. Кроме того,настоящее изобретение относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения настоящего изобретения в комбинации с терапевтически эффективным количеством другого агента, активного против РНК-зависимого РНК вируса, и в частности противHCV. Агенты, активные против HCV, включают (но этим не ограничиваются) рибавирин, левовирин,вирамидин, тимозин альфа-1, ингибитор HCV NS3 серинпротеазы, интерферон-, пэгилированный интерферон- (пэгинтерферон-), комбинацию интерферона- и рибавирина, комбинацию пэгинтерферона- и рибавирина, комбинацию интерферона- и левовирина и комбинацию пэгинтерферона- и левовирина. Интерферон- включает (но этим не ограничивается) рекомбинантный интерферон-2 а (такой,как Roferon интерферон, доступный от Hoffmann-LaRoche, Nutley, NJ), интерферон-2b (такой, как Intron-A интерферон, доступный от Schering Corp., Kenilworth, NJ), консенсусный интерферон и очищенный продукт интерферона-. Обсуждение рибавирина и его активности против HCV см.: J.O. Saunders and S.A. Raybuck,"Inosine Monophosphate Dehydrogenase: Consideration of Structure, Kinetics, and Therapeutic Potential", Ann.Rep. Med. Chem., 35: 201-210 (2000). В другом аспекте настоящего изобретения предложено использование нуклеозидных соединений, и их производных, и их фармацевтических композиций для изготовления лекарства для ингибирования РНК-зависимой РНК вирусной репликации, в частности HCV репликации, и/или для лечения РНКзависимой РНК вирусной инфекции, в частности HCV инфекции. Еще в одном аспекте настоящего изобретения нуклеозидные соединения, их производные и их фармацевтические композиции предложены в качестве лекарства для ингибирования РНК-зависимой РНК вирусной репликации, в частности HCV репликации, и/или для лечения РНК-зависимой РНК вирусной инфекции, в частности HCV инфекции. Фармацевтические композиции настоящего изобретения включают соединение структурной формулы I в качестве активного ингредиента или его фармацевтически приемлемую соль и могут также содержать фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты. Композиции изобретения включают композиции для перорального, ректального введения, наружного применения, парентерального (включая подкожное, внутримышечное и внутривенное), окулярного(глазного), легочного (через нос или с помощью ингаляции) или назального введения, хотя наиболее подходящий в данном случае способ будет зависеть от характера и тяжести подлежащего лечению состояния и от природы активного ингредиента. Они обычно могут быть представлены в единичной (стандартной) дозированной форме и приготовлены любым из известных специалистам-фармацевтам способов. На практике соединения структурной формулы I можно комбинировать в качестве активного ингредиента в однородной смеси с фармацевтическим носителем, используя обычную фармацевтическую методику смешивания. Носитель может принимать множество различных форм в зависимости от нужной для введения формы препарата, например перорального или парентерального (включая внутривенное). При приготовлении композиций для пероральных дозированных форм можно использовать любую обычную фармацевтическую среду, такую как, например, вода, гликоли, масла, спирты, вкусовые агенты, консерванты, красители и т.п. в случае жидких препаратов для перорального введения, например суспензий, эликсиров и растворов; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие вещества, разрыхлители и т.п. в случае твердых препаратов для перорального введения, таких как, например, порошки,твердые и мягкие капсулы и таблетки, причем твердые препараты для перорального введения предпочтительнее жидких препаратов. Благодаря простоте введения таблетки и капсулы являются наиболее удобными дозированными стандартными единичными формами для перорального введения, причем в этом случае обычно используют твердые фармацевтические носители. При желании на таблетки может быть нанесено покрытие с использованием стандартной водной или безводной методики. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Естественно, процент активного соединения в этих композициях может варьироваться и обычно может быть в интервале от около 2 до около 60 мас.% на единицу. Количество активного соединения в таких терапевтически подходящих композициях должно быть таким, чтобы достигалась величина эффективной дозы. Активные соединения можно также вводить интраназально, например, в форме жидких капель или спреев. Таблетки, пилюли, капсулы и т.п. могут также содержать связующее вещество, такое как трагакантовая камедь, акация, кукурузный крахмал или желатин; такие эксципиенты, как дикальцийфосфат; разрыхляющий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающий агент, такой как стеарат магния; и подслащивающий агент, такой как сахароза, лактоза или сахарин. Когда единичная доза приготовлена в форме капсулы, она может содержать, кроме материалов указанного выше типа, жидкий носитель, такой как жирное масло. Различные другие материалы могут присутствовать в виде покрытий или для модификации физической формы дозированной единицы. Например, таблетки могут быть покрыты шеллаком, сахаром или тем и другим вместе. Сироп или эликсир могут содержать в дополнение к активному ингредиенту сахарозу в качестве подслащивающего агента, метил- и пропилпарабены в качестве консервантов, красители и вкусовые агенты, такие как вишневый или апельсиновый аромат. Соединения структурной формулы I можно также вводить парентерально. Растворы или суспензии этих активных соединений можно приготовить в воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии можно также приготовить в глицерине, жидком полиэтиленгликоле и в их смесях в маслах. В обычных условиях хранения и использования эти препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, пригодные для использования в виде инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных растворов или дисперсий для инъекций непосредственно перед использованием. Во всех случаях форма должна быть стерильной и должна быть жидкой, чтобы ее можно было легко вводить через шприц. Она должна быть стабильной в условиях приготовления и хранения и должна быть предохранена от загрязнений, вызываемых действиями микроорганизмов, таких как бактерии и грибки. Носителем может быть растворитель или дисперсионная среда, содержащие, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.- 10007491 Любой подходящий способ введения можно использовать для обеспечения млекопитающего, особенно человека, эффективной дозой соединения настоящего изобретения. Например, можно использовать пероральный, ректальный, наружный, парентеральный, окулярный, легочный, назальный и т.п. способы. Дозированные формы включают таблетки, пастилки, дисперсии, суспензии, растворы, капсулы,кремы, мази, аэрозоли и т.п. Предпочтительно соединения структурной формулы I вводить перорально. Для перорального введения людям интервал доз составляет от 0,01 до 1000 мг/кг массы тела в раздельных дозах. В одном воплощении интервал доз составляет от 0,1 до 100 мг/кг массы тела в раздельных дозах. В другом воплощении интервал доз составляет от 0,5 до 20 мг/кг массы тела в раздельных дозах. Для перорального введения композиции предпочтительно приготавливают в форме таблеток или капсул, содержащих от 1,0 до 1000 мг активного ингредиента, в частности 1, 5, 10, 15, 20, 25, 50, 75, 100,150, 200, 250, 300, 400, 500, 600, 750, 800, 900 и 1000 мг активного ингредиента для симптоматического подбора дозы для подлежащего лечению пациента. Используемая эффективная доза активного ингредиента может меняться в зависимости от конкретно используемого соединения, способа введения, подлежащего лечению состояния и тяжести подлежащего лечению состояния. Такие дозы легко может определить специалист в данной области. Такую схему приема доз можно подобрать, чтобы обеспечить оптимальную терапевтическую реакцию. Соединения настоящего изобретения содержат один или более асимметричных центров и поэтому могут существовать в виде рацематов или рацемических смесей, отдельных энантиомеров, смесей диастереоизомеров и отдельных диастереоизомеров. Подразумевается, что настоящее изобретение включает нуклеозидные соединения, имеющие -D-стереохимическую конфигурацию пятичленного фуранозного кольца, как представлено структурной формулой далее, то есть нуклеозидные соединения, в которых заместители у С-1 и С-4 пятичленного фуранозного кольца имеют -стереохимическую конфигурацию(ориентация "вверх", как обозначено жирной линией). Некоторые из раскрытых здесь соединений содержат олефиновые двойные связи, и, если нет других указаний, подразумевается, что они включают как Е-, так и Z-геометрические изомеры. Некоторые из раскрытых здесь соединений могут существовать в таутомерной форме, такой как кетоенольные таутомеры. Отдельные таутомеры, а также их смеси включены в соединения структурной формулы I. Пример кетоенольных таутомеров, которые включены в соединения настоящего изобретения,проиллюстрирован далее: Соединения структурной формулы I можно разделить на отдельные диастереоизомеры, например, с помощью фракционной кристаллизации из подходящего растворителя, например метанола или этилацетата или их смеси, или с помощью хиральной хроматографии, используя оптически активную неподвижную фазу. Альтернативно, любой стереоизомер соединения структурной формулы I можно получить в результате стереоспецифического синтеза, используя оптически чистые исходные материалы или реагенты известной конфигурации. Стереохимию заместителей в положениях С-2 и С-3 фуранозного кольца соединений настоящего изобретения структурной формулы I обозначают волнистыми линиями, которые означают, что заместители R1, R2, R3 и R4 могут быть в - (заместитель "вниз") или - (заместитель "вверх") конфигурации независимо друг от друга. Обозначение стереохимии жирной линией, как у С-1 и С-4 фуранозного кольца,означает, что заместитель имеет -конфигурацию (заместитель "вверх"). Соединения настоящего изобретения можно вводить в форме фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические или органические основания и неорганические или органические кислоты. Соли основных соединений, охватываемые термином "фармацевтически приемлемая соль", относятся к нетоксичным солям соединений настоящего изобретения,которые обычно получают, осуществляя взаимодействие свободного основания с соответствующей органической или неорганической кислотой. Представительные соли основных соединений настоящего изобретения включают (но этим не ограничиваются) следующие: ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, камзилат (camsylate), карбонат, хлорид, клавуланат (clavulanate),цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, фумарат, глуцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, гидроксинафтоат, иодид,изотионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат,метилсульфат, мукат, напсилат, нитрат, аммониевая соль N-метилглюкамина, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, сульфат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Кроме того, если соединения настоящего изобретения содержат кислотную группу, тогда их фармацевтически приемлемые соли включают (но этим не ограничиваются) соли, полученные из неорганических оснований, включая алюминий, аммоний, кальций, медь, железо(III), железо(II), литий, магний, марганец(III), марганец(II), калий,натрий, цинк и т.п. Наиболее предпочтительны соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных,вторичных и третичных аминов, циклических аминов и основных ионообменных смол, таких как аргинин,бетаин, каффеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин,гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п. Кроме того, если в соединениях настоящего изобретения присутствует группа карбоновой кислоты(-СООН) или спиртовая группа, можно использовать фармацевтически приемлемые сложные эфиры производных карбоновой кислоты, такие как метиловый, этиловый или пивалоилоксиметиловый, или ацильные производные спиртов, такие как ацетат или малеат. Включены также те сложноэфирные и ацильные группы, которые известны специалистам в данной области для модификации растворимости или характеристик гидролиза для использования в качестве композиций пролонгированного действия или пролекарственных форм. Получение нуклеозидных соединений и производных настоящего изобретения Нуклеозидные соединения и их производные настоящего изобретения можно получить в соответствии с прочно установившимися способами синтеза, используемыми в химии нуклеозидов и нуклеотидов. Ссылка сделана на следующий текст для описания способов синтеза, используемых при получении соединений настоящего изобретения: "Chemistry of Nucleosides and Nucleotides", L.B. Townsend, ed., Vols. 1-3, Plenum Press, 1988, который включен в описание в качестве ссылки в полном объеме. Представительный общий способ получения соединений настоящего изобретения представлен далее на схеме 1. Эта схема иллюстрирует синтез соединений настоящего изобретения структурной формулы 1-7, где фуранозное кольцо имеет -D-рибоконфигурацию. Исходным материалом служит 3,5-бисО-защищенный алкилфуранозид, такой как метилфуранозид структурной формулы 1-1. С-2 гидроксильную группу окисляют, используя подходящий окисляющий агент, такой как трехокись хрома или реагент хромата, периодинан Dess-Martin, или используя окисление Сверна, получая С-2 кетон структурной формулы 1-2. Присоединение реагента Гриньяра, такого как алкил-, алкенил- или алкинилмагнийгалогенид (например, MeMgBr, EtMgBr, винил-МgВr, аллил-MgBr и этинил-MgBr) или алкил-, алкенил- или алкиниллитий, такого как MeLi, по карбонильной двойной связи 1-2 в подходящем органическом растворителе, таком как тетрагидрофуран, диэтиловый эфир и т.п., приводит к получению С-2 третичного спирта структурной формулы 1-3. Затем вводят подходящую защитную группу (такую как Cl, Br и I) в С 1 (аномерном) положении фуранозного сахарного производного, используя обработку фуранозида формулы 1-3 галогенводородом в подходящем органическом растворителе, таком как бромистый водород в уксусной кислоте, получая промежуточный фуранозилгалогенид 1-4. С-1 сульфонат, такой как метан- 12007491 сульфонат (MeSO2O-), трифторметансульфонат (CF3SO2O-) или п-толуолсульфонат (-OTs), также может служить подходящей отщепляемой группой в последующей реакции получения гликозидной (нуклеозидной) связи. Нуклеозидную связь создают, обрабатывая промежуточное соединение структурной формулы 1-4 солью металла (такого, как литий, калий или натрий) соответствующим образом замещенного 1 Н-пирроло[2,3-d]пиримидина 1-5, такого как соответствующим образом замещенный 4-гало-1 Нпирроло[2,3-d]пиримидин, который можно получить in situ в результате обработки гидридом щелочного металла (таким, как гидрид натрия), гидроксидом щелочного металла (таким, как гидроксид калия), карбонатом щелочного металла (таким, как карбонат натрия) или гексаметилдисилазидом щелочного металла (таким, как NaHMDS) в подходящем безводном органическом растворителе, таком как ацетонитрил,тетрагидрофуран, 1-метил-2-пирролидинон или N,N-диметилформамид (ДМФ). Реакцию замещения можно вести с катализатором, используя катализатор фазового переноса, такой как TDA-1 или триэтилбензиламмонийхлорид, в двухфазной системе (твердое-жидкость или жидкость-жидкость). Необязательные защитные группы в защищенном нуклеозиде структурной формулы 1-6 затем отщепляют, используя обычные способы удаления защитных групп, такие, как описаны в T.W. Greene and P.G.M. Wuts, "ProtectiveGroups in Organic Synthesis", 3rd ed., John WileySons, 1999. Необязательное введение аминогруппы в положение 4 пирроло[2,3-d]пиримидиновых ядер осуществляют, используя обработку 4-галогенного промежуточного соединения 1-6 соответствующим амином, таким как спиртовой аммиак или жидкий аммиак, для получения первичного амина в положении С-4 (-NН 2), алкиламином для получения вторичного амина (-NHR) или диалкиламином для получения третичного амина (-NRR'). Соединение 7 Нпирроло[2,3-d]пиримидин-4(3 Н)-он можно получить в результате гидролиза 1-6 водным основанием,таким как водный гидроксид натрия. В результате алкоголиза (такого, как метанолиз) 1-6 получают С-4 алкоксид (-OR), тогда как в результате обработки алкилмеркаптидом получают С-4 алкилтио (-SR) производное. Последующие химические манипуляции, хорошо известные специалистам в области органической/ медицинской химии, могут понадобиться для получения нужных соединений настоящего изобретения. Схема 1 В приводимых далее примерах имеются ссылки на литературные публикации, которые содержат подробные указания относительно способов получения целевых соединений или промежуточных соединений, используемых при получении целевых соединений настоящего изобретения. Нуклеозидные соединения настоящего изобретения были получены в соответствии со способами, подробно описанными в следующих примерах. Эти примеры никоим образом не ограничивают объем настоящего изобретения, и их не следует рассматривать с этой точки зрения. Специалистам в области синтеза нуклеозидов и нуклеотидов будет очевидно, что для получения этих и других соединений настоящего изобретения можно- 13007491 использовать известные варианты условий и процессов приводимых способов получения этих соединений. Все температуры указаны в градусах Цельсия, если нет других указаний. Пример 1. 4-Амино-7-(2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К трехокиси хрома (1,57 г, 1,57 ммоль) в дихлорметане (DCM) (10 мл) при 0 С добавляли уксусный ангидрид (145 мг, 1,41 ммоль) и затем пиридин (245 мг, 3,10 ммоль). Смесь перемешивали в течение 15 мин,затем добавляли раствор 7-[3,5-O-[1,1,3,3-тетракис(1-метилэтил)-1,3-дисилоксандиил]D-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин-4-амина (способ получения см.: J. Am. Chem. Soc. 105: 4059 (1983) 508 мг, 1,00 ммоль) в DCM (3 мл). Полученный раствор перемешивали в течение 2 ч, затем выливали в этилацетат (10 мл) и затем фильтровали через силикагель, используя этилацетат в качестве элюента. Объединенные фильтраты выпаривали в вакууме, помещали в смесь диэтиловый эфир/ТГФ (1:1) (20 мл),охлаждали до -78 С и по каплям добавляли метилмагнийбромид (3 М, в ТГФ) (3,30 мл, 10 ммоль). Смесь перемешивали при -78 С в течение 10 мин, затем оставляли охлаждаться до комнатной температуры и гасили, добавляя насыщенный водный аммонийхлорид (10 мл), и экстрагировали DCM (20 мл). Органическую фазу выпаривали в вакууме и сырой продукт очищали на силикагеле, используя 5% метанол в дихлорметане в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме. Полученное масло помещали в ТГФ (5 мл) и добавляли тетрабутиламмонийфторид (TBAF) на силикагеле (1,1 ммоль/г силикагеля) (156 мг). Смесь перемешивали при комнатной температуре в течение 30 мин,фильтровали и выпаривали в вакууме. Сырой продукт очищали на силикагеле, используя 10% метанол в дихлорметане в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме,получая нужное соединение (49 мг) в виде бесцветного твердого вещества. 1 Стадия А. 3,5-бис-О-(2,4-Дихлорфенилметил)-1-О-метилD-рибофураноза. Смесь 2-О-ацетил-3,5-бис-О-(2,4-дихлорфенилметил)-1-О-метилD-рибофуранозы (способ получения см. : Helv. Chim. Acta 78: 486 (1995 (52,4 г, 0,10 моль) в метанольном K2 СO3 (500 мл, насыщенный при комнатной температуре) перемешивали при комнатной температуре в течение 45 мин и затем концентрировали при пониженном давлении. Маслянистый остаток суспендировали в CH2Cl2 (500 мл),промывали водой (300 мл + 5x200 мл) и солевым раствором (200 мл), сушили (Na2O4), фильтровали и концентрировали, получая указанное в заголовке соединение (49,0 г) в виде бесцветного масла, которое использовали без дополнительной очистки на стадии В далее. 1H-ЯМР (ДМСО-d6):3,28 (с, 3 Н, ОСН 3), 3,53 (d, 2H, J5,4=4,5 Гц, Н-5 а, Н-5b), 3,72 (дд, 1 Н, J3,4=3,6 Гц,J3,2=6,6 Гц, Н-3), 3,99 (ддд, 1 Н, J2,1=4,5 Гц, J2,OH-2=9, 6 Гц, Н-2), 4,07 (м, 1 Н, Н-4), 4,50 (с, 2 Н, СH2 Рh), 4,52,4,60 (2 д, 2 Н, Jgem=13,6 Гц, СH2 Рh), 4,54 (д, 1 Н, ОН-2), 4,75 (d, 1H, Н-1), 7,32-7,45, 7,52-7,57 (2 м, 10 Н, 2Ph). 13 С-ЯМР (ДМСО-d6):55,40, 69,05, 69,74, 71,29, 72,02, 78,41, 81,45, 103,44, 127,83, 127,95, 129,05,129,28, 131,27, 131,30, 133,22, 133,26, 133,55, 133,67, 135,45, 135,92. Стадия В. 3,5-бис-О-(2,4-Дихлорфенилметил-1-O-метилD-эритропентофураноз-2-улоза. К охлажденной льдом суспензии Dess-Martin периодинана (50,0 г, 118 ммоль) в безводном СН 2 С 12(350 мл) в атмосфере аргона (Аr) добавляли по каплям раствор соединения стадии А (36,2 г, 75 ммоль) в безводном СН 2 С 12 (200 мл) в течение 0,5 ч. Реакционную смесь перемешивали при 0 С в течение 0,5 ч и затем при комнатной температуре в течение 3 суток. Смесь разбавляли безводным Et2O (600 мл) и выливали в охлажденную льдом смесь Na2S2O35H2O (180 г) в насыщенном водном NаНСО 3 (1400 мл). Слои разделяли и органический слой промывали насыщенным водным NаНСО 3 (600 мл), водой (800 мл) и солевым раствором (600 мл), сушили (МgSO4), фильтровали и выпаривали, получая указанное в заголовке соединение(34,2 г) в виде бесцветного масла, которое использовали без дополнительной очистки на стадии С далее. Н-ЯМР (CDCl3):3,50 (с, 3 Н, OСН 3), 3,79 (дд, 1 Н, J5a,5b=11,3 Гц, J5a,4=3,5 Гц, Н-5 а), 3,94 (дд, 1 Н,J5b,4=2,3 Гц, Н-5b), 4,20 (дд, 1 Н, J3,1=1,3 Гц, J3,4=8,4 Гц, Н-3), 4,37 (ддд, 1 Н, Н-4), 4,58, 4,69 (2 д, 2 Н, Jgem=13,0 Гц,СH2 Рh), 4,87 (д, 1 Н, Н-1), 4,78, 5,03 (2 д, 2 Н, Jgem=12,5 Гц, CH2Ph), 7,19-7,26, 7,31-7,42 (2 м, 10 Н, 2Ph). 13 С-ЯМР (ДMCO-d6):55,72, 69,41, 69,81, 69,98, 77,49, 78,00, 98,54, 127,99, 128,06, 129,33, 129,38,131,36, 131,72, 133,61, 133,63, 133,85, 133,97, 134,72, 135,32, 208,21. Стадия С. 3,5-бис-О-(2,4-Дихлорфенилметил)-2-С-метил-1-O-метилD-рибофураноза. К раствору MeMgBr в безводном Et2O (0,48 M, 300 мл) при -55 С добавляли по каплям раствор соединения стадии В (17,40 г, 36,2 ммоль) в безводном Et2O (125 мл). Реакционную смесь оставляли нагреваться до -30 С и перемешивали в течение 7 ч при температуре от -30 до -15 С, затем выливали в охлажденную льдом воду (500 мл) и смесь интенсивно перемешивали при комнатной температуре в течение 0,5 ч. Смесь фильтровали через слой целита (10x5 см), который тщательно промывали Et2O. Органический слой сушили (MgSO4), фильтровали и концентрировали. Остаток растворяли в гексане (30 мл),вводили в колонку с силикагелем (10x7 см, предварительно набитую в гексане) и элюировали гексаном и гексаном/EtOAc (9/1), получая указанное в заголовке соединение (16,7 г) в виде бесцветного сиропа. 1 Н-ЯМР (CDCl3):1,36 (д, 3 Н, JMe,OH=0,9 Гц, 2 С-Ме), 3,33 (к, 1 Н, ОН), 3,41 (д, 1 Н, J3,4=3,3 Гц), 3,46(с, 3 Н, ОСН 3), 3,66 (д, 2 Н, J5,4=3,7 Гц, Н-5 а, Н-5b), 4,18 (кажущийся кв, 1 Н, Н-4), 4,52 (с, 1 Н, Н-1), 4,60 (с,2 Н, CH2Ph), 4,63, 4,81 (2 д, 2 Н, Jgem=13,2 Гц, СH2 Рh), 7,19-7,26, 7,34-7,43 (2 м, 10 Н, 2Ph). 13 С-ЯМР (CDCl3):24,88, 55,45, 69,95, 70,24, 70,88, 77,06, 82,18, 83,01, 107,63, 127,32, 129,36,130,01, 130,32, 133,68, 133,78, 134,13, 134,18, 134,45, 134,58. Стадия D. 4-Хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии С (9,42 г, 19 ммоль) в безводном дихлорметане (285 мл) при 0 С по каплям добавляли НВr (5,7 М в уксусной кислоте, 20 мл, 114 ммоль). Полученный раствор перемешивали при 0 С в течение 1 ч и затем при комнатной температуре в течение 3 ч, выпаривали в вакууме и выпаривали совместно с безводным толуолом (3x40 мл). Маслянистый остаток растворяли в безводном ацетонитриле (50 мл) и добавляли к раствору натриевой соли 4-хлор-1 Н-пирроло[2,3-d]пиримидина в ацетонитриле (получали in situ из 4-хлор-1 Н-пирроло [2,3-d]пиримидина (способ получения см.: J. Chem. Soc.: 131 (1960 (8,76 г, 57 ммоль) в безводном ацетонитриле (1000 мл) и NaH (60% в минеральном масле, 2,28 г,57 ммоль) после 4 ч интенсивного перемешивания при комнатной температуре). Объединенную смесь перемешивали при комнатной температуре в течение 24 ч и затем выпаривали досуха. Остаток суспендировали в воде (250 мл) и экстрагировали EtOAc (2x500 мл). Объединенные экстракты промывали солевым раствором (300 мл), сушили над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали на колонке с силикагелем (10 х 10 см), используя этилацетат/гексан (1:3 и 1:2) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (5,05 г) в виде бесцветной пены. 1H-ЯМР (CDCl3):0,93 (с, 3 Н, СН 3), 3,09 (с, 1 Н, ОН), 3,78 (дд, 1 Н, J5',5"=10,9 Гц, J5',4=2,5 Гц, Н-5'),3,99 (дд, 1 Н, J5",4=2,2 Гц, Н-5"), 4,23-4,34 (м, 2 Н, Н-3', Н-4'), 4,63, 4,70 (2 д, 2 Н, Jgem=12,7 Гц, CH2Ph), 4,71,4,80 (2 д, 2 Н, Jgem=12,l Гц, СH2 Рh), 6,54 (д, 1 Н, J5,6=3,8 Гц, Н-5), 7,23-7,44 (м, 10 Н, 2Ph). 13 С-ЯМР (CDCl3):21,31, 69,10, 70,41, 70,77, 79,56, 80,41, 81,05, 91,11, 100,57, 118,21, 127,04, 127,46,127,57, 129,73, 129,77, 130,57, 130,99, 133,51, 133,99, 134,33, 134,38, 134,74, 135,21, 151,07, 151,15, 152,47. Стадия E. 4-Xлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии D (5,42 г, 8,8 ммоль) в дихлорметане (175 мл) при температуре -78 С по каплям добавляли треххлористый бор (1 M в дихлорметане, 88 мл, 88 ммоль). Смесь перемешивали при -78 С в течение 2,5 ч, затем при температуре от -30 до -20 С в течение 3 ча. Реакцию гасили, добавляя метанол/дихлорметан (1:1) (90 мл), и полученную смесь перемешивали при -15 С в течение 30 мин,затем нейтрализовали водным аммиаком при 0 С и перемешивали при комнатной температуре в течение 15 мин. Твердую часть фильтровали и промывали СН 2 Сl2/МеОН (1/1, 250 мл). Объединенный фильтрат выпаривали и остаток очищали, используя флеш-хроматографию на силикагеле, используя градиентное элюирование СН 2 Сl2 и СН 2 Сl2:МеОН (99:1, 98:2, 95:5 и 90:10), получая нужное соединениe (1,73 г) в виде бесцветной пены, которая превращалась в аморфное твердое вещество после обработки MeCN. 1(д, 1 Н, Н-6) , 8,65 (с, 1 Н, Н-2). 13 С-ЯМР (ДМСО-d6):20,20, 59,95, 72,29, 79,37, 83,16, 91,53, 100,17, 117,63, 128,86, 151,13, 151,19, 151,45. Стадия F. 4-Амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К соединению стадии Е (1,54 г, 5,1 ммоль) добавляли метанольный аммиак (насыщенный при 0 С; 150 мл). Смесь нагревали в автоклаве из нержавеющей стали при 85 С в течение 14 ч, затем охлаждали и выпаривали в вакууме. Сырую смесь очищали на колонке с силикагелем, используя СН 2 Сl2/МеОН (9/1) в качестве элюента и получая указанное в заголовке соединение в виде бесцветной пены (0,8 г), которое выделяли в виде аморфного твердого вещества после обработки MeCN. Аморфное твердое вещество перекристаллизовывали из метанола/ацетонитрила; т.пл. 222 С. 1 Н-ЯМР (ДМСО-d6):0,62 (с, 3 Н, СН 3), 3,57-3,67 (м, 1 Н, Н-5'), 3,75-3,97 (м, 3 Н, Н-5", Н-4', Н-3'),- 15007491 5,00 (с, 1 Н, 2"-ОН), 5,04 (д, 1 Н, J3'OH,3'=6,8 Гц, 3'-ОН), 5,06 (т, 1 Н, J5'OH,5',5"=5,1 Гц, 5'-ОН), 6,11 (с, 1 Н, Н-1'),6,54 (д, 1 Н, J5,6=3,6 Гц, Н-5), 6,97 (шир.с, 2 Н, NH2), 7,44 (д, 1 Н, Н-6), 8,02 (с, 1 Н, Н-2). 13 С-ЯМР (ДМСО-d6):20,26, 60,42, 72,72, 79,30, 82,75, 91,20, 100,13, 103,08, 121,96, 150,37, 152,33, 158,15. Жидкостная хроматография-масс-спектроскопия (ЖХ-МС): найдено: 279,10 (М-Н+); рассчитано для Стадия А. 3,5-бис-О-(2,4-Дихлорфенилметил)-2-С-этил-1-O-метилD-рибофураноза. К диэтиловому эфиру (300 мл) при -78 С медленно добавляли EtMgBr (3,0 M, 16,6 мл) и затем по каплям соединение стадии В примера 2 (4,80 г, 10,0 ммоль) в безводном Et2O (100 мл). Реакционную смесь перемешивали при -78 С в течение 15 мин, оставляли нагреться до -15 С и перемешивали в течение еще 2 ч, затем выливали в перемешиваемую смесь воды (300 мл) и Et2O (600 мл). Органическую фазу выделяли, сушили (MgSO4) и выпаривали в вакууме. Сырой продукт очищали на силикагеле, используя этилацетат/гексан (1:2) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (3,87 г) в виде бесцветного масла. Стадия В. 4-Xлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-этилD-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии А (1,02 мг, 2,0 ммоль) в дихлорметане (40 мл) по каплям добавляли НВr (5,7 М в уксусной кислоте) (1,75 мл, 10,0 ммоль) при 0 С. Полученный раствор перемешивали при комнатной температуре в течение 2 ч, выпаривали в вакууме и дважды выпаривали совместно с толуолом (10 мл). Маслянистый остаток растворяли в ацетонитриле (10 мл) и добавляли к интенсивно перемешиваемой смеси 4-хлор-1 Н-пирроло[2,3-d]пиримидина (307 мг, 2,0 ммоль), гидроксида калия (337 мг,6,0 ммоль) и трис[2-(2-метоксиэтокси)этил]амина (130 мг, 0,4 ммоль) в ацетонитриле (10 мл). Полученную смесь перемешивали при комнатной температуре в течение ночи и затем выливали в перемешиваемую смесь насыщенного аммонийхлорида (100 мл) и этилацетата (100 мл). Органический слой выделяли,промывали солевым раствором (100 мл), сушили над MgSO4, фильтровали и выпаривали в вакууме. Сырой продукт очищали на силикагеле, используя этилацетат/гексан (1:2) в качестве элюента и получая нужный продукт (307 мг) в виде бесцветной пены. Стадия C. 4-Xлор-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии В (307 мг, 0,4 5 ммоль) в дихлорметане (8 мл) добавляли треххлористый бор (1 М в дихлорметане) (4,50 мл, 4,50 ммоль) при -78 С. Смесь перемешивали при -78 С в течение 1 ч, затем при -10 С в течение 3 ч. Реакцию гасили, добавляя метанол/дихлорметан (1:1) (10 мл), перемешивали при -15 С в течение 30 мин и нейтрализовали, добавляя водный гидроксид аммония. Смесь выпаривали при пониженном давлении и полученное масло очищали на силикагеле, используя метанол/дихлорметан (1:9) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (112 мг) в виде бесцветной пены. Стадия D. 4-Aмино-7-(2-С-этилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К соединению стадии С (50 мг, 0,16 ммоль) добавляли насыщенный аммиак в метаноле (4 мл). Смесь перемешивали при 75 С в течение 72 ч в закрытом контейнере, охлаждали и выпаривали в вакууме. Сырую смесь очищали на силикагеле, используя метанол/дихлорметан (1:9) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (29 мг) в виде бесцветного порошка. 1- 16007491 Стадия А. 2-Амино-4-хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-метилD-рибофуранозил)7 Н-пирроло[2,3-d]пиримидин. К охлажденному льдом раствору продукта стадии С примера 2 (1,27 г, 2,57 ммоль) в СН 2 Сl2 (30 мл) по каплям добавляли НВr (5,7 М в уксусной кислоте; 3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали при пониженном давлении и выпаривали совместно с толуолом (2x15 мл). Полученное масло растворяли в ацетонитриле (MeCN) (15 мл) и добавляли по каплям к хорошо перемешиваемой смеси 2-амино-4-хлор-7 Н-пирроло[2,3-d]пиримидина (способ получения см.: Heterocycles 35: 825 (1993 (433 мг, 2,57 ммоль), KОН (85%, порошок) (0,51 г, 7,7 ммоль), трис[2-(2-метоксиэтокси)этил]амина(165 мкл, 0,51 ммоль) в ацетонитриле (30 мл). Полученную смесь перемешивали при комнатной температуре в течение 1 ч, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем, используя гексан/EtOAc,5/1, 3/1 и 2/1, в качестве элюента и получая указанное в заголовке соединение в виде бесцветной пены (0,65 г). Стадия В. 2-Aмино-4-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору продукта стадии А (630 мг, 1,0 ммоль) в СН 2 Сl2 (20 мл) при -78 С добавляли треххлористый бор (1 М в СН 2 Сl2) (10 мл, 10 ммоль). Смесь перемешивали при -78 С в течение 2 ч, затем при -20 С в течение 2,5 ч. Реакцию гасили, используя СН 2 Сl2/МеОН (1:1) (10 мл), перемешивали при -20 С в течение 0,5 ч и нейтрализовали при 0 С водным аммиаком. Твердую часть фильтровали, промывали СН 2 Сl2/МеОН (1:1) и объединенный фильтрат выпаривали в вакууме. Остаток очищали на колонке с силикагелем, используя СН 2 Сl2/МеОН,50/1 и 20/1, в качестве элюента, получая указанное в заголовке соединение в виде бесцветной пены (250 мг). Стадия С. 2-Aмино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он. Смесь продукта стадии В (90 мг, 0,3 ммоль) в водном NaOH (2 н, 9 мл) нагревали при температуре кипения с обратным холодильником в течение 5 ч, затем нейтрализовали при 0 С 2 н водной НСl и выпаривали досуха. В результате очистки на колонке с силикагелем с использованием СН 2 Сl2/МеОН, 5/1, в качестве элюента получали указанное в заголовке соединение в виде твердого вещества белого цвета (70 мг). 1 Раствор 2-амино-4-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина (пример 4,стадия В) (21 мг, 0,07 ммоль) в циклопропиламине (0,5 мл) нагревали при 70 С в течение 2 суток, затем выпаривали до маслянистого остатка и очищали на колонке с силикагелем, используя СН 2 Сl2/МеОН, 20/1, в качестве элюента, получая указанное в заголовке соединение в виде твердого вещества белого цвета (17 мг). 1 Это соединение получали по способу, описанному Y. Murai et al. в Heterocycles 33: 391-404 (1992). Пример 7. 4-Амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-5-карбоксамид. Это соединение получали по способу, описанному Y. Murai et al. в Heterocycles 33: 391-404 (1992). Пример 8. Общий способ получения SATE пролекарственного фрагмента."Pronucleotides: Toward the In Vivo Delivery of Antiviral and Anticancer Nucleotides", Med. Res. Rev., 20: 135 (2000), причем содержание включено в описание в качестве ссылки в полном объеме. SATE производные нуклеозидов раскрыты также в патентах США 5770725, 5849905 и 6020482, содержание которых также включено в описание в качестве ссылки в полном объеме. бис(S-Ацетил-2-тиоэтил)-N,N-диизопропилфосфорамидит. 2-Меркаптоэтанол (5 г, 64 ммоль) растворяли в CH2Cl2 (50 мл). К этому раствору добавляли триэтиламин (7,67 мл, 57,6 ммоль) и реакционную смесь охлаждали в бане со льдом до 0 С. За 10 мин по каплям добавляли уксусный ангидрид (4,54 мл, 48 ммоль) и реакционную смесь перемешивали в течение 1 ч при 0 С. Затем реакционной смеси давали нагреться до комнатной температуры в течение 2 ч. Реакционную смесь разбавляли СН 2 Сl2 (50 мл), промывали водой (75 мл), 5% водным NаНСО 3 (75 мл) и солевым раствором (75 мл). Органическую фазу сушили над безводным Na2SO4 и концентрировали в вакууме, получая масло. Затем это масло растворяли в безводном ТГФ (40 мл) и добавляли безводный триэтиламин (7,76 мл). К этой смеси добавляли активированные молекулярные сита (4 ) и выдерживали при комнатной температуре в течение 10 мин. Реакционную смесь охлаждали в бане со льдом до 0 С и добавляли диизопропилфосфорамидный дихлорид (6,47 г, 32,03 ммоль). Реакционную смесь перемешивали при 0 С в течение 2 ч в инертной атмосфере. К реакционной смеси добавляли гексан (40 мл) и образующийся осадок отфильтровывали. Полученный фильтрат концентрировали до четверти объема, очищали хроматографически на колонке с силикагелем и элюировали гексаном, содержащим 3% триэтиламина и возрастающее количество этилацетата (0-7%), получая указанное в заголовке соединение в виде масла (2,36 г). 1H-ЯМР (CDCl3):1,17 (с, 6 Н), 1,21 (с, 6 Н), 2,36 (с, 6 Н), 3,14 (т, J=6,44 Гц), 3,51-3,84 (м, 6 Н). 13 С-ЯМР (CDCl3):24,47, 24,61, 30,48, 42,85, 43,1, 61,88, 62,23, 195,26. 13 Р-ЯМР (CDCl3):146,96. Пример 9. 5'-Трифосфатные производные. Нуклеозидные 5'-трифосфаты настоящего изобретения получали по способам, описанным в Chem.Rev. 100: 2047 (2000). Пример 10. Очистка и анализ степени чистоты 5'-трифосфатных производных. Трифосфатные производные очищали с помощью анионообменной (АХ) хроматографии, используя колонку 30x100 мм Mono Q (Pharmacia) с буферной системой 50 мМ Tris, pH 8. Градиенты элюирования обычно составляли от 40 мМ NaCl до 0,8 М NaCl в двух объемах колонки при скорости 6,5 мл/мин. Соответствующие фракции, полученные с помощью анионообменной хроматографии, собирали и обессоливали с помощью хроматографии с обращенной фазой (RP), используя колонку Luna C18 250x21 мм (Phenomenex) при скорости потока 10 мл/мин. Градиенты элюирования обычно составляли от 1 до 95% метанола через 14 мин при постоянной концентрации 5 мМ триэтиламмонийацетата (ТЕАА). Масс-спектры очищенных трифосфатов получали, используя прибор, где скомбинированы последовательно ВЭЖХ и масс-спектрометр Hewlett-Packard (Palo Alto, CA) MSD 1100. Для ВЭЖХ с обращенной фазой использовали Phenomenex Luna (C18(2, 150x2 мм, плюс 30x2 мм guard колонку с размером частиц 3 мкм. Осуществляли градиентное элюирование с линейным градиентом от 0 до 50% (15 мин) ацетонитрила в 20 мМ ТЕАА (триэтиламмонийацетат) рН 7 с подключенным последовательно массспектральным детектором в режиме отрицательной ионизации. Для создания потока ионов использовали газообразный азот и пневматический распылитель. Получали массы в интервале 150-900. Молекулярные массы определяли, используя аналитические программы HP Chemstation analysis package. Степень чистоты очищенных трифосфатов определяли с помощью аналитической ВЭЖХ с обращенной фазой и анионообменной ВЭЖХ. ВЭЖХ с обращенной фазой с колонкой Phenomonex Luna илиJupiter (250x4,6 мм), с размером частиц 5 мкм обычно осуществляли с градиентом 2-70% ацетонитрила в 100 мМ ТЕАА, рН 7, через 15 мин. Анионообменную ВЭЖХ осуществляли на колонке 1,6x5 мм Mono Q(Pharmacia). Трифосфаты элюировали с градиентом от 0 до 0,4 М NaCl при постоянной концентрации 50 мМ Tris, рН 8. Степень чистоты трифосфатов обычно составляет более 80%. Пример 11. 5'-Монофосфатные производные. Нуклеозидные 5'-монофосфаты настоящего изобретения получали по способу, описанному вTetrahedron Lett. 50: 5065 (1967). Пример 12. Масс-спектральные характеристики 5'-трифосфатных производных. Масс-спектры 5'-трифосфатов соединений настоящего изобретения получали по способу примера 10. В следующей таблице перечислены расчетные и экспериментальные массы для представительных 5'-трифосфатов,полученных по способу примера 9. Номера примеров соответствуют исходному соединению 5'-трифосфата. К соединению стадии F примера 2 (14 мг, 0,05 ммоль) (высушенному в результате совместного испарения с пиридином и несколько раз с толуолом) добавляли триметилфосфат (0,5 мл). Смесь перемешивали в течение ночи в запаянном контейнере. Затем ее охлаждали до 0 С и через шприц добавляли хлорокись фосфора (0,0070 мл, 0,075 ммоль). Смесь перемешивали в течение 3 ч при 0 С, затем реакцию гасили,добавляя бикарбонат тетраэтиламмония (ТЕАВ) (1 М) (0,5 мл) и воду (5 мл). Реакционную смесь очищали и анализировали по способу примера 10. Масс-спектр с рассеянием электронов (ES-MS): найдено: 359,2 (М-Н+); рассчитано для С 12 Н 17N4 О 7 Р-Н+: 359,1. Пример 14. [4-Амино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-5'-дифосфат. К соединению стадии F примера 2 (56 мг, 0,20 ммоль) (высушенного совместным выпариванием с пиридином и несколько раз с толуолом) добавляли триметилфосфат (хранившийся над ситами) (1,0 мл). Смесь перемешивали в течение ночи в запаянном контейнере. Затем ее охлаждали до 0 С и через шприц добавляли хлорокись фосфора (0,023 мл, 0,25 ммоль). Смесь перемешивали в течение 2 ч при 0 С, затем добавляли трибутиламин (0,238 мл, 1,00 ммоль) и трибутиламмонийфосфат (полученный из фосфорной кислоты и трибутиламина в пиридине с последующей азеотропной перегонкой с пиридином и ацетонитрилом) (1,0 ммоль в 3,30 мл ацетонитрила). Смесь перемешивали в течение дополнительных 30 мин при 0 С, затем открывали запаянную ампулу и реакцию гасили, добавляя ТЕАВ (1 М) (1,0 мл) и воду (5 мл). Реакционную смесь очищали и анализировали по способу примера 10. К соединению стадии F примера 2 (20 мг, 0,07 ммоль) (высушенного совместным выпариванием с пиридином и несколько раз с толуолом) добавляли триметилфосфат (хранившийся над ситами) (0,4 мл). Смесь перемешивали в течение ночи в запаянном контейнере. Затем ее охлаждали до 0 С и через шприц добавляли хлорокись фосфора (0,0070 мл, 0,075 ммоль). Смесь перемешивали в течение 3 ч при 0 С, затем добавляли трибутиламин (0,083 мл, 0,35 ммоль), трибутиламмонийфосфат (127 мг, 0,35 ммоль) и ацетонитрил (хранившийся над ситами) (0,25 мл). Смесь перемешивали в течение дополнительных 30 мин при 0 С, затем открывали запаянную ампулу и реакцию гасили, добавляя ТЕАВ (1 М) (0,5 мл) и воду (5 мл). Реакционную смесь очищали и анализировали по способу примера 10.- 19007491 К соединению стадии Е примера 2 (59 мг, 0,18 ммоль) добавляли водный гидроксид натрия (1 М). Смесь нагревали при кипении с обратным холодильником в течение 1 ч, охлаждали, нейтрализовали водной НСl (2 М) и выпаривали в вакууме. Остаток очищали на силикагеле, используя дихлорметан/метанол (4:1) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (53 мг) в виде бесцветного масла. 1 Н-ЯМР (CD3CN):0,70 (с, 3 Н), 3,34-4,15 (перекрывающийся м, 7 Н), 6,16 (с, 1 Н), 6,57 (д, 3,6 Гц,1 Н), 7,37 (д, 3,6 Гц, 1 Н), 8,83 (с, 1 Н). Пример 17. 4-Амино-5-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К предварительно охлажденному (0 С) раствору соединения стадии F примера 2 (140 мг, 0,50 ммоль) в ДМФ (2,5 мл) по каплям добавляли N-хлорсукцинимид (0,075 г, 0,55 ммоль) в ДМФ (0,5 мл). Раствор перемешивали при комнатной температуре в течение 1 ч, реакцию гасили, добавляя метанол (4 мл), и выпаривали в вакууме. Сырой продукт очищали на силикагеле, используя метанол/дихлорметан (1:9) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (55 мг) в виде бесцветного твердого вещества. 1 Н-ЯМР (CD3CN):0,80 (с, 3 Н), 3,65-4,14 (перекрывающийся м, 7 Н), 5,97 (шир.с, 2 Н), 6,17 (с, 1 Н),7,51 (с, 1 Н), 8,16 (с, 1 Н). К предварительно охлажденному (0 С) раствору соединения стадии F примера 2 (28 мг, 0,10 ммоль) в ДМФ (0,5 мл) по каплям добавляли N-бромсукцинимид (0,018 г, 0,10 ммоль) в ДМФ (0,5 мл). Раствор перемешивали при 0 С в течение 20 мин, затем при комнатной температуре в течение 10 мин. Реакционный раствор гасили, добавляя метанол (4 мл), и выпаривали в вакууме. Сырой продукт очищали на силикагеле, используя метанол/дихлорметан (1:9) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (13,0 мг) в виде бесцветного твердого вещества. 1 Смесь 2-амино-4-хлор-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидина (пример 4,стадия В) (20 мг, 0,07 ммоль) в EtOH (1,0 мл), пиридина (0,1 мл) и 10% Pd/C (6 мг) в атмосфере Н 2 (давление атмосферное) перемешивали в течение ночи при комнатной температуре. Смесь фильтровали через слой целита, который тщательно промывали EtOH. Объединенный фильтрат выпаривали и очищали на колонке с силикагелем, используя СН 2 Сl2/МеОН, 20/1 и 10/1, в качестве элюента, получая указанное в заголовке соединение в виде твердого вещества белого цвета (16 мг). 1 Стадия А. 2-Амино-4-хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-метилD-рибофуранозил]-5 метил-7 Н-пирроло[2,3-d]пиримидин. К охлажденному льдом раствору продукта стадии С примера 2 (1,57 г, 3,16 ммоль) в CH2Cl2 (50 мл) по каплям добавляли НВr (5,7 М в уксусной кислоте; 3,3 мл). Реакционную смесь перемешивали при 0 С в течение 1 ч и затем при комнатной температуре в течение 2 ч, концентрировали в вакууме и выпаривали совместно с толуолом (2x20 мл). Полученное масло растворяли в MeCN (20 мл) и добавляли по каплям к раствору натриевой соли 2-амино-4-хлор-5-метил-1 Н-пирроло[2,3-d]пиримидина в ацетонитриле (получен in situ из 2 амино-4-хлор-5-метил-1 Н-пирроло[2,3-d]пиримидина (способ получения см.: Liebigs Ann. Chem. 1984: 708-721)(1,13 г, 6,2 ммоль) в безводном ацетонитриле (150 мл) и NaH (60% в минеральном масле, 248 мг, 6,2 ммоль),после 2 ч перемешивания при комнатной температуре). Объединенную смесь перемешивали при комнатной температуре в течение 24 ч и затем выпаривали досуха. Остаток суспендировали в воде (100 мл) и экстрагировали EtOAc (300+150 мл). Объединенные экстракты промывали солевым раствором (100 мл), сушили над (Na2SO4), фильтровали и выпаривали. Сырой продукт очищали на колонке с силикагелем (5x7 см), используя этилацетат/гексан (от 0 до 30% EtOAc; 5% ступенчатый градиент) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (0,96 г) в виде бесцветной пены. Стадия В. 2-Амино-4-хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С,2-O-диметилD-рибофуранозил]-5-метил-7 Н-пирроло[2,3-d]пиримидин. К охлажденной льдом смеси продукта стадии А (475 мг, 0,7 ммоль) в ТГФ (7 мл) добавляли NaH(60% в минеральном масле, 29 мг) и перемешивали при 0 С в течение 0,5 ч. Затем добавляли MeI (48 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Реакцию гасили МеОН и смесь выпаривали. Сырой продукт очищали на колонке с силикагелем (5x3,5 см), используя гексан/этилацетат (9/1, 7/1, 5/1 и 3/1) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали,получая нужное соединение (200 мг) в виде бесцветной пены. Стадия С. 2-Амино-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С,2-O-диметилD-рибофуранозил]-5 метил-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он. Смесь продукта стадии В (200 мг, 0,3 ммоль) в 1,4-диоксане (15 мл) и водного NaOH (2 н, 15 мл) в автоклаве нагревали в течение ночи при 135 С. Затем смесь охлаждали до 0 С, нейтрализовали 2 н водной НСl и выпаривали досуха. Сырой продукт суспендировали в МеОН, фильтровали и твердую часть тщательно промывали МеОН. Объединенный фильтрат концентрировали и остаток очищали на колонке с силикагелем (5x5 см), используя СН 2 Сl2/МеОН (40/1, 30/1 и 20/1) в качестве элюента, получая нужное соединение (150 мг) в виде бесцветной пены. Стадия D. 2-Амино-5-метил-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он. Смесь продукта стадии С (64 мг, 0,1 ммоль) в МеОН (5 мл), Et3N (0,2 мл) и 10% Pd/C (24 мг) гидрировали в аппарате для гидрирования Парра при давлении 50 пси при комнатной температуре в течение 1,5 суток, затем фильтровали через слой целита, который тщательно промывали МеОН. Объединенный фильтрат выпаривали и остаток очищали на колонке с силикагелем (3x4 см), используя CH2Cl2/MeOH(30/1, 20/1) в качестве элюента, получая 2-амино-5-метил-7-(5-O-бензил-2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин-4(3 Н)-он. Это соединение гидрировали далее в EtOH (2 мл), используя 10% Pd/C при атмосферном давлении водорода. После перемешивания в течение 2 дней при комнатной температуре реакционную смесь фильтровали через целит, фильтрат выпаривали и сырой продукт очищали на колонке с силикагелем (1x7 см), используя СН 2 Сl2/МеОН (30/1, 20/1 и 10/1) в качестве элюента, получая после сушки вымораживанием указанное в заголовке соединение (12 мг). 1- 21007491 Стадия А. 4-Хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-метилD-рибофуранозил]-5-метил 7 Н-пирроло[2,3-d]пиримидин. К охлажденному льдом раствору продукта стадии С примера 2 (1,06 г, 2,1 ммоль) в СН 2 Сl2 (30 мл) по каплям добавляли НВr (5,7 М в уксусной кислоте; 2,2 мл). Реакционную смесь перемешивали при 0 С в течение 1 ч и затем при комнатной температуре в течение 2 ч, концентрировали в вакууме и выпаривали совместно с толуолом (2x15 мл). Полученное масло растворяли MeCN (10 мл) и добавляли по каплям к раствору натриевой соли 4-хлор-5-метил-1 Н-пирроло[2,3-d]пиримидина в ацетонитриле (получен in situ из 4-хлор-5-метил-1 Н-пирроло[2,3-d]пиримидина (способ получения см.: J. Med. Chem. 33: 1984 (1990(0,62 г, 3,7 ммоль) в безводном ацетонитриле (70 мл) и NaH (60% в минеральном масле, 148 мг, 3,7 ммоль),после 2 ч интенсивного перемешивания при комнатной температуре). Объединенную смесь перемешивали при комнатной температуре в течение 24 ч, а затем выпаривали досуха. Остаток суспендировали в воде(100 мл) и экстрагировали EtOAc (250+100 мл). Объединенные экстракты промывали солевым раствором(50 мл), сушили над Na2SO4, фильтровали и выпаривали. Сырой продукт очищали на колонке с силикагелем(5x5 см), используя градиент гексан/этилацетат (9/1, 5/1, 3/1) в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (0,87 г) в виде бесцветной пены. Стадия В. 4-Хлор-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии А (0,87 г, 0,9 ммоль) в дихлорметане (30 мл) при -78 С по каплям добавляли треххлористый бор (1 M в дихлорметане, 9,0 мл, 9,0 ммоль). Смесь перемешивали при -78 С в течение 2,5 ч, затем при температуре от -30 до -20 С в течение 3 ч. Реакцию гасили, добавляя метанол/дихлорметан (1:1) (9 мл), и полученную смесь перемешивали при -15 С в течение 30 мин, затем нейтрализовали водным аммиаком при 0 С и перемешивали при комнатной температуре в течение 15 мин. Твердую часть фильтровали и промывали СН 2 Сl2/МеОН (1/1, 50 мл). Объединенный фильтрат выпаривали и остаток очищали на колонке с силикагелем (5x5 см), используя СН 2 Сl2 и градиент СН 2 Сl2/МеОН(40/1 и 30/1) в качестве элюента до получения нужного соединения (0,22 г) в виде бесцветной пены. Стадия С. 4-Амино-5-метил-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К соединению стадии В (0,2 г, 0,64 ммоль) добавляли метанольный аммиак (насыщенный при 0 С; 40 мл). Смесь нагревали в автоклаве из нержавеющей стали при 100 С в течение 14 ч, затем охлаждали и выпаривали в вакууме. Сырую смесь очищали на колонке с силикагелем (5x5 см), используя градиент СН 2 Сl2/МеОН (50/1, 30/1,20/1) в качестве элюента, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,12 г). 1 Соединение примера 6 (0,035 г, 0,11 ммоль) растворяли в смеси водного аммиака (4 мл, 30 мас.%) и насыщенного метанольного аммиака (2 мл) и добавляли раствор Н 2O2 в воде (2 мл, 35 мас.%). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Растворитель удаляли при пониженном давлении и полученный остаток очищали с помощью ВЭЖХ на колонке с обращенной фазой (Altech AltimaC-18, 10x299 мм, А=вода, В=ацетонитрил, 10-60% В в течение 50 мин, скорость потока 2 мл/мин), получая указанное в заголовке соединение (0,015 г, 41%) в виде твердого вещества белого цвета. 1H-ЯМР (CD3OD):0,85 (с, 3 Н, Me), 3,61 (м, 1 Н), 3,82 (м, 1 Н), 3,99-4,86 (м, 2 Н), 6,26 (с, 1 Н), 8,10 (с,2 Н), 8,22 (с, 1 Н). 13 С-ЯМР (CD3OD):20,13, 61,37, 73,79, 80,42, 84,01, 93,00, 102,66, 112,07, 130,07, 151,40, 152,74, 159,12, 169,30. Масс-спектр высокого разрешения (HRMS) (с бомбардировкой быстрыми атомами (FAB: рассчитано для C13H17N4O6+: 325,1148; найдено: 325,1143. Пример 23. 4-Амино-7-(2-С-винилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин.- 22007491 Стадия А. 3,5-бис-О-(2,4-Дихлорфенилметил)-2-С-винил-1-O-метилD-рибофураноза. Гептагидрат хлорида церия (50 г, 134,2 ммоль) тонко измельчают в предварительно нагретой ступке и переносят в круглодонную колбу, снабженную механической мешалкой. Колбу нагревали при высоком вакууме в течение ночи при 160 С. Вакуум заменяли атмосферой аргона и колбу охлаждали до комнатной температуры. В колбу через канюлю вводили безводный ТГФ (300 мл). Полученную суспензию перемешивали при комнатной температуре в течение 4 ч и затем охлаждали до -78 С. Добавляли винилмагнийбромид (1 M в ТГФ, 120 мл, 120 ммоль) и перемешивание продолжали при -78 С в течение 2 ч. К этой суспензии по каплям добавляли раствор 3,5-бис-О-(2,4-дихлорфенилметил)-1-О-метилDэритропентофураноз-2-улозы (14 г, 30 ммоль) (примера 2, стадии В) в безводном ТГФ (100 мл) при постоянном перемешивании. Реакционную смесь перемешивали при -78 С в течение 4 ч. Реакцию гасили насыщенным раствором аммонийхлорида и оставляли нагреваться до комнатной температуры. Смесь фильтровали через целит и остаток промывали Et2O (2x500 мл). Органический слой выделяли и водный слой экстрагировали Et2O (2x200 мл). Объединенные органические слои сушили над безводным Na2SO4 и концентрировали до вязкого масла желтого цвета. Это масло очищали, используя флеш-хроматографию(SiO2, 10% EtOAc в гексане). Указанное в заголовке соединение (6,7 г, 13,2 ммоль) получали в виде масла бледно-желтого цвета. Стадия В. 4-Хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-винилD-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии А (6,4 г, 12,6 ммоль) в безводном дихлорметане (150 мл) при -20 С по каплям добавляли НВr (30% раствор в АсОН, 20 мл, 75,6 ммоль). Полученный раствор перемешивали при температуре между -10 и 0 С в течение 4 ч, выпаривали в вакууме и выпаривали совместно с безводным толуолом (3x40 мл). Маслянистый остаток растворяли в безводном ацетонитриле (100 мл) и добавляли к раствору натриевой соли 4-хлор-1 Н-пирроло[2,3-d]пиримидина (5,8 г, 37,8 ммоль) в ацетонитриле(полученной in situ по способу примера 2) при -20 С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 24 ч. Затем смесь выпаривали досуха, помещали в воду и экстрагировали EtOAc (2x300 мл). Объединенные экстракты сушили надEtOAc в гексане), и указанное в заголовке соединение (1,75 г) выделяли в виде пены белого цвета. Стадия С. 4-Амино-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-винилD-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии В (80 мг) растворяли в минимальном количестве 1,4-диоксана и помещали в контейнер из нержавеющей стали. Контейнер охлаждали до -78 С и добавляли жидкий аммиак. Контейнер запаивали и нагревали при 90 С в течение 24 ч. Аммиаку давали испариться и остаток концентрировали до твердого вещества белого цвета, которое использовали на следующей стадии без дополнительной очистки. Стадия D. 4-Амино-7-(2-С-винилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К раствору соединения стадии С (60 мг) в дихлорметане при -78 С по каплям добавляли треххлористый бор (1 М в дихлорметане). Смесь перемешивали при -78 С в течение 2,5 ч, затем при температуре от -30 до -20 С в течение 3 ч. Реакцию гасили, добавляя метанол/дихлорметан (1:1), и полученную смесь перемешивали при -15 С в течение 0,5 ч, затем нейтрализовали водным аммиаком при 0 С и перемешивали при комнатной температуре в течение 15 мин. Твердую часть фильтровали и промывали метанолом/дихлорметаном (1:1). Объединенный фильтрат выпаривали и остаток очищали, используя флешхроматографию (SiO2, 10% метанол в EtOAc, содержащий 0,1% триэтиламина). Фракции, содержащие продукт, выпаривали, получая указанное в заголовке соединение в виде твердого вещества белого цвета Стадия А. 4-Хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-гидроксиметилD-рибофуранозил]7 Н-пирроло[2,3-d]пиримидин. К раствору соединения примера 23, стадии В (300 мг, 0,48 ммоль) в 1,4-диоксане (5 мл) добавлялиN-метилморфолин-N-оксид (300 мг, 2,56 ммоль) и четырехокись осмия (4% раствор в воде, 0,3 мл). Смесь перемешивали в темноте в течение 14 ч. Осадок удаляли фильтрованием через слой целита, разбавляли водой (3 х) и экстрагировали EtOAc. EtOAc слой сушили над Na2SO4 и концентрировали в вакууме. Маслянистый остаток помещали в дихлорметан (5 мл) и перемешивали над NaIO4 на силикагеле(3 г, 10% NaIO4) в течение 12 ч. Силикагель удаляли фильтрацией, а остаток выпаривали и помещали в абсолютный этанол (5 мл). Раствор охлаждали в бане со льдом и небольшими порциями добавляли боргидрид натрия (300 мг, 8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 4 ч и затем разбавляли EtOAc. Органический слой промывали водой (2x20 мл), солевым раствором(20 мл) и сушили над Na2SO4. Растворитель выпаривали и остаток очищали, используя флеш-хроматографию (SiO2, 2:1 гексан/EtOAc), получая указанное в заголовке соединение (160 мг, 0,25 ммоль) в виде хлопьев белого цвета. Стадия В. 4-Амино-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-гидроксиметилD-рибофуранозил]7 Н-пирроло[2,3-d]пиримидин. Соединение стадии А (150 мг, 0,23 ммоль) растворяли в минимальном количестве 1,4-диоксана (10 мл) и помещали в контейнер из нержавеющей стали. Бомбу охлаждали до -78 С и добавляли жидкий аммиак. Контейнер запаивали и затем нагревали при 90 С в течение 24 ч. Аммиаку давали испариться и остаток концентрировали до твердого вещества белого цвета, которое использовали на следующей стадии без дополнительной очистки. Стадия С. 4-Амино-7-(2-С-гидроксиметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии В (120 мг, 0,2 ммоль) растворяли в 1:1 метанол/дихлорметан, добавляли 10%Pd/C и полученную суспензию перемешивали в атмосфере Н 2 в течение 12 ч. Катализатор удаляли фильтрованием через слой целита и промывали большими количествами метанола. Объединенный фильтрат выпаривали в вакууме и остаток очищали, используя флеш-хроматографию (SiO2, 10% метанол в EtOAc, содержащем 0,1% триэтиламина), получая указанное в заголовке соединение (50 мг) в виде порошка белого цвета. 1 Стадия А. 4-Хлор-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-фторметилD-рибофуранозил]-7 Нпирроло[2,3-d]пиримидин. К раствору соединения примера 24, стадии А (63 мг, 0,1 ммоль) в безводном дихлорметане (5 мл) в атмосфере аргона добавляли 4-диметиламинопиридин (ОМАР) (2 мг, 0,015 ммоль) и триэтиламин (62 мкл,0,45 ммоль). Раствор охлаждали в бане со льдом и добавляли п-толуолсульфонилхлорид (30 мг, 0,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, промывали NaHCO3(2x10 мл), водой (10 мл), солевым раствором (10 мл), сушили над Na2SO4 и концентрировали в вакууме до твердого вещества розового цвета. Твердую часть растворяли в безводном ТГФ (5 мл) и охлаждали в бане со льдом. Добавляли тетрабутиламмонийфторид (1 М раствор в ТГФ, 1 мл, 1 ммоль) и смесь перемешивали при комнатной температуре в течение 4 ч. Растворитель удаляли в вакууме, остаток помещали в дихлорметан и промывали NаНСО 3 (2x10 мл), водой (10 мл) и солевым раствором (10 мл). Слой дихлорметана сушили над безводным Na2SO4, концентрировали в вакууме и очищали, используя флешхроматографию (SiO2, 2:1 гексан/EtOAc), получая указанное в заголовке соединение (20 мг) в виде твердого вещества белого цвета. Стадия В. 4-Амино-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С-фторметилD-рибофуранозил]-7 Нпирроло[2,3-d]пиримидин. Соединение стадии А (18 мг, 0,03 ммоль) растворяли в минимальном количестве 1,4-диоксана и помещали в контейнер из нержавеющей стали. Контейнер охлаждали до -78 С и добавляли жидкий аммиак. Контейнер запаивали и затем нагревали при 90 С в течение 24 ч. Аммиаку давали испариться и остаток концентрировали до твердого вещества белого цвета, которое использовали на следующей стадии без дополнительной очистки. Стадия С. 4-Амино-7-(2-С-фторметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии В (16 мг) растворяли в 1:1 метаноле/дихлорметане, добавляли 10% Pd/C и полу- 24007491 ченную суспензию перемешивали в атмосфере Н 2 в течение 12 ч. Катализатор удаляли фильтрованием через слой целита и промывали большими количествами метанола. Объединенный фильтрат выпаривали в вакууме и остаток очищали, используя флеш-хроматографию (SiO2, 10% метанол в EtOAc, содержащий 0,1% триэтиламина), получая указанное в заголовке соединение (8 мг) в виде порошка белого цвета. 1 Стадия A. 7-[2,5-бис-O-(трет-Бутилдиметилсилил)D-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин и 7-[3,5-бис-O-(трет-бутилдиметилсилил)D-рибофуранозил]-7 Н-пирроло[2,3-d]пиримидин. К перемешиваемому раствору туберцидина (5,0 г, 18,7 ммоль) в смеси пиридина (7,5 мл) и ДМФ(18,5 мл) добавляли нитрат серебра (6,36 г, 38,8 ммоль). Эту смесь перемешивали при комнатной температуре в течение 2 ч. Ее охлаждали в бане со льдом, добавляли ТГФ (37,4 мл) и трет-бутилдиметилсилилхлорид (5,6 г, 37 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь фильтровали через слой целита и промывали ТГФ. Фильтрат и промывки разбавляли простым эфиром,содержащим небольшое количество хлороформа. Органический слой промывали последовательно бикарбонатом натрия и водой (3x50 мл), сушили над безводным сульфатом натрия и концентрировали. Пиридин удаляли испарением совместно с толуолом и остаток очищали, используя флеш-хроматографию на силикагеле, используя 5-7% МеОН в СН 2 Сl2 в качестве элюента; выход 3,0 г. Стадия В. 7-[2,5-бис-О-(трет-Бутилдиметилсилил)D-рибофуранозил]-4-[ди(4-метоксифенил)фенилметил]амино-7 Н-пирроло[2,3-d]пиримидин и 7-[3,5-бис-О-(трет-бутилдиметилсилил)D-рибофуранозил]-4-[ди(4-метоксифенил)фенилметил]амино-7 Н-пирроло[2,3-d]пиримидин. К раствору смеси соединений стадии А (3,0 г, 6,0 ммоль) в безводном пиридине (30 мл) добавляли 4,4'-диметокситритилхлорид (2,8 г, 8,2 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем эту смесь тщательно растирали с водным пиридином и экстрагировали эфиром. Органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали до пены желтого цвета (5,6 г). Остаток очищали, используя флеш-хроматографию на силикагеле,используя 20-25% EtOAc в гексане в качестве элюента. Соответствующие фракции собирали и концентрировали, получая 2',5'-бис-О-(трет-бутилдиметилсилил)- и 3',5'-бис-О-(трет-бутилдиметилсилил)защищенные нуклеозиды в виде бесцветной пены (2,2 г и 1,0 г, соответственно). Стадия С. 7-[2,5-бис-О-(трет-Бутилдиметилсилил)-3-O-тозилD-рибофуранозил]-4-[ди(4-метоксифенил)фенилметил]амино-7 Н-пирроло[2,3-d]пиримидин. К охлажденному льдом раствору 2',5'-бис-О-(трет-бутилдиметилсилил)защищенному нуклеозиду стадии В (2,0 г, 2,5 ммоль) в пиридине (22 мл) добавляли п-толуолсульфонилхлорид (1,9 г, 9,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 суток. Затем ее тщательно растирали с водным пиридином (50%, 10 мл) и экстрагировали эфиром (3x50 мл), содержащим небольшое количество СН 2 Сl2 (10 мл). Органический слой промывали бикарбонатом натрия и водой (3x30 мл). Органический слой сушили над безводным Na2SO4 и концентрировали. Пиридин удаляли при совместном испарении с толуолом (3x25 мл). Оставшееся масло фильтровали через слой силикагеля, используя гексан:этилацетат (70:30) в качестве элюента; выход 1,4 г. Стадия D. 4-[Ди(4-метоксифенил)фенилметил]амино-7-[3-O-тозилD-рибофуранозил-7 Н-пирроло[2,3-d]пиримидин. Раствор соединения стадии С (1,0 г, 1,1 ммоль) и ТГФ (10 мл) перемешивали с тетрабутиламмонийфторидом (1 М раствор в ТГФ, 2,5 мл) в течение 0,5 ч. Смесь охлаждали и разбавляли простым эфиром (50 мл). Раствор промывали водой (3x50 мл), сушили над безводным Na2SO4 и концентрировали до масла. Остаток очищали, пропуская через слой силикагеля, используя гексан:этилацетат (1:1) в качестве элюента; выход 780 мг. Стадия Е. 4-Амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин и 4 амино-7-(3-дезокси-2-С-метилD-арабинофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Раствор CH3MgI (3,0 M раствор в простом эфире, 3,0 мл) в безводном толуоле (3,75 мл) охлаждали- 25007491 в бане со льдом. К этому добавляли раствор соединения стадии D (500 мг, 0,8 ммоль) в безводном толуоле (3,7 мл). Полученную смесь перемешивали при комнатной температуре в течение 3,5 ч. Ее охлаждали и обрабатывали водным раствором NH4Cl и экстрагировали простым эфиром (50 мл, содержащим 10 млCH2Cl2). Органический слой выделяли и промывали солевым раствором (2 х 30 мл) и водой (2x25 мл), сушили над безводным Na2SO4 и концентрировали до масла, которое очищали, используя флеш-хроматографию на силикагеле, используя 4% МеОН в СН 2 Сl2, получая соединение 2-Сметил (149 мг) и соединение 2-Сметил (34 мг). Эти производные отдельно обрабатывали 80% уксусной кислотой и реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч. Уксусную кислоту удаляли при повторных совместных испарениях с этанолом и толуолом. Остаток разделяли между хлороформом и водой. Водный слой промывали хлороформом и концентрировали. После выпаривания остаток очищали на силикагеле, используя 5-10% МеОН в СН 2 Сl2 в качестве элюента, получая нужные соединения в виде твердого вещества белого цвета. 4-Амино-7-(3-дезокси-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин (90 мг). 1(55,7 мл, 0,4 моль) растворяли в дихлорметане (300 мл). Добавляли п-толуолсульфонилхлорид (38,13 г,0,2 моль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь затем выливали в насыщенный водный раствор бикарбоната натрия (500 мл), и два слоя разделялись. Органический слой промывали водным раствором лимонной кислоты (20%, 200 мл), сушили(Na2SO4) и выпаривали, получая твердое вещество (70,0 г). Твердую часть растворяли в сухом ТГФ (300 мл) и порциями в течение 30 мин добавляли LiAlH4 (16,0 г, 0,42 моль). Смесь перемешивали при комнатной температуре в течение 15 мин. В течение 30 мин по каплям добавляли этилацетат (100 мл) и смесь фильтровали через слой силикагеля. Фильтрат концентрировали и полученное масло обрабатывали хроматографически на силикагеле (EtOAc/гексан 1/4), получая продукт в виде твердого вещества (32,5 г). Стадия В. 3,5-бис-О-(2,4-Дихлорфенилметил)-1-O-метил-4-метилD-рибофураноза. Оксид хрома (50 г, 0,5 моль), уксусный ангидрид (50 мл, 0,53 моль) и пиридин (100 мл, 1,24 моль) добавляли к дихлорметану (1 л) в бане со льдом и смесь перемешивали в течение 15 мин. Добавляли 5 дезокси-1,2-O-изопропилиден-D-ксилофуранозу (32 г, 0,18 моль) в дихлорметане (200 мл) и смесь перемешивали при той же температуре в течение 30 мин. Реакционный раствор разбавляли этилацетатом (1 л) и фильтровали через слой силикагеля. Фильтрат концентрировали, получая масло желтого цвета. Это масло растворяли в 1,4-диоксане (1 л) и формальдегиде (37%, 200 мл). Раствор охлаждали до 0 С и добавляли твердый KОН (50 г). Смесь перемешивали при комнатной температуре в течение ночи и затем экстрагировали этилацетатом (6x200 мл). После концентрирования остаток обрабатывали хроматографически на силикагеле (EtOAc), получая продукт в виде масла (1,5 г). Это масло растворяли в 1-метил-2 пирролидиноне (20 мл) и добавляли 2,4-дихлорфенилметилхлорид (4 г, 20,5 ммоль) и NaH (60%, 0,8 г). Смесь перемешивали в течение ночи и разбавляли толуолом (100 мл). Затем смесь промывали насыщенным водным бикарбонатом натрия (3 х 50 мл), сушили (Na2SO4) и выпаривали. Остаток растворяли в метаноле (50 мл) и добавляли НСl в диоксане (4 М, 2 мл). Раствор перемешивали в течение ночи и выпаривали. Остаток обрабатывали хроматографически на силикагеле (EtOAc/гексан: 1/4), получая нужный продукт в виде масла (2,01 г). Стадия С. 3,5-бис-О-(2,4-Дихлорфенилметил)-2,4-ди-С-метил-1-О-метилD-рибофураноза. Продукт (2,0 г, 4,0 ммоль) стадии В и периодинан Dess-Martin (2,0 г) в дихлорметане (30 мл) пере- 26007491 мешивали в течение ночи при комнатной температуре и затем концентрировали при пониженном давлении. Остаток тщательно растирали с простым эфиром (50 мл) и фильтровали. Фильтрат промывали раствором Na2S2O35H2O (2,5 г) в насыщенном водном растворе бикарбоната натрия (50 мл), сушили(MgSO4), фильтровали и выпаривали. Остаток растворяли в безводном Et2O (20 мл) и по каплям добавляли к раствору MeMgBr в Et2O (3 М, 10 мл) при -78 С. Реакционную смесь оставляли нагреваться до -30 С и перемешивали при температуре от -30 до -15 С в течение 5 ч, затем выливали в насыщенный водный раствор аммонийхлорида (50 мл). Два слоя разделяли и органический слой сушили (MgSO4), фильтровали и концентрировали. Остаток обрабатывали хроматографически на силикагеле (EtOAc/гексан: 1/9),получая указанное в заголовке соединение в виде сиропа (1,40 г). Стадия D. 4-Хлор-7-[3,5-бис-O-(2,4-дихлорфенилметил)-2,4-ди-С-метилD-рибофуранозил]-7 Нпирроло[2,3-d]пиримидин. К соединению стадии С (0,70 г, 1,3 ммоль) добавляли НВr (5,7 М в уксусной кислоте, 2 мл). Полученный раствор перемешивали при комнатной температуре в течение 1 ч, выпаривали в вакууме и выпаривали совместно с безводным толуолом (3x10 мл). 4-Хлор-7 Н-пирроло[2,3-d]пиримидин (0,5 г, 3,3 ммоль) и порошок KОН (85%, 150 мг, 2,3 ммоль) перемешивали в 1-метил-2-пирролидиноне (5 мл) в течение 30 мин и смесь выпаривали совместно с толуолом (10 мл). Полученный раствор выливали в вышеуказанный бромсахарный остаток и смесь перемешивали в течение ночи. Смесь разбавляли толуолом (50 мл), промывали водой (3x50 мл) и концентрировали при пониженном давлении. Остаток обрабатывали хроматографически на силикагеле, элюируя смесью EtOAc/гексан (15/85), получая твердое вещество (270 мг). Стадия Е. 4-Амино-7-(2,4-С-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии D (270 мг) растворяли в диоксане (2 мл) и добавляли жидкий аммиак (20 г) в автоклав из нержавеющей стали. Смесь нагревали при 100 С в течение 15 мин, затем охлаждали и выпаривали. Остаток обрабатывали хроматографически на силикагеле (EtOAc), получая твердое вещество (200 мг). Твердую часть (150 мг) и Pd/C (10%, 150 мг) в метаноле (20 мл) встряхивали в атмосфере Н 2 (30 пси) в течение 3 ч, фильтровали и выпаривали. Остаток обрабатывали хроматографически на силикагеле (МеОН/ СН 2 Сl2: 1/9), получая нужный продукт в виде твердого вещества (35 мг). 1 Стадия А. 3-Дезокси-3-фтор-1-О-метил-5-O-толуоилD-рибофураноза. 1,2-О-Изопропилиден-D-ксилофуранозу (9,0 г, 50 ммоль) и п-толуоилхлорид (7,0 мл, 50 ммоль) в пиридине (50 мл) перемешивали в течение 30 мин. Добавляли воду (10 мл) и смесь концентрировали при пониженном давлении. Остаток растворяли в толуоле (500 мл) и раствор промывали водой (200 мл) и насыщенным водным раствором бикарбоната натрия (200 мл). Два слоя разделяли и органический слой выпаривали. Остаток растворяли в метаноле (100 мл) и добавляли НСl в диоксане (4 М, 10 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем выпаривали при пониженном давлении. Полученное масло обрабатывали хроматографически на силикагеле (EtOAc/гексан: 1/1), получая масло (10,1 г). Это масло растворяли в дихлорметане (100 мл) и добавляли диэтиламиносульфуртрифторид (DAST) (5,7 мл). Смесь перемешивали в течение ночи и затем выливали в насыщенный водный раствор бикарбоната натрия (100 мл). Смесь экстрагировали толуолом (2x50 мл) и объединенные органические слои концентрировали. Остаток обрабатывали хроматографически на силикагеле (EtOAc/гексан: 15/85), получая указанное в заголовке соединение в виде масла (1,50 г). Стадия B. 3-Дезокси-3-фтор-2-С-метил-1-О-метил-5-O-толуоилD-рибофураноза. Продукт стадии А (1,0 г, 3,5 ммоль) и периодинан Dess-Martin (2,5 г) в дихлорметане (20 мл) перемешивали в течение ночи при комнатной температуре и затем концентрировали при пониженном давлении. Остаток тщательно растирали с простым диэтиловым эфиром (50 мл) и фильтровали. Фильтрат промывали раствором Na2S2O35H2O (12,5 г) в насыщенном водном бикарбонате натрия (100 мл), сушили(MgSO4), фильтровали и выпаривали. Остаток растворяли в безводном ТГФ (50 мл). При -78 С добавля- 27007491 ли TiCl4 (3 мл) и метилмагнийбромид в простом этиловом эфире (3 М, 10 мл) и смесь перемешивали при температуре от -50 до -30 С в течение 2 ч. Смесь выливали в насыщенный водный раствор бикарбоната натрия (100 мл) и фильтровали через целит. Фильтрат экстрагировали толуолом (100 мл) и выпаривали. Остаток обрабатывали хроматографически на силикагеле (EtOAc/гексан: 15/85), получая указанное в заголовке соединение в виде масла (150 мг). Стадия C. 4-Амино-7-(3-дезокси-3-фтор-2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Продукт стадии В (150 мг, 0,5 ммоль) растворяли в НВr (30%) в уксусной кислоте (2 мл). Через 1 ч смесь выпаривали при пониженном давлении и выпаривали совместно с толуолом (10 мл). 4-Хлор-7 Нпирроло[2,3-d]пиримидин (0,5 г, 3,3 ммоль) и KОН в виде порошка (85%, 150 мг, 2,3 ммоль) перемешивали в ДМФ (3 мл) в течение 30 мин и смесь выпаривали совместно с толуолом (2 мл). Полученный раствор выливали в вышеуказанную смесь бромсахара и перемешивали в течение ночи. Смесь разбавляли толуолом (50 мл), промывали водой (3x50 мл) и концентрировали при пониженном давлении. Остаток обрабатывали хроматографически на силикагеле (EtOAc/гексан: 15/85), получая масло (60 мг). Это масло растворяли в диоксане (2 мл) и жидкий аммиак (20 г) добавляли в автоклав из нержавеющей стали. Смесь нагревали при 85 С в течение 18 ч, затем охлаждали и выпаривали. Остаток обрабатывали хроматографически на силикагеле (метанол/дихлорметан: 1/9), получая указанное в заголовке соединение в виде твердого вещества (29 мг). 1 Н-ЯМР (ДMCO-d6):0,81 (с, 3 Н), 3,75 (м, 2 Н), 4,16 (м, 1 Н), 5,09 (дд, 1 Н, J=53,2, 7,8 Гц), 5,26 Стадия А. 4-Хлор-7-[3,5-бис-O-(2,4-дихлорфенилметил)-2-С,2-O-диметилD-рибофуранозил]-7 Нпирроло[2,3-d]пиримидин. К предварительно охлажденному (0 С) раствору соединения примера 2, стадии D (618 мг, 1,0 ммоль) в ТГФ (8 мл) добавляли метилиодид (709 мг, 5,0 ммоль) и NaH (60% в минеральном масле) (44 мг, 1,1 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре и затем выливали в перемешиваемую смесь насыщенного водного аммонийхлорида (50 мл) и дихлорметана (50 мл). Органический слой промывали водой (50 мл), сушили (MgSO4) и выпаривали в вакууме. Полученный сырой продукт очищали на силикагеле, используя этилацетат/гексан в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (735 мг) в виде бесцветной пены. Стадия В. 4-Амино-7-[3,5-бис-О-(2,4-дихлорфенилметил)-2-С,2-O-диметилD-рибофуранозил]7 Н-пирроло[2,3-d]пиримидин. К соединению стадии А (735 мг, 1,16 ммоль) добавляли метанольный аммиак (насыщенный при 0 С) (20 мл). Смесь нагревали в автоклаве из нержавеющей стали при 80 С в течение ночи, затем охлаждали и содержимое выпаривали в вакууме. Сырую смесь очищали на силикагеле, используя этилацетат/гексан в качестве элюента. Фракции, содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (504 мг) в виде бесцветной пены. Стадия С. 4-Амино-7-(2-С,2-O-диметилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Смесь продукта стадии С (64 мг, 0,1 ммоль), МеОН (5 мл), Et3N (0,2 мл) и 10% Pd/C (61 мг) гидрировали в аппарате Парра при 50 пси при комнатной температуре в течение ночи. Смесь фильтровали через целит, выпаривали в вакууме и фильтровали через слой силикагеля, используя 2% метанол в дихлорметане в качестве элюента. Нужный продукт собирали и выпаривали в вакууме. Это соединение снова растворяли в метаноле (10 мл) и добавляли 10% Pd/C (61 мг). Смесь гидрировали в аппарате Парра при 55 пси при комнатной температуре в течение 2 недель. Смесь фильтровали через целит, выпаривали в вакууме и очищали на силикагеле, используя 10% метанол в дихлорметане в качестве элюента. Фракции,содержащие продукт, объединяли и выпаривали в вакууме, получая нужный продукт (110 мг) в виде бесцветной пены. 13 С-ЯМР (ДMCО-d6):0,68 (с, 3 Н), 3,40 (с, 3 Н), 3,52-3,99 (перекрывающийся м, 4 Н), 4,92 (д, 1 Н),5,07 (т, 1 Н), 6,26 (с, 1 Н), 6,55 (д, 1 Н), 7,00 (шир.с, 2 Н), 7,46 (д, 1 Н), 8,05 (с, 1 Н). Соединение стадии Е примера 2 (200 мг, 0,67 ммоль) добавляли к метиламину (5 мл, конденсированный в небольшом автоклаве из нержавеющей стали) и нагревали при 85 С в течение 48 ч, затем охлаждали и выпаривали в вакууме. Сырую смесь очищали на силикагеле, используя этанол в качестве элюента и получая указанное в заголовке соединение, которое выделяли в виде аморфного твердого вещества после обработки MeCN. Аморфное твердое вещество растворяли в воде и лиофилизировали, получая бесцветный порошок (144 мг). 1 Н-ЯМР (ДMCO-d6):0,63 (с, 3 Н, СН 3), 3,32 (с, 3 Н, N-CH3), 3,58-3,67 (м, 1 Н, Н-5'), 3,79-3,39 (м, 3 Н,Н-5", Н-4', Н-3'), 5,03 (с, 1 Н, 2'-ОН), 5,04-5,11 (1 Н, 3'-ОН, 1 Н, 5'-ОН), 6,14 (с, 1 Н, Н-1'), 6,58 (д, 1 Н, J5,6=3,6 Гц,Н-5), 7,46 (д, 1 Н, Н-6), 7,70 (шир.с, 1 Н, NH), 8,14 (с, 1 Н, Н-2). ЖХ-МС: найдено: 295,1 (М-Н+); рассчитано для C13H18N4O4+H+: 294,3. Пример 32. 4-Диметиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии Е примера 2 (200 мг, 0,67 ммоль) добавляли к диметиламину (5 мл, конденсированному в небольшом автоклаве из нержавеющей стали) и нагревали при 85 С в течение 48 ч, затем охлаждали и выпаривали в вакууме. Сырую смесь очищали на силикагеле, используя этанол в качестве элюента и получая указанное в заголовке соединение, которое выделяли в виде аморфного твердого вещества после обработки MeCN. Аморфное твердое вещество растворяли в воде и лиофилизировали, получая бесцветный порошок (164 мг). 1 Н-ЯМР (ДМСО-d6):0,64 (с, 3 Н, СН 3), 3,29 (с, 3 Н, N-CH3), 3,32 (с, 3 Н, N-СН 3), 3,60-3,66 (м, 1 Н,Н-5'), 3,77-3,97 (м, 3 Н, Н-5", Н-4', Н-3'), 5,04 (с, 1 Н, 2'-ОН), 5,06-5,11 (1 Н, 3'-ОН, 1 Н, 5'-ОН), 6,21 (с, 1 Н,Н-1'), 6,69 (д, 1 Н, J5,6=3,6 Гц, Н-5), 7,55 (д, 1 Н, Н-6), 8,13 (с, 1 Н, Н-2). ЖХ-МС: найдено: 309,3 (М-Н+); рассчитано для C14H20N4O4+H+: 308,33. Пример 33. 4-Циклопропиламино-7-(2-С-метилD-рибофуранозил)-7 Н-пирроло[2,3-d]пиримидин. Соединение стадии Е примера 2 (200 мг, 0,67 ммоль) добавляли к циклопропиламину (5 мл, конденсированному в небольшом автоклаве из нержавеющей стали) и нагревали при 85 С в течение 48 ч, затем охлаждали и выпаривали в вакууме. Сырую смесь очищали на силикагеле, используя этанол в качестве элюента и получая указанное в заголовке соединение, которое выделяли в виде аморфного твердого вещества после обработки MeCN. Аморфное твердое вещество растворяли в воде и лиофилизировали, получая бесцветный порошок (148 мг). 1 Стадия A. 7-[2,5-бис-О-(трет-Бутилдиметилсилил)D-рибофуранозил]-4-[(4-метоксифенил)дифенилметил]амино-7 Н-пирроло[2,3-d]пиримидин и 7-[3,5-бис-О-(трет-бутилдиметилсилил)D-рибофуранозил]-4-[(4-метоксифенил)дифенилметил]амино-7 Н-пирроло[2,3-d]пиримидин. К раствору смеси соединений стадии А примеров 26 и 27 (0,32 г, 0,65 ммоль) в безводном пиридине(6 мл) добавляли монометокситритилхлорид (0,30 г, 0,98 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь затем концентрировали и остаток разделяли между СН 2 Сl2 (70 мл) и водой (20 мл). Органический слой промывали водой и солевым раствором, сушили (Na2SO4) и концентрировали. Остаток очищали на колонке с силикагелем, используя 5-13% EtOAc в гексане в качестве элюента. Соответствующие фракции собирали и концентрировали, получая 2',5'-бис-О-(трет-бутилдиметилсилил)- и 3',5'-бисО-(трет-бутилдиметилсилил)защищенные нуклеозиды в виде бесцветных пен (343 и 84 мг, соответственно). Стадия B. 7-[2,5-бис-О-(трет-Бутилдиметилсилил)D-эритропентофураноз-3-улозил]-4-[(4-метоксифенил)дифенилметил]амино-7 Н-пирроло[2,3-d]пиримидин. К тщательно перемешиваемой суспензии трехокиси хрома (91 мг, 0,91 ммоль) в СН 2 Сl2 (4 мл) при 0 С добавляли пиридин (147 мкл, 1,82 ммоль) и затем уксусный ангидрид (86 мкл, 0,91 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 ч. Затем добавляли 2',5'-бис-О-(третбутилдиметилсилил)защищенный нуклеозид стадии А (343 мг, 0,45 ммоль) в СН 2 Сl2 (2,5 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Затем смесь выливали в охлажденный льдомEtOAc (10 мл) и фильтровали через короткую колонку с силикагелем, используя EtOAc в качестве элюента. Фильтрат выпаривали и остаток очищали на колонке с силикагелем, используя гексан и гексан/EtOAc (7/1) в качестве элюента и получая указанное в заголовке соединение (180 мг). Стадия С. 7-[2,5-бис-О-(трет-Бутилдиметилсилил)-3-С-метилD-рибофуранозил]-4-[(4-метоксифенил)дифенилметил]амино-7 Н-пирроло[2,3-d]пиримидин и 7-[2,5-бис-О-(трет-бутилдиметилсилил)-3-СметилD-ксилофуранозил]-4-[(4-метоксифенил)дифенилметил]амино-7 Н-пирроло[2,3-d]пиримидин. К смеси MeMgBr (3,0 M раствор в простом эфире; 0,17 мл, 0,5 ммоль) в безводном гексане (1,5 мл) при комнатной температуре по каплям добавляли раствор соединения стадии В (78 мг, 0,1 ммоль) в безводном гексане (0,5 мл). После 2 ч перемешивания при комнатной температуре реакционную смесь выливали в охлажденную льдом воду (10 мл) и разбавляли EtOAc (20 мл), затем фильтровали через целит и затем тщательно промывали EtOAc. Слои разделяли и органический слой промывали солевым раствором, сушили (Na2SO4) и концентрировали. Остаток очищали на колонке с силикагелем, используя 8-25%EtOAc в гексане в качестве элюента и получая 3-С-метилксило- (60 мг) и 3-С-метилрибоизомер (20 мг). Стадия D. 4-Амино-7-(3-С-метилD-ксилофуранозил)-7 Н-пирроло[2,3-d]пиримидин. К охлажденному льдом раствору 3-С-метилксилоизомера стадии С (60 мг, 0,08 ммоль) в ТГФ (2 мл) добавляли TBAF (1 M в ТГФ; 0,32 мл, 0,32 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и затем разбавляли СН 2 Сl2 (50 мл), промывали водой (3x15 мл), сушили и выпаривали. Остаток растворяли в диоксане (0,3 мл) и добавляли 80% уксусную кислоту (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 суток и затем выпаривали. Остаток выпаривали совместно с диоксаном, помещали в воду (50 мл) и промывали CH2Cl2 (2x10 мл). Водный слой концентрировали и затем сушили вымораживанием. Остаток очищали на колонке с силикагелем,используя СН 2 Сl2/МеОН (20/1 и 10/1) в качестве элюента и получая указанное в заголовке соединение в виде белого пушистого соединения после сушки вымораживанием (10 мг). 1 Н-ЯМР (CD3CN):1,28 (с, 3 Н, СН 3), 3,56 (шир.с, 1 Н, ОН), 3,78 (м, 3 Н, Н-4', Н-5', Н-5"), 4,10
МПК / Метки
МПК: C07H 19/14, A61K 31/7064, A61P 31/14
Метки: производные, нуклеозидов, рнк-зависимой, полимеразы, рнк, качестве, ингибиторов, вирусной
Код ссылки
<a href="https://eas.patents.su/30-7491-proizvodnye-nukleozidov-v-kachestve-ingibitorov-rnk-zavisimojj-rnk-virusnojj-polimerazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные нуклеозидов в качестве ингибиторов рнк-зависимой рнк вирусной полимеразы</a>
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Гурвест Жан-Франсуа, Пейман Ануширван, Бодари Сара Кэтрин, Шойнеманн Карлхайнц, Гадек Томас, Карниато Дени, Кнолле Йохен, Вилл Дэвид Вильям, Макдауэлл Роберт, Катбертсон Роберт Эндрю
МПК: C07D 239/42, A61K 31/505, A61P 19/10...
Метки: клеток,способ, качестве, производные, костной, сульфонамидные, ткани, применение, ингибиторов, получения, фармацевтическая, композиция, адгезии, рассасывания
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Производные 6-фенилпиридил-2-амина, полезные в качестве ингибиторов nos

Номер патента: 2907
Опубликовано: 31.10.2002
Автор: Лоу Джон Адамс III
МПК: A61P 11/06, C07D 213/73, A61K 31/4418...
Метки: ингибиторов, качестве, 6-фенилпиридил-2-амина, полезные, производные
Формула / Реферат:
1. Соединение формулы где R1 и R2 независимо выбраны из водорода, гидрокси, метила и метокси; и G является группой формулы где n равно нулю или единице; Y представляет собой NR3R4, (С1-С6)алкил или аралкил, где арильная группировка указанного аралкила является фенилом или нафтилом, а алкильная группировка является нормальной или разветвленной и содержит от 1 до 6 атомов углерода и где указанный (С1-С6)алкил и арильная группировка указанного...
Сульфонамидные производные в качестве пролекарств, ингибиторов аспартилпротеаз

Номер патента: 3509
Опубликовано: 26.06.2003
Авторы: Кальдор Иштван, Спалтенштейн Эндрю, Ферфайн Эрик Стивен, Танг Роджер Д., Хейл Майкл Р., Бейкер Кристофер Т., Казмерский Веслав Мечислав
МПК: A61P 31/18, A61K 31/63, C07C 311/16...
Метки: аспартилпротеаз, сульфонамидные, пролекарств, производные, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы где R7 выбран из -PO32-Na2+, -PO32-K2+, -PO32-Mg2+, 2. Соединение по п.1, где R7 выбран из -PO32-Na2+, -PO32-K2+ или -PO32-Ca2+. 3. Соединение по п.2, где R7 представляет собой -PO32-Ca2+. 4. Фармацевтическая композиция, содержащая соединение, охарактеризованное в любом из пп.1-3, в количестве, эффективном для лечения инфекции, вызванной вирусом, характеризующимся наличием аспартилпротеазы; и фармацевтически приемлемый...
Производные ациклических нуклеозидов.

Номер патента: 1404
Опубликовано: 26.02.2001
Авторы: Хегберг Марита, Линдборг Бьерн, Энгельхардт Пер, Жоу Ксиао-Ксионг, Йоханссон Нильс Гуннар
МПК: A61P 31/12, A61K 31/52, C07D 473/18...
Метки: ациклических, производные, нуклеозидов
Формула / Реферат:
1. Соединение формулы I в которой a) R1 представляет собой -С(О)СН(СН(СН3)2NH2 или -С(O)СН(СН(СН3)СH2СН3)NН2, a R2 представляет собой -С(O)С3-С21 насыщенный или мононенасыщенный, необязательно замещенный гидроксигруппой, С1-6алкилом, С1-6алкокси-, С1-6алкоксиС1-6алкилом, С1-6алканоилом, амино-, галоид-, циано-, азидо-, меркапто- и нитрогруппой алкил; b) R1 представляет собой -С(O)С3-С21 насыщенный или мононенасыщенный, необязательно...
Производные пирролопирролона в качестве ингибиторов нейтрофильной эластазы

Номер патента: 1437
Опубликовано: 26.02.2001
Авторы: Джонсон Мартин Редпат, Доул Майкл Деннис, Ша Прайтом, Инглис Грэм Джордж Адам, Финч Гарри, Гаррисон Ли Эндрю, Макдоналд Саймон Джон Фосетт, Смит Робин Эндрю
МПК: A61K 31/407, A61P 11/00, C07D 487/04...
Метки: производные, пирролопирролона, ингибиторов, эластазы, качестве, нейтрофильной
Формула / Реферат:
1. Соединение общей формулы (I) где R1 представляет собой C1-6алкил, С2-6алкенил; арил, арил-С1-4алкил, арил-С2-4алкенил, гетероарил, гетероарил-С1-4алкил или гетероарил-С2-4-алкенил, или такую группу, в которой арильная или гетероарильная группировка замещена одной или более чем одной С1-4алкильной, галогено, тетразолильной, трифторметилсульфонамидной, NR9CO-C1-8алкильной, -(CH2)m-NR4R5, -CN, -COOR9, -CONR9R10, -NO2, -SO2-С1-6алкильной, -CF3...
Предыдущий патент: N-арил-2-оксазолидинон-5-карбоксамиды, их производные и их применение в качестве антибактериальных средств
Следующий патент: Соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат
Случайный патент: Регулируемый резонансный преобразователь напряжения и способ регулируемого резонансного преобразования постоянного напряжения в постоянное