Замещенные 8-арилхинолины в качестве ингибиторов фосфодиэстеразы-4
Номер патента: 6607
Опубликовано: 24.02.2006
Авторы: Макдональд Дуайт, Перрье Хелен, Квонг Элизабет, Тибер Рок, Клэс Софи-Дороти, Хо Гуо-Дзие, Вайлая Анант, Конлон Дэвид А.









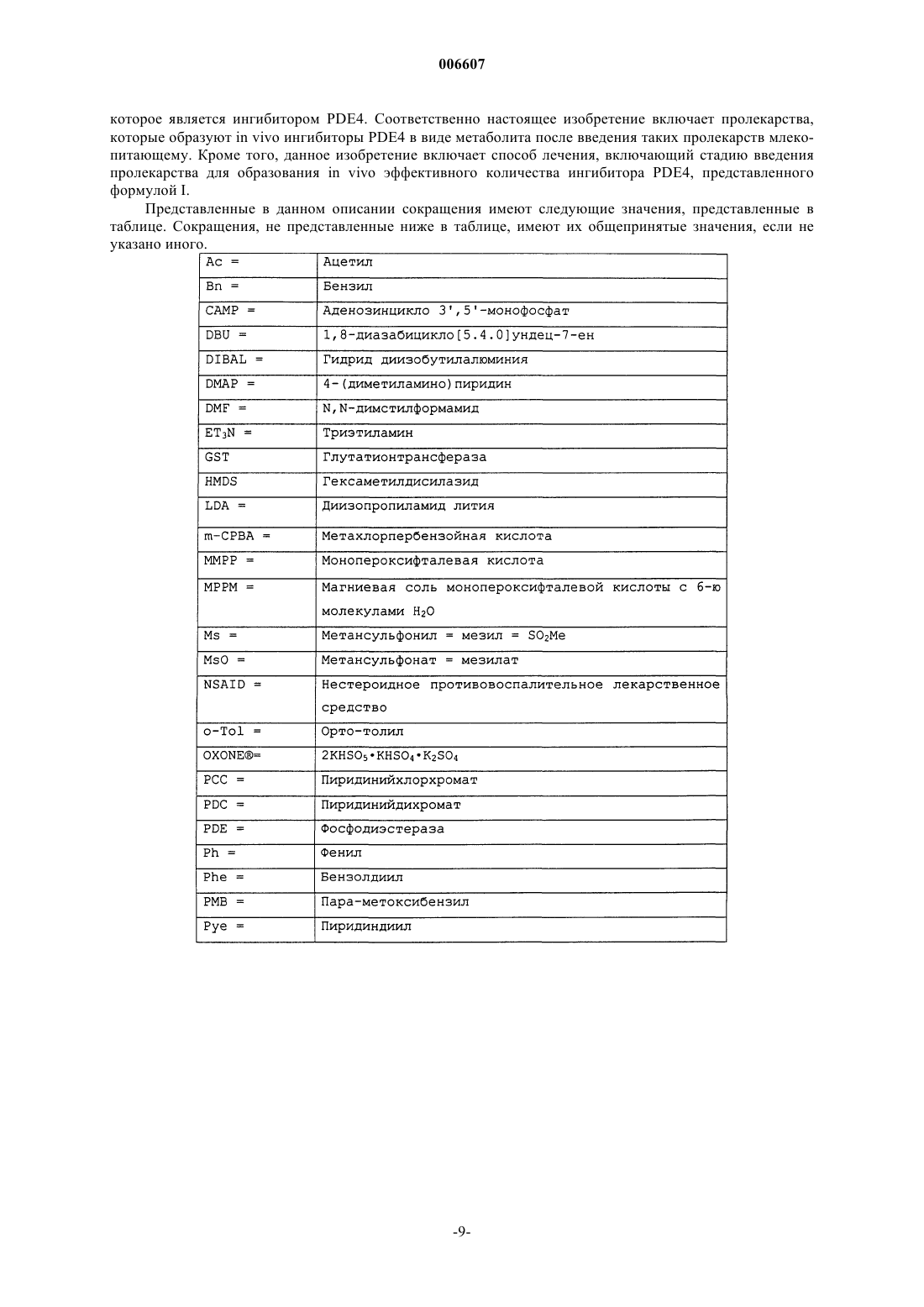








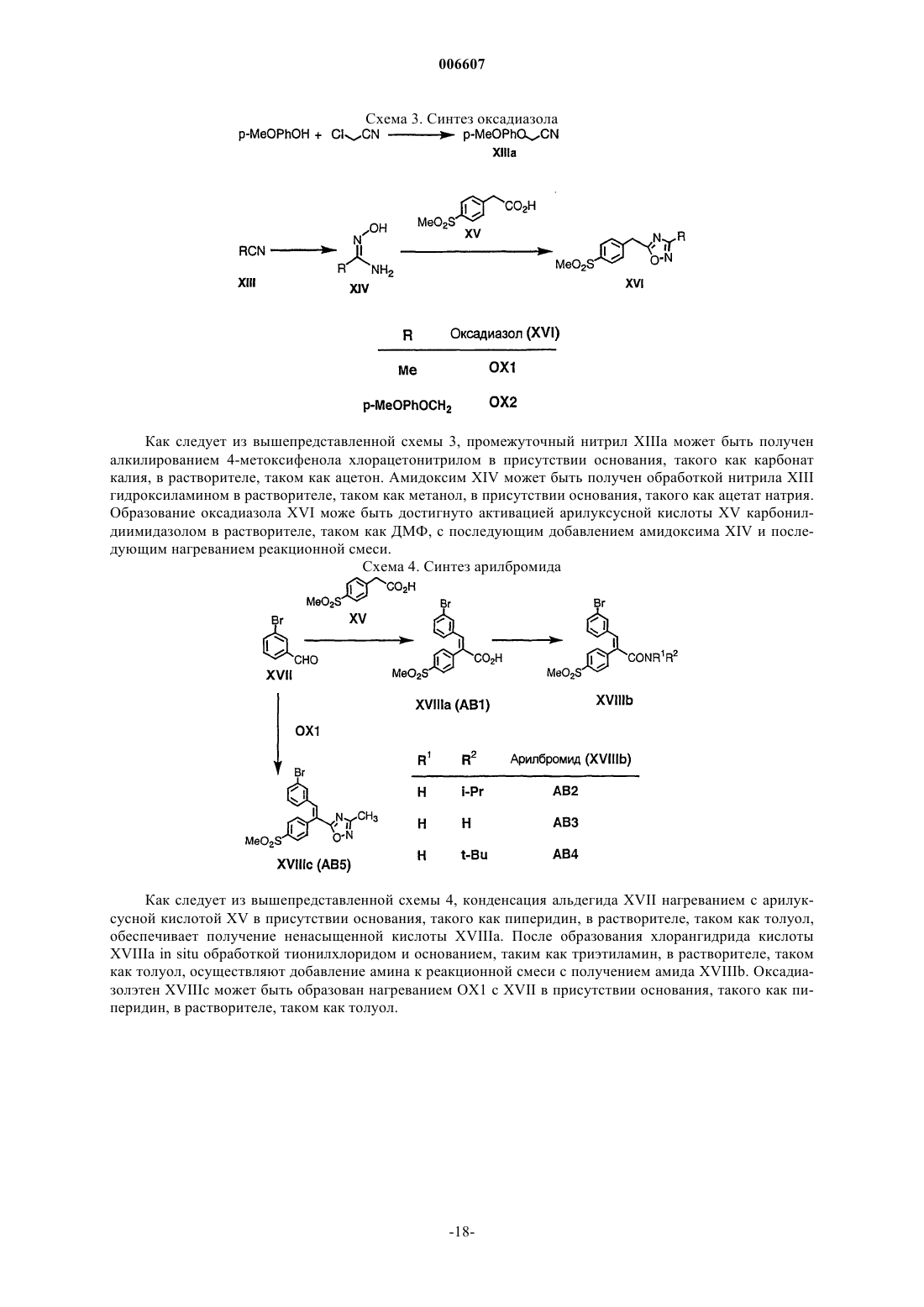






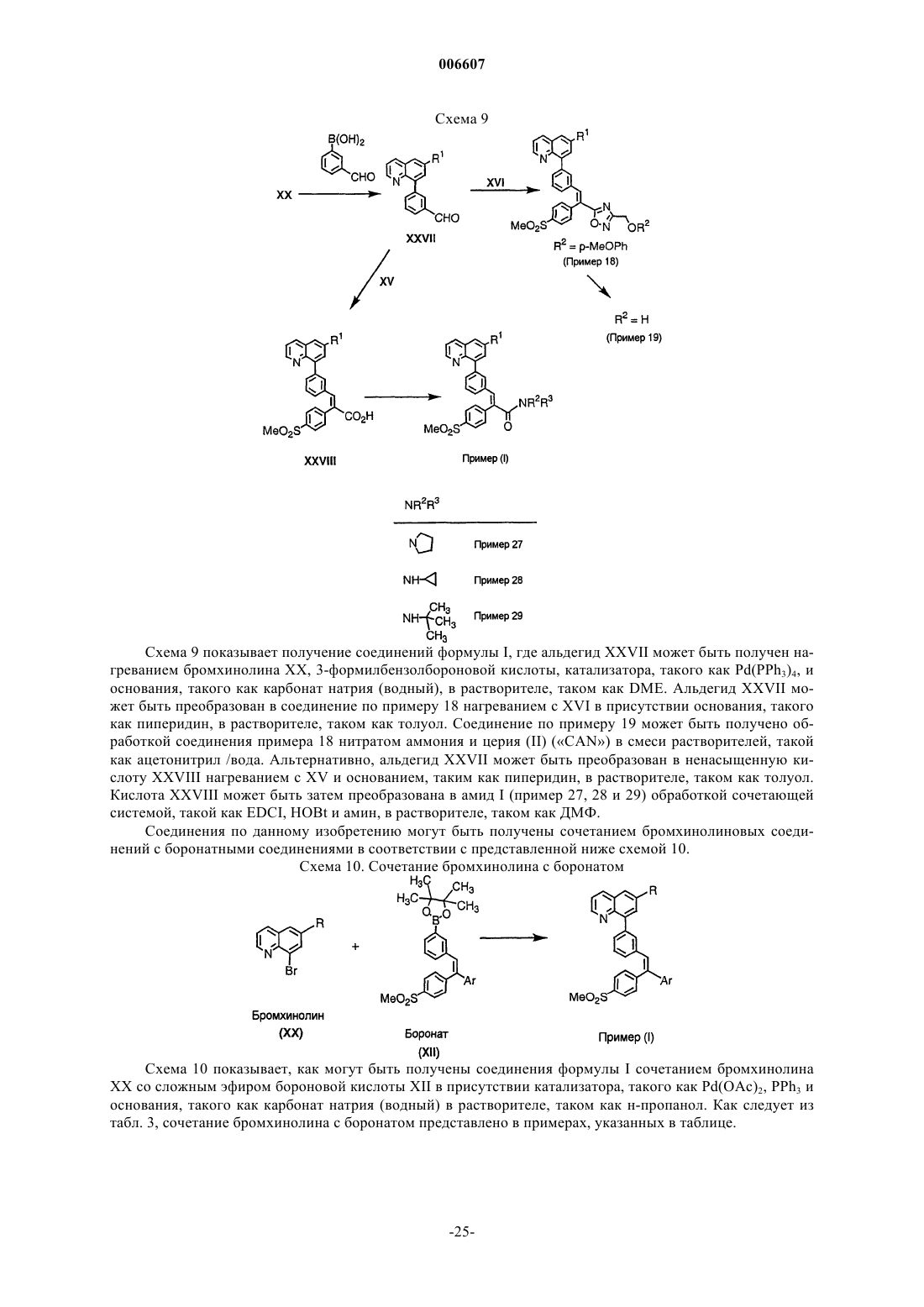




Формула / Реферат
1. Фармацевтически приемлемая соль серной, метансульфоновой, п-толуолсульфоновой, 2-нафталинсульфоновой, хлористо-водородной или бензолсульфоновой кислоты соединения, представленного формулой (I)

где R1 представляет собой -C1-C6алкильную группу, которая необязательно замещена одним или несколькими заместителями, выбранными из -C1-C6алкила, -CN и -SO2-(C1-C6алкил),
A представляет собой CH;
R2 и R3 независимо представляют собой H, гидроксиC1-C6алкильную группу, (C0-C6алкил)(C0-C6алкил)аминогруппу, карбонильную группу, -CN, фенильную группу, тиазолильную, имидазолильную, оксадиазолильную, пиридильную или пирролидинокарбонильную группу, которые необязательно замещены одним или несколькими заместителями, выбранными из галогена, -C1-C6алкила (который может быть замещен гидрокси, C1-C6алкоксифенокси, -CN), и -SO2-(C1-C6алкил);
один из R2 и R3 должен быть указанным выше фенилом или тиазолилом, имидазолилом, оксадиазолилом или пиридилом, необязательно замещенным.
2. Соединение по п.1, где
R2 представляет собой необязательно замещенный фенил и
R3 представляет собой необязательно замещенную тиазолильную, имидазолильную, оксадиазолильную или пиридильную группу.
3. Соединение по п.1, где
R2 представляет собой необязательно замещенный фенил и
R3 представляет собой необязательно замещенный фенил.
4. Соединение по п.1, где
R2 представляет собой карбонильную группу и
R3 представляет собой необязательно замещенный фенил.
5. Соединение по п 1, где
R2 представляет собой -CN и
R3 представляет собой необязательно замещенный фенил.
6. Соединение по п.1, где каждый из R2 и R3 независимо представляет собой фенил или гетероарил, каждый из которых необязательно замещен.
7. Фармацевтически приемлемая соль серной, метансульфоновой, п-толуолсульфоновой, 2-нафталинсульфоновой, хлористо-водородной или бензолсульфоновой кислоты соединения

или

8. Соль бензолсульфоновой кислоты соединения формулы

характеризующаяся идентифицирующими пиками

на спектрограмме рентгеновской порошковой дифрактометрии.
9. Соль бензолсульфоновой кислоты соединения формулы

характеризующаяся идентифицирующими пиками

в спектрограмме рентгеновской порошковой дифрактометрии.
10. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.7 и фармацевтически приемлемый носитель.
11. Фармацевтическая композиция по п.10, дополнительно содержащая антагонист рецептора лейкотриена, ингибитор биосинтеза лейкотриена, антагонист M2/M3, кортикостероид, антагонист H1 рецептора или агонист бета 2 адренорецептора.
12. Фармацевтическая композиция по п.10, дополнительно содержащая селективный ингибитор COX-2, статин или NSAID.
13. Способ лечения или профилактики астмы, хронического бронхита, хронического обструктивного легочного заболевания (COPD), эозинофильной гранулемы, респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, хронического обструктивного легочного заболевания у животных, аллергического ринита, аллергического конъюнктивита, весеннего конъюнктивита, кашля, включающий стадию введения терапевтически эффективного количества или профилактически эффективного количества фармацевтически приемлемой соли по п.1.
14. Способ лечения или профилактики астмы, хронического бронхита, хронического обструктивного легочного заболевания (COPD), эозинофильной гранулемы, респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, хронического обструктивного легочного заболевания у животных, аллергического ринита, аллергического конъюнктивита, весеннего конъюнктивита, кашля, включающий стадию введения терапевтически эффективного количества или профилактически эффективного количества фармацевтически приемлемой соли по п.7.
15. Способ лечения или профилактики астмы, хронического бронхита, хронического обструктивного легочного заболевания (COPD), эозинофильной гранулемы, респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, хронического обструктивного легочного заболевания у животных, аллергического ринита, аллергического конъюнктивита, весеннего конъюнктивита, кашля, включающий стадию введения терапевтически эффективного количества или профилактически эффективного количества фармацевтически приемлемой соли по п.8.
16. Способ лечения или профилактики астмы, хронического бронхита, хронического обструктивного легочного заболевания (COPD), эозинофильной гранулемы, респираторного дистресс-синдрома у взрослых, респираторного дистресс-синдрома у детей, хронического обструктивного легочного заболевания у животных, аллергического ринита, аллергического конъюнктивита, весеннего конъюнктивита, кашля, включающий стадию введения терапевтически эффективного количества или профилактически эффективного количества фармацевтически приемлемой соли по п.9.
Текст
006607 Предпосылки изобретения Область изобретения Настоящее изобретение относится к соединениям, которые представляют собой 8-арилхинолины. Данное изобретение, в частности, относится к солям замещенных 8-арилхинолинов, которые являются ингибиторами фосфодиэстеразы-4, где арильная группа в положении 8 содержит заместитель замещенную алкенильную группу. Предшествующий уровень техники Гормоны представляют собой соединения, которые по-разному влияют на клеточную активность. Во многих аспектах гормоны действуют в качестве мессенджеров запуска специфических клеточных реакций и активностей. Однако многие эффекты, производимые гормонами, не вызываются исключительным действием только гормонов. Вместо этого гормон сначала связывается с рецептором, вследствие чего запускается высвобождение второго соединения, которое продолжает действовать на клеточную активность. Согласно данному сценарию гормон известен как первичный мессенджер, тогда как второе соединение называют вторичным мессенджером. В качестве вторичного мессенджера гормонов,включающих эпинефрин, глюкагон, кальцитонин, кортикотропин, липотропин, лютеинизирующий гормон, норэпинефрин, паратиреоидный гормон, тиреотропный гормон и вазопрессин, известен циклический аденозинмонофосфат (аденозинцикло 3',5'-монофосфат, цАМФ (сАМР) или циклический АМФ (циклический АМР. Таким образом, цАМФ опосредует клеточные реакции на гормоны. Циклический АМФ опосредует также клеточные реакции на различные нейротрансмиттеры. Фосфодиэстеразы (PDE) представляют собой семейство ферментов, которые преобразуют в ходе обмена 3',5'-циклические нуклеотиды в 5'нуклеозидмонофосфаты, завершая таким образом активность вторичного мессенджера цАМФ. В качестве потенциальных мишеней для создаваемых новых противоастматических и противовоспалительных соединений повышенный интерес представляет конкретная фосфодиэстераза, которой является фосфодиэстераза-4 (PDE4, известная также как PDE-IV), которая представляет собой специфическую PDE IV типа, обладающую высоким сродством к цАМФ. Известно,что PDE4 существует в виде по меньшей мере четырех изоферментов, каждый из которых кодирован различным геном. Предполагается, что каждый из четырех известных генных продуктов PDE4 играет разные роли в аллергических и (или) воспалительных реакциях. Поэтому предполагается, что ингибирование PDE4, в частности, специфических изоформ PDE4, которые вызывают вредные для здоровья реакции, может оказывать благоприятное действие на симптомы аллергии и воспаления. Желательным является разработка новых соединений и композиций, которые ингибируют активность PDE4. Основная проблема, возникающая при использовании ингибиторов PDE4, состоит в побочном действии, выражающемся в появлении рвоты, которая наблюдается для нескольких соединений-кандидатов,представленных в: С. Burnouf et al., (Burnouf), Ann. Rep. In Med. Chem., 33: 91-109 (1998). В. Hughes etal., Br. J. Pharmacol., 118: 1183-1191 (1996); M.J. Perry et al., Cell Biochem. Biophys., 29: 113-132 (1998); S. В. Christensen et al., J. Med. Chem., 41: 821-835 (1988) и Burnouf описали широкое разнообразие серьезных нежелательных побочных действий, проявляемых различными соединениями. Как указывается в: М.(1998), имеется большая заинтересованность в терапевтических ингибиторах PDE4, поиск которых продолжается. В международной заявке на патент WO 9422852 описаны хинолины в качестве ингибиторов PDE4. А. Н. Cook, et al., J. Chem. Soc., 413-417 (1943) описаны гамма-пиридилхинолины. Другие производные хинолина представлены в Kei Manabe et al., J. Org. Chem., 58 (24): 6692-6700 (1993); Kei Manabeet al., J. Am. Chem. Soc., 115 (12): 5324-5325 (1993) и Kei Manabe et al., J. Am. Chem. Soc., 114 (17): 69406941 (1992). Соединения, включающие циклические системы, описаны различными исследователями как эффективные для различных терапий и применений. Так например, в международной заявке на патентWO 98/25883 в качестве ингибиторов калпаина описаны кетобензамиды, в Европейской заявке на патентЕР 811610 и в патентах США 5679712, 5693672 и 5747541 описан замещенный натрийбензоилгуанидин в качестве блокаторов каналов, в патенте США 5736297 описаны циклические системы,пригодные в качестве светочувствительной композиции. В патентах США 5491147, 5608070, 5622977, 5739144, 5776958, 5780477, 5786354, 5798373,5849770, 5859034, 5866593, 5891896 и в международной заявке на патент WO 95/35283 описаны ингибиторы PDE4, которые представляют собой тризамещенные производные арила или гетероарилфенила. В патенте США 5580888 описаны ингибиторы PDE4, которые представляют собой производные стирила. В патенте США 5550137 описаны ингибиторы PDE4, которые представляют собой производные фениламинокарбонила. В патенте США 5340827 описаны ингибиторы PDE4, которые представляют собой соединения фенилкарбоксамида. В патенте США 5780478 описаны ингибиторы PDE4, которые представляют собой тетразамещенные производные фенила. В международной заявке на патент WO 96/00215 описаны замещенные производные оксима, которые могут использоваться в качестве ингибиторов PDE4. В патенте США 5633257 описаны ингибиторы PDE4, которые представляют собой соединения цикло (алкил и алкенил)фенилалкенила (арила и гетероарила).-1 006607 Однако остается потребность в новых соединениях и композициях, которые терапевтически ингибируют PDE4 с минимальными побочными действиями. Сущность изобретения Настоящее изобретение относится к новым солям новых замещенных 8-арилхинолинов, которые являются ингибиторами PDE4, где арильная группа в положении 8 замещена замещенной алкенильной группой. Данное изобретение также относится к фармацевтической композиции, которая включает эффективное количество новых солей новых замещенных 8-арилхинолинов и фармацевтически приемлемый носитель. Данное изобретение также относится к способу лечения млекопитающих, например, таких заболеваний, как астма, хронический бронхит, хроническое обструктивное легочное заболевание(COPD), эозинофильная гранулема, псориаз и другие доброкачественные или злокачественные пролиферативные кожные заболевания, эндотоксиновый бактериально-токсический шок (и родственные состояния, такие как ламинит и кишечная колика у лошадей), септический шок, язвенный колит, болезнь Крона, нарушение реперфузии миокарда и головного мозга, воспалительный артрит, остеопороз, хронический гломерулонефрит, атопический дерматит, крапивница, респираторный дистресс-синдром у взрослых людей, респираторный дистресс-синдром у детей, хроническое обструктивное легочное заболевание у животных, несахарный диабет, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, артериальный рестеноз, атеросклероз, нейрогенное воспаление, боль, кашель, ревматоидный артрит, анкилозирующий спондилоартрит, отторжение трансплантата и гомологичная болезнь, повышенная секреция желудочного сока, сепсис или септический шок, вызванный грибами или вирусами,хроническая дегенерация тканей, опосредованная воспалением и цитокинами, остеоартрит, рак, общее истощение, мышечная гипотрофия, депрессия, ухудшение памяти, монополярная депрессия, острые и хронические нейродегенеративные расстройства с воспалительными компонентами, болезнь Паркинсона, болезнь Альцгеймера, травма спинного мозга, травмы головы, рассеянный склероз, рост опухоли и раковая инвазия нормальных тканей, введением эффективного количества новой соли замещенного 8 арилхинолина или соединения предшественника соли, который образует in vivo новый замещенный 8 арилхинолин. Краткое описание фигур Фиг. 1 представляет химическое схематическое изображение общей структуры соединений, из которых образованы новые соли согласно настоящему изобретению. Фиг. 2 представляет графическую зависимость счета от Тета для рентгеновской порошковой дифрактометрии полиморфа формы А соли бензолсульфоновой кислоты 6-[1-метил-1(метилсульфонил)этил]-8-[3-[(Е)-2-[3-метил-1,2,4-оксадиазол-5-ил]-2-[4-(метилсульфонил)фенил]этенил]фенил]хинолина. Фиг. 3 представляет графическую зависимость счета от Тета для рентгеновской порошковой дифрактометрии полиморфа формы В соли бензолсульфоновой кислоты 6-[1-метил-1(метилсульфонил)этил]-8-[3-[(Е)-2-[3-метил-1,2,4-оксадиазол-5-ил]-2-[4-(метилсульфонил)фенил]этенил]фенил]хинолина. Фиг. 4 представляет сравнение рентгеновской порошковой дифрактометрии полиморфа формы А(нижний чертеж) и формы В (верхний чертеж) соли бензолсульфоновой кислоты 6-[1-метил-1(метилсульфонил)этил]-8-[3-[(Е)-2-[3-метил-1,2,4-оксадиазол-5-ил]-2-[4-(метилсульфонил)фенил]этенил]фенил]хинолина. Фиг. 5 представляет графическую зависимость, показывающую различия в пиках рентгеновской порошковой дифрактометрии полиморфа формы А соли бензолсульфоновой кислоты 6-[1-метил-1(метилсульфонил)этил]-8-[3-[(Е)-2-[3-метил-1,2,4-оксадиазол-5-ил]-2-[4-(метилсульфонил)фенил]этенил]фенил]хинолина. Фиг. 6 представляет графическую зависимость, показывающую различия в пиках рентгеновской порошковой дифрактометрии полиморфа формы В соли бензолсульфоновой кислоты 6-[1-метил-1(метилсульфонил)этил]-8-[3-[(Е)-2-[3-метил-1,2,4-оксадиазол-5-ил]-2-[4-(метилсульфонил)фенил]этенил]фенил]хинолина. Подробное описание изобретения Соединение по данному изобретению представляет собой фармацевтически приемлемую соль соединения, представленного формулой (I)-2 006607 где S1, S2 и S3 независимо представляют собой Н, -ОН, галоген, -C1-С 6 алкил, -NO2, -CN или -C1-С 6 алкокси, где алкильная и алкоксильная группы необязательно замещены 1-5 заместителями; при этом каждый заместитель независимо представляет собой галоген или ОН;-(C1-С 6 алкил)-SOn-C1 С 6 алкильную)группу, где любая из групп необязательно замещена 1-5 заместителями; причем каждый заместитель независимо представляет собой галоген, -ОН, -CN, -C1-С 6 алкил, -циклоC3-С 6 алкил,-С(О)(гетероциклоC3-С 6 алкил), -С(О)-О-(C0-С 6 алкил), -С(О)арилокси, -C1-С 6 алкокси, -(C0-С 6 алкил)(C0 С 6 алкил)амино, циклоалкилокси, ацил, ацилокси, -циклоC3-С 6 алкил, гетероциклоC3-С 6 алкил, арил, гетероарил, карбонил, карбамоил или -SОn(C1-С 6 алкил); А представляет собой СН, С-сложный эфир или C-R4;R2 и R3 независимо представляют собой арил, гетероарил, Н, галоген, -CN, -C1-С 6 алкил, гетероциклоC3 С 6 алкил, -C1-С 6 алкокси, карбонил, карбамоил, -С(О)ОН, -(C1-С 6 алкил)-SOn-(C1-С 6 алкил), -С(О)N(C0 С 6 алкил)(C0-С 6 алкил) или -C1-С 6 алкилациламиногруппу, где любая из групп необязательно замещена 1-5 заместителями, причем каждый заместитель независимо представляет собой арил, гетероарил, галоген, -NO2,-С(О)ОН, карбонил, -CN, -C1-С 6 алкил, -SOn-(C1-С 6 алкил), -SOn(арил), арилокси, -гетероарилокси, -C1 С 6 алкокси, N-оксид, -С(О)-гетероциклоC3-С 6 акил, -NH-циклоC3-С 6 алкил, амино, -ОН или -(C0 С 6 алкил)(C0-С 6 алкил)амино, -С(O)-N(C0-С 6 алкил)(C0-С 6 алкил), где каждый заместитель, в свою очередь,независимо необязательно замещен -ОН, C1-С 6 алкокси, -C1-С 6 алкилом, -циклоC3-С 6 алкилом, арилокси,-С(О)ОН, -C(O)O(C1-С 6 алкилом), галогеном, -NO2, -CN, -SOn-(C1-С 6 алкилом) или -С(O)-N(C0 С 6 алкил)(C0-С 6 алкилом); один из R2 и R3 должен быть арилом или гетероарилом, необязательно замещенным; когда оба из R2 и R3 являются арилом или гетероарилом, тогда R2 и R3 могут быть необязательно соединены с помощью тио, окси или (С 1-С 4 алкил) мостика с образованием конденсированной трициклической структуры;R4 представляет собой арил, -C1-С 6 алкил, гетероарил, -CN, карбонил, карбамоил, -(C1-С 6 алкил)-SOn(C1-С 6 алкил), -C(O)N(C0-С 6 алкил)(C0-С 6 алкил) или -C1-С 6 алкилациламиногруппу, где любая из групп необязательно замещена 1-5 заместителями, при этом каждый заместитель независимо представляет собой карбонил, -CN, галоген, -С(О)(C0-С 6 алкил), -С(О)О(C0-С 6 алкил), -C1-С 6 алкил, -SOn-(C1-С 6 алкил),-ОН, -C1-С 6 алкокси или -(C0-С 6 алкил)(C0-С 6 алкил)аминогруппу;R2 или R3 может быть необязательно соединен связью с R4 с образованием кольца. В соответствии с одним аспектом соединение по данному изобретению представляет собой фармацевтически приемлемую соль серной,метансульфоновой,п-толуолсульфоновой,2 нафталинсульфоновой, хлористо-водородной или бензолсульфоновой кислоты соединения, представленного формулой (I), гдеS1, S2 и S3 независимо представляют собой Н, -ОН, галоген, -C1-С 6 алкил, -NO2, -CN или -C1-С 6 алкокси, где алкильная и алкоксильная группы необязательно замещены 1-5 заместителями; при этом каждый заместитель независимо представляет собой галоген или ОН;-(C1-С 6 алкил)-SOn-(C1 С 6 алкильную)группу, где любая из групп необязательно замещена 1-5 заместителями; причем каждый заместитель независимо представляет собой галоген, -ОН, -CN, -C1-С 6 алкил, -циклоC3-С 6 алкил,-С(О)(гетероциклоC3-С 6 алкил), -С(O)-O-(C0-С 6 алкил), -С(О)-арилокси, -C1-С 6 алкокси, -(C0-С 6 алкил)(C0 С 6 алкил)амино, циклоалкилокси, ацил, ацилокси, -циклоC3-С 6 алкил, гетероциклоC3-С 6 алкил, арил, гетероарил, карбонил, карбамоил или -SOn(C1-С 6 алкил); А представляет собой СН, С-сложный эфир или C-R4;R2 и R3 независимо представляют собой арил, гетероарил, Н, галоген, -CN, -C1-С 6 алкил, гетероциклоC3-С 6 алкил, -C1-С 6 алкокси, карбонил, карбамоил, -С(О)ОН, -(C1-С 6 алкил)-SOn-(C1-С 6 алкил),-С(О)N(C0-С 6 алкил)(C0-С 6 алкил) или -C1-С 6 алкилациламиногруппу, где любая из групп необязательно замещена 1-5 заместителями, причем каждый заместитель независимо представляет собой арил, гетероарил, галоген, -NO2, -С(О)ОН, карбонил, -CN, -C1-С 6 алкил, -SOn-(C1-С 6 алкил), -SOn(арил), арилокси,-гетероарилокси, -C1-С 6 алкокси, N-оксид, -С(О)-гетероциклоC3-С 6 акил, -NH-циклоC3-С 6 алкил, амино,-ОН или -(C0-С 6 алкил)(C0-С 6 алкил)амино, -С(O)-N(C0-С 6 алкил)(C0-С 6 алкил), где каждый заместитель, в свою очередь, независимо необязательно замещен -ОН, C1-С 6 алкокси, -C1-С 6 алкилом, -циклоC3 С 6 алкилом, арилокси, -С(О)ОН, -C(O)O(C1-С 6 алкилом), галогеном, -NO2, -CN, -SOn-(C1-С 6 алкилом) или-3 006607 один из R2 и R3 должен быть арилом или гетероарилом, необязательно замещенным; когда оба из R2 и R3 являются арилом или гетероарилом, тогда R2 и R3 могут быть необязательно соединены с помощью тио, окси или (С 1-С 4 алкил)мостика с образованием конденсированной трициклической структуры;R4 представляет собой арил, -C1-С 6 алкил, гетероарил, -CN, карбонил, карбамоил, -(C1-С 6 алкил)-SOn(C1-С 6 алкил), -C(O)N(C0-С 6 алкил)(C0-С 6 алкил) или -C1-С 6 алкилациламиногруппу, где любая из групп необязательно замещена 1-5 заместителями, при этом каждый заместитель независимо представляет собой карбонил, -CN, галоген, -С(О)(C0-С 6 алкил), -С(О)О(C0-С 6 алкил), -C1-С 6 алкил, -SOn-(C1 С 6 алкил), -ОН, -C1-С 6 алкокси или -(C0-С 6 алкил)(C0-С 6 алкил)аминогруппу;R2 или R3 может быть необязательно соединен связью с R4 с образованием кольца. Используемый в данном описании термин алкил, а также другие группы, имеющие префикс алк, такие как, например, алкокси, алканоил, алкенил, алкинил и подобные, означает углеродные цепи,которые могут быть линейными или разветвленными или их комбинацией. Примеры алкильных групп включают метил, этил, пропил, изопропил, бутил, втор- и трет-бутил, пентил, гексил, гептил и подобные группы. Алкенил, алкинил и другие подобные термины включают углеродные цепи, содержащие по меньшей мере одну ненасыщенную С-С связь. Термин циклоалкил означает карбоциклы, не содержащие гетероатомы, и включает моно-, би- и трициклические насыщенные карбоциклы, а также циклические системы. Указанные конденсированные циклические системы могут включать одно кольцо, такое как бензольное кольцо, которое является частично или полностью ненасыщенным для образования конденсированных циклических систем, таких как бензоконденсированные карбоциклы. Циклоалкил включает такие конденсированные циклические системы, как спироконденсированные циклические системы. Примеры циклоалкила включают циклопропил, циклобутил, циклопентил, циклогексил, декагидронафталин, адамантан, инданил, инденил,флуоренил, 1,2,3,4-тетрагидронафталин и подобные. Подобно, циклоалкенил означает карбоциклы, не содержащие гетероатомы и, по меньшей мере, одну неароматическую С-С двойную связь, и включает моно-, би- и трициклические частично насыщенные карбоциклы, а также бензоконденсированные циклоалкены. Примеры циклоалкенила включают циклогексенил, инденил и подобные. Термин циклоалкилокси, если не указанно иного, включает циклоалкильную группу, соединенную с соединяющим оксиатомом. Термин алкокси, если не указано иного, включает алкильную группу, соединенную с соединяющим оксиатомом. Термин арил, если не указано иного, включает полициклические системы, а также моноциклические системы, такие как, например, фенил или нафтил. Термин арилокси, если не указано иного, включает полициклические системы, а также моноциклические системы, такие как, например, фенил или нафтил, соединенные связывающим оксиатомом с местом связывания. Термин C0-С 6 алкил включает алкилы, содержащие 6, 5, 4, 3, 2, 1 атомов углерода или не содержащие атомы углерода. Алкил без атомов углерода представляет собой замещающий атом водорода или прямую связь, в зависимости от того какой группой является алкил: концевой или мостиковой группой. Термин гетеро, если не указано иного, включает один или несколько атомов О, S или N. Так например, гетероциклоалкил и гетероарил включает циклические системы, которые содержат в кольце один или несколько атомов О, S или N, включая смеси указанных атомов. Гетероатомы замещают кольцевые атомы углерода. Так например, гетероцикло С 5 алкил представляет собой пятичленное кольцо, содержащее от 5 до 0 атомов углерода. Примеры гетероарила включают, например, пиридинил, хинолинил, изохинолинил, пиридазинил,пиримидинил, пиразинил, хиноксалинил, фурил, бензофурил, дибензофурил, тиенил, бензтиенил, пирролил, индолил, пиразолил, индазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, имидазолил, бензомидазолил, оксадиазолил, тиадиазолил, триазолил, тетразолил. Термин гетероарилокси, если не указано иного, описывает гетероарильную группу, соединенную соединяющим оксиатомом с местом связывания. Примеры гетероарил(C1-6)алкила включают, например, фурилметил, фурилэтил, тиенилметил, тиенилэтил, пиразолилметил, оксазолилметил, оксазолилэтил, изоксазолилметил, тиазолилметил, тиазолилэтил, имидазолилметил, имидазолилэтил, бензимидазолилметил, оксадиазолилметил, оксадиазолилэтил,тиадиазолилметил, тиадиазолилэтил, триазолилметил, триазолилэтил, тетразолилметил, тетразолилэтил,пиридинилметил, пиридинилэтил, пиридазинилметил, пиримидинилметил, пиразинилметил, хинолинилметил, изохинолинилметил и хиноксалинилметил. Примеры гетероциклоС 3-7 алкила включают, например, азетидинил, пирролидинил, пиперидинил,пиперазинил, морфолинил, тетрагидрофуранил, имидазолинил, пирролидин-2-он, пиперидин-2-он и тиоморфолинил. Примеры арил(C1-6)алкила включают, например, фенил(C1-6)алкил и нафтил(C1-6)алкил.-4 006607 Примеры гетероцикло С 3-7 алкилкарбонил(C1-6)алкила включают, например, азетидинилкарбонил(C1-6)алкил, пирролидинилкарбонил(C1-6)алкил, пиперидинилкарбонил (C1-6)алкил, пиперазинилкарбонил(C1-6)алкил,морфолинилкарбонил(C1-6)алкил и тиоморфолинилкарбонил(C1-6)алкил. Термин амин, если не указано иного, включает первичные, вторичные и третичные амины. Если не указано иного, термин карбамоил используется для включения -NHC(O)ОС 1-С 4 алкила и-ОС(O)NНС 1-С 4 алкила. Термин галоген включает атомы фтора, хлора, брома и иода. Термин необязательно замещенный предназначен для включения как замещенных, так и незамещенных групп. Так например, необязательно замещенный арил может представлять пентафторфенильное или фенильное кольцо. Кроме того, замещение может быть осуществлено в любой из групп. Так, например, замещенный арил(C1-6)алкил включает замещение в арильной группе, а также замещение в алкильной группе. Представленные в данном описании соединения содержат одну или несколько двойных связей и поэтому могут давать цис-/трансизомеры, а также другие конформационные изомеры. Настоящее изобретение включает все указанные возможные изомеры, а также смеси таких изомеров. Представленные в данном описании соединения могут содержать один или несколько ассиметрических центров и поэтому могут давать диастереоизомеры и оптические изомеры. Настоящее изобретение включает все указанные возможные диастереоизомеры, а также их рацемические смеси, их по существу чистые разрешенные энантиомеры, все возможные геометрические изомеры и их фармацевтически приемлемые соли. Вышеуказанная формула I показана без определенной безусловной стереохимии в некоторых положениях. Настоящее изобретение включает все стереоизомеры формулы I и их фармацевтически приемлемых солей. Кроме того, включены также смеси стереоизомеров, а также отдельные выделенные конкретные стереоизомеры. В процессе осуществления способов синтеза, используемых для получения данных соединений, или при использовании методов рацемизации или эпимеризации, известных специалистам в данной области, продукты таких способов могут быть смесью стереоизомеров. Термин фармацевтически приемлемые соли относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот. Когда соединение по настоящему изобретению является кислотным, его соответствующая соль может быть обычно получена с фармацевтически приемлемыми нетоксичными основаниями, включающими неорганические основания и органические основания. Соли, полученные из таких неорганических оснований, включают соли алюминия, аммония, кальция,меди (III и II), железа (III), железа (II), лития, магния, марганца (III и II), калия, натрия, цинка и подобные соли. В особенности предпочтительными являются соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, а также циклических аминов и замещенных аминов, таких как амины, встречающиеся в природе, и полученные синтезом замещенные амины. Другие фармацевтически приемлемые органические нетоксичные основания, из которых могут быть образованы соли,включают ионообменные смолы, например, такие, как аргинин, бетаин, кофеин, холин, N,N'дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и подобные. Когда соединение по настоящему изобретению является основным, его соответствующая соль может быть обычно получена с фармацевтически приемлемыми нетоксичными кислотами, включающими неорганические и органические кислоты. Такие кислоты включают, например, уксусную, бензолсульфоновую, бензойную, камфорсульфоновую, лимонную, этансульфоновую, фумаровую, глюконовую, глутаминовую, бромисто-водородную, хлористо-водородную, изэтионовую, молочную, малеиновую, яблочную, миндальную, метансульфоновую, слизевую, азотную, памовую, пантотеновую, фосфорную, янтарную, серную, винную, п-толуосульфоновую кислоту и подобные кислоты. В особенности предпочтительными являются бензолсульфоновая, лимонная, бромисто-водородная, хлористо-водородная, малеиновая, фосфорная, серная и винная кислоты. Фармацевтические композиции по настоящему изобретению включают соединение, представленное формулой I (или его фармацевтически приемлемые соли), в качестве активного ингредиента, фармацевтически приемлемый носитель и необязательно другие терапевтические ингредиенты или адъюванты. Такие дополнительные терапевтические ингредиенты включают, например, i) антагонисты рецептора лейкотриена, ii) ингибиторы биосинтеза лейкотриена, iii) кортикостероиды, iv) антагонисты H1 рецептора, v) агонисты бета 2 адренорецептора, vi)селективные ингибиторы COX-2, vii) статины, viii) нестероидные противовоспалительные лекарственные средства (NSAID) и ix) антагонисты М 2/М 3. Композиции включают композиции, подходящие для перорального, ректального, местного и парентерального введения (включающего подкожное, внутримышечное и внутривенное), хотя наиболее подходящий способ введения в любом данном случае будет зависеть от конкретного хозяина и природы и тяжести заболеваний, для лечения которых вводят активный ингредиент. Фармацевтические композиции могут обыч-5 006607 но присутствовать в виде дозированной лекарственной формы и их получают любым из способов, хорошо известных в области фармации. Для местного применения могут быть использованы кремы, мази, желе, растворы или суспензии,содержащие соединение формулы I. Для целей по настоящему изобретению в область местного применения включены жидкости для полоскания рта и растворы или жидкости для полоскания горла. При лечении таких состояний, как астма, хронический бронхит, хроническое обструктивное легочное заболевание (COPD), эозинофильная гранулема, псориаз и другие доброкачественные или злокачественные пролиферативные кожные заболевания, эндотоксиновый бактериально-токсический шок (и родственные состояния, такие как ламинит и кишечная колика у лошадей), септический шок, язвенный колит, болезнь Крона, нарушение реперфузии миокарда и головного мозга, воспалительный артрит, остеопороз, хронический гломерулонефрит, атопический дерматит, крапивница, респираторный дистресссиндром у взрослых, респираторный дистресс-синдром у детей, хроническое обструктивное легочное заболевание у животных, несахарный диабет, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, артериальный рестеноз, атеросклероз, нейрогенное воспаление, боль, кашель,ревматоидный артрит, анкилозирующий спондилоартрит, отторжение трансплантата и гомологичная болезнь, повышенная секреция желудочного сока, сепсис или септический шок, вызванный грибами или вирусами, хроническая дегенерация тканей, опосредованная воспалением и цитокинами, остеоартрит,рак, общее истощение, мышечная гипотрофия, депрессия, ухудшение памяти, монополярная депрессия,острые и хронические нейродегенеративные расстройства с воспалительными компонентами, болезнь Паркинсона, болезнь Альцгеймера, травма спинного мозга, травмы головы, рассеянный склероз, рост опухоли и раковая инвазия нормальных тканей, которые восприимчивы к ингибированию PDE4, пригодными являются уровни дозы каждому больному от около 0,001 до 140 мг/кг массы тела в день или альтернативно от около 0,05 до около 7 г в день. Так например, воспаление можно эффективно лечить введением больному каждый день от около 0,01 до 50 мг соединения на килограмм массы тела или альтернативно от около 0,5 мг до около 2,5 г. Кроме того, следует иметь в виду, что соединения по данному изобретению, ингибирующие PDE4, можно вводить при профилактически эффективных уровнях доз для профилактики вышеуказанных состояний. Количество активного ингредиента, которое может быть объединено с материалами носителя для получения разовой лекарственной формы, будет изменяться в зависимости от подвергаемого лечению хозяина и от конкретного способа введения лекарственного средства. Так например, лекарственное средство, предназначенное для перорального введения людям, может обычно содержать от около 0,5 мг до около 5 г активного ингредиента, смешанного с соответствующим и общепринятым количеством материала носителя, которое может изменяться в диапазоне от около 5 до 95% в расчете на общее количество композиции. Дозированные лекарственные формы будут обычно содержать активный ингредиент в количестве между от около 0,01 до около 1000 мг, типично 0,01, 0,05, 0,25, 1, 5, 25, 50, 100, 200, 300, 400,500, 600, 800 или 1000 мг. Однако следует иметь в виду, что конкретный уровень дозы для каждого отдельного больного будет зависеть от множества факторов, включающих возраст, массу тела больного, общее состояние здоровья, пол, питание, время введения лекарственного средства, способ введения, скорость экскреции, лекарственная комбинация и тяжесть конкретного заболевания, подвергаемого лечению. На практике соединения по данному изобретению, представленные формулой I, или их фармацевтически приемлемые соли могут быть объединены в виде активного ингредиента, находящегося в однородной смеси с фармацевтически приемлемым носителем, в соответствии с традиционными способами смешивания в области фармации. Носитель может иметь широкое множество форм, зависящих от формы препарата, желательного для введения, например, перорального или парентерального (включая внутривенное). Таким образом, фармацевтические композиции по настоящему изобретению могут быть представлены в виде отдельных стандартных форм, подходящих для перорального введения, таких как капсулы, крахмальные облатки или таблетки, каждая из которых содержит заданное количество активного ингредиента. Кроме того, композиции могут быть представлены в виде порошка, в виде гранул, в виде раствора, в виде суспензии в водной жидкости, в виде неводной жидкости, в виде эмульсии масла в воде или в виде эмульсии воды в масле. Кроме обычных лекарственных форм, указанных выше, соединение,представленное формулой I, или его фармацевтически приемлемые соли, могут быть также введены с помощью средств, обеспечивающих регулируемое высвобождение активного ингредиента, и/или с помощью приспособлений для доставки лекарственного средства. Композиции могут быть приготовлены любым из способов, известных в области фармации. Обычно такие способы включают стадию приведения в ассоциацию активного ингредиента и носителя, который составляет один или несколько необходимых ингредиентов. Обычно композицию получают равномерным и однородным смешиванием активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями или с обоими из указанных носителей. Затем продукту может быть придана требуемая форма. Таким образом, фармацевтические композиции по данному изобретению могут включать фармацевтически приемлемый носитель и соединение или фармацевтически приемлемую соль формулы I. Соединения формулы I или их фармацевтически приемлемые соли могут быть также включены в фарма-6 006607 цевтические композиции в комбинации с одним или несколькими другими терапевтически активными соединениями. Используемый фармацевтический носитель может быть, например, твердым веществом, жидкостью или газом. Примеры твердых носителей включают лактозу, тера-альбу, сахарозу, тальк, желатин, агар,пектин, акацию, стеарат магния и стеариновую кислоту. Примеры жидких носителей включают сахарный сироп, арахисовое масло, оливковое масло и воду. Примеры газообразных носителей включают диоксид углерода и азот. При приготовлении композиций лекарственной формы для перорального введения может быть использована любая фармацевтическая среда. Так например, для образования жидких препаратов для перорального введения, таких как суспензии, эликсиры и растворы, могут быть использованы вода, гликоли,масла, спирты, корригенты, консерванты, красители и подобные вещества; тогда как для образования твердых препаратов для перорального введения, таких как порошки, капсулы и таблетки, могут быть использованы такие носители, как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители,гранулирующие вещества, смазывающие вещества, связывающие вещества, средства, способствующие распадаемости лекарственной формы, и подобные вещества. Таблетки и капсулы, вследствие простоты их введения, являются предпочтительными дозированными лекарственными формами, поэтому используются твердые носители. Таблетки необязательно могут быть покрыты стандартными методами с использованием водных или неводных средств. Таблетка, содержащая композицию по данному изобретению, может быть получена прессованием или литьем под давлением, необязательно с одним или несколькими дополнительными ингредиентами или адъювантами. Прессованные таблетки могут быть получены прессованием в подходящей машине активного ингредиента в свободнотекущей форме, такой как порошок или гранулы, необязательно смешанного со связывающим веществом, смазывающим веществом, инертным разбавителем, поверхностноактивным веществом или диспергатором. Отформованные литые таблетки могут быть получены литьем под давлением в подходящей машине смеси из порошкового соединения, увлажненного инертным жидким разбавителем. Каждая таблетка предпочтительно содержит от около 0,1 до около 500 мг активного ингредиента и каждая крахмальная облатка или капсула предпочтительно содержит от около 0,1 до около 500 мг активного ингредиента. Фармацевтические композиции по настоящему изобретению, подходящие для парентерального введения, могут быть получены в виде растворов или суспензий активных соединений в воде. Может быть включено подходящее поверхностно-активное вещество, такое как например, гидроксипропилцеллюлоза. Могут быть также приготовлены дисперсии в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Кроме того, для предотвращения вредного роста микроорганизмов может быть включен консервант. Фармацевтические композиции по настоящему изобретению, подходящие для инъекций, включают стерильные водные растворы или дисперсии. Кроме того, композиции могут быть в виде стерильных порошков, подходящих для импровизированного приготовления указанных стерильных инъецируемых растворов или дисперсий. Во всех случаях конечная инъецируемая форма должна быть стерильной и должна быть достаточно текучей для ее легкого введения в шприц. Фармацевтические композиции должны быть устойчивыми в условиях производства и хранения; поэтому они должны быть предпочтительно защищены против загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол(например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), растительные масла и их подходящие смеси. Фармацевтические композиции по настоящему изобретению могут быть в форме, подходящей для местного применения, например такой, как аэрозоль, крем, мазь, лосьон, присыпка, и в подобных формах. Кроме того, композиция может быть в форме, подходящей для использования при чрескожном введении. Такие композиции могут быть получены с использованием соединения по данному изобретению,представленного формулой I, или его фармацевтически приемлемых солей с помощью традиционных технологических способов. В качестве примера, крем или мазь получают смешиванием гидрофильного материала и воды с соединением в количестве от 5 до около 10 мас.% с получением крема или мази,имеющих требуемую консистенцию. Фармацевтические композиции по данному изобретению могут быть в форме, подходящей для ректального введения, где носитель является твердым веществом. Предпочтительно, чтобы смесь образовывала дозированные суппозитории. Подходящие носители включают масло какао и другие материалы,обычно применяемые в данной области. Суппозитории могут быть обычно образованы сначала смешиванием композиции с размягченным(и) или расплавленным(и) носителем(ями) с последующим охлаждением и формованием в формах. Кроме вышеуказанных ингредиентов, используемых в качестве носителей, представленные выше фармацевтические композиции могут включать, когда это необходимо, один или несколько дополнительных ингредиентов-носителей, таких как разбавители, буферы, корригенты, связывающие вещества,поверхностно-активные вещества, загустители, смазывающие вещества, консерванты (включающие ан-7 006607 тиоксиданты) и подобные вещества. Кроме того, могут быть включены другие адъюванты для того, чтобы сделать композицию изотонической с кровью конкретного реципиента. Композиции, содержащие соединение, представленное формулой I, или его фармацевтически приемлемые соли, могут быть также получены в форме порошка или жидкого концентрата. Было обнаружено, что соединения и фармацевтические композиции по данному изобретению показывают биологическую активность и могут использоваться в качестве ингибиторов PDE4. Соответственно, другой аспект изобретения касается способа лечения, например таких состояний у млекопитающих как астма, хронический бронхит, хроническое обструктивное легочное заболевание (COPD), эозинофильная гранулема, псориаз и другие доброкачественные или злокачественные пролиферативные кожные заболевания, эндотоксиновый бактериально-токсический шок (и родственные состояния, такие как ламинит и кишечная колика у лошадей), септический шок, язвенный колит, болезнь Крона, нарушение реперфузии миокарда и головного мозга, воспалительный артрит, остеопороз, хронический гломерулонефрит, атопический дерматит, крапивница, респираторный дистресс-синдром взрослых людей, респираторный дистресс-синдром детей, хроническое обструктивное легочное заболевание у животных, несахарный диабет, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, артериальный рестеноз, атеросклероз, нейрогенное воспаление, боль, кашель, ревматоидный артрит, анкилозирующий спондилоартрит, отторжение трансплантата и гомологичная болезнь, повышенная секреция желудочного сока, сепсис или септический шок, вызванный грибами или вирусами, хроническая дегенерация тканей, опосредованная воспалением и цитокинами, остеоартрит, рак, общее истощение, мышечная гипотрофия, депрессия, ухудшение памяти, монополярная депрессия, острые и хронические нейродегенеративные расстройства с воспалительными компонентами, болезнь Паркинсона, болезнь Альцгеймера,травма спинного мозга, травмы головы, рассеянный склероз, рост опухоли и раковая инвазия нормальных тканей, - заболеваний, которые поддаются уменьшению интенсивности симптомов за счет ингибирования изофермента PDE4 и образования повышенных уровней цАМФ - введением эффективного количества соединений по данному изобретению. Термин млекопитающие включает людей, а также других животных, например таких как собаки, кошки, лошади, свиньи и крупный рогатый скот. Соответственно следует иметь в виду, что лечение млекопитающих, иных, чем люди, является клиническим лечением болезней, аналогичных вышеуказанным примерам, которые являются болезнями человека. Кроме того, как указывалось выше, соединение по данному изобретению может быть использовано в комбинации с другими терапевтическими соединениями. В особенности комбинации соединения по данному изобретению, ингибирующего PDE4, могут быть выгодно использованы в комбинации с: i) антагонистами рецептора лейкотриена, ii) ингибиторами биосинтеза лейкотриена, iii) селективными ингибиторами COX-2, iv) статинами, v) NSAID, vi) антагонистами М 2/М 3, vii) кортикостероидами, viii) антагонистами H1 (гистаминового) рецептора и ix) агонистом бета 2 адренорецептора. В соответствии с другим аспектом было обнаружено, что соединение по данному изобретению может образоваться в виде метаболита в системе млекопитающего. Так например, когда вводят соединение по примеру 14 в виде метаболита образуется in vivo соединение по примеру 19 (5-(Е)-2(3-6-[1-метил-1(метилсульфонил)этил]-8-хинолинилфенил)-1-[4-(метилсульфонил)фенил]этенил-1,2,4-оксадиазол-3 ил)метанол-8 006607 которое является ингибитором PDE4. Соответственно настоящее изобретение включает пролекарства,которые образуют in vivo ингибиторы PDE4 в виде метаболита после введения таких пролекарств млекопитающему. Кроме того, данное изобретение включает способ лечения, включающий стадию введения пролекарства для образования in vivo эффективного количества ингибитора PDE4, представленного формулой I. Представленные в данном описании сокращения имеют следующие значения, представленные в таблице. Сокращения, не представленные ниже в таблице, имеют их общепринятые значения, если не указано иного. Анализы, показывающие биологическую активность Анализы TNF- и LTB4 в цельной человеческой крови, индуцированных LPS и fMLP Цельная кровь обеспечивает среду, обогащенную белками и клетками, подходящую для изучения биохимической эффективности противовоспалительных соединений, таких как селективные ингибиторыPDE4. Обычная нестимулированная человеческая кровь не содержит обнаружимые уровни TNF- иLTB4. При стимуляции LPS активированные моноциты выражают и секретируют TNF- в течение времени до 8 ч и уровни плазмы остаются устойчивыми в течение 24 ч. Опубликованные исследования показывают, что ингибированные TNF- увеличением внутриклеточного цАМФ, вследствие ингибирования PDE4 и/или повышенной активности аденилилциклазы, происходит на транскрипционном уровне. Синтез LTB4 является также чувствительным к уровням внутриклеточного цАМФ и может быть полностью ингибирован селективными ингибиторами PDE4. Поскольку во время 24-часовой стимуляции человеческой крови LPS продуцируется небольшое количество LTB4, дополнительная стимуляция LPS с последующим заражением цельной человеческой крови fMLP является необходимой для синтеза LTB4 активированными нейтрофилами. Таким образом, при использовании одной и той же пробы крови можно оценить эффективность соединения в цельной крови по двум суррогатным маркерам активности PDE4 следующим способом. Свежую кровь собирали в гепаринизованные пробирки венепункцией у здоровых людейдобровольцев (мужчин и женщин). Данные субъекты не имели явных воспалительных состояний и не-10 006607 принимали какие-либо NSAID в течение по меньшей мере 4 дней до сбора крови. 500 мкл аликвоты крови предварительно инкубировали или с 2 мкл наполнителя (DMCO) или с 2 мкл испытуемого соединения при различных концентрациях в течение 15 мин при 37 С. Затем осуществляли добавление или 10 мкл наполнителя (PBS) в качестве контроля или 10 мкл LPS (конечная концентрация 1 мкг/мл, NL-2630PBS. Через 24 ч инкубации при 37 С к крови добавляли другие 10 мкл PBS (контроль) или 10 мкл LPS(конечная концентрация 1 мкг/мл) и инкубировали в течение 30 мин при 37 С. Затем кровь заражали или 10 мкл PBS (контроль) или 10 мкл fMLP (конечная концентрация 1 мкМ, NF-3506 (Sigma); разбавленного в 1 мас./об.% BSA (в PBS) в течение 15 мин при 37 С. Пробы крови центрифугировали при 1500 х g в течение 10 мин при 4 С для получения плазмы. 50 мкл аликвоту плазмы смешивали с 200 мкл метанола для осаждения белков и центрифугировали, как указывалось выше. Супернатант анализировали на содержание LTB4 с использованием комплекта для иммуноферментного анализа ( 520111 от CaymanChemical Co., Ann Arbor, MI) в соответствии с методикой производителя. TNF- анализировали в разбавленной плазме (в PBS) с использованием комплекта ELISA (Cistron Biotechnology, Pine Brook, NJ) в соответствии с методикой производителя. Значения IC50 в примерах 1-42 обычно находились в диапазоне от 0,04 до 8,71 мкМ. Противоаллергическая активность in vivo Соединения изобретения испытывали на их действие на аллергическое воспаление легких, опосредованное IgE и вызванное вдыханием антигена сенсибилизованными морскими свинками. Морских свинок сначала сенсибилизировали к воздействию яичного альбумина в условиях мягкой иммунодепрессии,индуцированной циклофосфамидом, внутрибрюшинной инъекцией антигена в комбинациях с гидроксидом алюминия и вакциной против коклюша. Через 2 и 4 недели им давали бустер-дозы антигена. В течение 6 недель животных заражали яичным альбумином в форме аэрозоля под покровом внутрибрюшинно введенного антигистаминного средства (мепирамина). Еще через 48 ч осуществляли бронхиальноальвеолярный лаваж (BAL) и подсчитывали количество эозинофилов и других лейкоцитов в жидкостяхBAL. Кроме того, для осуществления гистологического обследования на воспалительное поражение удаляли легкие. Введение соединений примеров (0,001-10 мг/кг внутрибрюшинно или парентерально) до 3 раз в течение 48 ч с последующим заражением антигеном приводило к значительному уменьшению эозинофилии и аккумуляции других воспалительных лейкоцитов. Наблюдали также меньшее воспалительное поражение в легких животных, которых лечили соединениями примеров. Протокол анализа активности PDE на основе SPA Соединения, которые ингибируют гидролиз цАМФ в АМФ, вызываемый специфическими фосфодиэстеразами IV типа, засевами в 96-луночные планшеты следующим образом: В 96-луночные планшеты при 30 С добавляли испытуемое соединение (растворенное в 2 мкл ДМСО), 188 мл субстратного буфера, содержащего [2,8-3H]аденозинцикло 3',5'-фосфат (цАМФ, от 100 нМ до 50 мкМ), 10 мМ MgCl2, 1 мМ EDTA, 50 MMTris, рН 7,5. Реакцию инициировали добавлением 10 мл человеческого рекомбинанта PDE4 (количество регулировали таким образом, чтобы в течение 10 мин. образовалось 10% продукта). Реакцию завершали через 10 мин. добавлением 1 мг гранул PDE-SPA(Amersham Pharmacia Biotech, Inc., Piscataway, NJ). Полученный продукт АМФ определяли количественно с использованием счетчика 96-луночных планшетов Wallac Microbeta (EGG.Wallac Co.,Gaithersburg, MD). Сигнал в отсутствие фермента определяли как фон. 100% активность определяли в виде сигнала, обнаруженного в присутствии фермента и ДМСО, за вычетом фона. Соответственно вычисляли процентное значение ингибирования. Значение IC50 почти приближалось к нелинейной регрессии при использовании уравнения стандартный 4-параметр/множество участков связывания, исходя из десяти точек титрования. Значения IC50 в примерах 1-42 определяли со 100 нМ цАМФ с использованием очищенного синтезированного белка GST человеческого рекомбинанта фосфодиэстеразы IVa (met-248), полученного из системы экспрессии палочковидный виpyc/sf-9. Значения IC50 в примерах 1-42 обычно находились в диапазоне от 0,14 до 10,24 нМ, хотя в одном примере значение IC50 было равно 109 нМ. Следующие примеры представлены в качестве иллюстрации некоторых предпочтительных вариантов изобретения и они ни в коей мере не ограничивают изобретение. Если не указано иного, методики экспериментов осуществляли при следующих условиях. Все операции проводили при комнатной температуре или температуре окружающей среды, т.е. при температуре в диапазоне 18-25 С. Выпаривание растворителя осуществляли с использованием роторного испарителя при пониженном давлении (600-4000 паскалей: 4,5-30 мм Нg) с температурой бани до 60 С. Ход реакций контролировали тонкослойной хроматографией (ТСХ) (TLC) и время реакции дано только с целью иллюстрации. Точки плавления не скорректированы и d означает разложение. Данные точки плавления являются точками плавления, полученными для веществ, которые получены, как указано в описании. В некоторых получениях полиморфизм может привести к выделению веществ с различными точками плавления. Структуру и чистоту всех конечных продуктов гарантировали по меньшей мере одной из следующих методик: ТСХ, масс-спектрометрия, спектрометрия ядерного магнитного резонанса (ЯМР) или-11 006607 микроаналитические данные. Выходы даны только с целью иллюстрации. Если представлены данные ЯМР, то они даны в форме значений дельтадля основных диагностических протонов, представленных в частях на миллион (м.д.) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта,определенных при 300, 400 или 500 МГц с использованием вышеуказанного растворителя. Традиционные сокращения, использованные для формы сигналов, являются следующими: S=синглет; d=дублет;t=триплет; m=мультиплет; br=широкий; и т.д. Кроме того, Аr означает сигнал ароматического соединения. Химические символы имеют их обычные значения; использованы также следующие сокращения:(грамм(ы, мг (миллиграмм(ы, моль (моли), ммоль (миллимоли), экв. (эквивалент(ы. Методы синтеза Соединения по настоящему изобретению могут быть получены в соответствии со следующими методами. Заместители являются такими же, как и в формуле I, за исключением того, где они определены иным образом. Схема 1. Синтез кетонаY=галоген, Н А=4-(метилтио)бензальдегид Е=электрофил Аr=арил или гетероарил Как следует из вышеприведенной схемы и представленной ниже табл. 1, промежуточный спирт может быть получен взаимодействием арил- или гетероарилметаллических разновидностей III, таких как магнийорганический галогенид, с 4-(метилтио)бензальдегидом (А) в органическом растворителе, таким как ТГФ. Промежуточный спирт II может быть также получен обработкой арил- или гетероарилгидрида или бромида IV основанием или металлоорганическим соединением, таким как н-бутиллитий, в органическом растворителе, таком как ТГФ, и затем 4-(метилтио)бензальдегидом. Альтернативно, промежуточный спирт II может быть также получен следующими химическими преобразованиями: 1) обработкой арил- или гетероарилдигидрида, галогенидгидрида или дигалогенида V основанием или металлоорганическим соединением, таким как н-бутиллитий, в органическом растворителе, таком как ТГФ, и затем электрофилом, таким как ацетон или 4-(метилтио)бензальдегид; 2) последующей обработкой основанием или металлоорганическим соединением, таким как н-бутиллитий, в органическом растворителе, таком как ТГФ, и затем электрофилом, таким как ацетон или 4-(метилтио)бензальдегид, при этом в первом или втором преобразовании 4-(метилтио)бензальдегид должен использоваться в качестве электрофила. Сульфон-спирт VI может быть получен окислением сернистого спирта II окислителем, таким как оксон,в растворителе, таком как смесь ТГФ/МеОН/Н 2O. Кетоны VII и VIII могут быть получены окислением спиртов II и VI, соответственно окислителем, таким как MnO2 в растворителе, таком как CH2Cl2. Сульфон-кетон VIII может быть также получен окислением сернистого кетона VII окислителем, таким как оксон, в растворителе, таком как смесь ТГФ/МеОН/Н 2O. Кетон K1. (4-Фторфенил)[4-(метилсульфонил)]фенилкетон. Кетон K1 получали следующим способом. Стадия 1. (4-Фторфенил)[4-метилтио)фенил]кетон. К раствору 4-(метилтио)бензальдегида (2,5 г, 16,4 ммоль) в ТГФ (100 мл) при -78 С добавляли по каплям 4-фторфенилмагнийбромид (1,0 М в ТГФ, 19,7 мл, 19,7 ммоль). Полученный раствор перемешивали при -78 С в течение 3 ч и гасили насыщенным водным раствором NH4Cl. Затем смесь разбавлялиEtOAc и 10% НСl, экстрагировали и промывали (NaHCO3 (насыщ.), насыщенный раствор соли). Органическую фазу сушили над MgSO4 и концентрировали. Затем остаток обрабатывали MnO2 (28,6 г, 330 ммоль) в CH2Cl2 (150 мл) и реакционную смесь перемешивали при к.т. всю ночь. Смесь фильтровали через плаг из силикагеля (EtOAc) с получением 2,6 г (4-фторфенил)[4-метилтио)фенил]кетонового соединения. Стадия 2. (4-Фторфенил)[4-метилсульфонил)фенил]кетон. К раствору сульфида - иными словами, (4-фторфенил)[4-метилтио)фенил]кетона - с данной стадии 1 (2,0 г, 8,1 ммоль) в ТГФ/МеОН/Н 2O (80/40/40 мл) добавляли оксон (7,5 г, 12,2 ммоль). Смесь перемешивали при к.т. в течение 4 ч, гасили NaHCO3 (насыщ.), насыщенным раствором соли, сушили надNa2SO4, фильтровали и концентрировали. В результате кристаллизации (СН 2 Сl2/гексан) получали (4 фторфенил)[4-(метилсульфонил)фенил]кетон - кетоновое соединение K1 в виде белого твердого вещества. Кетон K2. (1-Метил-1 Н-имдазол-2-ил)[4-метилтио)фенил]кетон. Кетон K2 получали следующим способом. Стадия 1. (1-Метил-1 Н-имдазол-2-ил)[4-метилтио)фенил]метанол. К раствору N-метилимидазола (10,0 г, 122 ммоль) в 500 мл ТГФ при -78 С добавляли по каплям нбутиллитий (2,5 М в гексане, 48,7 мл, 118 ммоль) и образовавшийся раствор перемешивали при -78 С в течение 30 мин. Затем при -78 С добавляли 4-(метилтио)бензальдегид (14,73 мл, 110 ммоль) и смесь перемешивали до завершения реакции, которое определяли ТСХ, и гасили NH4Cl (насыщ.). Затем смесь разбавляли EtOAc, экстрагировали и промывали (NaHCO3 (насыщ.), насыщенный раствор соли). Органическую фазу сушили над MgSO4, фильтровали и концентрировали. В результате кристаллизации(EtOAc/гексан) получали (1-метил-1 Н-имидазол-2-ил)[4-метилтио)фенил]метанол. Стадия 2. (1-Метил-1 Н-имидазол-2-ил)[4-(метилтио)фенил]кетон. К раствору спирта с предыдущей стадии 1 (25,7 г, 111 ммоль) в EtOAc (250 мл) и СН 2 Сl2 (250 мл) добавляли МnO2 (140 г, 1,66 ммоль) и реакционную смесь перемешивали при к.т. всю ночь. Смесь фильтровали через плаг из силикагеля (EtOAc) с получением кетона K2.-13 006607 Кетон К 3. (4-Метилсульфонил)(фенил)кетон. Кетон К 3 получали следующим способом. Стадия 1. (4-Метилтио)(фенил)метанол. К раствору 4-(метилтио)бензальдегида (1,0 г, 6,5 ммоль) в ТГФ (20 мл) при 0 С добавляли фенилмагнийхлорид (2 М, ТГФ, 3,5 мл, 7,0 ммоль). Через 0,5 ч смесь нейтрализовали при к.т. насыщенным раствором NH4Cl, разбавляли водой и экстрагировали Et2O. Органические экстракты промывали (Н 2O), (насыщенным раствором соли), сушили (MgSO4), фильтровали и концентрировали. В результате очистки при энергичном перемешивании в смеси гексан/Et2O и фильтрации получали (4 метилтио)(фенил)метанол в виде белого твердого вещества. Стадия 2. (4-Метилтио)(фенил)кетон.(4-Метилтио)(фенил)кетон получали обработкой (4-метилтио)(фенил)метанола с данной стадии 1 МnO2, как на стадии 2 способа, представленного ниже для получения K4. Стадия 3. (4-Метилтио)(фенил)кетон. К раствору (4-метилтио)(фенил)кетона с данной стадии 2 (0,98 г, 4,3 ммоль) в СНСl3 (10 мл) при 0 С добавляли mСРВА (м-хлорпербензойную кислоту) (1,7 г, 10 ммоль). Через 0,5 ч при к.т. к смеси добавляли Са(ОН)2 (1,7 г, 23 ммоль) и перемешивали в течение 1 ч. В результате фильтрации на Celite и концентрирования получали кетон K3 в виде белого твердого вещества. Кетон K4. (1,3-Тиазол-2-ил)[4-(метилтио)фенил]кетон. Кетон K4 получали следующим способом. Стадия 1. (1,3-Тиазол-2-ил)[4-(метилтио)фенил]метанол. К раствору тиазола (5,0 г, 58,7 ммоль) в ТГФ (250 мл) при -78 С добавляли по каплям н-бутиллитий(2,5 М в гексане, 23,5 мл, 58,7 ммоль) и образовавшийся раствор перемешивали при -78 С в течение 10 мин. Затем при -78 С добавляли 4-(метилтио)бензальдегид (7,1 мл, 53,4 ммоль). Полученную смесь перемешивали до завершения реакции и гасили насыщенным водным раствором NH4Cl. Затем смесь разбавляли EtOAc и 10% НСl, экстрагировали и промывали (NaHCO3 (насыщ.), насыщенным раствором соли). Органическую фазу сушили над MgSO4 и концентрировали. Затем остаток очищали флэшхроматографией (80% CH2Cl2/20% EtOAc) с получением (1,3-тиазол-2-ил)[4-метилтио)фенил]метанола. Стадия 2. (1,3-Тиазол-2-ил)[4-метилтио)фенил]кетон. К раствору (1,3-тиазол-2-ил)[4-метилтио)фенил]метанола с данной стадии 1 (10,0 г, 42,1 ммоль) вEtOAc (250 мл) добавляли МnO2 (70 г, 843 ммоль) и реакционную смесь перемешивали при 25 С всю ночь. Смесь фильтровали через плаг из силикагеля (EtOAc) с образованием кетонового соединения K4. Кетон K5. (1,3-Тиазол-2-ил)[4-метилсульфонил)фенил]кетон. Кетон K5 получали следующим способом. К раствору K4 (1,3-тиазол-2-ил)[4-метилтио)фенил]кетона (8,2 г, 34,7 ммоль) в ТГФ/МеОН/Н 2O(350/175/175 мл) добавляли оксон (42,6 г, 69, 4 ммоль). Реакционную смесь перемешивали при 25 С в течение 3 ч и гасили насыщенным водным раствором NaHCO3. Затем полученную смесь разбавлялиEtOAc, экстрагировали и промывали (NaHCO3 (насыщ.), насыщенным раствором соли). Органическую фазу сушили над MgSO4 и концентрировали. Затем остаток очищали кристаллизацией (EtOAc/гексан) с получением (1,3-тиазол-2-ил)[4-метилсульфонил)фенил]кетона. Кетон K6. [5-(1-Гидрокси-1-метилэтил)-1,3-тиазол-2-ил][4-(метилсульфонил)фенил]кетон. Кетон K6 получали следующим способом. Стадия 1. [5-(1-Гидрокси-1-метилэтил)-1,3-тиазол-2-ил][4-(метилтио)фенил]кетон. К раствору тиазола (1,0 г, 12,0 ммоль) в ТГФ (100 мл) при -78 С добавляли по каплям н-бутиллитий(2,3 М в гексане, 5,3 мл, 12,3 ммоль) и образовавшийся раствор перемешивали при -78 С в течение 10 мин. Затем при -78 С добавляли 4-(метитио)бензальдегид (7,1 мл, 53,4 ммоль). Полученную смесь перемешивали 10 мин при к.т. и охлаждали при -78 С. Затем добавляли по каплям н-бутиллитий (2,3 М в гексане; 5,3 мл, 12,3 ммоль) и образовавшийся раствор перемешивали при 25 С в течение 10 мин и гасили ацетоном (3,0 мл). Затем смесь разбавляли EtOAc и 10% НСl, экстрагировали и промывали (NaHCO3 (насыщ.), насыщенным раствором соли). Органическую фазу сушили над МgSO4 и концентрировали. Затем остаток обрабатывали МnО 2 (20,4 г, 235 ммоль) в СН 2 Сl2 (250 мл) и реакционную смесь перемешивали при к.т. всю ночь. Затем образовавшуюся смесь фильтровали через плаг из силикагеля (EtOAc). В результате флэш-хроматографии (90% CH2Cl2/10% EtOAc) получали [5-(1-гидрокси-1-метилэтил)-1,3 тиазол-2-ил][4-(метилтио)фенил]кетон. Стадия 2. [5-(1-Гидрокси-1-метилэтил)-1,3-тиазол-2-ил][4-(метилсульфонил)фенил]кетон. К раствору сульфида, т.е. [5-(1-гидрокси-1-метилэтил)-1,3-тиазол-2-ил][4-(метилтио)фенил]кетона,с данной стадии 1 (1,7 г, 5,8 ммоль) в ТГФ/МеОН/Н 2O (100/50/50 мл) добавляли оксон (7,1 г, 11,5 ммоль). Реакционную смесь перемешивали при 25 С в течение 3 ч и гасили насыщенным водным раствором NaHCO3. Затем смесь разбавляли EtOAc, экстрагировали и промывали (NаНСО 3 (насыщ.), насыщенным раствором соли). Органическую фазу сушили над MgSO4 и концентрировали. Затем остаток очищали кристаллизацией (EtOAc/гексан) с получением кетона K6. Кетон K7. (6-Метил-3-пиридинил)[4-(метилсульфонил)фенил]кетон. Кетон K7 получали следующим способом.-14 006607 Стадия 1. (6-Метил-3-пиридинил)[4-(метилтио)фенил]метанол. К раствору 3-бром-6-метилпиридина (760 мг, 1 экв.) в ТГФ (20 мл) при -78 С медленно добавляли н-бутиллитий в гексане (1,1 экв.). Затем раствор перемешивали в течение 30 мин. После этого медленно добавляли 4-(тиометил)бензальдегид (738 мг, 1,1 экв.). Раствор нагревали до к.т. Добавляли NH4Cl (насыщ.), затем воду и EtOAc. Отделяли органическую фазу, сушили над MgSO4 и концентрировали. Осаждением с использованием смеси простой эфир/гексан получали (6-метил-3-пиридинил)[4(метилтио)фенил]метанол и использовали без дальнейшей очистки на следующей стадии. Стадия 2. (6-Метил-3-пиридинил)[4-(метилсульфонил)фенил]метанол. Следуя вышеуказанной методике стадии 2 получения кетона K1, но с заменой (4-фторфенил)[4(метилтио)фенил]кетона, используемого в качестве исходного материала, на сульфид (6-метил-3 пиридинил)[4-(метилтио)фенил]метанол с данной стадии 1, получали (6-метил-3-пиридинил)[4(метилсульфонил)фенил]метанол. Стадия 3. (6-Метил-3-пиридинил)[4-(метилсульфонил)фенил]кетон. Следуя вышеуказанной стадии 2 получения кетона K2, но с заменой (1-метил-1 Н-имидазол-2-ил)[4(метилтио)фенил]метанола, используемого в качестве исходного материала, на (6-метил-3-пиридинил)[4(метилсульфонил)фенил]метанол с данной стадии 2, получали кетон K7. Кетон K8. (5-Метил-2-пиридинил)[4-(метилсульфонил)фенил]кетон. Кетон 8 получали по способу, описанному для кетона K7, но при этом 3-бром-6-метилпиридин заменяли на 2-бром-5-метилпиридин. Кетон K9. бис-[(4-Метилсульфонил)фенил]кетон. Кетон K9 получали по способу, описанному для кетона K7, но при этом 3-бром-6-метилпиридин заменяли на 4-бромтиоанизол и на стадии окисления сульфида использовали двойное количество оксона. Кетон K10. (2-Пиридинил)[4-(метилсульфонил)фенил]кетон. Кетон K10 получали по способу, описанному для кетона K7, но при этом 3-бром-6-метилпиридин заменяли на 2-бромпиридин. Кетон K11. [5-(1-Гидрокси-1-метилэтил)-2-пиридинил][4-(метилсульфонил)фенил]кетон. Кетон K11 получали следующим способом. Стадия 1. [5-(1-Гидрокси-1-метилэтил)-2-пиридинил][4-(метилтио)фенил]метанол. К суспензии 2,5-дибромпиридина (5,12 г, 1 экв.) в простом эфире при -78 С медленно добавляли нбутиллитий в гексане (1,05 экв.). Образовавшийся желто-оранжевый осадок перемешивали 30 мин. Затем добавляли ацетон (1,54 мл, 1,05 экв.). Раствор поддерживали при -78 С в течение еще 30 мин. К полученной оранжевой суспензии медленно добавляли шприцом н-бутиллитий в гексане (1,1 экв.). Затем суспензию перемешивали 1 ч при -78 С. После этого добавляли 4-(метилтио)бензальдегид (2,85 мл, 1,1 экв.). Полученную суспензию нагревали до -35 С и гасили раствором NH4Cl (насыщ.). Добавляли воду иEtOAc и органический слой сушили над MgSO4, выпаривали и очищали флэш-хроматографией (EtOAc) с получением [5-(1-гидрокси-1-метилэтил)-2-пиридинил][4-(метилтио)фенил]метанола. Стадия 2. [5-(1-Гидрокси-1-метилэтил)-2-пиридинил][4-(метилсульфонил)фенил]метанол. Следуя вышеуказанной методике стадии 2 получения кетона К 1, но с заменой (4-фторфенил)[4(метилтио)фенил]кетона, используемого в качестве исходного материала, на сульфид, т.е. [5-(1 гидрокси-1-метилэтил)-2-пиридинил][4-(метилтио)фенил]-метанол с данной стадии 1, получали [5-(1 гидрокси-1-метилэтил)-2-пиридинил][4-(метилсульфонил)фенил]метанол. Стадия 3. [5-(1-Гидрокси-1-метилэтил)-2-пиридинил][4-(метилсульфонил)фенил]кетон. Следуя вышеуказанной методике стадии 2 получения кетона K2, но с заменой (1-метил-1 Нимидазол-2-ил)[4-(метилтио)фенил]метанола, используемого в качестве исходного материала, на [5-(1 гидрокси-1-метилэтил)-2-пиридинил][4-(метилсульфонил)фенил]метанол с данной стадии 2, получали кетон K11. Боронатные соединения, использованные для получения соединений по данному изобретению, могут быть получены в соответствии с представленной ниже схемой 2. Арилбромиды IX и X могут быть получены обработкой бензилфосфонийбромида XI основанием,таким как трет-ВuОK или LiHMDS, в органическом растворителе, таком как ТГФ, с последующим добавлением кетона VII или VIII к реакционной смеси. Сульфид IX может быть преобразован в сульфон Х обработкой оксоном в растворителе, таком как смесь ТГФ/МеОН/Н 2 О. Сложный эфир бороната XII может быть получен нагреванием арилбромида Х с пинакондибораном в присутствии основания, такого какCH3CN (50 мл) при 25 С добавляли по каплям трет-ВuОK (1,0 М в ТГФ, 19,9 мл, 19,9 ммоль) и образовавшийся красный раствор перемешивали при к.т. в течение 20 мин. К образовавшемуся илиду при 25 С затем добавляли кетон K2 (4,4 г, 18,9 ммоль). Полученную смесь перемешивали при 60 С в течение 2 дней и гасили NH4Cl (насыщ.). Затем смесь разбавляли EtOAc. Органическую фазу промывали NaHCO3(насыщ.), насыщенным раствором соли, сушили над МgSO4, фильтровали и концентрировали и непосредственно использовали на следующей данной стадии 2. Стадия 2. (Е)-2-(3-бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1[4-(метилсульфонил)фенил]этен. К раствору неочищенного сульфида, т.е. (E/Z)-2-(3-бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1-[4(метилтио)фенил]этена, с данной стадии 1 (18,9 ммоль) в ТГФ/МеОН/Н 2O (200/100/100 мл) добавляли оксон (23,2 г, 37,8 ммоль). Смесь перемешивали при к.т. в течение 4 ч, гасили NaHCO3 (насыщ.) и разбавляли EtOAc. Органическую фазу промывали NаНСО 3 (насыщ.), насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. В результате флэш-хроматографии (95% EtOAc/5% Et3N) получали (Е)-2-(3-бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1[4-(метилсульфонил)фенил]этен (одиночный изомер) в виде пены. Стадия 3. Пинакон 3-(Е)-2-(1-метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенилборонат. Суспензию бромида, т.е. (Е)-2-(3-бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1[4-(метилсульфонил)фенил]этена, с данной стадии 2 (2,0 г, 4,8 ммоль), пинакондиборана (1,5 г, 5,8 ммоль), KОАс (1,65-16 006607 г, 16,8 ммоль) и PdCl2 (dppf) (0,2 г, 0,24 ммоль) в 50 мл ДМФ перемешивали при 90 С в течение 4 ч. Полученную соль охлаждали до к.т., разбавляли EtOAc, промывали H2O (3 х), насыщенным раствором соли,сушили над Na2SO4, фильтровали и концентрировали. В результате флэш-хроматографии (95%(200 мл) при 0 С добавляли по каплям LiHMDS (1,0 М в ТГФ, 86,9 мл, 86,9 ммоль) и образовавшийся красный раствор перемешивали при к.т. в течение 20 мин. К полученному илиду затем добавляли при 0 С кетон K4 (18,6 г, 79,0 ммоль). Смесь перемешивали до завершения реакции, которое контролировали ТСХ, и гасили NH4Cl (насыщ.). Затем смесь разбавляли EtOAc. Органическую фазу промывали NаНСО 3(E/Z-2-(3-бромфенил)-1-(1,3-тиазол-2-ил)-1-[4(метилтио)фенил]этен (смесь изомеров при их соотношении 1,5 к 1). Стадия 2. (E/Z)-2-(3-бромфенил)-1-(1,3-тиазол-2-ил)-1-[4-(метилсульфонил)фенил]этен. К раствору сульфида, т.е. (E/Z)-2-(3-бромфенил)-1-(1,3-тиазол-2-ил)-1-[4-(метилтио)фенил]этена, с данной стадии 1 (24,8 г, 63, 9 ммоль) в ТГФ/МеОН/Н 2O (600/300/300 мл) добавляли оксон (78,5 г, 128 ммоль). Полученную реакционную смесь перемешивали при к.т. всю ночь. Смесь гасили NаНСО 3 (насыщ.) и разбавляли EtOAc. Органическую фазу промывали NaHCO3 (насыщ.), насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали с получением (E/Z)-2-(3-бромфенил)-1-(1,3 тиазол-2-ил)-1-[4-(метилсульфонил)фенил]этена (смесь изомеров при их соотношении 3 к 2). Стадия 3. Пинакон 3-(E/Z)-2-(1,3-тиазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенилборонат. Суспензию бромида (E/Z)-2-(3-бромфенил)-1-(1,3-тиазол-2-ил)-1-[4-(метилсульфонил)фенил]этена с данной стадии 2 (15,0 г, 35,7 ммоль), пинакондиборана (10,9 г, 42,8 ммоль), KОАс (12,3 г, 125 ммоль) иPdCl2 (dppf) (1,46 г, 1,78 ммоль) в 350 мл ДМФ перемешивали при 90 С в течение 4 ч. Полученную смесь охлаждали до к.т., разбавляли EtOAc, промывали Н 2O (3 х), насыщенным раствором соли, сушили надNa2SO4, фильтровали и концентрировали. В результате флэш-хроматографии (толуол/ацетон 9/1) получали боронат В 2 (смесь изомеров при их соотношении 3 к 1) в виде пены. Боронат В 3. Пинакон 3-(Е)-2-(5-метил-2-пиридиниил)-2-[4-(метилсульфонил)фенил]этенилфенилборонат. Боронат В 3 получали следующим способом. Стадия 1. (Е)-2-(3-бромфенил)-1-(5-метил-2-пиридинил)-1-[4-(метилсульфонил)фенил]этилен. Следуя способу, описанному для стадии 1 получения бороната В 1, но с заменой кетона K2, используемого в качестве исходного материала, на кетон K8, после разделения изомеров флэш-хроматографией получали (Е)-2-(3-бромфенил)-1-(5-метил-2-пиридинил)-1-[4-(метилсульфонил)фенил]этилен. Стадия 2. Пинакон 3-(Е)-2-(5-метил-2-пиридинил)-2-[4-(метилсульфонил)фенил]этенилфенилборонат. Следуя способу, описанному для стадии 3 получения бороната В 1, но с заменой (Е)-2-(3 бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1-[4-(метилсульфонил)фенил]этена, используемого в качестве исходного материала,на бромид(Е)-2-(3-бромфенил)-1-[5-(1-гидрокси-1-метилэтил)-2-пиридинил]-1-[4-(метилсульфонил)фенил]этен. Следуя способу, описанному для стадии 1 получения бороната В 1, но с заменой кетона K2, используемого в качестве исходного материала, на кетон K11, после разделения изомеров флэшхроматографией получали(Е)-2-(3-бромфенил)-1-[5-(1-гидрокси-1-метилэтил)-2-пиридинил]-1-[4(метилсульфонил)фенил]этен. Стадия 2. Пинакон 3-(Е)-2-(5-(1-гидрокси-1-метилэтил)-2-пиридинил)-2-[4-(метилсульфонил)фенил]этенилфенилборонат. Следуя способу, описанному для стадии 3 получения бороната В 1, но с заменой (Е)-2-(3 бромфенил)-1-(1-метил-1 Н-имидазол-2-ил]-1-[4-(метилсульфонил)фенил]этена, используемого в качестве исходного материала, на бромид (Е)-2-(3-бромфенил)-1-[5-(1-гидрокси-1-метилэтил)-2-пиридинил]-1[4-(метилсульфонил)фенил]этен с данной стадии 1, получали боронат В 4. Арилбромидные соединения, используемые для получения соединений по данному изобретению,могут быть получены в соответствии с представленными ниже схемами 3 и 4. Как следует из вышепредставленной схемы 3, промежуточный нитрил XIIIа может быть получен алкилированием 4-метоксифенола хлорацетонитрилом в присутствии основания, такого как карбонат калия, в растворителе, таком как ацетон. Амидоксим XIV может быть получен обработкой нитрила XIII гидроксиламином в растворителе, таком как метанол, в присутствии основания, такого как ацетат натрия. Образование оксадиазола XVI може быть достигнуто активацией арилуксусной кислоты XV карбонилдиимидазолом в растворителе, таком как ДМФ, с последующим добавлением амидоксима XIV и последующим нагреванием реакционной смеси. Схема 4. Синтез арилбромида Как следует из вышепредставленной схемы 4, конденсация альдегида XVII нагреванием с арилуксусной кислотой XV в присутствии основания, такого как пиперидин, в растворителе, таком как толуол,обеспечивает получение ненасыщенной кислоты XVIIIa. После образования хлорангидрида кислотыXVIIIa in situ обработкой тионилхлоридом и основанием, таким как триэтиламин, в растворителе, таком как толуол, осуществляют добавление амина к реакционной смеси с получением амида XVIIIb. Оксадиазолэтен XVIIIс может быть образован нагреванием OХ 1 с XVII в присутствии основания, такого как пиперидин, в растворителе, таком как толуол. Как следует из вышеуказанного приложения к схеме 4, обработка кислоты XVIIIа диазометаном в растворителе, таком как ТГФ, обеспечивает получение сложного метилового эфира XVIIId. Восстановление сложного эфира XVIIId с использованием DIBAL-H в растворителе, таком как ТГФ, дает аллиловый спирт XVIIIe. Преобразование спиртовой группы, имеющейся в XVIIIe, в удаляемую группу, такую как мезилат, с использованием реагентов, таких как метансульфонилхлорид и триэтиламин, в растворителе, таком как ТГФ, с последующим замещением нуклеофилом, таким как диметиламин, в растворителе, таком как ДМФ, обеспечивает получение соединения XVIIIf. Арилбромид АВ 1. (Е)-3-(3-Бромфенил)-2-[4-(метилсульфонил)фенил]-2-акриловая кислота. Арилбромид АВ 1 получали следующим способом. К раствору 3-бромбензальдегида (12,9 г, 70 ммоль) в толуоле (100 мл) добавляли 4(метилсульфонил)фенилуксусную кислоту (15 г, 70 ммоль) и пиперидин (2 мл). После кипячения с обратным холодильником в течение ночи смесь охлаждали до к.т. К образованной таким образом суспензии добавляли толуол (10 мл). В результате фильтрации получали (Е)-3-(3-бромфенил)-2-[4(метилсульфонил)фенил]-2-акриловую кислоту в виде белого твердого вещества. Арилбромид АВ 2. (Е)-N-Изопропил-3-(3-бромфенил)-2-[4-(метилсульфонил)фенил]-2-пропенамид. Арилбромид АВ 2 получали следующим способом. К раствору АВ 1 (24,9 г, 65 ммоль) в толуоле (250 мл) добавляли тионилхлорид (14,3 мл, 196 ммоль) и триэтиламин (34 мл, 245 ммоль). После перемешивания при к.т. в течение 0,5 ч добавляли изопропиламин (28 мл, 327 ммоль). Через 2 ч стояния при к.т. смесь охлаждали до 0 С и нейтрализовали насыщенным раствором NH4Cl, затем экстрагировали EtOAc. Органические экстракты промывали (Н 2O, насыщенный раствор соли), сушили (МgSO4), фильтровали и концентрировали. В результате очистки флэшхроматографией (от гексан:EtOAc, 1:1 до чистого EtOAc) получали (Е)-N-изопропил-3-(3-бромфенил)-2[4-(метилсульфонил)фенил]-2-пропенамид. Арилбромид АВ 3. (Е)-3-(3-Бромфенил)-2-[4-(метилсульфонил)фенил]-2-пропенамид. Арилбромид АВ 3 получали следующим способом, описанным для получения арилбромида АВ 2, но с заменой изопропиламина, используемого в качестве исходного материала, на гидроксид аммония. Арилбромид АВ 4. (Е)-N-(трет-Бутил)-3-(3-бромфенил)-2-[4-метилсульфонил)фенил]-2-пропенамид. Арилбромид АВ 4 получали по способу, описанному для получения арилбромида АВ 2, но с заменой изопропиламина, используемого в качестве исходного материала, на трет-бутиламин. Арилбромид АВ 5. (Е)-1-(3-Бромфенил)-2-(3-метил-1,2,4-оксадиазол-5-ил)-2-[метилсульфонил)фенил]этен. Арилбромид АВ 5 получали следующим способом. Стадия 1 (cхема 3, оксадиазол OХ 1). (3-Метил-1,2,4-оксадиазол-5-ил)[4-(метилсульфонил)фенил]метан. К раствору 4-(метилсульфонил)фенилуксусной кислоты (15 г, 70 ммоль) в ДМФ (300 мл) при к. т. добавляли карбонилдиимидазол (12,5 г, 77 моль). Через 0,5 ч при к.т. добавляли ацетамидоксим (5,7 г, 77 ммоль). После перемешивания образовавшейся смеси в течение ночи при к.т. смесь нагревали до 120 С в течение 6 ч. После охлаждения до к.т. смесь гасили Н 2O и экстрагировали EtOAc. Органические экстракты промывали (Н 2O, насыщенный раствор соли), сушили (МgSO4), фильтровали и концентрировали. В результате очистки флэш-хроматографией (гексан:EtOAc, 1:1) получали (3-метил-1,2,4-оксадиазол-5 ил)[4-(метилсульфонил)фенил]метан.-19 006607 Стадия 2 (схема 4). (Е)-1-(3-Бромфенил)-2-(3-(метил-1,2,4-оксадиазол-5-ил)-2-[4-(метилсульфонил)фенил]этен. К раствору 3-бромбензальдегида (2,2 г, 11,9 ммоль) в толуоле (30 мл) добавляли продукт со стадии 1 (ОХ 1) (3,0 г, 11,9 ммоль) и пиперидин (0,4 мл). После кипячения с обратным холодильником в течение ночи смесь охлаждали до к.т. К образовавшейся суспензии добавляли МеОН (30 мл). После дополнительного кипячения с обратным холодильником осуществляли охлаждение до 0 С и после фильтрации получали (Е)-1-(3-бромфенил)-2-(3-(метил-1,2,4-оксадиазол-5-ил)-2-[4-(метилсульфонил)фенил]этен в виде белого твердого вещества. Бромхинолины, используемые для получения соединений по данному изобретению, могут быть получены в соответствии с представленной ниже схемой 5. Схема 5. Получение бромхинолинов Как следует из представленной выше схемы 5 и представленной ниже таблицы к схеме 5, для получения соединений ХХа может быть использована обработка бромметилового соединения XIX нуклеофилом, таким как метансульфинат натрия или цианид калия, в растворителе, таком как ДМФ или смесь ДМФ и воды. Соединение ХХb может быть получено обработкой ХХа основанием, таким как третбутоксид калия (1,1 экв.), в растворителе, таком как ТГФ, с последующим добавлением образовавшейся смеси в раствор метилиодида в растворителе, таком как ТГФ. Соединение ХХс может быть получено обработкой ХХb основанием, таким как трет-бутоксид калия (1,1 экв.), в растворителе, таком как ТГФ, с последующим добавлением полученной смеси в раствор метилиодида в растворителе, таком как ТГФ. Соединение ХХс (где R1=CN) может быть также получено обработкой ХХа основанием, таким как третбутоксид калия (2,2 экв.), и метилиодидом в растворителе, таком как ТГФ. Соединение ХХс (гдеR1=SO2Me) может быть также получено обработкой ХХа основанием, таким как трет-бутоксид калия (1,3 экв.), и метилиодидом (1,6 экв.) в растворителе, таком как ТГФ, с последующим добавлением дополнительного количества метилиодида (1,6 экв.) и дополнительного количества указанного того же самого основания (1,0 экв.). Таблица к схеме 5 Бромхинолины-20 006607 Бромхинолин Q1. 6-(Метилсульфонил)метил-8-бромхинолин. Бромхинолин Q1 получали следующим способом. К 6-бромметил-8-бромхинолину (60 г, 200 ммоль) (описанному в международной заявке на патентWO 94/22852) и метансульфинату натрия (27,6 г, 270 ммоль) добавляли ДМФ (500 мл). После перемешивания в течение ночи при к.т. смесь гасили H2O (2000 мл), перемешивали в течение одного часа, выделяли фильтрацией и промывали Et2O с получением 6-(метилсульфонил)метил-8-бромхинолина. Бромхинолин Q2. 6-[1-(Метилсульфонил)этил]-8-бромхинолин. Бромхинолин Q2 получали следующим способом. К раствору бромхинолина Q1 (16,1 г, 54 ммоль) в ТГФ (500 мл) при -78 С добавляли трет-бутоксид калия (59 мл, 1 н. в ТГФ). После стояния при -78 С в течение 0,5 ч образовавшуюся смесь перемешивали при 0 С в течение 45 мин и затем переносили канюлей по каплям в раствор МеI (16,7 мл, 268,3 ммоль) в ТГФ (160 мл). После перемешивания в течение ночи при к.т. смесь нейтрализовали насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические экстракты промывали (Н 2 О), (насыщенным раствором соли), сушили (MgSO4), фильтровали и концентрировали. После перемешивания в простом эфире и последующего выделения фильтрацией получали 6-[1-(метилсульфонил)этил]-8-бромхинолин. Бромхинолин Q3. 6-[1-Метил-1-(метилсульфонил)этил]-8-бромхинолин. Бромхинолин Q3 получали следующим способом. К раствору бромхинолина Q2 (15,7 г, 50 ммоль) в ТГФ (500 мл) при -78 С добавляли трет-бутоксид калия (55 мл, 1 н. в ТГФ). После перемешивания в течение 0,5 ч при -78 С полученную смесь перемешивали при 0 С в течение 45 мин и затем переносили по каплям в раствор МеI (15,6 мл, 250 ммоль) в ТГФ(40 мл) при 0 С. После перемешивания в течение ночи при к.т. смесь нейтрализовали насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические экстракты промывали (Н 2O, насыщенным раствором соли), сушили (MgSO4), фильтровали и концентрировали. После перемешивания в простом эфире с последующим выделением фильтрацией получали 6-[1-метил-1-(метилсульфонил)этил]-8-бромхинолин. Бромхинолин Q4. 6-Цианометил-8-бромхинолин. Бромхинолин Q4 получали следующим способом. К 6-бромметил-8-бромхинолину (3 г, 10 ммоль), описанному в международной заявке на патент WO 94/22852, и цианиду калия (1,6 г, 25 ммоль) добавляли ДМФ (10 мл) и H2O (5 мл). После нагрева при 100 С в течение 1 ч образовавшуюся смесь гасили Н 2O и экстрагировали EtOAc. Органические экстракты промывали (Н 2O, насыщенный раствор соли), сушили (MgSO4), фильтровали и концентрировали. В результате очистки флэш-хроматографией (гексан:EtOAc, 3:1) получали 6-цианометил-8-бромхинолин. Бромхинолин Q5. 6-[1-Метил-1-цианоэтил]-8-бромхинолин. Брохинолин Q5 получали следующим способом. К раствору бромхинолина Q4 (3 г, 12,1 ммоль) в ТГФ (100 мл) при -78 С добавляли МеI (1,7 мл, 27 ммоль) и затем трет-бутоксид калия (27 мл, 27 ммоль). Через 2 ч стояния при -78 С смесь нагревали до 0 С и нейтрализовали насыщенным раствором NH4Cl и экстрагировали EtOAc. Органические экстракты промывали (Н 2O, насыщенный раствор соли), сушили (MgSO4), фильтровали и концентрировали. В результате очистки флэш-хроматографией (гексан:EtOAc, 3:1) получали 6-[1-метил-1-цианоэтил]-8 бромхинолин. Бензилфосфорные реагенты, используемые для получения соединений по данному изобретению,могут быть получены в соответствии с представленной ниже схемой 6. Схема 6. Получение бензилфосфорных реагентов Арилхинолины формулы XXII могут быть получены сочетанием бромхинолина XX с бороновой кислотой XXI при нагревании в присутствии катализатора, такого как Рd(РРh3)4, и основания, такого как карбонат натрия (водный раствор), в растворителе, таком как DME. Спирт XXII может быть преобразован в бромид XXIII обработкой НВr (водн.) в растворителе, таком как уксусная кислота. Спирт XXII может быть преобразован в сложный метиловый эфир сульфокислоты XXIV обработкой метансульфонилхлоридом в присутствии основания, такого как триэтиламин, в растворителе, таком как дихлорметан. Бензилфосфорные реагенты XXV могут быть получены или нагреванием XXIII в присутствии РРh3 в растворителе, таком как ацетонитрил, или обработкой XXIII или XXIV диэтилфосфитом и основанием,таким как трет-бутоксид калия, в растворителе, таком как ТГФ. Бензилфосфонийбромид Р 1. [3-(6-Изопропил-8-хинолинил)бензил](трифенил)фосфонийбромид. Бензилфосфонийбромид Р 1 получали следующим способом. Стадия 1. 6-Изопропил-8-[3-(гидроксиметил)фенил]хинолин. Смесь 6-изопропил-8-бромхинолина (11,1 г, 44,4 ммоль) (описанного в международной заявке на патент WO 94/22852), 3-(гидроксиметил)фенилбороновой кислоты (8,70 г, 57,2 ммоль), Nа 2 СО 3 (2 М, 71 мл, 142 ммоль) и Рd(РРh3)4 (2,51 мг, 2,17 ммоль) в 280 мл DME перемешивали при 80 С в течение 5 ч. Образовавшуюся смесь охлаждали до к.т., разбавляли EtOAc, промывали насыщенным раствором соли,сушили над Na2SO4, фильтровали и концентрировали. В результате флэш-хроматографии (гексан/EtOAc,1/1) и перемешивания в смеси СН 2 Сl2/гексан (1/9) получали 6-изопропил-8-[3-(гидроксиметил)фенил]хинолин в виде белого вещества. Стадия 2. 6-Изопропил-8-[3-(бромметил)фенил]хинолин. Суспензию гидроксиметилового соединения с настоящей стадии 1 (7,40 г, 26,7 ммоль) в АсОН (50 мл) и НВr (48% водн., 50 мл) перемешивали в течение 12 ч при 100 С. Смесь охлаждали до к.т., вливали в NaOH (2 н.) на льду, доводили рН до 8 и смесь разбавляли простым эфиром. Органическую фазу промывали насыщенным раствором соли, сушили над МgSO4, фильтровали и концентрировали с получением 6-изопропил-8-[3-(бромметил)фенил]хинолина в виде желтого твердого вещества. Стадия 3. [3-(6-Изопропил-8-хинолинил)бензил](трифенил)фосфонийбромид. К раствору бромметилового соединения с данной стадии 2 (3,807 г, 11,1 ммоль) в 40 мл CH3CN добавляли трифенилфосфин (3,22 г, 12,3 ммоль). Смесь перемешивали при 60 С в течение 12 ч, охлаждали до к.т., разбавляли простым эфиром, фильтровали и промывали простым эфиром с получением [3-(6 изопропил-8-хинолинил)бензил](трифенил)фосфонийбромида. Бензилфосфонат Р 2. Диэтил 3-(6-изопропил-8-хинолин)бензилфосфонат. Бензилфосфонат Р 2 получали следующим способом. Бромметиловое соединение с вышеуказанной стадии 2 синтеза Р 1 (11,34 г, 1 экв.) растворяли в ТГФ(170 мл). Добавляли диэтилфосфит (3,87 мл, 1,05 экв.) и раствор охлаждали до 0 С. Затем медленно добавляли трет-ВuОK (3,87 мл, 1 н. в ТГФ). Реакционную смесь перемешивали 2 ч и гасили добавлениемNH4Cl (насыщ.), воды и EtOAc. Отделяли органическую фазу и промывали насыщенным раствором соли,сушили над МgSО 4 и концентрировали. В результате очистки флэш-хроматографией на силикагеле (гексан:EtOAc, 1/9) получали диэтил 3-(6-изопропил-8-хинолинил)бензилфосфонат в виде прозрачного масла. Бензилфосфонат Р 3. Диэтил 3-[6-(1-циано-1-метилэтил)-8-хинолинил]бензилфосфонат. Бензилфосфонат Р 3 получали следующим способом. Стадия 1. 6-(1-Циано-1-метилэтил)-8-[3-(гидроксиметил)фенил]хинолин. Следуя способу вышеуказанной стадии 1 получения бензилфосфонийбромида Р 1, но с заменой 6 изопропил-8-бромхинолина, используемого в качестве исходного материала, на бромхинолин Q5, получали 6-(1-циано-1-метилэтил)-8-[3-(гидроксиметил)фенил]хинолин. Стадия 2. 3-[6-(1-Циано-1-метилэтил)-8-хинолинил]бензилметансульфонат. К раствору окси 6-(1-циано-1-метилэтил)-8-[3-(гидроксиметил)фенил]хинолина с данной стадии 1(5,15 г, 17 ммоль) в СН 2 Сl2 (150 мл) при -78 С добавляли Et3N (3,6 мл, 26 ммоль) и метансульфонилхлорид (MsCl) (1,6 мл, 21 ммоль). Через 0,5 ч стояния при -78 С смесь нейтрализовали насыщенным раствором NH4Cl, разбавляли водой и экстрагировали простым эфиром. Органические экстракты промывали-22 006607 Стадия 3. Диэтил 3-[6-(1-циано-1-метилэтил)-8-хинолинил]бензилфосфонат. К раствору диэтилфосфита (2,5 мл, 18 ммоль) в ТГФ (100 мл) при -78 С добавляли трет-бутоксид калия (1 М, ТГФ, 16 мл, 16 ммоль) и мезилатное соединение 3-[6-(1-циано-1-метилэтил)-8 хинолинил]бензилметансульфонат с данной стадии 2 (5,1 г, 13,5 ммоль). Через 0,5 ч стояния при -78 С и 12 ч при к.т. образовавшуюся смесь нейтрализовали насыщенным раствором NH4Cl, разбавляли водой и экстрагировали простым эфиром. Органические экстракты промывали (HzO, насыщенный раствор соли),сушили (MgSO4), фильтровали и концентрировали. В результате очистки флэш-хроматографией (гексан:EtOAc, от 1:4 до 1:10) получали диэтил 3-[6-(1-циано-1-метилэтил)-8-хинолинил]бензилфосфонат в виде масла. Схема 7. Сочетание бензилфосфорного реагента и кетона Соединения, соответствующие формуле I, могут быть получены с использованием путей реакций,показанных выше на схеме 7. Соединение XXVI может быть получено добавлением раствора кетона VII в растворителе, таком как ТГФ, к смеси бензилфосфорного реагента XXV и основания, такого как третбутоксид калия, в растворителе, таком как ТГФ. Соединения, соответствующие формуле I, могут быть затем получены обработкой XXVI оксоном в смеси растворителей, таком как ТГФ/МеОН/вода. Альтернативно, соединения формулы I могут быть получены взаимодействием кетона VIII с XXV в присутствии основания, такого как трет-бутоксид калия, в растворителе, таком как ТГФ. Как следует из представленной выше схемы 7 и представленной ниже табл. 1, сочетание кетонов с бензилфосфорными реагентами представлено в примерах, указанных в таблице. Таблица 1 Как следует из схемы 8, соединения, соответствующие формуле I, могут быть получены in situ преобразованием арилбромида XVIII в соответствующий сложный эфир бороновой кислоты нагреванием со сложным дибороновым эфиром пинакона, катализатором, таким как [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (II), и основанием, таким как ацетат калия, в растворителе, таком как ДМФ, с последующим добавлением бромхинолина XX, дополнительного количества этого же катализатора, дополнительного количества основания, такого как карбонат натрия (водный) при дополнительном нагреве. Как следует из вышепредставленной схемы 8, нижепредставленных табл. 2 и приложения к табл. 2,сочетание арилбромида с бромхинолином представлено в примерах, указанных в таблице. Таблица 2 Соединения по данному изобретению могут быть получены в соответствии с представленной ниже схемой 9. Схема 9 показывает получение соединений формулы I, где альдегид XXVII может быть получен нагреванием бромхинолина XX, 3-формилбензолбороновой кислоты, катализатора, такого как Рd(РРh3)4, и основания, такого как карбонат натрия (водный), в растворителе, таком как DME. Альдегид XXVII может быть преобразован в соединение по примеру 18 нагреванием с XVI в присутствии основания, такого как пиперидин, в растворителе, таком как толуол. Соединение по примеру 19 может быть получено обработкой соединения примера 18 нитратом аммония и церия (II) (CAN) в смеси растворителей, такой как ацетонитрил /вода. Альтернативно, альдегид XXVII может быть преобразован в ненасыщенную кислоту XXVIII нагреванием с XV и основанием, таким как пиперидин, в растворителе, таком как толуол. Кислота XXVIII может быть затем преобразована в амид I (пример 27, 28 и 29) обработкой сочетающей системой, такой как EDCI, HOBt и амин, в растворителе, таком как ДМФ. Соединения по данному изобретению могут быть получены сочетанием бромхинолиновых соединений с боронатными соединениями в соответствии с представленной ниже схемой 10. Схема 10. Сочетание бромхинолина с боронатом Схема 10 показывает, как могут быть получены соединения формулы I сочетанием бромхинолинаXX со сложным эфиром бороновой кислоты XII в присутствии катализатора, такого как Pd(OAc)2, РРh3 и основания, такого как карбонат натрия (водный) в растворителе, таком как н-пропанол. Как следует из табл. 3, сочетание бромхинолина с боронатом представлено в примерах, указанных в таблице. Примеры 1 и 2. 6-Изопропил-8-(3-(Z/E)-2-[4-(метилсульфонил)фенил]-2-фенилэтенилфенил)хинолин. Соединения по примерам 1 и 2 получали следующим способом. К смеси бензилфосфоната Р 2 (330 мг, 0,83 ммоль) и кетона K3 (200 мг, 0,77 ммоль) в ТГФ (6 мл) при к.т. добавляли трет-бутоксид калия (1 М, ТГФ, 0,83 мл, 0,83 ммоль). Через 1 ч стояния при к.т. смесь разбавляли водой и экстрагировали Et2O. Органические экстракты промывали (H2O), (насыщенным раствором соли), сушили (МgSO4), фильтровали и концентрировали. В результате очистки флэшхроматографией (гексан:ЕtOАс, 7:3) получали соединения по примерам 1 и 2 в виде белых пен, причем один продукт был менее полярным, чем другой продукт. Соединение по примеру 1 представляло собой менее полярный Z-изомер и соединение по примеру 2 представляло собой более полярный Е-изомер. Пример 1. ЯМР 1H (400 МГц, ацетон-d6):8,79 (кв., 1 Н), 8,28 (кв., 1 Н), 7,94 (д, 2 Н), 7,73 (д, 1 Н), 7,6-7,1 (м,14 Н), 3,14 (м, 1 Н), 2,97 (с, 3 Н), 1,34 (д, 6 Н). Пример 2. ЯМР 1H (400 МГц, ацетон-d6):8,78 (кв., 1 Н), 8,25 (кв., 1 Н), 7,89 (д, 2 Н), 7,71 (д, 1 Н), 7,6 (м, 3 Н),7,45 (м, 3 Н), 7,39-7,2 (м, 8 Н), 3,11 (м, 4 Н), 1,34 (д, 6 Н). Пример 3. 6-Изопропил-8-3-[(E/Z)-2-[4-(метилсульфонил)фенил]-2-(1,3-тиазол-2-ил)этенил]фенил]хинолин. Соединение по примеру 3 получали следующим способом. К суспензии бензилфосфонийбромида Р 1 (320 мг, 0,531 ммоль) в 2,5 мл ТГФ при -78 С добавляли по каплям трет-ВuОК (1,0 М в ТГФ, 0,55 мл, 0,55 ммоль) и образовавшийся красный раствор перемешивали 30 мин при 0 С. Затем к полученному илиду при -78 С добавляли по каплям кетон K5 (122 мг, 0,455 ммоль) в 2 мл ТГФ. Смесь нагревали до к.т., затем перемешивали в течение 1 ч, гасили NH4Cl (насыщ.) и разбавляли EtOAc. Органическую фазу промывали насыщенным раствором соли, сушили над Na2SO4,фильтровали и концентрировали. В результате флэш-хроматографии (кремнеземный картридж,гексан/EtOAc от 10 до 100% в течение 20 мин) получали соединение по примеру 3 (смесь изомеров при их соотношении 1,5 к 1). ЯМР 1H (500 МГц, ацетон-d6):8,79-8,78 (м, 1 Н), 8,26-8,23 (м, 1 Н), 8,01-7,92 (м, 3 Н), 7,84 (д, 0,4 Н,побочный), 7,78 (д, 0,6 Н, основной), 7,73-7,47 (м, 10 Н), 7,43 (дд, 1 Н), 7,34 (т, 0,6 Н, основной), 7,27 (т,0,4 Н, побочный), 7,18 (д, 0,6 Н, основной), 7,09 (д, 0,4 Н, побочный), 3,12 (м, 1 Н), 3,11 (с, 1,8 Н, основной),2,99 (с, 1,2 Н, побочный), 1,36-1,33 (м, 6 Н).MS (M+1): 511. Пример 4. 6-Изопропил-8-(3-(Е)-2-(1-(метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)хинолин. Соединение по примеру 4 получали следующим способом. Стадия 1. 6-Изопропил-8-(3-Е)-2-(1-(метил-1 Н-имидазол-2-ил)-2-[4-(метилтио)фенил]этенилфенил)хинолин. Следуя способу примера 3, но с заменой K5, используемого в качестве исходного материала, на кетон K2 получали 6-изопропил-8-3-(Е)-2-(1-(метил-1 Н-имидазол-2-ил)-2-[4-(метилтио)фенил]этенилфенил)хинолин. Стадия 2. 6-Изопропил-8-(3-(Е)-2-(1-(метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)хинолин. Следуя способу, используемому для получения бороната В 1 (стадия 2 схемы 2), но с заменой (E/Z)2-(3-бромфенил)-1-(1-метил-1 Н-имидазол-2-ил)-1-[4-(метилтио)фенил]этена, используемого в качестве исходного материала, на сульфид, полученный на данной стадии 1, получали соединение по примеру 4. ЯМР 1H (500 МГц, ацетон-d6):8,77 (дд, 1 Н), 8,24 (дд, 1 Н), 7,88 (д, 2 Н), 7,71 (д, 1 Н), 7,59 (д, 1 Н),7,53 (д, 2 Н), 7,48 (д, 2 Н), 7,41 (дд, 1 Н), 7,28 (т, 1 Н), 7,23 (с, 1 Н), 7,15 (д, 1 Н), 7,07 (д, 1 Н), 6,95 (д, 1 Н), 3,51MS (M+2): 509,4. Примеры 5 и 6. 6-Изопропил-8-(3-(Z/E)-2-(4-фторфенил)-2-[4-(метилсульфонил)фенил]этенилфенил)хинолин. Соединения по примерам 5 и 6 получали следующим способом. Следуя способу примера 1, но с заменой кетона K3, используемого в качестве исходного материала,на кетон K1 после очистки флэш-хроматографией (50% EtOAc/50% гексан) получали соединения по примерам 5 и 6. ЯМР 1H (500 МГц, ацетон-d6). Пример 5. Основной (Z) изомер,8,78 (дд, 1 Н), 8,25 (дд, 1 Н), 7,93 (д, 2 Н), 7,72 (д, 1 Н), 7,55-7,40 (м,6 Н), 7,35 (м, 2 Н), 7,25 (т, 1 Н), 7,23 (с, 1 Н), 7,11 (т, 2 Н), 7,05 (д, 1 Н), 3,12 (м, 1 Н), 2,96 (с, 3 Н), 1,34 (д, 6 Н). ЯМР 1H (500 МГц, ацетон-d6). Пример 6. Побочный (Е) изомер:8,78 (дд, 1 Н), 8,35 (дд, 1 Н), 7,93 (д, 2 Н), 7,72 (д, 1 Н), 7,65-7,55 (м,3 Н), 7,45 (дд, 1 Н), 7,35-7,15 (м, 9 Н), 3,12 (м, 4 Н), 1,34 (д, 6 Н). Пример 7. 2-(2-(E/Z)-2[3-(6-Изопропил-8-хинолинил)фенил]-1-[4-(метилсульфонил)фенил]этенил-1,3-тиазол-5-ил)-2-пропанол. Соединение по примеру 7 получали по способу примера 1, но с заменой кетона K3, используемого в качестве исходного материала, на кетон K6. В результате очистки флэш-хроматографией (100% EtOAc) получали соединение по примеру 7 в виде смеси изомеров. ЯМР 1H (400 МГц, ацетон-d6):8,80 (м, 1 Н), 8,30 (м, 1 Н), 8,05 (д(основной), 1,44 Н), 7,93 (д (побочный), 0,55 Н), 7,85 (с(основной), 0,72 Н 7,77 (с (побочный), 0,28 Н), 7,75-7,45 (м, 7 Н) 7,35 (т(побочный),0,28 Н), 7,28 (т (основной), 0,72 Н), 7,21 (д (побочный), 0,28 Н), 7,10 (д (основной), 0,72 Н 4,7 (м, 1 Н), 3,15MS (M+1): 569,6. Пример 8. 2-[8-(3-(E/Z)-2-[5-(1-Гидрокси-1-метилэтил)-1,3-тиазол-2-ил]-2-[4-(метилсульфонил)фенил]этенилфенил)-6-хинолинил]-2-метилпропаннитрил. Соединение по примеру 8 получали по способу примера 1, но с заменой кетона K3 и бензилфосфоната Р 2, используемых в качестве исходных материалов, на кетон K6 и бензилфосфонат Р 3. В результате очистки флэш-хроматографией (20% СН 2 Сl2/80% EtOAc) получали соединение по примеру 8 в виде смеси изомеров. ЯМР 1H (400 МГц, ацетон-d6):8,92 (м, 1 Н), 8,45 (м, 1 Н), 8,10 (м, 1 Н), 8,05 (м, 1 Н), 7,93 (м, 1 Н),7,85 (м, 2 Н), 7,77-7,55 (м, ХН), 7,40 (т (побочный), 0,43 Н), 7,28 (т (основной), 0,57 Н 7,21 (д (побочный),0,43 Н), 7,10 (д (основной), 0,57 Н), 4,67 (с (основной), 0,57 Н), 4,63 (с (побочный), 0,43 Н), 3,15 (с (побочный), 1,3 Н), 2,99 (с (основной), 1,7 Н), 1,90 (м, 6 Н), 1,65 (с (основной), 3,4 Н), 1,45 (с (побочный), 2,6 Н).MS (M+1): 594,6. Пример 9. 2-Метил-2-[8-(3-(Е)-2-(1-метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)-6-хинолинил]пропаннитрил. Соединение по примеру 9 получали следующим способом. Стадия 1. 2-Метил-2-[8-(3-(Е)-2-(1-метил-1 Н-имидазол-2-ил)-2-[4-(метилтио)фенил]этенилфенил)-6-хинолинил]пропаннитрил получали по способу примера 1, но с заменой кетона K3 и бензилфосфоната Р 2,используемых в качестве исходных материалов, на кетон K2 и бензилфосфонат Р 3. Стадия 2. Соединение по примеру 9 2-метил-2-[8-(3-(Е)-2-(1-метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)-6-хинолинил]пропаннитрил получали по способу, использованному для получения бороната В 1 (стадия 2 схемы 2), но с заменой (E/Z)-2-(3-бромфенил)-1-(1-метил-1 Нимидазол-2-ил)-1-[4-(метилтио)фенил]этена, используемого в качестве исходного материала, на сульфид,полученный на данной стадии 1. Соединение по примеру 9 получали после очистки флэшхроматографией (97% EtOAc/3% Et3N). ЯМР 1H (400 МГц, ацетон-d6):8,92 (дд, 1 Н), 8,45 (дд, 1 Н), 8,10 (д, 1 Н), 7,93 (д, 2 Н), 7,76 (д, 1 Н),7,60-7,50 (м, 5 Н), 7,38 (т, 1 Н), 7,35 (с, 1 Н), 7,19 (м, 1 Н), 7,10 (м, 1 Н), 6,95 (м, 1 Н), 3,55 (с, 3 Н), 3,00 (с, 3 Н),1,85 (с, 6 Н).MS (M+1): 533,3. Пример 10. 6-[1-(Метилсульфонил)этил]-8-3-[(Е)-2-[4-(метилсульфонил)фенил]-2-(1,3-тиазол-2 ил)этенил]фенилхинолин. Соединение по примеру 10 получали следующим способом. Смесь бромхинолина Q2 (105 мг, 0,33 ммоль), бороната В 2 (236 мг, 0,51 ммоль), Na2CO3 (2M, 0,65 мл, 1,3 ммоль), Pd(OAc)2 (6,3 мг, 0,028 ммоль) и РРh3 (28 мг, 0,11 ммоль) в 4 мл н-пропанола перемешивали при 90 С в течение 2 ч. Смесь охлаждали до к.т., разбавляли EtOAc, промывали насыщенным раствором соли, сушили над Na2SO4, фильтровали и концентрировали. В результате флэш-хроматографии(толуол/ацетон, 4:1) и перемешивания в смеси гексан/EtOAc получали соединение по примеру 10 (одиночный изомер) в виде белого твердого вещества.MS (М + 1) 576. Пример 11. 6-[1-Метил-1-(метилсульфонил)этил]-8-3-[(Е)-2-[4-(метилсульфонил)фенил]-2-(1,3 тиазол-2-ил)этенил]фенилхинолин. Соединение по примеру 11 получали следующим способом, описанным в примере 10, но с заменой бромхинолина Q2 на Q3 и с использованием бороната В 2. В результате флэш-хроматографии (толуол/ацетон, 9/1) и перемешивания в смеси EtOAc/гексан получали соединение по примеру 11 (одиночный изомер) в виде белого твердого вещества. ЯМР 1H (400 МГц, ацетон-d6):8,90 (дд, 1 Н), 8,41 (дд, 1 Н), 8,23 (с, 1 Н), 8,02-7,99 (д, 3 Н), 7,95 (с,1 Н), 7,86 (д, 1 Н), 7,70 (д, 2 Н), 7,60-7,54 (м, 4 Н), 7,32 (т, 1 Н), 7,13 (д, 1 Н), 3,00 (с, 3 Н), 2,69 (с, 3 Н), 1,96 (с,6 Н).MS (M+1): 523. Пример 12. 8-(3-(Z)-2-(1-Метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)6-[1-метилсульфонил)этил]хинолин. Соединение по примеру 12 получали по способу, описанному в примере 10, с использованием бромхинолина Q2, но с заменой бороната В 2 на боронат В 1. В результате очистки флэш-хроматографии(95% CH2Cl2/5% EtOH) получали соединение по примеру 12. ЯМР 1H (400 МГц, ацетон-d6):8,92 (дд, 1 Н), 8,45 (дд, 1 Н), 8,10 (с, 1 Н), 7,93 (д, 2 Н), 7,76-7,65 (м,4 Н), 7,59 (дд, 1 Н), 7,39 (т, 1 Н), 7,26 (с, 1 Н), 7,18 (с, 1 Н), 7,05 (м, 2 Н), 4,70 (кв., 1 Н), 3,40 (с, 3 Н), 3,13 (с,3 Н), 2,93 (с, 3 Н), 1,87 (д, 3 Н).MS (M+1): 572,4. Пример 13. 8-(3-(Z)-2-(1-Метил-1 Н-имидазол-2-ил)-2-[4-(метилсульфонил)фенил]этенилфенил)6-[1-метил-1-(метилсульфонил)этил]хинолин. Соединение по примеру 13 получали по способу, описанному в примере 10, но с заменой бромхинолина Q2 на бромхинолин Q3 и бороната В 2 на боронат В 1. В результате флэш-хроматографии (95%EtOAc/5% Et3N) получали соединение по примеру 13 (одиночный изомер) в виде пены. ЯМР 1H (400 МГц, ацетон-d6):8,92 (дд, 1 Н), 8,45 (дд, 1 Н), 8,37 (д, 1H), 8,05 (д, 1 Н), 7,93 (д, 2 Н),7,76 (д, 1 Н), 7,69 (д, 2 Н), 7,65 (д, 1H), 7,59 (дд, 1H), 7,38 (т, 1H), 7,31 (с, 1H), 7,18 (с, 1H), 7,05 (м, 2 Н),3,40 (с, 3 Н), 3,13 (с, 3 Н), 2,70 (с, 3 Н), 1,95 (с, 6 Н). Соединения по примерам 14 и 15 получали следующим способом. Раствор арилбромида АВ 5 (249 мг, 0,57 ммоль), сложного диборонового эфира пинакона (167 мг,0,66 ммоль), [1,1'-бис(дифенилфосфин)ферроцен]дихлорпалладий (II) (12 мг, 0,015 ммоль) и ацетата калия (176 мг, 1,8 ммоль) в ДМФ (N,N-диметилформамид) (10 мл) дегазировали и перемешивали при 80 С в течение 3 ч. Затем к образовавшейся смеси при 25 С добавляли бромхинолин Q3 (150 мг, 0,46 ммоль),[1,1'-бис(дифенилфосфин)ферроцен]дихлорпалладий (II) (12 мг, 0,015 ммоль) и карбонат натрия (0,6 мл,2 М). После дегазации смесь нагревали при 80 С всю ночь. Затем смесь охлаждали до к.т., гасили Н 2O и экстрагировали EtOAc. Органические экстракты промывали (Н 2 О, насыщенный раствор соли), сушили(гексан:EtOAc:Et3N, 22:68:10, затем гексан:ЕtOАс, 3:1) получали оба изомера (пример 14 и 15). ЯМР 1H (500 МГц, ацетон-d6):Основной (Е) изомер (пример 14):8,91 (дд, 1 Н), 8,42 (дд, 1 Н), 8,25MS (M+1): 588,2. Альтернативно, соединение по примеру 14 может быть получено следующим способом: К метансульфоновой кислоте (8-10 экв.) при 20 С добавляли м-нитробензолсульфонат натрия (0,60,8 экв.), затем гептагидрат сульфата железа (0,01-0,05 экв.). К полученной смеси добавляли 2-бром-4 метиланилин (1 экв.). Добавляли глицерин (2-3 экв.) и полученный раствор нагревали при 120-140 С и подвергали старению до завершения реакции. Смесь охлаждали до 70-90 С и разбавляли водой. Затем раствор охлаждали до около 20 С и нейтрализовали водным раствором NaOH и бикарбонатом натрия. Добавляли МТВЕ (простой метил третбутиловый эфир, смесь фильтровали и разделяли фазы (продукт находился в слое МТВЕ). Стадия 2. Бронирование Растворитель для раствора МТВЕ со стадии 1 представлял хлорбензол. После фильтрации через плаг из силикагеля и частичного концентрирования добавляли N-бромсукцинимид (NBS, 0,6-0,8 экв.) и 2,2'-азобисизобутилнитрил (AIBN, 0,01-0,1 экв.). Дегазированную смесь нагревали при 55-85 С. Полученную смесь разбавляли циклогексаном. Добавляли дополнительное количество NBS (0,3-0,5 экв.) иAIBN (0,01-0,05 экв.). Дегазированную смесь нагревали при 55-85 С до завершения реакции. Смесь охлаждали при 10-40 С и разбавляли циклогексаном и подвергали старению. Твердое вещество выделяли фильтрацией.
МПК / Метки
МПК: C07D 417/10, C07D 215/14, C07D 413/10, C07D 401/10, C07D 215/12, A61K 31/47, A61P 11/00
Метки: качестве, фосфодиэстеразы-4, замещенные, ингибиторов, 8-арилхинолины
Код ссылки
<a href="https://eas.patents.su/30-6607-zameshhennye-8-arilhinoliny-v-kachestve-ingibitorov-fosfodiesterazy-4.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные 8-арилхинолины в качестве ингибиторов фосфодиэстеразы-4</a>
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Норчини Габриеле, Мораццони Габриеле, Наполетано Мауро, Пеллачини Франко, Гранчини Джанкарло
МПК: C07D 237/30, A61K 31/502
Метки: фталазина, ингибиторов, фосфодиэстеразы, качестве, производные
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Кесицки Эдвард А., Оливер Эйми, Шлахтер Стивен, Джоунс Закари С., Фаулер Керри У., Годино Джон Дж., Бэрджесс Лоренс И., Ньюхаус Брэдли Дж., Одинго Джошуа, Мартинс Тимоти Дж.
МПК: C07D 295/20, A61K 31/40
Метки: специфичной, циклическому, фосфодиэстеразы, пирролидина, ингибиторов, производные, амф, качестве
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...
Производные замешенных индазолов, их применение в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продукции фактора некроза опухолей (фно)

Номер патента: 2272
Опубликовано: 28.02.2002
Автор: Марфэт Энтони
МПК: A61P 35/00, A61K 31/415, C07D 231/56...
Метки: замешенных, качестве, некроза, фосфодиэстеразы, опухолей, типа, ингибиторов, продукции, фдэ, фактора, фно, применение, индазолов, производные
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R представляет собой водород, -(СН2)n(С3-С7циклоалкил), где n равно 0, или фенил, возможно замещенный 1-3 галогено; R1 представляет собой С1-С7алкил; R2 выбран из группы, состоящей из где пунктирные линии в формулах (Iа) и (Iб) представляют простую связь; m равно от 0 до 1; R3 представляет собой циано или CH2CN; R4 представляет собой Н, CO2R14, C(Y)NR17R14, CN,...
Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)

Номер патента: 2113
Опубликовано: 24.12.2001
Автор: Марфат Энтони
МПК: C07D 401/12, A61P 11/06, A61K 31/415...
Метки: фосфодиэстеразы, ингибиторов, качестве, продуцирования, использование, индазола, производные, опухоли, фактора, фдэ, некроза, фно, типа
Формула / Реферат:
1. Соединение формулы (I) или их фармацевтически приемлемые соли, в которых R является Н, C1-C9 алкилом, -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,...
Призводные замещенного индазола и их применение в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и фактора некроза опухоли (фно)

Номер патента: 2274
Опубликовано: 28.02.2002
Автор: Марфэт Энтони
МПК: A61K 31/416, A61P 37/00, C07D 403/04...
Метки: фактора, типа, ингибиторов, применение, замещенного, некроза, индазола, опухоли, фно, призводные, качестве, фосфодиэстеразы, фдэ
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где R представляет собой циклопентил или циклогексил; R1 представляет собой С1-С2алкил; и R2 представляет собой группировку, выбранную из группы, состоящей из группировок формул с (1.1) по (1.12): 2. Соединение по п.1, в котором R2 представляет собой группировку формулы (1.1). 3. Соединение по п.1, в котором R2 представляет собой группировку формулы (1.10). 4. Соединение по п.1,...
Предыдущий патент: Гуманизированное антитело и его фрагмент, взаимодействующие с бета-амилоидным пептидом, и способы их применения
Следующий патент: Нафтотиазиновые позитивные аллостерические модуляторы амра-рецепторов
Случайный патент: Система аэрации жидкости