Ингибиторы трипептидил пептидазы
Номер патента: 6443
Опубликовано: 29.12.2005
Авторы: Бреслин Хенри Джозеф, Кукла Майкл Джозеф, Де Винтер Ханс Луи Йос



















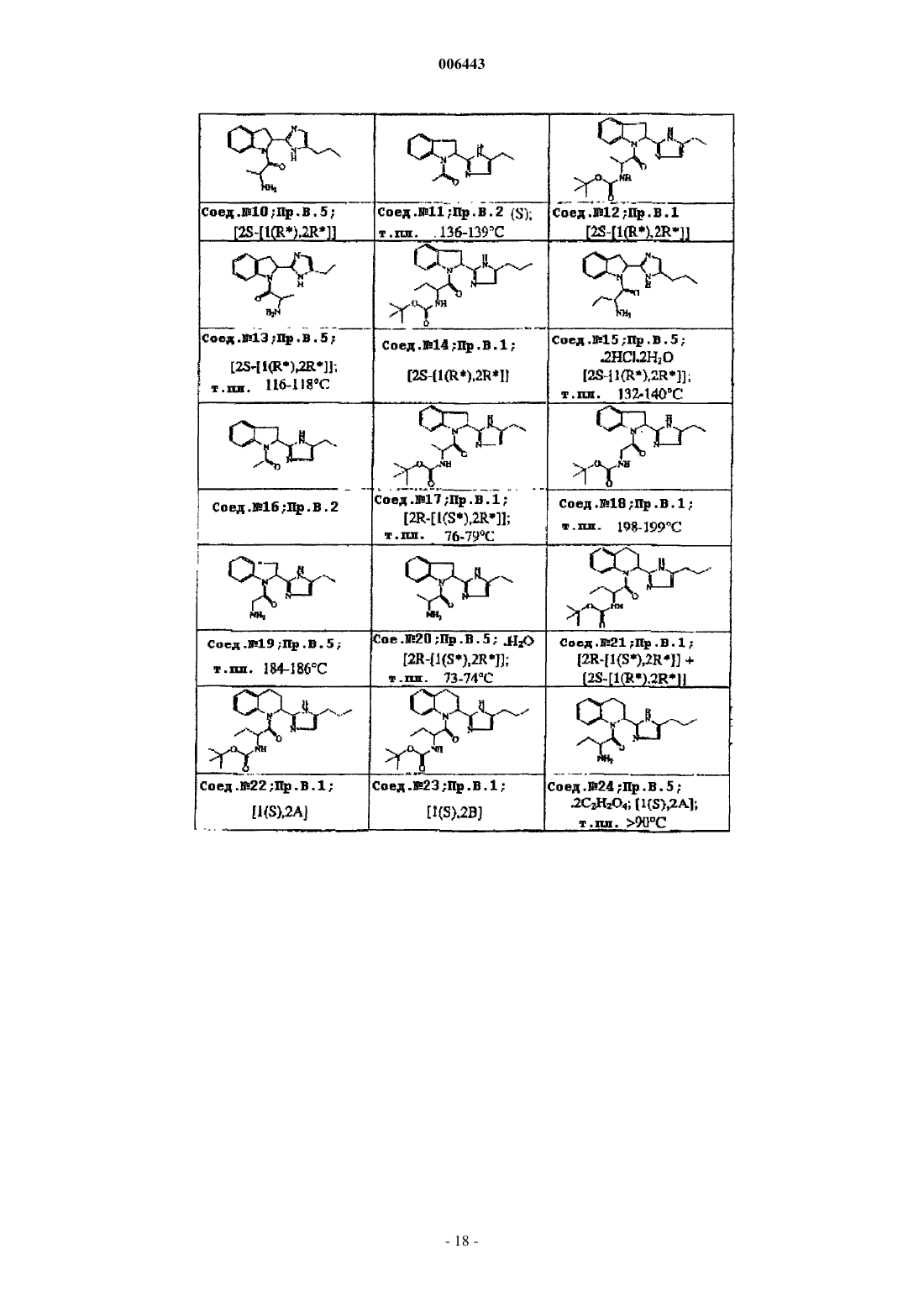
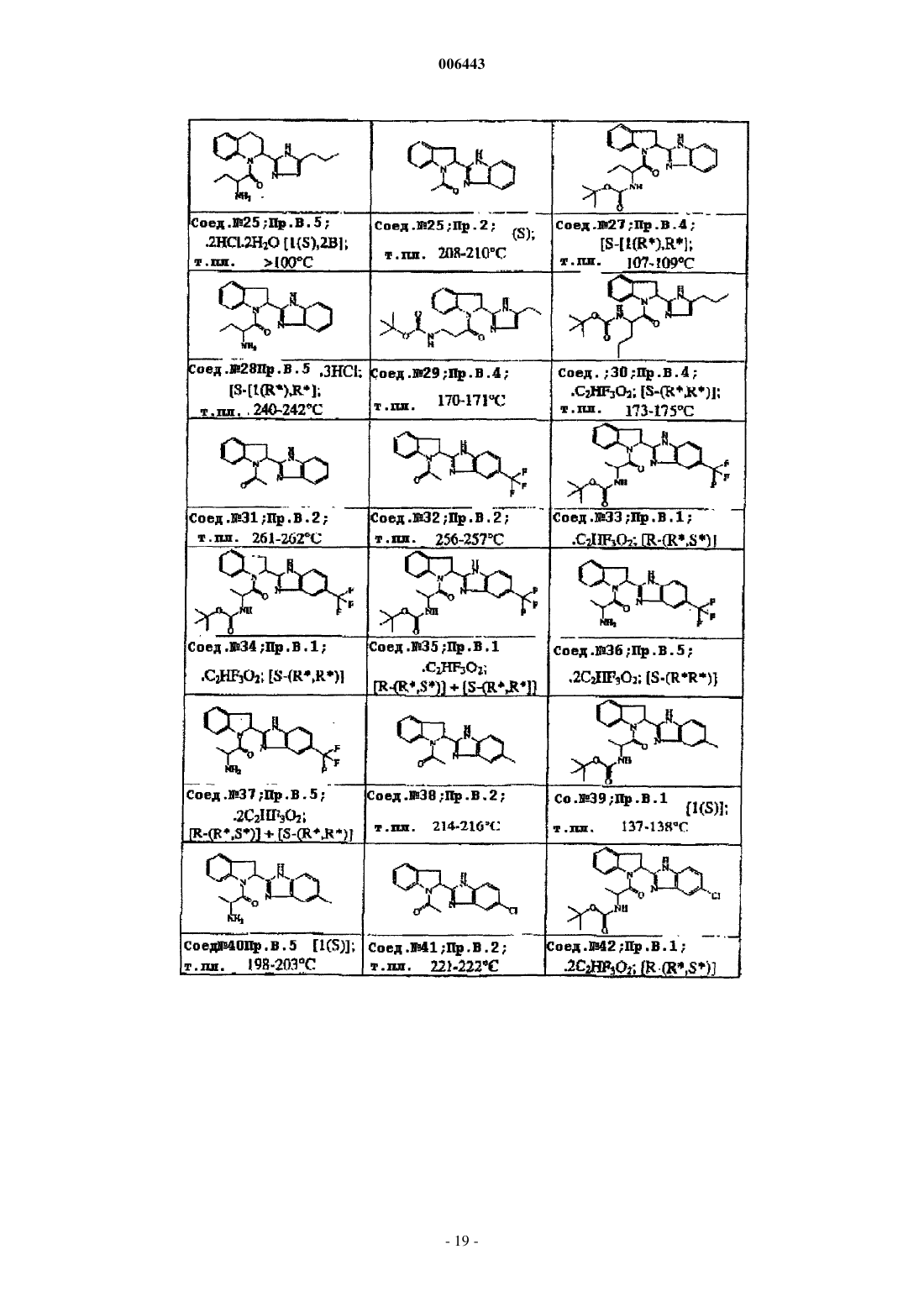
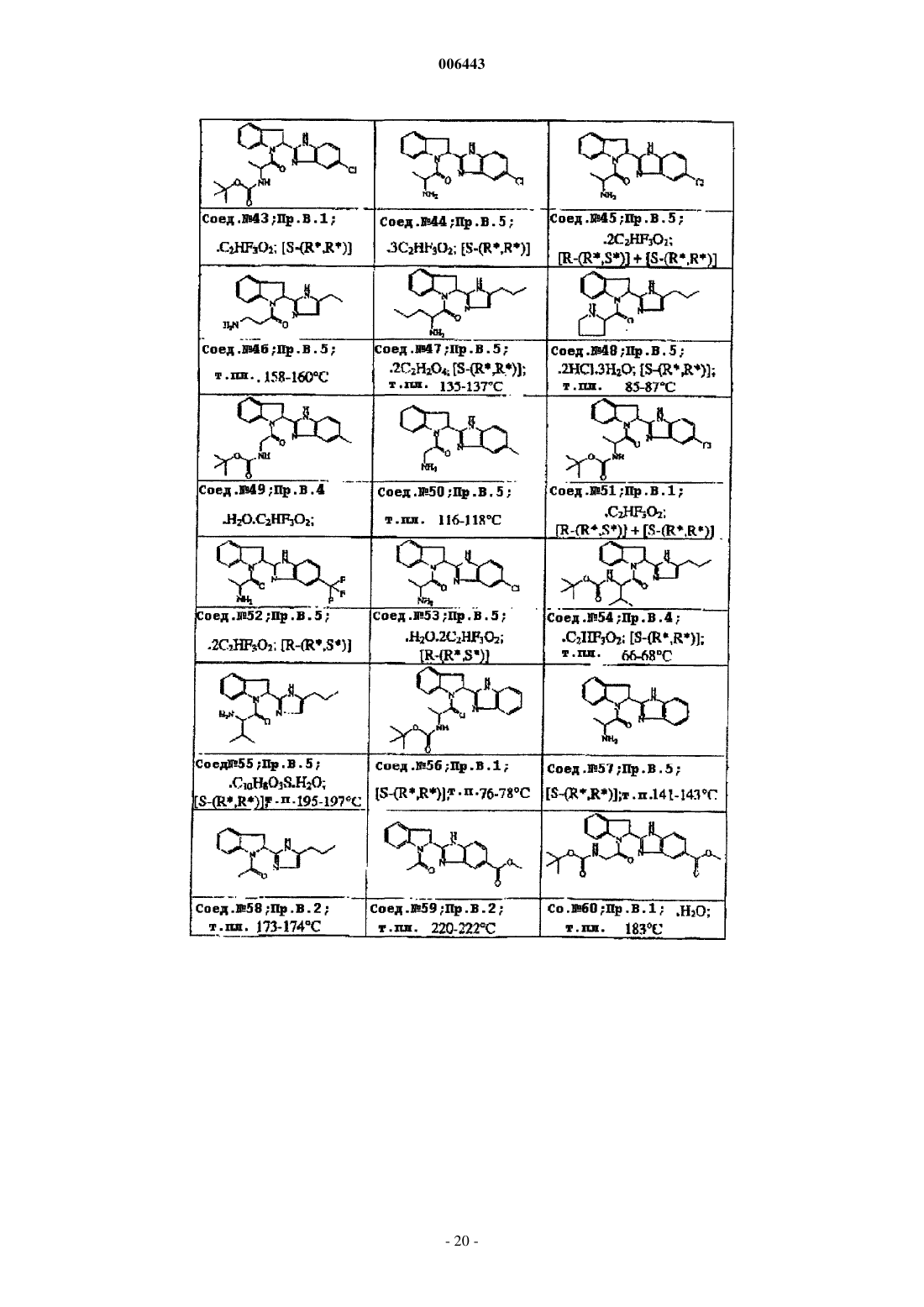
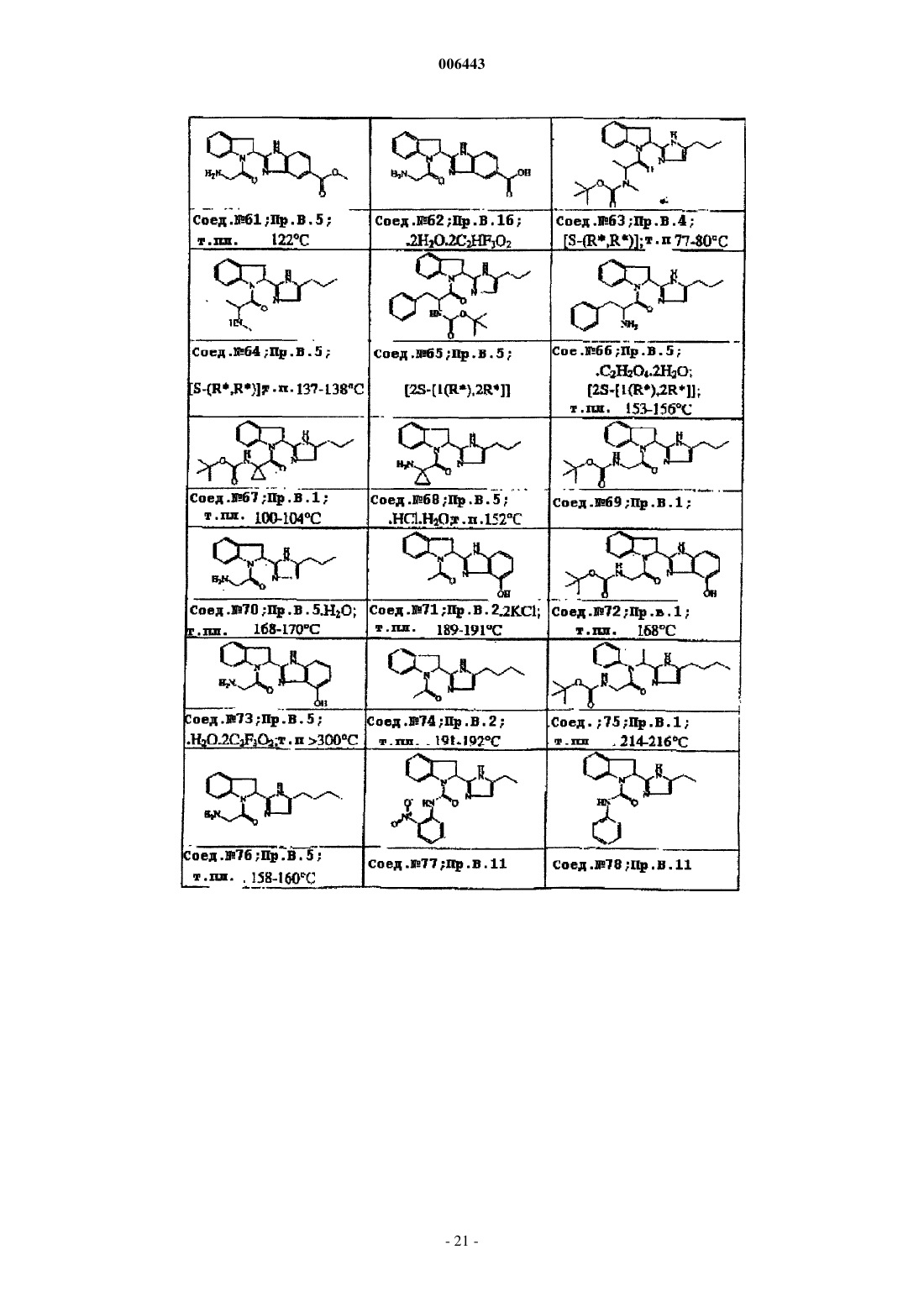

Формула / Реферат
1. Соединение формулы (I)

его стереохимически изомерная форма или его фармацевтически приемлемая аддитивная соль,
где n равно целому числу 0 или 1;
X представляет -(CR4R5)m-, где m равно целому числу 1 или 2;
каждый из R4 и R5 независимо представляет водород или C1-4алкил;
R1 представляет C1-6алкилкарбонил, необязательно замещенный гидрокси; C1-6алкилоксикарбонил; аминоC1-6алкилкарбонил, где C1-6алкил необязательно замещен C3-6циклоалкилом; моно- и ди(C1-4алкил)аминоC1-6алкилкарбонил; аминокарбонил, замещенный арилом; C1-6алкилкарбонилоксиC1-6алкилкарбонил; C1-6алкилоксикарбониламиноC1-6алкилкарбонил, где аминогруппа необязательно замещена C1-4алкилом; аминокислотный остаток, связанный через карбонильную группу; C1-6алкил, замещенный амино; или арилкарбонил;
R2 представляет 5-членный гетероцикл, выбранный из

где m' равно целому числу от 1 до 2;
R6 представляет водород или C1-4алкил;
R7 независимо друг от друга представляет водород; галоген; амино; гидрокси; трифторметил; C1-6алкил; C1-4алкил, замещенный гидрокси, гидроксикарбонилом, C1-4алкилоксикарбонилом, аминокарбонилом, моно- или ди(C1-4алкил)аминокарбонилом, амино, или моно- или ди(C1-4алкил)амино; фенил; аминокарбонил; гидроксикарбонил; C1-4алкилоксикарбонил; C1-4алкилкарбонил; или C1-4алкилоксикарбонилC1-4алкиламинокарбонил;
или R2 представляет бензимидазол или бензимидазол, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, трифторметила, C1-4алкила, гидрокси, гидроксикарбонила или C1-4алкилоксикарбонила;
R3 представляет двухвалентный радикал формулы

где (b-1) необязательно замещен галогеном или C1-6алкилоксилом;
арил представляет фенил или фенил, замещенный амино, нитро или гидроксикарбонилом.
2. Соединение по п.1, в котором n равно 0, а R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси.
3. Соединение по п.1, в котором n равно 0, R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси, а X представляет -CH2- или -CH2CH2-.
4. Соединение по любому из предыдущих пунктов, в котором R2 представляет радикал формулы (a-2), (a-4), (a-6) или (a-7).
5. Соединение по любому из предыдущих пунктов, в котором R1 представляет C1-6алкилкарбонил, аминоC1-6алкилкарбонил или аминокислотный остаток.
6. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-5.
7. Способ получения фармацевтической композиции по п.6, в котором терапевтически активное количество соединения по любому из пп.1-5 тщательно смешивают с фармацевтически приемлемым носителем.
8. Применение соединения по любому из пп.1-5 в качестве лекарственного препарата.
9. Способ получения соединения формулы (I), в котором a) промежуточное соединение формулы (II) подвергают взаимодействию с промежуточным соединением формулы (III) в реакционно-инертном растворителе и, необязательно, в присутствии подходящего основания, получая соединения формулы (I-a), указанные как соединения формулы (I), где R1a представляет все заместители R1, кроме C1-4алкила, замещенного амином; или

b) промежуточное соединение формулы (II) подвергают взаимодействию с промежуточным соединения формулы (IV), получая соединение формулы (I-a)

где в вышеприведенных реакционных схемах радикалы R1, R2, R3 и целое число n имеют значения, определенные в п.1;
c) или соединения формулы (I) превращают одно в другое в соответствии с известными реакциями превращения; или, при желании, соединение формулы (I) превращают в кислотно-аддитивную соль, или, наоборот, кислотно-аддитивную соль соединения формулы (I) превращают в свободное основание взаимодействием со щелочью; и, при желании, получают их стереохимически изомерные формы.
Текст
006443 Настоящее изобретение относится к новым соединениям формулы (I), которые являются ингибиторами мембранной трипептидил пептидазы, отвечающей за инактивацию эндогенных нейропептидов, таких как холецистокинины (CCKs). Данное изобретение также относится к способам получения таких соединений, фармацевтическим композициям, включающим указанные соединения, а также к применению указанных соединений в качестве лекарственного средства. Холецистокинины (CCKs) являются семейством гормональных и нейронных пептидов, оказывающих плейотропное, биологическое действие на кишечник и мозг. Действие ССK опосредовано рецепторами ССKA и ССKB. Известно, что ССK играет физиологическую роль, контролируя всасывание пищи,что усиливается агонистами ССKA (Smith G.P. et al., J. Ann. N.Y. Acad. Sci., 713, 236-241 (1994, а также контролируя беспокойство, что снижается антагонистами ССKB (Woodruff G. et al., Rev. Pharmac., 31,469-501 (1991. Трипептидил пептидаза II (ТПП II) представляет собой пептидазу, инактивирующую ССK. ТПП II найдена в нейронах, отвечающих за холецистокинин, а также в ненейронных клетках. ТПП II считают нейропептидазой, отвечающей за инактивацию ССK-8 (Rose С. et al., Nature, 380, 403-409 (1996. ТПП II может принимать участие в инактивации ССK-8 в желудочно-кишечном тракте. Экзогенный ССK снижает уровень всасывания пищи и выявляет другие поведенческие сопутствующие обстоятельства ситуации. Уровень всасывания пищи повышают путем системного введения агонистов рецептора ССK(Smith G.P. et al., J. Ann. N.Y. Acad. Sci., 713, 236-241 (1994. Контролируемое эндогенными ССK всасывание пищи, скорее всего, имеет нейронное, а не гормональное происхождение и действует на периферические рецепторы ССK на вагусных афферентных волокнах (Smith G.P. et al., Am. J. Physiol., 249, R638R641 (1985. Ингибиторы ТПП II являются полезными инструментами для исследования функций нейронов ССK и могут быть использованы в качестве лекарственных средств для лечения расстройств, таких как переедание, ожирение, проблемы с подвижностью желудочно-кишечного тракта и психотические синдромы.WO-96/35805, опубликованная 14 ноября 1996 г., описывает ингибиторы мембранной трипептидил пептидазы, отвечающие за инактивацию эндогенных нейропептидов, полученные для лечения желудочно-кишечных и психических расстройств. WO-99/33801, опубликованная 8 июля 1999 г., описывает соединения, ингибирующие инактивирующую ССK трипептидил пептидазу (ТПП II), полезные для лечения расстройств, связанных с приемом пищи, ожирения, психотических синдромов и связанных с ними психиатрических нарушений. Соединения в соответствии с настоящим изобретением отличаются от известных соединений структурно, природой заместителя R2. Настоящее изобретение относится к соединениям формулы (I) их стереохимически изомерной форме или их фармацевтически приемлемой аддитивной соли,где n равно целому числу 0 или 1; Х представляет -(СR4R5)m-, где m равно целому числу 1 или 2; каждый из R4 и R5 независимо представляет водород или С 1-4 алкил;R1 представляет C1-6 алкилкарбонил, необязательно замещенный гидрокси; C1-6 алкилоксикарбонил; аминоС 1-6 алкилкарбонил, где C1-6 алкил необязательно замещен С 3-6 циклоалкилом; моно- и ди(С 1-4 алкил) аминоС 1-6 алкилкарбонил; аминокарбонил, замещенный арилом; С 1-6 алкилкарбонилоксиС 1-6 алкилкарбонил; C1-6 алкилоксикарбониламиноС 1-6 алкилкарбонил, где аминогруппа необязательно замещена С 1-4 алкилом; аминокислотный остаток, связанный через карбонильную группу; C1-6 алкил, замещенный амино; или арилкарбонил; где m' равно целому числу от 1 до 2;R6 представляет водород или С 1-4 алкил;R7 независимо друг от друга представляет водород; галоген; амино; гидрокси; трифторметил; C1-6 алкил; С 1-4 алкил, замещенный гидрокси, гидроксикарбонилом, С 1-4 алкилоксикарбонилом, аминокарбонилом, моно- или ди(С 1-4 алкил)аминокарбонилом, амино, или моно- или ди(С 1-4 алкил)амино; фенил; аминокарбонил; гидроксикарбонил; С 1-4 алкилоксикарбонил; С 1-4 алкилкарбонил; или C1-4 алкилоксикарбонилС 1-4 алкиламинокарбонил;-1 006443 или R2 представляет бензимидазол или бензимидазол, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, трифторметила, С 1-4 алкила, гидрокси, гидроксикарбонила или С 1-4 алкилоксикарбонила;R3 представляет двухвалентный радикал формулы где (b-1) необязательно замещен галогеном или C1-6 алкилоксилом; арил представляет фенил или фенил, замещенный амино, нитро или гидроксикарбонилом. Термин аминокислотные остатки в данном описании означает глицин, аланин, валин, лейцин,изолейцин, метионин, пролин, фенилаланин, триптофан, серин, треонин, цистеин, тирозин, аспарагин,глутамин, аспарагиновую кислоту, сложные эфиры аспарагиновой кислоты, глутаминовую кислоту,сложные эфиры глутаминовой кислоты, лизин, аргинин и гистидин, которые связаны через их карбонильную группу с атомом азота остальной части молекулы и которые могут быть в целом представлены формулой R-CH(NH2)-CO-. В вышеуказанных определениях галоген означает фтор, хлор, бром и йод; С 1-4 алкил означает насыщенные, углеводородные радикалы с прямой и разветвленной цепью, имеющие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 2-метилпропил и подобные; C1-6 алкил означает С 1-4 алкил и его высшие гомологи, имеющие 5 или 6 атомов углерода, такие как, например, 2 метилбутил, пентил, гексил и подобные; С 3-6 циклоалкил означает циклопропил, циклобутил, циклопентил и циклогексил; С 3-6 алкенил означает ненасыщенные углеводородные радикалы с прямой и разветвленной цепью, имеющие от 3 до 6 атомов углерода, такие как пропенил, бутенил, пентенил или гексенил; С 1-2 алкандиил означает метилен или 1,2-этандиил; C1-5 алкандиил означает двухвалентные углеводородные радикалы с прямой или разветвленной цепью, содержащие от 1 до 5 атомов углерода, такие как, например, метилен, 1,2-этандиил, 1,3-пропандиил, 1,4-бутандиил, 1,5-пентандиил, а также их разветвленные изомеры; C1-6 алкандиил означает С 1-5 алкандиил и его высшие гомологи, имеющие 6 атомов углерода, такие как, например, 1,6-гександиил и подобные. Термин СО относится к карбонильной группе. Термин стереохимически изомерные формы в данном описании означает все возможные изомерные формы, которые могут иметь соединения формулы (I). Если не указано иначе, то химическое обозначение соединений означает смесь всех возможных стереохимически изомерных форм, при этом указанные смеси включают все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители на двухвалентных,циклических, (частично) насыщенных радикалах могут иметь цис- или транс-конфигурацию. Соединения, имеющие двойные связи, могут иметь Е- или Z-стереохимию по указанным двойным связям. Стереохимически изомерные формы соединений формулы (I) явно входят в объем данного изобретения. Фармацевтически приемлемые аддитивные соли включают фармацевтически приемлемые кислотно-аддитивные соли и терапевтически активные нетоксичные кислотно-аддитивные солевые формы, которые способны образовывать соединения формулы (I). Фармацевтически приемлемые кислотноаддитивные соли, как правило, могут быть получены обработкой основной формы соответствующей кислотой. Подобные кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная или бромисто-водородная кислота, серная, азотная,фосфорная и подобные кислоты; либо органические кислоты, такие как, например, уксусная, пропановая,гликолевая, молочная, пировиноградная, щавелевая (т.е. этандикислота), малоновая, янтарная (т.е. бутандикислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памоевая и подобные кислоты. Если соединения в соответствии с данным изобретением включают кислотный остаток, то возможно получение фармацевтически приемлемых основно-аддитивных солей, включающих соли щелочноземельных металлов, например соли натрия или калия; а также основно-аддитивных солей, образуемых с подходящими органическими лигандами, например первичные, вторичные, третичные или четвертичные аммониевые соли, такие как морфолинил, трет-бутиламино и подобные. И наоборот, указанные солевые формы могут быть превращены в свободную солевую форму обработкой соответствующим основанием. Термин аддитивная соль в данном описании также относится к сольватам, которые способны образовывать соединения формулы (I), а также их соли. Такие сольваты представляют собой, например,гидраты, алкоголяты и подобные. Интерес представляют те соединения формулы (I), к которым применимо одно или несколько из следующих ограничений:a) n равно 0, a R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси;b) n равно 0, R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси, а Х представляет -СН 2- или -СН 2 СН 2-;c) R2 представляет радикал формулы (а-2), (а-4), (а-6) или (а-7).d) R1 представляет C1-6 алкилкарбонил, аминоС 1-6 алкилкарбонил или аминокислотный остаток. Конкретными соединениями являются соединения формулы (I), в которых n равно 0, а R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси. Предпочтительными соединениями являются соединения формулы (I), в которых n равно 0, R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси, а Х представляет -СН 2-. Другими предпочтительными соединениями являются соединения формулы (I), в которых n равно 0, R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси, а Х представляет -СН 2 СН 2-. Следующими предпочтительными соединениями являются соединения формулы (I), в которых R1 представляет C1-6 алкилкарбонил, аминоС 1-6 алкилкарбонил или аминокислоту. Соединения формулы (I-а), определенные как соединения формулы (I), в которых R1a представляет все заместители R1, кроме С 1-4 алкила, замещенного амином, могут быть получены взаимодействием промежуточного соединения формулы (II) с промежуточным соединением формулы (III) в присутствии 4 метилморфолина, в реакционно-инертном растворителе, таком как, например, дихлорметан или хлороформ. Перемешивание может увеличить скорость реакции. Реакцию удобно проводить при температуре от комнатной до температуры кипения реакционной смеси и, при желании, реакцию можно проводить в автоклаве при повышенном давлении. После указанной реакции необязательно осуществляют стадию кислотного гидролиза для удаления кислотолабильных защитных групп, таких как третбутилоксикарбонил. Альтернативно, соединения формулы (I-а) также могут быть получены взаимодействием промежуточного соединения (II) с промежуточным соединением (IV) в присутствии подходящего активирующего агента, такого как, например, изобутилхлорформиат, в реакционно-инертном растворителе, таком как,например, дихлорметан, в присутствии подходящего основания, такого как, например, триэтиламин. После указанной реакции необязательно осуществляют стадию кислотного гидролиза для удаления кислотолабильных защитных групп, таких как трет-бутилоксикарбонил. Соединения формулы (I-b), определенные как соединения формулы (I), в которых R1 представляетC1-6 алкил, замещенный амином, могут быть удобно получены в результате подходящей реакции восстановления соответствующих исходных соединений (I-b'), в которых R1 представляет аминоС 1-5 алкилкарбонил. Подходящие реакции восстановления могут, например, включать обработку комплексом борантетрагидрофуран. Соединения формулы (I-с), определенные как соединения формулы (I), в которых R2 представляет радикал (а-2), где R6 представляет водород, а R7 расположен в позиции 3 имидазольной части, могут быть получены взаимодействием промежуточного соединения формулы (V) с промежуточным соединением формулы (IV) в присутствии ацетата калия в подходящем растворителе, таком как метанол. Соединения формулы (I) также могут быть получены превращением соединений формулы (I) одно в другое в соответствии с известными реакциями превращения групп. Исходные материалы и некоторые из промежуточных соединений, такие как, например, промежуточные соединения формул (III), (IV) и (VI), являются известными соединениями и доступны коммерчески или могут быть получены в соответствии с общепринятыми методиками реализаций, известными в данной области. Соединения формулы (I) и некоторые из промежуточных соединений, имеющие в своей структуре один или несколько стереогенных центров, присутствуют в R- или S-конфигурации, таких как, например,атом углерода, несущий заместитель R2. В соответствии с номенклатурой CAS, если в молекуле присутствуют два стереогенных центра известной абсолютной конфигурации, то R- или S-дескриптор приписывают (на основании правила последовательностей Cahn-Ingold-Prelog) хиральному центру с наименьшим номером, сравнительному центру. Конфигурацию второго стереогенного центра обозначают, применяя относительные дескрипторы[R,R] или [R,S], в которых R всегда определен как сравнительный центр, [R,R] обозначает центры с такой же хиральностыо, a [R,S] обозначает центры с иной хиральностью. Например, если в молекуле хиральный центр с наименьшим номером имеет S-конфигурацию, а второй центр имеет R, то стереодескриптор будет обозначен как S-[R,S]. Соединения формулы (I), полученные в соответствии с вышеописанными способами, могут быть синтезированы в виде рацемических смесей энантиомеров, которые могут быть отделены один от другого в соответствии с известными в данной области методиками разделения. Рацемические соединения формулы (I) могут быть превращены в соответствующие диастереомерные солевые формы взаимодействием с подходящей хиральной кислотой. Затем указанные диастереомерные солевые формы разделяют,например, селективной или фракционной кристаллизацией и энантиомеры высвобождают из них щелочью. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с применением хиральной неподвижной фазы. Указанные чистые, стереохимически изомерные формы также могут быть получены из соответствующих чистых, стереохимически изомерных форм соответствующих исходных материалов при условии, что взаимодействие происходит стереоспецифически. Предпочтительно, если желательно получение специфического стереоизомера, указанное соединение синтезируют при помощи стереоспецифических способов получения. В данных способах предпочтительно применяют энантиомерно чистые исходные материалы. Соединения формулы (I), их фармацевтически приемлемые соли и стереоизомерные формы являются ингибиторами мембранной трипептидил пептидазы, отвечающего за инактивацию эндогенных нейропептидов, таких как холецистокинины (CCKs), что подтверждает фармакологический пример С-1. Благодаря своим ТПП II-ингибирующим свойствам соединения настоящего изобретения могут быть использованы для лечения состояний или расстройств, связанных с активностью ТПП II, таких как,например, расстройства, связанные с приемом пищи, ожирение, психотические синдромы и связанные с ними психиатрические нарушения. Вследствие наличия полезных свойств у соединений формулы (I) настоящее изобретение также относится к способу лечения теплокровных животных, включая людей (в общем называемых в данном описании пациентами), страдающих от расстройств, связанных с приемом пищи, ожирения, психотических синдромов и связанных с ними психиатрических нарушений. Следовательно, способ лечения предусмотрен для ингибирования активности ТПП II и/или облегчения состояния пациентов, страдающих,например, от расстройств, связанных с приемом пищи, ожирения, психотических синдромов и связанных с ними психиатрических нарушений. Следовательно, предусмотрено применение соединения формулы (I) в качестве лекарственного препарата, действующего как ингибитор ССK-инактивирующей трипептидил пептидазы (ТПП II) и/или для лечения расстройств, связанных с приемом пищи, особенно ожирения, и/или для лечения психотических синдромов и связанных с ними психиатрических нарушений; включающего терапевтически эффективное количество соединения формулы (I). Также предусмотрено применение соединения формулы (I) для получения лекарственного препарата для ингибирования активности ТПП II и/или для лечения расстройств, связанных с приемом пищи, ожирения, психотических синдромов и связанных с ними психиатрических нарушений. Предусмотрено как профилактическое, так и терапевтическое лечение. Предполагается, что некоторые из соединений в соответствии с настоящим изобретением, в частности соединения (153)-(181), также могут проявлять опиоидную активность, такую как дельта-опиоидная, мю-опиоиднаяи/или каппа-опиоиднаяактивность. Опиоидная активность может быть измерена с использованием анализов, описанных в фармакологических примерах С.2 и С.3.-4 006443 Для получения фармацевтических композиций в соответствии с данным изобретением эффективное количество конкретного соединения в виде основания или кислотно-аддитивной соли объединяют в качестве активного ингредиента в однородной смеси самые разнообразные формы в зависимости от желаемой формы вводимого препарата. Данные фармацевтические композиции желательно имеют вид стандартной лекарственной формы, предпочтительно для перорального, ректального введения или парентеральных инъекций. Например, для получения композиций в виде дозированной формы, предназначенной для перорального введения, может быть использована любая из обычных фармацевтических сред, такая, например, как вода, гликоли, масла, спирты и подобные, при получении пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; либо твердые носители, такие как крахмалы, сахара, каолин, смазывающие агенты, связующие, разрыхляющие агенты и подобные, при получении порошков, пилюль, капсул и таблеток. Из-за легкости введения таблетки и капсулы являются наиболее предпочтительными пероральными стандартными лекарственными формами; в таком случае,безусловно, применяют твердые фармацевтические носители. В композициях, предназначенных для парентерального введения, носитель обычно включает стерильную воду, по меньшей мере, большая его часть, хотя могут быть включены и другие ингредиенты, например, улучшающие растворимость. Например, могут быть получены растворы для инъекций, в которых носитель включает солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть также получены суспензии для инъекций, в которых могут быть использованы соответствующие жидкие носители, суспендирующие агенты и подобные. В композициях, предназначенных для чрескожного введения, носитель необязательно включает агент, улучшающий проникновение, и/или подходящий смачивающий агент, необязательно объединенный в небольших пропорциях с подходящими добавками любой природы, не наносящими существенного вреда коже. Указанные добавки могут облегчить введение в кожу и/или могут быть полезными для получения желаемых композиций. Данные композиции могут быть введены различными способами, например в виде трансдермальных пластырей, нашлепок, мази. Из-за повышенной растворимости в воде по сравнению с соответствующими основными формами кислотно-аддитивные соли соединений формулы (I) очевидно являются более подходящими для получения водных композиций. Особенно предпочтительно получение вышеуказанных фармацевтических композиций в виде стандартной лекарственной формы с целью облегчения введения и равномерности дозировки. Термин стандартная лекарственная форма в данном описании и формуле изобретения означает физически дискретные единицы, приемлемые как стандартные дозы, причем каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанного таким образом, чтобы оказать желаемое терапевтическое действие, вместе с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечками или с покрытием), капсулы, пилюли, пакетики с порошками, облатки, растворы или суспензии для инъекций, чайные ложки,столовые ложки и подобные, а также объединенное вместе множество данных доз. Фармацевтические композиции, предназначенные для перорального введения, могут иметь вид твердых лекарственных форм, например таблеток (предназначенных как только для проглатывания, так и разжевывания), капсул или желатиновых капсул, получаемых обычными способами с фармацевтически приемлемыми эксципиентами, такими как связывающие агенты (например, предварительно желатинизированный маисовый крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители(например, лактоза, микрокристаллическая целлюлоза или фосфат кальция); смазывающие агенты (к примеру, стеарат магния, тальк или двуокись кремния); разрыхлители (к примеру, картофельный крахмал или натриевый гликолят крахмала); или смачивающие агенты (например, лаурилсульфат натрия). На таблетки может быть нанесено покрытие в соответствии со способами, хорошо известными в данной области. Жидкие препараты, предназначенные для перорального введения, могут иметь форму, например,растворов, сиропов или суспензий либо они могут иметь вид сухого продукта, смешиваемого перед введением с водой или другим подходящим носителем. Такие жидкие препараты могут быть получены известными способами, необязательно с фармацевтически приемлемыми добавками, такими как суспендирующие агенты (например, сироп сорбита, метилцеллюлоза, гидроксипропилметилцеллюлоза или гидрированные пищевые жиры); эмульгирующие агенты (например, лецитин или акация); неводные наполнители (например, миндальное масло, маслянистые сложные эфиры или этиловый спирт) и консерванты (к примеру, метил или пропил п-гидроксибензоаты или сорбиновая кислота). Фармацевтически приемлемые подсластители предпочтительно включают по меньшей мере один сильный подсластитель, такой как сахарин, сахарин натрия или кальция, аспартам, ацесульфам калия,цикламат натрия, алитам, дигидрохальконовый подсластитель, монеллин, стевиозид или сахаролоза(4,1',6'-трихлор-4,1',6'-тридезоксигалактосахароза), предпочтительно сахарин, сахарин натрия или кальция, и, необязательно, объемный подсластитель, такой как сорбит, маннит, фруктоза, сахароза, мальтоза,изомальт, глюкоза, гидрированный сироп глюкозы, ксилит, карамель или мед. Сильные подсластители обычно применяют в низких концентрациях. Например, при использовании сахарина натрия концентрация может изменяться от 0,04 до 0,1% (мас./об.) от общего объема готового состава и предпочтительно составляет около 0,06% в составах с низкой дозой и около 0,08% в составах с-5 006443 высокой дозой. Объемный подсластитель может быть эффективно использован в больших количествах,составляющих приблизительно от 10 до 35%, предпочтительно приблизительно от 10 до 15% (мас./об.). Фармацевтически приемлемые вкусовые добавки, способные маскировать ингредиенты с горьким вкусом в составах с низкой дозой, предпочтительно включают фруктовые добавки, такие как вишневые,малиновые, черносмородиновые или клубничные добавки. Сочетание двух вкусовых добавок может дать очень хорошие результаты. В составах с высокой дозой могут потребоваться более сильные вкусовые добавки, такие как карамельно-шоколадная мятно-прохладительная добавка, вкусовая добавка Фантазия и т.п. фармацевтически приемлемые сильные вкусовые добавки. Каждая вкусовая добавка может присутствовать в готовой композиции в концентрации, составляющей от 0,05 до 1% (мас./об.). Предпочтительно применять сочетания указанных сильных вкусовых добавок. Предпочтительно применение вкусовой добавки, не изменяющей или не теряющей вкус и цвет в кислотных условиях состава. Соединения в соответствии с данным изобретением также могут быть получены в виде препаратовдепо. Такие длительно действующие составы могут быть введены имплантацией (например, подкожно или внутримышечно) либо внутримышечной инъекцией. Таким образом, например, соединения могут быть сформулированы с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) либо ионообменными смолами или в виде слаборастворимых производных, например слаборастворимой соли. Соединения в соответствии с данным изобретением могут быть сформулированы для парентерального введения инъекциями, обычно, внутривенными, внутримышечными или подкожными инъекциями,например, путем болюсной инъекции или непрерывного внутривенного вливания. Составы для инъекций могут быть представлены в виде стандартных лекарственных форм, например в ампулах или контейнерах, содержащих многократную дозу с добавлением консерванта. Композиции могут иметь вид суспензий, растворов или эмульсий в масляных или водных наполнителях и могут содержать формулирующие агенты, такие как изотонизирующие, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный ингредиент может иметь вид порошка, смешиваемого перед введением с подходящим наполнителем, например стерильной, свободной от пирогена водой. Соединения в соответствии с данным изобретением также могут иметь вид композиций, предназначенных для ректального введения, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды. Соединения в соответствии с данным изобретением, предназначенные для интраназального введения, могут, например, иметь вид жидкого спрея, порошка или капель. Экспериментальная часть В описываемых ниже методиках использованы следующие сокращения: ACN означает ацетонитрил; ТГФ означает тетрагидрофуран; DCM означает дихлорметан; a MIK означает метилизобутилкетон. Некоторые химические вещества представлены химическими формулами, например СН 2 Сl2 - дихлорметан, СН 3 ОН - метанол, NH3 - аммиак, НСlхлористо-водородная кислота, NaOH - гидроксид натрия, NаНСО 3 - двууглекислый натрий и Na2CO3 - карбонат натрия. Первая выделенная стереохимическая изомерная форма обозначена буквой А, а вторая - В, без дальнейшей ссылки на действительную стереохимическую конфигурацию. Препаративную жидкостную хроматографию осуществляют на полупрепаративной установке для ВЭЖХ, применяя колонку YMC ODS-A (30100 мм, 5 мкм, температура окружающей среды, скорость потока: 35 мл/мин., подвижная фаза: а) 10/90 ацетонитрил/вода с 0,1% трифторуксусной кислотой, b) 90/10 ацетонитрил/вода с 0,1% трифторуксусной кислотой, градиент: линейный градиент от А до В в течение более 9 мин, УФ-детектирование при 254 нм. А. Получение промежуточных соединений. Пример А.1. а) 2,3-Дигидрокси-1H-индол-2-карбоксамид (0,030 моль) суспендируют в трихлорметане (400 мл). Смесь охлаждают до 0 С. Добавляют триэтиламин (0,045 моль). В течение более 2 мин добавляют ацетилхлорид (0,045 моль). Через 30 мин ТСХ показывает, что взаимодействие еще не завершено. В еще холодную колбу добавляют дополнительное количество триэтиламина (6,26 г), а через 15 мин - дополнительное количество ацетилхлорида (3,21 г). ТСХ показывает, что взаимодействие еще не завершено. Взаимодействие продолжают при перемешивании, охлаждают до 0 С и добавляют дополнительное количество триэтиламина (6,26 мл). В течение более 2 мин добавляют дополнительное количество неразбавленного ацетилхлорида (3,21 мл). Через 60 мин ТСХ показывает 80% завершение и никакого прогресса еще через 30 мин. Добавляют третью порцию ацетилхлорида и триэтиламина. Еще через 15 мин добавляют ледяную воду (200 мл). Смесь перемешивают в течение 10 мин, фильтруют и промывают водой(3100 мл) и трихлорметаном (275 мл). Образцу дают возможность сохнуть в течение ночи, получая 4,71 г (S)-1-ацетил-2,3-дигидро-1H-индол-2-карбоксамида (промежуточное соединение 1, т.пл. 260 С).b) Промежуточное соединение (1) (0,02022 моль) суспендируют в DCM (175 мл). Смесь охлаждают до 0 С. Добавляют неразбавленный триэтиламин (0,06066 моль). По каплям в течение более 20 мин добавляют трихлорацетилхлорид (0,03033 моль) в DCM (20 мл). Через 2 ч добавляют ледяную воду (200-6 006443 мл), фазы разделяют и органическую фазу подвергают повторному экстрагированию, применяя 3 н. НСl,а затем насыщенный водный раствор NaHCO3. Органическую фазу сушат, фильтруют и десорбируют,получая 4,61 г коричневого твердого вещества. Полученное твердое вещество растирают с ледяным диэтилэфиром (30 мл), фильтруют и промывают ледяным диэтилэфиром (дважды), получая 3,12 г (83%)c) Промежуточное соединение (2) (0,0151 моль) суспендируют в диэтилэфире (200 мл). Добавляют этанол (0,0214 моль) и смесь охлаждают до 0 С. НСl (газ) барботируют в течение 45 мин. Смесь удаляют с ледяной бани и перемешивают. Через 20 мин на стенках собирается осадок. Его соскребают, получая белый твердый осадок. Через 1 ч образец фильтруют, промывают диэтилэфиром и быстро сушат воздухом, получая 3,99 г (S)-этил 1-ацетил-2,3-дигидро-1H-индол-2-карбоксимидата моногидрохлорида (промежуточное соединение 3). Аналогичным способом получают этил 1-ацетил-2,3-дигидро-1H-индол-2-карбоксимидата моногидрохлорид (промежуточное соединение 6), исходя из 1-ацетил-2,3-дигидро-1 Н-индол-2-карбонитрила. Пример А.2. а) 5-Хлор-2,3-дигидрокси-1H-индол-2-карбоновую кислоту, метиловый эфир (0,00761 моль) растворяют в метаноле (25 мл) и охлаждают до 0 С. NН 3 барботируют в течение 10 мин. Колбу закупоривают и дают ей возможность нагреться до комнатной температуры. Смесь перемешивают в течение ночи. ТСХ показывает, что взаимодействие в основном завершено. Образец концентрируют до 1/3 объема, охлаждают и фильтруют, промывая полученное твердое вещество ледяным метанолом (2 мл), а затем сушат на воздухе, получая 0,74 г 5-хлор-2,3-дигидро-1 Н-индол-2-карбоксамида (промежуточное соединение 7,т.пл. 151-152 С).b) Триэтиламин (0,02080 моль) добавляют к промежуточному соединению (7) (0,09632 моль), растворенному в трихлорметане (700 мл). Смесь охлаждают до 5 С. В течение более 2 мин при перемешивании добавляют ацетилхлорид (0,2480 моль). Через 5 мин образуется осадок. Ледяную баню удаляют, а содержимому емкости дают возможность отстояться в течение 15 мин. Добавляют ледяную воду (250 мл) и смесь перемешивают в течение 10 мин. Образец фильтруют, промывают водой и трихлорметаном. Твердое вещество суспендируют в воде (200 мл) и перемешивают в течение 10 мин. Добавляют трихлорметан (200 мл) и смесь перемешивают, а затем фильтруют и промывают водой и трихлорметаном, потом сушат на воздухе в течение ночи, получая 19,71 г 1-ацетил-5-хлор-2,3-дигидро-1H-индол-2-карбоксамидаc) Триэтиламин (0,41291 моль) добавляют к промежуточному соединению (8) (0,08258 моль), суспендированному в дихлорметане (500 мл) при 0 С. В течение более 10 мин добавляют трихлорацетилхлорид (0,20645 моль). Когда взаимодействие замедляется, добавляют дополнительную порцию триэтиламина (20 мл), а затем дополнительное количество трихлорацетилхлорида (7,6 мл) и смесь перемешивают в течение 2 ч при низкой температуре. Ледяную баню удаляют, а смеси дают возможность отстояться в течение 2 ч, получая смесь более темного цвета, которую вновь охлаждают до 0 С. Медленно добавляют ледяную воду (150 мл) и смесь перемешивают в течение 5 мин. Слои разделяют, а органическую фазу промывают (ледяная 3 н. НСl, насыщенный NaHCO3), сушат, фильтруют и концентрируют. Остаток растирают с ледяным диэтилэфиром (40 мл). В результате фильтрации и промывания ледяным диэтилэфиром (10 мл) получают 15,13 г 1-ацетил-5-хлор-2,3-дигидро-1H-индол-2-карбонитрила (промежуточное соединение 9, т.пл. 140-142 С).d) HCl (в виде 2 М раствора) медленно добавляют к промежуточному соединению (9) до тех пор,пока не начнется выделение газа. Затем добавление прекращают и добавляют полученную HCl (2 н. в диэтилэфире), суспендированную в диэтилэфире (150 мл) и этанол (0,042 моль). Смесь охлаждают до 0 С и более часа добавляют HCl (газ), при этом масло осаждается. Смесь разбавляют до 1 л диэтилэфиром. Осаждается еще большее количество масла и после осаждения в течение часа не образуется никакого твердого вещества. Диэтиловый эфир сливают. Остаток разбавляют (диэтиловый эфир, 500 мл). Начинается образование твердого вещества, и смесь перемешивают в течение 2 ч. Образец фильтруют и промывают диэтилэфиром. Образец помещают в вакуум, получая 6,41 г этил 1-ацетил-5-хлор-2,3-дигидро 1H-индол-2-карбоксимидата моногидрохлорида (промежуточное соединение 10). Пример А.3.a) Бис (1,1-диметилэтил)эфир дикарбоновую кислоту (0,07615 моль) в DCM (50 мл) добавляют в течение более 5 мин к 2,3-дигидро-1H-индол-2-метанолу (0,07615 моль) в DCM (150 мл) при 0 С. Смеси дают возможность нагреться до комнатной температуры и перемешивают в течение ночи. Смесь концентрируют при пониженном давлении и подвергают дистилляции Kogel Rohr, получая 11,98 г 1,1 диметилэтил 2,3-дигидро-2-(гидроксиметил)-1 Н-индол-1-карбоксилата (промежуточное соединение 11).b) Неразбавленный реактив Dess-Martin (0,011 моль) добавляют в течение более 1 мин к промежуточному соединению (11) (0,010 моль), растворенному в DCM (35 мл). Через 15 мин ледяную баню удаляют, а смеси дают возможность нагреться до комнатной температуры. Добавляют дополнительное количество реактива Dess-Martin (0,33 г) и смесь перемешивают еще в течение 30 мин. Смесь вновь охлаждают до 0 С и медленно обрабатывают частичной суспензией/раствором Nа 2S2O3(25 г), который медленно растворяется в насыщенном водном растворе NaHCO3 (100 мл). Через 10 мин-7 006443 смесь снимают со льда и слои разделяют. Добавляют дополнительное количество DCM и смесь фильтруют. Органический слой отделяют от фильтрата и объединенные органические фазы сушат, фильтруют,концентрируют и очищают колоночной флэш-хроматографией (элюент: 10% этилацетат:гексан, растворяя образец в 3:1 этилацетате:гексане (5 мл, получая 1,1-диметилэтил 2-формил-2,3-дигидро-1H-индол 1-карбоксилат (промежуточное соединение 12, т.пл. 85-87 С). Пример А.4. Раствор этил 1-ацетил-2,3-дигидро-1 Н-индол-2-карбонитрила (0,00988 моль) и триэтиламина(0,0197 моль) в пиридине (50 мл) обрабатывают сероводородом (газ) при комнатной температуре в барботере в течение 2 ч, полученную насыщенную реакционную смесь закупоривают и дают ей возможность оседать в течение 16 ч. Реакционную смесь добавляют к 200 мл взвеси лед/вода. Образуется объемный осадок. Смесь вновь охлаждают на ледяной бане, осадок собирают при помощи фильтрации отсасыванием, промывают холодной водой и сушат на воздухе, получая 1,62 г 1-ацетил-2,3-дигидро-1Hиндол-2-карботиоамида (промежуточное соединение 13, т.пл. 194-195 С). Пример А.5. 1-Ацетил-2,3-дигидро-1H-индол-2-карбонитрил (0,0132 моль) обрабатывают водой (54 мл) и полученную суспензию последовательно обрабатывают, применяя Na2CO3 (0,00726 моль) и NH2OHHCl(0,0145 моль). Смесь обрабатывают этанолом (26 мл) и нагревают до 80-90 С. По достижении температуры взаимодействия смесь все еще представляет собой суспензию. Дополнительно добавляют 26 мл этанола, получая прозрачный раствор. Смесь нагревают в течение 2,5 ч и охлаждают до комнатной температуры при перемешивании. Образуется объемный осадок, который собирают при помощи фильтрации отсасыванием, промывают его холодной дистиллированной водой и сушат на воздухе, получая 2,23 г 1-ацетил-2,3-дигидро-N'-гидрокси-1 Н-индол-2-карбоксимидамида (промежуточное соединение 14, т.пл. 204-205 С). Пример А.6. 1-Ацетил-2-(4-этил-1H-имидазол-2-ил)-2,3-дигидро-1H-индол (0,0035 моль) и НСl, 6 н. (50 мл) объединяют в атмосфере азота. Реакционную смесь сразу же нагревают, продолжая нагревание в течение 3,5 ч. Смеси дают возможность охладиться до комнатной температуры, затем экстрагируют диэтилэфиром(360 мл). Объединенные органические слои сушат, фильтруют, а растворитель выпаривают, получая 0,79 г 2-(4-этил-1H-имидазол-2-ил)-2,3-дигидро-1H-индола (промежуточное соединение 15). Пример А.7.(600 мл) добавляют Мg (0,36 моль). Смесь находится в 3-горлой, круглодонной колбе в атмосфере аргона при комнатной температуре. Температуру взаимодействия тщательно контролируют. Приблизительно через 10 мин смесь начинает барботировать, вначале медленно, а затем более энергично. Температуру взаимодействия поддерживают на уровне 15-25 С, иногда применяя ледяную баню. Через 30 мин барботирование замедляется. Смесь перемешивают при комнатной температуре в течение трех дней. Смесь разделяют между 600 мл хлороформа и 500 мл насыщенного раствора NH4Cl. Органический слой сушат над МgSO4 и концентрируют до коричневого масла. Масло растворяют в простом эфире и экстрагируют,применяя 3 н. НСl. Водный слой промывают простым эфиром, превращают в основание, применяя 3 н.NaOH, и экстрагируют хлороформом. Экстракт сушат над МgSO4 и концентрируют, получая 13,91 г метил 5-фтор-2,3-дигидро-1H-индол-2-карбоксилат (промежуточное соединение 16).b) К 2 М NН 3 в метаноле (0,6 моль), охлажденному на ледяной бане в атмосфере Аr, добавляют промежуточное соединение (16) (0,0574 моль), растворенное в метаноле (150 мл). Смеси дают возможность нагреться до комнатной температуры и перемешивают в атмосфере аргона в течение 6 ч. Полученную смесь концентрируют до 150 мл и фильтруют. Твердое вещество промывают небольшим количеством холодного метанола и дают ему возможность высохнуть, получая 2,33 г 5-фтор-2,3-дигидро-1H-индол-2 карбоксамида (промежуточное соединение 17, т.пл. 197-199 С).c) К смеси промежуточного соединения (17) (0,0094 моль) в DCM (30 мл), охлажденной на ледяной бане в атмосфере аргона, добавляют триэтиламин (0,031 моль), а затем ацетилхлорид (0,031 моль). Полученной смеси дают возможность нагреться до комнатной температуры. После 6 ч перемешивания смесь охлаждают на ледяной бане и добавляют к ней 50 мл воды. Смесь перемешивают в течение приблизительно 20 мин, фильтруют, а твердое вещество подсушивают, получая 1,58 г 1-ацетил-5-фтор-2,3 дигидро-1H-индол-2-карбоксамида (промежуточное соединение 18, т.пл. 232-235 С).d) К суспензии промежуточного соединения (18) (0,0076 моль) в DCM (30 мл), охлажденной на ледяной бане в атмосфере аргона, добавляют триэтиламин (0,0228 моль), а затем трихлорацетилхлорид(0,0115 моль). Смеси дают возможность нагреться до комнатной температуры и перемешивают в течение 2 ч. Смесь промывают водой, 2 н. НСl и насыщенным NaHCO3. Органический слой сушат и концентрируют. Концентрат растирают с простым эфиром и очищают на колонке с силикагелем, элюируя 50% этилацетатом в гексане. Нужные фракции объединяют и концентрируют. Остаток растирают с простым-8 006443 эфиром, твердое вещество собирают фильтрацией и дают ему возможность высохнуть, получая 0,30 г 1 ацетил-5-фтор-2,3-дигидро-1H-индол-2-карбонитрила (промежуточное соединение 19, т.пл. 93-95 С).e) Раствор промежуточного соединения (19) (0,004 моль) и HCl/диэтилэфира (60 мл) охлаждают на ледяной бане в атмосфере аргона. Добавляют этанол (0,0075 моль). НСl барботируют в раствор в течение 50 мин до тех пор, пока смесь не станет гомогенной. Смеси дают возможность медленно нагреться до комнатной температуры и перемешивают в течение 4 ч. Эфир сливают и растворяют в метаноле. Раствор метанола концентрируют в вакууме, а остаток используют без обработки на следующей стадии, получая этил 1-ацетил-5-фтор-2,3-дигидро-1H-индол-2-карбоксимидата моногидрохлорид (промежуточное соединение 20). Пример А.8.a) Сложный метиловый эфир 2,3-дигидро-5-метокси-1H-индол-2-карбоновой кислоты (0,084 моль) и 2 М NН 3 в метаноле (500 мл) объединяют и перемешивают при комнатной температуре в атмосфере аргона в течение выходных дней. Раствор концентрируют до 100 мл, охлаждают на ледяной бане и фильтруют. Твердое вещество промывают небольшим количеством холодного метанола и сушат. Остаток растирают с метанолом/ACN и фильтруют, получая 4,56 г 2,3-дигидро-5-метокси-1 Н-индол-2 карбоксамида (промежуточное соединение 21, т.пл. 228-229 С).b) Триэтиламин (0,0106 моль), а затем ацетилхлорид (0,0106 моль) добавляют к раствору промежуточного соединения (21) (0,0032 моль) в DCM (40 мл), охлажденному на ледяной бане в атмосфере аргона. Смеси дают возможность медленно нагреться до комнатной температуры и перемешивают в течение ночи. Смесь охлаждают на ледяной бане и добавляют к ней ледяную воду (30 мл). После перемешивания в течение 10 мин смесь фильтруют и твердому веществу дают возможность высохнуть в течение ночи. Остаток суспендируют в 50 мл воды. Суспензию перемешивают в течение 30 мин, фильтруют и сушат в течение ночи, получая 0,40 г 1-ацетил-2,3-дигидро-5-метокси-1 Н-индол-2-карбоксамида (промежуточное соединение 22, т.пл. 196-197 С).c) К суспензии промежуточного соединения (22) (0,022 моль) в DCM (150 мл), охлажденной на ледяной бане в атмосфере аргона, добавляют триэтиламин (0,066 моль), а затем трихлорацетилхлорид(0,033 моль). Смеси дают возможность медленно нагреться до комнатной температуры в течение ночи. Смесь промывают водой, 2 н. НСl и насыщенным NaHCO3. Органическую фазу сушат, концентрируют и растирают с простым эфиром, а твердое вещество собирают, получая 1-ацетил-2,3-дигидро-5-метокси 1H-индол-2-карбонитрил (промежуточное соединение 23, т.пл. 108-110 С).d) В раствор промежуточного соединения (23) (0,0154 моль) и этанола (0,0231 моль) в 1 МHCl/диэтилэфире (200 мл), охлажденный на ледяной бане, в течение 60 мин барботируют НСl (газ). Ледяную баню поддерживают в течение 45 мин и смесь концентрируют при комнатной температуре в вакууме до получения 200 мл маслянистого осадка. Остаток растирают до коричневого твердого вещества,которое становится маслом после сливания диэтилэфира. Остаток дважды промывают диэтилэфиром,растворяют в метаноле и используют без дополнительной очистки для дальнейшего синтеза, получая 1 ацетил-2,3-дигидро-5-метокси-1 Н-индол-2-карбоксимидата моногидрохлорид (промежуточное соединение 24). Пример А.9.(S)-2-(Трет-бутоксикарбониламино)масляную кислоту (0,010 моль), растворенную в DCM (25 мл),помещают на охлаждающую баню при -10 С. Добавляют пиридин (0,010 моль), а затем 2,4,6-трифтор 1,3,5-триазин (0,0345 моль). Смесь перемешивают в атмосфере азота. Через час добавляют ледяную воду(75 мл). Добавляют дополнительное количество DCM (45 мл) и смесь встряхивают. Органическую фазу отделяют, вновь промывают ледяной водой (100 мл), а затем органическую фазу сушат, фильтруют и концентрируют, получая 2,29 г (S)-1,1-диметил[1-(фторкарбонил)пропил]карбамата (промежуточное соединение 25). Пример А.10. Соединение (8) (0,00170 моль) растворяют в НСl, 6 н. (20 мл) и сразу же нагревают на масляной бане при 100 С в атмосфере азота в течение 200 мин. Подачу тепла прекращают и образец охлаждают до 0 С. Медленно добавляют 3 н. NaOH (35 мл). Превращение в основание завершают, добавляя насыщенныйNaHCO3. Образец экстрагируют хлороформом. Объединенные органические фазы сушат, фильтруют, а полученный раствор используют без дополнительной очистки для дальнейшего синтеза, получая (S)-2,3 дигидро-2-(4-пропил-1 Н-имидазол-2-ил)-1H-индол (промежуточное соединение 5). Пример А.11. Смесь промежуточного соединения (13) (0,00844 моль) в этаноле (180 мл) обрабатывают 1-бром-2 бутаноном (0,0085 моль) в виде одной порции и кипятят с обратным холодильником в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры и экстрагируют между простым эфиром и холодным 1 М NaOH (водный). Органическую фракцию сушат над МgSO4 и концентрируют в вакууме,получая твердое вещество темного цвета, которое подвергают колоночной флэш-хроматографии с силикагелем (элюент 100% DCM к 97:3 DCM/простой диэтиловый эфир), получая 0,91 г 2-(4-этил-2 тиазолил)-2,3-дигидро-1H-индола (промежуточное соединение 4). 2-Трет-бутиловый эфир 3,4-дигидро-1 Н-изохинолин-2,3-дикарбоновой кислоты (2,77 г, 10 ммоль),2-амино-1-фенилэтанон (1,71 г, 10 ммоль) и НОВТ (1-гидроксибензотриазол) (2,70 г, 20 ммоль) растворяют в дихлорметане (100 мл). Раствор охлаждают до 0 С, а затем добавляют (4-диметиламинобутил)этилкарбодиимид (2,29 г, 12 ммоль) и NMM (N-метилморфолин) (1,31 г, 13 ммоль). Затем реакционную смесь нагревают до комнатной температуры. Через 72 ч реакционную смесь экстрагируют водой,а органическую фазу последовательно экстрагируют насыщенным NaHCO3, 2 н. лимонной кислотой иNaHCO3, сушат над МgSO4, фильтруют и концентрируют, получая указанный в заголовке продукт в виде желтой пены. Жидкостная хроматография (ЖХ) показывает, что чистота соединения составляет 86% (214 нм), поэтому его используют без дальнейшей очистки. Пример А.12 а. Дегидратация бензилового эфира 3-(2-оксо-2-фенил-этил-карбамоил)-3,4-дигидро-1 Н-изохинолин 2-карбоновой кислоты (получен таким же способом, как и трет-бутиловый эфир 3-(2-оксо-2 фенилэтилкарбамоил)-3,4-дигидро-1 Н-изохинолин-2-карбоновой кислоты в примере А.12) с применением РОСl3 приводит к получению следующего промежуточного соединения: Группу CBZ легко удаляют у получаемого оксазола обработкой иодотриметилсиланом. Получаемое промежуточное нораминовое соединение оксазола может быть превращено в соединение 170 в соответствии с методикой, описанной для его аналогичных промежуточных соединений имидазола. Пример А.13. Трет-бутиловый эфир 3-(4-фенил-1 Н-имидазол-2-ил)-3,4-дигидро-1 Н-изохинолин-2 карбоновой кислоты.(20,8 г, 270 ммоль) и АсОН (уксусная кислота) (30 мл) объединяют при комнатной температуре и реакционную смесь нагревают на паровой бане в течение приблизительно 3 ч. Затем реакционную смесь охлаждают до комнатной температуры и выливают в ледяную взвешенную смесь (400 г). К полученной смеси добавляют концентрированный гидроксид аммония (50 мл) и простой этиловый эфир. Слои разделяют и водную фазу промывают второй порцией простого этилового эфира. Органические фазы объединяют, сушат над МgSO4, фильтруют и концентрируют при пониженном давлении, получая коричневую пену. Полученный образец очищают с помощью препаративной ВЭЖХ, получая очищенное, указанное в заголовке соединение в виде белого порошка. ЖХ показывает, что чистота образца составляет 96% при 214 нм. Вычисленная мол. масса (МН+): 376 Трифторуксуную кислоту (ТФУ) (4 мл) охлаждают в пробирке до температуры приблизительно 0 С. Затем к холодному растворителю добавляют продукт, полученный в вышеописанном примере А.13(0,75 г, 2 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры в течение более 45 мин. Избыточную ТФУ удаляют под потоком газа N2. Остаток разделяют между дихлорметаном(15 мл) и насыщенным NаНСО 3. Затем водную фазу подвергают повторному экстрагированию второй порцией дихлорметана, органические фазы объединяют, сушат над MgSO4 и фильтруют, получая указанное в заголовке соединение в растворе дихлорметана. Фильтрат используют на следующей стадии (пример А.15) без дальнейшей очистки или выделения. Вычисленная мол. масса (МН+) 276. Пример А.15. трет-Бутиловый эфир [1-(4-трет-бутоксибензил)-2-оксо-2-[3-(4-фенил-1 Н-имидазол-2 ил)-3,4-дигидро-1 Н-изохинолин-2-ил]этил]карбаминовой кислоты. 2-трет-Бутоксикарбониламино-3-(4-трет-бутоксифенил)пропионовую кислоту (0,74 г, 2,2 ммоль) растворяют в дихлорметане (40 мл) и реакционную смесь охлаждают до температуры, составляющей приблизительно 0 С. Затем к раствору добавляют NMM (0,21 г, 2,1 ммоль) с последующим добавлением изобутилхлорформиата (0,27 г, 2 ммоль, 0,26 мл) и раствору дают возможность отстояться в течение приблизительно 1,25 ч. Затем к реакционной смеси добавляют продукт, полученный в соответствии с методикой примера А.14 (0,55 г, 2 ммоль), и реакционную смесь перемешивают в течение приблизительно 16 ч. Реакционную смесь затем экстрагируют водой, насыщенным NaHCO3, 2 н. лимонной кислотой,насыщенным NaHCO3, сушат над МgSO4, фильтруют и концентрируют, получая указанное в заголовке соединение в виде пены. Вычисленная моль. масса (МН+): 595. В положение 5 имидазольного остатка полученного промежуточного соединения может быть введен бром путем взаимодействия указанного промежуточного соединения с 1 эквивалентом Вr2 при 0 С в хлороформе. В положение 5 имидазольного остатка полученного промежуточного соединения может быть введен хлор путем взаимодействия указанного промежуточного соединения с N-хлорсукцинимидом. Пример А.16. трет-Бутиловый эфир 3-(5-метил-4-фенил-1 Н-имидазол-2-ил)-3,4-дигидро-1 Н-изохинолин-2-карбоновой кислоты. трет-Бутиловый эфир 3-формил-3,4-дигидро-1 Н-изохинолин-2-карбоновой кислоты (1,83 г, 7 ммоль) объединяют с АсОН (25 мл) и сразу же добавляют 1-фенилпропан-1,2-дион (3,11 г, 21 ммоль) иNH4OAC (13,49 г, 175 ммоль). Затем реакционную смесь помещают на паровую баню и нагревают в атмосфере аргона в течение 20 мин. Реакционную смесь охлаждают на ледяной бане, а затем добавляют к ледяной взвеси (44 г). Полученную смесь превращают в основную, добавляя концентрированный NH4OH(50 мл), а затем дважды экстрагируют простым диэтиловым эфиром (2150 мл). Объединенные органические фазы сушат над МgSO4, фильтруют и концентрируют, получая сырой продукт. Данный продукт очищают препаративной ВЭЖХ, получая указанное в заголовке соединение в виде белого твердого вещества. К раствору ТФУ (5 мл), охлажденному до температуры, составляющей около 0 С, добавляют соединение, полученное в соответствии с описанием, приведенным в примере А.16 (1,10 г, 2,82 ммоль), и реакционную смесь перемешивают в течение приблизительно 30 мин. Затем реакционную смесь удаляют с ледяной бани и дают ей возможность нагреться до комнатной температуры. Избыточную ТФУ удаляют в потоке N2. Остаток разделяют между насыщенным NаНСО 3 и дихлорметаном. Водную фазу промывают второй порцией дихлорметана и органические фазы объединяют. Объединенную органическую фазу сушат над Na2SO4, a затем фильтруют, получая указанное в заголовке соединение в виде раствора в дихлорметане, которое используют без дальнейшей очистки или выделения. Пример А.18. трет-Бутиловый эфир [1-(4-трет-бутоксибензил)-2-[3-(5-метил-4-фенил-1 Н-имидазол 2-ил)-3,4-дигидро-1 Н-изохинолин-2-ил]-2-оксоэтил]карбаминовой кислоты. 2-трет-Бутоксикарбониламино-3-(4-трет-бутоксифенил)пропионовую кислоту (0,74 г, 2,2 ммоль) растворяют в дихлорметане (60 мл) и охлаждают до температуры приблизительно 0 С. Затем к реакционной смеси добавляют NMM (0,30 г, 2,97 ммоль) с последующим добавлением изобутилхлорформиата(0,39 г, 2,82 ммоль, 0,37 мл). Раствору дают возможность отстояться при 0 С в течение приблизительно 90 мин. Затем к реакционной смеси добавляют продукт, полученный в соответствии с методикой примера А.17 (2,82 ммоль) в виде раствора в дихлорметане. Реакционную смесь затем нагревают до комнатной температуры. Через 16 ч реакционную смесь экстрагируют последовательно водой, насыщеннымNaHCO3, 2 н. лимонной кислотой, насыщенным NаНСО 3, затем сушат над МgSO4, фильтруют и концентрируют, получая указанный в заголовке сырой продукт. Данный продукт очищают с помощью препаративной ВЭЖХ, получая указанное в заголовке соединение в виде беловатой пены. Вычисленная мол. масса (МН+): 609. В. Получение готовых соединений. Пример В.1. 4-Метилморфолин (0,003 моль) добавляют к промежуточному соединению (5) (0,003 моль), растворенному в хлороформе (80 мл). После охлаждения до 0 С добавляют неразбавленное промежуточное соединение (25) (0,003 ммоль) в виде масла. Через 27 мин реакционную смесь промывают водой, насыщенным NaHCO3 и солевым раствором, сушат, фильтруют и концентрируют, получая [2S-[1(R),2R1,1-диметилэтил[1-2,3-дигидро-2-(4-пропил-1H-имидазол-2-ил)-1H-индол-1-ил]карбонил]пропил]карбамат (соединение 14). Пример В.2. К промежуточному соединению (3) (0,047 моль) в метаноле (200 мл) добавляют ацетат калия (0,199 моль). Смесь кипятят с обратным холодильником в атмосфере аргона. К ней медленно, в течение более 45 мин добавляют раствор 1-амино-2-пентанона гидрохлорида (0,094 моль) в метаноле (95 мл). Завершив добавление, смесь перемешивают в течение ночи при кипячении с обратным холодильником, а затем концентрируют. Концентрат помещают в DCM и промывают насыщенным NaHCO3. Водный слой экстрагируют, применяя DCM. Объединенные органические экстракты сушат и концентрируют до твердого остатка. Остаток очищают растиранием с простым диэтиловым эфиром и ACN и, необязательно, допол- 12006443 нительно очищают при помощи колоночной хроматографии, получая 5,83 г (S)-1-ацетил-2,3-дигидро-2(4-пропил-1H-имидазол-2-ил)-1H-индола (соединение 8, т.пл. 174-175 С). Пример В.3. Промежуточное соединение (12) (0,00101 моль), 2,3-гександион (0,004 моль) и ацетат аммония(0,025 моль) объединяют в уксусной кислоте (4 мл) и сразу же помещают на паровую баню на 15 мин. Через 2 ч при комнатной температуре реакционную смесь выливают в ледяную воду (100 мл), превращают в основание, применяя 3 н. NaOH, и экстрагируют диэтилэфиром (дважды). Органические фазы объединяют, сушат, фильтруют и концентрируют. Остаток помещают в диэтилэфир, концентрируют, а затем очищают при помощи препаративной ЖХ, получая 0,440 г 1,1-диметил 2,3-дигидро-2-(5-метил-4 пропил-1 Н-имидазол-2-ил)-1H-индол-1-карбоксилата (соединение 99). Аналогичным способом получают соединение (80) взаимодействием промежуточного соединенияN-[(1,1-Диметилэтокси)карбонил]-N-метил-L-аланин (0,00181 моль) растворяют в DCM и охлаждают до 0 С. Добавляют триэтиламин (0,00181 моль), затем изобутилхлорформиат (0,00181 моль) и смесь перемешивают при 0 С в течение 70 мин. Добавляют промежуточное соединение (5) (0,00181 моль) в DCM. Смеси дают возможность нагреться до комнатной температуры и перемешивают в течение ночи. Смесь экстрагируют (вода, насыщенный NaHCO3), сушат, фильтруют и концентрируют. Остаток очищают при помощи ВЭЖХ. Чистые фракции собирают и растворитель выпаривают, получая 0,380 г[2S-[1(R),2R-1,1-диметилэтил[2-[2,3-дигидро-2-(4-пропил-1 Н-имидазол-2-ил)-1H-индол-1-ил]метил 2-оксоэтил]метилкарбамата (соединение 63, т.пл. 77-80 С). Пример В.5. Соединение 14 (0,0073 моль) и трифторуксусную кислоту (5 мл), предварительно охлажденные на ледяной бане, объединяют и дают им возможность медленно нагреться до комнатной температуры в атмосфере азота. Через час смесь концентрируют. Концентрат растворяют в воде и экстрагируют простым диэтилэфиром. Водный слой превращают в основной, применяя насыщенный NaHCO3, и дважды экстрагируют хлороформом. Объединенные органические экстракты сушат над МgSO4 и концентрируют. Остаток растворяют в простом эфире и обрабатывают, применяя 3 мл 1 М НСl в простом эфире. Осадок фильтруют и сушат в вакууме. Остаток разделяют между насыщенным NaHCO3 и хлороформом. Органический слой сушат над MgSO4 и концентрируют. Концентрат очищают на колонке Biotage, используя в качестве элюента 5% МеОН в хлороформе. Остаток растворяют в простом эфире и обрабатывают, применяя 2 мл 1 М НСl в диэтиловом эфире. Твердое вещество собирают фильтрацией в атмосфере азота и сушат в вакууме в течение ночи, получая 0,364 г [2S-[1(R),2Rэтил-2,3-дигидрооксо-2-(4 пропил-1 Н-имидазол-2-ил)-1 Н-индол-1-этанамина дигидрохлорида дигидрата (соединение 15, т.пл. 132140 С). Пример В.6. Суспензию промежуточного соединения (13) (0,0102 моль) в н-бутаноле (200 мл) обрабатывают гидразидом бутановой кислоты (0,0254 моль), перемешивают в течение 10 мин, а затем кипятят с обратным холодильником в течение 10 дней. Реакционную смесь охлаждают, концентрируют в вакууме и распределяют между DCM и дистиллированной водой. Концентрированную органическую фазу подвергают препаративной, колоночной хроматографии с обращенной фазой, получая 1-ацетил-2,3-дигидро-2-(5 пропил-1 Н-1,2,4-триазол-3-ил)-1H-индол (соединение 91). Пример В.7.a) Раствор соединения 91 (0,42 г) в этаноле (25 мл) обрабатывают водным раствором NaOH (3 М, 25 мл) и реакционную смесь кипятят с обратным холодильником в течение 24 ч. Реакционную смесь охлаждают, разбавляют этилацетатом и обрабатывают холодной дистиллированной водой. Слои разделяют,водную фракцию экстрагируют 5 раз этилацетатом и объединенные органические фракции сушат, концентрируют и очищают с помощью препаративной колоночной хроматографии, получая 2,3-дигидро-2(5-пропил-1 Н-1,2,4-триазол-3-ил)-1H-индол.b) Раствор 2,3-дигидро-2-(5-пропил-1 Н-1,2,4-триазол-3-ил)-1H-индола (0,00017 моль) в DCM (5 мл) обрабатывают N-этил-N-(1-метил)-2-пропанамином (0,00072 моль), а затем сложным метиловым эфиром(2-фтор-2-оксоэтил)-9 Н-флуорен-9-ил-карбаминовой кислоты (0,00070 моль). Реакционную смесь перемешивают при комнатной температуре в течение 15 ч. Полученную смесь разбавляют, применяя DCM,дважды обрабатывают насыщенным NаНСО 3, сушат над Na2SO4 и концентрируют. Остаток подвергают препаративной, колоночной хроматографии с обращенной фазой, получая 0,02 г желаемого моноаддукта и 0,02 г бис-аддукта, который полностью превращают в желаемый моноаддукт обработкой элюентом для препаративной хроматографии (0,1% трифторуксусной кислоты в воде/ацетонитриле). Данные соединения объединяют, получая 0,03 г Н-флуорен-9-илметил[2-[2,3-дигидро-2-(5-пропил-1H-1,2,4-триазол-3 ил)-1 Н-индол-1-ил]-2-оксоэтил]карбамата.c) Раствор Н-флуорен-9-илметил[2-[2,3-дигидро-2-(5-пропил-1H-1,2,4-триазол-3-ил)-1H-индол-1 ил]-2-оксоэтил]карбамата (0,00006 моль) в DCM (10 мл) обрабатывают пиперидином (0,010 моль) и перемешивают при комнатной температуре в течение часа. Полученную реакционную смесь концентриру- 13006443 ют в вакууме и подвергают препаративной колоночной хроматографии с обращенной фазой, получая 0,02 г 2,3-дигидрооксо-2-(5-проипл-1 Н-1,2,4-триазол-3-ил)-1H-индол-1-этанамина трифторацетата(1:1) (соединение 92). Пример В.8. Смесь промежуточного соединения (14) (0,00898 моль) и бутаноилхлорида (0,0094 моль) в пиридине (140 мл) перемешивают при комнатной температуре в течение 40 ч, а затем кипятят с обратным холодильником. Через 21 ч реакционную смесь охлаждают и концентрируют в вакууме. Остаток экстрагируют между DCM и насыщенным водным NаНСО 3, а органическую фракцию сушат над Na2SO4, фильтруют и концентрируют. Остаток подвергают колоночной флэш-хроматографии на силикагеле (элюент: от 100% СН 2 Сl2 до 95/5 СН 2 Сl2/простого эфира), получая 1-ацетил-2,3-дигидро-2-(5-пропил-1,2,4 оксадиазол-3-ил)-1H-индол (соединение 89, т.пл. 93-94 С). Пример В.9. Раствор соединения 89 (0,0035 моль) в этаноле (60 мл) обрабатывают, применяя 3 М NaOH (60 мл),и реакционную смесь нагревают до 55-60 С в течение 5,5 ч. Полученную смесь быстро охлаждают на ледяной бане, разбавляют, применяя DCM, и обрабатывают холодной дистиллированной водой. Слои разделяют и водную фракцию экстрагируют трижды, применяя DCM. Органические фракции объединяют, один раз промывают 1 М NaOH, сушат над Na2SO4 и концентрируют в вакууме. Остаток очищают препаративной колоночной хроматографией, получая 0,45 г 2,3-дигидро-2-(5-пропил-1,2,4-оксадиазол-3 ил)-1H-индола.b) Раствор 2,3-дигидро-2-(5-пропил-1,2,4-оксадиазол-3-ил)-1H-индола (0,0011 моль) в DCM (10 мл) обрабатывают N-метил-N-1-метилэтил)-2-пропанамином (0,40 мл), а затем сложным метиловым эфиром (2-фтор-2-оксоэтил)-9H-флуорен-9-ил-карбаминовой кислоты (0,67 г). Реакционную смесь перемешивают при комнатной температуре в течение 40 ч и обрабатывают другой порцией N-метил-N-1 метилэтил)-2-пропанамина, а затем сложным метиловым эфиром (2-фтор-2-оксоэтил)-9H-флуорен-9-ил карбаминовой кислоты и перемешивают при комнатной температуре в течение двух дней. Реакционную смесь разбавляют, применяя DCM, дважды обрабатывают насыщенным NНСО 3, сушат над Na2SO4 и концентрируют. Остаток подвергают препаративной колоночной хроматографии с обращенной фазой,получая 0,35 г 9H-флуорен-9-илметил[2-[2,3-дигидро-2-(5-пропил-1,2,4-оксадиазол-3-ил]-2-оксоэтил] карбамата. с) 9H-флуорен-9-илметил[2-[2,3-дигидро-2-(5-пропил-1,2,4-оксадиазол-3-ил)-1H-индол-1-ил]-2 оксоэтил]карбамат (0,35 г) растворяют в DCM (40 мл), обрабатывают пиперидином (0,50 мл) и перемешивают при комнатной температуре в течение 18 ч. Полученную реакционную смесь концентрируют в вакууме и подвергают препаративной колоночной хроматографии с обращенной фазой, получая 0,13 г 2,3-дигидро-(3-оксо-2-(5-пропил-1,2,4-оксадиазол-3-ил)-1H-индол-1-этанамина трифторацетат (1:1) (соединение 90, т.пл. 160-162 С). Пример В.10. 2,3-Дигидро-2-(4-пропил-1H-имидазол-2-ил)-1H-индол (0,0024 моль) и 1,3-изобензофурандион(0,0026 моль) нагревают до 100 С в 25-мл грушевидной колбе в атмосфере аргона в течение 2 ч. Смесь растворяют в метаноле и кипятят с обратным холодильником в течение 15 ч. Реакционную смесь концентрируют и поглощают в DCM, промывают водой и 3 н. NaOH. Основный водный экстракт подкисляют 6 н. НСl и экстрагируют, применяя DCM. Органический экстракт сушат над MgSO4 и концентрируют. Концентрат растирают с простым эфиром и собирают. Полученный продукт подвергают дальнейшей очистке вместе с кислым водным раствором при помощи препаративной жидкостной хроматографии,получая 0,23 г трифторацетат 2-2-(4-этил-1 Н-имидазол-2-ил)-2,3-дигидро-1H-индол-1-ил]карбонил] бензойной кислоты (1:1) (соединение 85, т.пл. 98-103 С). Пример В.11. 1-Изоцианато-2-нитробензол (0,002 моль) добавляют к раствору промежуточного соединения (15)(0,016 моль) в ТГФ (10 мл). Смесь перемешивают при комнатной температуре в атмосфере аргона в течение 5 ч. Смесь разбавляют гексанами, фильтруют и дают ей возможность высохнуть, получая 0,34 г 2(4-этил-1H-имидазол-2-ил)-2,3-дигидро-N-(2-нитрофенил)-1 Н-индол-1-карбоксамида (соединение 77,т.пл. 208-209 С). Пример В.12. К смеси соединения 77 (0,0006 моль), никеля Ренея (0,02 г; 50% взвесь в воде) и метанола (20 мл) добавляют гидразин и воду (0,003 моль). Полученную смесь кипятят с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры смесь осторожно фильтруют через целит и фильтрат концентрируют. Остаток растирают с простым эфиром и фильтруют. Остаток очищают препаративной жидкостной хроматографией, получая 0,24 г трифторацетата N-(2-аминофенил-2-(4-этил-1Hимидазол-2-ил)-2,3-дигидро-1H-индол-1-карбоксамида (1:2) (соединение 79, т.пл. 106-108 С). Пример В.13. Смесь соединения 16 (0,00697 моль) в ТГФ (70 мл) обрабатывают одной порцией гидрида натрия(0,007 моль) и перемешивают при температуре окружающей среды в течение 16 ч. В виде одной порции вводят иодометан (0,0071 моль). После перемешивания при температуре окружающей среды в течение- 14006443 24 ч добавляют дополнительное количество гидрида натрия (0,007 моль) в виде одной порции в атмосфере аргона. Колбу вновь закупоривают после прекращения бурного выделения газа и смесь перемешивают в течение 16 ч. Полученную реакционную смесь охлаждают на ледяной бане, выливают в DCM и обрабатывают холодной водой. Слои разделяют и водный слой трижды экстрагируют, применяя DCM. Объединенные органические фракции промывают осажденным NaHCO3, сушат над Na2SO4 и концентрируют. Остаток подвергают колоночной флэш-хроматографии на силикагеле (отношение DCM к простому эфиру составляет до 9:1 простого эфира/ТГФ). Соответствующие фракции объединяют. Остаток помещают в простой эфир и помещают в морозильную камеру. Происходит кристаллизация с получением 0,55 г (29,3%) 1-ацетил-2-(4-этил-1-метил-1 Н-имидазол-2-ил)-2,3-дигидро-1 Н-индола (соединение 132,т.пл. 105-106 С). Объединяют вторые фракции. Остаток помещают в простой эфир и помещают в морозильную камеру. Мгновенно происходит кристаллизация с получением 0,38 г 1-ацетил-2-(4-этил-1 метил-1 Н-имидазол-2-ил)-2,3-дигидро-1H-индола (соединение 133, т.пл. 135-137 С). Пример В.14. Соединение 80 (0,001 моль) суспендируют в 1 н. NaOH (12 мл). Смесь энергично перемешивают и нагревают до 88 С в атмосфере азота в течение часа. После перемешивания при комнатной температуре в течение 3 ч смесь охлаждают до 0 С и медленно нейтрализуют, применяя 1 М НСl с целью осаждения некоторого количество твердого вещества. Полученное вещество фильтруют и дважды промывают ледяной водой. Водную фазу дважды экстрагируют, сушат, фильтруют, концентрируют и вновь сушат, получая 0,140 г 1,1-диметил 2-(4-карбокси-1H-имидазол-2-ил)-2,3-дигидро-1H-индол-1-карбоксилата (соединение 117). Пример В.15. 1-Гидроксибензотриазола гидрат (0,00036 моль), глицина метилэфира, гидрохлорид (0,00047 моль),4-метилморфин (0,00055 моль) и N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина моногидрохлорид (0,00047 моль) добавляют к соединению 117 (0,00036 моль), растворенному в DCM (30 мл) при 0 С. Смеси дают возможность нагреться до комнатной температуры в атмосфере азота и перемешивают в течение ночи. Смесь экстрагируют водой, насыщенным NaHCO3, 2 н. лимонной кислотой, затем насыщенным NaHCO3, сушат, фильтруют и концентрируют, получая 0,100 г (69%) 1,1-диметилэтил-2,3 дигидро-2-[4-2-метокси-2-оксоэтил)амино]карбонил]-1 Н-имидазол-2-ил]-1 Н-индол-1-карбоксилата (соединение 118). Пример В.16. Соединение 61 (0,00028 моль) обрабатывают, применяя 3 н. NaOH (3 мл) и перемешивая в течение 20 мин при комнатной температуре. Затем раствор обрабатывают, применяя 3 мл 3 н. НСl, и экстрагируют хлороформом. Продукт остается в водном слое. Водный слой очищают препаративной, жидкостной хроматографией, получая 0,12 г трифторацетата моногидрата 2-[1-(аминоацетил)-2,3-дигидро-1H-индол 2-ил]-1H-бензимидазол-5-карбоновой кислоты (1:2) (соединение 62, т.пл. 208-211 С). Пример В.17. Соединение 102 (0,00238 моль) растворяют в 40 мл метанола и объединяют с 1 н. КОН (50 мл). Реакционную смесь нагревают до 40 С в атмосфере аргона в течение ночи. Температуру повышают до 5560 С с целью дополнительного нагревания в течение ночи. Затем полученную смесь охлаждают до комнатной температуры, фильтруют и при 0 С медленно нейтрализуют, применяя 1 н. НСl. Образец экстрагируют 5 раз, применяя DCM, объединяют и сушат над Na2SO4. Полученный органический раствор фильтруют и используют для дальнейшего синтеза без дополнительной очистки, получая 1,1 диметилэтил 2-(4-карбокси-5-пропил-1 Н-имидазол-2-ил)-2,3-дигидро-1 Н-индол-1-карбоксилат (соединение 105). Пример В.18. 1-Гидроксибензотриазола гидрат (0,00318 моль) добавляют к раствору соединения 105 (0,00159 моль) в DCM (160 мл) при комнатной температуре. Добавляют неразбавленный N,N'метантетраилбисциклогексанамин (0,00206 моль) при комнатной температуре. Через 60 мин барботируют газ NH3 в течение 5 мин, вызывая осаждение твердого вещества. Смеси дают возможность отстояться в течение выходных дней. Полученную смесь фильтруют и фильтрат экстрагируют насыщеннымNaHCO3. Органические фазы сушат над МgSO4, фильтруют и концентрируют. Остаток очищают жидкостной хроматографией, получая 0,21 г 1,1-диметилэтил 2-[4-(аминокарбонил)-5-пропил-1H-имидазол-2 ил]-2,3-дигидро-1H-индол-1-карбоксилата (соединение 106). Пример В.19. 1-Гидроксибензотриазолгидрат (0,00158 моль) добавляют к раствору соединения 105 (0,00079 моль) в DCM (80 мл). Добавляют глицина метилэфиргидрохлорид (0,00103 моль), N'-(этилкарбонимидоил)N,N-диметил-1,3 пропандиамина моногидрохлорид (0,00103 моль) и 4-метилморфолин (0,00103 моль). Добавляют ТГФ (25 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Смесь экстрагируют водой. Органическую фазу промывают насыщенным NaHCO3, 2 н. лимонной кислотой, насыщенным NaHCO3, сушат над MgSO4, фильтруют и концентрируют, получая 0,20 г 1,1 диметилэтил-2,3-дигидро-2-[4-2-метокси-2-оксоэтил)амино]карбонил]-5-пропил-1H-имидазол-2-ил]1 Н-индол-1-карбоксилата (соединение 109).- 15006443 Пример В.20. Соединение 81 (0,0005 моль) суспендируют в 1 н. NaOH (6 мл) в атмосфере аргона. Смесь сразу же нагревают до 80 С в течение 60 мин. При комнатной температуре добавляют хлороформ (6 мл), а затем сложный эфир (2-фтор-2-оксоэтил)-1,1-диметилэтил карбамовой кислоты (0,001 моль). Смесь перемешивают в течение ночи. Слои разделяют. Водную фазу охлаждают, подкисляют и дважды экстрагируют хлороформом. Последние органические фазы объединяют, сушат, фильтруют и концентрируют. Образец очищают препаративной ВЭЖХ, получая 0,040 г 2-[1-(1,1-диметилэтокси)карбонил]амино]ацетил]-2,3 дигидро-1 Н-индол-2-ил]-1H-имидазол-4-карбоновой кислоты (соединение 138). Пример В.21. К соединению 145 (0,00097 моль), растворенному в этаноле (5 мл), добавляют несколько капель 21% NaOEt в этаноле. Смесь перемешивают при комнатной температуре в атмосфере аргона. Через 30 мин дополнительно добавляют 2 капли 21% NaOEt в этаноле. Через 16 ч вновь дополнительно добавляют 2 капли 21% NaOEt в этаноле. Через 30 мин смесь концентрируют и разделяют между водой и DCM. Водный слой промывают дополнительным количеством DCM. Объединенные органические вещества промывают водой, сушат и концентрируют, получая 0,193 г (66%) [2S-[1(R),2R-2,3-дигидрометил-оксо-2-(4-пропил-1 Н-имидазол-2-ил)-1 Н-индол-1-этанола (соединение 146). Соединение 148 получают аналогичным способом, используя в качестве исходного соединения соединение 147. Пример В.22. К суспензии соединения 58 (0,0019 моль) в ацетонитриле (15 мл) добавляют уксусную кислоту, ангидрид (0,074 моль). Перемешивают при комнатной температуре в атмосфере аргона в течение 4 ч. Дополнительно добавляют 1,0 мл уксусной кислоты, ангидрид и реакционную смесь перемешивают в течение ночи. После перемешивания еще в течение 6 ч реакцию завершают. Смесь концентрируют, а остаток разделяют между насыщенным NаНСО 3 и хлороформом. Органический слой сушат и концентрируют. Остаток очищают колоночной хроматографией. Нужные фракции объединяют, растирают с простым эфиром и собирают, получая 0,37 г 1-1-[(4-хлорфенил)ацетил]-4-пиперидинил]метил]-1,3-дигидро-2Hбензимидазол-2-она (соединение 149). Пример В.23. Раствор соединения 149 (0,0012 моль) и ТГФ (200 мл) помещают в фотохимический реактор и облучают УФ светом в течение 14 ч. Затем смеси дают возможность отстояться при комнатной температуре в атмосфере азота в течение 2 дней. Смесь концентрируют. Концентрат очищают на колонке Biotage,элюируя ТГФ в DCM и получая 0,077 г 1-[2-(1-ацетил-2,3-дигидро-1H-индол-2-ил)-5-пропил-1 Нимидазол-4-ил]этанона (соединение 150). Пример В.24. Соединение 13 (0,00106 моль), растворенное в 10 мл ТГФ, обрабатывают при комнатной температуре, применяя ВН 3.ТГФ (19 мл), представляющий собой раствор в ТГФ. Затем раствор помещают на масляную баню и нагревают до 60 С в течение ночи. После охлаждения до 0 С раствор осторожно обрабатывают 15 мл 3 н. НСl. Реакционную смесь потом нагревают до комнатной температуры и перемешивают в течение 4 ч. Затем смесь вновь охлаждают до 0 С и превращают в основную, применяя 12 мл 3 н.NaOH, после чего прекращают указанное превращение, применяя твердый Nа 2 СО 3. Слои разделяют, а водный слой вновь промывают хлороформом. Органические вещества объединяют, небольшое количество воды отделяют, а органические вещества сушат над Na2SO4. Смесь фильтруют, а фильтрат концентрируют при пониженном давлении. Остаток подвергают препаративной жидкостной хроматографии, получая 0,33 г трифторацетата [2S-[1(R),2R-2-(4-этил-1H-имидазол-2-ил)-2,3-дигидрометил-1H-индол 1-этанамина (1:1) (соединение 127). Пример В.25. 3-Амино-4-(4-гидроксифенил)-1-[3-(4-фенил-1 Н-имидазол-2-ил)-3,4-дигидро-1 Низохинолин-2-ил]бутан-1-он (соединение 155).- 16006443 ТФУ (4 мл) охлаждают до температуры, составляющей около 0 С, а затем добавляют продукт, полученный в примере А.15 (1,10 г, 1,85 ммоль). Реакционную смесь осаждают в течение приблизительно 0,5 ч. Затем в потоке N2 удаляют избыточную ТФУ, получая коричневое масло. Масло очищают при помощи препаративной ВЭЖХ, получая указанное в заголовке соединение в виде белого твердого вещества. Вычисленная мол. масса (МН+): 439. Пример В.26. 2-Амино-3-(4-гидроксибензил)-1-[3-(5-метил-4-фенилимидазол-2-ил)-3,4-дигидро-1 Низохинолин-2-ил]пропан-1-он (соединение 153). К раствору ТФУ (4 мл), охлажденному до температуры около 0 С, добавляют соединение, полученное в примере А.18 (0,24 г, 0,4 ммоль), и реакционную смесь перемешивают около 20 мин. Затем реакционную смесь удаляют с ледяной бани и дают ей возможность нагреться до комнатной температуры. Избыточную ТФУ удаляют в потоке N2, получая сырой продукт. Полученный продукт очищают препаративной ВЭЖХ, получая указанное в заголовке соединение в виде белого твердого вещества. Вычисленная мол. масса (МН+): 453. В табл. F-1 перечислены соединения, полученные в соответствии с одним из вышеописанных примеров. В таблице использованы следующие сокращения: С 2 НF3O2 означает трифторацетат, 2 С 2 Н 2O4 означает этандиоат, а С 10 Н 8 О 3S означает 2-нафталинсульфонат. В табл. F-1 указана структура соединений,номер примера, в соответствии с которым получают данные соединения, солевая форма, стереохимическое обозначение и температура плавления (если таковую определяли). Таблица F-1C.1. Ингибирование трипептидил пептидазы II (ТПП II). Ингибирование ТПП II измеряют, применяя методику, описанную С. Rose et al. in Nature, 380, 403409 (1996). Активность ТПП II определяют, применяя 15 мкМ AAF-AMC в качестве субстрата в 50 мМ калиево-фосфатного буфера, имеющего рН 7,5, с 1 мМ dTT и 1 мМ EGTA. К соединениям добавляют DMSO до конечной концентрации 1%. Флуоресценцию измеряют при 405 нм. Эффективность соединений формулы (I) выражена в виде величины IC50, т.е. концентрации, необходимой для 50% ингибирования. Соединения 6, 10, 13, 15, 19, 22, 24, 28, 30, 44, 47, 48, 54, 55, 57, 61, 62, 66, 68, 70, 73, 76, 82, 84, 88,90, 92, 95, 101, 104, 108, 111, 114, 116, 120, 122, 124, 126, 129, 131, 135, 142 и 144 имеют величину IC50,равную или ниже 110-5 М. С.2. Исследование связывания -опиоидного рецептора мозга крыс. Самцов крыс Wistar (150-250 г, VAF, Charles River, Kingston, NY) умерщвляют цервикальной дислокацией, удаляют их мозг и сразу же помещают в ледяной трис-HCl-буфер (50 мМ, pH 7,4). Передний мозг отделяют от остальной части мозга венечным поперечным разрезом, начиная с задней части в области холмика и продолжая вентрально через место соединения середины мозга и моста. После отсечения передний мозг гомогенизируют в трис-буфере в гомогенизаторе из стекла Teflon. Гомогенат разбавляют до концентрации 1 г ткани переднего мозга на 100 мл трис-буфера и центрифугируют при 39000 Хg в течение 10 мин. Осадок вновь суспендируют в таком же объеме трис-буфера при помощи нескольких коротких импульсов гомогенизатора Polytron. Полученный препарат в виде микрочастиц используют для исследований -опиоидного связывания. После инкубирования с -селективным пептидным лигандом[3H]DPDPE при 25 С содержимое пробирки фильтруют через листы ватманского фильтра GF/B в харвестере клеток марки Brandel. Пробирки и фильтры трижды промывают 4 мл 10 мМ HEPES (рН 7,4) и,применяя сцинтилляционную жидкость Formula 989 (New England Nuclear, Boston, MA), при помощи сцинтилляционного счетчика определяют радиоактивность, связанную с кругами на фильтре. Полученные данные используют для подсчета % ингибирования по сравнению с контрольным связыванием (при определении всего лишь одной концентрации исследуемого соединения) либо величины% Ингибирования подсчитывают следующим образом:- 28006443 Величину Ki подсчитывают, применяя программу анализа данных LIGAND (Munson, P.J. and Rodbard, D., Anal. Biochem. 107: 220-239, 1980). С.3. Исследование связывания -опиоидного рецептора мозга крыс. Самцов крыс Wistar (150-250 г, VAF, Charles River, Kingston, NY) умерщвляют цервикальной дислокацией, удаляют их мозг и сразу же помещают в ледяной трис-НСl-буфер (50 мМ, рН 7,4). Передний мозг отделяют от остальной части мозга венечным поперечным разрезом, начиная с задней части в области холмика и продолжая вентрально через место соединения середины мозга и моста. После отсечения передний мозг гомогенизируют в трис-буфере в гомогенизаторе из стекла Teflon. Гомогенат разбавляют до концентрации 1 г ткани переднего мозга на 100 мл трис-буфера и центрифугируют при 39000 Х g в течение 10 мин. Осадок вновь суспендируют в таком же объеме трис-буфера при помощи нескольких коротких импульсов гомогенизатора Polytron. Полученный препарат в виде микрочастиц используют для исследований -опиоидного связывания. После инкубирования с -селективным пептидным лигандом [3H]DAMGO при 25 С содержимое пробирки фильтруют через листы ватманского фильтра GF/B в харвестере клеток марки Brandel. Пробирки и фильтры трижды промывают 4 мл 10 мМ HEPES (рН 7,4) и, применяя сцинтилляционную жидкость. Formula 989 (New England Nuclear, Boston, MA), при помощи сцинтилляционного счетчика определяют радиоактивность, связанную с кругами на фильтре. Полученные данные используют для подсчета % ингибирования по сравнению с контрольным связыванием (при определении всего лишь одной концентрации исследуемого соединения) либо величины% Ингибирования подсчитывают следующим образом: Величину Ki подсчитывают, применяя программу анализа данных LIGAND (Munson, P.J. and Rodbard, D., Anal. Biochem. 107: 220-239, 1980). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) его стереохимически изомерная форма или его фармацевтически приемлемая аддитивная соль,где n равно целому числу 0 или 1; Х представляет -(CR4R5)m-, где m равно целому числу 1 или 2; каждый из R4 и R5 независимо представляет водород или С 1-4 алкил;R1 представляет C1-6 алкилкарбонил, необязательно замещенный гидрокси; C1-6 алкилоксикарбонил; аминоС 1-6 алкилкарбонил, где C1-6 алкил необязательно замещен С 3-6 циклоалкилом; моно- и ди(С 1-4 алкил) аминоС 1-6 алкилкарбонил; аминокарбонил, замещенный арилом; С 1-6 алкилкарбонилоксиС 1-6 алкилкарбонил; C1-6 алкилоксикарбониламиноС 1-6 алкилкарбонил, где аминогруппа необязательно замещена С 1-4 алкилом; аминокислотный остаток, связанный через карбонильную группу; C1-6 алкил, замещенный амино; или арилкарбонил; где m' равно целому числу от 1 до 2;R6 представляет водород или С 1-4 алкил;R7 независимо друг от друга представляет водород; галоген; амино; гидрокси; трифторметил;C1-6 алкил; С 1-4 алкил, замещенный гидрокси, гидроксикарбонилом, С 1-4 алкилоксикарбонилом, аминокарбонилом, моно- или ди(С 1-4 алкил)аминокарбонилом, амино, или моно- или ди(С 1-4 алкил)амино; фенил; аминокарбонил; гидроксикарбонил; С 1-4 алкилоксикарбонил; С 1-4 алкилкарбонил; или C1-4 алкилоксикарбонилС 1-4 алкиламинокарбонил; или R2 представляет бензимидазол или бензимидазол, замещенный одним или двумя заместителями, каждый из которых независимо выбран из галогена, трифторметила, С 1-4 алкила, гидрокси, гидроксикарбонила или С 1-4 алкилоксикарбонила;R3 представляет двухвалентный радикал формулы где (b-1) необязательно замещен галогеном или C1-6 алкилоксилом; арил представляет фенил или фенил, замещенный амино, нитро или гидроксикарбонилом. 2. Соединение по п.1, в котором n равно 0, а R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси. 3. Соединение по п.1, в котором n равно 0, R3 представляет радикал формулы (b-1), необязательно замещенный галогеном или метокси, а Х представляет -СН 2- или -СН 2 СН 2-. 4. Соединение по любому из предыдущих пунктов, в котором R2 представляет радикал формулы (а 2), (а-4), (а-6) или (а-7). 5. Соединение по любому из предыдущих пунктов, в котором R1 представляет C1-6 алкилкарбонил,аминоС 1-6 алкилкарбонил или аминокислотный остаток. 6. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-5. 7. Способ получения фармацевтической композиции по п.6, в котором терапевтически активное количество соединения по любому из пп.1-5 тщательно смешивают с фармацевтически приемлемым носителем. 8. Применение соединения по любому из пп.1-5 в качестве лекарственного препарата. 9. Способ получения соединения формулы (I), в котором а) промежуточное соединение формулы(II) подвергают взаимодействию с промежуточным соединением формулы (III) в реакционно-инертном растворителе и, необязательно, в присутствии подходящего основания, получая соединения формулы (Iа), указанные как соединения формулы (I), где R1a представляет все заместители R1, кроме С 1-4 алкила,замещенного амином; илиb) промежуточное соединение формулы (II) подвергают взаимодействию с промежуточным соединения формулы (IV), получая соединение формулы (I-а) где в вышеприведенных реакционных схемах радикалы R1, R2, R3 и целое число n имеют значения, определенные в п.1; с) или соединения формулы (I) превращают одно в другое в соответствии с известными реакциями превращения; или, при желании, соединение формулы (I) превращают в кислотно-аддитивную соль, или,наоборот, кислотно-аддитивную соль соединения формулы (I) превращают в свободное основание взаимодействием со щелочью; и, при желании, получают их стереохимически изомерные формы.
МПК / Метки
МПК: A61K 31/404, A61P 1/00, C07D 403/04
Метки: пептидазы, трипептидил, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-6443-ingibitory-tripeptidil-peptidazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы трипептидил пептидазы</a>
Замещенные амидазо [1, 2а] азины как селективные сох-2 ингибиторы

Номер патента: 3399
Опубликовано: 24.04.2003
Авторы: Фернандес Серрат Ана Мария, Фернандес Гарсия Андрес, Монсеррат Видаль Карлос, Фарреронс Гальеми Карлес, Хименес Гвасч Ферран, Микель Боно Игнасио-Хосе, Лагунас Арналь Кармен
МПК: C07D 471/04, A61K 31/53, A61P 35/00...
Метки: сох-2, азины, ингибиторы, замещенные, амидазо, селективные
Формула / Реферат:
1. Замещенный имидазо[1,2a]азин формулы (I) или его фармацевтически приемлемый сольват и его соль присоединения кислоты, где A и B независимо выбирают из N и CH, при условии, что если A представляет N, тогда B также представляет N; R1 выбирают из группы, состоящей из CH3 и NH2; R2 и R3 выбирают из группы, состоящей из H, CH3, Cl, Br, COCH3 и OCH3; и R4, R5 и R6, каждый независимо, выбирают из группы, состоящей из H, F, Cl, Br, (C1-C3)алкила,...
Диазепино-индоловые ингибиторы фосфодиэстераз iv

Номер патента: 1593
Опубликовано: 25.06.2001
Авторы: Паскаль Ив, Даль Свен Ж., Кальве Ален, Пен Адриан, Жакобелли Анри
МПК: C07D 487/06, A61K 31/551, A61P 11/06...
Метки: ингибиторы, фосфодиэстераз, диазепино-индоловые
Формула / Реферат:
1. Диазепино-индолы формулы (I) в которой А представляет собой фенил или нафтил или азотсодержащий гетероарил, причем каждый возможно замещен 1-3 группами, независимо выбранными из галогена, С1-4алкила, галоалкила, С1-4алкокси, циклоалкилокси, амино, С1-4алкилкарбониламино или С1-4алкилоксикарбониламино; В представляет собой: 1ш) -OR1, причем R1 является -Н или R4, 2ш) -NR2R3, причем R2 является -С(NН)NН2 и R3 является -Н, 3ш) -NR2R3, причем...
Ингибиторы тирозинкиназы

Номер патента: 5812
Опубликовано: 30.06.2005
Авторы: Хангейт Рэндалл В., Хартман Джордж Д., Ким Юнтае, Хоффман Вилльям Ф., Аррингтон Кеннет Л., Фрэли Марк Э., Билодо Марк Т.
МПК: A61K 31/495, C07D 401/14
Метки: тирозинкиназы, ингибиторы
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль или стереоизомер, где Z представляет группу a равно 0 или 1; b равно 0 или 1; m равно 0, 1 или 2; s равно 1 и t равно 1; R1 и R5 представляют H; R2 и R3 представляют H; R4 выбран из 1) OC1-C6алкилен-NR7R8, 2) (C=O)aC0-C6алкилен-Q, где Q представляет H, OH, CO2H или OC1-C6 алкил, 3) OC0-C6алкиленгетероциклила, необязательно, замещенного заместителями от одного до трех, выбранными...
Ингибиторы тромбина

Номер патента: 2767
Опубликовано: 29.08.2002
Авторы: Хорнбергер Вильфрид, Бём Ханс-Йоахим, Зайтц Вернер, Хёффкен Ханс Вольфганг, Цирке Томас, Козер Штефан, Пфайффер Томас, Мак Хельмут
МПК: A61P 7/02, A61K 38/55, C07K 5/06...
Метки: тромбина, ингибиторы
Формула / Реферат:
1. Ингибиторы тромбина формулы (I) где R1 - алкил с 1-20 атомами углерода, фенил- или нафтилалкил с 1-10 атомами углерода в алкильной части, группа R2OOC-(CH2), где R2 означает водород или алкил с 1-10 атомами углерода, А - остаток a-аминокислоты формулы (II) где R3 - водород, алкил с 1-8 атомами углерода, циклоалкил с 3-7 атомами углерода, фенил и нафтил, незамещенные или замещенные алкилом или алкоксилом, каждый с 1-4 атомами углерода, или...
Нестероидные ингибиторы воспалений

Номер патента: 5926
Опубликовано: 25.08.2005
Авторы: Леманн Манфред, Шоттелиус Арндт, Хеннекес Хартвиг, Шэкке Хайке, Ярох Штефан, Скубалла Вернер, Шмез Норберт, Кроликивич Конрад, Ревинкель Хартмут, Бухманн Бернд, Дрёшер Петер
МПК: C07D 265/02, A61P 29/00, A61K 31/536...
Метки: ингибиторы, нестероидные, воспалений
Формула / Реферат:
1. Применение соединений общей формулы I в которой R1 и R2 имеют идентичные или разные значения и обозначают атом водорода, C1-C5алкильную группу или вместе с C-атомом цепи образуют 3-7-членное кольцо, R3 обозначает прямоцепочечную или разветвленную C1-C5алкильную группу либо прямоцепочечную или разветвленную, частично или полностью фторированную C1-C5алкильную группу, A обозначает группу (прерывистая линия обозначает место присоединения), в...