Противовоспалительные и иммуносупрессорные соединения, ингибирующие клеточную адгезию
Номер патента: 5563
Опубликовано: 28.04.2005
Авторы: Гунавардана Индрани, Фримен Дженнифер С., Стаеджер Майкл А., Бойд Стивен А., Зу Гуи-Донг, Пей Зонгхуа, Ксин Зили, Вон Гелдерн Том, Уинн Мартин, Линк Джеймс, Линч Джон К., Дзае Хван-Соо, Ванг Шелдон, Лиу Ганг


























Формула / Реферат
1. Соединение формулы I

где R1 выбран из
a) водорода,
b) галогена,
c) C1-6 алкила,
d) галоген-C1-6 алкила,
e) циано,
f) нитро и
g) карбоксальдегида;
R2 выбран из
a) водорода,
b) галогена,
c) C1-6 алкила,
d) галоген-C1-6 алкила,
e) нитро и
f) карбоксальдегида;
R3 выбран из
a) водорода и
b) C1-6 алкила;
R4 выбран из
a) водорода,
b) гидрокси,
c) C1-6 алкила и
d) C1-6 алкокси,
R5 выбран из
a) водорода и
b) C1-6 алкила,
при условии, что один из R1 или R3 является цис-циннамидом или транс-циннамидом, определенным как

где каждый из R8 и R9 представляет водород и R10 и R11 независимо выбраны из
a) водорода
b) C1-6 алкила,
a) C3-12 циклоалкила,
d) C1-10 алкоксикарбонил-C1-10 алкила,
e) гидрокси-C1-10 алкила,
f) арила, который представляет моно- или бициклическую карбоциклическую кольцевую систему, имеющую один или два ароматических кольца, которые могут быть также конденсированы с циклогексановым, циклогексеновым, циклопентановым или циклопентеновым кольцом и необязательно замещены заместителями, такими как C1-10 алкил, галоген, гидрокси или C1-10 алкокси;
g) гетероциклила, который представляет 4-, 5-, 6- или 7-членное кольцо, содержащее один, два или три гетероатома, независимо выбранных из группы, состоящей из азота, кислорода и серы, которое может быть конденсировано с одним или двумя кольцами, независимо выбранными из арильных колец, определенных ниже, циклогексановым кольцом, циклогексеновым кольцом, циклопентановым кольцом, циклопентеновым кольцом или другим моноциклическим гетероциклическим кольцом;
h) гетероциклил-C1-10 алкила, где гетероциклил определен выше;
i) гетероциклиламино, где гетероциклил определен выше;
j) гетероциклила, определенного выше в g), имеющего заместители, независимо выбранные из C1-10 алкила, галогена, гидрокси, C1-10 алкокси, карбокси, карбокси-C1-10 алкила и C1-10 алкоксикарбонила;
k) гетероциклилалкила, определенного выше в h), имеющего заместители, независимо выбранные из C1-10 алкила, галогена, гидрокси, C1-10 алкокси, карбокси, карбокси-C1-10 алкила и C1-10 алкоксикарбонила;
либо где NR10R11 представляют гетероциклил или замещенный гетероциклил, где гетероциклил определен в g), а заместители независимо выбраны из
1) C1-10 алкила,
2) C1-10 алкокси,
3) C1-10 алкокси-C1-10 алкила,
4) C3-12 циклоалкила,
5) арила, который представляет моно- или бициклическую карбоциклическую кольцевую систему, имеющую один или два ароматических кольца, которые могут быть также конденсированы с циклогексановым, циклогексеновым, циклопентановым или циклопентеновым кольцом,
6) гетероциклила, определенного выше,
7) гетероциклилкарбонила, где гетероциклил определен выше,
8) гетероциклил-C1-10 алкиламинокарбонила, где гетероциклил определен выше,
9) гидрокси,
10) гидрокси-C1-10 алкила,
11) гидрокси-C1-10 алкокси-C1-10 алкила,
12) карбокси,
13) карбокси-C1-10 алкила,
14) карбоксикарбонила,
15) карбоксальдегида,
16) C1-10 алкоксикарбонила,
17) арил-C1-10 алкоксикарбонила,
18) амино-C1-10 алкила,
19) амино-C1-10 алканоила,
20) карбоксамидо,
21) C1-10 алкоксикарбонил-C1-10 алкила,
22) карбоксамидо-C1-10 алкила,
23) циано,
24) тетразолила,
25) замещенного тетразолила,
26) C1-10 алканоила,
27) гидрокси-C1-10 алканоила,
28) C1-10 алканоилокси,
29) C1-10 алканоиламино,
30) C1-10 алканоилокси-C1-10 алкила,
31) C1-10 алканоиламино-C1-10 алкила,
32) сульфоната,
33) C1-10 алкилсульфонила,
34) C1-10 алкилсульфониламинокарбонила,
35) арилсульфониламинокарбонила, где арил определен выше, и
36) гетероциклилсульфониламинокарбонила, где гетероциклил определен выше,
и где Ar означает замещенную арильную или замещенную гетероарильную группу, имеющую один или два ароматических кольца, которые могут быть также конденсированы с циклогексановым, циклогексеновым, циклопентановым или циклопентеновым кольцом, где заместители независимо выбраны из
a) гетероциклила, определенного выше, имеющего заместители,независимо выбранные из C1-10 алкила, галогена, гидрокси, C1-10 алкокси, карбокси, карбокси-C1-10 алкила и C1-10 алкоксикарбонила,
b) гетероциклил-C1-10 алкила, определенного выше, имеющего заместители, независимо выбранные из карбокси, карбокси-C1-10 алкила и C1-10 алкоксикарбонила,
c) карбокси-C1-10 алкокси,
d) карбокси-C1-10 тиоалкокси,
e) карбокси-C3-12 циклоалкокси,
f) C1-10 тиоалкила,
g) карбокси-C1-10 алкиламино,
или его фармацевтически приемлемая соль или пролекарство.
2. Соединение по п.1, где R1 представляет цис-циннамид или транс-циннамид, а R3 представляет водород.
3. Соединение по п.1, где R3 представляет цис-циннамид или транс-циннамид, а R1 представляет водород.
4. Соединение по п.1, где R3 представляет цис-циннамид или транс-циннамид, а R1, R8 и R9 представляют водород.
5. Соединение по п.4, где R3 представляет цис-циннамид.
6. Соединение по п.4, где R3 представляет транс-циннамид.
7. Соединение по п.1, где R3 представляет цис-циннамид или транс-циннамид; каждый из R1, R2 и R4 независимо представляет водород или C1-6 алкил.
8. Соединение по п.4, где каждый из R10 и R11 независимо выбран из водорода, C1-6 алкила, C3-12 циклоалкила, C1-10 алкоксикарбонил-C1-10 алкила, гидрокси-C1-10 алкила и гетероциклил-C1-10 алкила.
9. Соединение по п.4, где NR10R11 представляет гетероциклил или замещенный гетероциклил.
10. Соединение по п.8, где Ar выбран из замещенного фенила, 1,3-бензимидазол-2-она, 1,4-бензодиоксана, 1,3-бензодиоксола, 1-бензопир-2-ен-4-она, индола, изатина, 1,3-хиназолин-4-она и хинолина.
11. Соединение по п.1, где Ar выбран из метоксифенила и изопропилфенила.
12. Соединение по п.1, где Ar представляет собой замещенный бензодиоксан.
13. Соединение по п.1, где R3 представляет транс-циннамид, а Ar выбран из 1,3-бензимидазол-2-она, 1,4-бензодиоксана, 1,3-бензодиоксола, 1-бензопир-2-ен-4-она, индола, изатина, фенила, 1,3-хиназолин-4-она и хинолина.
14. Соединение по п.1, где R10 и R11 независимо выбраны из водорода, C1-6 алкила, C3-12 циклоалкила, C1-10 алкоксикарбонил-C1-10 алкила, гидрокси-C1-10 алкила и гетероциклил-C1-10 алкила.
15. Соединение по п.1, где NR10R11 представляют гетероциклил или замещенный гетероциклил.
16. Соединение по п.1, выбранное из группы, состоящей из
(3-(1-(3-карбоксипиперидинил)фенил))[2,3-дихлор-4-(E-((1,2,5,6-тетрагидропиридин-1-ил)карбонил)этенил)фенил]сульфида;
[3-(3-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-карбоксипиперидин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксилпиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(1,2,3,6-тетрагидропиридин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксилпиперидин)фенил][2,3-дихлор-4-(E-[(4-морфолинил)карбонил]этенил)фенил]сульфида;
[3-(3-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-морфолинил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-гидроксипиперидин-1-ил)карбонил]этенил)фенил]сульфида;
(2-гликоксифенил)[2,3-дихлор-4-(E-[(4-морфолино)карбонил]этенил)фенил]сульфида;
[2-(4-бутирокси)фенил][2,3-дихлор-4-(E-[(4-морфолино)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-гидроксиэтилпиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-фуроилпиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(пирролидин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-(диэтиламинокарбонил)этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-этилпиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-(аминокарбонил)пиперидин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[4-(2-(этоксиэтил)пиперидин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-[(4-морфолино)карбонил]этенил)фенил]сульфида;
[3-(4-бутирокси)фенил][2,3-дихлор-4-(E-[(4-морфолино)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((4-гидроксипиперидин-1-ил)карбонил)этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((1,2,5,6-тетрагидропиридин-1-ил)карбонил)этенил)фенил]сульфида;
[2-((4-карбокси)бутилокси)фенил][2,3-дихлор-4-(E-((4-морфолино)карбонил)этенил)фенил]сульфида;
(2-гликоксифенил)[2,3-бис(трифторметил)-4-(E-((4-морфолино)карбонил)этенил)фенил]сульфида;
[2-(4-бутирокси)фенил][2,3-бис(трифторметил)-4-(E-((4-морфолино)карбонил)этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((бис-(2-этоксиэтил)амино)карбонил)этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((бис-(2-гидроксипропил)амино)карбонил)этенил)фенил]сульфид;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-(трифторметил)-4-(E-((пиперазин-1-ил)карбонил)этенил)фенил]сульфида;
[3-(4-бутирокси)фенил][2,3-бис(трифторметил)-4-(E-((4-морфолино)карбонил)этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-(дихлор)-4-(E-[(4-(2-гидроксиэтил)пиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((1,2,5,6-тетрагидропиридин-1-ил)карбонил)этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((4-(2-гидроксиэтил)пиперазин-1-ил)карбонил)этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-((4-(2-(гидроксиэтокси)этил)пиперазин-1-ил)карбонил)этенил)фенил]сульфида и
(3-(3-пропиокси)фенил)[2,3-дихлор-4-(E-((4-морфолино)карбонил)этенил)фенил]сульфида.
17. Соединение по п.1, выбранное из группы, состоящей из
(2-метоксифенил)[2,3-дихлор-4-(E-[((4-гидроксиламинокарбонил)пиперидин-1-ил)карбонил]этенил)фенил]сульфида;
(2-метоксифенил)[2,3-дихлор-4-(E-[(N-карбоксиметил-N-фениламино)карбонил]этенил)фенил]сульфида;
(2-метоксифенил)[3-хлор-6-гидрокси-4-(E-[(3-карбоксипиперидин-1-ил)карбонил]этенил)фенил]сульфида;
(2-метоксифенил)[2,3-дихлор-4-(E-[(4-((1-(2-фенил-1-карбоксиэтил)амино)карбонил)пиперидин-1-ил)карбонил]этенил)фенил]сульфида;
(2-метоксифенил)[2,3-дихлор-4-(E-[(4-((1-(2-гидрокси-1-карбоксиэтил)амино)карбонил)пиперидин-1-ил)карбонил]этенил)фенил]сульфида;
((3-(4-пирролидин-1-ил)пиперидин-1-ил)фенил)[2,3-дихлор-4-(E-((3-(2-пирролидинон-1-ил)пропиламино)карбонил)этенил)фенил]сульфида;
[3-(4-(спиро-2,2-диоксоланил)пиперидин-1-ил)фенил][2,3-дихлор-4-(E-((4-морфолинил)карбонил)этенил)фенил]сульфида;
((2-(2-карбокси)этенил)фенил)[2,3-дихлор-4-(E-[(4-морфолинил)карбонил]этенил)фенил]сульфида;
[2-(4-ацетилпиперазин-1-ил)фенил][2,3-дихлор-4-(E-[(4-карбоксипиперидин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-(диметиламиносульфамоил)пиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-(трифторметилсульфонил)пиперазин-1-ил)карбонил]этенил)фенил]сульфида;
(2-метоксифенил)[2,3-дихлор-4-(E-((((4-карбоксифенил)метил)амино)карбонил)этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(4-(метилсульфонил)пиперазин-1-ил)карбонил]этенил)фенил]сульфида;
[3-(4-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-[(S-оксотиоморфолин-1-ил)карбонил]этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-дихлор-4-(E-(3-(2-пирролидинон-1-ил)пропиламинокарбонил)этенил)фенил]сульфида;
[2-(3-карбоксипиперидин-1-ил)фенил][2,3-бис(трифторметил)-4-(E-(3-(2-пирролидинон-1-ил)пропиламинокарбонил)этенил)фенил]сульфида.
18. Фармацевтическая композиция, содержащая соединение по п.1 в фармацевтически приемлемом носителе.
19. Способ ингибирования воспаления, предусматривающий введение соединения по п.1 млекопитающему, нуждающемуся в таком лечении.
20. Способ ингибирования воспаления, предусматривающий введение композиции по п.18 млекопитающему, нуждающемуся в таком лечении.
21. Способ подавления иммунного ответа, предусматривающий введение соединения по п.1 млекопитающему, нуждающемуся в таком лечении.
22. Способ подавления иммунного ответа, предусматривающий введение композиции по п.18 млекопитающему, нуждающемуся в таком лечении.
23. Соединение формулы II

где R1 и R2 независимо выбраны из группы, состоящей из
i) водорода,
j) галогена,
k) C1-6 алкила,
l) галоген-C1-6 алкила,
m) циано,
n) нитро и
o) карбоксальдегида.
24. Способ получения соединения формулы II

предусматривающий
a) взаимодействие соединения формулы II'

с гидроксидом лития и
b) отщепление полученного метилового эфира, где R1 и R2 определены в п.23.
25. Замещенный диарилсульфидциннамид, содержащий два ароматических кольца, связывающий сульфидный атом, циннамидную функциональную группу, находящуюся в пара-положении по отношению к указанному связывающему сульфидному атому, и по крайней мере один дополнительный заместитель в одном или нескольких ароматических кольцах, где указанный замещенный диарилсульфидциннамамид преимущественно связывает LFA-1 по сравнению с ICAM-1 или ICAM-3.
26. Соединение по п.25, где указанными двумя ароматическими кольцами являются фенильные кольца и где одно из указанных фенильных колец замещено 3-карбоксипиперидиновой функциональной группой, находящейся в мета-положении по отношению к связывающему сульфидному атому, а другое фенильное кольцо замещено двумя трифторметильными группами и N-морфолинозамещенным транс-циннамидом, находящимся в орто-, мета- и пара-положении соответственно по отношению к указанному связывающему сульфидному атому.
27. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель ш соединение по п.26.
28. Способ лечения клинического состояния у млекопитающего, для которого показан ингибитор LFA-1, предусматривающий введение указанному млекопитающему терапевтически эффективного количества диарилсульфидного соединения по п.26 или 28.
29. Применение диарилсульфидциннамида, содержащего два ароматических кольца, связывающий сульфидный атом, циннамидную функциональную группу, находящуюся в пара-положении по отношению к указанному связывающему сульфидному атому, и по крайней мере один дополнительный заместитель в одном или нескольких ароматических кольцах для преимущественного связывания LFA-1 по сравнению с ICAM-1 или ICAM-3.
Текст
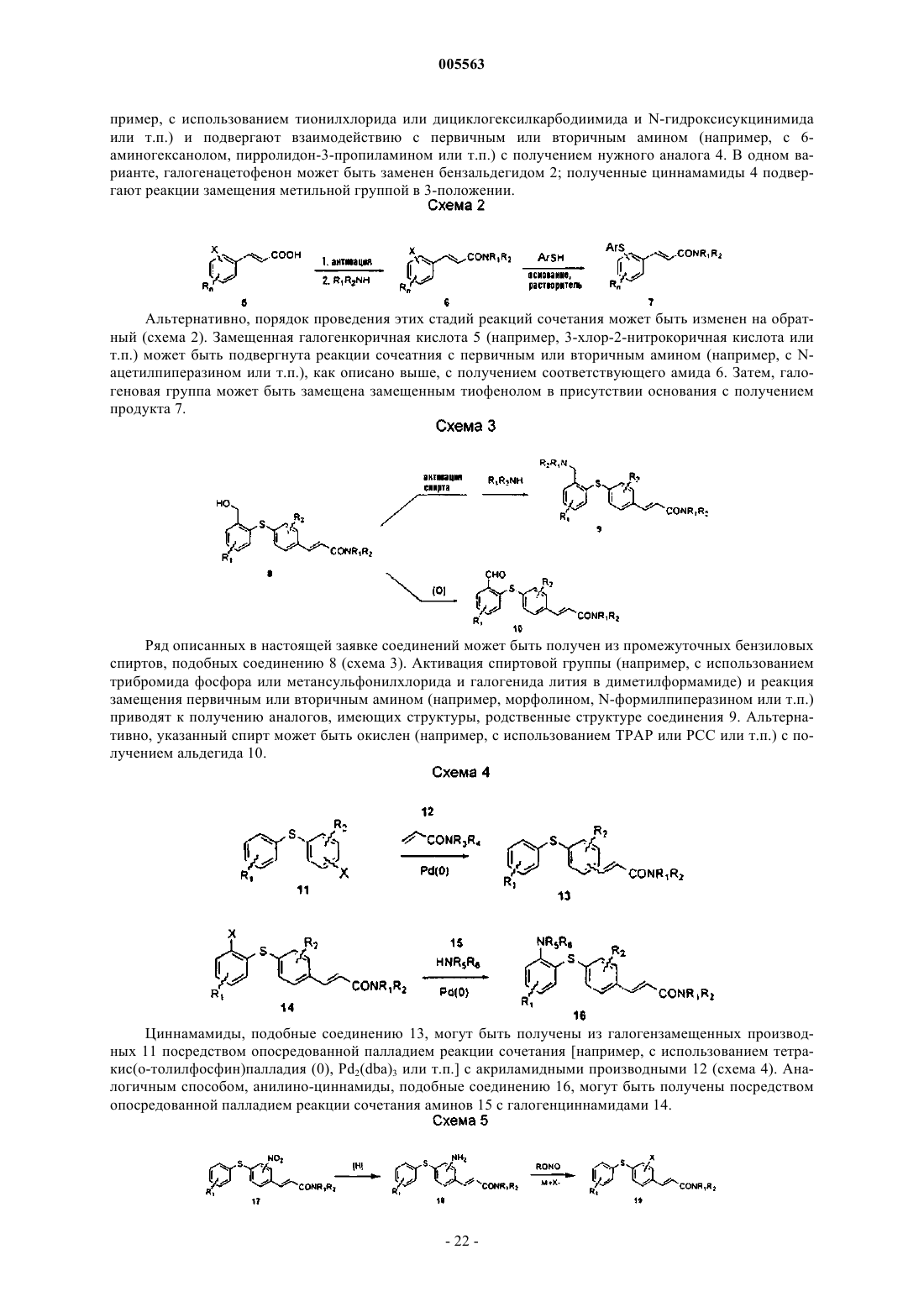
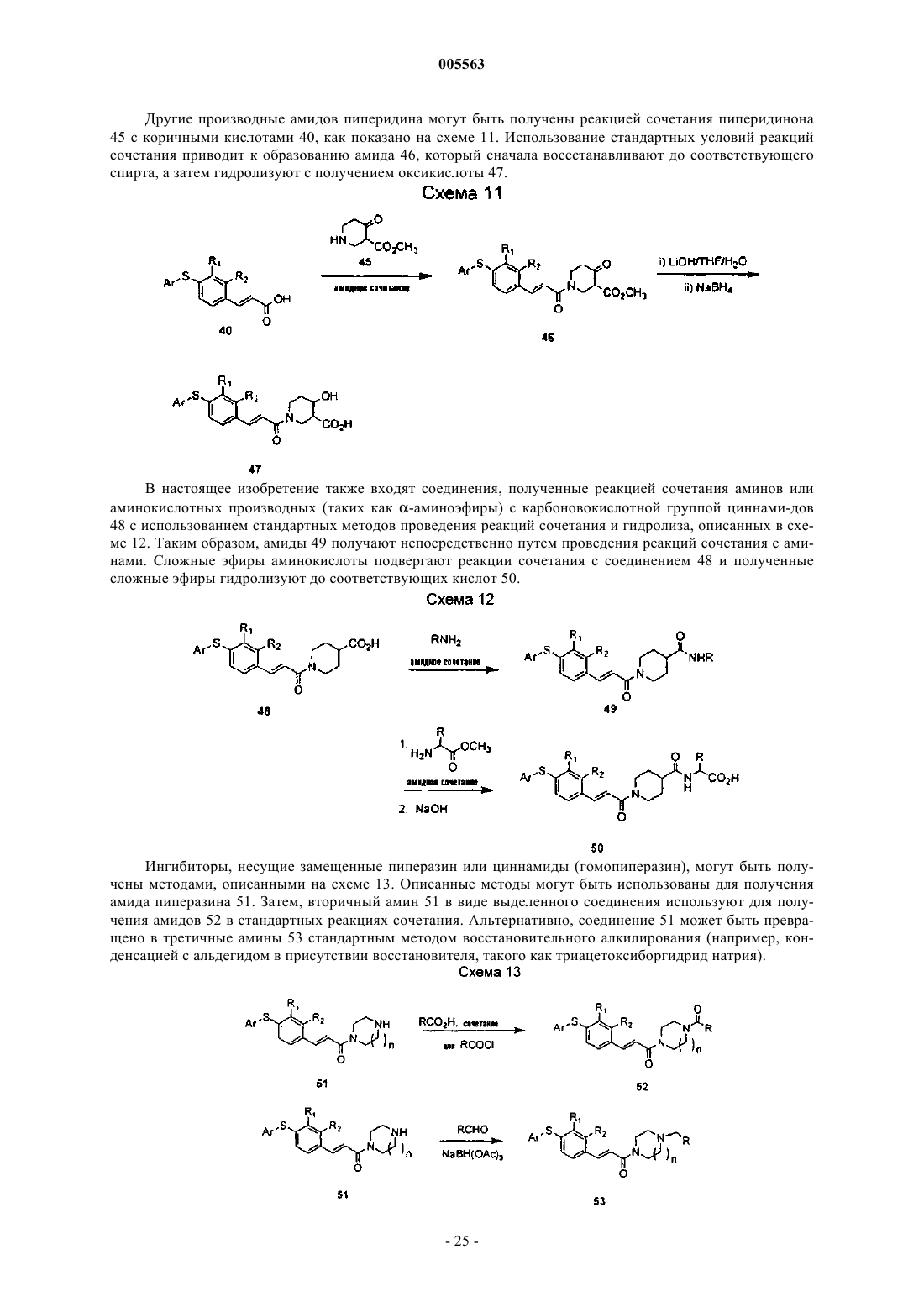
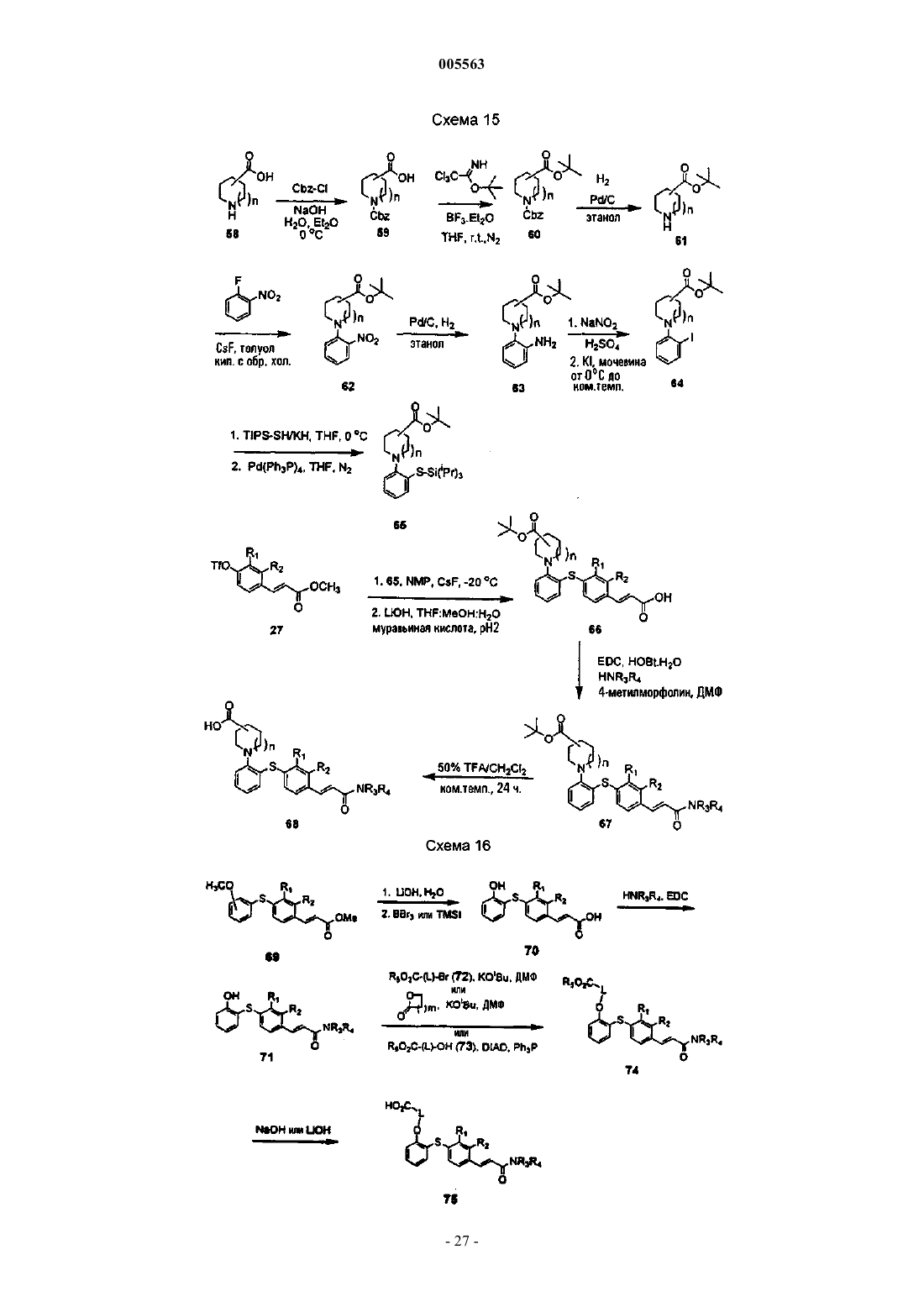
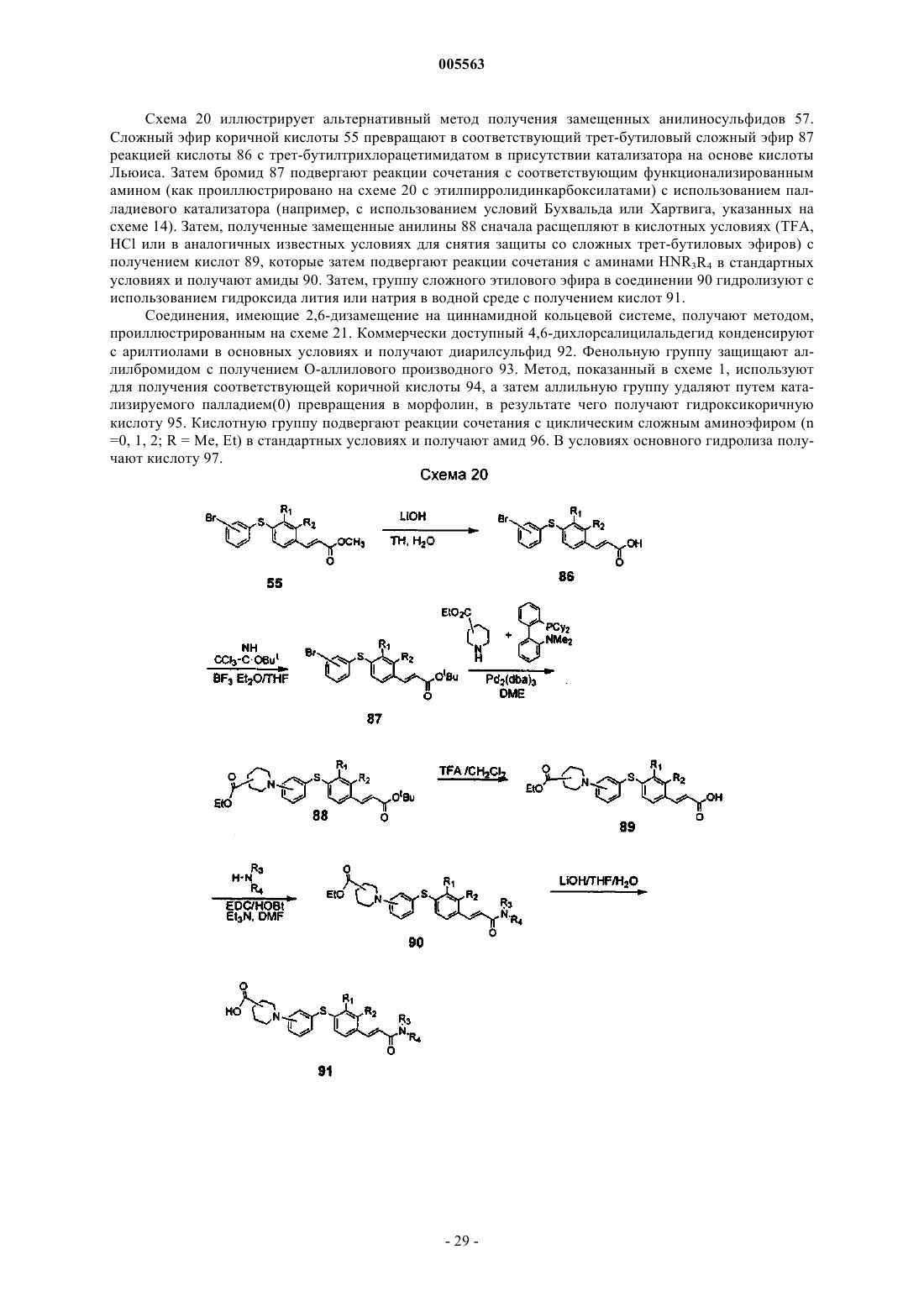
005563 Область, к которой относится изобретение Настоящее изобретение относится к соединениям, которые могут быть использованы для лечения воспалительных и иммунных заболеваний, к фармацевтическим композициям, содержащим эти соединения, и к способам ингибирования или подавления иммунного ответа у млекопитающих. Предпосылки создания изобретения Воспаления возникают в результате каскада событий, которые включают вазодилатацию, сопровождающуюся увеличением проницаемости сосудов и экссудацией белков жидкости и плазмы. Это нарушение целостности сосудов предшествует инфильтрации или сопровождается инфильтрацией затронутых воспалением клеток. Медиаторы воспаления, генерируемые в месте возникновения воспаления, служат для рекрутинга воспалительных клеток в зоне поражения. Указанные медиаторы (хемокины, такие как IL-8, МСР-1, MIP-1 и RANTES, фрагменты комплемента и липидные медиаторы) обладают хемотаксической активностью по отношению к лейкоцитам и притягивают воспалительные клетки в область поражения. Указанные медиаторы хемотаксиса, которые заставляют циркулирующие лейкоциты локализоваться в области воспаления, требуют, чтобы эти клетки пересекали сосудистый эндотелий в определенном участке. Такой рекрутинг лейкоцитов осуществляется благодаря процессу, называемому клеточной адгезией. Клеточная адгезия происходит посредством координационно регулируемой серии стадий, которые позволяют лейкоцитам сначала прикрепляться к определенному участку сосудистого эндотелия, а затем пересекать эндотелиальный барьер для миграции в воспалительную ткань (Springer T.A., 1994, TrafficSinger, M.S., Lasky, L.H.Rosen S.D. 1991, Lectin-Like Cell Adhesion Molecule 1 Mediates Rolling in Mesenteric Venules in vivo, Blood 77:2553-2555). Эти стадии опосредованы семействами адгезивных молекул,таких как интегрины, члены супергенного семейства Ig и селектины, которые экспрессируются на поверхности циркулирующих лейкоцитов и на клетках сосудистого эндотелия. Первая стадия состоит из"роллинга" лейкоцитов вдоль выстилки клеточного эндотелия сосудов в области воспаления. Стадия"роллинга" опосредуется взаимодействием между олигосахаридами поверхности лейкоцита, такими как сиалилированный антиген Lewis-X (SLex), и молекулой селектина, экспрессированной на поверхности эндотелиальной клетки в зоне воспаления. Молекула селектина обычно не экспрессируется на поверхности эндотелиальной клетки, а вместо этого индуцируется под действием воспалительных медиаторов,таких как TNF- и интерлейкин-1. "Роллинг" снижает скорость циркулирующего лейкоцита в зоне воспаления и позволяет клеткам более жестко прикрепляться к эндотелиальной клетке. Такая жесткая адгезия осуществляется посредством взаимодействия молекул интегрина, которые присутствуют на поверхности "катящихся" лейкоцитов и их контррецепторов (молекулы суперсемейства Ig) на поверхности эндотелиальной клетки. Суперсемейство молекул Ig или САМ (клеточно-адгезивные молекулы) не экспрессируются или экспрессируются на очень низком уровне на нормальных эндотелиальных клетках сосудов. САМ, подобно селектинам, индуцируются под действием медиаторов воспаления, таких какTNF- и IL-1. Конечным событием в процессе адгезии является экстравазация (прохождение) лейкоцитов через барьер эндотелиальных клеток и их миграция вдоль хемотаксического градиента в участок воспаления. Такая трансмиграция опосредуется превращением интегрина лейкоцита из низкого состояния авидности в высокое состояние авидности. Процесс адгезии основан на экспрессии селектинов и САМ на поверхности эндотелиальных клеток сосудов с опосредованием "роллинга" и жестким прикреплением лейкоцитов к эндотелию сосудов. Взаимодействие молекул межклеточной адгезии ICAM-1 (cd54) на эндотелиальных клетках с интегрином LFA-1 на лейкоцитах играет важную роль в контактировании эндотелия с лейкоцитом. Лейкоциты, несущие высокоаффинный LFA-1, прикрепляются к эндотелиальным клеткам посредством взаимодействия с ICAM-1, инициируя процесс их экстравазации из системы кровеносных сосудов в окружающие ткани. Таким образом, агент, который блокирует взаимодействие ICAM-1/LFA-1, будет подавлять эти ранние стадии воспалительного ответа. В соответствии с этими данными, являющимися предпосылкой настоящего изобретения, ICAM-1-дефектные мыши имеют множество аномалий в отношении их воспалительного ответа. В настоящем изобретении описаны соединения, которые связываются с участком взаимодействия(I-домен) LFA-1, и тем самым препятствуют адгезии лейкоцитов к эндотелиальной клетке путем блокирования взаимодействия LFA-1 с ICAM-1, ICAM-3 и другими адгезивными молекулами. Эти соединения могут быть использованы для лечения или профилактики заболеваний, в возникновении которых определенную роль играет транспорт лейкоцитов, а именно острых и хронических воспалительных заболеваний, аутоиммунных болезней, метастазов опухолей, отторжения трансплантата и повреждений при реперфузии. Соединения настоящего изобретения представляют собой диарилсульфиды, которые замеще-1 005563 ны циннамидной группой. Циннамидная функциональная группа может находиться либо в орто-, либо в пара-положении по отношению к связывающему атому серы, хотя предпочтительным является паразамещение. Соответствующее замещение у обоих ароматических колец является допустимым и может быть использовано для модуляции различных биохимических, физико-химических и фармакокинетических свойств. В частности, амидная группа легко модифицируется; ряд вторичных и третичных амидов являются активными, и альтернативно, в этом положении может быть присоединено гетероциклическое кольцо. Модификации амидной функциональной группы могут быть, в частности, использованы для модуляции физико-химических и фармакокинетических свойств. Краткое описание изобретения Настоящее изобретение относится к нижеуказанным соединениям формулы I: или к их фармацевтически приемлемой соли или их пролекарству, где R1, R2, R3, R4 и R5 независимо выбраны изR10 и R11 независимо выбраны из а. водорода,b. алкила,c. циклоалкила,d. алкоксикарбонилалкила,e. гидроксиалкила,f. замещенного арила,g. гетероциклила,h. гетероциклилалкила,i. гетероциклиламино,j. замещенного гетероциклила иk. замещенного гетероциклилалкила,либо где NR10R11 представляют гетероциклил или замещенный гетероциклил, где заместители независимо выбраны из 1) алкила,2) алкокси,3) алкоксиалкила,4) циклоалкила,5) арила,6) гетероциклила,7) гетероциклилкарбонила,8) гетероциклилалкиламинокарбонила,-2 005563 9) гидрокси,10) гидроксиалкила,11) гидроксиалкоксиалкила,12) карбокси,13) карбоксиалкила,14) карбоксикарбонила,15) карбоксальдегида,16) алкоксикарбонила,17) арилалкоксикарбонила,18) аминоалкила,19) аминоалканоила,20) карбоксамидо,21) алкоксикарбониалкила,22) карбоксамидоалкила,23) циано,24) тетразолила,25) замещенного тетразолила,26) алканоила,27) гидроксиалканоила,28) алканоилокси,29) алканоиламино,30) алканоилоксиалкила,31) алканоиламиноалкила,32) сульфоната,33) алкилсульфонила,34) алкилсульфониламинокарбонила,35) арилсульфониламинокарбонила и 36) гетороциклилсульфониламинокарбонила. и где Аr означает замещенную арильную или замещенную гетероарильную группу, где заместители независимо выбраны изjj. "транс-циннамида". Настоящее изобретение также относится к способам лечения или профилактики, для осуществления которых желательно ингибирование воспаления или супрессия иммунного ответа, где указанные способы предусматривают введение эффективного количества соединения формулы I. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединения формулы I. Подробное описание изобретения Используемый здесь термин "алканоил" означает алкильную группу, присоединенную к исходной молекулярной группе посредством карбонильной группы. Используемый здесь термин "алканоиламино" означает алканоильную группу, присоединенную к исходной молекулярной группе посредством аминогруппы. Используемый здесь термин "алканоиламиноалкил" означает алканоиламиногруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "алканоилокси" означает алканоильную группу, присоединенную к исходной молекулярной группе посредством кислородного радикала. Используемый здесь термин "алканоилоксиалкил" означает алканоилоксигруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "алкокси" означает алкильную группу, присоединенную к исходной молекулярной группе посредством атома кислорода. Используемый здесь термин "алкоксиалкокси" означает алкоксигруппу, присоединенную к исходной молекулярной группе посредством алкоксигруппы. Используемый здесь термин "алкоксиалкил" означает алкоксигруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "алкоксикарбонил" означает алкоксигруппу, присоединенную к исходной молекулярной группе посредством карбонильной группы. Используемый здесь термин "алкоксикарбонилалкил" означает алкоксикарбонильную группу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "алкил" означает насыщенную прямую или разветвленную группу,имеющую 1-10 атомов углерода и образующуюся из алкана при удалении одного атома водорода. Используемый здесь термин "алкил(алкоксикарбонил)амино" означает аминогруппу, замещенную одной алкильной группой и одной алкоксикарбонилалкильной группой. Используемый здесь термин "алкил(алкоксикарбонилалкил)аминоалкил" означает алкил(алкоксикарбонилалкил)аминогруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "алкилен" означает двухвалентную группу с 1-10 атомами углерода,образующуюся из алкана с прямой или разветвленной цепью при удалении двух атомов водорода. Используемый здесь термин "алкилсульфонил" означает алкильный радикал, присоединенный к исходной молекулярной группе через группу -SO2-. Используемый здесь термин "алкилсульфониламинокарбонил" означает алкилсульфонильную группу, присоединенную к исходной молекулярной группе посредством аминокарбонильной группы. Используемый здесь термин "амино" означает означает радикал, имеющий форму -NR18R19- или радикал, имеющий форму -NR18-, где R18 и R19 независимо выбраны из водорода, алкила или циклоалкила. Используемый здесь термин "аминоалканоил" означает аминогруппу, присоединенную к исходной молекулярной группе посредством алканоильной группы. Используемый здесь термин "аминоалкил" означает аминогруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "аминокарбонил" означает аминогруппу, присоединенную к исходной молекулярной группе посредством карбонильной группы. Используемый здесь термин "арил" означает моно- или бициклическую карбоциклическую кольцевую систему, имеющую один или два ароматических кольца. Арильная группа может быть также конденсирована с циклогексановым, циклогексеновым, циклопентановым или циклопентеновым кольцом. Арильные группы настоящего изобретения могут быть необязательно замещены заместителями, такими как алкил, галоген, гидрокси или алкокси. Используемый здесь термин "арилалкокси" означает арильную группу, присоединенную к исходной молекулярной группе через алкоксигруппу. Используемый здесь термин "арилалкоксикарбонил" означает арилалкоксигруппу, присоединенную к исходной молекулярной группе через карбонильную группу. Используемый здесь термин "арилсульфонил" означает арильный радикал, присоединенный к исходной молекулярной группе через группу -SO2-.-4 005563 Используемый здесь термин "арилсульфониламинокарбонил" означает арилсульфонильную группу,присоединенную к исходной молекулярной группе через аминокарбонильную группу. Используемый здесь термин "карбоксальдегид" означает радикал -СНО. Используемый здесь термин "карбоксальдегидгидразон" означает радикал -CH=N-NR20R21 где R20 иR21 независимо выбраны из водорода, алкила или циклоалкила. Используемый здесь термин "карбоксамид" означает аминогруппу, присоединенную к исходной молекулярной группе через карбонильную группу. Используемый здесь термин "карбоксамидоалкил" означает карбоксамидогруппу, присоединенную к исходной молекулярной группе через алкильную группу. Используемый здесь термин "карбокси" означает радикал -СООН. Используемый здесь термин "карбоксиалкил" означает карбоксигруппу, присоединенную к исходной молекулярной группе через алкильную группу. Используемый здесь термин "карбоксикарбонил" означает карбоксигруппу, присоединенную к исходной молекулярной группе через карбонильную группу. Используемый здесь термин "циано" означает радикал -CN. Используемый здесь термин "циклоалкил" означает моновалентную насыщенную циклическую или бициклическую углеводородную группу из 3-12 атомов углерода, образующуюся из циклоалкана при удалении одного атома водорода. Циклоалкильные группы настоящего изобретения могут быть необязательно замещены заместителями, такими как алкил, галоген, гидрокси или алкокси. Используемый здесь термин "галогено" или "галоген" означают F, Cl, Вr или I. Используемый здесь термин "галогеналкил" означает алкильную группу, замещенную одним или несколькими атомами галогена. Термины "гетероцикл" или "гетероциклил" означают 4-, 5-, 6- или 7-членное кольцо, содержащее один, два или три гетероатома, независимо выбранных из группы, состоящей из азота, кислорода и серы. 4- и 5-членные кольца имеют от нуля до двух двойных связей, а 6- и 7-членные кольца имеют от нуля до трех двойных связей. Используемый здесь термин "гетероцикл" или "гетероциклический", кроме того,относится к бициклической, трициклической и тетрациклической группам, в которых любое из вышеуказанных гетероциклических колец конденсировано с одним или двумя кольцами, независимо выбранными из арильного кольца, циклогексанового кольца, циклогексенового кольца, циклопентанового кольца,циклопентенового кольца или другого моноциклического гетероциклического кольца. Гетероциклами являются акридинил, бензимидазолил, бензфуил, бензотиазолил, бензотиенил, бензоксазолил, биотинил,циннолинил, дигидрофурил, дигидроиндолил, дигидропиранил, дигидротиенил, дитиазолил, фурил, гомопиперидинил, имидазолидинил, имидазолинил, имидазолил, индолил, изохинолил,изотиазолидинил,изотиазолил, изоксазолидинил, изоксазолил, морфолинил, оксадиазолил, оксазолидинил, оксазолил, пиперазинил, пиперидинил, пиранил, пиразолидинил, пиразинил, пиразолил, пиразилинил, пиридазинил,пиридил, пиримидинил, пиримидил, пирролидинил, пиррлидинил-2-онил, пирролинил, пирролил, хинолинил, хиноксалоил, тетрагидрофурил, тетрагидроизохинолил, тетрагидрохинолил, тетразолил, тиадиазолил, тиазолидинил, тиазолил, тиенил, тиоморфолинил, триазолил и т.п. Гетероциклы также включают мостиковые бициклические группы, где моноциклическая гетероциклическая группа связана мостиковой связью посредством алкиленовой группы, такой как и т.п. Гетероциклами являются также соединения формулы и где X и Z независимо выбраны из -СН 2-, -CH2NH-, -CH2O-, -NH-и -О- при условии, что по крайней мере один из X и Z не является -СН 2-, a Y выбран из -С(О) и (C(R")2)v-, где R" представляет водород или алкил с одним-четырьмя атомами углерода, а v равно 1-3. Указанными гетероциклами являются 1,3 бензодиоксонил, 1,4-бензодиоксалил, 1,3-бензимидазол-2-он и т.п. Гетероциклические группы настоящего изобретения могут быть необязательно замещены заместителями, такими как алкокси, алкил, галоген,гидрокси, карбокси, карбоксиалкил или алкоксикарбонил. Используемый здесь термин "гетероциклилалкил" означает гетероциклическую группу, присоединенную к исходной молекулярной группе через алкильную группу. Используемый здесь термин "гетероциклилалкиламино" означает гетероциклилалкильную группу,присоединенную к исходной молекулярной группе через аминогруппу.-5 005563 Используемый здесь термин "гетероциклилалкиламинокарбонил" означает гетероциклилалкиламино-группу, присоединенную к исходной молекулярной группе через карбонильную группу. Используемый здесь термин "гетероциклиламино" означает гетероциклильную группу, присоединенную к исходной молекулярной группе через аминогруппу. Используемый здесь термин "гетероциклилкарбонил" означает гетероциклильную группу, присоединенную к исходной молекулярной группе через карбонильную группу. Используемый здесь термин "гетероциклилсульфонил" означает гетероциклильный радикал, присоединенный к исходной молекулярной группе через группу -SO2-. Используемый здесь термин "гетероциклилсульфониламинокарбонил" означает гетероциклилсульфонильную группу, присоединенную к исходной молекулярной группе через аминокарбонильную группу. Используемый здесь термин "гидроксиалканоил" означает гидрокси-радикал, присоединенную к исходной молекулярной группе посредством алканоильной группы. Используемый здесь термин "гидроксиалкокси" означает гидрокси-радикал, присоединенный к исходной молекулярной группе посредством алкоксигруппы. Используемый здесь термин "гидроксиалкоксиалкил" означает гидроксиалкоксигруппу, присоединенную к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "гидроксиалкил" означает гидрокси-радикал, присоединенный к исходной молекулярной группе посредством алкильной группы. Используемый здесь термин "гидроксиалкиламинокарбонил" означает гидроксиалкильную группу,присоединенную к исходной молекулярной группе посредством аминокарбонильной группы. Используемый здесь термин "перфторалкил" означает алкильную группу, в которой все атомы водорода замещены атомами фтора. Используемый здесь термин "фенил" означает моноциклическую карбоциклическую кольцевую систему, имеющую одно ароматическое кольцо. Фенильная группа может быть также конденсирована с цик-логексановым или циклопентановым кольцом. Фенильная группа настоящего изобретения может быть необязательно замещена заместителями, такими как алкил, галоген, гидрокси или алкокси. Используемый здесь термин "фармацевтически приемлемые пролекарства" означает пролекарства соединений настоящего изобретения, которые, как установлено исходя из тщательной оценки специалистов-медиков, являются подходящими для их использования при контактировании с тканями человека и низших животных, не вызывают чрезмерной токсичности, раздражения, аллергических реакций или других побочных эффектов и соответствуют приемлемому отношению польза/риск, и являются эффективными при их применении в нужных целях, а также цвиттерионные формы соединений настоящего изобретения, если они возможны. Используемый здесь термин "пролекарство" означает соединения, которые быстро превращаются invivo в исходное соединение вышеуказанной формулы, например, путем гидролиза в кровотоке. Подробное обсуждение приводится в работах T.HiguchiV.Stella, Prodrugs as Novel Delivery Systems, Vol.14 ofPharmaceuticals Association and Pergamon Press, 1987, обе из которых вводятся в настоящее описание посредством ссылки. Используемый здесь термин "сульфонат" означает радикал -SO3H. Используемый здесь термин "тетразол" или "тетразолил" означает гетероциклический радикал-CN4H. Используемый здесь термин "тиоалкокси" означает алкильную группу, присоединенную к исходной молекулярной группе через атом серы. Соединения настоящего изобретения могут присутствовать в виде стереоизомеров, в которых присутствуют асимметрические или хиральные центры. Эти соединения обозначаются символами "R" или"S", в зависимости от конфигурации заместителей у хирального атома углерода. Настоящее изобретение относится к различным стереоизомерам и к их смесям. Стереоизомеры включают энантиомеры и диастереомеры, а смеси энантиомеров или диастереомеров обозначают . Отдельные стереоизомеры соединений настоящего изобретения могут быть получены путем синтеза из коммерчески доступных исходных веществ, которые содержат асимметрические или хиральные центры, или путем получения рацемических смесей с последующим разделением этих смесей методами, хорошо известными специалистам. Указанные способы разделения предусматривают: (1) присоединение смеси энантиомеров к хиральному вспомогательному веществу, разделение полученной смеси диастереомеров перекристаллизацией или хроматографией и выделение из указанного вспомогательного вещества оптически чистого продукта, (2) образование соли с применением оптически активного разделяющего агента или (3) прямое разделение смеси оптических энантиомеров на хиральных хроматографических колонках. Соединения настоящего изобретения могут также существовать в виде геометрических изомеров. В настоящем изобретении также рассматриваются различные геометрические изомеры и их смеси, образующиеся в результате распределения заместителей у углерод-углеродной двойной связи или распределения заместителей у карбоциклического кольца. Заместители у углерод-углеродной двойной связи на-6 005563 ходятся в Z- или Е-конфигурации, где термин " Z" означает заместители на одной и той же стороне углерод-углеродной двойной связи, а термин "Е" означает заместители на противоположных сторонах углерод-углеродной двойной связи. Расположение заместителей вокруг карбоциклического конца обозначают цис или транс, где термин "цис" означает, что заместители присутствуют на одной и той же стороне плоскости кольца, а термин "транс" означает, что заместители присутствуют на противоположных сторонах плоскости кольца. Смеси соединений, где заместители расположены как на одной и той же стороне, так и на противоположных сторонах плоскости кольца, называют цис/транс. Как очевидно из вышепривиденного описания, соединения формулы I могут быть использованы в различных формах, то есть, с различными указанными заместителями. Примерами особенно предпочтительных соединений являются абсолютно разные соединения и многие из них упомянуты в настоящем описании. Такими примерами являются соединения, в которых R1 представляет "цис-циннамид" или "транс-циннамид", a R3 представляет водород, или в которых R3 представляет "цис-циннамид" или "транс-циннамид", a R1 представляет водород либо каждый из R1, R2 и R4 независимо представляет водород или алкил, a R5 представляет галоген, галогеналкил или нитро. Другими предпочтительными соединениями являются соединения, указанные выше, где каждый из R10 и R11 независимо представляет водород, алкил, циклоалкил, алкоксикарбонилалкил, гидроксиалкил или гетероциклилалкил, либо где NR10R11 представляет гетероциклили или замещенный гетероциклил, и где Аr представляет арил, замещенный арил, гетероарил или замещенный гетероарил. Соединениями настоящего изобретения являются:(3-(3-пропиокси)фенил)[2,3-дихлор-4-(Е-4-морфолино)карбонил)этенил)фенил]сульфид. Фармацевтические композиции и методы лечения Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения настоящего изобретения вместе с одним или несколькими фармацевтически приемлемыми носителями. Эти фармацевтические композиции могут быть специально приготовлены для перорального введения в твердой или жидкой форме, для парентерального введения или для ректального введения. Указанные фармацевтические композиции настоящего изобретения могут быть введены человеку и другим животным перорально, ректально, парентерально, интрацистернально, интравагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), трансбуккально или в виде перорального или интраназального впрыскивания. Используемый здесь термин "парентеральное введение" означает способы введения, которые включают внутривенное, внутримышечное, внутрибрюшинное, внутригрудинное,подкожное и внутрисуставное введение, и введение путем инфузии. Фармацевтические композиции настоящего изобретения для парентерального введения содержат фармацевтически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для их разведения с получением стерильных инъецируемых растворов или дисперсий непосредственно перед использованием. Примерами подходящих водных и безводных носителей, разбавителей, растворителей или наполнителей являются: вода, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их подходящие смеси, растительные масла (такие как оливковое масло) и инъецируемые органические сложные эфиры, такие как этилолеат. Нужная текучесть может поддерживаться посредством использования материалов для покрытий, таких как лецитин, посредством сохранения нужного размера частиц в случае дисперсий и посредством использования поверхностно-активных веществ. Эти композиции могут также содержать адъюванты, такие как консерванты, смачивающие агенты,эмульгаторы и диспергирующие агенты. Для защиты от действия микроорганизмов могут быть включены различные противомикробные и противогрибковые агенты, например, парабен, хлорбутанол, фенолсорбиновая кислота и т.п. Может также оказаться желательным включение изотонических агентов, таких как сахара, хлорид натрия и т.п. Пролонгированная абсорбция инъецируемой фармацевтической формы может быть достигнута путем включения агентов, замедляющих абсорбцию, таких как моностеарат алюминия и желатин. В некоторых случаях, для продления действия лекарственных средств, желательно замедление абсорбции лекарственного средства после подкожной или внутримышечной инъекции. Это может быть осуществлено путем использования жидкой суспензии кристаллического или аморфного материала с плохой водорастворимостью. Кроме того, скорость абсорбции лекарственного средства зависит от скорости его растворения, которая, в свою очередь, может зависеть от размера и формы кристаллов. Альтернативно, замедленная абсорбция парентерально вводимого лекарственного средства достигается путем растворения или суспендирования указанного лекарственного средства в масляном носителе. Инъецируемые депо-формы изготавливают путем создания матриц для микрокапсулы, состоящих из лекарственного средства в биологически разлагаемых полимерах, таких как полилактидполигликолид. В зависимости от отношения лекарственного средства к полимеру и природы конкретно используемого полимера, скорость высвобождения лекарственного средства может регулироваться. Примерами других биологически разлагаемых полимеров являются поли(ортоэфиры) и поли(ангидриды). Инъецируемые композиции в виде депо-форм также получают путем включения лекарственного средства в липосомы или микроэмульсии, совместимые с тканями организма. Инъецируемые композиции могут быть стерилизованы, например, путем фильтрации через бактериальный фильтр, либо путем введения стерилизующих агентов в виде стерильных твердых композиций,которые могут быть растворены или диспергированы в стерильной воде или в другой стерильной инъецируемой среде непосредственно перед использованием. Твердыми лекарственными формами для перорального введения являются капсулы, таблетки, драже, порошки и гранулы. В таких твердых лекарственных формах, активное соединение смешивают, по крайней мере, с одним инертным фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или бифосфат кальция, и/или (а) с наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, (b) со связующими, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь, (с) с увлажнителями, такими как глицерин, (d) с дезинтеграторами, такими как агар-агар, карбо- 19005563 нат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, (е) с агентами замедляющими растворение, такими как парафин, (f) с ускорителями абсорбции, такими как четвертичные аммониевые соединения, (g) со смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, (h) с абсорбентами, такими как каолин и бентонитовая глина и (i) с замасливателями, такими как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси. В случае использования капсул, таблеток и драже,данная лекарственная форма может также содержать забуферивающие агенты. Твердые композиции аналогичного типа могут быть также использованы в качестве наполнителей в мягких и твердых желатиновых капсулах с использованием таких наполнителей, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.п. Твердые лекарственные формы в виде таблеток, драже, капсул, пилюль и гранул могут быть изготовлены с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные специалистам-фармацевтам. Указанные формы могут, но необязательно, содержать агенты, придающие непрозрачность, и могут быть также изготовлены в виде композиции, которая высвобождает активный ингредиент(ы) только или предпочтительно, в определенной части кишечного тракта, необязательно, пролонгированным образом. Примерами композиций, которые могут быть использованы для включения лекарственного препарата, являются полимерные вещества и воски. Активные соединения могут быть также изготовлены в микроинкапсулированной форме, если это необходимо, в сочетании с одним или несколькми из вышеупомянутых наполнителей. Жидкими лекарственными формами для перорального введения являются фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активных соединений, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые специалистами, такие как, например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт,бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, масло семян хлопчатника, арахисовое масло, кукурузное масло, масло из проросших семян, оливковое масло,касторовое масло и кунжутное масло), глицерин, тетрагидрофуриловый спирт, полиэтиленгликоли, эфиры, образованные жирной кислотой и сорбитаном, и их смеси. Помимо инертных разбавителей, композиции для перорального введения могут также включать адъюванты, такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, подсластители,отдушки и ароматизирующие агенты. Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, такие как,например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар, трагакант и их смеси. Композициями для ректального или вагинального введения являются, предпочтительно, суппозитории, которые могут быть изготовлены путем смешивания соединений настоящего изобретения с подходящими нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или воскообразная основа для суппозиториев, которые являются твердыми при комнатной температуре,но жидкими при температуре тела, а поэтому, при попадании в прямую кишку или во влагалище, они расплавляются и высвобождают активное соединение. Соединения настоящего изобретения могут быть введены в виде липосом. Как известно специалистам, липосомы обычно состоят из фосфолипидов или других липидных веществ. Липосомы образуются моно- или мультиламеллярными гидратированными жидкими кристаллами, которые диспергированы в жидкой среде. Может быть использован любой нетоксичный физиологически приемлемый и подверженный метаболизму липид, способный образовывать липосомы. Композиции настоящего изобретения, изготовленные в форме липосом, могут, помимо соединений настоящего изобретения, содержать стабилизаторы, консерванты, наполнители, и т.п. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины), которые могут быть натуральными и синтетическими. Методы получения липосом известны специалистам. См., например, Prescott, Ed., Methods in CellBiology, Volume XIV, Academic Press, New-York, N.Y. (1976), p 33 et seq. Соединения настоящего изобретения могут быть использованы в форме фармацевтически приемлемых солей, полученных из неорганических или органических кислот. Термин "фармацевтически приемлемая соль" означает такую соль, которая, согласно тщательной оценке специалистов-медиков, является подходящей для ее использования при контактировании с тканями человека и низших животных, и,при этом не вызывает чрезмерной токсичности, раздражения, аллергических реакций или других побочных эффектов и соответствует приемлемому отношению польза/риск. Фармацевтически приемлемые соли хорошо известны специалистам. См., например, S.M. Berge, et al., подробно описывают фармацевтически приемлемые соли в J. Pharmaceutical Sciences, 1977, 66: 1 et seq. Эти соли могут быть полученыin situ в процессе конечного выделения и очистки соединений настоящего изобретения или отдельно путем реакции свободного основания с соответствующей кислотой. Характериными примерами кислотноаддитивных солей являются ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, глицерофосфат, полусульфат, гептаноат,- 20005563 гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3 фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, фосфат, глутамат, бикарбонат, п-толуолсульфонат и ундеканоат. Кроме того, основные азотсодержащие группы могут быть кватернизованы такими агентами, как (низший алкил)галогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -иодиды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты; галогениды с длинной цепью, такие как децил-, лаурил-, миристил- и стеарилхлориды,-бромиды и -иодиды; арилалкилгалогениды, такие как бензил- и фенэтилбромиды и т.п. Таким образом,могут быть получены продукты, растворимые или диспергируемые в воде или масле. Примерами кислот,которые могут быть использованы для получения фармацевтически приемлемых кислотно-аддитивных солей, являются неорганические кислоты, такие как хлористо-водородная кислота, бромисто-водородная кислота, серная кислота и фосфорная кислота, и органические кислоты, такие как щавелевая кислота,малеиновая кислота, янтарная кислота и лимонная кислота. Основно-аддитивные соли могут быть получены in situ в процессе конечного выделения и очистки соединений настоящего изобретения путем реакции молекулы, содержащей группу карбоновой кислоты,с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, либо с аммиаком или с органическим первичным, вторичным или третичным амином. Фармацевтически приемлемыми основно-аддитивными солями являются катионы, образованные щелочными металлами или щелочно-земельными металлами, такие как соли лития, натрия, калия, кальция, магния и алюминия и т.п. и нетоксичные катионы четвертичного аммиака и амина, включая аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин,диэтиламин, этиламин и т.п. Другими характерными органическими аминами, которые могут быть использованы для получения основно-аддитивных соелй, являются этилендиамин, этаноламин, диэтаноламин, пиперидин, пиперазин и т.п. Лекарственными формами для местного введения являются порошки, спреи, мази и аэрозоли. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферами или распыляющими веществами, которые могут потребоваться для этой цели. Офтальмические композиции, глазные мази, порошки и растворы также входят в объем настоящего изобретения. Фактические уровни доз активных ингредиентов в фармацевтических композициях настоящего изобретения могут варьироваться в зависимости от их состава и способа введения, так, чтобы количество активного соединения(й) было эффективно для достижения нужного терапевтического ответа у конкретного пациента. Выбранное количество дозы будет зависеть от активности конкретного соединения, способа введения, тяжести состояния, подвергаемого лечению, и состояния здоровья и истории болезни пациента, подвергаемого лечению. Однако каждый специалист может назначить начальную дозу данного соединения в количестве, ниже того количества, которое необходимо для достижения нужного терапевтического эффекта, и постепенно увеличивать эту дозу до тех пор, пока не будет достигнут нужный эффект. Обычно при пероральном или внутривенном введении млекопитающему, доза составляет в пределах от около 0,1 до около 50 мг, а более предпочтительно, от около 5 до около 20 мг активного соединения на килограмм массы тела в день. Если это необходимо, то эффективная суточная доза может быть разделена на дробные дозы с введением, например, от двух до четырех отдельных доз в день. Получение соединений настоящего изобретения Для более подробного описания соединений и способов настоящего изобретения, ниже представлены схемы синтеза, иллюстрирующие способы, с использованием которых могут быть получены соединения настоящего изобретения. На схеме 1 показан синтез типичного замещенного циннамамидом диарилсульфида 4 через альдегидное промежуточное соединение 2. Альдегид 2 получают реакцией взаимодействия тиофенола (например, 2,4-дихлортиофенола, 2-бромтиофенола или т.п.) с галогензамещенным бензальдегидным производным 1 (например, с 2-хлорбензальдегидом, 3-хлор-4-фторбензальдегидом или т.п.) в присутствии основания (например, карбоната натрия, триэтиламина или т.п.) и полярного растворителя (например,диметилформамида, диметилсульфоксида или т.п.). Альдегидную группу подвергают гомологизации до соответствующей коричной кислоты 3 с использованием ацетатного эквивалента (например, малоновой кислоты, три-этоксифосфоноацетата или т.п.) в присутствии соответствующего основания и растворителя. В некоторых случаях может оказаться необходимым проведение гидролиза промежуточного сложного эфира (например, с использованием гидроксида натрия в спирте). Ацильную группу активируют (на- 21005563 пример, с использованием тионилхлорида или дициклогексилкарбодиимида и N-гидроксисукцинимида или т.п.) и подвергают взаимодействию с первичным или вторичным амином (например, с 6 аминогексанолом, пирролидон-3-пропиламином или т.п.) с получением нужного аналога 4. В одном варианте, галогенацетофенон может быть заменен бензальдегидом 2; полученные циннамамиды 4 подвергают реакции замещения метильной группой в 3-положении. Альтернативно, порядок проведения этих стадий реакций сочетания может быть изменен на обратный (схема 2). Замещенная галогенкоричная кислота 5 (например, 3-хлор-2-нитрокоричная кислота или т.п.) может быть подвергнута реакции сочеатния с первичным или вторичным амином (например, с Nацетилпиперазином или т.п.), как описано выше, с получением соответствующего амида 6. Затем, галогеновая группа может быть замещена замещенным тиофенолом в присутствии основания с получением продукта 7. Ряд описанных в настоящей заявке соединений может быть получен из промежуточных бензиловых спиртов, подобных соединению 8 (схема 3). Активация спиртовой группы (например, с использованием трибромида фосфора или метансульфонилхлорида и галогенида лития в диметилформамиде) и реакция замещения первичным или вторичным амином (например, морфолином, N-формилпиперазином или т.п.) приводят к получению аналогов, имеющих структуры, родственные структуре соединения 9. Альтернативно, указанный спирт может быть окислен (например, с использованием ТРАР или РСС или т.п.) с получением альдегида 10. Циннамамиды, подобные соединению 13, могут быть получены из галогензамещенных производных 11 посредством опосредованной палладием реакции сочетания [например, с использованием тетракис(о-толилфосфин)палладия (0), Pd2(dba)3 или т.п.] с акриламидными производными 12 (схема 4). Аналогичным способом, анилино-циннамиды, подобные соединению 16, могут быть получены посредством опосредованной палладием реакции сочетания аминов 15 с галогенциннамидами 14.- 22005563 В некоторых случаях функциональные группы на ароматических кольцах могут быть модифицированы с получением новых аналогов (схема 5). Так, например, нитрогруппа в соединениях, подобных соединению 17, может быть восстановлена (например, с использованием хлорида олова(II) или путем каталитического гидрирования или т.п.) до соответствующего амина 18. Затем этот амин может быть превращен в галоген, например, путем диазотирования с использованием азотистой кислоты или третбутилнитрила в присутствии галогенидной соли металла, такой как бромид меди(2), с получением аналога 19. Замещенные циннамиды диарилсульфидов могут быть также получены в обратном порядке (схема 6). Так, например, соединение 20, полученное как описано в схеме 1, может быть подвергнуто реакции снятия защиты путем обработки основанием (например, трет-бутоксидом калия или т.п.) с получением аниона тиолата 21, который может быть подвергнут реакции взаимодействия с активированным галогенареном (например, с 2,3-дихлорбензальдегидом, 3-хлор-4-фторбензальдегидом или т.п.) с получением соответствующего продукта 22. Альтернативно, тот же самый анион тиолата может быть подвергнут реакции сочетания с неактивированными арилгалогенидами (например, с арилбромидом или арилиодидами) с использованием катализируемой металлом реакции сочетания Уллмана (например, с использованием палладиевого или никелевого катализатора) с получением продукта 23. Другой способ получения циннамидов диарилсульфида показан на схеме 7, где указанный диарилсульфид получают в результате реакции сочетания соответствующим образом замещенного арилтиола 28 с образованием активированного сложного эфира коричной кислоты 27. Замещенный фенол 24 может быть подвергнут бромированию с получением бромфенола 25. Реакцию сочетания по Хеку бромида 25 с соответствующим олефиновым субстратом, например, с метилакрилатом, осуществляют в присутствии палладиевого катализатора, в результате чего получают сложный эфир коричной кислоты 26. Затем фенол активируют для последующей реакции, например, путем превращения в соответствующий трифлат 27 в стандартных условиях. Нужный защищенный тиол 28 может быть получен методом XXX (Tetrahedron Lett. 1994, 35, 3221-3224) в результате реакции сочетания арилгалогенида или трифлата с триизопропилсилилтиолом в присутствии палладиевого катализатора. Затем два соединения-партнера 27 и 28 подвергают реакции в присутствии фторидного источника, например, фторида цезия, с получением циннамата диарилсульфида 29. Гидролиз проводят в основной среде, такой как гидроксид лития или натрия в смеси воды/ТГФ, и полученную кислоту 30 подвергают реакции сочетания с аминами в стандартных условиях для образования амидной связи (например, EDC/HOBt) с получением амидов 31.- 23005563 Метод получения циннамидов, несущих две арилтиогруппы, показан на схеме 8. Коммерчески доступную дифторкоричнуго кислоту 32 подвергают реакции сочетания с амином в стандартных условиях и полученный амид 33 подвергают реакции взаимодействия с избытком арилтиола с получением биссульфида 34. Соединения, содержащие трифторметильные группы на циннамидной части ингибиторов, получают методом, показанным на схеме 9. В соответствии с методом XXX (см. ссылку), реакция ДильсаАльдера между 1,1,1,4,4,4-гексафтор-2-бутином и 2-метилфураном приводит к образованию бициклического эфира 35, который затем подвергают перегруппировке с кислотой Льюиса (например, с этератом трифторида бора), в результате чего получают фенол 36. Затем метильную группу превращают в соответствующий альдегид 37 путем бромирования с последующим проведением реакции взаимодействия с диметилсульфоксидом. С использованием аналогичной процедуры, описанной выше для схемы 1, фенол активируют и конденсируют с тиолами в основных условиях с получением альдегидов диарилсульфида 38, а затем их превращают в циннамиды 39 в соответствии с ранее описанными процедурами. Циннамиды, несущие более сложные замещенные пиперидинамиды, могут быть получены методами, описанными в схемах 10 и 11. Коричные кислоты 4 0 подвергают реакции сочетания со спирогидантоинпиперидином 41 и полученный амид 42 сначала подвергают реакции взаимодействия с активирующим реагентом (например, с ди-трет-бутилдикарбонатом), а затем гидролизуют с образованием аминокислоты 43. Затем полученная аминогруппа может быть подвергнута реакции, например, с ангидридами кислоты или хлорангидридами, с получением амидов 44.- 24005563 Другие производные амидов пиперидина могут быть получены реакцией сочетания пиперидинона 45 с коричными кислотами 40, как показано на схеме 11. Использование стандартных условий реакций сочетания приводит к образованию амида 46, который сначала воссстанавливают до соответствующего спирта, а затем гидролизуют с получением оксикислоты 47. В настоящее изобретение также входят соединения, полученные реакцией сочетания аминов или аминокислотных производных (таких как -аминоэфиры) с карбоновокислотной группой циннами-дов 48 с использованием стандартных методов проведения реакций сочетания и гидролиза, описанных в схеме 12. Таким образом, амиды 49 получают непосредственно путем проведения реакций сочетания с аминами. Сложные эфиры аминокислоты подвергают реакции сочетания с соединением 48 и полученные сложные эфиры гидролизуют до соответствующих кислот 50. Ингибиторы, несущие замещенные пиперазин или циннамиды (гомопиперазин), могут быть получены методами, описанными на схеме 13. Описанные методы могут быть использованы для получения амида пиперазина 51. Затем, вторичный амин 51 в виде выделенного соединения используют для получения амидов 52 в стандартных реакциях сочетания. Альтернативно, соединение 51 может быть превращено в третичные амины 53 стандартным методом восстановительного алкилирования (например, конденсацией с альдегидом в присутствии восстановителя, такого как триацетоксиборгидрид натрия).- 25005563 Способ получения аналогов с аминозамещениями в арильной части сульфидов проиллюстрирован на схеме 14. Промежуточный трифлат 27 подвергают реакции взаимодействия с галогензамещенными тиофенолами 54 (X=Br, Cl, OTf, OTs) в присутствии основного катализатора и получают сульфидное производное 55. Затем галоген или активированный гидроксил подвергают реакции замещения с амином методом Бухвальда (Old, D.W.; Wolfe, J.P. Buchwald, S.L. J.Am. Chera. Soc. 1998, 120, 9722-9723). Аналогичные реакции, катализируемые переходными металлами, могут быть проведены, например, методом Хартвига (Hamann, В.С. Hartwig J.F. J. Am. Chem. Soc. 1998, 120, 7369-7370). Группа NR3R4 может составлять циклическую или ациклическую группу, необязательно замещенную дополнительными функциональными группами, которые могут повышать активность этих соединений, и которые, кроме того,могут быть использованы для последующих превращений методами синтеза, известными специалистам. Так, например, сложноэфирные группы могут быть гидролизованы с образование соответствующих карбоновых кислот или амидов. Затем, полученные анилиносульфиды могут быть обработаны как описано выше с получением циннамидов 56. Схема 15 представляет синтез замещенных несущих карбоновую кислоту анилиновых производных конкретного класса. Циклическая аминокислота 58 может быть превращена в соответствующий третбутиловый эфир 61 через промежуточные соединения карбамата 59 и сложного эфира 60 стандартными методами синтеза. Затем сложный аминоэфир 61 подвергают реакции с 2-фторнитробензолом в присутствии слабого основного катализатора (например, фторида цезия, бикарбоната калия) с получением анилинового производного 62. Затем нитрогруппа может быть преобразована в иодзамещенное производное 64, сначала превращением в анилин 63 с последующим стандартным диазотированием, а затем реакцией диазониевой соли с иодидом калия (наряду с другими аналогичными методами, используемыми для этой реакции Зандмейера). Методом, описанным на схеме 7, иодид 64 может быть превращен в TIPSзащищенный арилтиол 65. В последовательности, показанной на схеме 7, силиловый тиоэфир 65 может быть подвергнут реакции с циннамидтрифлатом 27 в присутствии фторидного источника (например,фторида цезия), и таким образом, превращен в диарилсульфид 66. Стандартными методами синтеза посредством реакций превращения (гидролиз сложного эфира, амидное сочетание и отщепление сложного трет-бутилового эфира) получают нужную кислоту 68 через промежуточный сложный эфир 67. Соединения, несущие рассматриваемые эфирные группы на арилсульфидном кольце, получают в соответствии со схемой 16. Сложные эфиры коричной кислоты, содержащие группу простого эфира, такие как соединение 69, гидролизуют до соответствующих кислот, а затем метиловый эфир отщепляют трибромидом (или альтернативно, с использованием аналогичных отщепляющих эфир агентов, таких как триметилсилилиодид) с получением оксикислот 70. Стандартными методами сочетания получают амиды 71, которые затем алкилируют на фенольной группе с использованием соответствующего алкилгалогенида 72 (где L представляет связывающую группу, состоящую из ациклического или циклического алкила, либо гетероциклическую группу) или лактона (m =1,2) в присутствии основания (такого как третбутоксид калия, гидрид натрия или карбонат цезия). Альтернативно, фенольную группу алкилируют несущим сложноэфирную группу спиртом 73 с использованием условий Митцунобу. Затем эти несущие сложноэфирную группу простые эфиры 74 гидролизуют до соответствующих кислот 75 с использованием стандартных условий гидролиза. Альтернативно, сложный эфир 74 может быть трет-бутилом, и в этом случае должно быть осуществлено кислотное удаление защитных групп с получением кислоты 75(например, с использованием трифторуксусной кислоты в дихлорметане или хлористоводородной кислоты в диоксане).- 27005563 Родственные соединения, несущие рассматриваемые функционализированные аминозаместители,получают в соответствии со схемой 17. Трифлат 27 подвергают реакции с аминотиофенолом с получением диарилсульфидциннамида 76 способом, описанным в схемах 1, 2 и 7. Сложный эфир коричной кислоты гидролизуют с получением кислоты 77, которую подвергают реакции сочетания в стандартных условиях с получением амида 78. Затем аминогруппу 78 подвергают восстановительному алкилированию с несущим сложноэфирную группу алкилальдегидом в стандартных условиях (или альтернативно, с использованием триацетоксиборогидрида натрия) с получением вторичного амина 79. Сложноэфирную функциональную группу гидролизуют с получением соответствующей соли кислоты 80. Альтернативный способ получения промежуточного соединения 78 показан на схеме 18. Нитрозамещенное производное трет-бутилового сложного эфира 81 (полученного в соответствии со схемой 14 с использованием трет-бутилового аналога циннамата 27) расщепляют с получением карбоновой кислоты, превращают в циннамид с использованием стандартных условий, а затем нитрогруппу восстанавливают с использованием железного порошка в водном растворе хлорида аммония. Модифицированный метод получения аналогов, несущих 2,3-бис(трифторметил)циннамиды, проиллюстрирован на схеме 19. Коммерчески доступную акриловую кислоту 82 подвергают этерификации этилиодидом, а сложный эфир 83 конденсируют с 1,1,1,4,4,4-гексафтор-2-бутином при 110 С с получением бициклического аддукта 84. Затем указанный бициклический эфир превращают в соответствующий фенол 85 с использованием кислоты Льюиса (например, этерата трифторида бора). Фенол 85 используют в качестве иллюстрации на схеме 7 или на схеме 14 для получения нужных ингибиторов.- 28005563 Схема 20 иллюстрирует альтернативный метод получения замещенных анилиносульфидов 57. Сложный эфир коричной кислоты 55 превращают в соответствующий трет-бутиловый сложный эфир 87 реакцией кислоты 86 с трет-бутилтрихлорацетимидатом в присутствии катализатора на основе кислоты Льюиса. Затем бромид 87 подвергают реакции сочетания с соответствующим функционализированным амином (как проиллюстрировано на схеме 20 с этилпирролидинкарбоксилатами) с использованием палладиевого катализатора (например, с использованием условий Бухвальда или Хартвига, указанных на схеме 14). Затем, полученные замещенные анилины 88 сначала расщепляют в кислотных условиях (TFA,HCl или в аналогичных известных условиях для снятия защиты со сложных трет-бутиловых эфиров) с получением кислот 89, которые затем подвергают реакции сочетания с аминами HNR3R4 в стандартных условиях и получают амиды 90. Затем, группу сложного этилового эфира в соединении 90 гидролизуют с использованием гидроксида лития или натрия в водной среде с получением кислот 91. Соединения, имеющие 2,6-дизамещение на циннамидной кольцевой системе, получают методом,проиллюстрированным на схеме 21. Коммерчески доступный 4,6-дихлорсалицилальдегид конденсируют с арилтиолами в основных условиях и получают диарилсульфид 92. Фенольную группу защищают аллилбромидом с получением O-аллилового производного 93. Метод, показанный в схеме 1, используют для получения соответствующей коричной кислоты 94, а затем аллильную группу удаляют путем катализируемого палладием(0) превращения в морфолин, в результате чего получают гидроксикоричную кислоту 95. Кислотную группу подвергают реакции сочетания с циклическим сложным аминоэфиром (n=0, 1, 2; R = Me, Et) в стандартных условиях и получают амид 96. В условиях основного гидролиза получают кислоту 97. Примеры Более подробное описание соединений и способов настоящего изобретения приводится в нижеследующих примерах, которые представлены лишь в целях иллюстрации и не должны рассматриваться как ограничение объема изобретения. Пример 1. (2,4-Дихлорфенил)[2-(Е-6-гидроксигексиламино)карбонил)этенил)фенил]сульфид. Пример 1 А. 2-[(2,4-Дихлорфенил)тио]бензальдегид. К перемешанному раствору 2,4-дихлортиофенола (2,0 г, 11,2 ммоль) в 25 мл безводного ДМФ добавляют карбонат калия (3,09 г, 22,4 ммоль), а затем 2-хлорбензальдегид (1,26 мл, 11,3 ммоль). Затем смесь нагревают в атмосфере азота при 70 С в течение 5 ч. Затем реакционную смесь оставляют для охлаждения до комнатной температуры и распределяют между эфиром и водой. Водный слой один раз экстрагируют эфиром и объединенный органический слой промывают водой и насыщенным раствором соли, сушат над сульфатом натрия и конденсируют в вакууме. Неочищенный продукт очищают флешхроматографией на силикагеле, элюируя смесью 5-10% эфир/гексан, в результате чего получают 2,62 г(9,25 ммоль, 83%) нужного альдегида в виде бесцветного масла, которое медленно отверждается после отстаивания при комнатной температуре. Пример 1 В. транс-2-[(2,4-Дихлорфенил)тио]коричная кислота. Смесь альдегида (1,50 г, 5,3 ммоль) примера 1 А, малоновой кислоты (1,21 г, 11,6 ммоль), пиперидина (78,6 мкл, 0,80 ммоль) в 8,0 мл безводного пиридина нагревают при 110 С в течение 2 ч. В течение этого периода времени выделение газа прекращается. Затем пиридин удаляют в вакууме. После этого добавляют воду и 3 н. вод.HCl, перемешивая при этом. Затем нужную коричную кислоту собирают путем фильтрации, промывают холодной водой и сушат в вакуумной печи в течение ночи с получением 1,56 г(4,8 ммоль, 91%) белого твердого вещества. Пример 1 С. (2,4-Дихлорфенил)[2-(Е-6-гидроксигексиламино)карбонил)этенил)фенил]сульфид. Суспензию кислоты (284 мг, 0,87 ммоль) примера 1 В в 5 мл метиленхлорида перемешивают с(СОСl)2 (84 мкл, 0,97 ммоль) и с одной каплей ДМФ в атмосфере азота в течение 90 мин. Растворитель удаляют в вакууме. Остаток (СОСl)2 удаляют с бензолом (2 х) в вакууме. Затем в делительную колбу,предварительно заполненную 6-амино-1-гексанолом (12 мг, 0,10 ммоль), основанием Хьюнига (22,8 мкл,0,13 ммоль) и DMAP (1,1 мг, 0,008 ммоль) в 2,0 мл CH2Cl2, по каплям медленно добавляют хлорангидрид(30 мг, 0,087 ммоль) в 1,0 мл СН 2 Сl2. Через 30 мин, реакционную смесь выливают в 3 н. НСl и экстрагируют этилацетатом (EtOAc). Органический слой промывают водой, сушат Na2SO4 и конденсируют при пониженном давлении. Неочищенный продукт очищают препаративной ТСХ с получением 21,0 мг (90%) указанного в заголовке соединения в виде бесцветного масла. 1 Н-ЯМР (300 МГц, CDCl3) : 1,31-1,48
МПК / Метки
МПК: C07D 233/61, C07C 323/62, A61P 29/00, A61K 31/165
Метки: противовоспалительные, соединения, иммуносупрессорные, клеточную, адгезию, ингибирующие
Код ссылки
<a href="https://eas.patents.su/30-5563-protivovospalitelnye-i-immunosupressornye-soedineniya-ingibiruyushhie-kletochnuyu-adgeziyu.html" rel="bookmark" title="База патентов Евразийского Союза">Противовоспалительные и иммуносупрессорные соединения, ингибирующие клеточную адгезию</a>
Соединения, обладающие противовоспалительной и иммунносупрессорной активностью, ингибирующие клеточную адгезию

Номер патента: 5207
Опубликовано: 30.12.2004
Авторы: Бойд Стивен А., Ксин Зили, Уинн Мартин, Фримен Дженнифер С., Зху Гуи-Донг, Вон Гелдерн Том, Гунавардана Индрани В., Линк Джеймс, Линч Джон К., Лиу Ганг, Стаеджер Майкл А., Дзае Хван-Соо, Пей Зонгхуа
МПК: A61K 31/10, A61P 29/00, C07C 323/62...
Метки: активностью, ингибирующие, соединения, иммунносупрессорной, противовоспалительной, адгезию, обладающие, клеточную
Формула / Реферат:
1. Соединение формулы I где R1, R2, R3, R4 и R5 независимо выбраны из a) водорода, b) галогена, c) C1-C10алкила, d) галоген-C1-C10алкила, e) C1-C10алкокси, f) циано, g) нитро, h) карбоксальдегида и при условии, что по крайней мере один из R1 или R3 является "цис-циннамидом" или "транс-циннамидом", определенным как где R8 и R9 независимо выбраны из a) водорода, b) C1-C10алкила, c) карбокси-C1-C10алкила, d)...
Хинолиновые и хиноксалиновые соединения, ингибирующие тирозинкиназы тромбоцитарного фактора роста и/или p56 lck

Номер патента: 2600
Опубликовано: 27.06.2002
Авторы: Персонс Пол Э., Спада Альфред П., Магвир Мартин П., Майерс Майкл
МПК: A61P 35/00, C07D 241/44, A61K 31/47...
Метки: хиноксалиновые, ингибирующие, тромбоцитарного, фактора, роста, хинолиновые, тирозинкиназы, соединения
Формула / Реферат:
1. Соединение формулы I где R1a представляет необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный оксагетероциклилокси или галоген; R1b представляет водород, необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный...
Хинолиновые и хиноксалиновые соединения, ингибирующие тирозинкиназы тромбоцитарного фактора роста и/или р56 lck

Номер патента: 4103
Опубликовано: 25.12.2003
Авторы: Спада Альфред П., Хе Вей, Майерз Майкл Р.
МПК: A61K 31/47, C07D 241/44, A61P 29/00...
Метки: соединения, ингибирующие, фактора, тромбоцитарного, хиноксалиновые, роста, тирозинкиназы, хинолиновые
Формула / Реферат:
1. Соединение формулы I где X представляет L1OH или L2Z2; L1 представляет -Z3-(CR3'aR3'b)3; L2 представляет -Z4-(CR3'aR3'b)q; Z1 представляет CH или N; Z2 представляет C5-7гидроксициклоалкил, необязательно замещенный гидрокси, C1-6алкилом, C1-6алкокси, диметилкарбамидом, ацетилом или этиламидом; C5-7гидроксициклоалкенил, необязательно замещенный гидрокси, C1-6алкилом, C1-6алкокси, диметилкарбамидом, ацетилом или этиламидом; или пяти- или...
Пиримидины, ингибирующие репликацию вич

Номер патента: 4049
Опубликовано: 25.12.2003
Авторы: Де Жонж Марк Рене, Койманс Люсьен Мария Хенрикус, Андрис Конрад Йозеф Лодевийк Марсель, Хо Чих Юнг, Лудовики Дональд Вилльям, Каваш Роберт В., Ван Акен Кун Жанн Альфонс, Кукла Майкл Джозеф, Хэрес Ян, Жанссен Поль Адриан Ян, Де Корт Барт
МПК: C07D 239/48, C07D 239/46, A61K 31/505...
Метки: пиримидины, вич, ингибирующие, репликацию
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксид, аддитивная соль, четвертичный амин или стереохимически изомерная форма, где R1 представляет водород; арил; формил; C1-6алкилкарбонил; C1-6алкил, C1-6алкилоксикарбонил, C1-6алкил, замещенный формилом, C1-6-алкилкарбонилом, C1-6алкилоксикарбонилом, C1-6алкилкарбонилокси; C1-6алкилоксиC1-6алкилкарбонил, замещенный C1-6алкилоксикарбонилом; R2a представляет циано или аминокарбонил; L представляет...
Интерлейкин-5 ингибирующие производные 6-азаурацила

Номер патента: 4740
Опубликовано: 26.08.2004
Авторы: Дерос Фредерик Дирк, Кусеманс Эрвин, Лякрамп Жан Фернан Арман, Фрейн Эдди Жан Эдгар, Фортэн Жером Мишель Клод
МПК: A61K 31/53, A61P 11/06, C07D 417/10...
Метки: интерлейкин-5, 6-азаурацила, производные, ингибирующие
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксиды, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где p представляет целое число, являющееся 0, 1 или 2; X представляет прямую связь; Y представляет O, S, NR5 или S(O)2; каждый R1 независимо представляет хлор или трифторметил; R2 представляет Het1; R3 представляет водород, C1-6алкил или C3-7циклоалкил; R4 представляет водород, C1-6алкил или C3-7циклоалкил; каждый R6...
Предыдущий патент: Способ повышения селективности гербицида на основе 1,3 – циклогександиона
Следующий патент: Эффективные противоопухолевые лекарственные средства
Случайный патент: Роторно-поршневые двигатели внутреннего сгорания.