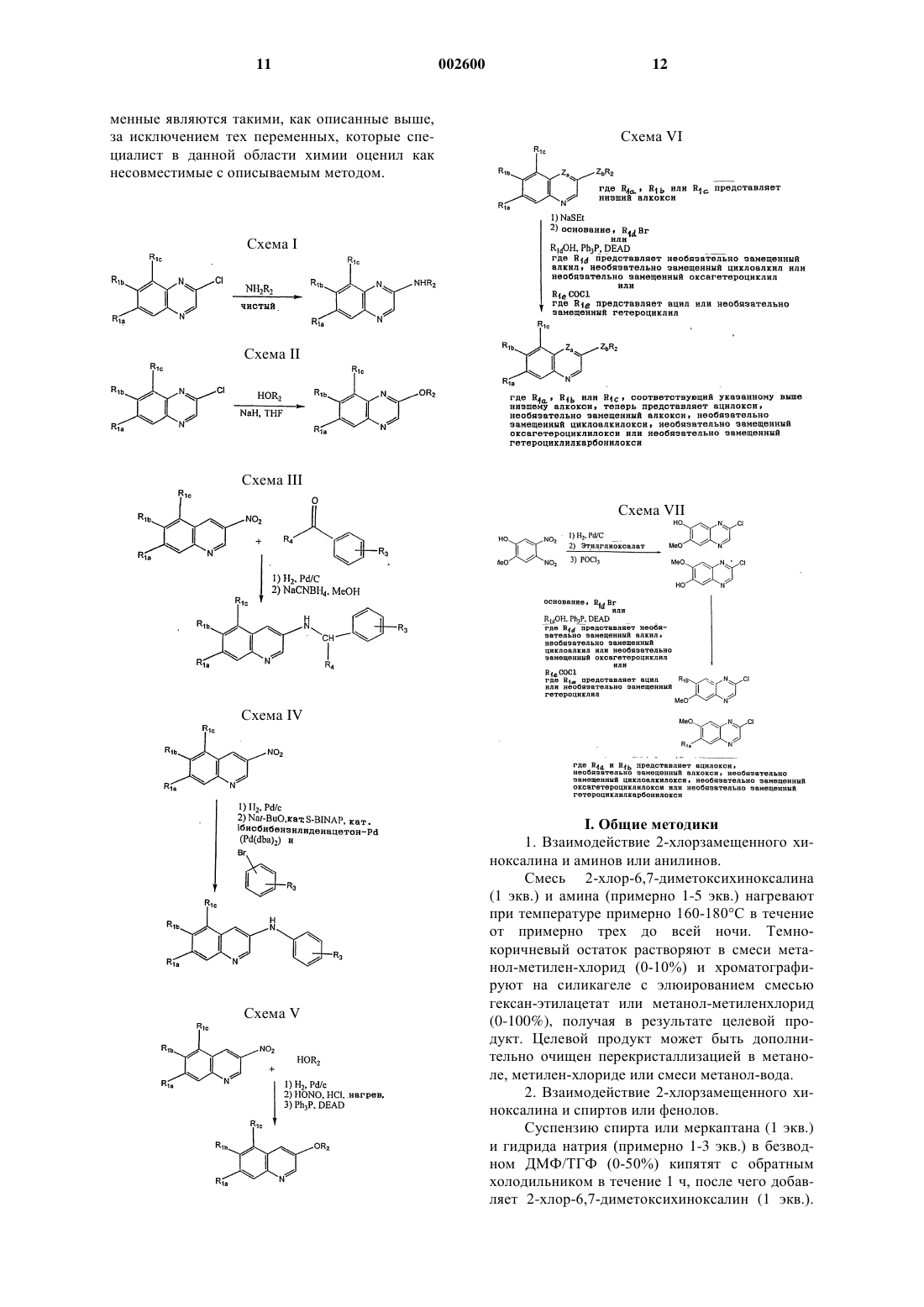
Хинолиновые и хиноксалиновые соединения, ингибирующие тирозинкиназы тромбоцитарного фактора роста и/или p56 lck
Номер патента: 2600
Опубликовано: 27.06.2002
Авторы: Персонс Пол Э., Майерс Майкл, Спада Альфред П., Магвир Мартин П.







Формула / Реферат
1. Соединение формулы I

где
R1a представляет необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный оксагетероциклилокси или галоген;
R1b представляет водород, необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный оксагетероциклилокси или галоген;
R1c представляет водород, необязательно замещенный C1-С10алкил или необязательно замещенный C1-С10алкокси,
R2 представляет

R3 представляет водород, орто- или парафтор или мета-С1-С3низший алкил, C1-С3низший алкокси, галоген или карбамоил;
Zd представляет N или СН; и
Zb представляет NH или О,
или его N-оксид, гидрат, сольват, пролекарство или соль, при условии, что R1a и R1b оба не являются необязательно замещенным C1-С10 алкилом.
2. Соединение формулы I по п.1, в котором R1a представляет необязательно замещенный C1-С3низший алкокси, необязательно замещенный моноциклический С1-С7циклоалкилокси, необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси.
3. Соединение по п.2, в котором R1a представляет необязательно замещенный C1-С3низший алкокси или необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси.
4. Соединение по п.3, в котором R1a представляет метокси, этокси, 2-(этокси)этокси, 2-(4-морфолинил)этокси или фуранилокси.
5. Соединение по п.1, в котором R1b представляет водород, необязательно замещенный C1-С3низший алкокси, необязательно замещенный моноциклический С3-С7циклоалкилокси или необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси.
6. Соединение по п.5, в котором R1b представляет водород или необязательно замещенный С1-С3низший алкокси.
7. Соединение по п.6, в котором R1b представляет метокси или этокси.
8. Соединение по п.1, в котором R1a и R1b представляют C1-С3низший алкокси.
9. Соединение по п.8, в котором R1a и R1b представляют метокси или этокси.
10. Соединение по п.1, в котором R1c представляет водород.
11. Соединение по п.1, в котором R3 представляет водород, орто- или парафтор или метаметил, трифторметил, метокси, фтор, хлор, бром или карбамоил.
12. Соединение по п.1, в котором Za представляет N.
13. Соединение по п.1, в котором Za представляет СН.
14. Соединение по п.1, в котором Zb представляет NH.
15. Соединение по п.1, в котором Zb представляет О.
16. Соединение по п.1, представляющее собой
2-анилино-6-хиноксалинол;
2-анилино-6-изопропоксихиноксалин;
2-фенокси-6-метоксихиноксалин;
2-(3-карбамоилфениламино)-6-метоксихиноксалин;
2-(2-фторфениламино)-6,7-диэтоксихиноксалин;
2-(3-трифторметилфениламино)-6,7-диэтоксихиноксалин;
фенил-[6-(тетрагидрофуран-3(R)илокси) хиноксалин-2-ил]амин;
(6-метоксихиноксалин-2-ил)-(3-метилфенил)амин;
6-метокси-2-фениламинохиноксалин;
2-анилино-6-этоксихиноксалин;
2-(3-метоксифениламино)-6,7-диэтоксихиноксалин;
2-(4-фторфениламино)-6,7-диэтоксихиноксалин;
6,7-диэтокси-2-феноксихиноксалин;
2-фениламино-6,7-диэтоксихиноксалин;
(6,7-диметоксихиноксалин-2-ил)-(3-фторфенил)амин;
2-(3-фторфениламино)-6,7-диэтоксихиноксалин;
(3-бромфенил)-(6,7-диметоксихиноксалин-2-ил)амин;
(6,7-диметоксихиноксалин-2-ил)фениламин; и
(3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин
или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемая соль.
17. Соединение по п.1, представляющее собой
фенил-[6-(тетрагидрофуран-3(R)-илокси)хиноксалин-2-ил]амин;
(6-метоксихиноксалин-2-ил)-(3-метилфенил)амин;
6-метокси-2-фениламинохиноксалин;
2-анилино-6-этоксихиноксалин;
2-(3-метоксифениламино)-6,7-диэтоксихиноксалин;
2-(4-фторфениламино)-6,7-диэтоксихиноксалин;
6,7-диэтокси-2-феноксихиноксалин;
2-фениламино-6,7-диэтоксихиноксалин;
(6,7-диметоксихиноксалин-2-ил)-(3-фторфенил)амин;
2-(3-фторфениламино)-6,7-диэтоксихиноксалин;
(3-бромфенил)-(6,7-диметоксихиноксалин-2-ил)амин;
(6,7-диметоксихиноксалин-2-ил)фениламин и
(3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин
или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемая соль.
18. Соединение по п.1, которое представляет собой фенил-[6-(тетрагидрофуран-3(R)-илокси)хиноксалин-2-ил]амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемую соль.
19. Соединение по п.1, которое представляет собой (6,7-диметоксихиноксалин-2-ил)-(3-фторфенил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемую соль.
20. Соединение по п.1, которое представляет собой (3-бромфенил)-(6,7-диметоксихиноксалин-2-ил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемую соль.
21. Соединение по п.1, которое представляет собой (3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемую соль.
22. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
23. Способ ингибирования активности тирозинкиназы фактора роста PDGF, включающий контактирование соединения по п.1 с композицией, содержащей тирозинкиназу PDGF.
24. Способ ингибирования активности тирозинкиназы Lck, включающий контактирование соединения по п.1 с композицией, содержащей тирозинкиназу Lck.
25. Способ ингибирования пролиферации клеток, дифференциации или высвобождения медиатора у пациента, страдающего от нарушения, характеризующегося такими пролиферацией, и/или дифференциацией, и/или высвобождением медиатора, включающий введение указанному пациенту фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
26. Способ по п.25, в котором нарушением является лейкоз, рак, глиобластома, псориаз, воспалительные заболевания,заболевания кости, фиброзные заболевания, артрит, фиброз легкого, фиброз почки, фиброз печени, атеросклероз или рестеноз после ангиопластики коронарной артерии, бедренной артерии или артерии почки.
27. Способ лечения пациента, подверженного патологии, связанной с гиперпролиферативным нарушением, включающий введение указанному пациенту фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
28. Способ по п.27, в котором патологией, связанной с гиперпролиферативным нарушением, является рак, восприимчивый к лечению путем ингибирования тирозинкиназы фактора роста PDGF.
29. Способ по п.28, в котором рак представляет собой рак головного мозга, рак яичника, рак толстой кишки, рак предстательной железы, рак легких, саркому Капоши или злокачественную меланому.
30. Способ по п.27, в котором указанной патологией является рестеноз.
31. Способ по п.30, в котором соединение по п.1 вводят посредством нанесения покрытия на расширительном устройстве (стенте), где покрытие содержит соединение по п. 1.
32. Способ лечения воспаления у пациента, страдающего от него, включающий введение указанному пациеэту фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
33. Способ лечения ревматоидного артрита, рассеянного склероза, системной красной волчанки, болезни "трансплантат против хозяина", астмы, воспалительного заболевания кишечника или панкреатита, включающий введение пациенту, нуждающемуся в таком лечении, ингибирующего тирозинкиназу Lck количества соединения по п.1.
Текст
1 Предпосылки создания изобретения 1. Область техники, к которой относится изобретение Настоящее изобретение относится к ингибированию пролиферации клеток и/или продуцирования клеточного матрикса, и/или передвижения клеток (хемотаксиса), и/или активации и пролиферации Т-клеток путем использования хинолиновых и хиноксалиновых соединений являвшихся полезными ингибиторами белковой тирозинкиназы (TKIs). Клеточная передача сигналов происходит посредством системы взаимодействий, включающей контакт клетки с клеткой, клетки с матриксом или внеклеточного рецептора с субстратом. Внеклеточный сигнал часто передается к другим частям клетки через опосредованное тирозинкиназой фосфорилирование, которое воздействует на белки субстрата за клеточной мембраной, связанной с передающим сигнал комплексом. Примером ферментов тирозинкиназ, вовлеченных в клеточную передачу сигналов, является конкретный ряд рецепторферментов, таких как инсулиновый рецептор,рецептор эпидермального фактора роста (EGFB) или рецептор тромбоцитарного фактора роста (PDGF-R). Для эффективного ферментативного фосфорилирования субстратных белков,содержащих тирозиновые остатки, требуется аутофосфорилирование фермента. Известно, что эти субстраты ответственны за разнообразные клеточные акты, включающие клеточную пролиферацию, продуцирования клеточного матрикса, клеточную миграцию, апоптоз и т.д. Понятно, что неуправляемое репродуцирование клеток, сверхпродуцирование матрикса или плохо регулируемая запрограммированная гибель клеток (апоптоз) вызывает много болезненных состояний. Эти болезненные состояния включают разнообразные типы клеток и представляют такие заболевания, как лейкоз, рак,глиобластома, псориаз, воспалительные заболевания, фиброзы, атеросклероз и рестеноз, возникающий после ангиопластики коронарных,бедренных или почечных артерий, или фибропролиферативное заболевание, такое как артрит,фиброз легкого, почки и печени. Kpoмe того,после коронарного шунтирования создаются условия для нерегулируемой клеточной пролиферации. Полагают, что ингибирование активности тирозинкиназы будет полезным для регулирования неуправляемого репродуцирования клеток, сверхпродуцирования матрикса или плохо регулируемой запрограммированной гибели клеток (апоптоза). Известно также, что определенные ингибиторы тирозинкиназы могут взаимодействовать с более чем одним типом фермента тирозинкиназы. Несколько ферментов тирозинкиназы очень важны для нормального функционирования организма. Например, было бы желательным ингибировать действие инсулина в боль 002600 2 шинстве обычных случаев. Таким образом, соединения, ингибирующие активность тирозинкиназы рецептора PDGF-R при концентрациях ниже концентраций, эффективных в ингибировании киназы инсулинового рецептора, могли бы давать ценные агенты для селективного лечения заболеваний, характеризующихся пролиферацией клеток, и/или продуцированием клеточного матрикса и/или передвижением клеток(хемотаксисом), таких как рестеноз. Настоящее изобретение относится к модулированию и/или ингибированию передачи сигналов в клетках, пролиферации клеток, продуцирования внеклеточного матрикса, хемотаксиса, регуляции аномального роста клеток и воспалительной реакции клеток. Более конкретно настоящее изобретение относится к применению замещенных хиноксалинов, обладающих способностью избирательно ингибировать дифференциацию, пролиферации или высвобождение медиатора путем эффективного ингибирования активности тирозинкиназы рецептора тромбоцитарного фактора роста (PDGF-R) и/или активности Lck тирозинкиназы. Уровень техники В ряде литературных источников описаны ингибиторы тирозинкиназы, избирательно действующие по отношению к ферментам рецепторов тирозинкиназы, таких как EGF-R или PDGFR, или нерецепторным цитозольным тирозинкиназам, таким как v-abl, p561ck или c-src. В недавних обзорах Spada and Myers (Exp. Opin.Opin. Ther. Patents 1995, 5(12), 1245) дано краткое описание литературы по ингибиторам тирозинкиназ и ингибиторам, избирательным к EGFR, соответственно. Кроме того. Law and Lydon дали краткое сообщение о противоопухолевой активности ингибиторов тирозинкиназ (Emerging Drugs: The Prospect For Improved Medicines 1996, 241-260). Известные ингибиторы активности тирозинкиназы PDGF-R включают ингибиторы на основе хинолина, описанные Maguire et al. (J.Med. Chem. 1994, 37, 2627). Класс ингибиторов на основе фениламинопиримидина был недавно описан Traxler et al. в ЕР 564409 и by Zimmerman, J.; and Traxler, P. et al. (Biorg.Med. Chem.al. (Prog. Nat. Acad. Sci. 1995, 92, 2558). Несмотря на прогресс в данной области, еще не существует средств из этих классов соединений, которые были бы одобрены для применения на людях для лечения пролиферативных заболевании. Взаимосвязь между многофакторным заболеванием рестенозом и PDGF и PDGF-R достаточно описана в научной литературе. Однако недавние разработки в изучении фиброзов легкого (Antoniades, H.N. et al. J. Clin. Invest. 1990,86, 1055), почки и печени (Peterson, T.С. Hepa 3tology, 1993, 17, 486) показали, что важную роль в этих заболеваниях также играют PDGF иPDGF-R. Например, гломерулонефрит является основной причиной почечной недостаточности,и, как показано Shultz et al. (Am. J. Physiol. 1988,255, F674) и Floege et al. (Clin. Exp. Immun. 1991,86, 334), PDGF был идентифицирован как мощный митоген in vitro для мезангиальных клеток. Как сообщалось Thornton, S.C.; et al. (Clin. Exp.Immun. 1981, 86, 79), TNP-альфа и PDGF (полученные от людей, больных ревматоидным артритом) являются основными цитокинами, вовлеченными в процесс пролиферации синовиальных клеток. Кроме того, уже идентифицированы конкретные типы опухолевых клеток (см.Silver, B.J., BioFactors, 1992, 3, 217), такие как глиобластома и саркома Капоши (Kaposi), которые осуществляют сверхэкспрессию белкаPDGF или рецептора, что ведет к неуправляемому росту опухолевых клеток через аутокринный или паракринный механизм. Таким образом, ожидается, что ингибитор PDGF тирозинкиназы будет полезен для лечения разнообразных, по-видимому неродственных болезненных состояний человека, которые можно характеризовать вовлечением в их этиологию PDGF и/илиPDGF-R. Роль различных нерецепторных тирозинкиназ, таких как р 56lск (далее "Lck"), в состояниях относящихся к воспалительным, включающих активацию и пролиферацию Т-клеток, уже описана Hanke, et al (Inflamm. Res. 1995, 44, 357) и Bolen and Brugge (Ann. Rev. Immunol., 1997,15, 371). Эти воспалительные состояния включают аллергию, аутоиммунную болезнь, ревматоидный артрит и отторжение трансплантата. В другом недавнем обзоре кратко описаны различные классы ингибиторов тирозинкиназ,включая соединения, обладающие ингибирующей активностью по отношению к Lck(Groundwater, et al Progress in Medicinal Chemistry, 1996, 33, 233). Ингибиторы активности Lck тирозинкиназ включают некоторые природные продукты, которые обычно являются неизбирательными ингибиторами тирозинкиназ, такие как ставроспорин, генистеин, некоторые флавоны и эрбстатин. Недавно сообщено дамнакантоле являющемся ингибитором Lck (Faltynek, et al.Biochemistry, 1995, 34, 12404). Примеры синтетических ингибиторов Lck включают: ряд дигидроксиизохинолиновых ингибиторов, которые, как сообщено (Burke, et al, J. Med. Chem. 1993, 36, 425), обладают активностью при низкой концентрации в пределах от микромолярной до субмикромолярной, и производное хинолина,которое, как обнаружено, является намного менее активным по отношению к Lck (IC50 = 610 мкМ). Исследователями был также раскрыт ряд 4-замещенных хиназолинов, ингибирующих Lck при низкой концентрации в пределах от микромолярной до субмикромолярной (Myers et al,WO 95/15758 и Myers et al, Bioorg. Med. Chem.(Hanke, et al, J. Biol. Chem. 1996, 271, 695) раскрыли два специфических пиразолопиримидиновых ингибитора, известных как РР 1 и РР 2,которые обладают активностью при низконаномолярной концентрации по отношению к Lck иFyn (другая киназа семейства Src). Не сообщалось об ингибиторах Lck, относящихся к соединениям на основе хинолина или хиноксалина. Поэтому ожидают, что хинолиновый или хиноксалиновый ингибитор активности тирозинкиназы Lck может быть полезным для лечения разнообразных неродственных болезненных состояний человека, которые можно характеризовать вовлечением в их этиологию передачи сигналов через тирозинкиназы Lck. Краткое описание изобретения Настоящее изобретение относится к соединению формулы IR1a представляет необязательно замещенный алкил, гидрокси, ацилокси, необязательно замещенный алкокси, необязательно замещенный циклоалкилокси, необязательно замещенный оксагетероциклилокси, необязательно замещенный гетероциклилкарбонилокси или галоген;R1b представляет водород, необязательно замещенный алкил, гидрокси, ацилокси, необязательно замещенный алкокси, необязательно замещенный циклоалкилокси, необязательно замещенный оксагетероциклилокси, необязательно замещенный гетероциклилкарбонилокси или галоген;R4 представляет водород или низший алкил;R5 и R6 независимо представляют водород или алкил или R5 и R6, взятые вместе с атомом азота, к которому R5 и R6 присоединены, образуют азагетроциклил;Zb представляет NH или О,или его N-оксиду, гидрату, сольвату, пролекарству или соли, при условии, что R1a и R1b не представляют оба необязательно замещенный алкил. Настоящее изобретение также относится к фармацевтической композиции, содержащей фармацевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель. Настоящее изобретение далее относится к промежуточным соединениям, пригодным для получения соединений формулы I, к способам получения промежуточных соединений и соединений формулы I и к применению соединения формулы I для лечения пациента,страдающего от или подверженного нарушениям или состояниям, включающим клеточную дифференциацию,пролиферацию,продуцирование точного матрикса или высвобождение медиатора. Подробное описание изобретения Использованные выше и во всем описании изобретения следующие термины имеют, если не указано иное, следующие значения. Определения"Эффективное количество" означает количество соединения по настоящему изобретению,эффективное в ингибировании активности тирозинкиназы PDGF-R и активности Lck тирозинкиназы, обеспечивающее требуемый терапевтический эффект."Aлкил" означает алифатическую углеводородную группу, которая может быть с разветвленной или неразветвленной, имеющую от примерно 1 до примерно 10 углеродных атомов. Предпочтительным алкилом является "низший алкил", имеющий от примерно 1 до примерно 3 углеродных атомов, причем более предпочтительным является метил. Разветвленная цепь означает, что к линейной алкильной цепи присоединены одна или несколько низших алкильных групп, таких как метил, этил или пропил. Алкильная группа является также необязательно замещенной алкокси, галогеном, карбокси, гидрокси или R5R6N- (где R5 и R6 независимо представляют водород или алкил или R5 и R6, взятые вместе с атомом азота, к которому R5 и R6 присоединены, образует азагетероциклил), а более предпочтительно необязательно замещенной фтором. Примеры алкила включают метил,фторметил, дифторметил, трифторметил, этил,н-пропил, изопропил, бутил, вторбутил, третбутил, амил и гексил."Циклоалкил" означает неароматическую моноциклическую кольцевую систему, имеющую от примерно 3 до примерно 7 углеродных атомов. Предпочтительные моноциклические циклоалкилы включают циклопентил, циклогексил и циклогептил; более предпочтительными являются циклогексил и циклопентил."Арил" означает ароматический карбоциклический радикал, содержащий от примерно 6 до примерно 10 углеродных атомов. Примером арила является фенил или нафтил или фенил или нафтил, замещенные одним или несколькими заместителями арильной группы, которые могут быть одинаковыми или разными, причем,заместитель арильной группы, включает водород, гидрокси, галоген, алкил, алкокси, карбокси, алкоксикарбонил или Y1 Y2 NCO-, где Y1 иY2 независимо представляют водород или алкил."Гетероарил" означает примерно 5-10 членную ароматическую моноциклическую или полициклическую углеводородную кольцевую систему, в которой один или несколько углеродных атомов является(ются) элементом(ами),отличным(и) от углерода, например азотом, кислородом или серой. Гетероарил может быть также замещенным одним или несколькими вышеупомянутыми "заместителями арильной группы". Пример гетероарильной группы включает замещенный пиразинил, фуранил, тиенил,пиридил, пиримидинил, изоксазолил, изотиазолил, оксазолил, тиазолил, пиразолил, фуразанил,пирролил, имидазо[2,1-b]тиазолил, бензофуразанил, индолил, азаиндолил, бензимидазолил,бензотиенил, хинолинил, имидазолил и изохинолинил."Гетероциклил" означает примерно 4-7 членную моноциклическую кольцевую систему,в которой один или несколько атомов в кольцевой системе является(ются) элементом(ами),отличным(и) от углерода и выбранным из азота,кислорода или серы. Наличие "аза" или "окса" в качестве приставки перед гетероциклилом означает, что в качестве кольцевого атома присутствует, по крайней мере, азот или кислород соответственно. Примеры моноциклических гетероциклильных групп включают пиперидил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,3-диоксоланил, 1,4 диоксанил, тетрагидрофуранил, тетрагидротиофенил, тетрагидротиопиранил и подобные. Примеры гетероциклильных фрагментов включают хинуклидил, пентаме-тиленсульфид, тетрагидропиранил, тетрагидротиофенил, пирролидинил, тетрагидрофуранил или 4-пиперидинопиперидин."Гетерoциклилкарбонилокси" означает гетероциклил-С(О)O-группу, в которой гетероциклил такой, как определено выше. Примером гетероциклилкарбонилокси группы является"Ацил" означает Н-СО- или алкил-COгруппу, в которой алкильная группа такая, как описано выше. Предпочтительные ацилы содержат низкий алкил. Примеры ацильных групп включают формил, ацетил, пропаноил, 2 метилпропаноил, бутаноил и капроил."Алкокси" означает алкил-O-группу, в которой алкильная группа такая, как описано выше. Предпочтительным алкокси является "низший алкокси", имеющий от примерно 1 до примepно 3 углеродных атомов; более предпочтительным является метокси. Алкокси может быть необязательно замещен одной или несколькими группами, включающими алкокси, карбокси,алкоксикарбонил, карбоксиарил или R5R6N- (гдеR5 и R6 такие, как определено выше). Примеры алкоксигрупп включают метокси, этокси, нпропокси, изопролокси, н-бутокси, гептокси, 2(морфолин-4-ил)этокси и 2-(этокси)этокси."Циклоалкилокси" означает циклоалкил-Огруппу, в которой циклоалкильная группа такая,как описано выше. Примерные циклоалкилоксигруппы включают циклопентилокси или циклогексилокси."Гетероциклилокси" означает гетероциклил-О-группу, в которой гетероциклильная группа такая, как описана выше. Примеры гетероциклилоксигруппы включают пентаметиленсульфидокси, тетрагидропиранилокси, тетрагидротиофенилокси, пирролидинилокси или тетрагидрофуранилокси."Арилокси" означает арил-О-группу, в которой арильная группа такая, как описанная выше."Гетероарилокси" означает гетероарил-Огруппу, в которой гетероарильная группа такая,как описано выше."Ацилокси" означает ацил-О-группу, в которой ацильная группа такая, как описана выше."R5R6N-" означает замещенную или незамещенную аминогруппу, в которой R5 и R6 такие, как определены выше. Примеры группы включают амино (H2N), метиламино, этилметиламино, диметиламино и диэтиламино."R5R6NCO-" означает замещенную или незамещенную карбамоильную группу, в которойR5 и R6 такие, как определены выше. Примерами группы являются карбамоил (H2NCO-) и диметиламинокарбамоил (Me2NCO-)."Галоген" означает фтор, хлор, бром или иод. Предпочтительными являются фтор, хлор или бром, а более предпочтительны фтор или хлор."Пролекарство" означает форму соединения формулы I, пригодную для введения пациенту, не вызывающую токсичности, раздражения, аллергической реакции и подобного и эффективную для определенного использования; такие формы включают кетальную, сложноэфирную и цвиттерионную формы. Пролекарство трансформируется in vivo в исходное соединение указанной выше формулы, например,гидролизом в крови. Подробное описание дано в Т. Higuchi and V. Stella, Prord-rugs as Novel De 002600Association and Pergamon Press, 1987; оба материала включены в данное описание в качестве ссылки."Сольват" означает физическую ассоциацию соединения по настоящему изобретению с одной или несколькими молекулами растворителя. Эта физическая ассоциация включает различные степени образования ионной и ковалентной связей, включая образование водородной связи. В некоторых случаях сольват может быть выделен, например, когда одна или несколько молекул растворителя внедрены в кристаллическую решетку кристаллического твердого вещества. "Сольват" включает как сольваты в виде раствора, так и выделяемые сольваты. Типичные сольваты включают этанолаты, метанолаты и подобные. "Гидрат" - это сольват, в котором молекула(ы) растворителя представляет(ют) собой Н 2O. Предпочтительные варианты воплощения изобретения Предпочтительным соединением по настоящему изобретений является соединение формулы I, в котором R1a представляет необязательно замещенный низший алкокси, необязательно замещенный моноциклический циклоалкилокси, необязательно замещенный гетероциклилкарбонилокси или необязательно замещенный моноциклический оксагетероциклилокси,более предпочтительно R1a представляет необязательно замещенный низший алкокси или необязательно замещенный моноциклический оксагетероциклилокси, а еще более предпочтительно R1a представляет метокси, этокси, 2(этокси)этокси, 2-(4-морфолинил)этокси или фуранилокси. Другим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором R1b представляет водород, необязательно замещенный низший алкокси, необязательно замещенный моноциклический циклоалкилокси, необязательно замещенный гетероциклилкарбонилокси или необязательно замещенный моноциклический оксагетероциклилокси, более предпочтительно R1b представляет водород или необязательно замещенный низший алкокси, а еще более предпочтительно R1b представляет метокси или этокси. Следующим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором R1a и R1b представляют низший алкокси, причем более предпочтительным низшим алкокси является метокси или этокси. Следующим предпочтительным соединением по настоящему изобретений является соединение формулы I, в котором R1c представляет водород или необязательно замещенный низ 9 ший алкокси, а более предпочтительно R1c представляет водород, метокси или этокси. Следующим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором R2 представляет Следующим предпочтительным соединением по настоящему изобретений является соединение формулы I, в которой R2 представляет Следующим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором R3 представляет водород, орто- или парафтор или метаметил,трифторметил, метокси, фтор, хлор, бром или карбамоил. Следующим предпочтительным соединением по настоящему изобретению является соединением формулы I, в котором R4 представляет водород или метил. Следующим предпочтительным соединением по настоящему изобретений является соединение формулы I, в котором Za представляетN. Следующим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором Za представляет СН. Следующим предпочтительным соединением по настоящему изобретений является соединение формулы I, в котором Zb представляетNH. Следующим предпочтительным соединением по настоящему изобретению является соединение формулы I, в котором Zb представляет О. Предпочтительные соединения по настоящему изобретению включают следующее: 2-анилино-6-хиноксалинол; 2-R)метилбензиламино)-6,7-диэтоксихиноксалин; 2-анилино-6-изопропоксихиноксалин; 2-фенокси-6-метоксихиноксалин;(3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин. Более предпочтительными соединениями является следующие: фенил-[6-(тетрагидрофуран-3(R)-илокси)хиноксалин-2-ил]амин; бензил-(6,7-диметоксихиноксалин-2-ил) амин; 2-S)метилбензиламино)-6,7-диэтоксихиноксалин; 2-бензиламино-6,7-диэтоксихиноксалин;(3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин. Следует понимать, что настоящее изобретение охватывает все подходящие комбинации отдельных предпочтительных групп, упомянутых в данном описании. Соединения по настоящему изобретению могут быть получены с использованием методик, известных из литературы, из известных соединений или легко получаемых промежуточных соединений. Примерные общие методики даны ниже. Кроме того, соединения формулы I получают в соответствии со схемами I-VI, где пере 11 менные являются такими, как описанные выше,за исключением тех переменных, которые специалист в данной области химии оценил как несовместимые с описываемым методом.(1 экв.) и амина (примерно 1-5 экв.) нагревают при температуре примерно 160-180 С в течение от примерно трех до всей ночи. Темнокоричневый остаток растворяют в смеси метанол-метилен-хлорид (0-10%) и хроматографируют на силикагеле с элюированием смесью гексан-этилацетат или метанол-метиленхлорид(0-100%), получая в результате целевой продукт. Целевой продукт может быть дополнительно очищен перекристаллизацией в метаноле, метилен-хлориде или смеси метанол-вода. 2. Взаимодействие 2-хлорзамещенного хиноксалина и спиртов или фенолов. Суспензию спирта или меркаптана (1 экв.) и гидрида натрия (примерно 1-3 экв.) в безводном ДМФ/ТГФ (0-50%) кипятят с обратным холодильником в течение 1 ч, после чего добавляет 2-хлор-6,7-диметоксихиноксалин (1 экв.). 13 Полученную смесь кипятят с обратным холодильником в течение примерно одного-четырех часов. Нейтрализует суспензию до примерно рН 5-8 и распределяют ее между метиленхлоридом и рассолом. Остаток после концентрирования метиленхлорида хроматографируют на силикагеле, элюируя смесью гексан-этилацетат или метанол-метиленхлорид (0-10%), с получением целевого продукта. 3. Реакция восстановительного аминирования с аминохинолинами и альдегидами или кетонами. Подходящим образом замещенный 3 аминохинолин (1 экв.) перемешивают с 1 экв. подходящего альдегида или кетона в метаноле(или другой подходящей смеси растворителей) до тех пор, пока ТСХ не покажет завершение образования имина. Добавляют избыточныйNaCNBH4 или NаВH4 или другой подходящий восстановитель и смесь перемешивают, пока ТСХ не покажет израсходование промежуточного имина. Смесь концентрируют и остаток хроматографируют на силикагеле смесью гексан-этилацетат (0-100%) или хлороформметанол (0-20%), с получением целевого продукта. 4. Взаимодействие 3-аминозамещенных хинолинов и бромфенильных соединений. Подходящим образом замещенный 3 аминохинолин (1 экв.) перемешивают с примерно 1,4 экв. сильного основания, такого как третбутоксид натрия, 1 экв. подходящего бромфенильного соединения и каталитическими количествами 2,2'-бис (дифенилфосфино)-1,1'-бинафтила (S-BINAP) и бис (дибинзилиденацетон)палладия (Pd(dba)2) в инертном органическом растворителе, таком как толуол, в атмосфере инертного газа такого как аргон, и нагревают до примерно 80 С в течение ночи. Смесь охлаждают, разбавляют растворителем, таким как эфир, фильтруют, концентрируют и хроматографируют смесью 50% EtOAc-гексан с получением целевого продукта. 5. Образование простого эфира из 3 гидроксизамещенных хинолинов в условиях реакции Мицунобу (Mitsunobu). Раствор подходящим образом замещенного гидроксихиноксалина (при примерно 0-25 С) в ТГФ обрабатывают 1 экв. каждого из желаемых спиртов, трифенилфосфином и наконец диэтилазодикарбоксилатом (DEAD) или подходящим эквивалентом. Ход реакции контролируют с помощью ТСХ и по окончании реакции(от примерно 1 до примерно 24 ч) смесь концентрируют и остаток хроматографируют на силикагеле с получением целевого продукта. 6. Деалкилирование хинолина или хиноксалина, замещенного низшим алкокси с последующим алкилированием. Подходящий хинолин или хиноксалин, замещенный низшим алкокси (1 экв.) в ДМФ обрабатывают избыточным количеством этантио 002600 14 лата натрия (обычно примерно 2 или более экв.) и реакционную смесь перемешивают при нагревании в течение примерно 1-24 ч. Смесь распределяют между водой и этилацетатом. Экстракционная обработка с последующей хроматографией, если необходимо, дает соответствующий целевой продукт: гидроксизамещенный хинолин или хиноксалин. Гидроксизамещенный хинолин или хиноксалин может быть алкилирован с использованием условий реакции Мицунобу, подробно описанных выше. Альтернативно простое алкилирование с использованием методов, хорошо известных в данной области химии, реакционноспособным алкил- или бензилгалогенидом с использованием NaH или другого подходящего основания в подходящем растворителе дает целевой алкилированный продукт. 7. Окисление азота в хинолине или хиноксалине до соответствующего N-оксида. Иминовый (=N-) фрагмент в хинолиновом или хиноксалиновом соединении формулы (I) может быть превращен в соответствующее соединение, в котором иминовый фрагмент окислен до N-оксида, предпочтительно взаимодействием с перкислотой, например перуксусной кислотой в уксусной кислоте или м-хлорпероксибензойной кислотой в инертном растворителе, таком как дихлорметан, при температуре в пределах от примерно комнатной температуры до температуры кипения, предпочтительно при повышенной температуре. Соединения по настоящему изобретению могут быть использованы в форме свободного основания или свободной кислоты или в форме их фармацевтически приемлемой соли. Все формы находятся в объеме настоящего изобретения. Когда соединения по настоящему изобретению замещенно-основной группой, образуются кислотно-аддитивные соли, являющиеся более удобной формой для применения; практически применение соединения в форме соли, по существу, равнозначно применению соединения в форме свободного основания. Кислоты, которые могут быть использованы для получения кислотно-аддитивных солей, включают предпочтительно те, которые при связывании со свободным основанием создают фармацевтически приемлемые соли, то есть соли, анионы которых не токсичны для пациента в фармацевтических дозах солей, благодаря чему полезны ингибирующие действия на PDGF, присущие свободному основанию, и не вызывают вредных побочных действий, обусловленных анионами. Хотя фармацевтически приемлемые соли указанных основных соединений являются предпочтительными, но все кислотно-аддитивные соли полезны в качестве источников формы свободного основания, даже если конкретная соль как таковая требуется лишь в качестве промежуточного продукта, как, например, в 15 случае, когда соль получают лишь для целей очистки и идентификации или когда ее используют в качестве промежуточного продукта для получения фармацевтически приемлемой соли ионообменными методами. Фармацевтически приемлемыми солями, включенными в объем настоящего изобретения, являются соли, производные от следующих кислот: минеральные кислоты, такие как хлороводородная кислота,серная кислота, фосфорная кислота и сульфаминовая кислота, и органические кислоты, такие как уксусная кислота, лимонная кислота,молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота,п-толуолсульфоновая кислота, циклогексилсульфаминовая кислота, хинная кислота и подобные. Соответствующие кислотно-аддитивные соли включают следующие: гидрогалогениды, например гидрохлорид и гидробромид,сульфат, фосфат, нитрат, сульфамат, ацетат,цитрат, лактат, тартарат, малонат, оксалат, салицилат, пропионат, сукцинат, фумарат, малеат,метилен-бисгидроксинафтоаты,гентизаты,мезилаты, изэтиноаты и ди-п-толуоилтартратесметансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, циклогексилсульфамат и хинат, соответственно. В соответствии с другим признаком настоящего изобретения кислотно-аддитивные соли соединений по настоящему изобретению получают взаимодействием свободного основания с соответствующей кислотой с применением или адаптацией известных методов. Например, кислотно-аддитивные соли соединений по настоящему изобретению получают либо растворением свободного основании в водном или водно-спиртовом растворе или других подходящих растворителях, содержащих соответствующую кислоту, и выделением соли выпариванием раствора, либо взаимодействием свободного основания и кислоты в органическом растворителе, причем в этом случае соль выделяется непосредственно или она может быть получена путем концентрирования раствора. Соединения по настоящему изобретению могут быть регенерированы из кислотноаддитивных солей путем применения или адаптации известных методов. Например, исходные(родоначальные) соединения по настоящему изобретению могут быть регенерированы из их кислотно-аддитивных солей обработкой щелочью, например водным раствором бикарбоната натрия или водным раствором аммиака. В случае соединения по настоящему изобретению, замещенного кислотной группой,могут быть образованы основно-аддитивные соли, являющиеся более удобной формой для применения; практически применение соединения в форме соли, по существу, равнозначно применению соединения в форме свободной кислоты. Основания, которые могут быть ис 002600 16 пользованы для получения основно-аддитивных солей, включают предпочтительно те, которые при соединении со свободной кислотой дают фармацевтически приемлемые соли, то есть соли, катионы которых не токсичны для организма животного в фармацевтических дозах солей,благодаря чему полезное ингибирующее действие на PDGF, присущие свободной кислоте, не сопровождается вредными побочными эффектами, обусловленными катионам. Фармацевтически приемлемыми солями, включающими,например, соли щелочных и щелочно-земельных металлов, в объеме настоящего изобретения являются соли, производные от следующих оснований: гидрид натрия, гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид алюминия, гидроксид лития, гидроксид магния,гидроксид цинка, аммиак, триметиламмиак,триэтиламмиак, этилендиамин, н-метилглюкамин, лизин, аргинин, орнитин, холин, N,N'дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, н-бензилфенетиламин, диэтиламин, пиперазин, трис(гидроксиметил) аминометан, гидроксид тетраметиламмония и подобные. Соли металлов соединений по настоящему изобретению могут быть получены контактированием гидрида, гидроксида, карбоната или подобного реакционноспособного соединения выбранного металла в водном или органическом растворителе с соединением в форме свободной кислоты. Используемым водным растворителем может быть вода или смесь воды с органическим растворителем, предпочтительно спиртом,таким как метанол или этанол, кетоном, таким как ацетон, алифатическим простым эфиром,таким как тетрагидрофуран, или сложным эфиром, таким как этилацетат. Такие реакции обычно проводят при температуре окружающей среды, но при необходимости их можно проводить при нагревании. Соли соединений с аминами по настоящему изобретению могут быть получены контактированием амина в водном или органическом растворителе с соединением в форме свободной кислоты. Подходящие водные растворители включают воду и смеси воды со спиртами, такими как метанол или этанол, простыми эфирами, такими как тетрагидрофуран, нитрилами,такими как ацетонитрил, или кетонами, такими как ацетон. Аналогичным образом могут быть получены соли с аминокислотами. Соединения по настоящему изобретению могут быть регенерированы из основноаддитивных солей путем применения или адаптации известных методов. Например, исходные(родоначальные) соединения по настоящему изобретению могут быть регенерированы из их основно-аддитивных солей обработкой кислотой, например хлороводородной кислотой. Кроме пользы, как таковых, в качестве активных соединений, соли соединений по на 17 стоящему изобретению полезны также для очистки соединений, например, с использованием разницы в растворимости между солями и родоначальными соединениями, побочными продуктами и/или исходными материалами, методами,хорошо известными специалистам в данной области химии. Соединения по настоящему изобретению могут содержать асимметрические центры. Эти асимметрические центры могут независимо иметь R или S конфигурацию. Для специалистов в данной области химии очевидно также, что некоторые соединения формулы I могут иметь геометрическую изомерию. Геометрические изомеры включают цис- и трансформы соединений по настоящему изобретению, т.е. соединения, имеющие алкенильные фрагменты или заместители в кольцевых системах. Кроме того,бициклические кольцевые системы включают эндо- и экзоизомеры. Настоящее изобретение включает отдельные геометрические изомеры,стереоизомеры, энантиомеры и их смеси. Такие изомеры могут быть выделены из их смесей путем применения или адаптации известных методов, например методов хроматографии и методов перекристаллизации, или могут быть получены в отдельности из соответствующих изомеров их промежуточных соединений, например, путем применения или адаптации методов, описанных в данном описании. Исходные материалы и промежуточные соединения получают путем применения или адаптации известных методов, например. таких,как описаны в ссылочных примерах, или их очевидных химических эквивалентов, или методами, описанными в соответствии с изобретением в данном описании. Настоящее изобретение далее проиллюстрировано примерами, которые не ограничивают его объем, описывающими получение соединений по настоящему изобретению. Кроме того, следующие далее примеры представляют способы, используемые для синтеза соединений по настоящему изобретению. Пример 1. 2-(3-Фторфениламино)-6,7-диэтоксихиноксалин. К 0,25 г (0,989 ммоль) 2-хлор-6,7 диэтоксихиноксалина добавляют 2 мл мфторанилина. Эту смесь нагревают в атмосфере в течение ночи до 120 С. Полученную смесь хроматографируют (CH2Cl2:EtOH=30:1) с получением частично очищенного продукта. Это твердое вещество растирают с этилацетатом с получением 0,175 г продукта в виде коричневато-желтого твердого вещества с выходом 54,1% 12,66. Найдено: С 65,30; Н 5,30; N 12,41. Пример 2. 2-Анилино-6-метоксихиноксалингидрохлорид. К 2-хлор-6-метоксихиноксалину (0,93 г,4,8 ммоль) в атмосфере аргон добавляют анилин(1,3 мл, 14,3 ммоль). Реакционную смесь нагревают при 120 С в течение 2 ч, а затем при 150 С в течение 1,5 ч. Смесь охлаждают и добавляютCH2Cl2. Полученную суспензию перемешивают и оранжевое твердое вещество отфильтровывают, промывают смесью CH2Cl2-Et2O и затем интенсивно перемешивают в H2O в течение 40 мин, фильтруют и промывают Et2O с получением светло-желтого твердого вещества. Следующие соединения получают аналогичным образом из соответствующего исходного материала. 2-(3-Карбамоилфениламино)-6-метоксихиноксалин, т.пл. 247 С. Элементный анализ. Вычислено для C16H14N4O20,25 Н 2O: С 64,31; Н 4,89; N 18,75. Найдено: С 64,24; Н 5,04; N 18,75; 2-(2-Фторфениламино)-6,7-диэтоксихиноксалин, т.пл. 184 С. Элементный анализ. Вычислено для C18H18FN3O2: С 66,04; Н 5,54; F 5,80; N 12,84. Найдено: С 65,75; Н 5,61; N 12,68; 2-(3-Трифторметилфениламино)-6,7-диэтоксихиноксалин, т.пл. 158 С. Элементный анализ. Вычислено для С 19 Н 18F3N3 О 2: С 60,47; Н 4,81; F 15,10; N 11,14. Найдено: С 60,27; Н 4,84;(3-Хлорфенил)-(6,7-диметоксихиноксалин 2-ил)амин, т.пл. 187-188 С. Элементарный анализ. Вычислено для С 18 Н 14 СlN3 О 2: С 60,86; Н 4,47; Сl 11,23; N 13,31. Найдено: С 60,85; Н 4,59,N 12,36. Пример 3. 2-Бензиламино-6,7-диэтоксихиноксалин. К 0,3 г (1,19 ммоль) 2-хлор-6,7 диэтоксихиноксалина добавляют 2 мл бензиламина. Эту смесь нагревают в атмосфере азота в течение ночи до 120 С. Полученную смесь распределяют между CH2Cl2 и насыщенным раствором NаНСО 3. Органический слой концентрируют и остаток хроматографируют (CH2Cl:EtOH=30:1) с получением 0,337 г продукта в виде желтого твердого вещества с выходом 87,6% (т.пл. 136 С). Элементарный анализ. Вычислено для C19H21N3O2: С 70,57; Н 6,54; N 12,99. Найдено: С 70,54; Н 6,66; N 12,80. Следующие соединения получают аналогичным образом из соответствующих исходных материалов.(3-Бромбензил)-(6,7-диметоксихиноксалин-2-ил)амин, т.пл. 199-206 С. Элементарный анализ. Вычислено для C17H16BrN3O2: С 54,56; Н 4,31; Вr 21,35; N 11,23. Найдено: С 49,90; Н 4,00; N 10,14; Бензил-(6,7-диметоксихиноксалин-2-ил) амин, т.пл. 210-214 С. Элементарный анализ. Вычислено для С 17 Н 17N3O2: С 69,14; Н 5,80; N 14,23. Найдено: С 61,78; Н 5,47; N 12,64; и 2-Бензиламино-6,7-диэтоксихиноксалин,т.пл. 136 С. Элементарный анализ. Вычислено для C19H21N3O2: С 70,57; Н 6,55; N 12,99. Найдено: С 70,54; Н 6,66; N 12,80. Пример 4. 2-R)Метилбензиламино)6,7-диэтоксихиноксалин. К 0,3 г (1,19 ммоль) 2-хлор-6,7-диэтоксихиноксалина добавляют 2 мл (R)-(+)-метилбензиламина. Эту смесь нагревают в атмосфере азота в течение трех дней до 120 С. Полученную смесь распределяют между СНСl3 и насыщенным раствором NаНСО 3. Органический слой концентрируют и остаток хроматографируют (CH2Cl2:EtOH=30:1) с получением 0,118 г продукта в виде желтого твердого вещества с выходом 29,4% (т.пл. 55-56 С). Элементный анализ. Вычислено для C20H23N3O20,25 Н 2 О: С 70,26; Н 6,93; N 12,29. Найдено: С 70,56; Н 6,80; N 12,35. Следующее соединение получают аналогичным образом из соответствующих исходных материалов. 2-S)Метилбензиламино)-6,7-диэтоксихиноксалин, т.пл. 55-58 С. Элементный анализ. Вычислено для C20H23N3O20,25 Н 2 О: С 70,26; Н 6,93; N 12,29. Найдено: С 70,49; Н 6,89; N 12,23. 20 Пример 5. 2,7-Бисциклогексилокси-6 метоксихиноксалин. К раствору NaH (0,32 г, 8 ммоль) в ДМФ (5 мл) в атмосфере добавляют по каплям циклогексанол (0,7 мл, 6,7 ммоль). Смесь перемешивают при комнатной температуре в течение 25 мин, после чего добавляют порциями 2-хлор 6,7-диметоксихиноксалин. Реакционную смесь перемешивают 15 мин при комнатной температуре, 2 ч при 90 С и 1 ч при 110 С. Смесь охлаждают, гасят добавлением Н 2 О. Органический слой промывают водой и рассолом, сушат(МgO4) и хроматографируют (10% EtOAc/ гексаны) с получением воскообразного белого твердого вещества (т.пл. 75-78 С). Элементный анализ. Вычислено для C21H28N2O3: С 70,76; Н 7,92; N 7,86. Найдено: С 70,81; Н 7,79; N 7,70. Следующее соединение получают аналогичным образом из соответствующих исходных материалов. 2-Фенокси-6-метоксихиноксалин, т.пл.7981 С; и 6,7-Диэтокси-2-феноксихиноксалин, т.пл. 130-131 С. Элементный анализ. Вычислено дляC18H18N2O3: С 69,66; Н 5,85; N 9,03. Найдено: С 69,53; Н 5,82; N 8,91. Пример 6. Циклогексил-(6,7-диметоксихиноксалин-2-илметил)амин. К 0,067 М раствору 6,7-диметокси-2 хиноксалинкарбоксальдегида в смеси (2:1) МеОН-1,2-дихлорэтан (7,5 мл, 0,5 ммоль) добавляют циклогексиламин (0,11 мл, 0,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, после чего добавляют NaBH4 (0,038 г, 1 ммоль) и перемешивают реакционную смесь в течение ночи. Затем смесь концентрируют и хроматографируютEtOAc/гексанах и обрабатывают НСl в EtOH). Полученный раствор концентрируют и твердый продукт растирают с получением белого твердого вещества после высушивания в вакууме при 60 С (т.пл. 185-190 С, разлож.). Элементный анализ. Вычислено для С 17 Н 23N3O2 НСl: С 60,44; Н 7,16; N 12,44. Найдено: С 60,48; Н 6,88;Buchwald, et al, J. Am. Сhеm. Soc., 1996, 118,7215. К толуоловому раствору 2-циклогексиламино-6-метокси-7-бромхиноксалина (0,1 г,0,3 ммоль) в атмосфере аргона добавляют морфолин (0,1 г, 0,3 ммоль), трет-бутоксид натрияPd(dba)2 (кат., 0,001 г). Реакционную смесь нагревают до 80 С в течение ночи. Смесь охлаждают, разбавляют Et2O, фильтруют, концентрируют и хроматографируют (50% EtOAc/ гексаны). Продукт перекристаллизовывают из смеси EtOAc/гексаны с получением (в двух сбо 21 рах) желтого твердого вещества (т.пл. 194196 С). Элементный анализ. Вычислено дляC19H26N4O2: С 66,64; Н 7,65; N 16,36. Найдено: С 66,60; Н 7,60; N 16,51. Пример 8. 3-Циклогексилокси-6,7-диметоксихинолин. К ТГФ раствору (30 мл) при 0 С добавляют 3-гидрокси-6,7-диметоксихинолин (0,237 г,1,15 ммоль), циклогексанол (0,347 г, 3,46 ммоль), Ph3P (0,908 г, 3,46 ммоль). Порциями добавляют диэтилазодикарбоксилат, пока раствор не приобретет темно-красный цвет (0,663 г,3,81 ммоль). Через 4 ч раствор концентрируют и остаток хроматографируют (50% EtOAc в гексанах). Продукт перекристаллизовывают из смеси изопропанол/гексаны с получением хлороводородной соли в виде белого твердого вещества (т.пл. 229-232 С, разлож). Пример 9. 2-Анилино-6-хиноксалинол. Методом, описанным в Feutrill, G.I.: Mirrington, R.N. Tet. Lett. 1970, 1327, превращают арилметиловый эфир в производное фенола. К 2-анилино-6-метоксихиноксалину (0,27 г, 1,07 ммоль) в атмосфере аргона в ДМФ добавляют натриевую соль этантиола (0,19 г, 2 ммоль). Реакционную смесь нагревают до 110 С в течение ночи. Смесь концентрируют и распределяют между EtOAc и смесью Н 2O/5% винная кислота,так что рН водного слоя становится равным приблизительно 4. Органический слой промывают водой (4 раза) и затем 2,5% NaOH (4 раза). Основные слои объединяют, промывают ЕТОАсEtOAc. Органические слои объединяют,пpoмывают рассолом, сушат (Na2SO4) и концентрируют. Полученное твердое вещество хроматографируют (50% EtOAc/гексаны). Путем растирания продукта с Et2O получают образец для анализа в виде желтого порошка (т.пл. 211213 С). Элементный анализ. Вычислено для С 14 Н 11N3 О: С 70,88; Н 4,67; N 17,71. Найдено: С 70,64; Н 4,85; N 17,58. Пример 10. Фенил-[6-(тетрагидрофуран 3(R)-илокси)хиноксалин-2-ил]амин. К ТГФ раствору при 0 С в атмосфере аргона добавляют 2-анилино-6-хиноксалинол (0,23 г, 0,97 ммоль). Добавляют порциями (3)-(+)-3 гидрокситетрагидрофуран (0,086 мл, 1,3 ммоль) и трифенилфосфин (0,31 г, 1,2 ммоль). Добавляют порциями DEAD (0,18 мл, 1,2 ммоль). Реакционной смеси дают согреться до комнатной температуры и перемешивают 1,5 ч. Смесь концентрируют и распределяют между EtOAc и Н 2 О. Органический слой промывают водой, рассолом, сушат (MgSO4) и концентрируют. Полученное желтое масло хроматографируют (50%EtOAc/гексаны) и растворяют в Et2O/IPA (изопропанол). Добавляют по каплям растворHCl/Et2O и полученный красно-оранжевый порошок высушивают в вакууме. Порошкообразный продукт превращают в свободноосновнуюH2O, 5 х МеОН) основной ионообменной смолой. Смесь перемешивают 30 мин, фильтруют,концентрируют и перекристаллизовывают из смеси EtOAc/гексаны с получением (в двух сборах) продукт (т.пл. 173-175 С). Элементный анализ. Вычислено для C18H17N3O2: С 70,35; Н 5,57; N 13,67. Найдено: С 70,19; Н 5,60; N 13,66. Пример 11. 2-Анилино-6-изопропоксихиноксалингидрохлорид. К NaH (0,033 г, 0,84 ммоль) в атмосфере аргона добавляют 1 мл ДМФ. Добавляют порциями 2-анилини-6-хиноксалинол (0,1 г, 0,42 ммоль) в 1,5 мл ДМФ. Через 30 минут добавляют по каплям 2-бромпропан и раствор нагревают до 50 С в течение 1,5 ч. Охлажденную реакционную смесь гасят водой и распределяют между EtOAc и Н 2O, промывают Н 2O (3 раза), рассолом, сушат (МgSO4) и концентрируют. Полученный остаток хроматографируют (30%EtOAc/гексаны) с получением 0,05 г диалкилированного продукта и 0,1 г указанного в заголовке соединения. Аналитический образец хлороводородной соли получают добавлениемIPA/HCl к Et2O/IPA раствору свободного основания с получением хлороводородной солиN 13,31. Найдено: С 64,51; Н 5,90; N 13,09. Пример 12. 3-Циклогексилокси-6,7-диметоксихиноксалина 1-оксид. Смесь 2-циклогексилокси-6,7-диметоксихиноксалина (110 мг, 0,38 ммоль) и метахлорбензойной перкислоты (70%, 113 мг, 0,46 ммоль) и 10 мл метиленхлорида перемешивают при комнатной температуре в течение дня. После фильтрования раствор концентрируют и остаток хроматографируют на силикагеле (20% этилацетатгексан) с получением целевого продукта (т.пл. 167-169 С). Аналогичным образом получают транс-4-(6,7-диметокси-4-оксихиноксалин-2-иламино)циклогексанол (т.пл. 220222 С). Элементный анализ. Вычислено для С 16 Н 21N3O40,2 Н 2 О: С 59,42; Н 6,69; N 12,99. Найдено: С 59,43; Н 6,64; N 12,95. Пример 1 получения промежуточного соединения. 4-Бром-5-метоксибензол-1,2-диамингидрохлорид. К раствору EtOAc (50 мл) и 5-бром-4 метокси-2-нитрофениламина (2,5 г, 10 ммоль) в атмосфере аргона добавляют 5% Pd/C (0,5 г). Реакционную смесь гидрируют при давлении 50 фунт/кв. дюйм (3,5 кг/см 2) в течение 1 ч. Смесь фильтруют через целит в раствор HCl/IPA/EtOAc и слой целита промывают дополнительным EtOAc. Полученный осадок отфильтровывают с получением белого твердого вещества. Пример 2 получения промежуточного соединения. 7-Бром-6-метоксихиноксалин-2-ол и 6 бром-7-метоксихиноксалин-2-ол. 23 К раствору МеОН (15 мл) в атмосфере аргона добавляют измельченные в порошок гранулы NaOH (0,86 г, 51 ммоль) и 4-бром-5 метоксибензол-1,2-диаминдигидрохлорид (2,7 г,9,3 ммоль). Смесь перемешивают 10 мин, после чего добавляют порциями раствор 45% этиленглиоксалата в толуоле (2,7 г, 12 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 1 ч, после чего охлаждают. Добавляют воду и затем суспензию фильтруют. Полученное твердое вещество промывают последовательно Н 2 О, МеОН, IPA и Et2O с получением желтого порошка. Пример 3 получения промежуточного соединения. 7-Бром-2-хлор-6-метоксихиноксалин и 6 бром-2-хлор-7-метоксихиноксалин. К смеси 7-бром-6-метоксихиноксалин-2 ола и 6-бром-7-метоксихиноксалин-2-ола (1 г,3,9 ммоль) добавляют РОСl3 (5 мл). Реакционную смесь кипятят с обратным холодильником в течение 1 ч, выливают в ледяную воду, фильтруют и затем промывают водой с получением светло-рыжевато-коричневого твердого вещества. Отношение 7-бром-2-хлор-6-метоксихиноксалин : 6-бром-2-хлор-7-метоксихиноксалин равно приблизительно 7:1 согласно анализу методом 1 Н ЯМР. Пример 4 получения промежуточного соединения. 5-Хлор-4-метокси-2-нитроанилин. К раствору N-(5-хлор-4-метокси-2-нитрофенил)ацетамида (2 г, 8,2 ммоль) в 5 н. НСl (20 мл) добавляют 1,4-диоксан (10 мл) и смесь перемешивают при 60 С в течение 1,5 ч. Реакционную смесь концентрируют и распределяют между EtOAc и 2 н. NaOH. Водные слои промывают EtOAc (3 раза), рассолом, сушат (MgSO4),адсорбируют на силикагель и хроматографируют (70% EtOAc/гексаны) с получением оранжевого порошка. Пример 5 получения промежуточного продукта. 4-Хлор-5-метоксибензол-1,2-диаминдигидрохлорид. К раствору EtOAc (25 мл) и 5-хлор-4 метокси-2-нитрофениламина (1,6 г, 7,9 ммоль) в атмосферу аргона добавляют 5% Pd/C (0,5 г). Реакционную смесь гидрируют при давлении 50 фунт/кв. дюйм (3,5 кг/см 2) в течение 1 ч. Смесь фильтруют в атмосфере азота через целит в раствор 1 н. HCl/Et2O в EtOAc и слой целита промывают дополнительным EtOAc. Полученный осадок отфильтровывают с получением белого твердого вещества. Пример 6 получения промежуточного соединения. 7-Хлор-6-метоксихиноксалин-2-ол и 6 хлор-7-метоксихиноксалин-2-ол. К раствору 4-хлор-5-метоксибензол-1,2 диаминдигидрохлорида (1,8 г, 7,2 ммоль) вTEA (триэтаноламин) (2,5 мл, 18 ммоль) при 0 С. Смесь перемешивают 20 мин, после чего добавляют порциями раствор 45% этиленглиоксалата в толуоле (2,1 г, 9,3 ммоль). Реакционную смесь нагревают до комнатной температуры, кипятят с обратным холодильником в течение 1,5 ч, после чего охлаждают. Добавляют воду и затем суспензию фильтруют и промывают последовательно H2O, IPA и Et2O с получением светло-желтого порошка. Продукт несколько раз азеотропируют с толуолом и высушивают в вакууме перед применением. Пример 7 получения промежуточного соединения. 2,7-Дихлор-6-метоксихиноксалин и 2,6 дихлор-7-метоксихиноксалин. К смеси 7-хлор-6-метоксихиноксалин-2 олa и 6-хлор-7-метоксихиноксалин-2-ола (1 г,4,7 ммоль) с использованием хлор-кальциевой(CaCl2) трубки добавляют РОСl3 (5 мл). Реакционную смесь кипятят с обратным холодильником в течение 30 мин, выливают в холодный насыщенный раствор NаНСО 3, фильтруют и затем промывают водой с получением твердого вещества. Соотношение 2,7-дихлор-6-метоксихиноксалин : 2,6-дихлор-7-метоксихиноксалин составляет приблизительно 6:1 согласно анализу методом 1H ЯМР. Соединения формулы I, как описано выше,ингибируют пролиферацию клеток и/или продуцирование клеточного матрикса, и/или передвижение клеток (хемотаксис) посредством ингибирования активности тирозинкиназы PDGFR. Неуправляемое размножение клеток, сверхпродуцирования матрикса или плохо регулируемая запрограммированная гибель клеток(апоптоз) вызывают много болезненных состояний. В эти болезненные состояния вовлечены самые разнообразные типы клеток и они включают такие заболевании, как лейкоз, рак, глиобластома, псориаз, воспалительные болезни, заболевания кости, фиброзы, атеросклероз и рестеноз, случающийся после ангиопластики коронарных, бедренных или почечных артерий, или фибропролиферативное заболевание, такое как артрит, фиброз легкого, почки и печени. В частности, как было сообщено, PDGF и PDGF-R принимают участие в некоторых типах раков и опухолей, таких как рак головного мозга, рак яичников, рак толстой кишки, рак предстательной железы, рак легкого, саркома Калоши и злокачественная меланома. Кроме того, после коронарного шунтирования создаются условия клеточной пролиферации с нарушенной регуляцией. Полагают, что ингибирование активности тирозинкиназы будет полезным в регуляции неуправляемой репродукции клеток, сверхпродуцирования матрикса или плохо регулируемой запрограммированной гибели клеток (апоптоза). Настоящее изобретение относится к модулированию и/или ингибированию передачи сигналов в клетках, пролиферации клеток, проду 25 цирования клеточного матрикса, передвижения клеток (хемотаксиса), регуляции аномального роста клеток и воспалительной реакции клеток. Более конкретно настоящее изобретение касается применения замещенных хинолинов и хиноксалинов, обладающих способностью селективно ингибировать дифференциацию, пролиферацию,продуцирование матрикса, хемотаксис или высвобождение медиатора путем эффективного ингибирования активности тирозинкиназы рецептора тромбоцитарного фактора роста (PDGFR). Инициирование аутофосфорилирования,т.е. фосфорилирования самого рецептора фактора роста, и фосфорилирования хозяина внутриклеточных субстратов является биохимическим процессом, который вовлекается в передачу сигналов в клетках, пролиферацию клеток,продуцирование матрикса, хемотаксис и высвобождение медиатора. Благодаря эффективному ингибированию активности тирозинкиназы Lck соединения по настоящему изобретению также полезны при лечении сопротивления трансплантации и аутоиммунных заболеваний, таких как ревматоидный артрит, рассеянный склероз и системная красная волчанка, при отторжении трансплантата, при состоянии, называемом "трансплантат против хозяина", при гиперпролиферативных нарушениях, таких как опухоли и псориаз и при заболеваниях, в которых клетки принимают провоспалительные сигналы, таких как астма,воспалительная болезнь кишечника и панкреатит. Для лечения сопротивления трансплантации соединения по настоящему изобретению могут быть использованы либо профилактически, либо в ответ на неблагоприятную реакцию человека на пересаженный орган или ткань. При профилактическом применении соединение по настоящему изобретению вводят пациенту или в пересаживаемую ткань или орган перед операцией трансплантации. Профилактическое лечение может также включать введение лекарственного средства после операции трансплантации, но до обнаружения каких-либо признаков неблагоприятной реакции на трансплантацию. При введении в ответ на неблагоприятную реакцию соединение по настоящему изобретению вводят непосредственно пациенту, чтобы противостоять сопротивлению трансплантации после обнаружения внешних признаков сопротивления. В соответствии с другим признаком настоящего изобретения предлагается способ лечения пациента, страдающего от или подверженного состояниям, которые могут быть ослаблены или предотвращены введением ингибитора активности тирозинкиназы PDCF-R и/или активности тирозинкиназы Lck, например,таких состояний, как описанные выше, который включает введение пациенту эффективного количества соединения формулы I или компози 002600 26 ции, содержащей соединение формулы I, или его фармацевтически приемлемой соли. Используемый в данном описании термин лечение включает как профилактическую терапию, так и лечение развившихся состояний. Настоящее изобретение включает в свой объем фармацевтические композиции, которые содержат фармацевтически приемлемое количество, по крайней мере, одного из соединений формулы I вместе с фармацевтически приемлемым носителем, например вспомогательным веществом, разбавителем, покрытием и наполнителем. На практике соединения или композиции для лечения в соответствии с настоящим изобретением могут быть введены любым из многих подходящих способов, например, путем ингаляции, местно, парентерально, ректально или перорально; более предпочтительным является пероральный способ введения. Более конкретные способы введения включают внутривенный,внутримышечный, подкожный, внутриглазный,внутрисуставный, в толстую кишку, перитонеальный, трансэпителиальный, включая чрескожный, офтальмологический, подъязычный,трансбуккальный, дермальный, окулярный, ингаляционный в нос путем инсуффляции и аэрозольный. Соединения формулы I могут быть представлены в формах, позволяющих введение наиболее подходящим способом, и изобретение касается также фармацевтических композиций,содержащих, по крайней мере, одно соединение по настоящему изобретению, которые пригодны для применения в качестве лекарственного средства для пациента. Эти композиции могут быть приготовлены обычными методами с использованием одного или нескольких фармацевтически приемлемых вспомогательных веществ или наполнителей. Вспомогательные вещества включают, кроме прочего, разбавители,стерильные водные среды и различные нетоксичные органические растворители. Композиции могут находиться в форме таблеток, пилюль, гранул, порошков, водных растворов или суспензий, растворов для инъекции, элексиров или сиропов и могут содержать одно или несколько веществ, выбранных из группы, содержащей подслащивающие агенты, такие как сахароза, лактоза, фруктоза, сахарин или Нутрасвит (Nutrasweet), корригенты, такие как мятное масло, винтергреновое масло или вишневые или апельсиновые корригенты, красители или стабилизаторы, такие как метил- или пропилпарабен, для получения фармацевтически приемлема препаратов. Выбор носителя и содержание активного вещества в носителе обычно определяют в соответствии с растворимостью и химическими свойствами продукта, конкретным способом введения и мерами предосторожности, применяемым в фармацевтической практике. Напри 27 мер, для изготовления таблеток, пастилок, пилюль, капсул и подобных могут быть использованы наполнители, такие как лактоза, цитрат натрия, карбонат кальция, фосфат дикальция и разрыхляющие вещества, такие как крахмал,альгиновые кислоты и некоторые комплексные силикагели, объединенные со смазывающими веществами, такими как стеарат магния, лаурилсульфат натрия и тальк. Для приготовления капсул преимущественно использовать лактозу и жидкий носитель, такой как высокомолекулярные полиэтиленгликоли. Различные другие материалы могут присутствовать в качестве покрытий или для какого-либо иного изменения физической формы дозированной единицы. Например, таблетки, пилюли или капсулы могут быть покрыты шеллаком, сахаром или тем и другим. При использовании водных суспензий они могут содержать эмульгирующие вещества или вещества, которые способствуют образованию суспензии. Могут быть также использованы разбавители, такие как сахароза, этанол, полиолы, такие как полиэтиленгликоль, пропиленгликоль и глицерин, и хлороформ или их смеси. Кроме того, активное соединение может быть включено в композиции и препаративные формы с продленным действием. При пероральном введении активное соединение может быть введено, например, с инертным разбавителем или с усвояемым съедобным носителем, может быть включено в твердые или мягкие желатиновые капсулы, может быть спрессовано в таблетки или может быть введено непосредственно с пищей или объединено с наполнителем и использовано в форме проглатываемых таблеток, буккальных таблеток, пастилок, капсул, элексиров, суспензий, сиропов, облаток и подобных. Для парентерального введения используют эмульсии, суспензии или растворы соединений по настоящему изобретению в растительном масле, например кунжутном, арахисовом или оливковом масле, или водно-органические растворы, такие как вода и пропиленгликоль, в используемых для инъекций органических сложных эфирах, таких как этилолеат, а также стерильные водные растворы фармацевтически приемлемых солей. Инъекционные формы должны быть настолько текучими, чтобы их можно было легко вводить с помощью шприца,причем надлежащая текучесть может быть обеспечена, например, путем применения оболочки, такой как лецитин, путем обеспечения требуемого размера частиц в случае дисперсии и путем применения поверхностно-активных веществ. Пролонгированное всасывание инъекционных композиций может быть обеспечено путем применения средств, задерживающих всасывание, например моностеарата алюминия и желатина. Растворы солей продуктов по настоящему изобретению особенно пригодны для введения путем внутримышечной или подкож 002600 28 ной инъекции. Растворы активного соединения в виде свободного основания или фармакологически приемлимой соли могут быть пригодны в воде, подходящим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Может быть также приготовлена дисперсия в глицерине, жидких полиэтиленгликолях и их смесях и в маслах. Водные растворы, содержащие также растворы солей в чистой дистиллированной воде, могут быть использованы для внутривенного введения при условии, что их рН должным образом отрегулирован, что им приданы надлежащие буферные свойства и они сделаны изотоническими с помощью достаточного количества глюкозы или хлорида натрия и что они стерилизованы путем нагревания, облучения, микрофильтрования и/или с помощью различных антибактериальных и противогрибковых средств, например парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и подобных. Стерильные инъекционные растворы готовят путем введения активного соединения в требуемом количество в подходящий растворитель с различными другими компонентами, перечисленными выше, когда они требуются, с последующей стeрилизацией фильтрованием. Обычно дисперсии готовят путем введения различных стерилизованных активных компонентов в стерильный носитель, который содержит основную среду дисперсии и другие необходимые компоненты из тех, что указаны выше. В случае стерильных порошков для приготовления стерильных инъекционных растворов предпочтительными методами приготовления являются вакуумная сушка и лиофильная сушка,которые даст порошок активного компонента плюс любой другой требуемый компонент из предварительно стерильно отфильтрованного раствора. Для местного введения могут быть использованы гели (на основе воды или спирта), кремы или мази, содержащие соединения по настоящему изобретению. Соединения по настоящему изобретению могут быть также введены в гелевую или матричную основу для нанесения на пластырь, которая могла бы обеспечить возможность регулируемого высвобождения соединения через чрескожный барьер. Для введения путем ингаляции соединения по настоящему изобретению могут быть растворены или суспендированы в подходящем носителе для применения и распылителе или для суспензии или растворов в аэрозольной форме или могут быть абсорбированы или адсорбированы на подходящем твердом носителе для применения в ингаляторе для сухого порошка. Твердые композиции для ректального введения включают суппозитории, изготовленные в соответствии с известными методами и содержащие, по крайней мере, одно соединение формулы I. 29 Композиции по настоящему изобретению могут быть также составлены с обеспечением сопротивления быстрому выведения через стенки сосуда (артерии или вены) путем конвекции и/или диффузии, благодаря чему увеличивается время пребывания вирусных частиц в желаемом месте действия. Для продленного высвобождения может быть использовано периадвентициальное депо, содержащее соединение по настоящему изобретению. Одним таким пригодным депо для введения соединения по настоящему изобретению может быть сополимерная матрица, такая как этилен-винилацетат, или гель поливинилового спирта, окруженный оболочкой из Силастика (Silastic). Альтернативно, соединение по настоящему изобретению может быть доставлено локально из силиконового полимера,имплантированного в адвентициальную оболочку. Альтернативный способ минимизации вымывания соединения по настоящему изобретению во время чрескожной трансваскулярной доставки включает применение недиффузионных микрочастиц, выделяющих лекарственное средство. Микрочастицы могут быть составлены из самых разных синтетических полимеров,таких как, например, полилактид, или природных веществ, включающих белки и полисахариды. Такие микрочастицы обеспечивают возможность оперативного манипулирования переменными, включающими суммарную дозу лекарственного средства и кинетику ее высвобождения. Микрочастицы можно эффективно вводить через артериальную или венозную стенку с помощью пористого баллонного катетера или баллона на стенте (расширители) и удерживать в стенке сосуда и периадвентициальной ткани в течение, по крайней мере, около двух недель. Препаративные формы и методики локальной доставки лекарственных средств в конкретное место внутри сосуда описаны в Reissen et al. (J. Am. Coll. Cardiol. 1994; 23; 12341244); который включен в данное описание в качестве ссылки. Композиция по настоящему изобретению может также содержать гидрогель, приготовленный из любого биосовместимого или нецитотоксического (гомо или гетеро) полимера,такого как гидрофильный полимер полиакриловой кислоты, который может действовать как губка, впитывающая лекарственное средство. Такие полимеры уже описаны, например, в заявке WO 93/08845, которая включена в данное описание в качестве ссылки. Некоторые из них,такие, в частности, как те, что получены из этилена и/или пропиленоксида, коммерчески доступны. При применении соединений по настоящему изобретению для лечения патологий, связанных с гиперпролиферативными нарушениями, эти соединения могут быть введены разными путями. Для лечения рестеноза соединения 30 по настоящему изобретению вводят непосредственно в стенку кровеносного сосуда с использованием баллона для ангиопастики, покрытого гидрофильной пленкой (например, гидрогелем),которую насыщают соединением, или посредством любого другого катетера, содержащего инфузионную камеру для соединения, которое,таким образом, может быть точно доведено до подлежащего лечению места и дает возможность эффективно высвободить соединение именно в месте расположения клеток, где необходимо лечение. Этот способ введения обеспечивает возможность быстрого введения соединения в контакт с клетками, нуждающимися в лечении. Способ лечения по настоящему изобретению предпочтительно состоит в введении соединения по настоящему изобретению в место,подлежащее лечению. Например, содержащая гидрогель композиция может быть наложена непосредственно на поверхность ткани, подлежащей лечению, например, при хирургическом вмешательстве. Удобно вводить гидрогель в требуемое место внутри сосуда, путем покрытия им катетера, например баллонного катетера, и доставки к стенке сосуда, предпочтительно во время ангиопластики. Является особенно целесообразным вводить насыщенный гидрогель в месте, подлежащем лечения, посредством баллонного катетера. При продвижении катетера в направлении к намеченному сосуду баллон может быть снабжен защитной оболочкой для минимизации смывания лекарственного средства после введения катетера в поток крови. Другой вариант осуществления настоящего изобретения относится к введению соединения с помощью перфузионных баллонов. Эти перфузионные баллоны, которые обеспечивают возможность поддерживать кровоток и тем самым уменьшать опасность возникновения ишемии миокарда при раздувании баллона, обеспечивают также возможность доставки соединения в нужное место при нормальном давлении в течение относительного длительного времени(более двадцати минут), которое может быть необходимым для его оптимального действия. Альтернативно можно использовать канализированный баллонный катетер ("канализированный баллонный катетер для ангиопластики",Mansfield Medical, Boston Scientific Corp., Watertown, MA). Последний состоит из традиционного баллона, покрытого слоем из 24 перфорированных каналов, которые заливаются благодаря независимому просвету через дополнительное инфузионное отверстие. Различные типы баллонных катетеров, такие как сдвоенный баллон, пористый баллон, микропористый баллон, баллон с каналами, баллон на стенте и гидрогелевый катетер, которые все могут быть использованы для практической реализации изобретения, описаны в Reissen et al. (1994); который включен в данное описание в качестве 31 ссылки. Особенно преимущественным является применение перфузионного балонного катетера,поскольку он обеспечивает как возможность сохранять баллон раздутым в течение длительного периода времени с сохранением свойств облегченного скольжения, так и конкретную локализацию гидрогеля одновременно. Другой аспект настоящего изобретения относится к фармацевтической композиции, содержащей соединение по настоящему изобретению и полоксамер, такой как Полоксамер(Poloxamer) 407-нетоксичный биосовместимый полиол, коммерчески доступный (BASF, Parsippany, NJ). Полоксамер, пропитанный соединением по настоящему изобретению, может быть наложен непосредственно на поверхность ткани, подлежащей лечению, например, во время хирургического вмешательства. Полоксамер имеет, по существу, такие же преимущества, как гидрогель, хотя имеет более низкую вязкость. Применение канального баллонного катетера с полоксамером, пропитанным соединением по настоящему изобретению, особенно предпочтительно. В этом случае одновременно обеспечиваются возможность сохранять баллон раздутым в течение длительного периода времени с сохранением свойств облегченного скольжения и конкретная локализация полоксамера. Процентное содержание активного компонента в композициях можно изменять, причем необходимо, чтобы он присутствовал в количестве, обеспечивающем необходимую дозу. Очевидно, что можно вводить несколько единичных дозированных форм примерно в одно и то же время. Используемая доза может быть определена врачом или квалифицированным медицинским профессионалом и зависит от требуемого терапевтического эффекта, способа введения,длительности лечения и состояния пациента. Для взрослых дозы составляют обычно от примерно 0,001 до примерно 50, предпочтительно от примерно 0,001 до примерно 5 мг/кг массы тела в сутки при ингаляции, от примерно 0,01 до примерно 100, предпочтительно 0,1-70, более предпочтительно 0,5-10 мг/кг массы тела в сутки при пероральном введении и от примерно 0,001 до 10, предпочтительно 0,01-10 мг/кг массы тела в сутки при внутривенном введении. В каждом конкретном случае дозы определяют в соответствии с характерными особенностями пациента, подлежащего лечению, такими как возраст, масса, общее состояние здоровья и другие характеристики, которые могут влиять на эффективность соединения по настоящему изобретению. Соединения композиции по настоящему изобретению можно вводить так часто, как нужно для достижений требуемого терапевтического эффекта. Некоторые пациенты могут быстро реагировать на более высокую или более 32 низкую дозу, причем для них могут оказаться достаточными намного более слабые поддерживающие дозы. Для других пациентов может оказаться необходимым длительное лечение при норме 1-4 дозы в сутки, в соответствии с физиологическими потребностями каждого конкретного пациента. Как правило, активный продукт можно вводить перорально 1-4 раза в сутки. Но,конечно, для других пациентов может быть необходимым предписывать не более чем одной или двух доз в сутки. Соединения по настоящему изобретению могут быть также использованы в сочетании с другими терапевтическими средствами или в сочетании с применением терапевтических методов к определенным фармакологическим состояниям, которые могут быть облегчены путем применения соединений формулы I, таким как следующие. Соединения по настоящему изобретению могут быть использованы для лечения рестеноза после ангиопластики с использованием любого устройства, такого как баллон, иссечения или лазерные процедуры. Соединения по настоящему изобретению могут быть использованы для лечения рестеноза после установки стента в кровеносной системе либо 1) для первичного лечения закупорки сосудов, либо 2) в случае,когда ангиопластика с использованием какоголибо приспособления не способна дать проходимую артерию. Соединение может быть использовано перорально, парентерально, путем местного применения при введении специального приспособления или в виде надлежащим образом составленного покрытия на расширителе (стенте). Соединения по настоящему изобретению могут быть использованы для лечения рестеноза в сочетании с любым противосвертывающим,антитромбоцитарным, антитромботическим или профибринолитическим средством. Часто пациентов лечат до, во время и после хирургического вмешательства средствами указанных классов, чтобы безопасно выполнить хирургическую операцию или предотвратить неблагоприятные эффекты тромбообразования. Некоторые примеры классов средств, известных в качестве противосвертывающих, антитромбоцитарных,антитромботических или профибринолитических средств, включают любые препараты гепарина, низкомолекулярные гепарины,пентасахариды, антагонисты фибриногеновых рецепторов, ингибиторы тромбина, ингибиторы фактора Ха или ингибиторы фактора VIIa. Соединения по настоящему изобретению могут быть использованы в сочетании с любым гипотензивным средством или средством, регулирующим холестерин или лилиды при лечении рестеноза илиатеросклероза параллельно с лечением высокого кровяного давления или атеросклероза. Некоторые примеры средств, пригодных для лечения высокого кровяного давле 33 ния, включают соединения следующих классов: бета-блокаторы, ингибиторы АСЕ (ангиотензинпревращающего фермента, АПФ), антагонисты кальциевых каналов и антагонисты альфарецепторов. Некоторые примеры средств, пригодных для лечения повышенных уровней холестерина или разрегулированных уровней липидов, включают соединения, известные как ингибиторы редуктазы HMGCoA, соединения класса фибратов. Соединения по настоящему изобретению могут быть использованы для лечения различных форм рака либо в отдельности или в сочетании с известными соединениями для лечения рака. Понятно, что настоящее изобретение охватывает комбинации соединений по настоящему изобретению с одним или несколькими из упомянутых выше лекарственных средств. Соединения в объеме настоящего изобретения показывают значительные фармакологические активности в тестах, описанных в литературе, результаты которых, как полагают, коррелируются с фармакологической активностью у людей и других млекопитающих. Следующие результаты фармакологических тестов in vitro иin vivo являются типичными для характеристики соединений по настоящему изобретению. Фармацевтические композиции и фармакологические тесты Соединения по настоящему изобретению проявляют значительную активность как ингибиторы белковых тирозинкиназ и обладают терапевтической ценностью в качестве клеточных антипролиферативных средств для лечения некоторых состояний, включающих псориаз, атеросклероз и рестеноз. Соединения по настоящему изобретению обеспечивают модулирование и/или ингибирование передачи сигналов в клетках и/или пролиферации клеток и/или продуцирование матрикса, хемотаксиса и/или воспалительной реакции клеток и могут быть использованы для предотвращения или задержки проявления или повторного проявления таких состояний или лечения состояния. Для определения эффективности соединений по настоящему изобретению используют описанные ниже фармакологические тесты, которые приняты в данной области медицины и,как известно, коррелируют с фармакологической активностью у млекопитающих. Соединения по настоящему изобретению были тестированы, и результаты, полученные в различных тестах, как полагают, соответствуют полезной активности в отношении медиатора дифференциации клеток. Результаты этих испытаний,надо полагать, дают достаточную информацию специалистам в области фармакологии и медицинской химии, чтобы определить параметры применения исследованных соединений в одном или нескольких терапевтических способах, описанных в данном описании. 34 1. Анализ аутофосфорилирования тирозинкиназы PDGF-R методом ELISA. Указанный в заголовке анализ выполняют так, как описано в Dolle et al. (J. Med. Chem. 1994, 37, 2627), который включен в данное описание в качестве ссылки, за исключением того,что используют клеточный лизат, полученный из клеток гладких мышц аорты человека(HAMSC), как описано ниже. 2. Общая методика анализа митогенеза. а. Культура клеток. Клетки гладких мышц аорты человека(пассах 4-9) помещают в лунки 96-луночного планшета в питательную среду с концентрацией 6000 клеток ячейка и позволяют мм расти 2-3 дня. При приблизительно 80%-ном слиянии рост клеток останавливают бессывороточной средой (SFM).b. Анализ митогенеза. Через 24 ч депривации сыворотки среду удаляют и заменяют тестированным соединением в носителе, SFM (200 мкл на лунку) . Соединения растворяют в клеточной культуре DMSO(ДМСО) при концентрации 10 мМ и делают дополнительные разведения в SFM. Через 30 мин предварительной инкубации с соединением клетки стимулируют PDGF в концентрации 10 нг/мл. Определения осуществляют дублированно со стимулированными и нестимулированными лунками при каждой концентрации соединения. Спустя четыре часа добавляют 1 мкКи 3H тимидина/ячейку. Культивирование заканчивают через 24 ч после добавления фактора роста. Клетки поднимают трипсином и собирают на фильтровальную бумагу, пользуясь автоматизированным сборщиком клеток (Wallac MachII96). Фильтровальную бумагу анализируют с помощью сцинтиляционного счетчика (Wallac Betaplate) для определения метки, введенной в ДНК. 3. Анализ хемотаксиса. Из АТСС получают клетки гладких мышц аорта человека (HASMC) при более ранних пассажах. Клетки культивируют в Clonetics SmGM 2 SingleQuots (используют среду и клетки при пассажах 4-10). При 80%-ном слиянии клеток в среду добавляют флуоресцентный зонд, кальцеин AM (5 мМ, Molecular Probe), и клетки инкубируют 30 мин. После промывки солевым раствором, с буфером HEPES клетки поднимают трипсином и нейтрализуют буфером MCDB 131(Gibco) с 0,1% BSA (бычий сывороточный альбумин), 10 мМ глутамином и 10% сывороткой плода коровы. После центрифугирования клетки промывают еще раз и ресуспендируют в том же самом буфере без сыворотки плода коровы при концентрации 30000 клеток на 50 мл. Клетки инкубируют при различных концентрациях соединения формулы I (конечная концентрация в ДМСО =1%) в течение 30 мин при 37 С. Для исследования хемотаксиса используют модифицированные камеры Бойдена с 96 ячейками(Neuroprobe, Inc.) и поликарбонатную мембрану с размером пор 8 мм (Poretics, СА). Мембрану покрывают коллагеном (Sigma C3657, 0,1 мг/мл). В нижнюю камеру помещают PDGF(3 нг/мл) в буфере с соединением формулы I и без него. В верхнюю камеру помещают клетки(30000) с ингибитором и без него. Клетки инкубируют в течение 4 ч. Фильтровальную мембрану снимают и удаляют клетки, находящиеся на верхней стороне мембраны. После высушивания определяют флуоресценцию на мембране, используя Cytofluor II (Milipore) при длине волны возбуждения и эмиссии 485 и 530 нм, соответственно. В каждом эксперименте среднюю миграцию клеток получают по шести повторениям. Процент ингибирования определяют по контрольным значениям с обработкой ДМСО. По пяти оценкам вычисляют зависимое от концентрации ингибирование, значение IC50. Результаты представляют как среднее значениеSEM(средняя квадратическая ошибка измерения) от пяти таких экспериментов. 4. Очистка EGF-рецептора. Очистка EGF-рецептора основана на методике Ярдена и Шлессингера (Yarden andSchlessinger). Клетки А 431 культивируют в емкостях площадью 80 см 2 до слияния (2 х 107 клеток на емкость). Клетки промывают два разаPBS (фосфатно-солевым буферным раствором) и собирают с помощью 11,0 ммоль EDTA содержащего PBS 1 ч при 37 С, и центрифугируют при 600 g в течение 10 мин. Клетки солюбилизируют в 1 мл на 2 х 107 клеток холодного солюбилизационного буфера (50 ммоль буфераHepes, pH 7,6, 1% Triton X-100, 150 ммоль NaCl,5 ммоль EGTA, 1 ммоль PMSF, 50 мг/мл апротинина, 25 ммоль бензамидина, 5 мг/мл ингибитора лейпепсина и 10 мг/мл ингибитора соевого трипсина) в течение 20 мин при 4 С. После центрифугирования при 100000 g в течение 30 мин супернатант загружают на WGA-агарозную колонку (100 мл упакованной смолы на 2 х 107 клеток) и встряхивают в течение 2 ч при 4 С. Неабсорбированный материал удаляют и смолу промывают два раза HTN буфером (50 ммоль буфера Нереs, pH 7,6, 0,1% Triton X-100, 150 ммоль NaCl), два раза HTN буфером, содержащим 1 М NaCl, и два раза HTNG буфером (50 ммоль буфера Hepes, pH 7,6, 0,1% Triton X-100,150 ммоль NaCl И 10% глицерин). EGFрецептор элюируют периодически HTNG буфером, содержащим 0,5 М N-ацетил-D-глюкозамина (200 мл на 2 х 107 клеток). Элюированный материал хранят аликвотами при -70 С и перед применением разводят TMTNG буфером (50 ммоль Tris-Mes буфера, pH 7,6, 0,1% Triton X100, 150 ммоль NaCl и 10% глицерин). 5. Ингибирование аутофосфорилированияEGF-R. Клетки А 431 культивируют до слияния на чашках Петри с культурой человеческой ткани,покрытой фибронектином. После двукратной 36 промывки охлажденным льдом раствором PBS клетки лизируют путем добавления 500 мл/чашку лизисного буфера (50 ммоль Hepes,pH 7,5, 150 ммоль NaCl, 1,5 ммоль MgCl, 1 ммоль EGTA, 10% глицерин, 1% Тритон X-100,1 ммоль PMSF, 1 мг/мл апротинина, 1 мг/мл лейпептина) и инкубируют в течение 5 мин при 4 С. После стимулирования EGF (500 мг/мл, 10 мин при 37 С) проводят иммуноосаждение с помощью анти EGF-R (Аb 108) и реакцию аутофосфорилирования образца (50 мл аликвоты, 3 мКи [g-32 Р]АТФ) в присутствии 2 или 10 мМ соединения по настоящему изобретению в течение 2 мин при 4 С. Реакцию останавливают путем добавления горячего электрофорезного буфера для образца. Проводят SDA-PAGE анализ(7,5% эл.) с последующей ауторадиографией и определяют количественные результаты реакции путем денситометрического сканирования рентгеновских пленок. а. Культура клеток. Клетки, обозначенные HER 14 и К 721 А получают путем трансфекции клеток NIH3T3(клон 2.2) (из С. Fryling, NCI, NIH), которые не содержат эндогенных EGF-рецепторов, с кДНКзвеньями дикого типа EGF-рецептора или мутантного EGF-рецептора, не обладающего тирозинкиназной активностью (в котором Лиз 721 в месте связывания АТФ заменен остатком Ала соответственно). Все клетки культивируют вDMEM с 10% сывороткой теленка (Hyclone,Logan, Utah). 6. Селективность по отношению к РКА и РКС определяют с использованием коммерческих наборов а. Колориметрический набор Pierce для анализа РКА, формат Spinzyme. Краткий протокол: 1U фермента РКА (бычье сердце)/аналитическая пробирка субстрат пептида (меченного красителем) Кемптид 45 мин при 30 С; спектральная поглощательная способность при 570 нм.(меченого красителем) Нейрогранин 30 мин при 30 С; поглощение при 570 нм. 7. Измерения ингибирующей активности в отношении тирозинкиназы р 56lck. Ингибирующая активность в отношении тирозинкиназы p56lck определяют в соответствии с методикой, раскрытой в патенте США 5714493, включенном в данное описание в качестве ссылки. Альтернативно, ингибирующую активность в отношении тирозинкиназы определяют следующим методом. Субстрат (тирозинсодержащий субстрат/ Biot- (Аlа)3-Lys-ValGlu-Lys-Ile-Gly-Glu-Gly-Thr-Tyr-Glu-Val-Val 37Tyr-Lys-(NH2), распознаваемый p56lck, 1 мкМ) сначала фосфорилируют в присутствии или в отсутствие заданной концентрации тестируемого соединения заданным количеством фермента(фермент продуцируют экспрессией гена р 56lck в дрожжевую конструкцию), очищенного от клонированных дрожжей (очистку фермента осуществляют следующими классическими методами), в присутствии АТФ (10 нкМ), MgCl2 (2,5 мМ), MnCl2 (2,5 мМ), NaCl (25 мМ), DTT (0,4 мМ) в Hepes 50 мМ, рН 7,5, в течение 10 мин при температуре окружающей среды. Общий объем реакционной смеси составляет 50 мкл и реакции проводят в черном флуропланшете с 96 ячейками. Реакцию останавливают добавлением 150 мкл останавливающего буфера (100 мМHepes, рН 7,5, KF 400 мМ, EDTA 133 мМ, BSA 1 г/л), содержащего антитело для тирозина, меченое криптатом европия (PY20-K), при 0,8 мкг/мл и меченый аллофикоцианином стрептавидин (XL665) при 4 мкг/мл. Мечение Стрептавидина и антител для тирозина проводили в CisBio International (Франция). Смесь обсчитывали с помощью счетчика Packard Discovery, способного измерять перенос однородной флуросценции с временным разрешением (возбуждение при 337 нм, считывание при 620 нм и 665 нм). Отношение сигнала при 665 нм к сигналу при 620 нм является показателем концентрации фосфорилированного тирозина. Контроль получают путем замены фермента буфером. Характерный сигнал представляет собой разницу между отношением, полученным без ингибитора,и отношением с контролем. Вычисляют характерный сигнал в процентах. Вычисляют IС 50 для 10 концентраций ингибитора дублированно,пользуясь программным обеспечением Xlfit. Ссылочным соединением является ставроспорин (Sigma), имеющий IС 50 = 306 нМ (n=20). Результаты, полученные описанными выше экспериментальными методами, доказывают,что соединения по настоящему изобретению обладают полезной ингибирующей активностью по отношению к белковой тирозинкиназе рецептора HDGF и ингибирующей способностью в отношении тирозинкиназы p56lck и потому обладают терапевтической ценностью. Результаты описанных выше фармакологических тестов могут быть использованы для определения дозы и способа введения для конкретного терапевтического метода. Настоящее изобретение может быть осуществлено в других конкретных видах в пределах его объема или существенных признаков. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы IR1a представляет необязательно замещенный C1-С 10 алкил, гидрокси, необязательно замещенный C1-С 10 алкокси, необязательно замещенный С 3-С 7 циклоалкилокси, необязательно замещенный 4-7-членный оксагетероциклилокси или галоген;R1c представляет водород, необязательно замещенный C1-С 10 алкил или необязательно замещенный C1-С 10 алкокси,R2 представляетR3 представляет водород, орто- или парафтор или мета-С 1-С 3 низший алкил, C1-С 3 низший алкокси, галоген или карбамоил;Zb представляет NH или О,или его N-оксид, гидрат, сольват, пролекарство или соль, при условии, что R1a и R1b оба не являются необязательно замещенным C1-С 10 алкилом. 2. Соединение формулы I по п.1, в которомR1a представляет необязательно замещенный C1 С 3 низший алкокси, необязательно замещенный моноциклический С 1-С 7 циклоалкилокси, необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси. 3. Соединение по п.2, в котором R1a представляет необязательно замещенный C1-С 3 низший алкокси или необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси. 4. Соединение по п.3, в котором R1a представляет метокси, этокси, 2-(этокси)этокси, 2-(4 морфолинил)этокси или фуранилокси. 5. Соединение по п.1, в котором R1b представляет водород, необязательно замещенныйC1-С 3 низший алкокси, необязательно замещенный моноциклический С 3-С 7 циклоалкилокси или необязательно замещенный 4-7-членный моноциклический оксагетероциклилокси. 6. Соединение по п.5, в котором R1b представляет водород или необязательно замещенный С 1-С 3 низший алкокси. 7. Соединение по п.6, в котором R1b представляет метокси или этокси. 8. Соединение по п.1, в котором R1a и R1b представляют C1-С 3 низший алкокси. 9. Соединение по п.8, в котором R1a и R1b представляют метокси или этокси. 10. Соединение по п.1, в котором R1c представляет водород. 11. Соединение по п.1, в котором R3 представляет водород, орто- или парафтор или мета 39 метил, трифторметил, метокси, фтор, хлор, бром или карбамоил. 12. Соединение по п.1, в котором Za представляет N. 13. Соединение по п.1, в котором Za представляет СН. 14. Соединение по п.1, в котором Zb представляет NH. 15. Соединение по п.1, в котором Zb представляет О. 16. Соединение по п.1, представляющее собой 2-анилино-6-хиноксалинол; 2-анилино-6-изопропоксихиноксалин; 2-фенокси-6-метоксихиноксалин; 2-(3-карбамоилфениламино)-6-метоксихиноксалин; 2-(2-фторфениламино)-6,7-диэтоксихиноксалин; 2-(3-трифторметилфениламино)-6,7-диэтоксихиноксалин; фенил-[6-(тетрагидрофуран-3(R)илокси) хиноксалин-2-ил]амин;(3-хлорфенил)-(6,7-диметоксихиноксалин 2-ил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемая соль. 17. Соединение по п.1, представляющее собой фенил-[6-(тетрагидрофуран-3(R)-илокси) хиноксалин-2-ил]амин;(3-хлорфенил)-(6,7-диметоксихиноксалин 2-ил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемая соль. 18. Соединение по п.1, которое представляет собой фенил-[6-(тетрагидрофуран-3(R)илокси)хиноксалин-2-ил]амин или его N-оксид,гидрат, сольват, пролекарство или фармацевтически приемлемую соль. 19. Соединение по п.1, которое представляет собой (6,7-диметоксихиноксалин-2-ил)-(3 фторфенил)амин или его N-оксид, гидрат, сольват, пролекарство или фармацевтически приемлемую соль. 20. Соединение по п.1, которое представляет собой(3-бромфенил)-(6,7-диметоксихиноксалин-2-ил)амин или его N-оксид, гидрат,сольват, пролекарство или фармацевтически приемлемую соль. 21. Соединение по п.1, которое представляет собой (3-хлорфенил)-(6,7-диметоксихиноксалин-2-ил)амин или его N-оксид, гидрат,сольват, пролекарство или фармацевтически приемлемую соль. 22. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель. 23. Способ ингибирования активности тирозинкиназы фактора роста PDGF, включающий контактирование соединения по п.1 с композицией, содержащей тирозинкиназу PDGF. 24. Способ ингибирования активности тирозинкиназы Lck, включающий контактирование соединения по п.1 с композицией, содержащей тирозинкиназу Lck. 25. Способ ингибирования пролиферации клеток, дифференциации или высвобождения медиатора у пациента, страдающего от нарушения, характеризующегося такими пролиферацией, и/или дифференциацией, и/или высвобождением медиатора, включающий введение указанному пациенту фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли. 26. Способ по п.25, в котором нарушением является лейкоз, рак, глиобластома, псориаз,воспалительные заболевания,заболевания кости,фиброзные заболевания, артрит, фиброз легкого, фиброз почки, фиброз печени, атеросклероз или рестеноз после ангиопластики коронарной артерии, бедренной артерии или артерии почки. 27. Способ лечения пациента, подверженного патологии, связанной с гиперпролиферативным нарушением, включающий введение указанному пациенту фармацевтически эффек 41 тивного количества соединения по п.1 или его фармацевтически приемлемой соли. 28. Способ по п.27, в котором патологией,связанной с гиперпролиферативным нарушением, является рак, восприимчивый к лечению путем ингибирования тирозинкиназы фактора роста PDGF. 29. Способ по п.28, в котором рак представляет собой рак головного мозга, рак яичника, рак толстой кишки, рак предстательной железы, рак легких, саркому Капоши или злокачественную меланому. 30. Способ по п.27, в котором указанной патологией является рестеноз. 31. Способ по п.30, в котором соединение по п.1 вводят посредством нанесения покрытия 42 на расширительном устройстве (стенте), где покрытие содержит соединение по п. 1. 32. Способ лечения воспаления у пациента,страдающего от него, включающий введение указанному пациенту фармацевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли. 33. Способ лечения ревматоидного артрита, рассеянного склероза, системной красной волчанки, болезни трансплантат против хозяина, астмы, воспалительного заболевания кишечника или панкреатита, включающий введение пациенту, нуждающемуся в таком лечении,ингибирующего тирозинкиназу Lck количества соединения по п.1.
МПК / Метки
МПК: C07D 241/44, A61P 35/00, A61K 31/47
Метки: тирозинкиназы, соединения, роста, хиноксалиновые, хинолиновые, тромбоцитарного, ингибирующие, фактора
Код ссылки
<a href="https://eas.patents.su/22-2600-hinolinovye-i-hinoksalinovye-soedineniya-ingibiruyushhie-tirozinkinazy-trombocitarnogo-faktora-rosta-i-ili-p56-lck.html" rel="bookmark" title="База патентов Евразийского Союза">Хинолиновые и хиноксалиновые соединения, ингибирующие тирозинкиназы тромбоцитарного фактора роста и/или p56 lck</a>
Лиофилизированная композиция морфогенетического фактора роста кости мр52 человека и способ её получения

Номер патента: 1579
Опубликовано: 25.06.2001
Авторы: Итикава Хидеки, Инагаки Митсуко
МПК: A61K 38/18
Метки: композиция, получения, кости, человека, лиофилизированная, роста, способ, фактора, морфогенетического, мр52
Формула / Реферат:
1. Лиофилизированная композиция морфогенетического фактора кости МР52 человека, содержащая смесь морфогенетического фактора кости МР52 человека с маннитом в весовом соотношении в пределах 1:5-50. 2. Композиция по п.1, отличающаяся тем, что морфогенетический фактор кости МР52 человека получен с применением технологий генетической инженерии. 3. Способ получения лиофилизированной композиции морфогенетического фактора кости МР52 человека,...
Способ селективной регуляции клеточного роста и клеточной дифференцировки и применение фармацевтической композиции, содержащей арил- и гетероарилхиназолиновые соединения для этой цели.

Номер патента: 840
Опубликовано: 24.04.2000
Авторы: Спада Альфред П., Майерз Майкл Р., Магир Мартин П., Персонз Пол Е.
МПК: A61K 31/535
Метки: способ, этой, регуляции, дифференцировки, клеточной, гетероарилхиназолиновые, клеточного, фармацевтической, роста, применение, композиции, цели, селективной, арил, соединения, содержащей
Формула / Реферат:
1. Способ селективной регуляции клеточного роста и клеточной дифференцировки, характеризующийся активностью рецептора типа 2 человеческого эпидермального фактора роста (HER2), причем указанный способ предусматривает введение пациенту, нуждающемуся в такой регуляции, эффективного HER2-ингибирующего количества соединения формулы где А представляет замещенную или незамещенную моно- или бициклическую арильную, гетероарильную, циклоалкильную или...
Хинолиновые антагонисты лейкотриенов

Номер патента: 1797
Опубликовано: 27.08.2001
Авторы: Балани Суреш К., Дюфресн Клод, Эрисон Байрон Х., Бэйлли Томас А.
МПК: A61P 11/06, A01N 43/42, A61K 31/47...
Метки: лейкотриенов, хинолиновые, антагонисты
Формула / Реферат:
1. Соединение формулы и его индивидуальные оптические изомеры или его фармацевтически приемлемое производное. 2. Выделенное и очищенное соединение по п.1, по существу свободное от других продуктов метаболизма монтелукаста натрия. 3. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 4. Способ предотвращения действия лейкотриена у млекопитающего, включающий...
Сочетание ингибитора тирозинкиназы и химической стерилизации для лечения рака предстательной железы

Номер патента: 2525
Опубликовано: 27.06.2002
Авторы: Дайонн Крэйг А., Вот Джеффри Л., Исаакс Джон
МПК: A61K 45/06, A61P 35/00
Метки: сочетание, тирозинкиназы, железы, рака, лечения, стерилизации, химической, предстательной, ингибитора
Формула / Реферат:
1. Способ ингибирования прогрессирования опухоли предстательной железы у млекопитающих, включающий введение указанному млекопитающему терапевтически эффективного количества ингибитора тирозинкиназы, выбранного из группы, состоящей из trkA ингибитора, trkB ингибитора и trkC ингибитора, и вместе с ним введение указанному млекопитающему терапевтически эффективного количества средства для химической стерилизации, выбранного из группы, состоящей из...
Производные 2-хинолона, ингибирующие фарнезилтрансферазу

Номер патента: 719
Опубликовано: 28.02.2000
Авторы: Вене Марк Гастон, Санз Жерар Шарль, Энд Девид Вилльям, Анжибо Патрик Рене
МПК: C07D 401/06, A61K 31/47
Метки: фарнезилтрансферазу, ингибирующие, 2-хинолона, производные
Формула / Реферат:
1. Соединение формулы (I) его стереоизомерная форма, фармацевтически приемлемая соль присоединения кислоты или основания, в которых пунктирная линия обозначает необязательную связь; Х представляет кислород или серу; R1 представляет водород, С1-12алкил, Аr1, Аr2C1-6алкил, хинолинилС1-6алкил, пиридилС1-6алкил, гидроксиС1-6алкил, C1-6-алкилоксиC1-6алкил, моно- или ди(C1-6алкил)аминоC1-6алкил, аминоС1-6алкил, или радикал формулы...
Предыдущий патент: Замещенные 2-бензоилциклогексан-1, 3-дионы
Следующий патент: Шлем
Случайный патент: Способ перехода между несовместимыми катализаторами полимеризации