Замещенные n-[1-циано-2-(фенил)этил]-2- азабицикло[2.2.1]гептан-3-карбоксамидные ингибиторы катепсина с
Номер патента: 24817
Опубликовано: 31.10.2016
Авторы: Ритер Дорис, Грундль Марк, Винен Вольфганг, Ост Торстен, Петерс Штефан, Пауч Александер





















Формула / Реферат
1. Соединения формулы I

в которой
n равно 0 или 1;
R1 обозначает F-, НО-;
R2 выбран из группы, включающей галоген, С1-С6-алкил-, С2-С6-алкенил-, С3-С6-циклоалкил-, С3-С6-циклоалкенил- или кольцевую систему, выбранную из группы, включающей
моноциклический С5-С7-гетероциклил-, в котором 1 или 2 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1;
бициклический C8-С10-гетероциклил-, в котором 1, 2, 3 или 4 атома углерода заменены гетероатомами, выбранными из группы, -S-, -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1;
арил-, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1;
С5-С10-гетероарил-, в котором 1, 2 или 3 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является ароматическим, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1;
R2.1 обозначает Me-, F2HC-H2C-, O=, Me(O)C-, Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Et(O)2S-, iPr(O)2S-, Me(O)2SO-, Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(О)С-, фенил-Н2С-, MeO(CH2)3-, NC-, F-, Me2N(O)2S-, MeHN(O)2S-, МеОН2С-, Ме2(НО)С-, циклопропил- или фенил-, необязательно замещенный с помощью МеО-;
или их соль.
2. Соединения формулы I по п.1, в которой
n равно 0 или 1;
R1 обозначает F-, НО-;
R2 выбран из группы, включающей галоген, С1-С4алкил-, С2-С4-алкенил-, С3-С6-циклоалкил-, С3-С6-циклоалкенил- или
моноциклический С5-С7-гетероциклил-, в котором 1 или 2 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей Me-, F2H-CH2C-, O=, Me(O)C-, Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Et(O)2S-, iPr(O)2S-, Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(O)C-, фенил-Н2С-;
бициклический С8-С10-гетероциклил-, в котором 1, 2, 3 или 4 атома углерода заменены гетероатомами, выбранными из группы -S-, -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей Me-, O=, МеО(СН2)3-;
фенил-, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, F-, Me(O)2S-, Et(O)2S-, Me(O)2SO-, Me2N(O)2S-, MeHN(O)2S-;
пиридинил, оксазолил или 1,2,3-триазол-, каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, МеОН2С-, Ме2(НО)С-, циклопропил- или фенил-, необязательно замещенный с помощью МеО-;
или их соль.
3. Соединения формулы I по п.1 или 2, в которой
n равно 0 или 1;
R1 обозначает F-, НО-;
R2 выбран из группы, включающей


или их соль.
4. Соединения формулы I'

в которой n, R1 и R2 обладают значениями, указанными в одном из пп.1-3.
5. Применение соединения формулы I по одному из пп.1-3 в качестве лекарственного средства.
6. Применение соединения формулы I по одному из пп.1-3 в качестве лекарственного средства для лечения астмы и аллергических заболеваний, желудочно-кишечных воспалительных заболеваний, эозинофильных заболеваний, хронического обструктивного заболевания легких, инфицирования патогенными микробами, ревматоидного артрита и атеросклероза.
7. Фармацевтическая композиция, отличающаяся тем, что она содержит одно или большее количество соединений формулы I по одному из пп.1-3 или их фармацевтически приемлемых солей.
8. Способ лечения или предупреждения заболеваний, при которых ингибиторы активности DPPI оказывают благоприятное терапевтическое воздействие, способ включает введение нуждающемуся в нем пациенту соединений формулы I по одному из пп.1-3 в терапевтически или профилактически эффективном количестве.
9. Фармацевтическая композиция, включающая соединение формулы I по одному из пп.1-3 и дополнительно одно или два активных вещества, выбранных из группы, включающей бета-миметики, антихолинергетики, кортикостероиды, ингибиторы PDE4, антагонисты LTD4, ингибиторы EGFR, ингибиторы CRTH2, ингибиторы 5-LO, антагонисты гистаминового рецептора, антагонисты CCR9 и ингибиторы SYK, ингибиторы NE, ингибиторы ММР9, ингибиторы ММР12.
Текст
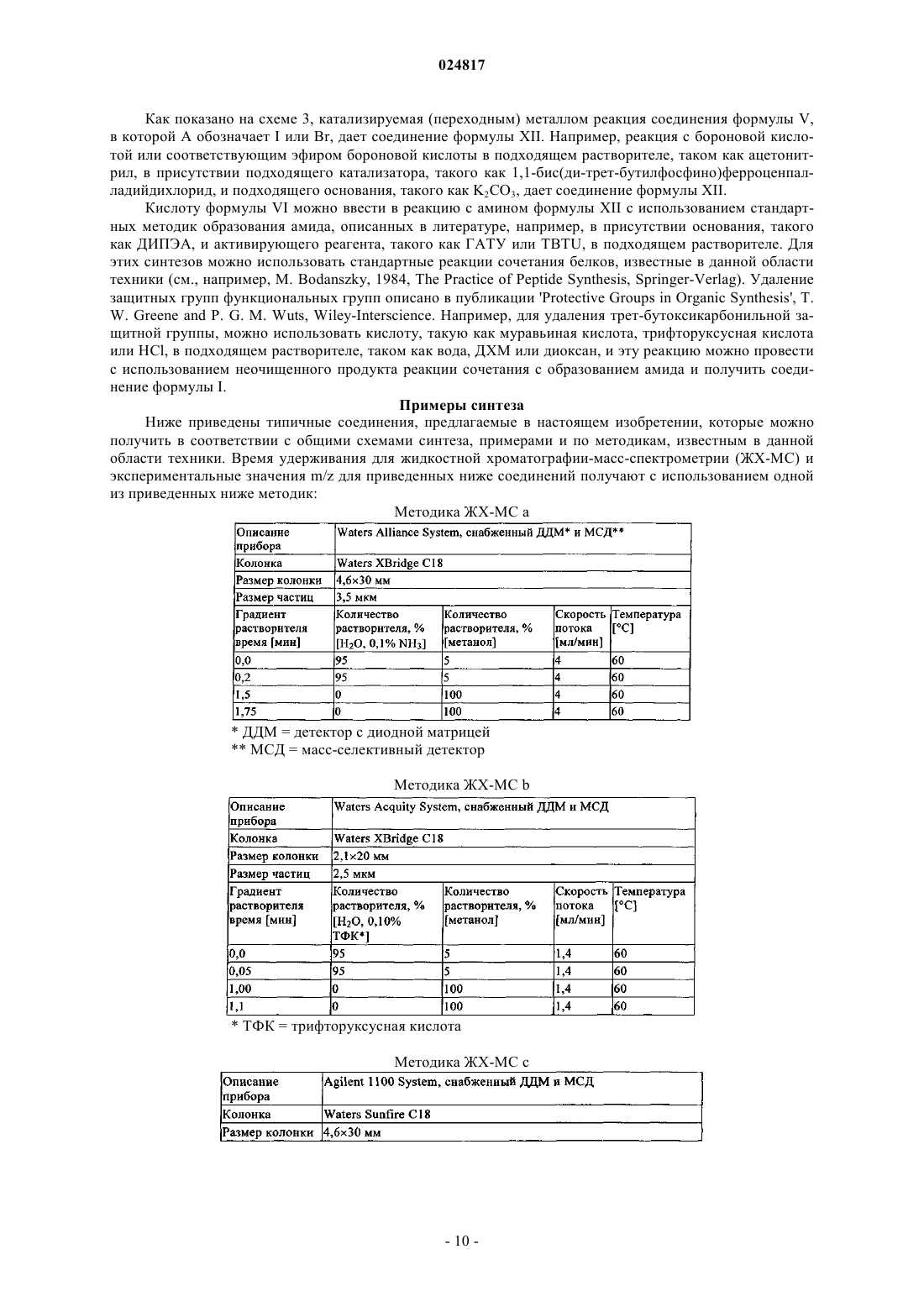
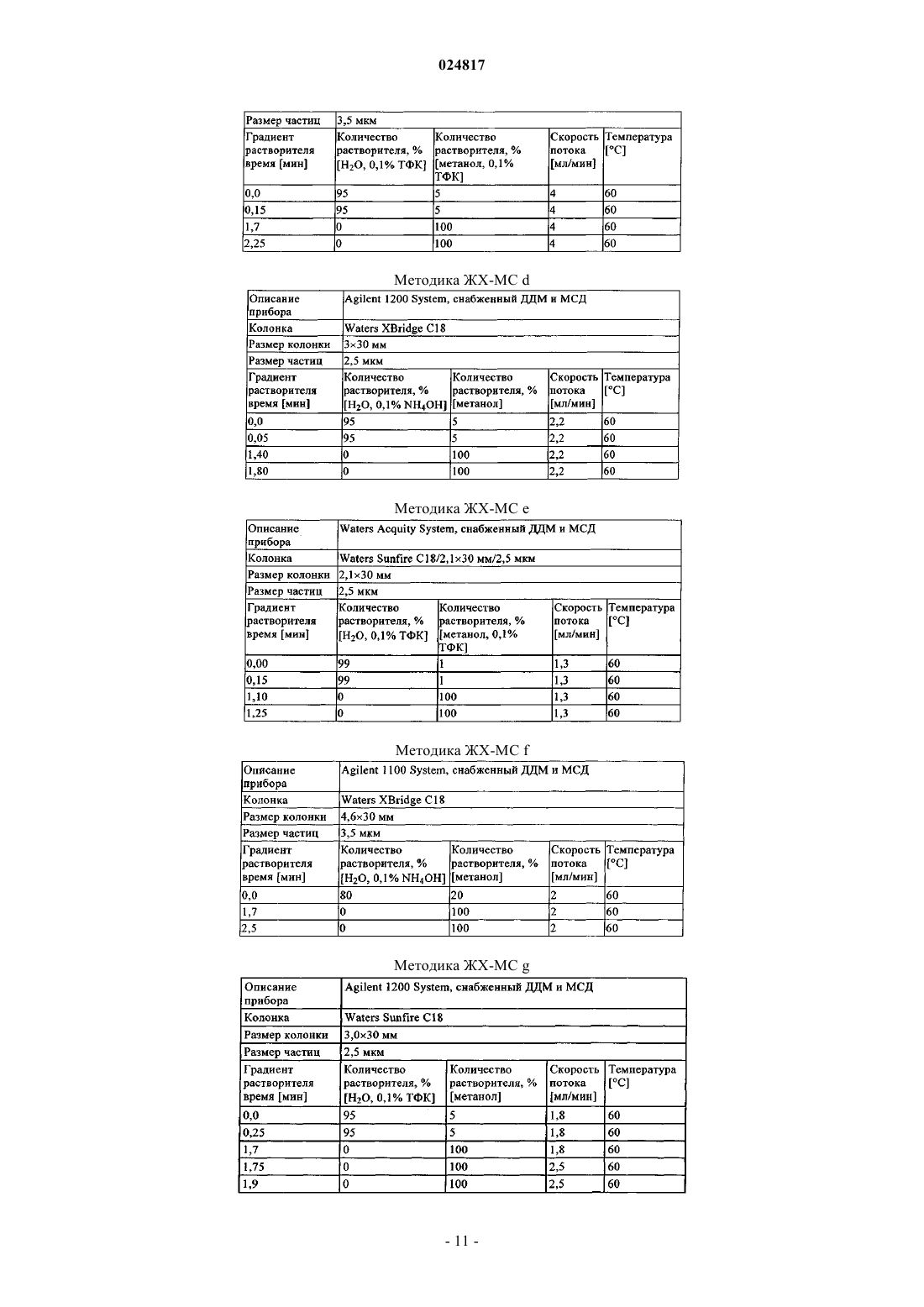
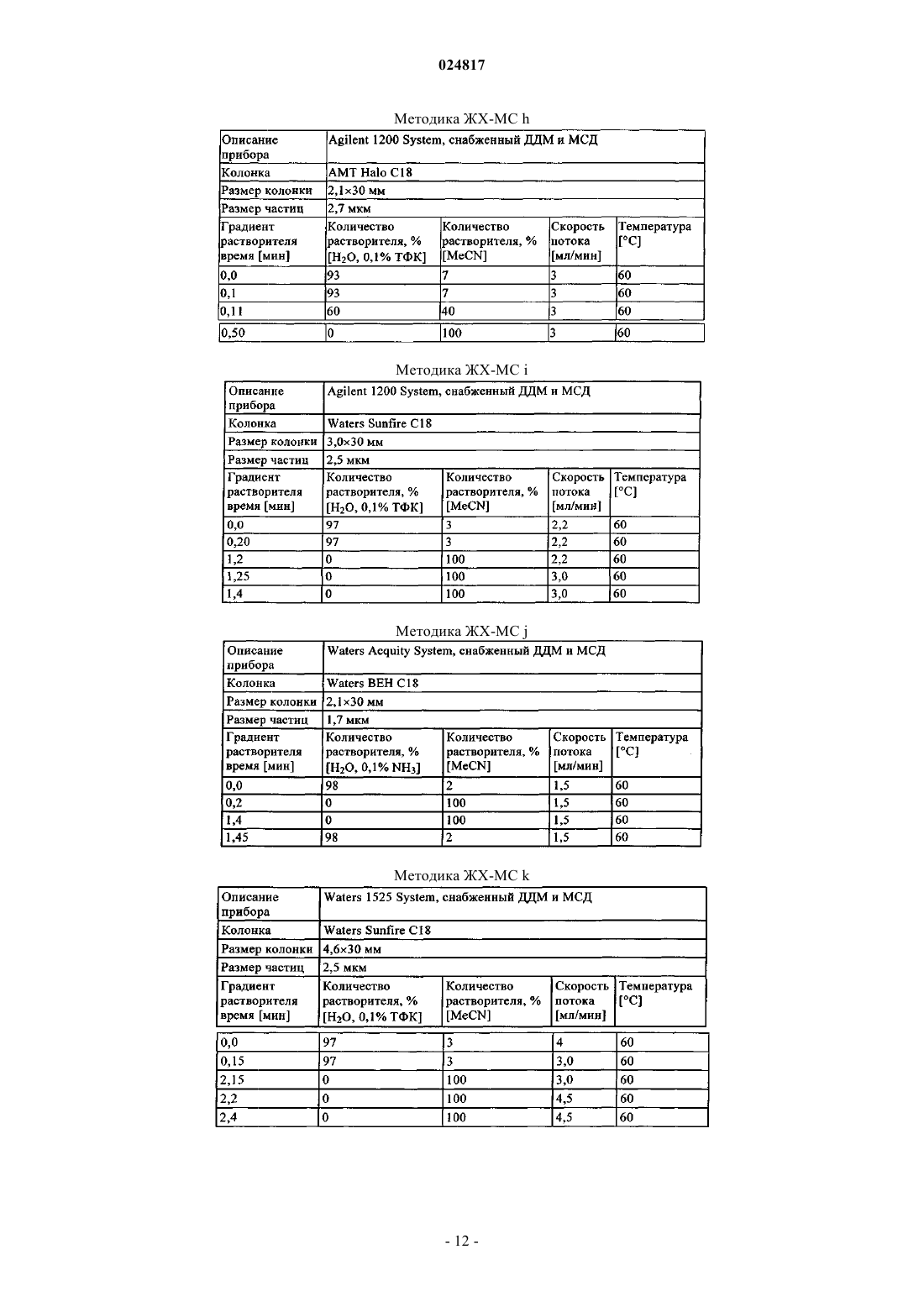
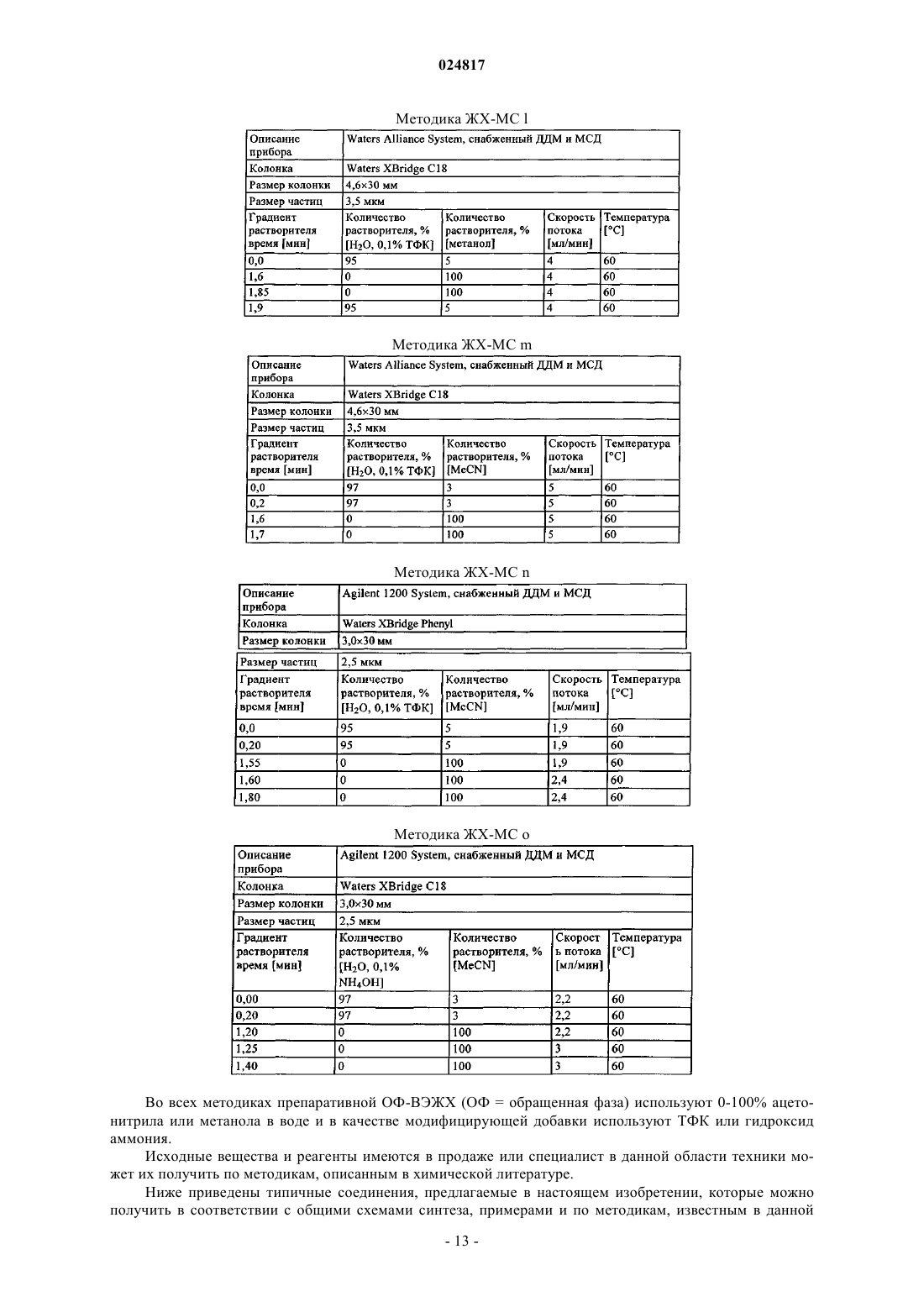
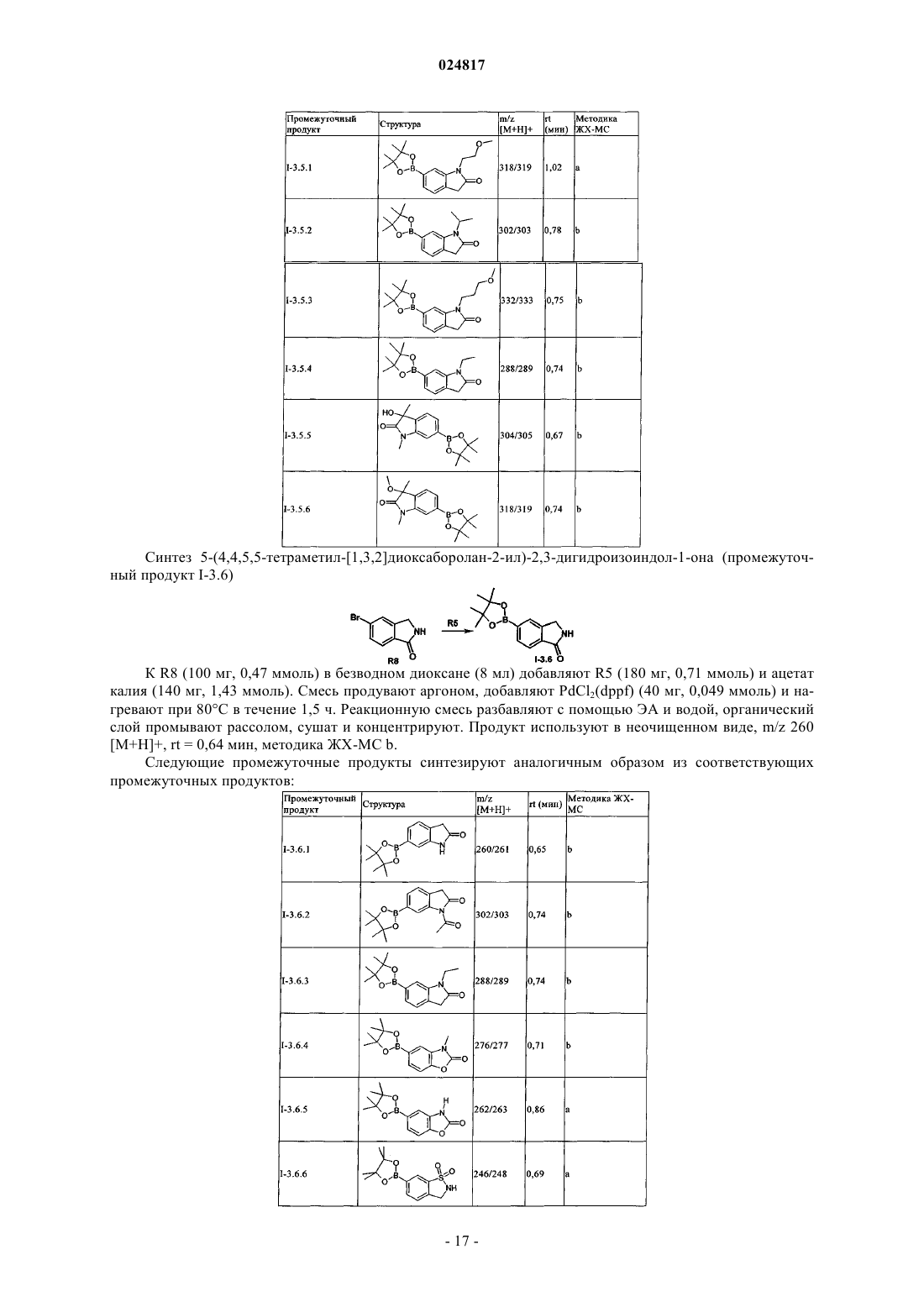
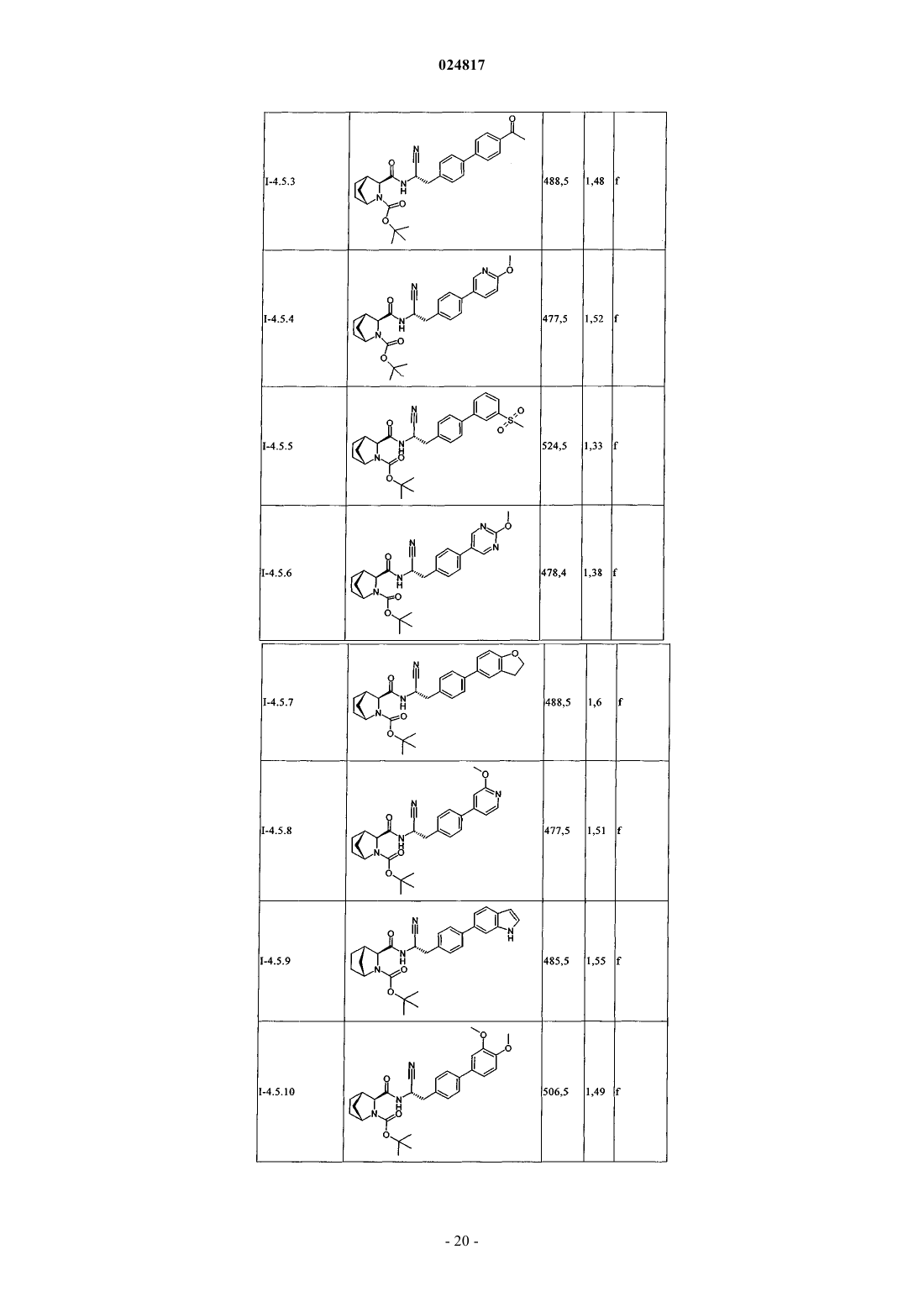
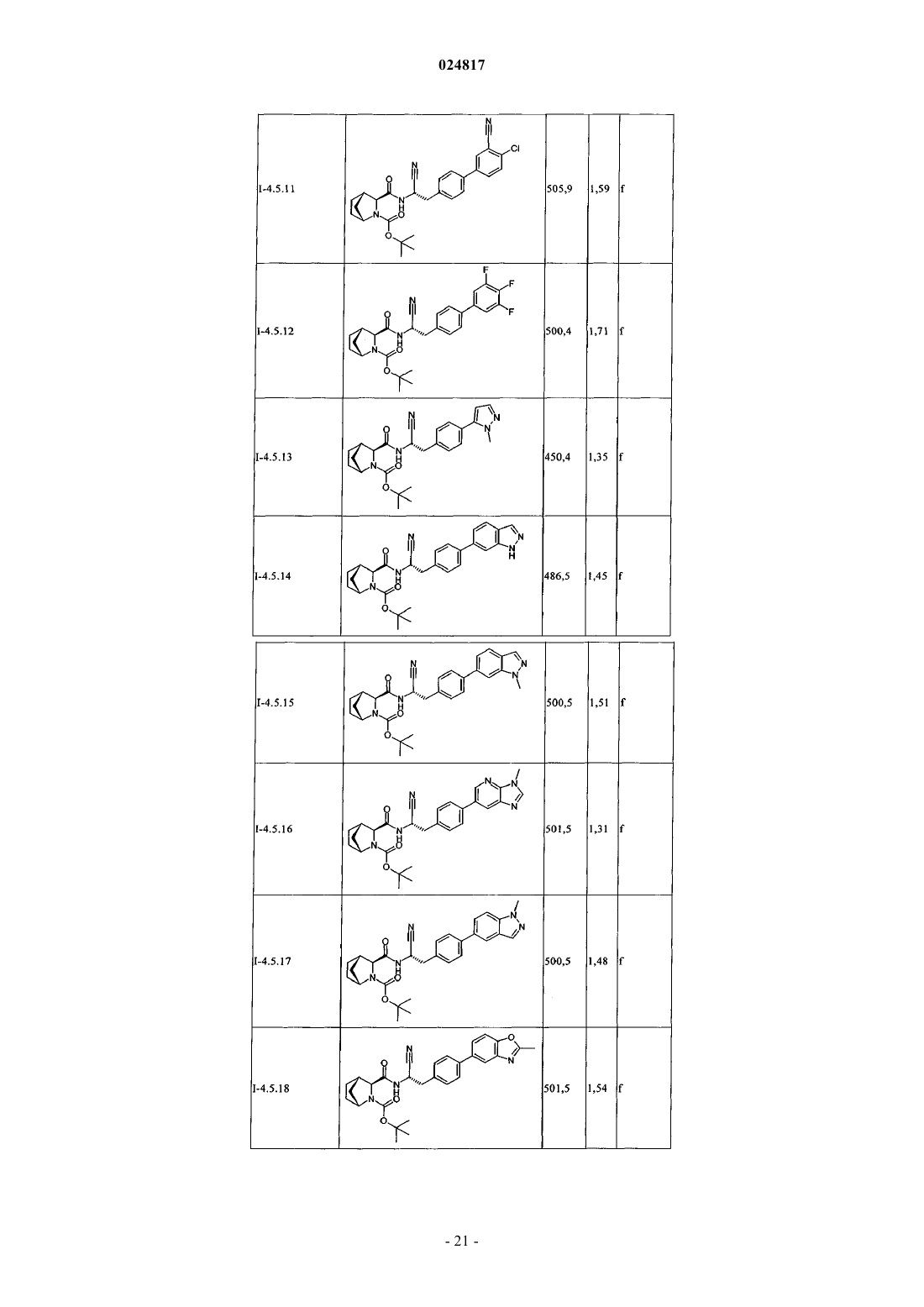
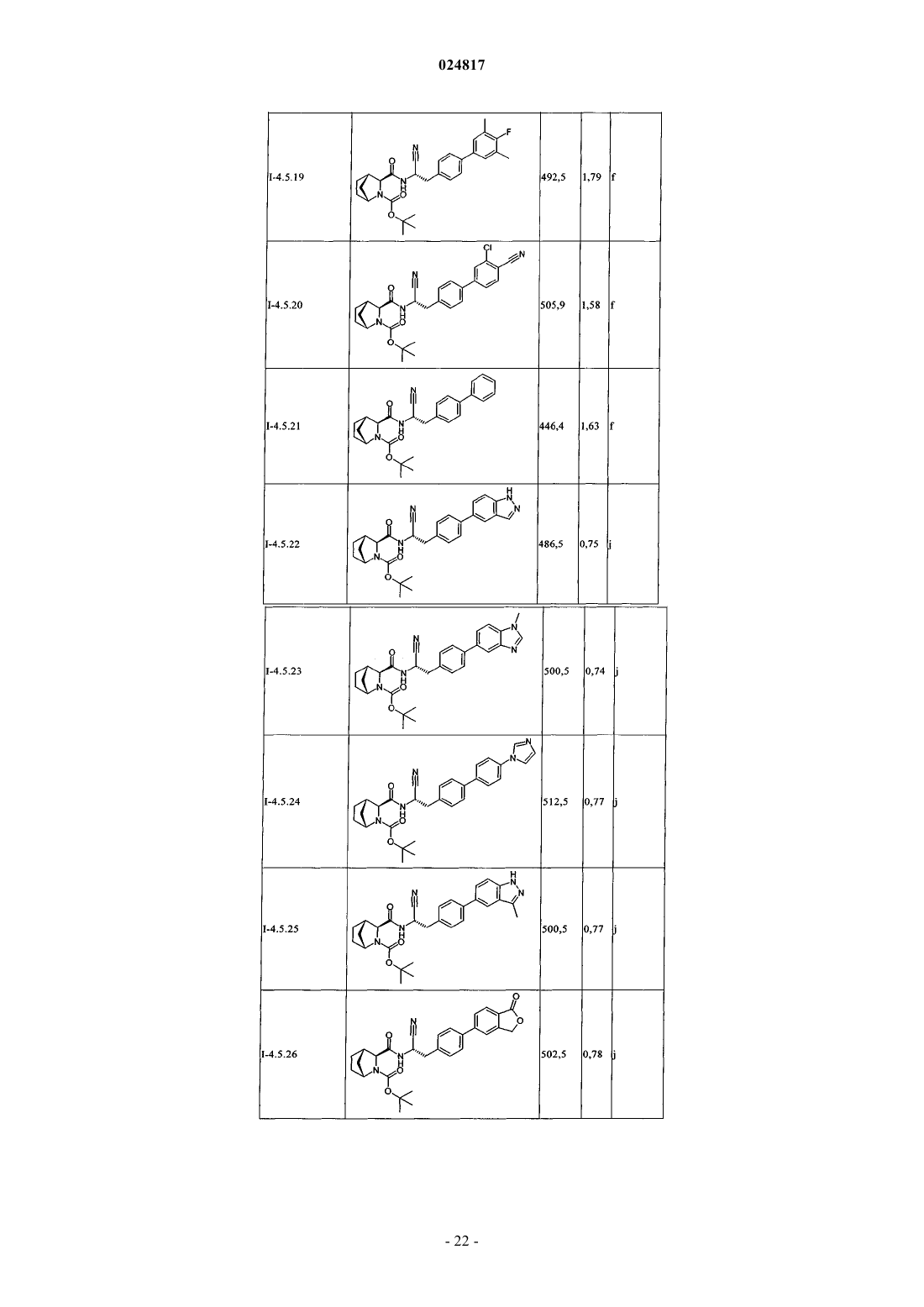
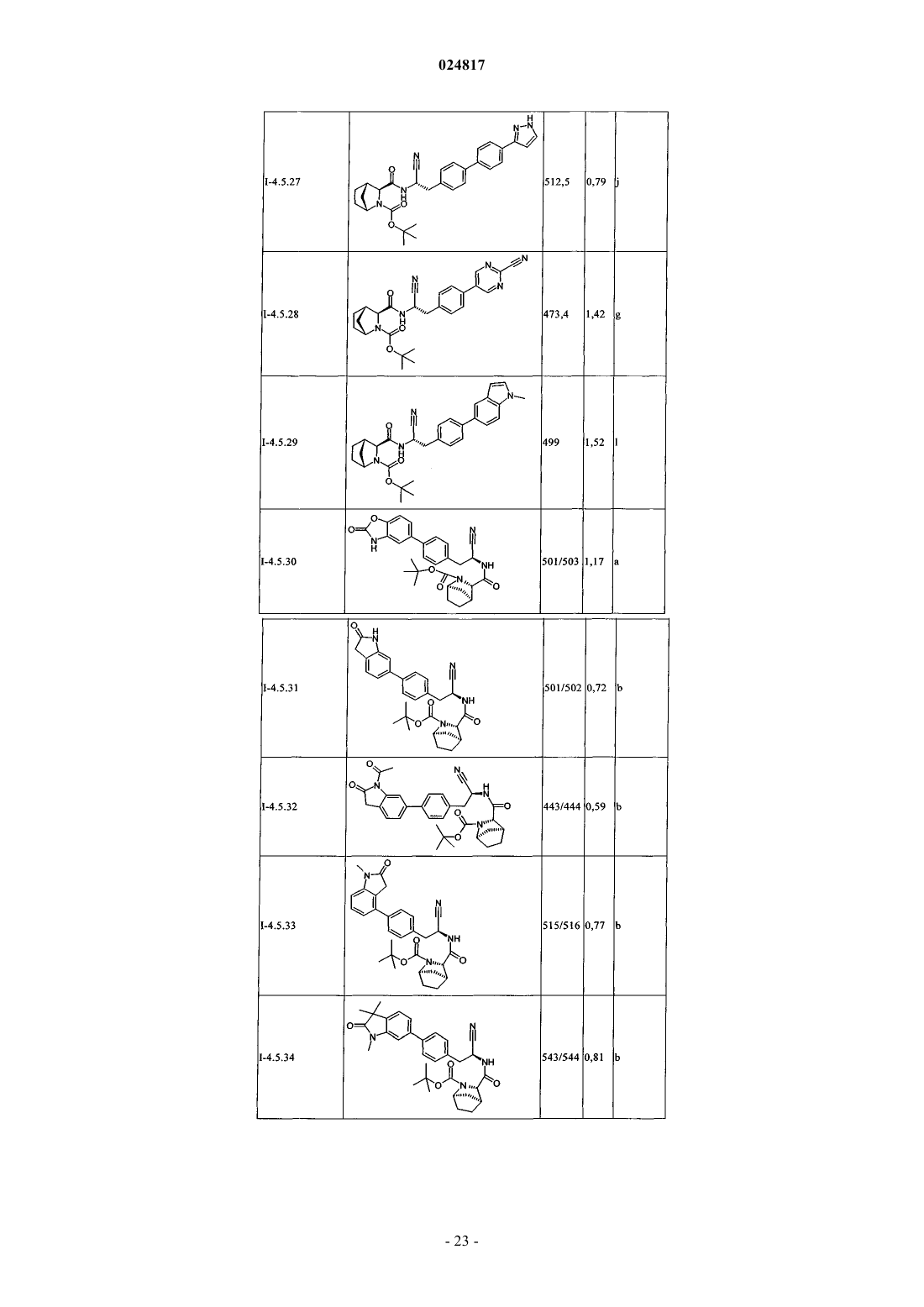
ЗАМЕЩЕННЫЕ N-[1-ЦИАНО-2-(ФЕНИЛ)ЭТИЛ]-2- АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3 КАРБОКСАМИДНЫЕ ИНГИБИТОРЫ КАТЕПСИНА С и их применение в качестве ингибиторов катепсина С, фармацевтические композиции, содержащие эти соединения, и способы применения этих соединений в качестве средств для лечения и/или предупреждения респираторных заболеваний.(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Область техники, к которой относится изобретение Настоящее изобретение относится к N-[1-циано-2-(фенил)этил]-2-азабицикло[2.2.1]гептан-3 карбоксамидам и их применению в качестве ингибиторов катепсина С, к фармацевтическим композициям, содержащим эти соединения, и к способам применения этих соединений в качестве средств для лечения и/или предупреждения заболеваний, связанных с активностью дипептидилпептидазы I, например,респираторных заболеваний. Уровень техники В WO 2004110988 раскрыты пептидилнитрилы, как ингибиторы дипептидилпептидазы I (DPPI),предназначенные для лечения ряда заболеваний. В WO 2009074829 и WO 2010142985 также раскрыты пептидилнитрилы, как ингибиторы дипептидилпептидазы I (DPPI), предназначенные для лечения астмы, ХОЗЛ (хроническое обструктивное заболевание легких) или аллергического ринита. Краткое изложение сущности изобретения Дипептидиламинопептидаза I (DPPI или катепсин С; ЕС 3.4.141) является лизосомальной цистеинпротеазой, которая способна удалять дипептиды из аминного конца белковых субстратов. Впервые DPPI обнаружили Gutman и Fruton в 1948 г. (J. Biol. Chem 174: 851-858, 1948). кДНК фермента человека описана в 1995 (Paris et al.; FEBS Lett 369: 326-330, 1995). Белок DPPI преобразуется в зрелый протеолитически активный фермент, состоящий из тяжелой цепи, легкой цепи и пропептида, который остается связанным с активным ферментом (Wolters et al.; J. Biol. Chem. 273: 15514-15520, 1998). В то время как другие цистеинкатепсины (например, В, Н, K, L и S) являются мономерами, DPPI представляет собой тетрамер,обладающий молекулярной массой, равной 200 кДа, содержащий 4 одинаковые субъединицы, каждая из которых состоит из 3 различных полипептидных цепей. DPPI конститутивно экспрессируется во многих тканях, причем самый высокий уровень экспрессии наблюдается в легких, почках, печени и селезенке(Kominami et al.; Biol. Chem. Hoppe Seyler 373: 367-373, 1992). В соответствии с ролью DPPI в активации серинпротеаз из гематопоэтических клеток, она также достаточно сильно экспрессируется в нейтрофилах, цитотоксических лимфоцитах, природных киллерах, альвеолярных макрофагах и мастоцитах. Недавние результаты исследования мышей с дефицитом DPPI показывают, что помимо того, что DPPI является важным ферментом в разрушении лизосомального белка, она также действует, как ключевой фермент в активации серинпротеаз-зерен в цитотоксических Т-лимфоцитах и природных киллерах (гранзимы А и В; Pham et al.; Proc. Nat. Acad. Sci 96: 8627-8632, 1999), мастоцитах (химаза и триптаза; Wolteret al.; J Clin. Invest. 109: 363.371, 2002). После активации эти протеазы способны разрушать различные компоненты внеклеточного матрикса, что может привести к повреждению тканей и хроническому воспалению. Таким образом, ингибиторы катепсина С, вероятно, могут быть применимы в качестве средств для лечения воспалительных заболеваний, в которых нейтрофилы играют главную роль, таких как хроническое обструктивное заболевание легких (ХОЗЛ). эмфизема легких, астма, рассеянный склероз и муковисцидоз (Guay et al.; Curr. Topics Med. Chem. 10: 708-716, 2010; Laine and Busch-Petersen; Expert Opin.Ther. Patents 20: 497-506, 2010). Обнаружено, что ревматоидный артрит является еще одним воспалительным заболеванием, в котором играет роль DPPI. Нейтрофилы поступают в место воспаления сустава и высвобождают катепсин G, эластазу и протеиназу 3, протеазы, которые предположительно вызывают разрушение хрящей, связанное с ревматоидным артритом. В действительности, мыши с дефицитом DPPI защищены от острого артрита, вызванного пассивным переносом моноклональных аттител к коллагену типа II (Adkison et al.; J Clin. Invest. 109: 363.371, 2002). С учетом роли, которую DPPI играет в активации некоторых провоспалительных серинпротеаз, желательно получить соединения, которые ингибируют ее активность, что тем самым ингибирует активность находящейся в прямом направлении серинпротеазы. Согласно изобретению неожиданно установлено, что бициклические соединения, предлагаемые в настоящем изобретении, обладают высокой активностью по отношению к катепсину С, высокой селективностью по отношению к другим катепсинам. например, катепсину K, и обычно подходящими фармакокинетическими характеристиками. Подробное описание изобретения Соединения формулы IR2 выбран из группы, включающей галоген, C1-С 6-алкил-, С 2-С 6-алкенил-, С 3-С 6-циклоалкил-, С 3 С 6-циклоалкенил- или кольцевую систему, выбранную из группы, включающей моноциклический С 5-С 7-гетероциклил-, в котором 1 или 2 атома углерода заменены гетероатомами,выбранными из группы, включающей -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1; бициклический С 8-С 10-гетероциклил-, в котором 1, 2, 3 или 4 атома углерода заменены гетероатомами, выбранными из группы, -S-, -О- или -N-, и кольцо является полностью или частично насыщенным,необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1; арил-, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1; С 5-С 10-гетероарил-, в котором 1, 2 или 3 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является ароматическим, необязательно независимо друг от друга замещенный с помощью 1 или 2 R2.1;R2.1 обозначает Me-, F2HC-H2C-, О=, Ме(О)С-, Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Eit(O)2S-,iPr(O)2S-, Me(O)2SO-, Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(О)С-, фенил-Н 2 С-, MeO(CH2)3-,NC-, F-, Me2N(O)2S-, MeHN(O)2S-, MeOH2C-, Me2(HO)C-, циклопропил- или фенил-, необязательто замещенный с помощью МеО-; или их соль. Предпочтительные варианты осуществления Предпочтительными являются указанные выше соединения формулы I, в которойR2 выбран из группы, включающей галоген, С 1-С 4-алкил-, С 2-С 4-алкенил-, С 3-С 6-циклоалкил-, C3 С 6-циклоалкенил- или моноциклический С 5-С 7-гетероциклил-, в котором 1 или 2 атома углерода заменены гетероатомами,выбранными из группы, включающей -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы,включающей Me-, F2H-CH2C-, O=, Me(O)C-, Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Et(O)2S-, iPr(O)2S-,Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(O)C-, фенил-Н 2 С-; бициклический С 8-С 10-гетероциклил-, в котором 1, 2, 3 или 4, предпочтительно 1 или 2 атома углерода заменены гетероатомами, выбранными из группы, -S-, -О- или -N-, и кольцо является полностью или частично насыщенным, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей Me-, О=, МеО(СН 2)3-; фенил-, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, F-, Ме(О)2S-, Et(O)2S-, Me(O)2SO-, Me2N(O)2S-, MeHN(O)2S-; пиридинил, оксазолил или 1,2,3-триазол-, каждый необязательно замещенный 1 или 2 остатками,независимо друг от друга выбранными из группы, включающей NC-, МеОН 2 С-, Ме 2(НО)С-, циклопропил- или фенил-, необязательно замещенный с помощью МеО-; или их соль. Предпочтительными являются указанные выше соединения формулы I, в которойR2 выбран из группы, включающей этил-, этенил-, изопропенил-, 2-метил-н-пропил-, 2-метил-н-1 пропенил-, циклогексил-, циклогексенил-, 1-, тетрагидропиранил-, 3,6-дигидропиранил-, октагидропирроло[1,2 а]пиразинил-, гексагидропирроло[1,2 а]пиразин-6-онил-, 4,5,6,7-тетрагидротиено[3,2 с]пиридинил-, или пиперидинил-, пиперазинил-, 1,4-диазепанил-, тетрагидропиранил-, тетрагидрофуранил-, диоксанил-, морфолинил-, пирролидинип-; предпочтительно пиперидинил-, пиперазинил-, 1,4-диазепанил-,каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы,включающей Me-, F2HC-H2C-, O=, Me(O)C-, Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Et(O)2S-, iPr(O)2S-,Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(О)С-, фенил-Н 2 С-; индолил-, индазолил-, хинолинил-, изохинолинил-, изохинолонил-, хинолонил-, индолин-2-онил-,изоиндолин-1-онил-, изатинил-, бензоксазол-2-онил-; пирролидинопиразинонил-, пирролидинопиразинил-,тетрагидротиенопиридинил-; предпочтительно индол-2-онил-, изоиндол-1-онил-, бензоксазол-2 онил, пирролидинопиразинонил-, пирролидиноииразинил-, тетрагидротиенопиридинил-, каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающейMe-, МеО(СН 2)3-; фенил-, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, F-, Me(O)2S-, Et(O)2S-, Me(O)2SO-, Me2N(O)2S-, MeHN(O)2S-; пирролил-, пиразолил-, имидазолил-, изоксазолил-, пиразинил-, пиридинил-, триазолил-, оксазолил, оксадиазолил-; предпочтительно пиридинил, 1,2,3-триазолил-, оксазолил-; предпочтительно пиридинил или 1,2,3-триазолил-, каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, МеОН 2 С-, Ме 2(НО)С-, циклопропил-или фенил-, необязательно замещенный с помощью МеО-; или их соль. Предпочтительными являются указанные выше соединения формулы I, в которой n равно 0 или 1;R2 выбран из группы, включающей этил-, этенил-, изопропенил-, 2-метил-н-пропил-, 2-метил-н-1 пропенил-, циклогексил-, циклогексенил-, 1-, тетрагидропиранил-, 3,6-дигидропиранил-, октагидропирроло[1,2 а]пиразинил-, гексагидропирроло[1,2 а]пиразин-6-онил-, 4,5,6,7-тетрагидротиено[3,2 с]пиридинил-, или пиперидинил-, пиперазинил-, 1,4-диазепанил-, каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей Me-, F2HC-H2C-, О=, Ме(О)С-,Et(O)C-, iPr(O)C-, nPr(O)C-, Me(O)2S-, Et(O)2S-, iPr(O)2S-, Me2N(O)C-, EtHN(O)C-, iPrHN(O)C-, циклопропил-(O)C-, фенил-Н 2 С-; индол-2-онил-, изоиндол-1-онил-, бензоксазол-2-онил-, каждый необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей Me-, МеО(СН 2)3-; фенил-, необязательно замещенный 1 или 2 остатками, независимо друг от друга выбранными из группы, включающей NC-, F-, Me(O)2S-, Et(O)2S-, Me(O)2SO-, Me2N(O)2S-, MeHN(O)2S-; пиридинил или 1,2,3-триазол-, оба необязательно замещенные 1 или 2 остатками, независимо друг от друга выбранными из группы, зключающей NC-, МеОН 2 С-, Ме 2(НО)С-, циклопропил- или фенил-,замещенный с помощью МеО-,или их соль. Предпочтительными являются указанные выше соединения формулы I, в которой или их соль. Из числа указанных выше значений группы R2 предпочтительными являются такие значения R2, когда она означает галоген предпочтительно Br, I; или выбраны из числа следующих групп, включающих: А 0: C1-C6-алкил-, С 2-С 6-алкенил-; предпочтительно метил-, этил-, этенил-, изопропил-, н-пропил-, изопропенил-, н-пропенил-, 2-метил-н-пропил-, 2-метил-н-1-пропенил-; предпочтительно этил-, этенил-,изопропенил-, 2-метил-н-пропил-, 2-метил-н-1-пропенил; каждый независимо друг от друга замещенный с помощью 1 или 2 R2.1, предпочтительно метил-, этил-, этенил-, изопропил-, н-пропил-, изопропенил-, н-3 024817 пропенил-, 2-метил-н-пропил-, 2-метил-н-1-пропенил-; предпочтительно этил-, этенил-, изопропенил-, 2 метил-н-пропил-, 2-метил-н-1-пропенил; или А 1: С 3-С 6-циклоалкил-, С 3-С 6-циклоалкенил-; предпочтительно циклопентил, циклопентенил, циклогексил-, циклогексенил; предпочтительно циклогексил-, циклогексенил; или А 2: моноциклический С 5-С 7-гетероциклил-, в котором 1 или 2 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является полностью или частично насыщенным; предпочтительно пиперидинил-, пиперазинил-, 1,4-диазепанил-, тетрагидропиранил-, тетрагидрофуранил-, диоксанил-, морфолинил-, пирролидинил-; предпочтительно пиперидинил-, пиперазинил-, 1,4-диазепанил-; илиA3: бициклический С 8-С 10-гетероциклил-, в котором 1, 2, 3 или 4 атома углерода заменены гетероатомами, выбранными из группы, включающей -S-, -О- или -N-, и кольцо является полностью или частично насыщенным; предпочтительно индолил-, индазолил-, хинолинил-, изохинолинил-, изохинолонил-,хинолонил-, индолин-2-онил-, изоиндолин-1-онил-, изатинил-, бензоксазол-2-онил-; пирролидинопиразинонил-, пирролидинопиразинил-, тетрагидротиенопиридинил- предпочтительно индол-2-онил-, изоиндол-1-онил-, бензоксазол-2-онил, пирролидинопиразинонил-, пирролидинопиразинил-, тетрагидротиенопиридинил-; или А 4: С 5-С 6-гетероарил-, в котором 1, 2 или 3 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является ароматическим; предпочтительно моноциклический C5-С 6-гетероарил-, в котором 1, 2 или 3 атома углерода заменены гетероатомами, выбранными из группы, включающей -О- или -N-, и кольцо является ароматическим; предпочтительно пирролил-, пиразолил-, имидазолил-, изоксазолил-, пиразинил-, пиридинил-, триазолил-, оксазолил-, оксадиазолил-; предпочтительно пиридинил, 1,2,3-триазолил-, оксазолил-; предпочтительно пиридинил или 1,2,3 триазолил-: или А 5: арил-, предпочтительно фенил-; или где каждый представитель групп А 0 - А 5 необязательно независимо друг от друга может быть замещен с помощью 1 или 2 R2.1. Предпочтительными являются указанные выше соединения формулы I в энантиомерно чистой форме, описывающиеся формулой I' в которой n, R1 и R2 обладают указанными выше значениями. Использующиеся термины и определения Терминам, специально не определенным в настоящем изобретении, следует придавать значения,которые им придал бы специалист в данной области техники с учетом описания и контекста. Однако,если не указано иное, то при использовании в описании приведенные ниже термины обладают указанными значениями и используются указанные ниже обозначения. В определенных ниже группах, радикалах или фрагментах, перед группой часто указано количество атомов углерода, например, C1-С 6-алкил означает алкильную группу или алкильный радикал, содержащий от 1 до 6 атомов углерода. Обычно в простых группах, таких как НО, H2N, OS, O2S, NC (цианогруппа), НООС. F3C и т. п., специалист в данной области техники может определить положение (положения) присоединения радикала к молекуле с учетом свободных валентных связей самой группы. В сложных группах, содержащих две или большее количество подгрупп, последняя названная группа является местом присоединения радикала,например, заместитель "арил-С 1-С 3-алкил-"означает арильную группу, которая присоединена к C1-С 3 алкильной группе, последняя присоединена к ядру или к группе, к которой присоединен заместитель. В случае, если соединение, предлагаемое в настоящем изобретении описано с помощью химического названия и в виде формулы, то случае любых различий определяющей является формула. Знак звездочки можно использовать в субформулах для обозначения связи, которая соединена с ядром молекулы,как это определено. Выражения "предупреждение", "профилактика", "профилактическое лечение" или "предупредительное лечение" при использовании в настоящем изобретении следует понимать, как синонимы и в том смысле, что риск развития патологического состояния, указанного выше в настоящем изобретении, снижается, особенно у пациента, для которого существует повышенная опасность возникновения указанных патологических состояний или соответствующий анамнез, например, повышенная опасность развития метаболического нарушения, такого как диабет или ожирение, или другого нарушения, указанного в настоящем изобретении. Таким образом, выражение "предупреждение заболевания" при использовании в настоящем изобретении означает лечение и уход за индивидуумом, для которого существует опасность развития заболевания, до появления клинических симптомов заболевания. Целью предупреждения является борьба с развитием заболевания, патологического состояния или нарушения, и оно включает введе-4 024817 ние активных соединений для предупреждения или задержки появления симптомов или осложнений, или для предупреждения или задержки развития родственных заболеваний, патологических состояний или нарушений. Успех указанного предупредительного лечения отражен статистически в уменьшении частоты возникновения указанного патологического состояния в группе пациентов, для которых существует опасность возникновения этого патологического состояния, по сравнению с аналогичной группой пациентов, не подвергающихся предупредительному лечению. Выражение "лечение" или "терапия" означает лекарственное лечение пациентов, у которых уже развилось одно или большее количество указанных патологических состояний в явной, острой или хронической форме, включая симптоматическое лечение, предназначенное для облегчения симптомов при конкретном показании, или этиотропное лечение, предназначенное для обращения или частичного обращения патологического состояния или для остановки или замедления прогрессирования заболевания настолько, насколько это возможно в зависимости от патологического состояния и его тяжести. Таким образом, выражение "лечение заболевания" при использовании в настоящем изобретении означает лечение и уход за пациентом, у которого развилось заболевание, патологическое состояние или нарушение. Целью лечения является борьба с заболеванием, патологическим состоянием или нарушением. Лечение включает введение активных соединений для устранения заболевания, патологического состояния или нарушения или борьбы с ними, а также облегчение симптомов или осложнений, связанных с заболеванием, патологическим состоянием или нарушением. Если специально не указано иное, то указанная в описании или в формуле изобретения структурная формула или химическое название соединения включает его таутомеры и все стереоизомеры, оптические и геометрические изомеры (например, энантиомеры, диастереоизомеры, E/Z-изомеры и т. п.) и рацематы,а также смеси отдельных энантиомеров в разных соотношениях, смеси диастереоизомеров, смеси любых описанных выше форм, в которых существуют такие изомеры и энантиомеры, а также его соли, включая его фармацевтически приемлемые соли и его сольваты, такие как, например гидраты, включая сольваты свободных соединений или сольваты соли соединения. Термин галоген обычно означает фтор, хлор, бром и йод. Выражение "фармацевтически приемлемое" используется в настоящем изобретении для указания таких соединений, материалов, композиций и/или дозированных форм, которые в соответствии с основными положениями медицины являются подходящими для использования при соприкосновении с тканями людей и животных без проявления какой-либо чрезмерной токсичности, раздражающего воздействия, аллергической реакции, или других затруднений или осложнений, и соответствуют приемлемому соотношению польза/риск. При использовании в настоящем изобретении "фармацевтически приемлемые соли" означают производные раскрытых соединений, в которых исходное соединение изменено путем образования его солей с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются только ими, соли неорганических или органических кислот с основными функциональными группами, такими как аминогруппы; соли щелочных металлов или органических соединений с кислотными функциональными группами, такими как карбоксигруппы и т. п. Например, такие соли включают соли, образованные с аммиаком, L-аргинином, бетаином, бенетамином, бензатином, гидроксидом кальция, холином, деканолом, диэтаноламином (2,2'-имино-бис-(этанол, диэтиламином, 2-(диэтиламино) этанолом, 2-аминоэтанолом, этилендиамином, N-этилглюкамином, гидрабамином, 1H-имидазолом, лизином, гидроксидом магния, 4-(2-гидроксиэтил)морфолином, пиперазином, гидроксидом калия, 1-(2 гидроксиэтил)пирролидином, гидроксидом натрия, триэтаноламином (2,2',2-нитрило-трис(этанол,трометамином, гидроксидом цинка, уксусной кислотой, 2,2-дихлоруксусной кислотой, адипиновой кислотой, альгиновой кислотой, аскорбиновой кислотой, L-аспарагиновой кислотой, бензолсульфоновой кислотой, бензойной кислотой, 2,5-дигидроксибензойной кислотой, 4-ацетамидобензойной кислотой,(+)-камфорной кислотой, (+)-камфор-10-сульфоновой кислотой, угольной кислотой, коричной кислотой,лимонной кислотой, цикламовой кислотой, декановой кислотой, додецилсерной кислотой, этан-1,2 дисульфоновой кислотой, этансульфоновой кислотой, 2-гидроксиэтансульфоновой кислотой, этилендиаминтетрауксусной кислотой, муравьиной кислотой, фумаровой кислотой, галактаровой кислотой, гентизиновой кислотой, D-глюкогептоновой кислотой, D-глюконовой кислотой, D-глюкуроновой кислотой,глутаминовой кислотой, глутаровой кислотой, 2-оксоглутаровой кислотой, глицерофосфорной кислотой,глицином, гликолевой кислотой, гексановой кислотой, гиппуровой кислотой, бромистоводородной кислотой, хлористоводородной кислотой, изомасляной кислотой, DL-молочной кислотой, лактобионовой кислотой, лауриновой кислотой, лизином, малеиновой кислотой, (-)-L-яблочной кислотой, малоновой кислотой, DL-миндальной кислотой, метансульфоновой кислотой, галактровой кислотой, нафталин-1,5 дисульфоновой кислотой, нафталин-2-сульфоновой кислотой, 1-гидрокси-2-нафтойной кислотой, никотиновой кислотой, азотной кислотой, октановой кислотой, олеиновой кислотой, оротовой кислотой, щавелевой кислотой, пальмитиновой кислотой, памоевой кислотой (эмбоновой кислотой), фосфорной кислотой, пропионовой кислотой, (-)-L-пироглутаминовой кислотой, салициловой кислотой, 4 аминосалициловой кислотой, себациновой кислотой, стеариновой кислотой, янтарной кислотой, серной кислотой, дубильной кислотой, (+)-L-винной кислотой, тиоциановой кислотой, п-толуолсульфоновой кислотой и ундециленовой кислотой. Другие фармацевтически приемлемые соли могут быть образованы с катионами металлов, таких как алюминий, кальций, литий, магний, калий, натрий, цинк и т. п. (см. также публикацию Pharmaceutical salts, Berge, S.M. et al., J. Pharm. Sci., (1977), 66, 1-19). Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно получить из исходного соединения, которое содержит основный или кислотный фрагмент, по обычным химическим методикам. Обычно такие соли можно получить по реакции этих соединений в форме свободной кислоты или основания с достаточным количеством соответствующего основания или кислоты в воде или органическом растворителе, таком как эфир, этилацетат, этанол, изопропанол или ацетонитрил, или в их смеси. Соли кислот, отличающиеся от указанных выше, которые например, применимы для очистки или выделения соединений, предлагаемых в настоящем изобретении (например, трифторацетаты), также являются частью настоящего изобретения. Термин "C1-Cn-алкил", где п является целым числом, равным от 4 до 6, по отдельности или в комбинации с другим радикалом, означает ациклический, насыщенный, обладающий разветвленной или линейной цепью углеводородный радикал, содержащий от 1 до 4 или 6 атомов С. Например, термин С 1-С 6 алкил включает радикалы Н 3 С-, Н 3 С-СН 2-, Н 3 С-СН 2-СН 2-, Н 3 С-СН(СН 3)-, Н 3 С-СН 2-СН 2-СН 2-, Н 3 С-СН 2 СН(СН 3)-, Н 3 С-СН(СН 3)-СН 2-, Н 3 С-С(СН 3)2-, Н 3 С-СН 2-СН 2-СН 2-СН 2-, Н 3 С-СН 2-СН 2-СН(СН 3)-, Н 3 С-СН 2 СН(СН 3)-СН 2-, Н 3 С-СН(СН 3)-СН 2-СН 2-, Н 3 С-СН 2-С(СН 3)2-, Н 3 С-С(СН 3)2-СН 2-, Н 3 С-СН(СН 3)-СН(СН 3)- и Н 3 С-СН 2-СН(СН 2 СН 3)-. Термин "C1-Cn-алкил" также включает такие радикалы, в которых один или большее количество атомов водорода могут быть заменены атомами фтора, поэтому примерами являются F3C, F2HC, F2HC-H2C, F3C-H2C. Термин "C2-Cn-алкенил" используют для обозначения группы, определенной в определении для "С 1Cn-алкила", содержащей по меньшей мере 2 атома углерода, если по меньшей мере два из этих атомов углерода указанной группы связаны друг с другом двойной связью. Термин "C2-Cn-алкинил" используют для обозначения группы, определенной в определении для"C1-Cn-алкила", содержащей по меньшей мере 2 атома углерода, если по меньшей мере два из этих атомов углерода указанной группы связаны друг с другом тройной связью. Термин "С 3-С 6-циклоалкил" по отдельности или в комбинации с другим радикалом означает циклический, насыщенный углеводородный радикал, содержащий 6 атомов С. Например, термин С 3-С 7 циклоалкил включает циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термин "С 3-С 6-циклоалкенил" по отдельности или в комбинации с другим радикалом означает циклический, ненасыщенный, но неароматический, неразветвленный углеводородный радикал, содержащий 6 атомов С, по меньшей мере 2 из которых связаны друг с другом двойной связью. Например, термин С 3 С 6-циклоалкенил включает циклопропенил, циклобутенил, циклопентенил, циклопентадиенил, циклогексенил и циклогексадиенил. Термин "арил" при использовании в настоящем изобретении по отдельности или в комбинации с другим радикалом, означает карбоциклическую ароматическую моноциклическую группу, содержащую 6 атомов углерода, которая может быть дополнительно сконденсирована со второй 5- или 6-членной карбоциклической группой, которая может являться ароматической, насыщенной или ненасыщенной. Арил включает, но не ограничивается только ими, фенил, инданил, инденил, нафтил, антраценил, фенантренил, тетрагидронафтил и дигидронафтил. Термин "моноциклический С 5-С 7-гетероциклил" означает насыщенные или ненасыщенные неароматические моноциклические кольцевые системы, содержащие один или большее количество гетероатомов, выбранных из группы, включающей N, О или S(O)r, где r = 0, 1 или 2, содержащие от 5 до 7 кольцевых атомов. Подразумевается, что термин "моноциклический С 5-С 7-гетероциклил" включает все возможные изомерные формы. Таким образом, термин "моноциклический С 5-С 7-гетероциклил" включает следующие типичные структуры, которые не изображены в виде радикалов, поскольку они в любой форме могут быть присоединены с помощью ковалентной связи к любому атому при условии, что сохраняются соответствующие валентности: Термин "С 5-С 10-гетероарил" означает моно- или бициклические кольцевые системы, содержащие один или большее количество гетероатомов, выбранных из группы, включающей N, О или S(O)r, содержащие от 5 до 10 кольцевых атомов, предпочтительно от 5 до 6 кольцевых атомов в случае моноциклических колец или от 7 до 10 кольцевых атомов в случае бициклических колец, где по меньшей мере один из гетероатомов является частью ароматического кольца. Подразумевается, что термин "С 5-С 10 гетероарил" включает все возможные изомерные формы. Таким образом, термин "С 5-С 10-гетероарил" включает следующие типичные структуры, которые не изображены в виде радикалов, поскольку они в любой форме могут быть присоединены с помощью ковалентной связи к любому атому при условии, что сохраняются соответствующие валентности: Термин "бициклический С 8-С 10-гетероциклил" означает частично насыщенные или ненасыщенные бициклические кольцевые системы, включая ароматические кольцевые системы, содержащие один или большее количество гетероатомов, выбранных из группы, включающей N, О или S(O)r, содержащие от 8 до 10 кольцевых атомов, где гетероатомы необязательно являются частью ароматического кольца. Подразумевается, что термин "бициклический С 8-С 10-гетероциклил" включает все возможные изомерные формы. Таким образом, термин "бициклический С 8-С 10-гетероциклил" включает следующие типичные структуры, которые не изображены в виде радикалов, поскольку они в любой форме могут быть присоединены с помощью ковалентной связи к любому атому при условии, что сохраняются соответствующие валентности: Получение Общие методики синтеза Настоящее изобретение также относится к способу получения соединения формулы I. Во всех методиках, если не указано иное, R1, R2 и п в приведенных ниже формулах обладают теми же значениями,что и R1, R2 и n в формуле I, предлагаемой в настоящем изобретении и описанной выше в настоящем изобретении. Оптимальные условия проведения реакций и длительность реакций могут меняться в зависимости от конкретных использующихся реагентов. Если не указано иное, то растворители, температуры, давления и другие условия проведения реакций могут быть легко выбраны специалистом с общей подготовкой в данной области техники. Конкретные методики приведены в разделе "Примеры синтеза". Обычно за протеканием реакции при необходимости можно следить с помощью тонкослойной хроматографии(ТСХ) или ЖХ-МС (жидкостная хроматография-масс-спектрометрия), и промежуточные продукты и конечные продукты можно очистить с помощью хроматографии на силикагеле, с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) и/или путем перекристаллизации. Приведенные ниже примеры являются иллюстративными и, как понятно специалисту в данной области техники, конкретные реагенты или условия можно менять в соответствии с тем, что необходимо для конкретных соединений,без проведения слишком большого объема исследований. В приведенных ниже методиках используемые исходные вещества и промежуточные продукты имеются в продаже или специалисты в данной области техники легко получат их из имеющихся в продаже веществ. Соединение формулы V, VII и IX можно получить по методике, представленной на схеме 1: Как показано на схеме 1, соединение формулы II, в которой PG обозначает защитную группу (например, трет-бутоксикарбонил), можно ввести в реакцию с водным раствором аммиака с использованием стандартных методик образования амида, описанных в литературе. Например, в присутствии основания,такого как N-метилморфолин или N-этилморфолин, и активирующего реагента, такого как О-(7 азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (ГАТУ) или O-(бензотриазол-1 -ил)N,N,N',N'-тетраметилуронийтетрафторборат (TBTU). Реакцию обычно проводят в подходящем растворителе, таком как N,N-диметилформамид. Для этих синтезов можно использовать стандартные реакции сочетания белков, известные в данной области техники (см., например, М. Bodanszky, 1984, The Practiceof Peptide Synthesis, Springer-Verlag). Дегидратацию амида, такого как соединение формулы III или формулы IX, с получением соответствующего нитрила формулы IV или VII можно провести с использованием дегидратирующего реагента,такого как гидроксид (метоксикарбонилсульфамоил)триэтиламмония, в подходящем растворителе, таком как дихлорметан (ДХМ). Реакция кислоты формулы VI с амином формулы V или VIII в подходящем растворителе, проводимая по стандартным методикам образования амида, описанным в литературе, например, в присутствии основания, такого как N,N-диизопропилэтиламин (ДИПЭА), и активирующего реагента, такого как ГАТУ или TBTU, дает соединение формулы VII или IX. Для этих синтезов можно использовать стандартные реакции сочетания белков, известные в данной области техники (см., например, М. Bodanszky, 1984,The Practice of Peptide Synthesis, Springer-Verlag). Введение и удаление защитных групп функциональных групп описано в публикации 'ProtectiveGroups in Organic Synthesis', Т. W. Greene and P. G. M. Wuts, Wiley-Interscience. Например, для удаления трет-бутоксикарбонильной защитной группы можно использовать кислоту, такую как муравьиная кислота, трифторуксусная кислота или НСl, в подходящем растворителе, таком как вода, ДХМ или диоксан. Как показано на схеме 2, катализируемая (переходным) металлом реакция соединения формулы VII или IX, в которой А обозначает I или Br, дает соединение формулы X или XI. Например, реакция с бороновой кислотой или соответствующим эфиром бороновой кислоты в подходящем растворителе, таком как ацетонитрил, в присутствии подходящего катализатора, такого как 1,1-бис(ди-трет-бутилфосфино) ферроценпалладийдихлорид, и подходящего основания, такого как K2CO3, дает соединение формулы X или XI. Альтернативно, реакция соединения формулы VII или IX, в которой А обозначает I или Br, с трибутил(винил)оловом в присутствии подходящего катализатора, такого как бис-(трифенилфосфин) палладийхлорид, в подходящем растворителе, таком как диметилформамид (ДМФ), и, если это является подходящим, в присутствии добавки, такой как тетраэтиламмонийхлорид, дает соединения формулы X или XI. Кроме того, реакция соединения формулы VII или IX, в которой А обозначает I или Br, с амином в присутствии подходящего катализатора, такого как Cu(I)I, и подходящего основания, такого как карбонат цезия, и подходящего промотора, такого как L-пролин, дает соединение формулы X или XI. Кроме того, как показано на схеме 2, реакция соединения формулы VII или IX, в которой А обозначает N3, с алкином в присутствии подходящего катализатора, такого как пентагидрат сульфата меди(И), и подходящего восстановительного реагента, такого как L-аскорбиновая кислота, в подходящем растворителе, таком как смесь диметилсульфоксид (ДМСО)/вода, дает соединение формулы X или XI. Дополнительные превращения соединений формулы X, XI и I, проводимые по методикам, известным в данной области техники и описанным в приведенных ниже примерах, можно использовать для получения дополнительных соединений, предлагаемых в настоящем изобретении. Дегидратацию амида формулы XI с получением соответствующего нитрила формулы X можно провести с использованием дегидратирующего реагента, такого как гидроксид (метоксикарбонилсульфамоил)триэтиламмония, в подходящем растворителе, таком как ДХМ. Как показано на схеме 3, катализируемая (переходным) металлом реакция соединения формулы V,в которой А обозначает I или Br, дает соединение формулы XII. Например, реакция с бороновой кислотой или соответствующим эфиром бороновой кислоты в подходящем растворителе, таком как ацетонитрил, в присутствии подходящего катализатора, такого как 1,1-бис(ди-трет-бутилфосфино)ферроценпалладийдихлорид, и подходящего основания, такого как K2CO3, дает соединение формулы XII. Кислоту формулы VI можно ввести в реакцию с амином формулы XII с использованием стандартных методик образования амида, описанных в литературе, например, в присутствии основания, такого как ДИПЭА, и активирующего реагента, такого как ГАТУ или TBTU, в подходящем растворителе. Для этих синтезов можно использовать стандартные реакции сочетания белков, известные в данной области техники (см., например, М. Bodanszky, 1984, The Practice of Peptide Synthesis, Springer-Verlag). Удаление защитных групп функциональных групп описано в публикации 'Protective Groups in Organic Synthesis', Т.W. Greene and P. G. M. Wuts, Wiley-Interscience. Например, для удаления трет-бутоксикарбонильной защитной группы, можно использовать кислоту, такую как муравьиная кислота, трифторуксусная кислота или HCl, в подходящем растворителе, таком как вода, ДХМ или диоксан, и эту реакцию можно провести с использованием неочищенного продукта реакции сочетания с образованием амида и получить соединение формулы I. Примеры синтеза Ниже приведены типичные соединения, предлагаемые в настоящем изобретении, которые можно получить в соответствии с общими схемами синтеза, примерами и по методикам, известным в данной области техники. Время удерживания для жидкостной хроматографии-масс-спектрометрии (ЖХ-МС) и экспериментальные значения m/z для приведенных ниже соединений получают с использованием одной из приведенных ниже методик: Методика ЖХ-МС а ТФК = трифторуксусная кислота Методика ЖХ-МС с Во всех методиках препаративной ОФ-ВЭЖХ (ОФ = обращенная фаза) используют 0-100% ацетонитрила или метанола в воде и в качестве модифицирующей добавки используют ТФК или гидроксид аммония. Исходные вещества и реагенты имеются в продаже или специалист в данной области техники может их получить по методикам, описанным в химической литературе. Ниже приведены типичные соединения, предлагаемые в настоящем изобретении, которые можно получить в соответствии с общими схемами синтеза, примерами и по методикам, известным в данной области техники. Получение промежуточных продуктов Синтез Стадия 1. Синтез промежуточного продукта I-1.1.TBTU (8,1 г, 25 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин. После охлаждения реакционной смеси до 0 С по каплям добавляют аммиак (35% водный раствор,2,6 мл, 46 ммоль). Реакционную смесь перемешивают в течение ночи, разбавляют водой (500 мл) и осадок отфильтровывают, промывают водой и сушат в сушильном шкафу при 50 С. Выход 95%. m/z 391[М+Н]+, m/z 389 [М+Н]-, время удерживания (rt) 1,40 мин, методика ЖХ-МС а. Стадия 2. Синтез промежуточного продукта I-1.2.I-1.1 (7,4 г, 19 ммоль) суспендируют в ДХМ (200 мл) и добавляют раствор R2 (9, 8 г, 41 ммоль) в ДХМ (39 мл) и реакционную смесь перемешивают в течение ночи. Реакционную смесь экстрагируют уксусной кислотой (1% раствор в воде, 170 мл), промывают рассолом и фильтруют. Органический слой сушат, концентрируют и очищают с помощью колоночной хроматографии (с использованием смеси растворителей циклогексан/этилацетат (ЭА) = 75/25) и получают I-1.2. Выход 81%. Стадия 3. Синтез промежуточного продукта I-1.3. К I-1.2 (1,7 г, 4,6 ммоль) добавляют раствор HCl в диоксане (4 М, 20 мл) и реакционную смесь перемешивают при комнатной температуре в течение 3 ч. За протеканием реакции следят с помощью ВЭЖХМС для того, чтобы регистрировать образование искомого продукта и гидролизованного побочного продукта (превращение нитрила в амид). В ходе реакции образуется белый осадок. К реакционной смеси добавляют диэтиловый эфир и твердый продукт I-1.3 отфильтровывают и промывают эфиром. Выход 75%. m/z 273/274 [М+Н]+, rt = 0,45 мин, методика ЖХ-МС b. Стадия 4. Синтез промежуточного продукта I-1.4. К R3 (452 мг, 1,87 ммоль) в ДХМ (20 мл) добавляют триэтиламин (1,1 мл, 99%, 7,84 ммоль) и ГАТУ(750 мг, 1,97 ммоль) и реакционную смесь перемешивают в течение 10 мин. Затем добавляют I-1.4 и смесь перемешивают в течение 1 ч. Полученную смесь промывают водным раствором NaHCO3 (10%),водой (50 мл) с добавлением 5 капель уксусной кислоты и рассолом, сушат, концентрируют и остаток очищают с помощью колоночной хроматографии (с использованием смеси растворителей циклогексан/ЭА = 75/25) и получают I-1.4. Выход 58%. m/z 486/487 [М+Н]+, rt = 0,80 мин, методика ЖХ-МС b. Синтез (1R,3S,4S)-трет-бутил-3-S)-1-амино-3-(4-йодфенил)-1-оксопропан-2-илкарбамоил)-2-азабицикло[2.2.1]гептан-2-карбоксилата (промежуточный продукт I-1.6) Стадия 1. Синтез промежуточного продукта I-1.5.I-1.1 (5,0 г, 12,79 ммоль), ДХМ (10 мл) и трифторуксусную кислоту (5 мл) перемешивают при комнатной температуре в течение 2 ч. Реакционную смесь концентрируют и получают I-1.5, выход 100%. Стадия 2. Синтез промежуточного продукта I-1.6. К R3 (716 мг, 2,97 ммоль) в ДМФ (5 мл) добавляют ДИПЭА (2,14 мл, 12,37 ммоль) и TBTU (874 мг,2,72 ммоль) и реакционную смесь перемешивают в течение 15 мин. Добавляют I-1.5 (1,0 г, 2,47 ммоль) и реакционную смесь перемешивают в течение ночи. Полученную смесь непосредственно очищают с помощью препаративной ВЭЖХ. Выход 79%. m/z 514 [М+Н]+, rt = 1,14 мин, методика ЖХ-МС d. Синтез (1R,3S,4S)-трет-бутил-3-S)-2-(4-азидофенил)-1-цианоэтилкарбамоил)-2-азабицикло[2.2.1] гептан-2-карбоксилата (промежуточный продукт I-2.4). Стадия 1. Синтез промежуточного продукта I-2.1. К R4 (500 мг, 1,63 ммоль) в ДМФ (5 мл) добавляют N-метилморфолин (0,270 мл, 2,46 ммоль) и ГАТУ (622 мг, 1,64 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. После охлаждения реакционной смеси до 0 С по каплям добавляют аммиак (32% водный раствор,0,180 мл, 2,98 ммоль). Реакционную смесь перемешивают в течение ночи, разбавляют с помощью ДХМ,органический слой промывают 1 М раствором HCl, водным раствором NaHCO3 (10%) и рассолом, сушат и концентрируют. Выход 89%. m/z 306 [М+Н]+, rt = 1,35 мин, методика ЖХ-МС а. Стадия 2. Синтез промежуточного продукта I-2.2. К I-2.1 (441 мг, 1,44 ммоль) добавляют раствор HCl в диоксане (4 М, 2 мл) и реакционную смесь перемешивают при комнатной температуре в течение 1 ч. К реакционной смеси добавляют диэтиловый эфир и твердый продукт I-2.2 отфильтровывают и промывают эфиром. Выход 91%. m/z 206 [М+Н]+, rt = 0,30 мин, методика ЖХ-МС b. Стадия 3. Синтез промежуточного продукта I-2.3. К R3 (320 мг, 1,33 ммоль) в ДМФ (2 мл) добавляют ДИПЭА (1,2 мл, 6,98 ммоль) и ГАТУ (600 мг,1,58 ммоль) и реакционную смесь перемешивают в течение 10 мин. Добавляют I-2.2 (319 мг, 1,32 ммоль) и реакционную смесь перемешивают в течение ночи. Полученную смесь разбавляют с помощью ДХМ и промывают водным раствором NaHCO3 (10%), 1 М раствором HCl и рассолом, сушат и концентрируют. Очистка с помощью колоночной хроматографии (ДХМ/МеОН = 96:4) дает I-2.3. Выход 100%. m/z 429[М+Н]+, rt = 0,76 мин, методика ЖХ-МС b. Стадия 4. Синтез промежуточного продукта I-2.4. К раствору I-2.3 (643 мг, 1,50 ммоль) в ДХМ (10 мл) добавляют R2 (750 мг, 3,15 ммоль) и реакционную смесь перемешивают в течение 3 ч, затем разбавляют с помощью ДХМ, промывают уксусной кислотой (1% раствор в воде) и рассолом. Органический слой сушат, концентрируют и очищают с помощью колоночной хроматографии (с использованием смеси растворителей циклогексан/ЭА = 2:1) и получают I-2.4. Выход 73%. m/z 411 [М+Н]+, rt = 0,77 мин, методика ЖХ-МС b. Синтез 1-метил-6-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1,3-дигидроиндол-2-она (промежуточный продукт I-3.3) Стадия 1. Синтез промежуточного продукта I-3.1. К R4 (500 мг, 2,21 ммоль) в ацетонитриле (15 мл) добавляют MeI (0,303 мл, 4,87 ммоль) и K2CO3(1,2 г, 8,68 ммоль) и реакционную смесь перемешивают при 60 С в течение 45 мин. Добавляют ДХМ и воду и водный слой дважды экстрагируют с помощью ДХМ, объединенные органические слои промывают рассолом, сушат и концентрируют. Выход 65%. m/z 240/242 [М+Н]+, rt = 0,49 мин, методика ЖХМС b. Стадия 2. Синтез промежуточного продукта I-3.2.I-3.1 (397 мг, 1,65 ммоль) и гидразингидрат (1 мл, 20,6 ммоль) нагревают при 100 С в течение 1 ч и при 125 С в течение 1 ч. К холодной реакционной смеси добавляют ДХМ и воду и водный слой дважды экстрагируют с помощью ДХМ. Объединенные органические слои промывают рассолом, сушат, концентрируют и остаток очищают с помощью колоночной хроматографии (с использованием смеси растворителей циклогексан/ЭА = 3:1). Выход 65%. m/z 226 [М+Н]+, m/z 224 [М+Н]-, rt = 0,58 мин, методика ЖХМС b. Стадия 3. Синтез промежуточного продукта I-3.4. К I-3.2 (91 мг, 0,40 ммоль) в безводном диоксане (8 мл) добавляют R5 (155 мг, 0,61 ммоль) и ацетат калия (120 мг, 1,22 ммоль). Смесь продувают аргоном, добавляют [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (PdCl2(dppf (33 мг, 0,040 ммоль) и нагревают при 80 С в течение 1,5 ч. Реакционную смесь разбавляют с помощью ЭА и водой, органический слой промывают рассолом, сушат и концентрируют. Остаток очищают с помощью колоночной хроматографии (циклогексан/ЭА = 1:1). Выход 100%. m/z 274 [М+Н]+, rt = 0,71 мин, методика ЖХ-МС b. Синтез 3-(3-метоксипропил)-5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3 Н-бензооксазол-2 она (промежуточный продукт I-3.5) Стадия 1. Синтез промежуточного продукта I-3.4.R6 (530 мг, 2,48 ммоль), R7 (473 мг, 2,81 ммоль) и K2CO3 (1 г, 7,24 ммоль) в ацетонитриле (10 мл) нагревают при 70 С в течение 3 ч. Холодную реакционную смесь разбавляют с помощью ЭА и водой,водный слой трижды экстрагируют с помощью ЭА, объединенные органические слои промывают рассолом, сушат и концентрируют. Остаток очищают с помощью колоночной хроматографии (циклогексан/ЭА = 3:1). Выход 30%. m/z 286/288 [М+Н]+, rt = 0,66 мин, методика ЖХ-МС b. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих промежуточных продуктов: Стадия 2. Синтез промежуточного продукта I-3.5. К I-3.4 (92 мг, 0,32 ммоль) в безводном диоксане (8 мл) добавляют R5 (130 мг, 0,51 ммоль) и ацетат калия (100 мг, 1,02 ммоль). Смесь продувают аргоном, добавляют PdCl2(dppf) (27 мг, 0,033 ммоль) и нагревают при 80 С в течение 3 ч. Реакционную смесь разбавляют с помощью ЭА и водой, органический слой промывают рассолом, сушат и концентрируют. Продукт используют в неочищенном виде, m/z 334[М+Н]+, rt = 0,78 мин, методика ЖХ-МС b. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих промежуточных продуктов: К R8 (100 мг, 0,47 ммоль) в безводном диоксане (8 мл) добавляют R5 (180 мг, 0,71 ммоль) и ацетат калия (140 мг, 1,43 ммоль). Смесь продувают аргоном, добавляют PdCl2(dppf) (40 мг, 0,049 ммоль) и нагревают при 80 С в течение 1,5 ч. Реакционную смесь разбавляют с помощью ЭА и водой, органический слой промывают рассолом, сушат и концентрируют. Продукт используют в неочищенном виде, m/z 260[М+Н]+, rt = 0,64 мин, методика ЖХ-МС b. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих промежуточных продуктов: Синтез промежуточного продукта I-3.7. К R8 (4,5 г, 17,2 ммоль) в безводном ТГФ (тетрагидрофуран) (130 мл) добавляют NaBH4 (6,8 г, 179 ммоль) и реакционную смесь охлаждают до -8 С. В течение 15 мин по каплям добавляют диэтилэфират трифторида бора (25 мл, 197 ммоль). После выдерживания при -8 С в течение еще 10 мин реакционную смесь кипятят с обратным холодильником в течение 2 ч, затем охлаждают до комнатной температуры и добавляют воду со льдом (30 мл). Добавляют 6 М раствор NaOH до щелочной реакции и раствор экстрагируют этилацетатом. Органический раствор 3 раза экстрагируют раствором NaOH. К охлажденным объединенным водным слоям добавляют 6 М раствор HCl до кислой реакции. Водный слой 3 раза экстрагируют этилацетатом, объединенные органические слои промывают рассолом, сушат и концентрируют. Продукт используют в неочищенном виде, m/z 246/148 [М+Н]+, rt = 0,76 мин, методика ЖХ-МС b. Синтез (1R,3S,4S)-трет-бутил-3-S)-2-(4-бромфенил)-1-цианоэтилкарбамоил)-2-азабицикло[2.2.1] гептан-2-карбоксилата I-12.4 и (1R,3S,4S)-трет-бутил-3-S)-1-циано-2-(4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)фенил)этилкарбамоил)-2-азабицикло[2.2.1]гептан-2-карбоксилата I-12.5 Стадия 1. Синтез промежуточного продукта I-12.1.(S)-3-(4-Бромфенил)-2-(трет-бутоксикарбониламино)пропановую кислоту R11 (20,0 г, 58,1 ммоль) растворяют в ДМФ (135 мл) и добавляют N-метилморфолин (9,59 мл, 87,1 ммоль) и TBTU (18,7 г, 58,1 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 45 мин. После охлаждения реакционной смеси до 0 С по каплям добавляют водный раствор аммиака (32%, 6,4 мл, 105,2 ммоль). Реакционную смесь перемешивают в течение 72 ч, разбавляют водой (700 мл) и осадок отфильтровывают, промывают водой и сушат в сушильном шкафу при 65 С. Выход 96%. m/z 343 [М+Н]+, время удерживания (rt) 1,39 мин, методика ЖХ-МС g. Стадия 2. Синтез промежуточного продукта I-12.2.I-12.1 (10,0 г, 29,1 ммоль) растворяют в ДХМ (60 мл) и добавляют водный раствор трифторуксусной кислоты (98%; 20 мл). Раствор перемешивают в течение 3 ч. Растворитель удаляют в вакууме и остаток растворяют в смеси вода/ацетонитрил и сушат вымораживанием. Выход 100%. Стадия 3. Синтез промежуточного продукта I-12.3. К R3 (7,28 г, 29,3 ммоль) в ДХМ (150 мл) добавляют диизопропилэтиламин (13,8 мл, 79,8 ммоль) и ГАТУ (11,1 г, 29,3 ммоль) и реакционную смесь перемешивают в течение 20 мин. Затем добавляют промежуточный продукт I-12.2 (9,5 г, 26,6 ммоль), растворенный в ДХМ (150 мл), и смесь перемешивают в течение 3 ч. Полученную смесь дважды промывают водным раствором KHSO4 (10%), водным растворомKHCO3 (10%), водой (50 мл). Органическую фазу сушат, концентрируют и остаток очищают с помощью колоночной хроматографии (с использованием смеси растворителей ДХМ/МеОН = 95/5) и получают промежуточный продукт I-12.3. Выход 78%, m/z 466 [М+Н]+, rt = 1,47 мин, методика ЖХ-МС g. Стадия 4. Синтез промежуточного продукта I-12.4.I-12.3 (13,0 г, 27,9 ммоль) суспендируют в ДХМ (200 мл) и добавляют раствор R2 (13,3 г, 55,9 ммоль) в ДХМ (100 мл) и реакционную смесь перемешивают в течение 3,5 ч. Органическую фазу дважды промывают водным раствором Na2CO3 (2 М) и насыщенным раствором NaCl, сушат и концентрируют в вакууме. К реакционной смеси добавляют диэтиловый эфир и твердый промежуточный продукт I-12.4 отфильтровывают и промывают эфиром. Выход 92%. m/z 448 [М+Н]+, rt = 1,52 мин, методика ЖХ-МС g. Стадия 5. Синтез промежуточного продукта I-12.5.I-12.4 (4,0 г, 8,9 ммоль), R5 (4,5 г, 17,8 ммоль) и KOAc (3,5 г, 35,6 ммоль) суспендируют в сухом ДМФ (70 мл) и дегазируют аргоном. Добавляют 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладийдихлорид (1,2 г, 1,8 ммоль) и реакционную смесь перемешивают при 100 С в течение 40 мин. Реакционную смесь выливают в воду и EtOAc, органическую фазу отделяют, сушат и концентрируют. Остаток очищают с помощью колоночной хроматографии (с использованием смеси растворителей EtOAc/циклогексан = 50/50) и получают промежуточный продукт I-12.5, выход 43%, m/z = 496 [М+Н]+, rt = 1,10 мин, методика ЖХ-МС i. Методика А. Синтез Стадия 1. Синтез промежуточного продукта I-4.1.I-1.4 (100 мг, 0,202 ммоль), I-3.6 (72 мг, 0,264 ммоль), 2 М раствор K2CO3 (0,40 мл, 0,400 ммоль) в ацетонитриле (8 мл) продувают аргоном и добавляют PdCl2(dppf) (14 мг, 0,021 ммоль) и реакционную смесь нагревают при 80 С в течение ночи. Реакционную смесь концентрируют, добавляют ДХМ и воду и водный слой экстрагируют с помощью ДХМ. Объединенные органические слои промывают рассолом,сушат и концентрируют. Остаток очищают с помощью колоночной хроматографии (циклогексан/ЭА = 3:1). Выход 57%. m/z 415 [М+Н-Вос]+, rt = 0,75 мин, методика ЖХ-МС b. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов: Для получения I-4.5 вместо эфира бороновой кислоты используют трифторборат калия, вместоK2CO3 используют 3 экв. Na2CO3 и в качестве катализатора используют 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II). Стадия 2. Синтез соединения примера 7.I-4.1 (59 мг, 0,115 ммоль), муравьиную кислоту (2 мл) и воду (0,2 мл) перемешивают при комнатной температуре в течение 2 ч. Добавляют аммиак и воду и водный слой экстрагируют с помощью ДХМ. Объединенные органические слои промывают рассолом, сушат и концентрируют. Остаток очищают с помощью ВЭЖХ. Выход 61%. Следующие соединения синтезируют аналогичным образом из соответствующих промежуточных продуктов: соединение примера 8 таблицы 1; соединение примера 13 таблицы 1; соединение примера 14 таблицы 1; соединение примера 16 таблицы 1; соединение примера 40 таблицы 1; соединение примера 41 таблицы 1; соединение примера 42 таблицы 1; соединения примеров 120-122 таблицы 1; соединение примера 125 таблицы 1; соединение примера 127 таблицы 1; соединение примера 129 таблицы 1; соединение примера 131 таблицы 1; соединение примера 134 таблицы 1; соединения примеров 139-140 таблицы 1. Для получения соединений примеров 40-42, 126, 128, 130, 132, 133, 135, 136, 138 таблицы 1 неочищенный продукт, полученный на стадии 1, непосредственно обрабатывают муравьиной кислотой для удаления защитной группы Boc, таким образом, продукт реакции сочетания, содержащий защитную группу Boc, не выделяют. Для получения соединений примеров 52-73, 96-102 таблицы 1 вместо I-1.4 используют и в качестве катализатора на стадии 1 используют 1,1'-бис(ди-трет-бутилфосфино)ферроценпалладийдихлорид. На стадии 2 реакцию проводят при 40 С в течение 10-15 мин. Для получения соединений примеров 123, 124 таблицы 1 соответствующий эфир бороновой кислоты получают в соответствии с методикой синтеза промежуточного продукта I-3.5, но его не выделяют из реакционной смеси. Вместо проведения обработки реакционную смесь охлаждают до комнатной температуры, к реакционной смеси в инертной атмосфере добавляют I-1.4 (1-1,1 экв.), PdCl2(dppf) (0,03-0,1 экв.) и Na2CO3 или K2CO3 (3,6-5 экв.) и нагревают при 80 С. Обработку проводят так, как описано в методике А, стадия 1, и окончательное превращение в соединения примеров проводят так, как описано в методике А, стадия 2. Методика В. Синтез Стадия 1. Синтез I-5.1. Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 1, с использованием соответствующих реагентов. Выход 65%. m/z 507 [М+Н]+, rt = 1,43 мин, методика ЖХ-МС а. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов:I-5.1 (96 мг, 0,19 ммоль) суспендируют в ДХМ (1 мл) и добавляют раствор R2 (113 мг, 0,47 ммоль) и реакционную смесь перемешивают в течение ночи. Полученную смесь концентрируют и неочищенный продукт используют без очистки, m/z 489 [М+Н]+, rt = 0,83 мин, методика ЖХ-МС b. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов: Стадия 3. Синтез соединения примера 1. Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 2, с использованием соответствующих реагентов. Выход 38%. Следующие соединения синтезируют аналогичным образом из соответствующих промежуточных продуктов: соединение примера 4 таблицы 1; соединение примера 5 таблицы 1; соединение примера 6 таблицы 1; соединение примера 11 таблицы 1. Методика С. Синтез (1R,3S,4S)-N-S)-1-циано-2-(4-(тетрагидро-2 Н-пиран-4-ил)фенил)этил)-2-азабицикло[2.2.1] гептан-3-карбоксамида (соединение примера 3 таблицы 1)I-5.2 (150 мг, 0,319 ммоль) и Pd/C (10%, 30 мг) в метаноле (10 мл) перемешивают в атмосфере водорода (50 фунт-сила/дюйм 2) при комнатной температуре в течение 2 ч. Реакционную смесь фильтруют и концентрируют. Продукт используют в неочищенном виде, m/z 372 [М+Н-Вос]+, rt = 1,45 мин, методика ЖХ-МС с. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов: При восстановлении I-5.7 в качестве основного продукта образуется I-5.12 (80%), тогда как второстепенным продуктом является I-5.11 (10%). Стадия 2. Синтез I-4.11. Эту стадию проводят в соответствии с процедурой, описанной в методике В, стадия 2, с использованием соответствующих реагентов. Продукт используют в неочищенном виде, m/z 354 [М+Н-Вос]+, rt = 1,50 мин, методика ЖХ-МС с. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов: Стадия 3. Синтез соединения примера 3. Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 2, с использованием соответствующих реагентов. Выход 25% (в пересчете на I-5.2). Следующие соединения синтезируют аналогичным образом из соответствующих промежуточных продуктов: соединение примера 2 таблицы 1; соединение примера 9 таблицы 1; соединение примера 30 таблицы 1.(0,071 мл, 0,246 ммоль) и ДМФ (2 мл) продувают аргоном и добавляют бис-(трифенилфосфин) палладийхлорид (8 мг, 0,011 ммоль) и реакционную смесь нагревают при 80 С в течение 1,5 ч. К охлажденной смеси добавляют воду и водный слой дважды экстрагируют с помощью ЭА, объединенные органические слои промывают рассолом, сушат и концентрируют. Остаток очищают с помощью колоночной хроматографии (циклогексан/ЭА = 3:1). Выход 100%. m/z 396 [М+Н]+, rt = 0,77 мин, методика ЖХ-МС b. Следующий промежуточный продукт синтезируют аналогичным образом из соответствующих реагентов: Стадия 2. Синтез соединения примера 10. Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 2, с использованием соответствующих реагентов. Выход 44%. Следующее соединение синтезируют аналогичным образом из соответствующих промежуточных продуктов: соединение примера 18 таблицы 1. Методика Е. Синтез (1R,3S,4S)-N-S)-1-циано-2-(4-этилфенил)этил)-2-азабицикло[2.2.1]гептан-3-карбоксамида Стадия 1. Синтез I-4.17. Эту стадию проводят в соответствии с процедурой, описанной в методике С, стадия 1, за исключением того, что реакцию проводят в смеси в метанол/тетрагидрофуран (ТГФ) (1:1). Продукт используют в неочищенном виде, m/z 398 [М+Н]+, rt = 0,80 мин, методика ЖХ-МС b. Следующий промежуточный продукт синтезируют аналогичным образом из соответствующих реагентов: Стадия 2. Синтез соединения примера 12. Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 2, с использованием соответствующих реагентов. Выход 50%. Следующее соединение синтезируют аналогичным образом из соответствующих промежуточных Эту стадию проводят в соответствии с процедурой, описанной в методике А, стадия 2, с использованием соответствующих реагентов. Выход 62%. Методика G. Синтез (1R,3S,4S)-N-S)-1 -циано-2-(4-(4-метилпиперазин-1-ил)фенил)этил)-2 азабицикло[2.2.1]гептан-3-карбоксамида (соединение примера 15 таблицы 1)I-1.6 (200 мг, 0,39 ммоль), L-пролин (13,5 мг, 0.117 ммоль) в ДМСО (1,5 мл) продувают аргоном и добавляют Cu(I)I (15,2 мг, 0,080 ммоль) и карбонат цезия (171 мг, 0,526 ммоль). Реакционную смесь нагревают при 90 С в течение ночи. К полученной смеси МеОН добавляют и смесь непосредственно очищают с помощью ВЭЖХ. Выход 26% m/z 486 [М+Н]+, rt = 0,99 мин, методика ЖХ-МС d. Следующие промежуточные продукты синтезируют аналогичным образом из соответствующих реагентов:
МПК / Метки
МПК: A61P 37/00, C07D 471/08, A61P 11/00, A61P 29/00, C07D 519/00, A61K 31/439
Метки: замещенные, n-[1-циано-2-(фенил)этил]-2, ингибиторы, катепсина, азабицикло[2.2.1]гептан-3-карбоксамидные
Код ссылки
<a href="https://eas.patents.su/30-24817-zameshhennye-n-1-ciano-2-feniletil-2-azabiciklo221geptan-3-karboksamidnye-ingibitory-katepsina-s.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные n-[1-циано-2-(фенил)этил]-2- азабицикло[2.2.1]гептан-3-карбоксамидные ингибиторы катепсина с</a>
N-замещённые гидроксипиримидинон-карбоксамидные ингибиторы вич интегразы

Номер патента: 7060
Опубликовано: 30.06.2006
Авторы: Пескаторе Джованна, Роули Майкл, Орвьето Федерика, Ници Эмануэла, Сумма Винченцо, Крешенци Бенедетта, Пома Марко, Гарделли Кристина, Муралья Эстер, Петрокки Алессия, Паче Паола, Скарпелли Рита
МПК: C07D 401/04, A61K 31/515, C07D 239/54...
Метки: n-замещённые, ингибиторы, вич, интегразы, гидроксипиримидинон-карбоксамидные
Формула / Реферат:
1. Соединение формулы (I) где R1 представляет собой: (1) -H, (2) -C1-4 алкил, который необязательно замещен 1-4 заместителями, каждый из которых независимо представляет собой галоген, -ОН, -CN, -O-C1-4 алкил, -O-C1-4 галогеналкил, -C(=O)Ra, -CO2Ra, -SRa, -S(=O)Ra, -N(RaRb), -С (=O)-C0-4алкил-N (RaRb), -N(Ra)-C(=O)-C0-4алкил-N (RbRc), -SO2Ra, -N(Ra)SO2Rb, -SO2N(RaRb), -N(Ra)-C(=O)Rb, N(Ra)-C(=O)Rk или -N (Ra) С (=O) С (=O) N (RaRb), (3) -Rk,...
(r)-(z)-1-азабицикло[2.2.1]гептан-3-он,о-[3-(3-метоксифенил)-2-пропинил] оксим малеинат в качестве фармацевтического средства

Номер патента: 381
Опубликовано: 24.06.1999
Авторы: Роуз Стивен Эдвард, Джэйн Хуан Карлос, Бэрретт Стифен Дуглас, Эндо Говард Йошихиса, Текле Хэйли
МПК: C07D 487/08, A61K 31/395
Метки: качестве, малеинат, оксим, средства, r)-(z)-1-азабицикло[2.2.1]гептан-3-он,о-[3-(3-метоксифенил)-2-пропинил, фармацевтического
Формула / Реферат:
1. (R)-(Z)-1-Азабицикло[2.2.1]гептан-3-он,O-[3-(3-метоксифенил)-2-пропинил]-оксим малеинат в качестве фармацевтического средства. 2. Соединение по п.1, у которого, по меньшей мере, 50% кристаллических частиц имеет размер более 10х10 мкм. 3. Фармацевтическая композиция, пригодная для облегчения боли у млекопитающего или для лечения расстройства познавательной способности, включающая анальгетически эффективное количество соединения по п.1 вместе...
Новые соединения и композиции как ингибиторы катепсина

Номер патента: 7335
Опубликовано: 25.08.2006
Авторы: Турайратнам Сукантини, Линк Джон О., Элдос Дэвид Дж., Тимм Эндрис П., Ли Джиайао, Зипфель Шейла, Грауп Майкл
МПК: A61P 33/06, A61P 19/02, A61K 31/16...
Метки: ингибиторы, соединения, композиции, катепсина, новые
Формула / Реферат:
1. Соединение формулы I в которой X1 является -NHC(R1)(R2)X3 или -NHX4; X2 представляет водород, фтор, -ОН, -OR4, -NHR15 или -NR17R18 и X7 является водородом, или X2 и X7, оба представляют фтор; X3 представляет циано, -С(R7)(R8)R16, -С(О)С(О)NR5R6; где R5 представляет водород, (C6-10)арил(С0-6)алкил, гетеро(C5-10)арил(С0-6)алкил; R6 представляет водород, гидрокси или (C1-6)алкил; R7 представляет водород или (C1-4)алкил и R8 представляет...
Способ получения (-)-(s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата

Номер патента: 6967
Опубликовано: 30.06.2006
Авторы: Степанкова Гана, Гайичек Йосеф, Симек Станислав
МПК: C07C 271/44, C07C 215/50, C07C 269/00...
Метки: получения, s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата, способ
Формула / Реферат:
1. Способ получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата, то есть ривастигмина формулы II или его гидротартрата формулы I отличающийся тем, что метоксиацетофенон формулы VI подвергают восстановительному аминированию с получением соединения формулы V которое затем О-дезалкилируют с получением рацемического амина формулы IV который далее разделяют путем взаимодействия с оптически активной кислотой, после чего...
Твердые формы (s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата и способы их применения

Номер патента: 17275
Опубликовано: 30.11.2012
Авторы: Де Поль Сьюзан, Чжан Хаймин, Канамарлапуди Раманаиах К., Беднарз Марк С., Перлберг Анетт
МПК: A61K 31/506, A61P 1/00, A61P 25/00...
Метки: применения, твердые, формы, способы, s)-этил-2-амино-3-(4-(2-амино-6-((r)-1-(4-хлор-2-(3-метил-1h-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноата
Формула / Реферат:
1. Кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой кристаллический (S)-этил-2-амино-3-(4-(2-амино-6-((R)-1-(4-хлор-2-(3-метил-1Н-пиразол-1-ил)фенил)-2,2,2-трифторэтокси)пиримидин-4-ил)фенил)пропаноат.3. Соединение по п.2, которое имеет температуру плавления,...
Предыдущий патент: Способы борьбы с паразитными сорняками при помощи смесей, включающих гербицидные ингибиторы ацетолактатсинтазы и регуляторы роста растений
Следующий патент: Подъемник для инвалидов
Случайный патент: Композиция для защиты сельскохозяйственных культур