Способ получения (-)-(s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата
Номер патента: 6967
Опубликовано: 30.06.2006
Авторы: Степанкова Гана, Симек Станислав, Гайичек Йосеф






Формула / Реферат
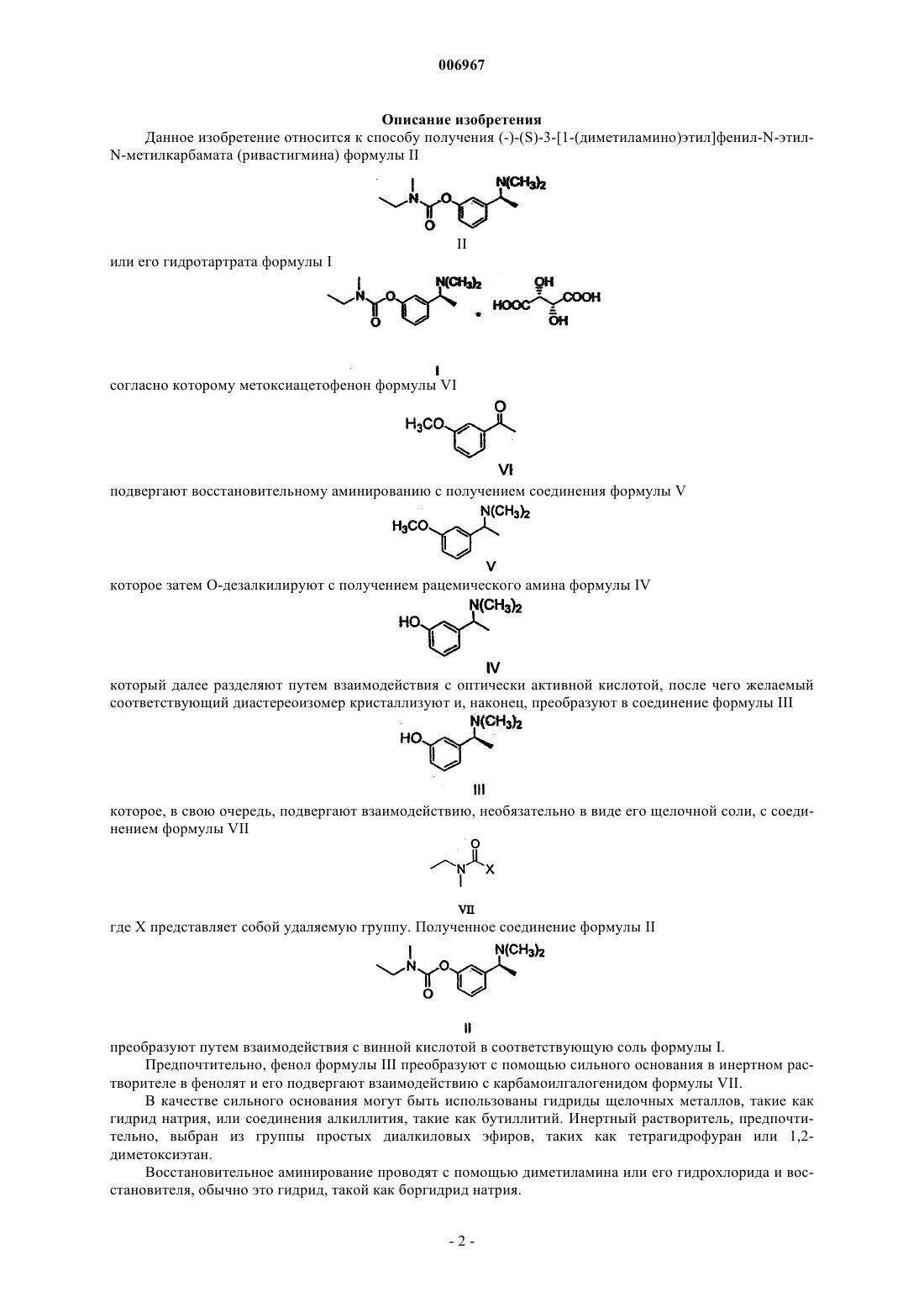
1. Способ получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата, то есть ривастигмина формулы II

или его гидротартрата формулы I

отличающийся тем, что метоксиацетофенон формулы VI

подвергают восстановительному аминированию с получением соединения формулы V

которое затем О-дезалкилируют с получением рацемического амина формулы IV

который далее разделяют путем взаимодействия с оптически активной кислотой, после чего желаемый соответствующий диастереоизомер кристаллизуют и, наконец, преобразуют в соединение формулы III

которое, в свою очередь, подвергают взаимодействию, необязательно в форме его щелочной соли, с соединением формулы VII

где X представляет собой удаляемую группу.
2. Способ по п.1, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с (S)-(+)-камфор-10-сульфокислотой.
3. Способ по п.2, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с 1 эквивалентом (S)-(+)-камфор-10-сульфокислоты.
4. Способ по п.2, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с 0,6 эквивалента (S)-(+)-камфор-10-сульфокислоты.
5. Способ по любому из предшествующих пунктов, отличающийся тем, что полученный желаемый соответствующий диастереоизомер дополнительно перекристаллизовывают.
6. Способ по п.5, отличающийся тем, что полученный желаемый соответствующий диастереоизомер дополнительно перекристаллизовывают из этилацетата, необязательно в смеси с этанолом.
Текст
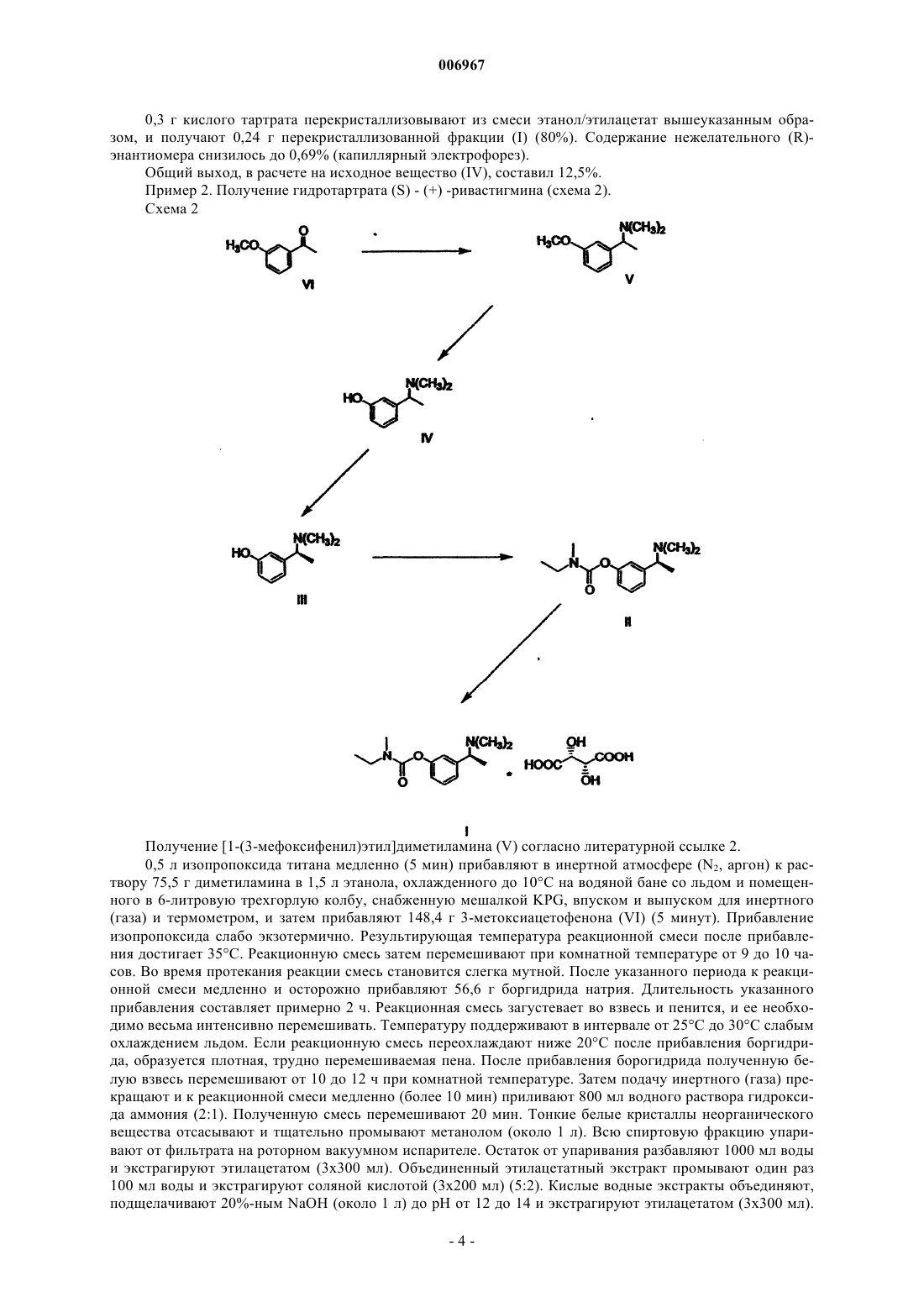
006967 Область техники Изобретение относится к способу получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-Nметилкарбамата и его гидротартрата. Предпосылки изобретения Тартрат (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата формулы I известный под названием INN ривастигмин, описан в патентной заявке CS No. PV 1991-4110 в качестве вещества, которое индуцирует селективное ингибирование активности ацетилхолинэстеразы в головном мозге. Указанное качество наряду с хорошей переносимостью организмом человека, возможностью выбора доставки в форме таблеток (пероральная эффективность) и продолжительным действием предопределяют возможность использования ривастигмина для лечения заболеваний, связанных с нарушением холинергической системы, в частности, болезни Альцгеймера. Рацемический 3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамат (далее рацемический ривастигмин) в качестве соединения, обладающего возможной активностью против болезни Альцгеймера,был описан в патенте ЕР 193926. Способ его получения был основан на взаимодействии мгидроксифенилэтилдиметиламина формулы IV где X представляет собой удаляемую группу. Способы получения этих рацемических промежуточных продуктов были описаны в более ранней литературе, но нигде не указывалась возможность их преобразования в оптически активные энантиомеры. В процитированной заявке CS PV 1991-4110 было продемонстрировано в экспериментах как "in vitro", так и "in vivo", что оптически активный (S)-изомер является намного более эффективным и селективным ингибитором ацетилхолинэстеразы, чем рацемическая смесь двух изомеров. В процитированной заявке описан способ получения ривастигмина из рацемической смеси, включающий получение диастереоизомерных солей с (+)-O,O-ди(пара-толуил)-D-винной кислотой и их разделение кристаллизацией. (S)-Энантиомер ривастигмина был высвобожден из полученной соли с помощью раствора гидроксида натрия. Основным технологическим недостатком этого способа является то, что оптическое разделение осуществляют только на конечной стадии синтеза. Это означает, что по меньшей мере 50% полученного рацемического ривастигмина (то есть, (R)-энантиомера) представляет собой бесполезный отход; на самом же деле, указанные отходы намного больше, поскольку при оптическом разделении энантиомеры никогда не разделяются количественно. Это делает общий выход синтеза низким, а весь способ экономически невыгодным. Другой недостаток состоит в наличии потерь на отдельных стадиях. Обычно потери промежуточного продукта на более поздних стадиях являются более дорогостоящими, чем на начальных стадиях. Возвращаясь к рассмотрению указанного способа, следует отметить, что расщепление не приводит к достижению удовлетворительной оптической чистоты, и вещество требуется дополнительно перекристаллизовать (ср. ссылочный пример 1). Другим недостатком является необходимость использования дорогого и канцерогенного соединения формулы VII (наиболее конкретно, N-этил-N-метилкарбамоилхлорида) примерно в 300%-ном избытке. Разделение на более ранней стадии синтеза кажется на первый взгляд желательным, но далеким от осуществимости. Остается вопрос, можно ли получать энантиомерно чистые промежуточные продукты и, в частности, могут ли указанные продукты быть использованы в дальнейшем синтезе без того, что они будут подвергаться рацемизации. Необходимость перекристаллизации ставит под сомнение преимущества указанного способа. В настоящее время возвращаются к тому, что оптическое разделение промежуточных продуктов (то есть, проведение операции на более ранней стадии получения) и проведение конечной стадии с оптически чистым веществом позволяет получать очень хороший выход (S)-ривастигмина с сохранением высокой аналитической чистоты.-1 006967 Описание изобретения Данное изобретение относится к способу получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этилN-метилкарбамата (ривастигмина) формулы IIII или его гидротартрата формулы I согласно которому метоксиацетофенон формулы VI подвергают восстановительному аминированию с получением соединения формулы V которое затем О-дезалкилируют с получением рацемического амина формулы IV который далее разделяют путем взаимодействия с оптически активной кислотой, после чего желаемый соответствующий диастереоизомер кристаллизуют и, наконец, преобразуют в соединение формулы III которое, в свою очередь, подвергают взаимодействию, необязательно в виде его щелочной соли, с соединением формулы VII где X представляет собой удаляемую группу. Полученное соединение формулы II преобразуют путем взаимодействия с винной кислотой в соответствующую соль формулы I. Предпочтительно, фенол формулы III преобразуют с помощью сильного основания в инертном растворителе в фенолят и его подвергают взаимодействию с карбамоилгалогенидом формулы VII. В качестве сильного основания могут быть использованы гидриды щелочных металлов, такие как гидрид натрия, или соединения алкиллития, такие как бутиллитий. Инертный растворитель, предпочтительно, выбран из группы простых диалкиловых эфиров, таких как тетрагидрофуран или 1,2 диметоксиэтан. Восстановительное аминирование проводят с помощью диметиламина или его гидрохлорида и восстановителя, обычно это гидрид, такой как боргидрид натрия.-2 006967 Агенты О-дезалкилирования могут быть выбраны из сильных кислот, таких как, например, бромистоводородная кислота, или из галогенидов бора, таких как бромид бора. Рацемический амин формулы IV предпочтительно разделяют путем взаимодействия с (S)-(+)камфор-10-сульфоновой кислотой. Полученный желаемый соответствующий диастереоизомер может быть затем перекристаллизован,предпочтительно, из этилацетата, необязательно, в смеси с этанолом. Как показано в примерах особенно предпочтительных вариантов выполнения, данный способ делает возможным получение продукта формулы I особенно высокой оптической чистоты. Воспроизведение ранее известного способа, даже с перекристаллизацией, не приводит к достижению столь высокой оптической чистоты. Дополнительным результатом сравнения является то, что использование оптически активного соединения формулы III приводит к уменьшению на 2/3 потребления дорогостоящего и канцерогенного Nэтил-N-метилкарбамоилхлорида (соответствующего общей формуле VII, где Х=Cl). Примеры Объект изобретения более подробно продемонстрирован в следующих примерах. Пример 1. Получение гидротартрата (S)-(-)-ривастигмина (I) (схема 1). Разделение рацемического соединения (IX) с помощью (+) -O,O'-дитолуилвинной кислоты. 49,6 г рацемического соединения (IX) растворяют в 100 мл метанола и к раствору прибавляют раствор 75,8 г ( + )-O,O'-дитолуилвинной кислоты в 200 мл метанола. К прозрачному раствору при перемешивании постепенно прибавляют 150 мл воды. Раствор становится мутным и начинает кристаллизоваться на холоде. Получают 81,3 г желаемой соли в виде белых кристаллов с т.пл.=145-147 С. Указанное количество первой фракции кристаллов перекристаллизовывают из смеси 200 мл метанола и 100 мл воды. Получают вторую фракцию кристаллов (49,6 г, т.пл.=155-156 С), которую снова перекристаллизовывают из смеси (метанол:вода (2:1), 200 мл). Получают третью фракцию с т.пл.=159-162 С. Ее перекристаллизовывают из 150 мл вышеуказанной смеси растворителей и выделяют четвертую конечную фракцию кристаллов (33,0 г, т.пл.=156-7 С), что соответствует выходу 26,9% от теоретического, [] D=+78,5(с=1,этанол). Высвобождение основания (II). Соль S-(-)-ривастигмина (II) с (+)-O,O'-дитолуилвинной кислотой (33,0 г) медленно прибавляют к хорошо перемешиваемой смеси 150 мл дихлорметана и 150 мл 1 н раствора NaOH. После растворения всего твердого вещества слои разделяют; дихлорметановый слой дважды экстрагируют 100 мл воды и сушат сульфатом магния. Растворитель упаривают в вакууме и остаток от упаривания перегоняют в вакууме (т.кип.=135-8 С, 40 Па). Получают 10,72 г бесцветного масла, что соответствует 85%-ному выходу от теоретического ( [] D =-24,5 С; с=3,5, метанол). Получение гидротартрата (S)-(+)-ривастигмина (I). 8,86 г основания (II) и 5,31 г L-(+)-винной кислоты растворяют при нагревании и перемешивании в 20 мл этанола, и к прозрачному раствору для осаждения прибавляют этилацетат (250 мл). Полученную смесь оставляют охлаждаться до +5 С. Выпавшие кристаллы отсасывают и промывают этилацетатом. Получают 9,7 г желаемого гидротартрата ривастигмина (I) (то есть выход 68,4% от теоретического) с т.пл.=124-126 С. Содержание нежелательного (R)-энантиомера в указанной фракции составляло 1% (по данным капиллярного электрофореза).-3 006967 0,3 г кислого тартрата перекристаллизовывают из смеси этанол/этилацетат вышеуказанным образом, и получают 0,24 г перекристаллизованной фракции (I) (80%). Содержание нежелательного (R)энантиомера снизилось до 0,69% (капиллярный электрофорез). Общий выход, в расчете на исходное вещество (IV), составил 12,5%. Пример 2. Получение гидротартрата (S) - (+) -ривастигмина (схема 2). Схема 2 Получение [1-(3-мефоксифенил)этил]диметиламина (V) согласно литературной ссылке 2. 0,5 л изопропоксида титана медленно (5 мин) прибавляют в инертной атмосфере (N2, аргон) к раствору 75,5 г диметиламина в 1,5 л этанола, охлажденного до 10 С на водяной бане со льдом и помещенного в 6-литровую трехгорлую колбу, снабженную мешалкой KPG, впуском и выпуском для инертного(газа) и термометром, и затем прибавляют 148,4 г 3-метоксиацетофенона (VI) (5 минут). Прибавление изопропоксида слабо экзотермично. Результирующая температура реакционной смеси после прибавления достигает 35 С. Реакционную смесь затем перемешивают при комнатной температуре от 9 до 10 часов. Во время протекания реакции смесь становится слегка мутной. После указанного периода к реакционной смеси медленно и осторожно прибавляют 56,6 г боргидрида натрия. Длительность указанного прибавления составляет примерно 2 ч. Реакционная смесь загустевает во взвесь и пенится, и ее необходимо весьма интенсивно перемешивать. Температуру поддерживают в интервале от 25 С до 30 С слабым охлаждением льдом. Если реакционную смесь переохлаждают ниже 20 С после прибавления боргидрида, образуется плотная, трудно перемешиваемая пена. После прибавления борогидрида полученную белую взвесь перемешивают от 10 до 12 ч при комнатной температуре. Затем подачу инертного (газа) прекращают и к реакционной смеси медленно (более 10 мин) приливают 800 мл водного раствора гидроксида аммония (2:1). Полученную смесь перемешивают 20 мин. Тонкие белые кристаллы неорганического вещества отсасывают и тщательно промывают метанолом (около 1 л). Всю спиртовую фракцию упаривают от фильтрата на роторном вакуумном испарителе. Остаток от упаривания разбавляют 1000 мл воды и экстрагируют этилацетатом (3x300 мл). Объединенный этилацетатный экстракт промывают один раз 100 мл воды и экстрагируют соляной кислотой (3x200 мл) (5:2). Кислые водные экстракты объединяют,подщелачивают 20%-ным NaOH (около 1 л) до рН от 12 до 14 и экстрагируют этилацетатом (3x300 мл).-4 006967 Органическую фракцию промывают 100 мл воды и 150 мл насыщенного солевого раствора. Сушат безводным сульфатом натрия. Осушитель отфильтровывают и фильтрат упаривают досуха на роторном вакуумном испарителе. Сырой продукт перегоняют и получают около 60% желаемого продукта в виде бесцветного масла. Т.кип.=68 С при 400 Па, 108 С при 800 Па. Получение 3-(1-диметиламиноэтил)фенола (IV) согласно литературной ссылке 3. 94 г [1-(3-метоксифенил)этил]диметиламина (V) растворяют в 285 мл азеотропной бромистоводородной кислоты и полученный раствор кипятят при перемешивании с обратным холодильником 12 ч(температура бани 145-150 С). В процессе кипячения реакционная смесь темнеет. Раствор затем оставляют остывать до комнатной температуры. Избыток бромистоводородной кислоты упаривают с использованием роторного вакуумного испарителя, и остаток от упаривания растворяют в 200 мл воды. Раствор экстрагируют этилацетатом (3x100 мл). Водную фракцию затем постепенно подщелачивают насыщенным раствором карбоната натрия при постоянном перемешивании (вспенивание). Раствор становится молочно мутным, и его экстрагируют этилацетатом (3x200 мл). Этилацетатную фракцию встряхивают 1 раз с водой, 1 раз с насыщенным раствором соли и сушат над безводным сульфатом магния. Перед фильтрацией осушителя прибавляют активированный уголь и осушитель отфильтровывают вместе с углем. Этилацетатный раствор соединения (IV) используют на следующей стадии. Получение S-(-)-3-(l-диметиламино)фенола (III). Разделение соединения (IV) 1 эквивалентным количеством S-(+)-камфор-10-сульфокислоты. Раствор соединения (IV) в этилацетате (0,505 моль) (содержание соединения (IV) определяли титрованием) в 500 мл этилацетата помещают в 1-литровую круглодонную колбу с магнитной мешалкой и прибавляют раствор S-(+)-камфор-10-сульфокислоты (117,4 г (0,505 моль) в 250 мл безводного этанола,полученный при нагревании. В раствор вносят затравку и оставляют стоять в холодильнике (+5 С) в течение ночи. Выпавшие кристаллы отсасывают через пористый стеклянный фильтр и оставляют сушиться на воздухе в течение ночи. 1) Получают 82,2 г белых кристаллов с т.пл.=165-171 С, которые растворяют в 190 мл абсолютного этанола при кипячении с обратным холодильником. Прибавляют 380 мл этилацетата и осуществляют кристаллизацию при нагревании согласно вышеуказанной методике. 2) Получают 64,1 г белых кристаллов с т.пл.=174-176 С, которые растворяют в 150 мл этанола(абс.) при кипячении с обратным холодильником и при нагревании прибавляют 300 мл этилацетата. 3) Получают 56,5 г белых кристаллов с т.пл.=177-179 С, которые растворяют в 130 мл этанола(абс.) при кипячении с обратным холодильником, и при нагревании прибавляют 260 мл этилацетата. 4) Получают 51,6 г белых кристаллов с т.пл. 179-181 С, то есть 25,7% от теоретического количества. Разделение соединения (IV) 0,6 эквивалентным количеством S-(+)-камфор-10-сульфокислоты. 100 г (0,605 моль) соединения (IV) растворяют в 600 мл этилацетата при перемешивании и кипячении с обратным холодильником. Прибавляют при перемешивании при 70 С раствор S-(+)-камфор-10 сульфокислоты (84,3 г (0,363 моль) в 125 мл безводного этанола. В раствор вносят затравку, оставляют остывать до комнатной температуры при перемешивании, охлаждают насыщенным солевым раствором до температуры от -10 до -15 С и оставляют кристаллизоваться по меньшей мере на 12 ч без доступа влаги воздуха. Выпавшую первую фракцию кристаллов отсасывают и сушат на воздухе. 1) Получают 95,0 г белых кристаллов с т.пл.=173-175 С, которые растворяют в 175 мл этанола при перемешивании и кипячении с обратным холодильником и прибавляют 350 мл этилацетата при температуре раствора от 60 до 70 С. Камфорсульфонат начинает кристаллизоваться, и его оставляют кристаллизоваться при температуре от -5 до -10 С по меньшей мере на 12 ч. Выпавшую фракцию отсасывают,промывают этилацетатом (2x50 мл) и сушат на воздухе. 2) Получают 79,5 г второй фракции с т.пл. 176-178 С, которую снова перекристаллизовывают из смеси этанол:этилацетат (150 мл:300 мл) согласно вышеописанному способу. После промывки этилацетатом (2x50 мл) продукт сушат на воздухе. 3) Получают 74,6 г третьей фракции с т.пл.=177-179 С, то есть выход 31,0% от теоретического. Высвобождение S-(-)-3-(1-диметиламино)фенола (III). 4 л воды помещают в 10-литровый тонкостенный химический стакан с KPG-мешалкой. Прибавляют и растворяют при перемешивании 250 г карбоната натрия. Частями при перемешивании прибавляют кристаллы камфорсульфоната (517,5 г). Когда прибавлена примерно половина всего количества, добавляют 2 литра дихлорметана. Остальной камфорсульфонат прибавляют при постоянном перемешивании. Период прибавления составлял около 0,5 ч. Полученную смесь перемешивают еще 0,5 ч. Затем слои разделяют в 10-литровой делительной воронке. Водную фракцию экстрагируют дихлорметаном (2x1,5 л). Объединенные органические фракции экстрагируют 1,5 л воды и сушат над 600 г безводного сульфата натрия. Осушитель отфильтровывают и фильтрат упаривают досуха. Полученный от упаривания остаток затем сушат на роторном вакуумном испарителе до постоянной массы при 50 С и 2,7 кПа. Образуется белое кристаллическое вещество (187,0 г; 87%), которое используют на следующей стадии без очистки,[]D=55,7; c=l,55, метанол).-5 006967 Содержание нежелательного (R)-энантиомера 0,4% определено путем газовой хроматографии на хиральной колонке. Получение S-(-)-ривастигмина (II). 300 мл тетрагидрофурана (ТГФ) помещают в 0,5-литровую трехгорлую колбу и медленно при перемешивании в инертных условиях (Ar или N2) прибавляют гидрид натрия в виде 60%-ной дисперсии в масле (11,3 г). Образуется суспензия, к которой при комнатной температуре прибавляют кристаллическое соединение (III) (46,5 г, 0,281 моль). Образуется раствор фенолята, к которому по каплям в течение 10 минут при слабом охлаждении до 15 С прибавляют 35,7 г (0,281 моль) карбамоилхлорида. Реакция слабо экзотермична. Скорость прибавления по каплям регулируют таким образом, чтобы температура реакционной смеси не превышала 30 С. После прибавления всего реагента охлаждающую систему убирают и реакционную смесь перемешивают 2 ч при комнатной температуре. После этого ТГФ упаривают в роторном вакуумном испарителе. Остаток от упаривания распределяют между 200 мл 1 н NaOH и 500 мл эфира. Органический слой отделяют, и водную фракцию встряхивают с дополнительным количеством эфира (2x200 мл). Объединенные эфирные слои встряхивают с водой (1x100 мл) и насыщенным солевым раствором (1x50 мл). Органическую фракцию сушат над безводным сульфатом натрия. Растворитель упаривают и сырой продукт перегоняют в вакууме. Т.кип.=135-140 С при 13 Па. Получают 45,6 г бесцветного вязкого масла, то есть выход 80,5%. Содержание по данным газовой хроматографии 99,6%. Получение гидротартрата S-(+)-ривастигмина (I). 45,6 г S-(-)-ривастигмина и 27,4 г L-(+)-винной кислоты растворяют в 125 (мл) безводного этанола при 60-70 С при перемешивании. К раствору при указанной температуре постепенно прибавляют 630 мл этилацетата. Раствору дают охладиться до комнатной температуры и оставляют кристаллизоваться при+5 С по меньшей мере на 12 ч. Выпавший белый кристаллический продукт отсасывают, промывают 100 мл этилацетата и сушат в вакууме при 40 С. Получают 67,5 г желаемого продукта с т.пл.=125-126 С (то есть выход 92,6% от теоретического)([]D=+5,5; с=5, этанол). Содержание нежелательного Rэнантиомера в образце было менее 0,2% (капиллярный электрофорез). Получение S-(-)-ривастигмина (II). 150 мл диэтилового эфира помещают в 0,5-литровую трехгорлую колбу и медленно при перемешивании в инертных условиях (Ar или N2) прибавляют гидрид натрия в виде 60%-ной дисперсии в масле. Образуется суспензия, к которой при комнатной температуре прибавляют кристаллическое соединениет(III) (2,0 г, 0,012 моль). После перемешивания в течение одного часа образуется слегка мутный раствор фенолята, к которому прибавляют по каплям при комнатной температуре 1,53 г (0,012 моль) N-этил-Nметилкарбамоилхлорида в 20 мл эфира. Полученную реакционную смесь перемешивают 3 ч при комнатной температуре. После этого ее разбавляют 100 мл воды. Органический слой отделяют и экстрагируют 0,1 н раствором NаОН (2x50 мл). Органическую фазу экстрагируют 50 мл воды, сушат безводным сульфатом магния и концентрируют в вакууме. Получают 2,6 г масла (выход 86,6% от теоретического). Получение S-(-)-ривастигмина (II). 50 мл 1,2-диметоксиэтана помещают в 0,25-литровую круглодонную колбу и растворяют в нем соединение (III) (2,0 г, 0,012 моль) при перемешивании и в инертной атмофере (Ar или N2) при комнатной температуре. Затем к полученному раствору прибавляют по каплям 1,6 М раствор н-бутиллития в гексане(7,5 мл). Образуется слегка мутный раствор фенолята, к которому прибавляют по каплям при комнатной температуре 1,53 г (0,012 моль) N-этил-N-метилкарбамоилхлорида в 20 мл 1,2-диметоксиэтана. Растворитель упаривают на роторном вакуумном испарителе. Остаток от упаривания распределяют между 20 мл 1 н NaOH и 50 мл эфира. Органический слой отделяют, а водную фракцию встряхивают с дополнительным количеством эфира (2x20 мл). Объединенные эфирные слои встряхивают с водой (1x20 мл) и насыщенным солевым раствором (1x20 мл). Органическую фракцию сушат над безводным сульфатом натрия и концентрируют в вакууме. Получают 1,56 г масла (выход 51,5% от теоретического). Получение гидротартрата S-(+)-ривастигмина (I). 2,0 г основания (II) и 1,2 г L-(+)-винной кислоты растворяют в 5 мл метанола при 60 С. Прозрачный раствор оставляют остывать до комнатной температуры и постепенно ацетоном (около 50 мл) вызывают осаждение. Полученную смесь оставляют кристаллизоваться при +5 С в течение ночи. Выпавшие кристаллы отсасывают, используя пористый стеклянный фильтр, и промывают ацетоном. Сушат в вакууме при 40 С и получают 2,4 г белых кристаллов (выход 80% от теоретического) с т.пл.=123-5 С. Полученные результаты отличались в зависимости от используемых способов разделения соединения IV. Способ с 1 эквивалентным количеством (S)-(+)-камфор-10-сульфокислоты дает общий выход 16,2% в расчете на соединение IV, а с 0,6 эквивалента дает общий выход 19,5% в расчете на соединение-6 006967 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата, то есть ривастигмина формулы II или его гидротартрата формулы I отличающийся тем, что метоксиацетофенон формулы VI подвергают восстановительному аминированию с получением соединения формулы V которое затем О-дезалкилируют с получением рацемического амина формулы IV который далее разделяют путем взаимодействия с оптически активной кислотой, после чего желаемый соответствующий диастереоизомер кристаллизуют и, наконец, преобразуют в соединение формулы III которое, в свою очередь, подвергают взаимодействию, необязательно в форме его щелочной соли, с соединением формулы VII где X представляет собой удаляемую группу. 2. Способ по п.1, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с (S)-(+)-камфор-10-сульфокислотой. 3. Способ по п.2, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с 1 эквивалентом (S)-(+)-камфор-10-сульфокислоты. 4. Способ по п.2, отличающийся тем, что рацемический амин формулы IV разделяют путем взаимодействия с 0,6 эквивалента (S)-(+)-камфор-10-сульфокислоты. 5. Способ по любому из предшествующих пунктов, отличающийся тем, что полученный желаемый соответствующий диастереоизомер дополнительно перекристаллизовывают. 6. Способ по п.5, отличающийся тем, что полученный желаемый соответствующий диастереоизомер дополнительно перекристаллизовывают из этилацетата, необязательно в смеси с этанолом.
МПК / Метки
МПК: C07C 271/44, C07C 215/50, C07C 269/00
Метки: способ, s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата, получения
Код ссылки
<a href="https://eas.patents.su/8-6967-sposob-polucheniya-s-3-1-dimetilaminoetilfenil-n-etil-n-metilkarbamata.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения (-)-(s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата</a>
Способ получения 17бета-гидрокси-11бета-{4-(диметиламино)фенил} 17альфа-(проп-1- инил)эстра-4,9-диен-3-она

Номер патента: 14
Опубликовано: 30.12.1997
Авторы: Густова Ольга Валериевна, Турчин Константин Федорович, Кочев Дмитрий Михайлович, Долгинова Елена Максовна, Гриненко Галина Семеновна, Климова Людмила Игоревна, Морозова Людмила Сергеевна, Ряховская Маргарита Игоревна
МПК: A61K 31/565, C07J 1/00
Метки: 17альфа-(проп-1, получения, инил)эстра-4,9-диен-3-она, 17бета-гидрокси-11бета-{4-(диметиламино)фенил, способ
Формула / Реферат:
Способ получения 17b -гидрокси-11b -[4-(диметиламино) фенил]-17a -(проп-1-инил)эстра-4,9-диен-3-она формулы I взаимодействием производного стероида с 4-диметиламинофенилмагнийбромидом в присутствии катализатора в среде тетрагидрофурана, выделением соответствующего арилкеталя с использованием насыщенного водного раствора хлористого аммония, дегидратацией и гидролизом в присутствии кислотного агента в среде растворителя при комнатной...
Способ получения (+/-) 3 – (3,4 – дихлорфенил) – 2 – диметиламино-2 – метилпропан-1-ола или церикламина (inn)

Номер патента: 4399
Опубликовано: 29.04.2004
Авторы: Никола Марк, Депернэ Доминик, Лабу Бландин
МПК: C07C 215/28, A61P 25/24
Метки: способ, метилпропан-1-ола, церикламина, дихлорфенил, inn, получения, диметиламино-2
Формула / Реферат:
1. Способ изготовления (+/-)3-(3,4-дихлорфенил)-2-диметиламин-2-метилпропан-1-ола (I) или церикламина, включающий i) арилирование метакриловой кислоты хлоридом диазония 3,4-дихлоранилина, приготовляемым in situ, с получением (+/-)2-хлор-3-(3,4-дихлорфенил)-2-метилпропионовой кислоты (IV) ii) аминирование кислоты (IV) амином HNR1R2, где R1 и R2 означают независимо водород или метил, с получением аминокислоты (III) где M означает щелочной или...
Способ получения [is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида.

Номер патента: 1989
Опубликовано: 22.10.2001
Авторы: Рейлли Лоренс В., Ванасс Бенуа Дж., Гарсиа Эрве, О'брайен Майкл К, Леон Патрик, Паунер Тори Х., Цуей Чинг Т., Томпсон Майкл Д., Вальтер Фрэнсис Л., Шах Харшавадан К.
МПК: C07D 409/12
Метки: получения, is-[1a,2b,3b,4a(s*)]]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3н-имидазо[4,5-b]пиридин-3-ил]-n-этил-2,3-дигидроксициклопентанкарбоксамида, способ
Формула / Реферат:
1. Способ получения [1S-[1a,2b,3b,4a(S*)]-4-[7-[[1-(3-хлор-2-тиенил)метил]пропил]амино]-3Н-имидазо[4,5-b]пиридин-3-ил]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (I)), включающий взаимодействие [1S-[1a,2b,3b,4a(S*)]]-4-[[3-амино-4-[[1-[(3-хлор-2-тиенил)метил]пропил]амино]-2-пиридинил]амино]-N-этил-2,3-дигидроксициклопентанкарбоксамида (соединение (IX)) со сложным эфиром ортоформиата, ацетатом формамидина или диметилацеталем...
Жидкая композиция этил-(z)-2-хлор-3-[2-хлор-5-(4,5,6,7-тетрагидро-1,3-диоксоизоиндолдион-2-ил)фенил]акрилата

Номер патента: 1620
Опубликовано: 25.06.2001
Авторы: Братц Маттиас, Бергхауз Райнер, Клойзер Дитер, Парг Адольф, Нуйкен Вессель
МПК: A01N 37/46
Метки: композиция, жидкая, этил-(z)-2-хлор-3-[2-хлор-5-(4,5,6,7-тетрагидро-1,3-диоксоизоиндолдион-2-ил)фенил]акрилата
Формула / Реферат:
1. Жидкая композиция этил-(Z)-2-хлор-3-[2-хлор-5-(4,5,6,7-тетрагидро-1,3-диоксоизоиндолдион-2-ил)фенил]акрилата, содержащая в основном наряду с этим активным веществом по защите растений а) ионный эмульгатор, б) неионный эмульгатор, в) ненуклеофильный и неосновный ароматический растворитель и г) необязательно еще одно обладающее гербицидным действием активное вещество по защите растений. 2. Композиция по п.1, содержащая в качестве ионного...
Новая кристаллическая форма n-[4- [2- ( 2-амино-4,7-дигидро-4-оксо-3h-пирроло[ 2,3-d]пиримидин-5-ил) этил] бензоил] -l-глутаминовой кислоты и способ ее получения

Номер патента: 4684
Опубликовано: 24.06.2004
Авторы: Ройтцель-Эденс Сюзн Мари, Снорек Шэрон Ван Ден Берг, Челиус Эрик Кристофер
МПК: C07D 487/04, A61P 35/00, A61K 31/519...
Метки: бензоил, 2-амино-4,7-дигидро-4-оксо-3h-пирроло, кислоты, новая, 2,3-d]пиримидин-5-ил, этил, форма, n-[4, l-глутаминовой, способ, получения, кристаллическая
Формула / Реферат:
1. Гидратная кристаллическая форма динатриевой соли N-[4-[2-(2-амино-4,7-дигидро-4-оксо-3H-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил]-L-глутаминовой кислоты ("гептагидратная кристаллическая форма"), характеризующаяся спектром дифракции рентгеновских лучей, который включает максимум, соответствующий межплоскостному расстоянию d: 7,78+ 0,04 Е, полученным измерением при 22+2шC и 20-80% относительной влажности с использованием медного источника...
Предыдущий патент: Эфиры флуоренкарбоновых кислот, способ их получения, а также их применение в качестве лекарственных средств
Следующий патент: Терморегулирующий материал, устройство и способ его изготовления
Случайный патент: Ингибиторы каспазы, содержащие изоксазолиновый цикл