Гетероциклические производные и их применение в лечении неврологических расстройств
Номер патента: 24059
Опубликовано: 31.08.2016
Авторы: Хурт Констанце, Фегтле Маркус, Жакье Себастьян, Венстра Сим Якоб, Люенд Райнер Мартин, Рюэгер Хайнрих, Бадигер Сангамеш, Тинтельнот-Бломлей Марина, Махауер Райнер, Чебролу Мурали









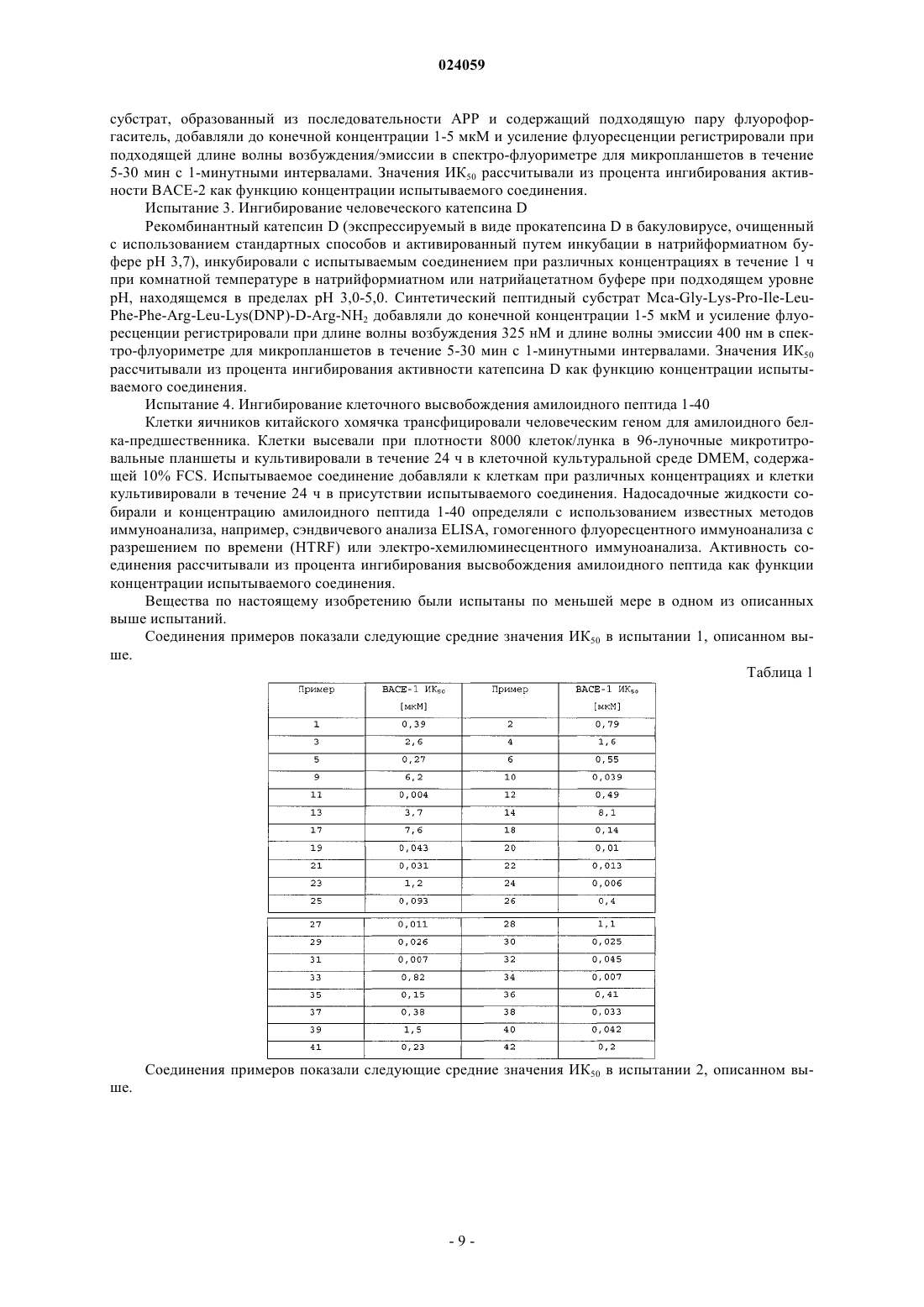



















Формула / Реферат
1. Соединение формулы (Ic) или его фармацевтически приемлемая соль

где X1 представляет собой СН или N;
Х3 представляет собой СН или N;
Х4 представляет собой CR4 или N;
где один и не более чем один из X1, X3 и Х4 представляет собой N;
R2 представляет собой пиридильную или пиразинильную группу, которая замещена 1, 2 или 3 заместителями, и где один из заместителей расположен в пара-положении пиридильной или пиразинильной группы относительно амидного линкера и заместители независимо выбраны из группы, включающей циано, амино, галоген, (С1-4)алкил, галоген-(C1-4)алкил, гидрокси, оксо, (C1-4)алкокси и галоген-(C1-4)алкокси;
R4 и R5 независимо представляют собой водород или галоген;
R6 представляет собой (C1-3)алкил или фтор-(C1-3)алкил;
каждый из R11 и R12 независимо выбран из группы, включающей водород, (C1-3)алкил или фтор-(C1-3)алкил.
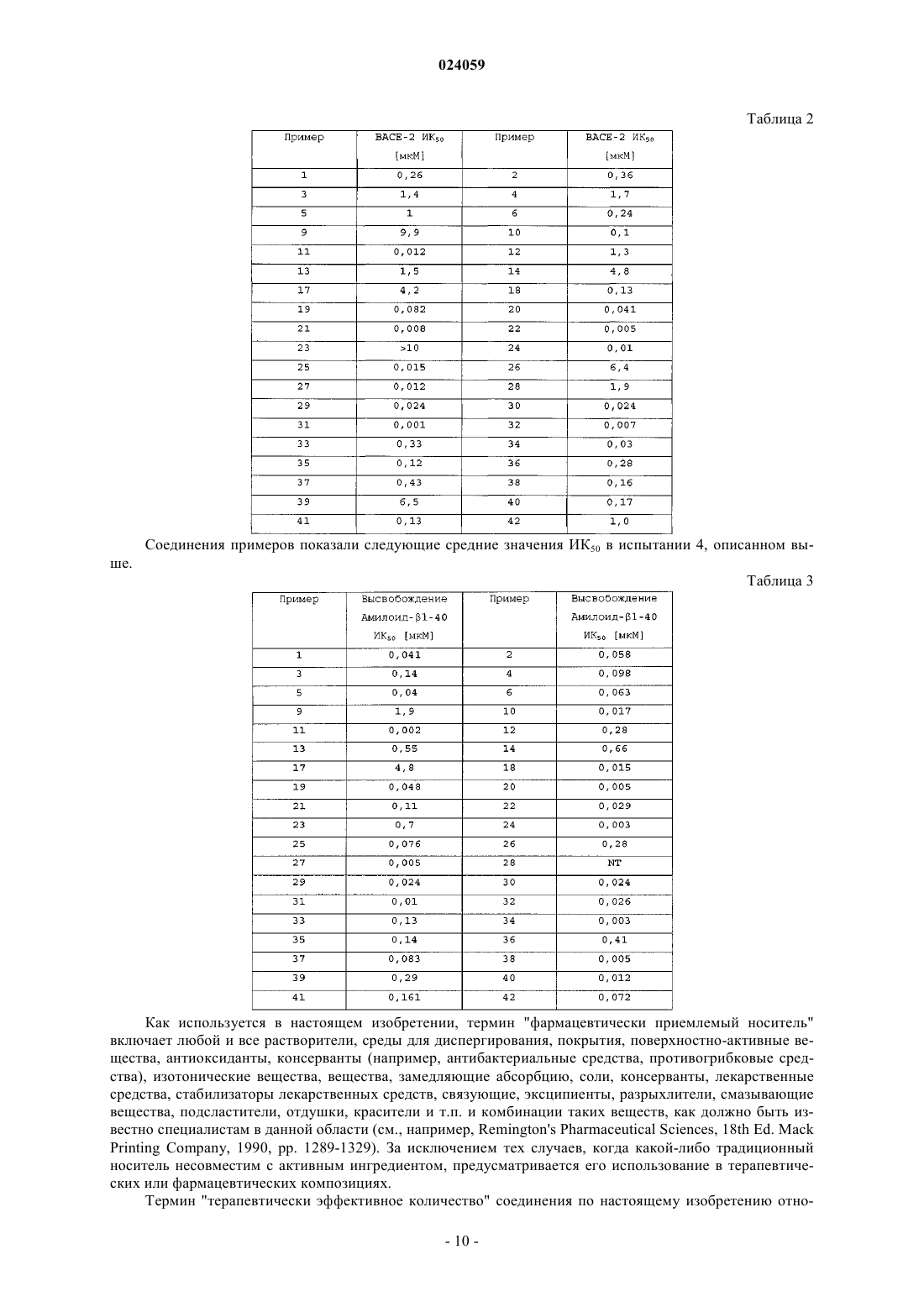
2. Соединение по п.1 формулы (Id) или его фармацевтически приемлемая соль:

где X1 представляет собой СН или N;
Х3 представляет собой СН или N;
Х4 представляет собой CR4 или N;
где один и не более чем один из X1, X3 и Х4 представляет собой N;
R2 представляет собой пиридильную или пиразинильную группу, которая замещена 2 или 3 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридильной или пиразинильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, галоген, (С1-4)алкил, галоген-(C1-4)алкил, гидрокси, оксо, (C1-4)алкокси и галоген-(C1-4)алкокси;
R4 и R5 независимо представляют собой водород или галоген;
R6 представляет собой метил, фторметил, дифторметил или трифторметил;
каждый из R11 и R12 независимо выбран из группы, включающей водород, метил, фторметил, дифторметил и трифторметил.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, где R2 представляет собой пиридин-2-ильную или пиразин-2-ильную группу, которая замещена 2 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридин-2-ильной или пиразин-2-ильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, фтор, бром, хлор, гидроксил, оксо, метил, фторметил, дифторметил, трифторметил, метокси, фторметокси, дифторметокси и трифторметокси.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где X1 представляет собой N; Х3 представляет собой СН и Х4 представляет собой CR4.
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где R4 представляет собой водород.
6. Соединение по п.1 формулы (Ie) или его фармацевтически приемлемая соль

где R2 представляет собой пиридин-2-ильную или пиразин-2-ильную группу, которая замещена 2 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридин-2-ильной или пиразин-2-ильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, фтор, бром, хлор, гидроксил, оксо, метил, фторметил, дифторметил, трифторметил, метокси, фторметокси, дифторметокси и трифторметокси;
R5 представляет собой водород или фтор;
R6 представляет собой метил, фторметил или дифторметил;
каждый из R11 и R12 независимо выбран из группы, включающей водород, метил, фторметил, дифторметил и трифторметил.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где R5 представляет собой фтор.
8. Соединение по п.1 или его фармацевтически приемлемая соль, которое выбрано из группы, включающей:
[6-((R)-5-амино-3-метил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-бромпиридин-2-карбоновой кислоты;
[6-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-хлорпиридин-2-карбоновой кислоты;
[6-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-бромпиридин-2-карбоновой кислоты;
[6-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 4,6-дидейтеро-5-хлор-3-тридейтерометилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((3S,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((3S,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[4-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 4,6-дидейтеро-5-хлор-3-тридейтерометилпиридин-2-карбоновой кислоты;
[4-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-хлорпиридин-2-карбоновой кислоты;
[4-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[4-((3S,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[5-(5-амино-3-фторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-6-хлорпиридин-3-ил]амид 5-бромпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 3-амино-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 3-хлор-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-хлор-4,6-дидейтерио-3-тридейтериометилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-бром-3-хлорпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 3-амино-5-(2,2,2-трифторэтокси)пиразин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-метокси-3-метилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-амино-5-(2,2,2-трифторэтокси)пиразин-2-карбоновой кислоты;
[6-((3S,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-амино-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-дифторметокси-3-метилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-дифторметоксипиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3,5-дихлорпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-фторметокси-3-метилпиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-метилпиразин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-трифторметилпиридин-2-карбоновой кислоты;
[4-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-цианопиридин-2-карбоновой кислоты;
[4-((3S,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-дифторметоксипиридин-2-карбоновой кислоты;
[4-((R)-5-амино-6,6-бис-фторметил-3-метил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((R)-5-амино-3-дифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((S)-5-амино-3-дифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты;
[6-((R)-5-амино-3-дифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-цианопиридин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3,5-диметилпиразин-2-карбоновой кислоты;
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-амино-5-(3-фторпропокси)пиразин-2-карбоновой кислоты и
[6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-амино-5-трифторметилпиразин-2-карбоновой кислоты.
9. Соединение по п.1, которое представляет собой [6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 5-циано-3-метилпиридин-2-карбоновой кислоты, имеющий следующую формулу:

или его фармацевтически приемлемая соль.
10. Соединение по п.1, которое представляет собой [6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-хлор-5-трифторметилпиридин-2-карбоновой кислоты, имеющий следующую формулу:

или его фармацевтически приемлемая соль.
11. Соединение по п.1, которое представляет собой [6-((3R,6R)-5-амино-3,6-диметил-6-трифторметил-3,6-дигидро-2Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амид 3-амино-5-трифторметилпиразин-2-карбоновой кислоты, имеющий следующую формулу:

или его фармацевтически приемлемая соль.
12. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли в качестве лекарственного средства.
13. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли в лечении или профилактике болезни Альцгеймера или незначительного ухудшения познавательных способностей.
14. Фармацевтическая композиция, содержащая соединение по любому из пп.1-11 или его фармацевтически приемлемую соль в качестве активного фармацевтического ингредиента в ассоциации по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем.
15. Применение соединения по любому из пп.1-11 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики болезни Альцгеймера или незначительного ухудшения познавательных способностей.
16. Фармацевтическая комбинация, включающая терапевтически эффективное количество соединения по любому из пп.1-11 или его фармацевтически приемлемой соли и второе лекарственное средство, выбранное из ингибиторов ацетилхолинэстеразы, антагонистов глутамата, препаратов антидепрессантов, анксиолитических средств, антипсихотических препаратов, стабилизаторов настроения, никотиновых альфа-7 агонистов, антагонистов mGluR5, антагонистов Н3, амилоидных терапевтических вакцин, антидиабетических средств, гиполипидемических средств, средств против ожирения, гипотензивных средств и агонистов рецепторов активатора пролиферации пероксисомы, для одновременного или последовательного введения.
Текст
Изобретение относится к новым гетероциклическим соединениям формулы (I), в которой все переменные имеют значения, определенные в описании изобретения, к фармацевтическим композициям на их основе, к их комбинациям и к их применению в качестве лекарственных средств,в частности, для лечения болезни Альцгеймера или диабета путем ингибирования ВАСЕ-1 или ВАСЕ-2. Область изобретения Изобретение относится к новым гетероциклическим производным и их фармацевтически приемлемым солям, к фармацевтическим композициям на их основе, к фармацевтическим комбинациям таких соединений и к их применению в качестве лекарственных средств, в частности, для лечения нейродегенеративного расстройства через ингибирование ВАСЕ-1 или диабета через ингибирование ВАСЕ-2. Предпосылки изобретения Болезнь Альцгеймера представляет собой разрушительное нейродегенеративное расстройство. Его спорадические формы поражают пожилых людей (резкое повышение случаев заболевания в возрасте 75 лет), кроме того, существуют различные семейные формы с началом развития заболевания в четвертой или пятой декаде жизни. Патологически, оно характеризуется присутствием внеклеточных сенильных бляшек и внутриклеточных нейрофибриллярных сплетений в головном мозге пациента. Основными составляющими элементами сенильных бляшек являются малые 4 кДа амилоидные пептиды. Они образуются в результате протеолитического процессинга большого трансмембранного белка, амилоидного белка-предшественника (АРР). Расщепление АРР бета-секретазой (ВАСЕ-1) высвобождает растворимый АРР-бета фрагмент, тогда как С-конец длиной 99-аминокислот остается связанным с мембраной. Этот Сконцевой фрагмент далее подвергается протеолитическому процессингу под действием гамма-секретазы(мембранный мультиферментный комплекс) с образованием амилоидных пептидов различной длины,преимущественно длиной 40 и 42 аминокислот (Hardy J., Selkoe D.J. (2002) Science; 297 (5580): 353-356). Если при патологических состояниях образование этих пептидов происходит с повышенной скоростью, или если их удаление из головного мозга нарушается, повышенные концентрации амилоидных пептидов в головном мозге приводят к образованию олигомеров, фибриллов и, в конечном счете, бляшек(Farris W., et al. (2007) Am. J. Pathol.; 171 (1): 241-251). Было показано, что отложение амилоидных пептидов и бляшек в головном мозге является первым измеримым событием в патогенезе болезни Альцгеймера и что это является пусковым механизмом для потери синапсов, синаптических контактов и нейронов (Grimmer T., et al. (2009) Neurobiology of Aging; 30 (12): 1902-1909). Атрофия головного мозга, вызванная массивной потерей нейронов, сопровождается ухудшением познавательной способности, памяти, ориентации и способности выполнять обычные бытовые задачи, т.е. клинически проявляется деменция (Okello A., et al. (2009) Neurology; 73 (10): 754-760). ВАСЕ-1, также известный как Asp2 или мемапсин 2, представляет собой трансмембранную аспарагинпротеазу, экспрессируемую в большом количестве в нейронах. Она локализуется совместно с ее субстратом АРР в Гольджи и эндоциотозных компартментах (Willem M., Lammich S., Haass С. (2009) Semin.Cell Dev. Biol; 20 (2): 175-182). Исследования мышей с "нокаутом" показали отсутствие образования амилоидного пептида, при этом животные были здоровыми и фертильными (Ohno М., et al. (2007) Neurobiol.Dis.; 26 (1): 134-145). Генетическое удаление ВАСЕ-1 у мышей, у которых имеет место гиперэкспрессия АРР, продемонстрировало отсутствие бляшкообразования и обратное развитие когнитивных нарушений (Ohno M., et al. (2004) Neuron; 41 (1): 27-33). Уровни ВАСЕ-1 в головном мозге спорадических пациентов с болезнью Альцгеймера являются повышенными (Hampel Н., Shen Y. (2009) Scand. J.Clin. Lab. Invest; 69 (1): 8-12). Взятые вместе, эти данные говорят о том, что ингибирование ВАСЕ-1 может представлять собой полезную терапевтическую стратегию для лечения болезни Альцгеймера. Расщепляющий бета-сайт амилоидного белка-предшественника фермент 2 (ВАСЕ-2) представляет собой трансмембранную аспарагинпротеазу, которая на высоком уровне экспрессируется в панкреатических -клетках и других периферических тканях (Brian D. Bennett, Safura Babu-Khan, Richard Loeloff,Jean-Claude Louis, Eileen Curran; Martin Citron, and Robert Vassar (2000) JJ. Biol. Chem. 275 (27) 2064720651). BACE-2 является близкородственным ВАСЕ-1 или бета-секретазе. Однако, не смотря на схожесть структур и последовательностей, оказалось, что специфичность в отношении субстрата у ВАСЕ-1 и ВАСЕ-2 разная. В то время как A или -амилоидный пептид является основным субстратом ВАСЕ-1,ВАСЕ-2 не образует ни одну из форм A (Vassar, R., Bennett, В.D., Babu-Khan, S., Kahn, S., Mendiaz, E.Citron, M. (1999) Science 286, 735-741). Трансмембранный белок 27 (TMEM27 или коллектрин) играет важную роль в -клеточной пролиферации и секреции инсулина (Pinar Akpinar, Satoru Kuwajima, Jan Krutzfeldt, and Markus Stoffel (2005)Tmem27: Cell Metabolism. 2(6) 385-397), и он был идентифицирован как субстрат для ВАСЕ-2 (WO 2010/063718). Tmem27 существует в виде димера, и внеклеточный домен расщепляется и выделяется из плазмы -клеточно-специфическим образом. Сверхэкспрессия непроцессированного Tmem27, но не усеченного или растворимого белка, повышает -клеточную пролиферацию, и это говорит о том, что непроцессированный белок необходим для этой биологической функции. Tcf1 (гепатоцитарный нуклеарный фактор-1, HNF-1) контролирует транскрипцию Tmem27. Мыши с прицельно направленной делецией панкреатических -клетках демонстрируют повышенную -клеточную массу по сравнению с мышами дикого типа. Эти данные показывают, что Tmem27 играет роль в контроле -клеточной массы и что ингибирование ВАСЕ-2, который расщепляет Tmem27, могло бы быть полезным для лечения потери клеточной массы и функции, причин, лежащих в основе диабета. Взятые вместе, эти данные говорят о том, что ингибирование ВАСЕ-2 может представлять собой полезную терапевтическую стратегию для лечения и профилактики метаболических расстройств, связанных с пониженной -клеточной массой и/или функцией, таких как диабет 2 типа. Краткое описание изобретения Настоящее изобретение относится к новым гетероциклическим производным, обладающим активностью ингибирования ВАСЕ, к их получению, к их медицинскому применению и к включающим их лекарственным средствам. Более конкретно, в первом аспекте изобретение относится к соединению формулы (Ic)X1 представляет собой СН или N; Х 3 представляет собой СН или N; Х 4 представляет собой CR4 или N; где один и не более чем один из X1, X3 и Х 4 представляет собой N;R2 представляет собой пиридильную или пиразинильную группу, которая замещена 1, 2 или 3 заместителями, и где один из заместителей расположен в пара-положении пиридильной или пиразинильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, галоген, (С 1-4)алкил, галоген-(С 1-4)алкил, гидрокси, оксо, (C1-4)алкокси и галоген-(C1-4)алкокси;R4 и R5 независимо представляют собой водород или галоген;R6 представляет собой (С 1-3)алкил или фтор-(C1-3)алкил; и каждый из R11 и R12 независимо выбраны из группы, включающей водород, (C1-3)алкил или фтор(C1-3)алкил,или его фармацевтически приемлемой соли. Во втором аспекте изобретение относится к применению соединения формулы Ic или его фармацевтически приемлемой соли в качестве лекарственного средства. В следующем аспекте изобретение относится к применению соединения формулы Ic или его фармацевтически приемлемой соли в лечении или профилактике болезни Альцгеймера или незначительного ухудшения познавательных способностей. В следующем аспекте изобретение относится к фармацевтической композиции, содержащей соединение формулы Ic или его фармацевтически приемлемую соль в качестве активного фармацевтического ингредиента в ассоциации по меньшей мере c одним фармацевтически приемлемым носителем или разбавителем. В еще одном аспекте изобретение относится к применению соединения формулы Ic или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики болезни Альцгеймера или незначительного ухудшения познавательных способностей. В следующем аспекте изобретение относится к фармацевтической комбинации, включающей терапевтически эффективное количество соединения формулы Ic или его фармацевтически приемлемой соли и второе лекарственное средство, выбранное из ингибиторов ацетилхолинэстеразы, антагонистов глутамата, препаратов антидепрессантов, анксиолитических средств, антипсихотических препаратов, стабилизаторов настроения, никотиновых альфа-7 агонистов, антагонистов mGluR5, антагонистов Н 3, амилоидных терапевтических вакцин, антидиабетических средств, гиполипидемических средств, средств против ожирения, гипотензивных средств и агонистов рецепторов активатора пролиферации пероксисомы, для одновременного или последовательного введения. Определения Галоген означает фтор, хлор, бром или йод. Галогенированная группа или фрагмент, такой как галогеналкил, может быть моно-, ди-, три-, полиили пер-галогенированным. Любая нециклическая углеродсодержащая группа или фрагмент с более чем 1 атомом углерода является линейной или разветвленной. Термин "алкокси" означает алкильную группу, когда она связана посредством кислорода. Подробное описание изобретения Настоящее изобретение обеспечивает соединения формулы (Ic), определенные выше, и фармацев-2 024059 тические композиции на их основе, которые могут быть полезными в лечении или профилактике заболеваний, состояний и/или расстройств, модулируемых путем ингибирования ВАСЕ. Благодаря одному или более чем одному асимметрическому атому углерода, который может присутствовать в соединении формулы Ic, соответствующее соединение формулы Ic может существовать в чистой оптически активной форме или в форме смеси оптических изомеров, например, в виде рацемической смеси. Все из таких чистых оптических изомеров и все из их смесей, включая рацемические смеси,являются частью настоящего изобретения. В одном варианте воплощения изобретение, соответственно, относится к соединению формулы Id или его фармацевтически приемлемой солиX1 представляет собой СН или N; Х 3 представляет собой СН или N; Х 4 представляет собой CR4 или N; где один и не более чем один из X1, X3 и Х 4 представляет собой N;R2 представляет собой пиридильную или пиразинильную группу, которая замещена 2 или 3 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридильной или пиразинильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, галоген, (C1-4)алкил, галоген-(C14)алкил, гидрокси, оксо, (C1-4)алкокси и галоген-(C1-4)алкокси;R4 и R5 независимо представляют собой водород или галоген;R6 представляет собой метил, фторметил, дифторметил или трифторметил; и каждый из R11 и R12 независимо выбран из группы, включающей водород, метил, фторметил, дифторметил и трифторметил. В одном варианте воплощения изобретение, соответственно, относится к соединению формулы Ic или Id или его фармацевтически приемлемой соли, где R2 представляет собой пиридин-2-ильную или пиразин-2-ильную группу, которая замещена 2 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридин-2-ильной или пиразин 2-ильной группы относительно амидного линкера, и где заместители независимо выбраны из группы,включающей циано, амино, фтор, бром, хлор, гидроксил, оксо, метил, фторметил, дифторметил, трифторметил, метокси, фторметокси, дифторметокси и трифторметокси. В другом варианте воплощения изобретение, соответственно, относится к соединению формулы Ic или Id или его фармацевтически приемлемой соли, где X1 представляет собой N; Х 3 представляет собой СН и Х 4 представляет собой CR4. В другом варианте воплощения изобретение, соответственно, относится к соединению формулы Ic или Id или его фармацевтически приемлемой соли, где R4 представляет собой водород. В одном варианте воплощения изобретение, соответственно, относится к соединению формулы Ie или его фармацевтически приемлемой солиR2 представляет собой пиридин-2-ильную или пиразин-2-ильную группу, которая замещена 2 заместителями, и где один из заместителей расположен в пара-положении и один из заместителей расположен в орто-положении пиридин-2-ильной или пиразин-2-ильной группы относительно амидного линкера, и где заместители независимо выбраны из группы, включающей циано, амино, фтор, бром,хлор, гидроксил, оксо, метил, фторметил, дифторметил, трифторметил, метокси, фторметокси,дифторметокси и трифторметокси;R5 представляет собой водород или фтор;R6 представляет собой метил, фторметил или дифторметил; и каждый из R11 и R12 независимо выбран из группы, включающей водород, метил, фторметил, дифторметил и трифторметил. В другом варианте воплощения изобретение, соответственно, относится к соединению формулы Ic,Id, Ie или его фармацевтически приемлемой соли, где R5 представляет собой фтор. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде вы-3 024059 деленного стереоизомера, где соединение содержит один стереоцентр, и стереоизомер находится в R конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде выделенного стереоизомера, где соединение содержит один стереоцентр, и стереоизомер находится в S конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде выделенного стереоизомера, где соединение содержит два стереоцентра, и стереоизомер находится в R-R конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде выделенного стереоизомера, где соединение содержит два стереоцентра, и стереоизомер находится в R-S конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде выделенного стереоизомера, где соединение содержит два стереоцентра, и стереоизомер находится в S-R конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами в виде выделенного стереоизомера, где соединение содержит два стереоцентра, и стереоизомер находится в S-S конфигурации. В одном варианте воплощения обеспечивается соединение в соответствии с примерами, где соединение содержит один или два стереоцентра, в виде рацемической смеси. Как используется в настоящем изобретении, термин "изомеры" относится к разным соединениям,которые имеют одинаковую молекулярную формулу, но различаются по расположению и конфигурации атомов. Также, как это используется в настоящем изобретении, термин "оптический изомер" или "стереоизомер" относится к любой из различных стереоизомерных конфигураций, которая может существовать для данного соединения по настоящему изобретению, и включает геометрические изомеры. Должно быть понятно, что заместитель может быть присоединен по хиральному центру атома углерода. Соответственно, изобретение включает энантиомеры, диастереомеры или рацематы соединения."Энантиомеры" представляют собой пару стереоизомеров, которые являются не совпадающими при наложении зеркальными отображениями друг друга. 1:1 смесь пары энантиомеров представляет собой"рацемическую" смесь. Этот термин используется для обозначения рацемической смеси, где это является подходящим. "Диастереоизомеры" представляют собой стереоизомеры, которые содержат, по меньшей мере, два асимметрических атома, но которые не являются зеркальными отображениями друг друга. Абсолютную стереохимию определяют в соответствии с R-S системой Cahn-Ingold-Prelog. Когда соединение представляет собой чистый энантиомер, стереохимия по каждому хиральному углероду может быть указана либо как R, либо как S. Разделенные соединения, абсолютная конфигурация которых неизвестна,могут быть обозначены как (+) или (-), в зависимости от направления (право- или левовращательное), в котором они вращают плоскость поляризованного света при длине волны D линии натрия. Некоторые из соединений, описанных в настоящей заявке, содержат один или несколько асимметрических центров или осей и, таким образом, могут обуславливать образование энантиомеров, диастереомеров и других стереоизомерных форм, которые могут быть определены, на основании абсолютной стереохимии, как (R)или (S)-. Предполагается, что настоящее изобретение включают все такие возможные изомеры, включая рацемические смеси, оптически чистые формы и промежуточные смеси. Оптически активные (R)- и (S)изомеры можно получить с использованием хиральных синтонов или хиральных реагентов, или путем разделения с использованием традиционных способов. Когда соединение содержит двойную связь, заместитель может иметь Е или Z конфигурацию. Соединение формулы Ic может существовать в таутомерной форме. Все такие таутомеры являются частью настоящего изобретения. Соединение формулы Ic может существовать в свободной форме или в форме соли, например, щелочное соединение может быть в форме кислотно-аддитивной соли, или кислотное соединение может быть в форме соли с основанием. Все такие свободные соединения и соли являются частью настоящего изобретения. В одном варианте воплощения изобретение относится к соединению формулы Ic, Id или Ie в свободной форме. В другом варианте воплощения изобретение относится к соединению формулы Ic, Id илиIe, определенному в настоящей заявке, в форме соли. В другом варианте воплощения изобретение относится к соединению формулы Ic, Id или Ie, определенному в настоящей заявке, в форме кислотноаддитивной соли. В следующем варианте воплощения изобретение относится к соединению формулы IIe,Id или Ie, определенному в настоящей заявке, в форме фармацевтически приемлемой соли. Еще в одном варианте воплощения изобретение относится к соединению формулы Ic, Id или Ie, определенному в настоящей заявке, в форме гидрохлоридной соли. Еще в одном варианте воплощения изобретение относится к любому из соединений примеров в свободной форме. Еще в одном варианте воплощения изобретение относится к любому из соединений примеров в форме соли. Еще в одном варианте воплощения изобретение относится к любому из соединений примеров в форме кислотно-аддитивной соли. Еще в одном варианте воплощения изобретение относится к любому из соединений примеров в форме фармацевтически приемлемой соли. Еще в одном варианте воплощения изобретение относится к любому из соедине-4 024059 ний примеров в форме гидрохлоридной соли. Как это используется в настоящем изобретении, термины "соль" или "соли" относятся к кислотноаддитивной или основно-аддитивной соли соединения по настоящему изобретению. "Соли" включают, в частности, "фармацевтически приемлемые соли". Термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению, и которые типично не являются биологически или иным образом нежелательными. Во многих случаях соединения по настоящему изобретению способны к образованию кислотных и/или основных солей, благодаря присутствию амино и/или карбоксильных групп или групп, подобных указанным выше группам. Фармацевтически приемлемые кислотно-аддитивные соли могут быть образованы с неорганическими кислотами и органическими кислотами, например, такие соли, как ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глуцептат, глюконат, глюкуронат, гиппурат, гидроиодид/иодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат,сульфосалицилат, тартрат, тозилат и трифторацетат. Неорганические кислоты, из которых могут быть образованы соли, включают, например, хлористо-водородную кислоту, бромисто-водородную кислоту,серную кислоту, азотную кислоту и фосфорную кислоту. Органические кислоты, из которых могут быть образованы соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту,щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту,винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту и сульфосалициловую кислоту. Фармацевтически приемлемые основно-аддитивные соли могут быть образованы с неорганическими и органическими основаниями. Неорганические основания, из которых могут быть образованы соли, включают,например, аммониевые соли и металлы из колонок I-XII Периодической таблицы. В некоторых вариантах воплощения соли образованы из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; в частности, подходящие соли включают аммониевые, калиевые, натриевые, кальциевые и магниевые соли. Органические основания, из которых могут быть образованы соли, включают, например, первичные, вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины и основные ионообменные смолы. Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и троматамин. Фармацевтически приемлемые соли по настоящему изобретению можно синтезировать из исходного соединения, щелочной или кислотной группы, традиционными химическими способами. Как правило,такие соли можно получить путем взаимодействия формы свободной кислоты этих соединений со стехиометрическим количеством подходящего основания (такого как Na, Ca, Mg или K гидроксид, карбонат, бикарбонат или т.п.) или путем взаимодействия формы свободного основания этих соединений со стехиометрическим количеством подходящей кислоты. Такие реакции типично осуществляют в воде или в органическом растворителе, или в смеси таких двух растворителей. Как правило, использование неводных сред, таких как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил, является желательным, где это практически возможно. Перечни дополнительных подходящих солей можно найти, например, в "Remington's Pharmaceutical Sciences", 20th ed., Mack Publishing Company, Easton, Pa., (1985); и в"Handbook of Pharmaceutical Salts: Properties, Selection and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Когда присутствует и щелочная группа, и кислотная группа в одной молекуле, соединения по настоящему изобретению также могут образовывать внутримолекулярные соли, например цвиттерионные молекулы. Кроме того, соединения по настоящему изобретению, включая их соли, также можно получить в форме их гидратов, или они включают другие растворители, используемые для их кристаллизации. Соединения по настоящему изобретению могут, по своей природе или, как это специально рассчитано, образовывать сольваты с фармацевтически приемлемыми растворителями (включая воду). Термин "сольват" относится к молекулярному комплексу соединения по настоящему изобретению (включая его фармацевтически приемлемые соли) с одной или несколькими молекулами растворителя. Такие молекулы растворителей представляют собой вещества, обычно используемые в фармацевтике, которые известны как безопасные для реципиента, например, вода, этанол и т.п. Термин "гидрат" относится к комплексу,где молекула растворителя представляет собой воду. Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченные соединения формулы Ic, где один или несколько атомов заменен/заменены одним или несколькими атомами,имеющими такой же атомный номер, но имеющими атомную массу, отличную от атомной массы, обычно наблюдаемой в природе. Примеры таких изотопов включают изотопы углерода, такие как 11 С, 13 С или 14 С, хлора, такие как 36Cl, фтора, такие как 18F, брома, такие как 76Br, водорода, такие как 2 Н или 3 Н, иода,-5 024059 такие как 123I, 124I, 125I или 131I, азота, такие как 13N или 15N, кислорода, такие как 15 О, 17 О или 18 О, фосфора, такие как 32 Р или серы, такие как 35S. Изотопно-меченное соединение формулы Ic можно получить способом, аналогичным тем, которые описаны в Примерах, или обычными способами, известными специалистам в данной области, с использованием подходящего изотопно-меченного реагента или исходного вещества. Включение более тяжелого изотопа, такого как 2 Н (дейтерий или D), может обеспечить лучшую метаболическую стабильность соединению формулы Ic, что может привести, например, более продолжительному периоду полужизни соединения in vivo или к более низким необходимым дозам. Некоторые изотопно-меченные соединения формулы Ic, например соединения, включающие радиоактивный изотоп, такой как 3 Н или 14 С, можно использовать в анализах распределения лекарственного средства или субстрата в тканях. Соединения формулы Ic с позитрон-испускающим изотопом, таким как 11 С,18F, 13N или 15O, могут быть полезны в анализах с использованием позитрон-эмиссионной томографии(PET) или однофотонной эмиссионной компьютерной томографии (SPECT), например, для определения заполненности субстрата рецептором. Фармацевтически приемлемые сольваты в соответствии с изобретением включают такие сольваты,где растворитель кристаллизации может быть изотопно-замещенным, например D2O, d6-ацетон, d6DMSO. Соединения по настоящему изобретению, т.е. соединения формулы Ic, Id или Ie, которые содержат группы, способные действовать как доноры и/или акцепторы для водородных связей, могут образовывать со-кристаллы с подходящими агентами образования со-кристаллов. Такие со-кристаллы можно получить из соединений формулы Ic, Id или Ie с использованием известных процедур образования сокристаллов. Такие процедуры включают измельчение, нагревание, совместное сублимирование, совместный расплав или контактирование в растворе соединений формулы Ic, Id или Ie с агентом образования со-кристаллов в условиях кристаллизации и выделение образованных таким образом со-кристаллов. Подходящие агенты образования со-кристаллов включают вещества, описанные в WO 2004/078163. В другом варианте воплощения изобретение относится к соединению по настоящему изобретению или его фармацевтически приемлемой соли, которое выбрано из группы, включающей: В одном варианте воплощения изобретение, соответственно, относится к [6-3R,6R)-5-амино-3,6 диметил-6-трифторметил-3,6-дигидро-2 Н-[1,4]оксазин-3-ил)-5-фтор-пиридин-2-ил]амиду 5-циано-3-метилпиридин-2-карбоновой кислоты следующей формулы: или его фармацевтически приемлемой соли. В следующем варианте воплощения изобретение, соответственно, относится к [6-3R,6R)-5-амино 3,6-диметил-6-трифторметил-3,6-дигидро-2 Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амиду 3-хлор-5 трифторметилпиридин-2-карбоновой кислоты следующей формулы: или его фармацевтически приемлемой соли. В следующем варианте воплощения изобретение, соответственно, относится к [6-3R,6R)-5-амино 3,6-диметил-6-трифторметил-3,6-дигидро-2 Н-[1,4]оксазин-3-ил)-5-фторпиридин-2-ил]амиду 3-амино-5 трифторметилпиразин-2-карбоновой кислоты следующей формулы: или его фармацевтически приемлемой соли. Соединения формулы Ic можно получить в соответствии с традиционными способами, например,как описано в примерах. Обработку реакционных смесей и очистку получаемых таким образом соединений можно осуществить в соответствии с известными процедурами. Соли могут быть получены из свободных соединений известным способом, и наоборот. Соединения формулы Ic, в свободной форме, в форме соли или в форме фармацевтически приемлемой соли, далее часто указанные как "вещества по настоящему изобретению", демонстрируют ценные фармакологические свойства при испытании in vitro или in vivo и, соответственно, могут быть полезными в лекарственных средствах, в терапии или для применения в качестве химических веществ для исследований, например, в качестве инструментов для исследований. Например, вещества по настоящему изобретению являются ингибиторами ВАСЕ-1 и ВАСЕ-2 и могут быть использованы для лечения или профилактики состояния, заболевания или расстройства, включающего процессинг таких ферментов, в частности, образование бета-амилоида и последующую агрегацию в олигомеры и фибриллы и потерю -клеточной массы и/или функции. Ингибирующие свойства вещества по настоящему изобретению в отношении протеаз можно оценить в испытаниях, описанных далее в настоящей заявке. Испытание 1. Ингибирование человеческого ВАСЕ-1 Рекомбинантный ВАСЕ-1 (внеклеточный домен, экспрессируемый в бакуловирусе и очищенный с использованием стандартных способов) при концентрации 0,1-10 нМ инкубировали с испытываемым соединением при различных концентрациях в течение 1 ч при комнатной температуре в 10-100 мМ ацетатном буфере, рН 4,5, содержащем 0,1% CHAPS. Синтетический флуоресцентный-гасимый пептидный субстрат, образованный из последовательности АРР и содержащий подходящую пару флуорофоргаситель, добавляли до конечной концентрации 1-5 мкМ и усиление флуоресценции регистрировали при подходящей длине волны возбуждения/эмиссии в спектро-флуориметре для микропланшетов в течение 5-30 мин с 1-минутными интервалами. Значения ИК 50 рассчитывали из процента ингибирования активности ВАСЕ-1 как функцию концентрации испытываемого соединения. Испытание 2. Ингибирование человеческого ВАСЕ-2 Рекомбинантный ВАСЕ-2 (внеклеточный домен, экспрессируемый в бакуловирусе и очищенный с использованием стандартных способов) при концентрации 0,1-10 нМ инкубировали с испытываемым соединением при различных концентрациях в течение 1 ч при комнатной температуре в 10-100 мМ ацетатного буфера, рН 4,5, содержащего 0,1% CHAPS. Синтетический флуоресцентный-гасимый пептидный субстрат, образованный из последовательности АРР и содержащий подходящую пару флуорофоргаситель, добавляли до конечной концентрации 1-5 мкМ и усиление флуоресценции регистрировали при подходящей длине волны возбуждения/эмиссии в спектро-флуориметре для микропланшетов в течение 5-30 мин с 1-минутными интервалами. Значения ИК 50 рассчитывали из процента ингибирования активности ВАСЕ-2 как функцию концентрации испытываемого соединения. Испытание 3. Ингибирование человеческого катепсина D Рекомбинантный катепсин D (экспрессируемый в виде прокатепсина D в бакуловирусе, очищенный с использованием стандартных способов и активированный путем инкубации в натрийформиатном буфере рН 3,7), инкубировали с испытываемым соединением при различных концентрациях в течение 1 ч при комнатной температуре в натрийформиатном или натрийацетатном буфере при подходящем уровне рН, находящемся в пределах рН 3,0-5,0. Синтетический пептидный субстрат Mca-Gly-Lys-Pro-Ile-LeuPhe-Phe-Arg-Leu-Lys(DNP)-D-Arg-NH2 добавляли до конечной концентрации 1-5 мкМ и усиление флуоресценции регистрировали при длине волны возбуждения 325 нМ и длине волны эмиссии 400 нм в спектро-флуориметре для микропланшетов в течение 5-30 мин с 1-минутными интервалами. Значения ИК 50 рассчитывали из процента ингибирования активности катепсина D как функцию концентрации испытываемого соединения. Испытание 4. Ингибирование клеточного высвобождения амилоидного пептида 1-40 Клетки яичников китайского хомячка трансфицировали человеческим геном для амилоидного белка-предшественника. Клетки высевали при плотности 8000 клеток/лунка в 96-луночные микротитровальные планшеты и культивировали в течение 24 ч в клеточной культуральной среде DMEM, содержащей 10% FCS. Испытываемое соединение добавляли к клеткам при различных концентрациях и клетки культивировали в течение 24 ч в присутствии испытываемого соединения. Надосадочные жидкости собирали и концентрацию амилоидного пептида 1-40 определяли с использованием известных методов иммуноанализа, например, сэндвичевого анализа ELISA, гомогенного флуоресцентного иммуноанализа с разрешением по времени (HTRF) или электро-хемилюминесцентного иммуноанализа. Активность соединения рассчитывали из процента ингибирования высвобождения амилоидного пептида как функции концентрации испытываемого соединения. Вещества по настоящему изобретению были испытаны по меньшей мере в одном из описанных выше испытаний. Соединения примеров показали следующие средние значения ИК 50 в испытании 1, описанном выше. Таблица 1 Соединения примеров показали следующие средние значения ИК 50 в испытании 2, описанном выше. Соединения примеров показали следующие средние значения ИК 50 в испытании 4, описанном выше. Таблица 3 Как используется в настоящем изобретении, термин "фармацевтически приемлемый носитель" включает любой и все растворители, среды для диспергирования, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные средства, противогрибковые средства), изотонические вещества, вещества, замедляющие абсорбцию, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие, эксципиенты, разрыхлители, смазывающие вещества, подсластители, отдушки, красители и т.п. и комбинации таких веществ, как должно быть известно специалистам в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. MackPrinting Company, 1990, pp. 1289-1329). За исключением тех случаев, когда какой-либо традиционный носитель несовместим с активным ингредиентом, предусматривается его использование в терапевтических или фармацевтических композициях. Термин "терапевтически эффективное количество" соединения по настоящему изобретению отно- 10024059 сится к количеству соединения по настоящему изобретению, которое будет вызывать биологический или медицинский ответ у субъекта, например, уменьшение или ингибирование активности фермента или белка или уменьшение тяжести симптомов, облегчение состояний, замедление или отсрочку развития заболевания или предотвращение развития заболевания, и т.п. В одном неограничивающем варианте воплощения, термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое, при введении субъекту, является эффективным для (1) по меньшей мере частичного облегчения, ингибирования, профилактики и/или уменьшения тяжести состояния или расстройства или заболевания, (i) опосредованного ВАСЕ-1 или (ii) связанного с активностью ВАСЕ-1 или(iii) характеризующегося активностью (нормальной или аномальной) ВАСЕ-1; или (2) снижения или ингибирования активности ВАСЕ-1. В другом неограничивающем варианте воплощения, термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое, при введении в клетку или ткань, или в не-клеточный биологический материал или среду, является эффективным для, по меньшей мере, частичного снижения или ингибирования активности ВАСЕ-1. Значение термина "терапевтически эффективное количество", проиллюстрированное в описанных выше вариантах воплощения для ВАСЕ-1, также применимо, при использовании таких же средств, к любым другим релевантным белкам/пептидам/ферментам, таким как ВАСЕ-2 или катепсин D. Как используется в настоящем изобретении, термин "субъект" относится к животному. Типично,животное представляет собой млекопитающее. Субъект также относится, например, к приматам (например, человеку мужского или женского пола), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т.п. В некоторых вариантах воплощения, субъектом является примат. В других вариантах воплощения, субъектом является человек. Как используется в настоящем изобретении, термин "ингибировать", "ингибирование" или "ингибирующий" относится к ослаблению или подавлению определенного состояния, симптома или расстройства или заболевания, или к существенному снижению базовой линии биологической активности или процесса. Как используется в настоящем изобретении, термин "лечить", "лечащий" или "лечение" какого-либо заболевания или расстройства относится, в одном варианте воплощения, к уменьшению тяжести заболевания или расстройства (т.е. замедлению или остановке или уменьшению развития заболевания или по меньшей мере одного из его клинических симптомов). Еще в одном варианте воплощения, "лечить", "лечащий" или "лечение" относится к модуляции заболевания или расстройства, либо физически (например,стабилизация различимого симптома), либо физиологически (например, стабилизация физического параметра), либо и тем и другим образом. Как используется в настоящем изобретении, термин "профилактика" любого конкретного заболевания или расстройства относится к введению соединения по настоящему изобретению субъекту до очевидного проявления симптомов такого заболевания или расстройства. Как это используется в настоящем изобретении, субъект "нуждается в" лечении, если этот субъект может получить пользу, с точки зрения биологической, медицинской или качества жизни, от такого лечения. Как используется в настоящем изобретении, термин "вещество" по настоящему изобретению используется взаимозаменяемым образом с термином "соединение" по настоящему изобретению и не отличается от него по смыслу. Как это используется в настоящем изобретении, неопределенные и определенные артикли и подобные термины, используемые в контексте настоящего изобретения (особенно в контексте формулы изобретения), следует рассматривать как охватывающие формы как единственного, так и множественного числа, если не указано иное, или если контекст явно не противоречит этому. Использование любого и всех примеров или слов для представления в качестве примера (например, "такой как"), представленных в настоящей заявке, предназначено просто для более ясного представления изобретения и не налагает ограничение на объем настоящего изобретения, заявленный в формуле изобретения. Благодаря их ингибирующим свойствам в отношении протеаз, и особенно ВАСЕ-1, вещества по настоящему изобретению могут быть полезными, например, для лечения или профилактики различных приводящих к нетрудоспособности психиатрических, психотических, неврологических или сосудистых состояний, например, состояния, заболевания или расстройства сосудистой системы или нервной системы, в котором играет роль образование и агрегация бета-амилоидов. Для указанных выше показаний подходящая доза будет варьироваться в зависимости от, например,соединения, используемого в качестве активного фармацевтического ингредиента, хозяина, способа введения, природы и тяжести состояния, заболевания или расстройства или желаемого эффекта. Однако, в основном, было показано, что удовлетворительные результаты у животных можно получить при суточной дозе от около 0,1 до около 100, предпочтительно от около 1 до около 50 мг/кг массы тела животного. Для более крупных млекопитающих, например человека, показанная суточная доза находится в пределах от около 0,5 до около 2000, предпочтительно от около 2 до около 200 мг средства по настоящему изобретению, которую удобным образом вводят, например, в виде дробных доз до четырех раз в день или в форме замедленного высвобождения. Средство по настоящему изобретению можно вводить любым традиционным путем, в частности энтерально, предпочтительно перорально, например, в форме таблетки или капсулы, или парентерально,например, в форме раствора или суспензии для инъекций. В следующем аспекте изобретение относится к фармацевтической композиции, включающей средство по настоящему изобретению в качестве активного фармацевтического ингредиента в ассоциации с,по меньшей мере, одним фармацевтически приемлемым носителем или разбавителем и, необязательно, в ассоциации с другими вспомогательными веществами, такими как ингибиторы ферментов цитохром Р 450, вещества, препятствующие разложению активных фармацевтических ингредиентов цитохромом Р 450, вещества, улучшающие или повышающие фармакокинетические свойства активных фармацевтических ингредиентов, вещества, улучшающие или повышающие биодоступность активных фармацевтических ингредиентов, и т.п., например, грейпфрутовый сок, кетоконазол или, предпочтительно, ритонавир. Такую композицию можно получить традиционным способом, например, путем смешивания ее компонентов. Стандартные лекарственные формы содержат, например, от около 0,1 до около 1000, предпочтительно от около 1 до около 500 мг вещества по настоящему изобретению. Кроме того, фармацевтические композиции по настоящему изобретению могут быть получены в твердой форме (включая, без ограничения, капсулы, таблетки, пилюли, гранулы, порошки или суппозитории) или в жидкой форме (включая, без ограничения, растворы, суспензии или эмульсии). Фармацевтические композиции могут подвергаться традиционным фармацевтическим процедурам, таким как стерилизация, и/или могут содержать традиционные инертные разбавители, смазывающие вещества или буферные вещества, а также адъюванты, такие как консерванты, стабилизаторы, смачивающие вещества,эмульгаторы и буферы, и т.п. Типично, фармацевтические композиции представляют собой таблетки или желатиновые капсулы,включающие активный ингредиент вместе с а) разбавителями, например лактозой, декстрозой, сахарозой, маннитом, сорбитом, целлюлозой и/или глицином;b) смазывающими веществами, например диоксидом кремния, тальком, стеариновой кислотой, ее магниевой или кальциевой солью и/или полиэтиленгликолем; для таблеток такжеc) связующими, например магний алюмосиликатом, крахмальной пастой, желатином, трагакантом,метилцеллюлозой, натрий карбоксиметилцеллюлозой и/или поливинилпирролидоном; если желательно,d) разрыхлителями, например крахмалами, агаром, альгиновой кислотой или ее натриевой солью,или шипучими смесями; и/илиe) абсорбентами, красителями, отдушками и подсластителями. Таблетки могут иметь либо пленочное покрытие, либо энтеросолюбильное покрытие, нанесенное в соответствии со способами, известными из уровня техники. Подходящие композиции для перорального введения включают эффективное количество соединения по настоящему изобретению в форме таблеток, лепешек, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсии, твердых или мягких капсул или сиропов или эликсиров. Композиции, предназначенные для перорального применения, получают в соответствии с любым способом,известным из уровня техники для получения фармацевтических композиций, и такие композиции могут содержать одно или несколько веществ, выбранных из группы, включающей подсластители, отдушки,красители и консерванты, для обеспечения фармацевтически привлекательных и имеющих приятный вкус препаратов. Таблетки могут содержать активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые являются подходящими для получения таблеток. Эти эксципиенты включают, например, инертные разбавители, такие как карбонат кальция, карбонат натрия,лактозу, фосфат кальция или фосфат натрия; гранулирующие вещества и разрыхлители, например, кукурузный крахмал или альгиновую кислоту; связующие, например, крахмал, желатин или аравийскую камедь; и смазывающие вещества, например, стеарат магния, стеариновую кислоту или тальк. Таблетки могут быть без покрытия или могут иметь покрытие, нанесенное известными способами, для замедления разложения и абсорбции в желудочно-кишечном тракте и обеспечения, таким образом, продолжительного действия в течение более длительного периода времени. Например, можно использовать замедляющие действие вещества, такие как глицерилмоностеарат или глицерилдистеарат. Композиции для перорального применения могут быть представлены в виде твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный ингредиент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом. Некоторые композиции для инъекций представляют собой водные изотонические растворы или суспензии, и суппозитории преимущественно получают из жирных эмульсий или суспензий. Указанные композиции могут быть стерилизованы и/или могут содержат адъюванты, такие как консерванты, стабилизаторы, смачивающие вещества или эмульгаторы, вещества, способствующие растворению, соли для регулирования осмотического давления и/или буферы. Кроме того, они также могут содержать другие терапевтически ценные вещества. Указанные композиции получают в соответствии с традиционными способами смешивания, гранулирования или нанесения покрытия, соответственно, и они содержат около 0,1-75% или содержат около 1-50% активного ингредиента. Подходящие композиции для чрескожного применения включают эффективное количество соединения по настоящему изобретению с подходящим носителем. Носители, подходящие для чрескожной доставки, включают абсорбируемые фармакологически приемлемые растворители, способствующие прохождению через кожу хозяина. Например, чрескожные устройства имеют форму повязки, включающей подложку, резервуар, содержащий соединение, необязательно с носителями, необязательно контролирующий скорость барьер для доставки соединения в кожу хозяина с контролируемой и предварительно установленной скоростью в течение продолжительного периода времени и средства для удерживания устройства на коже. Подходящие композиции для местного применения, например, для кожи и глаз, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, для доставки посредством аэрозоля или т.п. Такие системы местной доставки являются особенно подходящими для кожного применения, например, для лечения рака кожи, например, для профилактического применения в солнцезащитных кремах, лосьонах спреях и т.п. Они, таким образом, являются особенно подходящими для применения в местных, включая косметические, композициях, хорошо известных в данной области техники. Такие композиции могут содержать солюбилизаторы, стабилизаторы, усиливающие тоничность вещества, буферы и консерванты. Как это используется в настоящем изобретении, местное применение также может относиться к ингаляции или к интраназальному применению. Доставку средства удобно обеспечить в форме сухого порошка (либо отдельно, либо в виде смеси, например, сухой смеси с лактозой или смешиваемым компонентом в форме частиц, например, с фосфолипидами) из ингалятора сухого порошка или аэрозольного спрея из находящегося под давлением контейнера, насоса, спрея, распылителя или небулайзера, с или без использования подходящего пропеллента. Настоящее изобретение, кроме того, обеспечивает безводные фармацевтические композиции и лекарственные формы, включающие соединения по настоящему изобретению в качестве активных ингредиентов, поскольку вода может способствовать разложению некоторых соединений. Безводные фармацевтические композиции и лекарственные формы по настоящему изобретению можно получить с использованием безводных или содержащих незначительные количества влаги ингредиентов и условий низкой влажности. Безводную фармацевтическую композицию можно получить и хранить таким образом, чтобы сохранялась ее безводная природа. Соответственно, безводные композиции упаковывают с использованием веществ, известных как вещества, защищающие от воздействия воды, так чтобы их можно было включить в подходящие фармакологические наборы. Примеры подходящей упаковки включают, но не ограничиваются этим, герметичные упаковки из фольги, пластиков, однодозовые контейнеры (например, ампулы), блистерные упаковки и контурные упаковки. Изобретение, кроме того, обеспечивает фармацевтические композиции и лекарственные формы, которые включают одно или несколько веществ, которые уменьшают скорость, с которой соединение по настоящему изобретению в качестве активного ингредиента будет разлагаться. Такие вещества, которые указаны в настоящем изобретении как "стабилизаторы", включают, но не ограничиваются этим, антиоксиданты, такие как аскорбиновая кислота, рН буферы или солевые буферы и т.п. В соответствии с вышеизложенным, в следующем аспекте изобретение относится к веществу по настоящему изобретению для применения в качестве лекарственного средства, например, для лечения или профилактики неврологического или сосудистого состояния, заболевания или расстройства, в котором играет роль образование или агрегация бета-амилоидов, или для подавления процесса метастазирования,связанного с опухолевыми клетками, или для лечения или профилактики потери -клеточной массы и/или функции. В одном варианте воплощения изобретение относится к веществу по настоящему изобретению для применения в лечении заболевания или расстройства, опосредованного активностью ВАСЕ-1, ВАСЕ-2 или катепсина D. В другом варианте воплощения изобретение относится к веществу по настоящему изобретению для применения в лечении или профилактике болезни Альцгеймера или некоторого ухудшения познавательных способностей. В следующем варианте воплощения изобретение относится к веществу по настоящему изобретению для применения в лечении или профилактике инсулинорезистентности, непереносимости глюкозы, диабета 2 типа, ожирения, гипертензии или диабетических осложнений. Еще в одном варианте воплощения изобретение относится к соединению по настоящему изобретению для применения в лечении нарушенной толерантности к глюкозе или диабета 2 типа. В следующем аспекте изобретение относится к применению вещества по настоящему изобретению в качестве активного фармацевтического ингредиента в лекарственном средстве, например, для лечения или профилактики неврологического заболевания или расстройства, в котором играет роль образование или агрегация бета-амилоидов. В одном варианте воплощения изобретение относится к применению вещества по настоящему изобретению в качестве активного фармацевтического ингредиента в лекарственном средстве для лечения или профилактики болезни Альцгеймера или незначительного ухудшения познавательных способностей. В следующем аспекте изобретение относится к применению вещества по настоящему изобретению для получения лекарственного средства для лечения или профилактики неврологического заболевания или расстройства, в котором играет роль образование или агрегация бета-амилоидов. В одном варианте воплощения изобретение относится к применению вещества по настоящему изобретению для получения лекарственного средства для лечения или профилактики болезни Альцгеймера или незначительного ухудшения познавательных способностей. Средство по настоящему изобретению с можно вводить в виде отдельного активного фармацевтического ингредиента или в комбинации по меньшей мере с одним другим активным фармацевтическим ингредиентом, эффективным, например, для лечения или профилактики неврологического заболевания или расстройства, в котором играет роль образование или агрегация бета-амилоидов. Такая фармацевтическая комбинация может представлять собой стандартную лекарственную форму, где такая стандартная лекарственная форма включает предварительно определенное количество каждого по меньшей мере из двух активных компонентов в ассоциации по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем. Альтернативно, фармацевтическая комбинация может быть в виде упаковки, включающей по меньшей мере два активных компонента отдельно, например, упаковка или дозирующее устройство, адаптированное для одновременного или отдельного введения по меньшей мере двух активных компонентов, где эти активные компоненты расположены отдельно. В следующем аспекте изобретение относится к таким фармацевтическим комбинациям. В следующем аспекте изобретение, соответственно, относится к фармацевтической комбинации,включающей терапевтически эффективное количество вещества по настоящему изобретению и второе лекарственное вещество, для одновременного или последовательного введения. В одном варианте воплощения изобретение обеспечивает продукт, включающий средство по настоящему изобретению и по меньшей мере одно другое терапевтическое средство в виде комбинированного препарата для одновременного, отдельного или последовательного применения в терапии. В одном варианте воплощения терапия представляет собой лечение болезни Альцгеймера или незначительного ухудшения познавательных способностей. В одном варианте воплощения изобретение обеспечивает фармацевтическую композицию, включающую средство по настоящему изобретению и другое терапевтическое средство(средства). Необязательно, фармацевтическая композиция может включать фармацевтически приемлемый эксципиент, как описано выше. В одном варианте воплощения изобретение обеспечивает набор, включающий две или более отдельные фармацевтические композиции, по меньшей мере одна из которых содержит средство по настоящему изобретению. В одном варианте воплощения набор включает средства для отдельного содержания указанных композиций, такие как контейнер, разделенная на секции бутыль или разделенная на секции упаковка из фольги. Примером такого набора является блистерная упаковка, типично используемая для упаковки таблеток, капсул и т.п. Набор по настоящему изобретению можно использовать для введения различных лекарственных форм, например, перорально и парентерально, для введения отдельных композиций с разными интервалами дозирования или для титрования отдельных композиций против друг друга. Чтобы способствовать его соответствию, набор по настоящему изобретению типично включает указания по введению. В комбинированных терапиях по настоящему изобретению средство по настоящему изобретению и другое терапевтическое средство могут быть получены и/или сформулированы одним и тем же или разными изготовителями. Более того, соединение по настоящему изобретению и другое терапевтическое средство могут быть объединены вместе в виде комбинированной терапии: (i) до отпуска комбинированного продукта лечащим врачам (например, в случае набора, включающего соединение по настоящему изобретению и другое терапевтическое средство); (ii) самими лечащими врачами (или под руководством лечащего врача) непосредственно перед введением; (iii) в организме самих пациентов, например, при последовательном введении соединения по настоящему изобретению и другого терапевтического средства. Соответственно, изобретение обеспечивает средство по настоящему изобретению для применения в лечении болезни Альцгеймера, где лекарственное средство предназначено для введения с другим терапевтическим средством. Изобретение также обеспечивает применение другого терапевтического средства для лечения болезни Альцгеймера, где такое лекарственное средство вводят со средством по настоящему изобретению. В одном варианте воплощения изобретение относится к соединению по настоящему изобретению в комбинации с другим терапевтическим средством, где другое терапевтическое средство выбрано из следующих:(a) ингибиторы ацетилхолинэстеразы, такие как донепезил (Aricept, ривастигмин (Exelon) и галантамин (Razadyne);(b) антагонисты глутамата, такие как мемантин (Namenda);(c) препараты антидепрессантов от плохого настроения и раздражительности, такие как циталопрам(d) анксиолитические средства от беспокойства, возбужденного состояния, вербально-срывающегося поведения и резистентности, такие как лоразепам (Ativan) и оксазепам (Serax);(e) антипсихотические препараты для приема при галлюцинациях, состоянии бреда, агрессии, беспокойства, враждебности и конфликтном поведении, такие как арипипразол (Abilify), клозапин(f) стабилизаторы настроения, такие как карбамазепин (Tegretol) и дивалпроекс (Depakote);(j) амилоидные терапевтические вакцины. Таким образом, в одном варианте воплощения изобретение обеспечивает фармацевтическую композицию, включающую:i) соединение по настоящему изобретению или его фармацевтически приемлемую соль;ii) по меньшей мере одно соединение, выбранное из следующих:iii) один или несколько фармацевтически приемлемых носителей. В другом варианте воплощения изобретение относится к соединению по настоящему изобретению или его фармацевтически приемлемой соли в комбинации с другим терапевтическим средством, где другое терапевтическое средство выбрано из следующих: а) антидиабетические средства, такие как инсулин, производные и миметики инсулина; средства,усиливающие секрецию инсулина, такие как сульфонилмочевины, например, глипизид, глибурид и амарил; инсулинотропные лиганды сульфонилмочевинных рецепторов, такие как меглитиниды, например,натеглинид и репаглинид; ингибиторы протеинтирозинфосфатазы-1 В (РТР-1 В), такие как РТР-112; ингибиторы GSK3 (киназы-3 гликогенсинтазы), такие как SB-517955, SB-4195052, SB-216763, NN-57-05441 и NN-57-05445; RXR лиганды, такие как GW-0791 и AGN-194204; ингибиторы натрий-зависимых котранспортеров глюкозы, такие как Т-1095; ингибиторы гликогенфосфорилазы, такие как BAY R3401; бигуаниды такие как метформин; ингибиторы альфа-глюкозидазы, такие как акарбоза; GLP-1 (глюкагонподобный пептид-1), GLP-1 аналоги, такие как эксендин-4, и GLP-1 миметики; и ингибиторы DPPIV (дипептидилпептидазы IV), такие как вилдаглиптин;b) гиполипидемические средства, такие как ингибиторы 3-гидрокси-3-метилглутарил кофермент A(HMG-CoA) редуктазы, например, ловастатин, питавастатин, симвастатин, правастатин, церивастатин,мевастатин, велостатин, флувастатин, далвастатин, аторвастатин, розувастатин и ривастатин; ингибиторы скваленсинтазы; лиганды FXR (фарнезоидного X рецептора) и LXR (печеночного X рецептора); холестирамин; фибраты; смолы, связывающие никотиновую кислоту и желчную кислоту, такие как холестирамин; фибраты; никотиновая кислота и другие GPR109 агонисты; ингибиторы абсорбции холестерина,такие как эзетимиб; ингибиторы СЕТР (ингибиторы белков переноса сложных эфиров холестерина) и аспирин;c) средства против ожирения, такие как орлистат, сибутрамин и антагонисты каннабиноидного рецептора 1 (СВ 1), например, римонабант; иd) гипотензивные средства, например, сильные диуретики, ингибирующие повторное поглощение воды и натрия из петли Генле, такие как этакриновая кислота, фуросемид и торсемид; ингибиторы ангиотензин-превращающего фермента (АСЕ), такие как беназеприл, каптоприл, эналаприл, фосиноприл,лизиноприл, моексиприл, перинодоприл, квинаприл, рамиприл и трандолаприл; ингибиторы Na-KАТФазного мембранного насоса, такие как дигоксин; ингибиторы нейтральной эндопептидазы (NEP); ингибиторы ACE/NEP, такие как омапатрилат, сампатрилат и фасидотрил; антагонисты ангиотензина II,такие как кандесартан, эпросартан, ирбесартан, лосартан, телмисартан и валсартан, в частности, валсартан; ингибиторы ренина, такие как дитекирен, занкирен, терлакирен, алискирен, RO-66-1132 и RO-661168; блокаторы -адренергических рецепторов, такие как ацебутолол, атенолол, бетаксолол, бисопролол, метопролол, надолол, пропранолол, соталол и тимолол; инотропные средства, такие как дигоксин,добутамин и милринон; блокаторы кальциевых каналов, такие как амлодипин, бепридил, дилтиазем, фелодипин, никардипин, нимодипин, нифедипин, нисолдипин и верапамил; антагонисты рецепторов альдо- 15024059 стерона; и ингибиторы альдостеронсинтазы. е) агонисты рецепторов активатора пролиферации пероксисомы, такие как фенофибрат, пиоглитазон, розиглитазон, тезаглитазар, BMS-298585, L-796449, соединения, конкретно описанные в патентной заявке WO 2004/103995, т.е. соединения примеров 1-35 или соединения, конкретно перечисленные в пункте 21 формулы изобретения, или соединения, конкретно описанные в патентной заявке WO 03/043985, т.е. соединения примеров 1-7 или соединения, конкретно перечисленные в пункте 19 формулы изобретения, и особенно (R)-1-4-[5-метил-2-(4-трифторметилфенил)оксазол-4-илметокси]бензолсульфонил-2,3-дигидро-1 Н-индол-2-карбоновая кислота или ее соль. Таким образом, в одном варианте воплощения изобретение обеспечивает фармацевтическую композицию, включающую:i) соединение по настоящему изобретению или его фармацевтически приемлемую соль иii) по меньшей мере одно соединение, выбранное изa) антидиабетических средств,b) гиполипидемических средств,c) средств против ожирения,d) гипотензивных средств,e) агонистов рецепторов активатора пролиферации пероксисомы, иiii) один или несколько фармацевтически приемлемых носителей. Другие специфические антидиабетические соединения описаны в Patel Mona в Expert Opin InvestigDrugs, 2003, 12(4), 623-633, на фиг. 1-7. Структуру терапевтических средств, обозначенных кодовыми номерами, родовыми или торговыми названиями, можно найти в действующей редакции стандартного справочника "The Merck Index" или в базах данных, например, Patents International (например, IMS World Publications). Примеры Следующие примеры иллюстрируют настоящее изобретение, но не ограничивают его. Аббревиатуры водн. водный безв. безводный Вос трет-бутоксикарбонилESI ионизация электроспреем ЕТА этанол/конц. водн. раствор аммиака 95/5HOAt 1-гидрокси-7-азабензотриазол ВЭЖХ высоко-эффективная жидкостная хроматография ЖХ жидкостная хроматография МеОН метанол мин минут(минуты)NEt3 триэтиламин ЯМР спектрометрия ядерного магнитного резонанса орг. органическиеTBDMS третичный бутилдиметилсилил ТВМЕ трет-бутил-метиловый эфирTHF тетрагидрофуран ТСХ тонкослойная хроматографияXantphos 4,5-бис-(дифенилфосфино)-9,9-диметилксантен Методы ЯМР Протонные спектры регистрировали на ультраэкранирующем спектрометре Bruker 400 МГц, если не указано иное. Химические сдвиги указаны в миллионных долях (м.д.) относительно метанола ( 3,31),диметилсульфоксида ( 2,50) или хлороформа ( 7,29). Небольшое количество сухого образца (2-5 мг) растворяют в подходящем дейтерированном растворителе (0,7 мл). Регулировка является автоматической, и спектры получают в соответствии с обычной процедурой. Общая информация, касающаяся хроматографии ВЭЖХ способ H1 (RtH1):(4,27 г, 65,6 ммоль) в этаноле/воде (40,0/26,7 мл) добавляли карбонат аммония (21,02 г, 219,0 ммоль). Реакционную смесь перемешивали в автоклаве при 100 С в течение 17 ч, затем разбавляли при помощи Н 2 О, 1 М водного раствора NaHCO3 и EtOAc. Фазы разделяли и водную фазу повторно экстрагировали при помощи EtOAc, Et2O и DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде бледного белого вещества,которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 0,62 мин; ESIMS: 270, 272 [(М+Н)+]; 1 Н ЯМР (400 МГц, DMSO-d6):10,86 (шир.с, 1 Н), 8,48 (с, 1 Н), 7,81 (м, 1 Н), 7,64 (д,1 Н), 7,57 (д, 1 Н), 1,68 (с, 3 Н).b) ди-трет-Бутиловый эфир 4-(6-бромпиридин-2-ил)-4-метил-2,5-диоксоимидазолидин-1,3-дикарбоновой кислоты Раствор 5-(6-бромпиридин-2-ил)-5-метилимидазолидин-2,4-диона (22,8 г, 84,4 ммоль), Boc2O (58,8 мл, 55,3 г, 253,4 ммоль) и DMAP (0,516 г, 4,22 ммоль) в ТГФ (600 мл) перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали досуха, затем осуществляли поглощение смеси в EtOAc и фильтровали через диоксид кремния. Картридж с диоксидом кремния промывали при помощи EtOAc и ТГФ, объединенные фильтраты концентрировали с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 1,23 мин; ESIMS: 470, 472 [(М+Н)+]; 1 Н ЯМР (400 МГц, CD3OD):7,82 (м, 1 Н), 7,65 (м, 2 Н), 2,11 (с, 3 Н), 1,60 (с, 9 Н), 1,30 (с, 9 Н).c) 2-Амино-2-(6-бромпиридин-2-ил)пропионовая кислота Раствор ди-трет-бутилового эфира 4-(6-бромпиридин-2-ил)-4-метил-2,5-диоксоимидазолидин-1,3 дикарбоновой кислоты (31,53 г, 67,0 ммоль) в 2,5 М водном растворе NaOH (215 мл) нагревали при кипячении с обратным холодильником в течение 40 ч. Реакционную смесь разбавляли при помощи EtOAc(100 мл) и фильтровали. Фильтраты разделяли и органический слой промывали при помощи Н 2 О. Объединенные водные слои упаривали досуха с получением твердого вещества, которое суспендировали в МеОН (350 мл) и перемешивали в течение 30 мин. Суспензию фильтровали и белый осадок промывали при помощи МеОН. Фильтраты упаривали с получением бледно-оранжевого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 0,35-0,37 мин; ESIMS: 245, 247 [(М+Н)+]; 1 Н ЯМР (400 МГц, CD3OD):7,60-7,51 (м, 2 Н), 7,36 (дд, 1 Н), 1,62 (с, 3 Н).(33,8 мл, 31,8 г, 145,7 ммоль) в ацетонитриле (300 мл) и метаноле (150 мл) добавляли гидроксид тетраметиламмония (65,1 мл 25% водного раствора, 182 ммоль). Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 6,5 ч и фильтровали. Осадок промывали при помощи МеОН иCH3CN, затем упаривали с получением оранжевого твердого вещества, которое растирали в порошок сDCM. Объединенные органические фазы концентрировали с получением неочищенной 2-(6-бромпиридин-2-ил)-2-трет-бутоксикарбониламинопропионовой кислоты в виде бледно-коричневого пенистого вещества (ВЭЖХ RtH4= 0,96-0,97 мин, ESIMS: 345, 347 [(М+Н)+]). К суспензии 2-(6-бромпиридин-2-ил)-2-трет-бутоксикарбониламинопропионовой кислоты (14,1 г,40,8 ммоль) в ТГФ (150 мл) добавляли по порциям NaBH4 (3,45 г, 90,0 ммоль) при 0 С. Добавляли по каплям раствор BF3-Et2O (11,39 мл, 12,75 г, 90,0 ммоль) в течение 15 мин и реакционную смесь оставляли для перемешивания в течение 17 ч при комнатной температуре. Для взаимодействия оставшегося исходного вещества добавляли NaBH4 (1,0 г, 26,43 ммоль) и раствор BF3-Et2O (3,3 мл, 26,43 ммоль) при 0 С и реакционную смесь перемешивали при комнатной температуре еще в течение 23 ч. Добавляли МеОН и реакционную смесь перемешивали при 80 С в течение 30 мин, затем охлаждали до комнатной температуры и фильтровали. Фильтраты упаривали с получением белого пенистого вещества, которое брали для поглощения в EtOAc и 1 н водный раствор NaOH. Фазы разделяли и водную фазу экстрагировали три раза при помощи EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали с получением трет-бутилового эфира [1-(6-бромпиридин-2-ил)-2-гидрокси-1-метилэтил]карбаминовой кислоты в смеси с 2-амино-2-(6-бромпиридин-2-ил)пропан-1-олом. Эту смесь (7,5 г, 9,74 ммоль) снова подвергали процедуре введения Вос-защиты с использованием Boc2O (5,65 мл, 5,31 г, 24,34 ммоль) и гидроксида тетраметиламмония (65,1 мл 25% водного раствора, 182 ммоль) в ацетонитриле(100 мл). После перемешивания в течение 1,5 ч при комнатной температуре реакционную смесь гасили при помощи Н 2 О и разбавляли при помощи EtOAc. Фазы разделяли и водный слой два раза повторно экстрагировали при помощи EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали и растворитель удаляли с получением желтого твердого вещества, которое без дополнительной очистки подвергали процедуре удаления Вос-защиты в загрузке 8,1 г с использованием 100 мл 4 н водного раствора HCl. Реакционную смесь перемешивали при комнатной температуре в течение 17 ч, концентрировали и осуществляли поглощение остатка в Н 2 О и EtOAc. Фазы разделяли и органическую фазу промывали при помощи H2O. Объединенные водные фазы подщелачивали с использованием 2 н водного раствора NaOH и затем экстрагировали при помощи EtOAc. Фазы разделяли и водную фазу повторно экстрагировали два раза при помощи EtOAc. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали с получением 2-амино-2-(6-бром-пиридин-2-ил)-пропан-1-ола в виде бесцветного твердого вещества. ВЭЖХ RtH4= 0,35 мин; ESIMS: 231, 233 [(М+Н)+]; 1 Н ЯМР (400 МГц, DMSO-d6):7,73-7,63 (м, 2 Н), 7,45 (дд, 1 Н), 4,72-4,69 (м, 1 Н), 3,58 (дд, 1 Н), 3,40 (дд, 1 Н), 2,00 (шир.с, 2 Н), 1,26 (с,3 Н). е) N-[1-(6-Бромпиридин-2-ил)-2-гидрокси-1-метилэтил]-2-хлорацетамид К раствору 2-амино-2-(6-бромпиридин-2-ил)-пропан-1-ола (4,9 г, 21,2 ммоль) в DCM (50 мл) добавляли K2CO3 (5,86 г, 42,4 ммоль). Реакционную смесь охлаждали до 0 С и добавляли по каплям 2 хлорацетилхлорид (2,55 мл, 3,59 г, 31,8 ммоль). Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивания в течение 5 ч. Добавляли МеОН (20 мл) и перемешивание продолжали при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли при помощи Н 2 О иDCM, фазы разделяли и водную фазу два раза экстрагировали при помощи DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали и растворитель удаляли с получением N-[1-(6-бромпиридин-2-ил)-2-гидрокси-1-метилэтил]-2-хлорацетамида в виде оранжевого масла. ВЭЖХ RtH4= 0,730,77 мин; ESIMS: 307, 309 [(М+Н)+]; 1 Н ЯМР (400 МГц, DMSO-d6):8,35 (с, 1 Н), 7,70-7,66 (м, 1 Н), 7,48(90 мл) добавляли KOtBu и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь гасили при помощи Н 2 О и разбавляли при помощи EtOAc. Фазы разделяли и водную фазу два раза экстрагировали при помощи EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и растворитель удаляли с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества. ВЭЖХ RtH4= 0,73 мин;g) 5-(6-Бромпиридин-2-ил)-5-метилморфолин-3-тион Смесь 5-(6-бром-пиридин-2-ил)-5-метилморфолин-3-она (4,65 г, 17,15 ммоль) и P2S5 (4,57 г, 20,58 ммоль) в пиридине (60 мл) перемешивали при 80 С в атмосфере N2 в течение 6 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли 0,5 н водным раствором HCl и EtOAc. Фазы разделяли и водную фазу два раза экстрагировали при помощи EtOAc. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. Указанное в заголовке соединение получали в виде бледно-желтого твердого вещества после флэшхроматографии на силикагеле (циклогексан/EtOAc 100:0 до 75:25). ВЭЖХ RtH4= 0,89 мин; ESIMS: 287,289 [(М+Н)+]; 1 Н ЯМР (400 МГц, DMSO-d6):11,15 (с, 1 Н), 7,86-7,82 (м, 1 Н), 7,61 (дд, 1 Н), 7,39 (дд, 1 Н),4,44-4,34 (д, 2 Н), 4,13 (д, 1 Н), 3,74 (д, 1 Н), 1,52 (с, 3 Н).NH3/MeOH (20,89 мл, 146 ммоль) перемешивали при 50 С в течение 3 дней в автоклаве. Реакционную смесь упаривали досуха и очищали при помощи FC (флэш-хроматография) (градиент циклогексан:EtOAc 75:25 до 50:50, затем +10% Et3N, в завершение МеОН +10% Et3N) с получением неочищенного указанного в заголовке соединения, которое далее очищали путем промывки при помощи DCM. ВЭЖХ RtH4= 0,54 мин; ESIMS: 270, 272 [(М+Н)+]; 1H ЯМР (400 МГц, DMSO-d6):8,51 (шир.с, 2 Н), 7,83-7,79 (м, 1 Н), 7,637,61 (м, 2 Н), 4,45 (с, 2 Н), 4,11 (д, 1 Н), 3,84 (д, 1 Н), 1,51 (с, 3 Н).i) трет-Бутиловый эфир [5-(6-бромпиридин-2-ил)-5-метил-5,6-дигидро-2 Н-[1,4]оксазин-3-ил]карбаминовой кислоты Суспензию 5-(6-бромпиридин-2-ил)-5-метил-5,6-дигидро-2 Н-[1,4]оксазин-3-иламина (1,100 г, 4,07 ммоль), Boc2O (1,229 мл, 1,155 г, 5,29 ммоль) и DIPEA (1,067 мл, 0,789 г, 6,11 ммоль) в DCM (30 мл) перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь разбавляли при помощи Н 2 О и DCM. Фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде бесцветного твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 0,92 мин; ESIMS: 370, 372 [(М+Н)+].j) трет-Бутиловый эфир (+)- и (-)-5-(6-аминопиридин-2-ил)-5-метил-5,6-дигидро-2 Н-[1,4]оксазин-3 ил]карбаминовой кислоты Смесь трет-бутилового эфира [5-(6-бромпиридин-2-ил)-5-метил-5,6-дигидро-2 Н-[1,4]оксазин-3 ил]карбаминовой кислоты (986 мг, 2,66 ммоль), циклогександиметилдиамина (0,420 мл, 379 мг, 2,66 ммоль), аскорбата натрия (211 мг, 1,07 ммоль), NaN3 (1385 мг, 21,31 ммоль) и CuI (203 мг, 1,07 ммоль) в этаноле/воде (22,0/8,8 мл) дегазировали при помощи N2 на бане сухой лед/EtOH. Реакционную смесь затем перемешивали при 45 С в течение 4 ч. Реакционной смеси давали нагреться до комнатной температуры и фильтровали через hyflo, промывали при помощи EtOAc и концентрировали. Флэш-хроматография на силикагеле (циклогексан/EtOAc градиент 0-3 мин 100:0, 3-25 мин 60:40, 40-52 мин 50:50) давала указанное в заголовке соединение. ВЭЖХ RtH4= 0,60, 0,66 мин; ESIMS: 305 [(М-Н)+]; 1 Н ЯМР (400 МГц, DMSO-d6):7,77-7,73 (м, 1 Н), 6,81-6,76 (м, 2 Н), 4,71-4,63 (м, 1 Н), 4,70-4,56 (м, 2 Н), 4,06-3,96 (м,2 Н), 1,69 (с, 3 Н), 1,51 (с, 9 Н). Рацемический трет-бутиловый эфир 5-(6-аминопиридин-2-ил)-5-метил-5,6-дигидро-2 Н-[1,4]оксазин-3-ил]карбаминовой кислоты разделяли на чистые энантиомеры методом препаративной хиральной ВЭЖХ (колонка: Chiralpak AS; растворитель: н-гептан/этанол/изопропиламин = 80:12:8; скорость потока: 70 мл/мин; детекция при 220 нм). Энантиомер 1: []D = -138,5 (с=1,00, МеОН). Энантиомер 2: []D =+141,5 (с=1,03, МеОН). (-)-Энантиомер 1 использовали на следующих стадиях, его конфигурация была указана как (R) по аналогии с подобными структурами, конфигурация которых была определена методом рентгеновской кристаллографии.k) трет-Бутиловый эфир R)-5-6-[(5-бромпиридин-2-карбонил)амино]пиридин-2-ил-5-метил-5,6 дигидро-2 Н-[1,4]оксазин-3-ил)карбаминовой кислоты К раствору 5-бромпиридин-2-карбоновой кислоты (34,5 мг, 0,171 ммоль) в DCM (2 мл) добавляли 1-хлор-N,N,2-триметилпропениламин (0,045 мл, 45,7 мг, 0,342 ммоль) и реакционную смесь перемешивали при 0 С в течение 1 ч. Реакционную смесь затем добавляли по каплям к полученному на предыдущей стадии безводному раствору трет-бутилового эфира (-)-5-(6-аминопиридин-2-ил)-5-метил-5,6 дигидро-2 Н-[1,4]оксазин-3-ил]карбаминовой кислоты (энантиомер 1 со стадии j), 47,6 мг, 0,155 ммоль) иNEt3 (0,048 мл, 34,6 мг, 0,342 ммоль) в DCM (2 мл) при 0 С. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивания в течение 20 мин при комнатной температуре. Реакционную смесь разбавляли при помощи DCM и гасили при помощи Н 2 О. Фазы разделяли и водную фазу экстрагировали при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и два раза очищали при помощи ВЭЖХ (Alltech Groml) [6-R)-5-Амино-3-метил-3,6-дигидро-2 Н-[1,4]-оксазин-3-ил)пиридин-2-ил]амид 5-бромпиридин 2-карбоновой кислоты К раствору трет-бутилового эфира 5-6-[(5-бромпиридин-2-карбонил)амино]пиридин-2-ил-5 метил-5,6-дигидро-2 Н-[1,4]оксазин-3-ил)карбаминовой кислоты (43 мг, 0,088 ммоль) в DCM (270 мкл) добавляли TFA (270 мкл, 400 мг, 3,51 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь гасили 1 М водным раствором NaHCO3 и разбавляли при помощи DCM. Фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали и очищали флэшхроматографией с ручной загрузкой (NH3-дезактивированный силикагель, гексан:DCM:MeOH 10:10:1,затем DCM:MeOH 10:1, затем +0,1%NH3 и в завершение МеОН +1%NH3) с получением указанного в заголовке соединения в виде бесцветного твердого вещества. К раствору свободного основания в DCM добавляли 1 экв. раствора 2 н HCl/Et2O и полученную гидрохлоридную соль собирали. ВЭЖХ RtH4= 0,76 мин; ESIMS: 390, 392 [(М+Н)+]; 1 Н ЯМР (400 МГц, CD3OD):8,88 (д, 1 Н), 8,38 (д, 1 Н), 8,33 (дд, 1 Н),8,27-8,24 (м, 1 Н), 8,24-7,98 (м, 1 Н), 7,33 (д, 1 Н), 4,69 (д, 1 Н), 4,67 (д, 1 Н), 4,30 (д, 1 Н), 4,11 (д, 1 Н), 1,78 (с,3 Н). Пример 2. [6-(5-Амино-3-метил-3,6-дигидро-2 Н-[1,4]оксазин-3-ил)пиридин-2-ил]амид 5-бром-пиридин-2-карбоновой кислоты Рацемат примера 1 может быть получен с использованием процедуры, аналогичной той, которую использовали в примере 1, завершая синтез с использованием рацемической смеси, полученной на стадии j) примера 1, и имеет такие же аналитические данные. Пример 3. Трифторацетат 5-6-[(5-хлорпиридин-2-карбонил)амино]пиридин-2-ил-5-фторметил 5,6-дигидро-2 Н-[1,4]оксазин-3-ил-аммония а) Диэтиловый эфир 2-(6-бромпиридин-2-ил)малоновой кислоты Диизопропиламид лития (2,0 М раствор в гептане/ТГФ/этилбензоле, 581,3 мл) брали для поглощения в безводный ТГФ (400 мл) и охлаждали до -78 С. К LDA раствору медленно добавляли 2-бром-6 метилпиридин (50,0 г, 296,64 ммоль) при указанной выше температуре в течение 15 мин, оставляли для непрерывного перемешивания в течение 30 мин. Затем этилхлороформиат (94,62 г, 871,94 ммоль) в безводном ТГФ (50 мл) добавляли к перемешиваемому содержимому по каплям и реакционную массу оставляли для перемешивания при -78 С в течение 2 ч. Реакционную смесь гасили насыщенным раствором хлорида аммония и образовавшийся продукт экстрагировали этилацетатом с промывкой водой, насыщенным солевым раствором и сушили над безводным Na2SO4. Органический слой концентрировали при пониженном давлении и неочищенный продукт очищали колоночной хроматографией с использованием 10% этилацетата в гексане с получением указанного в заголовке соединения в виде коричневой жидкости. Выход = 65,0 г (71,4%). ТСХ (10% этилацетата в гексане) Rf = 0,31; LCMS: RtH8 = 1,866; [M+1]=315,8 и 317,9; ВЭЖХ: RtH9 = 4,636 мин (48%); 1 Н ЯМР (400 МГц, CDCl3):7,587 (т, 1 Н), 7,47 (д, 1 Н), 7,27 (д,1 Н), 3,91 (с, 1 Н), 4,25-4,09 (м, 4 Н), 1,24 (т, 6 Н).b) (6-Бромпиридин-2-ил)уксусная кислота Диэтиловый эфир 2-(6-бромпиридин-2-ил)малоновой кислоты (64,0 г, 202,4 ммоль) добавляли к раствору карбоната калия (279,8 г, 2024 ммоль) в воде (400 мл) при комнатной температуре и реакционную смесь нагревали до температуры кипения с обратным холодильником при 100 С в течение 36 ч. Реакционную смесь обрабатывали насыщенным раствором хлорида аммония и образовавшийся продукт экстрагировали этилацетатом (3800 мл), промывали насыщенным солевым раствором (10 мл). Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соедине- 22024059c) Этиловый эфир (6-бромпиридин-2-ил)уксусной кислоты К раствору (6-бромпиридин-2-ил)уксусной кислоты (34,0 г, 158,14 ммоль) в этаноле (300 мл) добавляли концентрированную H2SO4 (5,0 мл) и нагревали до температуры кипения с обратным холодильником в течение 12 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении досуха. К остатку добавляли воду и продукт экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного продукта. Очистка методом колоночной хроматографии давала указанное в заголовке соединение в виде коричневой жидкости. Выход = 31,2 г (82%). ТСХ (20% этилацетата в гексане) Rf = 0,51; LCMS: RtH7 = 0,996, [M+1]+ = 244,0 и 246,0; ВЭЖХ:d) Этиловый эфир 2-(6-бромпиридин-2-ил)-3-гидрокси-2-гидроксиметилпропионовой кислоты К раствору параформальдегида (9,6 г, 319,55 ммоль) и этоксида натрия (0,87 г, 12,784 ммоль) в безводном ТГФ (250 мл) добавляли этиловый эфир (6-бромпиридин-2-ил)-уксусной кислоты (31,2 г, 127,82 ммоль) при температуре в пределах от 0 до -10 С и реакционную смесь оставляли для перемешивания при этой температуре в течение 4 ч. Твердые частицы, образовавшиеся в реакционной смеси, фильтровали и промывали этилацетатом и фильтрат концентрировали с получением неочищенного продукта в виде коричневой жидкости. Выход = 30,0 г (неочищенный продукт). ТСХ (30% этилацетата в гексане) Rf = 0,28; LCMS: RtH8 = 0,702, M+1 = 304,0 и 306,0.e) Этиловый эфир 2-(6-бромпиридин-2-ил)-3-метоксиметокси-2-метоксиметоксиметилпропионовой кислоты К раствору этилового эфира 2-(6-бромпиридин-2-ил)-3-гидрокси-2-гидроксиметилпропионовой кислоты (30,0 г, 98,638 ммоль) в безводном ТГФ (250 мл) добавляли тетрабутиламмонийбромид (15,8 г,49,319 ммоль) и диизопропилэтиламин (127,47 г, 163,0 мл) с последующим добавлением метоксиметилхлорида по каплям при комнатной температуре. Полученное реакционное содержимое нагревали при кипячении с обратным холодильником при 65 С в течение 3 ч и охлаждали до комнатной температуры. Реакционную смесь концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле с использованием 10% этилацетата в гексане с получением указанного в заголовке соединения в виде коричневой жидкости. Выход = 20,4 г (52%). ТСХ (30% этилацетата в гексане) Rf = 0,55;f) 2-(6-Бромпиридин-2-ил)-3-метоксиметокси-2-метоксиметоксиметилпропионовая кислота Гидроксид лития (10,69 г, 254,95 ммоль) добавляли к раствору этилового эфира 2-(6-бромпиридин 2-ил)-3-метоксиметокси-2-метоксиметоксиметилпропионовой кислоты (20,0 г, 50,99 ммоль) в этаноле(100 мл) и воде (100 мл) при комнатной температуре и реакционную смесь оставляли для перемешивания в течение ночи. Реакционную массу концентрировали при пониженном давлении и подкисляли разбавленной HCl и при 0 С. Продукт экстрагировали этилацетатом, промывали минимальным количеством насыщенного солевого раствора. Органический слой концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде коричневой жидкости. Выход = 18,0 г. ТСХ (50% этилацетата в гексане) Rf = 0,05; LCMS: RtH8 = 1,383, [M+1] = 364,0 и 366,0; ВЭЖХ: RtH9 = 3,844 мин(14,97 г [20,6 мл], 148,27 ммоль) при комнатной температуре и перемешивали при 100 С в течение 15 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученный остаток растворяли в ТГФ (600 мл) и добавляли 20% раствор NaOH при комнатной температуре и перемешивали в течение 1 ч. Растворитель удаляли при пониженном давлении и образовавшийся продукт экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, с последующей сушкой над MgSO4. Органическую часть концентрировали при пониженном давлении и очистка методом колоночной хроматографии неочищенного продукта с использованием 35% этилацетата в гексане давала указанное в заголовке соединение в виде коричневой жидкости. Выход = 15,0 г (88% [2 стадии]). ТСХ (70% этилацетата в гексане) Rf = 0,51; LCMS: RtH7 = 0,28, [M+1]=335,0 и 337,0.h) N-[1-(6-Бромпиридин-2-ил)-2-метоксиметокси-1-метоксиметоксиметилэтил]-2-хлорацетамид К раствору 1-(6-бромпиридин-2-ил)-2-метоксиметокси-1-метоксиметоксиметилэтиламина (15,0 г,44,75 ммоль) в DCM (150 мл) добавляли водный раствор Na2CO3 (10,91 г, 102,925 ммоль в воде, 30 мл) при 0 С и перемешивали в течение 10 мин. К полученной реакционной смеси добавляли хлорацетилхлорид (5,56 г, 49,225 ммоль) при 0 С и продолжали перемешивание в течение 1 ч при температуре окру- 23024059 жающей среды. Реакционную смесь разбавляли при помощи DCM (1 л) и реакционную смесь обрабатывали путем промывки водой, насыщенным солевым раствором и сушили над безводным Na2SO4. Органический слой отделяли и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде коричневой жидкости. Выход = 9,0 г (48%). ТСХ (50% этилацетата в гексане)i) N-[1-(6-Бромпиридин-2-ил)-2-гидрокси-1-гидроксиметилэтил]-2-хлорацетамид К раствору N-[1-(6-бромпиридин-2-ил)-2-метоксиметокси-1-метоксиметоксиметилэтил]-2-хлорацетамида (9,0 г, 21,861 ммоль) в этантиоле (30 мл) добавляли BF3-Et2O (9,3 г, 141,93 ммоль) при 0 С и перемешивали в течение 10 мин. Перемешивание продолжали в течение 3 ч при комнатной температуре. Реакционную смесь гасили насыщенным раствором NaHCO3 и образовавшийся продукт экстрагировали этилацетатом. Органический слой отделяли и промывали насыщенным солевым раствором, с последующей сушкой над безводным Na2SO4. Органический слой концентрировали при пониженном давлении и очищали колоночной хроматографией с использованием 2% метанола в хлороформе, с получением указанного в заголовке соединения в виде коричневой жидкости. Выход = 5,5 г (77%). ТСХ (10% метанол в хлороформ) Rf = 0,51; LCMS: RtH8 = 0,916, [M+1] = 322,9 и 324,8; ВЭЖХ RtH9 = 5,931 мин (89%); 1 Н ЯМРj) 5-(6-Бромпиридин-2-ил)-5-гидроксиметилморфолин-3-он К раствору N-[1-(6-бромпиридин-2-ил)-2-гидрокси-1-гидроксиметилэтил]-2-хлорацетамида (5,4 г,16,69 ммоль) в t-BuOH (80 мл) добавляли t-BuOK (2,06 г, 18,38 ммоль) при комнатной температуре с последующим добавлением иодида натрия (0,25 г, 1,669 ммоль) и реакционную смесь оставляли для перемешивания при 90 С в течение 1 ч. Реакционную смесь концентрировали при пониженном давлении и полученный остаток обрабатывали водой. Соединение, присутствующее в остатке, экстрагировали этилацетатом (2100 мл). Органическую часть промывали насыщенным солевым раствором, сушили надNa2SO4 и концентрировали при пониженном давлении с получением смолистого соединения. Последующее растирание в порошок полученного продукта с н-пентаном (5,0 мл) и диэтиловым эфиром (5,0 мл) давало указанное в заголовке соединение в виде бледно-желтого смолистого соединения. Выход = 3,3 г (68,8%). ТСХ (50% этилацетата в гексане) Rf = 0,21; LCMS: RtH8 = 0,155, [M+1] = 286,9 и 288,8; ВЭЖХ RtH9 = 3,03 мин (69,8%); 1H ЯМР (400 МГц, CDCl3):8,40 (с, 1 Н), 7,79 (т, 1 Н), 7,57 (д, 2 Н), 5,11 (с,1 Н), 4,15 (д, 1 Н), 3,99 (д, 2 Н), 3,88 (д, 1 Н), 3,71-3,60 (м, 2 Н).k) 5-(6-Бромпиридин-2-ил)-5-фторметилморфолин-3-он К раствору 5-(6-бромпиридин-2-ил)-5-гидроксиметилморфолин-3-она (2,8 г, 9,756 ммоль) в безводном ТГФ (30 мл), добавляли при комнатной температуре трифторид диэтиламиносеры (4,72 г, 29,2 68 ммоль) и продолжали перемешивание в течение 4 ч. К полученной реакционной смеси затем добавлялиNa2CO3 и перемешивали еще в течение 30 мин. Реакционную смесь концентрировали при пониженном давлении и продукт экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного соединения в виде смолистого остатка. Очистка неочищенного продукта с использованием системы растворителей 45% этилацетата в гексане давала указанное в заголовке соединение в виде не совсем белого твердого вещества. Выход = 1,15 г (40%). ТСХ (70% этилацетата в гексане) Rf = 0,49;l) 5-Хлорпиридин-2-карбоновой кислоты [6-(3-фторметил-5-оксоморфолин-3-ил)пиридин-2 ил]амид Смесь 4,5-бис-(дифенилфосфино)-9,9-диметилксантена (0,04 г, 0,069 ммоль), трис-(дибензилиденацетон)дипалладия(0) (0,032 г, 0,035 ммоль) и карбоната цезия (0,678 г, 2,083 ммоль) добавляли в 1,4 диоксан и дегазировали при помощи аргона в течение 10 мин. К полученной реакционной смеси добавляли 5-(6-бромпиридин-2-ил)-5-фторметилморфолин-3-он (0,2 г, 0,694 ммоль) с последующим добавлением 5-хлорпиколинамида (0,119 г, 0,764 ммоль) и дегазировали при помощи аргона еще в течение 5 мин. Реакционную смесь затем нагревали при 80 С в течение 16 ч и охлаждали до комнатной температуры. Реакционную смесь обрабатывали водой и продукт экстрагировали этилацетатом, промывали насыщенным солевым раствором и сушили над безводным Na2SO4. Органический слой концентрировали при пониженном давлении с получением жидкости, которую растирали в порошок с н-пентаном с получением указанного в заголовке соединения в виде не совсем белого твердого вещества. Выход = 0,24 г (неочищенный продукт). ТСХ (50% этилацетата в гексане) Rf = 0,45; LCMS: RtH8 = 1,215, [M+1] = 365,1 и 366,9; ВЭЖХ RtH9 = 4,367 мин (53%). боновой кислоты (0,24 г, 0,658 ммоль) в ТГФ (10,0 мл) добавляли при комнатной температуре реагент Лоуссона (0,798 г, 1,974 ммоль) и нагревали при температуре кипения с обратным холодильником в течение 24 ч. Реакционную массу концентрировали при пониженном давлении. Неочищенное соединение непосредственно очищали колоночной хроматографией с использованием 23% этилацетата в гексане, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества. Выход = 0,19 г (72% [2 стадии]). ТСХ (50% этилацетата в гексане) Rf = 0,71; LCMS: RtH8 = 1,578, [M+1]+ = 381,1 и 382,9.(8,0 мл) при 0 С в герметично закрытой пробирке и перемешивали при комнатной температуре в течение 24 ч. Реакционную массу концентрировали при пониженном давлении и непосредственно очищали методом препаративной ВЭЖХ. Условия: колонка C18-ZORBAX 21,2150 мм; 5 мкм. Подвижная фаза: 0,1% TFA в воде (A)/ACN; скорость потока: 20 мл/мин. Выход: 86 мг (36%). Т.пл.: 216-218 С. ТСХ (20% метанола в хлороформе) Rf = 0,45; LCMS: RtH8 = 0,194 [M+1] = 364,0 и 366,1; ВЭЖХ RtH9 = 3,222 мин(98,7%); 1 Н ЯМР (400 МГц, DMSO-d6):10,85 (с, 1 Н), 10,37 (с, 1 Н), 9,37 (с, 1 Н), 8,50-8,79 (м, 2 Н), 8,318,23 (м, 3 Н), 8,06 (т, 1 Н), 7,37 (д, 1 Н), 4,96 (дд, 1 Н), 4,85 (дд, 1 Н), 4,62 (дд, 2 Н), 4,25-4,16 (м, 2 Н). Образование продукта также подтверждали при помощи 2D ЯМР-ROESY. Примеры 4-7. Соединения, перечисленные в табл. 4, были получены с использованием процедуры,аналогичной той, которую использовали в примере 3. Таблица 4 а) трет-Бутиловый эфир 4-(6-бромпиридин-2-ил)-4-метил-2-оксо-24-[1,2,3]оксатиазолидин-3-карбоновой кислоты К предварительно охлажденному при 0 С раствору тионилхлорида (3,42 мл, 5,57 г, 46,8 ммоль) в пиридине (9,46 мл, 9,25 г, 117,0 ммоль) добавляли по каплям раствор трет-бутилового эфира [1-(6-бромпиридин-2-ил)-2-гидрокси-1-метилэтил]карбаминовой кислоты (см. пример 1 стадия d) 7,75 г, 23,4 ммоль) в DCM (230 мл). Реакционную смесь оставляли для перемешивания в течение 1 ч при комнатной температуре, затем добавляли 0,5 н водный раствор HCl и DCM, фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (смесь диастереомеров) в виде оранжевого твердого вещества. ВЭЖХ RtH4= 1,16,1,20 мин (диастереомеры); ESIMS: 377, 379 [(М+Н)+].RuCl3 гидрат (0,971 г, 4,68 ммоль) и NaIO4 (10,01 г, 46,8 ммоль). Реакционную смесь перемешивали при 0 С в течение 2 ч. Добавляли Н 2 О и DCM, фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором,сушили над Na2SO4, фильтровали и концентрировали. Остаток растворяли в DCM и фильтровали через силикагель, фильтрат упаривали и остаток растирали в порошок с ТВМЕ (10 мл) и н-гексаном (100 мл). Полученный осадок фильтровали и промывали н-гексаном с получением указанного в заголовке соединения в виде бесцветного кристаллического твердого вещества. ВЭЖХ RtH4= 1,16 мин; ESIMS: 393, 395 пропионовой кислоты (2,54 г, 13,67 ммоль) в DMF (10 мл, раствор предварительно был высушен над 4 молекулярными ситами). Реакционную смесь оставляли для перемешивания при комнатной температуре в течение 30 мин, затем при 60 С в течение 17 ч. Реакционную смесь гасили при помощи H2O и разбавляли 1 н водным раствором HCl и EtOAc. Фазы разделяли и водную фазу два раза повторно экстрагировали при помощи EtOAc. Объединенные органические фазы промывали насыщенным солевым, сушили над Na2SO4, фильтровали и концентрировали. Флэш-хроматография на силикагеле (циклогексан: EtOAc,градиент 0-5 мин 100:0, 5-30 мин 90:10, 30-40 мин 90:10, 40-50 мин 80:20, 50-55 мин 80:20) давала указанное в заголовке соединение (диастереомерная смесь) в виде прозрачного масла. ВЭЖХ RtH4= 1,39 мин; ESIMS: 499, 501 [(М+Н)+].d) трет-Бутиловый эфир [(RS)-1-(6-бромпиридин-2-ил)-2-R)-1-карбамоил-2,2,2-трифтор-1-метилэтокси)-1-метилэтил]карбаминовой кислоты Раствор этилового эфира (R)-2-[(RS)-2-(6-бромпиридин-2-ил)-2-трет-бутоксикарбониламинопропокси]-3,3,3-трифтор-2-метилпропионовой кислоты (3,0 г, 6,01 ммоль) в 7 н NH3/MeOH (6,5 мл) перемешивали в герметично закрытом стеклянном флаконе при 55 С в течение 72 ч. Реакционную смесь концентрировали с получением указанного в заголовке соединения в виде бесцветного твердого вещества,которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 1/12, 1,14 минe) трет-Бутиловый эфир [(RS)-1-(6-бромпиридин-2-ил)-2-R)-1-циано-2,2,2-трифтор-1-метилэтокси)-1-метилэтил]карбаминовой кислоты К предварительно охлажденному при 0 С раствору трет-бутилового эфира [(RS)-1-(6-бромпиридин 2-ил)-2-R)-1-карбамоил-2,2,2-трифтор-1-метилэтокси)-1-метилэтил]карбаминовой кислоты (2,18 г, 4,64 ммоль) и NEt3 (1,615 мл, 1,173 г, 11,59 ммоль) в DCM (30 мл) добавляли по каплям TFAA (0,773 мл, 1,168 г, 5,56 ммоль). После перемешивания в течение 5 мин при 0 С, затем в течение 1 ч при комнатной температуре реакционную смесь разбавляли насыщенным водным раствором Na2CO3 и DCM. Фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали с получением бледно-желтого масла, которое перемешивали с 7 н NH3/MeOH в течение 5 мин. Смесь упаривали досуха и очищали флэш-хроматографиейf) (R)-2-[(RS)-2-Амино-2-(6-бромпиридин-2-ил)пропокси]-3,3,3-трифтор-2-метилпропионитрил Раствор трет-бутилового эфира [(RS)-1-(6-бромпиридин-2-ил)-2-R)-1-циано-2,2,2-трифтор-1-метилэтокси)-1-метилэтил]карбаминовой кислоты (0,456 г, 1,008 ммоль) и TFA (1,554 мл, 2,299 г, 20,17 ммоль) в DCM (5 мл) перемешивали при комнатной температуре в течение 30 мин, концентрировали и растирали в порошок с 7 н NH3/MeOH при комнатной температуре в течение 20 мин и снова концентрировали с получением указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки. ВЭЖХ RtH4= 0,69, 0,73 мин (диастереомеры); ESIMS: 352, 354 [(М+Н)+].g) (2R,5RS)-5-(6-Бромпиридин-2-ил)-2,5-диметил-2-трифторметил-5,6-дигидро-2 Н-[1,4]оксазин-3 иламин Суспензию (R)-2-[(RS)-2-амино-2-(6-бромпиридин-2-ил)пропокси]-3,3,3-трифтор-2-метилпропионитрила (0,688 г, 1,172 ммоль), N-ацетил-L-цистеина (0,383 г, 2,344 ммоль) и K2CO3 (0,356 г, 2,560 ммоль) в абсолютном EtOH (4 мл) перемешивали при 80 С в течение 18 ч. Реакционную смесь гасили при помощи 10% водного раствора K2CO3 и 3 экстрагировали при помощи ТВМЕ. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде бесцветного твердого вещества. ВЭЖХ RtH4= 0,68-0,70 мин; ESIMS: 352, 354 [(М+Н)+].(R)-5-(6-бромпиридин-2-ил)-2,5-диметил-2-трифторметил-5,6-дигидро-2 Н-[1,4]оксазин-3 иламина, Boc2O и DIPEA в DCM (4 мл) перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь гасили насыщенным водным раствором NaHCO3 и разбавляли при помощи DCM. Фазы разделяли и водную фазу два раза повторно экстрагировали при помощи DCM. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали. ВЭЖХ очистка (Alltech Grom Saphir 65 Si, 10 мкм, 25050 мм колонка, градиент Hept:EtOAc 01,6 мин 85:15, 1,6-16 мин 0:100, 16-21,2 мин 0:100, скорость потока: 100 мл/мин, детекция: 254 нм) давала желаемый (2R,5R), а также нежелательный (2R,5S) диастереомер. ВЭЖХ RtH4= 1,28 мин (2R,5S), 1,30 мин (2R,5R); ESIMS: 452, 454 [(М+Н)+]; 1H ЯМР (2R,5R) (400 МГц, CDCl3):10,98 (шир.с, 1 Н), 7,59 (т,1 Н), 7,43 (д, 1 Н), 7,34 (д, 1 Н), 4,39 (д, 1 Н), 4,08 (д, 1 Н), 1,62 (с, 3 Н), 1,55 (с, 12 Н); 1 Н ЯМР (2R, 5S) (400 МГц, CDCl3):11,01 (шир.с, 1 Н), 7,57 (т, 1 Н), 7,42 (д, 1 Н), 7,37 (д, 1 Н), 4,45 (д, 1 Н), 3,91 (д, 1 Н), 1,74 (с,3 Н), 1,65 (с, 3 Н), 1,55 (с, 19 Н).i) трет-Бутиловый эфир 2R,5R)-5-6-[(5-циано-3-метилпиридин-2-карбонил)амино]пиридин-2-ил 2,5-диметил-2-трифторметил-5,6-дигидро-2 Н-[1,4]оксазин-3-ил)карбаминовой кислоты Смесь трет-бутилового эфира [(2R,5R)-5-(6-бромпиридин-2-ил)-2,5-диметил-2-трифторметил-5,6 дигидро-2 Н-[1,4]оксазин-3-ил]карбаминовой кислоты (60,00 мг, 0,133 ммоль), 5-циано-3-метилпиколинамида (23,52 мг, 0,146 ммоль), Xantphos (6,91 мг, 0,012 ммоль) и Cs2CO3 (60,50 мг, 0,186 ммоль) в диоксане (0,611 мл) дегазировали при помощи аргона в течение 5 мин, затем добавляли Pd2dba3 (3,64 мг,3,98 мкмоль) и реакционную смесь перемешивали при 40 С в течение 18 ч. Реакционную смесь разбавляли при помощи Н 2 О и ТВМЕ. Фазы разделяли и водную фазу повторно экстрагировали при помощи ТВМЕ. Объединенные органические фазы промывали насыщенным солевым раствором, сушили надNa2SO4, фильтровали и концентрировали. ВЭЖХ очистка (Alltech Grom Saphir 65 Si 10-мкм колонка,15030 мм, градиент н-гептан:Е 1:ОАс 0-1,2 мин 75:25, 1,2-9 мин 0:100, 9-12 мин 0:100, скорость потока: 50 мл/мин, детекция: 254 нм) давала указанное в заголовке соединение в виде бесцветного твердого вещества. ВЭЖХ RtH4= 1,37 мин; ESIMS: 533 [(М+Н)+]; 1 Н ЯМР (400 МГц, CDCl3):11,22 (с, 1 Н), 10,47 (с,1 Н), 8,78 (д, 1 Н), 8,33 (д, 1 Н), 7,98 (д, 1 Н), 7,84-7,80 (м, 1 Н), 7,13 (д, 1 Н), 4,37 (д, 1 Н), 4,11 (д, 1 Н), 2,88 (с,3 Н), 1,64 (с, 3 Н), 1,57 (шир.с, 12 Н).(50,0 мг, 0,094 ммоль) в DCM (0,3 мл) добавляли TFA (0,289 мл, 428,0 мг, 3,760 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали, добавляли насыщенный водный раствор NaHCO3 и ТВМЕ, фазы разделяли и водную фазу повторно экстрагировали два раза при помощи ТВМЕ. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали и остаток промывали при помощи МеОН с получением указанного в заголовке соединения в виде бесцветного кристаллического твердого вещества. ВЭЖХ RtH4= 0,84 мин; ESIMS: 433 [(М+Н)+]; 1 Н ЯМР (400 МГц, CD3OD):8,85 (с, 1 Н), 8,21-8,18 (м, 2 Н), 7,82-7,78 (м, 1 Н), 7,23 (д, 1 Н), 4,18 (д, 1 Н),3,80 (д, 1 Н), 2,76 (с, 3 Н), 1,46-1,45 (2 с, 6 Н). Примеры 9 и 10. Соединения, перечисленные в табл. 5, можно получить с использованием процедуры, аналогичной той, которую использовали в примере 8. Гидрохлоридные соли получали из растворов соответствующего свободного основания путем добавления хлористо-водородной кислоты в диоксане или хлористо-водородной кислоты в диэтиловом эфире и выпаривания растворителей. Таблица 4a) 2-(6-Бром-3-фторпиридин-2-ил)пропан-2-ол К раствору 2-бром-5-фторпиридина (25 г, 142 ммоль) в диэтиловом эфире (600 мл) медленно добавляли н-бутиллитий (2,5 М в гексане, 56,8 мл, 142 ммоль) при -78 С в атмосфере азота. Полученную желтую реакционную смесь перемешивали при -78 С в течение 2 ч и добавляли безводный ацетон (11,47 мл,156 ммоль) в течение 30 мин. Перемешивание продолжали при -78 С в течение 1 ч. Добавляли HCl (2 н,50 мл) и реакционную смесь нагревали до 0 С. рН смеси доводили до 7 при помощи 2 н раствора HCl. Реакционную смесь разбавляли этилацетатом и промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенного продукт (29,36 г) хроматографировали на силикагеле (циклогексан:этилацетат 9:1): 22,3 г (67,1% выход). ТСХ (циклогексан/этилацетат 9:1) Rf=0,33; LCMS RtH5=0,89 мин (ES + 234, 236), 1 Н-ЯМР (360 МГц, DMSO-d6): 7,727,62 (м, 2 Н), 5,27 (с, 1 Н, ОН), 1,50 (с, 6 Н, 2 СН 3).b) 6-Бром-3-фтор-2-изопропенилпиридин К раствору 2-(6-бром-3-фторпиридин-2-ил)пропан-2-ола (22,3 г, 95 ммоль) и ангидрида метансульфоновой кислоты (49,8 г, 286 ммоль) в дихлорметане добавляли по каплям триэтиламин (53,1 мл, 381 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 20 ч. Реакционную смесь гасили водным раствором карбоната натрия и разбавляли дихлорметаном. Водную фазу экстрагировали дихлорметаном. Объединенные органические слои промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали в вакууме (летучие вещества). Неочищенное коричневое масло хроматографировали на диоксиде кремния (циклогексан:этилацетат 9:1) с получением указанного в заголовке соединения в виде прозрачной жидкости. 17,35 г (84% выход). ТСХc) 2-(6-Бром-3-фторпиридин-2-ил)пропан-1,2-диол К раствору 6-бром-3-фтор-2-изопропенилпиридина (17,35 г, 80 ммоль) в ацетоне (45 мл) и воде (90 мл) добавляли N-метилморфолин-N-оксид гидрат (11,4 г, 84 ммоль) и тетроксид осмия (5,04 мл, 0,402 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 44 ч. Добавляли дитионит натрия (2 г) в воде (70 мл) и реакционную смесь перемешивали в течение 15 мин и затем фильтровали и концентрировали в вакууме. Добавляли этилацетат и органический слой промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали. 18,29 г слегка желтого твердого вещества (91% выход). LCMS RtH5=0,64 мин (ES+ 250, 252); 1 Н-ЯМР (360 МГц,CDCl3): 7,46 (дд, 1 Н), 7,35 (дд, 1 Н), 5,09 (с, 1 Н, ОН), 3,96 (д, 1 Н), 3,78 (д, 1 Н), 2,45 (шир., 1 Н, ОН), 1, 53d) 2-(6-Бром-3-фторпиридин-2-ил)-2-гидроксипропиловый эфир метансульфоновой кислоты К раствору 2-(6-бром-3-фторпиридин-2-ил)-пропан-1,2-диола (18,29 г, 73,1 ммоль) в дихлорметане(350 мл) добавляли триэтиламин (20,39 мл, 146 ммоль). Добавляли метансульфонилхлорид (6,27 мл, 80 ммоль) по каплям при 0 С в течение 10 мин. Перемешивание продолжали при 0 С в течение 30 мин. Реакционную смесь промывали насыщенным раствором бикарбоната натрия, водой и насыщенным солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. 31,46 г(неочищенное вещество, используемое без дополнительной очистки на следующей стадии). LCMS RtH5 = 0,81 мин (ES+ 328, 330); 1H-ЯМР (360 МГц, CDCl3): 7,52 (дд, 1 Н), 7,41 (дд, 1 Н), 5,13 (с, 1 Н, ОН), 4,61 (д,1 Н), 4,45 (д, 1 Н), 3,05 (с, 3 Н, CH3SO2), 1,61 (с, 3 Н, СН 3).(5 г, 15,24 ммоль), хлорида аммония (4,08 г, 76 ммоль) и азида натрия (2,476 г, 38,1 ммоль) в этаноле (100 мл) перемешивали при 80 С в течение 20 ч. Реакционную смесь разбавляли этилацетатом и промывали водой и насыщенным солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и упаривали. 3,1 г (74% выход). ТСХ (циклогексан/этилацетат 9:1) Rf=0,35; LCMS RtH5= 0,97 минf) 6-Бром-3-фтор-2-(2-метилазиридин-2-ил)пиридин К раствору 1-азидо-2-(6-бром-3-фторпиридин-2-ил)пропан-2-ола (11,2 г, 40,7 ммоль) в ТГФ (60 мл) добавляли трифенилфосфин (10,68 г, 40,7 ммоль) и реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Растворитель удаляли в вакууме и полученный остаток растворяли в диэтиловом эфире и фильтровали через ватный тампон для удаления трифенилфосфиноксида. Фильтрат промывали лимонной кислотой (9,6 г в 20 мл воды) и органическую фазу отделяли. Водный слой подщелачивали при помощи 2 н раствора NaOH и экстрагировали диэтиловым эфиром. Органический слой сушили над сульфатом натрия, фильтровали и упаривали с получением указанного в заголовке соединения с не- 29
МПК / Метки
МПК: A61P 25/00, C07D 487/04, A61K 31/5377, C07D 413/14
Метки: лечении, применение, производные, расстройств, неврологических, гетероциклические
Код ссылки
<a href="https://eas.patents.su/30-24059-geterociklicheskie-proizvodnye-i-ih-primenenie-v-lechenii-nevrologicheskih-rasstrojjstv.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические производные и их применение в лечении неврологических расстройств</a>
Производные индола и их применение для лечения психических и неврологических расстройств

Номер патента: 6104
Опубликовано: 25.08.2005
Авторы: Мольтсен Айнер Кнуд, Роттлендер Марио, Руланд Томас, Миккельсен Иван, Крог-Енсен Кристиан, Андерсен Ким
МПК: A61K 31/497, C07D 401/12
Метки: производные, индола, психических, применение, неврологических, лечения, расстройств
Формула / Реферат:
1. Соединение, представленное общей формулой I где A представляет собой O или S; n равно 2, 3, 4, 5, 6, 7, 8, 9 или 10; m равно 2 или 3; W представляет собой N, C или CH; Q представляет собой N, C или CH; и пунктирная линия представляет возможную связь; R1 представляет собой водород, C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-8-циклоалкил-C1-6-алкил, арил-C1-6-алкил или ацил; R2, R3, R4, R5 и R6 независимо представляют собой водород, галоген,...
Производные 1,2,4-триазоло[4,3-a]пиридина и их применение для лечения или предупреждения неврологических и психиатрических расстройств

Номер патента: 20672
Опубликовано: 30.12.2014
Авторы: Тресадерн Гэри Джон, Макдональд Грегор Джеймс, Трабанко-Суарес Андрес Авелино, Вега-Рамиро Хуан Антонио, Оелрик Даниель, Сид-Нуньес Хосе Мария
МПК: A61P 25/28, A61K 31/437, C07D 471/04...
Метки: неврологических, лечения, расстройств, психиатрических, 1,2,4-триазоло[4,3-a]пиридина, предупреждения, применение, производные
Формула / Реферат:
1. Соединение формулы (I)или его стереохимически изомерная форма,где n выбран из группы, состоящей из 0 и 1;m выбран из группы, состоящей из 0 и 1;R представляет собой метил;R1 выбран из группы, состоящей из С1-6алкила; (С1-3алкилокси)С1-3алкила; [(С1-3алкилокси)С1-3алкилокси]С1-3алкила;C1-3алкила, замещенного 1-3 галогенозаместителями; незамещенного фенила; (бензилокси)С1-3алкила; незамещенного С3-7циклоалкила; С3-7циклоалкила, замещенного...
Производные имидазо[1,2-a]пиразина и их применение для профилактики или лечения неврологических, психиатрических и метаболических расстройств и заболеваний

Номер патента: 22012
Опубликовано: 30.10.2015
Авторы: Мартин-Мартин Мария Лус, Пастор-Фернандес Хоакин, Макдональд Грегор Джеймс, Ванхоф Грета Констанция Петер, Бартоломе-Небреда Хосе Мануэль, Конде-Сейде Сусана, Ван Гол Михиль Люк Мария
МПК: A61P 25/00, A61P 3/00, A61K 31/4985...
Метки: применение, неврологических, метаболических, расстройств, имидазо[1,2-a]пиразина, профилактики, заболеваний, производные, психиатрических, лечения
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма,где R1 выбран из группы, состоящей из радикалов формул (а-1), (а-2) и (а-3):где каждый из R6, R7 и R8 независимо выбран из группы, состоящей из фтора, С1-4алкила, С1-4алкилокси и С1-4алкила, замещенного 1, 2 или 3 атомами фтора;R9 представляет собой водород или С1-4алкил;каждый из m1, m2 и m3 независимо выбран из 0, 1, 2, 3 и 4;р2 выбран из 1, 2, 3 и 4;каждый из p1 и р3 независимо выбран из...
Аза-гетероциклические соединения, применяемые для лечения неврологических расстройств и выпадения волос

Номер патента: 8723
Опубликовано: 29.06.2007
Авторы: Норман Марк Х., Ву Йонг-Кайан, Гамильтон Грегори С.
МПК: C07D 207/12, A61K 31/40, A61K 31/41...
Метки: неврологических, лечения, выпадения, аза-гетероциклические, расстройств, соединения, применяемые, волос
Формула / Реферат:
1. Фармацевтическая композиция, содержащая: 1) соединение формулы (I) или его фармацевтически приемлемые соли, сложный эфир или сольват, где n представляет собой 1 или 2, X представляет собой либо О, либо S, R1 представляет собой С1-С9алкил с прямой или разветвленной цепью, D представляет собой связь или С1-С10алкилен с прямой или разветвленной цепью, R2 представляет собой тетразол, -СООН, -CONH2, -CN, -SO3H или -ОН; и 2) фармацевтически...
Гетероциклические, тетрациклические производные гидрофурана в качестве ингибиторов 5нт2 в терапии расстройств цнс

Номер патента: 13595
Опубликовано: 30.06.2010
Авторы: Мегенс Антониус Адрианус Хендрикус Петрус, Сид-Нуньес Хосе Мария, Трабанко-Суарес Андрес Авелино
МПК: A61K 31/381, A61K 31/38, A61K 31/55...
Метки: 5нт2, ингибиторов, тетрациклические, производные, цнс, гидрофурана, терапии, расстройств, качестве, гетероциклические
Формула / Реферат:
1. Соединение формулы (I)его N-оксидная форма, фармацевтически приемлемая аддитивная соль или его стереохимически изомерная форма, гдеn представляет собой целое число, равное 1;i, j представляют собой целые числа, независимо друг от друга равные 0 или 1;r равно 0;каждый из R1 и R2независимо друг от друга представляет собой водород или алкил, или R1 и R2, взятые вместе с атомом азота, к которому они присоединены, могут образовывать радикал...
Предыдущий патент: Одноразовое зажимное устройство для обрезания
Следующий патент: Способ получения огнеупорных зерен, содержащих оксид хрома (3)
Случайный патент: Способ получения олмесартан медоксомила