Способ получения олмесартан медоксомила
Номер патента: 14026
Опубликовано: 30.08.2010
Авторы: Зупанциц Сильво, Пецавар Аница, Осольник Рената, Врбинц Миха







Формула / Реферат
1. Способ получения олмесартан медоксомила, который включает:
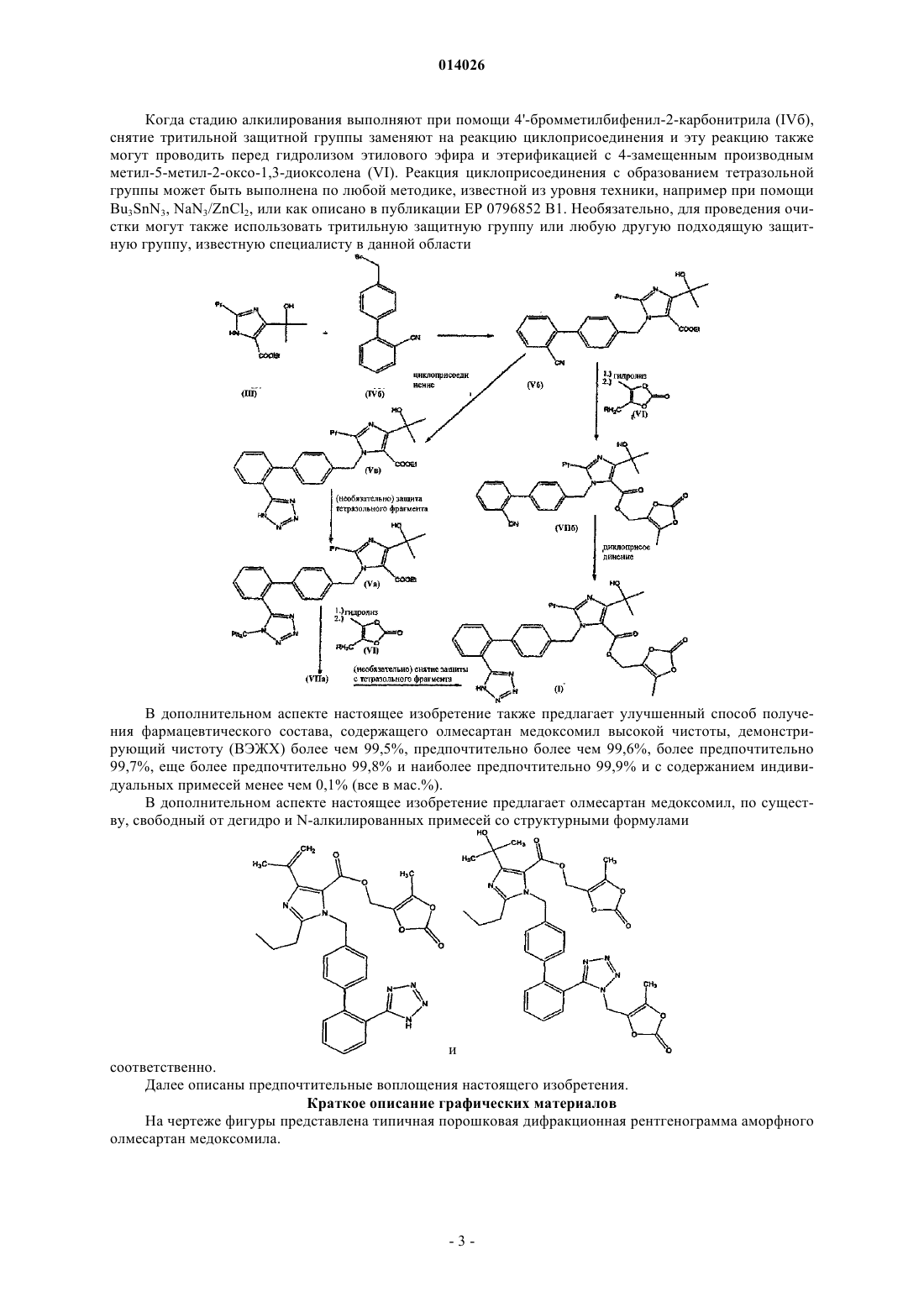
а) алкилирование этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) в органическом растворителе в присутствии основания с получением этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбоксилата (Va), кристаллизацию полученного соединения (Va) из реакционной смеси, причем ацетонитрил используют как растворитель при проведении реакции и как растворитель при кристаллизации; и
б) гидролиз соединения формулы (Va), этерификацию с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена формулы (VI)

где R представляет собой уходящую группу, выбранную из галогена, п-толуолсульфонилокси(тозилат), п-бромбензолсульфонилокси(брозилат), п-нитробензолсульфонилокси(нозилат) или метилсульфонилокси(мезилат) групп,
с получением соединения формулы (VIIa)

и последующее снятие тритильной защитной группы без осуществления каких-либо стадий выделения при осуществлении стадии (б), причем стадия (б) представляет собой однореакторный процесс.
2. Способ получения олмесартан медоксомила, который включает:
а) алкилирование этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе в присутствии основания с получением этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-цианобифенил}метилимидазол-5-карбоксилата (Vб), кристаллизацию полученного соединения (Vб), причем ацетонитрил используют как растворитель при проведении реакции и как растворитель при кристаллизации; либо
б) гидролиз соединения (Vб), этерификацию с соединением формулы (VI)

где R представляет собой уходящую группу, выбранную из галогена, п-толуолсульфонилокси (тозилат), п-бромбензолсульфонилокси(брозилат), п-нитробензолсульфонилокси(нозилат) или метилсульфонилокси (мезилат) групп, с получением соединения формулы (VIIб)

и последующую реакцию циклоприсоединения по цианогруппе с превращением ее в тетразольную группу; либо
в) реакцию циклоприсоединения по цианогруппе с превращением ее в тетразольную группу, приводящую к получению соединения формулы (Vв)

последующую защиту тетразольной группы с получением соединения формулы (Va)

однореакторный процесс, включающий гидролиз соединения формулы (Va), этерификацию с соединением формулы (VI) с получением соединения формулы (VIIa)

и снятие тритильной защитной группы без осуществления каких-либо стадий выделения по ходу выполнения указанного процесса.
3. Способ по п.1 или 2, где указанный органический растворитель, который используют в качестве растворителя при проведении реакции и в качестве растворителя при кристаллизации, частично отгоняют.
4. Способ по п.1, где соединение формулы (Va) после фильтрации суспендируют в воде и перекристаллизовывают из того же растворителя, что используют в реакции алкилирования.
5. Способ по п.2, где соединение формулы (Vb) после фильтрации суспендируют в воде и перекристаллизовывают из того же растворителя, что используют в реакции алкилирования.
6. Способ по любому из пп.1-5, в котором используемым основанием является карбонат калия.
7. Способ по п.1, где гидролиз соединения формулы (Va) и этерификацию с соединением формулы (VI) выполняют в диметилацетамиде.
8. Способ по п.2, где гидролиз соединения формулы (Vb) и этерификацию с соединением формулы (VI) выполняют в диметилацетамиде.
9. Способ по п.1 или 2, где снятие тритильной защитной группы с тритилированного олмесартан медоксомила проводят в присутствии кислоты, выбранной из органической кислоты, неорганической кислоты, их производных и их смесей, и сорастворителя.
10. Способ по п.9, где сорастворитель может быть выбран из спиртов, кетонов, нитрилов или воды.
11. Способ по п.10, где концентрация сорастворителя составляет до 30% (об./об.), предпочтительно до 20% (об./об.).
12. Способ по п.9, при котором снятие тритильной защитной группы с тритилированного олмесартан медоксомила проводят в этилацетате в присутствии соляной кислоты.
13. Способ по п.1 или 2, отличающийся тем, что после завершения снятия защиты с тетразольной группы в этилацетате реакционную смесь охлаждают предпочтительно до комнатной температуры и нейтрализуют водным раствором неорганического основания до значения рН не более 6, предпочтительно до значения рН от 3 до 5 и выделяют продукт.
14. Способ по п.13, отличающийся тем, что фазы растворителей разделяют, причем водную фазу могут повторно экстрагировать органическим растворителем, предпочтительно этилацетатом, объединенные органические фазы концентрируют и охлаждают и выпавший в осадок олмесартан медоксомил выделяют.
15. Способ по п.13, отличающийся тем, что трифенилкарбинол оставляют растворенным в реакционной смеси и не осаждают.
16. Способ по п.1 или 2, где реакционную смесь после снятия защитной группы нейтрализуют до значения рН не более 6.
Текст
СПОСОБ ПОЛУЧЕНИЯ ОЛМЕСАРТАН МЕДОКСОМИЛА Настоящее изобретение относится к улучшенному способу получения олмесартана и его фармацевтически приемлемых солей и эфиров в качестве активного ингредиента лекарственного средства для лечения гипертонии и связанных с ней болезней и состояний. 014026 Область изобретения Настоящее изобретение относится к улучшенному способу получения олмесартана и его фармацевтически приемлемых солей и эфиров в качестве активных ингредиентов лекарственного препарата для лечения гипертонии и связанных с ней болезней и состояний. Техническая проблема В медицине олмесартан медоксомил, химически описываемый как (5-метил-2-оксо-1,3-диоксолен 4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-(тетразол-5-ил)фенил]фенилметилимидазол-5 карбоксилат, широко применяют для лечения гипертонии и связанных с ней болезней и состояний из-за его способности ингибировать ангиотензин-превращающий фермент. Будучи антагонистом рецепторов ангиотензина II, олмесартан медоксомил лишен побочных эффектов антагонистов кальция, проявляет высокую стабильность и очевидные лечебные эффекты. Предшествующий уровень техники В публикации ЕР 0503785 В 1 раскрыт способ получения олмесартан медоксомила, включающий среди прочего реакцию (5-метил-2-оксо-1,3-диоксолен-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата и 4-[2-тритилтетразол-5-ил)фенил]бензил бромида в N,N-диметилацетамиде в присутствии карбоната калия или реакцию этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5 карбоксилата и 4-[2-тритилтетразол-5-ил)фенил]бензил бромида в N,N-диметилформамиде в присутствии гидрида натрия. В примере 70 раскрыто алкилирование этил 4-(1-гидрокси-1-метилэтил)-2 пропилимидазол-5-карбоксилата 4'-бромметилбифенил-2-карбонитрилом в N,N-диметилацетамиде в присутствии трет-бутоксида калия. Общим для всех раскрытых способов является то, что алкилированный продукт подвергают колоночной хроматографии для достижения приемлемой чистоты. Описано,что для получения эфира полученный продукт гидролизуют при помощи гидроксида щелочного металла,соль выделяют и далее этерифицируют. На последней стадии удаляют тритильную защитную группу путем реакции тритильного эфира медоксомила в уксусной кислоте. В публикации J. Med. Chem., 39 (1996), 323-338 описано, что стадию алкилирования 4-[2-тритилтетразол-5-ил)фенил]бензил бромида или его аналогов в имидазольное промежуточное соединение выполняют в N,N-диметилацетамиде в присутствии трет-бутоксида калия. К реакционной смеси добавляют этанол и воду и продукт экстрагируют в этанол. Очистку продукта выполняют с использованием колоночной флэш-хроматографии (этанол/гексан, 1:2) и, возможно, с дополнительной кристаллизацией из изопропилового эфира, гексана, этанола или их смесей. В публикации ЕР 0796852 В 1 авторы раскрывают более безопасное и более легкое получение 5-замещенных тетразолов без применения Bu3SnN3. Способ включает реакцию нитрила с неорганической солью азида в ароматическом углеводородном растворителе в присутствии соли амина. В публикации WO 2004/085428 описан новый способ получения олмесартан медоксомила. При этом способе производят раскрытие кольца в 4,4-диметил-2-пропил-1-4-[2-(трифенилметилтетразол-5 ил)фенил]фенилметил-4,6-дигидрофуран[3,4d]имидазол-6-оне и образующуюся в результате 4-(1 гидрокси-1-метилэтил)-2-пропил-1-4-[2-(трифенилметилтетразол-5-ил)фенил]фенилметилимидазол-5 карбоновую кислоту затем конденсируют с 4-бром(или -хлор)метил-5-метил-2-окси-1,3 диоксигетероциклопентеном под действием щелочи. После снятия трифенилметильной защитной группы получают олмесартан медоксомил. Публикация WO 2004/083213 относится к соединениям, представленным следующей формулой (II),и их фармацевтически приемлемым солям, а также к способу их получения. Они используются в качестве промежуточных соединений для получения антагонистов рецепторов ангиотензина II, например олмесартан медоксомила Общие недостатки способов предшествующего уровня техники заключаются в том, что предложенные способы, кроме применения колоночной хроматографии, включают дополнительные стадии выделения, которые, как признано, уменьшают выход и делают любой способ трудоемким. Также применение некоторых растворителей, например уксусной кислоты, на последних стадиях реакции требует дополнительных стадий кристаллизации/очистки, поскольку известно, что особенно уксусная кислота может приводить к образованию стойких примесей в процессе сушки и ее также трудно удалить из фармацевтически активного соединения, когда она присутствует в качестве остаточного растворителя. Ввиду недостатков предшествующего уровня техники цель настоящего изобретения заключается в обеспечении альтернативного способа получения олмесартан медоксомила, который может быть быстро осуществлен, является экономичным и дает желаемое соединение с высокой чистотой.-1 014026 Краткое описание изобретения Вышеупомянутую проблему решают созданием улучшенного способа синтеза для изготовления олмесартана и фармацевтически приемлемых солей и сложных эфиров, который включает стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2(тритилтетразол-5-ил)фенил]бензил бромида (IVa) или 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе и в присутствии основания, где ацетонитрил используют как в качестве растворителя в реакции, так и в качестве растворителя при кристаллизации. Согласно первому воплощению настоящее изобретение относится к улучшенному способу синтеза для изготовления олмесартан медоксомила, который включает стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) в органическом растворителе и в присутствии основания, где один и тот же растворитель используют как растворитель в реакции и как растворитель при кристаллизации; и однореакторный способ, состоящий из гидролиза этилового эфира V, этерификации с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI) и последующего снятия защитной тритильной группы без каких-либо стадий выделения в ходе способа. Согласно второму воплощению настоящее изобретение относится к улучшенному синтезу для изготовления олмесартан медоксомила, который включает стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе и в присутствии основания, где один и тот же растворитель используют как растворитель в реакции и как растворитель при кристаллизации; и способ, состоящий из гидролиза этилового эфира, этерификации с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI) и последующей реакции циклоприсоединения по цианогруппе с превращением ее в тетразольную группу. Было неожиданно обнаружено, что при получении олмесартан медоксомила стадия алкилирования приводит к гораздо более высоким выходам и более низкому содержанию примесей, если ее проводить в ацетонитриле в качестве растворителя и в присутствии основания, выбранного, например, из карбонатов или гидроксидов, например карбоната калия, карбоната натрия, гидроксида калия, гидроксида натрия и гидроксида лития, вместо применения N,N-диметилформамида в качестве растворителя, известного в уровне техники. Кроме того, оказалось, что ацетонитрил также превосходно подходит в качестве растворителя при кристаллизации, так что экстракция вторым, отличным растворителем, не смешивающимся с водой, может быть пропущена, так же как и очистка продукта колоночной хроматографией. Главным образом эта особенность ацетонитрила также может быть подходящей средой для кристаллизации, делает способ очень выгодным для промышленного производства, поскольку очистка с использованием колоночной хроматографии редко является применимой в промышленном масштабе. Схема 1-2 014026 Когда стадию алкилирования выполняют при помощи 4'-бромметилбифенил-2-карбонитрила (IVб),снятие тритильной защитной группы заменяют на реакцию циклоприсоединения и эту реакцию также могут проводить перед гидролизом этилового эфира и этерификацией с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI). Реакция циклоприсоединения с образованием тетразольной группы может быть выполнена по любой методике, известной из уровня техники, например при помощиBu3SnN3, NaN3/ZnCl2, или как описано в публикации ЕР 0796852 В 1. Необязательно, для проведения очистки могут также использовать тритильную защитную группу или любую другую подходящую защитную группу, известную специалисту в данной области В дополнительном аспекте настоящее изобретение также предлагает улучшенный способ получения фармацевтического состава, содержащего олмесартан медоксомил высокой чистоты, демонстрирующий чистоту (ВЭЖХ) более чем 99,5%, предпочтительно более чем 99,6%, более предпочтительно 99,7%, еще более предпочтительно 99,8% и наиболее предпочтительно 99,9% и с содержанием индивидуальных примесей менее чем 0,1% (все в мас.%). В дополнительном аспекте настоящее изобретение предлагает олмесартан медоксомил, по существу, свободный от дегидро и N-алкилированных примесей со структурными формулами и соответственно. Далее описаны предпочтительные воплощения настоящего изобретения. Краткое описание графических материалов На чертеже фигуры представлена типичная порошковая дифракционная рентгенограмма аморфного олмесартан медоксомила.-3 014026 Подробное описание изобретения Настоящее изобретение относится к улучшенному синтезу для изготовления олмесартан медоксомила, который включает алкилирование этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5 карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) или 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе и в присутствии основания,где один и тот же растворитель, ацетонитрил, используют и как растворитель в реакции, и как растворитель при кристаллизации. Во втором аспекте настоящего изобретения раскрыт однореакторный способ, который следует за стадией алкилирования, состоящий из гидролиза этилового эфира (Va), этерификации с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена и последующего снятия тритильной защитной группы без каких-либо стадий выделения в ходе способа. Если реакцию алкилирования выполняют при помощи 4'-бромметилбифенил-2-карбонитрила (IVб), второй аспект настоящего изобретения включает способ, состоящий из гидролиза этилового эфира, этерификации с 4-замещенным производным метил-5 метил-2-оксо-1,3-диоксолена (VI), и последующей реакциии циклоприсоединения по цианогруппе с образованием тетразольной группы. Первое воплощение настоящего изобретения относится к улучшенному синтезу для изготовления олмесартан медоксомила, который включает:i) стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) в органическом растворителе и в присутствии основания, где один и тот же растворитель используют как растворитель в реакции и как растворитель при кристаллизации; иii) однореакторный способ, включающий гидролиз этилового эфира, этерификацию с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI) и последующее снятие тритильной защитной группы без каких-либо стадий выделения в ходе способа. Второе воплощение настоящего изобретения относится к улучшенному синтезу для изготовления олмесартан медоксомила, который включает стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе и в присутствии основания, где один и тот же растворитель используют как растворитель в реакции и как растворитель при кристаллизации; и способ, включающий гидролиз этилового эфира, этерификацию с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI), и последующую реакцию циклоприсоединения по цианогруппе с образованием тетразольной группы. Когда стадию алкилирования выполняют при помощи 4'-бромметилбифенил-2-карбонитрила (IVб),снятие тритильной защитной группы может быть заменено реакцией циклоприсоединения, а также может быть проведено перед гидролизом этилового эфира и этерификацией при помощи 4-замещенного производного метил-5-метил-2-оксо-1,3-диоксолена (VI). Возможно, после завершения стадии алкилирования этил 4-(1-гидрокси-1-метилэтил)-2 пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида(IVa) или 4'-бром-метилбифенил-2-карбонитрила (IVб), органический растворитель частично испаряют,чтобы облегчить кристаллизацию продукта. Если требуется, алкилированный продукт (Va-в) можно также суспендировать в воде и перекристаллизовать из того же самого растворителя, который используют в реакции алкилирования. В предпочтительном воплощении настоящее изобретение относится к улучшенному синтезу для получения олмесартан медоксомила, который включает:i) стадию алкилирования этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) в ацетонитриле и в присутствии карбоната калия в качестве основания с получением соединения Va, где ацетонитрил используют как растворитель в реакции и как растворитель при кристаллизации; иii) однореакторный способ, состоящий из гидролиза этилового эфира, этерификации с 4-замещенным производным метил-5-метил-2-оксо-1,3-диоксолена (VI), предпочтительно 4-хлорметил 5-метил-2-оксо-1,3-диоксоленом, и последующее снятие тритильной защитной группы без каких-либо стадий выделения в ходе способа, где снятие тритильной защитной группы выполняют в EtOAc и в присутствии HCl и сорастворителя. Возможно, после завершения реакции алкилирования ацетонитрил частично испаряют, чтобы облегчить кристаллизацию продукта (Va). Если необходимо, продукт также можно суспендировать в воде и перекристаллизовать из ацетонитрила. Неожиданно оказалось, что применение одного и того же органического растворителя, ацетонитрила, в качестве растворителя в реакции и в качестве растворителя при кристаллизации в реакции алкилирования 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) привело к гораздо более высокому выходу (88%) и более низкому содержанию примесей, несмотря на то, что стадия экстракции вторым растворителем,-4 014026 несмешивающимся с водой, пропущена, также как очистка продукта колоночной хроматографией. Продукты, этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-(тритилтеразол-5-ил)фенил]фенил-метилимидазол-5-карбоксилат (Va) и этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-цианобифенилметилимидазол-5-карбоксилат (Vб), выделяют кристаллизацией. Возможно, реакционную смесь концентрируют приблизительно до 1/3 исходного объема и охлаждают до температуры ниже 25 С. После того как выпавший в осадок продукт отфильтрован, его суспендируют в воде для удаления избытка неорганического основания. Продукт можно перекристаллизовать из органического растворителя, например ацетонитрила. Во втором аспекте настоящего изобретения раскрыт однореакторный способ, который следует за стадией алкилирования, состоящий из гидролиза этилового эфира, этерификации с 4 замещенным производным метил-5-метил-2-оксо-1,3-диоксолена, и последующего снятия тритильной защитной группы без каких-либо стадий выделения в ходе способа. 4-Замещенное производное метил-5 метил-2-оксо-1,3-диоксолена (VI) является соединением, где R представляет собой подходящую уходящую группу, например галоген, такой как хлор, бром и йод, п-толуолсульфонилокси(тозилат),п-бромбензолсульфонилокси(брозилат), п-нитробензолсульфонилокси(нозилат) или метилсульфонилокси(мезилат)группу. В предпочтительном воплощении используют 4-хлорметил-5-метил-2-оксо-1,3 диоксолен. Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-(тритилтетразол-5-ил)фенил]фенилметилимидазол-5-карбоксилат (Va) растворяют в соответствующем растворителе и добавляют первое основание, и реакционную смесь перемешивают в течение 24 ч, предпочтительно в течение 4-12 ч при температуре между 15 и 30 С, предпочтительно при температуре окружающей среды. После завершения гидролиза этильного фрагмента 4-замещенное производное метил-5-метил-2 оксо-1,3-диоксолена, предпочтительно 4-хлорметил-5-метил-2-оксо-1,3-диоксолен могут просто добавлять к реакционной смеси вместе со вторым основанием без предварительного выделения образующейся соли. Перед добавлением смесь охлаждают предпочтительно до температуры 10 С или ниже, более предпочтительно до 5 С или ниже и оба реагента добавляют при выбранной температуре. Реакционную смесь нагревают в течение 5 ч, предпочтительно в течение 2 ч при температуре от комнатной температуры до 100 С, предпочтительно при температуре от 20 до 70 С, более предпочтительно от 30 до 40 С. В качестве растворителей для гидролиза и стадии этерификации можно использоватьN,N-диметилацетамид, другие амидные растворители, нитрилы или любой другой полярный и смешивающийся с водой растворитель. В предпочтительном воплощении этим растворителем является ДМА. В качестве первых оснований используют гидроксиды щелочных металлов, алкоксиды металлов или карбонаты в количестве 1-1,5 экв. В предпочтительном воплощении в качестве первого основания используют гидроксид натрия. В качестве вторых оснований используют гидроксиды щелочных или щелочно-земельных металлов, алкоксиды металлов или карбонаты, в количестве 0,5-1,5 экв. В предпочтительном воплощении в качестве второго основания используют карбонат калия. В предпочтительном воплощении, настоящее изобретение предлагает олмесартан медоксомил, по существу, свободный от дегидро- и N-алкилированных примесей. Данное изобретение также предлагает способ синтеза олмесартан медоксомила, включающего дегидро- и N-алкилированные примеси в количестве не более 0,2%, предпочтительно не более 0,10%, который включает анализ и отбор коммерческих партий 4-замещенного производного метил-5-метил-2-оксо-1,3 диоксолена или очистку 4-замещенного производного метил-5-метил-2-оксо-1,3-диоксолена и анализ очищенного продукта; применение партий производного диоксолена с чистотой более чем 90%, предпочтительно более чем 95%. После завершения стадии этерификации реакционную смесь охлаждают до температуры ниже 15 С, к реакционной смеси добавляют второй несмешивающийся с водой растворитель вместе с небольшим количеством солевого раствора и экстрагируют. Органические фракции собирают, промывают солевым раствором и сушат над осушителем, например, безводным сульфатом натрия или магния (VI). Экстракции проводят при температуре ниже 25 С. В качестве несмешивающихся с водой растворителей для экстракции могут быть выбраны растворители с низкой растворимостью олмесартан медоксомила, например сложные эфиры, простые эфиры,галогенированные углеводороды. Предпочтительно несмешивающимся с водой растворителем является этилацетат. На стадии снятия тритильной защитной группы второй несмешивающийся с водой растворитель,который добавляют после стадии этерификации, могут частично выпаривать и добавлять кислоту, выбранную из органической кислоты, неорганической кислоты, их производных и их смесей, и сорастворитель. Сорастворитель может быть выбран из спиртов, кетонов, нитрилов или воды. Концентрация сорастворителя составляет до 30% (об./об.), предпочтительно до 20% (об./об.). Предпочтительно сорастворителем является МеОН или EtOH. В качестве растворителей для стадии снятия тритильной защитной группы используют те же самые растворители, что и для упомянутой выше стадии экстракции. Предпоч-5 014026 тительно растворителем для стадии снятия тритильного фрагмента является этилацетат. Реакционную смесь нагревают до температуры от 15 до 30 С, предпочтительно реакцию проводят при комнатной температуре в течение времени до 5 ч, предпочтительно в течение 3 ч. Кислота может быть выбрана из HCl, HBr, HI, H2SO4, H3PO4 или других подходящих неорганических кислот. Предпочтительно добавляют HCl в виде раствора в воде или в органическом растворителе или в газообразной форме. После завершения процесса снятия защиты реакционную смесь охлаждают предпочтительно до комнатной температуры и нейтрализуют раствором неорганического основания до значения рН не более 6, предпочтительно до значения рН от 3 до 5. Фазы разделяют и водную фазу могут повторно экстрагировать органическим растворителем. Собранные органические фазы сушат, фильтруют и концентрируют. Смесь охлаждают и осаждают продукт. Конечный продукт (I) отфильтровывают и промывают свежим органическим растворителем и побочный продукт реакции (трифенилкарбинол) остается полностью растворенным в фильтрате. Подходящими неорганическими основаниями, используемыми для нейтрализации, являются NaOH,KOH, LiOH, Ca(OH)2, Na2CO3, NaHCO3, K2CO3, KHCO3, неорганические фосфаты. Предпочтительно используют водный раствор NaOH. Сырой продукт можно перекристаллизовать из органических растворителей, таких как ацетаты, кетоны, спирты, нитрилы и их смеси. Кристаллические формы продуктов, перекристаллизованные из вышеупомянутых растворителей, были теми же, как описано в публикации Annual Report of SankyoResearch Laboratories. Vol. 55 (2003). Если раствор олмесартан медоксомила медленно кристаллизуют из изобутанола или тетрагидрофурана (ТГФ), то получают новую форму олмесартан медоксомила, которая характеризуется интервалом температуры плавления 182-184 С и дифракционной рентгенограммой с пиками при 7,4, 9,0, 9,6, 11,6, 12,0, 13,4, 16,0, 17,9, 21,10,2 угла 2. Порошковые дифракционные рентгенограммы получали на порошковом дифрактометре Phillips PW3040/60 X'Pert PRO; излучение CuKa 1,541874A, 3231. Фраза "медленная кристаллизация" означает кристаллизацию, при которой раствор олмесартан медоксомила оставляют кристаллизоваться более чем на 8 ч. В течение процесса кристаллизации и при фильтровании могут образовываться сольваты олмесартан медоксомила. Аморфную форму олмесартан медоксомила получают, когда упаривают раствор олмесартан медоксомила в органическом растворителе, например эфирах, галогенированных углеводородах и спиртах,остаток сушат или лиофилизируют, и она характеризуется температурой стеклования приблизительно 120-140 С и порошковой дифракционной рентгенограммой, изображенной на фигуре. Когда олмесартан медоксомил кристаллизуют из органических растворителей при значении рН менее чем 2, выделяют соль олмесартан медоксомила. Это значение рН может быть достигнуто путем добавления неорганических кислот, например HCl, H2SO4, H3PO4, HBr, или сильных органических кислот,таких как CF3COOH, НСООН, СН 3 СООН, уксусного ангидрида и т.д. Важно контролировать размер частиц олмесартан медоксомила при его получении. Средний размер частиц, полученных и/или использованных в данной работе, составляет 1-80 мкм, предпочтительно меньше 30 мкм; такие частицы обычно получают кристаллизацией олмесартан медоксомила из органических растворителей или их смесей с водой при перемешивании. При отсутствии перемешивания кристаллизация из органических растворителей или их смесей с водой может также привести к образованию частиц большего размера, например со средним диаметром более 100 мкм, и такие частицы перед их применением в фармацевтических составах необходимо перемалывать или обрабатывать любым другим способом, который уменьшает размер частицы. Однако недостаточно контролировать только средний размер частиц, важно также контролировать и распределение частиц по размерам. Определены следующие параметры для контроля распределения частиц по размерам: 10% частиц имеют размер менее 20 мкм, предпочтительно менее 15 мкм; 50% частиц имеют размер менее 80 мкм, предпочтительно менее 50 мкм,90% частиц имеют размер менее 170 мкм, предпочтительно менее 140 мкм. Средний размер частиц и распределение частиц по размерам важны, чтобы гарантировать, что технологический процесс пригоден для осуществления в промышленном масштабе, т.е. не вызывает сегрегации компонентов смеси для таблетирования в случае, если ее не таблетируют/прессуют сразу же после получения смеси для таблетирования. Было неожиданно обнаружено, что добавление к фармацевтическому составу небольших количеств кислого вещества, приводящее к уменьшению значения рН по меньшей мере на 0,2 единицы рН по сравнению с фармацевтическим составом, не включающим кислое вещество, улучшает профили растворимости и улучшает стабильность продукта, поскольку образуется меньше продуктов распада за больший промежуток времени. Добавляемые кислые вещества могут иметь неорганическую или органическую природу, например можно применять кислые неорганические соли, например фосфаты или органические кислоты и/или их соли, например лимонную, аскорбиновую, винную, яблочную, стеариновую, пальмитиновую, молочную, глюконовую, пропионовую кислоты, аминокислоты и т.д.-6 014026 Составы олмесартан медоксомила могут быть получены согласно хорошо известным технологическим процессам, например путем прямого прессования или влажной грануляции (при помощи воды или органических растворителей, например МеОН, или их смесей), сухой грануляции или лиофилизации. Предпочтительно используют способ прямого прессования. Способ прямого прессования может быть осуществлен следующим образом:(а) активный ингредиент добавляют к смеси наполнителей и прессуют или(б) активный ингредиент смешивают вместе с наполнителями и прессуют. Твердую лекарственную форму (например, ядра таблеток), возможно, могут покрывать. Способ прямого прессования осуществляют благодаря низкому процентному содержанию активного ингредиента от общей массы таблетки. Подразумевается, что термин "процентное содержание" обозначает массовую долю активного ингредиента в процентах от общей массы таблетки. Подразумевается,что термин "низкое процентное содержание активного ингредиента" обозначает, что активный ингредиент составляет меньше 20% от общей массы таблетки. Необязательно, наполнители могут обрабатывать влажной грануляцией с использованием воды или органического растворителя или их смеси в качестве жидкости для грануляции. Обработка наполнителей влажной грануляцией означает гомогенизацию наполнителей и добавление жидкости для грануляции к их смеси. Необязательно, жидкость для грануляции может содержать связующее вещество или связующие вещества, по отдельности, или их смеси. Возможно, в твердый фармацевтический состав могут быть включены поверхностно-активные вещества. Поверхностно-активные вещества могут быть выбраны из группы неионогенных или ионных поверхностно-активных веществ или их смесей. Подходящие неионогенные поверхностно-активные вещества выбраны из группы алкилглюкозидов, алкилмальтозидов, алкилтиоглюкозидов, лаурил макроголглицеридов, полиоксиэтилен алкилфенолов, алкильных эфиров полиоксиэтилена, эфиров полиэтиленгликоля с жирными кислотами, эфиров полиэтиленгликоля и глицерина с жирными кислотами, эфиров полиоксиэтилен сорбитана с жирными кислотами, полиоксиэтилен-полиоксипропилен блок-сополимеров, эфиров полиглицерина с жирными кислотами, полиоксиэтилен глицеридов, соединений полиоксиэтилена с растительным маслом, соединений полиоксиэтилена с гидрогенизированным растительным маслом, стеринов и их смесей. Предпочтительными неионогенными поверхностно-активными веществами являются эфиры полиоксиэтиленсорбитана с жирными кислотами, которые продаются под торговыми наименованиями Полисорбат (Polysorbate) или Твин (Tween). Подходящие ионные поверхностно-активные вещества выбраны из группы солей жирных кислот,солей желчных кислот, фосфолипидов, эфиров фосфорных кислот, карбоксилатов, сульфатов, сульфонатов и их смеси. Предпочтительным ионным поверхностно-активным веществом является натрий лаурилсульфат. Фармацевтическая композиция согласно изобретению может включать 0,1-10%, предпочтительно 0,1-5 мас.% поверхностно-активного вещества. Подходящее перемешивающее устройство при прямом прессовании или, возможно, при влажной грануляции, как описано выше, представляет собой обычное оборудование, используемое для смешивания активных ингредиентов, наполнителей или комбинации активного(ых) ингредиента(ов) и наполнителей. Обычное оборудование представляет собой неподвижные (пассивные) смесители, смесители с псевдоожиженным слоем, смесители диффузионного смешения, двухконусные смесители диффузионного смешения, двухконусные смесители, турбулентные, кубические, планетарные, Y-, V-образные или высокоскоростные смесители, барабанные смесители т.д. В случае влажного гранулирования, как описано выше, оборудование выбирают из стандартного оборудования для сушки, т.е. сушилки с псевдоожиженным слоем, поддонов и т.д. Твердая лекарственная форма может быть, например, лекарственной формой немедленного высвобождения, легкоплавкой лекарственной формой, лекарственной формой контролируемого высвобождения, лиофилизированной лекарственной формой, лекарственной формой отсроченного высвобождения,лекарственной формой длительного высвобождения, лекарственной формой периодического высвобождения, смешанной лекарственной формой немедленного и контролируемого высвобождения или их комбинацией. Твердая лекарственная форма предпочтительно представляет собой таблетированный состав, который может, возможно, иметь покрытие. Твердая лекарственная форма предпочтительно представляет собой лекарственную форму немедленного высвобождения, имеющую преимущества, относящиеся к биодоступности активного соединения. Если выбрана лекарственная форма немедленного высвобождения, то для специалиста в этой области ясно, что количество вещества или веществ, контролирующих высвобождение, по отдельности или их смеси, которое используется для формирования внешней части, определяется на основе различных параметров, например желательных свойств доставки, включая количество активного ингредиента или вещества, которое будет доставлено, активный ингредиент или желательную скорость высвобождения вещества и размер частиц микроматрицы.-7 014026 Фармацевтическая композиция может состоять из: 1-99 мас.%, предпочтительно 5-50 мас.%, более предпочтительно 5-15 мас.% олмесартан медоксомила; 1-99 мас.%, предпочтительно 20-99 мас.%, более предпочтительно 50-99 мас.% разбавителя; 1-90 мас.%, предпочтительно 1-50 мас.% связующего вещества; 1-50 мас.%, предпочтительно 2-40 мас.% дезинтегриранта или супердезинтегриранта; 0,1-10% смазывающего вещества; 0,1-10 мас.%, предпочтительно 0,1-5 мас.% поверхностно-активного вещества и возможно, 0,1-10% покрывающего слоя в виде пленки Наполнители, присутствующие в композиции по изобретению, могут представлять собой разбавители, например микрокристаллическую целлюлозу, порошкообразную целлюлозу, лактозу (безводную или моногидрат), прессованный сахар, фруктозу, декстраты, иные сахара, например маннит, силиконизированную микрокристаллическую целлюлозу, гидрофосфат кальция, карбонат кальция, лактат кальция или смешанные разбавители. Предпочтительно наполнители включают по меньшей мере один разбавитель, выбранный из микрокристаллической целлюлозы и моногидрата лактозы. Композиция по настоящему изобретению может также включать связующие вещества, например повидон, микрокристаллическую целлюлозу, гидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, низкозамещенную гидроксипропилцеллюлозу (включающую от 5 до 16 мас.% гидроксипропильных групп),гидроксипропилметилцеллюлозу или другой эфир целлюлозы, крахмал, пептизированный крахмал или полиметакрилат или смесь связующих веществ. Предпочтительно, чтобы наполнители включали по меньшей мере одно связующее вещество, выбранное из микрокристаллической целлюлозы и низкозамещенной гидроксипропилцеллюлозы. Кроме того, могут присутствовать также дезинтегранты и/или супердезинтегранты, такие как крахмалы (например, кукурузный крахмал, картофельный крахмал), модифицированные крахмалы (натрия крахмалгликолят), модифицированная целлюлоза (кроскармеллоза, т.е. поперечно-связанная натрий карбоксиметилцеллюлоза), поперечно-связанный поливинилпирролидон (кросповидон), микрокристаллическая целлюлоза, натрия карбоксиметилцеллюлоза, Амберлит (Amberlite), альгиновая кислота, альгинат натрия, смола гуаровая, геллановая камедь, Ксантан SM (Xanthan SM). В качестве дезинтегранта микрокристаллическую целлюлозу предпочтительно применяют в количестве 5-15 мас.%. Предпочтительно,чтобы наполнители включали по меньшей мере один дезинтегрант или супердезинтегрант, выбранный из кроскармеллозы, кросповидона и микрокристаллической целлюлозы. Кроме того, в качестве наполнителей также могут присутствовать скользящие вещества, такие как стеариновая кислота, стеарат магния, стеарат кальция, натрий лаурилсульфат, гидрогенизированное растительное масло, гидрированное касторовое масло, натрий стеарил фумарат, тальк, макроголы. Предпочтительно, чтобы наполнители включали по меньшей мере одно скользящее вещество, выбранное из стеарата магния, талька и макроголов. Наполнители могут обладать несколькими функциями, т.е. один наполнитель может быть разбавителем и дополнительно связующим веществом, связующим и дезинтегрирующим веществом и т.д. Возможно, ядра таблеток могут быть покрыты обычными материалами, используемыми для пленочного покрытия. Составы пленочного покрытия обычно содержат следующие компоненты: полимер(ы), пластификатор(ы), краситель(и)/ вещество(а), делающее состав непрозрачным, наполнитель(и). В суспензии для пленочного покрытия могут использоваться незначительные количества ароматизаторов,поверхностно-активных веществ и восков. Огромное большинство полимеров, используемых для пленочного покрытия, является либо производными целлюлозы, например эфирами целлюлозы, либо акриловыми полимерами и сополимерами. Иногда встречаются высокомолекулярные полиэтиленгликоли,поливинилпирролидон, поливиниловый спирт и воскообразные материалы. Типичными эфирами целлюлозы являются гидроксиэтилцеллюлоза, гидроксипропилцеллюлоза,гидроксипропилметилцеллюлоза, метилцеллюлоза. Акриловые полимеры включают группу синтетических полимеров с разнообразными функциональными возможностями. Некоторые из них могут быть дополнительно модифицированы для увеличения набухаемости и проницаемости путем включения таких веществ, как водорастворимые эфиры целлюлозы и крахмалы, чтобы обеспечить полный распад/растворение пленки. Обычно используемые пластификаторы могут быть разделены на три группы: полиолы (глицерин,пропиленгликоль, макроголы); органические эфиры (эфиры фталевой кислоты, дибутилсебацинат, эфиры лимонной кислоты, триацетин) и масла/глицериды (касторовое масло, ацетилированные моноглицериды,фракционированное кокосовое масло). Красители/вещества, делающие состав непрозрачным, подразделяют на несколько групп: органические красители и лаки на их основе; неорганические красящие вещества; природные красящие вещества. Комбинацию различных материалов из каждой группы можно осуществить в определенном отношении. Суспензии для пленочного покрытия могут использоваться в виде готовых к применению препаратов, которые доступны на рынке.-8 014026 Дисперсию для пленочного покрытия можно получить при использовании различных растворителей, например воды, спиртов, кетонов, эфиров, хлорированных углеводородов, предпочтительно воды. В частности, предпочтительным является состав суспензии для покрытия (в пересчете на сухое вещество), который включает: 1-99 мас.% полимера, предпочтительно 1-95% полимера; 1-50 мас.% пластификатора, предпочтительно 1-40% пластификатора; 0,1-20% красителя/вещества, делающего состав непрозрачным, предпочтительно 0,1-10% красителя/ вещества, делающего состав непрозрачным. Лекарственная форма немедленного высвобождения может также включать вещество, которое облегчает обработку агентов, контролирующих высвобождение. Такие вещества обычно называются пластификаторами. Предпочтительные пластификаторы включают ацетилированные моноглицериды, бутилфталилбутил гликолят, дибутилтартрат, диэтилфталат, диметилфталат, этил фталилэтил гликолят,глицерин, этиленгликоль, пропиленгликоль, триэтилцитрат, триацетин, трипропионин, диацетин, дибутилфталат, ацетилмоноглицерид, полиэтиленгликоли, касторовое масло, многоатомные спирты, эфиры уксусной кислоты, триацетат глицерина, ацетил триэтилцитрат, дибензилфталат, дигексилфталат, бутилоктил фталат, диизононилфталат, утилоктил фталат, диоктилазелаинат, эпоксидированный таллат,триизоктилтримеллитат, ди(этилгексил)фталат, ди(н-октил)фталат, диоктилфталат, ди(изодецил)фталат,ди(н-ундецил)фталат, ди(н-тридецил)фталат, три(2-этилгексил)тримеллитат, ди(2-этилгексил)адипинат,ди(2-этилгексил)себацинат, ди(2-этилгексил)азелаинат, дибутилсебацинат, глицерил монокаприлат, глицерин дистеарат и глицерил монокапринат. Профили растворения определяли на приборе для определения растворимости Erweka DT80 с помощью спектрофотометра Agilent 8453 с диодной матрицей, в искусственном желудочном соке, при значении рН 2,0, скорости вращения шпинделя 50 об/мин. Предпочтительно однородность состава составляет менее чем приблизительно 7,5%, предпочтительно менее чем приблизительно 5% и более предпочтительно менее чем приблизительно 5%. Наиболее предпочтительно однородность состава составляет менее чем приблизительно 3%. Более низким пределом для однородности состава предпочтительно является ноль. Однородность состава определяют при помощи соответствующего теста фармакопеи США (Постоянство лекарственных средств, Общая глава 905 (Uniformity of dosage units, General Chapter 905), 2005), в котором 10 таблеток оценивают по отдельности, после чего вычисляют среднее арифметическое и относительное стандартное отклонение (ОСО). Критерии фармакопеи США лежат в пределах 85-115% от заявленного на этикетке содержания, и ОСО составляет не более 6%. Содержание олмесартан медоксомила в таблетках определяют при помощи ВЭЖХ, применяют метод внешнего стандарта и ультрафиолетовое детектирование. Значения рН 20% (масс./об.) суспензии измельченных таблеток в воде определяли при помощи калиброванного рН-метра при температуре 20-25 С. Настоящее изобретение иллюстрировано следующими примерами, но не ограничивается ими. Температуру плавления измеряли на аппарате Koffler для измерения температуры плавления и инфракрасные (ИК) спектры регистрировали на спектрометре Paragon 100 Perkin-Elmer FT-IR. Примеры Получение олмесартан медоксомила. Пример 1. 17,3 г (124,8 ммольь) K2CO3, 15 г (62,4 ммоль) этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол 5-карбоксилата (III) и 38,3 г (68,7 ммоль) 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) суспендировали в 750 мл ацетонитрила. Суспензию нагревали с обратным холодильником до завершения реакция (7 ч). Отгоняли 510 мл ацетонитрила и концентрат охлаждали до 23-25 С. Смесь перемешивали при этой температуре в течение ночи, затем суспензию охлаждали до 0 С и перемешивали при этой температуре в течение 1 ч. Сырой продукт (Va) отфильтровывали и промывали дважды по 20 мл охлажденного ацетонитрила. Влажный продукт суспендировали в 450 мл воды, перемешивали в течение 1,5 ч и после этого отфильтровывали. Масса высушенного продукта (Va) составила 39,5 г (89%). Т.пл.=165-169 С. ИК: 1666, 1525, 1291, 1446, 1177, 881, 756, 699, 640. Пример 2. 36,0 г (50,3 ммоль) этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-(тритилтетразол-5 ил)фенил]фенилметилимидазол-5-карбоксилата (Va) и 3,0 г (75,4 ммоль) NaOH суспендировали в 413 мл диметилацетамида. Затем суспензию перемешивали при комнатной температуре в течение 20 ч и после этого добавляли 6,9 г (50,3 ммоль) K2CO3. Смесь охлаждали до 0 С и медленно добавляли раствор 15,4 г (70,4 ммоль) 4-хлорметил-5-метил-2-оксо-1,3-диоксолена в 39 мл диметилацетамида. Смесь медленно нагревали до 50 С и перемешивали при этой температуре в течение 2 ч. После завершения этерификации смесь охлаждали до 10 С, выливали в смесь 625 мл этилацетата и 625 мл 10%-ного раствораNaCl и перемешивали при 25 С в течение 15 мин. Фазы разделяли, органическую фазу промывали дважды по 500 мл 10%-ного раствора NaCI, сушили над Na2SO4 и фильтровали. Фильтрат концентрировали-9 014026 до половины (приблизительно 270 г) при уменьшенном давлении. К образующемуся раствору добавляли 80 мл этанола и 8,3 мл (100 ммоль) концентрированной HCl и перемешивали при 24-26 С в течение 3 ч. К охлажденной смеси добавляли 600 мл воды и доводили значение рН суспензии до 5 путем добавления 5 М раствора NaOH. Фазы перемешивали в течение 15 мин и разделяли. Водную фазу повторно экстрагировали при помощи 150 мл этилацетата. Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали при уменьшенном давлении. Добавляли 560 мл этилацетата и смесь снова упаривали. После этого добавляли 300 мл этилацетата, смесь охлаждали до 20 С и перемешивали в течение 1 ч, отфильтровывали и промывали 20 мл свежего этилацетата. Выход продукта (I) составил 21 г (75%). Кристаллизация олмесартан медоксомила. Пример 3. 1,11 г олмесартан медоксомила растворяли в 12,5 мл 2-бутанона при температуре кипения. Раствор медленно охлаждали до комнатной температуры и перемешивали при этой температуре в течение 20 ч. В течение этого процесса медленно кристаллизовался олмесартан медоксомил. Продукт отфильтровывали и сушили в течение 18 ч при комнатных условиях. Получали 0,98 г олмесартан медоксомила. Кристаллическая форма продукта была той же самой, как описано в публикации Annual Report ofSankyo Research Laboratories. Vol. 55 (2003). Пример 4. 1,2 г олмесартан медоксомила растворяли в 8,5 мл 2-бутанона при температуре кипения. Раствор медленно охлаждали до комнатной температуры и перемешивали при этой температуре в течение 20 ч. В течение этого процесса медленно кристаллизовался олмесартан медоксомил. Затем суспензию охлаждали до 0 С и перемешивали при этой температуре в течение 2 ч. Продукт отфильтровывали и сушили при уменьшенном давлении при 30-40 С в течение 3 ч. Получали 0,98 г олмесартан медоксомила. Кристаллическая форма продукта была той же самой, как описано в публикации Annual Report ofSankyo Research Laboratories. Vol. 55 (2003). Пример 5. 0,5 г олмесартан медоксомила растворяли в 4 мл изобутанола при температуре кипения. Раствор медленно охлаждали до 0 С и перемешивали при этой температуре в течение 3 ч. В течение этого процесса медленно кристаллизовался олмесартан медоксомил. Продукт отфильтровывали и сушили в течение 18 ч при комнатных условиях. Получали кристаллическую форму олмесартан медоксомила (0,45 г),которая является новой полиморфной формой. Дифракционная рентгенограмма показывает пики при 7,4,9,0, 9,6, 11,6, 12,0, 13,4, 16,0, 17,9, 21,10,2 угла 2. Порошковые дифракционные рентгенограммы были получены на порошковом дифрактометре Phillips PW3040/60 X'Pert PRO; излучение CuKa 1,541874 А,3231. Т.пл.=182-184C. Пример 6. 2 г олмесартан медоксомила растворяли в 30 мл ТГФ при температуре кипения. Растворитель медленно испаряли при уменьшенном давлении до сухого остатка. В течение этого процесса медленно кристаллизовался олмесартан медоксомил. Продукт собирали и сушили в течение 18 ч при комнатных условиях. Получали кристаллическую форму олмесартан медоксомила (1,86 г). Т.пл.=182-184 С. Пример 7. 0,5 г олмесартан медоксомила растворяли в 18 мл метиленхлорида при температуре кипения. Растворитель медленно испаряли при уменьшенном давлении до сухого остатка. Получали аморфную форму олмесартан медоксомила (0,43 г). Пример 8. 2 г олмесартан медоксомила растворяли в 20 мл гептана при температуре кипения. Раствор медленно охлаждали до комнатной температуры и перемешивали при этой температуре в течение 3 ч. В течение этого процесса медленно выпадал в осадок олмесартан медоксомил. Продукт отфильтровывали и сушили в течение 18 ч при комнатных условиях. Получали аморфную форму олмесартан медоксомила (0,45 г). Т.пл.=120-140 С. Пример 9. 2 г олмесартан медоксомила растворяли в 45 мл изопропилового спирта при температуре кипения. Раствор медленно охлаждали до комнатной температуры и перемешивали при этой температуре в течение 3 ч. В течение этого процесса медленно выпадал в осадок олмесартан медоксомил. Продукт отфильтровывали и сушили в течение 18 при комнатных условиях. Получали 1,96 г олмесартан медоксомила. Средний размер частицы: 40 мкм. Кристаллическая форма продукта была той же самой, как описано в публикации Annual Report of- 10014026 Пример 10. 1,1 г олмесартан медоксомила растворяли в 15 мл ацетона при температуре кипения. Раствор концентрировали при уменьшенном давлении приблизительно до половины первоначального объема. Концентрат охлаждали до 0 С, отфильтровывали и сушили. Выделяли 0,9 г олмесартан медоксомила. Кристаллическая форма продукта была той же самой, как описано в публикации Annual Report ofSankyo Research Laboratories. Vol. 55 (2003). Пример 11. 12 г олмесартан медоксомила растворяли в 174 мл этанола при температуре кипения. Раствор медленно охлаждали до комнатной температуры без перемешивания. Смесь оставляли при комнатной температуре на ночь (18 ч). Продукт отфильтровывали и сушили в течение 3 ч в вакуумной сушилке. Получали 7,3 г олмесартан медоксомила. Средний размер частицы: 253 мкм. Кристаллическая форма продукта была той же самой, как описано в публикации Annual Report ofSankyo Research Laboratories. Vol. 55 (2003). Фармацевтический состав олмесартан медоксомила. Пример 12. Гомогенизировали 40 г олмесартан медоксомила, 104 г микрокристаллической целлюлозы, 230 г моногидрата лактозы и 40 г низкозамещенной гидроксипропилцеллюлозы. В заключение добавляли 6 г стеарата магния с получением смеси для прессования. Смеси прессовали до ядер с теоретической массой 210 мг. Ядра покрывали суспензией для пленочного покрытия, содержащей (в пересчете на сухую часть суспензии для пленочного покрытия) гидроксипропилцеллюлозу (43,75 мас.%), гидроксипропилцеллюлозу (37,5 мас.%), тальк (6,25 мас.%) и диоксид титана (12,5 мас.%). Теоретическая масса таблетки, покрытой пленкой, составляет 218 мг. Пример 13. Ядра из примера 1 покрывают при помощи готовой к применению суспензии для пленочного покрытия, содержащей (в пересчете на сухую часть суспензии для пленочного покрытия) частично гидролизованный поливиниловый спирт (40 мас.%), диоксид титана (25 мас.%), макрогол (20,2 мас.%) и тальк(14,8 мас.%). Теоретическая масса таблетки, покрытой пленкой, составляет 218 мг. Пример 14. 52 г микрокристаллической целлюлозы, 114 г моногидрата лактозы, 20 г низкозамещенной гидроксипропилцеллюлозы и 2 г натрий лаурилсульфата гомогенизируют и распыляют с очищенной водой в грануляторе с псевдоожиженным слоем. Гранулят просеивают. К грануляту добавляют 40 г олмесартан медоксомила, 52 г микрокристаллической целлюлозы, 114 г моногидрата лактозы и 20 г низкозамещенной гидроксипропилцеллюлозы и перемешивают. В заключение добавляют 6 г стеарата магния с получением смеси для прессования. Смесь для прессования прессуют до ядер с теоретической массой 210 мг. Ядра покрывают суспензией для покрытия из примера 12 или 13. Примеры 15 а-22 а (получение смеси для прессования). Компоненты (1-5) гомогенизируют в высокоскоростном смесителе. В заключение добавляют стеарат магния (6) с получением смеси для прессования. Размер частицы (т.е. средний размер частицы, 10% частиц меньше определенного размера, 10% частиц больше определенного размера, 50% частиц больше определенного размера) активного ингредиента (олмесартан медоксомила) относится к объемному диаметру частицы, определенному рассеиванием лазерного луча образцом, включающим 100-800 мг активного ингредиента, диспергированного в 5-8 мл растительного масла (т.е. подсолнечного масла) и не содержащим каких-либо солюбилизаторов или поверхностно-активных веществ, с использованием прибора Malvern Mastersizer MS2000. Потери при высушивании смеси для прессования измеряли с использованием галогенного анализатора влажности Mettler Toledo HR73 при 85 С в течение 20 мин. Результаты представлены в табл. 1. Использовали различные типы микрокристаллической целлюлозы (2). Кроме того, в различных примерах использовали различные типы моногидрата лактозы (3). Активный ингредиент со средним размером частиц 8 мкм, 10% частиц меньше 1,2 мкм, 10% частиц больше 7,2 мкм, 50% частиц больше 16,8 мкм.b Активный ингредиент со средним размером частиц 4 мкм, 10% частиц меньше 0,7 мкм, 10% частиц больше 2,6 мкм, 50% частиц больше 7,3 мкм.LH-111 Коммерчески доступная низкозамещенная гидроксипропилцеллюлоза. Примеры 15 б-22 б (получение ядер таблеток). Смеси для прессования (15 а-22 а) прессовали, используя круглые ядра таблеток (1 б-8 б) с теоретической массой 210 мг, на автоматической роторной таблеточной машине при определенном рабочем давлении. Твердость прессованных ядер таблеток и время их распада (в минутах) в очищенной воде при 37 С измеряли согласно Европейской фармакопее. Хрупкость всех образцов была менее 1%. Результаты представлены в табл. 2. Таблица 2 Кроме того, прессованные ядра таблеток (15 б-22 б) покрывали в автоматической ванне для покрытия основанной на воде суспензией смеси, готовой к использованию, для пленочного покрытия коммерчески доступной как Opacity F28751 II HP белый. Теоретическая масса покрытых таблеток, содержащих ядра таблеток (1 б-8 б), составляла 218 мг. Значения рН и содержание активного ингредиента в таблетках,покрытых пленкой, представлены в табл. 3. Таблица 3 Пример 23. Таблетки композиции согласно примеру 15 получали с использованием олмесартан медоксомила с различным размером частицы: средний размер частицы 34 мкм, 10% частиц меньше 13,1 мкм, 10% частиц больше 60,0 мкм, 50% частиц больше 30,3 мкм. Наблюдалось существенное различие в растворимости олмесартан медоксомила в кислых средах, используемых в примерах 15 и 22, причем активный ингредиент, используемый в примере 15, является предпочтительным и дает лучшую биодоступность по сравнению с композицией примера 22. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения олмесартан медоксомила, который включает: а) алкилирование этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4-[2-(тритилтетразол-5-ил)фенил]бензил бромида (IVa) в органическом растворителе в присутствии основания с получением этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-(тритилтетразол-5 ил)фенил]фенилметилимидазол-5-карбоксилата (Va), кристаллизацию полученного соединения (Va) из реакционной смеси, причем ацетонитрил используют как растворитель при проведении реакции и как растворитель при кристаллизации; и б) гидролиз соединения формулы (Va), этерификацию с 4-замещенным производным метил-5 метил-2-оксо-1,3-диоксолена формулы (VI) где R представляет собой уходящую группу, выбранную из галогена, п-толуолсульфонилокси(тозилат), п-бромбензолсульфонилокси(брозилат), п-нитробензолсульфонилокси(нозилат) или метилсульфонилокси(мезилат) групп,с получением соединения формулы (VIIa) и последующее снятие тритильной защитной группы без осуществления каких-либо стадий выделения при осуществлении стадии (б), причем стадия (б) представляет собой однореакторный процесс. 2. Способ получения олмесартан медоксомила, который включает: а) алкилирование этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата (III) при помощи 4'-бромметилбифенил-2-карбонитрила (IVб) в органическом растворителе в присутствии основания с получением этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-4-[2-цианобифенилметилимидазол-5 карбоксилата (Vб), кристаллизацию полученного соединения (Vб), причем ацетонитрил используют как растворитель при проведении реакции и как растворитель при кристаллизации; либо б) гидролиз соединения (Vб), этерификацию с соединением формулы (VI) где R представляет собой уходящую группу, выбранную из галогена, п-толуолсульфонилокси(тозилат), п-бромбензолсульфонилокси(брозилат), п-нитробензолсульфонилокси(нозилат) или метилсульфонилокси(мезилат) групп, с получением соединения формулы (VIIб) и последующую реакцию циклоприсоединения по цианогруппе с превращением ее в тетразольную группу; либо в) реакцию циклоприсоединения по цианогруппе с превращением ее в тетразольную группу, приводящую к получению соединения формулы (Vв) последующую защиту тетразольной группы с получением соединения формулы (Va) однореакторный процесс, включающий гидролиз соединения формулы (Va), этерификацию с соединением формулы (VI) с получением соединения формулы (VIIa) и снятие тритильной защитной группы без осуществления каких-либо стадий выделения по ходу выполнения указанного процесса. 3. Способ по п.1 или 2, где указанный органический растворитель, который используют в качестве растворителя при проведении реакции и в качестве растворителя при кристаллизации, частично отгоняют. 4. Способ по п.1, где соединение формулы (Va) после фильтрации суспендируют в воде и перекристаллизовывают из того же растворителя, что используют в реакции алкилирования. 5. Способ по п.2, где соединение формулы (Vb) после фильтрации суспендируют в воде и перекристаллизовывают из того же растворителя, что используют в реакции алкилирования. 6. Способ по любому из пп.1-5, в котором используемым основанием является карбонат калия. 7. Способ по п.1, где гидролиз соединения формулы (Va) и этерификацию с соединением формулы(VI) выполняют в диметилацетамиде. 8. Способ по п.2, где гидролиз соединения формулы (Vb) и этерификацию с соединением формулы(VI) выполняют в диметилацетамиде. 9. Способ по п.1 или 2, где снятие тритильной защитной группы с тритилированного олмесартан медоксомила проводят в присутствии кислоты, выбранной из органической кислоты, неорганической кислоты, их производных и их смесей, и сорастворителя. 10. Способ по п.9, где сорастворитель может быть выбран из спиртов, кетонов, нитрилов или воды. 11. Способ по п.10, где концентрация сорастворителя составляет до 30% (об./об.), предпочтительно до 20% (об./об.). 12. Способ по п.9, при котором снятие тритильной защитной группы с тритилированного олмесартан медоксомила проводят в этилацетате в присутствии соляной кислоты. 13. Способ по п.1 или 2, отличающийся тем, что после завершения снятия защиты с тетразольной группы в этилацетате реакционную смесь охлаждают предпочтительно до комнатной температуры и нейтрализуют водным раствором неорганического основания до значения рН не более 6, предпочтитель- 14014026 но до значения рН от 3 до 5 и выделяют продукт. 14. Способ по п.13, отличающийся тем, что фазы растворителей разделяют, причем водную фазу могут повторно экстрагировать органическим растворителем, предпочтительно этилацетатом, объединенные органические фазы концентрируют и охлаждают и выпавший в осадок олмесартан медоксомил выделяют. 15. Способ по п.13, отличающийся тем, что трифенилкарбинол оставляют растворенным в реакционной смеси и не осаждают. 16. Способ по п.1 или 2, где реакционную смесь после снятия защитной группы нейтрализуют до значения рН не более 6.
МПК / Метки
МПК: C07D 405/14
Метки: способ, олмесартан, получения, медоксомила
Код ссылки
<a href="https://eas.patents.su/16-14026-sposob-polucheniya-olmesartan-medoksomila.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения олмесартан медоксомила</a>
Производные 5-0-дезозаминил-6-0-метилэритронолида а, способ их получения и их применение для получения биологически активных продуктов

Номер патента: 575
Опубликовано: 29.12.1999
Авторы: Дельтиль Мишель, Мазюри Алан, Бонне Алан
МПК: C07H 17/08
Метки: получения, применение, биологически, способ, активных, производные, 5-0-дезозаминил-6-0-метилэритронолида, продуктов
Формула / Реферат:
1. Соединения формулы (I): в которой или R1 представляет радикал алкил, содержащий до 8 атомов углерода, замещенный одним или несколькими радикалами алкила, содержащими до 8 атомов углерода, или одним или несколькими радикалами арила, содержащими до 14 атомов углерода, или R1 представляет радикал арил, содержащий до 14 атомов углерода, который может быть замещен одним или несколькими радикалами алкил, алкенил или алкинил, содержащими до 8...
Цеолитный катализатор l-типа, способ его получения, способ получения ароматических углеводородов, способ получения бензина

Номер патента: 3559
Опубликовано: 26.06.2003
Авторы: Иннес Роберт А., Сугимото Митио, Фукунага Тецуя
МПК: B01J 29/61, C07C 5/41, C10G 35/095...
Метки: ароматических, цеолитный, углеводородов, l-типа, получения, катализатор, бензина, способ
Формула / Реферат:
1. Цеолитный катализатор L-типа, который получают при нанесении на цеолит L-типа платинового компонента, одного или более галогеновых компонентов и одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, при этом наносимое количество одного или более компонентов металлов, выбранных из Ib группы Периодической таблицы, находится в интервале от 0,001 до 3 мас.% из расчета на общую массу катализатора, молярное отношение...
Пирролидинил-, пиперидинил- или гомопиперидинилзамещенные производные, способ их получения, их применение в медицине, содержащая их фармацевтическая композиция и способ ее получения

Номер патента: 4648
Опубликовано: 24.06.2004
Авторы: Ван Эмелен Кристоф, Вигеринк Пит Том Берт Поль, Вирсхюэрен Вим Гастон, Де Брюин Марсель Франс Леопольд
МПК: C07D 405/14, A61P 9/00, A61K 31/505...
Метки: способ, получения, пиперидинил, применение, композиция, гомопиперидинилзамещенные, медицине, содержащая, фармацевтическая, пирролидинил, производные
Формула / Реферат:
1. Соединение формулы (I) его стереохимически изомерная форма или его фармацевтически приемлемая кислотно-аддитивная соль, в которых R1, R2 и R3 каждый представляет водород; -Z1-Z2- представляет двухвалентный радикал формулы -O-CH(R4)-CR2-CH2- (a-4), R4 представляет водород; Alk представляет C1-6алкандиил, необязательно замещенный гидроксигруппой; представляет двухвалентный радикал формулы в которой m равно 0 или 1; R6...
Катализатор для получения сложных эфиров,способ получения сложного эфира и способ получения сложного полиэфира с участием такого катализатора

Номер патента: 11171
Опубликовано: 27.02.2009
Авторы: Макинтош Кэлам Гарри, Партридж Мартин Грэхэм, Хэнратти Алан Джозеф
МПК: C08G 63/85, B01J 31/04
Метки: эфиров,способ, сложных, получения, сложного, участием, катализатора, полиэфира, катализатор, эфира, способ, такого
Формула / Реферат:
1. Катализатор для получения сложного эфира в реакции этерификации, состоящий из продукта взаимодействия: a) соединения титана, циркония или гафния; b) 2-оксикарбоновой кислоты; c) четвертичного аммониевого соединения, выбранного из группы, состоящей из гидроксида тетраэтиламмония и гидроксида тетраметиламмония, и d) соединения цинка. 2. Катализатор по п. 1, в котором соединением титана, циркония или гафния является алкоголят, имеющий формулу...
Способ получения дихлорпропанола, способ получения эпихлоргидрина и способ получения эпоксидных смол

Номер патента: 13681
Опубликовано: 30.06.2010
Авторы: Жильбо Патрик, Краффт Филипп
МПК: C07C 31/42, C07C 29/62, C07C 31/36...
Метки: эпоксидных, способ, получения, дихлорпропанола, смол, эпихлоргидрина
Формула / Реферат:
1. Способ получения дихлорпропанола, в котором вводят во взаимодействие глицерин, или сложный эфир глицерина, или их смесь, общее содержание металлов в которых, выраженное в расчете на элементы, выше или равно 0,1 мкг/кг и ниже или равно 1000 мг/кг, и агент хлорирования.2. Способ по п.1, в котором общее содержание металлов ниже или равно 500 мг/кг и который характеризуется по меньшей мере одним из следующих признаков:содержание железа в...
Предыдущий патент: Композиции антитела против cd3
Следующий патент: Способ получения четвертичных аммониевых солей с кислотой
Случайный патент: Способ выработки газа-фумиганта ( варианты ), композиция для выработки газа-фумиганта ( варианты ) и способ фумигации (варианты )