Аминохинолины в качестве ингибиторов киназ
Номер патента: 23998
Опубликовано: 31.08.2016
Авторы: Сингхаус Роберт Р., Бэри Майкл Джонатан, Романо Джозеф Дж., Ван Грен З., Касиллас Линда Н., Хейл Памела А., Чарнли Адам Кеннет, Марквис Роберт В., Мелманн Джон Ф.



























Формула / Реферат
1. Соединение формулы (I)

в которой R1 представляет собой Н;
R2 представляет собой -SORa или -SO2Ra, где Ra является (C1-C6)алкильной или 4-7-членной гетероциклоалкильной группой, где
указанный (C1-C6)алкил необязательно замещен заместителем, выбранным из гидроксила, (C1-C6)алкокси, (C1-C6)алкокси(C2-C6)алкокси, амино, (C1-C4-алкил)амино-, (C1-C4-алкил)-(C1-C4-алкил)амино-,
указанный 4-7-членный гетероциклоалкил необязательно замещен 1-2 группами, независимо выбранными из (C1-C4)алкила, и где указанный 4-7-членный гетероциклоалкил содержит один гетероатом, выбранный из группы, состоящей из N, О и S;
R3 представляет собой галоген, гидрокси, (C1-C3)алкил-, (C1-C3)алкокси-, галоген(C1-C2)алкил-, (C1-C3)алкокси(C1-C3)алкил-, (C1-C3)алкокси(C2-C3)алкокси-, гидрокси(C1-C3)алкил- или гидрокси(C2-C3)алкокси-;
Z представляет собой фенил формулы

где RZ1 представляет собой Н, галоген, -CF3, (C1-C4)алкил или (C1-C4)алкокси;
RZ2 представляет собой Н, галоген, -CF3, (C1-C4)алкил или (C1-C4)алкокси;
RZ3 представляет собой Н, галоген, циано, (C1-C4)алкил, галоген(C1-C4)алкил, (C1-C4)алкокси, фенокси, фенил(С1-С4)алкокси, гидроксил, гидрокси(C1-C4)алкил- или аминокарбонил, где фенильная группа указанного фенокси или фенил(C1-C4)алкокси- необязательно замещена 1-3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, (С1-С4)алкила и (C1-C4)алкокси; и
RZ4 представляет собой гидроксигруппу, гидрокси(C1-C4)алкил или (C1-C4)алкокси; или
Z представляет собой бензотиазолил, необязательно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, (C1-C4)алкила, -CF3 и (C1-C4)алкокси; или
Z представляет собой пиразолил формулы

в которой R12 и R13 независимо выбраны из группы, состоящей из метила или трифторметила; и
R14 представляет собой Н или метил; или
Z представляет собой индазолил или пиразоло[3,4-b]пиридинильную группу, каждая из которых необязательно замещена 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C4)алкила или (C1-C4)алкокси;
при условии, что соединение не представляет собой N-(4-хлор-2-фторфенил)-7-метокси-6-[(2-метоксиэтил)сульфинил]-4-хинолинамин и 3-[[7-бром-6-(метилсульфонил)-4-хинолинил]амино]-4-метилфенол,
или его фармацевтически приемлемая соль.
2. Соединение или его фармацевтически приемлемая соль по п.1, где Z представляет собой бензотиазол-6-ил, необязательно замещенный хлором, фтором, -CF3, метилом или метокси.
3. Соединение или его фармацевтически приемлемая соль по п.1, где Z представляет собой пиразолил; R12 представляет собой метил; R13 представляет собой метил или трифторметил и R14 представляет собой Н.
4. Соединение или его фармацевтически приемлемая соль по любому из пп.1-3, где R2 представляет собой -SO2Ra.
5. Соединение или его фармацевтически приемлемая соль по любому из пп.1-4, где Ra представляет собой (C1-C4)алкил, необязательно замещенный заместителем, выбранным из группы, состоящей из гидроксила и (C1-C2)алкокси; или Ra представляет собой 5-6-членный гетероциклоалкил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из (C1-C4)алкильных групп, где 5-6-членный гетероциклоалкил содержит 1 гетероатом, выбранный из N, О или S.
6. Соединение или его фармацевтически приемлемая соль по любому из пп.1-4, где Ra представляет собой -СН3, -СН(СН3)2, -С(СН3)3, -СН2СН2ОН или тетрагидро-2Н-пиран-4-ил.
7. Соединение или его фармацевтически приемлемая соль по любому из пп.1-6, где R3 представляет собой галоген, гидрокси, (С1-С3)алкил-, (C1-C3)алкокси-, гидрокси(C1-C3)алкил- или гидрокси(C2-C3)алкокси-.
8. Соединение, которое представляет собой
N-(4,5-диметил-1Н-пиразол-3-ил)-7-(метилокси)-6-(тетрагидро-2Н-пиран-4-илсульфинил)-4-хинолинамин;
6-[(1,1-диметилэтил)сульфинил]-N-(4,5-диметил-1Н-пиразол-3-ил)-7-(метилокси)-4-хинолинамин;
N-(4,5-диметил-1Н-пиразол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
2-((4-((4,5-диметил-1Н-пиразол-3-ил)амино)-7-метилхинолин-6-ил)сульфонил)этанол;
6-(трет-бутилсульфонил)-N-(5-фтор-1Н-пиразоло[3,4-b]пиридин-3-ил)-7-метоксихинолин-4-амин;
N-[4-хлор-3-(метилокси)фенил]-6-[(1,1-диметилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
6-[(1,1-диметилэтил)сульфонил]-N-(4,5-диметил-1Н-пиразол-3-ил)-7-(метилокси)-4-хинолинамин;
N-1,3-бензотиазол-5-ил-7-бром-6-(метилсульфонил)-4-хинолинамин;
N-[4-хлор-3-(метилокси)фенил]-7-(метилокси)-6-(тетрагидро-2Н-пиран-4-илсульфонил)-4-хинолинамин;
N-1,3-бензотиазол-5-ил-7-(метилокси)-6-(тетрагидро-2Н-пиран-4-илсульфонил)-4-хинолинамин;
2-{[4-{[4-хлор-3-(метилокси)фенил]амино}-7-(метилокси)-6-хинолинил]сульфонил}этанол;
N-(5-фтор-1Н-индазол-3-ил)-7-(метилокси)-6-(тетрагидро-2Н-пиран-4-илсульфонил)-4-хинолинамин;
2-({4-[(4,5-диметил-1Н-пиразол-3-ил)амино]-7-(метилокси)-6-хинолинил]сульфонил}этанол;
N-[4-хлор-3-(метилокси)фенил]-6-[(1-метилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
N-1,3-бензотиазол-5-ил-6-[(1-метилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
N-(4,5-диметил-1Н-пиразол-3-ил)-6-[(1-метилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
N-(5-фтор-1Н-индазол-3-ил)-6-[(1-метилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
2-{[4-(1,3-бензотиазол-5-иламино)-7-(метилокси)-6-хинолинил]сульфонил}этанол;
6-(изопропилсульфонил)-7-метокси-N-(4-метил-5-(трифторметил)-1Н-пиразол-3-ил)хинолин-4-амин;
6-(трет-бутилсульфонил)-7-метокси-N-(4-метил-5-(трифторметил)-1Н-пиразол-3-ил)хинолин-4-амин;
6-(трет-бутилсульфонил)-N-(4,5-диметил-1Н-пиразол-3-ил)-7-этоксихинолин-4-амин;
6-(трет-бутилсульфонил)-7-этокси-N-(5-фтор-1Н-индазол-3-ил)хинолин-4-амин;
7-хлор-N-(4,5-диметил-1Н-пиразол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(4,5-диметил-1Н-пиразол-3-ил)-7-метил-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
7-хлор-N-(5-фтор-1Н-индазол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(5-фтор-1Н-индазол-3-ил)-7-метил-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(5-фтор-1Н-индазол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)-7-(трифторметил)хинолин-4-амин;
6-(трет-бутилсульфонил)-N-(5-фтор-1Н-индазол-3-ил)-7-(трифторметил)хинолин-4-амин;
6-(трет-бутилсульфонил)-N-(4,5-диметил-1Н-пиразол-3-ил)-7-метилхинолин-4-амин;
6-(трет-бутилсульфонил)-N-(5-фтор-1Н-индазол-3-ил)-7-метилхинолин-4-амин;
6-(трет-бутилсульфонил)-N-(5-фтор-1Н-индазол-3-ил)-7-метоксихинолин-4-амин;
7-бром-6-(трет-бутилсульфонил)-N-(4,5-диметил-1Н-пиразол-3-ил)хинолин-4-амин;
7-бром-N-(4-метил-5-(трифторметил)-1Н-пиразол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-1,3-бензотиазол-5-ил-6-[(1,1-диметилэтил)сульфонил]-7-(метилокси)-4-хинолинамин;
6-(трет-бутилсульфонил)-4-((4,5-диметил-1Н-пиразол-3-ил)амино)хинолин-7-ол;
или его фармацевтически приемлемая соль.
9. Соединение, которое представляет собой
6-(трет-бутилсульфонил)-N-(4,5-диметил-1Н-пиразол-3-ил)-7-метоксихинолин-4-амин;
2-((4-((4,5-диметил-1Н-пиразол-3-ил)амино)-7-метоксихинолин-6-ил)сульфонил)-2-метилпропан-1-ол;
N-(4,5-диметил-1Н-пиразол-3-ил)-6-((2,2-диметилтетрагидро-2Н-пиран-4-ил)сульфонил)-7-метоксихинолин-4-амин;
N-(4,5-диметил-1Н-пиразол-3-ил)-7-метокси-6-((4-метилтетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(4,5-диметил-1Н-пиразол-3-ил)-7-метокси-6-((2-метоксиэтил)сульфонил)хинолин-4-амин;
N-(4,5-диметил-1Н-пиразол-3-ил)-7-метокси-6-(((3R,4R)-3-метилтетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(4,5-диметил-1Н-пиразол-3-ил)-6-(((2R,6S)-2,6-диметилтетрагидро-2Н-пиран-4-ил)сульфонил)-7-метоксихинолин-4-амин;
6-(трет-бутилсульфонил)-7-хлор-N-(5-фтор-1Н-индазол-3-ил)хинолин-4-амин;
(3-((6-(трет-бутилсульфонил)-7-метоксихинолин-4-ил)амино)-4-метилфенил)метанол;
7-этокси-N-(5-фтор-1Н-индазол-3-ил)-6-(изопропилсульфонил)хинолин-4-амин;
N-(7-хлор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
6-(трет-бутилсульфонил)-N-(7-фтор-1Н-индазол-3-ил)-7-метоксихинолин-4-амин;
6-(трет-бутилсульфонил)-N-(5,7-дифтор-1Н-индазол-3-ил)-7-метоксихинолин-4-амин;
6-(трет-бутилсульфонил)-N-(6,7-дифтор-1Н-индазол-3-ил)-7-метоксихинолин-4-амин;
6-(трет-бутилсульфонил)-N-(7-хлор-1Н-индазол-3-ил)-7-метоксихинолин-4-амин;
6-(трет-бутилсульфонил)-7-метокси-N-(5-метокси-1Н-индазол-3-ил)хинолин-4-амин;
7-метокси-N-(4-метил-5-(трифторметил)-1Н-пиразол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(5,7-дифтор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(4-хлор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(6-хлор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(6,7-дифтор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
7-метокси-N-(5-метокси-1Н-индазол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(5-хлор-1Н-индазол-3-ил)-7-метокси-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
N-(7-хлор-1Н-индазол-3-ил)-7-метокси-6-((4-метилтетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
7-бром-N-(4,5-диметил-1Н-пиразол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
7-бром-N-(4,5-диметил-1Н-пиразол-3-ил)-6-(изопропилсульфонил)хинолин-4-амин;
7-бром-N-(5-фтор-1Н-индазол-3-ил)-6-(изопропилсульфонил)хинолин-4-амин;
7-бром-N-(5-фтор-1Н-индазол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
2-((6-(трет-бутилсульфонил)-4-((4,5-диметил-1Н-пиразол-3-ил)амино)хинолин-7-ил)окси)этанол;
6-(трет-бутилсульфонил)-7-(дифторметокси)-N-(4,5-диметил-1Н-пиразол-3-ил)хинолин-4-амин;
7-(дифторметокси)-N-(4,5-диметил-1Н-пиразол-3-ил)-6-((тетрагидро-2Н-пиран-4-ил)сульфонил)хинолин-4-амин;
2-((4-(бензо[d]тиазол-5-иламино)-6-(трет-бутилсульфонил)хинолин-7-ил)окси)этанол,
или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении киназа рецептор-взаимодействующего протеина-2 (RIP2), включающая соединение по любому из пп.1-9 в терапевтически приемлемом количестве или его фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество.
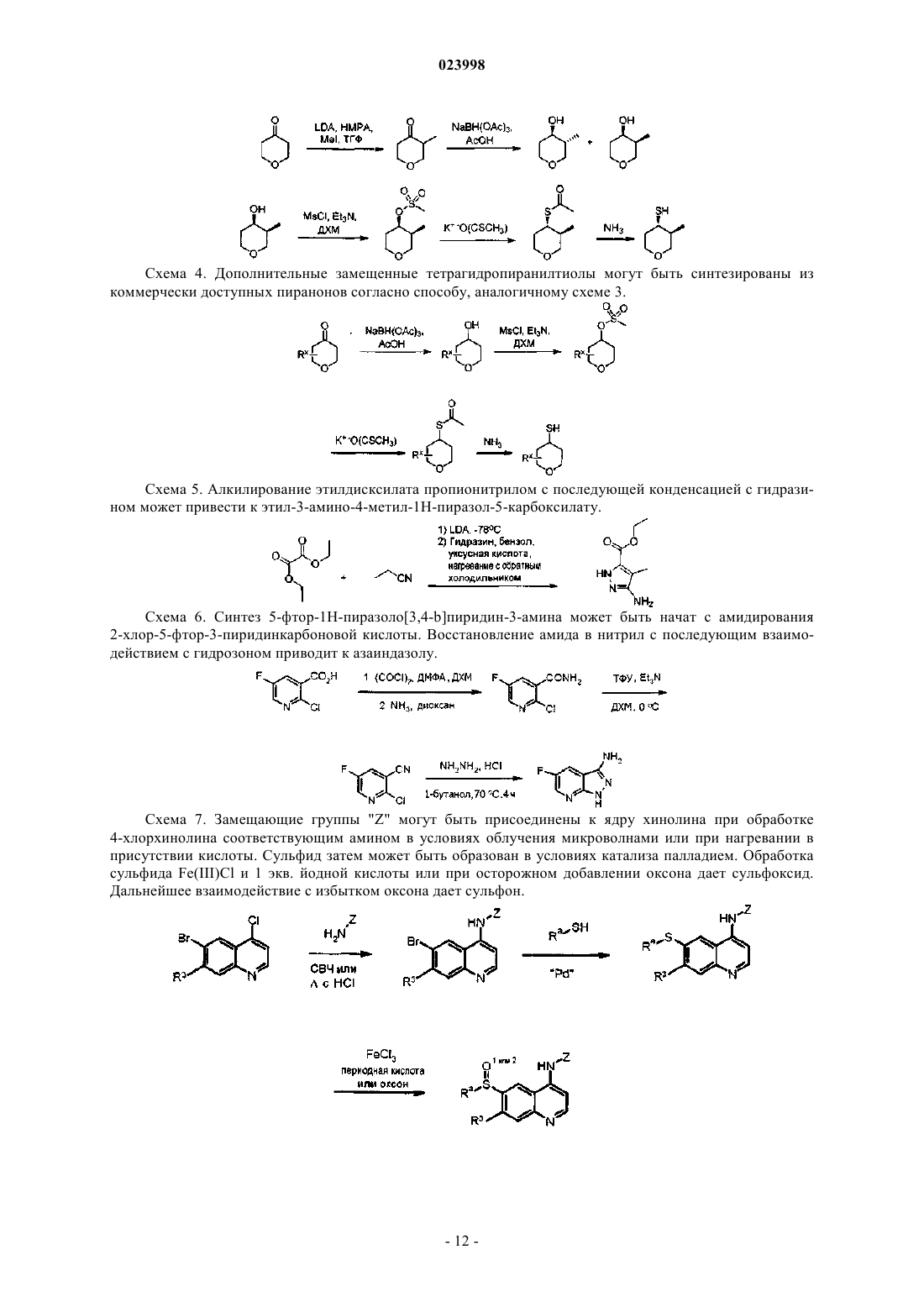
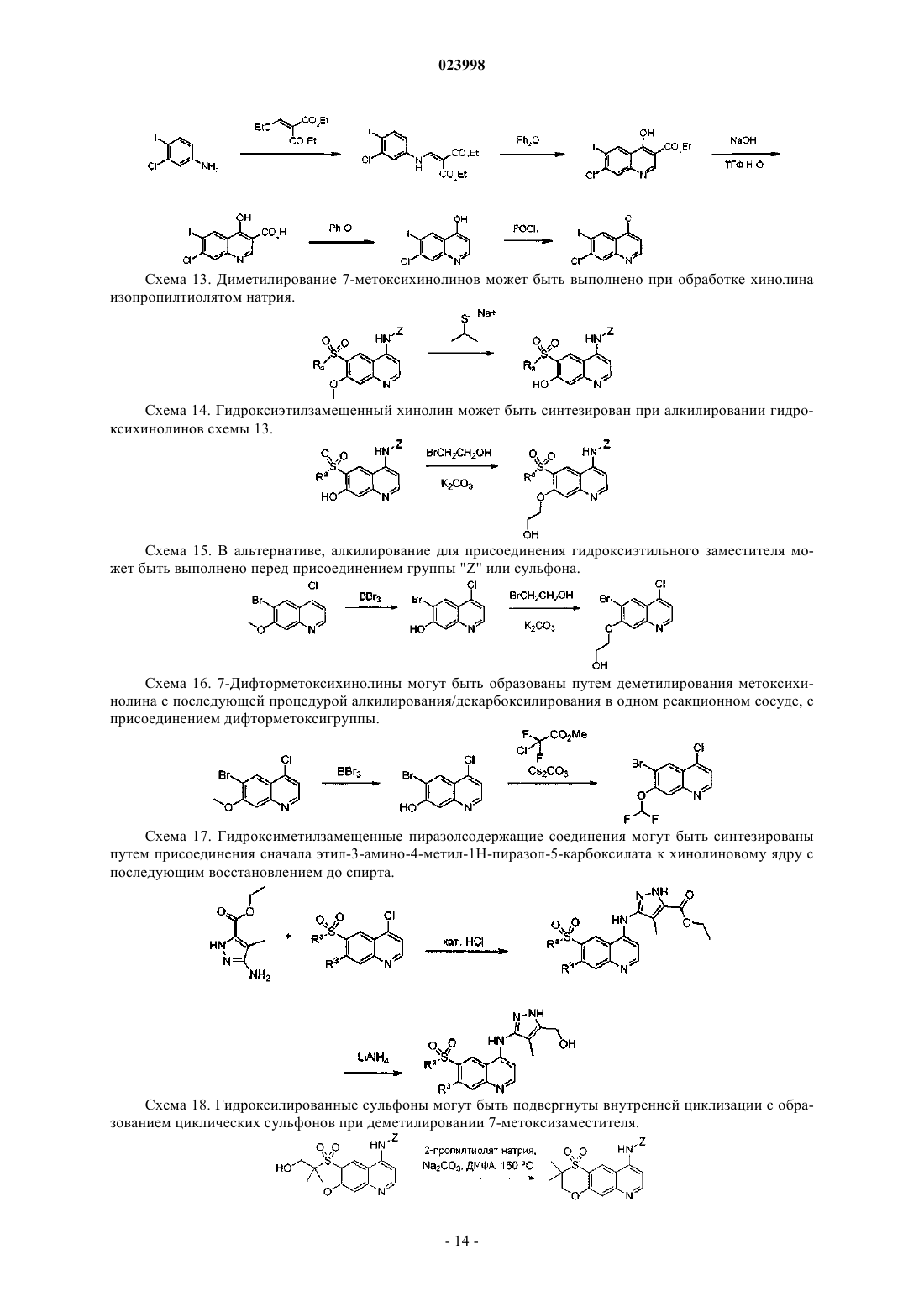
Текст
в которой R1, R2, R3 и Z определены в настоящем описании, а также способы их получения и применения.(71)(73) Заявитель и патентовладелец: ГЛЭКСОСМИТКЛАЙН ИНТЕЛЛЕКЧУАЛ ПРОПЕРТИ ДИВЕЛОПМЕНТ ЛИМИТЕД (GB) Область техники, к которой относится изобретение Настоящее изобретение относится к 4-аминохинолинам, которые ингибируют киназу RIP2, а также к фармацевтическим композициям их включающих. В частности, настоящее изобретение относится к замещенным 4-аминохинолинам в качестве ингибиторов RIP2 киназы. Уровень техники Киназа рецептор-взаимодействующего протеина-2 (RIP2), которую также называют CARD3, RICK,CARDIAK или RIPK2, является протеинкиназой из семейства TKL серин/треониновых протеинкиназ,участвующей в естественной иммунной сигнализации. Киназа RIP2 состоит из N-концевого киназного домена и С-концевого домена рекрутинга каспазы (CARD), связанные через промежуточную (IM) область 1998) J. Biol. Chem. 273, 12296-12300; (1998) Current Biology, 8, 885-889 и (1998) J. Biol. Chem. 273, 16968-16975). Домен CARD киназы RIP2 опосредует взаимодействие с другимиEMBO reports 2, 736-742). NOD1 и NOD2 являются цитоплазматическими рецепторами, которые играют ключевую роль в естественном иммунном надзоре. Они распознают грамположительные и грамотрицательные бактериальные патогены и активируются специфическими пептидогликановыми мотивами, диаминопимелиновой кислотой (т.е. DAP) и мурамилдипептидом (MDP), соответственно 2007) J.Immunol. 178, 2380-2386). После активации киназа RIP2 связывается с NOD1 или NOD2 и, по-видимому, функционирует преимущественно как молекулярный каркас, собирая другие киназы (TAK1, IKK//), участвующие вNF-В и митоген-активированной активации протеинкиназ 2006), Nature Reviews Immunology, 6, 9-20). Киназа RIP2 подвергается K63-связанному полиубиквитинилированию по лизину-209, что облегчает рекрутинг TAK1 2008) EMBO Journal, 27, 373-383). Данная посттрансляционная модификация требуется для сигнализации, поскольку мутация этого остатка препятствует NOD1/2 опосредованной активацииNF-В. Киназа RIP2 также подвергается аутофосфорилированию по серину-176 и, возможно, другим остаткам 2006) Cellular Signalling, 18, 2223-2229). Исследования с использованием мутантов с неактивной киназой (K47 А) и неселективных низкомолекулярных ингибиторов продемонстрировали, что активность киназы RIP2 важна для регуляции стабильности экспрессии и сигнализации киназы RIP2 2007)Biochem. J. 404, 179-190 и (2009) J. Biol. Chem. 284, 19183-19188). Дисрегуляция RIP2-зависимой сигнализации была связана с аутовоспалительными заболеваниями. Мутации с приобретением функции в NACHT-домене NOD2 вызывают синдром Блау, ювенильный саркоидоз, педиатрическую гранулематозную болезнь, характеризующуюся увеитом, дерматитом и артритом 2001), Nature Genetics, 29, 19-20; (2005) Journal of Rheumatology, 32, 373-375; (2005) CurrentBowel Diseases, 15, 1145-1154 и (2009) Microbes and Infection, 11, 912-918). Мутации в NOD1 были связаны с астмой 2005) Hum. Mol. Genet. 14, 935-918), а также ювенильной и внешнекишечной воспалительной болезнью кишечника 2005) Hum. Mol. Genet. 14, 1245-1250). Генетические и функциональные исследования также предположили роль RIP2-зависимой сигнализации в ряде других гранулематозных нарушений, таких как саркоидоз 2009) Journal of Clinical Immunology, 29, 78-89 и (2006) SarcoidosisVasculitis and Diffuse Lung Diseases, 23, 23-29) и гранулематоз Вегенера 2009) Diagnostic Pathology, 4,23). Мощный селективный низкомолекулярный ингибитор активности киназы RIP2 смог бы блокировать RIP2-зависимую провоспалительную сигнализацию и, таким образом, обеспечил бы благоприятный терапевтический эффект при аутовоспалительных заболеваниях, характеризуемых увеличенной и/или нарушенной активностью киназы RIP2. Сущность изобретения Изобретение направлено на 6,7-двузамещенные-4-аминохинолины. В частности, изобретение направлено на соединение формулы (I)R2 представляет собой -SORa или -SO2Ra, где Ra является (C1-C6)алкильной или 4-7-членной гетероциклоалкильной группой, где указанный (C1-C6)алкил необязательно замещен замещен заместителем, выбранным из гидроксила,(C1-C6)алкокси,(C1-C6)алкокси(C2-C6)алкокси,амино,(C1-C4-алкил)амино-,(C1-C4-алкил)-(C1-C4-алкил)амино-,указанный 4-7-членный гетероциклоалкил необязательно замещен 1-2 группами, независимо выбранными из (C1-C4)алкила, и указанный 4-7-членный гетероциклоалкил содержит один гетероатом, выбранный из группы, состоящей из N, О и S;Z представляет собой фенил формулыRZ3 представляет собой Н, галоген, циано, (C1-C4)алкил, галоген(C1-C4)алкил, (C1-C4)алкокси, фенокси, фенил(C1-C4)алкокси, гидроксил, гидрокси(C1-C4)алкил- или аминокарбонил, где фенильная группа указанного фенокси или фенил(C1-C4)алкокси- необязательно замещена 1-3 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, -CF3, (C1-C4)алкила и (C1-C4)алкокси; иZ представляет собой бензотиазолил, необязательно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, (C1-C4)алкила, -CF3 и (C1-C4)алкокси; илиZ представляет собой пиразолил формулы в которой R12 и R13 независимо выбраны из группы, состоящей из метила или трифторметила; иZ представляет собой индазолил или пиразоло[3,4-b]пиридинильную группу, каждая из которых необязательно замещена 1 или 2 заместителями, независимо выбранными из группы, состоящей из галогена, (C1-C4)алкила или (C1-C4)алкокси; при условии, что соединение не представляет собой N-(4-хлор-2-фторфенил)-7-метокси-6-[(2 метоксиэтил)сульфинил]-4-хинолинамин и 3-7-бром-6-(метилсульфонил)-4-хинолинил]амино]-4 метилфенол,или его фармацевтически приемлемую соль. Соединения формулы (I) или их фармацевтически приемлемые соли являются ингибиторами киназы RIP2 и пригодны для ингибирования киназы RIP2 путем контактирования клетки с соединением формулы (I) или его фармацевтически приемлемой солью. Соединения настоящего изобретения также могут быть пригодны для лечения опосредованногоRIP2 киназой заболевания или нарушения путем введения терапевтически эффективного количества соединения формулы (I) или фармацевтически приемлемой соли пациенту (человеку или другому млекопитающему, в особенности человеку), нуждающемуся в этом. Примеры опосредованных RIP2 киназой заболеваний или нарушений включают увеит, болезнь Крона, язвенный колит, ювенильную и внекишечную воспалительную болезнь кишечника, а также гранулематозные нарушения, такие как саркоидоз,синдром Блау, ювенильный саркоидоз и гранулематоз Вегенера. Настоящее изобретение также направлено на фармацевтическую композицию, включающую соединение формулы (I) или фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество. В частности, настоящее изобретение направлено на фармацевтическую композицию для лечения опосредованного RIP2 киназой заболевания или нарушения, где композиция включает соединение формулы (I) или фармацевтически приемлемую соль и фармацевтически приемлемое вспомогательное вещество. Краткое описание чертежей На фиг. 1 показан комбинированный цитокиновый ответ в образцах цельной крови крысы, полученных после предварительного введения крысам дозы соединения примера 1 с последующим введением дозы L18-MDP. На фиг. 2 показан комбинированный цитокиновый ответ в образцах цельной крови крысы, полученных после предварительного введения крысам дозы соединения примера 5 с последующим введением дозы L18-MDP. На фиг. 3 показан цитокиновый ответ IL-8 в образцах цельной крови крысы, полученных после предварительного введения крысам дозы соединения примера 23 с последующим введением дозыL18-MDP. На фиг. 4 показан цитокиновый ответ IL-8 в образцах цельной крови крысы, полученных после предварительного введения крысам дозы соединения примера 31 с последующим введением дозыL18-MDP. Подробное описание изобретения Альтернативные определения различных групп и замещающих групп формулы (I), представленные по всему тексту описания, предназначены для конкретного описания каждого индивидуального соединения, раскрытого в настоящем описании, в отдельности, а также групп из одного или нескольких индивидуальных соединений. Объем настоящего изобретения включают любую комбинацию таких определений групп и замещающих групп. Соединениями изобретения являются только те соединения, которые считаются "химически стабильными", как будет очевидно квалифицированным специалистам. Специалистам, квалифицированным в данной области, также будет очевидно, что в тех случаях, когда Z представляет собой пиразолил, соединения настоящего изобретения могут существовать в виде пиразольных изомеров, представленных формулой (I-A) и формулой (I-B): В случае, когда R14 представляет собой Н, соединения настоящего изобретения могут существовать в виде таутомеров (I-А) и (I-B) и могут быть представлены в виде формулы (I-C): В случае, когда R14 представляет собой метил, соединения настоящего изобретения могут существовать в виде одного из региоизомеров, представленных формулой (I-A) или формулой (I-B) или в виде их смеси. Кроме того, квалифицированным специалистам будет очевидно, что соединения настоящего изобретения, в зависимости от последующего замещения, могут существовать в других таутомерных формах. Необходимо понимать, что любая ссылка на названное соединение настоящего изобретения охватывает все таутомеры названного соединения и любые смеси таутомеров названного соединения. Используемый в настоящем описании термин "алкил" представляет собой насыщенную, нормальную или разветвленную углеводородную группу. Примеры алкилов включают, без ограничения, метил"галогеналкил", или "гидроксиалкил", или "арилалкил", термин "алкил" охватывает двухвалентный углеводородный радикал с нормальной или разветвленной цепью. Например, "арилалкил" означает радикал алкиларил, в котором его алкильная группа является двухвалентным углеродным радикалом с нормальной или разветвленной цепью, а его арильная группа является такой, как определено в настоящем описании, и представлена связывающей конфигурацией, присутствующей в бензильной группе (-СН 2-фенил);"галоген(C1-C4)алкил" или "(C1-C4)галогеналкил" означают радикал, содержащий один или большее количество атомов галогена, которые могут быть одинаковыми или различными, на одном или нескольких атомах углерода алкильной группы, содержащей от 1 до 4 атомов углерода, которая является углеродным радикалом с нормальной или разветвленной цепью, и представлены трифторметильной группой"Алкокси" относится к группе, содержащей алкильный радикал, присоединенный через связывающий атом кислорода. Термин "(C1-C4)алкокси" относится к углеводородному радикалу с нормальной или разветвленной цепью, содержащему по меньшей мере 1 и до 4 атомов углерода, присоединенных через связывающий атом кислорода. Примеры "(C1-C4)алкокси" групп, применимые в настоящем изобретении,включают, без ограничения, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, изобутокси и трет-бутокси."Арил" представляет собой фенил. Гетероциклические группы могут быть гетероарильными или гетероциклоалкильными группами."Гетероциклоалкил" представляет собой группу, включающую неароматический, одновалентный моноциклический или бициклический радикал, который является насыщенным или частично ненасыщенным и содержит 4-7 атомов в кольце, если не определено иное, и который включает 1 гетероатом,выбранных из азота, кислорода и серы. Иллюстративные примеры гетероциклоалкилов включают, без ограничения, азетидинил, оксетанил, пирролидил (или пирролидинил), пиперидинил, пиперазинил, морфолинил, тетрагидро-2 Н-1,4-тиазинил, тетрагидрофурил (или тетрагидрофуранил), дигидрофурил, оксазолинил,тиазолинил,пиразолинил,тетрагидропиранил,дигидропиранил,1,3-диоксоланил,1,3-диоксанил, 1,4-диоксанил, 1,3-оксатиоланил, 1,3-оксатианил, 1,3-дитианил, азабицикло[3.2.1]октил,азабицикло[3.3.1]нонил, азабицикло[4.3.0]нонил, оксабицикло[2.2.1]гептил и 1,5,9-триазациклододецил. В некоторых из соединений настоящего изобретения гетероциклоалкильные группы включают 4-членные гетероциклоалкильные группы, содержащие один гетероатом, такие как оксетанил, тиетанил и азетидинил. В других соединениях настоящего изобретения гетероциклоалкильные группы включают 5-членные гетероциклоалкильные группы, содержащие один гетероатом, выбранный из азота, кислорода и серы, и необязательно содержащие один или два дополнительных атома азота или необязательно содержащие один дополнительный атом кислорода или серы, такие как пирролидил (или пирролидинил),тетрагидрофурил (или тетрагидрофуранил), тетрагидротиенил, дигидрофурил, оксазолинил, тиазолинил,имидазолинил, пиразолинил, 1,3-диоксоланил и 1,3-оксатиолан-2-онил. В других соединениях настоящего изобретения гетероциклоалкильные группы являются 6-членными гетероциклоалкильными группами, содержащими один гетероатом, выбранный из азота,кислорода и серы, и необязательно содержащими один или два дополнительных атома азота или один дополнительный атом кислорода или серы, такими как пиперидил (или пиперидинил), пиперазинил,морфолинил, тиоморфолинил, 1,1-диоксоидотиоморфолин-4-ил, тетрагидропиранил, дигидропиранил,тетрагидро-2 Н-1,4-тиазинил, 1,4-диоксанил, 1,3-оксатианил и 1,3-дитианил. Следует понимать, что термины "гетероцикл", "гетероциклический", "гетероарил", "гетероциклоалкил" охватывают стабильные гетероциклические группы, в которых гетероатом азота в кольце необязательно окислен (например, гетероциклические группы, содержащие N-оксид, такие как пиридин-Nоксид) или в которых гетероатом серы в кольце необязательно окислен (например, гетероциклические группы, содержащие сульфоны или сульфоксидные группы, такие как тетрагидротиенил-1-оксид (тетрагидротиенилсульфоксид) или тетрагидротиенил-1,1-диоксид (тетрагидротиенилсульфон. Термины "галоген" и "галоген" обозначают заместители, включающие хлор, фтор, бром или йод."Гидрокси" или "гидроксил" означают радикал -ОН. Используемые в настоящем описании термины "соединение(я) изобретения" или "соединение(я) настоящего изобретения" означают соединение формулы (I), как определено выше, в любой форме, т.е. в его любой солевой или несолевой форме (например, в форме свободной кислоты или основания, или в виде соли, в особенности фармацевтически приемлемой соли) и его любой физической форме (например,включающей нетвердые формы (например, жидкие или полутвердые формы) и твердые формы (например, аморфные или кристаллические формы, определенные полиморфные формы, сольватные формы,включая гидратные формы (например, моно-, ди-и геми-гидраты, а также смеси различных форм. Используемый в настоящем описании термин "необязательно замещенный" означает незамещенные группы или кольца (например, циклоалкильные, гетероциклоалкильные и гетероарильные кольца) и группы или кольца, замещенные одним или более указанными заместителями. Изобретение также направлено на соединение формулы (I), в которой в еще одном варианте осуществления R2 представляет собой -SO2Ra; в другом варианте осуществления Ra представляет собой (C1-С 6)алкил, где указанный (C1-C6)алкил необязательно замещен заместителем, выбранным из гидроксила и (C1-C2)алкокси; илиRa представляет собой 5-6-членный гетероциклоалкил, необязательно замещенный 1 или 2 заместителями, независимо выбранными из (C1-C4)алкильных групп; где 5-6-членный гетероциклоалкил содержит 1 гетероатом, выбранный из N, О или S; в выбранных вариантах осуществления Ra представляет собой -СН 3, -СН(СН 3)2, -С(СН 3)3,-СН 2 СН 2 ОН или тетрагидро-2 Н-пиран-4-ил; в другом варианте осуществления R3 представляет собой галоген, гидрокси, (C1-C3)алкил-, гидрокси(C1-C3)алкил- или гидрокси(C2-C3)алкокси-; в одном варианте изобретения Z представляет собой бензотиазол-6-ил, который необязательно замещен хлором, фтором, -CF3, метилом или метокси. В определенном варианте осуществления Z представляет собой бензотиазол-6-ил; в другом варианте осуществления Z представляет собой пиразолил, R12 представляет собой метил,13R представляет собой метил или трифторметил и R14 представляет собой Н. Конкретными соединениями настоящего изобретения являются: или их фармацевтически приемлемые соли. Выбранными соединениями настоящего изобретения являются: или их соли, в особенности фармацевтически приемлемые соли. Конкретными соединениями настоящего изобретения являются: или их фармацевтически приемлемые соли. Репрезентативные соединения настоящего изобретения представлены в примерах 1-83. Соединения настоящего изобретения пригодны для ингибирования киназы RIP2 путем контактирования клетки с соединением изобретения. Соединения настоящего изобретения могут быть использованы для лечения опосредованного RIP2 киназой заболевания или нарушения путем введения терапевтически эффективного количества соединения изобретения человеку, нуждающемуся в этом. Соединения формулы (I) могут содержать один или большее количество асимметричных центров (также называемых хиральными центрами) и поэтому могут существовать в виде отдельных энантиомеров, диастереомеров или других стереоизомерных форм или в виде их смесей. Хиральные центры, такие как хиральный углерод или, в частности, хиральная группа -SO-, также могут присутствовать в соединениях настоящего изобретения. Если стереохимия хирального центра, присутствующего в соединении настоящего изобретения или в любой химической структуре, представленной в настоящем описании, не определена, то предполагается, что структура охватывает все индивидуальные стереоизомеры и все их смеси. Например,каждый из(R)-6-[(1,1-диметилэтил)сульфинил]-N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-4 хинолинамина и (S)-6-[(1,1-диметилэтил)сульфинил]-N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-4 хинолинамина включены в 6-[(1,1-диметилэтил)сульфинил]-N-(4,5-диметил-1 Н-пиразол-3-ил)-7(метилокси)-4-хинолинамин. Таким образом, соединения формулы (I), содержащие один или более хиральных центров, могут применяться в виде рацемических смесей, энантиомерно обогащенных смесей или в виде энантиомерно чистых индивидуальных стереоизомеров. Индивидуальные стереоизомеры соединения формулы (I), которые содержат один или более асимметричных центров, могут быть разделены методами, известными квалифицированным специалистам. Например, такое разделение может быть выполнено с помощью (1) формирования диастереоизомерных солей, комплексов или других производных; (2) селективной реакции со стереоизомер-специфичным реагентом, например посредством ферментативного окисления или восстановления; или (3) газожидкостной или жидкостной хроматографии в хиральной среде, например на хиральной подложке, такой как диоксид кремния со связанным хиральный лигандом, или в присутствии хирального растворителя. Квалифицированный специалист сумеет оценить, что, когда требуемый стереоизомер превращается в другую химическую структуру с помощью одной из методик разделения, описанных выше, требуется дополнительная стадия для выделения нужной формы. В альтернативе, определенные стереоизомеры могут быть синтезированы с помощью асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или посредством конверсии одного энантиомера в другой с помощью асимметричного превращения. Следует понимать, что твердая форма соединения изобретения может существовать в кристаллических формах, некристаллических формах или в виде их смесей. Такие кристаллические формы могут также демонстрировать полиморфизм (т.е. способность существовать в различных кристаллических формах). Такие различные кристаллические формы обычно известны как "полиморфы". Полиморфы имеют одинаковый химический состав, но отличаются упаковкой, геометрической конфигурацией и другими характерными свойствами кристаллического твердого тела. Полиморфы, таким образом, могут обладать различными физическими свойствами, такими как формой, плотностью, твердостью, деформируемостью, стабильностью и характеристиками растворения. Полиморфы обычно демонстрируют различные температуры плавления, ИК-спектры и порошковые дифрактограммы, которые могут использоваться для идентификации. Средний специалист в данной области сумеет оценить, что различные полиморфы могут быть получены, например, путем изменения или регулирования условий, используемых при кристаллизации/перекристаллизации соединения. Вследствие их потенциального применения в медицине соли соединений формулы (I) предпочтительно являются фармацевтически приемлемыми солями. Подходящие фармацевтически приемлемые соли включают соли, описанные в Berge, Bighley and Monkhouse J. Pharm. Sci (1977), 66, p. 1-19. Соли,включенные в рамки термина "фармацевтически приемлемые соли", относятся к нетоксичным солям соединений настоящего изобретения. В случае, когда соединение изобретения является основанием (содержит основную группу), требуемая форма соли может быть получена любым подходящим методом, известным в уровне техники,включая обработку свободного основания неорганической кислотой, такой как соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота, трифторуксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота,гликолевая кислота, салициловая кислота и т.п., или пиранозидиловой кислотой, такой как глюкуроновая кислота или галактуроновая кислота, или альфа-оксикислотой, такой как лимонная кислота или винная кислота, или аминокислотой, такой как аспарагиновая кислота или глутаминовая кислота, или ароматической кислотой, такой как бензойная кислота или коричная кислота, или сульфоновой кислотой, такой как п-толуолсульфоновая кислота, метансульфоновая кислота, этансульфоновая кислота или подобное. Подходящие соли присоединения сформированы из кислот, которые образуют нетоксичные соли, и примеры включают ацетат, п-аминобензоат, аскорбат, аспартат, бензолсульфонат, бензоат, бикарбонат,бисметиленсалицилат, бисульфат, битартрат, борат, кальция эдетат, камсилат, карбонат, клавуланат,цитрат, циклогексилсульфамат, эдетат, эдизилат, эстолат, эзилат, этандисульфонат, этансульфонат, формиат, фумарат, глюцептат, глюконат, глутамат, гликолят, гликолиларсенилат, гексилрезоцинат, гидрабамин, гидробромид, гидрохлорид, дигидрохлорид, гидрофумарат, гидрофосфат, гидройодид, гидромалеат,гидросукцинат, гидроксинафтоат, изетионат, итаконат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, монокалия малеат, мукат, напсилат, нитрат, N-метилглюкамин, оксалат,оксалоацетат, памоат (эмбонат), пальмат, пальмитат, пантотенат, фосфат/дифосфат, пируват, полигалактуронат, пропионат, сахарат, салицилат, стеарат, субацетат, сукцинат, сульфат, таннат, тартрат, теоклат,тозилат, триэтйодид, трифторацетат и валерат. Другие примеры солей присоединения кислот включают пиросульфат, сульфит, бисульфит, деканоат, каприлат, акрилат, изобутират, капроат, гептаноат, пропиолат, оксалат, малонат, суберат, себацинат,бутин-1,4-диоат, гексин-1,6-диоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, фенилацетат, фенилпропионат, фенилбутират, лактат, -гидроксибутират, манделат и сульфонаты, такие как ксиленсульфонат, пропансульфонат, нафталин-1-сульфонат и нафталин-2 сульфонат. Если основное соединение изобретения выделено в виде соли, соответствующая форма свободного основания данного соединения может быть получена любым подходящим способом, известным в уровне техники, включая обработку соли неорганическим или органическим основанием, предпочтительно неорганическим или органическим основанием, имеющим более высокий pKa, чем форма свободного основания данного соединения. В случае, когда соединение изобретения является кислотой (содержит кислотную группу), требуемая соль может быть получена любым подходящим способом, известным в уровне техники, включая обработку свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или щелочно-земельного металла или подобное. Иллюстративные примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака, первичных, вторичных и третичных аминов и циклических аминов, таких как N-метил-D-глюкамин, диэтиламин, изопропиламин, триметиламин, этилендиамин, дициклогексиламин, этаноламин, пиперидин, морфолин и пиперазин, а также неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития. Некоторые из соединений изобретения могут образовывать соли с одним или несколькими эквивалентами кислоты (если соединение содержит основную группу) или основания (если соединение содержит кислотную группу). Настоящее изобретение включает в свой объем все возможные стехиометриче- 10023998 ские и нестехиометрические формы солей. Соединения изобретения, имеющие и основную, и кислотную группу, могут находиться в форме цвиттерионов, соли присоединения кислоты по основной группе или солей основания по кислотной группе. Настоящее изобретение также предусматривает превращение одной фармацевтически приемлемой соли соединения настоящего изобретения, например гидрохлоридной соли, в другую фармацевтически приемлемую соль соединения настоящего изобретения, например сульфатную соль. В отношении сольватов соединений формулы (I), включая сольваты солей соединений формулы (I),которые находятся в кристаллической форме, квалифицированный специалист оценит, что могут быть получены фармацевтически приемлемые сольваты, в которых молекулы растворителя включаются в кристаллическую решетку в процессе кристаллизации. Сольваты могут включать неводные растворители, такие как этанол, изопропанол, ДМСО, уксусную кислоту, этаноламин и EtOAc, или они могут включать воду в качестве растворителя, который включен в кристаллическую решетку. Сольваты, в которых растворителем, включаемым в кристаллическую решетку, является вода, обычно называются "гидратами". Гидраты включают стехиометрические гидраты, а также композиции, содержащие переменные количества воды. Поскольку соединения формулы (I) предназначены для применения в фармацевтических композициях, будет очевидно, что каждое из них предпочтительно получают, по существу, в чистой форме, например, с чистотой по меньшей мере 60%, более соответственно с чистотой по меньшей мере 75% и предпочтительно по меньшей мере 85%, в особенности с чистотой по меньшей мере 98% (% указаны по весу в весовом отношении). Неочищенные препараты соединений могут применяться для получения более чистых форм, применяемых в фармацевтических композициях. Общие способы синтеза Соединения формулы (I) могут быть получены при использовании процедур синтеза, представленных на схемах ниже, или используя знания квалифицированного химика-органика. Синтезы, представленные на данных схемах, могут применяться для получения соединений изобретения, имеющих ряд различных замещающих групп, с использованием соответствующих соединений-предшественников, которые надлежащим образом защищены, при необходимости, для обеспечения совместимости с реакциями, приведенными в настоящем описании. После снятия защитных групп, в случае необходимости, получают соединения с раскрытыми в целом свойствами. Хотя на схемах показаны только соединения формулы (I), на них представлены примеры способов, которые могут применяться для получения соединений изобретения. Промежуточные соединения (соединения, используемые при получении соединений изобретения) также могут присутствовать в форме солей. Таким образом, в ссылке на промежуточные соединения,фраза "соединение(я) формулы (число)" означает соединение, имеющее данную структурную формулу,или его фармацевтически приемлемую соль. Схема 1. 6-Бром-4-хлор-7-метоксихинолины могут быть синтезированы путем конденсации анилина с кислотой Мельдрума, с последующей циклизацией в гидроксихинолин. Превращение гидроксихинолина в хлорхинолин может быть выполнено с использованием POCl3. Схема 2. 4-Метилтетрагидро-2 Н-пиран-4-тиол может быть получен посредством образовании эпоксида из дигидро-2 Н-пиран-4(3H)-она с последующим превращением в тииран и последующим восстановлением до тиола. Схема 3. -замещенный тетрагидропиранилтиол может быть синтезирован из дигидро-2 Н-пиран 4(3H)-она. Алкилирование по -положению может сопровождаться восстановлением до спирта. После разделения диастереомеров цис-изомер может быть подвергнут мезилированию и обработке тиолятом калия. Удаление ацетата в восстанавливающих условиях дает тиол. Схема 4. Дополнительные замещенные тетрагидропиранилтиолы могут быть синтезированы из коммерчески доступных пиранонов согласно способу, аналогичному схеме 3. Схема 5. Алкилирование этилдисксилата пропионитрилом с последующей конденсацией с гидразином может привести к этил-3-амино-4-метил-1 Н-пиразол-5-карбоксилату. Схема 6. Синтез 5-фтор-1 Н-пиразоло[3,4-b]пиридин-3-амина может быть начат с амидирования 2-хлор-5-фтор-3-пиридинкарбоновой кислоты. Восстановление амида в нитрил с последующим взаимодействием с гидрозоном приводит к азаиндазолу. Схема 7. Замещающие группы "Z" могут быть присоединены к ядру хинолина при обработке 4-хлорхинолина соответствующим амином в условиях облучения микроволнами или при нагревании в присутствии кислоты. Сульфид затем может быть образован в условиях катализа палладием. Обработка сульфида Fe(III)Cl и 1 экв. йодной кислоты или при осторожном добавлении оксона дает сульфоксид. Дальнейшее взаимодействие с избытком оксона дает сульфон. Схема 8. В альтернативе, группа "Z" может быть присоединена после катализируемого палладием образования сульфида и перед окислением сульфида. Схема 9. В другом способе группа "Z" может быть присоединена на последней стадии после катализируемого палладием образования сульфида и окисления сульфида. Однако окисление после присоединения группы "Z", как на схемах 3 и 4, может привести к образованию меньшего количества побочного N-оксида. Схема 10. 3-Бром-4-сульфониланилины могут быть синтезированы из 2-бром-1-фтор-4 нитробензола. Замещение фторгруппы арила тиолом с последующим окислением в сульфон приводит к сульфонилнитробензолу. Нитробензол может быть восстановлен до анилина с использованием Sn(II)Cl,как на данной схеме, или с использованием железа/уксусной кислоты, как на схеме 7. Схема 11. 7-Бромхинолины могут быть синтезированы через соответствующий нитробензол. Восстановление нитробензола в анилин с последующим взаимодействием с кислотой Мельдрума дает имин,который может быть циклизован в ядро гидроксихинолина. Функционализация в хлорид может быть проведена при взаимодействии с POCl3. Группа "Z" может быть затем присоединена на последней стадии. Схема 12. 4,7-Дихлор-6-йодхинолины могут быть синтезированы при конденсации анилина с диэтил[(этилокси)метилиден]пропандиоатом с последующей циклизацией в ариловый сложный эфир. После гидролиза сложного эфира и декарбоксилирования превращение гидроксихинолина в хлорхинолин может быть выполнено с использованием POCl3. Схема 13. Диметилирование 7-метоксихинолинов может быть выполнено при обработке хинолина изопропилтиолятом натрия. Схема 14. Гидроксиэтилзамещенный хинолин может быть синтезирован при алкилировании гидроксихинолинов схемы 13. Схема 15. В альтернативе, алкилирование для присоединения гидроксиэтильного заместителя может быть выполнено перед присоединением группы "Z" или сульфона. Схема 16. 7-Дифторметоксихинолины могут быть образованы путем деметилирования метоксихинолина с последующей процедурой алкилирования/декарбоксилирования в одном реакционном сосуде, с присоединением дифторметоксигруппы. Схема 17. Гидроксиметилзамещенные пиразолсодержащие соединения могут быть синтезированы путем присоединения сначала этил-3-амино-4-метил-1 Н-пиразол-5-карбоксилата к хинолиновому ядру с последующим восстановлением до спирта. Схема 18. Гидроксилированные сульфоны могут быть подвергнуты внутренней циклизации с образованием циклических сульфонов при деметилировании 7-метоксизаместителя. Схема 19. 6-Тетрагидропиранилсульфонилсодержащие хинолины могут быть непосредственно алкилированы с присоединением -метильной группы, в особенности, когда группа R3 является алкильной группой (Me или Et). Соединения настоящего изобретения могут, в частности, применяться для лечения опосредованных киназой RIP2 заболеваний или нарушений, в особенности увеита, лихорадочного синдрома, ассоциированного с интерлейкин-1 превращающим ферментом (ICE, также известным как Каспаза-1), дерматита,острого поражения легкого, сахарного диабета 2-го типа, артрита (в частности ревматоидного артрита),воспалительных нарушений кишечника (таких как язвенный колит и болезнь Крона), ювенильной и внекишечной воспалительной болезни кишечника, предотвращении реперфузионного ишемического повреждения в паренхиматозных органах (в частности, почке) в результате ишемии, вызванной операцией на сердце, пересадкой органа, сепсисом и другими повреждениями, болезней печени (неалкогольного стеатогепатита, алкогольного стеатогепатита и аутоиммунного гепатита), аллергических заболеваний (таких как астма), реакций при трансплантации (таких как реакция трансплантат против хозяина), аутоиммунных заболеваний (таких как системная красная волчанка и рассеянный склероз) и гранулематозных нарушений (таких как саркоидоз, синдром Блау, ювенильный саркоидоз, гранулематоз Вегенера и интерстициальная болезнь легких). Соединения настоящего изобретения могут быть особенно полезными при лечении увеита,ICE-лихорадки, синдрома Блау, ювенильного саркоидоза, язвенного колита, болезни Крона, гранулематоза Вегенера и саркоидоза. Лечение опосредованных киназой RIP2 заболеваний или нарушений, или более широко, лечение иммуноопосредованных заболеваний, включающих, без ограничения, аллергические заболевания, аутоиммунные заболевания, предотвращение отторжения трансплантата и т.п., может быть выполнено с применение соединения настоящего изобретения при монотерапии или двойной или множественной комбинированной терапии, в особенности в случае лечения рефракторных случаев, в комбинации с другими противовоспалительными средствами и/или анти-TNF средствами, которые можно вводить в терапевтически эффективных количествах, известных в уровне техники. Например, соединения настоящего изобретения можно вводить в комбинации с кортикостероидами и/или анти-TNF средствами для лечения синдрома Блау, ювенильного саркоидоза; или в комбинации с анти-TNF биопрепаратами или другими противовоспалительными биопрепаратами для лечения болезни Крона; или в комбинации с 5-ASA (месаламином) или сульфасалазином для лечения язвенного колита; или в комбинации с кортикостероидами в низких дозах и/или метотрексатом для лечения гранулематоза Вегенера или саркоидоза или интерстициальной болезни легких; или в комбинации с биопрепаратом(например, антителом против TNF, против IL-6 и т.д.) для лечения ревматоидного артрита; или в комбинации с антителом к IL6 и/или метотрексатом для лечения ICE-лихорадки. Примеры подходящих противовоспалительных средств включают кортикостероиды, в особенности кортикостероиды в низких дозах (такие как Дельтазон (преднизон и противовоспалительные биопрепараты (такие как Актемра (mAb против IL6R) и Ритуксимаб (mAb против CD20. Примеры подходящих анти-TNF средств включают анти-TNF биопрепараты (такие как Энбрел (этанерцепт,Хумира (адалимумаб), Ремикейд (инфликсимаб) и Симпони (голимумаб. В настоящем изобретении также предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии, в частности для применения в лечении опосредованногоRIP2 киназой заболевания или нарушения, например заболеваний и нарушений, указанных в настоящем описании. Изобретение также обеспечивает применение соединения формулы (I) или его соли, в особенности фармацевтически приемлемой соли, в производстве лекарственного средства для применения в лечении опосредованного RIP2 киназой заболевания или нарушения, например заболеваний и нарушений, указанных в настоящем описании. Терапевтически "эффективное количество" означает такое количество соединения, которое при введении пациенту, испытывающему потребность в таком лечении, является достаточным, чтобы осуществить такое лечение, как определено в настоящей заявке. Таким образом, например, терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли является количеством средства согласно изобретению, которое при введении человеку, нуждающемуся в этом, явля- 15023998 ется достаточным, чтобы модулировать или ингибировать активность киназы RIP2 таким образом, что патологическое состояние, которое опосредовано указанной активностью, уменьшается, облегчается или предотвращается. Количество данного соединения, которое будет соответствовать такому количеству,будет изменяться в зависимости от таких факторов, как свойства конкретного соединения (например,активность (pIC50), эффективность (ЕС 50) и полупериод существования конкретного соединения), патологического состояния и его тяжести, отличительных признаков (например, возраста, размера и веса) пациента, испытывающего потребность в лечении, но при этом может быть с помощью стандартного способа определено специалистом, квалифицированным в данной области. Аналогичным образом, продолжительность лечения и периода введения (период между введением доз и подбор времени введения доз, например, до/с/после приема пищи) соединения будет изменяться в соответствии с отличительными признаками млекопитающего, испытывающего потребность в лечении (например, вес), конкретным соединением и его свойствами (например, фармацевтическими характеристиками), заболеванием или нарушением и его тяжестью, а также определенной композицией и применяемым способом, но при этом может быть определена специалистом в данной области."Лечение" означает, по меньшей мере, уменьшение тяжести патологического состояния у пациента. Способы лечения для уменьшения тяжести патологического состояния включают применение соединений в настоящем изобретении любым стандартно применяемым способом, например, для предотвращения, замедления, профилактики, терапии или лечения опосредуемого заболевания или нарушения. Определенные заболевания и нарушения, которые могут в особенности поддаваться лечению с применением соединения настоящего изобретения, описаны в настоящей заявке. Соединения изобретения можно вводить любым подходящим путем введения, включая системное введение и наружное введение. Системное введение включает пероральное введение, парентеральное введение, трансдермальное введение, ректальное введение и введение ингаляцией. Парентеральное введение относится к путям введения, кроме энтерального, трансдермального или ингаляционного, и обычно осуществляется с помощью инъекции или инфузии. Парентеральное введение включает внутривенную, внутримышечную и подкожную инъекцию или инфузию. Ингаляция относится к введению в легкие пациента при вдохе либо через рот, либо через носовые проходы. Наружное введение включает нанесение на кожу. Соединения изобретения можно вводить однократно или согласно режиму дозирования, в котором множество доз вводят через переменные интервалы времени в течение данного периода времени. Например, дозы можно вводить один, два, три или четыре раза в день. Дозы можно вводить до тех пор, пока не будет достигнут требуемый терапевтический эффект, или неопределенно долго, чтобы поддерживать требуемый терапевтический эффект. Подходящие режимы дозирования для соединения изобретения зависят от фармакокинетических свойств соответствующего соединения, таких как абсорбция, распределение и полупериод существования, которые могут быть определены квалифицированным специалистом. Кроме того, подходящие режимы дозирования, включая продолжительность таких режимов, для соединения изобретения назначают в зависимости от заболевания или нарушения, подвергаемого лечению,тяжести заболевания или нарушения, подвергаемого лечению, возраста и физического состояния проходящего лечение пациента, анамнеза проходящего лечение пациента, природы сопутствующей терапии,требуемого терапевтического эффекта и подобных факторов в рамках знаний и опыта квалифицированного специалиста. Квалифицированным специалистам также будет понятно, что подходящие режимы дозирования могут требовать коррекции с учетом реакции индивидуального пациента на режим дозирования или в течение длительного времени при необходимости изменений для индивидуального пациента. Для применения в терапии соединения изобретения обычно, но не обязательно, будут включены в фармацевтическую композицию перед введением пациенту таким образом, изобретение также направлено на фармацевтические композиции, включающие соединение изобретения и фармацевтически приемлемое вспомогательное вещество. Фармацевтические композиции изобретения могут быть приготовлены и упакованы в недозированной форме, где эффективное количество соединения изобретения может быть извлечено и затем дано пациенту, как в случае порошков, сиропов и растворов для инъекции. В альтернативе, фармацевтические композиции изобретения могут быть приготовлены и упакованы в единичной дозированной форме. В случае перорального применения, например, можно принимать одну или несколько таблеток или капсул. Доза фармацевтической композиции содержит, по меньшей мере, терапевтически эффективное количество соединения настоящего изобретения (т.е. соединения формулы (I) или его соли, в особенности фармацевтически приемлемой соли). В случае, когда фармацевтические композиции приготовлены в единичной дозированной форме, они могут содержать от 1 до 1000 мг соединения настоящего изобретения. Фармацевтические композиции изобретения обычно содержат одно соединение изобретения. Впрочем, в некоторых вариантах осуществления фармацевтические композиции изобретения содержат больше одного соединения изобретения. Кроме того, фармацевтические композиции изобретения необязательно могут также включать одно или большее количество дополнительных фармацевтически активных соединений. В настоящем описании "фармацевтически приемлемое вспомогательное вещество" означает материал, композицию или среду, участвующие в придании формы или консистенции композиции. Каждое вспомогательное вещество должно быть совместимым с другими компонентами фармацевтической композиции при смешивании, чтобы исключить взаимодействия, которые могут существенно уменьшить эффективность соединения изобретения при введении пациенту, и взаимодействия, которые могут привести к получению фармацевтических композиций, которые не являются фармацевтически приемлемыми. Кроме того, каждое вспомогательное вещество конечно должно обладать достаточно высокой чистотой, чтобы оно могло быть фармацевтически приемлемым. Соединения изобретения и фармацевтически приемлемое вспомогательное вещество или вспомогательные вещества обычно будут включать в лекарственную форму, приспособленную для введения пациенту требуемым путем введения. Обычные лекарственные формы включают лекарственные формы,приспособленные для (1) перорального введения, такие как таблетки, капсулы, каплеты, пилюли, пастилки, порошки, сиропы, эликсиры, суспензии, растворы, эмульсии, пакетики и облатки; (2) парентерального введения, такие как стерильные растворы, суспензии и порошки для восстановления; (3) трансдермального введения, такие как трансдермальные пластыри; (4) ректального введения, такие как суппозитории; (5) ингаляции, такие как аэрозоли и растворы; и (6) наружного применения, такие как кремы, мази, лосьоны, растворы, пасты, спреи, пены и гели. Подходящие фармацевтически приемлемые вспомогательные вещества будут изменяться в зависимости от конкретной выбранной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые вспомогательные вещества могут быть выбраны в зависимости от конкретной функции, которую они могут иметь в композиции. Например, некоторые фармацевтически приемлемые вспомогательные вещества могут быть выбраны благодаря своей способности облегчать получение однородных лекарственных форм. Некоторые фармацевтически приемлемые вспомогательные вещества могут быть выбраны благодаря своей способности облегчать получение стабильных лекарственных форм. Некоторые фармацевтически приемлемые вспомогательные вещества могут быть выбраны благодаря своей способности облегчать перенос или транспортировку соединения или соединений изобретения, введенных пациенту, из одного органа или части тела в другой орган или часть тела. Некоторые фармацевтически приемлемые вспомогательные вещества могут быть выбраны благодаря своей способности увеличивать соблюдение пациентом схемы лечения. Подходящие фармацевтически приемлемые вспомогательные вещества включают следующие типы вспомогательных веществ: разбавители, наполнители, связующие вещества, разрыхлители, смазывающие вещества, скользящие вещества, гранулирующие добавки, вещества для нанесения покрытия, смачивающие вещества, растворители, вспомогательные растворители, суспендирующие добавки, эмульгаторы, подсластители, вкусовые добавки, маскирующие вкус добавки, красители, ингибиторы слеживания, влагоудерживающие вещества, хелатирующие вещества, пластификаторы, загустители, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные вещества. Квалифицированный специалист сумеет оценить, что некоторые фармацевтически приемлемые вспомогательные вещества могут иметь больше одной функции и могут выполнять альтернативные функции в зависимости от того, сколько вспомогательного вещества присутствует в лекарственной форме и какие еще компоненты присутствуют в лекарственной форме. Квалифицированные специалисты обладают знанием и навыками в данной области, которые позволяют им выбирать подходящие фармацевтически приемлемые вспомогательные вещества в подходящих количествах для применения в изобретении. Кроме того, существует множество ресурсов, доступных квалифицированному специалисту, которые описывают фармацевтически приемлемые вспомогательные вещества и могут быть полезными при выборе подходящих фармацевтически приемлемых вспомогательных веществ. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company),The Handbook of Pharmaceutical Additives (Gower Publishing Limited) и The Handbook of PharmaceuticalExcipients (the American Pharmaceutical Association and the Pharmaceutical Press). Фармацевтические композиции изобретения приготавливают с использованием методик и методов,известных специалистам в данной области техники. Некоторые из методов, стандартно применяемых в уровне техники, описаны в Remington's Pharmaceutical Sciences (Mack Publishing Company). В одном аспекте изобретение направлено на твердую лекарственную форму для перорального применения, такую как таблетка или капсула, включающие эффективное количество соединения изобретения и разбавителя или наполнителя. Подходящие разбавители и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и двухосновный фосфат кальция. Твердая лекарственная форма для перорального применения может дополнительно включать связующее вещество. Подходящие связующие вещества включают крахмал (например, кукурузный крахмал, картофельный крахмал и прежелатинизированный крахмал), желатин, гуммиарабик, альгинат натрия, альгиновую кислоту, трагакантовую камедь, гуаровую камедь, повидон, а также целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Твердая лекарственная форма для перорального применения может дополнительно включать раз- 17023998 рыхлитель. Подходящие разрыхлители включают кросповидон, натрий-крахмалгликолят, кроскармеллозу, альгиновую кислоту и натрий-карбоксиметилцеллюлозу. Твердая лекарственная форма для перорального применения может дополнительно включать смазывающее вещество. Подходящие смазывающие вещества включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк. Примеры Следующие примеры иллюстрируют изобретение. Данные примеры не следует рассматривать как ограничение объема настоящего изобретения, а скорее как руководство для квалифицированного специалиста по получению и применению соединений, композиций и способов настоящего изобретения. Хотя описываются конкретные варианты осуществления настоящего изобретения, квалифицированному специалисту будет очевидно, что могут быть внесены различные изменения и модификации без отступления от сущности и объема изобретения. Названия промежуточных и конечных соединений, описанных в настоящем изобретении, были получены с использованием программы, генерирующей названия соединений. Специалисту в данной области будет очевидно, что в некоторых случаях данная программа дает название структурно изображенному соединению как таутомеру данного соединения. Следует понимать, что любая ссылка на названное соединение или структурно изображенное соединение должна охватывать все таутомеры таких соединений и любые смеси соответствующих таутомеров. В следующих экспериментальных описаниях могут использоваться следующие сокращения: Стадия 1. 5-([4-Бром-3-(метилокси)фенил]аминометилиден)-2,2-диметил-1,3-диоксан-4,6-дион. 2,2-Диметил-1,3-диоксан-4,6-дион (8,5 г, 58 ммоль) в триметилортоформиате (50 мл, 450 ммоль) кипятили с обратным холодильником при 105C в течение 1 ч, затем добавляли 4-бром-3-метоксианилин(10,5 г, 50,4 ммоль) и кипячение с обратным холодильником продолжали еще 1 ч. Суспензию фильтровали, твердое вещество промывали МеОН и сушили в вакууме, получая 5-([4-бром-3-(метилокси)фенил]аминометилиден)-2,2-диметил-1,3-диоксан-4,6-дион (17 г, 49 ммоль,выход 96%). 1(68 мл,420 ммоль) при 230C добавляли 5-4-бром-3-(метилокси)фенил]аминометилиден)-2,2-диметил-1,3-диоксан-4,6-дион (15 г, 42 ммоль) и перемешивали смесь в течение 1 ч. Реакционную смесь вливали в гексан после охлаждения до комнатной температуры. Осадок отфильтровывали и промывали гексаном. Коричневое твердое вещество сушили в вакууме в течение ночи, получая 6-бром-7-(метилокси)-4-хинолинол (10 г, 33 ммоль, выход 79%). 1H-ЯМР (400 МГц, ДМСО-d)м.д.: 3,94 (с, 3H), 5,99 (дд, J=7,4, 1,2 Гц, 1 Н), 7,05 (с, 1 Н), 7,86 (дд,J=7,4, 5,8 Гц, 1 Н), 8,16 (с, 1 Н), 11,68 (шир.с, 1 Н). МС (m/z) 254, 256 (М+Н+). Стадия 3. 6-Бром-4-хлор-7-(метилокси)хинолин. 6-Бром-7-(метилокси)-4-хинолинол (4,17 г, 16,41 ммоль) в оксихлориде фосфора (7,73 мл,82 ммоль) перемешивали при 110C в течение 1 ч. Реакционную смесь охлаждали и медленно вливали в насыщенный раствор карбоната натрия со льдом при перемешивании. Полученную суспензию фильтровали, твердое вещество промывали водой и сушили в вакууме в течение ночи, получая 6-бром-4-хлор-7(метилокси)хинолин (4,6 г, 16 ммоль, выход 97%). 1 Следующие промежуточные соединения могут быть получены аналогичным способом:(0,17 г, 1,5 ммоль) нагревали в EtOH (3 мл) при 80C в закрытой пробирке в течение 16 ч. Реакционную смесь охлаждали и добавляли Et2O (10 мл). Осадок 6-бром-N-(4,5-диметил-1 Н-пиразол-3-ил)-7(метилокси)-4-хинолинамина отфильтровывали и сушили, получая коричневое твердое вещество. 1H-ЯМР (ДМСО-d6)м.д.: 12,63 (шир.с, 1 Н), 10,42 (шир.с, 1 Н), 9,10 (с, 1 Н), 8,47 (д, J=7 Гц, 1 Н),7,47 (с, 1 Н), 6,71 (д, J=6,8 Гц, 1 Н), 4,06 (с, 3H), 2,23 (с, 3H), 1,85 (с, 3H). МС (m/z) 347, 349 (М+Н+). Следующие соединения получали аналогичным способом. Изопропиловый спирт может использоваться в качестве растворителя в дополнение к этанолу. Стадия 1. 1,6-Диоксаспиро[2.5]октан. К суспензии йодида триметилсульфоксония (28,6 г, 130 ммоль) в ДМСО (200 мл) в двугорлой RBF(500 мл) порциями добавляли NaH (5,19 г, 130 ммоль, 60% в минеральном масле) в атмосфере N2 при комнатной температуре. Перемешивание продолжали в течение 1 ч, затем в течение 5 мин по каплям добавляли раствор дигидро-2 Н-пиран-4 (3H)-она (10 г, 100 ммоль) в ДМСО (10 мл). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре, затем вливали в воду со льдом (300 мл) и экстрагировали Et2O (2200 мл). Органическую фазу промывали водой и рассолом, сушили над MgSO4,фильтровали и выпаривали, получая 1,6-диоксаспиро[2.5]октан (4,9 г, 42,9 ммоль, выход 43,0%) в виде бесцветного масла. 1H-ЯМР (400 МГц, хлороформ-d) : 1,52-1,59 (м, 2 Н), 1,89 (ддд, J=13,20, 8,40, 4,67 Гц, 2 Н), 2,71 (с,2 Н), 3,79-3,95 (м, 4 Н). Стадия 2 6-Окса-1-тиаспиро[2.5]октан. К раствору 1,6-диоксаспиро[2.5]октана (200 мг, 1,752 ммоль) в МеОН (5 мл) добавляли тиомочевину (133 мг, 1,75 ммоль), реакционную смесь перемешивали и нагревали при 80C в течение 4 ч. Осадок,который образовался в ходе реакции, отфильтровывали. Фильтрат разбавляли Et2O (100 мл), промывали рассолом, сушили над MgSO4, фильтровали и выпаривали, получая бесцветное масло 6-окса-1-тиаспиро[2.5]октана (216 мг, 1,659 ммоль, выход 95%). 1H-ЯМР (хлороформ-d) : 3,97 (дт, J=11,3, 4,1 Гц, 2 Н), 3,76 (ддд, J=11,5, 9,2, 2,8 Гц, 2 Н), 2,49 (с, 2 Н),2,22 (ддд, J=13,4, 9,5, 3,9 Гц, 2 Н), 1,55 (д, J=13,4 Гц, 2 Н). Стадия 3. 4-Метилтетрагидро-2 Н-пиран-4-тиол. К кипящему под обратным холодильником раствору 6-окса-1-тиаспиро[2.5]октана (200 мг,1,54 ммоль) в ТГФ (5 мл) по каплям добавляли LiAlH4 в ТГФ (0,40 мл, 0,80 ммоль). Реакционную смесь перемешивали в течение 1 ч, затем охлаждали до 0C и приливали воду (1 мл). Смесь перемешивали в течение 10 мин и экстрагировали Et2O (210 мл). Органическую фазу промывали рассолом, сушили надMgSO4, фильтровали и выпаривали. Остаток очищали на колонке с силикагелем (10 г) и элюировали 10%EtOAc в гексане, получая требуемый продукт (94 мг, 46%) в виде бесцветного масла. 1 Стадия 1. 3-Метилтетрагидро-4 Н-пиран-4-он. К раствору LDA (2,0 М в гептане/ТГФ/этилбензоле, 12,0 мл, 24,0 ммоль) в ТГФ (100 мл), охлажденному до -78C, по каплям добавляли раствор дигидро-2 Н-пиран-4(3H)-она (2 г, 20,0 ммоль) и НМРА(3,5 мл, 20,0 ммоль) в ТГФ (70 мл). После перемешивания в течение 5 мин к вышеуказанному раствору добавляли MeI (6,25 мл, 100 ммоль) в ТГФ (30 мл), реакционную смесь нагревали до 0C и выдерживали в течение 2 ч, затем нагревали до комнатной температуры в течение 10 мин, а затем снова охлаждали до 0C. В реакционную смесь вливали NH4Cl (нас.) и экстрагировали Et2O (2200 мл). Органическую фазу промывали рассолом, сушили над MgSO4, фильтровали и выпаривали. Неочищенную смесь очищали с помощью колонки с силикагелем (100 г), используя 10-20% Et2O в ДХМ, получая оранжевое масло 3-метилдигидро-2 Н-пиран-4(3H)-она (2,2 г, 19,30 ммоль, выход 96%). 1H-ЯМР (хлороформ-d) : 4,12-4,32 (м, 2 Н), 3,67-3,81 (м, 2 Н), 2,60-2,74 (м, 1 Н), 2,54 (дт, J=17,6,1 Гц, 1 Н), 2,41 (дт, J=14, 2,7 Гц, 1 Н), 1,01 (д, J=6,8 Гц, 3H). Стадия 2. Транс-3-метилтетрагидро-2 Н-пиран-4-ол. К раствору 3-метилдигидро-2 Н-пиран-4(3H)-она (2,28 г, 20,0 ммоль) в ДХЭ (50 мл) добавляли триацетоксиборогидрид натрия (8,47 г, 40,0 ммоль), а затем уксусную кислоту (3,4 мл, 59,9 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 16 ч. В реакционную смесь вливали воду и экстрагировали Et2O (320 мл). Органические экстракты объединяли, промывали бикарбонатом натрия (нас.) и рассолом, сушили над MgSO4, фильтровали и выпаривали. Реакционную смесь очищали на колонке с силикагелем (100 г) с использованием 50-60% EtOAc в гексане, получая два продуктаH-ЯМР (400 МГц, хлороформ-d) : 0,96 (д, J=6,57 Гц, 3H), 1,58-1,66 (м, 1 Н), 1,92 (м, J=12,66, 4,64,2,46, 2,46 Гц, 2 Н), 2,96-3,07 (м, 1 Н), 3,35 (тд, J=9,85, 4,55 Гц, 1 Н), 3,44 (тд, J=11,87, 2,27 Гц, 1 Н), 3,86 (дд,J=12,25, 3,66 Гц, 1 Н), 3,97-4,03 (м, 1 Н). Стадия 3. цис-3-Метилтетрагидро-2 Н-пиран-4-ил-метансульфонат. К раствору цис-3-метилтетрагидро-2 Н-пиран-4-ола (780 мг, 6,71 ммоль) в ДХМ (20 мл) добавляли метансульфонилхлорид (0,63 мл, 8,06 ммоль) с последующим добавлением триметиламина (1,87 мл,13,43 ммоль) при 0C. Реакционную смесь перемешивали при 0C в течение 3 ч, затем вливали воду и экстрагировали ДХМ (230 мл). Органическую фазу промывали бикарбонатом натрия (нас.) и рассолом,сушили над MgSO4, фильтровали и выпаривали, получая бесцветное масло цис-3-метилтетрагидро-2 Нпиран-4-илметансульфоната (1,4 г, 7,21 ммоль, выход 107%), который использовали на следующей стадии без очистки. Стадия 4: Транс-3-3-метилтетрагидро-2 Н-пиран-4-ил)этантиоат. Тиоацетат калия (882 мг, 7,72 ммоль) добавляли к раствору цис-3-метилтетрагидро-2 Н-пиран-4 илметансульфоната (500 мг, 2,57 ммоль) в ДМА (8 мл) и реакционную смесь нагревали при 80C в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и экстрагировали Et2O (330 мл). Экстракты объединяли и промывали водой (220 мл) и рассолом, сушили над MgSO4, фильтровали и выпаривали, получая красное масло (одиночное пятно на ТСХ) требуемого продукта транс-S-3-метилтетрагидро-2 Н-пиран-4-ил)этантиоата (445 мг, 2,55 ммоль, выход 99%), который использовали на следующей стадии без очистки. Стадия 5. Транс-3-метилтетрагидро-2 Н-пиран-4-тиол. Аммиак (2,0 М в МеОН, 10,400 мл, 20,80 ммоль) добавляли к транс-S-3-метилтетрагидро-2 Н-пиран 4-ил)этантиоату (440 мг, 2,52 ммоль) и реакционную смесь нагревали при 40C в течение 12 ч. По окончании смесь выпаривали в вакууме, получая оранжевое твердое вещество. Твердое вещество очищали на колонке ISCO с силикагелем (25 г), используя 10-20% EtOAc в гексане, получая требуемый продукт транс-3-метилтетрагидро-2 Н-пиран-4-тиол (71 мг, 0,54 ммоль, выход 21,27%). 1 Стадия 1. 2,6-Диметилтетрагидро-2 Н-пиран-4-ол. К раствору 2,6-диметилдигидро-2 Н-пиран-4(3H)-она (3 г, 23,41 ммоль) в ДХЭ (60 мл) добавляли триацетоксиборогидрид натрия (14,88 г, 70,2 ммоль) с последующим добавлением уксусной кислоты(8,1 мл, 140 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 20 ч. В реакционную смесь вливали воду и экстрагировали Et2O (350 мл). Органическую фазу промывали рассолом, сушили над MgSO4, фильтровали и выпаривали, получая требуемый продукт 2,6-диметилтетрагидро-2 Н-пиран-4-ол в виде бесцветного масла (3 г, 23,04 ммоль, выход 98%). 1H-ЯМР (хлороформ-d) : 1,19-1,26 (м, 6 Н), 1,83 (д, J=12,13 Гц, 2 Н), 1,93 (дд, J=12,00, 4,67 Гц, 2 Н),3,58-3,68 (м, 1 Н), 3,75-3, 85 (м, 1 Н), 3,93 (м, 1 Н). Стадия 2. 2,6-Диметилтетрагидро-2 Н-пиран-4-илметансульфонат. К раствору 2,6-диметилтетрагидро-2 Н-пиран-4-ола (3 г, 23,04 ммоль) в ДХМ (100 мл) добавляли мезилхлорид (2,16 мл, 27,7 ммоль), а затем Et3N (6,42 мл, 46,1 ммоль). Реакционную смесь перемешивали при 0C в течение 1 ч и останавливали реакцию, приливая воду. Реакционную смесь экстрагировали ДХМ (250 мл) и промывали органическую фазу бикарбонатом натрия (нас.) и рассолом, сушили надMgSO4, фильтровали, выпаривали. Неочищенную смесь очищали на колонке ISCO с силикагелем (40 г) с использованием 50% EtOAc в гексане, получая белое твердое вещество 2,6-диметилтетрагидро-2 Нпиран-4-илметансульфонат (2,17 г, 10,42 ммоль, выход 45,2%). 1(25 мл) добавляли тиоацетат калия (2,380 г, 20,84 ммоль) и перемешивали реакционную смесь при 65C в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли Et2O (100 мл) и промывали органическую фазу водой (220 мл) и рассолом, сушили над MgSO4, фильтровали и выпаривали. Неочищенную смесь очищали на силикагеле (50 г) с использованием 10-20% EtOAc в гексане, получая требуемый продукт S-(2,6-диметилтетрагидро-2 Н-пиран-4-ил)этантиоат (1,93 г, 10,25 ммоль, выход 98%). 1H-ЯМР (хлороформ-d) : 4,03-4,09 (м, 1 Н), 3,65 (дд, J=6,6, 2,3 Гц, 2 Н), 2,34 (с, 3H), 1,65-1,71 (м,4 Н), 1,18 (д, J=6,3 Гц, 6 Н). МС (m/z) 189 (М+Н+). Стадия 4. (2R,6S)-2,6-Диметилтетрагидро-2 Н-пиран-4-тиол. Аммиак (2,0 М в МеОН, 6,37 мл, 12,75 ммоль) добавляли к S-(2,6-диметилтетрагидро-2 Н-пиран-4 ил)этантиоату (500 мг, 2,66 ммоль) и реакционную смесь перемешивали при 23C в течение 20 ч. Реакция протекала медленно, и реакционную смесь нагревали при 40C еще в течение 4 ч с последующим выпариванием в вакууме и очисткой на ISCO (колонка с силикагелем 25 г) с использованием 0-10%EtOAc в гексане, получая (2R,6S)-2,6-диметилтетрагидро-2 Н-пиран-4-тиол (308 мг, выход 79%). Структуру подтверждали экспериментом nOe. 1 Следующее промежуточное соединение синтезировали аналогичным способом с использованием на стадии 2 п-толуолсульфонилхлорида вместо метансульфонилхлорида.(55 мг, 0,06 ммоль) в 3,9 мл ДМФА нагревали при 100C в закрытой, продутой азотом, виале в течение 16 ч. Реакционную смесь разбавляли EtOAc и водой и разделяли слои. Органические фазы выпаривали и очищали неочищенный продукт с помощью колоночной хроматографии (ISCO CombiFlash, 0-10% 2 н.NH3/MeOH в ДХМ), получая N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-6-(тетрагидро-2 Н-пиран-4 илтио)-4-хинолинамин (80 мг, 35%). МС (m/z) 385 (М+Н+). В качестве растворителя также может использоваться 1,4-диоксан. В тех случаях, когда исходный хинолин является солью HCl, также может быть добавлен эквивалент триэтиламина. Способ В. В альтернативном варианте, реакции конденсации могут быть проведены следующим образом. К раствору хинолина (1 экв) в диксане (0,1 М) добавляли (оксидибензол-2,1-диил)-бис(дифенилфосфан) (0,1 экв), трис-(дибензилиденацетон)-дипалладий(0) (0,1 экв), трет-бутилат калия(1-2 экв), тиол (1,2 экв) и триэтиламин (1-3 экв). Колбу продували азотом и нагревали в атмосфере азота в течение 3 ч при 90C перед приливанием в EtOAc. Органический слой промывали насыщенным бикарбонатом натрия. Водный слой промывали 25% EtOH в метиленхлориде, затем метиленхлоридом. Органические фракции объединяли, сушили над MgSO4 и упаривали до коричневого масла. Остаток очищали с помощью ISCO CombiFlash. Следующие аналоги получали аналогичным способом. Стадия 1. 2-Хлор-5-фтор-3-пиридинкарбоксамид. 2-Хлор-5-фтор-3-пиридинкарбоновую кислоту (20 г, 110 ммоль) растворяли в ДХМ (400 мл) и затем добавляли ДМФА (88 мкл, 1,1 ммоль) при 0C. После добавления ДМФА по каплям при 0C добавляли оксалилхлорид (26 мл, 300 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и упаривали в вакууме. Полученную желтую жидкость растворяли в 1,4-диоксане (400 мл),охлаждали до 0C и пропускали через раствор NH3 (газ) (19,4 г, 1140 ммоль) в течение 30 мин. Смесь перемешивали при комнатной температуре в течение 16 ч. Полученную белую смесь фильтровали и фильтрат выпаривали, получая требуемый продукт в виде белого твердого вещества (18 г, выход 89%). МС (m/z) 175 (М+Н+). 1H-ЯМР (400 МГц, ДМСО-d6)м.д.: 8,53 (д, 1 Н), 8,10 (с, 1 Н), 8,00 (дд, 1 Н), 7,88 (с, 1 Н). Стадия 2. 2-Хлор-5-фтор-3-пиридинкарбонитрил. 2-Хлор-5-фтор-3-пиридинкарбоксамид (18 г, 102 ммоль) суспендировали в ДХМ (500 мл) и затем при 0C добавляли триэтиламин (31 мл, 220 ммоль). К реакционной смеси при 0C по каплям добавляли трифторуксусный ангидрид (ТФУА) (16 мл, 110 ммоль). Белый карбоксамид, используемый в качестве исходного материала, исчезал через 20 мин при 0C, что указывало на завершение реакции. Реакционную смесь перемешивали при 0C в течение 1 ч. Реакционную смесь разбавляли ДХМ, а затем промывали насыщенным NaHCO3 (водн.). Органический слой промывали рассолом, сушили над MgSO4, фильтровали и фильтрат выпаривали до коричневого остатка. Остаток очищали с помощью ISCO CombiFlash(8-20% EtOAc/гексан; колонка 330 г). Собранные фракции объединяли и выпаривали, получая требуемый продукт в виде белого твердого вещества (15 г, выход 96%). МС (m/z) 157 (М+Н+). 1H-ЯМР (400 МГц, ДМСО-d6)м.д.: 8,68 (дд, 1 Н), 8,83 (д, 1 Н). Стадия 3. 5-Фтор-1 Н-пиразоло[3,4-b]пиридин-3-амин. 2-Хлор-5-фтор-3-пиридинкарбонитрил(15,3 г, 98 ммоль) растворяли в 1-бутаноле (300 мл), а затем добавляли гидразин-моногидрат (16,82 мл, 293 ммоль) с последующим добавлением хлористоводородной кислоты (4 н. в диксане) (0,244 мл, 0,977 ммоль). Реакционную смесь выдерживали при 70C в течение 4 ч и полученное желтое кристаллическое твердое вещество собирали фильтрацией (12,5 г,выход 84%). МС (m/z) 153 (М+Н+). 1 Стадия 1. 2-Бром-1-[(1,1-диметилэтил)тио]-4-нитробензол. В круглодонную колбу, содержащую 2-бром-1-фтор-4-нитробензол (15 г, 68 ммоль) и 2-метил-2-пропантиол (8,4 мл, 75 ммоль) в ДМФА (45 мл), добавляли карбонат калия (10,37 г, 75 ммоль). Реакционную смесь нагревали до 50C в течение 3 дней и делили между EtOAc и водой. Водный слой экстрагировали EtOAc (2) и объединенные органические экстракты промывали водой (3) и рассолом Стадия 2. 2-Бром-4-нитрофенил-1,1-диметилэтилсульфон. В круглодонную колбу, содержащую 2-бром-4-нитрофенил-1,1-диметилэтилсульфид (15 г,53 ммоль) в МеОН (89 мл) и воду (89 мл), добавляли оксон (49 г, 80 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Затем добавляли дополнительное количество оксона (25 г), МеОН (30 мл) и воды (30 мл). Через 24 ч добавляли дополнительную порцию оксона (25 г) и реакцию перемешивали при комнатной температуре в течение 24 ч. Реакцию нейтрализовывали 1 н.NaOH и добавляли ДХМ. Водный слой экстрагировали ДХМ (1) и объединенные органические экстракты промывали рассолом (1), сушили над сульфатом магния и очищали с помощью колоночной хроматографии в 2 приема (ISCO-Rf, 120 г, 0-30% EtOAc/гексан), получая 2-бром-4-нитрофенил-1,1 диметилэтилсульфон (9,6 г, 30 ммоль, выход 56%). 1H-ЯМР (400 МГц, ДМСО-d6)м.д.: 8,61 (д, J=2,3 Гц, 1 Н), 8,43 (дд, J=8,6, 2,3 Гц, 1 Н), 8,27 (д,J=8,8 Гц, 1 Н), 1,35 (с, 9 Н). Стадия 3. 3-Бром-4-(трет-бутилсульфонил)анилин. Раствор дигидрата хлорида олова(II) (17 г, 73 ммоль) и конц. HCl (24 мл) в МеОН (49 мл) охлаждали до 0C и одной порцией добавляли 2-бром-1-(трет-бутилсульфонил)-4-нитробензол (4,7 г, 15 ммоль). Через 5 ч реакцию охлаждали до 0C и тщательно нейтрализовали 6 н. NaOH (приблизительно 75 мл). Добавляли этилацетат (350 мл) и порциями фильтровали смесь (белый осадок забивал фильтровальную бумагу). Слои фильтрата отделяли и водный слой экстрагировали EtOAc (2). Объединенные органические экстракты сушили над сульфатом магния и выпаривали досуха, получая 3-бром-4-(третбутилсульфонил) анилин (3,8 г, 13 ммоль, выход 90%). МС (m/z) 236, 238 (М-трет-бутил+Н+). 1H-ЯМР (400 МГц, ДМСО-d6)м.д.: 7,57 (д, J=8,8 Гц, 1 Н), 6,93 (д, J=2,3 Гц, 1 Н), 6,64 (дд, J=8,8,2,3 Гц, 1 Н), 6,41 (с, 2 Н), 1,25 (с, 9 Н). Следующее промежуточное соединение синтезировали аналогичным способом.(221 мл) порциями добавляли железо (11,13 г, 199 ммоль) при 0C. Реакционную смесь медленно нагревали до комнатной температуры в течение ночи и затем медленно вливали в воду (150 мл), EtOAc(600 мл) и 2 н. NaOH (450 мл) при перемешивании. Твердый карбонат натрия (приблизительно 300 г) медленно добавляли к коричневому раствору до прекращения образования пузырей, при этом рН раствора достигал приблизительно 10. Раствор переносили в делительную воронку, слои разделяли и водный слой экстрагировали EtOAc (1). Объединенные органические экстракты выпаривали досуха, получая 3-бром-4-(метилсульфонил)анилин (10,5 г, 42,0 ммоль, выход 63,2%). 1 Стадия 1. Диэтил[(3-хлор-4-йодфенил)амино]метилиденпропандиоат: 3-хлор-4-йоданилин (15 г,59 ммоль) растворяли в диэтил[(этилокси)метилиден]пропандиоате (19 мл, 95 ммоль) и нагревали до 160C в течение 4 ч при кипячении с обратным холодильником. Затем холодильник удаляли для выпаривания EtOH. Через 1 ч смесь охлаждали до комнатной температуры, при этом смесь твердела, разбивали и суспендировали твердое вещество в смеси гексанов. Смесь фильтровали и осадок на фильтре несколько раз промывали смесью гексанов, получая серое твердое вещество (23 г, 91%). МС (m/z) 424,0 (М+Н)+. Стадия 2. Этил-7-хлор-4-гидрокси-6-йод-3-хинолинкарбоксилат. К дифениловому эфиру (100 мл, 630 ммоль) при 240C частями добавляли диэтил[(3-хлор-4 йодфенил)амино]метилиденпропандиоат (18 г, 43 ммоль). Реакционную смесь нагревали в течение 5 ч прежде, чем охладить ее до комнатной температуры. После достижения комнатной температуры реакционную смесь разбавляли смесью гексанов (150 мл) и фильтровали суспензию. Осадок на фильтре промывали смесью гексанов (2100 мл), а затем сушили в вакууме (6,7 г, 41%). Стадия 3. 7-Хлор-4-гидрокси-6-йод-3-хинолинкарбоновая кислота. Этил-7-хлор-4-гидрокси-6-йод-3-хинолинкарбоксилат (6,7 г, 18 ммоль) и NaOH (3,5 г, 89 ммоль) суспендировали в ТГФ (50 мл) и воде (50 мл). Затем реакционную смесь нагревали до 70C в течение ночи. Смесь охлаждали до комнатной температуры, при этом ее частично упаривали для удаления ТГФ. Затем водный раствор подкисляли, используя конц. HCl. Полученную суспензию фильтровали и осадок на фильтре промывали водой (2100 мл), а затем сушили в вакууме в течение ночи, получая требуемый продукт (6,4 г, 93%). 1H-ЯМР (ДМСО-d6) : 14,78 (с, 1 Н), 13,47 (с, 1 Н), 8,97 (с, 1 Н), 8,70 (с, 1 Н), 7,99 (с, 1 Н). Стадия 4. 7-Хлор-6-йод-4-хинолинол. К дифениловому эфиру (44 мл, 276 ммоль) при 240C по частям добавляли 7-хлор-4-гидрокси-6 йод-3-хинолинкарбоновую кислоту (6,4 г, 18 ммоль). Смесь нагревали в течение 3 ч прежде, чем ее охлаждали до комнатной температуры в течение ночи. Реакционную смесь разбавляли смесью гексанов(200 мл) и обрабатывали ультразвуком. Суспензию фильтровали, осадок на фильтре промывали смесью гексанов (2100 мл) и сушили в вакууме, получая требуемый продукт (4,9 г, 71%). МС (m/z) 306,0 (М+Н)+. Стадия 5. 4,7-Дихлор-6-йодхинолин. 7-Хлор-6-йод-4-хинолинол (4,9 г, 16 ммоль) суспендировали в POCl3 (50 мл, 536 ммоль) и перемешивали при комнатной температуре в течение 72 ч. Затем смесь выпаривали, а остаток охлаждали до 0C и тщательно нейтрализовали добавлением нас. водн. раствора Na2CO3. Полученную суспензию фильтровали и осадок на фильтре промывали водой (250 мл). После сушки материала в вакууме, его растворяли в ДХМ и концентрировали на силикагеле. Сухой материал очищали с помощью флэш-хроматографии(2050% EtOAc в смеси гексанов). После выпаривания фракций требуемый продукт получали в виде белого твердого вещества (3,4 г, 63%). 1 К перемешиваемому раствору пропионитрила (1 г, 18,16 ммоль) в ТГФ (40 мл), охлажденному до 78C, по каплям добавляли LDA в гептане/ТГФ/этилбензоле (10,89 мл, 21,79 ммоль). Реакционную смесь перемешивали в течение 1 ч, затем добавляли к раствору диэтилоксалата (2,65 г, 18,16 ммоль) в ТГФ(40 мл), охлажденному до -78C. Полученный раствор перемешивали при -78C в течение 2 ч, позволяли нагреться до 0C, после чего добавляли водный NH4Cl. Затем добавляли 3 н. HCl до pH 5. Два слоя разделяли и водный слой экстрагировали EtOAc (2100 мл). Экстракты объединяли, промывали рассолом,сушили над MgSO4, фильтровали и выпаривали. Желтый осадок, образовавшийся после частичного упаривания, отфильтровывали. Оставшийся растворитель удаляли, получая коричневое масло. Оставшееся масло и гидразин (1,140 мл, 36,3 ммоль) растворяли в уксусной кислоте (3 мл) и бензоле (100 мл) и кипятили с обратным холодильником в течение 16 ч при использовании ловушки Дина-Старка. Собирали 1,5 мл воды. Реакционную смесь охлаждали до комнатной температуры и раствор снимали с осадка на дне колбы. Растворитель удаляли в вакууме, добавляли рассол (20 мл) и затем экстрагировали EtOAc(370 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и выпаривали,получая бесцветное масло. Осадок из реакции делили между EtOAc и насыщенным раствором бикарбоната натрия, и разделяли слои. Органический слой промывали рассолом, сушили над MgSO4, фильтровали, объединяли с полученным выше маслом и удаляли растворитель в вакууме, получая белый твердый этил-3-амино-4-метил-1 Н-пиразол-5-карбоксилат (1,92 г, 11,35 ммоль, выход 62,5%) в качестве требуемого продукта). 1 Стадия 1. 6-(трет-Бутилтио)-4-хлор-7-метоксихинолин. Смесь 6-бром-4-хлор-7-метоксихинолина (50 г, 183 ммоль), Pd(Ph3P)4 (5,30 г, 4,59 ммоль), карбоната натрия (48,6 г, 459 ммоль) и 1,4-диоксана (895 мл) барботировали азотом в течение 10 мин. Добавляли 2-метил-2-пропантиол (tBuSH; 22,75 мл, 202 ммоль) и реакционную смесь нагревали при 70C в течение 4 дней. Реакционную смесь охлаждали до комнатной температуры и пропускали через слой силикагеля,который был предварительно пропитан EtOAc, используя 100% EtOAc в качестве элюента. Содержащие продукт фракции растирали с MeOH и объединяли, получая 6-(трет-бутилтио)-4-хлор-7-метоксихинолин(с, 1 Н), 3,99 (с, 3H), 1,31 (с, 9 Н). МС (m/z) 282. Стадия 2. 6-(трет-Бутилсульфонил)-4-хлор-7-метоксихинолин. К раствору 6-(трет-бутилтио)-4-хлор-7-метоксихинолина (18,5 г, 63,0 ммоль) в EtOAc (315 мл) и воде (315 мл) добавляли оксон (44,6 г, 12,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Слои разделяли и водный слой экстрагировали EtOAc (2). Объединенные органические экстракты выпаривали досуха, растворяли в минимальном количестве 10% МеОН/ДХМ, наносили на колонку Biotage с силикагелем 340 г и очищали с помощью колоночной хроматографии (BiotageSP-1, 340 г, 100% EtOAc в течение 20 мин, затем 0-20% MeOH/EtOAc). Наиболее чистые фракции выпаривали досуха и растирали с EtOAc, получая 6-(трет-бутилсульфонил)-4-хлор-7-метоксихинолин (15,2 г). 1H-ЯМР (400 МГц, ДМСО-d6)м.д.: 8,95 (д, J=4,8 Гц, 1 Н), 8,65 (с, 1 Н), 7,71-7,79 (м, 2 Н), 4,04 (с,3H), 1,31 (с, 9 Н). МС (m/z) 314. Стадия 3. 6-(трет-Бутилсульфонил)-N-(4,5-диметил-1 Н-пиразол-3-ил)-7-метоксихинолин-4-амин. К раствору 6-(трет-бутилсульфонил)-4-хлор-7-метоксихинолина (4,7 г, 14,98 ммоль) и 4,5-диметил 1 Н-пиразол-3-амина (1,998 г, 17,97 ммоль) в EtOH (74,9 мл) добавляли конц. HCl (2 капли). Реакционную смесь нагревали при 70C в течение 42 ч. Реакционную смесь выпаривали досуха и делили между ДХМ и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали ДХМ (1) и объединенные органические экстракты промывали рассолом (1) и выпаривали досуха. Материал растирали с ацетонитрилом/водой 1:1 (60 мл) (2) и сушили в вакуумном сушильном шкафу в течение ночи, получая 6-(трет-бутилсульфонил)-N-(4,5-диметил-1 Н-пиразол-3-ил)-7-метоксихинолин-4-амин К раствору хлорида железа(III) (1 мг, 6 моль) и N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-6(тетрагидро-2 Н-пиран-4-илтио)-4-хинолинамина (80 мг, 0,21 ммоль) в ацетонитриле (1 мл), перемешиваемому в течение 5 мин, добавляли перйодную кислоту (52 мг, 0,23 ммоль). Через 4 ч в реакционную смесь вливали насыщенный водный раствор Na2S2O3 и экстрагировали ДХМ. Органический слой выпа- 27023998 ривали и очищали с помощью хроматографии на силикагеле (0-10% 2 н. NH3/MeOH в ДХМ). Очищенный материал содержал некоторый избыток окисленного сульфона и его повторно очищали с помощью обращенно-фазовой ВЭЖХ, получая N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-6-(тетрагидро-2 Нпиран-4-илсульфинил)-4-хинолинамин (10 мг, 12%). 1(0,29 мл, 2,6 ммоль) добавляли в пробирку для СВЧ-реактора и продували азотом в течение 10 мин. После нагревания при 80C в течение ночи реакция прошла только приблизительно на 50%, и добавляли дополнительную порцию тетракис-(трифенилфосфин)палладия(0) (150 мг). Реакционную смесь барботировали азотом в течение 10 мин, добавляли тиол (290 мкл) и нагревали при 100C в течение ночи. Реакционную смесь делили между EtOAc и водным раствором тиосульфата натрия/бикарбоната натрия (5:1,2 М). Водный слой экстрагировали EtOAc (1) и объединенные органические экстракты в сухую наносили на силикагель. Неочищенной продукт очищали с помощью колоночной хроматографии (ISCO-RfHCl и нагревали при 80C в течение 1 ч. Реакционную смесь выпаривали досуха, суспендировали в ДХМ и фильтровали, получая 6-[(1,1-диметилэтил)тио]-N-(4,5-диметил-1 Н-пиразол-3-ил)-7-(метилокси)-4 хинолинамин (175 мг, 0,45 ммоль, выход 50%) в виде соли HCl. 1(184 мг, 0,52 ммоль), ТГФ (4,9 мл), воды (246 мкл) и оксона (159 мг, 0,258 ммоль) перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь делили между EtOAc и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали EtOAc (1) и объединенные органические экстракты наносили всухую на силикагель и очищали с помощью колоночной хроматографии (ISCO-Rf,12 г, 0-20% МеОН/ДХМ), получая 96 мг требуемого продукта и 65 мг 1:1 SM/Pdt. 65 мг 1:1 SM/Pdt обрабатывали ТГФ (2 мл), водой (0,2 мл) и оксоном (30 мг). Реакционную смесь перемешивали в течение 1 ч. Реакционную смесь делили между EtOAc и насыщенным раствором бикарбоната натрия. Водный слой экстрагировали EtOAc (1) и объединенные органические экстракты объединяли с 96 мг чистого на 92% материала, наносили всухую на силикагель и очищали с помощью колоночной хроматографии (ISCO-Rf,4 г, 0-20% МеОН/ДХМ), получая 6-[(1,1-диметилэтил)сульфинил]-N-(4,5-диметил-1 Н-пиразол-3-ил)-7- 28023998H-ЯМР (400 МГц, ДМСО-d6)м.д.: 12,21 (с, 1 Н), 9,13 (с, 1 Н), 8,66 (с, 1 Н), 8,34 (д, J=5,6 Гц, 1 Н),7,32 (с, 1 Н), 6,40 (д, J=5,3 Гц, 1 Н), 3,86-3,98 (м, 3H), 2,20 (с, 3H), 1,78 (с, 3 Н), 1,17 (с, 9 Н). МС (m/z) 373 (М+Н). Сульфон может быть получен в три стадии при добавлении полного эквивалента оксона. Следующие примеры получали аналогичным способом, начиная с соответствующего хинолина из(150 мг, 0,39 ммоль) и оксон (240 мг, 0,39 ммоль) суспендировали в ТГФ (1 мл) и воде (1 мл) и перемешивали при комнатной температуре. После подтверждения завершения реакции согласно ЖХ/МС, реакционную смесь выпаривали, растворяли в МеОН и очищали с помощью обращенно-фазовой ВЭЖХ. Требуемые фракции нейтрализовали с использованием смолы MP-carbonate, которую затем удаляли фильтрованием и промывали МеОН. Фильтрат выпаривали, а остаток растворяли в 2 мл воды и 2 млMeCN. Раствор обрабатывали ультразвуком, полученную суспензию фильтровали и сушили в вакууме,получая соединение, указанное в заголовке (29 мг, 17%). 1H-ЯМР (ДМСО-d6) : 12,25 (шир.с, 1 Н), 9,29 (шир.с, 1 Н), 8,96 (с, 1 Н), 8,41 (д, J=5,3 Гц, 1 Н), 7,44 (с,1 Н), 6,46 (д, J=4,8 Гц, 1 Н), 4,04 (с, 3H), 3,88-3,98 (м, 2 Н), 3,74-3,88 (м, 1 Н), 3,32-3,41 (м, 2 Н), 2,20 (с, 3H),1,80 (с, 3H), 1,69 (м, 4 Н). МС (m/z) 417 (М+Н+). В альтернативе, EtOAc или МеОН могут использоваться в качестве органического компонента в смеси растворителей в отношениях, изменяющихся от 4:1 до 1:1 (органическая фаза:водная фаза). Следующие примеры получали аналогичным способом, начиная с соответствующего хинолина из"Получений" выше или коммерческих источников.
МПК / Метки
МПК: A61K 31/4709
Метки: киназ, качестве, ингибиторов, аминохинолины
Код ссылки
<a href="https://eas.patents.su/30-23998-aminohinoliny-v-kachestve-ingibitorov-kinaz.html" rel="bookmark" title="База патентов Евразийского Союза">Аминохинолины в качестве ингибиторов киназ</a>
Оксазолзамещенные индазолы в качестве ингибиторов pi3-киназ, содержащая их фармацевтическая композиция и их применение в лечении расстройств, опосредованных ненадлежащей активностью pi3-киназ

Номер патента: 21056
Опубликовано: 31.03.2015
Авторы: Парр Найджел Джеймс, Хэмблин Джули Николь, Ли Джоэль, Джоунс Пол Спенсер, Килинг Сюзанн Элейн, Митчелл Шарлотт Джейн
МПК: A61K 31/4439, A61P 11/00, A61K 31/422...
Метки: расстройств, качестве, содержащая, ненадлежащей, оксазолзамещенные, активностью, применение, опосредованных, лечении, фармацевтическая, pi3-киназ, индазолы, ингибиторов, композиция
Формула / Реферат:
1. Соединение формулы (I)где R1 представляет собой 9-членный бициклический гетероарил, причем указанный 9-членный бициклический гетероарил содержит один или два атома азота, или пиридинил, возможно замещенный одним или двумя заместителями, независимо выбранными из -OR6 и -NHSO2R7;R2 и R3 вместе с атомом азота, к которому они присоединены, связаны с образованием 6- или 7-членного гетероциклила, причем указанный 6- или 7-членный гетероциклил,...
Птеридиноны в качестве ингибиторов киназ

Номер патента: 5287
Опубликовано: 30.12.2004
Авторы: Рукасл Гордон Уильям, Крамер Джеймс Бернард, Добрусин Эллен Майра, Шоуолтер Хауард Дэниэл Холлис, Тугуд Питер Лоренс, Денни Уильям Эликзэндер, Мак Намара Деннис Джозеф
МПК: A61K 31/505, A61P 35/00, C07D 475/00...
Метки: качестве, киназ, птеридиноны, ингибиторов
Формула / Реферат:
1. Соединение формулы и его фармацевтически приемлемые соли, эфиры, амиды и пролекарства, где R2 представляет собой (CH2)nарил и (CH2)nгетероарил, где каждая из вышеупомянутых арильных и гетероарильных групп может быть не замещена или замещена 1-5 группами-заместителями, выбранными из (а) галогена; (б) амино, алкиламино и диалкиламино; (в) C1-C10алкокси, аминоалкокси, алкиламиноалкокси и диалкиламиноалкокси; (г) гидрокси; (д) алканоила,...
Замещенные 4-аминотиазолы в качестве ингибиторов циклинзависимых киназ

Номер патента: 3527
Опубликовано: 26.06.2003
Авторы: Чонг Уэсли К.М., Ксиао Вей, Ли Лин, Янг Йи, Чу Шао Сонг, Дувади Рохит К.
МПК: A61P 35/00, C07D 277/42, A61K 31/427...
Метки: ингибиторов, киназ, качестве, замещенные, 4-аминотиазолы, циклинзависимых
Формула / Реферат:
1. Соединение формулы I где R1 - замещенная или незамещенная группа, выбранная из C1-6-алкила; фенила; 5-9-членного гетероцикла, содержащего в качестве гетероатомов N, O, S; циклоалкила; (C1-6-алкил)карбонила; (C1-6-алкил)арила; и, в случае если R1 замещен, каждый заместитель независимо представляет собой галоген; галогеналкил; C1-6-алкил; C1-6-алкоксил; амино; нитро; простой тиоэфир; циано; амидо; карбоксил; сульфонамидную группу; кето-группу;...
Конденсированные производные тиазола в качестве ингибиторов киназ

Номер патента: 17187
Опубликовано: 30.10.2012
Авторы: Крепи Карен-Вивьян-Люсиль, Франклин Ричард Джереми, Фоли Анне Мари, Ауджла Павандип Сингх, Александер Рикки Питер
МПК: A61K 31/437, C07D 513/04, A61K 31/519...
Метки: тиазола, ингибиторов, производные, конденсированные, качестве, киназ
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль или сольватгде R11 обозначает С1-С6алкил,R12 обозначает С1-С6алкил,Т обозначает кислород или N-R25,R23 обозначает водород, цианогруппу, карбоксигруппу, С2-С6алкоксикарбонил, ди(C1-C6)алкиламинокарбонил, (С1-С6алкил)(циано(C1-C6)алкил)аминокарбонил, (С1-С6алкокси(С1-C6)алкил)(C1-С6алкил)аминокарбонил или азетидинилкарбонил иR25 обозначает С1-С6алкил.2. Соединение по п.1, где Т...
Производные 2-бифениламино-4-аминопиримидина в качестве ингибиторов киназ

Номер патента: 17252
Опубликовано: 30.11.2012
Авторы: Бурсалая Бадри, Чэнь Бэй, Ли Кристиан Чо-Хуа, Хе Сяосюй, Лю Вэньшу, Грей Натанаэл С., Марсилдже Томас Х.
МПК: A61K 31/506, A61P 31/00, A61P 35/00...
Метки: качестве, производные, ингибиторов, киназ, 2-бифениламино-4-аминопиримидина
Формула / Реферат:
1. Соединение формулы (1)или его фармацевтически приемлемая соль, где R1a обозначает галоген, С1-С6алкил или галогензамещенный С1-С6алкил;R1b обозначает Н;R2 обозначает С6-С10карбоциклическое или 5-10-членное гетероарильное или 5-7-членное гетероциклическое кольцо, каждое из которых содержит от 1 до 3 гетероатомов, выбранных из N, О и S, каждый из которых необязательно замещен 1-2 С1-С6алкилом, -L-Y, -L-C(O)O0-1-(CR2)q-R8 или -L-C(O)-NRR8;R3...
Предыдущий патент: Устройство для измерения деформаций грунта
Следующий патент: Ингибиторы csf-1r для лечения опухолей головного мозга
Случайный патент: Способ получения анилинов