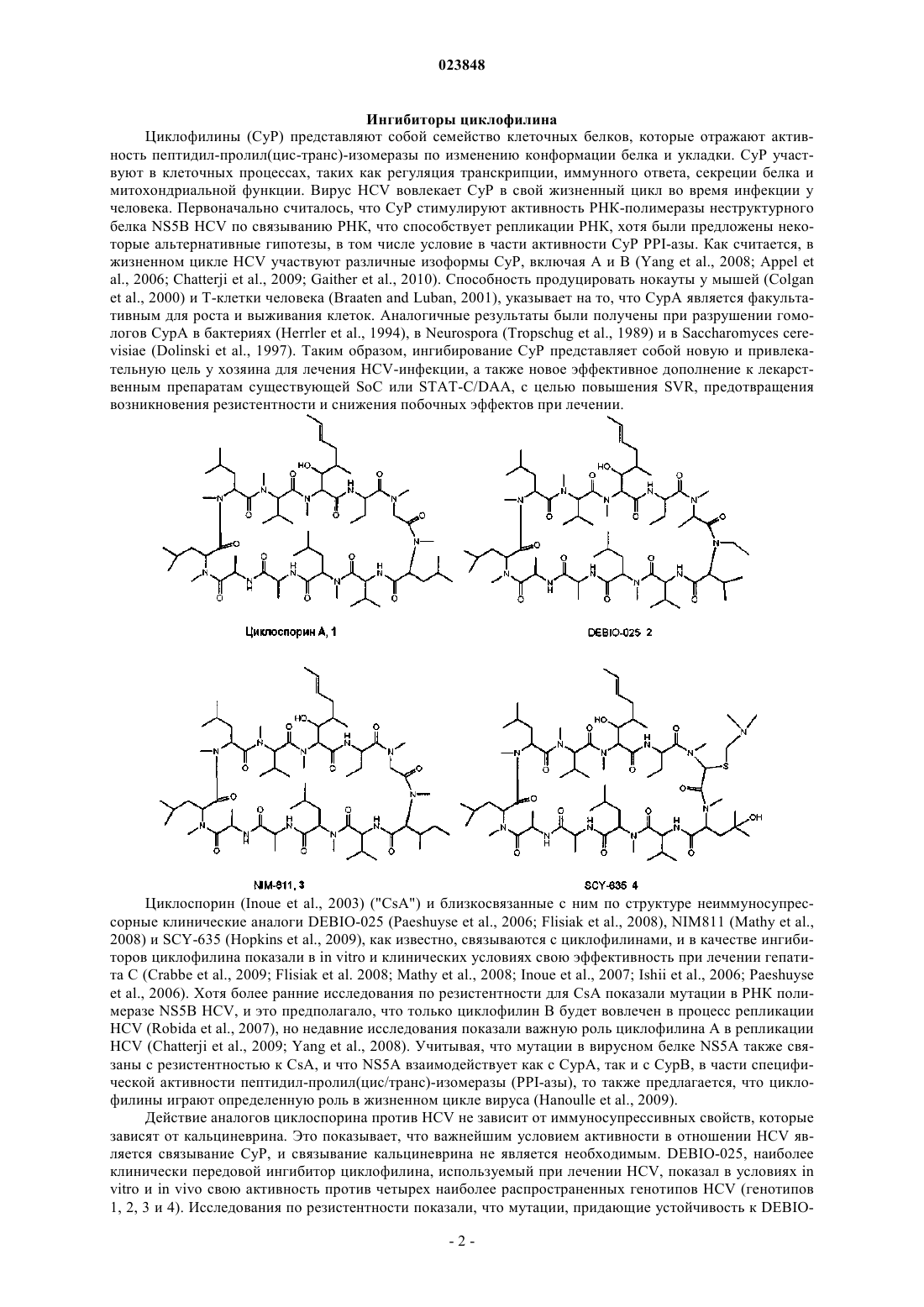
Макроциклическое соединение и способы его получения
Номер патента: 23848
Опубликовано: 29.07.2016
Авторы: Грегори Мэттью Алан, Уилкинсон Барри, Мосс Стивен Джеймс


























Формула / Реферат
1. Соединение формулы (I)

или его изомер, в котором С=С связь в положении С 26, 27, показанная в транс-форме, представляет собой цис-форму; или его фармацевтически приемлемая соль.
2. Твердая кристаллическая форма соединения формулы (I) по п.1.
3. Твердая кристаллическая форма по п.2, представляющая собой кристаллический полиморф формы I, пики на порошковой рентгеновской дифрактограмме (XPRD) которой имеют следующие характеристики:


4. Применение соединения по любому из пп.1-3 в качестве лекарственного средства для лечения вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ).
5. Применение соединения по любому из пп.1-3 в качестве иммунодепрессанта или противовоспалительного средства.
6. Фармацевтическая композиция, предназначенная для применения в качестве ингибитора циклофилина, содержащая соединение по любому из пп.1-3, в сочетании с фармацевтически приемлемым разбавителем или носителем.
7. Способ лечения вирусных инфекций, выбранных из вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-3.
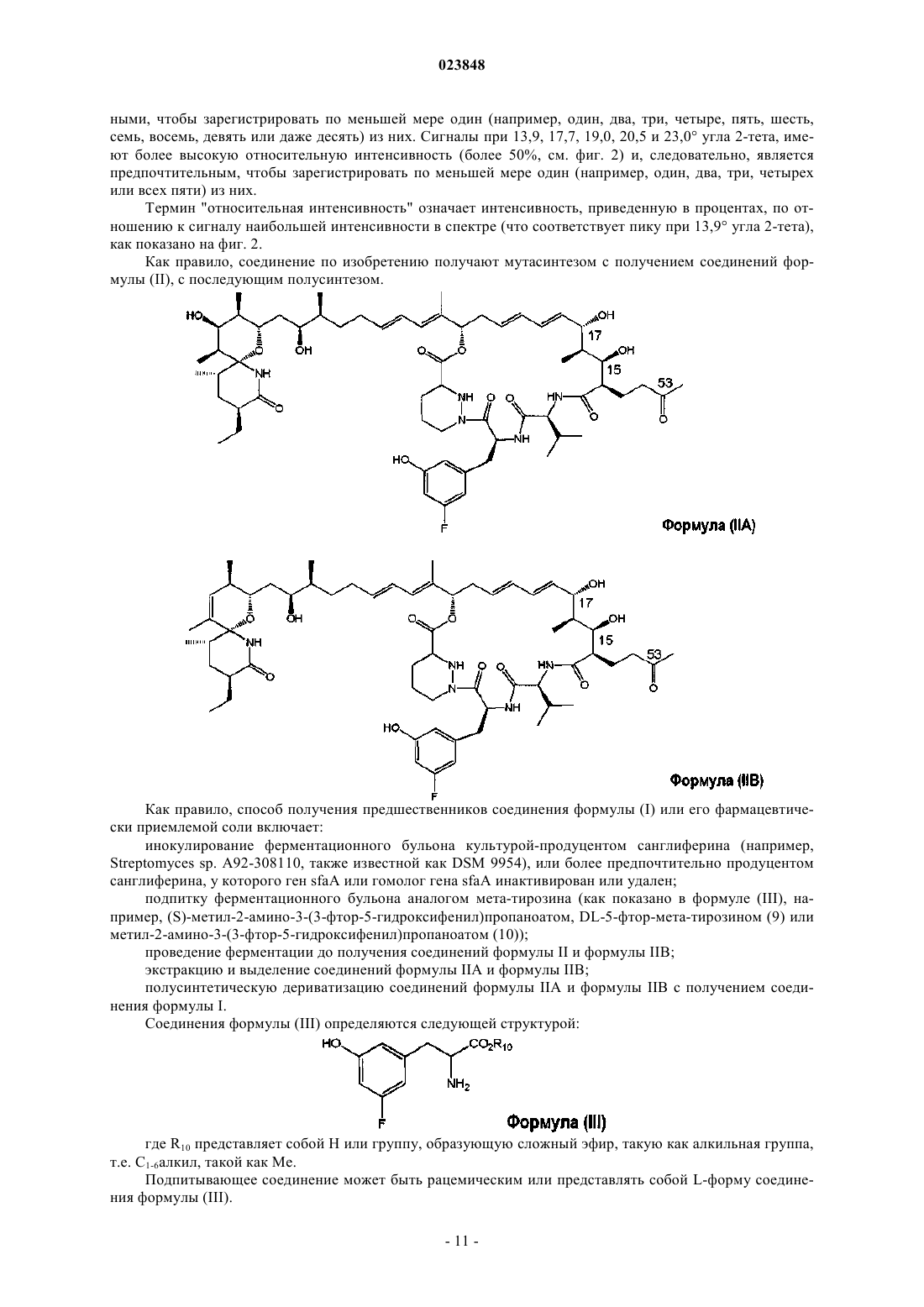
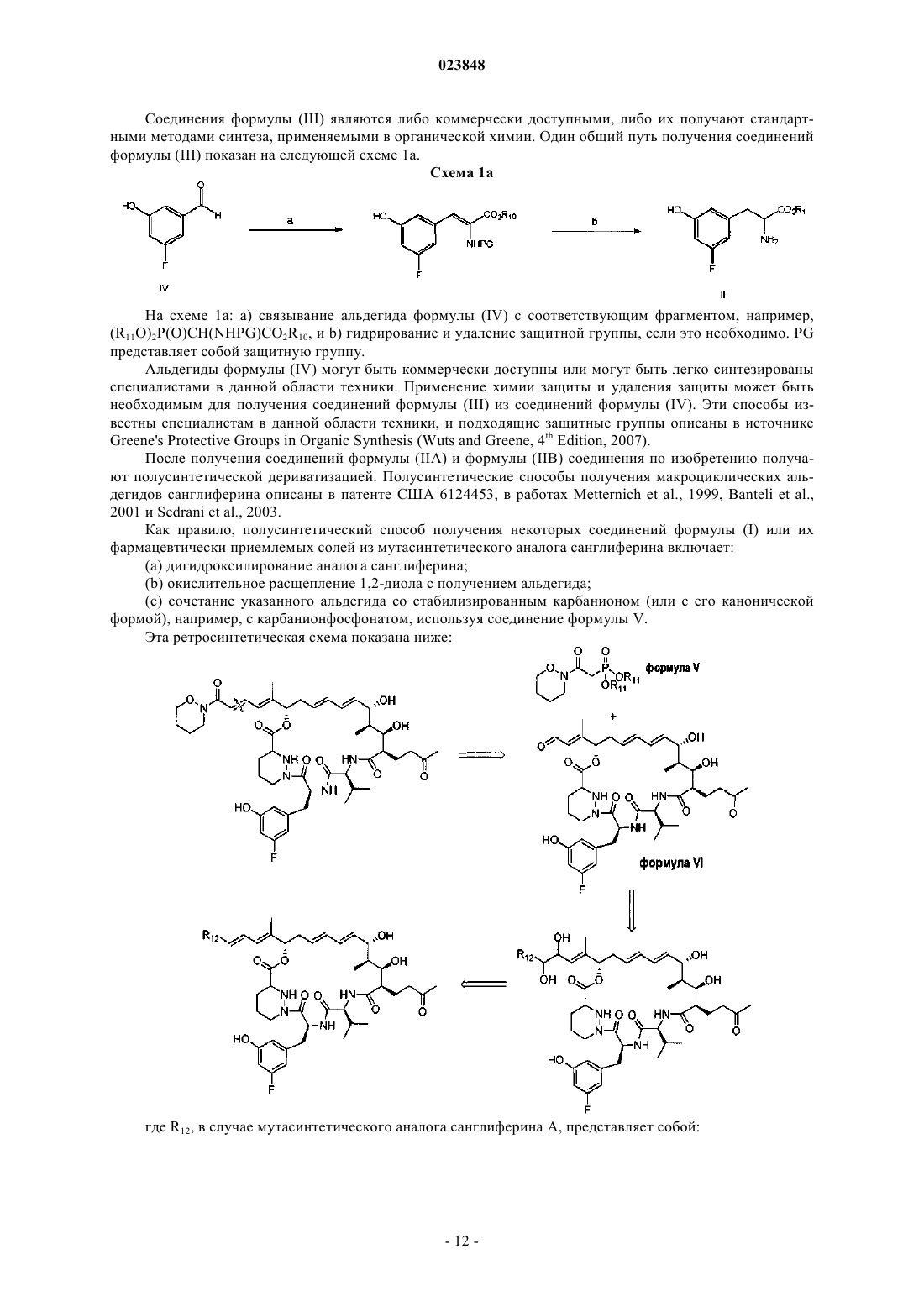
8. Способ получения соединения формулы (I) по п.1, заключающийся во взаимодействии соединения формулы (V)

где каждый R11 представляет собой независимо С1-4алкил или бензил;
с макроциклическим альдегидом (соединение формулы VI)

9. Способ получения соединения формулы (I) в виде кристаллического полиморфа формы I по п.3, заключающийся в кристаллизации соединения формулы (I) по п.1 из метилизобутилкетона.
Текст
Настоящее изобретение относится к соединению формулы (I), являющемуся аналогом санглиферина, которое полезно в качестве ингибитора циклофилина для лечения вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), а также в качестве иммунодепрессанта или противовоспалительного средства. Представлена также твердая кристаллическая форма указанного соединения, способ его получения, в частности в виде кристаллического полиморфа.(71)(73) Заявитель и патентовладелец: НЬЮРОУВИВ ФАРМАСЬЮТИКАЛ АБ (SE) Введение Настоящее изобретение относится к соединению - аналогу санглиферина, которое полезно в качестве ингибитора циклофилина. Настоящее изобретение также относится к применению указанного соединения в качестве лекарственного средства для лечения вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), а также в качестве иммунодепрессанта или противовоспалительного средства. Кроме того, настоящее изобретение относится к фармацевтической композиции, предназначенной для применения в качестве ингибитора циклофилина, которая содержит вышеуказанное соединение в сочетании с фармацевтических приемлемым разбавителем или носителем. Настоящее изобретение также относится к способу лечения вирусных инфекций, выбранных из вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), а также к способу получения указанного соединения, в частности к способу получения предложенного соединения в виде кристаллического полиморфа. Предпосылки создания изобретения Гепатит С Вирус гепатита С (HCV) представляет собой РНК-вирус с положительной цепью, и инфицирование им является основной причиной посттрансфузионного гепатита. HCV является наиболее распространенной хронической инфекцией, передаваемой через кровь, и основной причиной смерти от заболеваний печени в Соединенных Штатах. Всемирная организация здравоохранения считает, что в мире имеется более 170 млн хронических носителей вируса гепатита С, что составляет около 3% населения всего мира. Среди HCV-инфицированных пациентов, которые не были подвергнуты лечению, приблизительно у 7085% развивается хронический гепатит С, и, следовательно, у них имеется высокий риск развития цирроза печени и гепатоцеллюлярной карциномы. В развитых странах 50-76% всех случаев рака печени и две трети случаев трансплантации печени являются результатом хронической инфекции HCV (Manns et al.,2007). В дополнение к заболеваниям печени, у хронически инфицированных пациентов могут развиться другие хронические заболевания, связанные с HCV, а также они являются источником передачи инфекции другим людям. HCV инфекция вызывает осложнения, которые не связаны с печенью, такие как артралгии (боль в суставах), кожная сыпь и повреждения внутренних органов, преимущественно почек.HCV представляет важную глобальную проблему для здравоохранения, и в настоящее время не существует вакцины от гепатита С (Strader et al., 2004; Jacobson et al. 2007; Manns et al., 2007; Pawlotsky, 2005;ZeuzemHermann, 2002). Лечение гепатита С Существующий стандарт терапии (SoC) состоит в подкожных инъекциях ПЭГилированного интерферона- (pIFN) и пероральном приеме противовирусного препарата рибавирина в течение 24-48 недель. Успех в лечении определяется устойчивым вирусологическим ответом (SVR), который определяется как отсутствие РНК вируса гепатита С в сыворотке крови в конце периода лечения и через 6 месяцев. Общий уровень ответа на SoC в основном зависит от генотипа и от уровней РНК HCV до лечения. Пациенты с генотипами 2 и 3 более успешно отвечают на SoC, чем пациенты, инфицированные генотипом 1 (Melnikova, 2008; Jacobson et al., 2007). Значительное число больных гепатитом С неадекватно реагирует на лечение SoC или не может переносить лечение из-за побочных эффектов, что приводит к частым проблемам с завершением полного курса. Показатель общеклинического SVR на SoC составляет всего около 50% (Melnikova, 2008). Развитие резистентности представляет собой другой основной фактор неэффективности лечения (Jacobson etal., 2007). SoC также противопоказан некоторым пациентам, которые не считаются кандидатами на лечение, как пациенты, имевшие в прошлом значительные эпизоды депрессии или сердечных заболеваний. Побочные эффекты SoC, которые часто приводят к прекращению лечения, включают гриппоподобное заболевание, лихорадку, усталость, гематологическое заболевание, анемию, лейкопению, тромбоцитопению, алопецию и депрессию (Manns et al., 2007). Принимая во внимание побочные эффекты, связанные с длительным лечением с использованиемSoC, развитие резистентности и субоптимальный общий уровень успеха, для лечения HCV-инфекции крайне необходимы более эффективные и безопасные новые способы лечения. Цели новых способов лечения включают улучшение эффективности лечения, улучшение профиля токсичности, улучшение профиля резистентности, улучшение качества жизни и, в итоге, улучшение режима лечения пациента. HCV имеет короткий жизненный цикл и, следовательно, развитие лекарственной резистентности во время лекарственной терапии является распространенным явлением. В настоящее время разрабатывается новая, специфически нацеленная противовирусная терапия гепатита С (STAT-C), также известная как лечение с помощью лекарственных препаратов прямого противовирусного действия (DAA), направленная на целевые вирусные белки, такие как вирусная РНКполимераза NS5B или вирусная протеаза NS3 (Jacobson et al., 2007; Parfieniuk et al., 2007). Кроме того, в настоящее время также разрабатываются новые соединения, направленные на целевые белки человека(например, циклофилины), а не на вирусные цели, что, как можно ожидать, приведет к снижению случаев резистентности во время лекарственной терапии (Manns et al., 2007; Pockros, 2008; Pawlotsky J.-M.,2005). Ингибиторы циклофилина Циклофилины (CyP) представляют собой семейство клеточных белков, которые отражают активность пептидил-пролил(цис-транс)-изомеразы по изменению конформации белка и укладки. CyP участвуют в клеточных процессах, таких как регуляция транскрипции, иммунного ответа, секреции белка и митохондриальной функции. Вирус HCV вовлекает СуР в свой жизненный цикл во время инфекции у человека. Первоначально считалось, что СуР стимулируют активность РНК-полимеразы неструктурного белка NS5B HCV по связыванию РНК, что способствует репликации РНК, хотя были предложены некоторые альтернативные гипотезы, в том числе условие в части активности СуР PPI-азы. Как считается, в жизненном цикле HCV участвуют различные изоформы СуР, включая А и В (Yang et al., 2008; Appel etal., 2006; Chatterji et al., 2009; Gaither et al., 2010). Способность продуцировать нокауты у мышей (Colganet al., 2000) и Т-клетки человека (Braaten and Luban, 2001), указывает на то, что СурА является факультативным для роста и выживания клеток. Аналогичные результаты были получены при разрушении гомологов СурА в бактериях (Herrler et al., 1994), в Neurospora (Tropschug et al., 1989) и в Saccharomyces cerevisiae (Dolinski et al., 1997). Таким образом, ингибирование СуР представляет собой новую и привлекательную цель у хозяина для лечения HCV-инфекции, а также новое эффективное дополнение к лекарственным препаратам существующей SoC или STAT-C/DAA, с целью повышения SVR, предотвращения возникновения резистентности и снижения побочных эффектов при лечении. Циклоспорин (Inoue et al., 2003) ("CsA") и близкосвязанные с ним по структуре неиммуносупрессорные клинические аналоги DEBIO-025 (Paeshuyse et al., 2006; Flisiak et al., 2008), NIM811 (Mathy et al.,2008) и SCY-635 (Hopkins et al., 2009), как известно, связываются с циклофилинами, и в качестве ингибиторов циклофилина показали в in vitro и клинических условиях свою эффективность при лечении гепатита С (Crabbe et al., 2009; Flisiak et al. 2008; Mathy et al., 2008; Inoue et al., 2007; Ishii et al., 2006; Paeshuyseet al., 2006). Хотя более ранние исследования по резистентности для CsA показали мутации в РНК полимеразе NS5B HCV, и это предполагало, что только циклофилин В будет вовлечен в процесс репликацииHCV (Robida et al., 2007), но недавние исследования показали важную роль циклофилина А в репликацииHCV (Chatterji et al., 2009; Yang et al., 2008). Учитывая, что мутации в вирусном белке NS5A также связаны с резистентностью к CsA, и что NS5A взаимодействует как с СурА, так и с СурВ, в части специфической активности пептидил-пролил(цис/транс)-изомеразы (PPI-азы), то также предлагается, что циклофилины играют определенную роль в жизненном цикле вируса (Hanoulle et al., 2009). Действие аналогов циклоспорина против HCV не зависит от иммуносупрессивных свойств, которые зависят от кальциневрина. Это показывает, что важнейшим условием активности в отношении HCV является связывание СуР, и связывание кальциневрина не является необходимым. DEBIO-025, наиболее клинически передовой ингибитор циклофилина, используемый при лечении HCV, показал в условиях invitro и in vivo свою активность против четырех наиболее распространенных генотипов HCV (генотипов 1, 2, 3 и 4). Исследования по резистентности показали, что мутации, придающие устойчивость к DEBIO-2 023848 025, отличаются от известных мутаций для ингибиторов полимеразы и протеазы, и что не было какойлибо перекрестной устойчивости с вирусными репликонами, устойчивыми к STAT-C/DAA. Что еще более важно, DEBIO-025 также предотвращает развитие мутаций, которые придают устойчивость как к ингибиторам протеазы, так и к полимеразам (Crabbe et al., 2009). Тем не менее, в отношении ингибиторов циклофилина на основе CsA, находящихся на стадии клинической разработки, имеется ряд вопросов, которые, как считается, связаны с общими структурами этого класса, включая: наличие некоторых неблагоприятных событий, которые могут привести к прекращению лечения и ограничению уровней клинических доз; изменчивая фармакокинетика, которая может привести к различной эффективности и повышенным рискам лекарственных взаимодействий, что в итоге может привести к проблемам дозирования. Наиболее часто встречающиеся побочные эффекты (АЕ) у пациентов, получавших DEBIO-025,включают желтуху, боли в животе, рвоту, усталость, лихорадку. Наиболее клинически значимые ПЭ представляют собой гипербилирубинемию и уменьшение количества тромбоцитов (тромбоцитопению). ПЭГ-IFN может вызвать сильную тромбоцитопению, и в сочетании с DEBIO-025 может создавать значительные клинические проблемы. Увеличение билирубина и снижение тромбоцитов также были описаны в ранних клинических исследованиях с NIM-811 (Ke et al., 2009). Несмотря на то что наблюдавшаяся гипербилирубинемия при клинических исследованиях DEBIO-025 прекращалась по завершении лечения,это явилось причиной для прекращения лечения у 4 из 16 пациентов, а также для снижение уровней доз в последующих испытаниях. В отношении противовирусного эффекта ингибиторов циклофилина, в случаеHCV отмечена связь с дозой, при этом снижение дозы привело к уменьшению противовирусного эффекта, и ряд более поздних исследований с ингибиторами циклофилина на основе CsA также показали отсутствие или очень слабое снижение вирусной HCV нагрузки при введении дозы в виде монотерапии(Lawitz et al., 2009; Hopkins et al., 2009; Nelson et al., 2009). Как известно, DEBIO-025 и циклоспорин А представляют собой ингибиторы желчных транспортеров, таких как экспортирующая помпа желчных кислот и других печеночных транспортеров, в частности, ОАТ 1 В 1, ОАТ 1 В 3, MRP2, MRP3, сМОАТ иABCC2 (Crabbe et al., 2009). Было высказано предположение, что взаимодействие с желчными транспортерами, в частности с MRP2, может быть причиной гипербилирубинемии, наблюдаемой при высоких уровнях доз DEBIO-025 (Nelson et al., 2009, Wring et al., 2010). Лекарственные взаимодействия (DDI),связанные с классом CsA, за счет ингибирования другими лекарственными средствами, воздействующими на транспортеры, такими как Р-гликопротеин (Pgp/MDR1), BSEP, OAT1B1 и ОАТ 1 В 3 (Konig et al.,2010), также может быть проблемой, потенциально ограничивающей применение определенных комбинации лекарственных средств, и их применение для некоторых пациентов, проходящих лечение сопутствующих инфекций, таких как ВИЧ (Seden et al., 2010). Кроме того, DEBIO-025 и циклоспорин А являются субстратами для метаболизма цитохромом Р 450(особенно CYP3A4) и они известны как субстраты и ингибиторы Р-гликопротеина человека (MDR1)(Crabbe et al., 2009). Также было показано в условиях in vitro, что циклоспорин А является ингибитором для CYP3A4 (Niwa et al., 2007). Это означает, что в данном случае отсутствует повышенный риск лекарственного взаимодействия с другими препаратами, которые являются субстратами, индукторами или ингибиторами CYP3A4, такими как, например, кетоконазол, циметидин и рифампицин. Кроме того, также ожидается взаимодействие с препаратами, которые являются мишенями для транспорта Ргликопротеином (например, с дигоксином), что может привести к серьезным лекарственным взаимодействиям у пациентов с HCV, получающих медицинское лечение в отношении других сопутствующих заболеваний (Crabbe et al. 2009). Также известно, что CsA имеет значительные вариации в фармакокинетике, и ранее исследовавшиеся препараты показали биодоступность от 1 до 89% (Kapurtzak et al., 2004). Без выполнения дорогого мониторинга уровней в крови пациента, это может привести к развитию и росту побочных эффектов из-за увеличенных плазменных уровней, или к снижению клинического ответа за счет снижения плазменных уровней препарата. Учитывая, что ингибирование циклофилинов представляет собой новый перспективный подход к лечению гепатита С, существует необходимость в создании и разработке более эффективных и безопасных ингибиторов СуР для применения в комбинированной терапии против инфекции HCV. Санглиферины Санглиферин A (SfA) и его природные формы относятся к классу смешанных нерибосомальных пептидов/поликетидов, продуцируемых Streptomyces sp. A92-308110 (также известный как DSM 9954)(см. WO 97/02285), которые были первоначально обнаружены на основе их высокой аффинности к циклофилину А (CyPA). SfA является наиболее распространенным компонентом в ферментационных бульонах и проявляет приблизительно в 20 раз более высокую аффинность в отношении СуРА, чем CsA. Это привело к предположению, что санглиферины могут быть полезны для лечения инфекций HCV (WO 2006/138507). Также было показано, что санглиферины обладают более низкой иммуносупрессивной активностью, чем CsA, при испытании in vitro (Sanglier et al., 1999; Fehr et al., 1999). SfA связывается с высокой аффинностью с сайтом связывания CsA в CyPA (Kallen et al., 2005). Биосинтез санглиферинов Санглиферины биосинтезируют с использованием смешанной поликетидной синтазы(PKS)/нерибосомальной пептидной синтетазы (NRPS) (см. WO 2010/034243). 22-членная макролидная цепь состоит из поликетидной углеродной цепи и трипептидной цепи. Пептидная цепь состоит из одной природной аминокислоты, валина, и двух неприродных аминокислот: (S)-мета-тирозина и (S)пиперазиновой кислоты, связанных амидной связью. Гидроксилированием фенилаланина (либо на стадии NRPS in situ, либо до биосинтеза) получают(S)-мета-тирозин, что, как считается, происходит через продукт гена sfaA. Иммуносупрессивное действие санглиферинов Механизм иммуносупрессивного действия SfA отличается от механизма других известных иммунофилин-связывающих иммуносупрессивных лекарственных средств, таких как CsA, FK506 и рапамицин. SfA не ингибирует фосфатазную активность кальциневрина, мишени для CsA (Zenke et al., 2001), и вместо этого его иммуносупрессивная активность обусловлена ингибированием интерлейкина-6 (Hartelet al., 2005), интерлейкина-12 (Steinschulte et al., 2003) и ингибированием пролиферации зависимых от интерлейкина-2 Т-клеток (Zhang, Liu, 2001). Однако молекулярная мишень и механизм, посредством которого SfA оказывает свое иммуносупрессивное действие, до сих пор не известны. Молекулярная структура SfA является сложной и его взаимодействие с СурА, как полагают, опосредованно основной макроциклической частью молекулы. Действительно, макроциклическое соединение (гидроксимакроцикл), полученное окислительным расщеплением SfA, показало сильное сродство к СурА (Sedrani et al., 2003). Данные рентгеновского исследования кристаллической структуры показали,что гидроксимакроцикл связывается с тем же активным сайтом СурА, как и CsA. Также ранее было показано, что аналоги на основе макроцикла фрагмента SfA, лишенные иммуносупрессивных свойств (Sedrani et al., 2003), обеспечивают возможность получения неиммуносупрессорных ингибиторов СуР для возможного использования в терапии инфекций HCV. В противоположность этому, также существует возможность разработки иммуносупрессивных средств с низкой токсичностью для использования в таких областях, как профилактика отторжения трансплантата, лечения аутоиммунных, воспалительных и респираторных заболеваний, включая, но, не ограничиваясь ими, болезнь Крона, синдром Бехчета, увеит, псориаз, атопический дерматит, ревматоидный артрит, нефротический синдром, апластическую анемию, билиарный цирроз печени, астму, легочный фиброз, хроническую обструктивную болезнь легких (ХОБЛ) и целиакию. Как было показано, санглиферины имеют новый механизм иммуносупрессивной активности (Zenke et al., 2001), действуя, возможно, через дендритные клетки хемокинов (Immecke et al., 2011), и поэтому существует возможность разработки средств с механизмом действия, отличающимся от механизма известных клинических средств, таких как циклоспорин, рапамицин и FK 506. Было показано, что санглиферин А действует в 10 раз менее сильно, чем циклоспорин А, так что идеальное новое средство должно обладать улучшенной эффективностью и/или улучшенным терапевтическим окном. Другие терапевтические применения ингибиторов циклофилина Вирус иммунодефицита человека (ВИЧ) Ингибиторы циклофилина, такие как CsA и DEBIO-025, также показали потенциальную полезность в ингибировании репликации ВИЧ. Считается, что ингибиторы циклофилина влияют на функции СурА в ходе и/или завершении обратной транскрипции ВИЧ (Ptak et al., 2008). Тем не менее, при клинических испытаниях DEBIO-025 только снижал уровни РНК ВИЧ-1 на 0,5 и 1 log10 копий/мл у девяти и двух пациентов соответственно, в то время как 27 пациентов не показали снижения уровней РНК ВИЧ-1(Steyn et al., 2006). После этого DEBIO-025 был опробован для лечения HCV/ВИЧ инфицированных пациентов, показав при этом, более высокую эффективность против HCV, и клинические испытания в отношении ВИЧ были прекращены (см. Watashi et al., 2010). Лечение ВИЧ Во всем мире имеется более 30 млн человек, инфицированных ВИЧ-1, и каждый год возникает 3 млн новых случаев. С внедрением высокоактивной антиретровирусной терапии (HAART) варианты лечения значительно улучшились (Schopman et al., 2010), и к 2008 году для лечения ВИЧ-1 было лицензировано около 25 антиретровирусных лекарственных средств, включая девять нуклеозидных ингибиторов обратной транскриптазы (NRTI), четыре ненуклеозидных ингибиторов обратной транскриптазы(NNRTI), девять ингибиторов протеаз (PI), один ингибитор слияния, один ингибитор CCR5 и один ингибитор интегразы (Shafer and Schapiro, 2008). Тем не менее, ни один из этих известных режимов лечения не приводит к полной элиминации вируса, и они могут привести к серьезным побочным эффектам и устойчивости к противовирусным препаратам, что по-прежнему вызывает серьезную озабоченность. Таким образом, до сих пор существует потребность в новых противовирусных терапиях, особенно с использованием классов соединений с новым механизмом действия, где нет утвержденных лекарственных средств, в частности таких, как ингибиторы циклофилина. Вирус гепатита В Гепатит В является ДНК-содержащим вирусом из семейства Hepadnaviridae и является возбудителем гепатита В. В отличие от случаев инфекции HCV и ВИЧ, было очень мало публикаций, касающихся активности ингибиторов циклофилина против вируса гепатита В. В работе Ptak et al., 2008 описана слабая активность Debio-025 против HBV (IC50 4,1 мкМ), в то время как в работе Xie et al., 2007 описана некоторая активность CsA против HBV (IC50 1,3 мкг/мл). Это представляет собой отличие от случаев в отношении ВИЧ и HCV, где имеются многочисленные сообщения о противовирусной активности ингибиторов циклофилина в наномолярных количествах. Лечение HBV Вирусом гепатита В во всем мире заражено до 400 млн человек, и он является основной причиной хронического вирусного гепатита и гепатоцеллюлярной карциномы. В 2008 году было лицензировано шесть препаратов для лечения HBV: интерферон-альфа и пэгилированный интерферон-альфа, три нуклеозидных аналога (ламивудин, энтекавир и телбивудин) и один нуклеотидный аналог (дипивоксил адефовира). Однако из-за высоких уровней резистентности, плохой переносимости и возможных побочных эффектов, необходимы новые терапевтические возможности (Ferir et al., 2008). Ингибирование митохондриальных пор с транзиторной проницаемостью (mPTP) Открытие пор с транзиторной проницаемостью в митохондриях приводит к высокой проводимости этих пор и инициирует начало процесса мембранной проницаемости в митохондриях (MPT). Это событие является причиной, приводящей к некрозу и апоптозу гепатоцитов после окислительного стресса,Са 2+-токсичности и ишемии/реперфузии. Было показано, что ингибирование циклофилина D (также известного как циклофилин F) ингибиторами циклофилина блокирует открытие пор с транзиторной проницаемостью и защищает клетки от гибели после такого стрессового воздействия. Ингибиторы циклофилина D могут применяться при состояниях, когда имеет место открытие mPTP, таких как мышечная дистрофия, в частности, при врожденной мышечной дистрофии Ульриха и миопатии Бетлема (Millay et al.,2008, WO 2008/084368, Palma et al., 2009), при рассеянном склерозе (Forte et al., 2009), сахарном диабете(Fujimoto et al., 2010), боковом амиотрофическом склерозе (Martin, 2009), биполярном расстройстве (Kubota et al., 2010), болезни Альцгеймера (Du, Yan, 2010), болезни Хантингтона (Perry et al., 2010), восстановлении после инфаркта миокарда (Gomez et al., 2007) и хроническом потреблении алкоголя (King et al.,2010). Дополнительные терапевтические применения Ингибиторы циклофилина имеют потенциальную противовирусную активность и, следовательно,могут применяться в лечении инфекций других вирусов, таких как вирус ветряной оспы (Ptak et al.,2008), вирус гриппа A (Liu et al., 2009), коронавирус тяжелого острого респираторного синдрома и другие коронавирусы человека и кошачьих (Chen et al., 2005, Ptak et al., 2008), вирус Денге (Kaul et al., 2009),вирус желтой лихорадки (Qing et al., 2009), вирус западного Нила (Qing et al., 2009), вирус западного лошадиного энцефалита (Qing et al., 2009), цитомегаловирус (Kawasaki et al., 2007) и вирус коровьей оспы(Castro et al., 2003). Также имеются сообщения о полезности ингибиторов циклофилина и ингибирования циклофилина в других терапевтических областях, таких как лечение рака (Han et al., 2009). Общие замечания по санглиферинам Одной из проблем, возникающей в ходе разработки лекарственных средств, таких как санглиферины, является быстрый метаболизм и глюкуронидация, которые приводят к низкой биодоступности. Это может привести к увеличению вероятности возникновения эффекта от приема пищи, большей частоте неполного высвобождения из дозированной формы и высокой вариабельности показателей между пациентами. Поэтому сохраняется необходимость в идентификации новых ингибиторов циклофилина, которые могут быть полезны, в частности, при лечении инфекции HCV, а также при лечении других заболеваний,при которых ингибирование циклофилинов может быть полезно, например, в случае ВИЧ-инфекции,мышечной дистрофии, или для помощи при восстановлении после инфаркта миокарда, или в случаях,когда иммуносупрессия или противовоспалительное действие являются полезными. Предпочтительно,когда такие ингибиторы циклофилина обладают улучшенными свойствами по сравнению с применяемыми в настоящее время ингибиторами циклофилина, включая одно или более следующих свойств: более длительный период полувыведения или повышенную биодоступность, возможно через пониженный метаболизм Р 450 и/или сниженную глюкуронизацию, улучшенная растворимость в воде, повышенная эффективность против HCV, пониженная токсичность (включая гепатотоксичность), улучшенный фармакологический профиль, такой как направленное воздействие на органы-мишени (например, на печень,в случае HCV) и/или длительный период полувыведения (обеспечивающий меньшую частоту дозирования), снижение лекарственного взаимодействия, такого как протекающего через сниженные уровни метаболизма CYP3A4, и ингибирование и снижение ингибирования PGP (что облегчает случаи взаимодействия при множественной лекарственной комбинации), улучшение профиля побочных эффектов, таких как низкое связывание с MRP2, которое ведет к снижению вероятности возникновения гипербилирубинемии, снижение иммунодепрессивного действия, улучшенная активность против устойчивых видов вирусов, в частности, против видов вирусов, устойчивых к CsA и к аналогу CsA (например, DEBIO-025),высокий терапевтический индекс и/или индекс селективности. Настоящее изобретение раскрывает новый аналог санглиферина, который может обладать одним или более свойствами, указанными выше. В частности, настоящее изобретение раскрывает новый мутасинтетический аналог санглиферина, который, как ожидается, имеет сниженный метаболизм за счет Р 450 или глюкуронизацию, что, например, показано повышенным временем полужизни микросом, и/или уменьшенную активность против HCV, что, например, показано снижением ЕС 50 для репликона. Кроме того, также существует необходимость в разработке нового иммуносупрессивного средства,которое может быть полезно для профилактики отторжения трансплантата, или при лечении аутоиммунных, воспалительных и респираторных заболеваний. Предпочтительно, когда такой иммунодепрессант будет обладать улучшенными свойствами, по сравнению с известными природными санглиферинами,включая одно или более следующих свойств: более длительный период полувыведения или повышенную биодоступность, возможно через пониженный метаболизм Р 450 и/или сниженную глюкуронизацию,улучшенную растворимость в воде, повышенную эффективность в части иммуносупрессивной активности, такая как наблюдаемая в Т-клеточных анализах пролиферации, сниженную токсичность (включая гепатотоксичность), улучшенный фармакологический профиль, такой как направленное воздействие на органы-мишени и/или длительный период полувыведения (обеспечивающий меньшую частоту дозирования), снижение лекарственного взаимодействия, такого как протекающего через сниженные уровни метаболизма CYP3A4, и ингибирование и снижение ингибирования PGP (что облегчает случаи взаимодействия при множественной лекарственной комбинации), улучшение профиля побочных эффектов. Настоящее изобретение раскрывает новый аналог санглиферина, который может обладать одним или более свойствами, указанными выше. В частности, настоящее изобретение раскрывает новое производное, которое имеет сниженный метаболизм за счет Р 450 или глюкуронизации, что, например, показано повышенным временем полужизни микросом, и/или улучшенный иммуносупрессивный эффект, что, например, показано снижением IC50 для пролиферации Т-клеток. Таким образом, как можно видеть из примеров, соединение по настоящему изобретению обладает следующими положительными терапевтическими свойствами: улучшенной противовирусной активностью против ВИЧ и HCV, по сравнению с используемым в уровне техники ингибиторами циклофилина: циклоспорин A, DEBIO-025 (алиспоривир) и санглиферин А; уменьшенным клиренсом и увеличенным действием при пероральном приеме, по сравнению с используемым в уровне техники соединением санглиферин А; более мощным ингибированием СурА в части PPI-азной активности по сравнению с используемым в уровне техники ингибиторами циклофилина: циклоспорин A, DEBIO-025 (алиспоривир) и санглиферин А; улучшенным профилем побочных эффектов и снижением лекарственного взаимодействия, что показано пониженным ингибированием транспортеров билирубина (ОАТР-1 В 1, ОАТР-1 В 3, MRP2 и MRP3) и снижением ингибирования транспортеров ксенобиотиков (PGP и BSEP). Краткое изложение сущности изобретения Настоящее изобретение относится к новому макроциклическому аналогу санглиферина, в частности, к соединению формулы (I) или его изомеру, в котором С=С связь в положении С 26, 27, показанная в транс-форме, представляет собой цис-форму; или его фармацевтически приемлемой соли. Соединение формулы (I) может быть представлено в виде твердой кристаллической формы. Причем твердая кристаллическая форма соединения формулы (I) может представлять собой кристаллический полиморф формы I, пики на порошковой рентгеновской дифрактограмме (XPRD) которой имеют следующие характеристики: Еще одним аспектом настоящего изобретения является применение указанного соединения формулы (I) в качестве лекарственного средства для лечения вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), а также в качестве иммунодепрессанта или противовоспалительного средства. Еще одним другим аспектом настоящего изобретения является фармацевтическая композиция,предназначенная для применения в качестве ингибитора циклофилина, содержащая указанное соединение формулы (I) в сочетании с фармацевтически приемлемым разбавителем или носителем. Еще одним аспектом настоящего изобретения является способ лечения вирусных инфекций, выбранных из вируса гепатита С (HCV) или вируса иммунодефицита человека (ВИЧ), включающий введение пациенту терапевтически эффективного количества указанного соединения формулы (I). Еще одним аспектом настоящего изобретения является способ получения указанного соединения формулы (I), заключающийся во взаимодействии соединения формулы (V) где каждый R11 представляет собой независимо C1-4 алкил или бензил; с макроциклическим альдегидом (соединение формулы VI) Кроме того, предлагается способ получения указанного соединения формулы (I) в виде кристаллического полиморфа формы I, заключающийся в кристаллизации соединения формулы (I) из метилизобутилкетона. Определения Термины в единственном числе используются в данном описании для обозначения "один" или "более чем один" (например, по меньшей мере один) грамматический объект. В качестве примера, термин"аналог" означает один аналог или несколько аналогов. Используемый в данном описании термин "аналог(и)" относится к химическим соединениям, которые являются схожими друг с другом по структуре, но которые немного отличаются по составу (за счет замены одного атома на другой, или присутствия или отсутствия конкретной функциональной группы). Используемый в данном описании термин "санглиферин(ы)" относится к химическим соединениям,которые подобны по структуре санглиферину А, но немного отличаются по составу (за счет замены одного атома на другой, или присутствия или отсутствия конкретной функциональной группы), в частности, которые получают путем ферментации Streptomyces sp. A92-308110. Примеры соединений включают санглиферин-подобные соединения, раскрытые в WO 97/02285 и WO 98/07743, например санглиферин В. Используемый в данном описании термин "мутасинтетический санглиферин(ы)" или "мутасинтетический аналог(и) санглиферина" относится к химическим соединениям, которые подобны по структуре санглиферину А, В, С или D, но которые немного отличаются по составу (за счет замены одного атома на другой, или присутствия или отсутствия конкретной функциональной группы), в частности, которые получают путем ферментации Streptomyces sp. A92-308110 или его мутанта, где культура запитывается мета-тирозиновым аналогом. Используемый в данном описании термин "мета-тирозиновый аналог(и)" относится к химическим соединениям, которые подобны по структуре мета-тирозину, но которые немного отличаются по составу(за счет замены одного атома на другой, или присутствия или отсутствия конкретной функциональной группы), в частности, которые описываются формулой (III). Используемый в данном описании термин "макроциклический аналог", "макроциклический аналог санглиферина" или "макроциклический санглиферин" относится к соединению, указанному выше, как представляющие изобретение в его широком аспекте, например, к соединению формулы (I), представленной выше, или к его фармацевтически приемлемой соли. Эти соединения также называют "соединения по изобретению", или "производные санглиферина", или "аналоги санглиферина", при этом в настоящем описании эти термины используются взаимозаменяемо. Используемый в данном описании термин "HCV" относится к вирусу гепатита С, одноцепочечному,содержащему РНК, оболочечному вирусу в вирусном семействе Flaviviridae. Используемый в данном описании термин "ВИЧ" относится к вирусу иммунодефицита человека,возбудителя синдрома приобретенного иммунодефицита у человека. Используемый в данном описании термин "биодоступность" относится к показателю или скорости,с которой лекарственное средство или другое вещество поглощается или становится доступным на участке биологической активности после его введения. Это свойство зависит от ряда факторов, включая растворимость соединения, скорость абсорбции в кишечнике, степень связывания белка, метаболизма и т.п. Различные тесты по биологической доступности, которые должны быть известны специалисту в данной области техники, раскрыты в описании (также см. Egorin et al. 2002). Термин "растворимость в воде", используемый в данном описании, относится к растворимости в водной среде, например, в забуференном фосфатом физиологическом растворе (PBS) при рН 7,4 или в 5% растворе глюкозы. Тесты на растворимость в воде приведены ниже в примерах как "анализ растворимости в воде". Фармацевтически приемлемые соли соединений по изобретению, такие как соединения формулы(I), включают обычные соли, полученные с фармацевтически приемлемыми неорганическими или органическими кислотами или основаниями, а также кислотно-аддитивные соли четвертичного аммония. Более конкретные примеры подходящих кислот для получения солей включают хлористо-водородную,бромисто-водородную, серную, фосфорную, азотную, хлорную, фумаровую, уксусную, пропионовую,янтарную, гликолевую, муравьиную, молочную, малеиновую, винную, лимонную, пальмоиновую, малоновую, гидроксималеиновую, фенилуксусную, глутаминовую, бензойную, салициловую, фумаровую,толуолсульфоновую, метансульфоновую, нафталин-2-сульфоновую, гидроксинафтойную, бензолсульфоновую, иодисто-водородную, яблочную, стероиновую, танниновую кислоты и т.п. Соли хлористоводородной кислоты имеют особое значение. Другие соли, например соль щавелевой кислоты, которые сами по себе не являются фармацевтически приемлемыми, могут быть полезными в качестве промежуточных продуктов при получении соединений по изобретению и их фармацевтически приемлемых солей. Более конкретные примеры подходящих основных солей включают соли натрия, лития, калия, магния,алюминия, кальция, цинка, N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина и прокаина. Указание по тексту на "соединение по изобретению" включает соединение формулы (I) и его фармацевтически приемлемые соли. Используемый в данном описании термин "алкил" означает линейную или разветвленную алкильную группу, содержащую, как правило, 1-10 атомов углерода, например С 1-6 алкильная группа. Примеры алкильных групп включают С 1-4 алкильные группы, такие как метил, этил, н-пропил, изопропил и н-бутил. Термин "лечение" охватывает профилактическое и терапевтическое лечение. Термин "формула II" относится как к формуле IIA, так и к формуле IIB. Описание фигур На фиг. 1 представлен 1 Н-ЯМР спектр соединения 24. На фиг. 2 представлена порошковая рентгеновская дифрактограмма соединения 24 в твердой кристаллической форме (форма I). Подробное описание изобретения Настоящее изобретение относится к макроциклическим аналогам санглиферина, как указано выше,к способам получения данного соединения и к способам применения данного соединения в медицине. В одном варианте осуществления изобретения соединение представляет собой метанольный аддукт,в котором полукеталь образован комбинацией С-53 кетогруппы, С-15 гидроксильной группы и метанола. В другом варианте осуществления это не так. В другом варианте осуществления изобретения двойная связь в положении С 26, 27 представлена в цис-форме, как показано в следующей формуле: Такое соединение может быть получено путем химического синтеза. В дополнительном варианте осуществления изобретения предоставляется макроциклический аналог санглиферина формулы (I) в твердой кристаллической форме. В частности, предоставляется твердая кристаллическая форма (форма I) макроциклического аналога санглиферина формулы (I), который может быть получен (или был получен) путем кристаллизации аморфного макроциклического аналога санглиферина формулы (I) из метилизобутилкетона (MIBK). В одном варианте осуществления изобретения указанную аморфную форму суспендируют в MIBK и циклически изменяют температуру между максимальным и минимальным значением в течение общего периода времени, составляющем, например, 1, 2,5, 24 ч, 1 день, 2, 3, 4, 5, 6, 7 дней или 2 недели. В одном варианте осуществления изобретения температуру циклически изменяют в интервале от комнатной температуры до температуры 60 С, например, от комнатной температуры до 40 С. В одном варианте осуществления изобретения температурные циклы между величинами минимальной и максимальной температуры (и наоборот) выполняют каждые 2-8 ч,например каждые 3-5 ч или каждые 4 ч. В предпочтительном варианте осуществления изобретения температуру циклически изменяют от комнатной температуры до 40 С каждые 4 ч в течение, в общей сложности, 5 дней. Способ кристаллизации аморфной формы макроциклического аналога санглиферина формулы (I) подробно описано в примере 8. Макроциклический аналог санглиферина формулы (I) в виде кристаллической полиморфной формыI имеет порошковую рентгеновскую дифрактограмму (XRPD), по существу такую, как показано на фиг. 2. В табл. 2 (пример 8) указаны пики и их относительная интенсивность. Методика получения данныхXRPD описана в разделе "Общие способы". Таким образом, в изобретении предложен макроциклический аналог санглиферина формулы (I) в кристаллической форме (форма I), имеющий в дифрактограмме XRPD по меньшей мере один (например,один, два, три, четыре, пять, шесть, семь, восемь, девять, десять, одиннадцать, двенадцать, тринадцать,четырнадцать, пятнадцать, шестнадцать или даже семнадцать) сигналов при 8,3, 8,5, 11,1, 12,6, 13,9, 14,3,15,0, 16,9, 17,7, 18,6, 19,0, 20,1, 20,5, 20,9, 21,2, 21,7 и 23,0 градусах (0,2) угла 2-тета, при этом указанные сигналы представляют собой основные сигналы в дифрактограмме XRPD полиморфа формы I. Сигналы при 8,3, 8,5, 11,1, 13,9, 17,7, 18,6, 19,0, 20,5, 20,9 и 23,0 угла 2-тета обладают сравнительно высокой относительной интенсивностью (более 26%, см. фиг. 2) и, следовательно, являются предпочтитель- 10023848 ными, чтобы зарегистрировать по меньшей мере один (например, один, два, три, четыре, пять, шесть,семь, восемь, девять или даже десять) из них. Сигналы при 13,9, 17,7, 19,0, 20,5 и 23,0 угла 2-тета, имеют более высокую относительную интенсивность (более 50%, см. фиг. 2) и, следовательно, является предпочтительным, чтобы зарегистрировать по меньшей мере один (например, один, два, три, четырех или всех пяти) из них. Термин "относительная интенсивность" означает интенсивность, приведенную в процентах, по отношению к сигналу наибольшей интенсивности в спектре (что соответствует пику при 13,9 угла 2-тета),как показано на фиг. 2. Как правило, соединение по изобретению получают мутасинтезом с получением соединений формулы (II), с последующим полусинтезом. Как правило, способ получения предшественников соединения формулы (I) или его фармацевтически приемлемой соли включает: инокулирование ферментационного бульона культурой-продуцентом санглиферина (например,Streptomyces sp. А 92-308110, также известной как DSM 9954), или более предпочтительно продуцентом санглиферина, у которого ген sfaA или гомолог гена sfaA инактивирован или удален; подпитку ферментационного бульона аналогом мета-тирозина (как показано в формуле (III), например, (S)-метил-2-амино-3-(3-фтор-5-гидроксифенил)пропаноатом, DL-5-фтор-мета-тирозином (9) или метил-2-амино-3-(3-фтор-5-гидроксифенил)пропаноатом (10; проведение ферментации до получения соединений формулы II и формулы IIB; экстракцию и выделение соединений формулы IIA и формулы IIB; полусинтетическую дериватизацию соединений формулы IIA и формулы IIB с получением соединения формулы I. Соединения формулы (III) определяются следующей структурой: где R10 представляет собой Н или группу, образующую сложный эфир, такую как алкильная группа,т.е. С 1-6 алкил, такой как Me. Подпитывающее соединение может быть рацемическим или представлять собой L-форму соединения формулы (III). Соединения формулы (III) являются либо коммерчески доступными, либо их получают стандартными методами синтеза, применяемыми в органической химии. Один общий путь получения соединений формулы (III) показан на следующей схеме 1 а. Схема 1 а На схеме 1 а: а) связывание альдегида формулы (IV) с соответствующим фрагментом, например,(R11O)2P(О)СН(NHPG)CO2R10, и b) гидрирование и удаление защитной группы, если это необходимо. PG представляет собой защитную группу. Альдегиды формулы (IV) могут быть коммерчески доступны или могут быть легко синтезированы специалистами в данной области техники. Применение химии защиты и удаления защиты может быть необходимым для получения соединений формулы (III) из соединений формулы (IV). Эти способы известны специалистам в данной области техники, и подходящие защитные группы описаны в источникеGreene's Protective Groups in Organic Synthesis (Wuts and Greene, 4th Edition, 2007). После получения соединений формулы (IIA) и формулы (IIB) соединения по изобретению получают полусинтетической дериватизацией. Полусинтетические способы получения макроциклических альдегидов санглиферина описаны в патенте США 6124453, в работах Metternich et al., 1999, Banteli et al.,2001 и Sedrani et al., 2003. Как правило, полусинтетический способ получения некоторых соединений формулы (I) или их фармацевтически приемлемых солей из мутасинтетического аналога санглиферина включает:(c) сочетание указанного альдегида со стабилизированным карбанионом (или с его канонической формой), например, с карбанионфосфонатом, используя соединение формулы V. Эта ретросинтетическая схема показана ниже: где R12, в случае мутасинтетического аналога санглиферина А, представляет собой: где группы R11, которые могут быть одинаковыми или различными, независимо друг от друга представляют собой алкил (например, С 1-4 алкил) или бензил. Таким образом, способ получения соединения по изобретению включает взаимодействие соединения формулы (V) с альдегидным макроциклом (соединение формулы (VI. Получение соединений формулы (VI) может быть осуществлено способом, аналогично описанному ранее, для конверсии санглиферина А в соответствующий альдегидный макроцикл (Metternich et al. 1999). Кратко, соединение формулы (II) дигидроксилируют с использованием модифицированных условий Sharpless (катализ с четырехокисью осмия). Использование хиральных лигандов помогает обеспечить селективность. Затем полученный диол может быть расщеплен окислением, используя, например,периодат натрия. Полученное соединение формулы VI может быть далее использовано в качестве субстрата для дериватизации в гомологичный амид, сложный эфир или кетон. Обычно соединение формулы(V) растворяют в апротонном растворителе, охлаждают и обрабатывают основанием, например, гидридом натрия. Затем добавляют соединение формулы (VI), и реакционную смесь нагревают при повышенной температуре. После соответствующего периода времени реакцию останавливают, и соединение формулы I очищают стандартными методами (например, препаративной высокоэффективной жидкостной хроматографией, препаративной тонкослойной хроматографией, флэш-хроматографией с нормальными фазами и т.п.). Соединения формулы (V) могут быть известны или могут быть получены с использованием известных способов. Как показано на схеме 1 (см. ниже), для обработки хлорангидрида хлоруксусной кислоты может быть использован соответствующий амин или подобное соединение, чтобы получить альфа-хлорамид. Затем альфа-хлорамид подвергают реакции Арбузова с получением соединения формулы V. Другие пути получения соединений формулы V для специалистов в данной области очевидны. Схема 1 Дополнительные соединения формулы (V) могут быть известны или могут быть легко синтезированы из доступных производных карбоновых кислот (например, из R3COX), где R3 представляет собой 1,2 оксазинановое кольцо, показанное на схеме 2. Как показано на схеме 2 (см. ниже), производное карбоновой кислоты может быть соединено с метилфосфонатом после обработки фосфоната основанием. Это приводит к соединению формулы (V), хотя другие пути получения соединений формулы V также очевидны для специалистов в данной области. Схема 2 Если желательно или необходимо, то для защиты функциональности могут быть использованы защитные группы в альдегидном макроцикле или в макроцикле, или в соединении формулы V, как описано в T.W. Green, P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley-Interscience, New York, 1999. В дополнение к конкретным способам и цитированным в данном описании ссылкам, специалисты в данной области техники могут получить дополнительные сведения из стандартных справочников по методам синтеза, включая, но, не ограничиваясь ими, справочник Фогеля по практической органической химии (Furniss et al., 1989) и расширенный справочник по органической химии Марча (Smith and March,2001). Композиции, как правило, могут быть представлены в виде единичной дозированной формы и мо- 13023848 гут быть получены любым из способов, хорошо известных в области фармации. Такие способы включают стадию объединения активного ингредиента (соединения по изобретению) с носителем, который состоит из одного или более вспомогательных ингредиентов. Как правило, композиции получают путем равномерного и тщательного смешивания активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями, или с тем и другим типом носителей, и последующим, если необходимо, формованием продукта. Соединения по изобретению обычно вводят перорально в форме фармацевтической композиции,содержащей активный ингредиент, необязательно в форме нетоксичной органической или неорганической кислотно- или основно-аддитивной соли, в фармацевтически приемлемой дозированной форме. В зависимости от тяжести заболевания и состояния пациента, которого подвергают лечению, а также от пути введения, композиции могут вводиться в различных дозах. Например, соединения по изобретению могут быть введены перорально, трансбуккально или сублингвально в форме таблеток, капсул, каплет, эликсиров, растворов или суспензий, которые могут содержать ароматизаторы или красители, при этом формы могут быть предназначены для немедленного,замедленного или контролируемого высвобождения. Таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция и глицин; дезинтегранты, такие как крахмал (предпочтительно кукурузный, картофельный или тапиоки), натрийгликолят крахмала, натрийкроскармеллоза и некоторые комплексные силикаты; связующие для гранулирования, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза (НРС), сахароза, желатин и аравийская камедь. Кроме того, могут быть включены лубриканты, такие как стеарат магния,стеариновая кислота, глицерилбегенат и тальк. Сухие композиции подобного типа также могут быть использованы для наполнения желатиновых капсул. В этой связи, предпочтительные эксципиенты включают лактозу, крахмал, целлюлозу, молочный сахар или полиэтиленгликоли с высокой молекулярной массой. Для водных суспензии и/или эликсиров,соединения по изобретению могут быть объединены с различными подсластителями или ароматизаторами, окрашивающими веществами или красителями, с эмульгирующими и/или суспендирующими средствами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин, а также с их комбинациями. Таблетка может быть изготовлена прессованием или формованием, необязательно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены прессованием активного ингредиента в подходящем аппарате в свободно-текучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом (например, с повидоном, желатином, гидроксипропилметилцеллюлозой), лубрикантом, инертным разбавителем, консервантом, дезинтегрантом (например, натрийгликолята крахмала, поперечно сшитым повидоном, поперечно сшитой натрийкарбоксиметилцеллюлозы), поверхностно-активным веществом или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием смеси соединения в порошкообразной форме в подходящем аппарате, увлажненного инертным жидким разбавителем. На таблетки можно необязательно наносить покрытие или наносить бороздки, и они могут быть изготовлены таким образом, чтобы обеспечить замедленное или регулируемое высвобождение активного ингредиента, используя, например, гидроксипропилметилцеллюлозу в различных пропорциях для обеспечения желаемого профиля высвобождения. Композиции по изобретению, пригодные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, облатки или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или в неводной жидкости; или в виде жидкой эмульсии "масло-в-воде" или в виде жидкой эмульсии "вода-в-масле". Активный ингредиент также может быть предоставлен в виде болюса, электуария или пасты. Следует понимать, что в дополнение к ингредиентам, отмеченных, в частности, выше, композиции по изобретению могут содержать другие средства, обычно используемые в данной области в соответствии с типом композиции, например, ингредиенты, используемые в формах для перорального введения могут включать флаворанты. Средства, такие как консерванты и буферирующие агенты, преимущественно могут быть растворены в носителе. Для повышения стабильности, композиция может быть заморожена после заполнения ею сосуда, и вода может быть удалена в вакууме. Затем сухой лиофилизированный порошок запаивают в ампулы, при этом к ампуле может прилагаться флакон с водой для инъекций для восстановления в жидкую форму перед применением. Дозировка, в которой вводят соединение по изобретению, будет варьироваться в зависимости от конкретного соединения, заболевания, состояния пациента, характера и тяжести заболевания, физического состояния пациента и выбранного пути введения. Соответствующие дозы могут быть без труда определены специалистом в данной области. Композиции могут содержать от 0,1 мас.%, предпочтительно 5-60%, более предпочтительно 10-30 мас.%, соединения по изобретению, в зависимости от способа их введения. Специалистом в данной области следует принимать во внимание, что оптимальное количество и промежутки между введениями отдельных доз соединения по изобретению будут зависеть от характера и степени тяжести состояния, подлежащего лечению, формы, способа и места введения, а также от возраста и состояния конкретного пациента, подвергаемого лечению, и что врач, в конечном счете, будет определять соответствующие дозы, которые будут использоваться при лечении. Введение дозы может быть выполнено столько раз, сколько это необходимо. Если развиваются побочные эффекты, то количество доз и/или частота введения доз могут быть изменены или уменьшены, в соответствии с обычной клинической практикой. Общие способы Материалы и методы Бактериальные штаммы и условия роста Продуцентом санглиферина является Streptomyces sp. А 92-308110 (DSM 9954, который может быть приобретен в DSMZ, Брауншвейг, Германия), также обозначаемый как BIOT-4253 и BIOT-4370, или его производные, такие как BIOT-4585, рост которых поддерживается на овсяной агаровой среде, средахBIOT-4585 (для получения конструкта, см. пример 1) выращивали на овсяной агаровой среде при температуре 28 С в течение 7-10 дней. Споры с поверхности чашки с агаром собирали в 20% мас./об. стерильный раствор глицерина в дистиллированной воде, и хранили в 0,5-мл аликвотах при -80 С. Запас замороженных спор использовали для посева на среды SGS или SM25-3. Инокулированную среду для посева инкубировали при встряхивании в режиме от 200 до 300 об/мин при амплитуде 5,0 или 2,5 см, при температуре 27 С в течение 24 ч. Ферментационную среду SGP-2 или ВТб инокулировали 2,5-10% посевной культурой и инкубировали при встряхивании в режиме от 200 до 300 об/мин при амплитуде 5 или 2,5 см при температуре 24 С в течение 4-5 дней. Затем культуру собирали для экстракции. Аналог мета-тирозина(S)-Метил-2-амино-3-(3-фтор-5-гидроксифенил)пропаноат был приобретен у NetChem (США). (3 Бром-5-фторанизол (соединение 9-1) был приобретен у Accela ChemBio Co., Ltd., (Шанхай, Китай), а также он может быть приобретен у Amfinecom Inc (США) или у Apollo Scientific Ltd. (Великобритания). К раствору соединения 9-1 (20 г, 97,55 ммоль) в тетрагидрофуране (100 мл) добавляли по каплям нбутиллитий (43 мл, 2,5 М, 107,3 ммоль) при температуре -78 С. Смесь перемешивали в течение 30 мин и добавляли N,N-диметилформамид (15,1 мл, 195,1 ммоль) при этой же температуре. Смесь перемешивали еще в течение 30 мин и удаляли охлаждающую баню. Через 1 ч реакционную смесь гасили насыщенным водным раствором хлорида аммония. Органический слой промывали водой и насыщенным водным раствором хлорида натрия, затем сушили (над сульфатом натрия), фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле с получением соединения 9-2. К раствору соединения 9-2 (6 г, 38,9 ммоль) в сухом DCM (200 мл) по каплям добавляли BBr3 (4 M в дихлорметане, 30 мл, 116,8 ммоль) при температуре -70 С. После добавления BBr3 реакционную смесь перемешивали при температуре -20 С в течение 3 ч, осторожно добавляли воду со льдом и экстрагировали DCM. Органические слои промывали водой и насыщенным раствором соли, сушили над Na2SO4,фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силикагеле с получением желаемого соединения 9-3. К раствору метил-2-(бензилоксикарбониламино)-2-(диметоксифосфорил)ацетата (4,64 г, 14 ммоль) в DCM (150 мл) при комнатной температуре добавляли DBU (4,26 г, 28 ммоль). Через 10 мин добавляли соединение 9-3 (10,95 г, 14 ммоль) и полученную смесь перемешивали при комнатной температуре в течение ночи. Раствор разбавляли EtOAc (150 мл), разделяли и органический слой промывали 1 н. HCl, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали флэш-хроматографией на силика- 15023848 геле с получением соединения 9-4. Раствор соединения 9-4 (1 г) в метаноле (20 мл) подвергали гидрированию с использованием 200 мг 10% Pd/C при нормальном давлении в течение ночи. После удаления катализатора фильтрованием растворитель выпаривали с получением соединения 10. К раствору соединения 10 (300 мг, 1,4 ммоль) в EtOH (30 мл) добавляли водный раствор NaOH (2H,4 мл), реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Растворитель удаляли, остаток нейтрализовали до рН 6 с использованием 2 н. HCl и собирали фильтрованием образовавшиеся белые кристаллы с получением целевого соединения 9. Альтернативный путь получения метил-2-амино-3-(3-фтор-5-гидроксифенил)пропаноата (10). 3,5-Дифторбромбензол, соединение (9 а-1) был приобретен у Darui Fine Chemicals Co., Ltd., (Шанхай, Китай), а также это соединение может быть приобретено у Alfa Aesar или Sigma Aldrich. Получение соединения 9 а-2. К раствору BnOH (1,61 мл, 15,54 ммоль) в DMF (30 мл) добавляли NaH (622 мг, 60% дисперсия в минеральном масле, 15,54 ммоль) при температуре 0 С. Продолжали перемешивание при комнатной температуре в течение 0,5 ч с получением прозрачного раствора. Соединение 9 а-1 (10,79 мл, 15,54 ммоль) добавляли с такой скоростью, чтобы поддерживалась температура ниже 40 С. Смесь перемешивали при комнатной температуре в течение ночи с получением желтого раствора. Реакцию гасили водой и экстрагировали петролейным эфиром (35 мл 4). Объединенные органические слои концентрировали. Остаток очищали хроматографией на силикагеле, используя петролейный эфир в качестве элюента, с получением соединения 9 а-2 (2,544 г) в виде бесцветного масла. Получение соединения 9a-3. В сухую трехгорлую колбу в атмосфере азота добавляли Mg (170,1 мг, 7,10 ммоль), безводный THF(10 мл) и небольшое количества йода. Затем добавляли 1/3 количества соединения 9 а-2 (10,664 г,5,9192 ммоль) в THF (2 мл). Полученную смесь нагревали при кипячении с обратным холодильником. В это время желтая смесь постепенно становилась ярко-желтого цвета. Затем по каплям добавляли остальные 2/3 количества соединения 9 а-2 и реакционную смесь кипятили с обратным холодильником в течение 0,5 ч. К вышеуказанной смеси при температуре 0 С медленно добавляли DMF (0,504 мл, 6,51 ммоль). Перемешивание продолжали в течение 0,5 ч при комнатной температуре. Добавляли HCl (2 М, 10 мл) и выпаривали THF. Остаток экстрагировали этилацетатом (25 мл, трехкратно). Объединенные органические слои промывали насыщенным раствором соли и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле с использованием в качестве элюента градиента петролейный эфирпетролейный эфир/этилацетат=20/1 с получением соединения 9 а-3 (694 мг) в виде бесцветного масла. Получение соединения 9 а-5. К раствору метил-2-(бензилоксикарбониламино)-2-(диметоксифосфорил)ацетата, соединение 9 а-4(993 мг, 3,00 ммоль), в DCM (30 мл) добавляли DBU (832 мкл, 5,57 ммоль) при комнатной температуре. Через 10 мин добавляли соединение 9 а-3 (694 мг, 3,01 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 1 ч. Раствор промывали HCl (1 М, 10 мл) и объединенные органические слои сушили и концентрировали в вакууме. Остаток очищали флэш-хроматографией на силикагеле(элюирование смесью дихлорметан/этилацетат=10/1) с получением соединения 9 а-5 (10,11 г). Получение соединения 10. Раствор соединения 9 а-5 (100 мг) в метаноле (50 мл) подвергали гидрированию с использованием 20 мг 10% Pd/C при нормальном давлении в течение 2 ч. После удаления катализатора фильтрованием выпаривали растворитель с получением соединения 10 (33 мг). Составы сред Воду, используемую для изготовления сред, получают, используя систему очистки воды Millipore Примечания.Пеногаситель использовали только в посевных ферментерах, но не в посевных колбах.Конечный объем доводили до заданной величины с учетом посевного объема. Перед стерилизацией рН доводили до 7,0 с использованием 10 М NaOH/10 M H2SO4. Стерилизацию проводили нагреванием при температуре 121 С в течение 20-30 мин (автоклавирование). Раствор микроэлементов В. Примечания.Пеногаситель использовали только в ферментерах, но не в колбах.Конечный объем доводили до заданной величины с учетом посевного объема. Перед стерилизацией рН доводили до 6,8 с использованием 10 М NaOH. Стерилизацию проводили нагреванием при температуре 121 С в течение 20-30 мин (автоклавирование). Среда SM25-3 (также обозначаемая как SM25). Перед стерилизацией рН не регулировали (т.е. рН равен 7,0). Среда ISP4. Готовили пасту с крахмалом в небольшом объеме холодной воды и затем объем доводили до 500 мл. К раствору II добавляли другие ингредиенты в 500 мл воды, при этом значение рН должно находиться в пределах от 7,0 до 7,4 (рН 7,3). Смешивали два раствора вместе и затем добавляли агар. Раствор солей микроэлементов ISP. 20 г овсяной муки варили в 1 л воды на плите (или в микроволновой печи) в течение 20 мин. Приготовленную смесь фильтровали через муслин/марлю, рН доводили до 7,2 и объем доводили до 1 л. Добавляли 1 мл раствора солей микроэлементов ISP. Затем перед стерилизацией добавляли 18 г агара на 1 л раствора. Агар МАМ. Значение рН составляло 5,8 (до автоклавирования). Значение рН устанавливали 7,0 перед добавлением СаСО 3. Агаровая среда ISP2. рН устанавливали 7,3 перед добавлением агара и стерилизацией. Общие методы ферментации Криоконсервированные запасы спор BIOT-4585 (для получения конструкта, см. пример 1) оттаивали при комнатной температуре. Вегетативные культуры (посевные культуры) получали путем переноса 4,0 мл запаса спор в 400 мл среды SM25 в колбы Эрленмейера емкостью 2 л с противопенной вставкой. Культивирование проводили в течение 48 ч при температуре 27 С и 250 об/мин (амплитуда 5,0 см). Из посевной культуры 25 мл переносили в продукционную среду SGP2+5% HP20 объемом 250 мл в колбу Эрленмейера емкостью 2 л с противопенной вставкой. После 24 ч культивирования при температуре 24 С и 250 об/мин (амплитуда 2,5 см) добавляли 2 мл 250 мМ рацемического или 125 мМ энантиомерно чистого раствора желаемого предшественника(9) или метил-2-амино-3-(3-фтор-5 гидроксифенил)пропаноата (10, в 1 М хлористо-водородной кислоте и 2 мл 250 мМ метанольного раствора DL-пиперазиновой кислоты к каждой производственной колбе с получением конечной концентрации 1 мМ отдельных энантиомеров предшественников. При необходимости можно использовать ДМСО вместо 1 М хлористо-водородной кислоты. Добавление DL-пиперазиновой кислоты может быть необязательным. Культивирование продолжали еще в течение четырех дней при температуре 24 С и при 250 об/мин (амплитуда 2,5 см). Анализ культуральных бульонов при использовании жидкостной хроматографии с контролем в ультрафиолете (ЖХ-УФ) и жидкостной хроматографии с контролем в ультрафиолете и массспектрометрией (ЖХ-УФ-МС) К культуральному бульону (1 мл) добавляли этилацетат (1 мл) и перемешивали в течение 15-30 мин с последующим центрифугированием в течение 10 мин. Собирали 0,4 мл органического слоя, досуха выпаривали и затем повторно растворяли в 0,20 мл ацетонитрила. Условия ВЭЖХ: Колонка Hyperclone C18 BDS C18 Column 3u, 4,6 мм 150 мм, с подключенным картриджем Phenomenex Analytical C18 Security Guard (KJO-4282); Температура колонки 50 С; Скорость потока 1 мл/мин; Измерение УФ при длине волны 240 нм; Объем вводимой аликвоты 20 мкл. Градиент растворителя: 0 мин: 55% В; 1,0 мин: 55% В; 6,5 мин: 100% В; 10,0 мин: 100% В; 10,05 мин: 55% В; 13,0 мин: 55% В; Растворитель А: вода+0,1% муравьиной кислоты; Растворитель В: ацетонитрил+0,1% муравьиной кислоты. В этих условиях SfA элюируется на 5,5 мин. В этих условиях SfB элюируется на 6,5 мин. ЖХМС осуществляли на комплексной системе ВЭЖХ Agilent НР 1100 HPLC в сочетании с масс- 19023848 спектрометром с электрораспылением Bruker Daltonics Esquire 3000, работающим в режиме положительных ионов, используя хроматографию и растворители, описанные выше. Метод количественного контроля жидкостной хроматографией с масс-спектрометрией (QC ЖХ-МС) Условия ВЭЖХ: Колонка Hyperclone C18 BDS C18 Column 3u, 4,6 мм 150 мм, с подключенным картриджем Phenomenex Analytical C18 Security Guard (KJO-4282); Температура колонки 50 С; Скорость потока 1 мл/мин; Измерение УФ при длинах волн 210, 240 и 254 нм. Градиент растворителя: 0 мин: 10% В; 2,0 мин: 10% В; 15 мин: 100% В; 17 мин: 100% В; 17,05 мин: 10% В; 20 мин: 10% В; Растворитель А: вода+0,1% муравьиной кислоты. Растворитель В: ацетонитрил+0,1% муравьиной кислоты. Условия масс-спектрометрии (МС): МС осуществляли в режиме переключения (переключение между режимами положительных и отрицательных ионов), сканирование в диапазоне от 150 до 1500 а.е.м. Метод порошковой рентгеновской дифракции (XRPD) Около 2 мг образца аккуратно прессовали с один косым срезом на держатель образца для XRPD из кремнезема с нулевым потенциалом относительно земли. Затем образец помещали в дифрактометр Philips X-Pert MPD и проводили анализ в следующих условиях эксперимента: Анод трубки: Cu; Напряжение генератора: 40 кВ; Ток трубки: 40 мА; Длина волны альфа 1: 0,5406 ; Длина волны альфа 2: 1,5444 ; Начальный угол [2 тета]: 5; Конечный угол [2 тета]: 50; Режим: непрерывное сканирование. Анализ репликона in vitro для оценки противовирусной активности в отношении HCV Противовирусная эффективность против HCV генотипа 1 может быть определена следующим образом: за день до добавления тестируемых проб, клетки Huh5.2, содержащие репликон 1389luc-ubineo/NS3-3'/5.1 HCV генотипа 1b (Vrolijk et al., 2003), пересевали в клеточную среду для роста [DMEM(кат. номер 41965039), дополненную 10% FCS, 1% заменимых аминокислот (11140035), 1% пенициллина/стрептомицина (15140148) и 2% генетицина (10131027), Invitrogen] в соотношении 1,3-1,4, и выращивали в течение 3-4 дней в колбах для культуры ткани размером 75 см 2 (Techno Plastic Products), затем собирали и высевали в среду для анализа (DMEM, 10% FCS, 1% заменимых аминокислот, 1% пенициллина/стрептомицина) при плотности 6500 клеток на лунку (100 мкл/лунка) в 96-луночные планшеты для микротитрования культуры тканей (Falcon, Beckton Dickinson для оценки антиметаболического эффекта;CulturPlate, Perkin Elmer - для оценки противовирусного эффекта). Микропланшеты инкубировали в течение ночи (37 С, 5% CO2, относительная влажность 95-99%) с получением неконфлюентного монослоя клеток. Готовили серийные разведения, из расчета получения каждого разведения в серии, по меньшей мере, в повторе. После подготовки микропланшетов для анализа их инкубируют в течение 72 ч (37 С, 5% СО 2, относительная влажность 95-99%). Для оценки антиметаболических эффектов среду для анализа отсасывали, заменяя ее на 75 мкл 5% раствора MTS (Promega) в среде без фенолового красного, и микропланшеты инкубировали в течение 1,5 ч (37 С, 5% СО 2, относительная влажность 95-99%). Поглощение измеряли при длине волны 498 нм (Safire2, Tecan) и оптическую плотность (значения OD) пересчитывали в относительную величину, выраженную в процентах, по отношению к необработанному контролю. Для оценки противовирусных эффектов аналитическую среду отсасывали и клеточные монослои промывали PBS. Промывочный буфер отсасывали, добавляли 25 мкл буфера для лизиса (Glo Lysis Buffer,кат. номер Е 2661, Promega), E2661, Promega), затем проводили лизис в течение 5 мин при комнатной температуре. Затем добавляли 50 мкл аналитической люциферазной системы (Luciferase Assay System,катал, номер Е 1501, Promega) (кат. номер Е 1501, Promega) и немедленно количественно регистрировали сигнал люминесценции люциферазы (время интегрирования 1000 мс на лунку, Safire2, Tecan). Относительные единицы люминесценции пересчитывали в относительную величину, выраженную в процентах,- 20023848 по отношению к необработанному контролю. Показатели ЕС 50 и ЕС 90 (значения, полученные из кривой "доза-ответ") представляют собой концентрации, при которых соответственно можно наблюдать 50 и 90% ингибирования вирусной репликации. Показатель CC50 (значение, полученное из кривой "доза-ответ") представляет собой концентрацию,при которой метаболическая активность клетки снижена до величины 50% от метаболической активности необработанных клеток. Индекс селективности (SI), который указывает на терапевтическое окно соединения, рассчитывается как CC50/EC50. Концентрация соединения рассматривается как вызывающая реальное противовирусное действие в системе репликона HCV, когда при этой конкретной концентрации антирепликоновый эффект превышает 70%-ный порог, и при этом наблюдается не более чем 30%-ное снижение метаболической активности. Анализ репликона in vitro для оценки противовирусной активности в отношении HCV генотипов 1 а и 2 а Клетки-репликоны (субгеномные репликоны генотипа 1 (Н 77) и генотипа 2 а (JFH-1 выращивали в среде Игла, модифицированной по Дульбекко (DMEM), дополненной 10% фетальной телячьей сывороткой (FBS), 1% пенициллина/стрептомицина (pen/strep), 1% глутамина, 1% заменимых аминокислот, 250 мкг/мл G418, в инкубаторе с атмосферой, содержащей 5% СО 2 при температуре 37 С. Все реагенты клеточной культуры могут быть приобретены у Mediatech (Herndon, VA). Клетки-репликоны обрабатывали трипсином и высевали в количестве 5103 клеток на лунку в 96 луночные планшеты с указанной средой без G418. На следующий день культуральную среду заменяли средой DMEM, содержащей тестируемые соединения в серийном разведении в присутствии 5% FBS. Противовирусный анализ репликона HCV оценивали по эффекту действия соединений в последовательных разведениях. Кратко, клетки, содержащие репликон HCV, высевали в 96-луночные планшеты. Тестируемые образцы соединений серийно разводили средой DMEM, содержащей 5% FBS. Разведения соединений вводили в соответствующие лунки в планшетах. Клетки обрабатывали после 72 ч инкубации при температуре 37 С. Из каждой лунки экстрагировали внутриклеточную РНК, используя набор RNeasy 96 (Qiagen). Уровень РНК в HCV определяли с помощью анализа ПЦР с транскриптазой в реальном масштабе времени, с использованием набора реагентов TaqMan One-Step RT-PCR Master Mix Reagents(Applied Biosystems, Foster City, CA) и систему ABI Prism 7900 Sequence Detection (Applied Biosystems),как описано ранее (Vrolijk et al., 2003). Цитотоксические эффекты измеряли с использованием набора реагентов TaqMan Ribosomal RNA Control Reagents (Applied Biosystems) с регистрацией количества клеток в качестве показателя. Количество РНК вируса гепатита С и рибосомной РНК использовали для получения значений IC50 (концентрация ингибирования репликации репликона на 50%). Оценка метаболизма микросом (анализ стабильности микросом) Скорость метаболизма в микросомах может быть определена следующим образом. Микросомы печени мыши или человека разводили буфером С (0,1 М калий-фосфатный буфер, 1,0 мМ ЭДТА, рН 7,4) до концентрации 2,5 мг/мл. Затем готовили образцы микросом для оценки стабильности добавлением 50 мкл 5 мкМ раствора тестируемого соединения (0,5 мкл 10 мМ маточного раствора ДМСО в 9,5 мкл ACN, с добавлением 990 мкл буфера С) к 50 мкл раствора микросом (2,5 мг/мл) и 110 мкл буфера С, и затем все тщательно перемешивали. Все образцы были предварительно инкубированы в течение приблизительно 15 мин при температуре 37 С. После этого реакцию инициировали добавлением 40 мкл раствора NADPH (12,5 мМ) при осторожном перемешивании. Аликвоты (40 мкл) отбирали на 0,15, 30, 45 и 60 минутах и гасили ACN, содержащим внутренний стандарт (120 мкл). Белок удаляли центрифугированием (4000 об/мин, 15 мин) и планшеты с образцами анализировали для определения концентрации соединения с использованием ЖХ-МС/МС. Затем рассчитывали время полужизни стандартными методами, сравнивая концентрацию анализируемого вещества с количеством, которое присутствовало первоначально. Оценка стабильности гепатоцитов Замороженные гепатоциты, хранившиеся в жидком азоте, помещали на водяную баню со встряхиванием при температуре 371 С на время 2 мин 15 с. Затем добавляли гепатоциты к 10-кратному объему нагретого бикарбонатного буфера Кребса-Хенселейта (КНВ) (2000 мг/л глюкозы, не содержащего карбоната кальция и бикарбоната натрия, Sigma), осторожно перемешивали и центрифугировали при 500 об/мин в течение 3 мин. Супернатант после центрифугирования осторожно удаляли и добавляли 10 кратный объем предварительно подогретого буфера КНВ к ресуспендируемому осадку клеток. Полученную смесь осторожно перемешивали и центрифугировали при 500 об/мин в течение 3 мин. Затем супернатант удаляли. Жизнеспособность клеток и выход определяли подсчетом клеток, и эти значения использовали для получения необходимой плотности посева суспензий гепатоцитов человека (плотность жизнеспособных клеток=2106 клеток/мл). Двухкратный раствор (2 Х) для дозирования готовили в предварительно нагретом буфере KHB (с 1% ДМСО) (200 мкМ испытуемый раствор: 20 мкл исходного раствора субстрата (10 мМ) в 980 мкл ДМСО, дозируемый 2 Х раствор: 10 мкл 200 мкМ тестируемого раствора в 990 мкл КНВ (2 мкМ после разведения. В лунки вносили 50 мкл предварительно нагретого 2 Х раствора для дозирования, затем добавляли- 21023848 50 мкл предварительно нагретого раствора гепатоцитов раствор (2106 клеток/мл) и начинали отсчет времени. Затем планшет инкубировали при 37 С. После завершения времени инкубации (0, 15, 30, 60 и 120 мин) к каждой лунке добавляли 100 мкл ацетонитрила, содержащего внутренний стандарт, осторожно перемешивали и затем добавляли 50 мкл предварительно нагретого раствора гепатоцитов (2106 клеток/мл). В конце инкубации определяли жизнеспособность клеток. Образцы центрифугировали при 4000 об/мин в течение 15 мин при температуре 4 С, надосадочную жидкость разводили в 2 раза сверхчистой водой, и уровни соединений анализировали ЖХ-МС/МС. Оценка растворимости в воде Растворимость в воде может быть проверена следующим образом. Исходный 10 мМ раствор аналога санглиферина готовили в 100% ДМСО при комнатной температуре. Аликвоты объемом 0,01 мл в тройной повторности готовили в 0,5 мл или в растворе 0,1 М PBS, рН 7,3, или в 100% ДМСО в ампулах янтарного цвета. Полученные 0,2 мМ растворы встряхивали при комнатной температуре в шейкереIKA Vibrax VXR в течение 6 ч, с последующим переносом полученных растворов или суспензий в пробирки Эппендорфа емкостью 2 мл, и центрифугировали в течение 30 мин при 13200 об/мин. Аликвоты надосадочной жидкости анализировали методом ЖХМС, как описано выше. Также, растворимость в PBS при рН 7,4 может быть проверена следующим образом. Калибровочную кривую получали путем разведения исследуемых и контрольных соединений до концентрации 40; 16; 4; 1,6; 0,4; 0,16; 0,04 и 0,002 мкМ с использованием 50% метанола в Н 2 О. Стандартные точки затем дополнительно разводили в соотношении 1:20 в смеси MeOH:PBS 1:1. Конечные концентрации после разведения в соотношении 1:20 составляли 2000; 800; 200; 80; 20; 8; 2 и 1 нМ. Затем стандарты смешивали с таким же объемом (1:1) ACN, который содержал внутренний стандарт (гидроксимакроцикл, соединение 6). Образцы центрифугировали (5 мин, 12000 об/мин) и затем анализировали методом ЖХ/МС. Тестируемые соединения готовили в виде маточных растворов в ДМСО с концентрацией 10 мМ. Маточные растворы разводили в двух повторах в PBS, рН 7,4, в пробирках Эппендорфа емкостью 1,5 мл до целевой концентрации 100 мкМ с конечной концентрацией ДМСО, равной 1% (например, 4 мкл 10 мМ ДМСО маточного раствора в 396 мкл 100 мМ фосфатного буфера). Затем пробирки с образцами осторожно встряхивали в течение 4 ч при комнатной температуре. Образцы центрифугировали (10 мин,15000 об/мин) для осаждения нерастворенных частиц. Супернатанты переносили в новые пробирки и разводили PBS (коэффициент разведения для отдельного тестируемого образца подтверждался уровнем сигнала соединения в используемом аналитическом приборе). Затем разведенные образцы смешивали с таким же объемом (1:1) МеОН. Окончательно, образцы смешивали с равным объемом (1:1) ACN который содержал внутренний стандарт (гидроксимакроцикл, соединение 6), для проведения анализа методом ЖХ-МС/МС. Оценка клеточной проницаемости Клеточная проницаемость может быть проверена следующим образом. Тестируемое соединение растворяли до концентрации 10 мМ в ДМСО и далее разводили в буфере до получения конечной концентрации для дозирования, равной 10 мкМ. Маркер флуоресценции (люцифер желтый) также включали для контроля целостности мембран. Затем тестируемое соединение наносили на апикальную поверхность монослоя клеток Сасо-2 и измеряли проникновение соединения в базолатеральной компартмент. Эта оценка выполнялась в обратном направлении (от базолатеральной к апикальной поверхности) тестируемого активного транспорта. Для количественного определения уровней тестируемых и стандартных контрольных соединений (например, пропанолола и ацебутолола) использовали ЖХ-МС/МС. Оценка фармакокинетики in vivo Для измерения биологической доступности соединения также могут быть использованы анализы invivo. Как правило, соединение вводят тестируемому животному (например, мыши или крысе) внутривенно (в/в) и перорально (п/о), и затем через регулярные промежутки времени берут пробы крови для определения изменения концентрации лекарственного средства в плазме во времени. Изменения концентрации в плазме крови во времени могут быть использованы для расчета абсолютной биодоступности соединения в виде процентных долей с использованием стандартных моделей. Пример конкретного протокола описан ниже. Мышам вводили 1, 10 или 100 мг/кг соединения по изобретению или исходного соединения внутривенно или перорально. Образцы крови отбирали через 5, 10, 15, 30, 45, 60, 90, 120, 180, 240, 360, 420 и 2880 мин и концентрации соединения по изобретению или исходного соединения в образце определяли методом ВЭЖХ. Показатели изменения концентрации в плазме во времени могут быть использованы для получения ключевых параметров, таких как площадь под кривой зависимости концентрации в плазме крови от времени (AUC - величина, которая прямо пропорциональна общему количеству лекарственного средства, которое достигает системного кровотока в неизмененном виде), максимальная концентрация(пиковая) лекарственного средства в плазме, время, в течение которого достигается максимальная концентрация лекарственного средства в плазме (пиковое время), дополнительные факторы, используемые для точного определения биодоступности, которые включают конечное время полужизни лекарственного средства, общий клиренс, стационарный объем распределения и показатель F%. Затем эти параметры анализировали некомпартментными и компартментными методами, получая расчетную величину биодоступности, выраженную в процентах. Пример метода анализа такого типа описан в Egorin et al. 2002, и в цитированных в данном описании документах. Оценка фармакокинетики in vivo при пероральном и внутривенном введениях(специфический способ) Для оценки аналогов санглиферина анализу подвергали цельную кровь. Соединения для в/в и п/о введения растворяли в смеси 5% этанола, 5% кремофора EL и 90% физиологического раствора. Группам из 3 самцов мышей CD1 внутривенно вводили дозу 1 мг/кг или перорально вводили дозу 5 или 10 мг/кг. Пробы крови (40 мкл) забирали через подкожную вену лапы перед введением дозы и через 0,25, 0,5, 2, 8 и 24 ч после введения. Пробы крови разводили равным количеством дистиллированной воды и немедленно помещали в сухой лед. До проведения анализа образцы хранили при -70 С. Концентрацию соединения по изобретению или исходного соединения в образце определяли ЖХМС следующим образом: к пробе объемом 20 мкл, содержащей кровь и Н 2 О (1:1, об./об.), добавляли 20 мкл внутреннего стандарта(гидроксильный макроцикл, соединение 6) в концентрации 100 нг/мл, к 20 мкл рабочего раствора добавляли метанол и 150 мкл ACN, затем встряхивали в течение 1 мин при 1500 об/мин и центрифугировали при 12000 об/мин в течение 5 мин. Супернатант анализировали методом ЖХ-МС/МС. Строили график зависимости концентрации соединения в крови от времени, и использовали его для расчета площади под фармакокинетической кривой для случая цельной крови (AUC - величина, которая прямо пропорциональна общему количеству лекарственного средства, которое достигает системного кровотока в неизмененном виде). Полученные значения, когда это возможно, использовали для получения фармакокинетических параметров. Оценка цитотоксичности in vitro Клетки Huh-7 и HepG2, полученные из АТСС, выращивали в основной среде Игла, модифицированной по Дульбекко (DMEM), содержащей 10% фетальной телячьей сыворотки (FBS), 1% пенициллина/стрептомицина (pen/ctrep) и 1% глутамина; клетки СЕМ (лейкемические Т-клетки человека, полученные из АТСС) выращивали в среде RPMI 1640 с добавлением 10% FBS, 1% pen/ctrep и 1% глутамина. Свежи мононуклеарные клетки периферической крови (РВМС) человека выделяли из цельной крови,полученной по меньшей мере от двух здоровых скринированных доноров. Кратко, клетки периферической крови осаждали и промывали 2-3 раза с центрифугированием при низкой скорости, и затем ресуспендировали в PBS для удаления загрязняющих тромбоцитов. После этого промытые клетки крови разводили в соотношении 1:1 буферным фосфатным раствором по Дульбекко (D-PBS) и наслаивали на 14 мл среды для отделения лимфоцитов (среда для роста клеток LSM; Mediatech, Inc, плотность 1,0780,002 г/мл; кат. номер 85-072-CL) в центрифужной пробирке емкостью 50 мл, и затем центрифугировали в течение 30 мин при 600 g. Пластинчатые РВМС осторожно отсасывали из образованного межслойного пространства и затем их дважды промывали PBS и подвергали низкоскоростному центрифугированию. После последней промывки клетки подсчитывали путем эксклюзии красителя трипанового синего и ресуспендировали с получением концентрации 1107 клеток/мл в среде RPMI 1640 с добавлением 15% фетальной телячьей сыворотки (FBS), 2 мМ L-глутамина, 4 мкг/мл РНА-Р. Клетки инкубировали в течение 48-72 ч при 37 С. После инкубации РВМС центрифугировали и ресуспендировали в среде RPMI 1640, содержащей 15% FBS, 2 мМ L-глутамина, 100 ед./мл пенициллина, 100 мкг/мл стрептомицина, 10 мкг/мл гентамицина и 20 ед./мл рекомбинантного интерлейкина-2 человека. Цитотоксичность соединений по изобретению оценивали путем тестирования полулогарифмиче- 23023848 ских концентраций каждого соединения в трех повторах в отношении клеток, описанных выше. Клетки с чистой средой служили клеточным контролем (СС). Клетки Huh-7 и HepG2 высевали в 96-луночные планшеты при концентрации 5103 клеток на лунку. На следующий день среду отсасывали и добавляли 100 мкл соответствующей среды, содержащей 5% FBS. Разведения тестируемых лекарственных средств готовили в двухкратных концентрациях в микротитрационных пробирках и 100 мкл каждой концентрации помещали в соответствующие лунки в стандартном формате. Через 72 ч клетки подвергали обработке для оценки цитотоксичности. РВМС разводили в свежей среде и высевали во внутренние лунки 96-луночного планшета, имеющего лунки с круглым дном, в количестве 5104 клеток на лунку, в объеме 100 мкл. Аналогично, клетки СЕМ высевали в количестве 1104 клеток на лунку. Затем добавляли по 100 мкл растворов тестируемых лекарственных средств в двукратной концентрации в соответствующие лунки в стандартном формате. Культуры поддерживали в течение от шести до семи дней и затем подвергали обработке для определения цитотоксичности. Цитотоксичность определяли с использованием набора для анализа однородности целостности мембран CytoTox-ONE (Promega). При помощи анализа измеряли высвобождение лактатдегидрогеназы(LDH) из клеток с поврежденными мембранами в однородном флуорометрическом формате. Высвобождение LDH в культуральной среде измеряли с использованием количественного анализа ферментативного связывания, которое приводит к превращению резазурина во флуоресцентный продукт резоруфин. Продуцируемая флуоресценция пропорциональна количеству лизированных клеток. Для получения, где это применимо, значений ТС 50 (токсичные концентрации соединения, снижающие жизнеспособность клеток на 50%) и ТС 90 (токсичные концентрации соединения, снижающие жизнеспособность клеток на 90%), на клетки наносили шесть серийных разведений концентраций каждого соединения. Оценка ингибирования транспортеров MDR1 и MRP2 in vitro Для оценки ингибирования и активации транспортеров MDR1 (P-гликопротеина 1) и MRP2 in vitro может быть использован анализ для АТФазы производимый Solvo Biotechnology Inc. (Glavinas et al.,2003). Тестируемые соединения (0,1, 1, 10 и 100 мкМ) инкубировали с мембранными везикулами MDR1 или MRP2 при отсутствии и в присутствии ванадата для изучения потенциала активации АТФазы. Кроме того, аналогичные инкубации проводили в присутствии верапамила/сульфасалазина с целью выявления возможного ингибирования активности транспортера АТФазы. Активность АТФазы измеряли количественно, путем спектрофотометрии неорганического фосфата. Активацию вычисляли, исходя из чувствительности ванадата, которая увеличивается в случае АТФазной активности. Ингибирование определяли по снижению активности АТФазы, опосредованной верапамилом/сульфасалазином. Оценка ингибирования транспортеров Pgp с использованием клеток MDCK in vitro Для оценки ингибирования транспортера Р-гликопротеина (Pgp/MDR1) in vitro, использовали набор для анализа АТФазы, производимый Cyprotex. Для анализа использовали клетки MDR1-MDCK, полученные из NIH (Rockville, Мэриленд, США). После культивирования были подготовлены монослои базолатеральной и апикальной поверхностей путем двухкратной промывки буфером при рН 7,4 и температуре 37 С. Затем клетки инкубировали с буфером рН 7,4 и в апикальном и базолатеральном компартментах в течение 40 мин при температуре 37 С в атмосфере 5% CO2 с относительной влажностью от 95% до стабилизации физиологических параметров. Для исследования проникновения от апикальной к базолатеральной поверхности (А-В), буфер с рН 7,4 удаляли из апикального компартмента и заменяли на дозированный раствор лоперамида перед помещением в пластины-спутники. Растворы готовили путем разведения лоперамида в ДМСО буфером с получением конечной концентрации лоперамида, равной 5 мкМ (конечную концентрацию ДМСО доводили до 1%). Флуоресцентный маркер целостности мембран люцифер желтый также включали в дозирующий раствор. Эксперимент проводили в присутствии и при отсутствии тестируемого соединения (наносимого на апикальный и базолатеральный компартменты). Для исследования проникновения от базолатеральной к апикальной поверхности (В-А), субстрат Ргликопротеина, лоперамид (конечная концентрация=5 мкМ) помещали в базолатеральный компартмент. Эксперимент проводили в присутствии и при отсутствии тестируемого соединения (наносимого на апикальный и базолатеральный компартменты). Инкубацию проводили в атмосфере 5% CO2 с относительной влажностью 95% при 3 С в течение 60 мин. После инкубационного периода, пластины-спутники удаляли и образцы из апикального и базолатерального компартментов разводили для анализа методом ЖХ-МС/МС. Для каждой тестируемой концентрации соединений проводили одно определение в каждом тесте. На каждой пластине также подвергали скринированию положительный контроль ингибитора. Тестируемое соединение оценивали при концентрациях 0,1, 0,3, 1, 3, 10, 30 и 50 мкМ. Целостность монослоя в течение всего эксперимента контролировали путем мониторинга по проникновению красителя люцифера желтого с использованием флуориметрического анализа. После выполнения анализа рассчитывали значение IC50 (т.е. концентрацию ингибитора (тестируемого лекарственного средства), которая обеспечивает достижение половины максимального эффекта ингибирования). Оценка ингибирования поглощения транспортеров in vitro Для оценки ингибирования поглощения транспортеров ОАТ 1 В 1 и ОАТ 1 В 3 in vitro использовали набор для анализа, производимый Solvo Biotechnology Inc. Эксперименты по поглощению тестируемых образцов соединений (ТА) при концентрациях 0,068, 0,2, 0,62, 1,8, 5,5, 16,7 и 50 мкМ проводили на клетках СНО, стабильно экспрессирующих человеческие SLC транспортеры ОАТР 1 В 1 и ОАТР 1 В 3. Исходную клеточную линию СНО-K использовали в качестве отрицательного контроля. На стандартные 96 луночные планшеты для культуры ткани высевали клетки (1105 в 200 мкл смеси 1:1 среды Игла, модифицированную по Дульбекко, и среды Хэма DMEM F-12 (F-12, Lonza, Нью-Джерси, США) с добавлением 5 мМ бутирата натрия) и их инкубировали за 24 ч до начала эксперимента при температуре 37 С в атмосфере 5% СО 2 и 95% воздуха. Перед экспериментами среду удаляли вакуумным отсасыванием,клетки дважды промывали 100 мкл буфера Кребса-Хенселейта, рН 7,3 (изготовитель - Sigma Chemicals,Sigma-Aldrich, St Louis, Миссури, США). Эксперименты по поглощению проводили при температуре 37 С в 50 мкл буфера Кребса-Хенселейта (рН 7,3), содержащего, соответственно, субстрат для пробы и ТА или растворитель. Концентрация органического растворителя в каждой лунке была одинаковой, и не превышала 1% об/об. Субстрат для пробы в случае анализа OATP1 В 1 представлял собой E3S (0,1 мкМ),и в случае анализа OATP1 В 3, субстратом являлся Fluo-3 (10 мкМ). Для каждой лунки определяли количество перенесенного субстрата для пробы. Относительные активности рассчитывали по уравнению: Активность (%)=(A-B)/(C-D)100,где А = перемещенное количество субстрата в присутствии ТА на трансфицированных клетках; В = перемещенное количество субстрата в присутствии ТА на исходных клетках; С = перемещенное количество субстрата в присутствии растворителя на трансфицированных клетках;D = перемещенное количество субстрата в присутствии растворителя на исходных клеток. Значение IC50 определяли как концентрацию ТА, необходимую для ингибирования транспорта субстрата для пробы на 50%. Значение IC50 было получено из трехпараметрического логистического уравнения; кривая на графике была выровнена по величине относительной активности в зависимости от концентрации ТА путем нелинейной регрессии. Оценка ингибирования эффлюксных транспортеров in vitro Для оценки ингибирования эффлюксных транспортеров MRP2, MRP3 и BSEP in vitro использовали набор для везикулярного транспортерного анализа, производимый Solvo Biotechnology Inc. Тестируемые образцы соединений (ТА) (в концентрациях 0,068, 0,2, 0,62, 1,8, 5,5, 16,7 и 50 мкМ) инкубировали с везикулами мембранных эффлюксных транспортеров (Solvo Biotechnology Inc.) как при отсутствии, так и в присутствии 4 мМ АТФ для установления различия между поглощением, опосредованным транспортером, и пассивной диффузии ТА в везикулы. В случае использования транспортеров MRP2 и MRP3, реакции проводили в присутствии 2 мМ глутатиона. Реакционные смеси инкубировали в течение 10 мин при 37 С. Реакции инициировали добавлением 25 мкл раствора Mg-АТР с концентрацией 12 мМ (конечной концентрации в анализе составляла 4 мМ), или буфера, для контроля уровня фона. Реакцию останавливали добавлением 200 мкл ледяного промывочного буфера и после этого сразу выполняли фильтрацию на фильтрах из стекловолокна в 96-луночном формате (нанесение на фильтровальные пластины). На промытую и высушенную фильтровальную пластину добавляли сцинтилляционный буфер, и затем выполняли сцинтилляционный анализ с подсчетом импульсов. Субстратом для тестируемых соединений являлся таурохолат (2 мкМ) в случае везикул BSEP, и Е 217G (1 мкМ) в случае везикул MRP2 и MRP3. Для каждой лунки определяли количество перенесенного субстрата для тестируемых соединений в относительных единицах, выраженных в импульсах за минуту. Относительную активность рассчитывали по следующей формуле: Активность (%)=(A-B)/(CD)100,где А = перемещенное количество субстрата в присутствии ТА и АТФ; В = перемещенное количество субстрата в присутствии ТА; С = перемещенное количество субстрата в присутствии растворителя и АТФ;D = перемещенное количество субстрата в присутствии растворителя. Значение IC50 определяли как концентрацию ТА, необходимую для ингибирования транспорта субстрата для пробы на 50%. Значение IC50 было получено из трехпараметрического логистического уравнения; кривая на графике была выровнена по величине относительной активности в зависимости от концентрации ТА путем нелинейной регрессии. Анализ для оценки противовирусной активности против ВИЧ in vitro Противовирусная эффективность против ВИЧ может быть протестирована следующим образом. Как описано ранее (Bobardt et al., 2008), выделяли CD4+ Т-лимфоциты и макрофаги, полученные из крови. Кратко, РВМС человека очищали из свежей крови градиентным разделением на Ficoll-Hypaque (30 мин, 800 г, 25 С). Первичные человеческие CD4+ Т-клетки очищали от РВМС позитивной селекцией гранулами анти-CD4 Dynabeads с последующим их высвобождением с использованием системы Detachabead. Клетки культивировали в среде RPMI 1640 (Invitrogen) дополненной 10% FCS, MEM аминокисло- 25023848 тами, L-глутамином, MEM витаминами, пируватом натрия и пенициллином/стрептомицином, и затем их активировали бактериальным суперантигеном стафилококкового энтеротоксина В (SEB, 100 нг/мл) и клетками РВМС от другого донора, которые были обработаны митомицином С (соотношение РВМС:клетки CD4 составило 10:1). Через три дня после стимуляции клетки разделяли в соотношении 1:2 в среде, содержащей IL-2 (конечная концентрация 200 ед./мл). Затем культуры разделяли в соотношении 1:2 один раз в 2 дня в среде, содержащей IL-2, и инфицировали ВИЧ на 7 день после стимуляции. Для образования первичных человеческих макрофагов, моноциты очищали от РВМС человека путем негативной селекции и активировали, и затем их культивировали при концентрации 106 клеток/мл в средеDMEM, дополненной 10% FCS, MEM аминокислотами, L-глутамином, MEM витаминами, пируватом натрия, пенициллином (100 ед./мл), стрептомицином (100 мкг/мл) и 50 нг/мл рекомбинантного человеческого гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF), и выдерживали при 37 С в увлажненной атмосфере с 5% СО 2. Для получения макрофагов из моноцитов, клеткам давали возможность прилипнуть к полимерным пластинам, и затем их культивировали в течение 6 дней для дифференциации. Клетки CD4+ HeLa, клетки Jurkat, активированные CD4+ Т-лимфоциты и макрофаги периферической крови (500000 клеток/100 мкл) инкубировали с pNL4.3-GFP (вирус Х 4) или с pNL4.3-BAL-GFP (вирус R5) (из расчета 100 нг р 24) 48 ч в присутствии возрастающих концентраций образцов тестируемых соединений, заражение оценивали путем анализа процентной доли GFP-положительных клеток путемFACS, и рассчитывали значение ЕС 50. Анализ для оценки противовирусной активности против HBV in vitro Противовирусная эффективность против HBV может быть протестирована следующим образом. Клетки HepG2 2.2.15 высевали в 96-луночные планшеты для микротитрования. Через 16-24 ч конфлюентный монослой клеток HepG2 2.2.15 промывали и среду заменяли полной средой, содержащей различные концентрации тестируемого соединения в трех повторах (например, с шестью полулогарифмическими концентрациями). Через три дня культуральную среду заменяли свежей средой, содержащей тестируемые соединения, разведенные соответствующим образом. Через шесть дней после первоначального введения тестируемого соединения собирали супернатант культуры клеток, обрабатывали проназой и затем использовали для количественного анализа с помощью ПЦР TaqMan в режиме реального времени. Амплифицированную в ПЦР ДНК HBV обнаруживали в режиме реального времени путем мониторинга роста флуоресцентных сигналов, которые являются результатом экзонуклеолитической деградации погашенных молекул флуоресцентных зондов, которые гибридизуются с амплифицированной ДНКHBV. Для каждой ПЦР-амплификации получали стандартную кривую, используя разведения очищенной ДНК HBV. Противовирусную активность рассчитывали по снижению уровней ДНК HBV (IC50). Затем для оценки жизнеспособности клеток использовали анализ поглощения красителя, и результаты использовали для расчета токсичности (ТС 50). Терапевтический индекс (TI) рассчитывали как TC50/IC50. Анализ на основе смешанной реакции лимфоцитов (MLR) in vitro для оценки иммунодепрессантной активности Испытания по оценке иммунодепрессантной активности выполняли следующим образом. Популяции мононуклеарных клеток периферической крови (РВМС) выделяли из крови двух здоровых неродственных доноров-добровольцев (А и В), используя центрифугирование в градиенте плотности Histopaque. Клетки подсчитывали, и помещали их в количестве 1105 клеток на лунку в 96-луночные планшеты со средой RPMI с добавками, и вводили 2% сыворотки АВ человека. Условия культивирования включали следующее. Использовали отдельные популяции клеток А и В,смешанную популяцию клеток А и В, при отсутствии или в присутствии тестируемых соединений, применяя каждое в 6 различных концентрациях. Соединения тестировали в дозах от 10 до 0,0001 мкМ с инкрементом в 1 логарифм. Контрольные лунки содержали сопоставимые концентрации носителя (0,5% ДМСО), которые присутствуют в лунках, где было добавлено тестируемое соединение. Культуры были представлены в трех повторах в 96-луночных планшетах, и их инкубировали при 37 С в увлажненной атмосфере, содержащей 5% СО 2. На 6 день после начала анализа добавляли 3 Н-тимидин, и пробы собирали через 24 ч. После этого сравнивали уровни пролиферации клеток, культивируемых в различных условиях. Способность каждого разведения тестируемого соединения ингибировать пролиферацию в MLR рассчитывали как величину ингибирования, выраженную в процентах. Это дало возможность определить значение IC50 (концентрация тестируемого соединения, которая приводит к 50%-ному снижению количества импульсов в минуту). Для того чтобы вычислить значение IC50, ось X была представлена в логарифмической шкале. Для выравнивания средних точечных значений использовали нелинейную регрессию. При этом была выбрана сигмоидальная форма наклона кривой. Анализ ELISA для оценки Cyp-NS5A взаимодействия Данный анализ применяли для оценки разложения комплексов Cyp-NS5A и использовали для оценки активности взаимодействия с циклофилином D. Кратко, получение и очистку рекомбинантного GST,белков GST-CypD и Con1 NS5A-His проводили, как описано ранее (Chatterji et al., 2010). В 8-луночные узкие планшеты Nunc Maxisorb наносили GST или GST-CypD, инкубировали в течение 16 ч при температуре 4 С и блокировали. В лунки добавляли 50 мкл рекомбинантного NS5A-His (1 нг/мл) в связывающем буфере (20 мМ Трис, рН 7,9, 0,5 М NaCl, 10% глицерин, 10 мМ DTT и 1% NP-40), и инкубировали в течение 16 ч при температуре 4 С. Затем связанный NS5A-His определяли с использованием мышиных антител против His (1 мкг/мл) (anti-6His, Clontech) и кроличьих антител против мышиного антитела к пероксидазе-фосфатазе хрена (HRP) (разбавление 1:1000). Все эксперименты проводили дважды с использованием двух различных партий рекомбинантного CypD NS5A и белков. Анализ ингибирования циклофилина с использованием PPIA-азы Могут быть использованы альтернативные методики анализа взаимодействия с циклофилинами,состоящие в следующем. PPI-азная активность рекомбинантного СурА или CypD, который получен расщеплением GST-CypA или GST-CypD тромбином, определяют по скорости гидролиза N-сукцинил-AlaAla-Pro-Phe-п-нитроанилида под действием химотрипсина. Химотрипсин гидролизует только трансформу пептида, и гидролиз цис-формы, концентрация которой является максимальной при использовании исходного раствора в трифторэтаноле, содержащем 470 мМ LiCl, ограничена скоростью цис-трансизомеризации. СурА или CypD уравновешивали в течение 1 ч при температуре 5 С с выбранным тестируемым соединением, используя диапазон концентраций лекарственного средства от 0,1 до 20 нМ. Реакцию начинали добавлением пептида, и изменение поглощения контролировали спектрофотометрически со скоростью считывания 10 измерений в секунду. Начальную скорость гидролиза (при отсутствии СурА или CypD) вычитали из скорости, определенной в присутствии СурА или CypD. Начальные скорости ферментативной реакции анализировали с использованием регрессионного анализа для уравнения первого порядка изменения поглощения света во времени. Примеры Пример 1. Получение делеционного мутанта sfaA Sfreptomyces sp. A92-308110 (DSM9954). 1.1. Получение делеционного конструкта sfaA. Фрагмент EcoRV-StuI, размером приблизительно 7 т.п.о., из космиды TL3006 (SEQ ID NO:3), охватывающий sfaA (в положении нуклеотидов 14396-21362, последовательность под номером доступа вNCBI FJ809786), вырезали расщеплением под действием EcoRV и StuI, затем полученный выделенный фрагмент непосредственно лигировали в вектор pKC1139, который ранее был расщеплен с помощьюEcoRV, и обрабатывали щелочной фосфатазой креветки (Roche). Полученная плазмида была обозначена как pSGK268. Делецию в рамке гена sfaA, содержащегося в данном клоне, проводили с использованием набора для рекомбинации Red/ET, который производится Gene Bridges (катал, номер K006). Два олигонуклеотида, SfaA17161f и SfaA17825r использовали для амплификации маркера неомицина FRT-PGK-gb2-neo-FRT из матрицы ДНК, которая поставляется в наборе, где используется ДНКполимераза KOD. Полученный амплифицированный продукт, размером приблизительно 1,7 т.п.о., выделяли с использованием гель-электрофореза и очищали из геля с использованием смолы QiaEX. Плазмиду pSGK268 трансформировали в E.coli DH10B с применением стандартных методов и отбор проводили на чашках, содержащих апрамицин (50 мкг/мл). Включение делеционных конструкций осуществляли в основном по протоколу для набора Gene Bridges. Единичные колонии выращивали в течение ночи в присутствии раствора апрамицина (50 мкг/мл) в среде 2TY и трансформировали с помощью плазмиды pRedET (tet) и затем подвергали селекции по апрамицину (50 мкг/мл) и тетрациклину (3 мкг/мл) при 30 С. Единичную колонию использовали для получения ночной культуры этого штамма в 3 мл среды 2TY с апрамицином (50 мкг/мл) и тетрациклином (3 мкг/мл) при 30 С. 0,5 мл полученной культуры использовали для инокуляции 10 мл среды 2TY с апрамицином (50 мкг/мл) и тетрациклином (3 мкг/мл) при температуре 30 С, затем культуру выращивали до величины OD600 нм, приблизительно равной 0,5. 1,4 мл полученной культуры переносили в каждую из двух пробирок Эппендорфа и в одну из пробирок добавляли 50 мкл 10% арабинозы для индукции экспрессии рекомбинантных белков Red/ET. Пробирки встряхивали приблизительно 1 ч при температуре 37 С. Индуцированные и неиндуцированные клетки дважды осаждали центрифугированием на верхнем ярусе ротора центрифуги и промывали охлажденной стерильной водой, каждый раз подвергая клетки ресуспендированию и центрифугированию для их осаждения. Полученные осадки суспендировали в 30-40 мкл воды и хранили на льду. Выделенный фрагмент размером 1,7 т.п.о., полученный ранее вырезанием, добавляли в пробирки с индуцированными и неиндуцированными клетками и пробы переносили 1 мм электрокюветы Biorad на льду. Образцы подвергали электропорации (Bio-Rad Micropulser, напряжение 1,8 кВ, результирующая постоянная времени 4 мс), добавляли 1 мл среды 2TY (без антибиотиков) и перемешивали для удаления клеток из кюветы. Клетки инкубировали в течение 3 ч при температуре приблизительно 37 С со встряхиванием(1100 об/мин, компактный термомиксер Eppendorf), затем их высевали на чашки со средой 2TY, содержащей апрамицин (50 мкг/мл) и 25 мкг/мл канамицина, и инкубировали в течение ночи при температуре 37 С. Колонии из чашек с индуцированными пробами переносили штрихом на чашки со средой 2TY,содержащей канамицин (50 мкг/мл), для очистки и подтверждения включения кассеты устойчивости к канамицину. Для подтверждения введения кассеты применяли ПЦР для индивидуальных бактериальных колоний. Из этих культур были получены плазмиды и их подвергали расщеплению для подтверждения экспрессии плазмиды pSGK270. Плазмиды затем подвергали расщеплению с использованием NsiI для удаления маркерного фрагмента и остаток снова лигировали с получением конструкта pSGK271 с рамочной делецией sfaA. 1.2. Коньюгирование Streptomyces sp. A92-308110 (DSM9954) и введение делеции sfaA. Клетки Е.coli ET12567 pUZ8002 трансформировали плазмидой pSGK271 с использованием стандартных методов, и клетки подвергали отбору на чашках со средой 2TY, содержащей апрамицин (50 мкг/мл), канамицин (25 мкг/мл) и хлорамфеникол (10 мкг/мл). Полученный штамм инокулировали в 3 мл жидкой среды 2TY, содержащей апрамицин (50 мкг/мл), канамицин (25 мкг/мл) и хлорамфеникол (10 мкг/мл), и инкубировали в течение ночи при 37 С, 250 об/мин. 0,8 мл полученной культуры использовали для инокуляции 10 мл жидкой среды 2TY, содержащий апрамицин (50 мкг/мл), канамицин (25 мкг/мл) и хлорамфеникол (10 мкг/мл), в 50 мл пробирках Falcon, и инкубировали при 37 С, 250 об/мин,до достижения оптической плотности OD600 нм, равной 0,5. Полученную культуру центрифугировали при 3500 об/мин в течение 10 мин при 4 С, дважды промывали 10 мл среды 2TY с использованием центрифугирования для осаждения клеток после каждой промывки. Полученный осадок ресуспендировали в 0,5 мл среды 2TY и сохраняли на льду до использования. Этот процесс был выполнен к моменту полной подготовки спор Streptomyces, как описано ниже. Споры Streptomyces sp. A92-308110 (DSM9954) (Biot-4370) собирали из чашки с конфлюентным слоем в возрасте 1-2 недели путем ресуспендирования приблизительно в 3 мл 20% глицерина. Споры центрифугировали (5000 об/мин, 10 мин, при комнатной температуре) и дважды промывали 50 мМ буфером TES перед суспендированием в 1 мл 50 мМ буфера TES и распределением их в 2 пробирки Эппендорфа. Пробирки были подвергнуты тепловому шоку при температуре 50 С в течение 10 мин на водяной бане перед добавлением 0,5 мл среды 2TY и инкубации в компактном термомиксере Eppendorf при температуре 37 С в течение 4-5 ч. Подготовленные клетки Е.coli ET12567 pUZ8002 pSGK271 и Biot-4370 смешивали в соотношении 1:1 (250 мкл каждого штамма) и 1:3 (100 мкл Е.coli), немедленно распределяли на чашки R6 и переносили в инкубатор с температурой 37 С. Примерно через 2 ч инкубации чашки покрывали 2 мл стерильной воды, содержащей налидиксовую кислоту, с конечной концентрацией в чашке 25 мкг/л. Чашки возвращали в инкубатор с температурой 37 С и инкубировали в течение ночи перед нанесением 2 мл стерильной воды, содержащей апрамицин, с конечной концентрацией в чашке 20-25 мкг/л. Эксконъюгатные колонии, появившиеся приблизительно через 4-7 дней, наносили на среду ISP4, содержащую апрамицин(25 мкг/л) и налидиксовую кислоту (25 мкг/л), и инкубировали при температуре 37 С. После приемлемого роста мицелия наблюдаемые штаммы переносили на среду ISP4, содержащую апрамицин (25 мкг/л),при температуре 37 С и оставляли до споруляции. Затем штаммы пересевали в три раза (с целью удаления чувствительных к температуре плазмид), с нанесением их на среду ISP4 (без антибиотиков) и инкубированием при температуре 37 С в течение 3-4 дней. Затем штаммы наносили на среду ISP4 и инкубировали при температуре 28 С для обеспечения полного спорообразования (5-7 дней). Споры собирали и серийно разводили на чашках со средой ISP4 при температуре 28 С, чтобы обеспечить отбор отдельных колоний. Единичные спорулированные колонии были дважды пересеяны на чашках со средой ISP4 с или без апрамицина (25 мкг/л), чтобы подтвердить потерю плазмиды, и им давали расти приблизительно 7 дней до тестирования на продуцирование санглиферинов. 1.3. Скрининг штаммов для продукции санглиферинов в пробирках Falcon. Одну агаровую пробку, размером приблизительно 7 мм, полностью спорулированного штамма использовали для инокуляции 7 мл стерильной среды SM25-3, которую затем инкубировали при температуре 27 С при 200 об/мин в шейкере с двухдюймовым (50,8 мм) ходом. После 48 ч роста 0,7 мл полученной культуры переносили в стерилизованную пробирку Falcon, содержащую 7 мл среды SGP2 с 5% смолы НР 20. Культуры выращивали при температуре 24 С и 300 об/мин в инкубаторе с шейкером с однодюймовым (25,4 мм) ходом в течение 5 дней до сбора культуры. Отбирали 0,8 мл бактериальной культуры, и аликвоты переносили в пробирку Эппендорфа объемом 2 мл, обеспечивая при этом равномерное диспергирование смолы в культуре до отбора аликвот. Добавляли 0,8 мл ацетонитрила и 15 мкл муравьиной кислоты, после чего пробирки перемешивали в течение приблизительно 30 мин. Смесь осветляли центрифугированием и 170 мкл экстракта переносили во флакон для ВЭЖХ с последующим анализом пробы высокоэффективной жидкостной хроматографией. 1.4. Анализ штаммов на реверсию в дикий тип или в sfaA фенотип. Экстракты штаммов были проанализированы с помощью ВЭЖХ. Штаммы, которые продуцируют санглиферины А и В, не подвергали дополнительному анализу, поскольку они реверсируются в дикий тип. Штаммы, которые не продуцируют санглиферины А и В, показали небольшие уровни (приблизительно 1-2 мг/л) пика со временем удерживания, равным 6,5 мин, который отображает санглиферин как хромофор. Анализ ЖХМС показал, что этот пик имел величину m/z, равную 1073, т.е. -16 единиц от ожидаемого значения m/z для санглиферина. Было сделано предположение, данный пик обусловлен включением фенилаланина на место отсутствующей группы мета-окситирозина. Восемь штаммов, которые показали отсутствие продукции санглиферина, впоследствии использовали для оценки того, насколько потенциальная мутация sfaA могла быть дополнена химическими модификациями, чтобы обеспечить возможность мутасинтетического процесса получения новых санглиферинов. Штаммы были выращены в посевной среде SM25-3 в течение 48 ч перед переносом в продукционную среду SGP2, содержащую 5% смолы. После дополнительных 24 ч роста штаммы трехкратно подпитывали 2 мМ DL-мета-окситирозином (добавлением 100 мкл 0,16 М раствора в 1 М HCl) или 2 мМ Lфенилаланином, при этом в качестве контроля использовали штаммы без подпитки. Для повышения выхода продукта штаммы также подпитывали пипеколиновой кислотой (2 мМ в метаноле). Штаммы собирали после дополнительного роста в течение 4 дней и подвергали экстракции с анализом проб методом ВЭЖХ. Было показано, что мета-гидрокситирозин полностью дополняет мутации sfaA, и добавление Lфенилаланина повышает уровни соединения с -16 а.е.м. Для дальнейшего изучения был выбран штаммBiot-4585, как делеционный мутант по sfaA. Пример 2. Другие способы получения конструкта с делецией sfaA. Могут быть использованы и другие способы получения мутантов с делецией sfaA. Примеры этого включают инсерционные мутанты с инактивацией sfaA (см., например, пример 12 в WO 2010/034243). Данный штамм был получен способом, описанным в WO 2010/034243, и данный штамм был обозначен как BIOT-4452. Для получения делеции sfaA по альтернативной методике использовали два олигонуклеотида: для амплификации области гомологии в положении апстрим, с использованием космиды TL3006(SEQ ID NO:3) в качестве матрицы, и ДНК-полимеразы KOD. Продукт амплификации обрабатывали полинуклеотидкиназой Т 4 (NEB) и клонировали в вектор pUC18, который дефосфорилировали под воздействием щелочной фосфатазы креветки (Roche). Полученный конструкт тщательно проверяли рестрикционным расщеплением и секвенировали для определения того, была ли получена желаемая последовательность, и что во время полимеразной амплификации в последовательность не были включены ошибки. Полученный конструкт расщепляли с помощью EcoRI и NsiI, и продукты анализировали с использованием гель-электрофореза. Полосы, содержащие нужную последовательность (т.е. 2 т.п.н. участок гомологии в положении апстрим) вырезали из геля и очищали с использованием стандартных методик (со смолой QiaEX). Второй набор олигонуклеотидов: использовали для амплификации области гомологии в положении даунстрим с использованием космиды TL3006 (SEQ ID NO:3) в качестве матрицы и ДНК-полимеразы KOD. Продукт амплификации обрабатывали полинуклеотидкиназой Т 4 (NEB) и клонировали в вектор pUC18, который дефосфорилировали под воздействием щелочной фосфатазы креветки (Roche). Полученный конструкт тщательно проверяли рестрикционным расщеплением и секвенировали для определения того, была ли получена желаемая последовательность, и что во время полимеразной амплификации в последовательность не были
МПК / Метки
МПК: A61P 31/14, A61P 31/18, C07D 231/54, C07K 5/06, A61P 31/12
Метки: получения, способы, соединение, макроциклическое
Код ссылки
<a href="https://eas.patents.su/30-23848-makrociklicheskoe-soedinenie-i-sposoby-ego-polucheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Макроциклическое соединение и способы его получения</a>
Макроциклическое хиноксалиновое соединение в качестве ингибитора протеазы вгс ns3

Номер патента: 19327
Опубликовано: 28.02.2014
Авторы: Ливертон Найджел Дж., Харпер Стивен, Сумма Винченцо, Макколи Джон А.
МПК: A61K 38/08, A61K 38/07, A61K 38/06...
Метки: ингибитора, соединение, протеазы, макроциклическое, качестве, хиноксалиновое, вгс
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль2. Фармацевтическая композиция для лечения пациента, инфицированного ВГС, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.3. Фармацевтическая композиция по п.2, дополнительно содержащая второй терапевтический агент, выбранный из группы, состоящей из ингибитора протеазы ВГС и ингибиторов полимеразы ВГС NS5B.4. Применение соединения по п.1...
Арилвинилазациклоалкановое соединение, фармацевтическая композиция на его основе и способы его применения

Номер патента: 14714
Опубликовано: 28.02.2011
Авторы: Шев Мишель, Женевуа-Борелла Ариель, Капе Марк, Далл Гари Морис, Шмитт Джеффри Дэниел
МПК: A61K 31/505, A61K 31/506, A61K 31/44...
Метки: применения, фармацевтическая, соединение, композиция, способы, арилвинилазациклоалкановое, основе
Формула / Реферат:
1. Арилвинилазациклоалкановое соединение формулыили его фармацевтически приемлемая соль или его геометрия относительно двойной связи.2. Соединение по п.1, в котором геометрия относительно двойной связи является Е-геометрией.3. Фармацевтическая композиция, включающая эффективное количество соединения по п.1 или 2 и фармацевтически приемлемый носитель.4. Способ лечения или предотвращения a4b2 nAChR-опосредованных, включающий введение субъекту...
Гетероциклическое соединение, лекарственное средство, включающее его, и способы его применения

Номер патента: 20724
Опубликовано: 30.01.2015
Авторы: Миязаки Тору, Огуро Юя, Хомма Мисаки, Курасава Осаму
МПК: A61K 31/519, A61K 31/541, A61K 31/5377...
Метки: включающее, применения, гетероциклическое, лекарственное, его, соединение, средство, способы
Формула / Реферат:
1. Соединение, представленное формулойгде X представляет собой атом серы,Y представляет собой СН,R1 представляет собой C1-6 алкильную группу, необязательно замещенную 1-3 атомами галогена,R2 представляет собой(1) C1-6 алкильную группу, необязательно замещенную 1-3 заместителями, выбранными из(а) неароматической гетероциклической группы, необязательно замещенной 1-3 заместителями, выбранными из(i) атома галогена,(ii) гидроксигруппы,(iii) C1-6...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Чандер Мадхави С., Свинделл Чарльз С., Систи Николас Дж.
МПК: C07D 305/14
Метки: соединение, способ, паклитаксела, промежуточное, промежуточного, соединения, получения
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Cимметричное соединение для модификации полимерной композиции, полимерная композиция, способный к вулканизации кабель и способы вулканизации

Номер патента: 21922
Опубликовано: 30.09.2015
Авторы: Густафссон Билль, Нильссон Даниэль, Смедберг Анника
МПК: C08K 5/14
Метки: модификации, композиции, полимерная, кабель, соединение, вулканизации, способы, композиция, способный, cимметричное, полимерной
Формула / Реферат:
1. Симметричное соединение для модификации полимерной композиции, представляющее собой органический пероксид формулы (I)где R1 и R1' одинаковые или различные и каждый представляют собой (С6-С30)алкил с линейной или разветвленной цепью или метил;R2, R2', R3 и R3', каждый, представляют собой моноциклический (С4-С14)циклоалкил; (С1-С30)алкил с линейной или разветвленной цепью или моноциклический (С5-С14)арил илиR2 и R3 совместно с атомом углерода...
Предыдущий патент: Способ производства углеводородного масла
Следующий патент: Биотехнологический хондроитинсульфат, сульфатированный в положении 4 или 6 на его полисахаридной цепи, и способ его получения
Случайный патент: Способ получения мочевины