Азабензимидазолы в качестве противовирусных средств в отношении респираторного синцитиального вируса
Номер патента: 21613
Опубликовано: 30.07.2015
Авторы: Коиманс Людвиг Поль, Демэн Самюэль Доминик, Тахри Абделлах, Ху Лили, Рабуассон Пьер Жан-Мари Бернар, Йонкерс Тим Хьюго Мария, Вендевилль Сандрин Мари Элен




















Формула / Реферат
1. Соединение формулы I или его аддитивная соль

где каждый X независимо представляет собой С или N, по меньшей мере один X = N;
каждый Y независимо представляет собой С или N;
R1 присутствует в тех случаях, когда X = С, и R1 выбран из группы Н, галогена, N(R5)2, CO(R6), C(=NH)NH2, CF3 и B(OH)2;
R1 отсутствует в тех случаях, когда X = N;
R2 представляет собой -(CR7R8)n-R9;
R3 выбран из группы, состоящей из Н, C1-C10-алкила, C3-C7-циклоалкила, CH2CF3 или 4-6-членного насыщенного кольца, содержащего атом кислорода;
R4 присутствует в тех случаях, когда Y представляет собой С и выбран из группы, состоящей из Н, COO(R7) и галогена;
R5 выбран из группы, состоящей из Н и C1-C6-алкила;
R6 выбран из группы, состоящей из ОН и О-(C1-C6-алкила);
R7 и R8, каждый независимо, выбраны из Н, C1-C10-алкила или C3-C7-циклоалкила;
R9 выбран из группы, состоящей из Н, R10, C1-C6-алкила, ОН, CN, F, CF2H, CF3, COOR7, NR7COOR8, OCOR7, SO2R7, OCONR7R10, N(R7)CON(R7R8), N(R7)COOR10, фталимидо или 2-метилбензотиофен(1,1)диоксида;
n представляет собой целое число от 2 до 6;
R10 выбран из группы, состоящей из C1-C6-алкила, C3-C7-циклоалкила, фенила, пиридина или пиразола, необязательно замещенного одним или несколькими заместителями, выбранными из группы, включающей CF3, CH3, OCH3, OCF3 или галоген.
2. Соединение по п.1, где один X представляет собой N, указанный N находится в любом из двух орто-положений по отношению к имидазольному кольцу.
3. Соединение по п.1 или 2, где R1 выбран из группы, состоящей из Н и галогена.
4. Соединение по любому из предшествующих пунктов, где R1 в пара-положении по отношению к C-N-R2 выбран из группы, состоящей из Н и галогена, а все остальные R1 представляют собой Н.
5. Соединение по любому из предшествующих пунктов, где R7 и R8 представляют собой Н и n равно 2-4.
6. Соединение по любому из предшествующих пунктов, где R9 выбран из группы, состоящей из ОН, F, CF2H, CF3, C1-C6-алкила и SO2R7.
7. Соединение по любому из предшествующих пунктов, где R3 выбран из группы, состоящей из C3-C7-циклоалкила, и 4-членного насыщенного углеводорода, содержащего атом кислорода.
8. Соединение по любому из предшествующих пунктов, где R3 представляет собой циклопропил или CH2CF3.
9. Соединение по любому из предшествующих пунктов, где один Y представляет собой N, а другие Y представляют собой С, один Y, который представляет собой N, предпочтительно находится в пара-положении по отношению к N-R3.
10. Соединение по любому из предшествующих пунктов, где R4 на одном Y, который находится в пара-положении по отношению к N-R3, представляет собой F.
11. Соединение по пп.1-9, где все R4 представляют собой Н.
12. Лекарственное средство для ингибирования репликации RSV, включающее соединение по любому из пп.1-11.
13. Фармацевтическая композиция для ингибирования репликации RSV, содержащая в качестве активного ингредиента терапевтически эффективное количество соединения по любому из пп.1-11 и фармацевтически приемлемый носитель.
14. Способ получения фармацевтической композиции по п.13, включающий равномерное перемешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения по любому из пп.1-11.
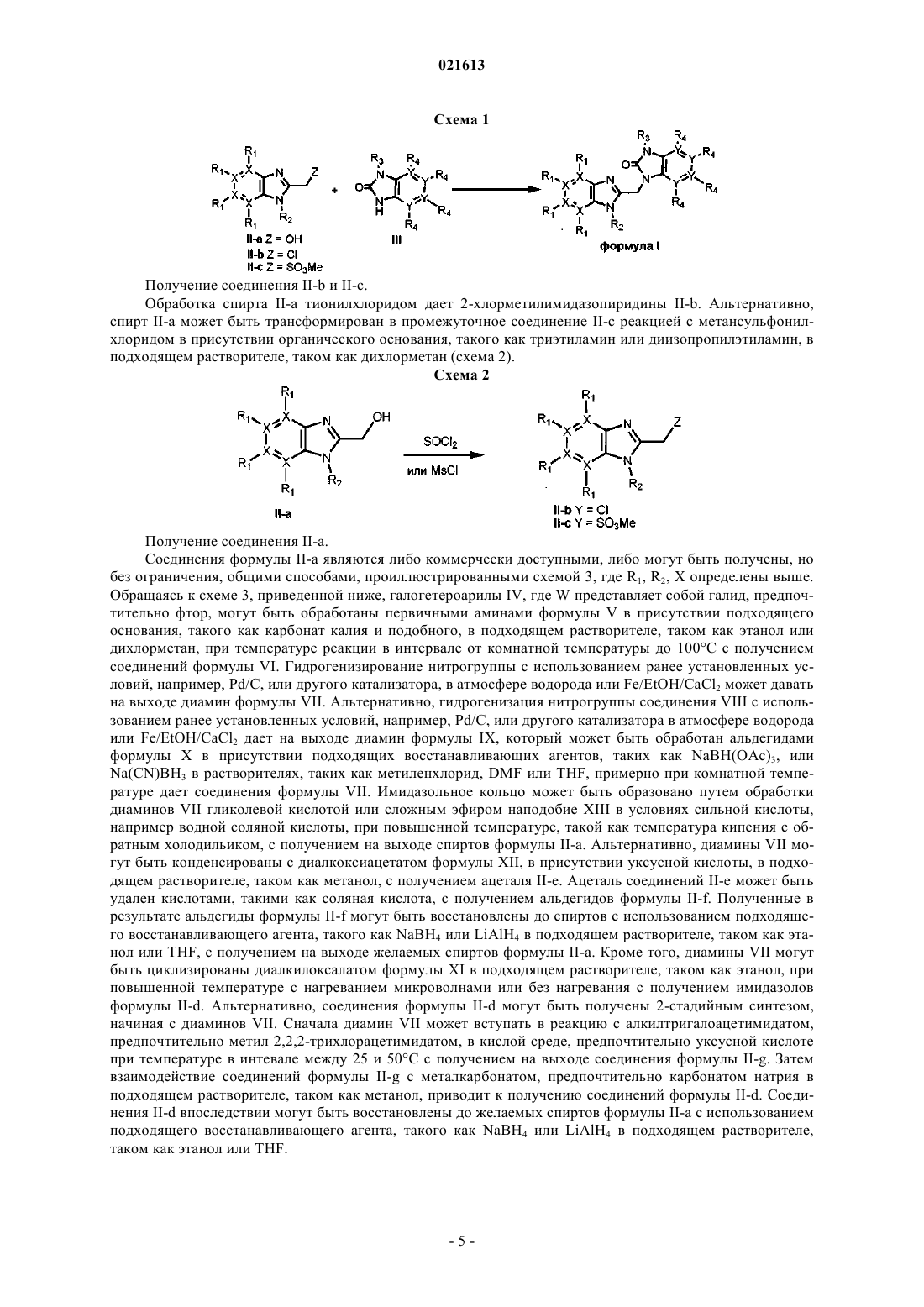
15. Способ получения соединения по любому из пп.1-11, включающий присоединение соединения, выбранного из группы, состоящей из II-a, II-b и II-с, к соединению III в соответствии со схемой 1

приводя к получению производных формулы I со всеми заместителями R и X, имеющими значения в соответствии с п.1.
16. Применение соединения по любому из пп.1-11 для производства лекарственного средства для ингибирования репликации RSV.
Текст
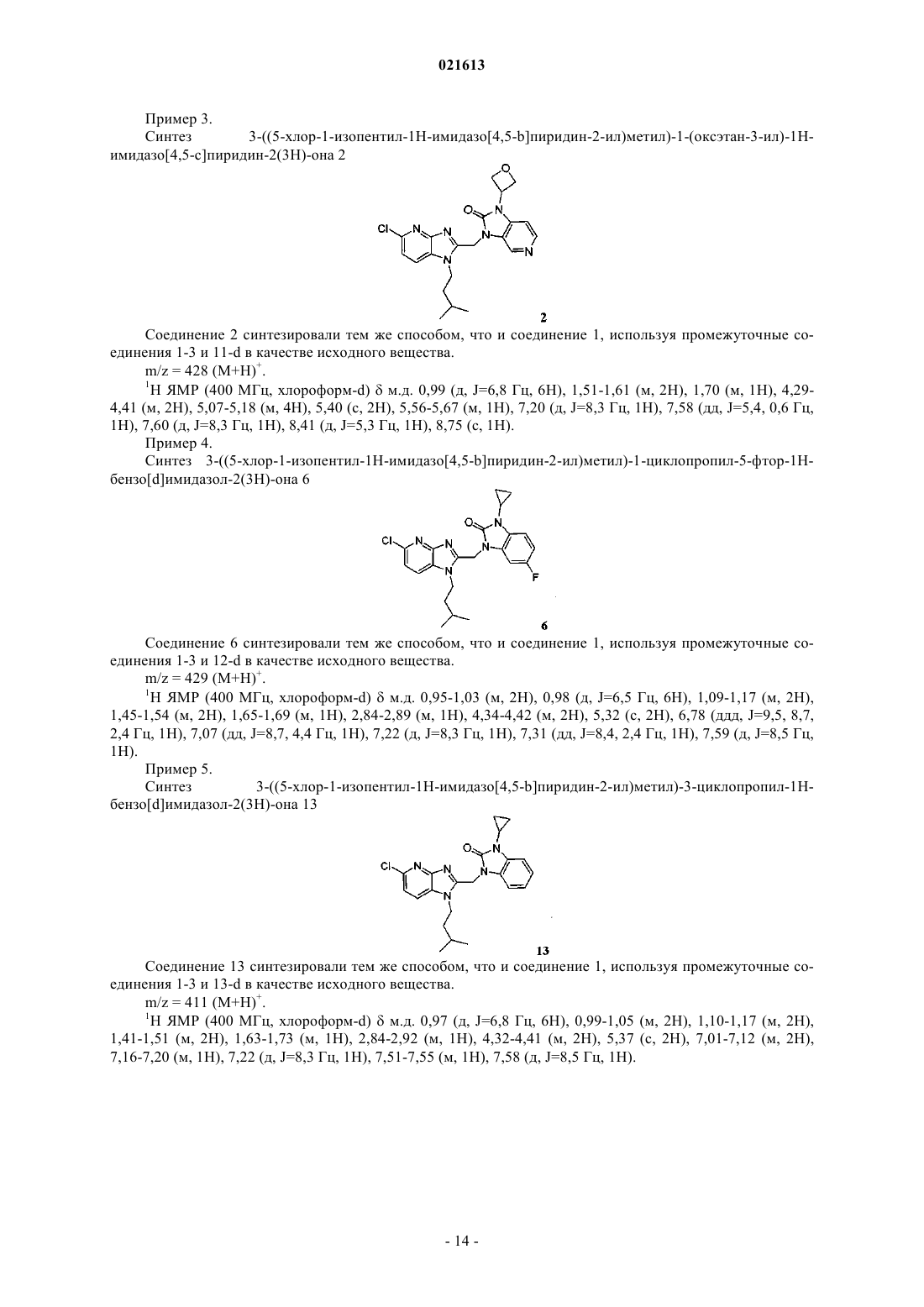
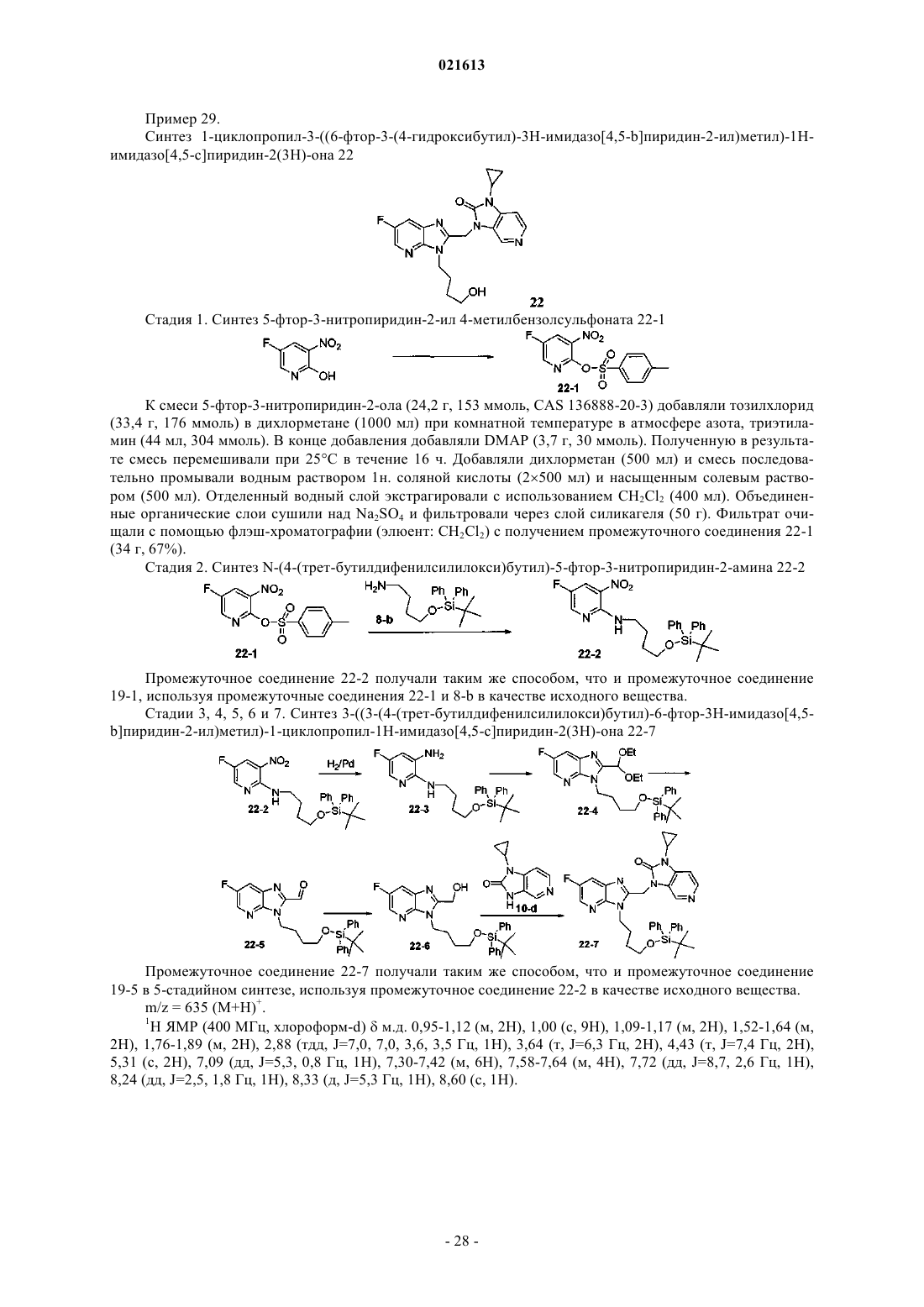
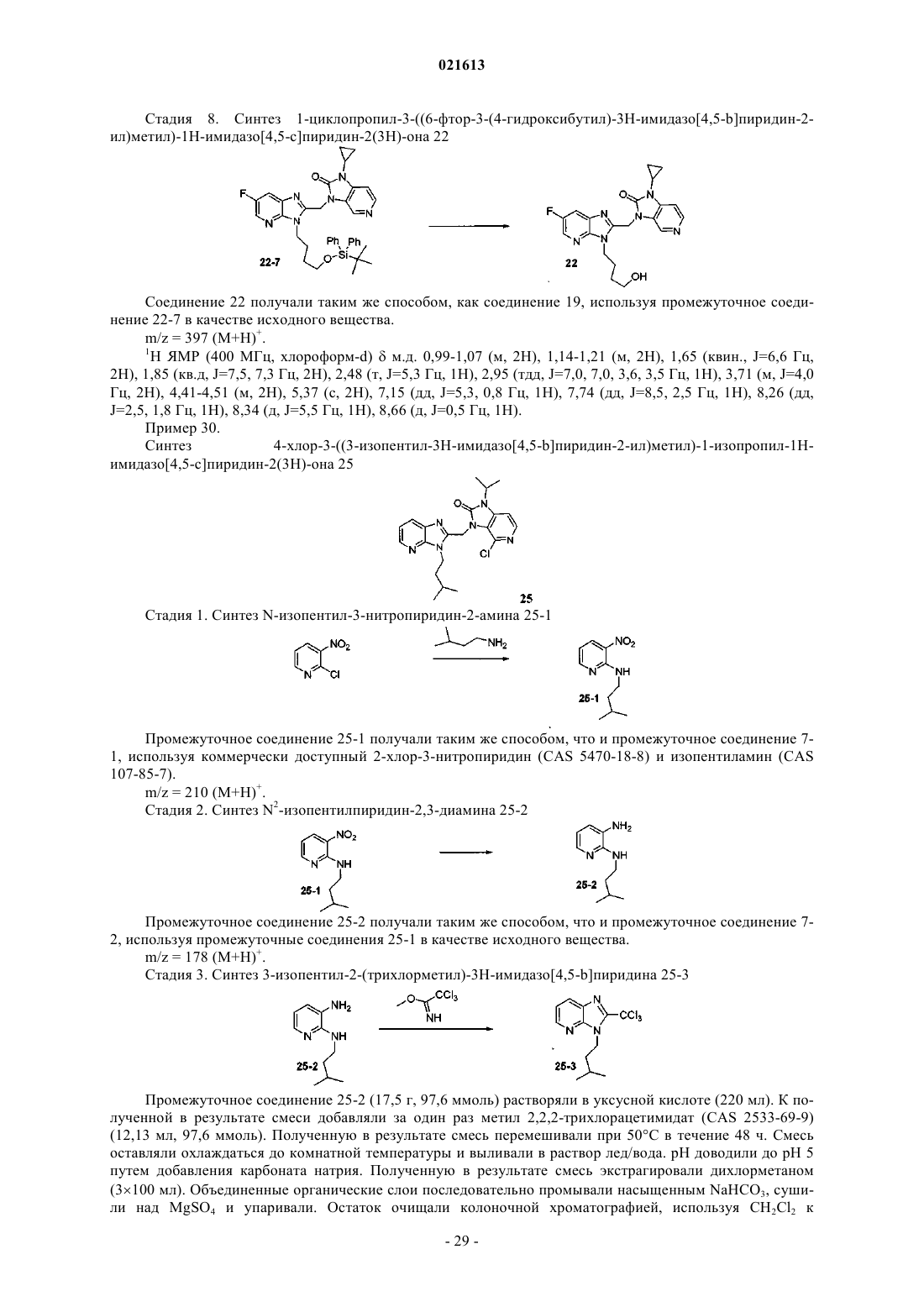
АЗАБЕНЗИМИДАЗОЛЫ В КАЧЕСТВЕ ПРОТИВОВИРУСНЫХ СРЕДСТВ В ОТНОШЕНИИ РЕСПИРАТОРНОГО СИНЦИТИАЛЬНОГО ВИРУСА Описаны соединение формулы I и его аддитивная соль Коиманс Людвиг Поль, Демэн Самюэль Доминик, Ху Лили, Йонкерс Тим Хьюго Мария, Рабуассон Пьер Жан-Мари Бернар, Тахри Абделлах,Вендевилль Сандрин Мари Элен (BE) где R1, R2, R3 и R4 раскрыты в формуле изобретения, а также композиции, содержащие эти соединения в качестве активного ингредиента для лечения инфекции, вызванной респираторным синцитиальным вирусом, и способы получения этих соединений и композиций.(71)(73) Заявитель и патентовладелец: ЯНССЕН Ар ЭНД Ди АЙРЛЭНД (IE) Область, к которой относится изобретение Настоящее изобретение относится к азабензимидазолам, обладающим противовирусной активностью, в частности обладающим ингибирующим действием на репликацию респираторного синцитиального вируса (RSV). Настоящее изобретение дополнительно относится к получению таких азабензимидазолов, композициям, содержащим такие соединения, и к соединениям для использования в лечении инфекции, вызванной респираторным синцитиальным вирусом. Предпосылки к созданию изобретенияRSV человека или респираторный синцитиальный вирус является крупным РНК вирусом, представителем семейства парамиксовирусов, подсемейства пневмовирусов вместе с коровьим RSV вирусом.RSV человека вызывает спектр заболеваний дыхательных путей у людей всех возрастов во всем мире. Он является главной причиной заболеваний нижних дыхательных путей в период младенчества и детства. Свыше половины всех грудных детей встречаются с RSV в первый год жизни и почти все в течение первых двух лет. Эта инфекция у детей младшего возраста может вызывать повреждение легких, которое персистирует годами и может вносить вклад в хроническое заболевание легких в последующей жизни(хронические обструктивные заболевания, астма). Дети более старшего возраста и взрослые зачастую страдают (тяжело) от вирусной инфекции верхних дыхательных путей при инфицировании RSV. В пожилом возрасте подверженность этой инфекции снова возрастает, и RSV вовлекается в целый ряд вспышек пневмонии у пожилых людей, приводя в результате к значительной смертности. Инфицирование вирусом из данной подгруппы не защищает от последующего инфицирования изолятом RSV из той же подгруппы в следующем зимнем сезоне. Повторное инфицирование RSV, таким образом, является обычным, несмотря на существование только двух подтипов, А и В. На сегодняшний день только три лекарственных средства были одобрены к применению противRSV инфекции. Первым средством является рибавирин, нуклеозидный аналог, который обеспечивает аэрозольное лечение тяжелой RSV инфекции у госпитализированных детей. Аэрозольный путь введения,токсичность (риск тератогенности), стоимость и высокая вариабельность эффективности ограничивают его применение. Два других лекарственных средства, RespiGam (RSV-IG) и Synagis (паливизумаб),поликлональные и моноклональные иммуностимуляторы антител, предназначены для использования в профилактике. Они оба являются очень дорогими, и для них требуется парентеральное введение. Другие попытки разработать безопасную и эффективную RSV вакцину на сегодняшний день оказались неудачными. Инактивированные вакцины не защищали от заболевания и действительно в некоторых случаях усугубляли заболевание во время последующего инфицирования. Живые аттенуированные вакцины были опробованы с ограниченным успехом. Ясно, что существует необходимость в эффективном нетоксичном и простом для введения лекарственном средстве против репликации RSV. Особенно было бы предпочтительным предоставить лекарственные средства против репликации RSV, которые можно было бы вводить перорально. Ссылка под названием "имидазопиридиновые и имидазопиримидиновые противовирусные средства" представляет собой публикацию международной заявки WO 01/95910, которая, по сути, относится к бензоимидазольным противовирусным средствам. В ней представлены соединения, обладающие противовирусной активностью, еще со значениями ЕС 50 в широком диапазоне от 0,001 до 50 мкМ (которые обычно не обеспечивают желаемой биологической активности). Другой ссылкой, относящейся к замещенным 2-метилбензимидазольным противовирусным средствам в отношении RSV, в том же самом диапазоне активности, является WO 03/053344. Другой релевантной ссылкой из предшествующего уровня техники в отношении соединений в том же диапазоне активностей является публикация международной заявки WO 02/26228, касающейся бензимидазолоновых противовирусных средств. Ссылкой на взаимосвязь структуры и активности в отношении ингибирования RSV, 5-замещенных бензимидазольных соединений, является X.А. Wang et al., Bioorganic and Medicinal Chemistry Letters 17 (2007), 4592-4598. Является желательным предоставить новые лекарственные средства, которые обладают противовирусной активностью. В частности, было бы желательным предоставить новые лекарственные средства,которые обладают ингибирующей активностью в отношении репликации RSV. Кроме того, было бы желательным выбрать структуры соединений, которые дают возможность получить противовирусные биологические активности порядка величины в более эффективных диапазонах предшествующего уровня техники (т.е. на нижней границе указанного выше интервала до 50 мкМ) и предпочтительно на уровне примерно максимальной активности, более предпочтительно даже более высокой активности, чем соединения, описанные в уровне техники. Дополнительным требованием является поиск соединений, обладающих противовирусной активностью при пероральном введении. Краткое изложение сущности изобретения Обращаясь конкретно к одной или нескольким указанным выше целям, настоящее изобретение в одном аспекте представляет противовирусные азабензоимидазольные соединения, представленные формулой I, или их аддитивную соль где каждый X независимо представляет собой С или N, по меньшей мере один X = N; каждый Y независимо представляет собой С или N;R1 присутствует в тех случаях, когда X = С, и R1 выбран из группы Н, галогена, N(R5)2, CO(R6),C(=NH)NH2, CF3 и B(OH)2;R6 выбран из группы, состоящей из ОН и О-(C1-C6-алкила);n представляет собой целое число от 2 до 6;R10 выбран из группы, состоящей из C1-C6-алкила, C3-C7-циклоалкила, фенила, пиридина или пиразола, необязательно замещенного одним или несколькими заместителями, выбранными из группы, включающей CF3, CH3, OCH3, OCF3 или галоген. В другом аспекте настоящее изобретение относится к указанным выше соединениям для применения при лечении RSV инфекций у теплокровных животных, предпочтительно у людей. Еще в одном аспекте настоящее изобретение относится к способу лечения вирусных инфекций, вызванных RSV, у пациента, нуждающегося в этом, включающему введение указанному пациенту эффективного количества соединения, определенного выше. Еще в одном аспекте настоящее изобретение относится к применению соединения, определенного выше, для производства лекарственного средства для лечения RSV инфекций. Еще в одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение, определенное выше, и фармацевтически приемлемый эксципиент. Еще в одном аспекте настоящее изобретение относится к способам получения соединений, определенных выше. Подробное описание изобретения Молекулы формулы I, в отличие от молекул предшествующего уровня техники, имеют на одной стороне (левой стороне изображенной формулы) замещенную азабензимидазольную группу. Настоящее изобретение в широком смысле основано на обоснованном установлении, что эти замещенные азабензимидазольные соединения в основном обладают интересующей ингибирующей активностью в отношенииRSV. Более того, эти соединения обеспечивают доступ к анти-RSV активностям в более высоких областях (т.е. нижней границе значений ЕС 50) доступного диапазона в указанных выше ссылках. В частности,на основе этих соединений могут быть раскрыты молекулярные структуры, которые даже превосходят эталонные соединения в смысле биологической активности. Настоящее изобретение дополнительно будет описано в отношении конкретных вариантов осуществления и со ссылкой на конкретные примеры, но настоящее изобретение этим не ограничивается, а ограничивается только формулой изобретения. Там, где термин "содержащий" используется в настоящем описании и формуле изобретения, он не исключает другие элементы или стадии. Использование существительного в единственном числе включает существительное во множественном числе, если иное не указано особо. В контексте настоящего изобретения C1-C6-алкил как группа или часть группы означает насыщен-2 021613 ные углеводородные радикалы с прямой или разветвленной цепью, имеющие от 1 до 6 атомов углерода,такие как метил, этил, пропил, 1-метилэтил, бутил, пентил, гексил, 2-метилбутил и подобные.C1-C10-алкил как группа или часть группы означает насыщенные углеводородные радикалы с прямой или разветвленной цепью, имеющие от 1 до 10 атомов углерода, такие как группы, определенные для C1-6 алкила, и гептил, октил, нонил, 2-метилгексил, 2-метилгептил, децил, 2-метилнонил и подобные. Необязательно, C1-10 алкил включает циклоалкильную группу, предпочтительно циклопропильную группу, например метилциклопропил, этилциклопропил и подобные. Считается, что термин "C2-C10-алкенил", используемый в контексте настоящего изобретения как группа или часть группы, включает ненасыщенные углеводородные радикалы с прямой или разветвленной цепью, имеющие по меньшей мере одну двойную связь и предпочтительно имеющие одну двойную связь и от 2 до 10 атомов углерода, такие как этенил, пропенил, бутен-1-ил, бутен-2-ил, пентен-1-ил, пентен-2-ил, гексен-1-ил, гексен-2-ил, гексен-3-ил, 2-метилбутен-1-ил, гептен-1-ил, гептен-2-ил, гептен-3-ил,гептен-4-ил, 2-метилгексен-1-ил, октен-1-ил, октен-2-ил, октен-3-ил, октен-4-ил, 2-метилгептен-1-ил,нонен-1-ил, нонен-2-ил, нонен-3-ил, нонен-4-ил, нонен-5-ил, 2-метилоктен-1-ил, децен-1-ил, децен-2-ил,децен-3-ил, децен-4-ил, децен-5-ил, 2-метилнонен-1-ил и подобные. Во всех случаях, когда C2-C10-алкенильная группа соединена с гетероатомом, она предпочтительно соединена через насыщенный атом углерода.C1-C6-алкокси как группа или часть группы означает O-C1-C6-алкильный радикал, где C1-C6-алкил имеет, независимо, значения, данные выше.C3-C7-циклоалкил является обобщением циклопропила, циклобутила, циклопентила, циклогексила или циклогептила. Термин "-(CR7R8)n" в контексте настоящего изобретения означает n повторов подгруппы CR7R8, где каждая из этих подгрупп независимо определена. Термин "галоген" является обобщением фтора, хлора, брома и йода. Следует отметить, что положения радикалов на любой молекулярной группе, используемой в определениях, могут быть в любом месте на такой группе при условии, что это является химически стабильным. Радикалы, используемые в определениях переменных, включают все возможные изомеры, если иное не указано. Например, пентил включает 1-пентил, 2-пентил и 3-пентил. В тех случаях, когда переменные появляются более одного раза в любом составном элементе, такое определение является независимым. Во всех случаях, используемых в дальнейшем, считается, что термин "соединения формулы I", или"соединения по настоящему изобретению", или аналогичный термин включает соединения общей формулы I и их аддитивные соли. Для терапевтического использования соли соединений формулы I представляют собой соли, в которых противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые не являются фармацевтически приемлемыми, также могут найти применение, например, в получении или очистке фармацевтически приемлемого соединения. Все соли, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения. Считается, что фармацевтически приемлемые кислотно- и основно-аддитивные соли, как упоминается выше в настоящем описании, включают терапевтически активные нетоксичные формы кислотно- и основно-аддитивных солей, которые способны образовывать соединения формулы I. Фармацевтически приемлемые кислотно-аддитивные соли удобно получать путем обработки основной формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородная или бромисто-водородная кислота, серную,азотную, фосфорную и подобные кислоты; или органические кислоты, например, такие как уксусная,пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.е. этандионовая), малоновая,янтарная (т.е. бутандионовая кислота), малеиновая, фумаровая, яблочная (т.е. гидроксибутандионовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, птолуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. Наоборот, указанные формы солей могут быть преобразованы обработкой соответствующим основанием в форму свободного основания. Соединения формулы I, содержащие кислотную часть, также могут быть преобразованы в формы их нетоксичных аддитивных солей металлов или аминов путем обработки соответствующими органическими и неорганическими основаниями. Подходящие формы основных солей включают, например, аммонийные соли, соли щелочных и щелочно-земельных металлов, например соли лития, натрия, калия,магния, кальция и подобные, соли с органическими основаниями, например соли бензатина, N-метил-Dглюкамина, гидрабамина и соли с аминокислотами, например, такими как аргинин, лизин и подобные. Термин "аддитивная соль", используемый выше в контексте настоящего изобретения, также включает сольваты, которые соединения формулы I, а также их соли, способны образовывать. Такие сольваты, например, представляют собой гидраты, алкоголяты и подобное. Некоторые соединения формулы I также могут существовать в их таутомерной форме. Такие фор-3 021613 мы, хотя не указаны явным образом в приведенной выше формуле, предназначены для включения в объем настоящего изобретения. Будет понятно, что соединения по настоящему изобретению со ссылкой на указанные выше левые и правые части формулы I находятся в большом разнообразии модификаций. Не приуменьшая общий объем изобретения, некоторые варианты осуществления рассматриваются ниже более подробно. В предпочтительном варианте осуществления максимально два X представляют собой N. В предпочтительном варианте осуществления один X представляет собой N. В более предпочтительном варианте осуществления один X, который представляет собой N, расположен в мета-положении по отношению к N-R2 группе имидазольного кольца, а указанный N расположен в орто-положении по отношению к=N- атому имидазольного кольца. В одном предпочтительном варианте осуществления R1 выбран из группы, состоящей из Н и галогена. В другом предпочтительном варианте осуществления R1 в пара-положении по отношению к C-N-R2 выбран из группы, состоящей из Н и галогена, и все другие R1 представляют собой Н. В другом предпочтительном варианте осуществления галоген представляет собой бром или хлор. В наиболее предпочтительном варианте осуществления максимально один R1 представляет собой хлор, а все другие R1 представляют собой Н. Еще в одном более предпочтительном варианте осуществления R1 в пара-положении по отношению к C-N-R2 представляет собой хлор. В другом предпочтительном варианте осуществления R2 включает -(CR7R8)n-R9 цепь, где R7 и R8 предпочтительно представляют собой Н и n составляет 2-4. Предпочтительно R9 выбран из группы, состоящей из ОН, C1-C6-алкила, более предпочтительно 2-пропила, C1-C6-алкокси, более предпочтительно метокси, SO2R7, с R7 предпочтительно представляющим собой метил. Наиболее предпочтительно R9 представляет собой фтор или CF3. В предпочтительном варианте осуществления R3 выбран из группы, состоящей из C3-C7 циклоалкила, более предпочтительно циклопропила и 4-членного насыщенного углеводорода, содержащего атом кислорода. В предпочтительном варианте осуществления и более предпочтительно в совокупности с другими предпочтительными вариантами осуществления, один Y представляет собой N, а другие Y представляют собой С. В наиболее предпочтительном варианте осуществления один Y, который представляет собой N,представляет собой Y в пара-положении по отношению к N-R3. Предпочтительно максимально один R4 представляет собой галоген, предпочтительно фтор. Наиболее предпочтительно все R4 представляют собой Н. Предпочтительные соединений перечислены ниже. Более предпочтительными являются соединения с номерами 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 16, 17, 18, 19, 20, 21, 31, 32, 33, 34, 35 и 36. Наиболее предпочтительными являются соединения с номерами 1, 2, 16, 31, 32 и 33. Соединения формулы I могут быть получены способами, описанными ниже, с использованием способов синтеза, известных в области органической химии, или модификаций и производных, которые знакомы специалистам в данной области. Исходные вещества, использованные в контексте настоящего изобретения, являются коммерчески доступными или могут быть получены рутинными способами, известными в данной области, такими как способы, описанные в обычных справочниках. Предпочтительные способы включают те, которые описаны ниже, но не только их. Во время любого из следующих последовательностей синтеза может быть необходимо и/или желательно защитить чувствительные или реакционноспособные группы на любой из рассматриваемых молекул. Это может достигаться с помощью общепринятых защитных групп, таких как описано у Т.W.Greene и P.G.M. Wuts, Protective Groups in Organic Chemistry, John WileySons, 1999, что включено, таким образом, посредством ссылки. Соединения формулы I или их фармацевтически приемлемые соли могут быть получены в соответствии со схемами реакций, рассмотренными ниже в настоящем описании. Если не указано иное, заместители на схемах определены выше. Выделение и очистка продуктов осуществляется стандартными способами, которые известны химикам. Схема 1 иллюстрирует способ получения соединений формулы I, где R1-R4, X и Y определены выше. Обращаясь к схеме 1, соединение формулы I может быть синтезировано путем присоединения 2 гидроксиметиленимидазопиридинов II-а к N3-замещенному 2-оксоимидазопиридину или к N3 замещенному 2-оксоимидазобензолу III в способе, известном из уровня техники, таком как реакция Мицунобу, в которой используется азадиизопропилдикарбоксилат и трифенилфосфин в подходящем растворителе, таком как DMF или THF. Альтернативно, соединение формулы I может быть получено путем замещения Z, который представляет собой галид, предпочтительно хлор II-b, или сульфонат, такой как мезилат II-с, в присутствии основания, такого как гидрид натрия, карбонат калия или карбонат цезия в подходящем растворителе, таком как DMF или THF. Получение соединения II-b и II-с. Обработка спирта II-а тионилхлоридом дает 2-хлорметилимидазопиридины II-b. Альтернативно,спирт II-а может быть трансформирован в промежуточное соединение II-с реакцией с метансульфонилхлоридом в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин, в подходящем растворителе, таком как дихлорметан (схема 2). Схема 2 Получение соединения II-а. Соединения формулы II-а являются либо коммерчески доступными, либо могут быть получены, но без ограничения, общими способами, проиллюстрированными схемой 3, где R1, R2, X определены выше. Обращаясь к схеме 3, приведенной ниже, галогетероарилы IV, где W представляет собой галид, предпочтительно фтор, могут быть обработаны первичными аминами формулы V в присутствии подходящего основания, такого как карбонат калия и подобного, в подходящем растворителе, таком как этанол или дихлорметан, при температуре реакции в интервале от комнатной температуры до 100 С с получением соединений формулы VI. Гидрогенизирование нитрогруппы с использованием ранее установленных условий, например, Pd/C, или другого катализатора, в атмосфере водорода или Fe/EtOH/CaCl2 может давать на выходе диамин формулы VII. Альтернативно, гидрогенизация нитрогруппы соединения VIII с использованием ранее установленных условий, например, Pd/C, или другого катализатора в атмосфере водорода или Fe/EtOH/CaCl2 дает на выходе диамин формулы IX, который может быть обработан альдегидами формулы X в присутствии подходящих восстанавливающих агентов, таких как NaBH(OAc)3, илиNa(CN)BH3 в растворителях, таких как метиленхлорид, DMF или THF, примерно при комнатной температуре дает соединения формулы VII. Имидазольное кольцо может быть образовано путем обработки диаминов VII гликолевой кислотой или сложным эфиром наподобие XIII в условиях сильной кислоты,например водной соляной кислоты, при повышенной температуре, такой как температура кипения с обратным холодильиком, с получением на выходе спиртов формулы II-а. Альтернативно, диамины VII могут быть конденсированы с диалкоксиацетатом формулы XII, в присутствии уксусной кислоты, в подходящем растворителе, таком как метанол, с получением ацеталя II-е. Ацеталь соединений II-е может быть удален кислотами, такими как соляная кислота, с получением альдегидов формулы II-f. Полученные в результате альдегиды формулы II-f могут быть восстановлены до спиртов с использованием подходящего восстанавливающего агента, такого как NaBH4 или LiAlH4 в подходящем растворителе, таком как этанол или THF, с получением на выходе желаемых спиртов формулы II-а. Кроме того, диамины VII могут быть циклизированы диалкилоксалатом формулы XI в подходящем растворителе, таком как этанол, при повышенной температуре с нагреванием микроволнами или без нагревания с получением имидазолов формулы II-d. Альтернативно, соединения формулы II-d могут быть получены 2-стадийным синтезом,начиная с диаминов VII. Сначала диамин VII может вступать в реакцию с алкилтригалоацетимидатом,предпочтительно метил 2,2,2-трихлорацетимидатом, в кислой среде, предпочтительно уксусной кислоте при температуре в интевале между 25 и 50 С с получением на выходе соединения формулы II-g. Затем взаимодействие соединений формулы II-g с металкарбонатом, предпочтительно карбонатом натрия в подходящем растворителе, таком как метанол, приводит к получению соединений формулы II-d. Соединения II-d впоследствии могут быть восстановлены до желаемых спиртов формулы II-а с использованием подходящего восстанавливающего агента, такого как NaBH4 или LiAlH4 в подходящем растворителе,таком как этанол или THF. Альтернативный путь получения соединений типа II-а изображен на схеме 4. Диамин IX сначала может быть присоединен к алкилгликолевой кислоте или сложному эфиру наподобие XIII в условиях сильной кислоты, например водной соляной кислоты, при повышенной температуре, такой как температура кипения с обратным холодильником, с получением на выходе спиртов формулы XIV. Этот спирт может быть защищен с использованием PG, где PG представляет собой защитную группу, такую как, но без ограничения указанным, тритил, что впоследствии приводит к получению соединений XV. Подходящим растворителем для этого типа реакций может быть, но без ограничения указанным, дихлорметан. Обработка соединения XV соединением XVI, где LG представляет собой уходящую группу, такую как галид, предпочтительно бром, или сульфонат, в присутствии основания, такого как гидрид натрия, карбонат калия или карбонат цезия в подходящем растворителе, таком как DMF или THF, дает соединениеII-h. Удаление PG в соединении II-h может быть выполнено в присутствии кислоты, такой как соляная кислота, в присутствии растворителя, не ограниченного указанным, такого как диоксан, с получением на выходе соединения II-а. Схема 4 Соединения III могут быть синтезированы способом, изображенным на схеме 5. Замещение W, который представляет собой галид, предпочтительно фтор или алкоксигруппу, предпочтительно метокси,нитропиридина или нитроарила XVII амином, в подходящем растворителе, таком как THF или DMF, в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин, дает соединение XVIII. Восстановление нитрогруппы до амина XIX может быть выполнено каталитическим путем с использованием водорода в присутствии катализатора, такого как палладий или платина, в подходящем растворителе, таком как метанол, или стехиометрическим путем, с использованием железа в присутствии хлорида аммония или хлорида олова в присутствии концентрированной соляной кислоты. Циклизация полученного в результате диамина XIX, с использованием CDI, фосгена или трифосгена, в растворителе,таком как ацетонитрил или THF, дает N3-замещенный 2-оксоимидазопиридин или N3-замещенный 2 оксоимидазобензол III. Альтернативно, соединение типа III может быть получено, начиная с коммерчески доступных дианилинов XX, которые могут быть циклизованы путем закрытия кольца с использованием CDI, фосгена или трифосгена, и дает на выходе промежуточные соединения типа XXI. Алкилиро-6 021613 вание азота мочевины соединения XXI может быть выполнено с помощью реакции Мицунобу с коммерчески доступными спиртами и сульфонилирования путем замены хлора в соединениях типа XXII с получением на выходе соединений формулы III. Схема 5 Соединения формулы I могут быть превращены в соответствующие формы N-оксида в соответствии со способами, известными из уровня техники для превращения трехвалентного азота в форму его Nоксида. Указанная реакция N-окисления в основном может быть выполнена путем взаимодействия исходного вещества формулы I с соответствующим органическим или неорганическим пероксидом. Соответствующие неорганические пероксиды включают, например, перекись водорода, пероксиды щелочных или щелочно-земельных металлов, например пероксид натрия, пероксид калия; соответствующие органические пероксиды могут включать пероксикислоты, например, такие как бензолкарбопероксоевая кислота или галогензамещенная бензолкарбопероксоевая кислота,например 3 хлорбензолкарбопероксоевая кислота, пероксоалкановые кислоты, например пероксоуксусная кислота,алкилгидропероксиды, например т-бутилгидропероксид. Подходящими растворителями, например, являются вода, низшие спирты, например этанол и подобные, углеводороды, например толуол, кетоны,например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей. Чистые стереохимически изомерные формы соединений формулы I могут быть получены путем использования способов, известных из уровня техники. Диастереомеры могут быть разделены физическими методами, такими как селективная кристаллизация и хроматографические методики, например противоточное распределение, жидкостная хроматография и подобные. Соединения формулы I, полученные в описанных выше способах, обычно представляют собой рацемические смеси энантиомеров, которые могут быть отделены один от другого в соответствии со способами разрешения, известными из уровня техники. Рацемические соединения формулы I, которые являются достаточно щелочными или кислыми, могут быть превращены в соответствующие формы диастереомерных солей, с использованием реакции с подходящей хиральной кислотой, соответственно хиральным основанием. Указанные формы диастереомерных солей впоследствии разделяют, например,селективной или фракционной кристаллизацией, и энантиомеры высвобождают щелочью или кислотой. Альтернативный способ разделения энантиомерных форм соединений формулы I включает жидкостную хроматографию, в частности жидкостную хроматографию с использованием хиральной стационарной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ, при условии,что реакция происходит стереоспецифически. Предпочтительно, если желателен конкретный стереоизомер, указанное соединение будет синтезировано стереоспецифическими способами получения. В этих способах предпочтительно будут использоваться энантиомерно чистые исходные вещества. Еще в одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы I, описанного в настоящем изобретении, или соединения любого из вариантов осуществления соединений формулы I, описанных в настоящем изобретении, и фармацевтически приемлемый носитель. Терапевтически эффективное количество в данном контексте представляет собой количество, достаточное для профилактического действия, для стабилизации или уменьшения вирусной инфекции, в частности RSV вирусной инфекции, у инфицированных пациентов или пациентов с риском заражения. Еще в одном аспекте настоящее изобретение относится к способу получения фармацевтической композиции, описанной в настоящем изобретении, который включает тщательное перемешивание фармацевтически приемлемого носителя с терапевтически эффективным количеством соединения формулы I, описанного в настоящем изобретении, или соединения любого из вариантов осуществления соединений формулы I, описанных в настоящем изобретении. Следовательно, соединения по настоящему изобретению или любой вариант их осуществления могут быть составлены в целый ряд фармацевтических форм с целью введения. В качестве подходящих композиций здесь могут быть процитированы все композиции, обычно используемые для лекарственных средств системного введения. Для получения фармацевтических композиций по настоящему изобрете-7 021613 нию эффективное количество конкретного соединения, необязательно, в форме аддитивной соли или комплекса с металлом, в качестве активного ингредиента, объединяют в равномерной смеси с фармацевтически приемлемым носителем, который может принимать широкое разнообразие форм в зависимости от формы препарата, желаемого для введения. Эти фармацевтические композиции желательно получать в единичной лекарственной форме, подходящей, в частности, для перорального введения, ректального введения, чрескожного введения или путем парентеральной инъекции. Например, при получении композиций в пероральной лекарственной форме могут быть использованы любые обычные фармацевтические среды, например, такие как вода, гликоли, масла, спирты и подобные в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, дезинтегрирующие агенты и подобные в случае порошков, пилюль, капсул и таблеток. Благодаря легкому введению таблетки и капсулы представляют наиболее предпочтительные пероральные формы - единиц дозирования, и в этом случае очевидно использовать твердые фармацевтические носители. Для парентеральных композиций носитель обычно будет включать стерильную воду, по меньшей мере, в значительной степени, хотя могут быть включены другие ингредиенты, например, способствующие растворимости. Например, могут быть получены инъецируемые растворы, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены инъецируемые суспензии, и в этом случае могут быть использованы соответствующие жидкие носители, суспендирующие агенты и подобные. Также включены формы твердых препаратов, которые предназначены для превращения, непосредственно перед использованием, в форму жидкого препарата. В композициях, подходящих для чрескожного введения, носитель необязательно содержит вещество, усиливающее проникновение и/или подходящий увлажнитель, необязательно объединенный с подходящими добавками любой природы в минимальных пропорциях, которые не оказывают значительного неблагоприятного воздействия на кожу. Соединения по настоящему изобретению также можно вводить посредством пероральной ингаляции или вдувания с помощью способов и составов, используемых в данной области для введения этим путем. Таким образом, в основном соединения по настоящему изобретению можно вводить в легкие в форме раствора, суспензии или сухого порошка, предпочтительно раствора. Любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством пероральной ингаляции или вдувания, подходит для введения соединений по настоящему изобретению. Таким образом, настоящее изобретение также относится к фармацевтической композиции, адаптированной для введения путем ингаляции или вдувания через рот, содержащей соединение формулы I и фармацевтически приемлемый носитель. Предпочтительно соединения по настоящему изобретению вводят посредством ингаляции раствора в распыляемых или аэрозольных дозах. Особенно предпочтительно составлять указанные выше фармацевтические композиции в единичную лекарственную форму для легкого введения или равномерного дозирования. Единичная лекарственная форма в контексте настоящего изобретения относится к физически дискретным единицам, подходящим в качестве единичных доз, каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное для получения желаемого терапевтического эффекта вместе с требуемым фармацевтическим носителем. Примерами таких единичных лекарственных форм являются таблетки (в том числе таблетки с риской или таблетки с покрытием), капсулы, пилюли, суппозитории, пакеты для порошкообразных продуктов, вафли, инъекционные растворы или суспензии и подобное и их сегрегированные кратные части. Соединения формулы I демонстрируют противовирусные свойства. Вирусные инфекции, которые лечатся с использованием соединений и способов по настоящему изобретению, включают инфекции,вызванные орто- и парамиксовирусами и, в частности, респираторным синцитиальным вирусом человека или коров (RSV). Целый ряд соединений по настоящему изобретению помимо этого являются активными против мутантных штаммов RSV. Дополнительно, многие из соединений по настоящему изобретению демонстрируют благоприятный фармакокинетический профиль и обладают привлекательными свойствами в понятиях биодоступности, в том числе подходящим периодом полужизни, AUC и максимальными величинами, и лишены неблагоприятных явлений, таких как недостаточно быстрое начало действия и удержание в тканях. Противовирусная активность in vitro в отношении RSV соединений по настоящему изобретению была протестирована в тесте, описанном в экспериментальной части этого описания, и также может быть продемонстрирована в анализе снижения урожая вируса. Противовирусная активность in vivo соединений по настоящему изобретению в отношении RSV может быть продемонстрирована в тест-модели с использованием хлопковых хомяков, как описано у Wyde et al. (Antiviral Research (1998), 38, 31-42). Благодаря их противовирусным свойствам, в частности их анти-RSV свойствам, соединения формулы I или любой вариант их осуществления, их пролекарства, N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы, применимы при лечении индивидуумов, подвергающихся вирусной инфекции, в частности RSV инфекции, и для профилактики этих инфекций. В целом соединения по настоящему изобретению могут быть использованы при лечении теп-8 021613 локровных животных, инфицированных вирусами, в частности респираторным синцитиальным вирусом. Соединения по настоящему изобретению или любой вариант их осуществления, следовательно, могут быть использованы в качестве лекарственных средств. Такое применение в качестве лекарственного средства или способ лечения включает системное введение пациентам, инфицированным вирусом, или пациентам, подверженным вирусным инфекциям, эффективного количества для лечения состояний, связанных с вирусной инфекцией, в частности RSV инфекцией. Настоящее изобретение также относится к применению соединений по настоящему изобретению или любого варианта их осуществления, в производстве лекарственного средства для лечения или профилактики вирусных инфекций, в частности RSV инфекции. Кроме того, настоящее изобретение относится к способу лечения теплокровного животного, инфицированного вирусом, или имеющего риск инфицирования вирусом, в частности RSV, указанный способ включает введение количества, эффективного против вируса, соединения формулы I, описанного в настоящем изобретении, или соединения любого из вариантов осуществления соединений формулы I, описанных в настоящем изобретении. В основном предполагается, что ежедневное количество, эффективное против вирусов, составило бы от 0,01 до 500 мг/кг массы тела, более предпочтительно от 0,1 до 50 мг/кг массы тела. Может быть целесообразным вводить требуемую дозу в виде двух, трех, четырех или более частей дозы с соответствующими интервалами в течение всего дня. Указанные части дозы могут быть составлены в виде единичных лекарственных форм, например, содержащих от 1 до 1000 мг и, в частности, от 5 до 200 мг активного ингредиента на единичную лекарственную форму. Точная доза и частота введения зависят от конкретного используемого соединения формулы I, конкретного состояния, подвергаемого лечению, тяжести состояния, подвергаемого лечению, возраста, массы тела, пола, продолжительности заболевания и общего физического состояния пациента, а также другого лекарственного средства, которое может принимать пациент, как хорошо известно специалистам в данной области. Более того, очевидно, что указанное эффективное суточное количество может быть уменьшено или увеличено в зависимости от реакции пациента, подвергающегося лечению, и/или в зависимости от оценки лечащего врача, назначающего соединения по настоящему изобретению. Эффективное суточное количество в указанных выше интервалах, следовательно, является только руководством. Также, сочетание другого противовирусного средства и соединения формулы I может быть использовано в качестве лекарственного средства. Таким образом, настоящее изобретение также относится к продукту, содержащему (а) соединение формулы I, и (b) другое противовирусное соединение, в виде комбинированного препарата для одновременного, раздельного или последовательного использования в противовирусном лечении. Различные лекарственные средства могут быть объединены в один препарат вместе с фармацевтически приемлемыми носителями. Например, соединения по настоящему изобретению могут быть объединены с интерфероном бета или фактором некроза опухоли альфа для лечения или профилактики RSV инфекций. Настоящее изобретение далее будет проиллюстрировано ссылками на следующие не ограничивающие примеры. Пример 1. Синтез промежуточных соединений. Все промежуточные соединения, необходимые для синтеза целевых соединений формулы I, синтезируют, как описано на следующих схемах 6-14. Схема 6. Синтез 3-(метилсульфонил)пропан-1-амингидрохлорид 6-е. Стадия 1. Синтез 3-(метилсульфонил)пропан-1-ола 6-b. 3-(Метилтио)пропан-1-ол 6-а (200 г, 1900 ммоль, CAS 505-10-2) растворяли в CH2Cl2 (2000 мл). Смесь охлаждали до 0 С. Добавляли в воду м-СРВА 85% (970 г, 5700 ммоль, CAS 937-14-4) частями,поддерживая температуру между 0 и 5 С. После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь фильтровали через слой целита. Фильтрат очищали с помощью флэшколонки (элюент:петролейный эфир:этилацетат=3:1, а затем этилацетат:метанол=10:1) с получением на выходе промежуточного соединения 6-b (75 г, 29%). Стадия 2. Синтез 1-бром-3-(метилсульфонил)пропана 6-с. Промежуточное соединение 6-b (75 г, 543 ммоль) растворяли в CH2Cl2 (750 мл). Смесь охлаждали до 0 С. Трибромид фосфора (53,6 мл, 570 ммоль) добавляли по каплям, поддерживая температуру между 0 и 5 С. После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Эту смесь выливали в воду со льдом. Отделенный органический слой промывали насыщенным солевым раствором (2500 мл), сушили над Na2SO4, фильтровали и упаривали под вакуумом с получением на выходе названного соединения 6-с (77 г, 71%). 1H ЯМР (400 МГц, хлороформ-d)м.д. 2,25-2,40 (м, 2 Н), 2,91 (с, 3 Н), 3,1-3,2 (м, 2 Н), 3,5-3,6 (м, 2 Н). Стадия 3. Синтез N-(дифенилметилен)-3-(метилсульфонил)пропанаминов 6-d. Промежуточное соединение 6-с (27 г, 134 ммоль) растворяли в CH3CN (60 мл). Добавляли дифенилметанимин (27 г, 148 ммоль) и DIEA (19,6 г, 152 ммоль). Смесь нагревали с обратным холодильником в течение 4 ч, а затем охлаждали до комнатной температуры. Смесь нейтрализовали с использованием 50% водной уксусной кислоты при 25 С. Добавляли воду (80 мл). Смесь экстрагировали этилацетатом(2300 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили надNa2SO4, фильтровали и упаривали под вакуумом. Остаток промывали петролейным эфиром (4100 мл). Смесь обрабатывали метиловым трет-бутиловым эфиром. Твердое вещество собирали и промывали петролейным эфиром. Фильтрат сушили под вакуумом. Остаток очищали колоночной хроматографией(элюент: CH2Cl2:этилацетат от 1:0 к 10:1). Названное соединение 6-d получали в виде твердого вещества белого цвета (34 г, 85%). Стадия 4. Синтез 3-(метилсульфонил)пропан-1-амингидрохлорида 6-е. Промежуточное соединение 6-d (34 г, 113 ммоль) растворяли в диоксане (600 мл). Смесь охлаждали до 0-5 С и добавляли по каплям раствор 4 н. HCl/диоксан (120 мл, 480 ммоль). После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь фильтровали. Твердое вещество собирали и промывали диоксаном. Названный продукт 6-е получали в виде порошка желтого цвета (11,5 г, 50%). Схема 7. Синтез трет-бутил(4-хлорбутокси)диметилсилана 7-b. 4-Хлорбутан-1-ол 7-а (100 г, 920 ммоль, CAS 928-51-8) растворяли в CH2Cl2 (1000 мл) при комнатной температуре. Смесь охлаждали до 0 С, затем добавляли имидазол (81,5, 1200 ммоль) и TBDMS-Cl(152 г, 1010 ммоль). Полученную в результате смесь перемешивали в течение 4 ч при комнатной температуре, затем отфильтровывали. Фильтрат промывали последовательно водным раствором 10% HCl и насыщенным солевым раствором. Полученный в результате раствор сушили над MgSO4, фильтровали,затем концентрировали с получением на выходе названных соединений 7-b в виде бесцветного масла Схема 8. Синтез 4-(трет-бутилдифенилсилилокси)бутан-1-амина 8-b. Смесь 4-аминобутан-1-ола 8-а (50 г, 561 ммоль, CAS 13325-10-5), имидазола (167 г, 2450 ммоль) и трет-бутилхлордифенилсилана (170 г, 618 ммоль, CAS 58479-61-1) в CH2Cl2 (1500 мл) перемешивали при 25 С в течение 15 ч. Полученную в результате смесь промывали последовательно насыщенным NaHCO3(2800 мл), водой (2800 мл) и насыщенным солевым раствором (2500 мл). Органический слой отделяли, сушили над Na2SO4, фильтровали и упаривали под вакуумом. Продукт 8-b получали в виде масла Схема 9. Синтез 1-бром-4-(метилсульфонил)бутана 9-с. Стадия 1. Синтез 4-(метилтио)бутан-1-ола 9-а. 4-Хлорбутан-1-ол 7-а (180 г, 1658 ммоль, CAS 928-51-8) добавляли к тиометоксиду натрия (656 г,1965 ммоль, 21% раствор в воде) при 0-5 С. После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 48 ч. Смесь экстрагировали с использованием CHCl3. Отделенный органический слой сушили над Na2CO3, фильтровали и упаривали под вакуумом. Остаток перегоняли с получением спирта 9-а в виде масла (144,2 г, 72%). Стадия 2. Синтез 4-(метилсульфонил)бутан-1-ола 9-b. Промежуточное соединение 9-а (141 г, 1173 ммоль) растворяли в CH2Cl2 (9000 мл). Смесь охлаждали до 0-5 С. м-СРВА (483 г, 85% чистота, 2375 ммоль, CAS 937-14-4) добавляли частями при 0-5 С. После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь фильтровали через слой целита. Фильтрат очищали с помощью флэш-колонки (элюент:петролейный эфир:этилацетат=3:1, а затем этилацетат:метанол=10:1). На выходе получали продукт 9-b (98 г, 65%). Стадия 3. Синтез 1-бром-4-(метилсульфонил)бутана 9-с. Промежуточное соединение 9-b (98 г, 645 ммоль) растворяли в CH2Cl2 (1100 мл). Смесь охлаждали до 0-5 С. PBr3 (64 мл, 674 ммоль) добавляли по каплям при 0-5 С. После добавления смесь оставляли нагреваться до 25 С и перемешивали в течение 15 ч. Смесь выливали в ледяную воду. Отделенный органический слой промывали насыщенным солевым раствором (2500 мл), сушили над Na2SO4, фильтровали и упаривали под вакуумом. Получали продукт 9-с (84,5 г, 80%).(185,5 г, 3250 ммоль) и диизопропилэтиламина (336 г, 2600 ммоль) в сухом этаноле (800 мл) нагревали с обратным холодильником в течение 3 ч. Смесь охлаждали до 0 С. Твердое вещество собирали фильтрованием. Фильтровальный осадок промывали холодным этанолом (150 мл). Твердое вещество сушили с получением названного соединения 10-b в виде порошка белого цвета (167 г, 72%). Стадия 2. Синтез N4-циклопропилпиридин-3,4-диамина 10-с. Промежуточное соединение 10-b (167 г, 932 ммоль) в этаноле (1400 мл) гидрогенизировали (50 фунтов на квадратный дюйм) при 20 С с мокрым 10% Pd/C (34 г) в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток промывали метил трет-бутиловым эфиром с получением названного соединения 10-с в виде порошка желтого цвета (133 г, 95%). Стадия 3. Синтез 1-циклопропил-1H-имидазо[4,5-с]пиридин-2(3H)-она 10-d. Карбонилдиимидазол (151,8 г, 936 ммоль) добавляли к раствору промежуточного соединения 10-с(133 г, 891,4 ммоль) в CH3CN (1800 мл) при 0 С. Реакционную смесь оставляли нагреваться до 10 С и перемешивали в течение 1 ч. Твердое вещество собирали фильтрованием и промывали с использованиемCH3CN (200 мл) с получением названного соединения 10-d в виде порошка белого цвета (101 г, 65%). Схема 11. Синтез 1-(оксэтан-3-ил)-1H-имидазо[4,5-с]пиридин-2(3H)-она 11-d. Соединение 11-d получали таким же способом, как соединение 10-d, используя 3-аминооксэтан в качестве исходного вещества. Схема 12. Синтез 1-циклопропил-5-фтор-1H-бензо[d]имидазол-2(3H)-она 12-d. Стадия 1. Синтез N-циклопропил-4-фтор-2-нитроанилина 12-b. 1,4-Дифтор-2-нитробензол 12-а (CAS 364-74-9) (15 г, 94,3 ммоль) растворяли в DMF (500 мл). Добавляли циклопропиламин (7 мл, 100 ммоль) с последующим добавлением триэтиламина (30 мл, 217 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду и экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Твердое вещество оранжевого цвета очищали колоночной хроматографией, используя дихлорметан и метанол с получением на выходе промежуточного соединения 12-b в виде твердого вещества оранжевого цветаH ЯМР (400 МГц, хлороформ-d)м.д. 0,63-0,68 (м, 2 Н), 0,88-0,95 (м, 2 Н), 2,54-2,55 (м, 1 Н), 7,277,34 (м, 2 Н), 7,84-7,90 (м, 1 Н), 7,93-8,02 (м, 1 Н). Стадия 2. Синтез N1-циклопропил-4-фторбензол-1,2-диамина 12-с. Промежуточное соединение 12-b (16 г, 82 ммоль) в этаноле (200 мл) гидрогенизировали при комнатной температуре с мокрым 10% Pd/C в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток промывали этанолом с получением названного соединения 9-с в виде твердого вещества белого цвета (12,8 г, 94%).m/z = 167 (М+Н)+. Стадия 3. Синтез 1-циклопропил-5-фтор-1H-бензо[d]имидазол-2(3H)-она 12-d. Карбонилдиимидазол (13,15 г, 81 ммоль) добавляли к раствору промежуточного соединения 12-с(12,8 г, 77,3 ммоль) в CH3CN (150 мл) при 0 С. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 4 ч. Растворитель удаляли, затем остаток очищали колоночной хроматографией, используя CH2Cl2/метанол с получением на выходе светло-коричневого твердого вещества, которое растирали в диэтиловом простом эфире с получением на выходе соединения 12-d в виде твердого вещества белого цвета (7,4 г, 50%). Схема 13. Синтез 1-циклопропил-1H-бензо[d]имидазол-2(3H)-она 13-d. Соединение 13-d получали таким же способом, как соединение 12-d, используя 2-фторнитробензол 13-а в качестве исходного вещества. Схема 14. Синтез 4-хлор-1-изопропил-1H-имидазо[4,5-с]пиридин-2(3H)-она 13-d. Соединение 14-d получали таким же способом, как соединение 12-d, используя 2,4-дихлор-3 нитропиридин 14-а и изопропиламин в качестве исходных веществ. Пример 2. Синтез 3-5-хлор-1-изопентил-1H-имидазо[4,5-b]пиридин-2-ил)метил)-1-циклопропил-1Hимидазо[4,5-с]пиридин-2(3H)-она 1(CAS 27048-04-0) (15 г, 86,42 ммоль), дегидрат хлорида олова (CAS 10025-69-1) (97,5 г, 432,1 ммоль). Полученную в результате смесь перемешивали при 60 С в течение 1 ч. Добавляли боргидрид натрия(1,63 г, 43,21 ммоль) и смесь перемешивали дополнительно при 60 С в течение еще 3 ч. Смесь охлаждали и десорбировали из EtOAc на роторном испарителе. Полученный в результате остаток разбавляли водой (350 мл) и нейтрализовали до рН 9-10 путем добавления водного раствора карбоната калия. Полученную в результате смесь экстрагировали с использованием EtOAc (3250 мл), сушили над Na2SO4 и упаривали. Остаток перемешивали в течение 72 ч в смеси EtOAc/гептана 1/1. Осадок фильтровали и сушили в вакууме в течение 2 ч. Промежуточное соединение 1-1 собирали в виде зеленоватого порошка Промежуточное соединение 1-1 (5 г, 34,82 ммоль) растворяли в дихлорметане (200 мл), добавляли уксусную кислоту (20 капель) и 4-метилпентанал (3 г, 34,8 ммоль, CAS 1119-16-0). Полученную в результате смесь перемешивали в течение 30 мин, а затем добавляли триацетоксигидроборат натрия (22,14 г, 104,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи и раствор 50% Na2CO3 добавляли по каплям, пока не прекратится выделение газа. Органический слой отделяли,сушили на MgSO4, фильтровали и упаривали досуха. Остаток очищали колоночной хроматографией используя гептан/EtOAc 7/3 до чистого EtOAc. Соединение 1-2 выделяли в виде твердого вещества белого цвета и сушили в вакууме в течение ночи (4,8 г, 65%). Смесь промежуточного соединения 1-2 (4,8 г, 22,46 ммоль) и 2-гидроксиуксусной кислоты (4,27 г,56,2 ммоль) перемешивали при 150 С в течение 4 ч. Смесь оставляли охлаждаться до комнатной температуры и осторожно обрабатывали 3 н. соляной кислотой. Полученную в результате смесь делали основной с использованием водного раствора аммиака и экстрагировали с использованием CH2Cl2 (300 мл). Органический слой сушили над MgSO4 и упаривали досуха. Остаток очищали колоночной хроматографией на силикагеле, используя CH2Cl2-EtOAc. Продукт 1-3 выделяли в виде твердого вещества коричневого цвета (3,5 г, 61%). К перемешанному раствору промежуточного соединения 1-3 (0,29 г, 1,14 ммоль), трифенилфосфинаDIAD (94%, 0,287 мл, 1,37 ммоль) по каплям при комнатной температуре. Реакционную смесь перемешивали в течение ночи. После завершения реакции смесь концентрировали досуха, остаток очищали колоночной хроматографией, элюировали с использованием этилацетата/CH2Cl2, затем CH2Cl2/метанол с получением на выходе названного соединения 1 в виде твердого вещества белого цвета (233 мг, 50%). Соединение 2 синтезировали тем же способом, что и соединение 1, используя промежуточные соединения 1-3 и 11-d в качестве исходного вещества. Соединение 6 синтезировали тем же способом, что и соединение 1, используя промежуточные соединения 1-3 и 12-d в качестве исходного вещества. Соединение 13 синтезировали тем же способом, что и соединение 1, используя промежуточные соединения 1-3 и 13-d в качестве исходного вещества. Смесь промежуточного соединения 1-1 (14,5 г, 101 ммоль) и 2-гидроксиуксусной кислоты (16 г, 210 ммоль) перемешивали при 150 С в течение 4 ч. Полученную в результате смесь охлаждали до 60 С и обрабатывали водным раствором 3 н. HCl (70 мл), затем ощелачивали до рН 7-8 путем добавления водного раствора аммиака. Смесь фильтровали и твердое вещество собирали, промывали водой и метил третбутиловым эфиром. Продукт 3-1 собирали в виде порошка желтого цвета (17,5 г, 94%). Промежуточное соединение 3-1 (17,5 г, 95,3 ммоль) и триэтиламин (28 мл, 190,6 ммоль) растворяли в дихлорметане (300 мл). Затем добавляли тритилхлорид (40 г, 143 ммоль). Полученную в результате смесь перемешивали при 25 С в течение 1,5 ч. Реакционную смесь промывали водным раствором 1 н. соляной кислоты и фильтровали. Твердое вещество собирали и промывали дихлорметаном (500 мл). Фильтрат промывали водным раствором 1 н. соляной кислоты (200 мл) и насыщенным водным растворомNaHCO3 (200 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали почти досуха под вакуумом. Остаток фильтровали. Твердое вещество собирали и промывали дихлорметаном. Продукт 3-2 собирали (27 г, 68%). К промежуточному соединению 3-2 (27 г, 63,4 ммоль), 4-хлорбутилпивалату (19 г, 83,8 ммоль) добавляли карбонат цезия (40 г, 122 ммоль) и йодид калия (3 г, 18 ммоль). Смесь растворяли в DMF при 25 С, а затем нагревали до 80 С и перемешивали в течение 2 ч. Реакционную смесь охлаждали до 25 С,фильтровали и фильтрат выливали в ледяную воду. Смесь экстрагировали этилацетатом (2500 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над Na2SO4,фильтровали и упаривали под вакуумом. Остаток очищали колоночной хроматографией (элюент: этилацетат:петролейный эфир=1:3). Собирали два изомера: соединение 3-3 (5 г) и соединение 3-4 (20 г).HCl/диоксана (20 мл, 80 ммоль) при 0 С. Смесь перемешивали при 25 С в течение 2 ч. Реакционную смесь упаривали под вакуумом при 40-45 С. Остаток упаривали совместно с CH2Cl2 (70 мл). К остатку добавляли дихлорметан (70 мл). Смесь фильтровали, твердое вещество собирали и промывали, используя метил трет-бутиловый эфир. Хлористо-водородную соль продукта 3-5 собирали в виде порошка белого цвета (2,83 г, 86%). Этот порошок растворяли в смеси воды (50 мл) и дихлорметана (50 мл). Затем добавляли бикарбонат натрия (1/02 г, 12 ммоль) частями при 25 С и смесь перемешивали при 25 С в течение ночи. Полученную в результате смесь экстрагировали с использованием дихлорметана, сушили надMgSO4 и концентрировали. Продукт 3-5 собирали в виде твердого вещества белого цвета. К перемешиваемому раствору промежуточного соединения 3-5 (0,4 г, 1,16 ммоль), трифенилфосфина (0,35 г, 1,34 ммоль) и соединения 10-d (0,214 г, 1,22 ммоль) в сухом THF (30 мл) добавляли DIAD(94%, 0,264 мл, 1,34 ммоль) по каплям при комнатной температуре. Реакционную смесь перемешивали в течение ночи. После завершения реакции смесь концентрировали досуха, остаток очищали колоночной хроматографией, элюировали с использованием этилацетата/CH2Cl2, затем CH2Cl2/метанола с получением на выходе названного соединения 3 в виде твердого вещества белого цвета (360 мг, 60%). Соединение 3 (0,29 г, 0,58 ммоль) растворяли в THF (15 мл) и добавляли гидроксид лития (40 мг,1,6 ммоль), растворенный в воде (5 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду и экстрагировали с использованием этилацетата. Органический слой сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией, используя дихлорметан и метанол. Названное соединение 14 выделяли в виде порошка белого цвета (200 мг, 80%). Промежуточное соединение 4-3 получали тем же способом, что и промежуточное соединение 1-3,используя пиридин-2,3-диамин 4-1 в качестве исходного вещества. Стадия 2. 1-Циклопропил-3-1-изопентил-1H-имидазо[4,5-b]пиридин-2-ил)метил)-1Hбензо[d]имидазол-2(3H)-он 4. Соединение 4 получали таким же способом, как соединение 1, используя промежуточные соединения 4-3 и 13-d в качестве исходного вещества. Соединение 5 получали таким же способом, как соединение 4, используя промежуточное соединение 10-d в качестве исходного вещества. Соединение 8 получали таким же способом, как соединение 4, используя промежуточное соединение 12-d в качестве исходного вещества. Соединение 9 получали таким же способом, как соединение 4, используя промежуточное соединение 11-d в качестве исходного вещества. 3-Фтор-2-нитропиридин (0,7 г, 4,92 ммоль, CAS 54231-35-5) растворяли в DMF (30 мл). Затем добавляли 3-(метилсульфонил)пропан-1-амин гидрохлорид 6-е (0,9 г, 5,2 ммоль) с последующим добавлением триэтиламина (1,5 мл, 11,3 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в воду и экстрагировали с использованием дихлорметана,сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией, используя этилацетат, с получением на выходе промежуточного соединения 7-1 в виде твердого вещества оранжевого цвета (1,2 г, 93%). Промежуточное соединение 7-1 (1,2 г, 4,62 ммоль) в THF (300 мл) гидрогенизировали при 20 С с мокрым 10% Pd/C (0,5 г) в качестве катализатора в течение ночи. После поглощения Н 2 (3 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток промывали метил трет-бутиловым эфиром с получением названного соединения 7-2 в виде порошка светло-желтого цвета (1 г, 94%). Смесь промежуточного соединения 7-2 (1 г, 4,36 ммоль) и метил 2-гидроксиацетата (2 мл, 26 ммоль) перемешивали при 130 С в течение ночи. Полученную в результате смесь оставляли охлаждаться до комнатной температуры и разбавляли дихлорметаном. Полученную в результате смесь выливали в воду и экстрагировали с использованием дихлорметана. Органический слой сушили над MgSO4, фильтровали и концентрировали. Водный слой упаривали, затем оба остатка перемешивали и очищали колоночной хроматографией дихлорметан/метанол. Продукт 7-3 собирали в виде порошка белого цвета (0,43 г, 36%).m/z = 270 (М+Н)+. Стадия 4. Синтез 1-циклопропил-3-1-(3-(метилсульфонил)пропил)-1H-имидазо[4,5-b]пиридин-2 ил)метил)-1H-имидазо[4,5-с]пиридин-2(3H)-она 7. Соединение 7 получали таким же способом, как соединение 1, используя промежуточные соединения 7-3 и 10-d в качестве исходного вещества. Промежуточное соединение 10-2 получали таким же способом, что и промежуточное соединение 72, используя 3-метоксипропан-1-амин в качестве исходного вещества. Стадия 2. Синтез 2-(диэтоксиметил)-1-(3-метоксипропил)-1H-имидазо[4,5-b]пиридина 10-3 Промежуточное соединение 10-2 (10 г, 34,43 ммоль) растворяли в этаноле (70 мл). Затем добавляли этил 2,2-диэтоксиацетат (7,39 мл, 41,3 ммоль) и этанолят натрия (14,14 мл, 37,8 ммоль). Полученную в результате смесь нагревали с обратным холодильником в течение 4 дней. Темный раствор оставляли охлаждаться до комнатной температуры, затем растворитель удаляли под вакуумом. Остаток растворяли в воде (300 мл) и добавляли дихлорметан. Смесь экстрагировали дихлорметаном. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали. Остаток очищали колоночной хроматографией, элюируя этилацетатом/дихлорметаном. Промежуточное соединение 10-3 собирали (5,15 г, 48%). Раствор промежуточного соединения 10-3 (5,15 г, 17,55 ммоль) в водном растворе 1 н. соляной кислоты (79 мл, 79 ммоль) перемешивали при 60 С в течение 2 дней. Полученную в результате смесь оставляли охлаждаться до комнатной температуры, затем добавляли этилацетат и воду. Добавляли насыщенный раствор Na2CO3 для доведения рН до щелочного и смесь экстрагировали с использованием этилацетата. Органический слой промывали насыщенным солевым раствором, сушили над Na2SO4, фильтровали и упаривали под вакуумом. Соединение 10-4 собирали в виде темного коричневого масла (3 г, 76%). Стадия 4. Синтез 1-3-метоксипропил)-1H-имидазо[4,5-b]пиридин-2-ил)метанола 10-5 К раствору промежуточного соединения 10-4 (3 г, 10,4 ммоль) в THF (40 мл) и метанола (40 мл) добавляли боргидрид натрия (0,8 г, 21 ммоль) по частям при 0 С. Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли, затем остаток растворяли в этилацетате (50 мл), добавляли воду (100 мл). Полученную в результате смесь экстрагировали этилацетатом (350 мл). Отделенный органический слой промывали насыщенным солевым раствором, сушили надNa2SO4, фильтровали и упаривали под вакуумом. Остаток очищали колоночной хроматографией, используя дихлорметан/метанол. Названное соединение собирали в виде оранжевого масла (1 г, 42%).m/z = 222 (М+Н)+. Стадия 5. Синтез 1-циклопропил-3-(1-(3-(метоксипропил)-1H-имидазо[4,5-b]пиридин-2-ил)метил)1H-имидазо[4,5-с]пиридин-2(3H)-она 10. Соединение 10 получали таким же способом, как соединение 1, используя промежуточные соединения 10-5 и 10-d в качестве исходного вещества. Соединение 12 получали таким же способом, как соединение 10, используя промежуточное соединение 11-d в качестве исходного вещества. Соединение 15 получали таким же способом, как соединение 10, используя промежуточное соединение 12-d в качестве исходного вещества. Промежуточное соединение 11-5 получали таким же способом, что и промежуточное соединение 10-5, используя 3-фторпропан-1-амин гидрохлорид (CAS 64068-31-1) и 3-фтор-2-нитропиридин (CAS 54231-35-35) в качестве исходных веществ. Соединение 11 получали таким же способом, как соединение 10, используя промежуточные соединения 11-5 и 10-d в качестве исходного вещества. Промежуточное соединение 16-5 получали в соответствии с 5 стадиями синтеза, описанными для промежуточного соединения 10-5, используя 5-бром-2-хлор-3-нитропиридин (CAS 67443-38-3) и 3 метилбутан-1-амин (CAS 107-85-7) в качестве исходного вещества. Соединение 16 получали таким же способом, как соединение 10, используя промежуточные соединения 16-5 и 10-d в качестве исходного вещества. Соединение 18 получали таким же способом, как соединение 16, используя промежуточные соединения 16-5 и 11-d в качестве исходного вещества. Соединение 30 получали таким же способом, как соединение 16, используя промежуточные соединения 16-5 и 13-d в качестве исходного вещества.(0,128 г, 1,3 ммоль), тиофенол (0,5 мл) и мокрый 10% Pd/C (0,2 г). Реакционную смесь перемешивали при 25 С в атмосфере водорода. После поглощения Н 2 (1 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток растворяли в воде и дихлорметане. Полученную в результате смесь последовательно экстрагировали дихлорметаном, сушили над MgSO4 и концентрировали. Остаток очищали колоночной хроматографией, используя дихлорметан/метанол. Названное соединение 24 собирали в виде порошка белого цвета (333 мг, 81%). Смесь соединения 16 (1 г, 2,15 ммоль), ацетата палладия (9,8 мг, 0,043 ммоль), 1,3 бис(дифенилфосфино)пропана (35,4 мг, 0,086 ммоль), ацетата калия (316 мг, 3,22 ммоль) и метанола (10 мл) в THF (100 мл) помещали в автоклав в атмосфере азота. Автоклав закрывали и создавали давление до 20 бар монооксида углерода, реакцию проводили в течение 16 ч при 125 С. Реакционную смесь оставляли охлаждаться до комнатной температуры и фильтровали через акродиск. Растворитель упаривали и остаток очищали колоночной хроматографией, используя этилацетат/метанол. Названное соединение 26 собирали в виде порошка белого цвета (870 мг,91%). Соединение 26 (0,84 г, 1,89 ммоль) растворяли в THF (15 мл) и добавляли гидроксид лития (544 мг,22,7 ммоль), растворенный в воде (10 мл). Полученную в результате смесь перемешивали при комнатной температуре в течение ночи. рН полученной в результате смеси доводили до рН 4 путем добавления 1 М раствора соляной кислоты. Затем смесь экстрагировали с использованием этилацетата. Органический слой сушили над MgSO4 и концентрировали. Названное соединение 28 выделяли в виде порошка белого цвета (690 мг, 84%). Смесь соединения 16 (0,5 г, 1 ммоль), дихлор(дифенилфосфиноферроцен)палладия (78,7 мг, 0,108 ммоль), дицианоцинка (0,505 г, 4,3 ммоль) и триэтиламина (0,6 мл, 4,3 ммоль) в диоксане (10 мл) в атмосфере азота подвергали воздействию излучения в течение 1 ч в микроволновом реакторе при 125 С. Полученную в результате смесь оставляли охлаждаться до комнатной температуры, затем фильтровали через дикалит. Фильтрат упаривали досуха. Остаток очищали колоночной хроматографией, используяEtOAc/MeOH 8-2. Названное соединение 27 выделяли в виде твердого вещества белого цвета (200 мг,45%). Соединение 27 (125 мг, 0,31 ммоль) в метаноле/NH3 (100 мл) гидрогенизировали при 20 С в течение ночи, используя в качестве катализатора скелетный никелевый катализатор гидрирования (50 мг). После поглощения Н 2 (2 экв.) катализатор отфильтровывали и фильтрат упаривали. Остаток очищали колоночной хроматографией, используя дихлорметан/МеОН/NH3. Названное соединение 17 выделяли в виде твердого вещества белого цвета (25,5 мг, 20%).(0,382 г, 1,5 ммоль) и ацетата калия (0,16 г, 1,6 ммоль) в диоксане (20 мл) в атмосфере аргона перемешивали при комнатной температуре в течение 10 мин. К полученной в результате смеси добавляли дихлор(дифенилфосфиноферроцен)палладий (39 мг, 0,05 ммоль). Полученную в результате смесь нагревали до 115 С в течение 3 ч. Смесь оставляли охлаждаться до комнатной температуры, затем удаляли растворитель. Остаток (23-1) растворяли в ацетонитриле (40 мл) и добавляли водный раствор соляной кислоты 6 М (1,7 мл, 10 ммоль). Полученную в результате смесь перемешивали при 110 С в течение 2 ч. Смесь оставляли охлаждаться до комнатной температуры и добавляли воду (30 мл), рН доводили до рН 7 путем добавления 7 н. аммиачного раствора в метаноле. Полученную в результате смесь концентрировали и остаток очищали препаративной HPLC. Названное соединение 23 выделяли в виде твердого вещества белого цвета (309 мг, 67%). Смесь 5-бром-2-хлор-3-нитропиридина (CAS 67443-38-3) (33 г, 101 ммоль), 4-(третбутилдифенилсилилокси)бутан-1-амина 8-b (20 г, 84,2 ммоль), карбоната калия (23,3 г, 168 ммоль) и йодида калия (1,4 г, 8,4 ммоль) в CH3CN (200 мл) перемешивали при 20 С в течение 15 ч. Полученную в результате смесь обрабатывали с использованием CH2Cl2 (400 мл) и воды (400 мл). Отделенный водный слой экстрагировали с использованием CH2Cl2 (2200 мл). Объединенные органические слои промывали насыщенным солевым раствором (400 мл), сушили над Na2SO4, фильтровали и упаривали под вакуумом. Получали промежуточное соединение 19-1 (44 г, 90%).(25 мл). Полученную в результате смесь нагревали до 50 С. Железо (Fe) (36,1 г, 647 ммоль) очень медленно добавляли в смесь через 20 мин. Смесь перемешивали при 50 С в течение 2 ч, и оставляли охлаждаться до комнатной температуры. Добавляли воду (400 мл) и смесь фильтровали через слой целлита. Остаток собирали на фильтре, промывали водой. Фильтрат обрабатывали этилацетатом (2300 мл). Органический слой отделяли и промывали водой (2400 мл) и насыщенным солевым раствором (500 мл). Органический слой сушили над Na2SO4, фильтровали и упаривали под вакуумом. Остаток упаривали совместно с толуолом под вакуумом с получением промежуточного соединения 19-2 (40 г, 90%). Стадии 3, 4 и 5. Синтез (6-бром-3-(4-(трет-бутилдифенилсилилокси)бутил)-3H-имидазо[4,5b]пиридин-2-ил)метанола 19-5 Промежуточное соединение 19-5 получали таким же способом, что и промежуточное соединение 10-5 в 3-стадийном синтезе, начиная с промежуточного соединения 19-2. Стадия 6. Синтез 3-6-бром-3-(4-(трет-бутилдифенилсилилокси)бутил)-3H-имидазо[4,5-b]пиридин 2-ил)метил)-1-циклопропил-1H-имидазо[4,5-с]пиридин-2(3H)-она 19-6 Промежуточное соединение 19-6 получали таким же способом, как соединение 10, используя промежуточные соединения 19-5 и 10-d в качестве исходного вещества. Промежуточное соединение 19-6 (1,65 г, 2,32 ммоль) растворяли в метаноле (40 мл), затем добавляли фторид аммония (0,206 г, 5,58 ммоль). Полученную в результате смесь перемешивали при нагревании с обратным холодильником в течение 56 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры, затем растворитель удаляли. Остаток очищали колоночной хроматографией, используя дихлорметан/метанол с получением на выходе продукта в виде твердого вещества белого цвета (1 г,92%). Промежуточное соединение 20-1 получали таким же способом, что и промежуточное соединение 19-6, используя промежуточные соединения 19-5 и 11-d в качестве исходного вещества. Соединение 20 получали таким же способом, как соединение 19, используя промежуточные соединения 20-1 в качестве исходного вещества. Промежуточное соединение 21-1 получали таким же способом, что и промежуточное соединение 19-5 в 5-стадийном синтезе, используя 2,5-дихлор-3-нитропиридин (CAS 21427-62-3) в качестве исходного вещества. Промежуточное соединение 21-2 получали таким же способом, что и промежуточное соединение 19-6, используя промежуточные соединения 21-1 и 10-d в качестве исходного вещества. Соединение 21 получали таким же способом, как соединение 19, используя промежуточные соединения 21-2 в качестве исходного вещества.(33,4 г, 176 ммоль) в дихлорметане (1000 мл) при комнатной температуре в атмосфере азота, триэтиламин (44 мл, 304 ммоль). В конце добавления добавляли DMAP (3,7 г, 30 ммоль). Полученную в результате смесь перемешивали при 25 С в течение 16 ч. Добавляли дихлорметан (500 мл) и смесь последовательно промывали водным раствором 1 н. соляной кислоты (2500 мл) и насыщенным солевым раствором (500 мл). Отделенный водный слой экстрагировали с использованием CH2Cl2 (400 мл). Объединенные органические слои сушили над Na2SO4 и фильтровали через слой силикагеля (50 г). Фильтрат очищали с помощью флэш-хроматографии (элюент: CH2Cl2) с получением промежуточного соединения 22-1 Промежуточное соединение 22-2 получали таким же способом, что и промежуточное соединение 19-1, используя промежуточные соединения 22-1 и 8-b в качестве исходного вещества. Стадии 3, 4, 5, 6 и 7. Синтез 3-3-(4-(трет-бутилдифенилсилилокси)бутил)-6-фтор-3H-имидазо[4,5b]пиридин-2-ил)метил)-1-циклопропил-1H-имидазо[4,5-с]пиридин-2(3H)-она 22-7 Промежуточное соединение 22-7 получали таким же способом, что и промежуточное соединение 19-5 в 5-стадийном синтезе, используя промежуточное соединение 22-2 в качестве исходного вещества. Соединение 22 получали таким же способом, как соединение 19, используя промежуточное соединение 22-7 в качестве исходного вещества. Промежуточное соединение 25-1 получали таким же способом, что и промежуточное соединение 71, используя коммерчески доступный 2-хлор-3-нитропиридин (CAS 5470-18-8) и изопентиламин (CAS 107-85-7). Промежуточное соединение 25-2 получали таким же способом, что и промежуточное соединение 72, используя промежуточные соединения 25-1 в качестве исходного вещества. Промежуточное соединение 25-2 (17,5 г, 97,6 ммоль) растворяли в уксусной кислоте (220 мл). К полученной в результате смеси добавляли за один раз метил 2,2,2-трихлорацетимидат (CAS 2533-69-9)(12,13 мл, 97,6 ммоль). Полученную в результате смесь перемешивали при 50 С в течение 48 ч. Смесь оставляли охлаждаться до комнатной температуры и выливали в раствор лед/вода. рН доводили до рН 5 путем добавления карбоната натрия. Полученную в результате смесь экстрагировали дихлорметаном(3100 мл). Объединенные органические слои последовательно промывали насыщенным NaHCO3, сушили над MgSO4 и упаривали. Остаток очищали колоночной хроматографией, используя CH2Cl2 к
МПК / Метки
МПК: A61P 31/12, C07D 471/04, A61K 31/437
Метки: респираторного, средств, вируса, синцитиального, качестве, противовирусных, отношении, азабензимидазолы
Код ссылки
<a href="https://eas.patents.su/30-21613-azabenzimidazoly-v-kachestve-protivovirusnyh-sredstv-v-otnoshenii-respiratornogo-sincitialnogo-virusa.html" rel="bookmark" title="База патентов Евразийского Союза">Азабензимидазолы в качестве противовирусных средств в отношении респираторного синцитиального вируса</a>
Морфолинилсодержащие бензимидазолы в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 9876
Опубликовано: 28.04.2008
Авторы: Мейер Кристоф, Тиммерман Филип Мария Марта Берн, Виллебрордс Руди Эдмонд, Бонфанти Жан-Франсуа, Андрис Кунрад Йозеф Лодевейк, Фортэн Жером Мишель Клод, Мюллер Филипп, Дубле Фредерик Марк Морис, Жевер Том Валериус Жозефа
МПК: C07D 413/14, C07D 401/06, A61K 31/4184...
Метки: ингибиторов, морфолинилсодержащие, качестве, респираторно-синцитиального, вируса, бензимидазолы, репликации
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксиды, аддитивные соли, четвертичные амины, комплексы с металлами и стереохимически изомерные формы, где G представляет собой прямую связь или C1-10алкандиил, необязательно замещенный одним или несколькими заместителями, индивидуально выбранными из группы заместителей, состоящей из гидрокси, C1-6алкилокси, Ar1C1-6алкилокси, C1-6алкилтио, Ar1C1-6алкилтио, НО(-CH2-CH2-O)n-, C1-6алкилокси(-CH2-CH2-O)n- или...
Производные бензимидазола и имидазопиридина в качестве ингибиторов репликации респираторно-синцитиального вируса

Номер патента: 4746
Опубликовано: 26.08.2004
Авторы: Янссенс Франс Эдуард, Андрис Кунрад Йозеф Лодевейк Марсель, Лякрамп Жан Фернан Арман, Гийемон Жером Эмиль Жорж, Вене Марк Гастон
МПК: A61K 31/4709, A61P 11/00, C07D 401/14...
Метки: репликации, вируса, ингибиторов, бензимидазола, производные, качестве, имидазопиридина, респираторно-синцитиального
Формула / Реферат:
1. Соединение формулы его аддитивная соль или стереохимически изомерная форма, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- (a-2); -CH=N-CH=CH- (a-3); -CH=CH-N=CH- (a-4) или -CH=CH-CH=N- (a-5); где каждый атом водорода в...
Ингибиторы репликации респираторно-синцитиального вируса

Номер патента: 4939
Опубликовано: 28.10.2004
Авторы: Янссенс Франс Эдуард, Лякрамп Жан Фернан Арман, Гийемон Жером Эмиль Жорж, Соммен Франсуа Мария, Андрис Кунрад Йозеф Лодевейк Марсель, Мерсман Катлен Петрус Мари-Жозе
МПК: C07D 401/06, A61K 31/501, A61P 31/14...
Метки: респираторно-синцитиального, вируса, репликации, ингибиторы
Формула / Реферат:
1. Применение соединения для получения лекарственного средства для лечения вирусных инфекций, где указанное соединение представляет собой соединение формулы его пролекарство, N-оксид, аддитивную соль, четвертичный амин, комплекс с металлом или стереохимически изомерную форму, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- ...
Ингибиторы репликации респираторно-синцитиального вируса

Номер патента: 5027
Опубликовано: 28.10.2004
Авторы: Андрис Кунрад Йозеф Лодевейк Марсель, Мерсман Катлен Петрус Мари-Жозе, Соммен Франсуа Мария, Янссенс Франс Эдуард
МПК: A61K 31/437, A61P 11/00, C07D 401/12...
Метки: ингибиторы, вируса, респираторно-синцитиального, репликации
Формула / Реферат:
1. Соединение формулы его пролекарство, N-оксид, аддитивная соль, четвертичный амин, комплекс с металлом или стереохимический изомер, где в указанной формуле -a1=a2-a3=a4- представляет двухвалентный радикал формулы -CH=CH-CH=CH- (a-1); -N=CH-CH=CH- (a-2); -CH=N-CH=CH- (a-3); -CH=CH-N=CH- (a-4) или ...
Хинолинкарбоксамиды в качестве противовирусных агентов

Номер патента: 3945
Опубликовано: 30.10.2003
Авторы: Шнуте Марк Э., Такер Джон Алан, Таисривонгс Сувит, Вэйлланкорт Валери А., Стробач Джозеф Волтер, Тернер Стивен Рональд
МПК: C07D 215/16, A61K 31/47, A61P 31/12...
Метки: противовирусных, хинолинкарбоксамиды, агентов, качестве
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где X представляет a) O или b) S; W представляет a) R2, b) NR7R8, c) OR9 или d) SOiR9; R1 представляет a) Cl, b) F, c) Br, d) CN или e) NO2; R2 представляет a) (CH2CH2O)mR10, b) het, где указанный het связан через атом углерода, c) C1-7алкил, который может быть частично ненасыщенным и необязательно замещенным одним или несколькими заместителями, выбранными из группы,...