Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)




























Формула / Реферат
1. Соединение формулы (I)

или их фармацевтически приемлемые соли,
в которых R является Н, C1-C9 алкилом, -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12;
и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, гидрокси, C1-C5 алкил, С2-С5 алкенил, C1-C6 алкокси, трифторметил, нитро, -CO2R12, -С(O)NR12R13, -NR12R13 и -SO2NR12R13;
R1 является Н или C1-C9 алкилом, необязательно замещенным от 1 до 3 заместителей, независимо выбранных из группы, включающей метил, этил, трифторметил и галоген;
R2 является R19; -С(О)NR12(CHR12)mC(O)NR12O(CH2)q(C6-C10 арилом); -C(O)NR8(CHR12)mC(O)NR12-(CH2)pOR12; -C(O)NR12(CHR12)mS(C1-C4 алкилом) или -Z3-R7, где р равно от 0 до 2; m равно от 1 до 6 и q равно 1 или 2;
R7 является С6-С10 арилом или 5-10 членным гетероциклилом, где указанные R7 группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, трифторметил, циано, нитро, -CO2R12, C1-C4 алкокси, -OC(O)(C1-C4 алкил), -NR12C(O)(C1-C4 алкил), -C(O)NH2, -C(O)NHOH, -С(О)О(C1-C4 алкил), C1-C4 алкил, -S(O)nR12, где n равно от 0 до 2, бензоил, -NR12R13, -OR12, C1-C6 алканоил, -Y1-(C6-C10 арил), -С(O)O(С6-С10 арил), -NН(С6-С10 арил), -С(О)NН(С6-С10 арил), -С(O)NR12O(CH2)n(C6-C10 арил), где n равно от 1 до 3 и -SO2NН(С6-С10 арил);
R8 является Н или C1-C6 алкилом;
R12 и R13 каждый независимо являются Н или C1-C4 алкилом;
R19 является фенилом, нафтилом, пирролилом, фуранилом, тиенилом, оксазолилом, пиридинилом, пиримидинилом, пиридазинилом, хинолинилом, изохинолинилом, 5,6,7,8-тетрагидрохинолинилом или 5,6,7,8-тетрагидроизохинолинилом;
где указанные R19 группы, за исключением указанного фенила, необязательно замещены от 1 до 3 группами заместителей R23 и где указанный фенил необязательно замещен от 1 до 3 группами заместителей, независимо выбранных из R23 и R24;
R23 каждый независимо выбирают из группы, включающей галоген, C1-C6 алкил, C1-C7 алкокси, C2-C6 алкилендиокси, трифторметил, -NR12R13, нитро, -С(NR12)NR12R13, -С(O)NR12R13C(О)R12, -C(NOR12)R13, -С(NCN)NR12R13, -C(NCN)SR12, -(CH2)m(CN), где m равно от 0 до 3, гидрокси, -С(О)R12, -С(O)NR12OR13, -C(O)NR12NR12R13, -ОС(О)NR12R13, -NR12C(О)R12, -С(О)С(О)NR12R13,
-CO2R12, -SO2R12, -SO2NR12R13, -C(O)NR12R13, -NR12SO2R13 и -NR12C(O)NR12R13;
R24 каждый независимо выбирают из группы, включающей имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, тиазолил, оксазолидинил, тиазолидинил и имидазолидинил, где каждый R24 необязательно замещен от 1 до 3 группами заместителей R23;
Y1 является О или S; и
Z3 выбирают из группы, включающей, в сущности, -NR12-, -(CH2)m-, -CH2C(O)NH-, -NHCH2C(O)-, -CH2C(Y1)CH2-, -СН=СН-, -С(О)С-, -CH(Y1)H-, -CH(Y1)-, -CH2C(Y1)-, -C(Y1)CH2-, -C(Y1)C(Y1)-, -CH2NR12-, -CH2-Y1-, -C(Y1)NR8(CHR12)n-, -NR8C(Y1)(CHR12)n-, -NHCH2-, -Y1-CH2-, -SOCH2-, -CH2SO-, -SO2CH2-, -CH2-SO2-, -OC(Y1)-, -N=N-, -NHSO2-, -SO2NH-, -C(Y1)C(Y1)NH-, -NHC(O)O-, -OC(O)NH- и -NHC(O)NH-, где для указанных Z3 групп n равно от 0 до 4 и m равно от 1 до 3;
при условии, что
(1) R1 и R не могут быть оба одновременно -Н;
(2) если R2 определен как -Z3-R7, где Z3 является -NHC(O)- и R7 является изоксазолилом, необязательно замещенным (C1-C4) алкилом или незамещенным бензимидазолилом, R1 должен быть этилом; и
(3) если R2 определен как -Z3-R7, где Z3 является -C(O)NH(CH2)n-, где n равно от 0 до 4, или -NHC(O)O-, и где R7 является необязательно замещенным фенилом, R отличен от -(CH2)2-тетразолила и необязательный заместитель на группе R отличен от -CO2R12.
2. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(Y1)NH-, -C(O)CH2-, -NHC(Y1)-, -Y1-CH2-, -ОС(О)-, -СН=СН- или -C(Y1)C(Y1)-; и R7 является необязательно замещенным арильной или гетероарильной группой, выбранной из группы, включающей фенил, пиридил, пиразинил, тиенил, пиримидинил, 2,4-диоксопиримидин-5-ил, изоксазолил, изотиазолил, пиридазинил и 1,2,4-триазинил.
3. Соединение по п.2, в котором R7 является замещенным фенилом, 2,6-дигалогензамещенным фенилом или 3,5-дигалопирид-4-илом.
4. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -С(O)NH(CH2)n- или -NНС(О)(CH2)n-, где n равно от 0 до 1 и R7 является фенилом или пиридилом, каждый из которых необязательно замещен от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, нитро, трифторметил, -СО2СН3, метил, метокси и -С(O)NН2.
5. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(O)NH- и R7 является фенилом или пиридилом, каждый из которых независимо замещен от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, C1-C4 алкил, C1-C4 алкокси, циано, карбокси и -ОС(О)(C1-C4 алкил).
6. Соединение по п.1, в котором R2 является R19, где R19 необязательно замещхэ тиенилом, пиримидинилом или пиридазинилом.
7. Соединение по п.1, в котором R2 является фенилом, замещенным R24, где указанный R24 является оксадиазолилом, тиадиазолилом или тетразолилом, где указанные R24 группы могут быть необязательно замещены C1-C2 алкилом или где R2 является фенилом, замещенным цианометилом, гидрокси или формилом.
8. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(Y1)NH- и R7 является фенилом, тиенилом, пиразинилом, пиримидинилом, изоксазолилом или пиридилом, где каждая из указанных R7 групп может быть необязательно замещена от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, метоксикарбонил, трифторметил, бензоил, ацетил, диметиламино, гидрокси, нитро, метил, циано, метилсульфонил и метилтио.
9. Соединение по п.1, в котором R2 является -C(O)NR8(CHR12)mC(O)NR12(CH2)POR12 или -С(O)NR12(CHR12)mS(C1-C4-алкилом), где R8, R12, m и р такие, как определены в п.1.
10. Соединение по п.1, выбранное из группы соединений, включающей
(3,5-дихлорпиридин-4-ил)амид 1-циклопентил-3-этил-1Н-индазол-6-карбоновой кислоты;
(2,6-дихлорфенил)амид 1-циклопентил-3-этил-1H-индазол-6-карбоновой кислоты;
(3,5-дихлорпиридин-4-ил)амид 1-циклобутил-3-этил-1Н-индазол-6-карбоновой кислоты;
(3,5-дихлорпиридин-4-ил)амид 3-этил-1-изопропил-1Н-индазол-6-карбоновой кислоты;
(3,5-дихлорпиридин-4-ил)амид 1-циклопропилметил-3-этил-1Н-индазол-6-карбоновой кислоты;
(3,5-дихлорпиридин-4-ил)амид 1-циклогексил-3-этил-1Н-индазол-6-карбоновой кислоты;
(3,5-дихлорпиридин-4-ил)амид 3-этил-1-(4-фторфенил)-1Н-индазол-6-карбоновой кислоты;
гидроксикарбамоилметиламид 1-циклопентил-3-этил-1Н-индазол-6-карбоновой кислоты;
(метилсульфанилэтил)амид 1-циклопентил-3-этил-1Н-индазол-6-карбоновой кислоты;
гидроксикарбамоилметилметиламид 1-циклопентил-3-этил-1H-индазол-6-карбоновой кислоты;
(1-бензилоксикарбамоилэтил)амид S-1-циклопентил-3-этил-1H-индазол-6-карбоновой кислоты;
(1-гидроксикарбамоилэтил)амид R-1-циклопентил-3-этил-1H-индазол-6-карбоновой кислоты;
1-циклопентил-3-этил-6-тиофен-2-ил-1Н-индазол;
1-циклопентил-3-этил-6-фенил-1Н-индазол; и
фармацевтически приемлемая соль любого из перечисленных выше соединений.
11. Соединение формулы (ХХХХ)

или их фармацевтически приемлемые соли,
где R является Н; C1-C9 алкилом; -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2; или (Z1)b(Z2)c(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или С2-С8 алкениленом и Z2 является О, S, SО2 или NR12;
и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген, гидрокси, C1-C5 алкил, С2-С5 алкенил, C1-C6 алкокси, трифторметил, нитро, -CO2R12, -С(O)NR12R13, -NR12R13 и -SO2NR12R13;
R1 является Н или C1-C9 алкилом, необязательно замещенным от 1 до 3 заместителями, независимо выбранными из группы, включающей метил, этил, трифторметил и галоген;
R12 и R13 каждый независимо являются Н или C1-C4 алкилом; и
X является бромом, -C(O)O(C1-C6 алкилом), карбокси, -CH2OH или -С(O)Сl.
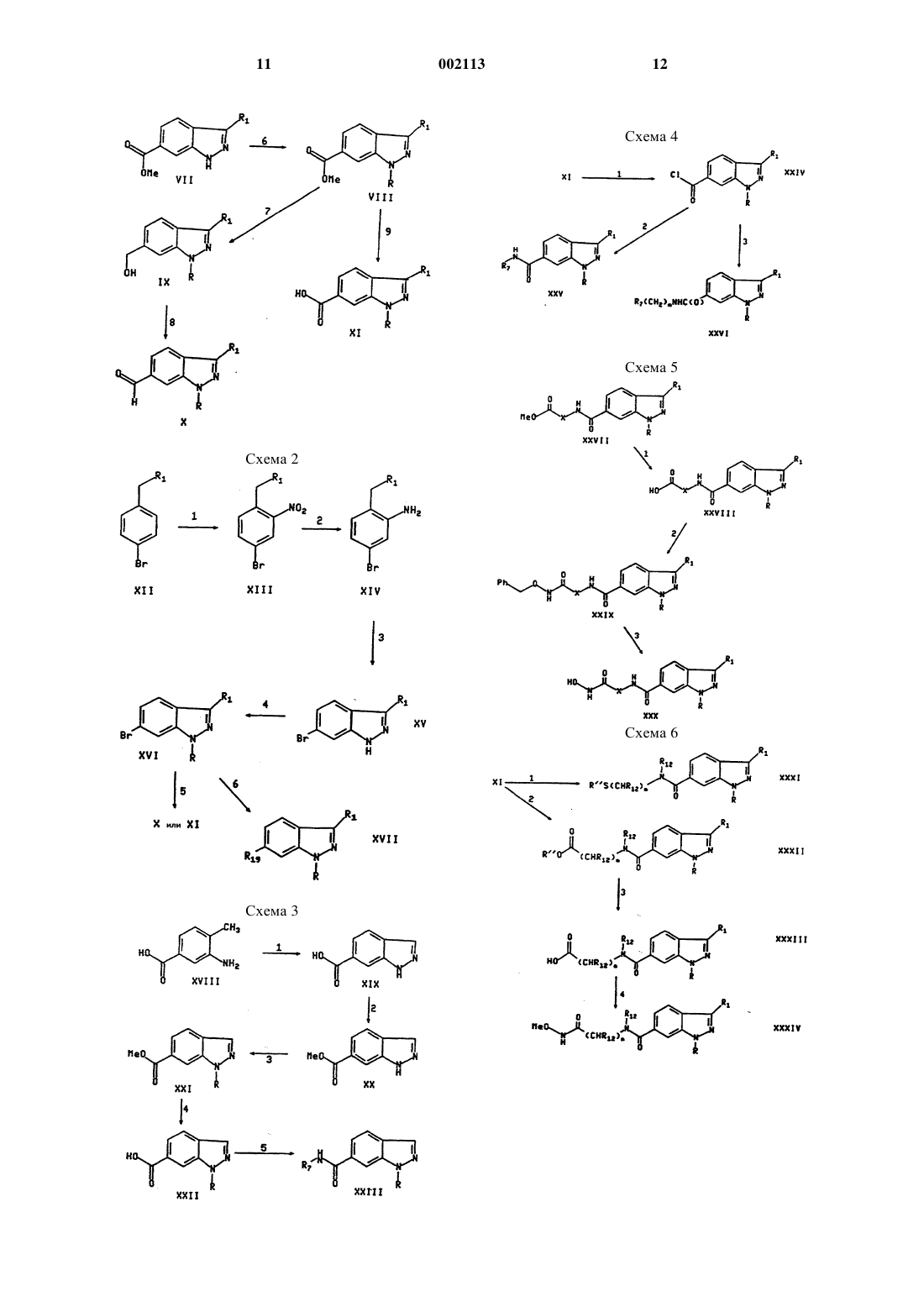
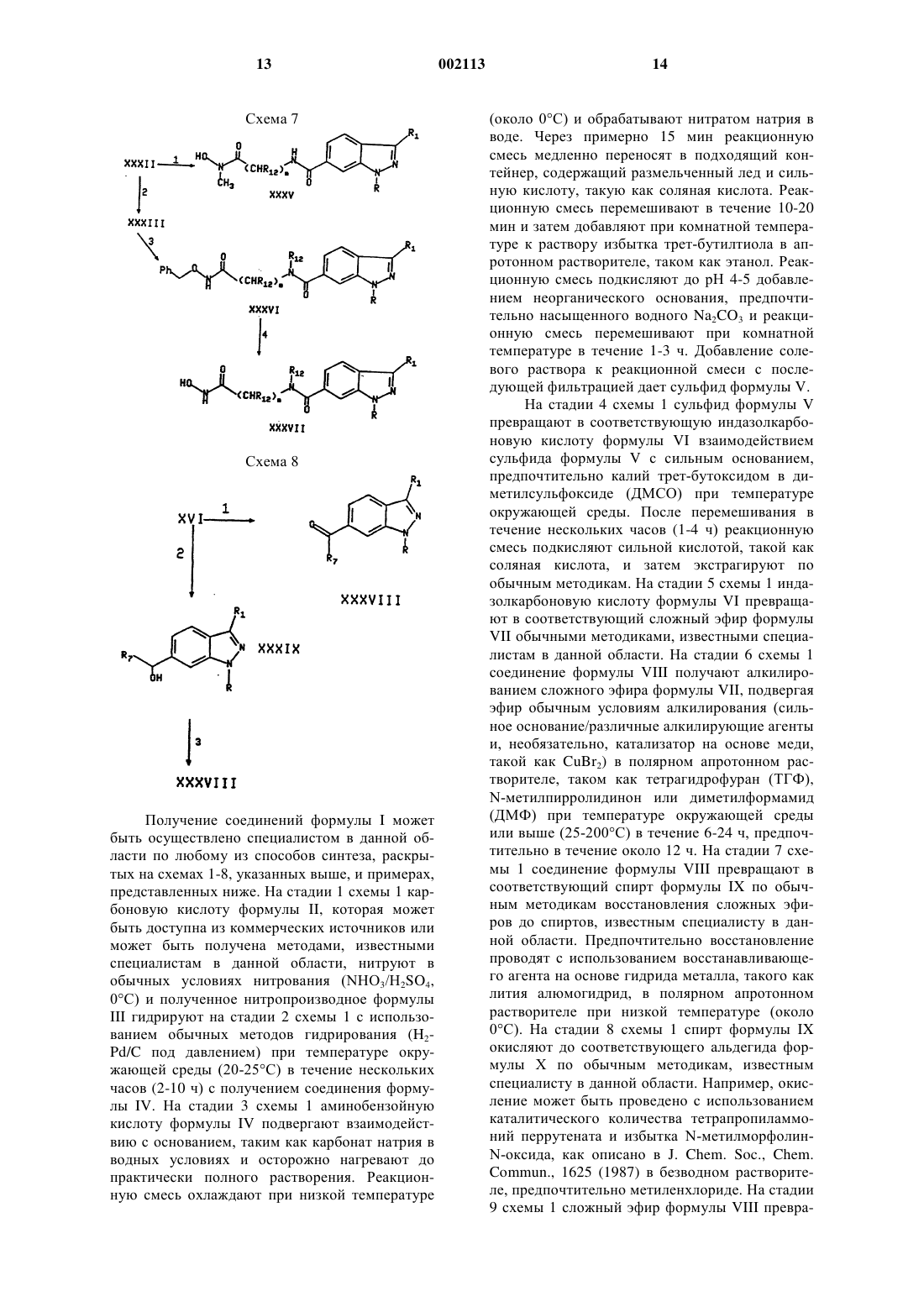
Текст
1 Данное изобретение относится к аналогам индазола. Соединения являются селективными ингибиторами фосфодиэстеразы (ФДЭ) типа IV и продуцирования фактора некроза опухоли(ФНО) и таким образом используются при лечении астмы, артритов, бронхитов, хронической обструкции дыхательных путей, псориаза, аллергических ринитов, дерматитов и других воспалительных заболеваний, расстройств центральной нервной системы, таких как депрессии и мультиинфарктное слабоумие, СПИД, септический шок и других заболеваний, включающих образование ФНО. Изобретение также относится к способу использования таких соединений при лечении указанных выше заболеваний у млекопитающих, особенно у человека, и к фармацевтическим композициям, содержащим такие соединения. В следующих ниже заявках на патенты США также раскрыты и заявлены производные индазола, которые являются селективными ингибиторами ФДЭ типа IV и продукции ФНО: заявка США 60/021,072 от 27 июня 1996; заявка США 60/020,385 от 25 июня 1996 и заявка США 60/016,861 от 3 мая 1996. Вышеуказанные заявки на патент США включены сюда в качестве ссылок. С тех пор как было открыто, что циклический аденозинфосфат (АМФ) является вторичным внутриклеточным мессенджером (E.W.Sutherland and Т.W. Rall, Pharmacol Rev., 12, 265,(1960, ингибирование фосфодиэстеразы стало целью для модулирования и соответственно терапевтического вмешательства в ряд болезненных процессов. Не так давно были открыты определенные классы ФДЭ (J.A. Beavo et al.,Trends in Pharm. Sci (TIPS), 11, 150, (1990, и их селективное ингибирование привело к улучшению лекарственной терапии (C.D. Nicholson,M.S. Hahid, TIPS, 12, 19, (1991. Более конкретно, было обнаружено, что ингибирование ФДЭ типа IV может привести к подавлению высвобождения медиаторов воспаления (M.W.Verghese et al., J. Mol. Cell Cardiol, 12 (Suppl II),S61, (1989 и способствовать расслаблению гладких мышц дыхательных путей (T.J. Torphy вO'Donnell and C.G.A. Persson, 1988, 37 Birkhauser-Vertag). Таким образом, соединения, которые ингибируют ФДЭ типа IV, но которые имеют незначительную активность в отношении других типов ФДЭ, могут ингибировать высвобождение медиаторов воспаления и способствовать расслаблению гладких мышц дыхательных путей, не оказывая влияния на сердечнососудистую систему и не оказывая антитромбоцитного действия. Также обнаружено, что ингибиторы ФДЭ типа IV полезны при лечении несахарного диабета (Kidney Int. 37:362, 1990;Kidney Int. 35:494) и расстройств центральной нервной системы, таких как депрессии и мультиинфарктное слабоумие (международная заяв 002113 2 ка РСТ WO 92/19594 (дата публикации 12 ноября 1992. Обнаружено, что ФНО вовлечен во многих инфекционных и аутоиммунных заболеваниях(W. Friers, Fed. of Euro. Bio. Soc. (FEBS) Letters,285, 199 (1991. Более того, показано, что ФНО является основным медиатором воспалительных реакций, имеющих место при сепсисе и септическом шоке (С.Е. Spooner et al., Clinical Immunology and Immunopatology, 62, S11, (1992). Описание изобретения Данное изобретение относится к соединениям формулы I и их фармацевтически приемлемым солям, в которыхb и с независимо равны 0 или 1, Z является С 1 С 6 алкиленом или С 2-С 6 алкениленом и Z2 является О, S, SO2 или NR12 и в котором R группы необязательно замещены 1-3 заместителями,независимо выбранными из группы, включающей галоген, гидрокси С 1-С 5 алкил, С 2-С 5 алкенил, С 1-С 6 алкокси, трифторметил, нитро,-CO2R12, -C(O)NR12R13, -NR12R13 и -SO2NR12R13;R1 является Н, С 1-С 9 алкилом, С 2-С 3 алкенилом или фенилом, где указанные алкильная,алкенильная и фенильная группы R1 не обязательно замещены 1-3 заместителями, независимо выбранными из группы, включающей метил,этил, трифторметил и галоген; где в указанных формулах (Iа-Ii) структуры формул (If) и (Ig) присоединены к формуле I через атомы углерода в 5, 6 и 7 положениях указанных групп формул (If) и (Ig), пунктирные линии в формулах (Ic) и (Id) означают одинарную связь или двойную связь, за исключением того, что R6 отсутствует в формулах (Iс) и (Id),когда указанные пунктирные линии означают двойную связь, n равно 0-2, р равно 0-6 и m равно 0-1;R4 и R5 независимо выбирают из группы,включающей Н, этил, -СО 2 Н и -C(O)NHOH;R7 группы необязательно замещены 1-3 заместителями, независимо выбранными из галогена,трифторметила, циано, нитро, -CO2R12, С 1-С 4 алкокси, -OC(O)(С 1-С 4 алкила), -NR12C(O)(С 1-С 4 алкила), -C(O)NH2, -C(O)NHOH, -C(O)O(C1-C4 алкила), C1-C4 алкила, -S(O)nR12, где n равно 0-2,бензоила, -NR12R13, -OR12, С 1-С 5 алканоила, -Y1(C6-C10 арила), -С(О)О(С 6-С 10 арила), -NH(C6-C10 арила), -С(О)NH(C6-C10 арила), -С(О)NR12R14 является метилом или фенилом;NR12R13, при условии, что если R17 является гидрокси, то R13 является Н или C1-C4 алкилом; каждый R18 независимо является Н, фтором, циано или C1-C4 алкилом, где указанный метил необязательно замещен 1-3 фторзаместителями; или R17 и R18 вместе образуют оксо (=O) часть;R19 является фенилом, нафтилом, пирролилом, фуранилом, тиенилом, оксазолилом, пиридинилом, пиримидинилом, пиридазинилом, хинолинилом, изохинолинилом, 5,6,7,8-тетрагидрохинолинилом или 5,6,7,8-тетрагидроизохинолинилом, где указанные R19 группы, за исключением указанного фенила, необязательно замещены 1-3 заместителями R23 и где указанная фенильная группа R19 необязательно замещена 1-3 заместителями, независимо выбранными из R23 и R24;NR12R13; каждый R24 независимо является имидазолилом, пиразолилом, триазолилом, тетразолилом, оксазолилом, изоксазолилом, оксадиазолилом, тиадиазолилом, тиазолилом, оксазолидинилом, тиазолидинилом или имидазолидинилом, где каждый из вышеупомянутых R24 замес 5 тителей необязательно замещен 1-3 R23 заместителями;-(CH2)mY4(фенилом), где m равно 0-4 и фенильная часть указанной -(CH2)mY4(фенильной) R26 группы необязательно замещена галогеном,-OR12, C1-C6 алканоилокси, C6-C10 арилокси,-NR12R13, -NН(C6-C10 арилом) или -NHC(O)(C1C4 алкилом); каждый R27 независимо является галогеном, -(CH2)pNR12C(O)CH3, где р равно 1-5, нит-CO2R12,-OR12,ро,циано,-NR12R13,-С(Y1)NR12R13, -NR12C(NCN)S(C1-C3 алкилом),-NR12C(NCN)NR12R13, -NR12C(O)NR12R13,-NR12C(O)C(O)NR12R13, -C(=NR12)NR12R13,-S(O)mСН 3, где m равно 0-2, -C(=NR12)S(C1-С 3 алкилом), -NR12SO2 (C1-С 3 алкилом), -OC(O)R12,-ОС(О)NR12R13, -NR12SO2CF3,-NR12C(O)C(O)OR12, -NR12C(O)R12,-NR12C(O)OR12, имидазолилом, тиазолилом,оксазолилом, пиразолилом, триазолилом или тетразолилом;R28 является Н, фтором, циано или C1-C2 алкилом, где указанный алкил необязательно замещен 1-3 заместителями, независимо выбранными из галогена, -С(О)NR12R13 иR29 является фенилом, необязательно замещенным 1-2 заместителями, независимо выбранными из -NR12R13, нитро, галогена, -OR12,-NНR30, -NR30R31 и -C(O)OR12; каждый R30 и R31 независимо является C1 С 3 алкилом или С 2-С 3 алкенилом;R32 является пиридин-4-илом, необязательно замещенным 1 или 2 заместителями, независимо выбранными из галогена и C1-C4 алкила; каждый А независимо является C1-C6 алкилом, пиридилом, морфолинилом, пиперидинилом, имидазолилом, тиенилом, пиримидилом,тиазолилом, триазолилом, хинолинилом, фенилом или нафтилом, где вышеуказанные А группы необязательно замещены 1-3 заместителямиY3 является связью или -СН=СН-;-NHC(O)NH-, где в указанных Z1 частях n равно 0-4 и m равно 1-3. Данное изобретение также относится к соединениям формулы и их фармацевтически приемлемым солям, гдеR и R1 такие как определено выше и Х является-С(O)Сl. Соединения, описываемые указанной выше формулой, являются промежуточными соединениями и используются при получении соединений формулы I. Определенные соединения формулы I включают те, в которых R2 является -Z3-R7, гдеR7 является необязательно замещенным арильной или гетероарильной группой, выбранной из фенила, пиридила, пиразинила, тиенила, пиримидила, 2,4-диоксопиримидин-5-ила, изоксазолила, изотиазолила, пиридазинила и 1,2,4 триазинила. Более конкретно R7 является замещенным фенилом, 2,6-дигалозамещенным фенилом или 3,5-дигалопирид-4-илом. Другие конкретные соединения формулы I включают те, в которых R2 является -Z3-R7, где(СН 2)n-, где n равно 0-1 и R7 является фенилом или пиридилом, необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, нитро, трифторметила, -СО 2 СН 3, метила,метокси и -C(O)NH2. Другие конкретные соединения формулы I включают те, в которых R2 является -Z3-R7, гдеZ3 является -C(O)NH- и R7 является фенилом или пиридилом, необязательно замещенным 1-3 заместителями, независимо выбранными из галогена, C1-C4 алкила, C1-C4 алкокси, циано, карбокси и -ОС(О)(C1-C4 алкила). Другие конкретные соединения формулы I включают те, в которых R2 является R19, где R19 является необязательно замещенным пиримидинилом или необязательно замещенным пиридазинилом. Другие конкретные соединения формулы I включают те, в которых R2 является заместителем формулы (Ih), где R20 является -C(O)R21,-C(O)C(O)R21 или 7 где R21 является -OR22, -NHR22 или -NHOH и R22 является Н, C1-С 3 алкилом, бензилом или пиридилметилом. Другие конкретные соединения формулы I включают те, в которых R2 является фенилом,замещенным R24, где указанный R24 является оксадиазолилом, тиадиазолилом или тетразолилом, где указанные R24 группы необязательно замещены C1-C2 алкилом, или где R2 является фенилом, замещенным цианометилом, гидрокси или формилом. Другие конкретные соединения формулы I включают те, в которых R2 является -Z3-R7, гдеZ3 является -C(Y1)NH-, и R7 является фенилом,пиразинилом, пиримидинилом, изоксазолилом или пиридилом, где указанные R7 группы необязательно замещены 1-3 заместителями, независимо выбранными из галогена, метоксикарбонила, трифторметила, бензоила, ацетила, диметиламино, гидрокси, нитро, метила, циано,метилсульфонила и метилтио. Другие конкретные соединения формулы I включают те, в которых R2 является-(СН 2)m(фенилом), где m равно 1-4 и указанная фенильная часть необязательно замещена галогеном, гидрокси, ацетокси, амино или ацетамидо. Другие конкретные соединения формулы I включают те, в которых R2 являетсяR10 является амино, гидрокси, метокси или гидроксиамино. Другие конкретные соединения формулы I включают те, в которых R2 являетсяC1-C4 алкилом. Другие конкретные соединения формулы I включают те, в которых R2 является заместителем формулы (Ic), (Id) или (Iе), где в формулах(Ic) и (Id) R6 является Н, гидрокси, C1-C4 алкилом или C1-C4 алкокси. Другие конкретные соединения формулы I включают те, в которых R2 является-С(О)NR12(CHR12)mS(C1-C4 алкилом), где R8, R12,m и р такие, как определено выше. Конкретные соединения формулы I включают выбранные из группы, включающейR-1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты; 1-циклопентил-3-этил-6-тиофен-2-ил-1 Ниндазол; 1-циклопентил-3-этил-6-фенил-1 Н-индазол; и фармацевтически приемлемые соли вышеуказанных соединений. Данное изобретение далее относится к фармацевтической композиции для ингибирования фосфодиэстеразы (ФДЭ) типа IV или продукции фактора некроза опухоли (ФНО),включающей фармацевтически эффективное количество соединения формулы I, как определено выше, или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель. Данное изобретение также относится к способу ингибирования фосфодиэстеразы(ФДЭ) типа IV или продукции фактора некроза опухоли (ФНО) введением пациенту эффективного количества соединения формулы I, как определено выше, или его фармацевтически приемлемой соли. Данное изобретение также относится к фармацевтической композиции для профилактики или лечения астмы, воспаления суставов,ревматоидного артрита, подагрического артрита, ревматоидного спондилита, остеоартрита и других состояний артрита; сепсиса, септического шока, эндотоксического шока, грамотрицательного сепсиса, синдрома токсического шока, острого респираторного дистресссиндрома, церебральной малярии, хронического воспаления легких, силикоза, саркоидоза лег 9 ких, резорбции костей, реперфузионных повреждений, реакции на введение имплантата, отторжения аллотрансплантантов, лихорадки и миалгии, возникающих вследствие инфекции,такой как грипп, кахексии вследствие инфекции или злокачественного развития болезни, кахексии вследствие синдрома приобретенного иммунодефицита (СПИД), СПИДа, ВИЧ, ССК(СПИД связанного комплекса), образования келоидов, рубцов на тканях, болезни Крона, язвенного колита, пиреза, рассеянного склероза,сахарного диабета типа I, несахарного диабета,аутоиммунного диабета, системной красной волчанки, бронхитов, хронической обструкции дыхательных путей, псориаза, болезни Бекета,нефрита с аллергической пурпурой, хронического гломерулонефрита, воспаления кишечника, лейкемии, аллергического ринита, дерматита, депрессии или мультиинфарктного слабоумия, включающей эффективное количество соединения формулы I, как определено выше,или его фармацевтически приемлемой соли вместе с фармацевтически приемлемым носителем. Данное изобретение также относится к способу лечения или профилактики вышеуказанных определенных заболеваний и состояний введением пациенту эффективного количества соединения формулы I, как определено выше,или его фармацевтически приемлемой соли. Термин "галоген" в данном случае, если не указано иначе, означает фтор, хлор, бром или йод. Предпочтительными группами галогена являются фтор, хлор и бром. Термин "алкил" в данном случае, если не указано иначе, означает насыщенный одновалентный углеводородный радикал, имеющий прямую, циклическую или разветвленную структуру. Понятно, что если предполагается циклическая структура, в указанном алкиле должно присутствовать, по крайней мере, три атома углерода. Такие циклические структуры включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Термин "алкокси" в данном случае, если не указано иначе, означает -O-алкильную группу, в которой алкил такой, как определено выше. Термин "алканоил" в данном случае, если не указано иначе, означает -С(О)-алкильную группу, в которой алкил такой, как определено выше. Термин "арил" в данном случае, если не указано иначе, означает органический радикал,полученный из ароматического углеводорода удалением одного атома водорода, такой как фенил или нафтил. Термин "5-10-членный гетероциклил" в данном случае, если не указано иначе, означает ароматические и неароматические гетероциклические группы, содержащие один или более гетероатомов, каждый выбранный из О, S и N, где каждая гетероциклическая группа имеет от 5 до 10 10 атомов в кольце. Гетероциклические группы включают бенз-конденсированные кольца и кольца, замещенные одной или более оксогруппами. Примером 5-членной гетероциклической группы является тиазолил и примером 10 членной гетероциклической группы является хинолинил. Примерами неароматических гетероциклических групп являются пирролидинил,пиперидино, морфолино, тиоморфолино и пиперазинил. Примерами ароматических гетероциклических групп являются пиридинил, имидазолил, пиримидинил, пиразолил, тиазолил,пиразинил, тетразолил, фурил, тиенил, изоксазолил и тиазолил. Гетероциклические группы,имеющие конденсированное бензольное кольцо включают бензимидазолил. Термин "гетероарил" в данном случае, если не указано иначе, означает ароматические гетероциклические группы, в которых гетероциклы такие как определено выше. Фраза "фармацевтически приемлемая соль(соли)" в данном случае, если не указано иначе,означает соли кислотных и основных групп,которые могут присутствовать в соединениях формулы I. Определенные соединения формулы I могут иметь асимметрические центры и поэтому существовать в различных энантиомерных формах. Данное изобретение относится к использованию всех оптических изомеров и стереоизомеров соединений формулы I и их смесей. Соединения формулы I могут также существовать в виде таутомеров. Данное изобретение относится к использованию всех таких таутомеров и их смесей. Подробное описание изобретения Следующие схемы реакций 1-8 иллюстрируют получение соединений данного изобретения. В следующих схемах, если не указано иначе, R, R1, R7, R8 и R12 такие как определено выше. В следующих схемах "Me" означает метил и Получение соединений формулы I может быть осуществлено специалистом в данной области по любому из способов синтеза, раскрытых на схемах 1-8, указанных выше, и примерах,представленных ниже. На стадии 1 схемы 1 карбоновую кислоту формулы II, которая может быть доступна из коммерческих источников или может быть получена методами, известными специалистам в данной области, нитруют в обычных условиях нитрования (NНО 3/Н 2SO4,0 С) и полученное нитропроизводное формулыIII гидрируют на стадии 2 схемы 1 с использованием обычных методов гидрирования (H2Pd/C под давлением) при температуре окружающей среды (20-25 С) в течение нескольких часов (2-10 ч) с получением соединения формулы IV. На стадии 3 схемы 1 аминобензойную кислоту формулы IV подвергают взаимодействию с основанием, таким как карбонат натрия в водных условиях и осторожно нагревают до практически полного растворения. Реакционную смесь охлаждают при низкой температуре(около 0 С) и обрабатывают нитратом натрия в воде. Через примерно 15 мин реакционную смесь медленно переносят в подходящий контейнер, содержащий размельченный лед и сильную кислоту, такую как соляная кислота. Реакционную смесь перемешивают в течение 10-20 мин и затем добавляют при комнатной температуре к раствору избытка трет-бутилтиола в апротонном растворителе, таком как этанол. Реакционную смесь подкисляют до рН 4-5 добавлением неорганического основания, предпочтительно насыщенного водного Na2CO3 и реакционную смесь перемешивают при комнатной температуре в течение 1-3 ч. Добавление солевого раствора к реакционной смеси с последующей фильтрацией дает сульфид формулы V. На стадии 4 схемы 1 сульфид формулы V превращают в соответствующую индазолкарбоновую кислоту формулы VI взаимодействием сульфида формулы V с сильным основанием,предпочтительно калий трет-бутоксидом в диметилсульфоксиде (ДМСО) при температуре окружающей среды. После перемешивания в течение нескольких часов (1-4 ч) реакционную смесь подкисляют сильной кислотой, такой как соляная кислота, и затем экстрагируют по обычным методикам. На стадии 5 схемы 1 индазолкарбоновую кислоту формулы VI превращают в соответствующий сложный эфир формулыVII обычными методиками, известными специалистам в данной области. На стадии 6 схемы 1 соединение формулы VIII получают алкилированием сложного эфира формулы VII, подвергая эфир обычным условиям алкилирования (сильное основание/различные алкилирующие агенты и, необязательно, катализатор на основе меди,такой как CuBr2) в полярном апротонном растворителе, таком как тетрагидрофуран (ТГФ),N-метилпирролидинон или диметилформамид(ДМФ) при температуре окружающей среды или выше (25-200 С) в течение 6-24 ч, предпочтительно в течение около 12 ч. На стадии 7 схемы 1 соединение формулы VIII превращают в соответствующий спирт формулы IX по обычным методикам восстановления сложных эфиров до спиртов, известным специалисту в данной области. Предпочтительно восстановление проводят с использованием восстанавливающего агента на основе гидрида металла, такого как лития алюмогидрид, в полярном апротонном растворителе при низкой температуре (около 0 С). На стадии 8 схемы 1 спирт формулы IX окисляют до соответствующего альдегида формулы Х по обычным методикам, известным специалисту в данной области. Например, окисление может быть проведено с использованием каталитического количества тетрапропиламмоний перрутената и избытка N-метилморфолинN-оксида, как описано в J. Chem. Soc., Chem.Commun., 1625 (1987) в безводном растворителе, предпочтительно метиленхлориде. На стадии 9 схемы 1 сложный эфир формулы VIII превра 15 щают в соответствующую кислоту формулы XI по методикам, известным специалисту в данной области, таким как обработка исходного соединения гидроксидом натрия в метаноле и нагревание смеси с обратным холодильником в течение нескольких часов (2 или более часа). Кислота формулы XI, как и альдегид формулы Х являются полезными промежуточными соединениями для получения различных соединений формулы I. Схема 2 иллюстрирует альтернативный способ получения альдегида формулы Х и кислоты формулы XI, а также способ получения соединения формулы XVII. На стадии 1 схемы 2 соединение формулы XII нитруют с использованием обычных условий нитрования (азотная и серная кислоты) с получением соединения формулы XIII. На стадии 2 схемы 2 нитропроизводное соединение формулы XIII восстанавливают до соответствующего амина формулы XIV по обычным методикам, известным специалисту в данной области. Предпочтительно соединение формулы XIII восстанавливают до амина формулы XIV с использованием безводного двухлористого олова в безводном апротонном растворителе, таком как этанол. На стадии 3 схемы 2 амин формулы XIV превращают в соответствующий индазол формулы XV, получая соответствующий диазоний тетрафторборатов как описано в A. Roe, Organic Reactions, Vol. 5,Wiley, New York, 1949, pp. 198-206, с последующей циклизацией с использованием катализатора фазового переноса как описано в R.A. Ваtrisch and I/W/Yanq, J. Het. Chem. 21, 1063(1984). На стадии 4 схемы 2 алкилирование соединения формулы XV проводят с использованием стандартных методик, известных специалисту в данной области (например, сильное основание, полярный апротонный растворитель и галоидный алкил) с получением N-алкилированного соединения формулы XVI. На стадии 5 схемы 2 соединение формулы XVI подвергают обмену металл-галоген с использованием алкиллития, такого как н-бутиллитий, в полярном апротонном растворителе, таком как ТГФ, при низкой температуре (-50-100 С (предпочтительно -78 С с последующим гашением ДМФ при низкой температуре и нагреванием до температуры окружающей среды с получением альдегидного промежуточного соединения формулы Х или смесь, содержащую соединение формулы XVI, гасят СО 2, нагревают до температуры окружающей среды и затем гасят кислотой, такой как соляная кислота с получением кислоты формулы XI. На стадии 6 схемы 2 соединение формулы XVI превращают в соединение формулы I, в которой R2 является R19, который, как определено выше, представляет собой арил, гетероарил или гетероциклический фрагмент. На стадии 6 схемы 2 соединение формулыXVII получают взаимодействием соединения формулы XVI с соединением формулы R19 002113B(OH)2, где R19 такой, как определено выше, в присутствии Рd(РРh3)4 в водном Na2CO3 при нагревании с обратным холодильником в течение 4 ч. Схема 3 иллюстрирует получение соединения формулы I, где R1 является Н и R2 является -C(O)NHR7, где R7 такой, как определено выше. На стадии 1 схемы 3 соединение формулы XVIII обрабатывают эфиратом трифторида бора в хлороформе, не содержащем этанола, при температуре около -20 С. Через короткий период времени, примерно 5 мин, к смеси добавляют трет-бутилнитрит и реакционную смесь перемешивают при температуре около 0 С в течение 2 ч. Затем добавляют ацетат калия с последующим добавлением 18-краун-6 с получением соединения формулы(1 Н-индазол-6 карбоновой кислоты). На стадии 2 схемы 3 соединение формулы XIX обрабатывают концентрированной серной кислотой в метаноле при нагревании с обратным холодильником в течение около 8 ч с последующим перемешиванием при температуре окружающей среды в течение 18 ч с получением соединения формулы XX. На стадии 3 схемы 3 соединение формулы XX подвергают взаимодействию с соединением формулы R-X, в которой R такой как определено выше и Х является уходящей группой, такой как хлор,бром или йод, предпочтительно бром, в присутствии гидрида натрия в ДМФ в течение 10-24 ч,предпочтительно 24 ч, при температуре окружающей среды с получением сложного эфира формулы XXI. На стадии 4 схемы 3 сложный эфир формулы XXI превращают в кислоту формулы XXII по методике стадии 9 схемы 1. На стадии 5 схемы 3 соединение формулы XXII обрабатывают тионилхлоридом и ДМФ (в качестве катализатора) в безводном толуоле при нагревании с обратным холодильником в течение 3 ч с получением соответствующего хлорангидрида. Отдельно соединение формулы R7-NH2,где R7 такой, как определено выше, добавляют к смеси гидрида натрия в безводном ТГФ, который охлаждают до температуры 0 С. К этой второй смеси добавляют упомянутый выше хлорангидрид в ТГФ и смесь перемешивают при температуре окружающей среды в течение 4-24 ч с получением соединения формулы XXIII. Схема 4 иллюстрирует получение соединений формулы I, в которых R2 является-C(O)NHR7 или -С(О)NH(СН 2)nR7, где n равно 14 и R7 такой, как определено выше. На стадии 1 схемы 4 кислоту формулы XI превращают в соответствующий хлорангидрид формулы XXIV обработкой исходного соединения тионилхлоридом и ДМФ (в качестве катализатора) в безводном толуоле при нагревании с обратным холодильником в течение 3 ч. Хлорангидрид формулы XXIV может быть превращен в соединение формулы XXV взаимодействием исходного соединения с соединением формулы R7NH2, где R7 такой, как определено выше, в при 17 сутствии гидрида натрия в безводном ТГФ при температуре около 0 С с последующим нагреванием до температуры окружающей среды в течение 4-24 ч. Этот метод предпочтителен, когда R7 является замещенным или незамещенным пиридинилом. Альтернативно, соединение формулы XXIV может быть превращено в соединение формулы XXV взаимодействием исходного соединения с соединением формулы R7-NH2, гдеR7 такой как определено выше, в присутствии гидрида натрия в безводном ДМФ при температуре окружающей среды в течение 4-24 ч. Этот метод предпочтителен, когда R7 является замещенным или незамещенным пиримидинилом. Альтернативно, соединение формулы XXIV может быть превращено в соединение формулыXXV добавлением соединения формулы R7NH2, в котором R7 такой, как определено выше,к реакционной смеси, в которой получают хлорангидрид, и затем нагреванием смеси до температуры около 200 С за короткий период времени, например около 15 мин. На стадии 3 схемы 4 соединение формулыXXVI, в котором n равно 1-4 и R7 такой, как определено выше, взаимодействием исходного соединения с соединением формулы H2NC(О)NH(СН 2)nR7, где n равно 1-4 и R7 такой, как определено выше, в присутствии триэтиламина и необязательно диметиламинопиридина(ДМАП) в метиленхлориде при температуре окружающей среды в течение 10-48 ч. Альтернативно, соединение формулы XXIV может быть превращено в соединение формулы XXVI взаимодействием исходного соединения с соединением формулы H2N-(CH2)nR7, где n равно 1-4 и R7 такой, как определено выше, в безводном пиридине при температуре 40 С в течение 1 ч. Такая альтернативная методика предпочтительна на стадии 3 схемы 4, когда R7 является азотсодержащей гетероциклической частью,такой как пиридинил. Схема 5 иллюстрирует получение соединений формулы I, в которых R2 является -Z3-R7,где Z3 является -C(O)NH- и R7 является арилом,таким как фенил или нафтил, замещенными-C(O)NHOH. На схеме 5 исходным является соединение формулы XXVII, где Х является арильной частью. Соединение формулы XXVII получают по методике, проиллюстрированной на схеме 4. На стадии 1 схемы 5 соединение формулы XXVII гидриролизуют до соответствующей кислоты формулы XXVIII, что может быть проведено по методикам, известным специалисту в данной области, таким как обработка соединения формулы XXVII гидроксидом натрия в метаноле при нагревании с обратным холодильником в течение от 30 мин до 1 ч. На стадии 2 схемы 5 кислоту формулы XXVIII превращают в соединение формулы XXIX обработкой кислоты гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида и гидрохло 002113 18 ридом O-бензилгидроксиламина в метиленхлориде при температуре окружающей среды в течение 4-24 ч. На стадии 3 схемы 5 соединение формулы XXIX превращают в соединение формулы XXX обработкой исходного соединения 10% Pd/C в этилацетате и метаноле в атмосфереH2 (около 2,109 кг/см 2 (30 пси при температуре окружающей среды в течение от 30 мин до 1 ч. Схема 6 иллюстрирует получение соединений формулыH2N-(CHR12)mSR", где R", R12 и m такие, как определено выше,гидрохлоридом 1-(3 диметиламинопропил)-3-этилкарбодиимида и триэтиламином в метиленхлориде при температуре окружающей среды в течение 18 ч с получением соединения формулы XXXI. На стадии 2 схемы 6 соединение формулы XXXII получают по методике, проиллюстрированной на схеме 4. На стадии 3 схемы 6 соединение формулыXXXIII получают нагреванием с обратным холодильником соединения формулы XXXII в этаноле или метаноле и гидрохлорида натрия в течение 1 ч. На стадии 4 схемы 6 соединение формулы XXXIV получают обработкой соединения формулы XXXIII метоксиламин гидрохлоридом, гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида 1-гидроксибензотриазолгидрата и триэтиламином в метиленхлориде при температуре окружающей среды в течение 18 ч. Схема 7 иллюстрирует получение соединений формулы I, в которых R2 является(соединение формулы XXXVII), где R12 такой,как определено выше, и m равно 1-6. На стадии 1 схемы 7 соединение формулы XXXV получают обработкой соединения формулы XXXII натрием и гидрохлоридом N-метилгидроксиламина в метаноле при температуре окружающей среды в течение 16 ч. На стадии 2 схемы 7 соединение формулы XXXIII получают по методике стадии 3 схемы 6. На стадии 3 схемы 7 соединение формулы XXXVI получают обработкой соединения формулыOбензилгидроксиламином, 1-гидроксибензтриазолгидратом, гидрохлоридом 1-(3-диметиламинопропил)-3-этилкарбодиимида и триэтиламином в метиленхлориде при температуре окружающей среды в течение 18 ч. На стадии 4 схемы 7 соединение формулы XXXVII получают обработкой соединения формулы XXXVI 10% Pd/C в метаноле и этилацетатом в атмосфе 19 ре Н 2 (около 2,109 кг/см 2 (30 пси при температуре окружающей среды в течение от 30 мин до 1 ч. Схема 8 иллюстрирует получение соединений формулы XXXVIII. На стадии 1 схемы 8 соединение формулы XVI, которое получают по методике схемы 2,обрабатывают нбутиллитием в безводном ТГФ при низкой температуре, например -78 С, в течение 30 мин с последующим гашением смеси соединением формулы R7-CN, где R7 такой, как определено выше, и нагреванием смеси до комнатной температуры в течение от 30 мин до 1 ч с получением соединения формулы XXXVII. Этот способ получения соединения формулы XXXVIII предпочтителен для соединений, в которых R7 является азотсодержащей гетероарильной частью. Стадии 2 и 3 схемы 8 иллюстрируют альтернативный способ получения соединения формулы XXXVIII, который предпочтителен для соединений, в которых R7 является замещенным или незамещенным арилом. На стадии 2 схемы 8 соединение формулы XVI обрабатывают, как описано на стадии 1 схемы 8, за исключением того, что соединением формулы R7C(О)H заменяют соединение формулы R7-CN,где R7 такой, как определено выше. На стадии 3 схемы 8 соединение формулы XXXIX окисляют с получением соединения формулы XXXVIII по методикам, известным специалисту в данной области, как описано на стадии 8 схемы 1. Соединения формулы I могут также быть получены по одному или более способам синтеза, которые раскрыты в опубликованных патентах или заявках на патенты. В частности, используя промежуточные соединения, описанные на схемах 1-8, указанные выше, а именно, промежуточные соединения формул VIII, X, XI,XVI и XXIV, специалист в данной области может получить соединения формулы I, используя аналогичные способы синтеза, которые описаны для соединений, в которых фенильное кольцо заменено индазольным кольцом в соединениях формулы I. Такие аналогичные способы синтеза описаны в следующих патентах и заявках на патенты: патент США 5,449,676 (выдан 12 сентября 1995); патент США 5,459,151 (выдан 17 октября 1995); патент США 5,491,147 (выдан 13 февраля 1996); заявка на европейский патент ЕР 470,805 (опубликована 12 февраля 1992); заявка на европейский патент ЕР 497,564 (опубликована 5 августа 1992); заявка на европейский патент ЕР 723,962 (опубликована 15 июля, 1996);WO 96/00218 (опубликована 4 января 1996); WO 96/21435 (опубликована 18 июля 1996). Вышеупомянутые патенты и опубликованные европейские заявки и заявки РСТ включены сюда в качестве ссылок. Конкретно соединения формулы I, в которой R2 является -Z3-R7, могут быть получены по аналогичным способам синтеза, представленным в WO 94/02465, WO 95/01338, WO 93/25517, WO 95/20578, WO 96/00218 и ЕР 497,564, каждый из которых указан выше. Соединения формулы I, в которой R2 является R18,могут быть получены по аналогичным способам синтеза, описанным в патенте США 5,491,147,WO 95/27692 и WO 95/22520, каждый из которых указан выше. Соединения формулы I, где R2 является заместителем формулы (If) или (Ig) могут быть получены по аналогичным способам синтеза, раскрытым в WO 95/22520, который указан выше. Соединения формулы I, в которыхR2 является заместителем формулы (Ih), могут быть получены по аналогичным способам синтеза, описанным в патенте США 5,459,151, который указан выше. Соединения формулы I, в которых R2 является -С(=NOC(О)R28)R28, могут быть получены по аналогичным способам синтеза, описанным в ЕР 470,805, который указан выше. Соединения формулы I, в которых R2 является -(CR17R18)mNR9(С(ОqR10, могут быть получены по аналогичным способам синтеза,раскрытым в WO 92/00968, WO 95/05386, WO 93/15044 и WO 93/15045, которые указаны выше. Соединения формулы I, в которых R2 является -CR17R18CHR28NR9SO2(CH2)pA, -CR17R18-СR17R18 СНR28NR9 Р(О)(C1-C4 алкокси)2, могут быть получены по аналогичным способам синтеза, раскрытым в WO 95/05386, который указан выше. Соединение формулы I, в которой R2 является заместителем формулы (Ic), (Id) или (Iе),могут быть получены по аналогичным способам синтеза, раскрытым в WO 93/18024, который указан выше. Соединения формулы I, в которойR2 является заместителем формулы (Ii), могут быть получены по аналогичным способам синтеза, раскрытым в ЕР 723,962, который указан выше. Соединения формулы I, в которых R2 является -C(=NR32)NH(CH2)p(C6-C10 арилом), могут быть получены по аналогичным способам синтеза, раскрытым в WO 96/21435, указанном выше. Соединения формулы I могут быть разделены на отдельные энантиомеры при помощи методики хиральной ЖХ при следующих условиях: колонка: Chiralcel OD (2504,6 мм); подвижная фаза: 50:50:0,1 (гексан:2-пропанол: диэтиламин); скорость потока: 1 мл/мин; проявление: УФ (230 нм); температура окружающей 21 среды (20-25 С); объем впрыскивания: 20 мкл. Соединения формулы I могут также быть разделены на отдельные энантиомеры по методикам,хорошо известным специалисту в данной области, включая описанную в J. March, AdvancedOrganic Chemistry (4th Edition, J. WileySons),1992, стр. 118-125. Соединения формулы I, которые по природе являются основаниями, способны образовывать широкий спектр различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми солями для введения животным, на практике часто желательно изначально выделять соединение формулы I из реакционной смеси в виде фармацевтически неприемлемой соли и затем просто превращать последнюю обратно в свободное основание обработкой щелочным реагентом с последующим превращением такого основания в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли оснований данного изобретения легко получают обработкой основания эквивалентным количеством выбранной минеральной или органической кислоты в водном растворителе или в подходящем органическом растворителе, таком как метанол или этанол. При выпаривании растворителя легко получить желаемую соль в виде твердого вещества. Желаемая кислотно-аддитивная соль может быть легко осаждена из раствора свободного основания в органическом растворителе добавлением к раствору подходящей минеральной или органической кислоты. Катионные соли соединений формулы I получают подобным образом только реакцией карбоксильной группы с подходящим реагентом катионной соли, таким как натрий, калий, кальций, магний, аммоний,N,N'-дибензилэтилендиамин, N-метилглюкамин(меглумин), этаноламин, трометамин или диэтаноламин. Для введения человеку при лечении или профилактике воспалительных заболеваний могут быть использованы различные обычные способы введения, включая пероральное, парентеральное, местное и ректальное (суппозитории) введение одной или разделенными дозами. Дозы соединения формулы I или его фармацевтически приемлемой соли ("активного соединения") для перорального введения обычно находятся в интервале от 0,1 до 1000 мг ежедневно для среднестатистического взрослого пациента(70 кг) одной или раздельными дозами. Отдельные таблетки или капсулы должны обычно содержать от 0,1 до 100 мг активного соединения в подходящем фармацевтически приемлемом наполнителе или носителе. Дозы для внутривенного введения обычно составляют от 0,1 до 10 мг на одну дозу, по требованию. Для интраназального введения или ингаляции доза обычно составляет от 0,1 до 1% (вес./об.) раствора. На практике терапевт сам может определить 22 необходимую дозу, которая необходима для отдельного пациента и которая зависит от возраста, веса и реакции конкретного пациента. Указанные выше дозы являются примерными для среднестатистического случая и, конечно,могут быть отдельные случаи, когда необходимы более низкие или более высокие дозы, причем все такие дозы включены в область данного изобретения. Для введения человеку с целью ингибирования ФНО может быть использовано множество обычных способов введения, включая пероральное, парентеральное, местное и ректальное(суппозитории) введение, одной или несколькими дозами. Обычно активное соединение вводится перорально или парентерально в дозах от 0,1 до 25 мг/кг веса тела пациента в день, предпочтительно от 0,3 до 5 мг/кг одной или несколькими дозами. Однако могут быть необходимы некоторые изменения доз в зависимости от состояния пациента. Врач, отвечающий за лечение, в любом случае, определяет подходящую дозу для каждого отдельного пациента. Для использования у человека, активное соединение данного изобретения может быть введено в чистом виде, но обычно оно вводится в смеси с фармацевтическим разбавителем или носителем, выбранным в соответствии с применяемым способом введения и обычной фармацевтической практикой. Например, активное соединение может быть введено перорально в виде таблеток, содержащих такие наполнители,как крахмал или лактоза, или в капсулах, либо в чистом виде, либо в смеси с наполнителями или в виде эликсиров или суспензий, содержащих вкусовые добавки и красители. Активные соединения могут быть инъецированы парентерально, например, внутривенно, внутримышечно или подкожно. Для парентерального введения активное соединение наиболее удобно вводить в виде стерильного водного раствора, который может содержать другие вещества: например, достаточное количество солей или глюкозы, чтобы сделать раствор изотоническим. Кроме того, активные соединения могут вводиться местно при лечении воспалительных состояний кожи, такое введение можно осуществлять при помощи кремов, желе, гелей, паст и мазей в соответствии с обычной фармацевтической практикой. Терапевтические соединения могут также вводиться млекопитающим, отличным от человека. Дозы вводимых соединений у млекопитающих зависят от вида животного и болезни или расстройства. Терапевтические соединения могут также вводиться животным в виде капсул,шариков, таблеток или жидких примочек. Терапевтические соединения могут также вводиться животным при помощи инъекций или имплантатов. Такие дозированные формы готовят обычными способами в соответствии со стандартной ветеринарной практикой. Альтернатив 23 но, терапевтические соединения могут вводиться животным вместе с кормом и для этих целей могут быть приготовлены концентрированные пищевые добавки или примеси для смешивания с обычным кормом для животных. Способность соединений формулы I или их фармацевтически приемлемых солей ингибировать ФДЭ IV может быть определена при помощи следующего теста. 30-40 г ткани человеческого легкого помещают в 50 мл буферного раствора трис/фенилметилсульфонил фторида (ФМСФ)/ сахарозы с рН 7,4 и гомогенизируют с использованием Tekmar Tissumizer (Tekmar Co., 7143Kemper Road, Cincinnati, Ohio 45249) при полной скорости в течение 30 с. Гомогенат центрифугируют при 48000g течение 70 мин при температуре 4 С. Надосадочную жидкость фильтруют дважды через 0,22 мкм фильтр и вводят в колонку для ВЭЖХ Mono-Q (PharmaciaLKB Biotechnology, 800 Centennial Avenue, Piscataway, New Jersey 08854) предварительно уравновешенную буфером трис/ФМСФ с рН 7,4. Образец вводят в колонку со скоростью потока 1 мл/мин с последующим увеличением скорости потока до 2 мл/мин для последующего промывания и элюирования. Образец элюируют, используя ступенчато возрастающий градиентNaCl в буфере трис/ФМСФ с рН 7,4. Собирают 8 мл фракций. Фракции исследуют на специфическую ФДЭIV активность, определяемую при помощи [3H]цАМФ гидролиза и способность известного ингибитора ФДЭIV (например, ролипрама) ингибировать этот гидролиз. Соответствующие фракции объединяют, разбавляют этиленгликолем (2 мл этиленгилколя/5 мл ферментного препарата) и хранят при -20 С до использования. Соединения растворяют в диметилсульфоксиде (ДМСО) с концентрацией 10 мМ и разбавляют 1:25 водой (400 мкМ соединения, 4% ДМСО). Дальнейшее серийное разведение осуществляют 4% ДМСО для получения желаемой концентрации. Конечная концентрация ДМСО в экспериментальной пробирке составляет 1%. Следующие соединения добавляют, дважды,последовательно, в 1275 мм стеклянные пробирки (все концентрации даны как конечные концентрации в экспериментальной пробирке).i) 25 мкл соединения или ДМСО (1% для контроля и контрольной пробы);ii) 25 мкл трис буфера с рН 7,5;iv) 25 мкл ферментов ФДЭIV (для контроля ферменты предварительно инкубируют в кипящей воде в течение 5 мин). Реакционные пробирки встряхивают и помещают на водяную баню (37 С) на 20 мин, после чего реакцию останавливают помещением пробирок в кипящую воду на 4 мин. В каждую пробирку на ледяной бане добавляют промы 002113 24 вочный буфер (0,5 мл, 0,1 М 4-(2-гидроксиэтил)1-пиперазинэтансульфоновой кислоты (HEPES)/ 0,1M naci, рН 8,5). Содержимое каждой пробирки вводят в колонку AFF-Gel 601 (Biorad Laboratories, P.O.Box 1229, 85A, Marcus Drive, Melvile, New York 11747) (гель, имеющий сродство к боронату (боронатно-аффинный) 1 мл слой) предварительно уравновешенную промывочным буфером. [3 Н]цАМФ промывают 26 мл промывочным буфером и [3H]5'AMФ затем элюируют 4 мл 0,25 М уксусной кислотой. После интенсивного перемешивания к 3 мл сцинтилляционной жидкости добавляют 1 мл элюата в подходящей пробирке, интенсивно перемешивают и подсчитывают для [3H].IC50 определяют как концентрацию соединения, которая подавляет 50% специфического гидролиза [3H]цАМФ до [3H]5'AМФ. Способность соединений формулы I или их фармацевтически приемлемых солей ингибировать образование ФНО и, следовательно,демонстрация их эффективности при лечении заболеваний, включающих образование ФНО,показаны в следующем исследовании in vitro. Периферийную кровь (100 мл), взятую у добровольных доноров, собирают в этилендиаминтетрауксусной кислоте (ЭДТК). Одноядерные клетки отделяют при помощи FICOLL/Hypaque и промывают три раза в неполном HBSS (сбалансированный солевой раствор Хенкса). Клетки повторно суспендируют с конечной концентрацией 1106 клеток на мл предварительно разогретого RPMI (содержащего 5% FCS, глутамин, pen/step и нистатин). Моноциты помещают при концентрации 1106 клеток в 1,0 мл 24-ячеечные планшеты. Клетки инкубируют при температуре 37 С (5% двуокись углерода) и оставляют для адгезии к планшетам в течение 2 ч, после чего неприклеившиеся клетки удаляют осторожным промыванием. Затем к клеткам добавляют тестируемое соединение (10 мкл) при 3-4 концентрациях каждое и инкубируют в течение 1 ч. ДобавляютLPS (10 мкл) в соответствующие ячейки. Планшеты инкубируют в течение ночи (18 ч) при температуре 37 С. В конце инкубационного периода ФНО анализируют при помощи sandwichELISA (RD Quatikine Kit). Определение IC50 проводят для каждого соединения на основе анализа линейной регрессии. Следующие примеры иллюстрируют данное изобретение. В следующих примерах "мин" означает минуту(минуты), "пси" означает "фунты на квадратный дюйм", "ч" означает час (часы), "экв." означает эквивалент(ы) и "конц." означает концентрированный. Способ получения 1. Метиловый эфир 1-циклопентил-3-этил 1 Н-индазол-6-карбоновой кислоты. 25 А. 3-Нитро-4-пропилбензойная кислота. 9,44 г (57,5 ммоль, 1,0 экв.) 4-пропилбензойной кислоты частично растворяют в 50 мл концентрированной H2SO4 и охлаждают на ледяной бане. По каплям добавляют раствор 4,7 мл (74,7 ммоль, 1,3 экв.) концентрированной НNО 3 в 10 мл концентрированной H2SO4 в течение 1-2 мин. После перемешивания в течение 1 ч при температуре 0 С реакционную смесь выливают в 1 л химический стакан, наполовину заполненный льдом. После перемешивания в течение 10 мин образуется белое твердое вещество, которое фильтруют, промывают 1 Н 2 О и сушат с получением 12,01 г (100%) целевого соединения: т.пл. 106-109 С; ИК (КВr) 32003400, 2966, 2875, 2667, 2554, 1706, 1618, 1537,1299, 921 см-1; 1H ЯМР (300 МГц, ДМСО-d6)0,90 (т, 3 Н, J=7,4 Гц), 1,59 (м, 2 Н), 2,82 (м, 2 Н),7,63 (д, 1 Н, J=8,0 Гц), 8,12 (дд, 1 Н, J=1,7, 8,0 Гц), 8,33 (д, 1 Н, J=1,7 Гц); 13 С ЯМР (75,5 МГц,ДMCO-d6)14,2, 23,7, 34,2, 125,4, 130,5, 132,9,133,6, 141,4, 149,5, 165,9; Элем. анал. рассч. дляC10H11NO41/4 H2O: С, 56,20; Н, 5,42; N, 6,55. Найдено: С, 56,12; Н, 5,31; N, 6,81. В. 3-Амино-4-пропилбензойная кислота. Смесь 11,96 г (57,2 ммоль) 3-нитро-4 пропилбензойной кислоты и 1,5 г 10% Pd/C,50% влажности в 250 мл СН 3 ОН помещают в аппарат Парра для гидрирования и встряхивают при давлении Н 2 1,758 кг/см 2 (25 пси) при температуре окружающей среды (20-25 С). Через 1 ч реакционную смесь фильтруют через CeliteH2O осторожно нагревают при помощи фена до практически полного растворения. Реакционную смесь охлаждают на ледяной бане и по каплям добавляют раствор 3,73 г (54,0 ммоль, 1,0 экв.) нитрита натрия в 27 мл H2O. Через 15 мин реакционную смесь переносят в капельную воронку и добавляют в течение более 10 мин в химический стакан, содержащий 55 г раскрошенного льда и 10,6 мл концентрированной НСl. После перемешивания в течение 10 мин содержимое химического стакана переносят в 26 капельную воронку и добавляют в течение более 5 мин к раствору 5,31 мл (47,1 ммоль, 0,96 экв.) трет-бутилтиола в 130 мл этанола при комнатной температуре. рН доводят до 4-5 добавлением насыщенного водного раствора Na2CO3 и реакционную смесь перемешивают в течение 1 ч при температуре окружающей среды (2025 С). Добавляют 200 мл солевого раствора и смесь фильтруют. Твердое вещество промывают 1H2O и сушат в течение ночи с получением 12,25 г (89%) коричнево-ржавого порошка (осторожно - неприятный запах): т.пл. 102 СD. 3-Этил-1 Н-индазол-6-карбоновая кислота. Раствор 12,0 г (42,8 ммоль, 1,0 экв) 3 карбокси-6-пропилбенздиазо-трет-бутилсульфида в 150 мл диметилсульфоксида (ДМСО) добавляют по каплям в течение более 15 мин к раствору 44,6 г (398 ммоль, 9,3 экв.) третбутоксида калия в 200 мл ДМСО при температуре окружающей среды. После перемешивания в течение 2 ч при температуре окружающей среды, реакционную смесь вливают в 1,5 л 1N(дд, 1 Н, J=1,1, 8,4 Гц), 7,81 (д, 1 Н, J=8,4 Гц),8,06 (д, 1 Н, J=1,1 Гц), 12,95 (шс, 1 Н); MC (Cl,NH3) m/z 191 (M+H+, основание). Элем. анал. рассч. для C10H10N2O2: С, 63,14; Н, 5,30; N,14,73. Найдено: С, 62,66; Н, 5,42; N, 14,80. Е. Метиловый эфир 3-этил-1 Н-индазолкарбоновой кислоты. 8,78 г (45,8 ммоль, 1,1 экв.) 1-(3 диметиламинопропил)-3-этилкарбодиимид гидрохлорида добавляют одной порцией в раствор 7,92 г (41,6 ммоль, 1,0 экв.) 3-этил-1 Н-индазол 6-карбоновой кислоты, 16,9 мл (416 ммоль, 10 экв.) метанола и 5,59 г (45,8 ммоль, 1,1 экв.) диметиламинопиридина (ДМАП) в 250 млCH2Cl2 при температуре окружающей среды. Через 18 ч при комнатной температуре реакционную смесь концентрируют до 150 мл, разбав 27 ляют 500 мл этилацетата, промывают 2100 мл 1N HCl, 1100 мл H2O, 1100 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 7,8 г коричневого твердого вещества, которое очищают на колонке с силикагелем (30-50% этилацетат/гексан градиент) с получением 6,41 г (75%) рыжевато-коричневого вещества: т.пл. 107108 С; ИК (КВr) 3100-2950, 1723, 1222 см-1; 1H ЯМР (300 МГц, CDCl3)8,19 (м, 1 Н), 7,7-7,8 (м,2 Н), 3,96 (с, 3 Н), 3,05 (кв, 2H, J=7,7 Гц), 1,43 (т,3 Н, J=7,7 Гц); MC (Cl, NН 3) m/z 205 (M+H+, основание). Элем. анал. рассч. для C11H12N2O2: С,64,70; Н, 5,92; N, 13,72. Найдено: С, 64,88; Н,6,01; N, 13,96.F. Метиловый эфир 1-циклопентил-3-этил 1 Н-индазол-6-карбоновой кислоты. 1,17 г (29,4 ммоль, 1,05 экв.) гидрида натрия, 60% дисперсия в масле, добавляют одной порцией при температуре окружающей среды к раствору 5,7 г (27,9 ммоль, 1,0 экв.) метилового эфира 3-этил-1 Н-индазол-6-карбоновой кислоты в 125 мл безводного ДМФ. Через 20 мин по каплям добавляют 3,89 мл (36,6 ммоль, 1,3 экв.) циклопентилбромида и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь затем вливают в 1 л Н 2 О и экстрагируют 3450 мл этилацетата. Органические экстракты объединяют, промывают 3400 мл H2O, 1200 мл солевого раствора и сушат над Na2SО 4. Фильтрация, концентрация фильтрата и сушка дают янтарное масло, которое очищают на колонке с силикагелем (10%,этилацетат/гексан, сила тяжести) с получением 5,48 г (72%) чистого масла: 1H ЯМР (300 МГц,CDCl3)8,16 (д, 1 Н, J=1,0 Гц), 7,7 (м, 2 Н), 5,00G. (1-Циклопентил-3-этил-1 Н-индазол-6 ил)-метанол. 7 мл (7,0 ммоль, 1,0 экв.) алюмогидрида лития, 1,0 М раствор в тетрагидрофуране (ТГФ) добавляют к раствору 1,02 г (7,05 ммоль, 1,0 экв.) метилового эфира 1-циклопентил-3-этил 1 Н-индазол-6-карбоновой кислоты в 50 мл безводного ТГФ при температуре 0 С. Через 20 мин осторожно добавляют 1 мл метанола, затем реакционную смесь вливают в 500 мл 5% H2SO4 и экстрагируют 350 мл этилацетата. Органические экстракты объединяют, промывают 240 мл H2O, 140 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 1,58 г чистого масла, которое очищают на колонке с силикагелем с получением 1,53 г (89%) чистого масла: ИК (СНСl3) 3606,3411, 3009, 2972, 2875, 1621, 1490 см-1; 1H ЯМРJ=7,6 Гц), 2,2 (м, 4 Н), 2,0 (м, 2H), 1,7 (м, 3 Н),1,38 (т, 3 Н, J=7,6 Гц); МС (термоспрей,NH4OAc) m/z 245 (М+Н+, основание); ВРМС рассч. для C15H20N2O+H: 245.1654. Найдено: 245.1675. Н. 1-Циклопентил-3-этил-1 Н-индазол-6 карбальдегид. 106 мг (0,301 ммоль, 0,05 экв.) тетрапропиламмоний перрутената (VII) добавляют при комнатной температуре к суспензии 1,47 г (6,02 ммоль, 1,0 экв.) (1-циклопентил-3-этил-1 Ниндазол-6-ил)-метанола, 1,06 г (9,03 ммоль, 1,5 экв.) N-оксида N-метилморфолин и 3,01 г 4 А молекулярных сит в 12 мл безводного CH2Cl2. Через 20 мин реакционную смесь фильтруют через короткую колонку силикагеля (элюированную CH2Cl2). Фракции, содержащие продукт,концентрируют и остаток хроматографиpуют на колонке с силикагелем (15% этилацетат, гексан,флэш) с получением 924 мг (63%) бледножелтого твердого вещества: т.пл. 41 С; ИК(M+H+, основание). Элем. анал. рассч. для С 16H18N2 О: С, 74,35; Н, 7,49; N, 11,56. Найдено: С, 74,17; Н, 7,58; N, 11,79. Способ получения 2. 1-Циклопентил-3-этил-1 Н-индазол-6 карбальдегид. А. 4-Бром-2-нитро-1-пропилбензол. 125 г (628 ммоль, 1,0 экв.) 1-бром-4 пропилбензола добавляют при температуре 10 С одной порцией к раствору 600 мл конц.H2SO4 и 200 мл Н 2 О. При энергичном механическом перемешивании при температуре окружающей среды 43,2 мл (691 ммоль, 1,1 экв.) конц. HNO3 (69-71%, 16 М) в 150 мл конц. H2SO4 и 50 мл H2O добавляют по каплям в течение более 30 мин. Ледяную баню нагревают до температуры окружающей среды и реакционную смесь перемешивают при комнатной температуре в течение 68 ч. Реакционную смесь вливают в 4 л химический стакан, плотно заполненный раскрошенным льдом. После перемешивания в течение 1 ч смесь переносят в 4 л делительную воронку и экстрагируют 4800 мл изопропиловым эфиром. Органические экстракты объединяют, промывают 3800 мл H2O, 1500 мл солевым раствором и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 150 мл желтой жидкости, которую очищают хроматографией на колонке с силикагелем (2 колонки, 3 кг силикагеля каждая, 2% этилацетат/гексан) с получением 63,9 г (42%) желтой жидкости. Желаемый региоизомер является менее полярным, чем два, которые образуются в соотношении 1:1. Т.кип. 108 С, 2,0 мм; ИКH ЯМР (300 МГц, CDCl3)8,01 (д, 1H, J=2,1 Гц), 7,62 (дд, 1H, J=2,1, 8,3 Гц), 7,23 (д, 1H,J=8,3 Гц), 2,81 (м, 2 Н), 1,67 (м, 2 Н), 0,98 (т, 3 Н,J=7,4 Гц); 13 С ЯМР (75,5 МГц, СDСl3)13,94,23,74, 34,43, 119,6, 127,4, 133,3, 135,7, 136,4,149,8; ГХМС (ЭИ) m/z 245/243 (М+), 147 (основание); ВРМС рассч. для C9H10NO2Br+H: 243,9973. Найдено: 243,9954. В. 5-Бром-2-пропилфениламин. 121 г (639 ммоль, 3,0 экв.) двухлористого олова (безводного) добавляют при комнатной температуре одной порцией к раствору 51,9 г(213 ммоль, 1,0 экв.) 4-бром-2-нитро-1-пропилбензола в 1200 мл абсолютного этанола и 12 мл(6 экв.) Н 2 О. Через 24 ч при комнатной температуре основную часть этанола удаляют на роторном испарителе. Остаток вливают в 4 л химический стакан, заполненный на 3/4 размельченным льдом и Н 2O. Порциями добавляют 150 г гранулNaOH при перемешивании до тех пор, пока рН не станет 10 и основная часть гидроксида олова не растворится. Смесь делят пополам и каждую половину экстрагируют 2750 мл этилацетатом. Все четыре экстракта этилацетата объединяют, промывают 1500 мл каждый 1N NaOH,H2O и солевым раствором, затем сушат надNa2SO4. Фильтрация, концентрация и сушка дают желтую жидкость, которую очищают на колонке с 1,2 кг силикагеля (1:12 этилацетат/гексан) с получением 41,83 г (92%) бледножелтой жидкости: ИК (СНСl3) 3490, 3404, 3008,2962, 2933, 2873, 1620, 1491 см-1; 1H ЯМР (300 МГц, CDCl3)6,8-6,9 (м, 3 Н), 3,90 (шс, 2 Н),2,42 (м, 2 Н), 1,62 (м, 2 Н), 0,99 (т, 3 Н, J=7,3 Гц); ГХМС (ЭИ) m/z 215/213 (М+), 186/184 (основание); Элем. анал. рассч. для C9H12NBr: С, 50,49; Н, 5,65; N, 6,54. Найдено: С, 50,77; Н, 5,70; N,6,50. С. 6-Бром-3-этил-1 Н-индазол. 49,22 г (230 ммоль, 1,0 экв.) 5-бром-2 пропилфениламина помещают в 3 л колбу и охлаждают на ледяной бане. Добавляют при температуре 0 С раствор 57,5 мл (690 ммоль, 3,0 экв.) конц. НСl в 165 мл Н 2 О, и полученную твердую массу, которая образуется, измельчают до получения тонкоизмельченной белой суспензии. Добавляют еще 100 мл Н 2O, затем раствор 15,9 г (230 ммоль, 1,0 экв.) нитрита натрия в 75 мл Н 2 О добавляют по каплям в течение свыше 10 мин. Ледяную баню удаляют и реакционную смесь перемешивают при комнатной температуре в течение 30 мин. Реакционную смесь затем фильтруют через воронку с фильтром из спекшегося стекла, предварительно охлаждая до температуры 0 С. Фильтрат охлаждают на ледяной бане и при механическом перемешивании в течение более 10 мин по каплям добавляют при температуре 0 С раствор/суспензию 32,8 г(313 ммоль, 1,36 экв.) тетрафторбората аммония в 110 мл H2O. Образовавшуюся густую белую 1 30 суспензию (соль тетрафторборат арилдиазония) перемешивают в течение 1,5 ч при температуре 0 С. Смесь затем фильтруют и твердое вещество промывают 1200 мл 5% водн. NH4BF4 (охлажденным, при температуре 0 С), 1150 мл СН 3 ОН (охлажденным до 0 С), затем 1200 млEt2O. Сушка в высоком вакууме при температуре окружающей среды в течение 1 ч дает 54,47 г(76%) соли диазония, бесцветного твердого вещества. 1500 мл свободного от этанола хлороформа помещают в 3-горлую колбу, затем добавляют 34,16 г (348 ммоль, 2,0 экв.) ацетата калия(размельченного до порошка и высушенного) и 2,3 г (8,7 ммоль, 0,05 экв.) 18-краун-6. Через 10 мин одной порцией добавляют соль диазония и реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 18 ч. Смесь затем фильтруют, твердое вещество промывают 2 раза СНСl3 и фильтрат концентрируют с получением 47 г неочищенного продукта(коричневых кристаллов). Хроматография на колонке с силикагелем (1,2 кг силикагеля, этилацетат/гексан градиент 15%, 20%, 40%) дает 21,6 г (55% на второй стадии, 42% всего) рыжевато-коричневых кристаллов: Т.пл. 112-114 С; ИК (КВr) 3205, 3008, 2969, 2925, 1616, 1340,1037 см-1; 1H ЯМР (300 МГц, СDСl3)9,86 (шc,1H), 7,61 (д, 1H, J=1,3 Гц), 7,57 (д, 1 Н, J=8,4 Гц),7,24 (дд, 1H, J=1,5, 8,6 Гц), 2,99 (кв, 2H, J=7,6 Гц), 1,41 (т, 3 Н, J=7,6 Гц); МС (Сl, NН 3) m/z 227/225 (М+Н+, основание). Элем. анал. рассч. для C9H9N2Br: С, 48,02; Н, 4,03; N, 12,45. Найдено: С, 48,08; Н, 3,87; N, 12,45.D. 6-Бром-1-циклопентил-3-этил-1 Н-индазол. 2,46 г (61,4 ммоль, 1,05 экв.) гидрида натрия (60% масляная суспензия) добавляют при температуре 10 С порциями по 0,5 г в раствор 13,17 г (58,5 ммоль, 1,0 экв.) 6-бром-3-этил-1 Ниндазола в 500 мл безводного ДМФ. Смесь перемешивают при температуре окружающей среды в течение 20 мин, затем по каплям добавляют раствор 8,8 мл (81,9 ммоль, 1,4 экв.) циклопентил бромида в 10 мл безводного ДМФ. Через 18 ч реакционную смесь вливают в 2 л Н 2 О и экстрагируют 21 л этилацетатом. Органические экстракты объединяют, промывают 2750 мл H2O, 1500 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 20,7 г неочищенного продукта, который очищают на колонке с силикагелем 31 анал. рассч. для C14H17N2Br: С, 57,35; Н, 5,84; N,9,55. Найдено: С, 57,48; Н, 5,83; N, 9,90. Е. 1-Циклопентил-3-этил-1 Н-индазол-6 карбальдегид. 11,6 мл (28,4 ммоль, 1,0 экв.) н-BuLi, 2,45 М в гексане добавляют при температуре -78 С к раствору 8,32 г (28,4 ммоль, 1,0 экв.) 6-бром-1 циклопентил-3-этил-1 Н-индазола в 200 мл безводного ТГФ. Через 30 мин по каплям добавляют 8,8 мл (114 ммоль, 4,0 экв.) безводного ДМФ при температуре -78 С и реакционную смесь перемешивают еще 30 мин при температуре-78 С. Смесь нагревают до комнатной температуры в течение 1 ч, затем добавляют 125 мл 1NHCl. После перемешивания в течение 10 мин основную часть ТГФ удаляют на роторном испарителе. Остаток разбавляют 500 мл Н 2 О и экстрагируют 2250 мл этилацетатом. Органические экстракты объединяют, промывают 1100 мл Н 2 О, 1100 мл солевого раствора и сушат над Nа 2SO4. Фильтрация, концентрация фильтрата и сушка дают желтое масло, которое очищают на колонке с силикагелем (15% этилацетат/гексан, гравитационная) с получением 4,70 г (68%) желтого кристаллического твердого вещества: 1H ЯМР (300 МГц, CDCl3) идентичен спектру целевого соединения из способа получения 1. Способ получения 3. 1-Циклопентил-3-этил-1 Н-индазол-6 карбоновая кислота. А. 3-Нитро-4-пропилбензойная кислота. 9,44 г (57,5 ммоль, 1,0 экв.) 4 пропилбензойной кислоты частично растворяют в 50 мл конц. H2SO4 и охлаждают на ледяной бане. По каплям добавляют раствор 4,7 мл (74,7 ммоль, 1,3 экв.) конц. HNO3 в мл конц. H2SO4 в течение 1-2 мин. После перемешивания в течение 1 ч при температуре 0 С реакционную смесь вливают в 1 л химический стакан наполовину заполненный льдом. После перемешивания в течение 10 мин фильтруют образовавшееся твердое вещество, промывают 1H2O и сушат с получением 12,01 г (100%) целевого соединения: т.пл. 106-109 С; ИК (КВr) 3200-3400, 2966,2875, 2667, 2554, 1706, 1618, 1537, 1299, 921 см-1; 1H ЯМР (300 МГц, ДМСО-d6)0,90 (т, 3 Н,J=7,4 Гц), 1,59 (м, 2 Н), 2,82 (м, 2 Н), 7,63 (д, 1 Н,J=8,0 Гц), 8,12 (дд, 1 Н, J=1,7, 8,0 Гц), 8,33 (д,1 Н, J=1,7 Гц); 13 С ЯМР (75,5 МГц, ДМСО-d6)14,2, 23,7, 34,2, 125,4, 130,5, 132,9, 133,6, 141,4,149,5, 165,9. Элем. анал. рассч. для C10H11NO4H2O: С, 56,20; Н, 5,42; N, 6,55. Найдено: С,56,12; Н, 5,31; N, 6,81. В. 3-Амино-4-пропилбензойная кислота. Смесь 11,96 г (57,2 ммоль) 3-нитро-4 пропилбензойной кислоты и 1,5 г 10% Pd/C,50% воды в 250 мл СН 3 ОН помещают в аппарат для гидрирования Парра и встряхивают в атмосфере H2 1,758 кг/см 2 (25 пси) при температуре окружающей среды. Через 1 ч реакционную 32 смесь фильтруют через Целит и фильтрат концентрируют и сушат с получением 9,80 г(22,1 ммоль, 0,45 экв.) карбоната натрия в 55 мл Н 2 О осторожно нагревают феном до практически полного растворения. Реакционную смесь охлаждают на ледяной бане и по каплям добавляют раствор 3,73 г (54,0 ммоль, 1,0 экв.) нитрита натрия в 27 мл Н 2O. Через 15 мин реакционную смесь переносят в капельную воронку и добавляют в течение более 10 мин в химический стакан, содержащий 55 г раскрошенного льда и 10,6 мл конц. НСl. После перемешивания в течение более 10 мин содержимое химического стакана переносят в капельную воронку и добавляют свыше 5 мин к раствору при комнатной температуре 5,31 мл (47,1 ммоль, 0,96 экв.) третбутилтиола в 130 мл этанола. рН доводят до 4-5 добавлением насыщенного водного раствораNа 2 СО 3 и реакционную смесь перемешивают в течение 1 ч при температуре окружающей среды. Добавляют 200 мл солевого раствора и смесь фильтруют. Твердое вещество промывают 1 Н 2 О и сушат в течение ночи с получением 12,25 г (89%) коричнево-ржавого порошка (осторожно - запах): т.пл. 102 С (разл) ; ИК (КВr) 3200-2400, 2962, 2872, 2550, 1678, 1484, 1428,1298, 1171 см-1; 1H ЯМР (300 МГц, ДМСО-d6)0,84 (т, 3 Н, J=7,3 Гц), 1,48 (м, 2 Н), 1,55 (с, 9 Н),2,42 (м, 2 Н), 7,29 (д, 1 Н, J=1,6 Гц), 7,50 (д, 1 Н,J=8,0 Гц), 7,86 (дд, 1 Н, J=1,7, 7,9 Гц), 13,18 (шс,1 Н); MC (термоспрей, NH4OAc) m/z 281 (М+Н+,основание). Элем. анал. рассч. для C14H20N2O2S: С, 59,96; Н, 7,19; N, 9,99. Найдено: С, 59,71; Н,7,32; N, 10,02.D. 3-Этил-1 Н-индазол-6-карбоновая кислота. Раствор 12,0 г (42,8 ммоль, 1,0 экв.) 3 карбокси-6-пропилбенздиазотрет-бутилсульфида в 150 мл ДМСО добавляют по каплям в течение 15 мин к раствору 44,6 г (398 ммоль, 9,3 экв.) трет-бутоксида калия в 200 мл ДМСО при комнатной температуре. После перемешивания в течение 2 ч при комнатной температуре реакционную смесь вливают в 1,5 л 1N НСl пpи температуре 0 С, перемешивают в течение 5 мин затем экстpагиpуют 2350 мл этилацетата.(М+Н+, основание). Элем. анал. рассч. для С 10 Н 10N2 О 2: С, 63,14; Н, 5,30; N, 14,73. Найдено: С, 62,66; Н, 5,42; N, 14,80. Е. Метиловый эфир 3-этил-1 Н-индазол-6 карбоновой кислоты. 8,78 г (45,8 ммоль, 1,1 экв.) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида добавляют одной порцией к раствору 7,92 г(41,6 ммоль, 1,0 экв.) 3-этил-1 Н-индазол-6 карбоновой кислоты, 16,9 мл (416 ммоль, 10 экв.) метанола и 5,59 (45,8 ммоль, 1,1 экв.) ДМАП в 250 мл CH2Cl2 при комнатной температуре. Через 18 ч при комнатной температуре реакционную смесь концентрируют до примерно 150 мл, разбавляют 500 мл этилацетата, промывают 2100 мл 1N HCl, 1100 мл H2O, 1100 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 7,8 г коричневого твердого вещества, которое очищают на колонке с силикагелем (градиент 30-50% этилацетат/гексан) с получением 6,41 г (75%) рыжевато-коричневого твердого вещества: т.пл. 107-108 С; ИК (КВr) 3100-2950,1723, 1222 см-1; 1H ЯМР (300 МГц, СDСl3)8,19F. Метиловый эфир 1-циклопентил-3-этил 1 Н-индазол-6-карбоновой кислоты. 1,17 г (29,4 ммоль, 1,05 экв.) гидрида натрия, 60% дисперсия в масле, добавляют одной порцией к раствору при комнатной температуре 5,7 г (27,9 ммоль, 1,0 экв.) метилового эфира 3 этил-1 Н-индазол-6-карбоновой кислоты в 125 мл безводного ДМФ. Через 20 мин по каплям добавляют 3,89 мл (36,6 ммоль, 1,3 экв.) циклопентилбромида и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь затем вливают в 1 л Н 2 О и экстрагируют 3400 мл H2O, 1200 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают янтарное масло,которое очищают на колонке с силикагелемG. 1-Циклопентил-3-этил-1 Н-индазол-6 карбoновая кислота. Смесь 5,24 г (19,2 ммоль, 1,0 экв.) метилового эфира 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты в 120 мл метанола и 60 мл 1N NaOH нагревают с обратным холодильником. Через 1 ч реакционную смесь охлаждают до комнатной температуры, концентрируют до 75 мл, подкисляют до рН 1 1N НСl и экстрагируют 2200 мл этилацетата. Органические слои объединяют, промывают 1150 мл H2O, 1150 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 4,79 г (96%) белого твердого вещества. Небольшой образец перекристаллизовывают из этилацетата/гексана для получения аналитических данных: т.пл. 157-159 С; ИК (КВr) 31002500, 1683, 1298 см-1; 1H ЯМР (300 МГц,ДМСО-d6)13,0 (шс, 1 Н), 8,21 (с, 1 Н), 7,79 (д,1 Н, J=7,9 Гц), 7,62 (сдд, 1 Н, J=1,2, 8,4 Гц), 5,18C15H18N2O2: С, 69,74; Н, 7,02; N, 10,85. Найдено: С, 69,77; Н, 7,02; N, 10,85. Пример 1. 2-Циклопентил-2 Н-индазол-6 карбоновой кислоты (3,5-дихлорпиридин-4 ил)амид и 1-циклопентил-2 Н-индазол-6-карбоновой кислоты (3,5-дихлорпиридин-4-ил)амид. 1 А. 1H-Индазол-6-карбоновая кислота. Парциальный раствор 15,1 г (100 ммоль,1,0 экв.) 3-амино-4-метилбензойной кислоты в 150 мл безводного ТГФ добавляют по каплям к раствору 18 мл (146 ммоль, 1,46 экв.) эфирата трифторида бора в 450 мл свободного от этанола хлороформа при температуре -20 С. Через 5 мин по каплям добавляют 14 мл (106 ммоль,1,06 экв.) 90% трет-бутилнитрита и реакционную смесь перемешивают при температуре 0 С в течение 2 ч. Порциями добавляют 49 г (500 ммоль, 5,0 экв.) ацетата калия с последующим добавлением 2,65 г (10 ммоль, 0,1 экв.) 18 краун-6 одной порцией. Реакционную смесь перемешивают при комнатной температуре в течение 48 ч, затем концентрируют на роторном испарителе. Добавляют 500 мл 3:7 ацетон/этилацетата и 150 мл 1N HCl и смесь перемешивают в течение 2 ч. Добавляют 150 мл солевого раствор и смесь фильтруют. Фильтрат переносят в делительную воронку, слои разделяют и водный слой экстрагируют 2100 мл 3:7 ацетон/ацетонитрилом. Органические слои объединяют и сушат над MgSO4. Фильтрация и концентрация фильтрата дает твердое вещество,к которому добавляют 250 мл уксусной кислоты. Суспензию нагревают на паровой бане до 35 практически полного растворения, затем вынимают из паровой бани и медленно добавляют 300 мл эфирной HCl (получают пропусканиемHCl (г) через 350 мл Et2O, охлажденного на ледяной бане, в течение 10 мин) к еще горячему раствору уксусной кислоты. Добавляют 250 млEt2O и смесь перемешивают при комнатной температуре в течение 1 ч. Фильтрация и сушка дают золотисто-коричневый порошок. Порошок суспендируют в 500 мл 3:7 ацетон/этилацетата,добавляют 100 мл солевого раствора и смесь перемешивают в течение 1 ч при комнатной температуре. Слои разделяют и водный слой экстрагируют 1100 мл этилацетатом. Объединенные органические слои сушат над МgSО 4. Фильтрация, концентрация фильтрата и сушка при сильном разрежении при комнатной температуре в течение 18 ч дают 7,81 г (48%) коричневого порошка. Т.пл. 275 С; МС (Сl, NН 3) m/z 180 (М+18+, основание). 1 В. Метиловый эфир 1H-индазол-6 карбоновой кислоты. Смесь 7,59 г (46,8 ммоль, 1,0 экв.) 1Hиндазол-6-карбоновой кислоты в 500 мл СН 3 ОН и 1 мл конц. H2SO4 нагревают с обратным холодильником в течение 8 ч, затем перемешивают при комнатной температуре в течение 18 ч. Смесь концентрируют до примерно 200 мл, разбавляют 1 л этилацетата и промывают 1250 мл насыщенным водным NаНСО 3, 1250 мл Н 2 О, 1250 мл солевым раствором и сушат надNa3SO4. Водные промывки экстрагируют двумя порциями этилацетата для регенерации продуктов присоединения. Органические слои объединяют, концентрируют и сушат с получением 6,75 г (82%) желто-оранжево-красно-коричневого твердого вещества: 1 Н ЯМР (300 МГц,CDCl3)10,8 (шс, 1 Н), 8,28 (дд, 1 Н, J=0,9, 1,9 Гц), 8,15 (д, 1 Н, J=1,0 Гц), 7,8 (м, 2 Н), 3,97 (с,3 Н); МС (Сl, NН 3) m/z 177 (М+Н+, основание). 1 С. Метиловый эфир 1-циклопентил-1 Ниндазол-6-карбоновой кислоты и метиловый эфир 2-циклопентил-2 Н-индазол-6-карбоновой кислоты. 1,60 г (39,9 ммоль, 1,05 экв.) гидрида натрия (60% дисперсия в масле), добавляют одной порцией к раствору 6,70 г (38,0 ммоль, 1,0 экв.) метилового эфира 1H-индазол-6-карбоновой кислоты в 150 мл безводного ДМФ при комнатной температуре. Через 30 мин по каплям добавляют 4,5 мл (41,8 ммоль, 1,1 экв.) циклопентил бромида и смесь перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь разбавляют 1,2 л этилацетата, промывают 3350 мл Н 2 О, 1250 мл солевым раствором и сушат над Na2SO4. Фильтрация,концентрация фильтрата и сушка дают 12 г янтарного масла, которое очищают на колонке с 700 г силикагеля (20% этилацетат/гексан, флэш) с получением 4,26 г метилового эфира 1 циклопентил-1 Н-индазол-6-карбоновой кислоты(46% выход, менее полярный изомер) и 3,66 г метилового эфира 2-циклопентил-2 Н-индазол-6 карбоновой кислоты (39% выход, более полярный изомер). Оба соединения являются оранжевыми маслами: данные для 1H-индазольного региоизомера: ИК (СНСl3) 2996, 2955, 2874,1717, 1249 см-1; ВРМС рассч. для С 14 Н 16N2O2: 244,1213; найдено 244,1209; данные для 2 Ниндазольного региоизомера: ИК (СНСl3) 2972,2955, 2876, 1714, 1242 см-1; ВРМС рассч. дляNa2OH нагревают с обратным холодильником в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры и большую часть СН 3 ОН удаляют на роторном испарителе. Остаток разбавляют 325 мл Н 2 О и подкисляют до рН 1 2N НСl. После перемешивания в течение 5 мин смесь фильтруют и твердое вещество промывают 2 Н 2 О и сушат в течение ночи с получением 3,42 г (92%) желтого порошка: т.пл. 172175 С. Элем. анал. рассч. для C13H14N2O2: С,67,79; Н, 6,13; N, 12,16; найдено: С, 67,62; Н,5,82; N, 12,19. 1 Е. 2-Циклопентил-2 Н-индазол-6-карбоновая кислота. Это соединение получают по методике примера 1D, исходя из 3,28 г (13,4 ммоль) метилового эфира 2-циклопентил-2 Н-индазол-6 карбоновой кислоты, 100 мл СН 3 ОН и 40 мл 1N(3,5-Дихлорпиридин-4-ил)амид 1 циклопентил-1 Н-индазол-6-карбоновой кислоты. Суспензию 495 мг (2,15 ммоль, 1,0 экв.) 1 циклопентил-1 Н-индазол-6-карбоновой кислоты, 204 мкл (2,79 ммоль, 1,3 экв.) тионилхлорида и 10 мкл ДМФ в 10 мл безводного толуола нагревают с обратным холодильником в течение 3 ч, затем охлаждают до температуры окружающей среды и концентрируют досуха на роторном испарителе. В отдельной колбе раствор 333 мг (2,04 ммоль, 0,95 экв.) 3,5-дихлор-4 аминопиридина в 10 мл безводного ТГФ добавляют по каплям к суспензии при температуре 0 С 198 мг (4,95 ммоль, 2,3 экв.) гидрида натрия(60% дисперсия в масле) в 10 мл безводного ТГФ. Смесь перемешивают в течение 15 мин при комнатной температуре, затем повторно охлаждают до температуры 0 С. По каплям добавляют раствор хлорангидрида (приготовленный ранее) в 10 мл безводного ТГФ и реакционную смесь перемешивают при комнатной температуре в течение ночи. К реакционной смеси по каплям добавляют 4,5 мл 1N НС 1, реакцион 37 ную смесь paзбавляют 200 мл СН 2 Сl2, промывают 130 мл каждый Н 2O, 10%-ным воднымNa2 СО 3, Н 2 О, затем сушат над Na2SO4. Очистка на колонке с силикагелем (2% СН 3 ОН/СН 2 Сl2,флэш) дает 0,76 г (94%) продукта в виде желтой аморфной пены: 1H ЯМР (300 МГц, CDCl3)8,58 (с, 2 Н), 8,19 (д, 1 Н, J=0,9 Гц), 8,09 (д, 1 Н,J=0,7 Гц), 7,93 (шс, 1 Н), 7,84 (дд, 1 Н, J=0,7, 8,4 Гц), 7,63 (дд, 1 Н, J=1,4, 8,4 Гц), 5,08 (квинтет,1 Н), 2,2 (м, 4 Н), 2,0 (м, 2 Н), 1,75 (м, 2 Н); МСN, 14,93; найдено: С, 57,68; Н, 4,55; N, 14,55. 1G. (3,5-Дихлорпиридин-4-ил)амид 2 циклопентил-2 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 1F, используя 424 мг 2-циклопентил 2 Н-индазол-6-карбоновой кислоты в качестве исходного соединения с получением 406 мгC18H16N4OCl2: С, 57,62; Н, 4,30; N, 14,93; найдено: С, 57,39; Н, 4,59; N, 14,56. Пример 2. 3-Нитро-4-пропилбензойная кислота. 9,44 г (57,5 ммоль, 1,0 экв.) 4-пропилбензойной кислоты частично растворяют в 50 мл конц. H2SO4 и охлаждают на ледяной бане. По каплям добавляют раствор 4,7 мл (74,7 ммоль, 1,3 экв.) конц. НNО 3 в 10 мл конц. H2SO4 в течение 1-2 мин. После перемешивания в течение 1 ч при температуре 0 С реакционную смесь вливают в 1 л химический стакан наполовину наполненный льдом. После перемешивания в течение 10 мин, фильтруют образовавшееся твердое вещество, промывают 1 Н 2 О и сушат с получением 12,01 г (100%) целевого соединения: т.пл. 106-109 С; ИК (КВr) 3200-3400,2966, 2875, 2667, 2554, 1706, 1618, 1537, 1299,921 см-1; 1H ЯМР (300 МГц, ДМСО-d6)0,90 (т,3 Н, J=7,4 Гц), 1,59 (м, 2 Н), 2,82 (м, 2 Н), 7,63 (д,1 Н, J=8,0 Гц), 8,12 (дд, 1 Н, J=1,7, 8,0 Гц), 8,33(д, 1 Н, J=1,7 Гц); 13 С ЯМР (75,5 МГц, ДМСО-d6)14,2, 23,7, 34,2, 125,4, 130,5, 132,9, 133,6,141,4, 149,5, 165,9. Элем. анал. рассч. для С 10 Н 11NO41/4 Н 2O: С, 56,20; Н, 5,42; N, 6,55. Найдено: С, 56,12; Н, 5,31; N, 6,81. Пример 3. 3-Амино-4-пропилбензойная кислота. Смесь 11,96 г (57,2 ммоль) 3-нитро-4 пропилбензойной кислоты и 1,5 г 10% Pd/C,50% воды в 250 мл СН 3 ОН помещают в аппарат для гидрирования Парра и встряхивают в атмосфере Н 2 1,758 кг/см 2 (25 пси) при температуре окружающей среды. Через 1 ч реакционную 38 смесь фильтруют через Целит и фильтрат концентрируют и сушат с получением 9,80 гH2O осторожно нагревают феном до практически полного растворения. Реакционную смесь охлаждают на ледяной бане и по каплям добавляют раствор 3,73 г (54,0 ммоль, 1,0 экв.) нитрита натрия в 27 мл Н 2 О. Через 15 мин реакционную смесь переносят в капельную воронку и добавляют в течение свыше 10 мин в химический стакан, содержащий 55 г раскрошенного льда и 10,6 мл конц. НСl. После перемешивания в течение 10 мин содержимое химического стакана переносят в капельную воронку и добавляют в течение свыше 5 мин к раствору при комнатной температуре 5,31 мл (47,1 ммоль,0,96 экв.) трет-бутил тиола в 130 мл этанола. рН доводят до 4-5 добавлением насыщенного водного раствора Nа 2 СО 3 и реакционную смесь перемешивают в течение 1 ч при температуре окружающей среды. Добавляют 200 мл солевого раствора и смесь фильтруют. Твердое вещество промывают 1 Н 2 О и сушат в течение ночи с получением 12,25 г (89%) коричнево-ржавого порошка (осторожно - запах): т.пл. 102 С (разл); ИК (КВr) 3200-2400, 2962, 2872, 2550, 1678,1484, 1428, 1298, 1171 см-1; 1H ЯМР (300 МГц,ДМСО-d6)0,84 (т, 3 Н, J=7,3 Гц), 1,48 (м, 2 Н),1,55 (с, 9 Н), 2,42 (м, 2 Н), 7,29 (д, 1 Н, J=1,6 Гц),7,50 (д, 1 Н, J=8,0 Гц), 7,86 (дд, 1 Н, J=1,7, 7,9 Гц), 13,18 (шс, 1 Н); МС (термоспрей, NH4OAc)m/z 281 (M+H+, основание). Элем. анал. рассч. для C14H20N2O2S: С, 59,96; Н, 7,19; N, 9,99. Найдено: С, 59,71; Н, 7,32; N, 10,02. Пример 5. 3-Этил-1 Н-индазол-6-карбоновая кислота. Раствор 12,0 г (42,8 ммоль, 1,0 экв.) 3 карбокси-6-пропилбенздиазотрет-бутилсульфида в 150 мл ДМСО добавляют по каплям в течение 15 мин к раствору при комнатной температуре 44,6 г (398 ммоль, 9,3 экв.) трет-бутоксида калия в 200 мл ДМСО. После перемешивания в течение 2 ч при комнатной температуре реакционную смесь вливают в 1,5 л 1N НСl при температуре 0 С, перемешивают в течение 5 мин затем экстрагируют 2350 мл этилацетата. Экс 39 тракты этилацетата (осторожно - запах) объединяют, промывают 2250 мл Н 2 О и сушат надC10H10N2O2: С, 63,14; Н, 5,30; N, 14,73. Найдено: С, 62,66; Н, 5,42; N, 14,80. Пример 6. Метиловый эфир 3-этил-1 Ниндазол-6-карбоновой кислоты. 8,78 г (45,8 ммоль, 1,1 экв.) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида добавляют одной порцией к раствору 7,92 г(41,6 ммоль, 1,0 экв.) 3-этил-1 Н-индазол-6 карбоновой кислоты, 16,9 мл (416 ммоль, 10 экв.) метанола и 5,59 (45,8 ммоль, 1,1 экв.) ДМАП в 250 мл CH2Cl2 при комнатной температуре. Через 18 ч при комнатной температуре реакционную смесь концентрируют до примерно 150 мл, разбавляют 500 мл этилацетата, промывают 2100 мл 1N HCl, 1100 мл Н 2 О, 1100 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 7,8 г коричневого твердого вещества, которое очищают на колонке с силикагелем (градиент 30-50% этилацетат/гексан) с получением 6,41 г (75%) рыжевато-коричневого твердого вещества: т.пл. 107-108 С; ИК (КВr) 3100-2950,1723, 1222 см-1; 1H ЯМР (300 МГц, CDCl3)8,19m/z 205 (М+Н+, основание). Элем. анал. рассч. для C11H12N2O2: С, 64,70; Н, 5,92; N, 13,72. Найдено: С, 64,88; Н, 6,01; N, 13,96. Пример 7. Метиловый эфир 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 1,17 г (29,4 ммоль, 1,05 экв.) гидрида натрия (60% дисперсия в масле) добавляют одной порцией к раствору 5,7 г (27,9 ммоль, 1,0 экв.) метилового эфира 3-этил-1 Н-индазол-6 карбоновой кислоты в 125 мл безводного ДМФ при комнатной температуре. Через 20 мин по каплям добавляют 3,89 мл (36,6 ммоль, 1,3 экв.) циклопентилбромида и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь затем вливают в 1 л Н 2 О и экстрагируют 3400 мл H2O, 1200 мл солевого раствора и сушат над Na2SO4. Фильтрация,концентрация фильтрата и сушка дают янтарное масло, которое очищают на колонке с силикагелем (10% этилцетат/гексан, гравитационный) с получением 5,48 г (72%) чистого масла: 1H ЯМР(300 МГц, СDСl3)8,16 (д, 1 Н, J=1,0 Гц), 7,7 (м,2 Н), 5,00 (квинтет, 1 Н, J=7,5 гц), 3,97 (с, 3 Н),3,01 (кв, 2H, J=7,6 Гц), 2,2 (м, 4 Н), 2,0 (м, 2H),1,8 (м, 2H), 1,39 (т, 3 Н, J=7,6 Гц); ВРМС рассч. для C16H20N2O2: 272,1526. Найдено: 272,15078. Пример 8. 1-Циклопентил-3-этил-1 Ниндазол-6-карбоновая кислота. Смесь 5,24 г (19,2 ммоль, 1,0 экв.) метилового эфира 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты в 120 мл метанола и 60 мл 1N NaOH нагревают с обратным холодильником. Через 1 ч реакционную смесь охлаждают до комнатной температуры, концентрируют до 75 мл, подкисляют до рН 1 1N HCl и экстрагируют 2200 мл этилацетата. Органические слои объединяют, промывают 1150 мл H2O, 1150 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 4,79 г (96%) белого твердого вещества. Небольшой образец перекристаллизовывают из этилацетата/гексана для получения аналитических данных: т.пл. 157-159 С; ИК (КВr) 31002500, 1683, 1298 см-1; 1 Н ЯМР (300 МГц,ДМСО-d6)13,0 (шс, 1 Н), 8,21 (с, 1 Н), 7,79 (д,1 Н, J=7,9 Гц), 7,62 (сдд, 1 Н, J=1,2, 8,4 Гц), 5,18C15H18N2O2: С, 69,74; Н, 7,02; N, 10,85. Найдено: С, 69,77; Н, 7,02; N, 10,85. Пример 9. (3,5-Дихлорпиридин-4-ил)амид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 9 А. 1-Циклопентил-3-этил-1 Н-индазол-6 карбоновая кислота. 7,73 мл (18,9 ммоль, 1,0 экв.) нбутиллития, 2,45 М в гексане добавляют по каплям к раствору 5,55 г (18,9 ммоль, 1,0 экв.) 6 бром-1-циклопентил-3-этил-1 Н-индазола в 100 мл безводного ТГФ при температуре -78 С. Через 30 мин в реакционную смесь барботируют СО 2 (г) в течение 15 мин. Реакционную смесь нагревают до комнатной температуры в течение нескольких часов, затем вливают в 600 мл Н 2 О,подкисляют до рН 1 и экстрагируют 2250 мл этилацетатом. Органические экстракты объединяют, промывают 1150 мл Н 2 О, 1100 мл солевого раствора и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 4,90 г (100%) белых кристаллов: т.пл. 153155 С; 1H ЯМР (300 МГц, ДМСО-d6) идентичен спектру продукта из примера 12. 9 В. 1-Циклопентил-3-этил-1 Н-индазол-6 карбонилхлорид. 791 мкл (10,8 ммоль, 1,4 экв.) тионил хлорида добавляют к раствору 2,00 г (7,74 ммоль,1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты и 100 мкл ДМФ в 100 мл безводного толуола при комнатной температуре. Реакционную смесь нагревают с обратным хо 41 лодильником в течение двух часов, затем охлаждают до комнатной температуры, концентрируют на роторном испарителе и сушат в высоком вакууме при комнатной температуре с получением 2,16 г (100 %) коричневых кристаллов: т.пл. 46-48 С; МС (Сl, NH3) m/z 279 (М+Н+,37Cl), 277 (M+H+, 36Cl). 9 С. (3,5-Дихлорпиридин-4-ил)амид 1 циклопентил-3-этил-1H-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 1F, используя 1,08 г (3,87 ммоль) 1 циклопентил-3-этил-1 Н-индазол-6-карбонил хлорида в качестве исходного материала с получением 1,327 г (85%) бледно-желтого кристаллического твердого вещества: т. пл. 174176 С; МС (Сl, NН 3) m/z 405 (М+Н+, 37Cl), 403(М+Н+, 35Cl). Элем. анал. рассч. для С 20 Н 20N4 ОСl2: С, 59,56; Н, 5,00; N, 13,89. Найдено: С, 60,23; Н, 5,42; N, 14,09. Пример 10. (4,6-Дихлорпиримидин-5-ил) амид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 50 мкл (0,680 ммоль, 1,3 экв.) тионилхлорида добавляют к раствору 135 мг (0,523 ммоль,1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты и 10 мкл ДМФ в 5 мл безводного толуола при комнатной температуре. Реакционную смесь нагревают с обратным холодильником в течение 2 ч, затем охлаждают до комнатной температуры, концентрируют досуха на роторном испарителе и сушат в высоком вакууме при комнатной температуре в течение нескольких часов. В отдельную колбу, содержащую раствор 86 мг (0,523 ммоль, 1,0 экв.) 5 амино-4,6-дихлорпиримидина в 5 мл безводного ДМФ при комнатной температуре добавляют 21 мг (0,523 ммоль, 1,0 экв.) гидрида натрия (60% дисперсия в масле). Через 10 мин добавляют раствор хлорангидрида (приготовленный ранее) в 5 мл безводного ДМФ и смесь перемешивают в течение 24 ч при комнатной температуре. Реакционную смесь затем нагревают до температуры 70 С в течение 1,5 ч, охлаждают до комнатной температуры, разбавляют 75 мл этилацетата, промывают 215 мл Н 2O, 116 мл солевого раствора и сушат над МgSO4. Неочищенный продукт очищают на колонке с силикагелем (20% этилацетат/гексан) с получением 31 мг (15%) белого кристаллического твердого вещества: т.пл. 171-172 С. Элем. анал. рассч. для C19H19N5OCl2: С, 56,44; Н, 4,74; N,17,32. Найдено: С, 56,38; Н, 4,76; N, 17,33. Пример 11. Фениламид 1-циклопентил-3 этил-1 Н-индазол-6-карбоновой кислоты. 46 мкл (0,629 ммоль, 1,3 экв.) тионилхлорида при комнатной температуре добавляют к раствору 125 мг (0,484 ммоль, 1,0 экв.) 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 5 мкл ДМФ в 5 мл безводного толуола. Реакционную смесь нагревают с обратным холодильником в течение 2 ч, затем охлаждают 42 до комнатной температуры, концентрируют досуха на роторном испарителе и сушат в высоком вакууме при комнатной температуре в течение нескольких часов. Неочищенный хлорангидрид растворяют в 5 мл CH2Cl2 и добавляют к раствору при комнатной температуре 49 мкл (0,532 ммоль, 1,1 экв.) анилина и 67 мкл (0,484 ммоль,1,0 экв.) триэтиламина в 5 мл CH2Cl2. Через 18 ч при комнатной температуре реакционную смесь разбавляют 75 мл этилацетата, промывают 115 мл каждый 1N HCl, H2O, солевым раствором и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 180 мг янтарного масла, которое очищают на колонке с силикагелем с получением 160 мг белого твердого вещества. Кристаллизация из этилацетата/гексана дает 130 мг (81%) белое кристаллическое твердое вещество: т.пл. 146-147 С. Элем. анал. рассч. для С 21 Н 23N3 О: С, 75,65; Н, 6,95; N, 12,60. Найдено: С, 75,65; Н, 7,02; N, 12,55. Пример 12. Метиловый эфир 4-[(1 циклопентил-3-этил-1 Н-индазол-6-карбонил) амино]бензойной кислоты. Это соединение получают по методике примера 11, используя 300 мг (1,16 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 193 мг (1,28 ммоль, 1,1 экв.) метил-4-амино-бензоата в качестве исходных материалов с получением 415 мг (91%) белых кристаллов: т.пл. 129-132 С. Элем. анал. рассч. для С 23 Н 25N3 О 3: С, 70,56; Н, 6,44; N, 10,73. Найдено: С, 70,36; Н, 6,43; N, 10,61. Пример 13. (3-Хлорфенил)-амид 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 11, используя 150 мг (0,581 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 68 мкл (0,639 ммоль, 1,1 экв.) 3-хлоранилина в качестве исходных соединений с получением 211 мг (99%) чистого масла: ВРМС рассч. для C21N22OCl+H: 368,1532. Найдено: 368,1567. Пример 14. (3-Метоксифенил)-амид 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 87 мг (0,708 ммоль, 1,0 экв.) ДМАП добавляют к раствору 196 мг (0,708 ммоль, 1,0 экв.) 1 циклопентил-3-этил-1 Н-индазол-6-карбонил хлорида, 99 мкл (0,708 ммоль, 1,0 экв.) триэтиламина и 80 мкл (0,708 ммоль, 1,0 экв.) манизидина в 5 мл CH2Cl2 при комнатной температуре. Через 48 ч реакционную смесь разбавляют 75 мл этилацетата, промывают 215 мл 1N НСl, 115 мл Н 2 О, 115 мл солевым раствором и сушат над MgSO4. Фильтрация, концентрация фильтрата и сушка дают 0,27 г янтарного твердого вещества, которое очищают хроматографией на колонке с силикагелем (градиент этилацетат/гексан 10%, 20%) с получением 118 мг чистого масла. Кристаллизация из 43 петролейного эфира дает 75 мг (29%) белого порошка: т.пл. 91-93 С. Элем. анал. рассч. дляC22H25N3O2: С, 72,71; Н, 6,95; N, 11,56. Найдено: С, 72,35; Н, 7,15; N, 11,47. Пример 15. Пиридин-4-иламид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 46 мкл (0,629 ммоль, 1,3 экв.) тионилхлорида добавляют к раствору 125 мг (0,484 ммоль,1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты и 5 мкл ДМФ в 5 мл безводного толуола при комнатной температуре. Реакционную смесь нагревают с обратным холодильником в течение 2 ч, затем охлаждают до комнатной температуры, концентрируют досуха на роторном испарителе и сушат в высоком вакууме, комнатной температуре в течение нескольких часов. Неочищенный хлорангидрид растворяют в 5 мл безводного пиридина, добавляют 50 мг (0,532 ммоль, 1,1 экв.) 4 аминопиридина и смесь нагревают до 40 С в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры и выстаивают в течение ночи. Добавляют 10 мл Н 2 О и смесь концентрируют досуха на роторном испарителе. Остаток помещают в 50 мл Н 2 О и 25 мл CH2Cl2 и слои разделяют. Водный слой экстрагируют 125 млCH2Cl2. Органические экстракты объединяют,промывают 110 мл каждый Н 2 О, солевым раствором и сушат над Na2SO4. Фильтрация,концентрация фильтрата и сушка дают 133 мг белой пены, которую очищают на колонке с силикагелем (25% CH3OH/CH2Cl2) с получением 122 мг белых игольчатых кристаллов. Перекристаллизация из этилацетата/гексана дает 101 мг(62%) белых блестящих хлопьев: т.пл. 144146 С. Элем. анал. рассч. для C20H22N4O: С,71,83; Н, 6,63; N, 16,75. Найдено: С, 72,00; Н,7,03; N, 16,16. Пример 16. Пиридин-3-иламид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 15, используя 58 мг (0,210 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбонилхлорида и 22 мг (0,231 ммоль, 1,1 экв.) 3 аминопиридина в качестве исходных материалов с получением 24 мг (34%) белых кристаллов: т. пл. 133-135 С; ВРМС рассч. дляC20H22N4O+H: 335,1872. Найдено: 335,1900. Пример 17. Пиридин-2-иламид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 15, используя 49 мг (0,177 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбонилхлорида и 18 мг (0,195 ммоль, 1,1 экв.) 2 аминопиридина в качестве исходных материалов с получением 17 мг (34%) желтой аморфной пены: ВРМС рассч. для C20H22N4O+H: 335,1872. Найдено: 335,1874. 44 Пример 18. (Пиридин-4-илметил)-амид 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 15, используя 51 мг (0,184 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбонилхлорида и 20 мкл (0,193 ммоль, 1,05 экв.) 4-(аминометил)пиридина в качестве исходных материалов с получением 13 мг (20%) белых кристаллов: т.пл. 147-149 С; ВРМС рассч. дляC21H24N4O+H: 349,2028. Найдено: 349,2031. Пример 19. (2-Пиридин-4-ил-этил)-амид 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 15, используя 78 мг (0,282 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбонилхлорида и 36 мкл (0,295 ммоль, 1,05 экв.) 4-(2-аминоэтил)пиридина в качестве исходных материалов с получением 35 мг (35%) белых кристаллов: т.пл. 123-126 С. Элем. анал. рассч. для С 22 Н 26N4O1/4 Н 2O: С, 72,01; Н, 7,28; N,15,27. Найдено: С, 71,77; Н, 7,45; N, 15,23. Пример 20. Хинолин-5-иламид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 15, используя 91 мг (0,329 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбонилхлорида и 52 мг (0,362 ммоль, 1,1 экв.) 5 аминохинолина в качестве исходных материалов с получением 38 мг (30%) бледно-желтого порошка: т.пл. 176-178 С. Элем. анал. рассч. дляC24H24N4O: С, 74,96; Н, 6,29; N, 14,57. Найдено: С, 74,33; Н, 6,53; N, 14,31. Пример 21. (2,6-Дихлорфенил)-амид 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. 1,1 мл (15,1 ммоль, 1,3 экв.) тионилхлорида добавляют к раствору 3,00 г (11,6 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 150 мкл ДМФ в 60 мл безводного толуола при комнатной температуре. Реакционную смесь нагревают с обратным холодильником в течение двух часов, затем охлаждают до комнатной температуры, концентрируют на роторном испарителе и сушат в высоком вакууме, при комнатной температуре с получением 3,40 г желто-коричневых кристаллов. Добавляют 1,88 г (11,6 ммоль, 1,0 экв.) 2,6 дихлоранилина и смесь нагревают на масляной бане с температурой 200 С в атмосфере азота. Через 15 мин реакционную смесь охлаждают до комнатной температуры. Добавляют 75 мл этилацетата и 50 мл насыщенного водного NаНСО 3,и смесь перемешивают в течение 10 мин. Слои разделяют и органический слой промывают 120 мл насыщенным NaHCO3, 120 мл Н 2 О, 120 мл солевым раствором и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают коричневое твердое вещество, которое 45 перекристаллизовывают из этилацетата/гексана с получением 3,78 г (81%) рыжеватокоричневого кристаллического твердого вещества: т.пл. 177-179 С. Элем. анал. рассч. дляC21H21N3OCl2: С, 62,69; Н, 5,26; N, 10,45. Найдено: С, 62,67; Н, 5,20; N, 10,43. Пример 22. 4-[(1-Циклопентил-3-этил-1 Ниндазол-6-карбонил)-амино]-бензойная кислота. Смесь 380 мг (0,971 ммоль, 1,0 экв.) метилового эфира 4-[(1-циклопентил-3-этил-1 Ниндазол-6-карбонил)-амино]-бензойной кислоты, 4 мл 1N NaOH и 20 мл метанола нагревают с обратным холодильником в течение 40 мин. После охлаждения до комнатной температуры реакционную смесь концентрируют на роторном испарителе и остаток разбавляют 150 мл Н 2 О, подкисляют до рН 1 и экстрагируют 250 мл этилацетата. Органические экстракты объединяют, промывают каждый 125 мл Н 2 О, солевым рассолом и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка в высоком вакууме, комнатной температуре дают 298 мг (81%) белого кристаллического твердого соединения: т.пл. 249-251 С. Элем. анал. рассч. для С 22 Н 23N3 О 3: С, 70,00; Н, 6,14; N, 11,13. Найдено: С, 69,66; Н, 6,13; N, 11,08. Пример 23.(4-Бензилоксикарбамоилфенил)-амид 1-циклопентил-3-этил-1H-индазол 6-карбоновая кислота. 140 мг (0,728 ммоль, 1,1 экв.) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида добавляют одной порцией к раствору 250 мг (0,662 ммоль, 1,0 экв.) 4-[(1-циклопентил-3 этил-1 Н-индазол-6-карбонил)-амино]-бензойной кислоты, 106 мг (0,662 ммоль, 1,0 экв.) Oбензилгидроксиламин гидрохлорида, 101 мг(0,662 ммоль, 1,0 экв.) 1-гидроксибензтриазол гидрата и 194 мкл (1,39 ммоль, 2,1 экв.) триэтиламина в 25 мл CH2Cl2 при комнатной температуре. Через 18 ч реакционную смесь разбавляют 150 мл этилацетата, промывают 125 мл каждый 1N HCl, Н 2 О, солевым раствором и сушат над Na2SO4. Неочищенный продукт очищают на колонке с силикагелем (2% CH3OH/CH2Cl2,флэш) с получением 212 мг (66%) белого кристаллического твердого вещества: т.пл. 194198 С. Элем. анал. рассч. для С 29 Н 30N4 О 3: С,72,17; Н, 7,10; N, 11,61. Найдено: С, 71,62; Н,6,47; N, 11,85. Пример 24. (4-Гидроксикарбамоилфенил)амид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Смесь 187 мг (0,387 ммоль, 1,0 экв.) (4 бензилоксикарбамоилфенил)амида 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 200 мг 10% Pd/C в 10 мл этилацетата и 10 мл метанола помещают в аппарат Парра для гидрирования и встряхивают при давлении Н 2 2,109 кг/см 2 (30 пси) при комнатной температуре в течение 1 ч. Реакционную смесь фильтруют через Целит и фильтрат концентрируют и сушат 46 с получением рыжевато-коричневого твердого вещества. Очистка на колонке с силикагелем(градиент СН 3 ОН/СН 2 Сl2 4%, 10%, 20%, флэш) дает 74 мг (49%) рыжевато-коричневого твердого вещества: т.пл. 175 С (разл); ВРМС рассч. для C22H24N4O3+H: 393,1928. Найдено: 393,1949. Пример 25. 25 А. Метиловый эфир 1-циклобутил-3 этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике способа получения 3, используя 750 мг (3,67 ммоль, 1,0 экв.) метилового эфира 3-этил-1Hиндазол-6-карбоновой кислоты и 0,38 мл (4,04 ммоль, 1,1 экв.) циклобутилбромида в качестве исходных материалов с получением 307 мгC15H18N2O2+H: 259,1447. Найдено: 259,14550. 25 В. Метиловый эфир 3-этил-1-изопропил 1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике способа получения 3, используя 750 мг (3,67 ммоль, 1,0 экв.) метилового эфира 3-этил-1Hиндазол-6-карбоновой кислоты и 0,38 мл (4,04 ммоль, 1,1 экв.) 2-бромпропана в качестве исходных материалов с получением 359 мг (40%) чистого масла: ВРМС рассч. для C14H18N2O2+H: 247,1448. Найдено: 247,14530. 25 С. Метиловый эфир 1-циклопропилметил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике способа получения 3, используя 750 мг (3,67 ммоль, 1,0 экв.) метилового эфира 3-этил-1Hиндазол-6-карбоновой кислоты и 0,39 мл (4,04 ммоль, 1,1 экв.) циклопропилметилбромида в качестве исходных материалов с получением 338 мг (36%) чистого масла: ВРМС рассч. дляC15H18N2O2+H: 259,1447. Найдено: 259,1435. 25D. Метиловый эфир 1-циклогекс-2-енил 3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике способа получения 3, используя 750 мг (3,67 ммоль, 1,0 экв.) метилового эфира 3-этил-1Hиндазол-6-карбоновой кислоты и 0,46 мл (4,04 ммоль, 1,1 экв.) 3-бромциклогексена в качестве исходных материалов с получением 467 мгC17H20N2O2: 284,1525. Найдено: 284,1512. 25 Е. Метиловый эфир 3-этил-1-[6-(4 фенилбутокси)-гексил]-1H-индазол-6-карбоновой кислоты. Это соединение получают по методике способа получения 3, используя 137 мг (0,671 ммоль, 1,0 экв.) метилового эфира 3-этил-1 Ниндазол-6-карбоновой кислоты и 273 мг (0,872 ммоль, 1,3 экв.) 6-(4-фенилбутокси)-гексил бромида в качестве исходных материалов с получением 163 мг (56%) желтого масла: ВРМС рассч. для C27H36N2O3+H: 437,2804. Найдено: 437,2833. 47 Пример 26. 26 А. 1-Циклобутил-3-этил-1 Н-индазол-6 карбоновая кислота. Это соединение получают по методике способа получения 3, используя 225 мг (0,906 ммоль, 1,0 экв.) метилового эфира 1 циклобутил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 168 мг (76%) белого кристаллического твердого вещества: т.пл. 148-150 С; ВРМС рассч. для C14H16N2O2+H: 245,1290. Найдено: 245,1302. 26 В. 3-Этил-1-изопропил-1 Н-индазол-6 карбоновая кислота. Это соединение получают по методике способа получения 3, используя 300 мг (1,22 ммоль, 1,0 экв.) метилового эфира 3-этил-1 изопропил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 260 мг (92%) бледно-желтого кристаллического твердого вещества: т.пл. 160-163 С. Элем. анал. рассч. для С 13 Н 16N2O2: С, 67,21; Н, 6,94; N,12,05. Найдено: С, 67,07; Н, 7,04; N, 12,16. 26 С. 1-Циклопропилметил-3-этил-1 Ниндазол-6-карбоновая кислота. Это соединение получают по методике способа получения 3, используя 294 мг (1,14 ммоль, 1,0 экв.) метилового эфира 1 циклопропилметил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 261 мг (94%) бледно-желтого кристаллического твердого вещества: т.пл. 126130 С. Элем. анал. рассч. для C14H18N2O2: С,68,83; Н, 6,60; N, 11,46. Найдено: С, 68,39; Н,6,67; N, 11,41. 26D. 3-Этил-1-[6-(4-фенилбутокси)гексил]-1H-индазол-6-карбоновая кислота. Это соединение получают по методике способа получения 3, используя 147 мг (0,337 ммоль, 1,0 экв.) метилового эфира 3-этил-1-[6(4-фенилбутокси)-гексил]-1H-индазол-6-карбоновой кислоты в качестве исходного материала с получением 137 мг (96%) бледно-желтого масла: ВРМС рассч. для С 26 Н 34N2 О 3+Н: 423,2648. Найдено: 423,26795. Пример 27. 27 А. Метиловый эфир 1-циклогексил-3 этил-1 Н-индазол-6-карбоновой кислоты. Смесь 417 мг (1,47 ммоль, 1,0 экв.) метилового эфира 1-циклогекс-2-енил-3-этил-1 Ниндазол-6-карбоновой кислоты и 50 мг 10%Pd/C, 50% воды в 20 мл этилацетата помещают в аппарат для гидрирования Парра и встряхивают при давлении Н 2 3,164 кг/см 2 (45 пси) в течение 45 мин. Реакционную смесь фильтруют через Целит и фильтрат концентрируют на роторном испарителе и сушат в высоком вакууме, комнатной температуре с получением 399 мг 48 Это соединение получают по методике способа получения 3, используя 366 мг (1,28 ммоль, 1,0 экв.) метилового эфира 1 циклогексил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 325 мг (93%) бледно-желтого твердого вещества: т.пл. 196-197 С; ВРМС рассч. дляC16H20N2O2+H: 273,1603. Найдено: 273,1596. Пример 28. 28 А. Метиловый эфир 3-этил-1-(4-фторфенил)-1 Н-индазол-6-карбоновой кислоты. 0,245 мл (2,24 ммоль, 2,0 экв.) 1-бром-4 фторбензола добавляют при комнатной температуре к суспензии 0,23 г (1,12 ммоль, 1,0 экв.) метилового эфира 3-этил-1 Н-индазол-6 карбоновой кислоты, 0,23 г (1,67 ммоль, 1,5 экв.) карбоната калия и около 100 мг (0,348 ммоль, 0,3 экв.) Сu2 Вr2 в 6 мл Nметилпирролидинона. Реакционную смесь нагревают до 175 С в течение 28 ч, затем охлаждают до комнатной температуры, вливают в 100 моль/л H2O и экстрагируют 350 моль/л этилацетата. Органические экстракты объединяют,промывают 250 мл H2O, 150 мл солевым раствором. Водные промывки повторно экстрагируют 175 мл этилацетата. Все экстракты этилацетата затем объединяют и сушат надNa2SO4. Фильтрация, концентрация фильтрата и сушка дают 0,6 г коричневого масла, которое очищают на колонке с силикагелем (10% этилацетат/гексан) с получением 96 мг (29%) белого кристаллического твердого вещества: т.пл. 7274 С; МС (Сl, NН 3) m/z 299 (М+Н+, основание). 28 В. 3-Этил-1-(4-фторфенил)-1H-индазол 6-карбоновая кислота. Это соединение получают по методике способа получения 3, используя 96 мг (0,322 ммоль, 1,0 экв.) метилового эфира 3-этил-1-(4 фторфенил)-1H-индазол-6-карбоновой кислоты в качестве исходного материала с получением 84 мг (92%) белого твердого вещества: т.пл. 204-205 С; ВРМС рассч. для C16H13N2O2F+H: 285,1040. Найдено: 285,10257. Пример 29. (3,5-Дихлорпиридин-4-ил)амид 1-циклобутил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 10, используя 144 мг (0,589 ммоль, 1,0 экв.) 1-циклобутил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 44 мг (18%) белого кристаллического твердого вещества: т.пл. 166-168 С; ВРМС рассч. для C19H18N4OCl2+H: 389,0936. Пример 30. (3,5-Дихлорпиридин-4-ил)амид 3-этил-1-изопропил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 10, используя 232 мг (1,00 ммоль, 1,0 экв.) 3-этил-1-изопропил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 73 мг (20%) белого кристалличе 49 ского твердого вещества: т.пл. 145-148 С; ВРМС рассч. для C18H18N4OCl2+H: 377,0936. Найдено: 377,0938. Пример 31. (3,5-Дихлорпиридин-4-ил)амид 1-циклопропилметил-3-этил-1H-индазол 6-карбоновой кислоты. Это соединение получают по методике примера 10, используя 224 мг (0,917 ммоль, 1,0 экв.) 1-циклопропилметил-3-этил-1 Н-индазол-6 карбоновой кислоты в качестве исходного материала с получением 51 мг (14%) белого кристаллического твердого вещества: т.пл. 148150 С; ВРМС рассч. для C19H18N4OCl2+H: 389,0936. Найдено: 389,091. Пример 32. (3,5-Дихлорпиридин-4-ил)амид 1-циклогексил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 10, используя 300 мг (1,10 ммоль, 1,0 экв.) 1-циклогексил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала с получением 83 мг (18%) белого кристаллического твердого вещества: т.пл. 124-127 С; МС(Cl, NH3) m/z 421 (М+Н+, 37 Сl+37 Сl), 419 (М+Н+,35 Сl+37 Сl, основание), 417 (М+Н+, 35 Сl+35 Сl). Пример 33. (3,5-Дихлорпиридин-4-ил)амид 3-этил-1-[6-(4-фенилбутокси)-гексил]-1Hиндазол-6-карбоновой кислоты. Это соединение получают по методике примера 1, используя 127 мг (0,301 ммоль, 1,0 экв.) 3-этил-1-[6-(4-фенилбутокси)-гексил]-1Hиндазол-6-карбоновой кислоты в качестве исходного материала с получением 119 мг (70%) чистого масла, которое кристаллизуют из эфира/гексана с получением 72 г (42%) белых кристаллов: т.пл. 76-79 С; ВРМС рассч. для С 31 Н 36N4O2 Сl2+Н: 567,2294. Найдено: 567,2288. Пример 34. (3,5-Дихлорпиридин-4-ил)амид 3-этил-1-(4-фторфенил)-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 10, используя 80 мг (0,281 ммоль, 1,0 экв.) 3-этил-1-(4-фторфенил)-1 Н-индазол-6 карбоновой кислоты в качестве исходного материала с получением 22 мг (18%) белого кристаллического твердого вещества: т.пл. 197199 С; ВРМС рассч. для C21H15N4OCl2F+H: 429,0688. Найдено: 429,0704. Пример 35. Метиловый эфир [(1 циклопентил-3-этил-1 Н-индазол-6-карбонил)амино]-уксусной кислоты. 134 мг (0,697 ммоль, 1,0 экв.) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида добавляют при комнатной температуре одной порцией к раствору 180 мг (0,697 ммоль,1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты, 87,5 мг (0,697 ммоль, 1,0 экв.) гидрохлорида метилового эфира глицина,107 мг (0,697 ммоль, 1,0 экв.) 1 гидроксибензотиазолгидрата и 194 мкл (1,39 ммоль, 2,0 экв.) триэтиламина в 5 мл CH2Cl2. Через 18 ч реакционную смесь разбавляют 150Na2SO4. Неочищенный продукт очищают на колонке с силикагелем (40% этилацетат/гексан,флэш) с получением 187 мг (66%) белого парафинообразного твердого вещества: т.пл. 8993 С. Элем. анал. рассч. для С 18 Н 23N3 О 3: С,65,63; Н, 7,04; N, 12,76. Найдено: С, 65,74; Н,7,02; N, 12,74. Пример 36. [(1-Циклопентил-3-этил-1 Ниндазол-6-карбонил)-амино]-уксусная кислота. Это соединение получают по методике способа получения 3, используя 170 мг (0,516 ммоль, 1,0 экв.) метилового эфира [(1 циклопентил-3-этил-1 Н-индазол-6-карбонил)амино]-уксусной кислоты в качестве исходного материала с получением 155 мг (95%) белых кристаллов: т.пл. 182-184 С. Элем. анал. рассч. для С 17 Н 21N3 О 3: С, 64,73; Н, 6,71; N, 13,32. Найдено: С, 64,73; Н, 6,80; N, 12,81. Пример 37. (Бензилоксикарбамоилметил)амид 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 23, используя 144 мг (0,457 ммоль, 1,0 экв.)[(1-циклопентил-3-этил-1 Н-индазол-6 карбонил)-амино]-уксусной кислоты в качестве исходного материала с получением 125 мг(65%) белого аморфного твердого вещества: 1 Н ЯМР (300 МГц, CDCl3, частичный)4,93 (с,2 Н), 3,00 (кв, 2 Н, J=7,6 Гц), 2,2 (м, 4 Н), 2,0 (м,2 Н), 1,7 (м, 2 Н), 1,39 (т, 3 Н, J=7,6 Гц); МС (Cl,NH3) m/z 421 (M+H+, основание). Пример 38. Гидроксикарбамоилметиламид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Смесь 120 мг (0,285 ммоль, 1,0 экв.) (бензилоксикарбамоилметил)-амида 1-циклопентил 3-этил-1 Н-индазол-6-карбоновой кислоты и 0,08 г 10% Pd/C, 50% воды в 10 мл метанола и 10 мл этилацетата помещают в аппарат для гидрирования Парра и встряхивают при давлении Н 2 2,109 кг/см 2 (30 пси) при комнатной температуре в течение 40 мин. Реакционную смесь фильтруют через Целит и фильтрат концентрируют и сушат с получением 104 мг рыжеватокоричневого твердого вещества, растирание в порошок с гексаном дает 69 мг (73%) рыжеватокоричневого кристаллического порошка: т.пл. 105 С (разл.); ВРМС рассч. для C17H22N4O3+H: 331,1772. Найдено 331,1769 Пример 39. (2-Метилсульфонилэтил)-амид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 35, используя 57 мг (0,221 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 20 мг (0,221 ммоль, 1,0 экв.) 2-(метилтио)этиламина в качестве исходных материалов. Хроматография на колонке с силикагелем (30% этилацетат/гексан) дает 53 мг(73%) белых кристаллов: т.пл. 81-83 С. Элем. анал. рассч. для C18H25N3O3: С, 65,21; Н, 7,60; N,12,68. Найдено: С, 65,26; Н, 7,49; N, 12,81. Пример 40. (Метоксикарбамоилметил)амид 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 37, используя 222 мг (0,704 ммоль, 1,0 экв.)(0,704 ммоль, 1,0 экв.) метоксиламингидрохлорида в качестве исходных материалов с получением 39 мг чистого масла, которое кристаллизуют из эфира/гексана с получением 34 мг(14%) белых кристаллов: т.пл. 135-136 С. Элем. анал. рассч. для С 18 Н 24N4 О 3: С, 62,77; Н, 7,02; N,16,27. Найдено: С, 62,64; Н, 6,87; N, 16,47. Пример 41. Этиловый эфир 3-[(1 циклопентил-3-этил-1 Н-индазол-6-карбонил)амино]-пропионовой кислоты. Это соединение получают по методике примера 35, используя 297 мг (1,15 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 177 мг (1,15 ммоль, 1,0 экв.) гидрохлорида -аланин этилового эфира в качестве исходных материалов с получением 372 мг(90%) белых кристаллов: т.пл. 74-76 С. Элем. анал. рассч. для C20H27N3O3: С, 67,21; Н, 7,61; N,11,76. Найдено: С, 67,40; Н, 7,56; N, 11,99. Пример 42. 3-[(1-Циклопентил-3-этил-1Hиндазол-6-карбонил)-амино]-пропионовой кислоты. Смесь 330 мг (0,923 ммоль, 1,0 экв.) этилового эфира 3-[(1-циклопентил-3-этил-1 Н-индазол-6-карбонил)-амино]-пропионовой кислоты в 10 мл этанола и 4 мл 1N NaOH нагревают с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры реакционную смесь концентрируют, разбавляют 75 мл Н 2 О, подкисляют до рН 1 и экстрагируют 335 мл этилацетатом. Органические экстракты объединяют, промывают 125 мл Н 2 О, 125 мл солевого раствора и сушат Na2SO4. Фильтрация,концентрация фильтрата на роторном испарителе и сушка в высоком вакууме, комнатной температуре с получением 297 мг (98%) белого твердого вещества: т.пл. 151-153 С. Элем. анал. рассч. для С 18 Н 23N3 О 3: С, 65,63; Н, 7,04; N,12,76. Найдено: С, 65,75; Н, 7,12; N, 12,91. Пример 43.(2-Бензилоксикарбамоилэтил)-амид 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 37, используя 250 мг (0,759 ммоль, 1,0 экв.) 3-[(1-циклопентил-3-этил-1 Н-индазол-6 карбонил)-амино]-пропионовой кислоты и 121 мг (0,759 ммоль, 1,0 экв.) O-бензилгидроксиламин гидрохлорида в качестве исходных материалов с получением 237 мг (72%) белых кристаллов: т.пл. 134-136 С. Элем. анал. рассч. 52 для C25H30N4O3: С, 69,10; Н, 6,96; N, 12,89. Найдено: С, 69,36; Н, 6,75; N, 12,85. Пример 44. (2-Гидроксикарбамоилэтил)амид 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 38, используя 203 мг (0,467 ммоль, 1,0 экв.) (2-бензилоксикарбамоилэтил)-амида 1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала и 50 мг[(1 циклопентил-3-этил-1 Н-индазол-6-карбонил)метиламино]уксусной кислоты. Это соединение получают по методике примера 35, используя 284 мг (1,10 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 169 мг (1,10 ммоль, 1,0 экв.) гидрохлорида этилового эфира саркозина в качестве исходных материалов с получением 220 мг (56%) чистого масла: Элем. анал. рассч. дляC20H27N3O3: С, 67,21; Н, 7,61; N, 11,76. Найдено: С, 66,93; Н, 7,73; N, 11,77. Пример 46. [(1-Циклопентил-3-этил-1 Ниндазол-6-карбонил)-метиламино]-уксусная кислота. Это соединение получают по методике примера 42, используя 201 мг (0,562 ммоль, 1,0 экв.) этилового эфира [(1-циклопентил-3-этил 1 Н-индазол-6-карбонил)-метиламино]-уксусной кислоты в качестве исходного материала с получением 185 мг (100%) белого аморфного твердого вещества: Элем. анал. рассч. дляC18H23N3O31/4H2O: С, 64,74; Н, 7,09; N, 12,58. Найдено: С, 64,73; Н, 7,55; N, 12,47. Пример 47. (Бензилоксикарбамоилметил)метиламид 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 37, используя 160 мг (0,486 ммоль, 1,0 экв.)[(1-циклопентил-3-этил-1 Н-индазол-6 карбонил)-метиламино]-уксусной кислоты в качестве исходного материала с получением 134 мг (64%) чистого масла: ВРМС рассч. для С 25 Н 30N4O3+Н: 435,2398. Найдено: 435,2376. Пример 48. Гидроксикарбамоилметилметиламид 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 38, используя 126 мг (0,290 ммоль, 1,0 экв.) (бензилоксикарбамоилметил)метиламида 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала и 40 мг Рd(ОН)2/С (катализатор Пэрлмена) в качестве катализатора с получением 78 мг (78%) светлого рыжевато-коричневого порошка: т.пл. 63 С 53 Пример 49. [(Гидроксиметилкарбамоил)метил]-амид 1-циклопентил-3-этил-1 Н-индазол 6-карбоновой кислоты. 390 мг (16,9 ммоль, 20 экв.) натрия, 3-8 мм сферы, добавляют к 10 мл метанола порциями в течение 30 мин. По каплям добавляют раствор 707 мг (8,47 ммоль, 10 экв.) N-метилгидроксиламин гидрохлорида в 10 мл метанола и реакционную смесь перемешивают в течение 10 мин. По каплям добавляют раствор 279 мг (0,847 ммоль, 1,0 экв.) метилового эфира [(1 циклопентил-3-этил-1H-индазол-6-карбонил)амино]-уксусной кислоты в 10 мл метанола и реакционную смесь перемешивают в течение 16 ч при комнатной температуре. Смесь затем концентрируют до 1/2 от ее первоначального объема, разбавляют 150 мл H2O, подкисляют до рН 2 и экстрагируют 250 мл этилацетатом. Органические экстракты объединяют, промывают 220 мл H2O, 120 мл солевым раствором и сушат над Na2SO4. Фильтрация, концентрация фильтрата и сушка дают 0,33 г чистого масла,которое очищают на колонке с силикагелем(10% CH3OH/CH2Cl2, флэш) с получением 236 мг белой пены. Растирание с пентаном дает 150 мг (51%) белого аморфного твердого соединения: т.пл. 60 С (разл). Элем. анал. рассч. для С 18 Н 24N4 О 3 Н 2 О: С, 59,65; Н, 7,23; N, 15,46. Найдено: С, 62,04; Н, 7,39; N, 15,86. Пример 50. 50 А. Трет-бутиловый эфирS-(1 бензилоксикарбамоилэтил)-карбаминовой кислоты. Это соединение получают по методике примера 44, используя 500 мг (2,64 ммоль, 1,0 экв.) N-(трет-бутоксикарбонил)-L-аланина и 422 мг (2,64 ммоль, 1,0 экв.) O-бензилгидроксиламин гидрохлорида с получением 583 мг(75%) белого масляного твердого вещества: 1H ЯМР (300 МГц, СDСl3)9,02 (шс, 1 Н), 7,37 (м,5 Н), 4,95 (м, 1 Н), 4,90 (с, 2 Н), 4,03 (м, 1 Н), 1,41(с, 9 Н), 1,33 (д, 3 Н, J=7,0 Гц); МС (Cl, NH3) m/z 295 (М+Н+, основание). 50 В. Гидрохлорид S-2-амино-N-бензилоксипропионамида НСl (г) барботируют в раствор при температуре 0 С 561 г (1,91 ммоль, 1,0 экв.) трет-бутилового эфира S-(1-бензилоксикарбамоилэтил)-карбаминовой кислоты в 10 мл безводного 1,4-диоксана в течение 1-2 мин. Реакционную смесь перемешивают при комнатной температуре в течение 45 мин, затем концентрируют на роторном испарителе и сушат в высоком вакууме с получением 492 мг (100%) белого гигроскопичного аморфного твердого вещества: 1H ЯМР (300 МГц, ДМСО-d6)11,8 54 Это соединение получают по методике примера 37, используя 200 мг (0,774 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 180 мг (0,774 ммоль, 1,0 экв.) S-2-амино-N-бензилоксипропионамид гидрохлорида в качестве исходных материалов с получением 322 мг (96%) чистого масла: ВРМС. рассч. для С 25 Н 30N4O3+Н: 435,2396. Найдено: 435,2424. Пример 52. (1-Гидроксикарбамоилэтил)амидS-1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. Это соединение получают по методике примера 38, используя 288 мг (0,663 ммоль, 1,0 экв.) (1-бензилоксикарбамоилэтил)-амида S-1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала и 90 мг(разл.); ВРМС. рассч. для C18H24N4O3+H: 345,1927. Найдено: 345,1923. Пример 53. Трет-бутиловый эфир R-(1 бензилоксикарбамоилэтил)-карбаминовой кислоты. Это соединение получают по методике примера 37, используя 500 мг (2,64 ммоль, 1,0 экв.) N-(трет-бутоксикарбонил)-D-аланина и 422 мг (2,64 ммоль, 1,0 экв.) O-бензилгидроксиламин гидрохлорида с получением 592 мг (76%) белого масляного твердого вещества: 1R-1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты. 54 А. Гидрохлорид R-2-амино-N-бензилоксипропионамида НСl (г) барботируют при температуре 0 С в раствор 570 г (1,94 ммоль, 1,0 экв.) трет-бутилового эфира R-(1-бензилоксикарбамоилэтил)-карбаминовой кислоты в 10 мл безводного 1,4-диоксана в течение 1-2 мин. Реакционную смесь перемешивают при комнатной температуре в течение 45 мин, затем концентрируют на роторном испарителе и сушат в высоком вакууме с получением 512 мг (100%) белого гигроскопичного аморфного твердого вещества: 1H ЯМР (300 МГц, ДМСО-d6)11,8R-1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 37, используя 200 мг (0,774 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты и 180 мг (0,774 ммоль, 1,0 экв.)R-2-амино-N-бензилоксипропионамид гидрохлорида в качестве исходных материалов с получением 330 мг (98%) чистого масла: ВРМС 55 рассч. для С 25 Н 30N4 О 3+Н: 435,2396. Найдено: 435,2414. 54 С. (1-Гидроксикарбамоилэтил)-амид R1-циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты. Это соединение получают по методике примера 30, используя 295 мг (0,679 ммоль, 1,0 экв.) (1-бензилоксикарбамоилэтил)-амида R-1 циклопентил-3-этил-1 Н-индазол-6-карбоновой кислоты в качестве исходного материала и 90 мг(разл.); ВРМС. рассч. для С 18 Н 24N4O3+Н: 345,1927. Найдено: 345,1927. Пример 55. 1-Циклопентил-3-этил-6 тиофен-2-ил-1 Н-индазол. Раствор 76 мг (0,598 ммоль, 1,2 экв.) тиофен-2-борной кислоты в 0,5 мл метанола добавляют при комнатной температуре к суспензии 146 мг (0,498 ммоль, 1,0 экв.) 6-бром-1 циклопентил-3-этил-1 Н-индазола и 17 мг(0,0149 ммоль, 0,03 экв.) Рd(РРh3)4 в 2 мл толуола и 0,5 мл 2 М водного Na2 СО 3. Смесь нагревают с обратным холодильником в течение 4 ч,затем охлаждают до комнатной температуры,реакционную смесь разбавляют 50 мл этилацетата, промывают 110 мл каждый H2O, солевым раствором и сушат над MgSO4. Неочищенный продукт очищают на колонке с силикагелем(3% этилацетат/гексан, флэш) с получением 81 мг (55%) чистого масла, которое кристаллизуется при выстаивании: т.пл. 60-64 С (разл.); ВРМС. рассч. для C18H20N2S+H: 297,1427. Найдено: 297,1484. Пример 56. 1-Циклопентил-3-этил-6 фенил-1 Н-индазол. Это соединение получают по методике примера 55, используя 128 мг (0,437 ммоль, 1,0 экв.) 6-бром-1-циклопентил-3-этил-1 Н-индазола и 75 мг (0,612 ммоль, 1,4 экв.) фенил борной кислоты в качестве исходных материалов с получением 98 мг (77%) белых кристаллов: т.пл. 72-74 С. Элем. анал. рассч. для C20H22N2: С,82,77; Н, 7,64; N, 9,65. Найдено: С, 81,95; Н,7,82; N, 9,75. Пример 57. (3,5-Дихлорпиридин-4-ил)метиламид 1-циклопентил-3-этил-1H-индазол-6 карбоновой кислоты. 5,2 мг (0,130 ммоль, 1,05 экв.) гидрида натрия, 60% дисперсия в масле, добавляют при комнатной температуре к раствору 50 мг (0,124 ммоль, 1,0 экв.) (3,5-дихлорпиридин-4-ил)амида 1-циклопентил-3-этил-1 Н-индазол-6 карбоновой кислоты в 3 мл безводного ДМФ. Через 30 мин добавляют 7,7 мкл (0,124 ммоль,1,0 экв.) йодметана и смесь перемешивают при комнатной температуре в течение 4 ч реакционную смесь разбавляют 50 мл Н 2 О и экстрагируют 220 мл этилацетата. Экстракты этилацетата объединяют, промывают 225 мл Н 2 О, 15 мл солевым раствором и сушат над Na2SO4. 56 Фильтрация, концентрация фильтрата и сушка дают желтое масло, которое очищают на колонке с силикагелем (25% этилацетат/гексан,флэш), с получением 27 мг (52%) белого кристаллического твердого вещества: т.пл. 118119 СC21H22N4OCl3+H: 417,12519. Найдено: 417,12270. Пример 58. Диметиламид 1-циклопентил 3-этил-1 Н-индазол-6-карбоновой кислоты. 47 мг (0,246 ммоль, 1,1 экв.) 1-(3 диметиламинопропил)-3-этилкарбодиимид гидрохлорида добавляют при комнатной температуре одной порцией к раствору 57,8 мг (0,224 ммоль, 1,0 экв.) 1-циклопентил-3-этил-1 Ниндазол-6-карбоновой кислоты, 18 мг (0,224 ммоль, 1,0 экв.) диметиламин гидрохлорида, 34 мг (0,224 ммоль, 1,0 экв.) гидрата гидроксибензтриазола и 66 мкл (0,470 ммоль, 2,1 экв.) триэтиламина в 5,0 мл безводного метиленхлорида. После перемешивания реакционной смеси в течение 18 ч в атмосфере N2, реакционную смесь разбавляют 40 мл этилацетата, промывают 10 мл 1N НСl, водой и солевым раствором и сушат над Nа 2SO4. Неочищенный продукт очищают на колонке с силикагелем (50% или их фармацевтически приемлемые соли,в которых R является Н, C1-C9 алкилом,-(СН 2)m (5-10-членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С 6-С 10 арилом),где b и с независимо равны от 0 до 1, Z1 является C1-С 6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,гидрокси, C1-C5 алкил, С 2-С 5 алкенил, C1-C6 алкокси,трифторметил,нитро,-CO2R12,-С(O)NR12R13, -NR12R13 и -SO2NR12R13;R1 является Н; или C1-C9 алкилом, необязательно замещенным от 1 до 3 заместителями,независимо выбранными из группы, включающей метил, этил, трифторметил и галоген;R7 является С 6-С 10 арилом; или 5-10 членным гетероциклилом, где указанные R7 группы необязательно замещены от 1 до 3 за 57 местителями, независимо выбранными из группы, включающей галоген, трифторметил, циано,нитро, -CO2R12, C1-C4 алкокси, -OC(O)(C1-C4 алкил), -NR12C(O)(C1-C4 алкил), -C(O)NH2,-C(O)NHOH, -С(О)О(C1-C4 алкил), C1-C4 алкил,-S(O)nR12, где n равно от 0 до 2, бензоил,-NR12R13, -OR12, C1-C6 алканоил, -Y1-(C6-C10 арил), -С(O)O(С 6-С 10 арил), -NН(С 6-С 10 арил),-С(О)NН(С 6-С 10 арил), -С(O)NR12O(CH2)n(C6-C10 арил), где n равно от 1 до 3 и -SO2NН(С 6-С 10 арил);R12 и R13 каждый независимо являются Н или C1-C4 алкилом;R19 является фенилом, нафтилом, пирролилом, фуранилом, тиенилом, оксазолилом, пиридинилом, пиримидинилом, пиридазинилом, хинолинилом, изохинолинилом, 5,6,7,8-тетрагидрохинолинилом или 5,6,7,8-тетрагидроизохинолинилом; где указанные R19 группы, за исключением указанного фенила, необязательно замещены от 1 до 3 группами заместителей R23 и где указанный фенил необязательно замещен от 1 до 3 группами заместителей, независимо выбранных из R23 и R24;R23 каждый независимо выбирают из группы, включающей галоген, C1-C6 алкил, C1-C7 алкокси, C2-C6 алкилендиокси, трифторметил,-NR12R13, нитро, -С(NR12)NR12R13, -С(O)NR12R24 каждый независимо выбирают из группы, включающей имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, оксадиазолил, тиадиазолил, тиазолил, оксазолидинил, тиазолидинил и имидазолидинил, где каждый R24 необязательно замещен от 1 до 3 группами заместителей R23;-NHC(O)NH-, где для указанных Z3 групп n равно от 0 до 4 и m равно от 1 до 3; при условии, что(1) R1 и R не могут быть оба одновременно(3) если R2 определен как -Z3-R7, где Z3 является -C(O)NH(CH2)n-, где n равно от 0 до 4,или -NHC(O)O-, и где R7 является необязательно замещенным фенилом, R отличен от -(CH2)2 тетразолила и необязательный заместитель на группе R отличен от -CO2R12. 2. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(Y1)NH-, -C(O)CH2-,-NHC(Y1)-, -Y1-CH2-, -ОС(О)-, -СН=СН- или-C(Y1)C(Y1)-; и R7 является необязательно замещенным арильной или гетероарильной группой, выбранной из группы, включающей фенил,пиридил, пиразинил, тиенил, пиримидинил, 2,4 диоксопиримидин-5-ил, изоксазолил, изотиазолил, пиридазинил и 1,2,4-триазинил. 3. Соединение по п.2, в котором R7 является замещенным фенилом, 2,6-дигалогензамещенным фенилом или 3,5-дигалопирид-4 илом. 4. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -С(O)NH(CH2)n- или-NНС(О)(CH2)n-, где n равно от 0 до 1 и R7 является фенилом или пиридилом, каждый из которых необязательно замещен от 1 до 3 заместителями, независимо выбранными из группы,включающей галоген, нитро, трифторметил,-СО 2 СН 3, метил, метокси и -С(O)NН 2. 5. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(O)NH- и R7 является фенилом или пиридилом, каждый из которых независимо замещен от 1 до 3 заместителями,независимо выбранными из группы, включающей галоген, C1-C4 алкил, C1-C4 алкокси, циано,карбокси и -ОС(О)(C1-C4 алкил). 6. Соединение по п.1, в котором R2 является R19, где R19 необязательно замещен тиенилом,пиримидинилом или пиридазинилом. 7. Соединение по п.1, в котором R2 является фенилом, замещенным R24, где указанный R24 является оксадиазолилом, тиадиазолилом или тетразолилом, где указанные R24 группы могут быть необязательно замещены C1-C2 алкилом или где R2 является фенилом, замещенным цианометилом, гидрокси или формилом. 8. Соединение по п.1, в котором R2 является -Z3-R7, где Z3 является -C(Y1)NH- и R7 является фенилом, тиенилом, пиразинилом, пиримидинилом, изоксазолилом или пиридилом, где каждая из указанных R7 групп может быть необязательно замещена от 1 до 3 заместителями,независимо выбранными из группы, включающей галоген, метоксикарбонил, трифторметил,бензоил, ацетил, диметиламино, гидрокси, нитро, метил, циано, метилсульфонил и метилтио. 9. Соединение по п.1, в котором R2 является -C(O)NR8(CHR12)mC(O)NR12(CH2)pOR12 или-С(O)NR12(CHR12)mS(C1-C4 алкилом), где R8, R12,m и p такие, как определены в п.1. 10. Соединение по п.1, выбранное из группы соединений, включающей 60 1-циклопентил-3-этил-6-тиофен-2-ил-1 Ниндазол; 1-циклопентил-3-этил-6-фенил-1 Н-индазол; и фармацевтически приемлемая соль любого из перечисленных выше соединений. 11. Соединение формулы (ХХХХ) или их фармацевтически приемлемые соли,где R является Н; C1-C9 алкилом; -(СН 2)m(5-10-членным гетероциклилом), где m равно от 0 до 2; или (Z1)b(Z2)c(С 6-С 10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С 6 алкиленом или С 2-С 8 алкениленом и Z2 является О, S, SО 2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,гидрокси, C1-C5 алкил, С 2-С 5 алкенил, C1-C6 алкокси,трифторметил,нитро,-CO2R12,-С(O)NR12R13, -NR12R13 и -SO2NR12R13;R1 является Н или C1-C9 алкилом, необязательно замещенным от 1 до 3 заместителями,независимо выбранными из группы, включающей метил, этил, трифторметил и галоген;R12 и R13 каждый независимо являются Н или C1-C4 алкилом; и
МПК / Метки
МПК: C07D 401/12, A61P 11/06, A61K 31/415
Метки: опухоли, индазола, фдэ, производные, продуцирования, ингибиторов, типа, фосфодиэстеразы, использование, некроза, фактора, фно, качестве
Код ссылки
<a href="https://eas.patents.su/30-2113-proizvodnye-indazola-i-ih-ispolzovanie-v-kachestve-ingibitorov-fosfodiesterazy-fde-tipa-iv-i-producirovaniya-faktora-nekroza-opuholi-fno.html" rel="bookmark" title="База патентов Евразийского Союза">Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)</a>
Бензо[с]хинолизиновые производные, их получение и использование в качестве ингибиторов 5&alpha-редуктаз

Номер патента: 975
Опубликовано: 28.08.2000
Авторы: Серио Марио, Гуарна Антонио
МПК: A61P 13/08, C07D 455/04, A61K 31/435...
Метки: ингибиторов, производные, использование, получение, бензо[с]хинолизиновые, качестве, 5&alpha-редуктаз
Формула / Реферат:
1. Бензо[с]-хинолизиновые соединения формулы (I) (I) в которой R1, R2, R3, R4, R6 одинаковые или различные выбирают из группы, состоящей из Н, C1-8-алкила, С2-8-алкенила, C2-8-алкинила, циклопропана, циклобутана, циклопентана, циклогексана, циклогептана, циклооктана, норборнана, канфана, адамантана, фенила, нафтила, насыщенного или ароматического гетероцикла, содержащего один или более атомов N, галогена, CN, азида, NRR',...
Производные пиридазин-3-она, их использование в качестве гербицидов и промежуточные соединения для их получения.

Номер патента: 868
Опубликовано: 26.06.2000
Авторы: Мине Йоко, Санемицу Юзуру, Кавамура Синити, Катаяма Тадаси
МПК: C07D 237/14, C07C 251/76, A01N 43/58...
Метки: пиридазин-3-она, качестве, соединения, производные, использование, получения, гербицидов, промежуточные
Формула / Реферат:
1. Соединение формулы в которой R1 представляет C1-С3 галогеналкил; R2 и R3 являются одинаковыми или различными и представляют водород, C1-С3алкил, C1-С3галогеналкил или C1-С3алкокси C1-С3алкил; и Q представляет [Q-1], [Q-2], [Q-3], [Q-4] формулы где в формуле Q-1 Х представляет Н, F или Сl, Y представляет Сl, F или Вr, R1 представляет СF3, R2 представляет Н или СН3, R3 представляет Н или СН3, В представляет водород, нитро,...
Производные спиропиперидина и их использование в качестве терапевтических агентов

Номер патента: 1574
Опубликовано: 25.06.2001
Авторы: Свейн Кристофер Джон, Холлингворт Грегори Джон, Харрисон Тимоти, Эллиотт Джейсон Мэттью, Сьюард Эйлин Мэри, Бейкер Рэймонд, Кулаговский Януш Йозеф, Кертис Нил Рой, Вилльямс Брайан Джон, Джексон Филип Стефен
МПК: A61P 1/08, C07D 491/10, A61K 31/4355...
Метки: использование, производные, терапевтических, спиропиперидина, агентов, качестве
Формула / Реферат:
1. Соединение формулы (I) где R1 означает водород, гидрокси, C1-6-алкил, С2-6-алкенил, С3-7-циклоалкил, С3-7-циклоалкил-С1-4-алкил, C1-6-алкокси, фтор-С1-6-алкокси, C1-6-алкокси-С1-4-алкил, С1-б-алкокси-С1-4-алкокси, фтор-С1-6-алкокси-С1-4-алкил, С2-6-алкенилокси, С3-7-циклоалкокси, С3-7-циклоалкил-С1-4-алкокси, фенокси, бензилокси, циано, галоген, NRaRb, SRa, SORa, SO2Ra, OSO2Ra, NRaCOR14, CORa, CO2Ra или CONRaRb, где Ra и Rb независимо друг...
Способ получения ингибиторов фосфодиэстеразы iv

Номер патента: 877
Опубликовано: 26.06.2000
Авторы: Волант Ральф П., Молина Одри, Хоупис Иоаннис
МПК: A61K 31/4409, A61P 11/06, C07D 213/30...
Метки: фосфодиэстеразы, способ, получения, ингибиторов
Формула / Реферат:
1. Способ получения соединения структурной формулы 1 который включает стадии: 1) обработки соединения структурной формулы 2 катализатором, Ni(acac)2, в простом эфирном растворителе при -35 - -15шС с последующей обработкой цинкатом формулы R13M и выдерживанием в течение 20-30 ч, с получением аддукта 4 2) обработки аддукта 4 в простом эфирном растворителе и органической кислоте металлическим Zn, с получением продукта 1, где R1...
Производные пирролопирролона в качестве ингибиторов нейтрофильной эластазы

Номер патента: 1437
Опубликовано: 26.02.2001
Авторы: Макдоналд Саймон Джон Фосетт, Смит Робин Эндрю, Гаррисон Ли Эндрю, Инглис Грэм Джордж Адам, Финч Гарри, Джонсон Мартин Редпат, Ша Прайтом, Доул Майкл Деннис
МПК: A61P 11/00, C07D 487/04, A61K 31/407...
Метки: пирролопирролона, качестве, ингибиторов, нейтрофильной, эластазы, производные
Формула / Реферат:
1. Соединение общей формулы (I) где R1 представляет собой C1-6алкил, С2-6алкенил; арил, арил-С1-4алкил, арил-С2-4алкенил, гетероарил, гетероарил-С1-4алкил или гетероарил-С2-4-алкенил, или такую группу, в которой арильная или гетероарильная группировка замещена одной или более чем одной С1-4алкильной, галогено, тетразолильной, трифторметилсульфонамидной, NR9CO-C1-8алкильной, -(CH2)m-NR4R5, -CN, -COOR9, -CONR9R10, -NO2, -SO2-С1-6алкильной, -CF3...
Предыдущий патент: Носитель данных и способ его изготовления
Следующий патент: Новые ингибиторы фарнезилтрансферазы, способы их получения, содержащие их фармацевтические композиции и их применение для получения медикаментов.
Случайный патент: Световозвращающее покрытие из стеклянных микросфер, в котором предусмотрено изображение, имеющее визуальную направленность