Гетероциклические сульфонамиды, их применения и фармацевтические композиции
Номер патента: 21057
Опубликовано: 31.03.2015
Авторы: Флири Антон Франц Джозеф, Шварц Джейкоб Брэдли, Сигелстейн Барбара Эйлин, Галлащун Рэндэлл Джеймс, О'доннелл Кристофер Джон







Формула / Реферат
1. Соединение формулы I или его фармацевтически приемлемая соль

где каждый R1 и каждый R2 независимо представляет собой галоген, гидроксил, -CN, -(NR8)-(C=O)-R8,
-(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -N(R8)2, -SO2-N(R8)2 или (C1-C6)алкил, где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9;
m равен 0 или 1;
n равен 0, 1, 2 или 3;
s равен 1 и t равен 1; или один из s или t равен 1, а другой из s или t равен 2;
R3 представляет собой водород;
R5 представляет собой водород;
R6 представляет собой (C1-C6)алкил-SO2-;
R8 независимо представляет собой водород, (C1-C6)алкил или (C3-C10)циклоалкил; где указанный (C1-C6)алкил возможно может быть замещен одним, двумя или тремя галогено;
каждый R9 независимо представляет собой галоген или -(NR10)-SO2-R10;
R10 независимо представляет собой водород или (C1-C6)алкил;
кольцо "A" представляет собой (C6-C10)арил, гетероарил, (С4-C10)циклоалкил или гетероциклоалкил; где два из указанных заместителей, представляющих собой R1, указанного (С4-C10)циклоалкила и указанного гетероциклоалкила возможно могут быть присоединены к одному и тому же атому углерода и возможно могут быть взяты вместе с образованием оксо, где (C6-C10)арил представляет собой фенил, нафтил, тетрагидронафтил или инданил; где гетероарил представляет собой 5-10-членный гетероарил, содержащий от одного до четырех гетероатомов, независимо выбранных из О, S и N; и где гетероциклоалкил представляет собой азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил, тетрагидродиазинил, оксазинил, оксатиазинил, индолинил, изоиндолинил, хинуклидинил, хроманил, изохроманил или бензоксазинил;
кольцо "В" представляет собой фенил или пиридил;
"X" представляет собой -O- или >CH2.
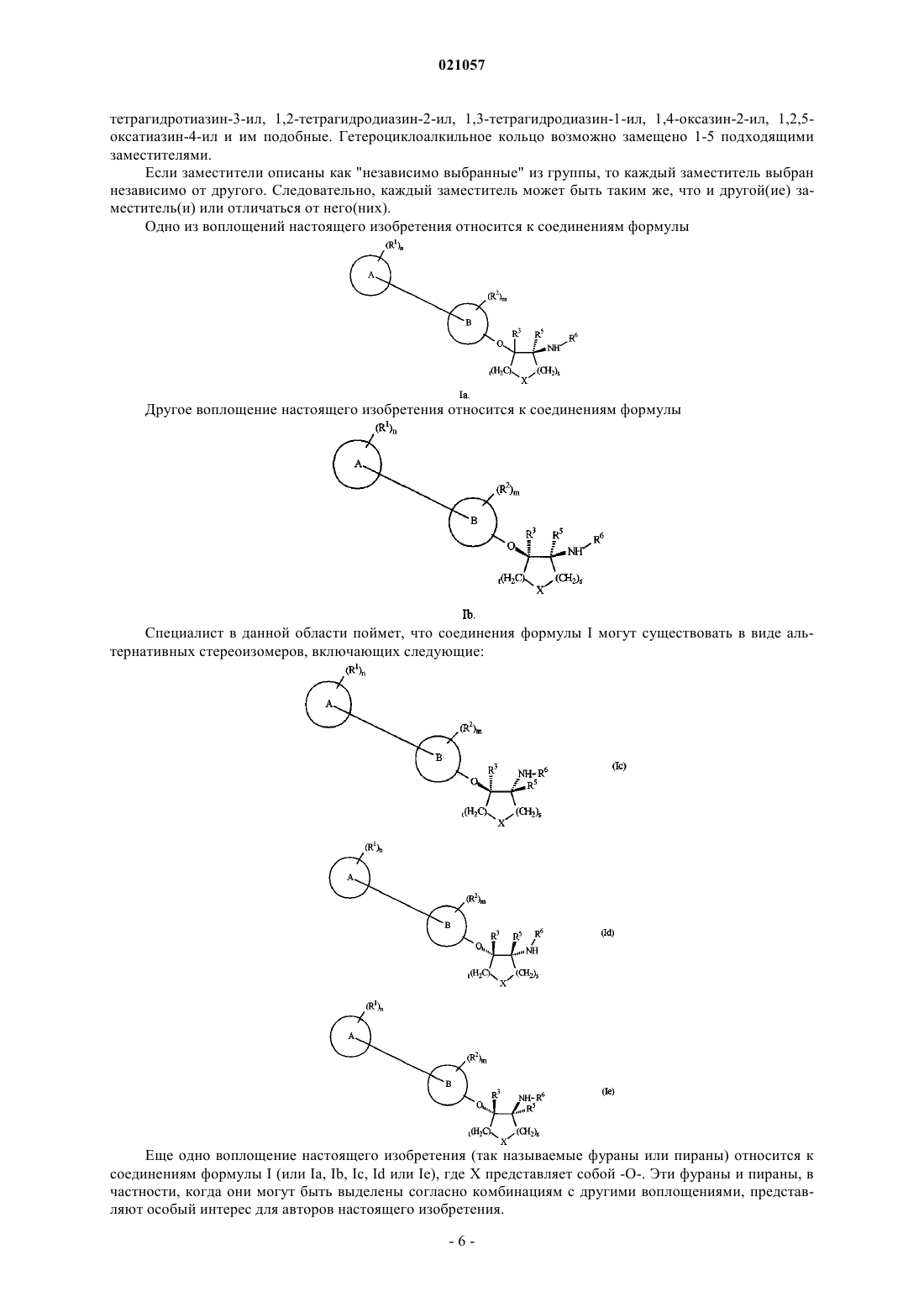
2. Соединение по п.1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой соединение формулы Ia

3. Соединение по п.1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой соединение формулы Ib

4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, где X представляет собой
-O-.
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где кольцо "A" представляет собой фенил; n равен 0, 1 или 2; R1 представляет собой галоген, гидроксил, -CN, -(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -N(R8)2, -SO2-N(R8)2 или (С1-C6)алкил; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9.
6. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, где кольцо "A" представляет собой гетероарил; n равен 0, 1 или 2 и где R1 представляет собой галоген, гидроксил, -CF3, -CN, -OR8,
-N(R8)2 или (C1-C6)алкил.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, где кольцо "В" представляет собой фенил; m равен 0 или 1 и R2 представляет собой галоген.
8. Соединение по любому из пп.1-7 или его фармацевтически приемлемая соль, где m равен 0.
9. Соединение по любому из пп.1-8 или его фармацевтически приемлемая соль, где R6 представляет собой (С1-С5)алкил-SO2-.
10. Соединение по п.1 или его фармацевтически приемлемая соль, где указанное соединение представляет собой


11. Соединение по п.1, представляющее собой N-{(3S,4S)-4-[4-(5-циано-2-тиенил)фенокси]тетрагидрофуран-3-ил}пропан-2-сульфонамид или его фармацевтически приемлемая соль.
12. Соединение по п.11, представляющее собой N-{(3S,4S)-4-[4-(5-циано-2-тиенил)фенокси]тетрагидрофуран-3-ил}пропан-2-сульфонамид.
13. Способ лечения или предупреждения у млекопитающего состояния, выбранного из группы, состоящей из острых неврологических и психиатрических расстройств, удара, церебральной ишемии, травмы спинного мозга, травмы головы, перинатальной гипоксии, остановки сердца, гипогликемического нейронного повреждения, деменции, болезни Альцгеймера, хореи Хантингтона, бокового амиотрофического склероза, повреждения глаз, ретинопатии, когнитивных нарушений, идиопатической и индуцированной лекарством болезни Паркинсона, мышечных спазмов и расстройств, ассоциированных с мышечной спастичностью, включающих тремор, эпилепсии, конвульсий, мигрени, недержания мочи, толерантности к веществу, синдрома отмены вещества, психоза, шизофрении, тревоги, расстройств настроения, невралгии тройничного нерва, тугоухости, шума в ушах, дегенерации желтого пятна глаза, рвоты, отека мозга, боли, поздней дискинезии, расстройств сна, синдрома дефицита внимания/гиперактивности, синдрома дефицита внимания и кондуктивного расстройства, включающий введение млекопитающему соединения по любому из пп.1-12 или его фармацевтически приемлемой соли.
14. Способ по п.13, где состояние выбрано из группы, состоящей из когнитивного нарушения, шизофрении и тугоухости.
15. Способ по п.13, где состоянием является когнитивное нарушение.
16. Способ по п.13, где состоянием является шизофрения.
17. Способ по п.13, где состоянием является тугоухость.
18. Фармацевтическая композиция, имеющая AMPA (рецептор α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты)-потенцирующую активность, содержащая соединение по любому из пп.1-12 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
19. Фармацевтическая композиция по п.18, содержащая соединение по п.12 и фармацевтически приемлемый носитель.
Текст
Это изобретение относится к классу соединений, включающему фармацевтически приемлемые соли соединений, имеющих структуру формулы I, определенную в описании изобретения. Изобретение также относится к применениям соединений формулы I и композициям, содержащим их. Флири Антон Франц Джозеф,Галлащун Рэндэлл Джеймс,О'Доннелл Кристофер Джон, Шварц Джейкоб Брэдли, Сигелстейн Барбара Эйлин (US) Поликарпов А.В. (RU) Область изобретения Настоящее изобретение относится к новому классу соединений, имеющих структуру формулы I,определенную в данном описании изобретения, и фармацевтическим композициям, содержащим соединение формулы I или его фармацевтически приемлемую соль. Настоящее изобретение также включает способы лечения субъекта путем введения субъекту терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Эти соединения полезны для состояний, раскрытых в данном описании. Кроме того, настоящее изобретение включает способы получения соединений формулы I и соответствующих промежуточных соединений. Предшествующий уровень техники Первичным возбуждающим нейротрансмиттером в центральной нервной системе млекопитающих(ЦНС) является аминокислота глутамат, чья сигнальная трансдукция опосредована либо ионотропными,либо метаботропными глутаматными рецепторами (GluR). Ионотропные глутаматные рецепторы (iGluR) состоят из трех подтипов, дифференцированных вследствие их уникальных ответов на три селективных агониста iGluR -амино-3-гидрокси-5-метилизоксазол-4-пропионовую кислоту (AMPA), N-метил-Dаспартат (NMDA) и каинат (Parsons С. G., Danysz W. and Lodge D. (2002) в: Ionotropic Glutamate Receptorsas Therapeutic Targets (Danysz W., Lodge D. and Parsons С. G. eds), pp 1-30, F.P. Graham Publishing Co.,Tennessee). AMPA рецепторы, белковоподобные гомо- или гетеротетрамеры, состоящие из любой комбинации четырех из приблизительно 900 аминокислотных мономерных субъединиц, кодируемых, каждая, отдельным геном (GluA1-A4) С каждым субъединичным белком, существующим в виде одного из двух сплайс-вариантов, принимаемых как "flip" и "flop", опосредуют огромное большинство возбуждающих синаптических трансмиссий в головном мозге млекопитающего и были предложены в течение длительного времени в качестве интегрального компонента нервной цепи, которая опосредует когнитивные процессы (Bleakman D. and Lodge D. (1998) Neuropharmacology of AMPA and Kainate Receptors. Neuropharmacology 37:1187-1204). Комбинация различных гетеротетрамерных вариантов, двух сплайс-форм для каждого из четырех iGluR мономеров и РНК-редактирование рецепторной субъединицы с гетерогенным распределением AMPA рецепторов во всем головном мозге подчеркивают огромное количество потенциальных AMPA рецепторных ответов в пределах этого органа (Black M. D. (2005) Therapeutic PotentialData. Psychopharmacology 179:154-163). В настоящее время модуляторы AMPA стали активной мишенью для разработки лекарств (см. Rogers В. and Schmidt С. (2006) Novel Approaches for the Treatment of Schizophrenia, Annual Reports in Medicinal Chemistry 3-21). Краткое изложение сущности изобретения Настоящее изобретение относится к соединениям, включая фармацевтически приемлемые соли соединений, имеющим структуру формулы где каждый R1 и каждый R2 независимо представляет собой галоген, гидроксил, -CN, -(NR8)-(C=O)R , -(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -N(R8)2, -SO2-N(R8)2 или (C1-C6)алкил, где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9;s равен 1 и t равен 1; или один из s или t равен 1, а другой из s или t равен 2;R8 независимо представляет собой водород, (C1-C6)алкил или (C3-C10)циклоалкил; где указанный(C1-C6)алкил возможно может быть замещен одним, двумя или тремя галогено; каждый R9 независимо представляет собой галоген или -(NR10)-SO2-R10;R10 независимо представляет собой водород или (C1-C6)алкил; кольцо "А" представляет собой (C6-C10)арил, гетероарил, (С 4-C10)циклоалкил или гетероциклоалкил; где два из указанных заместителей, представляющих собой R1, указанного (С 4-C10)циклоалкила и указанного гетероциклоалкила, возможно могут быть присоединены к одному и тому же атому углерода и воз 8 можно могут быть взяты вместе с образованием оксо, где (C6-C10)арил представляет собой фенил, нафтил, тетрагидронафтил или инданил; где гетероарил представляет собой 5-10-членный гетероарил, содержащий от одного до четырех гетероатомов, независимо выбранных из О, S и N; и где гетероциклоалкил представляет собой азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил,пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил, тетрагидродиазинил, оксазинил, оксатиазинил, индолинил,изоиндолинил, хинуклидинил, хроманил, изохроманил или бензоксазинил; кольцо "B" представляет собой фенил или пиридил;"X" представляет собой -О- или CH2. Предпочтительными являются соединения формулы Также предпочтительными являются соединения формулы Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где X представляет собой -O-. Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где кольцо "A" представляет собой фенил; n равен 0, 1 или 2; R1 представляет собой галоген,гидроксил, -CN, -(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -N(R8)2, -SO2-N(R8)2 или (C1-C6)алкил; где указанный(C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где кольцо "A" представляет собой гетероарил; n равен 0, 1 или 2; и где R1 представляет собой галоген, гидроксил, -CF3, -CN, -OR8, -N(R8)2 или (С 1-С 6)алкил. Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где кольцо "В" представляет собой фенил; m равен 0 или 1 и R2 представляет собой галоген. Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где m равен 0. Также предпочтительными являются соединения по изобретению или их фармацевтически приемлемые соли, где R6 представляет собой (C1-C5)алкил-SO2-. Также предпочтительными являются следующие соединения по изобретению или их фармацевтически приемлемые соли: Также предпочтительным является соединение по изобретению, представляющее собой N-(3S,4S)4-[4-(5-циано-2-тиенил)фенокси]тетрагидрофуран-3-илпропан-2-сульфонамид или его фармацевтически приемлемая соль. Более предпочтительным является соединение по изобретению, представляющее собой N-(3S,4S)4-[4-(5-циано-2-тиенил)фенокси]тетрагидрофуран-3-илпропан-2-сульфонамид. Настоящее изобретение также относится к способу лечения или предупреждения у млекопитающего состояния, выбранного из группы, состоящей из острых неврологических и психиатрических расстройств, удара, церебральной ишемии, травмы спинного мозга, травмы головы, перинатальной гипоксии, остановки сердца, гипогликемического нейронного повреждения, деменции, болезни Альцгеймера,хореи Хантингтона, бокового амиотрофического склероза, повреждения глаз, ретинопатии, когнитивных нарушений, идиопатической и индуцированной лекарством болезни Паркинсона, мышечных спазмов и расстройств, ассоциированных с мышечной спастичностью, включающих тремор, эпилепсии, конвульсий, мигрени, недержания мочи, толерантности к веществу, синдрома отмены вещества, психоза, шизофрении, тревоги, расстройств настроения, невралгии тройничного нерва, тугоухости, шума в ушах, дегенерации желтого пятна глаза, рвоты, отека мозга, боли, поздней дискинезии, расстройств сна, синдрома дефицита внимания/гиперактивности, синдрома дефицита внимания и кондуктивного расстройства,включающему введение млекопитающему соединения по изобретению или его фармацевтически приемлемой соли. Предпочтительным является способ по изобретению, где состояние выбрано из группы, состоящей из когнитивного нарушения, шизофрении и тугоухости. Более предпочтительным является способ по изобретению, где состоянием является когнитивное нарушение. Также более предпочтительным является способ по изобретению, где состоянием является шизофрения. Также более предпочтительным является способ по изобретению, где состоянием является тугоухость. Кроме того, настоящее изобретение относится к фармацевтической композиции, имеющей AMPA(рецептор -амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты)потенцирующую активность,содержащую соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, где предпочтительно соединение по изобретению представляет собой N(3S,4S)-4-[4-(5-циано-2-тиенил)фенокси]тетрагидрофуран-3-илпропан-2-сульфонамид. Подробное описание изобретения Термин "алкил", используемый в данном описании, относится к насыщенному, мононенасыщенному и полиненасыщенному гидрокарбильному заместителю с линейной или разветвленной цепью (то есть,заместителю, полученному путем удаления водорода из углеводорода), содержащему от одного до шести атомов углерода; а в другом воплощении от одного до четырех атомов углерода. Моно- и полиненасыщенные заместители, так называемые алкенильные, имеют от 2 до 6 атомов углерода. Алкенильная группа может существовать в чистой Е- (entgegen (напротив форме, чистой Z- (zusammen (вместе форме или любой их смеси. Полиненасыщенная содержит множественные двойные связи и одну или более тройных связей. Такие алкильные группы, содержащие тройную связь, так называемые алкинильная группа, имеют от 2 до 6 атомов углерода. Примеры таких насыщенных заместителей включают метил,этил, пропил (в том числе н-пропил и изопропил), бутил (в том числе н-бутил, изобутил, втор-бутил иmpem-бутил), пентил, изоамил, гексил и им подобные. Примеры ненасыщенного алкила включают этенил, 1-пропенил, 2-пропенил (аллил), изопропенил, 2-метил-1-пропенил, 1-бутенил, 2-бутенил и им подобные. Примеры алкинила включают этинил, пропинил, бутинил, 3,3-диметилбутинил и им подобные. В некоторых случаях количество атомов углерода в гидрокарбильном заместителе (например, алкиле, алкениле, циклоалкиле, циклоалкениле, ариле и так далее) указано с помощью префикса "Cx-Cy-", где х является минимальным, а у является максимальным количеством атомов углерода в заместителе. Так,например, "C1-C6-алкил" относится к алкильному заместителю, содержащему от 1 до 6 атомов углерода. В качестве следующего примера: C3-C6-циклоалкил относится к насыщенному циклоалкилу, содержащему от 3 до 6 кольцевых атомов углерода. Термин "гидрокси" или "гидроксил" относится к -OH. Префикс "гидрокси", когда используется в комбинации с другим(и) термином(ами), указывает на то, что заместитель, к которому присоединен этот префикс, замещен одним или более заместителями, представляющими собой гидрокси. Соединения,имеющие углерод с одним или более заместителями, представляющими собой гидрокси, включают, например, спирты, енолы и фенол. Термин "циано" (также имеющий название "нитрил") означает -CN, который также можно изобразить как -CN. Термин "карбонил" означает -C(O)-, С=О, -(С=О)-, который также можно изобразить как Термин "оксо" относится к=O. Термин "алкокси" относится к алкилу, связанному с кислородом, который также можно представить как -O-R, где R представляет собой алкильную группу. Примеры алкокси включают метокси, этокси, пропокси и бутокси. Термин "сульфонил" относится к -S(O)2-, который также можно изобразить как Таким образом, например, "алкил-сульфонил-алкил" относится к группе алкил-S(O)2-алкил. Примеры алкилсульфонила включают метилсульфонил, этилсульфонил и пропилсульфонил. Термин "циклоалкил", используемый в данном описании, определен как включающий насыщенные или ненасыщенные (неароматические), мостиковые, полициклические, спироциклические или конденсированные полициклические 3-10-членные углеводородные кольца (например, циклопропил, циклобутил,циклопентил, циклогексил, циклогептил, циклооктил, циклононил, бицикло[2.2.1]гептанил, бицикло[3.2.1]октанил и бицикло[5.2.0]нонанил и так далее); возможно замещенные 1-5 подходящими заместителями. Предпочтительно, циклоалкильная группа имеет от 3 до 6 атомов углерода. В одном из воплощений циклоалкил возможно может содержать одну, две или более некумулированных неароматических двойных или тройных связей. Спироциклические кольца являются одним из конкретных видов циклоалкила, который имеет место, когда кольцо образовано вокруг одного атома углерода, по сравнению с конденсированным кольцом, когда кольцо образовано посредством двух общих атомов углерода. Термин "арил", используемый в данном описании, определен как включающий полностью углеродные моноциклические или полициклические с конденсированным кольцом (то есть, кольцами, которые имеют общие, расположенные рядом пары атомов углерода) группы, имеющие полностью конъюгированную пи-электронную систему. Арильная группа имеет 6, 8, 9 или 10 атомов углерода в кольце(ах). Более предпочтительно, арильная группа имеет 6 или 10 атомов углерода в кольце(ах). Наиболее предпочтительно, арильная группа имеет 6 атомов углерода в кольце(ах). Например, термин "(C6-C10)арил",используемый в данном описании, означает ароматические радикалы, содержащие от 6 до 10 атомов углерода, такие как фенил, нафтил, тетрагидронафтил, антраценил, инданил и им подобные. Арильная группа возможно замещена 1-5 подходящими заместителями. Термин "гетероарил", используемый в данном описании, определен как включающий моноциклические или полициклические с конденсированным кольцом ароматические гетероциклические группы с одним или более гетероатомами, выбранными из О, S и N в одном или более чем одном из указанных колец. Гетероарильная группа имеет от 5 до 12 кольцевых атомов, в том числе от одного до пяти гетероатомов, независимо выбранных из О, S и N. Одно или более чем одно из указанных колец указанной гетероциклической группы может не содержать гетероатомов. Предпочтительно гетероарильная группа имеет от 5 до 10 кольцевых атомов, в том числе от одного до четырех гетероатомов. Более предпочтительно, гетероарильная группа имеет от 5 до 8 кольцевых атомов, в том числе один, два или три гетероатома. Наиболее предпочтительно гетероарильная группа имеет от 6 до 8 кольцевых атомов, в том числе один или два гетероатома. Например, термин "гетероарил", используемый в данном описании, означает ароматические радикалы, содержащие по меньшей мере один кольцевой гетероатом, независимо выбранный из О, S и N, и от 1 до 9 атомов углерода, такие как пиридил, пиразинил, пиримидинил, пиридазинил, тиенил, фурил, имидазолил, пирролил, оксазолил (например, 1,3-оксазолил, 1,2-оксазолил), тиазолил (например, 1,2-тиазолил, 1,3-тиазолил), пиразолил, тетразолил, триазолил (например, 1,2,3 триазолил, 1,2,4-триазолил), оксадиазолил (например, 1,2,3-оксадиазолил), тиадиазолил (например, 1,3,4 тиадиазолил), хинолил, изохинолил, бензотиенил, бензофурил, индолил и им подобные. Гетероарильная группа возможно замещена 1-5 подходящими заместителями. Термин "гетероциклоалкил", используемый в данном описании, определен как включающий моноциклическое, мостиковое, полициклическое, спироциклическое или конденсированное полициклическое насыщенное или ненасыщенное неароматическое 3-20-членное кольцо, содержащее 1 или более гетероатомов, независимо выбранных из О, S и N. Одно или более чем одно из указанных колец указанной мостиковой, полициклической или конденсированной гетероциклической группы может не содержать гетероатомов. Примеры таких гетероциклоалкильных колец включают азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил, пиразолидинил,тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил, тетрагидродиазинил, оксазинил, оксатиазинил, индолинил, изоиндолинил, хинуклидинил, хроманил, изохроманил, бензоксазинил и им подобные. Дополнительными примерами указанных гетероциклоалкильных колец являются тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, имидазолидин-1-ил, имидазолидин-2-ил, имидазолидин-4-ил, пирролидин-1-ил, пирролидин-2-ил, пирролидин-3-ил, пиперидин-1-ил, пиперидин-2-ил,пиперидин-3-ил, пиперазин-1-ил, пиперазин-2-ил, пиперазин-3-ил, 1,3-оксазолидин-3-ил, изотиазолидин,1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,3-пиразолидин-1-ил, 1,2-тетрагидротиазин-2-ил, 1,3-5 021057 тетрагидротиазин-3-ил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, 1,4-оксазин-2-ил, 1,2,5 оксатиазин-4-ил и им подобные. Гетероциклоалкильное кольцо возможно замещено 1-5 подходящими заместителями. Если заместители описаны как "независимо выбранные" из группы, то каждый заместитель выбран независимо от другого. Следовательно, каждый заместитель может быть таким же, что и другой(ие) заместитель(и) или отличаться от него(них). Одно из воплощений настоящего изобретения относится к соединениям формулы Другое воплощение настоящего изобретения относится к соединениям формулы Специалист в данной области поймет, что соединения формулы I могут существовать в виде альтернативных стереоизомеров, включающих следующие: Еще одно воплощение настоящего изобретения (так называемые фураны или пираны) относится к соединениям формулы I (или Ia, Ib, Ic, Id или Ie), где X представляет собой -O-. Эти фураны и пираны, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения (так называемые циклопентилы или циклогексилы) относится к соединениям формулы I (или Ia, Ib, Ic, Id или Ie), где X представляет собой CH2. Эти циклопентилы или циклогексилы, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, также представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "A" представляет собой фенил; более конкретно, где n равен 0, 1 или 2; более конкретно, где R1 выбран из группы, состоящей из галогена, гидроксила, -CN, -(NR8)-(C=O)-R8, -(C=O)-OR8,-(C=O)-N(R8)2, -OR8, -SO2-N(R8)2 и (C1-C6)алкила; где указанный (C1-C6)алкил возможно замещен одним,двумя, тремя или четырьмя R9. Эти "A" фенильные соединения, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на"циклопентилы" или "циклогексилы", также представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "A" представляет собой гетероарил; более конкретно, где n равен 0, 1 или 2; и более конкретно, где R1 выбран из группы, состоящей из галогена, гидроксила, -CN, -(NR8)-(C=O)-R8, -(C=O)OR8, -(C=O)-N(R8)2, -OR8, -SO2-N(R8)2 и (C1-C6)алкила; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Эти "A" гетероарильные соединения, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на воплощения "X" "циклопентилы" или "циклогексилы", также представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "A" представляет собой гетероциклоалкил; более конкретно, где n равен 0, 1 или 2; и более конкретно, где R1 выбран из группы, состоящей из галогена, гидроксила, -CN, -(NR8)-(C=O)-R8,-(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -SO2-N(R8)2 и (C1-C6)алкила; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Эти "A" гетероциклоалкильные соединения, в частности,когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на воплощения "X" "циклопентилы" или "циклогексилы", также представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "A" представляет собой (С 4-C10)циклоалкил; более конкретно, где n равен 0, 1 или 2; и более конкретно, где R1 выбран из группы, состоящей из галогена, гидроксила, -CN, -(NR8)-(C=O)-R8,-(C=O)-OR8, -(C=O)-N(R8)2, -OR8, -SO2-N(R8)2 и (C1-C6)алкила; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Эти "A" (С 4-C10)циклоалкильные соединения, в частности,когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на воплощения "X" "циклопентилы" или "циклогексилы", также представляют особый интерес для авторов настоящего изобретения. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где R1 представляет собой (C1-C6)алкил, циано или галоген и находится в орто- или параположении относительно "В". Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "В" представляет собой фенил; более конкретно, где m равен 0 или 1; более конкретно, где R2 представляет собой галоген, гидроксил, -CN, -(NR8)-(C=O)-R8, -(C=O)-OR8, -(C=O)-N(R8)2,-OR8, -SO2-N(R8)2 и (C1-C6)алкил; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Эти "В" фенильные соединения, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на воплощения"X" "циклопентилы" или "циклогексилы", представляют особенно повышенный интерес для авторов настоящего изобретения. Каждое из этих воплощений также образует дополнительные, представляющие интерес воплощения с воплощениями кольца "A", описанными выше. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где кольцо "В" представляет собой пиридил; более конкретно, где m равен 0 или 1; и более конкретно, где R2 представляет собой галоген, гидроксил, -CN, -(NR8)-(C=O)-R8, -(C=O)-OR8, -(C=O)-N(R8)2,-OR8, -SO2-N(R8)2 и (C1-C6)алкил; где указанный (C1-C6)алкил возможно замещен одним, двумя, тремя или четырьмя R9. Эти "В" пиридильные соединения, в частности, когда они могут быть выделены согласно комбинациям с другими воплощениями, среди которых особенное внимание обращают на воплощения "X" "циклопентилы" или "циклогексилы", также представляют особенно повышенный интерес для авторов настоящего изобретения. Каждое из этих воплощений также образует дополнительные,представляющие интерес воплощения с воплощениями кольца "A", описанными выше. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где R2 представляет собой (C1-C6)алкил, циано или галоген. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id или Ie), где m равен 0. Еще одно воплощение настоящего изобретения относится к соединению формулы I (или Ia, Ib, Ic, Id-7 021057 или Ie), где R6 представляет собой (С 1-С 5)алкил-SO2-. Еще одно воплощение изобретения также относится к каждому из индивидуальных соединений,описанных в примерах 1-54 в разделе "Примеры" из этого описания изобретения, и их фармацевтически приемлемым солям. Конкретные предпочтительные соединения по изобретению включают Когда в соединении формулы I (которая, как подразумевают в этом описании в дальнейшем, означает формулу I, Ia, Ib, Ic, Id или Ie), ниже называемом как "соединение по изобретению", присутствует асимметрический центр, соединение может существовать в форме оптических изомеров (энантиомеров). В одном из воплощений настоящее изобретение включает энантиомеры и смеси, в том числе рацемические смеси соединений формулы I. В другом воплощении, касающемся соединений формулы I, содержащих более одного асимметрического центра, настоящее изобретение включает диастереомерные формы (индивидуальные диастереомеры и их смеси) соединений. Когда соединение формулы I содержит алкенильную группу или группировку, могут быть геометрические изомеры. Настоящее изобретение включает таутомерные формы соединений формулы I. В тех случаях, когда структурные изомеры являются взаимопревращаемыми посредством низкого энергетического барьера,может иметь место таутомерная изомерия ("таутомерия"). Она может принимать форму протонной таутомерии в соединениях формулы I, содержащих, например, группы имино, кето или оксим, или так называемой валентной таутомерии в соединениях, которые содержат ароматическую группировку. Отсюда следует, что единичное соединение может проявлять более одного типа изомерии. Разные соотношения для таутомеров в твердой и жидкой форме зависят от разнообразных заместителей в молекуле, а также от конкретной методики кристаллизации, используемой для выделения соединения. Подходящие фармацевтически приемлемые соли присоединения кислоты соединений по настоящему изобретению, когда они возможны, включают соли, полученные из неорганических кислот, таких как соляная, бромисто-водородная, фтористо-водородная, борная, фторборная, фосфорная, метафосфорная,азотная, угольная, сульфоновая и серная кислоты, и органических кислот, таких как уксусная, бензолсульфоновая, бензойная, лимонная, этансульфоновая, фумаровая, глюконовая, гликолевая, изотионовая,молочная, лактобионовая, малеиновая, яблочная, метансульфоновая, трифторметансульфоновая, янтарная, толуолсульфоновая, винная и трифторуксусная кислоты. Подходящие органические кислоты обычно включают, например, алифатический, циклоалифатический, ароматический, аралифатический, гетероциклический, карбоновый и сульфоновый классы органических кислот. Кроме того, в случаях, когда соединения по изобретению имеют кислотную группировку, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например соли натрия или калия; соли щелочно-земельных металлов, например соли кальция или магния; и соли, образованные с подходящими органическими лигандами, например соли четвертичного аммония. В другом воплощении основные соли образованы из оснований, которые образуют нетоксичные соли, в том числе соли алюминия, аргинина, бензатина, холина, диэтиламина, диоламина, глицина, лизина, меглумина,оламина, трометамина и цинка. В одном из воплощений также могут быть образованы гемисоли кислот и оснований, например гемисульфат и гемикалыдиевые соли. Соединения по настоящему изобретению могут быть меченны изотопом, то есть они являются такими же, что и перечисленные в формуле I, за исключением того, что один или более атомов заменены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, например 2H, 3H, 13C, 11C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl соответственно. Соединения по настоящему изобретению и фармацевтически приемлемые соли указанных соединений, которые содержат упомянутые выше изотопы и/или другие изотопы других атомов, находятся в пределах объема этого изобретения. Некоторые соединения по настоящему изобретению, меченные изотопом, например соединения, в которые включены такие радиоактивные изотопы, как 3H и 14C, полезны в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, то есть 3H, и углерода-14, то есть 14C, являются особенно предпочтительными вследствие легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2H, может предоставить некоторые терапевтические преимущества, вытекающие из большей метаболической стабильности, например увеличенный период полувыведения in vivo или потребность в сниженной дозировке, и,следовательно, в некоторых случаях может быть предпочтительным. Соединения формулы I по этому изобретению, меченные изотопом, обычно могут быть получены путем осуществления методик, раскрытых ниже на схемах и/или в примерах и подготовительных примерах, посредством замены немеченного изотопом реагента на легкодоступный меченный изотопом реагент. Соединения формулы I и их фармацевтически приемлемые соли полезны для лечения множества неврологических и психиатрических расстройств, ассоциированных с глутаматной дисфункцией, включая: острые неврологические и психиатрические расстройства, такие как церебральные недостаточности после операции шунтирования на сердце и трансплантации, удар, церебральная ишемия, травма спинного мозга, травма головы, перинатальная гипоксия, остановка сердца, гипогликемическое нейронное повреждение, деменция (в том числе деменция, индуцированная СПИДом), болезнь Альцгеймера, хорея Хантингтона, боковой амиотрофический склероз, повреждение глаз, ретинопатия, когнитивные расстройства, идиопатическая и индуцированная лекарством болезнь Паркинсона, мышечные спазмы и расстройства, ассоциированные с мышечной спастичностью, в том числе треморы, эпилепсия, конвульсии,мигрень (в том числе головная боль при мигрени), недержание мочи, толерантность к веществу, синдром отмены вещества (в том числе таких веществ, как опиаты, никотин, табачные продукты, алкоголь, бензодиазепины, кокаин, седативные средства, снотворные средства и так далее), психоз, шизофрения, тревога(в том числе генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство и обсессивно-компульсивное расстройство),расстройства настроения (в том числе депрессия, мания, биполярные расстройства), невралгия тройничного нерва, тугоухость, шум в ушах, дегенерация желтого пятна глаза, рвота, отек мозга, боль (в том числе острые и хронические болевые состояния, страшная боль, неустранимая боль, невропатическая боль и посттравматическая боль), поздняя дискинезия, расстройства сна (в том числе нарколепсия), расстройство дефицита внимания/гиперактивности, расстройство дефицита внимания и кондуктивное расстройство. Соответственно, в одном из воплощений изобретения предложен способ осуществления лечения состояния у млекопитающего, такого как человек, выбранного из состояний, упомянутых выше,включающий введение млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Млекопитающим предпочтительно является млекопитающее, нуждающееся в таком лечении или предупреждении. Термин "осуществление лечения" ("treating"), используемый в данном описании, если не указано иное, означает реверсирование, облегчение, модулирование, ингибирование прогрессирования или предупреждение расстройства или состояния, к которому такой термин применяется, или одного или более симптомов такого расстройства или состояния. Термин "лечение" ("treatment"), используемый в данном описании, если не указано иное, относится к акту осуществления лечения, такого как оно ("осуществление лечения") определено непосредственно выше. Например, согласно изобретению предложен способ осуществления лечения состояния, выбранного из мигрени, тревожных расстройств, шизофрении и эпилепсии. Типичными тревожными расстройствами являются генерализованное тревожное расстройство, социальное тревожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство и обсессивно-компульсивное расстройство. В качестве другого примера, согласно изобретению предложен способ осуществления лечения депрессии, выбранной из глубокой депрессии, хронической депрессии (дистимии), сезонной депрессии (сезонного аффективного расстройства), психотической депрессии и послеродовой депрессии. В качестве еще одного примера, согласно изобретению предложен способ осуществления лечения расстройства сна, выбранного из бессонницы и депривации сна. В другом воплощении изобретение включает способы осуществления лечения состояния млекопитающего, такого как человек, путем введения млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли, где состояние выбрано из группы, состоящей из атеросклеротических сердечно-сосудистых заболеваний, цереброваскулярных заболеваний и заболеваний периферических артерий. Млекопитающим предпочтительно является млекопитающее, нуждающееся в таком лечении или предупреждении. Другие состояния, которые можно лечить согласно настоящему изобретению, включают гипертензию и ангиогенез. В еще одном воплощении настоящего изобретения предложены способы осуществления лечения неврологических и психиатрических расстройств, ассоциированных с глутаматной дисфункцией, включающие введение млекопитающему, предпочтительно млекопитающему, нуждающемуся в этом, соединения формулы I или его фармацевтически приемлемой соли в количестве, эффективном при лечении таких расстройств. Соединение формулы I или его фармацевтически приемлемую соль возможно применяют в комбинации с другим активным агентом. Таким активным агентом может быть, например, атипичное антипсихотическое средство или AMPA-потенцирующее средство. Соответственно, в другом воплощении изобретения предложены способы осуществления лечения неврологических и психиатрических расстройств,ассоциированных с глутаматной дисфункцией, включающие введение млекопитающему эффективного количества соединения формулы I или фармацевтически приемлемой соли и дополнительно включающие введение другого активного агента. Термин "другой активный агент", используемый в данном описании, относится к любому терапевтическому агенту, отличающемуся от соединения формулы (I) или его соли, который полезен для лечения расстройства субъекта. Примеры дополнительных терапевтических агентов включают антидепрессанты, антипсихотические средства, противоболевые средства, средства против болезни Альцгеймера и успокаивающие средства. Примеры конкретных классов антидепрессантов, которые можно применять в комбинации с соединениями по изобретению, включают ингибиторы обратного захвата норэпинефрина,селективные ингибиторы обратного захвата серотонина (SSRI), антагонисты NK-1 (нейрокинин-1) рецептора, ингибиторы моноаминоксидазы (MAOI), обратимые ингибиторы моноаминоксидазы (RIMA),ингибиторы обратного захвата серотонина и норадреналина (SNRI), антагонисты кортикотропинвысвобождающего фактора (CRF), антагонисты -адренорецептора и атипичные антидепрессанты. Подходящие ингибиторы обратного захвата норэпинефрина включают четвертичные аминные трициклические соединения и вторичные аминные трициклические соединения. Примеры подходящих четвертичных аминных трициклических соединений и вторичных аминных трициклических соединений включают амитриптилин, кломипрамин, доксепин, имипрамин, тримипрамин, дотиепин, бутриптилин, иприндол,лофепрамин, нортриптилин, протриптилин, амоксапин, дезипрамин и мапротилин. Примеры подходящих селективных ингибиторов обратного захвата серотонина включают флуоксетин, флувоксамин, пароксетин и сертралин. Примеры ингибиторов моноаминоксидазы включают изокарбоксазид, фенелзин и транилциклопрамин. Примеры подходящих обратимых ингибиторов моноаминоксидазы включают моклобемид. Примеры применения в настоящем изобретении подходящих ингибиторов обратного захвата серотонина и норадреналина включают венлафаксин. Примеры подходящих атипичных антидепрессантов включают бупропион, литий, нефазодон, тразодон и вилоксазин. Примеры средств против болезни Альцгеймера включают димебон, антагонисты NMDA рецептора, такие как мемантин; и ингибиторы холинестеразы, такие как допенезил и галантамин. Примеры подходящих классов успокаивающих средств, которые можно применять в комбинации с соединениями по изобретению, включают бензодиазепины и агонисты или антагонисты серотонина 1A (5-HT1 А), особенно частичные агонисты 5-HT1 А, и антагонисты кортикотропин-высвобождающего фактора (CRF). Подходящие бензодиазепины включают алпразолам, хлордиазепоксид, клоназепам, хлоразепат, диазепам, галазепам, лоразепам, оксазепам и празепам. Подходящие агонисты или антагонисты 5-HT1 А рецептора включают буспирон, флезиноксан, гепирон и ипсапирон. Подходящие атипичные антипсихотические средства включают палиперидон, бифепрунокс,зипрасидон, рисперидон, арипипразол, оланзапин и кветиапин. Подходящие никотиновые ацетилхолиновые агонисты включают изпрониклин, варениклин и MEM 3454. Противоболевые средства включают прегабалин, габапентин, клонидин, неостигмин, баклофен, мидазолам, кетамин и зиконотид. Изобретение также относится к фармацевтическим композициям, содержащим соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Соединения формулы I могут быть получены способами, описанными ниже, вместе со способами синтеза, известными в области органической химии, или модификациями и способами получения производных, которые хорошо знакомы специалистам в данной области. В процессе осуществления любой из следующих ниже последовательностей синтеза может быть необходимой и/или желательной защита чувствительных или реакционноспособных групп любой из молекул, имеющих отношение к этому. Ее можно достичь посредством традиционных защитных групп,таких как группы, описанные в книге Т. W. Greene, Protective Groups in Organic Chemistry, John WileySons, 1999, которая включена в данное описание путем ссылки. Как понятно специалисту, применение формулы I имеет преимущество, и следует полагать, что изобретение включает все до единого разновидности, подпадающие под него, несмотря на индивидуально изложенные в данном описании. Таким образом, в изобретении предполагается каждая разновидность по отдельности и в любой и во всех комбинациях таких разновидностей. В частности, R1, R2, R3, R5, R6,R8, R9, R10, m, n, s, t, А, В и X на следующей ниже схеме являются такими, как определено выше. Схема 1 Схема 1 относится к получению соединений формулы I. Следуя схеме 1, арилгалогенид формулы II,где L1 представляет собой йодо, бромо или трифлат, можно подвергнуть реакции сочетания с подходящим образом замещенной арилбороновой кислотой, имеющей структуру (R1)n-ArB(OH)2, где Ar представляет собой подходящим образом замещенную арильную или гетероарильную группу, и В представляет собой бор, в стандартных условиях реакции кросс-сочетания, катализируемой палладием, хорошо известных специалисту в данной области, с получением соединения формулы I. [Suzuki, A., Journal of(1995).] Соединения формулы II могут быть получены из соединений формулы III путем замещения L2,где L2 может представлять собой галогено, -OSO2CH3 (-OMs) или -OSO2CF3 (-OTf), реагентом Типичные условия включают проведение реакции в органическом растворителе, таком как ацетонитрил, в присутствии основания, такого как карбонат цезия, при повышенной температуре, такой как 150C. Альтернативно, соединение формулы III можно превратить в соединение формулы II, где L2 представляет собой OH, путем реакции нуклеофильного ароматического замещения (такой как реакция с арилоловом, таким как SnAr) с подходящим образом замещенным арильным реагентом где L2 представляет собой галогено или -OSO2CF3 (-OTf), в соответствии со способами, аналогичными способам, описанным Withbroe, G. J.; Singer, R. A.; Sieser, J. E. в "Streamlined Synthesis of the Bippyphos Family of Ligands and Cross-Coupling Applications" Org. Process Res. Dev. 2008, 12, 480-489. Типичные условия включают проведение реакции в органическом растворителе, таком как этанол, в присутствии основания, такого как гидроксид калия, катализатора, такого как палладиевый (такой какPd2(dba)3), и лиганда, такого как 1-[2-[бис(трет-бутил)фосфино]фенил]-3,5-дифенил-1 Н-пиразол (bippyphos), при повышенной температуре, такой как 80C. Альтернативно, соединение формулы I может быть получено из соединения формулы II, где L1 представляет собой синильную группу (такую как триметилсилил), путем превращения сначала силильной группы до галогенида, например путем взаимодействия с галогенирующим реагентом, таким как бромид калия/N-хлорсукцинимид (NCS), в присутствии кислоты (такой как уксусная кислота), с последующим арилированием, как описано выше. Подходящие растворители для галогенирования включают спирты, такие как метанол или этанол. Реакцию можно проводить при температуре от примерно 10C до примерно 60C в течение от примерно 10 до примерно 120 мин. Соединение формулы II может быть получено из соединения формулы III путем сочетания с подходящим образом замещенным арильным реактивом Гриньяра в эфирном растворителе, таком как THF(тетрагидрофуран), при от примерно -30C до примерно комнатной температуры. Протеканию реакции может способствовать катализатор, такой как палладий или медь. Соединения формулы III имеются в продаже или могут быть получены способами, хорошо известными специалистам в данной области, или могут быть получены стандартными способами, известными в данной области (такими как способы, раскрытые в обычных справочниках, таких как COMPENDIUM OFORGANIC SYNTHETIC METHODS, Vol. I-VI (опубликованном Wiley-Interscience. Соединения формулы I можно разделить на энантиомерно чистые изомеры в соответствии со способами, хорошо известными специалистам в данной области и подробно описанными в разделе "Примеры" из этого описания изобретения. Соединения по этому изобретению можно применять в форме солей, полученных из неорганических или органических кислот. Соли присоединения кислоты к соединениям по этому изобретению,представляющим собой основание, легко получают путем обработки соединения, представляющего собой основание, по существу, эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. Желаемую твердую соль получают после осторожного выпаривания растворителя. Соли оснований можно легко получить путем обработки соответствующих кислотных соединений водным раствором, содержащим желаемые фармацевтически приемлемые катионы, а затем путем выпаривания полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно, их также можно получить путем смешивания вместе низших алканольных растворов кислотных соединений и алкоксида желаемого щелочного металла, а затем путем выпаривания полученного раствора досуха таким же образом, как описано выше. В любом случае для обеспечения полноты реакции и максимальных выходов продукта предпочтительно применяют стехиометрические количества реагентов. В тех случаях, когда соль предназначена для введения пациенту (в отличие от, например, применения в in vitro контексте), соль предпочтительно является фармацевтически приемлемой. Термин "фармацевтически приемлемая соль" относится к соли, полученной путем сочетания соединения формулы I с кислотой, чей анион, или основанием, чей катион, обычно считается подходящим для потребления человеком. Фармацевтически приемлемые соли являются особенно полезными в качестве продуктов способов по настоящему изобретению вследствие их более высокой растворимости в воде по сравнению с исходным соединением. Для применения в медицине солями соединений по этому изобретению являются нетоксичные "фармацевтически приемлемые соли". Обычно соединение по изобретению вводят в количестве, которое эффективно для лечения или предупреждения состояния, описанного здесь. Соединения по изобретению вводят любым подходящим путем в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для намеченного лечения или предупреждения. Терапевтически эффективные дозы соединений, требуемые для лечения или предупреждения прогрессирования медицинского состояния, легко устанавливаются специалистом в данной области, использующим преклинические и клинические подходы, которые хорошо знакомы специалистам в области медицины. Соединения по изобретению можно вводить перорально. Пероральное введение может включать глотание, так что соединение поступает в желудочно-кишечный тракт, или можно применять трансбуккальное или подъязычное введение, посредством которого соединение поступает в кровоток непосредственно из полости рта. В другом воплощении соединения по изобретению можно вводить непосредственно в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенный, внутриартериальный, внутрибрюшинный, интратекальный, интравентрикулярный, интрауретральный, интрастернальный, внутричерепной, внутримышечный и подкожный. Подходящие устройства для парентерального введения включают игольчатые (включая микроигольчатые) инжекторы, безыгольные инжекторы и оборудование для инфузии. В другом воплощении соединения по изобретению можно вводить местно на кожу или слизистую оболочку, то есть дермально или трансдермально. В еще одном воплощении соединения по изобретению можно вводить интраназально или путем ингаляции. В еще одном воплощении соединения по изобретению можно вводить ректально или вагинально. В еще одном воплощении соединения по изобретению можно вводить непосредственно в глаз или ухо. Схема приема соединений и/или композиций, содержащих соединения, основана на множестве факторов, включающих тип, возраст, массу, пол и медицинское состояние пациента; тяжесть состояния; путь введения и активность конкретного применяемого соединения. Таким образом, схему приема можно широко варьировать. Уровни дозирования примерно от примерно 0,01 до примерно 100 мг на килограмм массы тела в сутки являются полезными при лечении или предупреждении указанных выше состояний. В одном из воплощений суммарная суточная доза соединения по изобретению (вводимого в однократной или разделенных дозах) обычно составляет от примерно 0,01 до примерно 100 мг/кг. В другом воплощении суммарная суточная доза соединения по изобретению составляет от примерно 0,1 до примерно 50 мг/кг, а в еще одном воплощении от примерно 0,5 до примерно 30 мг/кг (то есть, мг соединения по изобретению на кг массы тела). В одном из воплощений дозирование составляет от 0,01 до 10 мг/кг/сутки. В другом воплощении дозирование составляет от 0,1 до 1,0 мг/кг/сутки. Единицы дозирования композиций могут содержать такие количества или их дольные единицы, чтобы составить в итоге суточную дозу. Во многих случаях введение соединения будут повторять множество раз в сутки (обычно не больше, чем 4 раза). При желании, для увеличения суммарной суточной дозы, обычно можно применять многократные дозы в сутки. Для перорального введения композиции могут быть предоставлены в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 75,0, 100, 125, 150, 175, 200, 250 или 500 мг активного ингредиента для симптоматической корректировки дозирования пациенту. Лекарственное средство обычно содержит от примерно 0,01 до примерно 500 мг активного ингредиента или, в другом воплощении, от примерно 1 до примерно 100 мг активного ингредиента. Внутривенные дозы могут находиться в диапазоне от примерно 0,1 до примерно 10 мг/кг/мин при постоянной скорости инфузии. В соответствии с настоящим изобретением подходящие субъекты включают млекопитающих субъектов. В соответствии с настоящим изобретением млекопитающие включают собачьих, кошачьих, коров,коз, лошадей, овец, свиней, грызунов, зайцеобразных, приматов и им подобных, но не ограничиваются ими, и охватывают млекопитающих в утробе. В одном из воплощений подходящими субъектами являются люди. Субъекты, представляющие собой людей, могут быть любого пола и на любой стадии развития. В другом воплощении изобретение включает применение одного или более соединений по изобретению для изготовления лекарственного средства для лечения или предупреждения состояний, перечисленных в данном описании. Для лечения или предупреждения вышеуказанных состояний соединение по изобретению можно вводить в виде соединения как такового. Альтернативно, для медицинских применений подходят фармацевтически приемлемые соли соединений вследствие их более высокой растворимости в воде по сравнению с исходными соединениями. В другом воплощении настоящее изобретение включает фармацевтические композиции. Такие фармацевтические композиции содержат соединение по изобретению или его фармацевтически приемлемую соль, представленную с фармацевтически приемлемым носителем. Носитель может представлять собой твердое вещество, жидкость или и то и другое, и может быть приготовлен вместе с соединением в виде стандартной лекарственной композиции, например таблетки, которая может содержать от 0,05 до 95 мас.% активных соединений. Соединение по изобретению можно соединять с подходящими полимерами в качестве имеющих цель лекарственных носителей. Также могут присутствовать другие фармакологи- 13021057 чески активные вещества. Соединения по настоящему изобретению и их фармацевтически приемлемые соли можно вводить любым подходящим путем, предпочтительно в форме фармацевтической композиции, адаптированной к такому пути, и в дозе, эффективной для намеченного лечения или предупреждения. Активные соединения, их фармацевтически приемлемые соли и композиции, например, можно вводить перорально, ректально, парентерально или местно. Пероральное введение твердой лекарственной формы может быть, например, представлено дискретными единицами, такими как твердые или мягкие капсулы, пилюли, облатки, пастилки или таблетки,каждая из которых содержит заранее определенное количество по меньшей мере одного соединения по настоящему изобретение или его фармацевтически приемлемой соли. В другом воплощении пероральное введение может быть в порошковой или гранульной форме. В еще одном воплощении пероральная лекарственная форма является подъязычной, такой как, например, пастилка. В таких твердых лекарственных формах соединения формулы I или их фармацевтически приемлемая соль обычно объединены с одним или более адъювантами. Такие капсулы или таблетки могут содержать композицию с контролируемым высвобождением. В случае капсул, таблеток и пилюль лекарственные формы также могут содержать буферные агенты или могут быть изготовлены с энтеросолюбильными покрытиями. В другом воплощении пероральное введение может быть в жидкой лекарственной форме. Жидкие лекарственные формы для перорального введения включают, например, фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры, содержащие инертные разбавители, широко используемые в данной области (например, воду). Такие композиции также могут содержать адъюванты, такие как увлажняющие агенты, эмульгаторы, суспендирующие агенты, корригенты (например, подсластители) и/или придающие запах агенты. В другом воплощении настоящее изобретение включает парентеральную лекарственную форму."Парентеральное введение" включает, например, подкожные инъекции, внутривенные инъекции, внутрибрюшинное введение, внутримышечные инъекции, интрастернальные инъекции и инфузию. Согласно сведениям, известным из данной области, инъецируемые препараты (например, стерильные инъецируемые водные или масляные суспензии) могут быть изготовлены с использованием подходящих диспергирующих, увлажняющих агентов и/или суспендирующих агентов. В другом воплощении настоящее изобретение включает местную лекарственную форму. "Местное введение" включает, например, трансдермальное введение, такое как посредством трансдермальных пластырей или устройств для ионтофореза, внутриглазное введение или интраназальное введение либо введение путем ингаляции. Композиции для местного введения также включают, например, местные гели,спреи, мази и кремы. Местная композиция может включать соединение, которое усиливает поглощение или проникновение активного ингредиента через кожу или другие пораженные области. Когда соединения по этому изобретению и их фармацевтически приемлемые соли вводят с помощью трансдермального устройства, введение будут осуществлять с использованием пластыря либо из емкости с мембраной пористого типа, либо из множества твердых матриц. Типичные композиции для этой цели включают гели,гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пены, пленки, кожные пластыри, пластинки, имплантаты, губки, волокна, бандажи и микроэмульсии. Также можно применять липосомы. Типичные носители включают спирт, воду, минеральное масло, вазелиновое масло, медицинский вазелин,глицерин, полиэтиленгликоль и пропиленгликоль. Могут быть включены усилители проникновения, см.,например, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999). Композиции, подходящие для местного введения в глаз, включают, например, глазные капли, когда соединение по этому изобретению или его фармацевтически приемлемую соль растворяют или суспендируют в подходящем носителе. Типичная композиция, подходящая для глазного или ушного введения,может быть в форме капель микронизированной суспензии или раствора в изотоническом, стерильном солевом растворе с подходящим pH. Другие композиции, подходящие для глазного и ушного введения,включают мази, биоразлагаемые (например, абсорбируемые гелевые губки, коллаген) и небиоразлагаемые (например, силиконовые) имплантаты, пластинки, линзы и корпускулярные или везикулярные системы, такие как ниосомы или липосомы. Полимер, такой как поперечно-сшитая акриловая кислота, поливиниловый спирт, гиалуроновая кислота, целлюлозный полимер, например гидроксипропилметилцеллюлоза, гидроксиэтилцеллюлоза или метилцеллюлоза, или гетерополисахаридный полимер, например гелановая смола, может быть включен вместе с консервантом, таким как хлорид бензалкония. Такие композиции также можно доставлять путем ионтофореза. Для интраназального введения или введения путем ингаляции активные соединения по изобретению или их фармацевтически приемлемые соли удобно доставлять в виде раствора или суспензии из контейнера пульверизатора, который пациент сжимает или в отношении которого осуществляет откачивание, или путем предоставления аэрозольной струи из находящегося под давлением контейнера или небулайзера с использованием подходящего пропеллента. Композиции, подходящие для интраназального введения, обычно вводят в виде сухого порошка (либо одного, как в смеси, например в сухой смеси с лактозой, либо в виде смешанной компонентной частицы, например, в смеси с фосфолипидами, такими как фосфатидилхолин) из ингалятора сухого порошка или в виде аэрозольной струи из находящегося под давлением контейнера, насоса, распыляемого раствора, атомайзера (предпочтительно атомайзера с использованием электрогидродинамики с целью получения мелкодисперсного тумана) или небулайзера с использованием подходящего пропеллента, такого как 1,1,1,2-тетрафторэтан или 1,1,1,2,3,3,3 гептафторпропан, или без использования его. Для интраназального применения порошок может содержать биоадгезивный агент, например хитозан или циклодекстрин. В другом воплощении настоящее изобретение включает ректальную лекарственную форму. Такая ректальная лекарственная форма может быть в виде, например, суппозитория. Традиционной суппозиторной основой является масло какао, однако в соответствующих случаях можно применять многочисленные альтернативы. Также можно применять другие материалы, являющиеся носителями, и пути введения, известные в фармацевтике. Фармацевтические композиции по изобретению могут быть изготовлены любым из способов, хорошо известных в фармации, таких как способы эффективного приготовления и методики введения. Приведенные выше рассуждения в отношении способов эффективного приготовления и методик введения хорошо известны в данной области и описаны в обычных пособиях. Изготовление лекарственных средств обсуждается, например, в Hoover, John E., Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pennsylvania, 1975; Liberman, et al., Eds., Pharmaceutical Dosage Forms, Marcel Decker, NewYork, N.Y., 1980; и Kibbe, et al., Eds., Handbook of Pharmaceutical Excipients (3rd Ed.), American Pharmaceutical Association, Washington, 1999. Соединения по настоящему изобретению и их фармацевтически приемлемые соли можно применять по отдельности или в комбинации с другими терапевтическими агентами при лечении или предупреждении различных условий или болезненных состояний. Соединение(я) по настоящему изобретению,его(их) фармацевтически приемлемые соли и другой(ие) терапевтический(е) агент(ы) можно вводить совместно (либо в одной и той же лекарственной форме, либо в раздельных лекарственных формах) или последовательно в любом порядке. Типичным терапевтическим агентом может быть, например, агонист метаботропного глутаматного рецептора. Введение двух или более соединений "в комбинации" означает, что два соединения вводят достаточно близко во времени, так что присутствие одного вносит изменения в биологические эффекты другого. Два или более соединений можно вводить совместно, одновременно или последовательно в любом порядке. Кроме того, совместное введение можно осуществлять путем смешивания соединений до введения или путем введения соединения в один и тот же момент времени, но в разные анатомические области или используя разные пути введения. Фразы "одновременное введение", "со-введение", "совместное введение" и "введенные совместно" означают, что соединения вводят в комбинации. Кроме того, настоящее изобретение включает наборы, которые являются подходящими для применения при осуществлении способов лечения или предупреждения, описанных выше. В одном из воплощений набор содержит первую лекарственную форму, содержащую одно или более соединений по настоящему изобретению или их фармацевтически приемлемую соль, и контейнер для доз в количествах,достаточных для осуществления способов по настоящему изобретению. В другом воплощении набор по настоящему изобретению содержит одно или более соединений по изобретению или их фармацевтически приемлемую соль. Три соединения по изобретению подвергали рентгеновскому определению структуры монокристалла с целью объяснения их абсолютной стереохимии. Кристаллографические данные приведены ниже в данном описании. Типичные кристаллы были тщательно изучены (см. индивидуальные соединения ниже в отношении определения характеристик для наборов данных и используемых дифрактометров). Для облегчения определения абсолютной конфигурации собирали пары Фриделя (Friedel pairs). Атомные факторы рассеяния получали из International Tables for Crystallography, Vol. С, pp. 219, 500, Kluwer Academic Publishers,1992. Все кристаллографические расчеты облегчали путем использования SHELXTL system, Version 5.1,Bruker AXS, 1997. Все дифрактометрические данные собирали при комнатной температуре. Относящийся к делу кристалл, совокупность данных и уточнение структуры суммированы в табл. 1 для каждого соединения. Опытную структуру получали прямыми способами для каждого соединения. Эти опытные структуры уточняли обычным путем. Положения водорода рассчитывали всегда, когда было возможно. Метальные водороды локализовали с помощью разностных методов Фурье, а затем идеализировали. Любые водороды на азоте локализовали с помощью разностных методов Фурье и оставляли для уточнения. Водородные параметры добавляли к расчетам структурных факторов, но не уточняли. Все сдвиги, рассчитанные в конечных циклах уточнения по методу наименьших квадратов, составили меньше чем 0,1 от соответствующих стандартных отклонений. Конечные R-индексы приведены для каждой структуры. Конечное применение разностного Фурье выявило отсутствие недостающей или несоответствующей электронной плотности для любой из этих структур. Абсолютные конфигурации определяли способом, описанным Флаком в Flack, Acta Crystallogr.,1983 А 39, 876. Координаты, температурные анизотропные коэффициенты, расстояния и углы приведены ниже (табл. 1-5) для каждой структуры. Экспериментальные методики Эксперименты обычно проводили в инертной атмосфере (азота или аргона), в частности в случаях,когда использовали реагенты или промежуточные соединения, чувствительные к кислороду или влаге. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки, в том числе, в соответствующих случаях, безводные растворители (обычно продукты Sure-Seal от Aldrich Chemical Company, Milwaukee, Wisconsin). Представленные масс-спектрометрические данные получены путем использования приборов для жидкостной хроматографии - масс-спектрометрии (ЖХ-МС),химической ионизации при атмосферном давлении (ХИАД) или газовой хроматографии - массспектрометрии (ГХ-МС). Химические сдвиги в данных ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (м.д., ) со ссылкой на остаточные пики от используемых дейтерированных растворителей. Реакционные условия (продолжительность реакции и температуру) синтезов, ссылающихся на методики в других примерах, подготовительных примерах или способах, можно варьировать. В общем, за реакциями следовали тонкослойная хроматография или масс-спектрометрия и, в соответствующих случаях, придание продукту окончательного вида. Если в неочищенной реакционной смеси присутствовали твердые вещества, не являющиеся продуктом, можно было использовать фильтрование через Celite. Очистку можно менять от эксперимента к эксперименту: в общем, растворители и соотношения растворителей, используемые для элюантов/градиентов, выбирали для обеспечения подходящих Rf или времен удерживания. Подготовительный пример 1. Синтез цис-N-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3 илпропан-2-сульфонамида Стадия 1. Синтез транс-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3-ола. Соединение, указанное в заголовке стадии 1, получали согласно общей методике синтеза транс-4(4-бромфенокси)тетрагидрофуран-3-ола из примера 2, за исключением того, что вместо 4-бромфенола использовали 6-бромпиридин-3-ол, и неочищенный продукт очищали хроматографией на силикагелеH ЯМР (500 МГц, CDCl3)3.79 (dd, J=9,9, 1,9 Гц, 1H), 3.87 (dd, J=10,4, 1,8 Гц, 1H), 4.00 (dd, J=9,9,4,3 Гц, 1H), 4.19 (dd, J=10,4, 4,7 Гц, 1H), 4.38 (br m, 1H), 4.59 (br s, 1H), 4.68 (br d, J=4,4 Гц, 1H), 7.14 (dd,J=8,7, 3,2 Гц, 1H), 7.32 (dd, J=8,7, 0,5 Гц, 1H), 8.01 (br d, J=3,1 Гц, 1H). Стадия 2. Синтез транс-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3-ил-метансульфоната. Соединение, указанное в заголовке стадии 2, получали согласно общей методике синтеза транс-2(4-бромфенокси)циклопентил-метансульфоната из примера 5, за исключением того, что вместо транс-2(4-бромфенокси)циклопентанола использовали транс-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3 ол. Продукт получали в виде твердого вещества. Выход: 5,95 г, 17,6 ммоль, 87%. 1H ЯМР (400 МГц, CDCl3)3.07 (s, 3H), 3.94 (br dd, J=10,5, 1,8 Гц, 1H), 4.00 (m, 1H), 4.11 (dd,J=11,1, 4,1 Гц, 1H), 4.18 (dd, J=10,6, 4,5 Гц, 1H), 4.97 (br d, J=4,4 Гц, 1H), 5.13 (br d, J=3,8 Гц, 1H), 7.19 (dd,J=8,7, 3,2 Гц, 1H), 7.35 (dd, J=8,7, 0,5 Гц, 1H), 8.04 (dd, J=3,2, 0,5 Гц, 1H). Стадия 3. Синтез цис-N-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3-илпропан-2 сульфонамида. транс-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3-ил-метансульфонат (591,7 мг, 1,75 ммоль),пропан-2-сульфонамид (647 мг, 5,25 ммоль) и карбонат цезия (855 мг, 2,62 ммоль) объединяли в ацетонитриле (8 мл) и подвергали воздействию микроволнового излучения в течение 55 мин при 150C. Неочищенную реакционную смесь объединяли с рядом из нескольких аналогичных реакционных смесей,полученных в тех же самых условиях (суммарное количество использованного исходного материала: 1,527 г, 4,515 ммоль), и встряхивали с насыщенным водным раствором бикарбоната натрия (100 мл). Водный слой экстрагировали этилацетатом и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагелеH ЯМР (400 МГц, CDCl3)1.34 (d, J=6,8 Гц, 3H), 1.36 (d, J=6,8 Гц, 3H), 3.17 (септет, J=6,8 Гц, 1H),3.74 (dd, J=9, 9 Гц, 1H), 3.94 (dd, J=10,9, 1,5 Гц, 1H), 4.14 (dd, J=8, 8 Гц, 1H), 4.19 (dd, J=10,9, 4,3 Гц, 1H),4.27 (m, 1H), 4.84 (m, 1H), 5.66 (d, J=9,9 Гц, 1H), 7.20 (dd, J=8,8, 3,2 Гц, 1H), 7.40 (d, J=8,8 Гц, 1H), 8.01 (d,J=3,2 Гц, 1H). Подготовительный пример 2. Синтез метил-3-циано-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)бензоата. Стадия 1. Синтез метил-3-циано-4-[(трифторметил)сульфонил]оксибензоата. Раствор метил-3-циано-4-гидроксибензоата [см. P. Madsen et al., J. Medicinal Chemistry 2002, 45,5755-5775] (4,18 г, 23,6 ммоль) в дихлорметане (81 мл) обрабатывали 4-(диметиламино)пиридином (432 мг, 3,54 ммоль) и охлаждали до 0C. После добавления триэтиламина (4,93 мл, 35,4 ммоль) раствор обрабатывали трифторметансульфоновым ангидридом (по каплям, 5,96 мл, 35,4 ммоль) и оставляли нагре- 16021057 ваться до комнатной температуры. Через 2 ч реакционную смесь концентрировали в вакууме и повторно обрабатывали дихлорметаном и концентрировали до тех пор, пока не осталось 17 г вещества. Его подвергали хроматографии на силикагеле (градиент: от 0 до 10% этилацетата в гептане) с получением продукта в виде бесцветного масла. Выход: 6,50 г, 21,0 ммоль, 89%. 1H ЯМР (500 МГц, CDCl3)4.00 (s, 3H), 7.60 (d, J=8,8 Гц, 1H), 8.39 (dd, J=8,8, 2,1 Гц, 1H), 8.45 (d,J=2,2 Гц, 1H). Стадия 2. Синтез соединения, представляющего собой метил-3-циано-4-(4,4,5,5-тетраметил-1,3,2 диоксаборолан-2-ил)бензоат. 4,4,4',4',5,5,5',5'-Октаметил-2,2'-би-1,3,2-диоксаборолан (бис(пинаколато)дибор, 5,81 г, 22,9 ммоль),метил-3-циано-4-[(трифторметил)сульфонил]оксибензоат (5,90 г, 19,1 ммоль), ацетат калия (99%, 9,46 г, 95,4 ммоль) и 1,1'-[бис(дифенилфосфино)ферроцен]дихлорпалладий(II) (1,40 г, 1,91 ммоль) объединяли в дегазированном диоксане (83 мл) в толстостенной реакционной колбе. Реакционную колбу герметично закрывали и содержимое нагревали при 100C в течение 18 ч, затем обрабатывали дихлорметаном(100 мл), хорошо перемешивали и фильтровали через Celite. Осадок на фильтре ополаскивали дихлорметаном (2100 мл) и объединенные фильтраты концентрировали в вакууме и подвергали хроматографии на силикагеле (градиент: от 0% до 30% этилацетата в гептане). Фракции, содержащие продукт, концентрировали и подвергали перекристаллизации из 2-пропанола с получением указанного в заголовке соединения в виде белого твердого вещества. Выход: 3,395 г, 11,82 ммоль, 62%. 1H ЯМР (400 МГц, CDCl3)1.41 (s, 12H), 3.97 (s, 3H), 7.98 (d, J=7,8 Гц, 1H), 8.20 (dd, J=7,8, 1,6 Гц,1H), 8.35 (br d, J=1,6 Гц, 1H). Подготовительный пример 3. Синтез N-[(1S,2R)-2-(4-бром-3-фторфенокси)циклопентил]пропан-2 сульфонамида. Стадия 1. Синтез транс-2-(4-бром-3-фторфенокси)циклопентанола. 4-Бром-3-фторфенол (8,00 г, 41,9 ммоль) и 6-оксабицикло[3.1.0]гексан (8,25 мл, 95,2 ммоль) объединяли в бутиронитриле (5,0 мл) и обрабатывали карбонатом натрия (4,04 г, 38,1 ммоль). Реакционную смесь подвергали воздействию микроволнового излучения в течение 2 ч при 175C, затем фильтровали через Celite. Осадок на фильтре промывали этилацетатом, затем дихлорметаном и объединенные фильтраты концентрировали при пониженном давлении с получением продукта в виде темно-коричневого масла. Это вещество использовали без дополнительной очистки. Выход: 11,59 г, больше 41,9 ммоль, полагали, что количественный. 1H ЯМР (400 МГц, CDCl3) 1.61-1.89 (m, 5H), 2.02-2.24 (m, 2H), 4.30 (m, 1H), 4.46 (m, 1H), 6.63 (ddd,J=8,9, 2,8, 1,1 Гц, 1H), 6.73 (dd, J=10,5, 2,8 Гц, 1H), 7.40 (dd, J=8,9, 8,0 Гц, 1H). Стадия 2. Синтез (1R,2R)-2-(4-бром-3-фторфенокси)циклопентилацетата. Соединение, указанное в заголовке стадии 2, получали согласно общей методике синтеза (1R,2R)-2(4-бромфенокси)циклогексилацетата из примера 7, за исключением того, что вместо транс-2-(4 бромфенокси)циклогексанола использовали транс-2-(4-бром-3-фторфенокси)циклопентанол. Менее полярный материал, полученный в результате хроматографической очистки на силикагеле, предоставил(m, 1H), 5.14 (m, 1H), 6.66 (ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.78 (dd, J=10,5, 2,8 Гц, 1H), 7.40 (dd, J=8,8, 8,1 Гц,1H). Стадия 3. Синтез (1R,2R)-2-(4-бром-3-фторфенокси)циклопентанола. Соединение, указанное в заголовке стадии 3, получали согласно общей методике синтеза (1R,2R)-2(4-бромфенокси)циклогексанола из примера 7, за исключением того, что вместо (1R,2R)-2-(4 бромфенокси)циклогексилацетата использовали (1R,2R)-2-(4-бром-3-фторфенокси)циклопентилацетат. Продукт получали в виде желтого масла. Выход: 5,29 г, 19,2 ммоль, 95%. 1H ЯМР (500 МГц, CDCl3)1.62-1.68 (m, 2H), 1.71-1.90 (m, 3H), 2.04-2.11 (m, 1H), 2.15-2.22 (m, 1H),4.30 (m, 1H), 4.47 (m, 1H), 6.63 (ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.73 (dd, J=10,5, 2,8 Гц, 1H), 7.40 (dd, J=8,9, 8,1 Гц, 1H). Стадия 4. Синтез (1R,2R)-2-(4-бром-3-фторфенокси)циклопентилметансульфоната. Соединение, указанное в заголовке стадии 4, получали согласно общей методике синтеза транс-2(4-бромфенокси)циклопентил-метансульфоната из примера 5, за исключением того, что вместо транс-2(4-бромфенокси)циклопентанола использовали (1R,2R)-2-(4-бром-3-фторфенокси)циклопентанол. Продукт получали в виде масла, которое переносили на следующую стадию без очистки. МС (ГХ-МС) m/z 352, 354 (M+1). 1H ЯМР (500 МГц, CDCl3)1.81-2.00 (m, 4H), 2.16-2.26 (m, 2H), 3.04 (s, 3H), 4.77 (m, 1H), 5.07 (m,1H), 6.65 (ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.74 (dd, J=10,2, 2,8 Гц, 1H), 7.43 (dd, J=8,9, 8,0 Гц, 1H). Стадия 5. Синтез (1R,2S)-2-азидоциклопентил-4-бром-3-фторфенилового эфира. Соединение, указанное в заголовке стадии 5, получали согласно общей методике синтеза цис-2 азидоциклопентил-4-бромфенилового эфира из примера 5, за исключением того, что вместо транс-2-(4- 17021057 бромфенокси)циклопентил-метансульфоната применяли (1R,2R)-2-(4-бром-3-фторфенокси)циклопентилметансульфонат. Продукт выделяли в виде коричневого масла, которое использовали на следующей стадии без очистки. 1(ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.75 (dd, J=10,4, 2,8 Гц, 1H), 7.42 (dd, J=8,8, 8,0 Гц, 1H). Стадия 6. Синтез (1S,2R)-2-(4-бром-3-фторфенокси)циклопентанамина. Соединение, указанное в заголовке стадии 6, получали согласно общей методике синтеза цис-2-(4 бромфенокси)циклопентанамина из примера 5, за исключением того, что вместо цис-2 азидоциклопентил-4-бромфенилового эфира использовали (1R,2S)-2-азидоциклопентил-4-бром-3 фторфениловый эфир, и цис-2-(4-бромфенокси)циклопентанамин переносили на следующую стадию без очистки. ЖХ-МС m/z 276,2 (M+1). 1H ЯМР (500 МГц, CDCl3)1.5 (v br s, 2H), 1.57-1.66 (m, 2H), 1.80-1.87 (m, 2H), 1.93-2.01 (m, 2H),3.36 (m, 1H), 4.41 (m, 1H), 6.63 (ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.71 (dd, J=10,5, 2,8 Гц, 1H), 7.40 (dd, J=8,8, 8,0 Гц, 1H). Стадия 7. Синтез N-[(1S,2R)-2-(4-бром-3-фторфенокси)циклопентил]пропан-2-сульфонамида. Соединение, указанное в заголовке стадии 7, получали согласно общей методике синтеза цис-N-[2(4-бромфенокси)циклопентил]пропан-2-сульфонамида из примера 5, за исключением того, что вместо цис-2-(4-бромфенокси)циклопентанамина использовали(1S,2R)-2-(4-бром-3-фторфенокси)циклопентанамин и хроматографическую очистку осуществляли с использованием от 0% до 10% метанола в дихлорметане в качестве градиента, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества. Выход: 4,15 г, 10,9 ммоль, 54% из (1R,2R)-2-(4-бром-3 фторфенокси)циклопентилацетата (5 стадий). 1(m, 3H), 1.92-2.00 (m, 1H), 2.10-2.15 (m, 1H), 3.14 (септет, J=6,8 Гц, 1H), 3.86 (m, 1H), 4.56-4.60 (m, 2H),6.61 (ddd, J=8,9, 2,8, 1,1 Гц, 1H), 6.70 (dd, J=10,3, 2,8 Гц, 1H), 7.43 (dd, J=8,8, 8,1 Гц, 1H). Абсолютную конфигурацию N-[(1S,2R)-2-(4-бром-3-фторфенокси)циклопентил]пропан-2-сульфонамида устанавливали аналогично стереохимии соединений в примере 7 и подготовительном примере 6. Подготовительный пример 4. Синтез (2-циано-4-фторфенил)бороновой кислоты 2-Бром-5-фторбензонитрил (6,00 г, 30,0 ммоль) и триизопропилборат (8,28 мл, 36,0 ммоль) растворяли в смеси толуола (48 мл) и тетрагидрофурана (12 мл) и раствор охлаждали в бане сухой лед/ацетон. По каплям в течение 1 ч добавляли раствор н-бутиллития в гексанах (2,5 М, 14,4 мл, 36,0 ммоль) и затем реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании в течение 18 ч. Смесь охлаждали в ледяной бане и обрабатывали 2 н. водным раствором соляной кислоты до достижения значения pH 1, затем оставляли нагреваться до комнатной температуры; за этот период времени произошло разделение слоев, и водный слой дважды экстрагировали этилацетатом. Объединенные органические слои промывали дважды водой, один раз насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное твердое вещество перекристаллизовывали из этилацетата-гептана с получением (2-циано-4-фторфенил)бороновой кислоты в виде белого твердого вещества. Выход: 2,20 г, 13,3 ммоль, 44%. 1N-2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенил]этилметансульфонамида Стадия 1. Синтез трет-бутил-(метилсульфонил)2-[4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2 ил)фенил]этилкарбамата. 4,4,5,5-Тетраметил-1,3,2-диоксаборолан (4,29 мл, 29,6 ммоль) медленно добавляли к смеси третбутил-[2-(4-йодфенил)этил](метилсульфонил)карбамата (см. J.P. Gardner and W.D. Miller, РСТ публ. заявки на патент WO 2001090055, 2001) (8,39 г, 19,7 ммоль), триэтиламина (9,64 мл, 69,1 ммоль) и 1,1'[бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (217 мг, 0,296 ммоль) в ацетонитриле (50 мл) реакционную смесь нагревали при 75C в течение 4 ч. После удаления растворителя остаток смешивали с водой (240 мл) и экстрагировали метил-трет-бутиловым эфиром. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия и водой, затем сушили над сульфатом магния,фильтровали и концентрировали в вакууме, обеспечивая синтез трет-бутил-(метилсульфонил) 2-[4(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил]этилкарбамата, который использовали без дополнительной очистки. Выход: полагали, что количественный. ЖХ-МС m/z 326,1 (M+1). 1 что 19,7 ммоль) в дихлорметане (100 мл). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение 18 ч. Затем ее охлаждали до 0C и 4 н. водным раствором гидроксида натрия устанавливали значение pH 10,5. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (градиент: от 0 до 5% метанола в дихлорметане) с получением указанного в заголовке соединения в виде не совсем белого твердого вещества. Выход: 2,5 г, 7,7 ммоль, 39% за две стадии. ЖХ-МС m/z 326,1 (M+1). 1H ЯМР (400 МГц, CDCl3)1.35 (s, 12H), 2.84 (s, 3H), 2.90 (t, J=6,8 Гц, 2H), 3.42 (dt, J=6,6, 6,6 Гц,2H), 4.18 (br t, J=6 Гц, 1H), 7.23 (d, J=8,1 Гц, 2H), 7.78 (d, J=8,1 Гц, 2H). Подготовительный пример 6. Синтез N-[(1S,2R)-2-(4-бром-3-фторфенокси)циклогексил]пропан-2 сульфонамида. Указанное в заголовке соединение получали согласно общей методике синтеза цис-N-[2-(4 бромфенокси)циклопентил]пропан-2-сульфонамида из примера 5, за исключением того, что вместо цис 2-(4-бромфенокси)циклопентанамина использовали (1S,2R)-2-(4-бром-3-фторфенокси)циклогексанамин и в качестве градиента для хроматографической очистки применяли от 0 до 1% метанола в дихлорметане. (1S,2R)-2-(4-Бром-3-фторфенокси)циклогексанамин синтезировали согласно общим методикам, описанным в примере 7 для синтеза (1S,2R)-2-(4-бромфенокси)циклогексанамина, за исключением использования 4-бром-3-фторфенола вместо 4-бромфенола. Указанное в заголовке соединение получали в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3)1.33-1.53 (m, 4H), 1.35-1.38 (m, 6H), 1.78-1.89 (m, 3H), 2.04-2.10 (m, 1H),3.12 (септет, J=6,8 Гц, 1H), 3.54 (m, 1H), 4.44 (d, J=9,6 Гц, 1H), 4.55 (m, 1H), 6.67 (br dd, J=8,9, 2,8 Гц, 1H),6.75 (dd, J=10,2, 2,7 Гц, 1H), 7.43 (dd, J=8,5, 8,5 Гц, 1H). Абсолютную конфигурацию указанного в заголовке соединения устанавливали посредством рентгеновской кристаллографии. Подготовительный пример 7. СинтезN-(1S,2R)-2-[(6-бромпиридин-3-ил)окси]циклогексилпропан-2-сульфонамида Стадия 1. Синтез транс-2-[(6-бромпиридин-3-ил)окси]циклогексанола. Соединение, указанное в заголовке стадии 1, получали согласно общей методике синтеза транс-2(4-бром-3-фторфенокси)циклопентанола из подготовительного примера 3, за исключением того, что вместо 4-бром-3-фторфенола использовали 6-бромпиридин-3-ол, а вместо 6-оксабицикло[3.1.0]гексана 7 оксабицикло[4.1.0]гептан. Неочищенный продукт (экспериментально получили четыре партии) перекристаллизовывали из гептана с получением транс-2-[(6-бромпиридин-3-ил)окси]циклогексанола в виде не совсем белого твердого вещества. Выход: 11,09 г, 40,75 ммоль, 46%. 1H ЯМР (500 МГц, CDCl3)1.27-1.46 (m, 4H), 1.76-1.80 (m, 2H), 2.09-2.13 (m, 2H), 2.41 (d, J=2,6 Гц,1H), 3.74 (m, 1H), 4.00 (m, 1H), 7.17 (dd, J=8,7, 3,1 Гц, 1H), 7.37 (d, J=8,5 Гц, 1H), 8.10 (d, J=3,0 Гц, 1H). Стадия 2. Синтез цис-2-[(6-бромпиридин-3-ил)окси]циклогексанамина. цис-5-[(2-Азидоциклогексил)окси]-2-бромпиридин, полученный из транс-2-[(6-бромпиридин-3 ил)окси]циклогексанола по общим методикам, описанным в примере 5 в отношении превращения транс 2-(4-бромфенокси)циклопентанола в цис-2-азидоциклопентил-4-бромфениловый эфир) (13,5 г, 45,4 ммоль) растворяли в тетрагидрофуране (292 мл) и воде (23 мл) и раствор обрабатывали трифенилфосфином (23,8 г, 90,7 ммоль). Реакционную смесь перемешивали в течение 18 ч при комнатной температуре,затем распределяли между этилацетатом (500 мл) и водой (200 мл). Водный слой экстрагировали этилацетатом и объединенные органические слои промывали водой (2200 мл) и насыщенным водным раствором хлорида натрия (200 мл). Затем органический слой экстрагировали водной 1 н. соляной кислотой(4 х 150 мл), и объединенные водные слои промывали этилацетатом (150 мл). Затем водный слой охлаждали в ледяной бане и медленно обрабатывали водным 2 н. раствором гидроксида натрия, пока смесь была мутно-белой; затем ее экстрагировали этилацетатом (3150 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (150 мл), сушили над сульфатом натрия,фильтровали и концентрировали в вакууме. цис-2-[(6-Бромпиридин-3-ил)окси]циклогексанамин получали в виде желтого масла. Выход: 8,00 г, 29,5 ммоль, 65%. 1(m, 3H), 2.02 (m, 1H), 3.11 (септет, J=6,8 Гц, 1H), 3.53 (m, 1H), 4.59 (br s, 1H), 4.68 (m, 1H), 7.19 (dd, J=8,7,3,1 Гц, 1H), 7.38 (d, J=8,7 Гц, 1H), 8.07 (d, J=3,2 Гц, 1H). Стадия 3. Синтез цис-N-2-[(6-бромпиридин-3-ил)окси]циклогексилпропан-2-сульфонамида. Соединение, указанное в заголовке стадии 3, получали согласно общей методике синтеза цис-N-[2(4-бромфенокси)циклопентил]пропан-2-сульфонамида из примера 5, за исключением того, что вместо цис-2-(4-бромфенокси)циклопентанамина использовали цис-2-[(6-бромпиридин-3-ил)окси]циклогексанамин и пренебрегали 4-(диметиламино)пиридином. В этом случае хроматографию на силикагеле проводили с элюированием 2% метанола в дихлорметане, получая цис-N-2-[(6-бромпиридин-3 ил)окси]циклогексилпропан-2-сульфонамид в виде бежевой пены. Выход: 7,96 г, 21,1 ммоль, 72%. 1N-(1S,2R)-2-[(6-бромпиридин-3-ил)окси]циклогексилпропан-2 сульфонамида. Разделение энантиомеров, содержащих цис-N-2-[(6-бромпиридин-3-ил)окси]циклогексилпропан 2-сульфонамид (7,96 г, 21,1 ммоль), осуществляли посредством хиральной хроматографии. Колонка: Chiralpak AD-H, 2,125 см, 5 мкм; подвижная фаза: 70:30 диоксид углерода: метанол; скорость потока: 65 г/мин. Соединение, элюирующееся первым, представляло собой энантиомер [N-(1R,2S)-2-[(6 бромпиридин-3-ил)окси]циклогексилпропан-2-сульфонамид]; а соединение, соответствующее пику,элюирующемуся вторым,предоставило целевой продуктN-(1S,2R)-2-[(6-бромпиридин-3 ил)окси]циклогексилпропан-2-сульфонамид после удаления растворителя в вакууме. Выход: 3,13 г, 8,30 ммоль, 39%. Абсолютную стереохимию этих энантиомеров устанавливали аналогично примеру 5. Подготовительный пример 8. Синтез цис-N-[4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2 сульфонамида Указанное в заголовке соединение получали согласно общей методике синтеза цис-N-4-[(6 бромпиридин-3-ил)окси]тетрагидрофуран-3-илпропан-2-сульфонамида из подготовительного примера 1, за исключением того, что вместо транс-4-[(6-бромпиридин-3-ил)окси]тетрагидрофуран-3-илметансульфоната использовали транс-4-(4-бромфенокси)тетрагидрофуран-3-ил-метансульфонат, и хроматографическую очистку осуществляли с использованием градиента: от 15% до 35% ацетона в гептане. Выход: 238 мг, 0,65 ммоль, 31%. 1 Стадия 1. Синтез транс-N-(4-гидрокситетрагидрофуран-3-ил)пропан-2-сульфонамида. 3,6-Диоксабицикло[3.1.0]гексан (1,90 г, 22,1 ммоль), пропан-2-сульфонамид (полученный в соответствии со способом, описанным D.C. Johnson, II и T.S. Widlanski в Tetrahedron Letters 2004, 45, 84838487) (3,13 г, 25,4 ммоль), карбонат калия (584 мг, 4,23 ммоль) и хлорид бензилтриэтиламмония (963 мг,4,23 ммоль) суспендировали в диоксане (10 мл) и кипятили с обратным холодильником в течение 120 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 40 до 80% этилацетата в гептане) с получением транс-N-(4-гидрокситетрагидрофуран-3-ил)пропан-2-сульфонамида в виде твердого вещества. Выход: 3,76 г, 18,0 ммоль, 81%. 1(1,99 г, 9,52 ммоль) в дихлорметане (20 мл) добавляли триэтиламин (1,99 мл, 14,3 ммоль). Затем добавляли метансульфонилхлорид (0,885 мл, 11,4 ммоль) и реакционную смесь перемешивали при 0C в течение 50 мин. Добавляли насыщенный водный раствор бикарбоната натрия (10 мл) и водный слой экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 10% до 50% этилацетата в гептане) с получением транс-4-[(изопропилсульфонил)амино]тетрагидрофуран-3-илметансульфоната. Выход: 2,27 г, 7,90 ммоль, 83%. 1H ЯМР (400 МГц, CDCl3)1.40 (d, J=6,8 Гц, 3H), 1.41 (d, J=6,8 Гц, 3H), 3.14 (s, 3H), 3.25 (септет,J=6,8 Гц, 1H), 3.77 (dd, J=9,5, 2,4 Гц, 1H), 3.96 (dd, J=11,3, 2,2 Гц, 1H), 4.08-4.16 (m, 2H), 4.21 (dd, J=11,3,5,1 Гц, 1H), 4.77 (d, J=8,0 Гц, 1H), 5.15 (m, 1H). 13 С ЯМР (100 МГц, CDCl3)16.48, 16.54, 38.28, 54.28,59.49, 71.83, 71.87, 83.97. Стадия 3. Синтез транс-N-[4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2-сульфонамида. Раствор транс-4-[(изопропилсульфонил)амино]тетрагидрофуран-3-ил-метансульфоната (546 мг,1,90 ммоль) в ацетонитриле (8 мл) объединяли с 4-бромфенолом (97%, 407 мг, 2,28 ммоль) и карбонатом цезия (929 мг, 2,85 ммоль) во флаконе для микроволновой обработки. Реакционную смесь облучали в микроволновом реакторе при 160C в течение 2 ч, затем охлаждали до комнатной температуры и обрабатывали насыщенным водным раствором бикарбоната натрия (10 мл). Реакционную смесь экстрагировали этилацетатом и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (градиент: от 20 до 50% этилацетата в гептане) с получением транс-N-[4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2-сульфонамида. Выход: 626 мг, 1,72 ммоль, 91%. ЖХ-МС m/z 361,9 (М-1). 1(3,2 мг, 0,008 ммоль) и трис(дибензилиденацетон)дипалладий(0) (2,7 мг, 0,003 ммоль). Затем фиолетовую реакционную смесь дегазировали в течение 20 мин и добавляли транс-N-[4-(4 бромфенокси)тетрагидрофуран-3-ил]пропан-2-сульфонамид (95 мг, 0,26 ммоль) и N-пирролидин-3 илацетамид (67 мг, 0,52 ммоль). Потом добавляли гидроксид калия (32 мг, 0,57 ммоль) и реакционную смесь дегазировали в течение еще 20 мин. Коричневую реакционную смесь под азотом нагревали до образования флегмы, и смесь становилась желтой. Протекание реакции контролировали посредством ГХМС, и когда реакция завершилась, добавляли насыщенный водный раствор бикарбоната натрия (5 мл). Водный слой экстрагировали этилацетатом и объединенные органические вещества сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и очищали хроматографией на силикагеле (градиент: от 50% до 70% этилацетата в гептане) с получением диастереомерной N-1-[4-транс-(4[(изопропилсульфонил)амино]тетрагидрофуран-3-илокси)фенил]пирролидин-3-илацетамидной смеси в виде смолы Выход: 97,5 мг, 0,236 ммоль, 91%. 1H ЯМР (400 МГц, CDCl3)1.31 (d, J=6,8 Гц, 3H), 1.32 (d, J=6,8 Гц, 3H), 1.93 (s, 3H), 2.22 (m, 1H),2.64 (br s, 1H), 3.07-3.13 (m, 2H), 3.19 (m, 1H), 3.34-3.43 (m, 2H), 3.74 (dd, J=9,0, 1,8 Гц, 1H), 3.85 (dd,J=10,3, 1,7 Гц, 1H), 4.00-4.13 (m, 3H), 4.53 (m, 1H), 4.69 (br s, 1H), 5.67 (d, J=8,0 Гц, 1H), 6.46 (d, J=8,8 Гц,2H), 6.46 (1H, профиль заслонен ароматическим сигналом), 6.84 (d, J=9,0 Гц, 2H). 13 С ЯМР (100 МГц,CDCl3)16.37, 16.46, 22.98, 31.44, 46.32, 49.31, 53.61, 53.88, 58.67, 71.45, 71.87, 82.80, 112.72, 117.00,143.13, 147.98, 170.23. Пример 2. N-[(3S,4S)-4-(бифенил-4-илокси)тетрагидрофуран-3-ил]пропан-2-сульфонамид(492 г, 1,51 моль) и хлорид бензилтриэтиламмония (52,9 г, 0,23 моль) суспендировали в диоксане (1 л) и кипятили с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом, затем промывали насыщенным водным раствором карбоната натрия. Водный слой экстрагировали этилацетатом и объединенные органические части сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением неочищенного продукта, который затвердевал при стоянии. Этот продукт использовали без очистки на следующей стадии. Выход: 354 г, больше 100%, полагали, что количественный. Вещество, полученное после проведения эксперимента в меньшем масштабе аналогичным образом, очищали хроматографией на силикагеле для определения характеристик (градиент: от 10 до 35% этилацетата в гептане), получая транс-4-(4 бромфенокси)тетрагидрофуран-3-ола в виде твердого вещества. 1(dd, J=10,1, 4,0 Гц, 1H), 4.26 (dd, J=10,4, 4,9 Гц, 1H), 4.41 (br s, 1H), 4.67 (m, 1H), 6.81 (d, J=9,0 Гц, 2H),7.40 (d, J=9,1 Гц, 2H). Стадия 2. Синтез транс-4-(4-бромфенокси)тетрагидрофуран-3-ил-метансульфоната. К раствору транс-4-(4-бромфенокси)тетрагидрофуран-3-ола с предыдущей стадии (354 г, полагали,что 300,6 г, 1,16 моль) в метиленхлориде (2 л) добавляли триэтиламин (181,9 мл, 1,31 моль), и реакционную смесь охлаждали до 0C в ледяной бане. Затем по каплям добавляли метансульфонилхлорид (101,3 мл, 1,31 моль), поддерживая температуру реакционной смеси ниже 5C, и реакционную смесь перемешивали при комнатной температуре в течение 18 ч. Добавляли воду (1,5 л), и водный слой экстрагировали метиленхлоридом. Органические вещества объединяли и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта в виде коричневого масла. Выход: 399,6 г, 1,18 моль,количественный. Вещество, полученное после проведения эксперимента в меньшем масштабе аналогичным образом, растирали с этанолом для определения характеристик.H ЯМР (400 МГц, CDCl3)3.10 (s, 3H), 4.00 (br dd, J=10,4, 1,9 Гц, 1H), 4.07 (m, 1H), 4.16 (dd,J=11,1, 3,9 Гц, 1H), 4.23 (dd, J=10,5, 4,6 Гц, 1H), 4.98 (br d, J=4,6 Гц, 1H), 5.20 (m, 1H), 6.85 (d, J=9,1 Гц,2H), 7.42 (d, J=9,0 Гц, 2H). Стадия 3. Синтез цис-3-азидо-4-(4-бромфенокси)тетрагидрофурана. К раствору транс-4-(4-бромфенокси)тетрагидрофуран-3-ил-метансульфоната (133,2 г, 0,395 моль) в диметилформамиде (3 л) добавляли азид натрия (192,6 г, 2,96 моль) и реакционную смесь нагревали при 110C в течение 66 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (12 л). Эту реакцию проводили всего три раза в одном и том же масштабе и объединенные партии экстрагировали трет-бутилметиловым эфиром. Затем органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением продукта в виде красно-коричневого масла, загрязненного 18% диметилформамида. Уточненный выход: 286,7 г, 1,01 моль, 85%. 1(1,25 л) охлаждали до 0C и обрабатывали трифенилфосфином (278 г, 1,06 моль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 18 ч. Добавляли воду (53 мл) и реакционную смесь перемешивали при комнатной температуре в течение 66 ч. При пониженном давлении удаляли растворитель и остаток смешивали с диэтиловым эфиром. Надосадочную жидкость декантировали и концентрировали в вакууме, получая остаток, который фильтровали через небольшой слой силикагеля (градиент: от 0 до 5% метанола в метиленхлориде) с получением цис-4-(4 бромфенокси)тетрагидрофуран-3-амина (155 г) и 366 г загрязненного продукта. Загрязненное вещество подвергали кислотной/основной экстракции, получая дополнительное количество продукта (48,5 г). Суммарный выход: 203,5 г, 0,788 моль, 68% за четыре стадии. 1H ЯМР (300 МГц, CDCl3)3.6 (m, 1H), 3.7 (m, 1H), 3.9 (m, 1H), 4.0 (m, 1H), 4.1 (m, 1H), 4.6 (m, 1H),6.8 (m, 2H), 7.3 (m, 2H). Стадия 5. Синтез (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина. Смесь цис-4-(4-бромфенокси)тетрагидрофуран-3-амина (191 г, 0,74 моль) и (1S)-(+)-10 камфорсульфоновой кислоты (154,2 г, 0,66 моль) растворяли в 2-пропаноле (2,4 л) и воде (100 мл) при кипячении с обратным холодильником. Прозрачный раствор оставляли охлаждаться до комнатной температуры в течение 18 ч и полученные в результате кристаллы отделяли, промывали и сушили с получением соли (+)-камфорсульфоновой кислоты и (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина (139,2 г, 0,284 моль) с е.е. (энантиомерный избыток) 85%. Перекристаллизация из 2-пропанола (1,2 л) и водыH ЯМР (300 МГц, DMSO(диметилсульфоксид)-d6)0.74 (s, 3H), 1.05 (s, 3H), 1.23 (d, половина АВ квартета, J=10 Гц, 1H), 1.29 (d, половина АВ квартета, J=10 Гц, 1H), 1.76-1.94 (m, 2H), 2.19-2.28 (m, рассчитано, что 2H), 2.35 (d, J=14,7 Гц, 1H), 2.66-2.73 (m, рассчитано, что 1H), 2.85 (d, J=14,7 Гц, 1H), 3.783.84 (m, 2H), 3.96-4.10 (m, 3H), 5.04 (m, 1H), 6.99 (d, J=8,8 Гц, 2H), 7.52 (d, J=9,0 Гц, 2H), 8.23 (br s, 3H). Добавляли дополнительное количество соли (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина и(+)-камфорсульфоновой кислоты (13,6 г, 27,7 ммоль) из другого эксперимента и объединенную партию(126,8 г, 0,258 моль) промывали 2 н. водным раствором гидроксида натрия и три раза экстрагировали метиленхлоридом. Органические слои объединяли и концентрировали в вакууме, получая (3S,4S)-4-(4 бромфенокси)тетрагидрофуран-3-амин в виде белого твердого вещества с е.е. 99%. Выход: 65,6 г, 0,254 ммоль, 98% вследствие нейтрализации. Объединенные маточные растворы,полученные выше,обогащенные(3R,4R)-4-(4 бромфенокси)тетрагидрофуран-3-амином, промывали 2 н. гидроксидом натрия и экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением остатка (156,3 г, 0,606 моль продукта и его энантиомера). Это вещество объединяли с (1R)-(-)-10-камфорсульфоновой кислотой (126,2 г, 0,54 моль) и растворяли в 2-пропаноле (1,65 л) и воде (100 мл) при кипячении с обратным холодильником. Прозрачный раствор оставляли охлаждаться до комнатной температуры в течение 18 ч и полученные в результате кристаллы отделяли, промывали и сушили. Это привело к соли(3R,4R)-4-(4 бромфенокси)тетрагидрофуран-3-амина (152,6 г, 0,311 моль) с е.е. 90%. Перекристаллизация, такая как описано выше,привела к соли(3R,4R)-4-(4 бромфенокси)тетрагидрофуран-3-амина в виде белого твердого вещества с е.е. 99%. Выход: 132,0 г, 0,27 моль, 36%. Маточный раствор концентрировали в вакууме, промывали 2 н. гидроксидом натрия и экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении с получением смеси (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина и его энантиомера (67,7 г, 0,26 моль). Это вещество, вместе с (15)-(+)-10-камфорсульфоновой кислотой(54,6 г, 0,24 моль), растворяли в 2-пропаноле (425 мл) и воде (17,5 мл) при кипячении с обратным холодильником. Прозрачный раствор оставляли достигать комнатной температуры в течение 18 ч и полученные в результате кристаллы отделяли, промывали и сушили. Это привело к дополнительному количеству соли (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина и (+)-камфорсульфоновой кислоты (48,0 г, 97,9 ммоль) с е.е. 89-93%. Перекристаллизация привела к соли (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3 амина и (+)-камфорсульфоновой кислоты (40,0 г, 81,6 ммоль, дополнительно 11%) с е.е. 99+%. Стадия 6. Синтез N-[(3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2-сульфонамида. К раствору (3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-амина (65,6 г, 0,254 моль) в метиленхлориде (500 мл) добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU, 53 мл, 0,35 моль). Реакционную смесь охлаждали до 0C и по каплям добавляли пропан-2-сульфонилхлорид (31,2 мл, 0,28 моль). Затем смесь перемешивали при комнатной температуре в течение 18 ч. Протекание реакции контролировали посредством протонного ЯМР: добавляли дополнительное количество пропан-2-сульфонилхлорида (2,8 мл, 25 ммоль) и смесь перемешивали при комнатной температуре в течение еще 18 ч. Реакцию снова контролировали с помощью ЯМР и добавляли дополнительное количество пропан-2-сульфонилхлорида (2,8 мл,25 ммоль). Согласно данным ЯМР анализа реакция завершилась через 2,5 ч. Добавляли воду (500 мл) и разделяли слои. Водный слой экстрагировали метиленхлоридом и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток растворяли в метиленхлориде и промывали водной соляной кислотой (1 н., 2300 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением N-[(3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3 ил]пропан-2-сульфонамида в виде оранжевого/коричневого масла. Выход: 92,5 г, 0,254 моль, 100%. 1(dd, J=8,5, 8,5 Гц, 1H), 3.93 (dd, J=10,6, 1,5 Гц, 1H), 4.10-4.28 (m, 3H), 4.72-4.81 (m, 2H), 6.77 (d, J=9,1 Гц,2H), 7 41 (d, J=9,1 Гц, 2H). Абсолютную конфигурацию этого вещества устанавливали посредством рентгеновского кристаллографического анализа кристаллаN-[(3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2 сульфонамида, выросшего из гептанового/этилацетатного раствора. Результаты представлены ниже. Набор данных 0,90(максимум sin /=0,56) получали на дифрактометре Bruker APEX. Конечный индекс R составил 3,61%. Таблица 1. Кристаллографические данные и уточнение структуры Таблица 2. Атомные координаты (104) и эквивалентные параметры изотропного смещения(2103). U(eq) определен как одна треть следа ортогонализированного Uij тензора. Преобразования симметрии, используемые для генерирования эквивалентных атомов Таблица 4. Параметры анизотропного смещения (2103). Показатель коэффициента анизотропного смещения имеет вид: -22 [h2a2U11 ++ 2hkabU12] Таблица 5. Водородные координаты (104) и параметры изотропного смещения (2103). Стадия 7. Синтез N-[(3S,4S)-4-(бифенил-4-илокси)тетрагидрофуран-3-ил]пропан-2-сульфонамида. Во флакон для микроволновой обработки загружалиN-[(3S,4S)-4-(4 бромфенокси)тетрагидрофуран-3-ил]пропан-2-сульфонамид (124 мг, 0,340 ммоль), фенилбороновую кислоту (63,5 мг, 0,521 ммоль), дициклогексил(2',4',6'-триизопропилбифенил-2-ил)фосфин (XPhos, 16,2 мг,0,034 ммоль), ацетат палладия(II) (5,2 мг, 0,023 ммоль) и фторид калия (99,6 мг, 1,71 ммоль). Флакон закрывали и три раза продували азотом/вакуумировали. Добавляли дегазированную смесь метанол/толуол(1:1, 1,5 мл) и реакционную смесь подвергали воздействию микроволнового излучения при 130C в течение 30 мин. В вакууме удаляли растворитель и остаток распределяли между этилацетатом и насыщенным водным раствором хлорида натрия. Водный слой дважды экстрагировали этилацетатом, и органические слои объединяли, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на силикагеле (градиент: от 10% до 25% этилацетата в гептане) с получением указанного в заголовке соединения в виде твердого вещества. Выход: 90 мг, 0,25 ммоль, 73%. 1 Н ЯМР (400 МГц, CDCl3)1.37 (d, J=6,8 Гц, 3H), 1.40 (d, J=6,8 Гц, 3H), 3.17 (септет, J=6,8 Гц, 1H),3.76 (dd, J=8,6, 8,6 Гц, 1H), 4.01 (dd, J=10,6, 1,6 Гц, 1H), 4.16-4.30 (m, 3H), 4.84 (m, 1H), 5.09 (d, J=9,4 Гц,1H), 6.97 (d, J=8,7 Гц, 2H), 7.34 (t, J=7,4 Гц, 1H), 7.44 (dd, J=7,6, 7,6 Гц, 2H), 7.55 (m, 4H). 13 С ЯМР (100 МГц, CDCl3)16.48, 16.55, 54.27, 55.35, 70.29, 71.96, 75.87, 115.85, 126.65, 126.88,128.34, 128.68, 135.11, 140.25, 155.93. Пример 3. N-(3S,4S)-4-[(2'-цианобифенил-4-ил)окси]тетрагидрофуран-3-илпропан-2-сульфонамид Указанное в заголовке соединение получали согласно общей методике синтеза примера 2, за исключением того, что вместо фенилбороновой кислоты использовали (2-цианофенил)бороновую кислоту,получая продукт в виде твердого вещества. Выход: 675,3 мг, 1,75 ммоль, 85%. ЖХ-МС m/z 387,0 (M+1). 1 Указанное в заголовке соединение получали согласно общей методике синтеза примера 2, за исключением того, что вместо фенилбороновой кислоты использовали (5-циано-2-тиенил)бороновую кислоту, получая продукт в виде твердого вещества. Выход: 365 мг, 0,93 ммоль, 58%. ЖХ-МС m/z 393,5(99%, 8,93 г, 27,1 ммоль) и хлорид бензилтриэтиламмония (99%, 1,09 г, 4,74 ммоль) суспендировали в диоксане (65 мл) и кипятили с обратным холодильником в течение 18 ч. Добавляли дополнительное количество 6-оксабицикло[3.1.0]гексана (0,50 мл, 5,8 ммоль) и нагревание продолжали в течение 66 ч. Опять добавляли 6-оксабицикло[3.1.0]гексан (0,50 мл, 5,8 ммоль) и реакционную смесь кипятили с обратным холодильником в течение дополнительных 18 ч. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме и распределяли между насыщенным водным раствором бикарбоната натрия и этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом кальция, фильтровали и концентрировали при пониженном давлении с получением масла золотистого цвета, которое очищали хроматографией на силикагеле (градиент: от 0% до 20% этилацетата в гептане), получая продукт в виде масла. Выход: 3,21 г, 12,5 ммоль, 48%. ГХ-МС m/z 256, 258 (М+). 1H ЯМР (400 МГц, CDCl3)1.64 (d, J=3,7 Гц, 1H), 1.60-1.68 (m, 1H), 1.70-1.88 (m, 3H), 2.07 (m, 1H),2.17 (m, 1H), 4.30 (m, 1H), 4.48 (m, 1H), 6.80 (d, J=9,0 Гц, 2H), 7.37 (d, J=9,0 Гц, 2H). Стадия 2. Синтез транс-2-(4-бромфенокси)циклопентилметансульфоната. Соединение, указанное в заголовке стадии 2, получали согласно общей методике синтеза транс-4(4-бромфенокси)тетрагидрофуран-3-ил-метансульфоната из примера 2, за исключением того, что вместо транс-4-(4-бромфенокси)тетрагидрофуран-3-ола использовали транс-2-(4-бромфенокси)циклопентанол и реакционную смесь гасили путем добавления насыщенного водного раствора хлорида аммония. Затем органический слой промывали насыщенным водным раствором хлорида аммония, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта в виде коричневого масла. Выход: 3,86 г, 11,5 ммоль, 98%. 1H ЯМР (400 МГц, CDCl3)1.79-2.00 (m, 4H), 2.14-2.26 (m, 2H), 3.03 (s, 3H), 4.78 (m, 1H), 5.07 (m,1H), 6 82 (d, J=9,0 Гц, 2H), 7.39 (d, J=9,1 Гц, 2H). Стадия 3. Синтез цис-2-азидоциклопентил-4-бромфенилового эфира. К раствору транс-2-(4-бромфенокси)циклопентил-метансульфоната (3,52 г, 10,5 ммоль) в диметилформамиде (22 мл) добавляли азид натрия (897 мг, 13,7 ммоль) и реакционную смесь нагревали при 100C в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом и 1 н. водным раствором хлорида лития. Водный слой экстрагировали этилацетатом и объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом кальция, фильтровали и концентрировали в вакууме с получением продукта в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки. Выход: 2,59 г, 9,18 ммоль, 87%. 1 Н ЯМР (400 МГц, CDCl3)1.65-1.73 (m, 1H), 1.89-2.05 (m, 5H), 3.74 (m, 1H), 4.66 (m, 1H), 6.83 (d,- 27021057J=9,0 Гц, 2H), 7.39 (d, J=9,0 Гц, 2H). Стадия 4. Синтез цис-2-(4-бромфенокси)циклопентанамина. Раствор цис-2-азидоциклопентил-4-бромфенилового эфира с предыдущей стадии (2,59 г, 9,18 ммоль) в тетрагидрофуране (63 мл) и воде (5,0 мл) обрабатывали трифенилфосфином на полимерном носителе (3 ммоль/г, 7,15 г, 21,5 ммоль). Реакционную смесь перемешивали в течение 18 ч, затем фильтровали через Celite. Осадок на фильтре ополаскивали тетрагидрофураном, затем смесью метиленхлорида и метанола и объединенные фильтраты концентрировали в вакууме и подвергали азеотропной перегонке с этанолом. Остаток очищали хроматографией на силикагеле (градиент: от 0% до 10% метанола в этилацетате) с получением продукта в виде светло-коричневого масла. Выход: 1,43 г, 5,58 ммоль, 61%. МС (ХИАД) m/z 257,9 (M+1). 1(915 мг, 7,49 ммоль). Реакционную смесь охлаждали до 0C и по каплям добавляли пропан-2 сульфонилхлорид (0,937 мл, 8,38 ммоль). Затем смесь оставляли нагреваться до комнатной температуры и перемешиваться в течение 18 ч. Реакционную смесь обрабатывали 1 н. водной соляной кислотой и органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом кальция, фильтровали и концентрировали в вакууме. Полученный остаток очищали хроматографией на силикагеле (градиент: от 0% до 15% этилацетата в гептане) с получением продукта в виде бесцветной смолы. Выход: 1,586 г, 4,38 ммоль, 78%. Стадия 6. Выделение N-[(1S,2R)-2-(4-бромфенокси)циклопентил]пропан-2-сульфонамида. Разделение энантиомеров,содержащих цис-N-[2-(4-бромфенокси)циклопентил]пропан-2 сульфонамид (1,586 г, 4,38 ммоль) осуществляли посредством хиральной хроматографии. Колонка: Chiralpak AD-H, 2,125 см, 5 мкм; подвижная фаза: 75:25 диоксид углерода: метанол; скорость потока: 65 г/мин. Соединение, элюирующееся первым, представляло собой энантиомер N-[(1R,2S)-2-(4 бромфенокси)циклопентил]пропан-2-сульфонамид (767 мг, 2,12 ммоль, 48%), а соединение, соответствующее пику, элюирующемуся вторым, предоставило целевой продукт N-[(1S,2R)-2-(4 бромфенокси)циклопентил]пропан-2-сульфонамид после удаления растворителя в вакууме. Выход: 758 мг, 2,09 ммоль, 48%. Абсолютную стереохимию этих энантиомеров устанавливали аналогично их более высоким гомологам (см. Пример 7). Указанное в заголовке соединение, синтезированное на следующей стадии, оказалось значительно более действенным, чем его энантиомер (полученный таким же путем из(m, 4H), 2.12 (m, 1H), 3.13 (септет, J=6,8 Гц, 1H), 3.86 (m, 1H), 4.59 (m, 1H), 4.64 (d, J=9,5 Гц, 1H), 6.78 (d,J=9,1 Гц, 2H), 7.39 (d, J=9,0 Гц, 2H). Стадия 7. Синтез соединения, представляющего собой N-(1S,2R)-2-[(2'-цианобифенил-4 ил)окси]циклопентилпропан-2-сульфонамид. Соединение, указанное в заголовке стадии 7, получали согласно общей методике синтеза примера 2,за исключением того, что вместо N-[(3S,4S)-4-(4-бромфенокси)тетрагидрофуран-3-ил]пропан-2 сульфонамида использовали N-[(1S,2R)-2-(4-бромфенокси)циклопентил]пропан-2-сульфонамид и вместо фенилбороновой кислоты добавляли (2-цианофенил)бороновую кислоту. В этом случае реакционную смесь после концентрирования в вакууме непосредственно очищали хроматографией на силикагеле(элюант: 25% этилацетата в гептане) с получением продукта в виде липкой белой пены. Растирание с гексанами привело к продукту в виде белого порошка. Выход: 53 мг, 0,14 ммоль, 82%. МС (ХИАД) m/z 382,9 (М-1). 1 Во флакон для микроволновой обработки загружали(150,0 мг,0,414 ммоль),(5-циано-2 тиенил)бороновую кислоту (95,0 мг, 0,621 ммоль), дициклогексил(2',4',6'-триизопропилбифенил-2 ил)фосфин (20,2 мг, 0,0410 ммоль), ацетат палладия(II) (7,4 мг, 0,033 ммоль) и фторид калия (120 мг, 2,07 ммоль). Добавляли диметоксиэтан (1,5 мл) и реакционную смесь три раза продували азотом/вакуумировали. Реакционную смесь подвергали воздействию микроволнового излучения при 120C в течение 2 ч, затем в вакууме удаляли растворитель и остаток распределяли между этилацетатом и насыщенным водным раствором хлорида натрия. Водный слой экстрагировали этилацетатом и органические слои объединяли, сушили над сульфатом кальция, фильтровали и концентрировали в вакууме. Остаток очищали препаративной тонкослойной хроматографией на силикагеле (элюант: 40% этилацетата в гептане) с получением указанного в заголовке соединения в виде желтого масла, которое впоследствии затвердело. Выход: 104 мг, 0,266 ммоль, 64%. ЖХ-МС m/z 391,0 (M+1). 1 Стадия 1. Синтез транс-2-(4-бромфенокси)циклогексанола. Металлический натрий (2,58 г, 112 ммоль) объединяли с абсолютным этанолом (200 мл) и оставляли взаимодействовать до завершения реакции. Добавляли 4-бромфенол (19,4 г, 112 ммоль) и реакционную смесь перемешивали в течение 20 мин, после которых добавляли 7-оксабицикло[4.1.0]гептан (10,0 г,102 ммоль) и раствор кипятили с обратным холодильником в течение 15 ч. После удаления в вакууме растворителя остаток распределяли между водой (300 мл) и этилацетатом (100 мл). Водный слой экстрагировали этилацетатом (2100 мл) и объединенные органические слои промывали водой (2200 мл), сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученное светлое желто-коричневое твердое вещество перекристаллизовывали из гептана (приблизительно 200 мл) с получением транс-2-(4-бромфенокси)циклогексанола в виде белого пушистого твердого вещества. Выход: 12,5 г, 46,1 ммоль, 45%. 1H ЯМР (400 МГц, CDCl3)1.26-1.46 (m, 4H), 1.75-1.79 (m, 2H), 2.08-2.14 (m, 2H), 2.52 (d, J=2,1 Гц,1H), 3.72 (m, 1H), 3.96 (ddd, J=10,3, 8,6, 4,4 Гц, 1H), 6.84 (d, J=9,0 Гц, 2H), 7.37 (d, J=9,0 Гц, 2H). Стадия 2. Синтез (1R,2R)-2-(4-бромфенокси)циклогексилацетата. транс-2-(4-Бромфенокси)циклогексанол (5,305 г, 19,56 ммоль) растворяли в этилацетате (196 мл) и обрабатывали винилацетатом (3,37 г, 39,1 ммоль), а затем липазным ферментом от Candida antarctica(Novozyme 435, Sigma L4777, липаза, иммобилизованная на акриловой смоле, 5,3 г). Реакционную смесь закрывали и перемешивали в течение 18 ч, затем фильтровали через Celite и ополаскивали этилацетатом (500 мл). Концентрирование фильтрата в вакууме привело к бледно-желтому маслу, которое очищали хроматографией на силикагеле (градиент: от 0% до 10% этилацетата в гептане) с получением (1R,2R)2-(4-бромфенокси)циклогексилацетата, менее полярного продукта, в виде бесцветного масла. Выход: 2,047 г, 6,54 ммоль, 33%. Данные для (1R,2R)-2-(4-бромфенокси)циклогексилацетата: 1 Н ЯМР (400 МГц, CDCl3)1.32-1.59 (m, 4H), 1.71-1.80 (m, 2H), 1.95 (s, 3H), 2.02-2.14 (m, 2H), 4.17(ddd, J=9,6, 8,1, 4,4 Гц, 1H), 4.96 (m, 1H), 6.84 (d, J=9,0 Гц, 2H), 7.36 (d, J=9,1 Гц, 2H). Энантиомерный спирт (1S,2S)-2-(4-бромфенокси)циклогексанол, более полярный продукт, получали в виде белого твердого вещества (3,57 г). Данные для (1S,2S)-2-(4-бромфенокси)циклогексанола: 1H ЯМР (400 МГц, CDCl3)1.26-1.46 (m, 4H), 1.74-1.79 (m, 2H), 2.08-2.15 (m, 2H), 2.50 (br s, 1H),3.72 (ddd, J=10,6, 8,5, 4,6 Гц, 1H), 3.96 (m, 1H), 6.84 (d, J=9,0 Гц, 2H), 7.38 (d, J=9,0 Гц, 2H). Абсолютные конфигурации этих соединений устанавливали на основании рентгеновской кристаллической структуры энантиомера N-[(1S,2S)-2-(4-бромфенокси)циклогексил]пропан-2-сульфонамида (см. стадию 7 ниже). Стадия 3. Синтез (1R,2R)-2-(4-бромфенокси)циклогексанола. Раствор (1R,2R)-2-(4-бромфенокси)циклогексилацетата (2,047 г, 6,54 ммоль) в метаноле (12,2 мл) и воде (0,32 мл) охлаждали до 0C и обрабатывали гидратом гидроксида лития (95%, 1,73 г, 39,2 ммоль). Реакционную смесь перемешивали при 0C в течение 15 мин, затем оставляли нагреваться и перемешиваться при комнатной температуре в течение 18 ч. При пониженном давлении удаляли метанол и водный остаток распределяли между этилацетатом (200 мл) и водой (100 мл). Объединенные органические вещества после экстрагирования водного слоя этилацетатом (100 мл) промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над сульфатом магния, фильтровали и концентрировали в ва- 29
МПК / Метки
МПК: C07C 311/07, C07D 213/65, A61K 31/341, A61K 31/402, C07D 295/096, C07D 409/12, C07D 405/12, A61K 31/443, A61K 31/4453, A61K 31/4709, C07D 307/42, C07D 409/14, C07D 307/22, C07D 333/38, A61K 31/381
Метки: композиции, сульфонамиды, гетероциклические, применения, фармацевтические
Код ссылки
<a href="https://eas.patents.su/30-21057-geterociklicheskie-sulfonamidy-ih-primeneniya-i-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические сульфонамиды, их применения и фармацевтические композиции</a>
Азотсодержащие гетероциклические производные и их фармацевтические применения

Номер патента: 11402
Опубликовано: 27.02.2009
Авторы: Мартин Мэттью В., Ким Тай-Сенг, Чаффе Стюарт К., Хэбгуд Грегори Дж., Макгован Дэвид К., Чоквитте Дебора, Мессе Крейг И., Виттингтон Дуглас А., Поташман Мишель, Патель Винод Ф., Ходоус Брайан Л., Буханан Джон Л., Арман Жан-Кристоф, Ченг Юан, Ла Дэниэл, Букнер Вилльям Х., Грацеффа Рассел, Кси Нинг, Ксу Шимин, Беллон Стивен, Ким Джозеф Л., Дипьетро Лусиан В., Букер Шон, Нанес Джозеф Дж., Герман Джули, Борг Джордж, Тэскер Эндрю, Вейс Мэтью
МПК: A61K 31/47, C07D 217/24, A61K 31/505...
Метки: гетероциклические, производные, применения, азотсодержащие, фармацевтические
Формула / Реферат:
1. Соединение формулы I' где R выбрана из а) замещенной или незамещенной арилгруппы, где арилгруппа является возможно замещенной фенилгруппой или возможно замещенной нафтилгруппой, б) замещенной или незамещенной гетероциклической группы, где гетероциклическая группа является замещенным или незамещенным гетероциклическим кольцом, выбранным из пирролидинил-, пирролил-, имидазолил-, пиразолил-, пиразинил-, пиримидинил-, пиридил-, хинолинил-,...
Новые гетероциклические карбоксамиды и фармацевтические композиции, содержащие их

Номер патента: 18502
Опубликовано: 30.08.2013
Авторы: Густафссон Давид, Полла Магнус, Брональт Йонас, Нильссон Ингемар
МПК: A61K 31/4155, A61K 31/422, A61P 7/02...
Метки: новые, содержащие, фармацевтические, карбоксамиды, гетероциклические, композиции
Формула / Реферат:
1. Соединение, представляющее собой (5S)-N-[5-хлор-2-(1Н-тетразол-1-ил)бензил]-1-[(2R)-2-(4-фторфенил)-2-гидроксиацетил]-4,5-дигидро-1Н-пиразол-5-карбоксамидили фармацевтически приемлемая соль указанного соединения.2. Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль по п.1 в смеси по меньшей мере с одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.3. Соединение, представляющее...
Замещенные сульфоны и сульфонамиды и фармацевтические композиции на их основе, применимые для лечения ожирения, сахарного диабета ii типа и расстройств центральной нервной системы

Номер патента: 8835
Опубликовано: 31.08.2007
Авторы: Бейерлейн Катарина, Йоханссон Гари, Йенмальм Йенсен Анника
МПК: C07D 211/40, C07D 401/04, C07D 207/12...
Метки: основе, расстройств, сульфоны, замещенные, центральной, применимые, типа, фармацевтические, нервной, ожирения, системы, лечения, диабета, композиции, сахарного, сульфонамиды
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, где кольцо В представляет собой каждый W независимо представляет собой -N-, -(СН)- или -С-, при условии, что всего не более трех групп W представляют собой -N- в обоих кольцах А и В одновременно, Р представляет собой любую из групп формулы (а), (b) или (с) где х=0, 1 или 2 и y=0, 1 или 2; и Р и R3 могут быть присоединены к любому атому углерода, который допускает замещение в...
Гетероциклические соединения, содержащие их композиции и способы их применения

Номер патента: 16495
Опубликовано: 30.05.2012
Авторы: Бо Саймон Д.П., Джессоп Теодор К., Карсон Кеннет Г., Оджери Дэвид Дж., Багданофф Джеффри, Тарвер Джеймс Э.
МПК: A61K 31/427, A61K 31/422, A61K 31/4196...
Метки: композиции, применения, содержащие, соединения, способы, гетероциклические
Формула / Реферат:
1. Соединение формулы I(a)или формулы I(b)или его фармацевтически приемлемая соль или сольват, гдеA представляет 5- или 6-членный гетероцикл, содержащий по меньшей мере один атом азота и необязательно замещенный C1-10-алкокси, C1-10-алкилфенилом, C1-10-алкилом, фенилом или гидроксилом;R1 представляет водород или C1-4-алкил;R5 представляет OR5a, OC(O)R5a или водород;R6 представляет OR6A, OC(O)R6A или водород;R7 представляет OR7A, OC(O)R7A или...
Новые гетероциклические соединения оксима, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 14317
Опубликовано: 29.10.2010
Авторы: Кторза Ален, Кэньяр Даньель-Анри, Дакю Катрин, Пиллар Кристелль, Сюзене Франк, Бассен Карине, Гилломе Жеральд
МПК: A61K 31/437, A61P 3/06, A61P 3/04...
Метки: содержат, получения, которые, композиции, новые, соединения, гетероциклические, фармацевтические, способ, оксима
Формула / Реферат:
1. Соединения формулы (I)в которой R1 представляет собой арильную, гетероарильную или (С3-С8)циклоалкильную группу,R2 представляет собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу, арильную группу или арил-(С1-С6)алкильную группу, в которой алкильная часть может быть линейной или разветвленной,X представляет собой атом водорода или галогена или линейную или разветвленную (С1-С6)алкильную группу,R3 и R4, которые могут...