Серосодержащее гетероциклическое производное, обладающее активностью ингибитора бета-секретазы
Номер патента: 20740
Опубликовано: 30.01.2015
Авторы: Хори Акихиро, Уено Тадзухико, Йонедзава Сюдзи, Като Терукацу, Коорияма Юудзи, Фудзикоси Тиаки, Мацумото Сае







Формула / Реферат
1. Соединение, представленное формулой (I)

где кольцо А представляет собой замещенный фенил или замещенный пиридил, каждый из которых замещен одним или более заместителем, выбранным из галогена или замещающей группы Y;
R1 представляет собой C1-C3-алкил;
R3a и R3c независимо друг от друга представляют собой водород, возможно замещенный C1-C3-алкил, где заместители от одного до трех выбраны из гидрокси и галогена, возможно замещенный карбамоил, где заместители выбраны из C1-C2-алкила и C1-C2-алкокси(C1-C2)алкила, фенил или 5-6-членную гетероарильную группу, содержащую один-три гетероатома, выбранных из азота и кислорода, или R3a и R3c могут быть соединены вместе, образуя 5-членное карбоциклическое кольцо или 6-членное гетероциклическое кольцо, содержащее один атом азота;
замещающая группа Y представляет собой следующие группы:

где кольцо В представляет собой возможно замещенный 5-6-членный гетероарил или возможно замещенную 8-9-членную бициклическую группу, каждый из которых содержит от одного до трех гетероатомов, выбранных из азота и кислорода и который возможно замещен одним или более заместителем, выбранным из замещающей группы X;
замещающая группа X представляет собой галоген; C1-C2-алкил, возможно замещенный 1-3 галогенами; C1-C4-алкоксикарбонил; амино; C1-C2-алкиламино; C1-C2-алкилкарбамоил; C1-C4-алкилсульфониламино; C1-C2-алкилсульфонил-C1-C2-алкиламино; C1-C4-алкилсульфонилимино; циано; 5-членную гетероарильную группу, содержащую один-три гетероатома, выбранных из азота и кислорода, которая возможно замещена C1-C2-алкилом; С2алкинил; C1-C2-алкокси, возможно замещенный одним-двумя заместителями, выбранными из галогена и C1-C2-алкокси; C2-C4-алкинилокси, возможно замещенный гидроксигруппой; 5-членный гетероарил-C1-C2-алкокси, возможно замещенный одним или более C1-C2-алкилом,
или его фармацевтически приемлемая соль, или его сольват.
2. Соединение по п.1, где оба R3a или R3c представляют собой водород, или его фармацевтически приемлемая соль, или его сольват.
3. Соединение по любому из пп.1, 2, где кольцо А представляет собой

где кольцо А' представляет собой фенил или пиридил;
G представляет собой

где кольцо В представляет собой пиразинил, пиридил, пиримидинил, оксазолил, пиразолил, тетрагидропиразолопиримидинил, тетрагидропиразолопиразинил, дигидроимидазопиразолил или бензофурил, каждый из которых возможно замещен одним или более заместителем, выбранным из замещающей группы X;
R4 представляет собой галоген;
n представляет собой 0 или 1,
или его фармацевтически приемлемая соль, или его сольват.
4. Соединение по п.3, где кольцо А' представляет собой пиридил, или его фармацевтически приемлемая соль, или его сольват.
5. Соединение по любому из пп.3, 4, где кольцо В представляет собой пиразинил, пиридил, пиримидинил, оксазолил или пиразолил, каждый из которых возможно замещен одним или более заместителем, выбранным из замещающей группы X, или его фармацевтически приемлемая соль, или его сольват.
6. Фармацевтическая композиция, обладающая активностью ингибитора β-секретазы, содержащая в качестве активного компонента соединение по любому из пп.1-5, или его фармацевтически приемлемую соль, или его сольват.
7. Способ ингибирования активности β-секретазы, согласно которому пациенту вводят соединение по любому из пп.1-5, или его фармацевтически приемлемую соль, или его сольват.
8. Применение соединения по любому из пп.1-5, или его фармацевтически приемлемой соли, или его сольвата при изготовлении лекарства для ингибирования активности β-секретазы.
9. Применение соединения по любому из пп.1-5, или его фармацевтически приемлемой соли, или его сольвата для ингибирования активности β-секретазы.
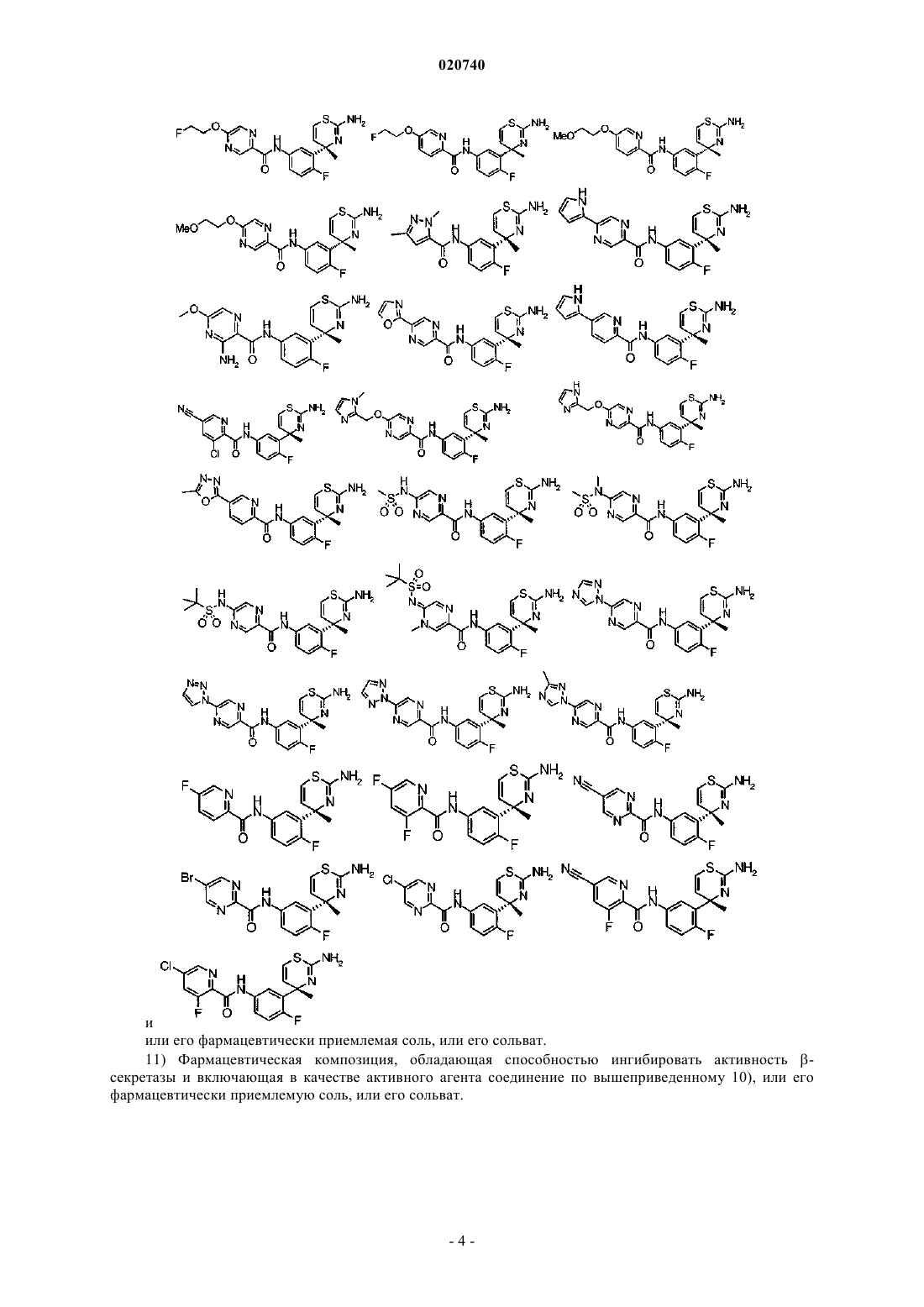
10. Соединение, выбранное из группы, состоящей из следующих соединений:



или его фармацевтически приемлемая соль, или его сольват.
11. Фармацевтическая композиция, обладающая способностью ингибировать активность β-секретазы и включающая в качестве активного агента соединение по п.10, или его фармацевтически приемлемую соль, или его сольват.
12. Соединение по п.1 формулы (I')

где кольцо А' представляет собой фенил;
R1 представляет собой C1-C3-алкил;
R4 представляет собой галоген;
кольцо В представляет собой пиридил или пиразинил, каждый из которых возможно замещен одним-двумя заместителями, выбранными из группы, состоящей из галогена; C1-C2-алкила, возможно замещенного одним-тремя галогенами; C1-C4-алкоксикарбонила; амино; C1-C4-алкилсульфониламино, циано; 5-членной гетероарильной группы, содержащей один-три гетероатома, выбранных из азота и кислорода; C1-C2-алкокси, возможно замещенного одним-двумя заместителями, выбранными из галогена и C1-C2-алкокси,
или его фармацевтически приемлемая соль, или его сольват.
Текст
СЕРОСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ АКТИВНОСТЬЮ ИНГИБИТОРА БЕТА-СЕКРЕТАЗЫ где кольцо А представляет собой замещенный фенил или замещенный пиридил; R1 представляет собой C1-3 алкил; R3a и R3c независимо друг от друга представляют собой водород, возможно замещенный C1-3 алкил, где заместители от одного до трех выбраны из гидрокси и галогена, или возможно замещенный карбамоил, где заместители выбраны из C1-С 2-алкила и С 1-С 2-алкокси(С 1 С 2)алкила, фенил или 5-6-членную гетероарильную группу, содержащую один-три гетероатома,выбранных из азота и кислорода, или R3a и R3c могут быть соединены вместе, образуя 5-членное карбоциклическое кольцо или 6-членное гетероциклическое кольцо, содержащее один атом азота,или его фармацевтически приемлемая соль, или его сольват, или его фармацевтически приемлемая соль. Оно может быть использовано в качестве терапевтического агента для лечения заболевания,вызванного продуцированием, секрецией и/или депонированием -амилоидного белка. Область изобретения Настоящее изобретение относится к соединению, которое обладает активностью ингибитора продуцировать -амилоид, и применяется в качестве средства для лечения заболевания, вызванного продуцированием, секрецией и/или депонированием -амилоидного белка. Предшествующий уровень техники В головном мозге пациентов с болезнью Альцгеймера наблюдается в значительной степени накопление в форме нерастворимых бляшек (сенильных бляшек) вне нервных клеток пептида, состоящего из приблизительно 40 аминокислотных остатков, так называемого -амилоидного белка. В связи с тем, что сенильные бляшки убивают нервные клетки, вызывая болезнь Альцгеймера, проводятся исследования терапевтических средств от болезни Альцгеймера, таких как агенты разложения -амилоидного белка и амилоидная вакцина. Секретаза представляет собой фермент, который расщепляет белок, называемый предшественником-амилоидного белка (ПБА, АРР от amyloidprecursor protein), в клетке и продуцирует -амилоидный белок. Фермент, который контролирует продуцирование N-конца -амилоидного белка, называется секретазой (бета-сайт ПБА-расщепляющий фермент 1, ВАСЕ-1 от beta-site АРР-cleaving enzyme 1). Считается, что ингибирование данного фермента приводит к снижению продуцирования -амилоидного белка, и терапевтическое средство от болезни Альцгеймера будет разработано благодаря ингибированию. В патентной литературе 1 описаны соединения, которые похожи на соединения по настоящему изобретению, и эти соединения обладают активностью ингибитора фермента NO-синтазы и применяются при слабоумии. В патентной литературе 2-5 и непатентной литературе 1 и 2 описаны соединения, которые похожи на соединения по настоящему изобретению, и используются в качестве гипертонического средства, морфинно-подобного болеутоляющего средства, транквилизаторов, промежуточного соединения для лекарства, NPYY5 антагониста, обезболивающего средства или подобного соответственно. Из патентной литературы 6-24 известны ингибиторы -секретазы, однако все соединения из этой литературы имеют структуры, отличные от структур соединений по настоящему изобретению. Литература предшествующего уровня техники Патентная литература. Патентная литература 1. Публикация международной заявки на патент WO 96/014842. Патентная литература 2. Патент США 3235551. Патентная литература 3. Патент США 3227713. Патентная литература 4. Публикация японской заявки JP09-067355. Патентная литература 5. Публикация международной заявки на патент WO 2005/111031. Патентная литература 6. Публикация международной заявки на патент WO 02/96897. Патентная литература 7. Публикация международной заявки на патент WO 04/043916. Патентная литература 8. Публикация международной заявки на патент WO 2005/058311. Патентная литература 9. Публикация международной заявки на патент WO 2005/097767. Патентная литература 10. Публикация международной заявки на патент WO 2006/041404. Патентная литература 11. Публикация международной заявки на патент WO 2006/041405. Патентная литература 12. Публикация патента США 2007/0004786. Патентная литература 13. Публикация патента США 2007/0004730. Патентная литература 14. Публикация патента США 2007/27199. Патентная литература 15. Публикация международной заявки на патент WO 2007/049532. Патентная литература 16. Публикация международной заявки на патент WO 2007/146225. Патентная литература 17. Публикация международной заявки на патент WO 2007/114771. Патентная литература 18. Публикация международной заявки на патент WO 2007/073284. Патентная литература 19. Публикация международной заявки на патент WO 2007/058583. Патентная литература 20. Публикация международной заявки на патент WO 2007/058580. Патентная литература 21. Публикация международной заявки на патент WO 2006/138217. Патентная литература 22. Публикация международной заявки на патент WO 2006/138192. Патентная литература 23. Публикация международной заявки на патент WO 2006/065277. Патентная литература 24. Публикация международной заявки на патент WO 2005/058311. Патентная литература 25. Публикация международной заявки на патент WO 2008/103351. Непатентная литература. Непатентная литература 1. Journal of Heterocyclic Chemistry, 14, 717-723 (1977). Непатентная литература 2. Journal of Organic Chemistry, 33, 8, 3126-3132 (1968). Непатентная литература 3. Journal of Medicinal Chemistry, 50, 24, 5912-5925 (2007). Раскрытие изобретения Задачи, которые решаются данным изобретением Согласно настоящему изобретению созданы соединения, которые обладают эффектами снижения продуцирования -амилоидного белка, особенно активностью ингибитора -секретазы, и применяются в качестве средства для лечения заболевания, вызванного продуцированием, секрецией и/или депонированием -амилоидного белка. Решение задач Согласно настоящему изобретению создано следующее. 1) Соединение, представленное формулой (I) где кольцо А представляет собой замещенный фенил или замещенный пиридил, каждый из которых замещен одним или более заместителем, выбранным из галогена или замещающей группы Y;R3a и R3c независимо друг от друга представляют собой водород, возможно замещенный C1-C3 алкил, где заместители от одного до трех выбраны из гидрокси и галогена, возможно замещенный карбамоил, где заместители выбраны из C1-C2-алкила и C1-C2-алкокси(C1-C2)алкила, фенил или 5-6-членную гетероарильную группу, содержащую один-три гетероатома, выбранных из азота и кислорода, или R3a иR3c могут быть соединены вместе, образуя 5-членное карбоциклическое кольцо или 6-членное гетероциклическое кольцо, содержащее один атом азота; замещающая группа Y представляет собой следующие группы: где кольцо В представляет собой возможно замещенный 5-6-членный гетероарил или возможно замещенную 8-9-членную бициклическую группу, каждый из которых содержит от одного до трех гетероатомов, выбранных из азота и кислорода, и который возможно замещен одним или более заместителем,выбранным из замещающей группы X; замещающая группа X представляет собой галоген; C1-C2-алкил, возможно замещенный 1-3 галогенами;C1-C4 алкилсульфониламино; C1-C2-алкилсульфонил-C1-C2-алкиламино; C1-C4-алкилсульфонилимино; циано; 5-членную гетероарильную группу, содержащую один-три гетероатома, выбранных из азота и кислорода,которая возможно замещена C1-C2-алкилом; С 2 алкинил; C1-C2-алкокси, возможно замещенный однимдвумя заместителями, выбранными из галогена и C1-C2-алкокси; C2-C4-алкинилокси, возможно замещенный гидроксигруппой; 5-членный гетероарил-C1-C2-алкокси, возможно замещенный одним или более C1C2-алкилом,или его фармацевтически приемлемая соль, или его сольват. 2) Соединение по вышеприведенному 1), где оба R3a или R3c представляют собой водород, или его фармацевтически приемлемая соль, или его сольват. 3) Соединение по вышеприведенным 1) или 2), где кольцо А представляет собой где кольцо А' представляет собой фенил или пиридил; где кольцо В представляет собой пиразинил, пиридил, пиримидинил, оксазолил, пиразолил, тетрагидропиразолопиримидинил, тетрагидропиразолопиразинил, дигидроимидазопиразолил или бензофурил,каждый из которых возможно замещен одним или более заместителем, выбранным из замещающейn представляет собой 0 или 1,или его фармацевтически приемлемая соль, или его сольват. 4) Соединение по вышеприведенному 3), где кольцо А' представляет собой пиридил, или его фармацевтически приемлемая соль, или его сольват. 5) Соединение по вышеприведенному 3) или 4), где кольцо В представляет собой пиразинил, пиридил, пиримидинил, оксазолил или пиразолил, каждый из которых возможно замещен одним или более заместителем, выбранным из замещающей группы X, или его фармацевтически приемлемая соль, или его сольват. 6) Фармацевтическая композиция, обладающая активностью ингибитора -секретазы, содержащая в качестве активного компонента соединение по любому из вышеприведенных 1)-5), или его фармацевтически приемлемую соль, или его сольват. 7) Способ ингибирования активности -секретазы, согласно которому пациенту вводят соединение по любому из вышеприведенных 1)-5), или его фармацевтически приемлемую соль, или его сольват. 8) Применение соединения по любому из вышеприведенных 1)-5), или его фармацевтически приемлемой соли, или его сольвата при изготовлении лекарства для ингибирования активности -секретазы. 9) Применение соединения по любому из вышеприведенных 1)-5), или его фармацевтически приемлемой соли, или его сольвата для ингибирования активности -секретазы. 10) Соединение, выбранное из группы, состоящей из следующих соединений: и или его фармацевтически приемлемая соль, или его сольват. 11) Фармацевтическая композиция, обладающая способностью ингибировать активность секретазы и включающая в качестве активного агента соединение по вышеприведенному 10), или его фармацевтически приемлемую соль, или его сольват.R4 представляет собой галоген; кольцо В представляет собой пиридил или пиразинил, каждый из которых возможно замещен одним-двумя заместителями, выбранными из группы, состоящей из галогена; C1-C2-алкила, возможно замещенного одним-тремя галогенами; C1-C4-алкоксикарбонила; амино; C1-C4-алкилсульфониламино; циано; 5-членной гетероарильной группы, содержащей один-три гетероатома, выбранных из азота и кислорода; C1-C2-алкокси, возможно замещенного одним-двумя заместителями, выбранными из галогена и C1C2-алкокси,или его фармацевтически приемлемая соль, или его сольват. Результат изобретения Соединения по настоящему изобретению пригодны в качестве средства для лечения заболевания,вызванного продуцированием, секрецией или депонированием -амилоидного белка (болезни Альцгеймера или подобной). Наилучший способ выполнения изобретения Как используется здесь, "галоген" включает фтор, хлор, бром и йод. В другом воплощении кольцо А может быть замещено одним или более чем одним заместителем, выбранным из галогена и следующих заместителей: кольцо В представляет собой возможно замещенный 5-6-членный гетероарил или возможно замещенную 8-9-членную бициклическую группу. В качестве заместителя кольца В предпочтительной является одна или более чем одна группа, выбранная из X группы заместителей. Формула (I), где "R3a и R3c могут быть соединены вместе, образуя 5-членное карбоциклическое кольцо или 6-членное гетероциклическое кольцо, содержащее один атом азота", включает следующую формулу (I): где кольцо С представляет собой 5-членную карбоциклическую группу или 6-членную гетероциклическую группу, содержащую один атом азота;R2a и R2b, каждый, являются водородом; другие обозначения являются такими, как определено в формуле (I), и примеры предпочтительного воплощения кольца С включают пиридин и тетрагидропиридин. В настоящем описании "сольват" включает, например, сольват с органическим растворителем и гидрат. При образовании гидрата может быть скоординировано произвольное число молекул воды. Соединение, представленное формулой (I), включает фармацевтически приемлемую соль. Примеры включают соли щелочных металлов, таких как литий, натрий или калий; щелочно-земельных металлов,таких как магний или кальций; аммония; органических оснований и аминокислот; или соли с неорганическими кислотами, такими как соляная кислота, серная, азотная кислота, бромисто-водородная кислота,фосфорная кислота или йодисто-водородная кислота; и органическими кислотами, такими как уксусная кислота, трифторуксусная кислота, лимонная кислота, молочная кислота, винная кислота, щавелевая ки-5 020740 слота, малеиновая кислота, фумаровая кислота, миндальная кислота, глутаровая кислота, яблочная кислота, бензойная кислота, фталевая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота,метансульфоновая кислота или этансульфоновая кислота. Особенно предпочтительной является соляная кислота, фосфорная кислота, винная кислота или метансульфоновая кислота. Эти соли могут быть получены общепринятыми способами. Кроме того, соединение, представленное формулой (I), не ограничено конкретным изомером, а включает все возможные изомеры, такие как кето-енольные изомеры, имин-енаминные изомеры, диастереоизомеры, оптические изомеры и вращающие изомеры; и рацемат. Например, соединение, представленное формулой (I), в которой R2a представляет собой водород, включает следующие таутомеры. Соединения, представленные формулами (II), (III) и (IV), включают подобные таутомеры. Настоящее соединение, представленное формулой (I), (II), (III) или (IV), может быть получено, например способом, описанным в патентной литературе 15 или непатентной литературе 1, или следующим способом. На всех следующих стадиях, когда содержится заместитель, который препятствует реакции, например гидрокси, меркапто, амино, формил, карбонил, карбокси, заместитель защищают способом, описанным ранее в Protective Groups in organic Synthesis, Theodora W Greene (John WileySons), и защитная группа может быть удалена на желательной стадии. Кроме того, для всех стадий порядок осуществляемых стадий может быть соответствующим образом изменен и каждое промежуточное соединение может быть выделено и использовано на следующей стадии. А. Получение соединения, представленного формулой (I). Соединение, представленное формулой (I), может быть получено, например, согласно способу синтеза соединения х или соединения аб, показанному ниже. А-1) Синтез соединения х. где кольцо А представляет собой возможно замещенную карбоциклическую группу или возможно замещенную гетероциклическую группу;R1 представляет собой возможно замещенный низший алкил, возможно замещенный низший алкенил, возможно замещенный низший алкинил, возможно замещенную карбоциклическую группу или возможно замещенную гетероциклическую группу;R2a представляет собой водород, возможно замещенный низший алкил или возможно замещенный ацил; каждый R3c независимо представляет собой водород, галоген, гидрокси, возможно замещенный низший алкил, возможно замещенный низший алкенил, возможно замещенный ацил, возможно замещенный низший алкокси, возможно замещенный аралкил, возможно замещенный гетероарилалкил, возможно замещенный аралкилокси, возможно замещенный гетероарилалкокси, возможно замещенный низший алкилтио, карбокси, возможно замещенный низший алкоксикарбонил, возможно замещенный амино, возможно замещенный карбамоил, возможно замещенную карбоциклическую группу или возможно замещенную гетероциклическую группу. Первая стадия. Соединение р можно диастереоселективно получить, добавляя титановый реагент, такой как триизопропоксид хлортитана, к еноляту, который получают в ходе реакции соответствующего эфира, такого как трет-бутилпропионат, в присутствии основания, такого как диизопропиламид лития, в растворителе,таком как толуол, дихлорметан и тетрагидрофуран или смеси этих растворителей, добавляя соединение п, которое может быть получено известным способом, и проводя реакцию при -80-30 С, предпочтительно -80-0 С, в течение 0,1-24 ч, предпочтительно 0,1-12 ч. Вторая стадия. Соединение с можно получить в ходе реакции соединения р при 0-80 С, предпочтительно 0-30 С, в течение 0,5-48 ч, предпочтительно 1-24 ч, в присутствии кислоты, такой как соляная кислота, бромистоводородная кислота, серная кислота и трифторуксусная кислота, в растворителе, таком как диоксан, метанол и дихлорметан или смеси этих растворителей. Третья стадия. Соединение т можно получить, добавляя восстановитель, такой как боргидрид, гидрид натрия и алюмогидрид лития, к соединению с и проводя реакцию при -80-80 С, предпочтительно -20-30 С, в течение 0,5-48 ч, предпочтительно 1-12 ч, в растворителе, таком как диоксан, тетрагидрофуран и толуол или смеси этих растворителей. Четвертая стадия. Соединение ф можно получить, добавляя окислитель, такой как 2-йодоксибензойная кислота, к соединению т и проводя реакцию при 0-80 С, предпочтительно 10-40 С, в течение 0,5-48 ч, предпочтительно 1-12 ч, в растворителе, таком как диметилсульфоксид и дихлорметан. На третьей стадии и четвертой стадии аминную и/или альдегидную группы соединения т и соединения ф можно защитить по методу, описанному в Protective Groups in Organic Synthesis, Theodora WGreen (John WileySons), и удалить защитную группу, если необходимо, в соответствующее время. Пятая стадия. Соединение х можно получить, добавляя изотиоцианат, имеющий защитную группу, например бензоилизотиоцианат, который имеется в продаже или получен известным способом, к соединению ф, проводя реакцию при -30-50 С, предпочтительно -10-25 С, в течение 0,1-12 ч, предпочтительно 0,1-3 ч, в растворителе, таком как диоксан, тетрагидрофуран, толуол и ацетон или смеси этих растворителей, и затем после добавления концентрированной серной кислоты или концентрированной азотной кислоты,проводя реакцию при 0-100 С, предпочтительно 0-60 С, в течение 0,5-24 ч, предпочтительно 1-12 ч. А-2) Синтез соединения аб. где Rp представляет собой защитную группу для амина; каждый R3a независимо представляет собой водород, галоген, гидрокси, возможно замещенный низший алкил, возможно замещенный низший алкенил, возможно замещенный ацил, возможно замещенный низший алкокси, возможно замещенный аралкил, возможно замещенный гетероарилалкил, возможно замещенный аралкилокси, возможно замещенный гетероарилалкокси, возможно замещенный низший алкилтио, карбокси, возможно замещенный низший алкоксикарбонил, возможно замещенный амино, возможно замещенный карбамоил, возможно замещенную карбоциклическую группу или возможно замещенную гетероциклическую группу; и другие обозначения являются такими, как определено выше. Первая стадия. Соединение ч можно получить в ходе реакции соединения ц, которое может быть получено при защите аминогруппы соединения т с помощью защитной группы, при 0-80 С, предпочтительно 10-40 С, в течение 0,5-48 ч, предпочтительно 1-12 ч, в условиях реакции окисления Сверна, в которой используется оксалилхлорид-диметилсульфоксид, или добавляя окислитель спиртовой группы, такой как 2 йодоксибензойная кислота, в растворителе, таком как толуол, дихлорметан и тетрагидрофуран или смеси этих растворителей. Вторая стадия. Соединение ш можно получить, добавляя реактив Гриньяра, соответствующий требуемому соединению, такой как метилмагнийбромид, к соединению ч и проводя реакцию при -80-50 С, предпочтительно -20-20 С, в течение 0,2-48 ч, предпочтительно 1-24 ч, в растворителе, таком как толуол, эфир и тетрагидрофуран или смеси этих растворителей. По реакции выход может быть улучшен путем добавления тетрахлорида титана. Третья стадия. Соединение аа можно получить, добавляя окислитель спиртовой группы, такой как оксалилхлориддиметилсульфоксид или 2-йодоксибензойная кислота, к соединению ш и проводя реакцию при 0-80 С,предпочтительно 10-40 С, в течение 0,5-48 ч, предпочтительно 1-6 ч, в растворителе, таком как диметилсульфоксид. Четвертая стадия. Соединение аб можно получить, добавляя изотиоцианат, имеющий защитную группу, например бензоилизотиоцианат, который имеется в продаже или получен известным способом, к соединению аа,проводя реакцию при -30-50 С, предпочтительно -10-25 С, в течение 0,1-12 ч, предпочтительно 0,1-3 ч, в растворителе, таком как диоксан, тетрагидрофуран, толуол и ацетон или смеси этих растворителей, и затем после добавления концентрированной серной кислоты или концентрированной азотной кислоты,проводя реакцию при 0-100 С, предпочтительно 0-60 С, в течение 0,5-12 ч, предпочтительно 1-6 ч. В-2) Синтез соединения ал. где R12 представляет собой возможно замещенный арил или возможно замещенный гетероарил; и другие обозначения являются такими, как определено выше. Первая стадия. Соединение ай можно стереоселективно получить, добавляя соединение п, которое может быть получено известным способом, к еноляту, полученному в ходе реакции соответствующего фенилалкилкетона, например ацетофенона, в присутствии основания, такого как диизопропиламид лития и гексаметилдисилазид калия, и проводя реакцию при -80-30 С, предпочтительно -80-0 С, в течение 0,1-24 ч,предпочтительно 0,1-12 ч, в растворителе, таком как толуол, дихлорметан и тетрагидрофуран или смеси этих растворителей. Вторая стадия. Соединение ак можно получить, добавляя соляную кислоту, бромисто-водородную кислоту, трифторуксусную кислоту или подобную, к соединению ай, полученному на первой стадии, и проводя реакцию при 0-60 С, предпочтительно 0-30 С, в течение 0,1-24 ч, предпочтительно 0,5-12 ч. Третья стадия. Соединение ал можно получить, добавляя изотиоцианат, имеющий защитную группу, например бензоилизотиоцианат, который имеется в продаже или получен известным способом, к соединению ак и проводя реакцию при -30-70 С, предпочтительно -20-50 С, в течение 0,1-12 ч, предпочтительно 0,1-6 ч, в растворителе, таком как диоксан, тетрагидрофуран, толуол и ацетон или смеси этих растворителей, затем отгоняя растворитель, добавляя концентрированную серную кислоту, концентрированную азотную кислоту или подобную, и проводя реакцию при -30-70 С, предпочтительно -20-50 С, в течение 1-12 ч,предпочтительно 1-6 ч. Г-3) Синтез соединения бз. где соответствующие обозначения являются такими, как определено выше. Первая стадия. Соединение бе можно получить, добавляя этилакрилат и реагент Граббса к соединению бб, в котором аминогруппа соответствующим образом защищена защитной группой, и проводя реакцию метатезиса олефинов в растворителе, таком как толуол, дихлорметан и тетрагидрофуран или смеси этих растворителей. Температура реакции составляет -20-60 С, предпочтительно 0-30 С, и время реакции составляет 0,5-24 ч, предпочтительно 1-12 ч. Вторая стадия. Соединение бж можно получить, добавляя изотиоцианат, имеющий защитную группу, например бензоилизотиоцианат, который имеется в продаже или получен известным способом, к соединению бе и проводя реакцию при -30-70 С, предпочтительно -20-50 С, в течение 0,1-12 ч, предпочтительно 0,1-6 ч, в растворителе, таком как дихлорметан, диоксан, тетрагидрофуран, толуол и ацетон или смеси этих растворителей. Третья стадия. Соединение бз можно получить, добавляя гидрид диизобутилалюминия, алюмогидрид лития или гидрид натрия к соединению бж, подвергая реакции восстановления и проводя реакцию при -80-0 С,предпочтительно от -80 до -20 С, в течение 0,1-12 ч, предпочтительно 0,1-3 ч, в растворителе, таком как диоксан, тетрагидрофуран и толуол или смеси этих растворителей. Кроме того, соединение бз можно подвергнуть соответствующей реакции по спиртовой группе. Д. Превращение заместителя (1). Синтез соединения аб-2, полученного превращением заместителя, описан ниже. где Pa и Pb представляют собой аминозащитную группу; другие обозначения являются такими, как определено выше. Соединение аб-2 можно получить, добавляя трис-(дибензилиденацетон)дипалладия, ацетат палладия, Pd(0), полученные in situ, или подобные, и фосфиновый лиганд, такой как три-трет-бутилфосфин и дициклогексилбифенилфосфин, к соединению аб-1 в растворителе, таком как тетрагидрофуран, толуол и ксилол, добавляя реагент, имеющий заместитель, соответствующий требуемому соединению, такой как гексаметилдисилазид лития и бензофенонимин, при -10-30 С, и проводя реакцию при 30-120 С, предпочтительно 50-100 С, в течение 0,5-48 ч, предпочтительно 3-20 ч. Аминозащитная группа может представлять собой заместитель, который можно удалить способом,описанным в Protective Groups in Organic Synthesis, Theodora W Green (John WileySons), и примеры включают низший алкоксикарбонил, низший алкенилоксикарбонил, триалкилсилил, ацил, метансульфонил, трифторэтансульфонил, толуолсульфонил и подобные. где соответствующие обозначения являются такими, как определено выше. Соединение аб-4 можно получить, добавляя железо к соединению аб-3 в смешанном растворителе уксусной кислоты и воды с последующей реакцией при 20-120 С, предпочтительно 50-80 С, в течение 0,5-48 ч, предпочтительно 6-20 ч. Кроме того, соединение аб-4 также можно получить, добавляя восстанавливающий катализатор, такой как 10% палладий/углерод, к соединению аб-3 в растворителе, таком как тетрагидрофуран, этилацетат и метанол, и проводя реакцию при 30-120 С, предпочтительно 50-80 С, в течение 0,5-48 ч, предпочтительно 6-20 ч, в атмосфере водорода при нормальном давлении до 5 атм, предпочтительно нормальном давлении до 2 атм, или согласно способу, описанному в Comprehensive Organic Transformations, Richard CLarock (Mcgraw-Hill). Соединения х, аб, ал, ао, бд и бз можно получить путем разделения оптических изомеров каждого промежуточного соединения и конечного продукта, или следующим способом, например согласно способу, описанному в (1) Т. Fujisawa et al., Tetrahedron Lett., 37, 3881-3884 (1996), (2) D.H. Hua et al., SulfurReports, vol. 21, p. 211-239 (1999), (3) Y. Koriyama et al., Tetrahedron, 58, 9621-9628 (2002) или (4) Т. Vilavan et al., Current Organic Chemistry, 9, 1315-1392 (2005). В качестве процедуры разделения оптических изомеров существуют способ отделения оптического изомера с использованием оптически активной колонки; кинетическое разделение оптических изомеров с использованием ферментативной реакции и т.п.; кристаллизационное разделение диастереомера в ходе солеобразования с использованием хиральной кислоты или хирального основания; предпочтительно кристаллизационный способ и т.п. Что касается соединения по настоящему изобретению, то следующие соединения являются предпочтительными. Соединение по формуле (I')R4 представляет собой галоген; кольцо В представляет собой пиридил или пиразинил, каждый из которых возможно замещен одним-двумя заместителями, выбранными из группы, состоящей из галогена; C1-C2-алкила, возможно замещенного одним-тремя галогенами; C1-C4-алкоксикарбонила; амино; C1-C4-алкилсульфониламино; циано; 5-членной гетероарильной группы, содержащей один-три гетероатома, выбранных из азота и кислорода; C1-C2-алкокси, возможно замещенного одним-двумя заместителями, выбранными из галогена и C1C2-алкокси,или его фармацевтически приемлемая соль, или его сольват. Настоящие соединения используются при заболевании, вызванном продуцированием, секрецией или депонированием -амилоидного белка, и являются эффективными при лечении и/или предотвращении и облегчении симптомов, таких как слабоумие альцгеймеровского типа (болезнь Альцгеймера, старческое слабоумие альцгеймеровского типа), синдром Дауна, нарушение памяти, прионная болезнь (болезнь Крейтцфельдта-Якоба), умеренные когнитивные нарушения (УКН), наследственная церебральная геморрагия с амилоидозом голландского типа, церебральная амилоидная ангиопатия, другие виды дегенеративного слабоумия, смешанное слабоумие альцгеймеровского и васкулярного типа, слабоумие с болезнью Паркинсона, слабоумие с прогрессирующим надъядерным параличом, слабоумие с кортикобазальной дегенерацией, болезнь Альцгеймера с болезнью диффузных телец Леви, возрастная макулярная дегенерация, болезнь Паркинсона, амилоидная ангиопатия и прочие. Поскольку настоящее соединение обладает высокой активностью ингибитора -секретазы и/или обладает высокой селективностью относительно других ферментов, то оно может представлять собой лекарственное средство с уменьшенным побочным эффектом. Более того, поскольку соединение оказывает значительное влияние на снижение продуцирования -амилоида в клеточной системе, особенно оказывает значительное влияние на снижение продуцирования -амилоида в головном мозге, то оно может представлять собой превосходное лекарственное средство. Кроме того, при преобразовании соединения в оптически активную основу, обладающую подходящей стереохимией, соединение может представлять собой лекарственное средство, имеющее широкий спектр безопасности в отношении побочного эффекта. Кроме того, настоящее соединение также обладает такими преимуществами, как высокая устойчивость к метаболизму, высокая растворимость, высокая пероральная всасываемость, проявление хорошей биодоступности, хорошее выведение, значительный перенос к головному мозгу, высокое время полужизни, высокая скорость небелкового связывания, низкое ингибирование канала hERG, низкое ингибированиеCYP (цитохрома Р 450) и/или отрицательный результат по тесту Эймса. Настоящие соединения можно вводить в сочетании с другими фармацевтическими агентами, такими как другие терапевтические лекарства от болезни Альцгеймера, например ингибиторы ацетилхолинэстеразы и подобные. Настоящие соединения можно вводить одновременно с другими агентами от слабоумия, такими как донепезила гидрохлорид, такрин, галантамин, ривастигмин, занапезил, мемантин и винпоцетин. При введении настоящего соединения человеку оно может быть введено перорально в виде порошков, гранул, таблеток, капсул, пилюль, микстур или т.п., или парентерально в виде инъекций, суппозиториев, трансдермальных всасываемых агентов, абсорбируемых агентов или т.п. Кроме того, настоящее соединение можно разработать в виде фармацевтических препаратов, добавляя фармацевтические вспомогательные вещества, такие как эксципиенты, связующие вещества, смачивающие вещества, дезинтегрирующие вещества, скользящие вещества и подобные, которые пригодны для препаратов и эффективного количества настоящего соединения. Дозировка различается в зависимости от состояния заболевания, способа введения и возраста и веса пациента, и обычно составляет 0,1 мкг-1 г/день, предпочтительно 0,01-200 мг/день при пероральном введении взрослому, и обычно составляет 0,1 мкг-10 г/день, предпочтительно 0,1-2 г/день при парентеральном введении. Примеры Следующие примеры и исследуемые примеры иллюстрируют настоящее изобретение более подробно, но настоящее изобретение не ограничено этими примерами. В примерах значение каждой аббревиатуры следующее.mCPBA - метахлорпербензойная кислота; Катализатор Граббса второго поколения - бензилиден[1,3-бис-(2,4,6-триметилфенил)-2 имидазолидинилиден]дихлор(трициклогексилфосфин)рутений. ЖХ/МС данные настоящего соединения измеряли при любом из следующих условий (способы А и Б), и показаны время удерживания и [М+Н]+. Способ А. Колонка: Waters XBridge C18 5 мкм Размер: 4,650 мм Скорость потока: 3 мл/мин Термостат колонки: 50 С. Длина волны УФ-детектирования: PDA (фотодиодная матрица) (254 мм) Осуществляли линейный градиент от 10 до 100% растворителя (ацетонитрильный раствор, содер- 11020740 жащий 0,1% муравьиную кислоту) в течение 3 мин, и 100% растворитель (ацетонитрильный раствор,содержащий 0,1% муравьиную кислоту) поддерживали в течение 0,5 мин. Способ Б. Колонка: Shimadzu Shim pack XR-ODS 50L3,0 Размер: 503,0 мм Скорость потока: 1,6 мл/мин Термостат колонки: 50 С. Длина волны УФ-детектирования: PDA (фотодиодная матрица) (254 мм). Осуществляли линейный градиент от 10 до 100% растворителя (ацетонитрильный раствор, содержащий 0,1% муравьиную кислоту) в течение 3 мин, и 100% растворитель (ацетонитрильный раствор,содержащий 0,1% муравьиную кислоту) поддерживали в течение 0,5 мин. Справочный пример 1. Синтез промежуточного соединения (21) Первая стадия. Соединение (10) (7,17 г) растворяли в тетрагидрофуране (70,0 мл), добавляли 1-этинил-4 метоксибензол (5,63 г), тетракис-трифенилфосфин палладия (4,10 г), иодид меди (338 мг) и диизопропиламин (70,0 мл) и смесь перемешивали при нагревании с обратным холодильником в течение 30 мин. Растворитель выпаривали при пониженном давлении, добавляли дихлорметан и нерастворимые вещества удаляли. Остаток очищали с помощью колоночной хроматографии, добавляли дихлорметан и н-гексан и выпавшее в осадок твердое вещество собирали в ходе фильтрации, что давало соединение (11) (8,99 г). 1 Н-ЯМР (ДМСО-d6) : 3,81 (3 Н, s), 7,02 (2 Н, d, J=8,6 Гц), 7,57 (2 Н, d, J=8,6 Гц), 7,71 (1H, t, J=8,0 Гц),7,96 (1H, d, J=7,6 Гц), 8,22 (1 Н, d, J=8,1 Гц), 8,30 (1H, s). Вторая стадия. Соединение (11) (1,00 г) растворяли в диметилсульфоксиде (10,0 мл), добавляли йод (501 мг), смесь нагревали до 165 С и перемешивали в течение 2 ч. Реакционный раствор экстрагировали этилацетатом и органический слой промывали водным раствором тиосульфата натрия и дистиллированной водой. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. К остатку добавляли дихлорметан и диизопропиловый эфир и выпавшее в осадок твердое вещество собирали в ходе фильтрации, что давало соединение (12) (532 мг). 1 Н-ЯМР (ДМСО-d6) : 3,89 (3 Н, s), 7,16 (2 Н, d, J=8,6 Гц), 7.91 (1 Н, t, J=8,0 Гц), 7,98 (2 Н, d, J=8,8 Гц),8,33 (1H, d, J=7,3 Гц), 8,58-8,61 (2 Н, m). Третья стадия. Соединение (12) (2,00 г) растворяли в этаноле (10,0 мл), добавляли гидроксид калия (7,87 г) и дистиллированную воду (10,0 мл) и смесь перемешивали при нагревании с обратным холодильником в течение 3 ч. Реакционный раствор экстрагировали дистиллированной водой и водным раствором гидроксида калия и водный слой промывали хлороформом. Водный слой нейтрализовали соляной кислотой и экстрагировали в органический слой смесью хлороформа и метанола. Органический слой высушивали над безводным сульфатом натрия и при пониженном давлении, что давало соединение (13) (2,02 г). К полученному в результате соединению (13) (2,02 г) добавляли 2-хлорацетонитрил (10,1 мл), уксусную кислоту (12,8 мл) и концентрированную серную кислоту (12,8 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор выливали в ледяную воду и выпавшее в осадок твердое вещество собирали в ходе фильтрации, что давало соединение (14) (2,34 г). 1 Н-ЯМР (ДМСО-d6) : 3,74 (30 Н, s), 4,26 (2 Н, dd, J=20,5, 13,1 Гц), 6,94 (2 Н, d, J=8,8 Гц), 7,21 (2 Н, d,J=8,8 Гц), 7,62 (1 Н, t, J=8,0 Гц), 7,77 (1H, d, J=7,3 Гц), 8,14 (1 Н, d, J=7,6 Гц), 8,25 (1 Н, s), 9,22 (1 Н, s). Четвертая стадия. К соединению (14) (2,34 г) добавляли тиомочевину (564 мг), этанол (12,0 мл) и уксусную кислоту(2,40 мл), смесь нагревали при 80 С и перемешивали в течение ночи. Растворитель выпаривали при пониженном давлении, к остатку добавляли дистиллированную воду и выпавшее в осадок твердое вещество собирали в ходе фильтрации, что давало соединение (15) (4,76 г). 1H-ЯМР (ДМСО-d6) : 3,75 (3 Н, s), 3,89 (2 Н, s), 6,89 (2 Н, d, J=8,8 Гц), 7,22 (2 Н, br s), 7,30 (2 Н, d,J=8,8 Гц), 7,58 (1 Н, t, J=8,0 Гц), 7,81 (1 Н, d, J=8,3 Гц), 8,11 (1H, d, J=8,1 Гц), 8,34 (1H, s). Пятая стадия. Соединение (15) (455 мг) растворяли в тетрагидрофуране (5,00 мл) и добавляли 0,99 моль/л комплекс борана/тетрагидрофурана (10,0 мл). После перемешивания при 0 С в течение 1 ч смесь перемешивали при комнатной температуре в течение дополнительных 3 ч. К реакционному раствору добавляли 2 моль/л соляную кислоту (6,8 мл), растворитель концентрировали и нерастворимые вещества удаляли в ходе фильтрации. Полученный в результате фильтрат экстрагировали дистиллированной водой и промывали дихлорметаном. Полученный в результате водный слой нейтрализовали соляной кислотой, экстрагировали смесью дихлорметана и метанола и растворитель выпаривали при пониженном давлении, что давало соединение (16) (300 мг). 1H-ЯМР (ДМСО-d6) : 3,71 (3 Н, s), 3,83 (1 Н, d, J=9,9 Гц), 4,04 (1 Н, d, J=6,6 Гц), 5,10 (1 Н, s), 5,75 (2 Н,s), 6,84 (2 Н, d, J=8,6 Гц), 7,33 (2 Н, d, J=8,6 Гц), 7,55 (1H, t, J=8,0 Гц), 7,83 (1 Н, d, J=8,1 Гц), 8,04 (1 Н, d,J=7,1 Гц), 8,34 (1 Н, s). Шестая стадия. Соединение (16) (300 мг) растворяли в ацетоне (3,00 мл), добавляли бензоилизотиоцианат (187 мг) и смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью хроматографии, что давало соединение (17) (470 мг). 1(1H, d, J=8,1 Гц), 8,22 (1H, s), 8,31 (2H, s), 11,20 (1H, s), 12,15 (1H, s). Седьмая стадия. Соединение (17) (31,2 мг) растворяли в дихлорметане (1,00 мл), добавляли 1-хлор-2 триметилпропениламин (0,0183 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Реакционный раствор экстрагировали дихлорметаном, органический слой промывали дистиллированной водой и растворитель выпаривали при пониженном давлении, что давало соединение (18) (15,6 мг). 1 Н-ЯМР (ДМСО-d6) : 3,74 (3 Н, s), 3,97 (2 Н, dd, J=20,2, 11,6 Гц), 6,94 (2 Н, d, J=8,3 Гц), 7,43 (2 Н, d,J=8,6 Гц), 7,49 (2 Н, t, J=7,5 Гц), 7,59 (1 Н, t, J=7,1 Гц), 7,68 (1H, t, J=8,0 Гц), 7,94 (1 Н, d, J=7,3 Гц), 8,08(2 Н, d, J=7,3 Гц), 8,15 (1 Н, d, J=8,6 Гц), 8,36 (1H, s). Восьмая стадия. Соединение (18) (320 мг) растворяли в метаноле (3,00 мл), добавляли моногидрат гидразина (0,107 мл), смесь перемешивали при 40 С в течение 3 ч и перемешивали при комнатной температуре в течение дополнительных 14 ч. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (19) (128 мг). Н-ЯМР (ДМСО-d6) : 3,71 (3 Н, s), 3,93 (2 Н, s), 6,78 (2 Н, s), 6,85 (2 Н, d, J=8,8 Гц), 7,40 (2 Н, d, J=8,8 Гц), 7,58 (1 Н, t, J=8,2 Гц), 7,89 (1 Н, d, J=7,6 Гц), 8,04 (1 Н, d, J=8,8 Гц), 8,32 (1H, s). Девятая стадия. Соединение (19) (120 мг) растворяли в дихлорметане (1,20 мл), добавляли ди-трет-бутил дикарбонат (564 мг) и смесь перемешивали при комнатной температуре в течение 6 ч. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (20) (105 мг). 1 Н-ЯМР (ДМСО-d6) : 1,41 (9 Н, s), 3,72 (3 Н, s), 3,90 (2 Н, dd, J=22,7, 12,6 Гц), 6,89 (2 Н, d, J=7,8 Гц),7,37 (2 Н, d, J=8,8 Гц), 7,64 (1 Н, s), 7,88 (1 Н, d, J=6,6 Гц), 8,10 (1 Н, s), 8,30 (1H, d, J=9,1 Гц). Десятая стадия. Соединение (20) (106 мг) растворяли в метаноле, добавляли 10% палладированный уголь (52,5 мг) и смесь перемешивали в течение 5 ч в атмосфере водорода. Нерастворимые вещества удаляли в ходе фильтрации через целит и растворитель выпаривали при пониженном давлении, что давало соединение (21)(98,7 мг). 1 Н-ЯМР (ДМСО-d6) : 1,40 (9 Н, s), 3,70-3,73 (4 Н, m), 3,84 (1H, d, J=11,4 Гц), 6,85-6,89 (3 Н, m), 6,966,99 (2 Н, m), 7,20 (1H, t, J=7,2 Гц), 7,33 (2 Н, d, J=8,8 Гц). Справочный пример 2. Синтез промежуточного соединения (31). Первая стадия. К перемешиваемому раствору 2,0 моль/л диизопропиламида лития в нормальном гептане/этилбензоле/тетрагидрофуране (16,2 мл) при -78 С по каплям в течение 10 мин добавляли раствор трет-бутилпропионата (4,73 мл) в тетрагидрофуране (10 мл). После перемешивания при -78 С в течение 1 ч и 20 мин раствор триизопропоксида хлортитана (12,0 г) в тетрагидрофуране (20 мл) добавляли по каплям в течение 40 мин. После перемешивания при -78 С в течение 1 ч раствор соединения (22) (3,00 г) в тетрагидрофуране (10 мл) добавляли по каплям в течение 30 мин. После перемешивания при -78 С в течение 1 ч смесь частями добавляли к насыщенному водному раствору хлорида аммония на бане со льдом, и полученные в результате нерастворимые вещества отделяли в ходе фильтрации. Нерастворимые вещества промывали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (23) (5,00 г) в виде неочищенного продукта. Вторая стадия. К соединению (23) (5,00 г), полученному на первой стадии, добавляли 4 моль/л раствор соляной кислоты/диоксана (39 мл) и метанол (1,2 мл) и смесь перемешивали при комнатной температуре в течение ночи. Добавляли диэтиловый эфир (60 мл) и осадок собирали в ходе фильтрации, что давало соединение(24) (2,37 г) в виде неочищенного продукта. Третья стадия. Соединение (24) (2,37 г), полученное на второй стадии, суспендировали в тетрагидрофуране (8 мл) и добавляли 1 моль/л раствор борана/тетрагидрофурана (24 мл) при 0 С. После перемешивания при комнатной температуре в течение 3,5 ч смесь выливали в ледяную воду и добавляли 1 моль/л водный раствор гидроксида натрия, чтобы получить щелочной раствор. Смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (25) (1,67 г) в виде неочищенного продукта. Четвертая стадия. К раствору соединения (25) (1,67 г), полученному на третье стадии, в тетрагидрофуране (16 мл) добавляли воду (8 мл), гидрокарбонат натрия (1,74 г) и имид N-(флуоренилметоксикарбонилокси)янтарной кислоты (1,74 г), смесь перемешивали при комнатной температуре в течение 2 ч и 20 мин, после чего экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, что давало соединение (26) (3,72 г) в виде неочищенного продукта. Пятая стадия. К раствору соединения (26) (3,72 г), полученному на четвертой стадии, в диметилсульфоксиде (15 мл) добавляли 2-йодоксибензойную кислоту (1,93 г) и смесь перемешивали при комнатной температуре в течение 2 ч и 30 мин. Добавляли воду и этилацетат, полученные в результате нерастворимые вещества удаляли в ходе фильтрации, органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, что давало соединение (27) (3,77 г) в виде неочищенного продукта. Шестая стадия. К раствору соединения (27) (3,77 г), полученному на пятой стадии, в метаноле (60 мл) добавляли метилортоформиат (38 мкл) и моногидрат п-толуолсульфоновой кислоты (66 мг) и смесь перемешивали и нагревали с обратным холодильником в течение 30 мин. Растворитель выпаривали при пониженном давлении, и остаток разбавляли этилацетатом. Смесь промывали насыщенным водным раствором гидрокарбоната натрия и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (28) (3,55 г) в виде неочищенного продукта. Седьмая стадия. К раствору соединения (28) (3,55 г), полученному на шестой стадии, в диметилформамиде (14 мл) добавляли пиперидин (1 мл) и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли воду, смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, и остаток пропускали через хроматографическую колонку, что давало смесь диастереомеров (29) (985 мг) при соотношении приблизительно 2:3, рассчитанному из протонного соотношения 1 Н-ЯМР. 1 Н-ЯМР (диастереомерная смесь, CDCl3) : 0,80 (d, J=7,2 Гц), 1,06 (d, J=7,2 Гц), 1,48 (d, J=1,4 Гц),1,62 (d, J=1,7 Гц), 2,40-2,60 (m), 3,17 (s), 3,21 (s), 3,36 (s), 3,41 (s), 3,72 (d, J=2,6 Гц), 4,34 (d, J=3,8 Гц),7,11-7,20 (m), 8,10-8,19 (m), 8,59 (dd, J=6,9, 3,0 Гц), 8,71 (dd, J=6,9, 2,9 Гц). Восьмая стадия. К раствору соединения (29) (985 мг) в ацетоне (5 мл) добавляли бензоилизотиоцианат (497 мкл) при 0 С, смесь перемешивали при 0 С в течение 20 мин и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную серную кислоту (6 мл), смесь перемешивали при 50 С в течение ночи, добавляли лед и смесь подщелачивали 28% водным раствором аммиака, перемешивая при охлаждении льдом. Смесь экстрагировали этилацетатом, промывали водой и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (30) (968 мг). 1 Н-ЯМР (CDCl3) : 1,77 (3 Н, d, J=1,1 Гц), 1,82 (1 Н, d, J=1,4 Гц), 6,07 (1 Н, d, J=1,4 Гц), 7,15 (1 Н, dd,J=10,3, 8,9 Гц), 8,16 (1 Н, ddd, J=8,9, 4,0, 2,9 Гц), 8,30 (1 Н, dd, J=6,9, 2,9 Гц). Девятая стадия. К раствору соединения (30) (968 мг) в уксусной кислоте (8 мл) и воде (1 мл) добавляли железо (769 мг) и смесь перемешивали при 60 С. Смесь подщелачивали 28% водным аммиаком, полученные в результате нерастворимые вещества удаляли в ходе фильтрации, фильтрат экстрагировали этилацетатом и высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (31) (968 мг). 1 Н-ЯМР (CDCl3) : 1,66 (3 Н, s), 1,78 (3 Н, d, J=1,3 Гц), 3,51 (2 Н, br), 5,94 (1 Н, d, J=4,7, 1,5 Гц), 6,51 Справочный пример 3. Синтез промежуточного соединения (39). Первая стадия. К раствору соединения (32) (5,14 г) в тетрагидрофуране (50 мл) добавляли воду (40 мл), гидрокарбонат натрия (7,57 г) и имид N-(флуоренилметоксикарбонилокси)янтарной кислоты (8,36 г), смесь перемешивали при комнатной температуре в течение 2,5 ч и экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, что давало соединение (33) (11,0 г) в виде неочищенного продукта. Вторая стадия. К раствору соединения (33) (11,0 г), полученному на первой стадии, в диметилсульфоксиде (40 мл) добавляли 2-йодоксибензойную кислоту (6,94 г) и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли воду и этилацетат и полученные в результате нерастворимые вещества удаляли в ходе фильтрации. Органический слой промывали водой, высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (34) (6,06 г). МС (М + 1) 449,00. Третья стадия. К диэтиловому эфиру (60 мл) добавляли тетрахлорид титана (3,1 мл) при -78 С и к полученной в результате желтой суспензии добавляли по каплям раствор 3 моль/л метилмагнийбромида в диэтиловом эфире (9,5 мл). Затем смесь нагревали до -40 С, раствор соединения (34) (3,18 г) в диэтиловом эфире (15 мл) добавляли по каплям в течение 30 мин. После перемешивания в течение 2,5 ч, добавляли лед, и смесь нагревали до комнатной температуры. Смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (35) (3,4 г) в виде неочищенного продукта. Четвертая стадия. К раствору соединения (35) (3,4 г), полученному на третьей стадии, в диметилсульфоксиде (16 мл) добавляли 2-йодоксибензойную кислоту (1,98 г) и смесь перемешивали при комнатной температуре в течение 2 дней. Добавляли воду и этилацетат, и полученные в результате нерастворимые вещества удаляли в ходе фильтрации. Органический слой промывали водой, высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (36) (2,34 г). МС(М + 1) 463,16. Пятая стадия. К раствору соединения (36) (2,34 г) в диметилформамиде (10 мл) добавляли пиперидин (600 мкл) и смесь перемешивали при комнатной температуре в течение 20 мин. Добавляли воду и смесь экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния, растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (37) (928 мг). 1 Н-ЯМР (CDCl3) : 1,51 (3 Н, s), 2,08 (3 Н, s), 2,93 (1H, d, J=18 Гц), 3,44 (1H, d, J=18 Гц), 7,13 (1H, dd,- 16020740J=11,0, 8,9 Гц), 8,13 (1H, ddd, J=8,9, 4,0, 2,9 Гц), 8,59 (1H, dd, J=7,2, 2,9 Гц). Шестая стадия. К раствору соединения (37) (928 мг) в ацетоне (4 мл) добавляли бензоилизотиоцианат (497 мкл) при 0 С, смесь перемешивали в течение 20 мин и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную серную кислоту (6 мл) и смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли лед и смесь подщелачивали 28% водным раствором аммиака, перемешивая при охлаждении льдом. Смесь экстрагировали этилацетатом, органический слой промывали водой и высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, полученный в результате остаток растворяли в этаноле (6 мл) и добавляли моногидрат гидразина(290 мг). После перемешивания при 50 С в течение 2 ч добавляли воду, смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток пропускали через хроматографическую колонку, что давало соединение(415 мг), смесь перемешивали при 50 С в течение 2 ч. Смесь подщелачивали 28% водным аммиаком,полученные в результате нерастворимые вещества удаляли в ходе фильтрации и фильтрат экстрагировали этилацетатом. Затем органический слой высушивали над безводным сульфатом магния, растворитель выпаривали при пониженном давлении, что давало соединение (39) (407 мг). 1 Н-ЯМР (CDCl3) : 1,66 (3 Н, d, J=1,1 Гц), 1,96 (3 Н, d, J=1,5 Гц), 3,51 (2 Н, br), 5,95 (2 Н, dd, J=4,7, 1,5 Гц), 6,48 (1 Н, ddd, J=8,5, 3,6, 2,9 Гц), 6,73 (1H, dd, J=6,7, 2,9 Гц), 6,81 (1H, dd, J=11,4, 8,5 Гц). Справочный пример 4. Синтез промежуточного соединения (53) Первая стадия. К охлажденной концентрированной серной кислоте (61 мл) на бане со льдом добавляли по каплям азотную кислоту (20 мл) для получения реагента. При -38 С к соединению (40) (50 г) добавляли по каплям концентрированную серную кислоту (137 мл) с последующим растворением. К этому раствору добавляли по каплям предварительно полученный реагент при -25 С в течение 1 ч. После этого внутреннюю температуру поддерживали от -15 до -20 С. После перемешивания в течение 1 ч реакционный рас- 17020740 твор добавляли к ледяной воде, энергично перемешивали в течение 40 мин при охлаждении льдом и смесь экстрагировали диэтиловым эфиром. Органический слой промывали водой, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (41) (72,3 г). 1 Н-ЯМР (смесь с гидратом, CDCl3) : 8,81 (1 Н, s), 8,51 (1 Н, d, J=8,1 Гц), 8,44 (1,5H, s), 8,33 (1 Н, d,J=7,6 Гц), 8,20 (1,5 Н, d, J=6,8 Гц), 7,93 (1,5 Н, d, J=7,8 Гц), 7,77 (1 Н, t, J=8,1 Гц), 7,56 (1,5 Н, t, J=16,8 Гц). Вторая стадия. Соединение (41) (10 г) растворяли в метаноле (200 мл), добавляли железо (15 г) и по каплям добавляли концентрированную соляную кислоту (23 мл) при охлаждении льдом. После 5 ч перемешивания полученные в результате нерастворимые вещества удаляли в ходе фильтрации через целит и к полученному в результате фильтрату добавляли 4 моль/л водный раствор гидроксида натрия (69 мл) при охлаждении льдом. Затем смесь фильтровали через целит, фильтрат экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (42) (8,63 г). 1 Н-ЯМР (CDCl3) : 7,43 (1H, d, J=7,6 Гц), 7,32-7,28 (2 Н, m), 6,99 (1 Н, d, J=8,1 Гц), 3,93 (2 Н, brs). Третья стадия. Соединение (42) (37,2 г) растворяли в этилацетате (200 мл), добавляли уксусный ангидрид (19,5 мл), смесь перемешивали при комнатной температуре в течение 2 ч и растворитель выпаривали при пониженном давлении. К остатку добавляли изопропиловый эфир, полученное в результате твердое вещество собирали в ходе фильтрации и промывали изопропиловым эфиром, что давало соединение (43) (15,8 г). Кроме того, к полученному в результате фильтрату добавляли воду и этилацетат и смесь экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (43) (10,5 г). 1 Н-ЯМР (смесь с гидратным соединением, ДМСО-d6) : 10,33 (1 Н, s), 10,01 (0,7 Н, s), 8,41 (1 Н, s),7,97 (1 Н, d, J=7,8 Гц), 7,78 (0,7 Н, s), 7,73-7,70 (1,7 Н, m), 7,59 (1 Н, t, J=8,0 Гц), 7,52 (1,4 Н, s), 7,32-7,27(1,4 Н, m), 2,08 (3 Н, s), 2,04 (2,1 Н, s). Четвертая стадия. Соединение (43) (2,0 г) растворяли в толуоле (100 мл), добавляли 2-амино-2-фенилэтанол (1,2 г) и пиридиния пара-толуолсульфонат (0,22 г) и смесь нагревали при 130 С в сосуде, оборудованном аппаратом Дина-Старка для дегидратации. Смесь перемешивали при этой температуре в течение 10 ч, охлаждали до комнатной температуры, добавляли водный раствор гидрокарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на аминосиликагеле, что давало соединение (44) (1,8 г). 1 Н-ЯМР (ДМСО-d6) : 10,07 (1 Н, s), 7,84 (1 Н, s), 7,73 (1 Н, d, J=8,3 Гц), 7,38-7,27 (7 Н, m), 4,68-4,61(1H, m), 4,52 (1 Н, t, J=7,1 Гц), 4,08 (1 Н, d, J=8,8 Гц), 3,59 (1H, t, J=8,3 Гц), 2,04 (3H, s). Пятая стадия. К тетравинилолову (10 мл) добавляли по каплям бутиллитий (42,2 мл, 2,6 моль/л) в потоке азота,добавляли пентан к полученному в результате белому твердому веществу, надосадочную жидкость сливали и растворяли в ТГФ (25 мл). В то же время соединение (44) (3,2 г) растворяли в ТГФ (32 мл) и предварительно полученный раствор виниллития (22,1 мл) добавляли по каплям при -78 С. После 1,5 ч перемешивания смесь добавляли к охлажденному льдом насыщенному водному раствору хлорида аммония и экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на аминосиликагеле, что давало соединение (45)(1H, m), 2,86-2,84 (1H, m), 2,10 (3 Н, s). Шестая стадия. Соединение (45) (1,1 г) растворяли в этаноле (11 мл), добавляли соляную кислоту (5,5 мл, 6 моль/л),смесь перемешивали при 90 С в течение 5 ч и добавляли гидроксид натрия (8,7 мл, 4 моль/л). Смесь экстрагировали этилацетатом, органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало неочищенный продукт (46)(0,42 мл) и бензилхлорформиат (0,308 мл) в потоке азота при охлаждении льдом. После 1 ч перемешива- 18020740 ния добавляли насыщенный водный раствор хлорида аммония при охлаждении льдом и смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором хлорида натрия,высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (47) (839 мг). 1 Н-ЯМР (ДМСО-d6) : 9,79 (1 Н, s), 7,79 (1 Н, s), 7,48-7,14 (13 Н, m), 5,80 (1 Н, dd, J=18,1, 11,2 Гц),5,42-5,38 (2 Н, dd, J=18,1, 11,2 Гц), 5,15 (2 Н, s), 5,08 (1 Н, s), 3,89-3,87 (1 Н, m), 3,35-3,25 (2 Н, m), 3,03-3,00(15,3 мл, 2 моль/л) в потоке азота при охлаждении льдом и смесь перемешивали при этой температуре в течение 30 мин. Смесь нагревали до комнатной температуры и перемешивали в течение 30 мин. Затем смесь нагревали до 40 С, перемешивали в течение 2,5 ч и охлаждали льдом. Потом добавляли воду (4,8 мл) к реакционному раствору, добавляли водный раствор гидроксида натрия (25,5 мл, 4 моль/л) и водный пероксид водорода (10,4 мл, 30%). После перемешивания при комнатной температуре в течение ночи смесь экстрагировали этилацетатом. Органический слой промывали водным раствором хлорида натрия,высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (48) (1,7 г). 1 Н-ЯМР (ДМСО-d6) : 9,77 (1 Н, s), 7,81 (1 Н, s), 7,49-7,12 (13 Н, m), 5,16 (2 Н, s), 4,98-4,96 (1H, m),4,52-4,49 (1H, m), 3,86-3,84 (1 Н, m), 3,37-3,18 (4 Н, m), 2,90-2,87 (1H, m), 2,28-2,25 (1 Н, m), 2,12-2,10 (1 Н,m). Девятая стадия. Соединение (48) (50 мг) растворяли в метаноле (0,5 мл), добавляли 20% Pd(OH)2 (28 мг, влажность 50%) и смесь перемешивали в течение 8 ч в потоке водорода. Растворитель фильтрата, полученного в ходе фильтрации через целит, выпаривали, что давало неочищенный продукт (49) (33 мг). Десятая стадия. Соединение (49) (24 мг) растворяли в ТГФ (0,5 мл), добавляли трифторуксусный ангидрид (0,029 мл) при охлаждении льдом, смесь перемешивали при комнатной температуре в течение 5,5 ч и добавляли водный раствор гидрокарбоната натрия. Затем смесь экстрагировали этилацетатом, органический слой промывали водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (50) (12 мг). 1 Н-ЯМР (CDCl3) : 8,53 (1 Н, s), 7,76 (1 Н, s), 7,68 (1 Н, d, J=8,1 Гц), 7,45 (1 Н, t, J=8,0 Гц), 7,40 (1 Н, d,J=7,8 Гц), 3,61-3,58 (1H, m), 3,46-3,39 (1H, m), 2,22 (2 Н, t, J=5,2 Гц). Одиннадцатая стадия. Соединение (50) (68 мг) растворяли в ацетоне (1 мл) и добавляли бензоилизотиоцианат (0,043 мл) при охлаждении льдом. После перемешивания в течение 15 мин смесь нагревали до комнатной температуры, перемешивали в течение 2,5 ч и затем перемешивали при 40 С в течение 4 ч. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, что давало соединение (51) (80 мг). 1 Н-ЯМР (ДМСО-d6) : 12,40 (1 Н, s), 11,69 (1 Н, s), 11,30 (1 Н, s), 8,03-8,01 (2 Н, m), 7,84-7,81 (2 Н, m),7,68-7,66 (1 Н, m), 7,56-7,54 (2 Н, m), 7,45-7,43 (1 Н, m), 7,34-7,33 (1H, m), 4,81-4,79 (1 Н, m), 3,73-3,62 (2 Н,m), 3,48-3,45 (1 Н, m), 2,67-2,65 (1 Н, m). Двенадцатая стадия. Соединение (51) (80 мг) растворяли в дихлорметане (2 мл) и добавляли 1-хлор-2 триметилпропениламин (0,043 мл) в потоке азота. Затем смесь перемешивали при комнатной температуре в течение 1 ч, добавляли водный раствор гидрокарбоната натрия и смесь экстрагировали этилацетатом. Органический слой промывали водой и водным раствором хлорида натрия, высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток очищали с помощью тонкослойной хроматографии на силикагеле, что давало соединение (52) (58 мг). 1(5 Н, m), 3,10-2,99 (1 Н, m), 2,92-2,82 (1 Н, m), 2,65-2,59 (1 Н, m), 2,12-2,05 (1 Н, m). Тринадцатая стадия. Соединение (52) (57 мг) растворяли в этаноле (1 мл) и добавляли 2 моль/л водный раствор гидроксида натрия (0,6 мл). После нагревания и перемешивания при 70 С в течение 7,5 ч смесь охлаждали до комнатной температуры и экстрагировали этилацетатом. Растворитель выпаривали при пониженном давлении, что давало соединение (53) (7,7 мг). 1 Н-ЯМР (ДМСО-d6) : 7,01 (1 Н, t, J=7,8 Гц), 6,67 (1 Н, s), 6,59 (1 Н, d, J=7,6 Гц), 6,51 (1H, d, J=7,8 Гц),6,29 (2 Н, s), 5,07 (2 Н, s), 2,90 (1 Н, d, J=10,6 Гц), 2,59-2,49 (2 Н, m), 1,74 (1 Н, t, J=13,4 Гц). Справочный пример 5. Синтез промежуточного соединения (58).(30 мл) и медленно по каплям добавляли ацетофенон (407 мкл), перемешивая при -78 С. После перемешивания при -78 С в течение 1 ч добавляли по каплям раствор соединения (54) (500 мг) в тетрагидрофуране (3 мл). После перемешивания смеси при -78 С в течение 1 ч добавляли насыщенный водный раствор хлорида аммония и смесь нагревали до комнатной температуры. Смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (55) (380 мг) в виде смеси диастереомеров. Вторая стадия. К смеси соединения (55) (380 мг), полученной на первой стадии, добавляли 4 моль/л раствор соляной кислоты/диоксана (1,8 мл) и метанол (45 мкл) и смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли диэтиловый эфир и воду, водный слой подщелачивали 28% водным аммиаком и смесь экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, что давало соединение (56) (181 мг). 1 Н-ЯМР (CDCl3) : 1,62 (3H, s, J=1,7 Гц), 3,43 (1 Н, d, J=18,8 Гц), 4,02 (1 Н, d, J=18,8 Гц), 7,08 (1 Н, dd,J=11,1, 9,0 Гц), 7,41-7,59 (3 Н, m), 7,85-7,90 (2 Н, m), 8,11 (1H, ddd, J=9,0, 4,0, 2,8 Гц), 8,69 (1 Н, dd, J=7,2,2,8 Гц). Третья стадия. К раствору соединения (56) (181 мг) в ацетоне (1,5 мл) добавляли бензоилизотиоцианат (81 мкл) при 0 С, смесь перемешивали при 0 С в течение 30 мин и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную серную кислоту (1,1 мл), смесь перемешивали при комнатной температуре в течение ночи, добавляли воду (500 мкл) и смесь перемешивали при 50 С в течение 6 ч. Смесь подщелачивали 28% водным раствором аммиака, перемешивая при охлаждении льдом. Смесь экстрагировали этилацетатом, органический слой промывали водой и высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (57) (112 мг). 1 Н-ЯМР (CDCl3) : 1,81 (3 Н, d, J=0,9 Гц), 4,80 (2 Н, s), 6,46 (1 Н, d, J=5,5 Гц), 7,17 (1 Н, dd, J=10,5, 8,9 Гц), 7,34-7,49 (5 Н, m), 8,13 (1 Н, ddd, J=8,9, 4,0, 3,0 Гц), 8,45 (1 Н, dd, J=6,8, 3,0 Гц). Четвертая стадия. К раствору соединения (57) (112 мг) в уксусной кислоте (1 мл) и воде (0,1 мл) добавляли железо (91 мг) и смесь перемешивали при 50 С в течение 2,5 ч. Полученные в результате нерастворимые вещества удаляли в ходе фильтрации, фильтрат подщелачивали 28% водным аммиаком, экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (58) (98,7 мг) в виде неочищенного продукта. Справочный пример 6. Синтез промежуточного соединения (63). Первая стадия. К раствору 0,5 моль/л гексаметилдисилазида калия в толуоле (8 мл) добавляли диэтиловый эфир (30 мл) и медленно по каплям добавляли 3-метил-2-бутанон (376 мкл), перемешивая при -78 С. После перемешивания при -78 С в течение 1 ч добавляли по каплям раствор соединения (59) (500 мг) в диэтиловом эфире (8 мл). После перемешивания при -78 С в течение 1,5 ч добавляли насыщенный водный раствор хлорида аммония и смесь нагревали до комнатной температуры. Смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (60)(307 мг). 1 Н-ЯМР (CDCl3) : 1,02 (3 Н, d, J=6,9 Гц), 1,06 (3 Н, d, J=6,9 Гц), 1,34 (9 Н, s), 1,77 (3 Н, s), 2,55 (1H,септет, J=6,9 Гц), 3,41 (1 Н, d, J=18,8 Гц), 3,72 (1 Н, d, J=18,8 Гц), 5,46 (1 Н, s), 7,12 (1H, dd, J=11,3, 8,9 Гц),8,13 (1H, ddd, J=8,9, 4,0, 2,8 Гц), 8,56 (1 Н, dd, J=6,9, 2,8 Гц). Вторая стадия. К соединению (60) (307 мг), полученному на первой стадии, добавляли 4 моль/л раствор соляной кислоты/диоксана (1,65 мл) и метанол (40 мкл) и смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли диэтиловый эфир и воду, водный слой подщелачивали 28% водным аммиаком и смесь экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении, что давало соединение (61) (209 мг). Третья стадия. К раствору соединения (61) (209 мг) в ацетоне (1 мл) добавляли бензоилизотиоцианат (105 мкл) при 0 С, смесь перемешивали при 0 С в течение 30 мин и растворитель выпаривали при пониженном давлении. К остатку добавляли концентрированную серную кислоту (1,6 мл), смесь перемешивали при комнатной температуре в течение ночи и смесь подщелачивали 28% водным раствором аммиака, перемешивая при охлаждении льдом. Смесь экстрагировали этилацетатом, органический слой промывали водой и высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении и остаток очищали с помощью колоночной хроматографии, что давало соединение (62) (96 мг). 1 Н-ЯМР (CDCl3) : 1,57 (3 Н, s), 1,68 (3 Н, s), 1,77 (3 Н, s), 2,70 (1 Н, d, J=14,2 Гц), 2,87 (1H, d, J=14,2 Гц), 4,80 (2 Н, s), 6,46 (1 Н, d, J=5,5 Гц), 7,14 (1 Н, dd, J=11,0, 8,9 Гц), 8,12 (1H, ddd, J=8,9, 4,0, 3,0 Гц), 8,44(1 Н, dd, J=6,9, 3,0 Гц). Четвертая стадия. К раствору соединения (62) (96 мг) в уксусной кислоте (1 мл) и воде (0,1 мл) добавляли железо (87 мг) и смесь перемешивали при 50 С в течение 2 ч. Смесь подщелачивали 28% водным аммиаком, полученные в результате нерастворимые вещества удаляли в ходе фильтрации. Фильтрат экстрагировали этилацетатом и высушивали над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, что давало соединение (63) (78,3 мг) в виде неочищенного продукта. Справочный пример 7. Синтез промежуточного соединения (70) Первая стадия. Соединение (64) (12 г) растворяли в тетрагидрофуране (240 мл) и по каплям добавляли 1 моль/л раствор аллилмагнийбромида/эфира при -78 С в течение 1 ч при перемешивании. После дополнительного перемешивания при -78 С в течение 1 ч смесь переносили в насыщенный водный раствор хлорида аммония, экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом натрия. Неорганическое вещество удаляли в ходе фильтрации, растворитель выпаривали при пониженном давлении и остаток пропускали через хроматографическую колонку, что давало соединение (65) (9,7 г). 1 Н-ЯМР (CDCl3) : 1,21 (9 Н, s), 1,68 (3 Н, s), 2,79 (1 Н, dd, J=13,4, 7,3 Гц), 2,92 (1 Н, dd, J=13,4, 6,8 Гц),4,16 (1 Н, s), 5,10 (1 Н, d, J=9,6 Гц), 5,13 (1 Н, d, J=17,2 Гц), 5,52-5,66 (1 Н, m), 6,95 (1H, dd, J=10,6, 10,1 Гц),7,55-7,62 (1 Н, m), 7,67-7,72 (1H, m), 9,96 (1 Н, s). Вторая стадия. Соединение (65) (3,99 г) растворяли в этаноле (20 мл), добавляли 1 моль/л раствор соляной кислоты-этанола при комнатной температуре при перемешивании и смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении, остаток разбавляли этилацетатом и смесь экстрагировали 2 моль/л соляной кислотой. Полученный в результате водный слой делали основным с помощью карбоната калия (рН 8-9), смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом натрия. Неорганическое вещество удаляли в ходе фильтрации и растворитель выпаривали при пониженном давлении, что давало соединение (66). Все его количество использовали в следующей реакции без очистки. 1 Н-ЯМР (CDCl3) : 1,51 (3 Н, s), 1,75-1,89 (2 Н, br), 2,47 (1H, dd, J=13,1, 8,1 Гц), 2,76 (1H, dd, J=13,1,7,1 Гц), 5,03-5,11 (2 Н, m), 5,46-5,58 (1H, m), 7,01-7,08 (1 Н, m), 7,52-7,60 (2H, m), 8,25-8,36 (1 Н, br). Третья стадия. Полученное в результате соединение (66) растворяли в метиленхлориде (20 мл) и бензоилизотиоцианат (1,43 мл) добавляли по каплям, перемешивая при охлаждении льдом. Смесь перемешивали в течение 5 мин при охлаждении льдом и перемешивали при комнатной температуре в течение 45 мин. Растворитель выпаривали при пониженном давлении и остаток пропускали через хроматографическую колонку, что давало соединение (67) (4,34 г). 1 Н-ЯМР (CDCl3) : 2,00 (3 Н, s), 2,73 (1 Н, dd, J=13,1, 6,6 Гц), 3,18 (1 Н, dd, J=13,1, 7,6 Гц), 5,24 (1 Н, d,J=10,6 Гц), 5,29 (1 Н, J=17,2 Гц), 5,67-5,79 (1 Н, m), 6,99-7,07 (1 Н, dd, J=10,4, 10,4 Гц), 7,45-7,53 (3 Н, m),7,57-7,65 (2 Н, m), 7,83 (2 Н, d J=7,07 Гц), 8,39 (1 Н, s), 8,86 (1 Н, s), 11,36 (1 Н, s). Четвертая стадия. Соединение (67) (4,34 г) растворяли в метиленхлориде (220 мл), добавляли йод (3,64 г), одновременно перемешивая при охлаждении льдом и смесь дополнительно перемешивали в течение 30 мин при охлаждении льдом. После разбавления водой оставшийся йод восстанавливали с гипосульфитом натрия,водный слой нейтрализовали (рН 8) с гидрокарбонатом натрия. Смесь экстрагировали этилацетатом и органический слой высушивали над сульфатом натрия. Неорганическое вещество удаляли в ходе фильтрации и растворитель выпаривали при пониженном давлении, что давало соединение (68). Все его количество использовали в следующей реакции без очистки. Пятая стадия. Соединение (68) растворяли в тетрагидрофуране (130 мл), добавляли пирролидин (4 мл), смесь нагревали с обратным холодильником в течение 2 ч. Добавляли воду, смесь экстрагировали этилацетатом и органический слой высушивали над сульфатом натрия. Неорганическое вещество удаляли в ходе фильтрации и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (69) (3,44 г). 1 Н-ЯМР (CDCl3) : 1,71 (3 Н, s), 2,71 (1 Н, d, J=14,1 Гц), 3,39 (1 Н, d, J=14,1 Гц), 5,01 (1 Н, s), 5,09 (1 Н,s), 7,10 (1 Н, dd, J=10,6, 10,1 Гц), 7,31-7,40 (4 Н, m), 7,47 (1 Н, t, J=7,0 Гц), 8,05 (2 Н, d, J=7,6 Гц), 8,08-8,13(1H, m). Шестая стадия. Соединение (69) (459 мг) растворяли в этаноле (9,2 мл), добавляли моногидрат гидразина (0,986 мл) и смесь перемешивали при 50 С в течение 4,5 ч. Добавляли воду, смесь экстрагировали хлороформом и органический слой высушивали над сульфатом натрия. Неорганическое вещество удаляли в ходе фильтрации и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (70) (158 мг). 1 Н-ЯМР (CDCl3) : 1,54 (3 Н, s), 2,53 (1H, d, J=13,6 Гц), 2,89 (1 Н, d, J=13,6 Гц), 4,97 (1 Н, s), 5,09 (1 Н,s), 6,45-6,51 (1 Н, m), 6,73-6,84 (2 Н, m). Справочный пример 8. Синтез промежуточного соединения (76). Первая стадия. Соединение (71) (8,00 г) растворяли в метиленхлориде (20 мл), добавляли этилакрилат (40 мл) и катализатор Граббса второго поколения (0,412 г) и смесь перемешивали при комнатной температуре в течение 2 ч в атмосфере азота. Растворитель выпаривали при пониженном давлении и остаток пропускали через хроматографическую колонку, что давало соединение (72) (8,34 г). 1 Н-ЯМР (CDCl3) : 1,26 (3 Н, t, J=6,6 Гц), 1,53-1,78 (3 Н, br), 2,72-2,92 (1H, br), 3,14-3,38 (1 Н, br), 4,23(2 Н, q, J=6,6 Гц), 4,25-4,50 (2 Н, br), 5,44-5,67 (1 Н, br), 5,82-5,98 (1 Н, br), 6,73-6,93 (1 Н, br), 6,97 (1 Н, dd,J=9,6, 9,6 Гц), 7,19-7,85 (11H, m), 8,67-8,82 (1H,br). Вторая стадия. Соединение (72) (8,32 г) растворяли в N,N-диметилформамиде, добавляли пиперидин (0,125 мл) и смесь перемешивали при комнатной температуре в течение 15 ч. К реакционному раствору добавляли воду, смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении и остаток пропускали через хроматографическую колонку, что давало соединение (73) (4,81 г). 1 Н-ЯМР (CDCl3) : 1,26 (3 Н, t, J=7,1 Гц), 1,55 (3 Н, s), 1,65-1,88 (2 Н, br), 2,66 (1 Н, dd, J=14,1, 8,1 Гц),2,89 (1 Н, dd, J=14,1, 8,1 Гц), 4,15 (2 Н, q, J=7,1 Гц), 5,86 (1 Н, d, J=15,2 Гц), 6,69 (1 Н, ddd, J=15,2, 8,1, 8,1 Гц), 7,07 (1H, dd, J=10,1, 8,1 Гц), 7,56-7,64 (2 Н, m), 8,14-8,26 (1 Н, br). Третья стадия. Соединение (73) (0,950 г) растворяли в метиленхлориде (14 мл), по каплям добавляли бензоилизотиоцианат (0,370 мл) в течение 10 мин, охлаждая льдом, и смесь перемешивали в течение 30 мин при охлаждении льдом. После этого добавляли йод (0,732 г), одновременно охлаждая льдом и смесь переме- 23020740 шивали при комнатной температуре в течение 45 мин. Смесь опять охлаждали льдом и раствор 1,8 бицикло[5.4.0]ундек-7-ена (1,581 мл), растворенного в метиленхлориде (10 мл), добавляли по каплям в течение 5 мин. Смесь нагревали до комнатной температуры и смесь перемешивали в течение 4 ч. Реакционный раствор разбавляли этилацетатом и водой, добавляли сульфит натрия, смесь нейтрализовали гидрокарбонатом натрия и экстрагировали этилацетатом. Органический слой высушивали над безводным сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (74) (0,939 г). 1 Н-ЯМР (CDCl3) : 1,22 (3 Н, t, J=7,1 Гц), 1,73 (3 Н, s), 2,93 (1H, d, J=14,7 Гц), 3,58 (1H, d, J=14,7 Гц),4,09 (2 Н, q, J=7,1 Гц), 5,83 (1 Н, s), 7,14 (1 Н, dd, J=11,1, 9,1 Гц), 7,31-7,45 (3 Н, m), 7,58 (1 Н, t, J=7,1 Гц),8,00 (2 Н, d, J=7,1 Гц), 8,10-8,16 (1 Н, m), 9,62-9,76 (1H, br). Четвертая стадия. Соединение (74) (0,700 мг) растворяли в толуоле и внутреннюю температуру поддерживали ниже-50 С. 1 моль/л раствор диизобутилалюмогидрида в толуоле добавляли по каплям в течение 5 мин, смесь охлаждали на бане с ацетоном-сухим льдом и дополнительно перемешивали в течение 30 мин. После гашения метанолом смесь нагревали до комнатной температуры, фильтровали и растворитель выпаривали при пониженном давлении. Остаток пропускали через хроматографическую колонку, что давало соединение (75) (0,183 г). 1 Н-ЯМР (CDCl3) : 1,52-1,98 (1 Н, br), 1,78 (3H, s), 2,75 (1H, d, J=13,9 Гц), 3,40 (1H, d, J=13,9 Гц),3,44-3,87 (2H, br), 4,07 (1 Н, dd, J=13,1, 5,1 Гц), 4,14 (1H, dd, J=13,1, 3,5 Гц), 5,63 (1H, dd, J=5,1, 3,5 Гц),6,46-6,59 (2H, m), 6,87 (1 Н, dd, J=11,1, 9,1 Гц), 7,46 (2H, dd, J=7,1, 7,1 Гц), 7,53 (1H, t, J=7,1 Гц), 8,25 (2H,d, J=7,1 Гц). Пятая стадия. Соединение (75) (60,4 мг) растворяли в этаноле (600 мкл), добавляли моногидрат гидразина (38 мкл) и смесь перемешивали при комнатной температуре в течение ночи. Реакционный раствор разбавляли насыщенным водным раствором хлорида натрия, экстрагировали хлороформом и органический слой высушивали над безводным сульфатом натрия. После фильтрации растворитель выпаривали при пониженном давлении, что давало соединение (76) в виде смеси с бензоилгидразином. Соединение (76) использовали в следующей реакции без очистки. 1 Н-ЯМР (CDCl3) : 1,45 (3 Н, s), 2,41 (1 Н, d, J=14,2 Гц), 2,82 (1 Н, d, J=14,2 Гц), 3,29-3,80 (4 Н, br),3,98 (2 Н, d, J=6,6 Гц), 5,49 (1 Н, m), 6,36-6,41 (1 Н, m), 6,60-6,65 (1H, m), 6,70 (1 Н, dd, J=12,1, 9,1 Гц). Справочный пример 9. Синтез промежуточного соединения (79) Первая стадия. Соединение (77) (500 мг) растворяли в диметилацетамиде (7,5 мл), добавляли карбонат калия (440 мг) и 1 Н-1,2,4-триазол (200 мг) и смесь перемешивали при 130 С в течение 10 мин в условиях микроволнового излучения. Добавляли воду, смесь экстрагировали этилацетатом, органический слой промывали водой и насыщенным водным раствором хлорида натрия. Органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток промывали диизопропиловым спиртом, что давало соединение (78) (451 мг). 1H-ЯМР (ДМСО-d6) : 3,96 (3 Н, s), 8,48 (1H, s), 9,16 (1 Н, d, J=1,5 Гц), 9,29 (1 Н, d, J=1,5 Гц), 9,56 (1H,s). Вторая стадия. Соединение (78) (50 мг) растворяли в тетрагидрофуране (1 мл) и метаноле (1 мл), добавляли 2 моль/л водный раствор гидроксида натрия (0,12 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли 2 моль/л соляную кислоту (0,12 мл), растворитель выпаривали при пониженном давлении и к остатку добавляли хлороформ, что в ходе фильтрации давало соединение (79) (27 мг) (содержащее хлорид натрия). 1 Справочный пример 10. Синтез промежуточного соединения (84).(0,66 г) и карбонат калия (1,6 г) и смесь перемешивали при 120 С в течение 1 ч. Добавляли 0,2 моль/л соляную кислоту, чтобы довести рН до 4, и смесь экстрагировали этилацетатом. Органический слой промывали сначала водой и затем насыщенным водным раствором хлорида натрия, органический слой высушивали над безводным сульфатом магния и растворитель выпаривали при пониженном давлении. Остаток промывали диэтиловым эфиром, что давало соединение (81) (1,1 г). 1(1 Н, s). Вторая стадия. Соединение (81) (200 мг) растворяли в тетрагидрофуране (4 мл) и метаноле (4 мл), добавляли 2 моль/л водный раствор гидроксида натрия (1,3 мл) и смесь перемешивали при комнатной температуре в течение 3 ч. Добавляли 2 моль/л соляную кислоту (1,3 мл) и органический растворитель выпаривали при пониженном давлении. Добавляли воду и остаток собирали в ходе фильтрации и промывали тетрагидрофураном, что давало соединение (82) (161 мг). 1(1H, s). Третья стадия. Соединение (81) (300 мг) и карбонат калия (197 мг) растворяли в диметилформамиде (3 мл), добавляли метилиодид (203 мг) и смесь перемешивали при 60 С в течение 2 ч. Добавляли воду, смесь экстрагировали этилацетатом, органический слой промывали водой и насыщенным водным раствором хлорида натрия. Органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток хроматографировали на силикагеле, что давало соединение (83) (162 мг). 1H-ЯМР (ДМСО-d6) : 3,39 (3 Н, s), 3,42 (3 Н, s), 3,91 (3 Н, s), 8,87 (1 Н, d, J=1,5 Гц), 8,99 (1 Н, d, J=1,5 Гц). Четвертая стадия. Соединение (83) (153 мг) растворяли в тетрагидрофуране (3 мл) и метаноле (3 мл), добавляли 2 моль/л водный раствор гидроксида натрия (0,31 мл) и смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли 2 моль/л соляную кислоту (0,31 мл), органический растворитель выпаривали при пониженном давлении и добавляли воду. Остаток собирали в ходе фильтрации и промывали водой, что давало соединение (84) (121 мг). 1 Справочный пример 11. Синтез промежуточного соединения (88). Первая стадия. Соединение (85) (2,50 г) растворяли в пиридине (25 мл), добавляли тритилхлорид (5,47 г) и диметиламинопиридин (2,40 г), смесь перемешивали при 100 С в течение 22 ч. Добавляли воду, смесь экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле,что давало соединение (86) (2,81 г). 1 Н-ЯМР (CDCl3) : 0,94 (3 Н, t, J=7,1 Гц), 3,82 (2 Н, q, J=7,1 Гц), 6,97 (1 Н, d, J=1,0 Гц), 7,12 (7 Н, m),7,28-7,30 (9H, m). Вторая стадия. К охлаждаемому льдом и перемешиваемому тетрагидрофурану (25 мл) добавляли алюмогидрид лития (333 мг) и добавляли по каплям раствор соединения (86) (2,80 г) в тетрагидрофуране (40 мл). После проведения реакции при комнатной температуре в течение 1 ч добавляли воду (0,35 мл), 2 моль/л водный раствор гидроксида натрия (0,35 мл) и воду (1,05 мл), перемешивая при охлаждении льдом и смесь перемешивали при комнатной температуре в течение 1 ч. Полученные в результате нерастворимые вещества удаляли через целит и нерастворимые вещества на целите промывали горячим хлороформом и метанолом. Эти промывочные растворы выпаривали при пониженном давлении, что давало соединение (87)(9 Н, m). Третья стадия. Соединение (87) (681 мг) и 5-хлорпиразин-2-карбоновую кислоту (317 мг) растворяли в ДМФА (20 мл), добавляли гидрид натрия (240 мг), перемешивая при охлаждении льдом и смесь перемешивали при 75 С в течение 3 ч. После проведения реакции добавляли насыщенный водный раствор хлорида аммония(50 мл) и реакционный раствор концентрировали при пониженном давлении. К остатку добавляли горячий этанол, смесь экстрагировали и растворитель выпаривали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, что давало соединение (88) (497 мг, содержащее примеси). 1 Н-ЯМР (CDCl3) : 4,85 (2 Н, s), 6,88 (1 Н, s), 7,00-7,42 (16 Н, m), 7,84 (1 Н, s), 8,59 (1H, s). Справочный пример 12. Синтез промежуточного соединения (95) Первая стадия. Когда n представляет собой 1. Соединение (89) (10,0 г) растворяли в ацетонитриле (200 мл), добавляли бензил 2-бромэтиловый эфир (9,5 мл) и карбонат калия (11,3 г) и смесь перемешивали при 85 С в течение 1,5 ч. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении и добавляли воду. Смесь экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении, что давало соединение (90) (17,0 г). 1 Н-ЯМР (CDCl3) : 3,84 (3 Н, d, J=2,5 Гц), 3,86 (2 Н, t, J=5,8 Гц), 3,94 (3 Н, s), 4,47 (2 Н, s), 4,90 (2 Н, t,J=5,8 Гц), 7,19-7,36 (6 Н, m). Когда n представляет собой 2. Соединение (89) (10,0 г) растворяли в ацетонитриле (100 мл), добавляли бензил 3-бромпропиловый эфир (11,2 мл) и карбонат калия (11,3 г) и смесь перемешивали при 85 С в течение 1,5 ч. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении и добавляли воду. Смесь экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении, что давало соединение (90) (18,0 г). 1 Н-ЯМР (CDCl3) : 2,16-2,23 (2 Н, m), 3,50 (2 Н, t, J=6,1 Гц), 3,87 (3 Н, s), 3,93 (3 Н, s), 4,48 (2 Н, s), 4,77(2 Н, t, J=7,4 Гц), 7,26-7,35 (6 Н, m). Вторая стадия. Когда n представляет собой 1. Соединение (90) (17,0 г) растворяли в МеОН и добавляли 0,7 моль/л водный раствор гидроксида натрия (32 мл), перемешивая при охлаждении льдом. После перемешивания при комнатной температуре в течение 18 ч реакционный раствор концентрировали при пониженном давлении. Добавляли 0,2 моль/л соляную кислоту. Смесь экстрагировали этилацетатом и органический слой высушивали над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, что давало соединение (91)(16,0 г) в виде неочищенного продукта. Когда n представляет собой 2, соединение синтезировали подобным образом. Спектр неочищенного продукта не измеряли. Третья стадия. Когда n=1. Соединение (91) (16,0 г) растворяли в трет-бутиловом спирте, добавляли дифенилфосфорилазид (14 мл) и триэтиламин (10 мл), смесь перемешивали и нагревали с обратным холодильником в течение 4 ч. Реакционный раствор концентрировали при пониженном давлении. Добавляли воду, смесь экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле,что давало соединение (92) (15,0 г). 1 Н-ЯМР (CDCl3) : 1,44 (9 Н, s), 3,85 (2 Н, t, J=4,3 Гц), 3,92 (3 Н, s), 4,37 (2 Н, t, J=4,3 Гц), 4,52 (2 Н, s),6,82 (1H, s), 7,28 (5 Н, m), 7,90 (1 Н, s). Когда n представляет собой 2, соединение синтезировали подобным образом. 1 Н-ЯМР (CDCl3) : 1,48 (9 Н, s), 2,08-2,14 (2 Н, m), 3,35 (2 Н, t, J=5,6 Гц), 3,90 (3 Н, s), 4,23 (2 Н, t, J=6,1 Гц), 4,57 (2 Н, s), 6,80 (1H, s), 7,35 (5 Н, m, Гц), 7,75 (1 Н, s). Четвертая стадия. Когда n=1. Соединение (92) (15,0 г) растворяли в метаноле (150 мл), добавляли гидроксид палладия (1,40 г) и смесь энергично перемешивали при комнатной температуре в потоке водорода. Через 5 ч нерастворимые вещества удаляли в ходе фильтрации через целит и фильтрат концентрировали при пониженном давлении, что давало соединение (93) (11,4 г) в виде неочищенного продукта. 1(1H, s). Когда n представляет собой 2, соединение синтезировали подобным образом. 1(12,1 г) и трибутилфосфин (13 мл) и смесь перемешивали при 75 С в течение 15 мин. После проведения реакции смесь охлаждали до комнатной температуры. Выпавшие в осадок нерастворимые вещества удаляли в ходе фильтрации и фильтрат концентрировали при пониженном давлении. К остатку добавляли воду, смесь экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, что давало соединение (94) (7,60 г). 1 Н-ЯМР (CDCl3) : 1,57 (9 Н, s), 3,91 (3 Н, s), 4,39 (4 Н, s), 6,17 (1 Н, s). Когда n представляет собой 2, соединение синтезировали подобным образом. Н-ЯМР (CDCl3) : 1,58 (9 Н, s), 2,20 (2 Н, m), 3,84 (2 Н, t, J=5,6 Гц), 3,91 (3 Н, s), 4,24 (2 Н, t, J=6,1 Гц),6,80 (1 Н, s). Шестая стадия. Когда n=1. Соединение (94) (1,01 г) растворяли в ТГФ (10 мл) и МеОН (10 мл), добавляли 4 моль/л водный раствор гидроксида лития (1,9 мл) и смесь перемешивали при комнатной температуре в течение 4,5 ч. К реакционному раствору добавляли 0,1 моль/л соляную кислоту, смесь экстрагировали хлороформом и органический слой высушивали над безводным сульфатом натрия. Растворитель выпаривали при пониженном давлении, что давало соединение (95) (844 мг) в виде неочищенного продукта. 1 Н-ЯМР (CDCl3) : 1,59 (9 Н, s), 4,41 (4 Н, s), 6,21 (1 Н, s). Когда n представляет собой 2, соединение синтезировали подобным образом. 1 Н-ЯМР (CDCl3) : 1,56 (9 Н, s), 2,18-2,24 (2 Н, m), 3,85 (2 Н, t, J=5,8 Гц), 4,25 (2H, t, J=6,1 Гц), 6,82(1H, s). Справочный пример 13. Синтез промежуточного соединения (101). 1 Первая стадия. Соединение (96) (15 г) растворяли в дихлорметане (150 мл), добавляли mCPBA (21,4 г) и смесь перемешивали при комнатной температуре в течение 24 ч. Добавляли насыщенный водный раствор гидрокарбоната натрия, смесь экстрагировали дихлорметаном. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, высушивали над сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, что давало соединение (97) (14,3 г). 1 Н-ЯМР (ДМСО-d6) : 8,60-8,58 (1,0 Н, m), 7,85-7,83 (2,0 Н, m), 4,40-4,32 (4,0 Н, m), 1,33-1,31 (6,0 Н,m). Вторая стадия. Соединение (97) (14,3 г) растворяли в этаноле (72 мл) и воде (72 мл), добавляли концентрированную соляную кислоту (14,3 мл) и смесь перемешивали при 60 С в течение 14,5 ч. После охлаждения льдом полученное в результате твердое вещество собирали в ходе фильтрации и промывали водой, что давало соединение (98) (9,8 г). 1 Н-ЯМР (ДМСО-d6) : 9,00-8,97 (1,0 Н, m), 8,41-8,39 (1,0 Н, m), 8,28-8,26 (1,0 Н, m), 4,40 (2,0 Н, q,J=6,91 Гц), 1,36 (3,0 Н, t, J=6,95 Гц). Третья стадия. Тионилхлорид (13,5 мл) растворяли в дихлорметане (200 мл), добавляли ДМФА (4,7 мл) при охлаждении льдом, добавляли соединение (98) (9,7 г) при этой температуре и смесь перемешивали при 65 С в течение 2,5 ч. После охлаждения до комнатной температуры добавляли воду (31 мл), охлаждая льдом. Дихлорметан выпаривали при пониженном давлении, выпавшее в осадок твердое вещество собирали в ходе фильтрации и промывали водой, что давало соединение (99) (9,3 г). 1 Н-ЯМР (ДМСО-d6) : 8,99 (1,0 Н, d, J=1,68 Гц), 8,42 (1,0 Н, d, J=1,68 Гц), 4,36 (2,0 Н, q, J=7,07 Гц),1,34 (3,0 Н, t, J=7,09 Гц). Четвертая стадия. К соединению (99) (6 г) добавляли 28% водный аммиак (40,2 мл), смесь перемешивали при комнатной температуре в течение 2 ч и по каплям добавляли концентрированную соляную кислоту (44,1 мл) при охлаждении льдом. Выпавшее в осадок твердое вещество собирали в ходе фильтрации и промывали водой, что давало соединение (100) (4,6 г). 1 Н-ЯМР (ДМСО-d6) : 8,97-8,95 (1,0 Н, m), 8,43-8,40 (1,0 Н, m), 8,33 (1,0H, br s), 7,84 (1,0 Н, brs). Пятая стадия. Соединение (100) (1,5 г) растворяли в ДМФА (15 мл), добавляли оксалилхлорид (1,9 мл) при охлаждении льдом, смесь перемешивали при этой температуре в течение 1,5 ч и добавляли 4 моль/л гидроксид натрия (9,4 мл). Выпавшее в осадок твердое вещество собирали в ходе фильтрации и промывали водой, что давало соединение (6) (706 мг). Кроме того, фильтрат экстрагировали этилацетатом, органический слой высушивали над безводным сульфатом натрия и растворитель выпаривали при пониженном давлении. Остаток промывали водой и собирали в ходе фильтрации, что давало соединение (101) (305 мг). 1 Н-ЯМР (ДМСО-d6) : 9,03-9,00 (1,0 Н, m), 8,75-8,72 (1,0 Н, m). Справочный пример 14. Синтез промежуточного соединения (105). Первая стадия. К суспензии гидрида натрия (3,90 г) в тетрагидрофуране (30 мл) добавляли соединение (102) (9,8 мл) и этилформиат (10,8 мл) при 15 С и смесь перемешивали при комнатной температуре в течение 5 дней. Полученное в результате твердое вещество собирали в ходе фильтрации, промывали эфиром и естественным образом сушили, что давало соединение (103) (11,7 г). Вторая стадия. Соединение (103) (1,05 г) растворяли в этаноле (3 мл) и добавляли гидрохлорид ацетамидина (500 мг), смесь нагревали с обратным холодильником в течение 7 ч. Затем растворитель выпаривали при пониженном давлении, добавляли воду и смесь экстрагировали этилацетатом. Потом растворитель выпаривали при пониженном давлении, остаток подвергали колоночной хроматографии на силикагеле, что давало соединение (104) (294 мг). 1 Н-ЯМР (CDCl3) : 2,84 (3 Н, s), 8,90 (2 Н, s). Третья стадия. К раствору соединения (104) (2,15 г) в пиридине (20 мл) добавляли диоксид селена (4,01 г) и смесь перемешивали при 80 С в течение 4 ч. Смесь оставляли при комнатной температуре на ночь, полученные в результате нерастворимые вещества удаляли в ходе фильтрации и растворитель выпаривали при пониженном давлении. Остаток растворяли в воде, смесь пропускали через хроматографическую колонку HP20SS и растворитель выпаривали при пониженном давлении. Остаток промывали ацетоном, что давало соединение (105) (491 мг). 1 Н-ЯМР (ДМСО-d6) : 9,44 (2 Н, s). Справочный пример 15. Синтез промежуточного соединения (107). Первая стадия. Раствор соединения (106) (1,0 г) в концентрированной серной кислоте (5,4 мл) и воде (600 мкл) перемешивали при 110 С в течение 22 ч, смесь выливали в ледяную воду и выпавшее в осадок твердое вещество собирали в ходе фильтрации. Твердое вещество промывали водой и естественным образом сушили, что давало соединение (107) (1,02 г). 1 Н-ЯМР (ДМСО-d6) : 8,08 (1H, m), 8,60 (1H, m).
МПК / Метки
МПК: A61K 31/497, C07D 417/14, A61K 31/541, A61P 25/28, A61P 43/00, C07D 417/10, C07D 417/12, A61K 31/4439
Метки: ингибитора, обладающее, производное, гетероциклическое, серосодержащее, бета-секретазы, активностью
Код ссылки
<a href="https://eas.patents.su/30-20740-serosoderzhashhee-geterociklicheskoe-proizvodnoe-obladayushhee-aktivnostyu-ingibitora-beta-sekretazy.html" rel="bookmark" title="База патентов Евразийского Союза">Серосодержащее гетероциклическое производное, обладающее активностью ингибитора бета-секретазы</a>
Полициклическое карбамоилпиридоновое производное, обладающее ингибиторной активностью в отношении интегразы вич

Номер патента: 14162
Опубликовано: 29.10.2010
Авторы: Таиши Терухико, Кавасуджи Такаши, Таода Иошиюки, Джонс Брайан Элвин
МПК: A01N 43/58, A61K 31/495, A01N 43/60...
Метки: интегразы, вич, активностью, карбамоилпиридоновое, полициклическое, обладающее, производное, ингибиторной, отношении
Формула / Реферат:
1. Соединение формулыгде Z1представляет собой NR4;R4 представляет собой С1-10алкил, замещенный группой С1-6алкокси;Z2 представляет собой С1-6алкилен или С2-6алкенилен, каждый из которых возможно замещен С1-10алкилом, возможно замещенным группой С1-6алкокси;R1 представляет собой водород;X представляет собой С1-6алкилен;R2 представляет собой С6-14арил, возможно замещенный атомами галогена в количестве от 1 до 3;R3 представляет собой водородили R4и...
Производное триазола в качестве ингибитора hsp90

Номер патента: 15366
Опубликовано: 30.06.2011
Авторы: Зирренберг Кристиан, Бухшталлер Ханс-Петер, Эггенвайлер Ханс-Михаэль, Вольф Михаэль
МПК: A61P 35/00, C07D 249/12, A61K 31/4196...
Метки: триазола, hsp90, ингибитора, качестве, производное
Формула / Реферат:
1. Соединение 5-[4-(2-метилфенил)-3-гидрокси-4H-1,2,4-триазол-5-ил]-2,4-дигидрокси-N-метил-N-бутилбензамид, и его фармацевтически приемлемые производные, соли, сольваты, таутомеры и стереоизомеры, включая их смеси во всех соотношениях.2. Соединение по п.1, где фармацевтически приемлемые производные выбирают из группы производных моно- и дифосфорной кислоты, тиоксопроизводных, производных моно- и диглюкуроновой кислоты.3. Лекарственное средство,...
Производное гидрокси-6-гетероарилфенантридина и его применение в качестве ингибитора pde4

Номер патента: 17282
Опубликовано: 30.11.2012
Авторы: Клей Ханс-Петер, Хатцельманн Армин, Шмидт Беате, Каутц Ульрих, Барзиг Иоганнес, Флоккерци Дитер, Цитт Кристоф, Маркс Дегенхард
МПК: A61K 31/473, C07D 401/04, C07D 401/14...
Метки: гидрокси-6-гетероарилфенантридина, ингибитора, применение, качестве, производное
Формула / Реферат:
1. Применение 5-((2R,4aR,10bR)-9-этокси-2-гидрокси-8-метокси-1,2,3,4,4а,10b-гексагидрофенантридин-6-ил)-1-метил-1Н-пиридин-2-она или его фармацевтически приемлемой соли для получения фармацевтических композиций, предназначенных для лечения сахарного диабета.2. Способ лечения сахарного диабета у пациента, включающий введение указанному пациенту терапевтически эффективного количества...
Вещество, обладающее антидепрессивной активностью

Номер патента: 9044
Опубликовано: 26.10.2007
Авторы: Александровский Юрий Анатольевич, Ахапкина Валентина Ивановна, Нестерук Владимир Викторович, Ахапкин Роман Витальевич, Аведисова Алла Сергеевна, Воронина Татьяна Александровна
МПК: A61P 25/24, A61P 25/28, A61K 31/40...
Метки: вещество, активностью, обладающее, антидепрессивной
Формула / Реферат:
Применение N-карбамоил-метил-4-фенил-2-пирролидона в качестве вещества, обладающего антидепрессивной активностью.
Средство, обладающее геропротекторной активностью, и способ его получения

Номер патента: 10737
Опубликовано: 30.10.2008
Авторы: Хавинсон Владимир Хацкелевич, Рыжак Галина Анатольевна, Малинин Владимир Викторович
МПК: A61K 35/30, A61P 35/00, A61P 15/12...
Метки: геропротекторной, получения, обладающее, активностью, средство, способ
Формула / Реферат:
1. Средство, обладающее геропротекторной активностью, представляющее собой лекарственную форму для парентерального введения в виде раствора пептидного комплекса с содержанием низкомолекулярной фракции от 70 до 90% с молекулярной массой входящих в него пептидных компонентов в пределах от 70 до 212 Да, с концентрацией полипептидов 2,5-2,9 мг/мл, и полученное из эпифиза (шишковидной железы) телят не старше 12-месячного возраста или свиней путем...
Предыдущий патент: Способы и композиции для замедления набора веса, связанного с применением атипичных антипсихотических препаратов
Следующий патент: Способ диагностики деталей двигателя внутреннего сгорания
Случайный патент: Способ получения 1,2-пропандиола