Производные пиколинамида в качестве ингибиторов киназы
Номер патента: 20136
Опубликовано: 29.08.2014
Авторы: Лань Цзион, Хан Усок, Бергер Маттью Т., Нишигучи Джизель





























Формула / Реферат
1. Соединение формулы II или его стереоизомер, таутомер или фармацевтически приемлемая соль

в которой Y обозначает циклогексил, содержащий 1-3 заместителя, выбранных из гидроксигруппы, аминогруппы, C1-C4-алкила и C1-C4-галогеналкила;
R1 обозначает водород, NH2 или галоген;
R12 в каждом случае независимо выбран из группы, включающей водород и галоген; и
R5 выбран из группы, включающей циклогексил, фенил и пиридил, где указанный циклогексил, указанный фенил и указанный пиридил, каждый независимо, содержат до 3 заместителей, выбранных из группы, включающей галоген, гидроксигруппу, C1-C4-алкил и OC1-C4-алкил.
2. Соединение по п.1, в котором Y содержит 1-3 заместителя, выбранных из группы, включающей метил, гидроксигруппу, аминогруппу и CF3.
3. Соединение по п.1, в котором R1 обозначает водород, аминогруппу или фтор.
4. Соединение по п.3, в котором R1 обозначает аминогруппу.
5. Соединение по п.1, в котором R5 обозначает пиридил или фенил.
6. Соединение по п.5, в котором R5 обозначает фенил, содержащий до 3 заместителей, выбранных из группы, включающей галоген, гидроксигруппу, OC1-C4-алкил и C1-C4-алкил.
7. Соединение по п.5, в котором
Y содержит 1-3 заместителя, выбранных из группы, включающей метил, гидроксигруппу, аминогруппу и CF3;
R1 обозначает водород и
R5 обозначает фенил, содержащий до 3 заместителей, выбранных из группы, включающей фтор, гидроксигруппу, метил, этил, метоксигруппу и пропоксигруппу.
8. Соединение по п.7, в котором R5 обозначает 2,6-дифторфенил.
9. Соединение по п.1, выбранное из группы, включающей
N-(4-((3S,5S)-3-амино-5-метилциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид;
3-амино-N-(4-((1R,3R,4S,5S)-3-амино-4-гидрокси-5-метилциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)пиколинамид;
N-(4-((3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид;
3-амино-N-(4-((1R,3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид и
N-(4-((3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид,
или его стереоизомер, таутомер или фармацевтически приемлемая соль.
10. Соединение по п.1, выбранное из группы, включающей

11. Соединение по п.1, формулы

или его фармацевтически приемлемая соль.
12. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
13. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
14. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
15. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
16. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
17. Соединение по п.1 формулы

или его фармацевтически приемлемая соль.
18. Соединение по п.1, выбранное из группы, включающей





или его фармацевтически приемлемая соль.
19. Фармацевтическая композиция, содержащая соединение по любому из пп.1-18 и фармацевтически приемлемый носитель или инертный наполнитель.
20. Фармацевтическая композиция, содержащая соединение по любому из пп.1-18 и фармацевтически приемлемый носитель или инертный наполнитель, где указанная фармацевтическая композиция содержит дополнительное средство для лечения рака.
21. Фармацевтическая композиция по п.20, в которой дополнительное средство выбрано из группы, включающей иринотекан, топотекан, гемцитабин, 5-фторурацил, лейковорин карбоплатин, цисплатин, таксаны, тезацитабин, циклофосфамид, алкалоиды барвинка, иматиниб (Глеевек), антрациклины, ритуксимаб и трастузумаб.
22. Применение соединения по любому из пп.1-18 для приготовления лекарственного средства, предназначенного для лечения патологического состояния путем модуляции активности провирусной интеграции киназы Мэлони (киназы PIM).
23. Применение по п.22, где патологическим состоянием является рак, выбранный из группы, включающей карциному легких, поджелудочной железы, щитовидной железы, яичников, мочевого пузыря, молочной железы, предстательной железы или толстой кишки, меланому, миелолейкоз, множественную миелому и эритролейкоз, ворсинчатую аденому толстой кишки и остеосаркому.
24. Применение соединения по пп.1-18 для лечения патологического состояния путем модуляции активности провирусной интеграции киназы Мэлони (киназы PIM).
25. Применение по п.24, где патологическим состоянием является рак, выбранный из группы, включающей карциному легких, поджелудочной железы, щитовидной железы, яичников, мочевого пузыря, молочной железы, предстательной железы или толстой кишки, меланому, миелолейкоз, множественную миелому и эритролейкоз, ворсинчатую аденому толстой кишки и остеосаркому.
26. Соединение формулы II или его стереоизомер, таутомер или фармацевтически приемлемая соль

в которой Y обозначает пиперидинил, одновременно замещенный метилом, гидроксигруппой и аминогруппой;
R1 выбран из группы, включающей водород, NH2 и фтор;
R12 в каждом случае независимо выбран из группы, включающей водород и галоген; и
R5 выбран из группы, включающей пиридил, фторпиридил, циклогексил и фенил, где указанный фенил содержит до 3 заместителей, выбранных из группы, включающей фтор, гидроксигруппу и метил.
27. Соединение по п.26, в котором Y обозначает 3-амино-4-гидрокси-5-метилпиперидин-1-ил.
28. Соединение по п.26 или 27, в котором R1 обозначает водород.
29. Соединение по п.28, в котором R5 обозначает дифторфенил.
30. Соединение по п.26 или 27, в котором R5 обозначает 2,6-дифторфенил.
31. Соединение по п.26, выбранное из группы, включающей
N-(4-((3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид,
3-амино-N-(4-((3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид и
N-(4-((3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифтор-3-метилфенил)-5-фторпиколинамид.
32. Фармацевтическая композиция, содержащая соединение по п.26 или 31 и фармацевтически приемлемый носитель или инертный наполнитель, где указанная фармацевтическая композиция содержит по меньшей мере одно дополнительное средство для лечения рака.
33. Фармацевтическая композиция по п.32, в которой дополнительное средство выбрано из группы, включающей иринотекан, топотекан, гемцитабин, 5-фторурацил, лейковорин карбоплатин, цисплатин, таксаны, тезацитабин, циклофосфамид, алкалоиды барвинка, иматиниб (Глеевек), антрациклины, ритуксимаб и трастузумаб.
34. Применение соединения по п.26 для приготовления лекарственного средства, предназначенного для лечения патологического состояния путем модуляции активности провирусной интеграции киназы Мэлони (киназы PIM).
35. Применение по п.34, где патологическим состоянием является рак, выбранный из группы, включающей карциному легких, поджелудочной железы, щитовидной железы, яичников, мочевого пузыря, молочной железы, предстательной железы или толстой кишки, меланому, миелолейкоз, множественную миелому и эритролейкоз, ворсинчатую аденому толстой кишки и остеосаркому.
36. Применение по пп.26-31 для лечения патологического состояния путем модуляции активности провирусной интеграции киназы Мэлони (киназы PIM).
37. Применение по п.36, где патологическим состоянием является рак, выбранный из группы, включающей карциному легких, поджелудочной железы, щитовидной железы, яичников, мочевого пузыря, молочной железы, предстательной железы или толстой кишки, меланому, миелолейкоз, множественную миелому и эритролейкоз, ворсинчатую аденому толстой кишки и остеосаркому.
38. Соединение формулы II, выбранное из группы, включающей















или его фармацевтически приемлемая соль.
39. Фармацевтическая композиция, содержащая соединение по п.38 и фармацевтически приемлемый носитель.
40. Способ лечения рака, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения по п.38.
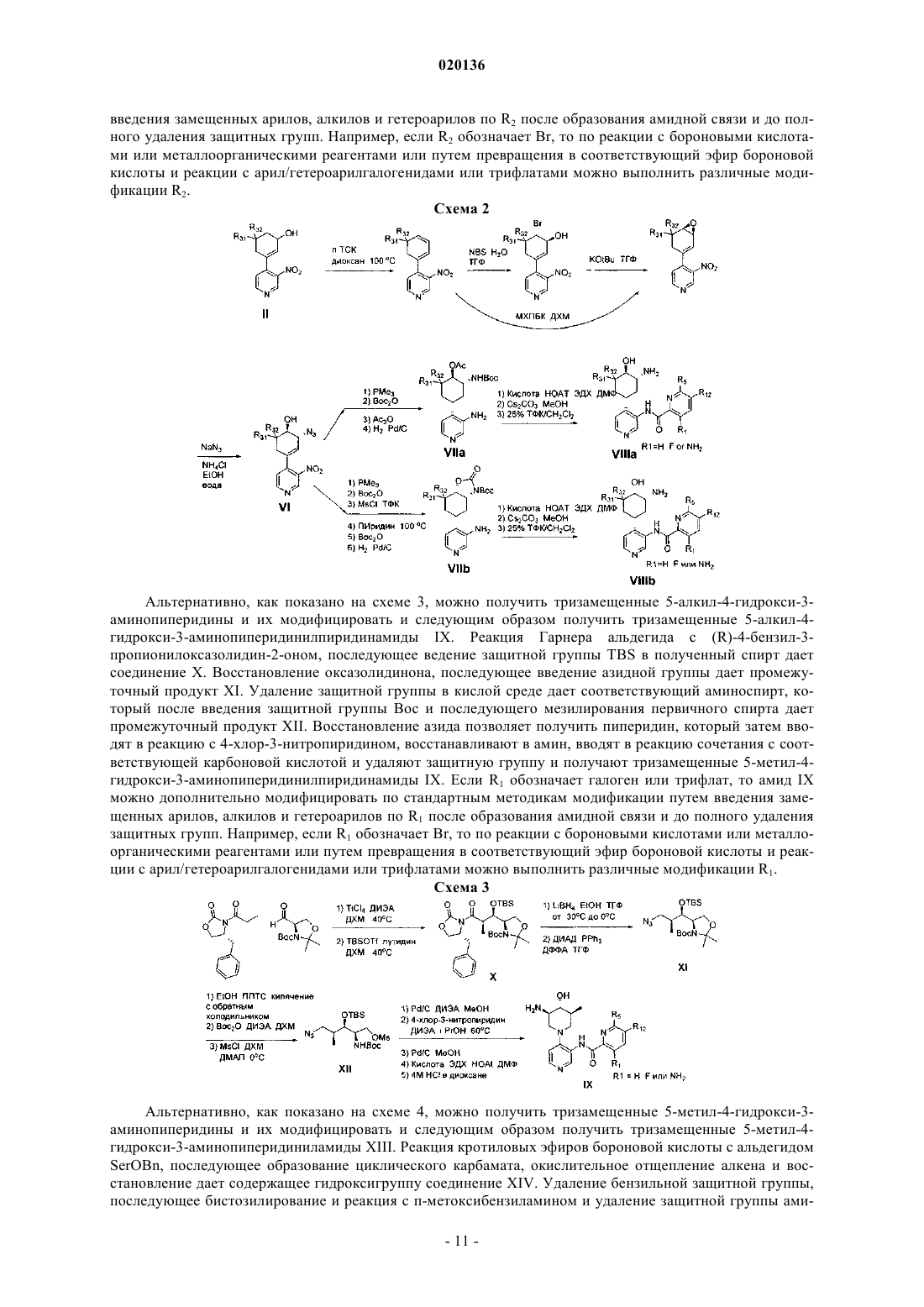
Текст
Изобретение относится к новым соединениям, композициям и способам ингибирования активности провирусной интеграции киназы Мэлони (киназы PIM), связанным с онкогенезом у человека или животного. В некоторых вариантах осуществления соединения и композиции эффективны для ингибирования активности по меньшей мере одной киназы PIM. Новые соединения и композиции можно использовать по отдельности или в комбинации по меньшей мере с одним дополнительным средством, предназначенным для лечения нарушения, опосредуемого серин/треонинкиназой или рецепторной тирозинкиназой, такого как рак. Перекрестная ссылка на родственные заявки По заявке на данное изобретение в соответствии с 35 U.S.С. 119(е) испрашивается приоритет по предварительной заявке U.S.61/093666, поданной 2 сентября 2008 г., и по предварительной заявкеU.S.61/225660, поданной 15 июля 2009 г., которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Область техники, к которой относится изобретение Настоящее изобретение относится к новым соединениям, их таутомерам и стереоизомерам и их фармацевтически приемлемым солям, сложным эфирам, метаболитам или пролекарствам, композициям новых соединений вместе с фармацевтически приемлемыми носителями и к применению новых соединений по отдельности или в комбинации по меньшей мере с одним дополнительным терапевтическим средством для профилактики или лечения рака. Уровень техники Инфицирование ретровирусом Мэлони и интеграция генома в геном клетки-хозяина приводят к развитию лимфом у мышей. Провирусная интеграция киназы Мэлони (киназы PIM) идентифицирована как один из частых протоонкогенов, способный транскрипционно активироваться этим явлением ретровирусной интеграции (Cuypers H.T. et al., "Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in distinct chromosomal region", Cell. 37(1):141-50 (1984); Selten G., et al., "Proviralactivation of the putative oncogene Pim-1 in MuLV induced T-cell lymphomas" EMBO. J. 4(7):1793-8 (1985,и тем самым установлена корреляция между сверхэкспрессированием этой киназы и ее онкогенным потенциалом. Анализ гомологии последовательностей показал, что существуют 3 сильно гомологичные киназы Pim (Pim 1, 2 и 3), Pim 1 является протоонкогеном, впервые идентифицированным с помощью ретровирусной интеграции. Кроме того, у трансгенных мышей, сверхэкспрессирующих Pim 1 или Pim 2,обнаруживается повышенная частота Т-клеточных лимфом (Breuer M. et al., "Very high frequency of lymphoma induction by chemical carcinogen in pim-1 transgenic mice". Nature. 340(6228):61-3 (1989, а сверхэкспрессирование в сочетании с c-myc связывают с частотой В-клеточных лимфом (Verbeek S. et al.,"Mice bearing the E mu-myc и Е mu-pim-1 transgenes develop pre-B-cell leukemia prenatally". Mol. Cell. Biol. 11(2):1176-9 (1991. Таким образом, эти модели на животных приводят к сильной корреляции между сверхэкспрессированием Pim и онкогенезом при гематопоэтических злокачественных новообразованиях. В дополнение к данным для этих моделей на животных сверхэкспрессирование Pim обнаружено для многих других новообразований человека. Сверхэкспрессирование Pim 1, 2 и 3 часто наблюдается для многих гематопоэтических злокачественных новообразований (Amson R. et al., "The human protooncogene product p33pim is expressed duringchronic lymphocytic leukaemia prognostic subgroups defined by ZAP-70 и CD38 expression status", Leukemia. 20:1774-1782 (2006 и при раке предстательной железы (Dhanasekaran S.M., et al., "Delineation of prognostic biomarkers in prostate cancer", Nature. 412(6849):822-6 (2001); Cibull T.L., et al., "Overexpression of Pim-1during progression of prostatic adenocarcinoma", J. Clin. Pathol. 59(3):285-8 (2006, а сверхэкспрессирование Pim 3 часто наблюдается при гепатоцеллюлярной карциноме (Fujii С., et al., "Aberrant expression ofhepatoma cell lines", Int. J. Cancer. 114:209-218 (2005 и раке поджелудочной железы (Li Y.Y. et al., "Pim3, proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer andPim 1, 2 и 3 являются серин/треонинкиназами, нормально функционирующими при жизнедеятельности и пролиферации гематопоэтических клеток в ответ на факторы роста и цитокины. Сигналы цитокинов по пути Jak/Stat приводят к активации транскрипции генов Pim и синтезу белков. Для обеспечения активности киназы Pim не требуются дополнительные посттрансляционные модификации. Таким образом, передача сигналов в прямом направлении в первую очередь контролируется на уровне транскрипции/трансляции и метаболизма белков. Субстраты киназ Pim включают регуляторы апоптоза, такие какnecessary for mitosis", Chromosoma. 111(2):80-95 (2002 и регулятор синтеза белка 4 ЕВР 1 (HammermanP.S. et al., "Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival",Blood. 105(11):4477-83 (2005. Влияние киназ Pim на эти регуляторы согласуется с ролью в защите от апоптоза и стимулировании пролиферации и роста клеток. Поэтому предполагается, что сверхэкспресси-1 020136 рование киназ Pim при раке играет роль при стимулировании выживаемости и пролиферации раковых клеток, и поэтому их ингибирование должно представлять собой эффективный путь лечения раковых заболеваний, при которых они сверхэкспрессируются. В действительности, в нескольких публикациях отмечают, что нарушение экспрессирования киназ Pim с помощью si-РНК приводит к ингибированию пролиферации и гибели клеток (Dai J.M., et al., "Antisense oligodeoxynucleotides targeting the serine/threonine kinase Pim-2 inhibited proliferation of DU-145 cells", Acta Pharmacol. Sin. 26(3):364-8 (2005);Fujii et al. 2005; Li et al. 2006). Кроме того, полагают, что мутационная активация некоторых хорошо известных онкогенов при гематопоэтических злокачественных новообразованиях, по меньшей мере частично, оказывает свои воздействия посредством киназ Pim. Например, направленная понижающая регуляция экспрессирования Pim нарушает жизнедеятельность гематопоэтических клеток, трансформированных с помощью Flt3 и BCR/ABL (Adam et al. 2006). Таким образом, ингибиторы Pim 1, 2 и 3 должны быть применимы для лечения этих злокачественных новообразований. В дополнение к потенциальной применимости для лечения рака и миелопролиферативных заболеваний, такой ингибитор должен быть полезен для регулирования распространения иммунных клеток при других патологических состояниях,таких как аутоиммунные заболевания, аллергические реакции и синдромы при отторжении трансплантатов органов. В пользу такого заключения свидетельствуют данные о том, что дифференциация Th1 хелперных Т-клеток с помощью IL-12 и IFN- приводит к индукции экспрессирования обеих Pim 1 и 2 (Ahotype 1, but not T helper type 2, cell differentiation", Immunology, 116:82-88 (2005. Кроме того, экспрессирование киназ Pim подавляется в обоих типах клеток иммуносупрессорными TGF-(Aho et al., 2005). Эти результаты показывают, что киназы Pim участвуют в раннем процессе дифференциации хелперных Т-клеток, что координирует иммунные ответы при аутоиммунных заболеваниях, аллергических реакциях и отторжении трансплантатов тканей. Сохраняется необходимость в соединениях, которые подавляют пролиферацию капилляров, подавляют рост опухолей, лечат рак, модулируют остановку клеточного цикла и/или ингибируют такие молекулы, как Pim 1, Pim 2, Pim 3, и в фармацевтических препаратах и лекарственных средствах, которые содержат такие соединения. Также необходимы способы введения таких соединений, фармацевтических препаратов и лекарственных средств нуждающимся в них пациентам или субъектам. Краткое изложение сущности изобретения Настоящее изобретение относится к новым соединениям формулы II и к их стереоизомерам, таутомерам и фармацевтически приемлемым солямR12 в каждом случае независимо выбран из группы, включающей водород и галоген; иR5 выбран из группы, включающей циклогексил, фенил и пиридил, где указанный циклогексил,указанный фенил и указанный пиридил, каждый независимо, содержат до 3 заместителей, выбранных из группы, включающей галоген, гидроксигруппу, C1-C4-алкил и OC1-C4-алкил. В типичном варианте осуществления соединения формулы II или их стереоизомер, таутомер или фармацевтически приемлемая соль выбраны из группы, включающейN-(4-1R,3R,4R,5S)-3-амино-4-гидрокси-5-метилциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)5-фторпиколинамид,N-(4-1R,3S,5S)-3-амино-5-метилциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5 фторпиколинамид,N-(4-3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифторфенил)5-фторпиколинамид,3-амино-N-(4-3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6 дифторфенил)-5-фторпиколинамид,N-(4-1R,3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид и 3-амино-N-(4-1R,3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5 фторпиколинамид. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных с провирусной интеграцией киназы Мэлони (киназы PIM), у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы II в количестве, эф-2 020136 фективном для ингибирования активности PIM у субъекта. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных сPIM, у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы II в количестве, эффективном для уменьшения или предупреждения роста опухоли у субъекта. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных сPIM, у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы II в количестве, эффективном для уменьшения или предупреждения роста опухоли у субъекта, в комбинации по меньшей мере с одним дополнительным средством, предназначенным для лечения рака. В других объектах настоящее изобретение относится к терапевтической композиции, содержащей по меньшей мере одно соединение формулы II в комбинации с одним или большим количеством дополнительных средств, предназначенных для лечения рака, которые обычно используются для лечения рака. Соединения, предлагаемые в настоящем изобретении, применимы для лечения рака, включая гематопоэтические злокачественные новообразования, карциномы (например, легких, печени, поджелудочной железы, яичников, щитовидной железы, мочевого пузыря или толстой кишки), меланомы, миелоидные нарушения (например, миелолейкоза, множественной миеломы и эритролейкоза), аденомы (например, ворсинчатой аденомы толстой кишки), саркомы (например, остеосаркомы), аутоиммунных заболеваний, аллергических реакций и синдромов отторжения трансплантатов органов. Настоящее изобретение также относится к композициям, способам применения и способам получения, описанным в подробном описании изобретения. Описание чертежей Указанные выше объекты и многие другие преимущества настоящего изобретения будет легче оценить, когда они будут лучше поняты из приведенного ниже подробного описания вместе с прилагаемыми чертежами, на которых представлено следующее. На фиг. 1 - зависимость для эффективности соединения примера 99 по данным модели ксеротрансплантата KMS11-luc, описанной в примере 144. На фиг. 2 - зависимость для эффективности соединения примера 70 по данным модели ксеротрансплантата KMS11-luc, описанной в примере 144. На фиг. 3 - зависимость для эффективности соединения примера 96 по данным модели ксеротрансплантата KMS11-luc, описанной в примере 144. Подробное описание изобретения Варианты осуществления относятся к новым соединениям и их стереоизомерам, таутомерам и фармацевтически приемлемым солям формулы IIR12 в каждом случае независимо выбран из группы, включающей водород и галоген; иR5 выбран из группы, включающей циклогексил, фенил и пиридил, где указанный циклогексил,указанный фенил и указанный пиридил каждый независимо содержит до 3 заместителей, выбранных из группы, включающей галоген, гидроксигруппу, C1-C4-алкил и OC1-C4-алкил. В типичном варианте осуществления предпочтительные соединения формулы II или их стереоизомер, таутомер или фармацевтически приемлемая соль выбраны из группы, включающейN-(4-1R,3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5-фторпиколинамид; 3-амино-N-(4-3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6 дифторфенил)-5-фторпиколинамид и 3-амино-N-(4-1R,3S)-3-аминоциклогексил)пиридин-3-ил)-6-(2,6-дифторфенил)-5 фторпиколинамид. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных с провирусной интеграцией киназы Мэлони (киназы PIM), у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы II в количестве, эффективном для ингибирования активности PIM у субъекта. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных сPIM, у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы II в количестве, эффективном для уменьшения или предупреждения роста опухоли у субъекта. В других объектах настоящее изобретение относится к способам лечения нарушений, связанных с PIM, у человека или животного, нуждающегося в таком лечении, включающим введение указанному субъекту соединения формулы I или II в количестве, эффективном для уменьшения или предупреждения роста опухоли у субъекта, в комбинации по меньшей мере с одним дополнительным средством, предназначенным для лечения рака. В другом объекте настоящее изобретение относится к терапевтической композиции, содержащей по меньшей мере одно соединение формулы II в комбинации с одним или большим количеством дополнительных средств, предназначенных для лечения рака, которые обычно используются для лечения рака. Таким образом, настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы II. Предпочтительным вариантом осуществления этого объекта является фармацевтическая композиция, содержащая соединение, выбранное из группы, включающейN-(4-3R,4R,5S)-3-амино-4-гидрокси-5-метилпиперидин-1-ил)пиридин-3-ил)-6-(2,6-дифтор-3 метилфенил)-5-фторпиколинамид. Другой предпочтительный вариант осуществления относится к фармацевтической композиции, дополнительно содержащей дополнительное средство, предназначенное для лечения рака, причем предпочтительное дополнительное средство выбрано из группы, включающей иринотекан, топотекан, гемци-4 020136 табин, 5-фторурацил, лейковорин карбоплатин, цисплатин, таксаны, тезацитабин, циклофосфамид, алкалоиды барвинка, иматиниб (Глеевек), антрациклины, ритуксимаб и трастузумаб. Соединения, предлагаемые в настоящем изобретении, применимы для лечения рака, включая гематопоэтические злокачественные новообразования, карциномы (например, легких, печени, поджелудочной железы, яичников, щитовидной железы, мочевого пузыря или толстой кишки), меланомы, миелоидные нарушения (например, миелолейкоза, множественной миеломы и эритролейкоза), аденомы (например, ворсинчатой аденомы толстой кишки), саркомы (например, остеосаркомы), аутоиммунных заболеваний, аллергических реакций и синдромов отторжения трансплантатов органов. В другом объекте настоящее изобретение относится к применению соединения формулы II для приготовления лекарственного средства, предназначенного для лечения патологического состояния путем модуляции активности провирусной интеграции киназы Мэлони (киназы PIM). В предпочтительном варианте осуществления этого объекта настоящего изобретения патологическим состоянием является рак,выбранный из группы, включающей карциному легких, поджелудочной железы, щитовидной железы,яичников, мочевого пузыря, молочной железы, предстательной железы или толстой кишки, меланому,миелолейкоз, множественную миелому и эритролейкоз, ворсинчатую аденому толстой кишки и остеосаркому. В другом объекте настоящее изобретение относится к способам ингибирования активности по меньшей мере одной киназы, выбранной из группы, включающей Pim 1, Pim 2 и Pim 3, у субъекта или лечения биологического патологического состояния, опосредуемого по меньшей мере одной из Pim 1,Pim 2 и Pim 3, у человека или животного, нуждающегося в таком лечении, включающим введение субъекту по меньшей мере одного соединения формулы I или II в количестве, эффективном для ингибирования киназы у субъекта. Терапевтические соединения применимы для лечения пациентов, нуждающихся в таких ингибиторах (например, страдающих от рака, опосредуемого аномальной передачей сигналов рецептора серин/треонинкиназы). Определения."Ингибитор PIM" используется в настоящем изобретении для обозначения соединения, которое обладает значением IC50 для активности киназы PIM, равным не более примерно 100 мкМ и чаще не более примерно 50 мкМ, полученным при исследованиях расходования при воздействии PIM, описанных ниже в настоящем изобретении. Термин "алкил" означает алкильные группы, которые не содержат гетероатомы. Этот термин включает алкильные группы с линейной цепью, такие как метил, этил, пропил, бутил, пентил, гексил, гептил,октил, нонил, децил, ундецил, додецил и т.п. Термин также включает обладающие разветвленной цепью изомеры алкильные группы с линейной цепью, включая, но не ограничиваясь только ими, следующие,которые приведены в качестве примера: -CH(CH3)2, -CH(CH3)(CH2CH3), -CH(CH2CH3)2, -С(CH3)3,-С(CH2CH3)3, -CH2CH(CH3)2, -CH2CH(CH3)(CH2CH3), -CH2CH(CH2CH3)2, -CH2C(CH3)3, -CH2C(CH2CH3)3,-CH(CH3)CH(CH3)(CH2CH3), -CH2CH2CH(CH3)2, -CH2CH2CH(CH3)(CH2CH3), -CH2CH2CH(CH2CH3)2,-CH2CH2C(CH3)3,-CH2CH2C(CH2CH3)3,-CH(CH3)CH2CH(CH3)2,-CH(CH3)CH(CH3)CH(CH3)2,-CH(CH2CH3)CH(CH3)CH(CH3)(CH2CH3) и др. Таким образом, выражение "алкильные группы" включает первичные алкильные группы, вторичные алкильные группы и третичные алкильные группы. Предпочтительные алкильные группы включают обладающие линейной и разветвленной цепью алкильные группы, содержащие от 1 до 12 атомов углерода. Предпочтительное определение "алкила" относится к обладающим линейной цепью C1-C4-алкильным группам, таким как метил, этил, н-пропил и н-бутил. Предпочтительное определение "алкила" также включает разветвленные C3-C5-алкильные группы, включаяCH(CH3)2, CH2CH(CH3)2, CH(CH3)CH2CH3, С(CH3)3, CH(CH3)CH2CH2CH3, CH(CH3)CH(CH3)2,CH2CH(CH3)CH2CH3, CH2CH2CH(CH3)2, CH(CH2CH3)2 и т.п. Термин "алкенил" означает алкильные группы, определенные выше, в которых содержится по меньшей мере один ненасыщенный фрагмент, т.е. в котором два соседних атома углерода связаны двойной связью. Термин "алкинил" означает алкильные группы, в которых два соседних атома углерода связаны тройной связью. Термин "алкоксигруппа" означает OR, где R обозначает алкил. При использовании в настоящем изобретении термин "галоген" означает хлоридную, бромидную,фторидную и йодидную группы. "Галогеналкил" означает алкильный радикал, замещенный одним или большим количеством атомов галогенов. Таким образом, термин "галогеналкил" включает моногалогеналкил, дигалогеналкил, тригалогеналкил и т.п. Типичные моногалогеналкильные группы включают -CH2F, -CH2Cl, -CH2CH2F, -CH2CH2Cl, -CH(F)CH3, -CH(Cl)CH3; типичные дигалогеналкильные группы включают CHCl2, -CHF2, -CCl2CH3, -CH(Cl)CH2Cl, -CH2CHCl2, -CH2CHF2; типичные тригалогеналкильные группы включают -CCl3, -CF3, -CCl2CH2Cl, -CF2CH2F, -CH(Cl)CHCl2, -CH(F)CHF2 и типичные пергалогеналкильные группы включают -CCl3, -CF3, -CCl2CCl3, -CF2CF3."Аминогруппа" в настоящем изобретении означает группу -NH2, которая может быть замещенной с образованием -NRR'. Термин "алкиламиногруппа" в настоящем изобретении означает группу -NRR', в которой R и R' все независимо выбраны из группы, включающей водород и низш. алкил. Термин"ариламиногруппа" в настоящем изобретении означает группу -NRR', в которой R обозначает арил и R' обозначает водород, низш. алкил или арил. Термин "арилалкиламиногруппа" в настоящем изобретении означает группу -NRR', в которой R обозначает низш. арилалкил и R' обозначает водород, низш. алкил,арил или низш. арилалкил. Термин цианогруппа означает группу -CN. Термин "нитрогруппа" означает группу -NO2. Термин "алкоксиалкил" означает группу -alk1-O-alk2, в которой alk1 обозначает алкил или алкенил иalk2 обозначает алкил или алкенил. Термин "низш. алкоксиалкил" означает алкоксиалкил, где alk1 обозначает низш. алкил или низш. алкенил и alk2 обозначает низш. алкил или низш. алкенил. Термин"арилоксиалкил" означает группу -алкил-О-арил. Термин "арилалкоксиалкил" означает группу алкиленил-О-арилалкил, в которой арилалкил означает низш. арилалкил. Термин "аминокарбонил" в настоящем изобретении означает группу -С(О)-NH2. "3 амещенный аминокарбонил" в настоящем изобретении означает группу -C(O)-NRR', в которой R обозначает низш. алкил и R' обозначает водород или низш. алкил. В некоторых вариантах осуществления R и R' вместе с присоединенным к ним атомом N могут образовать "гетероциклоалкилкарбонильную" группу. Термин "ариламинокарбонил" в настоящем изобретении означает группу C(O)-NRR', в которой R обозначает арил и R' обозначает водород, низш. алкил или арил. "арилалкиламинокарбонил" в настоящем изобретении означает группу -C(O)-NRR', в которой R обозначает низш. арилалкил и R' обозначает водород, низш. алкил,арил или низш. арилалкил."Аминосульфонил" в настоящем изобретении означает группу -S(O)2-NH2. "3 амещенный аминосульфонил" в настоящем изобретении означает группу -S(O)2-NRR', в которой R обозначает низш. алкил и R' обозначает водород или низш. алкил. Термин "арилалкиламиносульфониларил" в настоящем изобретении означает группу -арил-S(О)2-NH-арилалкил, в которой арилалкил представляет собой низш. арилалкил."Карбонил" означает двухвалентную группу -С(О). "Карбоксигруппа" означает -С(=О)-ОН. "Алкоксикарбонил" означает сложный эфир -C(=O)-OR, в котором R обозначает алкил. "Низш. алкоксикарбонил" означает сложный эфир -C(=O)-OR, в котором R обозначает низш. алкил. "Циклоалкоксикарбонил" означает -C(=O)-OR, в котором R обозначает циклоалкил."Циклоалкил" означает моно- или полициклический, гетероциклический или карбоциклический алкильный заместитель. Карбоциклоалкильные группы являются циклоалкильными группами, в которых все кольцевые атомы являются атомами углерода. Типичные циклоалкильные заместители содержат от 3 до 8 входящих в главную цепь (т.е. в кольцо) атомов, в которых каждый атом главной цепи является атомом углерода или гетероатомом. Термин "гетероциклоалкил" в настоящем изобретении означает циклоалкильные заместители, которые содержат в кольцевой структуре от 1 до 5 и чаще от 1 до 4 гетероатомов. Подходящими гетероатомами, использующимися в соединениях, предлагаемых в настоящем изобретении, являются азот, кислород и сера. Типичные гетероциклоалкильные фрагменты включают, например, морфолиновую группу, пиперазинил, пиперидинил и т.п. Карбоциклоалкильные группы представляют собой циклоалкильные группы, в которых все кольцевые атомы являются атомами углерода. При использовании в связи с циклоалкильными заместителями термин "полициклический" в настоящем изобретении означает конденсированные и неконденсированные циклоалкильные структуры. Термины"частично ненасыщенный циклоалкил", "частично насыщенный циклоалкил" и "циклоалкенил", все, означают циклоалкильную группу, в которой содержится по меньшей мере один ненасыщенный фрагмент,т.е. в которой два соседних атома углерода связаны двойной связью или тройной связью. Иллюстративные примеры включают циклогексенил, циклогексинил, циклопропенил, циклобутинил и т.п. Термины "замещенный гетероцикл", или "гетероциклическая группа", или "гетероцикл" при использовании в настоящем изобретении означают любое 3- или 4-членное кольцо, содержащее гетероатом, выбранный из группы, включающей азот, кислород и серу, или 5- или 6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из группы, включающей азот, кислород и серу; где 5-членное кольцо содержит 0-2 двойных связей и 6-членное кольцо содержит 0-3 двойных связей; где атомы азота и серы необязательно могут быть окислены; где атомы азота и серы необязательно могут кватернизованы; и включает любую бициклическую группу, в которой любое из указанных выше гетероциклических колец сконденсировано с бензольным кольцом или другим 5- или 6-членным гетероциклическим кольцом,независимо определенным выше. Термин "гетероциклоалкил" при использовании в настоящем изобретении означает 5- или 6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из группы, включающей азот, кислород и серу, причем кольцо не содержит двойных связей. Например, термин гетероцикло-С 5-алкил означает 6-членное кольцо, содержащее 5 атомов углерода и гетероатом, такой как N. Таким образом, термин"гетероцикл" включает кольца, в которых азот является гетероатомом, а также частично и полностью насыщенные кольца. Предпочтительные гетероциклы включают, например, диазепинил, пиррил, пирролинил, пирролидинил, пиразолил, пиразолинил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиперидинил, пиразинил, пиперазинил, N-метилпиперазинил, азетидинил,N-метилазетидинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, бензотиазолил, бензоксазолил, фурил, тиенил, триазолил и бензотиенил. Гетероциклические фрагменты могут быть незамещенными или монозамещенными или дизамещенными различными заместителями, независимо выбранными из группы, включающей гидроксигруппу, галоген, оксогруппу (С=О), алкилиминогруппу (RN=, в которой R обозначает низш. алкил или низш. алкоксигруппу), аминогруппу, алкиламиногруппу, диалкиламиногруппу, ациламиноалкил, алкоксигруппу, тиоалкоксигруппу, полиалкоксигруппу, низш. алкил, циклоалкил или галогеналкил. Гетероциклические группы могут быть присоединены в разных положениях, как это должно быть очевидно специалистам в области органической и медицинской химии в связи с раскрытием в настоящем изобретении. Типичные гетероциклилы включают, например, имидазолил, пиридил, пиперазинил, пиперидинил,азетидинил, тиазолил, фуранил, триазолил бензимидазолил, бензотиазолил, бензоксазолил, хинолинил,изохинолинил, хиназолинил, хиноксалинил, фталазинил, индолил, нафтпиридинил, индазолил и хинолизинил."Арил" означает необязательно замещенные моноциклические и полициклические ароматические группы, содержащие в главной цепи от 3 до 14 атомов углерода или гетероатомов и включают карбоциклические арильные группы и гетероциклические арильные группы. Карбоциклические арильные группы представляют собой арильные группы, в которых все кольцевые атомы ароматического кольца являются атомами углерода. Термин "гетероарил" в настоящем изобретении означает арильные группы, содержащие от 1 до 4 гетероатомов в качестве кольцевых атомов ароматического кольца, а остальные кольцевые атомы являются атомами углерода. При использовании в связи с арильными заместителями термин "полициклический арил" в настоящем изобретении означает конденсированные и неконденсированные циклические структуры, в которых по меньшей мере одна циклическая структура является ароматической,такой как, например, бензодиоксозоловая группа (которая содержит гетероциклическую структуру,сконденсированную с фенильной группой, т.е. нафтил и т.п). Типичные арильные фрагменты, использующиеся в качестве заместителей в соединениях, предлагаемых в настоящем изобретении, включают фенил, пиридил, пиримидинил, тиазолил, индолил, имидазолил, оксадиазолил, тетразолил, пиразинил,триазолил, тиофенил, фуранил, хинолинил, пуринил, нафтил, бензотиазолил, бензопиридил и бензимидазолил и т.п."Необязательно замещенный" или "замещенный" означает замещение одного или большего количества атомов водорода одновалентным или двухвалентным радикалом. Подходящие замещающие группы включают, например, гидроксигруппу, нитрогруппу, аминогруппу, иминогруппу, цианогруппу, галоген,тиогруппу, сульфонил, тиоамидную группу, амидиновую группу, имидино, оксогруппу, оксамидиновую группу, метоксамидиновую группу, имидино, гуанидиновую группу, сульфонамидную группу, карбоксигруппу, формил, низш. алкил, галоген-низш. алкил, низш. алкиламиногруппу, галоген-низш. алкиламиногруппу, низш. алкоксигруппу, галоген-низш. алкоксигруппу, низш. алкоксиалкил, алкилкарбонил,аминокарбонил, арилкарбонил, арилалкилкарбонил, гетероарилкарбонил, гетероарилалкилкарбонил, алкилтиогруппу, аминоалкил, цианоалкил, арил и т.п. 3 амещающая группа сама может быть замещенной. 3 аместителем замещающей группы может быть карбоксигруппа, галоген, нитрогруппа, аминогруппа, цианогруппа, гидроксигруппа, низш. алкил, низш. алкоксигруппа, аминокарбонил, -SR, тиоамидная группа, -SO3H, -SO2R или циклоалкил, в которых R обычно обозначает водород, гидроксигруппу или низш. алкил. Если замещенный заместитель включает группу, обладающую линейной цепью, то замещение может происходить внутри цепи (например, 2-гидроксипропил, 2-аминобутил и т.п.) или в конце цепи (например, 2-гидроксиэтил, 3-цианопропил и т.п.). 3 амещенные заместители могут представлять собой обладающую линейной цепью, разветвленную или циклическую группировку ковалентно связанных атомов углерода или гетероатомов. Следует понимать, что приведенные выше определения не включают недопустимые схемы замещения (например, метил, замещенный 5 фторидными группами, или атом галогена, замещенный другим атомом галогена). Такие недопустимые схемы замещения хорошо известны специалисту в данной области техники. Специалистам в данной области техники должно быть понятно, что соединения, предлагаемые в настоящем изобретении, или их стереоизомеры, а также фармацевтически приемлемые соли, сложные эфиры, метаболиты и пролекарства любого из них могут подвергаться таутомерным превращениям и поэтому могут существовать в разных таутомерных формах, в которых протон одного атома молекулы перемещается к другому атому и химические связи между атомами молекулы соответствующим образом перестраиваются. См., например, March, Advanced Organic Chemistry: Reactions, Mechanisms and Structures, Fourth Edition, John WileySons, pages 69-74 (1992). При использовании в настоящем изобретении термин "таутомер" означает соединения, образованные путем перемещения протона, и следует понимать,что все таутомерные формы, если они могут существовать, входят в объем настоящего изобретения. Соединения, предлагаемые в настоящем изобретении, а также фармацевтически приемлемые соли,сложные эфиры, метаболиты и пролекарства любого из них могут содержать асимметрично замещенные атомы углерода. Такие асимметрично замещенные атомы углерода могут привести к соединениям, предлагаемым в настоящем изобретении, существующим в энантиомерных, диастереоизомерных и других стереоизомерных формах, которые с помощью обозначений абсолютных стереохимических конфигура-7 020136 ций можно обозначить как (R)- или (S)-формы. Поэтому все такие возможные изомеры, отдельные стереоизомеры в их оптически чистых формах, их смеси, рацемические смеси (или "рацематы"), смеси диастереоизомеров, а также отдельные диастереоизомеры соединений, предлагаемых в настоящем изобретении, входят в объем настоящего изобретения. Термины "S" и "R" конфигурация при использовании в настоящем изобретении обладают значениями, определенными в публикации IUPAC 1974Recommendations for Section E, Fundamental Stereochemistry, Pure Appl. Chem. 45:13-30 (1976). Обозначенияииспользуются для определения положений циклов в циклических соединениях. -Положение в рассматриваемой плоскости является положением, в котором предпочтительный заместитель расположен в положении с меньшим номером. 3 аместители, расположенные с противоположной стороны рассматриваемой плоскости, отмечают символом . Следует отметить, что эти обозначения отличаются от обозначений циклических стереоизомеров, для которых означает "ниже плоскости" и обозначает абсолютную конфигурацию. Термины - и -конфигурация при использовании в настоящем изобретении обладают значениями, определенными в публикации Chemical Abstracts Index Guide-Appendix IV (1987),paragraph 203. При использовании в настоящем изобретении термин "фармацевтически приемлемые соли" означает соли нетоксичной кислоты или соли нетоксичного щелочно-земельного металла соединений формулыI или II. Эти соли можно получить in situ при окончательном выделении и очистке соединений формулы I или II или путем раздельных реакций основных или кислотных групп с подходящей органической или неорганической кислотой или основанием соответственно. Типичные соли включают, но не ограничиваются только ими, следующие: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат,этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат,2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат и ундеканоат. Кроме того, основные азотсодержащие группы можно кватернизировать такими реагентами, как низш. алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды; диалкилсульфаты, такие как диметил-,диэтил-, дибутил- и диамилсульфаты, галогениды с длинными цепями, такие как децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -йодиды, арилалкилгалогениды, такие как бензил- и фенетилбромиды и др. Таким образом, получают растворимые или диспергирующиеся в воде или масле продукты. Примеры кислот, которые можно использовать для получения фармацевтически приемлемых солей присоединения с кислотами, включают такие неорганические кислоты, как хлористо-водородная, азотная, серная и фосфорная кислоты, и такие органические кислоты, как щавелевая, малеиновая, метансульфоновая, янтарная и лимонная кислоты. Соли присоединения с основаниями можно получить in situ при окончательном выделении и очистке соединений формулы (I) или путем раздельных реакций кислотных групп карбоновых кислот с подходящим основанием, таким как гидроксид, карбонат или бикарбонат фармацевтически приемлемого катиона металла, или с аммиаком или с первичным, вторичным или третичным органическим амином. Фармацевтически приемлемые соли включают, но не ограничиваются только ими, соли, содержащие катионы щелочных и щелочно-земельных металлов, такие как соли натрия, лития, калия, кальция, магния, алюминия и т.п., а также нетоксичные соли аммония, четвертичного аммония и аминов, включая, но не ограничиваясь только ими, соли аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и т.п. Другие типичные органические амины, применяющиеся для получения солей присоединения с основаниями, включают диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п. При использовании в настоящем изобретении термин "фармацевтически приемлемый сложный эфир" означает сложные эфиры, которые гидролизуются in vivo, и включают такие, которые легко разрушаются в организме человека с высвобождением исходного соединения или его соли. Подходящие сложноэфирные группы включают, например, образованные из фармацевтически приемлемых алифатических карбоновых кислот, предпочтительно алканкарбоновых, алкенкарбоновых, циклоалканкарбоновых и алкандикарбоновых, в которых каждый алкильный или алкенильный фрагмент предпочтительно содержит не более 6 атомов углерода. Типичные примеры предпочтительных сложных эфиров включают, но не ограничиваются только ими, формиаты, ацетаты, пропионаты, бутираты, акрилаты и этилсукцинаты. Термин "фармацевтически приемлемые пролекарства" при использовании в настоящем изобретении означает такие пролекарства соединений, предлагаемых в настоящем изобретении, которые с медицинской точки зрения применимы для взаимодействия с тканями человека и низших животных без проявления нежелательной токсичности, раздражения, аллергической реакции и т.п., характеризуются разумным соотношением польза/риск и эффективны для применения по назначению, а также, когда это возможно, цвиттерионные формы соединений, предлагаемых в настоящем изобретении. Термин "пролекарство" означает соединения, которые быстро подвергаются превращению in vivo и образуют исходные соединения указанной выше формулы, например, путем гидролиза в крови. Подробное обсуждение при-8 020136Association and Pergamon Press, 1987, которые включены в настоящее изобретение в качестве ссылки. Любая формула, приведенная в настоящем изобретении, также характеризует немеченые формы, а также изотопно-меченые формы соединений. Изотопно-меченые соединения обладают структурами,описывающимися формулами, приведенными в настоящем изобретении, за исключением того, что один или большее количество атомов заменены на атомы, обладающими указанными атомными массами или массовыми числами. Примеры изотопов, которые можно вводить в соединения, предлагаемые в настоящем изобретении, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора и хлора, такие как 2 Н, 3 Н, 11 С, 13 С, 14 С, 15N, 18F, 31P, 32P, 35S, 36Cl, 125I соответственно. В объем настоящего изобретения входят различные изотопно-меченые соединения, предлагаемые в настоящем изобретении, например, в которые включены радиоактивные изотопы, такие как 3 Н, 13 С и 14 С. Такие изотопно-меченые соединения применимы для изучения метаболизма (предпочтительно содержащие 14 С), кинетики реакций (содержащие, например, 2 Н или 3 Н), в методологиях детектирования и визуализации, таких как позитронная эмиссионная томография (ПЭТ) или однофотонная эмиссионная компьютерная томография(ОФЭКТ), включая исследование распределения лекарственного средства или субстрата в тканях или лучевую терапию пациентов. В частности, содержащее 18F или меченое им соединение может быть особенно предпочтительным для исследований с помощью ПЭТ или ОФЭКТ. Изотопно-меченые соединения, предлагаемые в настоящем изобретении, и их пролекарства обычно можно получить по методикам,раскрытым на схемах или в примерах и синтезах, описанных ниже, путем замены не содержащего изотопа реагента на легко доступный изотопно-меченый реагент. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2 Н или D), может обеспечить некоторые терапевтические преимущества, обусловленные их более высокой метаболической стабильностью, например увеличенной длительностью полувыведения in vivo или возможностью использования меньших доз, или улучшением терапевтического индекса. Следует понимать, что в этом контексте дейтерий рассматривается в качестве заместителя в соединении формулы (I). Концентрацию такого более тяжелого изотопа, в частности дейтерия, можно определить с помощью коэффициента изотопного обогащения. Термин "коэффициент изотопного обогащения" при использовании в настоящем изобретении означает отношение содержания изотопа к содержанию конкретного изотопа в природе. Если заместитель в соединении, предлагаемом в настоящем изобретении, означает дейтерий, то такое соединение характеризуется коэффициентом изотопного обогащения для каждого обозначенного атома дейтерия, равным не менее 3500 (содержание дейтерия для каждого обозначенного атома дейтерия равно 52,5%), не менее 4000 (содержание дейтерия равно 60%), не менее 4500 (содержание дейтерия равно 67,5%), не менее 5000 (содержание дейтерия равно 75%), не менее 5500 (содержание дейтерия равно 82,5%), не менее 6000 (содержание дейтерия равно 90%), не менее 6333,3 (содержание дейтерия равно 95%), не менее 6466,7 (содержание дейтерия равно 97%), не менее 6600 (содержание дейтерия равно 99%) или не менее 6633,3 (содержание дейтерия равно 99,5%). Изотопно-меченые соединения формулы (I) обычно можно получить по обычным методикам, известным специалистам в данной области техники, или по методикам, аналогичным описанным в прилагающихся примерах и синтезах с использованием изотопно-меченого реагента вместо использовавшегося ранее немеченого реагента. Специалистам в данной области техники должно быть очевидно, что соединения, предлагаемые в настоящем изобретении, или их таутомеры, пролекарства и стереоизомеры, а также фармацевтически приемлемые соли, сложные эфиры и пролекарства любого из них могут подвергнуться преобразованиямin vivo путем метаболизма в организме человека или животного или в клетке с образованием метаболитов. Термин "метаболит" при использовании в настоящем изобретении означает любое производное, образовавшееся в субъекте после введения исходного соединения. Производные могут образоваться из исходного соединения посредством различных биохимических превращений, происходящих в субъекте,таких как, например, окисление, восстановление, гидролиз или конъюгация, и включают, например, оксиды и деметилированные производные. Метаболиты соединения, предлагаемого в настяощем изобретении, можно идентифицировать по стандартным методикам, известным в данной области техники. См.,например, Bertolini, G. et al., J. Med. Chem. 40:2011-2016 (1997); Shan, D. et al., J. Pharm. Sci. 86(7):765767; Bagshawe K., Drug Dev. Res. 54:220-230 (1995); Bodor, N., Advances in Drug Res. 13:224-331 (1984);Bundgaard, H., Design of Prodrugs (Elsevier Press 1985) и Larsen, I.K., Design and Application of Prodrugs,Drug Design and Development (Krogsgaard-Larsen et al., eds., Harwood Academic Publishers, 1991). Следует понимать, что индивидуальные химические соединения, которые представляют собой метаболиты соединений формулы I или II, или их таутомеры, пролекарства и стереоизомеры, а также фармацевтически приемлемые соли, сложные эфиры и пролекарства любого из них входят в объем настоящего изобретения. Термин "рак" означает раковые заболевания, которые можно эффективно лечить путем ингибирования киназы Pirn, включая, например, солидные раковые заболевания, такие как карциномы (например,легких, поджелудочной железы, щитовидной железы, яичников, мочевого пузыря, молочной железы,-9 020136 предстательной железы или толстой кишки), меланомы, миелоидные нарушения (например, миелолейкоз, множественную миелому и эритролейкоз), аденомы (например, ворсинчатую аденому толстой кишки) и саркомы (например, остеосаркому). Методики синтеза. Соединения, предлагаемые в настоящем изобретении, можно получить по методикам, известным специалистам в данной области техники. Например, как показано на схеме, циклогександионы через монотрифлаты можно превратить в соответствующие эфиры циклогексенонбороновой кислоты, которые при катализе палладием образуют связь с атомом углерода 4-хлор,3-нитропиридина с получением замещенных нитропиридином циклогексенонов I. Восстановление еноновой группы может дать циклогексенол II, который после защиты гидроксигруппы, восстановления нитрогруппы и алкена, реакции сочетания с образованием амида и удаления защитной группы может дать циклогексаноламиды III. Циклогексенол II также можно ввести в реакцию Мицунобу с фталимидом и получить защищенный аминоциклогексен IV. После восстановления нитрогруппы и алкена содержащий фталимидную защитную группу аминоциклогексилпиридиланилин Va можно ввести в реакцию сочетания с образованием амида и удалить защитную группу и получить аминоциклогексанамиды VI. Соответствующий содержащий защитную группу Boc аминоциклогексанпиридиланилин Vb также можно получить из циклогексенола II следующим образом: защитить гидроксигруппу, восстановить алкен и нитрогруппу, ввести в пиридиламин защитную группу Cbz, удалить силильную защитную группу, провести окисление по Дессу-Мартину в циклогексанон, провести восстановительное аминирование бензиламином, удалить защитные группыCbz и Bn и удалить защитную группу Boc из первичного алифатического амина. В случае амидов III иVI, если R2 обозначает галоген или трифлат, то амиды III и VI можно дополнительно модифицировать по стандартным методикам модификации путем введения замещенных арилов, алкилов и гетероарилов поR2. Например, если R2 обозначает Br, то по реакции с бороновыми кислотами или металлоорганическими реагентами или путем превращения в соответствующий эфир бороновой кислоты и реакции с арил/гетероарилгалогенидами или трифлатами можно выполнить различные модификации R2. Схема 1 Альтернативно, как показано на схеме 2, циклогексенол II можно дегидратировать и получить циклогексадиен, который после эпоксидирования (путем образования бромгидрина и отщепления HBr или непосредственно из МХПБК (м-хлорпероксибензойная кислота и раскрытия эпоксидного кольца азидом дает циклогексенилазидоспирт VI. Циклогексенилазидоспирт VI можно превратить в трансзащищенный аминогидроксианилин VIIa путем восстановления азидной группы, защиты гидроксигруппы и восстановления алкена и нитрогруппы. Альтернативно, циклогексенилазидоспирт VI можно превратить в цис-защищенный аминогидроксианилин VIIb путем восстановления азидной группы и введения защитной группы Boc, мезилирования спирта и внутримолекулярной циклизации в цис-циклический карбамат, последующего введения защитной группы Boc и восстановления алкена и нитрогруппы. Полученные циклогексилпиридиланилины VIIa и VIIb можно превратить в соответствующие пиридинамидыVIIIa и VIIIb с помощью реакции сочетания с образованием амида, расщепления ацетата или циклического карбамата и удаления защитной группы Boc. Если R2 обозначает галоген или трифлат, то амидыVIIIa и VIIIb можно дополнительно модифицировать по стандартным методикам модификации путем введения замещенных арилов, алкилов и гетероарилов по R2 после образования амидной связи и до полного удаления защитных групп. Например, если R2 обозначает Br, то по реакции с бороновыми кислотами или металлоорганическими реагентами или путем превращения в соответствующий эфир бороновой кислоты и реакции с арил/гетероарилгалогенидами или трифлатами можно выполнить различные модификации R2. Схема 2 Альтернативно, как показано на схеме 3, можно получить тризамещенные 5-алкил-4-гидрокси-3 аминопиперидины и их модифицировать и следующим образом получить тризамещенные 5-алкил-4 гидрокси-3-аминопиперидинилпиридинамиды IX. Реакция Гарнера альдегида с (R)-4-бензил-3 пропионилоксазолидин-2-оном, последующее ведение защитной группы TBS в полученный спирт дает соединение X. Восстановление оксазолидинона, последующее введение азидной группы дает промежуточный продукт XI. Удаление защитной группы в кислой среде дает соответствующий аминоспирт, который после введения защитной группы Boc и последующего мезилирования первичного спирта дает промежуточный продукт XII. Восстановление азида позволяет получить пиперидин, который затем вводят в реакцию с 4-хлор-3-нитропиридином, восстанавливают в амин, вводят в реакцию сочетания с соответствующей карбоновой кислотой и удаляют защитную группу и получают тризамещенные 5-метил-4 гидрокси-3-аминопиперидинилпиридинамиды IX. Если R1 обозначает галоген или трифлат, то амид IX можно дополнительно модифицировать по стандартным методикам модификации путем введения замещенных арилов, алкилов и гетероарилов по R1 после образования амидной связи и до полного удаления защитных групп. Например, если R1 обозначает Br, то по реакции с бороновыми кислотами или металлоорганическими реагентами или путем превращения в соответствующий эфир бороновой кислоты и реакции с арил/гетероарилгалогенидами или трифлатами можно выполнить различные модификации R1. Схема 3 Альтернативно, как показано на схеме 4, можно получить тризамещенные 5-метил-4-гидрокси-3 аминопиперидины и их модифицировать и следующим образом получить тризамещенные 5-метил-4 гидрокси-3-аминопиперидиниламиды XIII. Реакция кротиловых эфиров бороновой кислоты с альдегидомSerOBn, последующее образование циклического карбамата, окислительное отщепление алкена и восстановление дает содержащее гидроксигруппу соединение XIV. Удаление бензильной защитной группы,последующее бистозилирование и реакция с п-метоксибензиламином и удаление защитной группы ами- 11020136 на дает пиперидин XV. Реакция замещенного пиперидина XV с галогеннитрозамещенными аренами или гетероаренами, последующее введение карбаматной защитной группы, восстановление нитрогруппы,реакция сочетания с образованием амида, раскрытие циклического карбамата и удаление защитной группы дает тризамещенные 5-метил-4-гидрокси-3-аминопиперидиниламиды XIII. Если R3 обозначает галоген или трифлат, то амид XV можно дополнительно модифицировать по стандартным методикам модификации путем введения замещенных арилов, алкилов и гетероарилов по R3. Например, если R3 обозначает Br, то по реакции с бороновыми кислотами или металлоорганическими реагентами или путем превращения в соответствующий эфир бороновой кислоты и реакции с арил/гетероарилгалогенидами или трифлатами можно выполнить различные модификации R3. Схема 4 Соединения, предлагаемые в настоящем изобретении, применимы in vitro или in vivo для подавления роста раковых клеток. Соединения можно применять по отдельности или в композициях вместе с фармацевтически приемлемым носителем или инертным наполнителем. Подходящие фармацевтически приемлемые носители или инертные наполнители включают, например, технологические добавки и вещества, модифицирующие и улучшающие доставку лекарственного средства, такие как, например, фосфат кальция, стеарат магния, тальк, моносахариды, дисахариды, крахмал, желатин, целлюлоза, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, декстроза, гидроксипропилциклодекстрин, поливинилпирролидинон, низкоплавкие воски, ионообменные смолы и т.п., а также комбинации любых двух или большего количества из них. Другие подходящие фармацевтически приемлемые инертные наполнители описаны в публикации "Remington's Pharmaceutical Sciences", Mack Pub. Co., New Jersey(1991), которая включена в настоящее изобретение в качестве ссылки. Эффективные количества соединений, предлагаемых в настоящем изобретении, обычно представляют собой любые количества, достаточные для регистрируемого ингибирования активности Pim, обнаруживаемого с помощью любой из методик исследования, описанных в настоящем изобретении, с помощью других методик исследования активности киназы Pim, известных специалистам с общей подготовкой в данной области техники, или обнаруживаемого по подавлению или ослаблению симптомов рака. Количество активного ингредиента, которое можно объединить с носителями с получением разовой дозированной формы, будет меняться в зависимости от подвергающегося лечению реципиента и конкретного пути введения. Однако следует понимать, что точная доза для любого конкретного пациента будет зависеть от множества факторов, включая активность конкретного использующегося соединения,возраст, массу тела, общее состояния здоровья, пол, диету, время введения, путь введения, скорость выведения, комбинацию лекарственных средств и тяжесть конкретного заболевания, подвергающегося лечению. В каждом случае терапевтически эффективное количество можно легко определить с помощью стандартных исследований, и на основании своей профессиональной подготовки решение принимает лечащий врач. Для задач настоящего изобретения терапевтически эффективная доза обычно представляет собой полную суточную дозу, вводимую реципиенту в виде одной или разделенных доз в количествах, которые могут составлять, например, от 0,001 до 1000 мг/(кг массы тела) в сутки и более предпочтительно от 1,0 до 30 мг/(кг массы тела) в сутки. Разовая доза композиции может составлять доли таких количеств, так чтобы в сумме они образовывали суточную дозу. Соединения, предлагаемые в настоящем изобретении, можно вводить перорально, парентерально,сублингвально, с помощью аэрозоля или ингаляции спрея, ректально или местно в виде разовых доз композиций, при необходимости содержащих обычные нетоксичные фармацевтически приемлемые носители, вспомогательные вещества и растворители. Местное введение также может включать применение средств чрескожного введения, таких как чрескожные пластыри или устройства для ионофореза. Термин "парентерально" при использовании в настоящем изобретении включает подкожные инъекции,внутривенные, внутримышечные, надчревные инъекции и вливание. Препараты для инъекции, например стерильные водные или масляные суспензии для инъекции,можно приготовить с использованием известных в данной области техники подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный препарат для инъекции также может представлять собой стерильный раствор, суспензию или эмульсию для инъекции в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-пропандиоле. В число приемлемых носителей и разбавителей, которые можно использовать, входят вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла. Для этой цели можно использовать любое жидкое нелетучее масло, включая синтетические моно- или диглицериды. Кроме того, для приготовления средств для инъекции используют жирные кислоты, такие как олеиновая кислота. Суппозитории для ректального введения лекарственного средства можно приготовить путем смешивания лекарственного средства с подходящим не оказывающим раздражающее воздействие инертным наполнителем, таким как масло какао или полиэтиленгликоли, которые являются твердыми при температуре окружающей среды, но жидкими при температуре прямой кишки и поэтому плавятся в прямой кишке и высвобождают лекарственное средство. Твердые дозированные формы для перорального введения могут включать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых дозированных формах активное соединение можно смешать по меньшей мере с одним инертным разбавителем, таким как сахароза, лактоза или крахмал. Такие твердые дозированные формы могут включать, что является обычной практикой, дополнительные вещества,не являющиеся инертными разбавителями, например смазывающие агенты, такие как стеарат магния. В случае капсул, таблеток и пилюль дозированные формы также могут включать буферные агенты. Таблетки и пилюли также можно изготовить с энтеросолюбильными покрытиями. Жидкие дозированные формы для перорального введения могут включать фармацевтически приемлемые эмульсии, растворы,суспензии, сиропы и эликсиры, содержащие инертные разбавители, обычно использующиеся в данной области техники, такие как вода. Такие композиции также могут содержать вспомогательные вещества,такие как смачивающие агенты, эмульгирующие и суспендирующие агенты, циклодекстрины и подсластители, вкусовые добавки и отдушки. Соединения, предлагаемые в настоящем изобретении, также можно вводить в виде липосом. Как известно в данной области техники, липосомы обычно получают из фосфолипидов или других липидов. Липосомы формируются моно- или многослойными гидратированными жидкими кристаллами, которые диспергированы в водной среде. Можно использовать любой нетоксичный физиологически приемлемый и подвергающийся метаболизму липид, способный образовывать липосомы. Композиции, предлагаемые в настоящем изобретении, в липосомной форме в дополнение к соединению, предлагаемому в настоящем изобретении, могут содержать стабилизаторы, консерванты, инертные наполнители и т.п. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины), натуральные и синтетические. Методики приготовления липосом известны в данной области техники. См., например, публикацию Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N.W., p. 33 et seq.(1976). Хотя соединения, предлагаемые в настоящем изобретении, можно вводить в качестве единственного лекарственного средства, их также можно применять в комбинации с одним или большим количеством других средств, применяющихся для лечения рака. Соединения, предлагаемые в настоящем изобретении, также применимы в комбинации с известными лекарственными средствами и противораковыми средствами и комбинации соединений, предлагаемых в настоящем изобретении, с другими противораковыми средствами или химиотерапевтическими средствами и входят в объем настоящего изобретения. Примеры таких средств приведены в публикации Cancer Principles and Practice of Oncology, V.T. Devitaand S. Hellman (editors), 6th edition (Feb. 15, 2001), Lippincott WilliamsWilkins Publishers. Специалист с общей подготовкой в данной области техники должен быть способен определить, какие комбинации средств будут применимы в соответствии с характеристиками конкретных лекарственных средств и подвергающимся лечению раком. Такие противораковые средства включают, но не ограничиваются только ими, следующие: модуляторы эстрогенного рецептора, модуляторы андрогенного рецептора, модуляторы ретиноидного рецептора, цитотоксические/цитостатические средства, антипролиферативные средства, ингибиторы пренилпротеинтрансферазы, ингибиторы HMG-CoA редуктазы и другие ингибиторы ангиогенеза, ингибиторы пролиферации клеток и передачи сигналов жизнеспособности, средства, индуцирующие апоптоз, и средства, которые оказывают мешающее воздействие на ключевых стадиях клеточно- 13020136 го цикла. Соединения, предлагаемые в настоящем изобретении, также применимы совместно с лучевой терапией. Поэтому в одном варианте осуществления настоящего изобретения соединения, предлагаемые в настоящем изобретении, также применяются в комбинации с известными противораковыми средствами,включая, например, модуляторы эстрогенного рецептора, модуляторы андрогенного рецептора, модуляторы ретиноидного рецептора, цитотоксические средства, антипролиферативные средства, ингибиторы пренилпротеинтрансферазы, ингибиторы HMG-CoA редуктазы, ингибиторы протеазы ВИЧ (вирус иммунодефицита человека), ингибиторы обратной транскриптазы и другие ингибиторы ангиогенеза. В некоторых в настоящее время предпочтительных вариантах осуществления настоящего изобретения типичные средства, применимые в комбинации с соединениями, предлагаемыми в настоящем изобретении, для лечения рака включают, например, иринотекан, топотекан, гемцитабин, 5-фторурацил,лейковорин карбоплатин, цисплатин, таксаны, тезацитабин, циклофосфамид, алкалоиды барвинка, иматиниб (Глеевек), антрациклины, ритуксимаб, трастузумаб, а также противораковые химиотерапевтические средства. Указанные выше соединения, применяющиеся в комбинации с соединениями, предлагаемыми в настоящем изобретении, можно использовать в терапевтических количествах, как это указано в публикации Physicians' Desk Reference (PDR) 47th Edition (1993), которая включена в настоящее изобретение в качестве ссылки, или в таких терапевтически применимых количествах, которые должны быть известны специалисту с общей подготовкой в данной области техники. Соединения, предлагаемые в настоящем изобретении, и другие противораковые средства можно вводить в рекомендованной максимальной клинической дозировке или в меньших дозах. Величины доз активных соединений в композициях, предлагаемых в настоящем изобретении, могут меняться так, чтобы обеспечить необходимое терапевтическое воздействие в соответствии с путем введения, тяжестью заболевания и реакцией пациента. Комбинацию можно вводить в виде отдельных композиций или в виде разовой дозированной формы, содержащей оба средства. При введении в виде комбинации лекарственные средства можно приготовить в виде отдельных композиций, которые вводят одновременно или в разные моменты времени, или лекарственные средства можно вводить в виде одной композиции. В одном варианте осуществления настоящее изобретение относится к способу ингибирования Pim 1, Pim 2 или Pim 3 у человека или животного. Способ включает введение нуждающемуся в нем субъекту соединения или его фармацевтически приемлемой соли, соответствующей любому из вариантов осуществления соединений формулы I или II, в эффективном количестве. Настоящее изобретение будет легче понять с использованием приведенных ниже примеров, которые представлены только для иллюстрации и не предназначены для ограничения настоящего изобретения. Примеры В приведенных ниже примерах соединения, соответствующие предпочтительным вариантам осуществления, синтезировали по методикам, описанным в настоящем изобретении, или по другим методикам, которые известны в данной области техники. Соединения и/или промежуточные продукты характеризовали с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) с использованием хромагографической системы Waters Millenium с модулем разделения 2695 (Milford, MA). Использовали аналитические колонки с обращенной фазой Phenomenex Luna C18 - 5 мкм, 4,650 мм, выпускающиеся фирмой Alltech (Deerfield, IL). Использовали элюирование в градиентном режиме (скорость потока 2,5 мл/мин), обычно начиная от смеси 5% ацетонитрил/95% вода и заканчивая 100% ацетонитрилом, в течение 10 мин. Все растворители содержали 0,1% трифторуксусной кислоты (ТФК). Соединения детектировали по поглощению в ультрафиолетовой(УФ) области спектра при длине волны 220 или 254 нм. Использовали растворители для ВЭЖХ, выпускающиеся фирмами Burdick and Jackson (Muskegan, MI) или Fisher Scientific (Pittsburgh, PA). В некоторых случаях чистоту определяли с помощью тонкослойной хроматографии (ТСХ) с использованием стеклянных или пластмассовых пластин, покрытых силикагелем, таких как, например,гибкие пластины Baker-Flex Silica Gel 1B2-F. Данные ТСХ легко получали путем визуального детектирования при ультрафиолетовом освещении или путем использования хорошо известной методики окрашивания парами йода или различных других методик окрашивания. Масс-спектрометрические исследования проводили на одном из трех приборов для ЖХМС (жидкостная хроматография-масс-спектроскопия): Waters System (Alliance HT HPLC и Micromass ZQ массспектрометр; колонка: Eclipse XDB-C18, 2,150 мм; градиентный режим: 5-95% (или 35-95%, или 65-95%, или 95-95%) ацетонитрил в воде с добавлением 0,05% ТФК в течение 4 мин; скорость потока 0,8 мл/мин; диапазон молекулярных масс 200-1500; разность потенциалов на конусе 20 В; температура колонки 40 С), другого Waters System (ACQUITY UPLC system и ZQ 2000 system; колонка: ACQUITYUPLC HSS-C18, 1,8 мкм, 2,150 мм; градиентный режим: 5-95% (или 35-95%, или 65-95%, или 95-95%) ацетонитрил в воде с добавлением 0,05% ТФК в течение 1,3 мин; скорость потока 1,2 мл/мин; диапазон молекулярных масс 150-850; разность потенциалов на конусе 20 В; температура колонки 50C) или Hew- 14020136lett Packard System (Series 1100 HPLC; колонка: Eclipse XDB-C18, 2,150 мм; градиентный режим: 5-95% ацетонитрил в воде с прибавлением 0,05% ТФК в течение 4 мин; скорость потока 0,8 мл/мин; диапазон молекулярных масс 150-850; разность потенциалов на конусе 50 В; температура колонки 30C). Все приведенные массы относятся к протонированным исходным ионам. Исследования с помощью ядерного магнитного резонанса (ЯМР) проводили для некоторых соединений на приборе Varian 400 МГц ЯМР (Palo Alto, CA). В качестве стандарта использовали ТМС (триметилсилан) или известный химический сдвиг растворителя. Препаративные разделения проводили с помощью хроматографической системы Flash 40 и KPSil,60A (Biotage, Charlottesville, VA), или с помощью колоночной флэш-хроматографии с использованием силикагеля (230-400 меш) в качестве насадки, или с помощью ВЭЖХ с использованием Waters 2767Sample Manager, C-18 колонки с обращенной фазой, 3050 мм, скорость потока 75 мл/мин. Типичными растворителями, использованными для системы Flash 40 Biotage и колоночной флэш-хроматографии,являлись дихлорметан, метанол, этилацетат, гексан, ацетон, водный раствор аммиака (или гидроксид аммония) и триэтиламин. Типичными растворителями, использованными для ВЭЖХ с обращенной фазой, являлись ацетонитрил в разных концентрациях и вода с добавлением 0,1% трифторуксусной кислоты. Следует понимать, что органические соединения, соответствующие предпочтительным вариантам осуществления, могут обладать таутомерией. Поскольку химические структуры в настоящем изобретении могут представлять только одну из возможных таутомерных форм, следует понимать, что предпочтительные варианты осуществления включают любую таутомерную форму изображенной структуры. Следует понимать, что настоящее изобретение не ограничивается вариантами осуществления, представленными в настоящем изобретении для иллюстрации, а включает все его формы, соответствующие объему приведенного выше описания. В примерах, приведенных ниже, а также во всем описании представленные ниже аббревиатуры обладают указанными значениями. Термины, для которых не приведены определения, обладают общепринятыми значениями. К раствору циклогексан-1,3-диона (1 экв.) в ДХМ (0,4 М) добавляли Na2CO3 (1,0 экв.) и охлаждали до 0C. По каплям в течение 1 ч при комнатной температуре в атмосфере азота добавляли Tf2O (1,0 экв.) в ДХМ (5 М). После добавления реакционную смесь перемешивали в течение 2 ч (темно-красный раствор). Раствор фильтровали и к фильтрату добавляли насыщенный раствор NaHCO3 (осторожно), затем экстрагировали органические вещества, промывали рассолом, затем сушили над Na2SO4 и концентрировали. Неочищенное вещество использовали на следующей стадии без дополнительной очистки. 3 Оксоциклогекс-1-енилтрифторметансульфонат получали с выходом 67%. Трифлат разлагается при хранении и его необходимо сразу использовать в следующей реакции. ЖХ/МС=244,9/286,0 (М+Н и M+CH3CN); Rt=0,88 мин.(0,3 М) добавляли бис-(пинаколято)дибор (2,0 экв.), KOAc (3,0 экв.) и Pd(dppf)Cl2-ДХМ (0,05 экв.). Реакционную смесь нагревали при 80 С в течение 2 ч, затем фильтровали. Раствор в диоксане использовали на следующей стадии без дополнительной очистки. ЖХ/МС = 140,9 (М+Н бороновой кислоты). Синтез 3-(3-нитропиридин-4-ил)циклогекс-2-енона(0,05 экв.). Реакционную смесь нагревали на масляной бане при 110 С в течение 2 ч. Охлаждали до комнатной температуры, затем разбавляли с помощью EtOAc, добавляли Н 2 О - образовывался темный раствор, много эмульгированного вещества. Фильтровали для удаления твердых веществ, затем органическую фазу экстрагировали, сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали с помощью хроматографии на силикагеле при элюировании этилацетатом и гексанами (1:1) и получали 3-(3-нитропиридин-4-ил)циклогекс-2-енон (55%, 2 стадии). ЖХ/МС = 219 (М+Н), ЖХ = 2,294 мин. Синтез 3-(3-нитропиридин-4-ил)циклогекс-2-енолаCeCl37H2O (1,3 экв.). Реакционную смесь охлаждали до 0 С, затем порциями добавляли NaBH4 (1,3 экв.). Перемешивали в течение 2 ч при 0 С, затем реакцию останавливали путем добавления воды, концентрировали для удаления EtOH, добавляли EtOAc, экстрагировали органические вещества, промывали рассолом, затем сушили над Na2SO4 и концентрировали и получали 3-(3-нитропиридин-4-ил)циклогекс-2-енол К раствору 3-(3-нитропиридин-4-ил)циклогекс-2-енола (1,0 экв.), трифенилфосфин (1,5 экв.) и фталимид (1,5 экв.) в ТГФ (0,3 М) при 0 С по каплям добавляли (Е)-ди-трет-бутилдиазен-1,2-дикарбоксилат(1,5 экв.). Реакционную смесь перемешивали при 0 С в течение 2 ч. Концентрировали досуха в вакууме,затем неочищенное вещество очищали с помощью колоночной хроматографии на силикагеле при элюировании с помощью EtOAc и гексанов (1:1 с добавлением 5% метанола) и получали 2-(3-(3-нитропиридин-4-ил)циклогекс-2-енил)изоиндолин-1,3-дион (выход 63%). ЖХ/МС = 350,3 (М+Н), ЖХ = 3,936 мин.(0,38 М) добавляли Fe (6,0 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Фильтровали, затем промывали метанолом и фильтрат концентрировали. К неочищенному веществу добавляли эгилацетат и насыщенный раствор NaHCO3, органические вещества сушили над Na2SO4 и концентрировали и получали 2-(3-(3-аминопиридин-4-ил)циклогекс-2-енил)изоиндолин-1,3-дион в виде вязкой желтой смолы с выходом 96%. ЖХ/МС = 320,0 (М+Н), ЖХ = 2,410 мин. Синтез 2-(3-(3-аминопиридин-4-ил)циклогексил)изоиндолин-1,3-диона К раствору 2-(3-(3-нитропиридин-4-ил)циклогекс-2-енил)изоиндолин-1,3-диона (1,0 экв.) в уксусной кислоте (0,1 М) добавляли 10% Pd/C (0,2 экв.) и реакционную смесь перемешивали в атмосфере H2,подаваемого из баллона. Через 3 дня реакционную смесь фильтровали через целит, промывали этилацетатом и метанолом, фильтрат разбавляли этилацетатом и дважды промывали 2 М раствором Na2CO3. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Неочищенное вещество растирали с гексанами и этилацетатом и получали 2-(3-(3-аминопиридин-4 ил)циклогексил)изоиндолин-1,3-дион с выходом 73%. ЖХ/МС = 322,2 (М+Н), Rt=0,64 мин. Синтез 5,5-диметил-3-оксоциклогекс-1-енилтрифторметансульфоната(0,2 М). Добавляли карбонат натрия (1,1 экв.) и смесь в атмосфере N2 при перемешивании магнитной мешалкой в бане лед/раствор соли охлаждали примерно до -5 С. Ангидрид трифторметансульфоновой кислоты (1,05 экв.), разбавленный с помощью ДХМ, по каплям добавляли через капельную воронку в течение 90 мин. После завершения добавления реакционную смесь перемешивали примерно при 0 С в течение 1 ч. По данным ЖХМС и 1 Н ЯМР еще оставалось исходное вещество. Дополнительно добавляли карбонат натрия (0,51 экв.) и ангидрид трифторметансульфоновой кислоты (0,50 экв.). Через 2 ч смесь фильтровали через воронку с грубым фильтром из пористого стекла (осадок на фильтре промывали с помощью ДХМ), переносили в колбу Эрленмейера, реакционную смесь нейтрализовывали до рН 7 путем осторожного добавления насыщенного водного раствора бикарбоната натрия при энергичном перемешивании, переносили в делительную воронку и слои разделяли. Органический слой промывали рассолом,сушили над MgSO4, фильтровали и концентрировали и получали 5,5-диметил-3-оксоциклогекс-1 енилтрифторметансульфонат, который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=273,1, Rt=1,03. Синтез 5,5-диметил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-2-енона(3,0 экв.) и бис-(пинаколято)дибор (2,0 экв.) добавляли к 1,4-диоксану (0,2 М) в круглодонной колбе и дегазировали путем пропускания N2 через смесь в течение 10 мин. Добавляли аддукт PdCl2(dppf) - ДХМ(0,03 экв.) и реакционную смесь нагревали в атмосфере N2 в течение ночи при 80 С на масляной бане в колбе, снабженной обратным холодильником. Смесь охлаждали до комнатной температуры, фильтровали через воронку с грубым фильтром из пористого стекла, осадок на фильтре промывали 1,4-диоксаном и получали 5,5-диметил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-2-енон в 1,4-диоксане, который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+ (бороновая кислота) = 169,1, Rt=0,50. Синтез 5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енона Эфир бороновой кислоты 5,5-диметил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-2 енона (1,0 экв.) растворяли в 1,4-диоксане в круглодонной колбе и дегазировали путем пропускания N2 через раствор в течение 30 мин. Добавляли 4-хлор-3-нитропиридин (1,3 экв.) и 2 М водный раствор карбоната натрия (2,0 экв.) и в течение 10 мин N2 пропускали и затем добавляли PdCl2(dppf)-ДХМ(0,05 экв.). Реакционную смесь перемешивали при 110 С в течение 2 ч. Смесь добавляли к EtOAc и воде. Полученную смесь фильтровали через целит, осадок на фильтре промывали с помощью EtOAc. Органический слой отделяли и водный слой экстрагировали с помощью EtOAc. Объединенные органические слои промывали рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на силикагеле (элюировали смесью EtOAc:гексаны = от 1:10 до 2:1) и получали 5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енон (46,7% за три стадии). ЖХ/МС (m/z): MH+=247,2, Rt=0,79. Синтез 5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енола(1,2 экв.) в МеОН (0,2 М) при 0C добавляли NaBH4 (1,0 экв.). Раствор перемешивали в течение 1 ч и затем реакцию останавливали путем добавления 5 мл воды. Летучие вещества удаляли в вакууме и остаток подвергали распределению между EtOAc и Н 2 О. Органический слой отделяли и промывали рассолом. Объединенный водный слой подвергали обратной экстракции с помощью EtOAc и органическое вещество промывали рассолом. Объединенные органические вещества сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали 5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енол (74%). ЖХ/МС (m/z): MH+=249,2, Rt=0,76. Синтез 2-(5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енил)изоиндолин-1,3-диона К гомогенному раствору 5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енола (1,0 экв.) добавляли трифенилфосфин (1,5 экв.) и фталимид (1,5 экв.) в ТГФ (0,2 М), охлажденные до 0C, к раствору добавляли ди-трет-бутилазодикарбоксилат (1,5 экв.) в ТГФ. Смесь перемешивали при 0C в течение 2 ч. Реакционную смесь концентрировали в вакууме. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали 2-(5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2 енил)изоиндолин-1,3-дион (99%). ЖХ/МС (m/z): МН+=378,2, Rt=1,10. Синтез 2-(3-(3-аминопиридин-4-ил)-5,5-диметилциклогекс-2-енил)изоиндолин-1,3-диона Раствор 2-(5,5-диметил-3-(3-нитропиридин-4-ил)циклогекс-2-енил)изоиндолин-1,3-диона (1 экв.) в уксусной кислоте (0,1 М) продували азотом в течение 10 мин. Затем добавляли 10% Pd/C (0,10 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи в атмосфере водорода. Твердые вещества удаляли фильтрованием через целит, затем промывали с помощью EtOAc и МеОН.Na2CO3. Органический слой сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали 2-(3-(3-аминопиридин-4-ил)-5,5 диметилциклогекс-2-енил)изоиндолин-1,3-дион (89%). ЖХ/МС (m/z): МН+=348,3, Rt=0,79. Синтез 2-(5-(3-аминопиридин-4-ил)-3,3-диметилциклогексил)изоиндолин-1,3-диона Раствор 2-(3-(3-аминопиридин-4-ил)-5,5-диметилциклогекс-2-енил)изоиндолин-1,3-диона (1,0 экв.) в уксусной кислоте (0,1 М) продували азотом в течение 10 мин. Затем 10% Pd/C (0,1 экв.) добавляли. Реакционную смесь перемешивали при 45C при давлении водорода, равном 300 фунт-сила/дюйм, в стальном автоклаве в течение ночи и при 65C при 300 фунт-сила/дюйм в течение 5 ч. Твердые вещества удаляли фильтрованием через целит, затем промывали с помощью EtOAc и МеОН. Фильтрат концентрировали, разбавляли с помощью EtOAc и 2 промывали 2 М водным раствором Na2CO3. Органический слой сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали 2-(5-(3-аминопиридин-4-ил)-3,3 диметилциклогексил)изоиндолин-1,3-дион (53%). ЖХ/МС (m/z): МН+=350,3, Rt=0,78. Энантиомерно чистые 2-1R,5R)-5-(3-аминопиридин-4-ил)-3,3-диметилциклогексил)изоиндолин 1,3-дион и 2-1S,5S)-5-(3-аминопиридин-4-ил)-3,3-диметилциклогексил)изоиндолин-1,3-дион выделяли с помощью хиральной ВЭЖХ (для анализа Rt=7,526 мин и 13,105 мин соответственно; гексаны:этанол= 80:20 (об.:об.), Chiralcel OJ-H 1004,6 мм при 1 мл/мин. Для препаративного разделения, гексаны:этанол К раствору 3-(3-нитропиридин-4-ил)циклогекс-2-енола (1,0 экв.) добавляли диоксан (0,18 М) и пТСК (п-толуолсульфоновая кислота) (1,1 экв.). Раствор нагревали при 100C в течение 4 ч. Охлаждали до комнатной температуры, обрабатывали насыщенным раствором NaHCO3 и этилацетатом, органическую фазу сушили над Na2SO4 и концентрировали. Неочищенное вещество очищали с помощью колоночной хроматографии на силикагеле при элюировании с помощью 100% ДХМ и получали 4-(циклогекса-1,3 диенил)-3-нитропиридин в виде желтого масла (выход 27%). ЖХМС (m/z): 203,1 (МН+), ЖХ Rt=3,53 мин,1 Н-ЯМР (CDCl3): 9,02 (s, 1 Н), 8,70 (d, J=5,3 Гц, 1H), 7,30 (d, J=5,3 Гц, 1H), 6,15-6,17 (m, 1H), 6,026,11 (m, 2H), 2,35-2,38 (m, 4H). Синтез (+/-)-2-азидо-4-(3-нитропиридин-4-ил)циклогекс-3-енолаNaHCO3 (1,2 экв.) и получали желтый раствор. Охлаждали до 0C, затем к раствору одной порцией добавляли МХПБК (1,0 экв.) в виде твердого вещества. Реакционную смесь перемешивали при 0C в течение 3,5 ч. За протеканием реакции следили с помощью ТСХ и ЖХ/МС. Продукт ионизируется в виде М+Н = 237 (диол); Rt=0,41 мин в UPLC. Реакцию останавливали насыщенным раствором NaHCO3, затем экстрагировали с помощью ДХМ (3 раза). Органическую фазу дополнительно промывали рассолом, затем сушили над Na2SO4, фильтровали и концентрировали и получали неочищенный эпоксид в виде желтого масла, которое использовали без дополнительной очистки. К раствору полученного выше неочищенного вещества в EtOH и воде (3:1) (мутный желтый раствор) добавляли NaN3 (2,0 экв.) и NH4Cl (2,0 экв.) и получали прозрачный оранжевый раствор. Реакционную смесь перемешивали в течение 16 ч, затем концентрировали. EtOAc и добавляли воду, органическую фазу дополнительно сушили над MgSO4 и концентрировали и получали коричневое масло. Масло загружали в силикагель и очищали с помощью колоночной хроматографии (ISCO, 0-50% EtOAc) и получали (+/-)-2-азидо-4-(3-нитропиридин-4-ил)циклогекс-3-енол в виде желтого масла (44% за 2 стадии). ЖХМС (m/z): 262 (МН+), ЖХ Rt=2,35 мин. Синтез (+/-)-4-(3-азидо-4-(трет-бутилдиметилсилилокси)циклогекс-1-енил)-3-нитропиридина К раствору (+/-)-2-азидо-4-(3-нитропиридин-4-ил)циклогекс-3-енола (1,0 экв.) в ДХМ (0,15 М) при комнатной температуре добавляли TBSC1 (2,0 экв.), имидазол (2,0 экв.) и ДМАП (0,1 экв.). Через 18 ч добавляли воду, органические вещества промывали рассолом, затем сушили над Na2SO4 и концентрировали. Неочищенное вещество загружали в силикагель и очищали с помощью колоночной хроматографии(ISCO) при элюировании этилацетатом и гексанами(+/-)-4-(3-Азидо-4-(третбутилдиметилсилилокси)циклогекс-1-енил)-3-нитропиридин получали в виде желтого масла с выходом 60%. ЖХМС (m/z): 376,3 (МН+), ЖХ Rt=5,848 мин. Синтез В круглодонную колбу добавляли (+/-)-4-(3-азидо-4-(трет-бутилдиметилсилилокси)циклогекс-1 енил)-3-нитропиридин (1,0 экв.) и пиридин (0,1 М) и получали желтый раствор. Добавляли гидроксид аммония (10:1 пиридин:гидроксид аммония), затем РМе 3 (3,0 экв.). Через 10 мин реакционная смесь становилась темно-коричневой. Перемешивали при комнатной температуре в течение 1,5 ч. Реакцию останавливали путем добавления EtOH и концентрировали. Повторяли еще 2 раза. К неочищенному веществу добавляли насыщенный раствор NaHCO3 и диоксан (1:1, 0,1 М). Добавляли Вос 2 О (1,0 экв.). Перемешивали в течение 1 ч при комнатной температуре. Промывали с помощью Н 2 О и EtOAc, органическую фазу сушили над MgSO4, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (ISCO, 5:1 Нех/EtOAc). Чистые фракции собирали и концентрировали и получали К раствору (+/-)-трет-бутил-6-(трет-бутилдиметилсилилокси)-3-(3-нитропиридин-4-ил)циклогекс-2 енилкарбамата (1,0 экв.) в АсОН (0,18 М) добавляли Fe (6,0 экв.) и реакционную смесь перемешивали в течение 20 ч. Обрабатывали путем разбавления реакционной смеси метанолом, фильтровали и фильтрат концентрировали. К неочищенному веществу добавляли этилацетат и насыщенный раствор NaHCO3,органические вещества сушили над сульфатом натрия и концентрировали и получали (+/-)-трет-бутил-3(3-аминопиридин-4-ил)-6-(трет-бутилдиметилсилилокси)циклогекс-2-енилкарбамат в виде желтого масла с выходом 94%. ЖХМС (m/z): 420,3 (МН+), ЖХ Rt=3,88 мин.(+/-)-трет-бутил-5-(3-аминопиридин-4-ил)-2-(трет-бутилдиметилсилилокси)циклогексил К раствору (+/-)-трет-бутил-3-(3-аминопиридин-4-ил)-6-(трет-бутилдиметилсилилокси)циклогекс-2 енилкарбамата (1,0 экв.) в МеОН (0,1 М) добавляли Pd/C (20 мас.%) и реакционную смесь перемешивали в атмосфере водорода, подаваемого из баллона, в течение 18 ч. ЖХ/МС реакционной смеси указывала на наличие диастереоизомеров, реакционную смесь фильтровали, промывали с помощью EtOAc и фильтрат концентрировали. Неочищенное вещество очищали с помощью препаративной ВЭЖХ (в ДМСО - диметилсульфоксид) и чистые фракции объединяли, нейтрализовывали твердым NaHCO3, экстрагировали этилацетатом, промывали рассолом, сушили над Na2SO4 и концентрировали и получали продукт А (выход 8%) и продукт В (выход 51%). Продукт А: ЖХМС (m/z): 422,4 (МН+), ЖХ Rt=3,75 мин. Продукт В: ЖХМС (m/z): 422,4 (МН+), ЖХ Rt=3,94 мин. Синтез 1,4-диоксаспиро[4,5]дец-7-ен-8-илтрифторметансульфоната(1,05 экв.) и реакционной смеси давали медленно нагреться до КТ. Смесь перемешивали в течение 28 ч,промывали насыщенным водным раствором NaHCO3 и затем водой. Водные слои объединяли и экстрагировали эфиром. Органические слои объединяли, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке (этиловый эфир:гексаны = 1:4) и получали 1,4-диоксаспиро[4,5]дец-7-ен-8 илтрифторметансульфонат (65%). ЖХ/МС (m/z): MH+=289,0, Rt=0,97, ВЭЖХ Rt=3,77. Синтез 4,4,5,5-тетраметил-2-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)-1,3,2-диоксаборолана Раствор 1,4-диоксаспиро[4,5]дец-7-ен-8-илтрифторметансульфоната (1,0 экв.) в диоксане (0,5 М) продували азотом в течение 30 мин. Затем добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2 диоксаборолан) (1,0 экв.), KOAc (3,0 экв.), Pd(dppf)Cl2-ДХМ (0,2 экв.) и раствор перемешивали в закрытом автоклаве при 80C. Реакционную смесь фильтровали через слой целита, затем к фильтрату добавляли этилацетат и промывали рассолом, сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке Раствор ДМЭ (0,2 М) и 2 М водный раствор карбоната натрия (1,7 экв.) продували азотом в течение 20 мин. Затем добавляли 4-хлор-3-нитропиридин(1,6 экв.),4,4,5,5-тетраметил-2-(1,4 диоксаспиро[4,5]дец-7-ен-8-ил)-1,3,2-диоксаборолан (1,0 экв.), Pd(dppf)Cl2-ДХМ (0,05 экв.) и перемешивали в закрытом автоклаве при 110C. Реакционную смесь перемешивали при этой температуре в течение 3,5 ч. Реакционную смесь разбавляли этилацетатом, промывали водой, сушили над MgSO4, фильтро- 22020136 вали и концентрировали. Остаток очищали на колонке (этилацетат:гексаны = 1:1 с добавлением 10% метанола) и получали 3-нитро-4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)пиридин (83%). ЖХ/МС (m/z): МН+=263,2, Rt=0,71. Синтез 4-(3-нитропиридин-4-ил)циклогекс-3-енона(0,2 М) перемешивали при комнатной температуре в течение ночи. Растворители удаляли при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали насыщенным раствором NaHCO3(30 мл) и насыщенным раствором NaCl (30 мл). Органические вещества сушили над MgSO4, фильтровали и концентрировали и получали 4-(3-нитропиридин-4-ил)циклогекс-3-енон (85%). Неочищенный продукт использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=218,9, Rt=0,60 Синтез 4-(3-нитропиридин-4-ил)циклогекс-3-енола К раствору 4-(3-нитропиридин-4-ил)циклогекс-3-енона (1,0 экв.) в метаноле (0,2 М) при 0C добавляли борогидрид натрия (1,8 экв.). Реакционную смесь перемешивали при 0C в течение 2 ч. Метанол удаляли при пониженном давлении. Остаток растворяли в этилацетате (200 мл) и промывали насыщенным раствором NaCl (30 мл). Органические вещества сушили над MgSO4, фильтровали и концентрировали и получали 4-(3-нитропиридин-4-ил)циклогекс-3-енол (85%). Неочищенный продукт использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=221,0, Rt=0,55. Синтез 4-(3-нитропиридин-4-ил)циклогекс-3-енилметансульфоната(2,5 экв.) в CH2Cl2 (0,15 М) при 0C добавляли метансульфонилхлорид (1,8 экв.). Реакционную смесь перемешивали при 0C в течение 1 ч. Реакционную смесь разбавляли этилацетатом (200 мл) и промывали насыщенным раствором NaCl (30 мл). Органические вещества сушили над MgSO4, фильтровали и концентрировали и получали 4-(3-нитропиридин-4-ил)циклогекс-3-енилметансульфонат (93%). Остаток использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=299,0, Rt=0,70. Синтез 4-(циклогекса-1,3-диенил)-3-нитропиридина К раствору 4-(3-нитропиридин-4-ил)циклогекс-3-енилметансульфоната (1,0 экв.) в тетрагидрофуране (0,1 М) при комнатной температуре добавляли ДБУ (1,8-диазабикло[5.4.0]ундец-7-ен) (1,8 экв.). Реакционную смесь перемешивали при КТ в течение ночи. Реакционную смесь разбавляли этилацетатом (200 мл) и промывали насыщенным раствором NaCl (30 мл). Органические вещества сушили над MgSO4,фильтровали и концентрировали. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали 4-(циклогекса-1,3-диенил)-3-нитропиридин. ЖХ/МС (m/z): MH+=203,2, Rt=0,85.(8:1, 0,23 М) при комнатной температуре добавляли триметилфосфин (3,0 экв.). Смесь перемешивали при комнатной температуре в течение 3 ч. Растворители удаляли. К остатку добавляли этанол. Затем этанол удаляли в вакууме для обеспечения полного удаления аммиака. Остаток растворяли в 1,4-диоксане и к смеси добавляли насыщенный водный раствор бикарбоната натрия и затем Вос 2 О (1,0 экв.) в ТГФ. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором NaCl. Органические вещества сушили над MgSO4,фильтровали и концентрировали. Остаток очищали на колонке (5% метанола в смеси 1:1 этилацетата и гексанов) и получали (+/-)-трет-бутил-6-гидрокси-3-(3-нитропиридин-4-ил)циклогекс-2-енилкарбамат(1,0 экв.) и триэтиламина (1,5 экв.) в CH2Cl2 (0,2 М) при 0C добавляли метансульфонилхлорид (1,2 экв.). Смесь перемешивали в течение 2 ч при этой температуре. Реакционную смесь разбавляли этилацетатом и промывали насыщенным раствором NaCl. Органические вещества сушили над MgSO4, фильтровали и концентрировали и получали (+/-)-2-(трет-бутоксикарбониламино)-4-(3-нитропиридин-4-ил)циклогекс-3 енилметансульфонат (85%), который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=414,0, Rt=0,82. Синтез (+/-)-5-(3-нитропиридин-4-ил)-3,3 а,7,7 а-тетрагидробензо[d]оксазол-2(6 Н)-она(+/-)-2-(трет-бутоксикарбониламино)-4-(3-нитропиридин-4-ил)циклогекс-3 енилметансульфоната (1,0 экв.) с пиридином (0,21 М) перемешивали в микроволновой печи при 110C в течение 10 мин. Пиридин удаляли при пониженном давлении. Остаток растворяли в этилацетате и промывали насыщенным раствором NaCl. Органические вещества сушили над MgSO4, фильтровали и концентрировали и получали (+/-)-5-(3-нитропиридин-4-ил)-3,3 а,7,7 а-тетрагидробензо[d]оксазол-2(6 Н)-он(85%), который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=262,1, Rt=0,49. Синтез К раствору (+/-)-5-(3-нитропиридин-4-ил)-3,3 а,7,7 а-тетрагидробензо[d]оксазол-2(6 Н)-она (1,0 экв.),ТЭА (1,8 экв.) и каталитического количества ДМАП в CH2Cl2 (0,19 М) при комнатной температуре добавляли ди-трет-бутилдикарбонат (1,2 экв.). Реакционную смесь перемешивали в течение 1 ч. Реакционную смесь разбавляли этилацетатом (100 мл) и промывали насыщенным раствором NaCl (30 мл). Орга- 24020136 нические вещества сушили над MgSO4, фильтровали и концентрировали. Остаток очищали на колонке К раствору (+/-)-трет-бутил-5-(3-нитропиридин-4-ил)-2-оксо-3 а,6,7,7 а-тетрагидробензо[d]оксазол 3(2 Н)-карбоксилата (1,0 экв.) в метаноле и этилацетат (1:1, 0,1 М) добавляли 10% Pd/C (0,1 экв.). Полученную смесь перемешивали в атмосфере Н 2 в течение 6 ч. Твердое вещество удаляли фильтрованием. Фильтрат концентрировали при пониженном давлении и получали (+/-)-трет-бутил-5-(3-аминопиридин 4-ил)-2-оксогексагидробензо[d]оксазол-3(2 Н)-карбоксилат (87%), который использовали на следующей стадии без дополнительной очистки. ЖХ/МС (m/z): MH+=334,1, Rt=0,51. Синтез 5-метил-3-оксоциклогекс-1-енилтрифторметансульфоната К раствору 5-метилциклогексан-1,3-диона (1,0 экв.) в ДХМ (0,5 М) добавляли Na2CO3 (1,1 экв.) и охлаждали до 0C. В течение 1 ч при 0C в атмосфере азота по каплям добавляли Tf2O (1,0 экв.) в ДХМ(5,0 М). После добавления реакционную смесь перемешивали в течение 1 ч при комнатной температуре(темно-красный раствор). Раствор фильтровали и фильтрат нейтрализовывали до pH 7 путем осторожного добавления насыщенного раствора NaHCO3 при энергичном перемешивании. Раствор переносили в делительную воронку и слои разделяли. Органический слой промывали рассолом, сушили над Na2SO4,фильтровали, концентрировали в вакууме и сушили в высоком вакууме в течение 15 мин и получали 5-метил-3-оксоциклогекс-1-енилтрифторметансульфонат в виде светло-желтого масла с выходом 78%. Трифлат разлагается при хранении, и его необходимо сразу использовать в следующей реакции. ЖХ/МС=259,1/300,1 (М+Н и M+CH3CN); Rt=0,86 мин, ЖХ = 3,84 мин. 1 Н-ЯМР (400 МГц, CDCl3)част./млн: 6,05 (s, 1H), 2,70 (dd, J=17,2, 4,3 Гц, 1 Н), 2,53 (dd, J=16,6,3,7 Гц, 1H), 2,48-2,31 (m, 2H), 2,16 (dd, J=16,4, 11,7 Гц, 1H), 1,16 (d, J=5,9 Гц, 3H). Синтез 5-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)циклогекс-2-енона(0,03 экв.). Реакционную смесь нагревали при 80C в течение 10 ч (сильное начальное нагревание приводит к образованию оранжевой пены в верхней части раствора с разогревом, нагревающую баню следует удалить до оседания пены, повторно нагреть до 80C и при этой температуре пена не образуется), затем охлаждали до комнатной температуры и фильтровали через воронку с грубым фильтром из пористого стекла. Осадок на фильтре дополнительно промывали диоксаном и фильтрат использовали на следующей стадии без дополнительной очистки. ЖХ/МС (жидкостная хроматография/масс-спектроскопия) = 155,1 (М+Н бороновой кислоты);Pd(dppf)Cl2-AXM (0,05 экв.). Реакционную смесь помещали в колбу, снабженную обратным холодильником, и нагревали на масляной бане при 110C в течение 1 ч. Охлаждали до комнатной температуры,фильтровали через слой целита, слой промывали этилацетатом и фильтрат концентрировали в вакууме. Остаток дополнительно выпаривали при 80C в роторном испарителе в течение 1 ч для удаления борсодержащих промежуточных продуктов (М+Н=101) путем возгонки. Остаток подвергали распределению между рассолом и этилацетатом и слои разделяли, водную фазу дополнительно экстрагировали этилацетатом (4), органические вещества объединяли, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали с помощью хроматографии на силикагеле в ДХМ и при элюировании смесью 2-50% этилацетата и гексанов. Чистые фракции концентрировали в вакууме и получали оранжевое масло. Масло выдерживали в высоком вакууме (500 мторр) с затравочными кристаллами в течение ночи и получали оранжевое твердое вещество. Твердое вещество дополнительно очищали путем растирания с гексанами и получали 5-метил-3-(3-нитропиридин-4-ил)циклогекс-2-енон (48%, 2 стадии). ЖХ/МС = 233,2 (М+Н); Rt=0,69 мин, ЖХ = 2,70 мин. 1 Н-ЯМР (400 МГц, CdCl3)част./млн: 9,31 (s, 1 Н), 8,88 (d, J=5,1 Гц, 1 Н), 7,30 (d, J=5,1 Гц, 1H), 6,00CeCl37H2O (1,2 экв.). Реакционную смесь охлаждали до 0C, затем порциями добавляли NaBH4 (1,2 экв.). Перемешивали в течение 1 ч при 0C, затем реакцию останавливали путем добавления воды, концентрировали для удаления EtOH, добавляли EtOAc, экстрагировали органические вещества, промывали рассолом, затем сушили над Na2SO4, фильтровали и концентрировали и получали цис-(+/-)-5-метил-3-(3 нитропиридин-4-ил)циклогекс-2-енол (94%). ЖХ/МС = 235,2 (М+Н), ЖХ = 2,62 мин. Синтез 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогекс-1-енил)-3-нитропиридина К раствору 5-метил-3-(3-нитропиридин-4-ил)циклогекс-2-енола (1,0 экв.) в ДМФ (0,5 М) добавляли имидазол (4,0 экв.) и TBDMSCl (2,5 экв.). После перемешивания в течение 18 ч раствор подвергали распределению между EtOAc и Н 2 О и разделяли. После дополнительной промывки с помощью Н 2 О (2) иNaCl(sat), сушки над MgSO4, фильтрования и удаления растворителей получали 4-(3-(третбутилдиметилсилилокси)-5-метилциклогекс-1-енил)-3-нитропиридин (85%). ЖХ/МС = 349,2 (М+Н), ЖХ = 5,99 мин. Синтез 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогекс-1-енил)пиридин-3-амина Гетерогенный раствор 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогекс-1-енил)-3 нитропиридина (1,0 экв.) и железо (6,0 экв.) в уксусной кислоте при концентрации, равной 0,4 М, энергично перемешивали в течение 2 ч. Смесь затем пропускали через слой целита, при элюировании с помощью МеОН. После удаления летучих веществ в вакууме остаток растворяли в EtOAc, промывали с помощью Na2CO3(sat), NaCl(sat), сушили над MgSO4, фильтровали и летучие вещества удаляли в вакууме и получали 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогекс-1-енил)пиридин-3-амин (78%). ЖХМС (m/z): 319,3 (МН+); ЖХ Rt=3,77 мин. Синтез 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогексил)пиридин-3-амина(1,0 экв.) в метаноле при концентрации, равной 0,1 М, добавляли 10% палладий на угле (0,1 экв.). Полученный гетерогенный раствор помещали в атмосферу водорода и перемешивали в течение 15 ч. Затем смесь фильтровали через слой целита при элюировании метанолом. Летучие вещества удаляли в вакууме и получали 4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогексил)пиридин-3-амин (90%). ЖХМС (m/z): 321,3 (MH+); ЖХ Rt=3,85 мин. Синтез цис-(+/-)-бензил-4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогексил)пиридин-3 илкарбамата(1,1 экв.) и ДМАП (0,05 экв.). После перемешивания в течение 16 ч при КТ добавляли еще бензил-2,5 диоксопирролидин-1-илкарбонат (0,55 экв.) и ДМАП (0,03 экв.). После перемешивания в течение еще 24 ч при КТ добавляли еще бензил-2,5-диоксопирролидин-1-илкарбонат (0,1 экв.) и ДМАП (0,03 экв.). После перемешивания в течение еще 18 ч раствор подвергали распределению между EtOAc и Na2CO3(sat) и разделяли. После дополнительной промывки с помощью Na2CO3(sat) (2) и NaCl(sat), сушки над MgSO4,фильтрования и удаления растворителей получали цис-(+/-)-бензил-4-(3-(трет-бутилдиметилсилилокси)5-метилциклогексил)пиридин-3-илкарбамат. Неочищенное вещество использовали без обработки. ЖХ/МС = 455,3 (М+Н), ЖХ = 4,39 мин. Синтез цис-(+/-)бензил-4-(3-гидрокси-5-метилциклогексил)пиридин-3-илкарбамата Раствор цис-(+/-)-бензил-4-(3-(трет-бутилдиметилсилилокси)-5-метилциклогексил)пиридин-3 илкарбамата в смеси 1:2:1 6 н. HCl/ТГФ/МеОН при концентрации, равной 0,1 М, перемешивали при КТ в течение 6 ч. Затем значение рН доводили до 7 путем добавления 6 н. NaOH и летучие вещества удаляли в вакууме. Водный слой экстрагировали с помощью EtOAc и органическое вещество промывали с помощью NaCl(sat), сушили над MgSO4, фильтровали и после удаления летучих веществ в вакууме получали цис-(+/-)бензил-4-(3-гидрокси-5-метилциклогексил)пиридин-3-илкарбамат. Неочищенное вещество использовали без обработки. ЖХ/МС = 341,2 (М+Н), ЖХ = 2,38 мин. Синтез цис-(+/-)-бензил-4-(3-метил-5-оксоциклогексил)пиридин-3-илкарбамата При 0C к раствору цис-(+/-)-бензил-4-(3-гидрокси-5-метилциклогексил)пиридин-3-илкарбамата во влажном CH2Cl2 при концентрации, равной 0,16 М, добавляли перйодинан Десса-Мартина (1,5 экв.) и раствор перемешивали в течение 18 ч, пока он нагревался до КТ. Раствор подвергали распределению между EtOAc и 1:1 10% Na2S2O3/NaHCO3(sat) и разделяли. После дополнительной промывки с помощью 1:1 10% Na2S2O3/NaHCO3(sat) (2) и NaCl(sat), сушки над MgSO4, фильтрования, удаления растворителей и очистки с помощью хроматографии на силикагеле (75-100% EtOAc/гексаны) получали цис-(+/-)-бензил 4-(3-метил-5-оксоциклогексил)пиридин-3-илкарбамат в виде белого твердого вещества (53%, 5 стадий). ЖХ/МС = 339,2 (М+Н). Синтез цис-(+/-)-бензил-4-(-3-(бензиламино)-5-метилциклогексил)пиридин-3-илкарбамата Раствор цис-(+/-)-бензил-4-(3-метил-5-оксоциклогексил)пиридин-3-илкарбамата (1,0 экв.) и бензиламина (3,0 экв.) в МеОН при концентрации, равной 0,25 М, перемешивали при КТ в течение 2 ч. При охлаждении в бане до -78C добавляли LiBH4 (1,1 экв., 2,0 М в ТГФ) и раствору при перемешивании давали нагреваться до КТ в течение 16 ч. Раствор подвергали распределению между EtOAc и NaHCO3(sat),разделяли, дополнительно промывали с помощью NaHCO3(sat) и NaCl(sat), сушили над MgSO4, фильтровали и после удаления летучих веществ в вакууме получали цис-(+/-)-бензил-4-(3-(бензиламино)-5 метилциклогексил)пиридин-3-илкарбамат в виде 4:1 смеси изомеров с преобладанием полностью цисизомера. ЖХ/МС = 430,3 (М+Н), ЖХ = 0,62 мин. Синтез цис-(+/-)-трет-бутил-(3-(3-аминопиридин-4-ил)-5-метилциклогексилкарбамата(1,0 экв.) в метаноле при концентрации, равной 0,07 М, добавляли 20% гидроксид палладия на угле(0,2 экв.). Полученный гетерогенный раствор помещали в атмосферу водорода и перемешивали в течение 14 ч. Затем реакционную смесь продували с помощью Ar, добавляли Вос 2 О (1,0 экв.) и раствор перемешивали в течение 8 ч. Добавляли еще Вос 2 О (1,0 экв.) и раствор перемешивали в течение еще 16 ч. Затем смесь фильтровали через слой целита при элюировании метанолом. После удаления летучих веществ в вакууме очистка с помощью хроматографии на силикагеле (2,5-2,5 MeOH/CH2Cl2 с добавлением 0,1% ДИЭА) и перекристаллизация из 10% EtOAc/гексаны давала цис-(+/-)-трет-бутил-(3-(3-аминопиридин-4 ил)-5-метилциклогексилкарбамат (49%). ЖХМС (m/z): 306,3 (МН+), ЖХ Rt=2,59 мин. Чистые энантиомеры можно было получить с помощью хиральной хроматографии. Синтез (+/-)-4-(5-метилциклогекса-1,3-диенил)-3-нитропиридина К раствору (+/-)-5-метил-3-(3-нитропиридин-4-ил)циклогекс-2-енола (1,0 экв.) в диоксане (0,1 М) добавляли п-ТСК (1,0 экв.) и реакционную смесь перемешивали при 100C в течение 3 ч. Раствор охлаждали до комнатной температуры, затем пропускали через слой нейтрального оксида алюминия при элюировании с помощью EtOAc и получали (+/-)-4-(5-метилциклогекса-1,3-диенил)-3-нитропиридин в виде желтого масла с выходом 68%. ЖХ/МС = 217,1 (М+Н), ЖХ = 3,908 мин. Синтез (+/-)-2-азидо-6-метил-4-(3-нитропиридин-4-ил)циклогекс-3-енола К раствору (+/-)-4-(5-метилциклогекса-1,3-диенил)-3-нитропиридина (1,0 экв.) в ДХМ (0,1 М) при 0C добавляли МХПБК (1,1 экв.) и реакционной смеси давали нагреться до комнатной температуры. Через 3 ч реакцию останавливали насыщенным раствором NaHCO3, экстрагировали с помощью ДХМ и органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали и получали желтое масло. Неочищенное вещество растворяли в этаноле и воде (3:1, 0,1 М) и добавляли азид натрия (2,0 экв.) и хлорид аммония (2,0 экв.). Реакционную смесь перемешивали в течение 4 ч, затем концентрировали в вакууме. К неочищенному веществу добавляли этилацетат и воду, органическую фазу промывали рассолом, сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали с помощью колоночной хроматографии на силикагеле при элюировании этилацетатом и гексанами (1:1) и получали (+/-)-2-азидо-6-метил-4-(3-нитропиридин-4-ил)циклогекс-3-енол в виде масла с выходом 49%. ЖХ/МС = 276,1 (М+Н), Rt=0,71 мин. Синтез трет-бутил-(+/-)-6-гидрокси-5-метил-3-(3-нитропиридин-4-ил)циклогекс-2-енилкарбамата К раствору (+/-)-2-азидо-6-метил-4-(3-нитропиридин-4-ил)циклогекс-3-енола (1,0 экв.) в пиридине и гидроксиде аммония (8:1, 0,08 М) добавляли триметилфосфин (3,0 экв.) и коричневый раствор перемешивали при комнатной температуре в течение 2 ч. К смеси добавляли этанол и раствор концентрировали в вакууме (2). Затем неочищенную смесь растворяли в диоксане и добавляли насыщенный растворNaHCO3 (1:1, 0,08 М) и Вос 2 О (1,0 экв.). Раствор перемешивали при комнатной температуре в течение 2 ч, затем подвергали распределению между этилацетатом и водой. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле при элюировании этилацетатом и гексанами (1:1) и получали трет-бутил-(+/-)-6-гидрокси-5-метил-3-(3-нитропиридин-4-ил)циклогекс-2-енилкарбамат с выходом 69%. ЖХ/МС = 350,1 (М+Н), Rt=0,76 мин. Синтез К раствору трет-бутил-(+/-)-6-гидрокси-5-метил-3-(3-нитропиридин-4-ил)циклогекс-2 енилкарбамата (1,0 экв.) в ДХМ (0,09 М) добавляли триэтиламин (1,5 экв.). Реакционную смесь охлаждали до 0C и к реакционной смеси добавляли MsCl (1,2 экв.) и перемешивали в течение 3 ч. К раствору добавляли воду, органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали с помощью колоночной хроматографии на силикагеле при элюировании этилацетатом и гексанами (1:1) и получали (+/-)-2-(трет-бутоксикарбониламино)-6-метил-4-(3 нитропиридин-4-ил)циклогекс-3-енилметансульфонат в виде белого вспененного вещества с выходом 65%. ЖХ/МС = 428,2 (М+Н), Rt=0,88 мин. Синтез(+/-)-2-(трет-бутоксикарбониламино)-6-метил-4-(3-нитропиридин-4-ил)циклогекс-3 енилметансульфонат (1,0 экв.) в пиридине (0,2 М) в сосуде для микроволновой печи нагревали при 110C в течение 10 мин. Затем оранжевый раствор концентрировали досуха и обрабатывали путем распределения между этилацетатом и водой. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество растворяли в ДХМ (0,2 М) и к реакционной смеси добавляли триэтиламин (1,8 экв.), затем Вос 2 О (1,2 экв.). После перемешивания при комнатной температуре в течение 40 мин реакционную смесь концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле при элюировании этилацетатом и гексанами (1:1) и получали (+/-)-трет-бутил-7 метил-5-(3-нитропиридин-4-ил)-2-оксо-3 а,6,7,7 а-тетрагидробензо[d]оксазол-3(2 Н)-карбоксилат в виде белого вспененного вещества с выходом 66%. ЖХ/МС = 376,0 (М+Н), Rt=0,87 мин. Синтез (+/-)-трет-бутил-5-(3-аминопиридин-4-ил)-7-метил-2-оксогексагидробензо[d]оксазол-3(2 Н)карбоксилата(+/-)-трет-бутил-7-метил-5-(3-нитропиридин-4-ил)-2-оксо-3 а,6,7,7 атетрагидробензо[d]оксазол-3(2 Н)-карбоксилата (1,0 экв.) в МеОН и этилацетате (1:1, 0,07 М) добавляли 10% Pd/C (0,1 экв.) и реакционную смесь перемешивали при комнатной температуре в атмосфере водорода. После завершения реакции раствор фильтровали через слой целита, промывали с помощью МеОН
МПК / Метки
МПК: C07D 401/14, A61P 35/00, C07D 401/12, A61K 31/444
Метки: ингибиторов, качестве, пиколинамида, киназы, производные
Код ссылки
<a href="https://eas.patents.su/30-20136-proizvodnye-pikolinamida-v-kachestve-ingibitorov-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиколинамида в качестве ингибиторов киназы</a>
Производные хинолина в качестве ингибиторов pi3 киназы

Номер патента: 19540
Опубликовано: 30.04.2014
Авторы: Дарси Майкл Джерард, Адамс Николас Д., Донателли Карла А., Сарпонг Марта, Шмидт Стэнли Дж., Берджесс Жоэлль Лоррейн, Ньюландер Кеннет Аллен, Риджерс Ланс, Найт Стивен Дэвид
МПК: A61K 31/47
Метки: хинолина, киназы, производные, ингибиторов, качестве
Формула / Реферат:
1. Соединение, представляющее собой 2,4-дифтор-N-{2-(метилокси)-5-[4-(4-пиридазинил)-6-хинолинил]-3-пиридинил}бензолсульфонамид, следующей структуры:или его фармацевтически приемлемая соль.2. Соединение по п.1, которое представляет собой 2,4-дифтор-N-{2-(метилокси)-5-[4-(4-пиридазинил)-6-хинолинил]-3-пиридинил}бензолсульфонамид.3. Применение соединения по п.1 или 2 или его фармацевтически приемлемой соли в производстве лекарственного средства...
Производные аминоизоксазола в качестве ингибиторов киназы

Номер патента: 6769
Опубликовано: 28.04.2006
Авторы: Вульпетти Анна, Кавиккьоли Марчелло, Салом Барбара, Певарелло Паоло
МПК: A61K 31/4439, A61K 31/422, A61K 31/427...
Метки: производные, качестве, ингибиторов, аминоизоксазола, киназы
Формула / Реферат:
1. Способ лечения заболеваний, вызванных и/или ассоциированных с изменением активности протеинкиназы, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества производного аминоизоксазола, представленного формулой (I) где R представляет 5- или 6-членную гетероарильную группу, содержащую 1-3 гетероатома, выбранных из азота, кислорода или серы, необязательно дополнительно конденсированную с 5-7-членным ароматическим...
Производные триазолопиридинсульфанила в качестве ингибиторов p38 мар киназы

Номер патента: 12119
Опубликовано: 28.08.2009
Авторы: Матиас Джон Пол, Льютуайт Рассел Эндрю, Филлипс Кристофер, Миллан Дэвид Саймон
МПК: A61K 31/4745, C07D 471/04, A61P 29/00...
Метки: триазолопиридинсульфанила, киназы, качестве, мар, ингибиторов, производные
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль и/или сольват (включая гидрат), где R1 представляет CH3, SCH3, SCH2CH3, CH2CH3, Н или CH2SCH3; R1a представляет CH3 или CH2CH3; R2 представляет пиридил, тетрагидронафтил или арил, причем указанные пиридил, тетрагидронафтил и арил, каждый, необязательно были замещены одним или более из заместителей, независимо выбранных из группы, состоящей из галогена, -CN, -CO2H, OH, CONR5R6,...
Производные хиназолина в качестве ингибиторов киназы

Номер патента: 5809
Опубликовано: 30.06.2005
Авторы: Ирие Дзунко, Цукуда Еидзи, Скарборо Роберт М., Итимура Митио, Иде Синити, Ода Содзи, Номото Юдзи, Мацуно Кендзи, Панди Анджали
МПК: C07D 403/12
Метки: киназы, производные, качестве, ингибиторов, хиназолина
Формула / Реферат:
1. Азотсодержащее гетероциклическое соединение формулы в которой R1 обозначает радикал, выбранный из группы, в которую входят -CN, -O-метил, -O-этил, -O-пропил, -O-изопропил, -O-бутил, -O-трет-бутил, -O-изоамил, 1-нафтилокси, 2-нафтилокси, 4-индолилокси, 5-индолилокси, 5-изохинолилокси и их пространственные изомеры и гомологи; R2 и R4 каждый представляют собой -O-CH3 или -O(-CH2)n-R3; причем, когда R2 представляет собой -O-CH3, R4 представляет...
Производные пиразолизохинолинмочевины в качестве ингибиторов p38 киназы

Номер патента: 15123
Опубликовано: 30.06.2011
Авторы: Лопес Де Уралде-Гармениа Беатрис, Ших Чуан, Де Дьес Альфонсо, Чжун Боюй, Побанс Марк Эндрю, Мэйдер Мэри Маргарет, Гарсиа-Паредес Кристина
МПК: A61P 29/00, A61K 31/4725, A61P 25/00...
Метки: пиразолизохинолинмочевины, производные, киназы, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы Iгде X выбран из группы, включающейR1 представляет собой C1-C4 алкил, C3-C4 циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, включающей C1-C4 алкокси, метил и трифторметил; или C1-C4 алкилгалоген;R2 представляет собой фенил, необязательно замещенный C1-C4 алкилом, или пиридинил, необязательно замещенный C1-C4 алкилом;R3 представляет собой амино, C1-C4алкил, C1-C4алкокси,...
Предыдущий патент: Гидроксиметилпирролидины в качестве агонистов адренергических рецепторов бета 3
Следующий патент: Способ наплавки
Случайный патент: Способ и устройство для формовки верхней и нижней полуформ, не имеющих опок