Пуриновые соединения
Номер патента: 18386
Опубликовано: 30.07.2013
Авторы: Тидуэлл Майкл Вэйд, Эстлз Питер Чарльз, Холлиншед Шон Патрик, Гвидетти Росселла











Формула / Реферат
1. Соединение формулы

где R1 выбран из Н, F, Cl, C1-C2-алкила, CF3, циклопропила, ОСН3, OCF3 и CN;
R2 выбран из тетрагидрофуранила, тетрагидропиранила, метилового эфира азетидин-1-карбоновой кислоты и тетрагидротиофен-1,1-диоксида;
R3 представляет собой Н или совместно с R4 образует конденсированный пирролидин-2-он;
R4 выбран из C1-C2-алкила, C1-С2-фторалкила, циклопропила и СОСН3;
R5 выбран из Н, СН3 и CF3;
n равно 0 или 1;
X1 и X3 независимо выбраны из N, СН и CR6;
X2 выбран из СН и CR6;
при условии, что только один из X1, X2 и X3 может быть отличным от СН;
R6 выбран из F, Cl, CF3, OCH3 и OCF3;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что R1 выбран из Cl, C1-C2-алкила, CF3, циклопропила и OCF3.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль, отличающееся тем, что R1 представляет собой Cl.
4. Соединение по любому из пп.1-3 или его фармацевтически приемлемая соль, отличающееся тем, что R2 выбран из тетрагидрофуранила и тетрагидропиранила.
5. Соединение по любому из пп.1-4 или его фармацевтически приемлемая соль, отличающееся тем, что n равно 0.
6. Соединение по любому из пп.1-5 или его фармацевтически приемлемая соль, отличающееся тем, что R3 представляет собой Н.
7. Соединение по любому из пп.1-6 или его фармацевтически приемлемая соль, отличающееся тем, что R4 выбран из C1-С2-алкила, C1-C2-фторалкила или циклопропила.
8. Соединение по любому из пп.1-7 или его фармацевтически приемлемая соль, отличающееся тем, что R4 представляет собой C1-C2-алкил.
9. Соединение по любому из пп.1-8 или его фармацевтически приемлемая соль, отличающееся тем, что R5 представляет собой СН3.
10. Соединение по любому из пп.1-9 или его фармацевтически приемлемая соль, отличающееся тем, что X1, X2 и X3 независимо выбраны из СН и CR6, где R6 выбран из Cl, CF3, OCH3 и OCF3.
11. Соединение по любому из пп.1-10 или его фармацевтически приемлемая соль, отличающееся тем, что каждый из X1, X2 и X3 представляет собой СН.
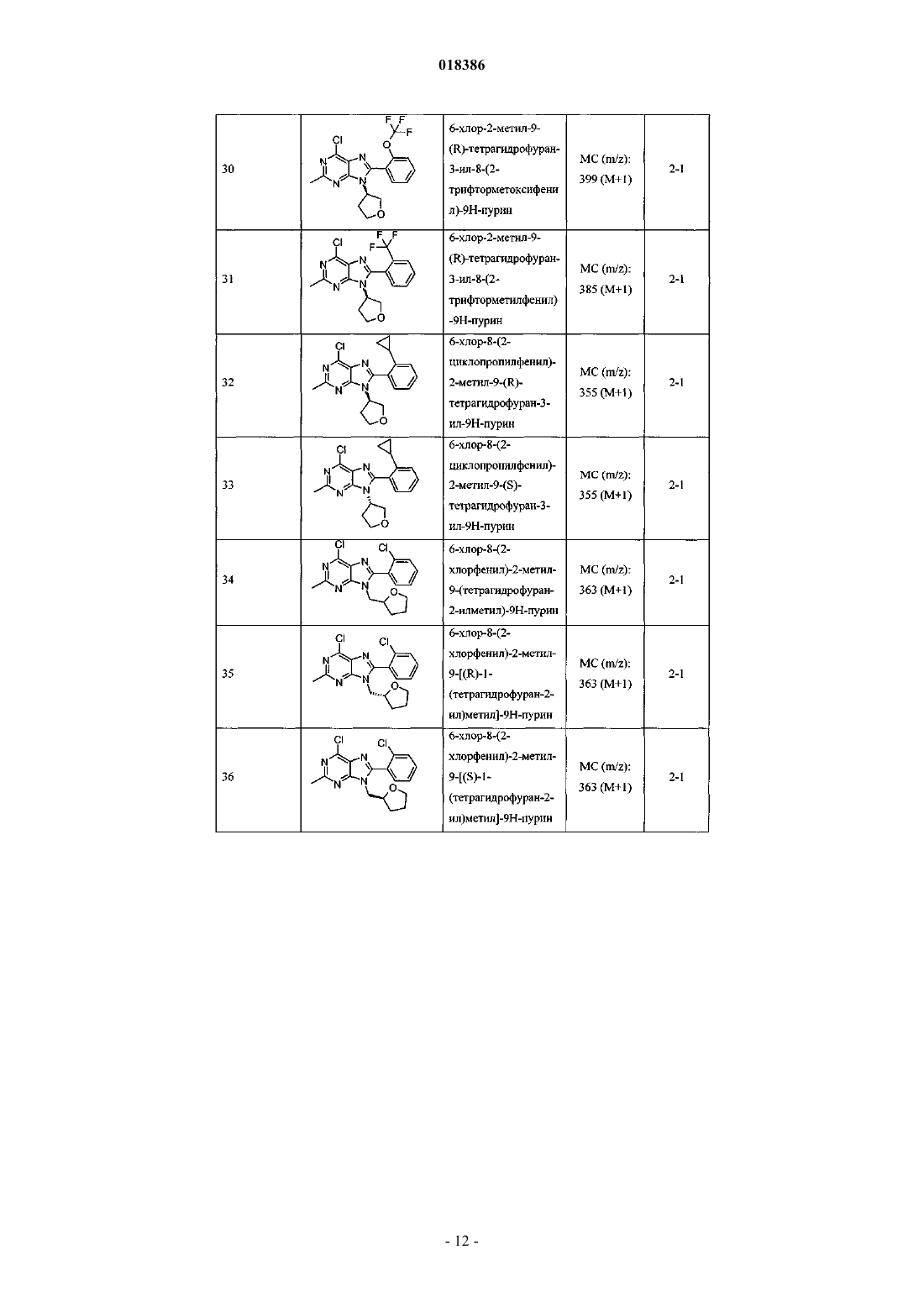
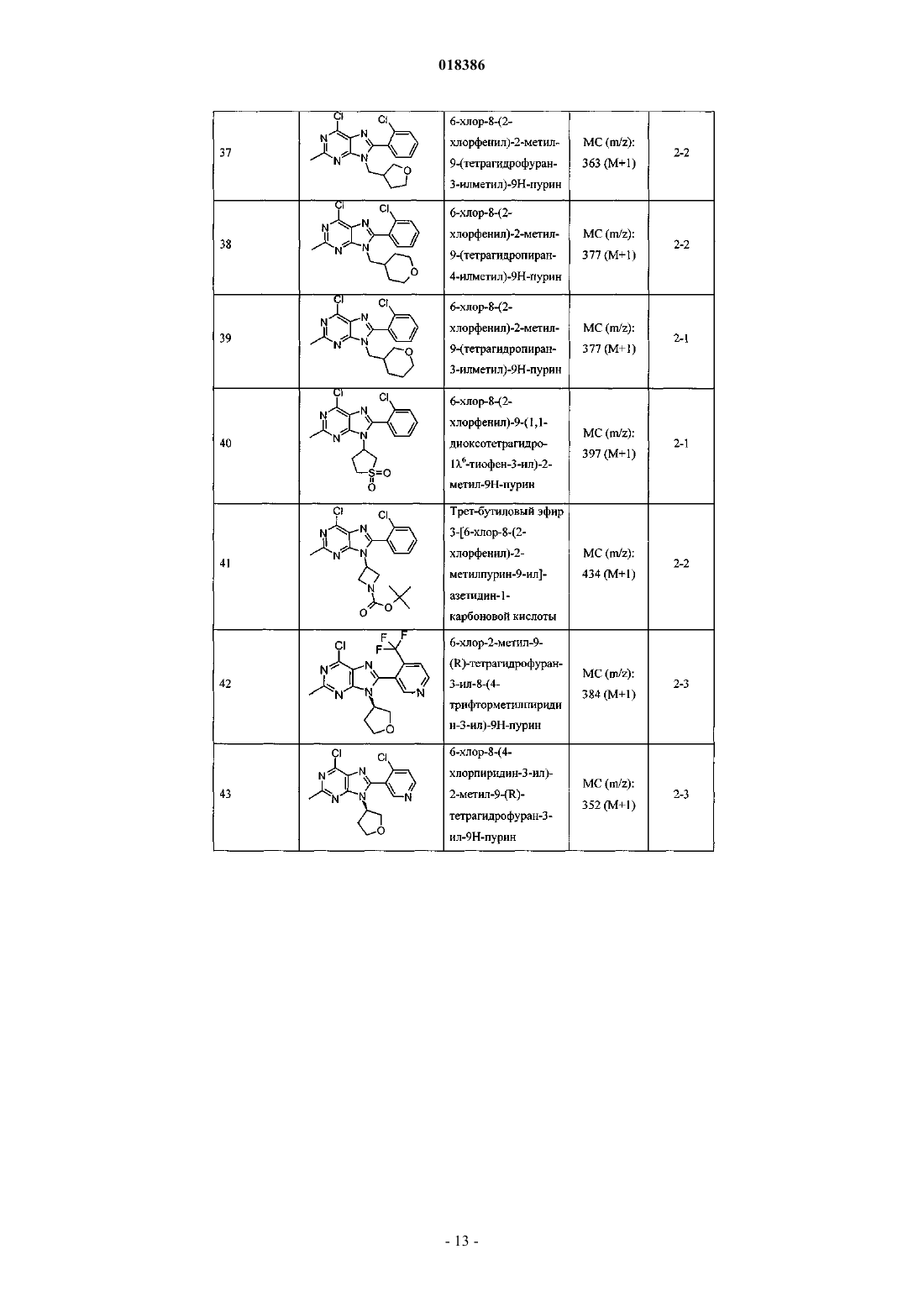
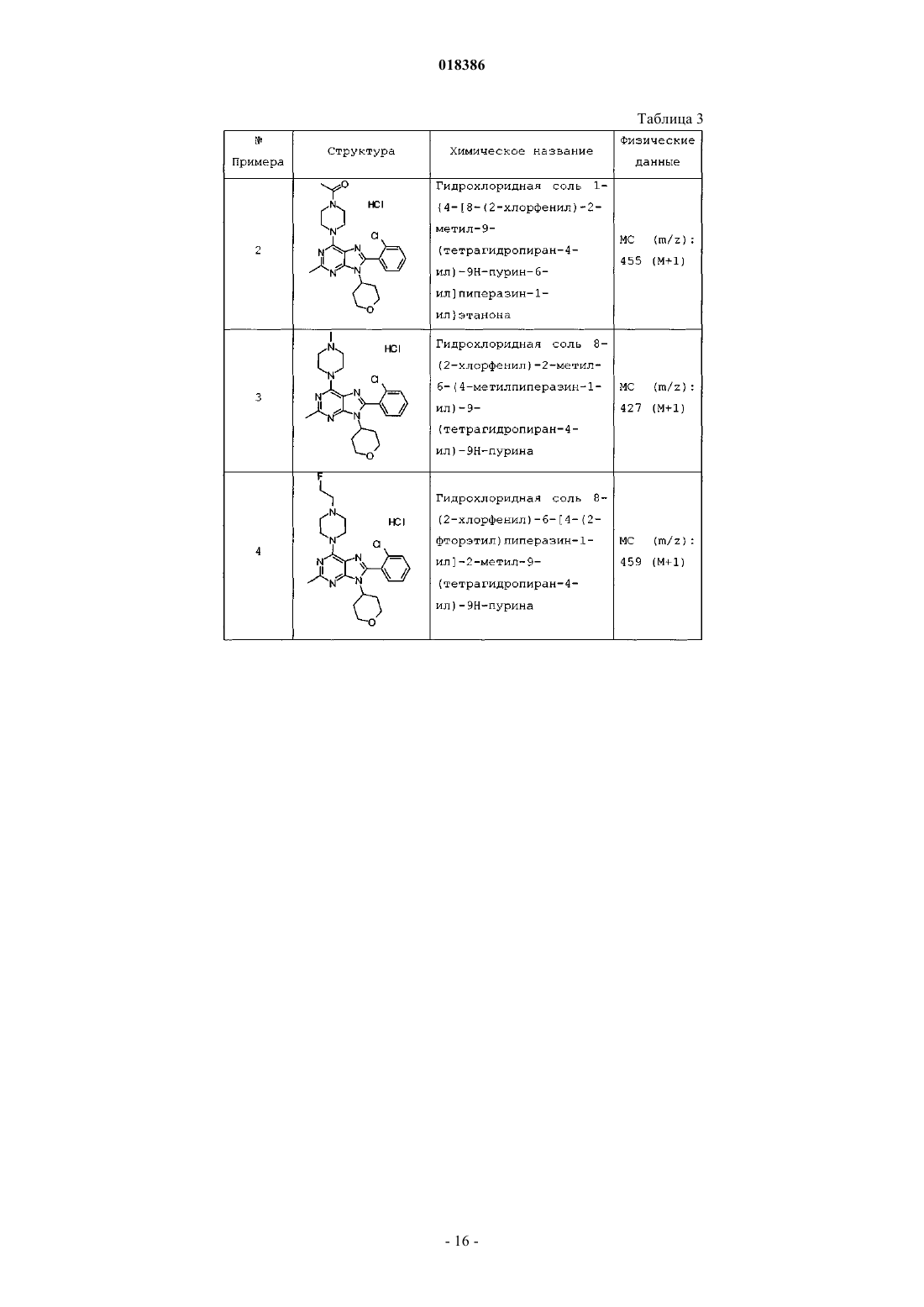
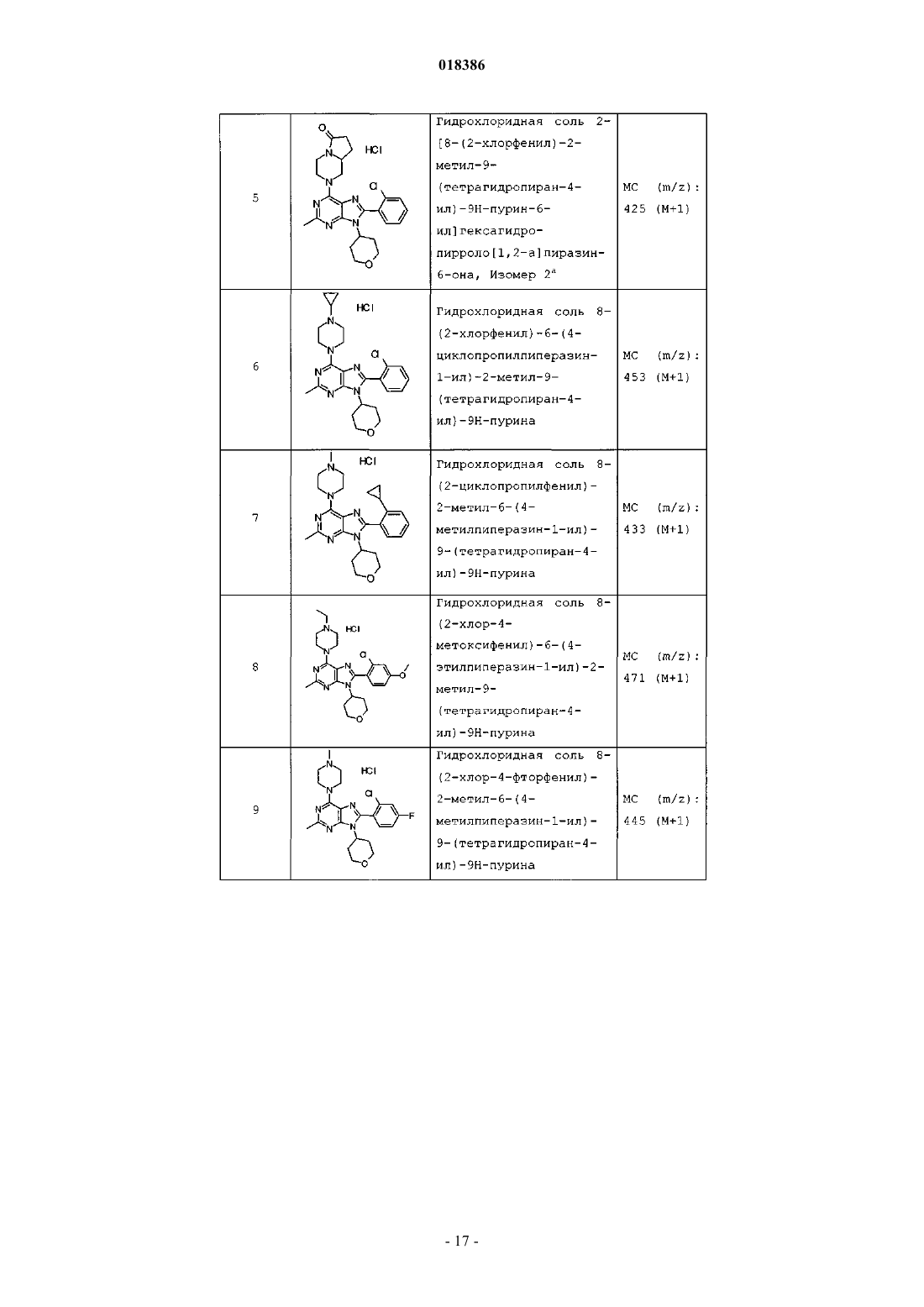
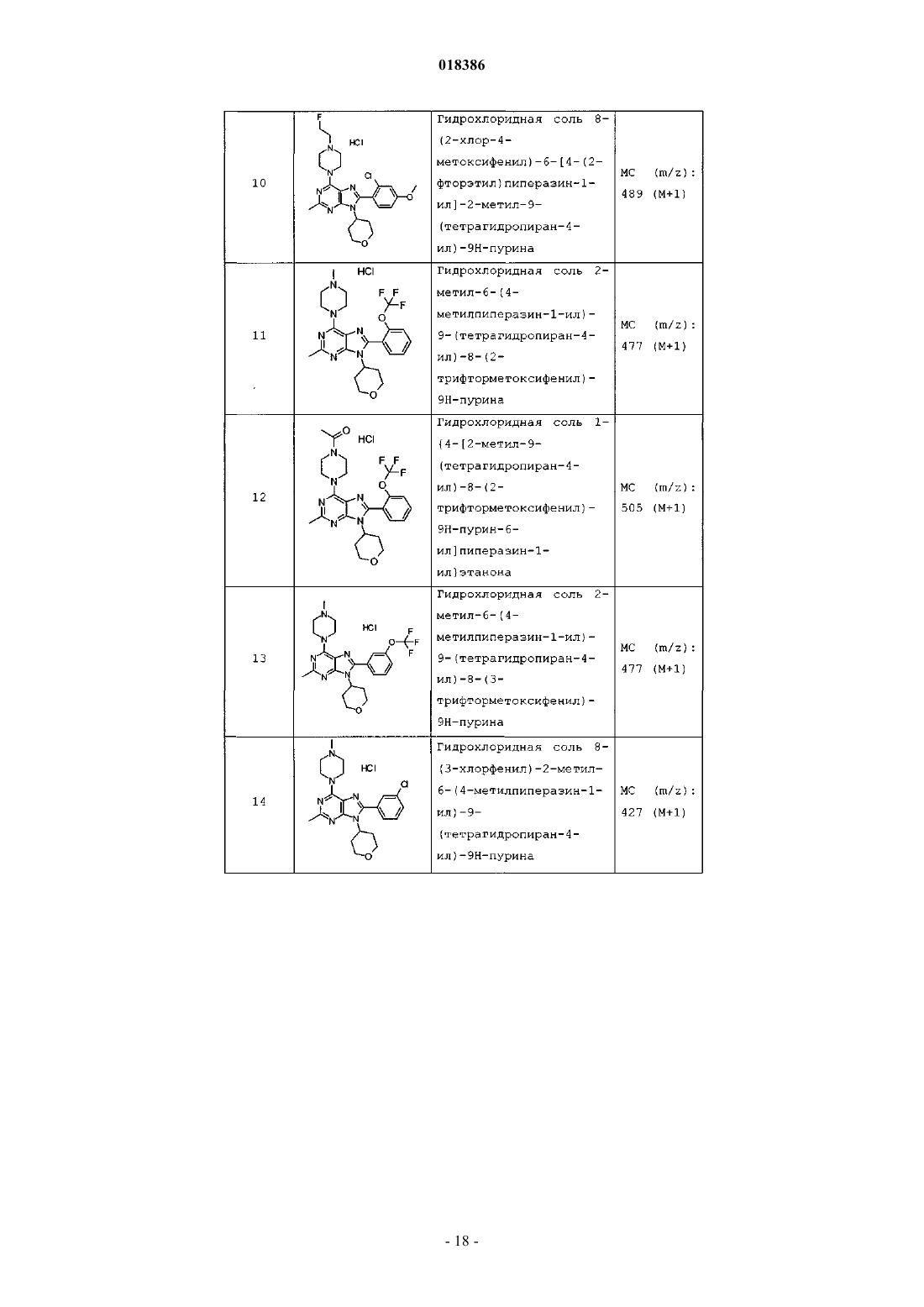
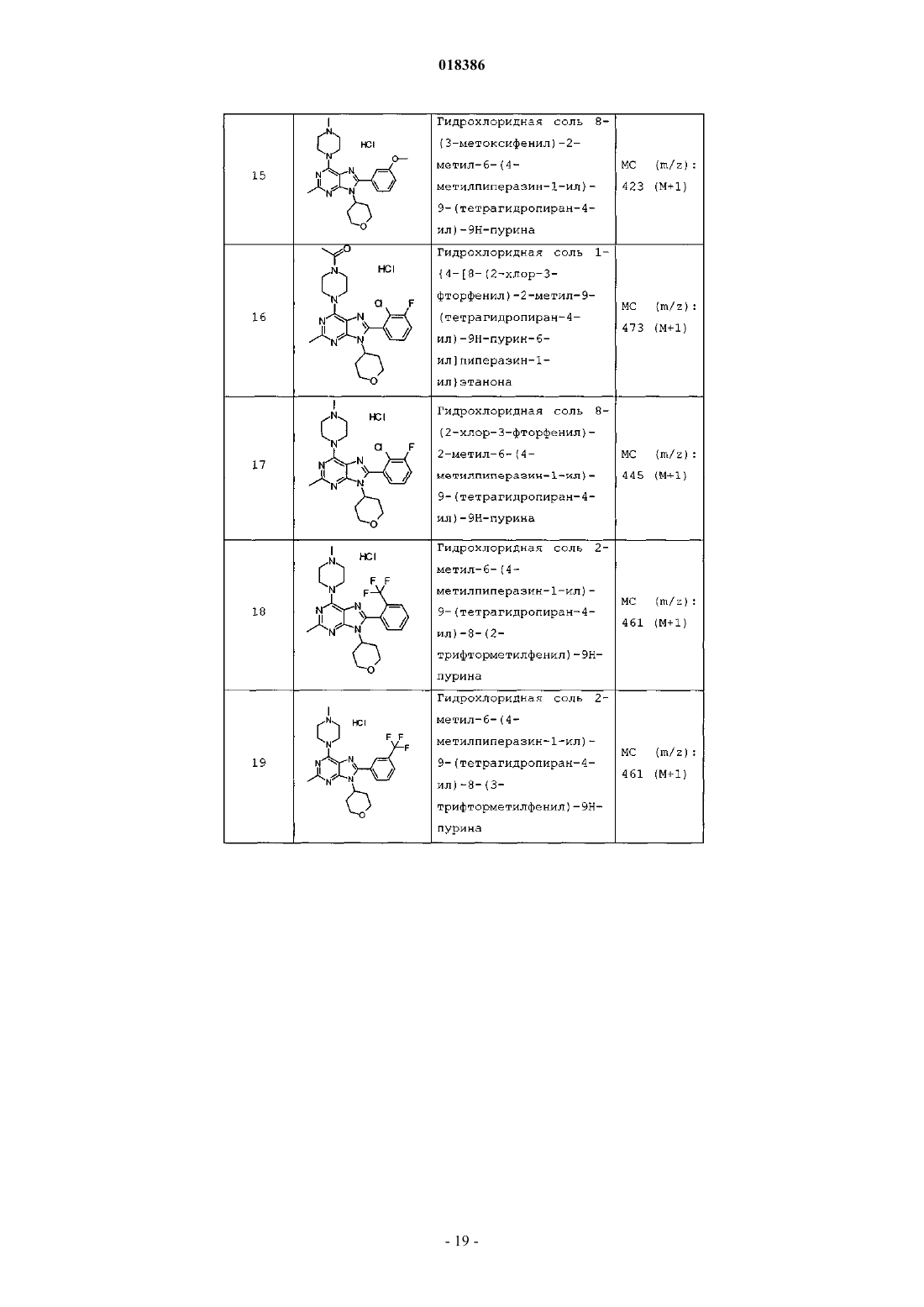
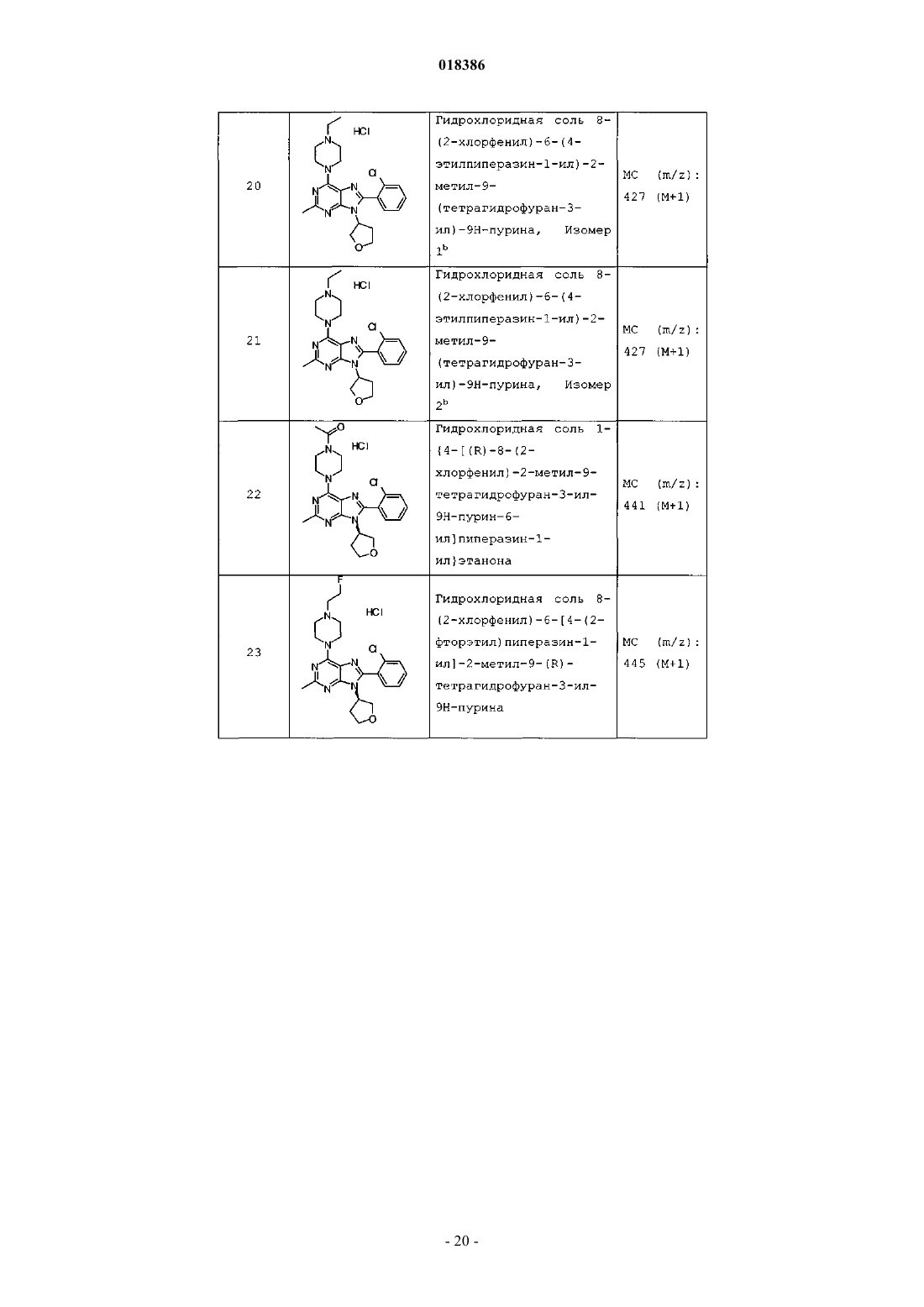
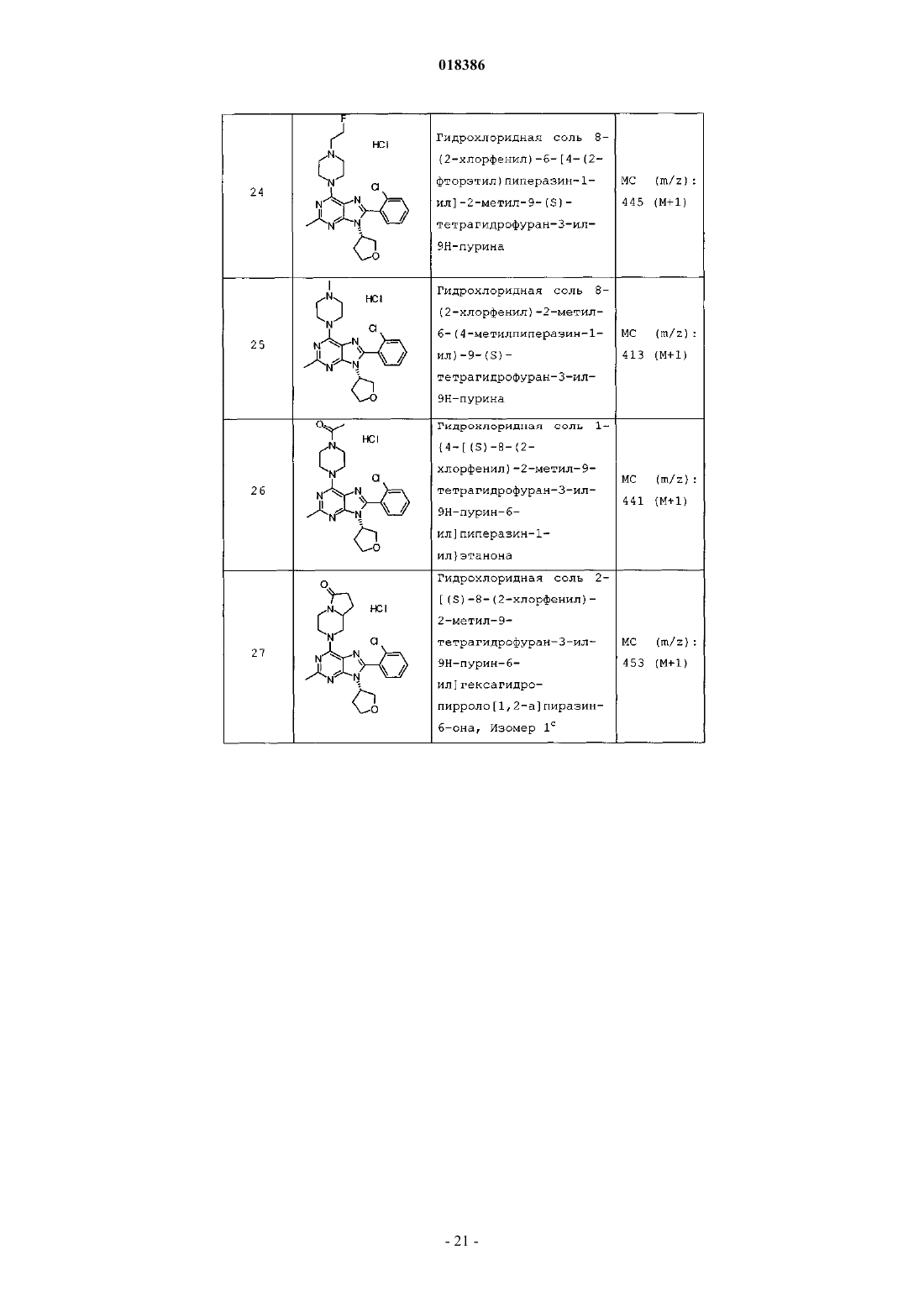
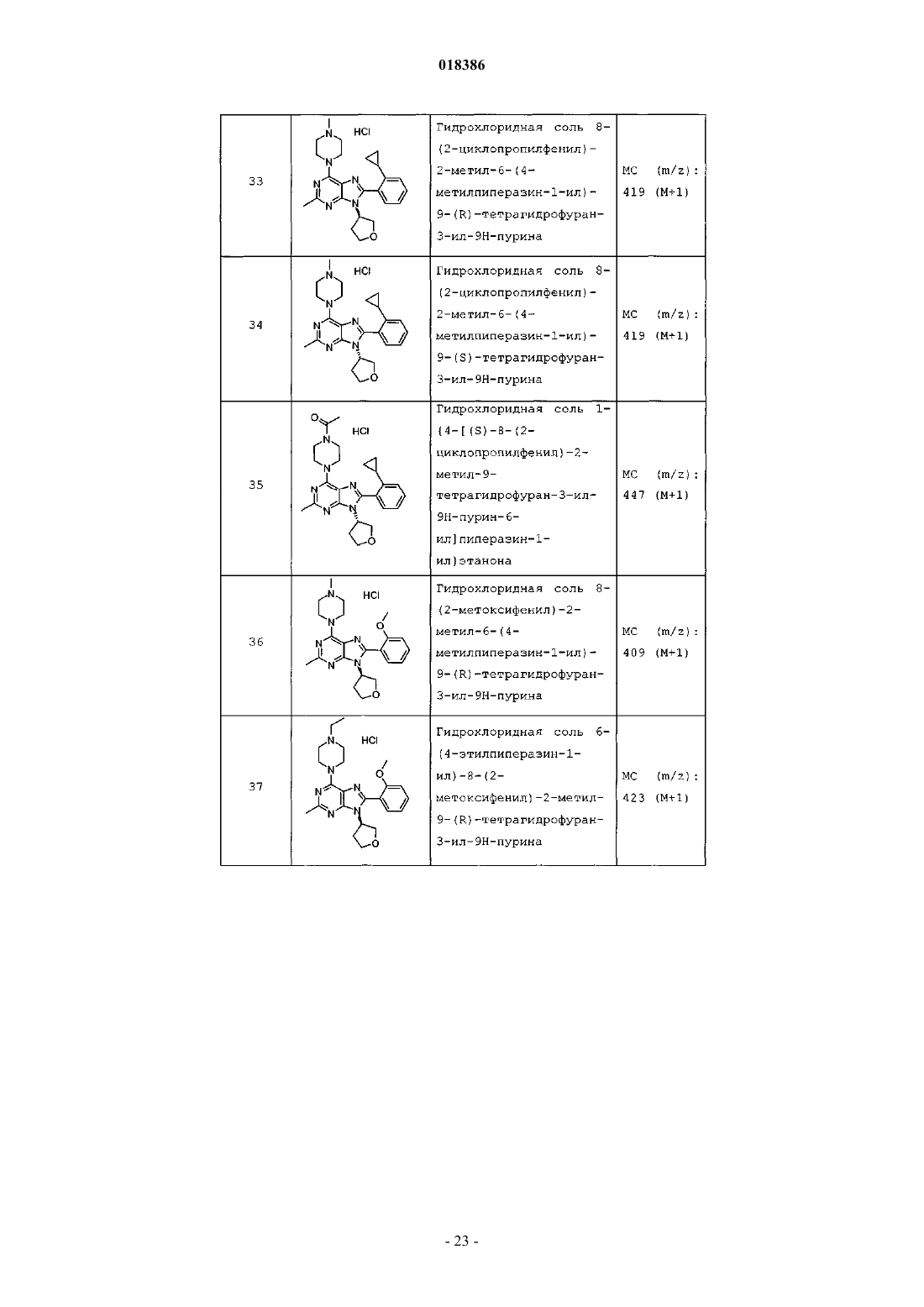
12. Соединение по п.1, выбранное из
8-(2-хлорпиридин-3-ил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-9Н-пурина;
2-метил-6-(4-метилпиперазин-1-ил)-9-(тетрагидропиран-4-ил)-8-(2-трифторметилфенил)-9Н-пурина;
2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-8-(2-трифторметилфенил)-9Н-пурина;
2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-8-о-толил-9Н-пурина;
8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(S)-тетрагидрофуран-3-ил-9Н-пурина;
8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-9Н-пурина;
8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(тетрагидропиран-4-ил)-9Н-пурина и
8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метил-9-(тетрагидропиран-4-илметил)-9Н-пурина или
их фармацевтически приемлемой соли.
13. Соединение по п.1, представляющее собой 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(тетрагидропиран-4-ил)-9Н-пурин или его фармацевтически приемлемую соль.
14. Фармацевтическая композиция, содержащая соединение по любому из пп.1-13 или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель.
15. Применение соединения по любому из пп.1-13 или его фармацевтически приемлемой соли в терапии.
16. Применение соединения по любому из пп.1-13 или его фармацевтически приемлемой соли для лечения боли.
17. Применение по п.16 для лечения остеоартритной боли.
Текст
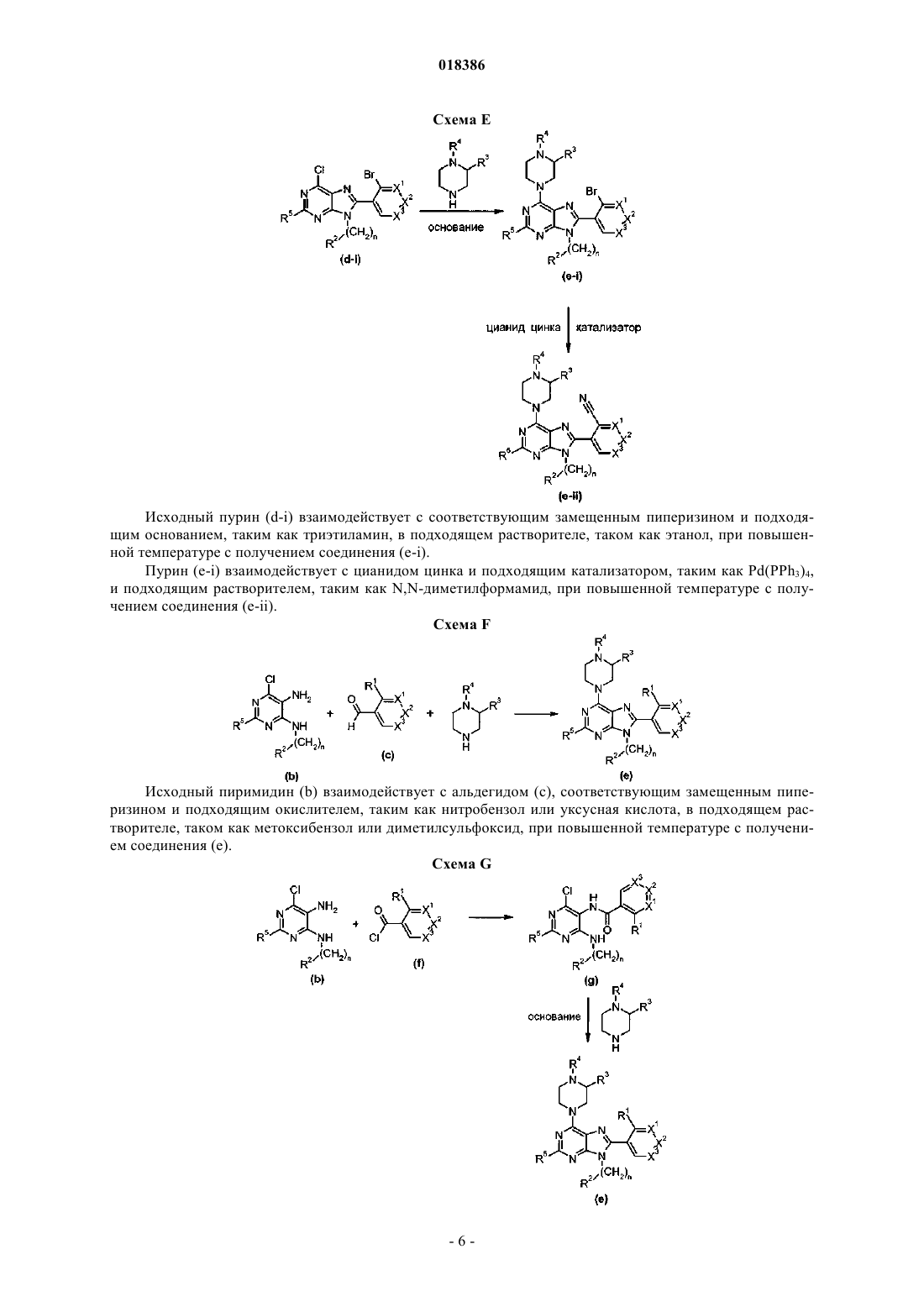
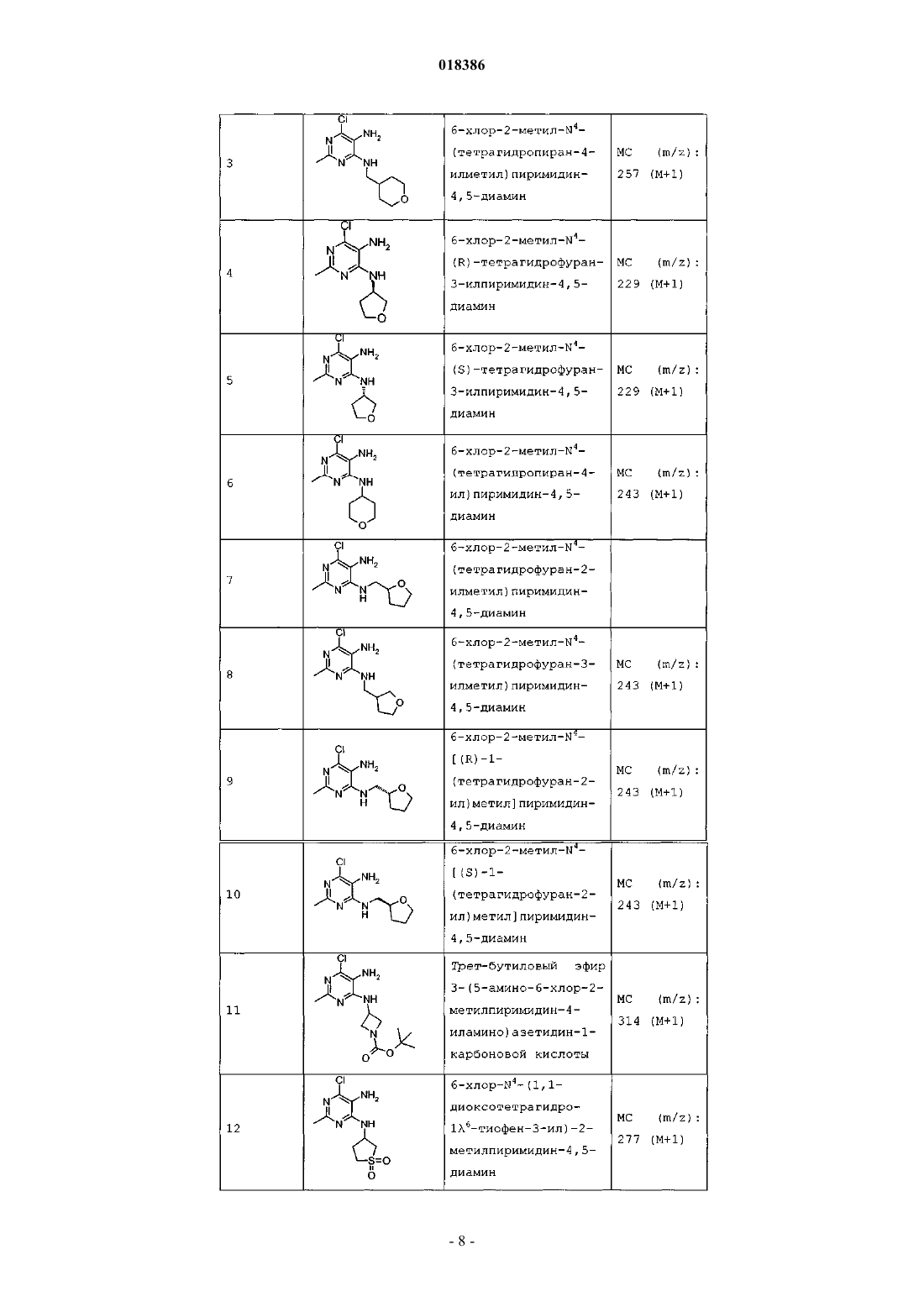
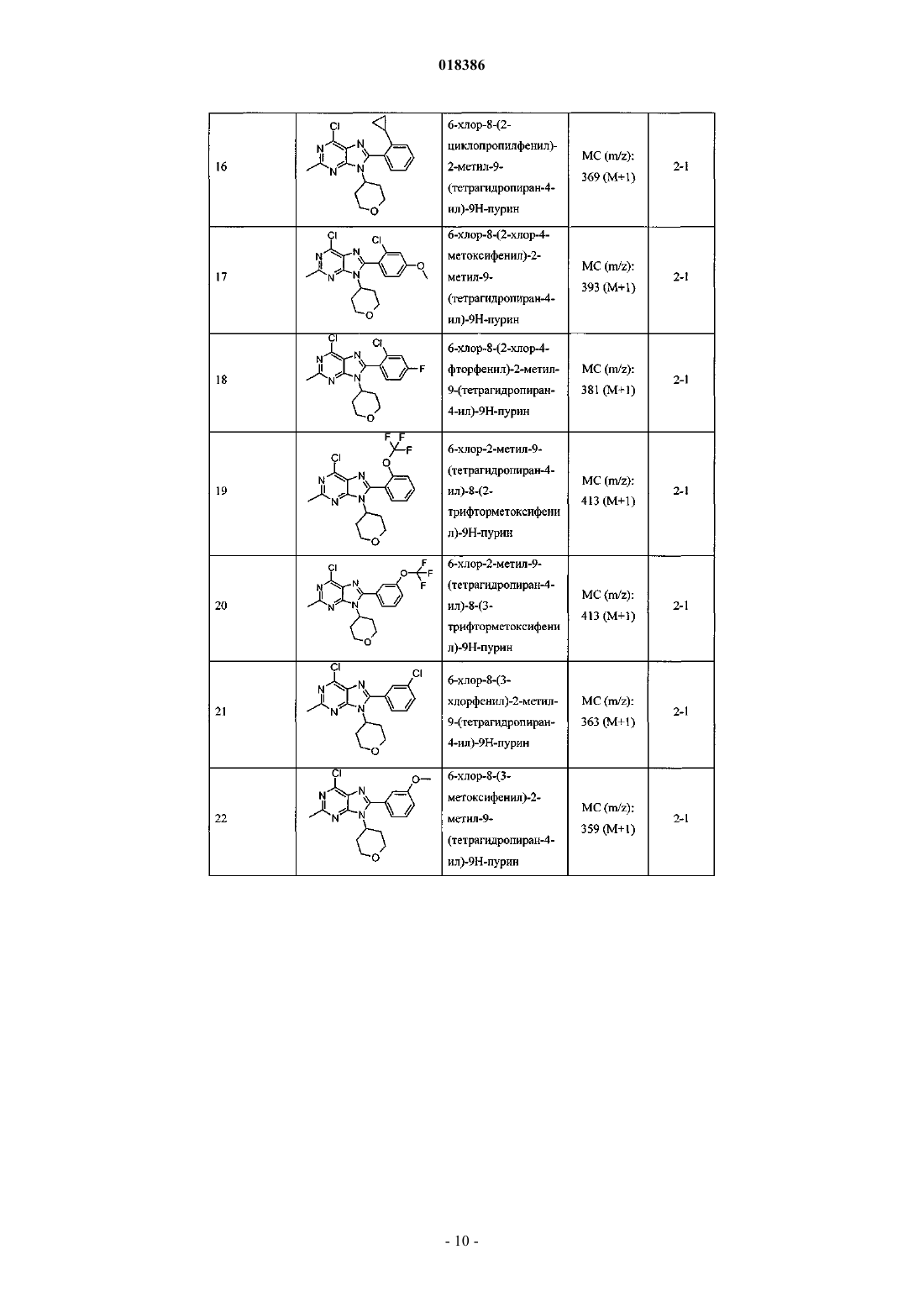
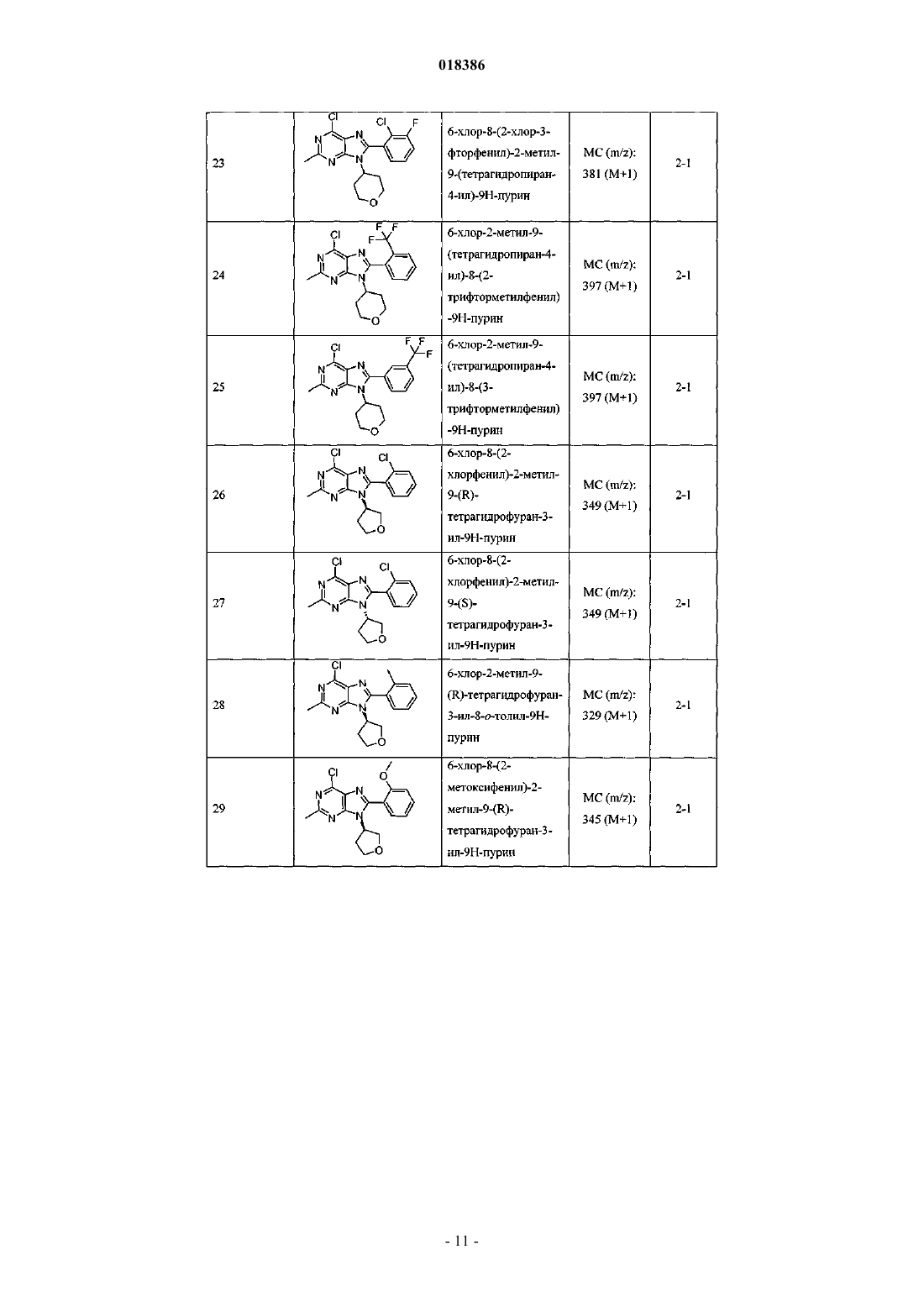
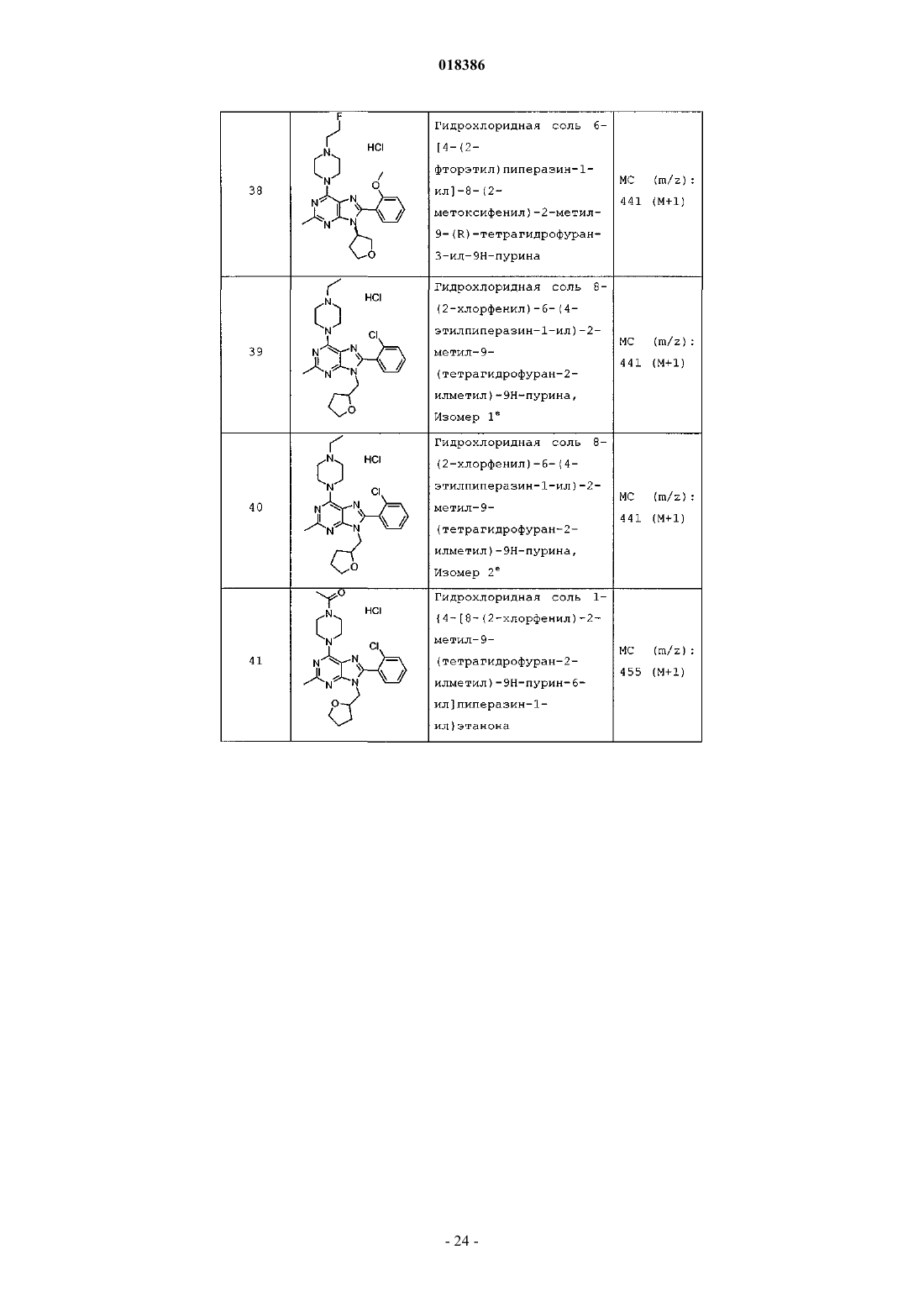
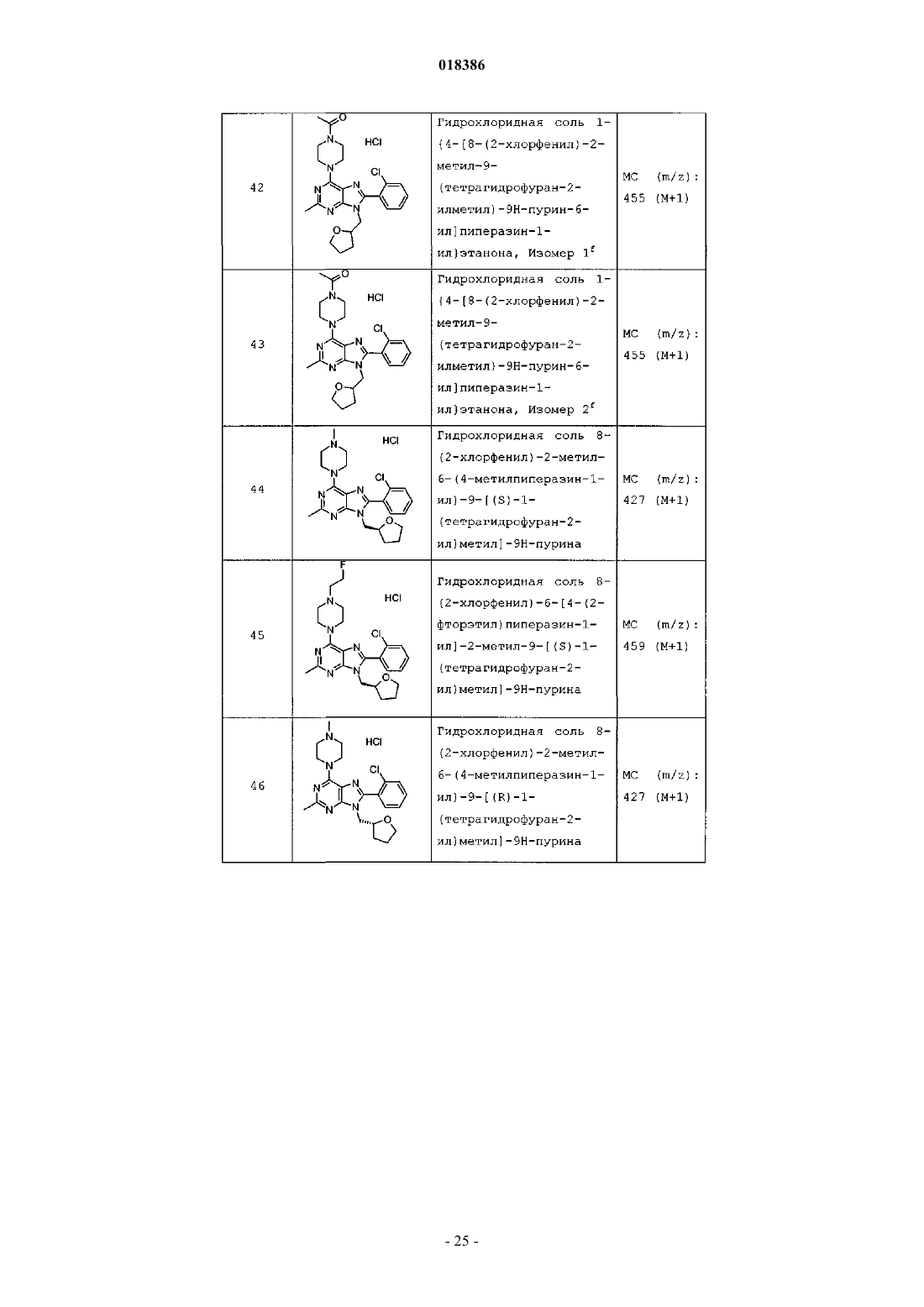
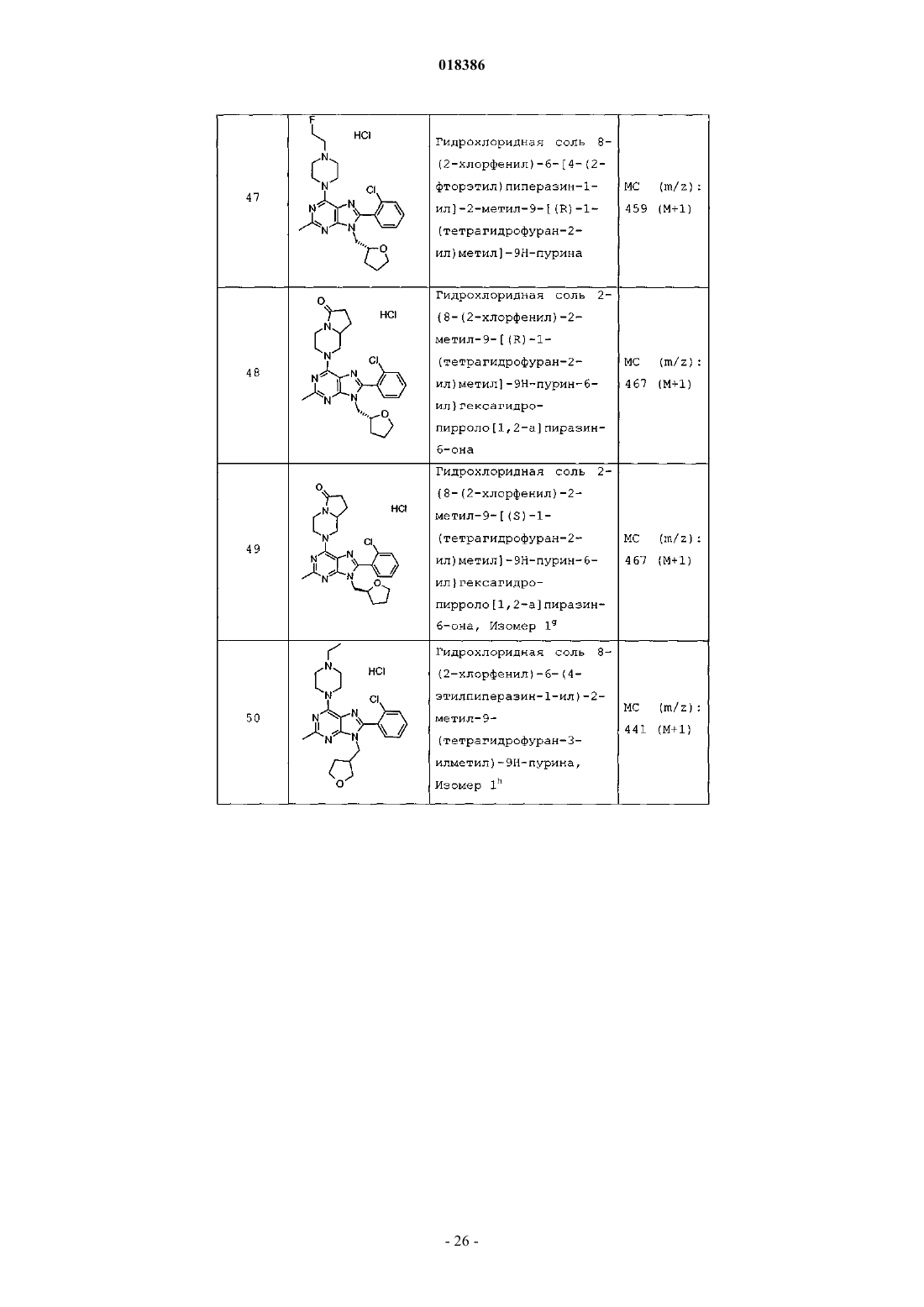
(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Эстлз Питер Чарльз, Гвидетти Росселла (GB), Холлиншед Шон Патрик, Тидуэлл Майкл Вэйд (US) и фармацевтические композиции для лечения боли. Вследствие побочных эффектов, связанных с имеющимися в настоящее время фармакологическими агентами для перорального введения, по-прежнему существует необходимость в разработке альтернативных видов терапии для лечения боли. Каннабиноидные рецепторы CB1 и СВ 2 принадлежат к классу рецепторов, сопряженных с G-белком(GPCR). Рецепторы CB1 экспрессированы как в центральной, так и в периферических областях нервной системы, тогда как рецепторы СВ 2 главным образом экспрессированы в периферических областях, в основном в иммунных клетках и тканях. Согласно недавно проведенному обзору, посвященному фармакологическому и терапевтическому потенциалу рецептора СВ 2 (Br. J. Prarmacol. (2008) 153, 319-334), СВ 2 указан в качестве терапевтической мишени для лечения боли, в частности воспалительной и невропатической боли. Агонисты СВ 2, в частности селективные к СВ 2 агонисты, обеспечивают мишень для лечения боли при ограниченных побочных эффектах, опосредуемых в центральной нервной системе.WO 2004/037823 относится к пуриновым соединениям и их применению в качестве лигандов каннабиноидного рецептора, в частности антагонистов рецептора CB1. В настоящем изобретении предложено соединение формулыX1 и X3 независимо выбраны из N, СН и CR6;X2 выбран из СН и CR6; при условии, что только один из X1, X2 и X3 может отличаться от СН;R6 выбран из F, Cl, CF3, OCH3 и OCF3; или его фармацевтически приемлемая соль. Было обнаружено, что соединения согласно настоящему изобретению являются агонистами рецептора СВ 2 in vitro. Предпочтительные соединения согласно настоящему изобретению проявляют большую активность по сравнению с существующими агонистами СВ 2. Более предпочтительные соединения согласно настоящему изобретению являются агонистами, селективными к СВ 2. Наиболее предпочтительные соединения согласно настоящему изобретению проявляют большую селективность к СВ 2 по сравнению с существующими агонистами СВ 2. В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый разбавитель или носитель. В настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии. В настоящем изобретении также предложено соединение формулы(I) или его фармацевтически приемлемая соль для применения для лечения боли, в частности остеоартритной боли. Согласно другому аспекту настоящего изобретения предложено применение соединения формулы(I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения боли,в частности остеоартритной боли. В настоящем изобретении предложен способ лечения боли, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом человеку или животному. В настоящем изобретении также предложен способ лечения остеоартритной боли, включающий введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли нуждающемуся в этом человеку или животному. В настоящем изобретении предложена фармацевтическая композиция для применения в терапии,-1 018386 содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль. В настоящем изобретении предложена фармацевтическая композиция для применения при боли, в частности остеоартритной боли, содержащая соединение согласно настоящему изобретению или его фармацевтически приемлемую соль. Предпочтительно соединения согласно настоящему изобретению применяют для лечения боли, в частности воспалительной боли, более конкретно боли в суставах, наиболее конкретно остеоартритной боли. Предпочтительные примеры соединений согласно настоящему изобретению представляют собой соединения формулы где R1, R2, R4, X1, X2 и X3 определены в настоящем описании,или их фармацевтически приемлемые соли. Предпочтительные примеры соединений согласно настоящему изобретению представляют собой соединения формулы где R1, R2 и R4 определены в настоящем описании,или их фармацевтически приемлемые соли. Конкретные классы соединений формул (I), (II) или (III) являются предпочтительными. Следующая пронумерованная выборка описывает указанные предпочтительные классы: 1) R1 представляет собой Cl, C1-С 2-алкил, CF3, циклопропил или OCF3; 2) R1 представляет собой Cl, метил или этил; 3) R1 представляет собой Cl; 4) R2 представляет собой тетрагидрофуранил или тетрагидропиранил; 5) R2 представляет собой тетрагидрофуранил; 6) R2 представляет собой тетрагидропиранил; 7) R3 представляет собой Н; 8) R4 представляет собой C1-С 2-алкил, C1-C2-фторалкил или циклопропил; 9) R4 представляет собой метил, этил, 2-фторэтил или циклопропил; 10) R4 представляет собой метил или этил; 11) R5 представляет собой СН 3; 12) X1, X2 и X3 независимо выбраны из СН и CR6, где R6 выбран из Cl, CF3, OCH3 или OCF3; 13) X1, X2 и X3 представляют собой СН; 14) n равно 0; 15) R3 представляет собой Н, a R5 представляет собой СН 3; 16) R1 представляет собой Cl, метил или этил; R4 представляет собой метил, этил, 2-фторэтил или циклопропил; 17) R1 представляет собой Cl, метил или этил; R2 представляет собой тетрагидрофуранил или тетрагидропиранил; R4 представляет собой метил, этил, 2-фторэтил или циклопропил; 18) R1 представляет собой Cl; R2 представляет собой тетрагидрофуранил или тетрагидропиранил; R4 представляет собой метил, этил, 2-фторэтил или циклопропил; 19) R1 представляет собой Cl; R2 представляет собой тетрагидрофуранил или тетрагидропиранил; R4 представляет собой метил или этил; 20) R1 представляет собой Cl; R2 представляет собой тетрагидрофуранил или тетрагидропиранил; R4 представляет собой метил или этил; X1, X2 и X3 независимо выбраны из СН и CR6, где R6 выбран из Cl,CF3, OCH3 или OCF3. Фармацевтически приемлемые соли каждого из соединений согласно настоящему изобретению находятся в рамках настоящего патента. Предпочтительные соединения согласно настоящему изобретению включают 8-(2-хлорпиридин-3-ил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-9 Н-пурин; 2-метил-6-(4-метилпиперазин-1-ил)-9-(тетрагидропиран-4-ил)-8-(2-трифторметилфенил)-9 Н-пурин; 2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-8-(2-трифторметилфенил)-9 Нпурин; 2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-8-о-толил-9 Н-пурин; 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(S)-тетрагидрофуран-3-ил-9 Н-пурин; 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(R)-тетрагидрофуран-3-ил-9 Н-пурин; 8-(2-хлорфенил)-2-метил-6-(4-метилпиперазин-1-ил)-9-(тетрагидропиран-4-ил)-9 Н-пурин и 8-(2-хлорфенил)-6-(4-этилпиперазин-1-ил)-2-метил-9-(тетрагидропиран-4-илметил)-9 Н-пурин или их фармацевтически приемлемые соли. Следует понимать, что, если группа, используемая в настоящем описании, определена как "определенная в настоящем описании", указанная группа включает первое встречающееся и наиболее широкое определение, а также каждое из всех конкретных определений указанной группы. Используемые ранее и далее в описании настоящего изобретения термины, если не указано иное,имеют следующие значения: Используемый в настоящем описании термин C1-С 2-алкил относится к метилу или этилу. Используемый в настоящем описании термин C1-С 2-фторалкил относится к C1-С 2-алкильной группе, определенной в настоящем описании, где один или более водородов замещены на фтор, и включающей трифторметил, 2-фторэтил, 2,2-дифторэтил и 2,2,2-трифторэтил. Предпочтительная C1-С 2 фторалкильная группа представляет собой 2-фторэтил. Используемый в настоящем описании термин "фармацевтически приемлемая соль" относится к солям соединений согласно настоящему изобретению, которые являются, по существу, нетоксичными для живых организмов. Указанные соли и общие способы их получения хорошо известны в данной области техники. См., например, P. Stahl et al., Handbook of Pharmaceutical Salts: Properties Selection and Use(VCHA/Wiley-VCH, 2002) и J. Pharm. Sci. 66, 2-19 (1977). Предпочтительные фармацевтически приемлемые соли представляют собой гидрохлорид и фосфат. Варианты реализации настоящего изобретения включают примеры, приведенные в настоящем описании, и хотя предложенный пример может представлять собой одну из хиральных или конформационных форм или их соли, дополнительные варианты реализации настоящего изобретения включают все другие стереоизомерные и конформационные формы описанных примеров, а также их фармацевтически приемлемые соли. Используемый в настоящем описании термин "агонисты, селективные к СВ 2" или "селективность к СВ 2" относится к соединениям, которые проявляют большую активность к СВ 2 по сравнению с активностью к СВ 1. Предпочтительные соединения согласно настоящему изобретению проявляют 100-кратную селективность к СВ 2. Более предпочтительные соединения согласно настоящему изобретению проявляют 500-кратную селективность к СВ 2. Наиболее предпочтительные соединения согласно настоящему изобретению проявляют 1000-кратную селективность к СВ 2. Соединения согласно настоящему изобретению предпочтительно входят в состав фармацевтических композиций, вводимых различными способами. Предпочтительно указанные композиции предназначены для перорального введения. Указанные фармацевтические композиции и способы их получения хорошо известны в данной области техники. См., например, Remington: The Science and Practice of Pharmacy (A. Gennaro et al., eds., 19th ed., Mack Publishing Co., 1995). Представленные далее схемы, методики и примеры предложены для лучшего пояснения осуществления настоящего изобретения на практике. Подходящие условия проведения реакции на стадиях, указанных в схемах, методах и примерах, хорошо известны в данной области техники, и подходящие модификации условий проведения реакции, включая замену растворителей и совместно применяемых реагентов, могут быть выбраны специалистом в данной области техники. Кроме того, для специалиста в данной области техники очевидно, что в некоторых случаях очередность введения фрагментов не имеет определяющего значения. Конкретная очередность проведения стадий, требуемая для получения соединений формулы (I), зависит от конкретного получаемого соединения,исходного соединения и относительной подвижности замещенных фрагментов, что вполне очевидно для специалиста-химика. Для специалиста в данной области техники очевидно, что не все заместители подходят для всех условий проведения реакции. Указанные соединения могут быть защищены или модифи-3 018386 цированы в подходящем месте синтеза при помощи способов, хорошо известных в данной области техники. Подходящие защитные группы включают группы, описанные в работе Т.В. Грина (T.W. Greene),"Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N.Y., 1991, далее называемойGreene. У Greene представлены подходящие условия для "защиты" и "снятия защиты" подходящих защитных групп, применяемых специалистами в данной области техники. Промежуточные соединения и конечные продукты согласно настоящему изобретению могут быть при необходимости дополнительно очищены при помощи общепринятых способов, таких как перекристаллизация или хроматография на твердофазной подложке, такой как силикагель или оксид алюминия. Названия соединений согласно настоящему изобретению даны с применением AutoNom 2000. Сокращения, используемые в настоящем описании, представлены далее."Солевой раствор" означает насыщенный водный раствор хлорида натрия;"БСА" означает бычий сывороточный альбумин;"SCX" означает колонку, сменный картридж или их эквивалент, содержащие сильную катионообменную смолу на оксиде кремния;"СКФХ" означает сверхкритическую флюидную хроматографию. Схема А Исходный пиримидин (а) взаимодействует с соответствующим замещенным амином и подходящим основанием, таким как диизопропилэтиламин или триэтиламин, в подходящем растворителе, таком как изопропанол, при повышенной температуре с получением соединения (b). Схема В Исходный бромид (c-i) взаимодействует с сильным основанием, таким как н-бутиллитий, при пониженной температуре и N,N-диметилформамидом в подходящем растворителе, таком как безводный диэтиловый эфир, с получением соединения (с). Исходный альдегид (c-ii) взаимодействует с производным бороновой кислоты R1, подходящим катализатором, таким как хлорид (1,1'-бис-(дифенилфосфино)ферроцен)палладия(II) или Pd(OAc)2, и подходящим основанием, таким как фторид цезия, в подходящем растворителе, таком как 1,4-диоксан или толуол, при повышенной температуре в условиях реакции сочетания по Сузуки с получением соединения (с). Схема С Исходный пиримидин (b) взаимодействует с альдегидом (с) (где Х 1-Х 4 независимо выбраны из СН и CR ) и подходящей кислотой, такой как п-толуолсульфоновая кислота или 15% хлорид железа на оксиде кремния, в подходящем растворителе, таком как 1,4-диоксан или толуол, при повышенной температу 6 ре. Реакционную смесь фильтруют и концентрируют перед взаимодействием с ДДХ в подходящем растворителе, таком как дихлорметан, при пониженной температуре с получением пурина (d). Исходный пиримидин (b) взаимодействует с альдегидом (с) (где один из Х 1-Х 4 представляет собойN) и подходящей кислотой, такой как п-толуолсульфоновая кислота, в подходящем растворителе, таком как толуол, при повышенной температуре. Реакционную смесь фильтруют и концентрируют перед реакцией с тионилхлоридом при повышенной температуре с получением пурина (d). Общая методика 2-1. Смесь пиримидина (b) (1,0 экв., лабораторный реактив (L.R., альдегида (с) (2,0 экв.) и 15% хлорида железа на оксиде кремния (200 мас.% по L.R.) в 1,4-диоксане нагревали до 100 С в течение 16 ч. Охлаждали и отфильтровывали оксид кремния через диатомитовую землю, концентрировали и фильтровали при пониженном давлении с получением остатка. Растворяли остаток в сухом дихлорметане и добавляли ДДХ (1,0 экв.) при 0 С. Оставляли нагреваться до комнатной температуры при перемешивании. После завершения реакции разбавляли реакционную смесь дихлорметаном, промывали 15% водным раствором гидроксида натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Очищали остаток при помощи флэш-хроматографии на силикагеле с получением пурина (d). Общая методика 2-2. Раствор пиримидина (b) (1,0 экв., L.R.), альдегида (с) (2 экв.), п-толуолсульфоновой кислоты(10 мас.% по L.R.) и молекулярные сита (200 мас.% по L.R.) в толуоле кипятили с обратным холодильником в течение 16 ч. Охлаждали и отфильтровывали молекулярные сита через диатомитовую землю, концентрировали и фильтровали при пониженном давлении с получением остатка. Растворяли остаток в сухом дихлорметане и добавляли ДДХ (1,0 экв.) при 0 С. Оставляли нагреваться до комнатной температуры и перемешивали. После завершения реакции разбавляли реакционную смесь дихлорметаном, промывали 1 н. раствором гидроксида натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Очищали остаток при помощи флэш-хроматографии на силикагеле с получением пурина (d). Общая методика 2-3. В реакционный сосуд помещали пиримидин (b) (1,0 экв., L.R.), альдегид (с) (1,1 экв.), толуол и моногидрат п-толуолсульфоновой кислоты (0,05 экв.). Перемешивали при 100 С в атмосфере азота в течение 1 ч. Охлаждали до комнатной температуры, фильтровали через диатомитовую землю и концентрировали при пониженном давлении. Далее, к неочищенной маслянистой жидкости (имину) при комнатной температуре в атмосфере азота медленно добавляли тионилхлорид (в массе/в растворителе). Кипятили при перемешивании с обратным холодильником в течение 30 мин. Охлаждали до комнатной температуры и концентрировали при пониженном давлении. Добавляли толуол и дважды удаляли при пониженном давлении. Растворяли в дихлорметане и медленно подщелачивали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очищали при помощи флэш-хроматографии на силикагеле с получением пурина (d). Схема D Исходный пурин (d) взаимодействует с соответствующим замещенным пиперизином и подходящим основанием, таким как триэтиламин, в подходящем растворителе, таком как этанол, при повышенной температуре с получением соединения (е). Исходный пурин (d-i) взаимодействует с соответствующим замещенным пиперизином и подходящим основанием, таким как триэтиламин, в подходящем растворителе, таком как этанол, при повышенной температуре с получением соединения (e-i). Пурин (e-i) взаимодействует с цианидом цинка и подходящим катализатором, таким как Pd(PPh3)4,и подходящим растворителем, таким как N,N-диметилформамид, при повышенной температуре с получением соединения (e-ii). Схема F Исходный пиримидин (b) взаимодействует с альдегидом (с), соответствующим замещенным пиперизином и подходящим окислителем, таким как нитробензол или уксусная кислота, в подходящем растворителе, таком как метоксибензол или диметилсульфоксид, при повышенной температуре с получением соединения (е). Схема G Исходный пиримидин (b) в подходящем растворителе, таком как диметилацетамид, взаимодействует с соответствующим замещенным хлорангидридом (f) при пониженной температуре с получением соединения (g). Из пиримидина (g) в присутствии соответствующего замещенного пиперизина и подходящего основания, такого как диизопропилэтиламин, в подходящем растворителе, таком как изопропанол, при повышенной температуре и давлении получают соединение (е). Пример получения 1. 6-Хлор-2-метил-N4-(тетрагидропиран-4-ил)пиримидин-4,5-диамин Нагревали раствор 4,6-дихлор-2-метилпиримидин-5-иламина (0,008 моль, 1,5 г, 1,0 экв.), 4 аминотетрагидропирана (0,012 моль, 1,27 г, 1,5 экв.) и N,N-диизопропилэтиламина (0,0092 моль, 1,1 г,1,1 экв.) в 2-пропаноле (80 мл) при 150 С в герметичной трубке в течение 16 ч. Охлаждали реакционную смесь до комнатной температуры и удаляли 2-пропанол при пониженном давлении с получением остатка. Растворяли остаток в дихлорметане и промывали водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Очищали остаток на колонке с силикагелем, элюируя смесью дихлорметан:метанол 96:4, с получением указанного в заголовке соединения. МС (m/z): 243,41 (М+1). Примеры получения 2-12 в табл. 1 могут быть реализованы, по существу, в соответствии с описанием, представленным в примере получения 1, с применением соответствующего амина в соответствии со схемой А. Таблица 1 Реакционную ампулу вместимостью 60 мл заполняли 2-бромбензальдегидом (10,810 ммоль,1,264 мл), циклопропилбороновой кислотой (14,053 ммоль, 1,207 г), N-гидратом трехосновного фосфата калия (37,834 ммоль, 8,031 г), трициклогексилфосфином (1,081 ммоль, 303,139 мг), толуолом(283,654 ммоль, 30,000 мл) и водой (83,263 ммоль, 1,500 мл). Затем смесь полностью дегазировали. Затем добавляли Pd(OAc)2 (540,482 мкмоль, 121,343 мг) и смесь помещали в атмосферу азота и нагревали до 100 С. Через 2 ч охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл) и солевым раствором (50 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очищали при помощи хроматографии на силикагеле, элюируя 2050% смесями гексан:дихлорметан, с получением указанного в заголовке соединения. 1 Н ЯМР (400,31 МГц, CDCl3): 10,57 (с, 1 Н), 7,78 (дд, J=1,3, 7,9 Гц, 1 Н), 7,45 (тд, J=7,5, 1,3 Гц, 1 Н),7,28 (т, J=7,5 Гц, 1 Н), 7,09 (д, J=7,9 Гц, 1 Н), 2,63-2,56 (м, 1 Н), 1,07-1,02 (м, 2 Н), 0,77-0,73 (м, 2 Н). Пример получения 14. 2-Циклопропилпиридин-3-карбальдегид В реакционную ампулу вместимостью 40 мл помещали 3 мл 1,4-диоксана и мешалку. Дегазировали азотом в течение 5 мин. Затем в ампулу помещали 2-бромникотинальдегид (645,134 мкмоль, 120,000 мг),циклопропилбороновую кислоту (1,290 ммоль, 110,831 мг) и фторид цезия (1,935 ммоль, 293,995 мг). Затем ампулу снова дегазировали азотом. Затем добавляли хлорид(1,1'-бис(дифенилфосфино)ферроцен)палладия(II) (32,257 мкмоль, 26,342 мг) и реакционную смесь нагревали до 100 С в атмосфере азота. После завершения реакции смесь охлаждали до комнатной температуры,фильтровали через прокладку диатомитовой земли с применением этилацетата с получением указанного в заголовке соединения. ГХ-МС (m/z): 146 (М). Примеры получения 15-47 в табл. 2 могут быть реализованы с применением соответствующего замещенного пиримидина в соответствии со схемой C и с применением соответствующего общего способа от 2-1 до 2-3, что отмечено в табл. 2. Таблица 2 В реакционный сосуд помещали R-6-хлор-2-метил-N4-(тетрагидрофуран-3-ил)пиримидин-4,5 диамин (2,186 ммоль, 500,000 мг), 2-бромникотинальдегид (3,280 ммоль, 610,046 мг), толуол(94,551 ммоль, 10,000 мл) и моногидрат ПТСК (109,323 мкмоль, 20,795 мг). Нагревали при 100 С в атмосфере азота в течение 1 ч. Охлаждали до комнатной температуры, фильтровали и концентрировали при пониженном давлении. Затем к неочищенной маслянистой жидкости (имину) при комнатной температуре в атмосфере азота медленно добавляли тионилхлорид (68,630 ммоль, 5,000 мл). Нагревали до 80 С в течение 30 мин. Концентрировали при пониженном давлении. Добавляли толуол (10 мл) и дважды удаляли при пониженном давлении. Растворяли остаток в дихлорметане и медленно подщелачивали насыщенным водным раствором бикарбоната натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очищали при помощи хроматографии на силикагеле, элюируя смесью гексан:ацетон, с получением указанного в заголовке соединения Стадия 1. В реакционный сосуд помещали N-трет-бутоксикарбонилпиперазин (8,590 ммоль, 1,600 г), карбонат калия (25,771 ммоль, 3,562 г), йодид натрия (кат.) (66,714 мкмоль, 10,000 мг), 1,4-диоксан(234,262 ммоль, 20,000 мл), 1-бром-2-фторэтан (9,449 ммоль, 704,029 мкл) и мешалку. Кипятили при перемешивании с обратным холодильником в течение ночи. После завершения реакции охлаждали до комнатной температуры и концентрировали при пониженном давлении. Разделяли в этилацетате и воде. Отделяли органический слой и сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением чистого трет-бутилового эфира 4-(2-фторэтил)пиперазин-1 карбоновой кислоты. ГХ-МС (m/z): 232 (М). Стадия 2. К перемешиваемому раствору трет-бутилового эфира 4-(2-фторэтил)пиперазин-1-карбоновой кислоты (8,610 ммоль, 2,000 г) в сухом дихлорметане (60 мл) при комнатной температуре в атмосфере азота добавляли 4 н. HCl в 1,4-диоксане (86,096 ммоль, 21,524 мл). Перемешивали в течение ночи в атмосфере азота. Концентрировали при пониженном давлении с получением указанного в заголовке соединения(0,0005 моль, 0,2 г), N-этилпиперазина (0,0006 моль, 0,069 г, 1,1 экв.) и триэтиламина (0,0006 моль,0,061 г, 1,1 экв.) в этаноле (5,0 мл) с обратным холодильником в течение 20 ч. В качестве альтернативы,нагревали реакционную смесь при помощи микроволнового излучения. После завершения реакции концентрировали реакционную смесь при пониженном давлении. Растворяли остаток в сухом дихлорметане и промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Очищали при помощи хроматографии на силикагеле, элюируя 90:10 смесью гексан:ацетон, с получением свободного основания. К свободному основанию (0,23 г, 0,500 ммоль) в диэтиловом эфире (5 мл) при 0 С добавляли HCl (2 М раствор в этаноле) и перемешивали в течение 2 ч при комнатной температуре. Фильтровали и промывали осадок диэтиловым эфиром. Сушили осадок в вакууме с получением указанного в заголовке соединения (0,2 г). МС (m/z): 441,28 (М+1). Альтернативно получали HCl соль в результате растворения свободного основания в ацетоне, 1:1 ацетонитрил вода или другом подходящем органическом растворителе, затем при перемешивании добавляли водный или эфирный раствор HCl. Затем лиофилизировали с получением гидрохлоридной соли. Примеры соединений 2-72 в табл. 3 могут быть получены по существу в соответствии с описанием примера 1 с применением соответствующего замещенного пурина и соответствующего замещенного пиперазина в соответствии со схемой D.
МПК / Метки
МПК: C07D 473/34, A61P 25/04, A61K 31/52
Метки: соединения, пуриновые
Код ссылки
<a href="https://eas.patents.su/30-18386-purinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Пуриновые соединения</a>
Пуриновые соединения и их применение в качестве лигандов каннабиноидных рецепторов

Номер патента: 8176
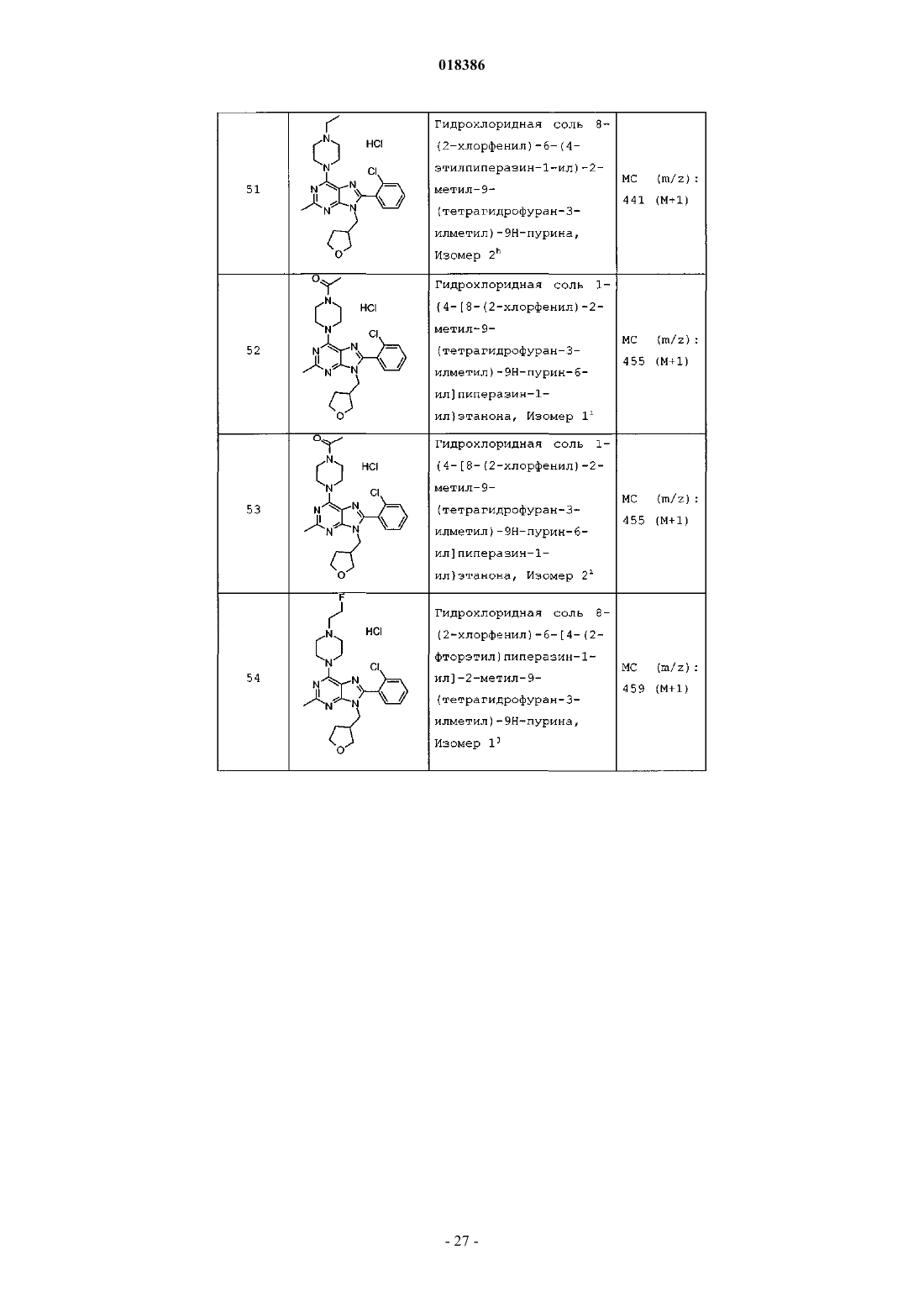
Опубликовано: 27.04.2007
Автор: Гриффит Дейвид Эндрю
МПК: A61K 31/52, A61P 3/04, C07D 239/48...
Метки: качестве, пуриновые, лигандов, применение, соединения, каннабиноидных, рецепторов
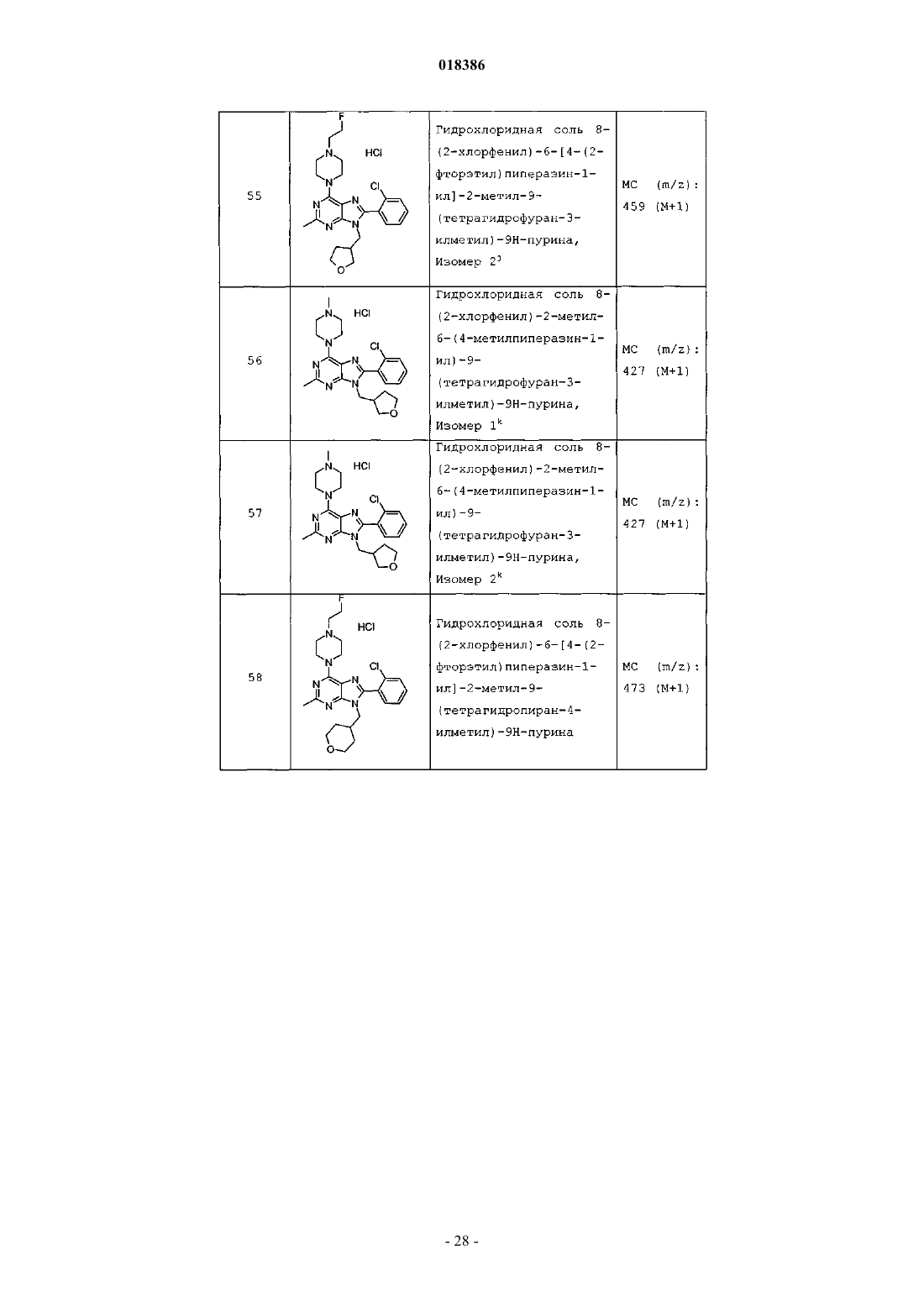
Формула / Реферат:
1. Соединение формулы (I) где А представляет собой возможно замещенный фенил или возможно замещенный гетероарил; В представляет собой возможно замещенный фенил или возможно замещенный гетероарил; R1 представляет собой водород, (С1-С4)алкил, галогенозамещенный (С1-С4)алкил или (С1-С4)алкокси; R4 представляет собой (1) группу, имеющую формулу (IA) или формулу (IB) где R4a представляет собой водород или (С1-С3)алкил; R4b и R4b', каждый...
Пуриновые производные

Номер патента: 4655
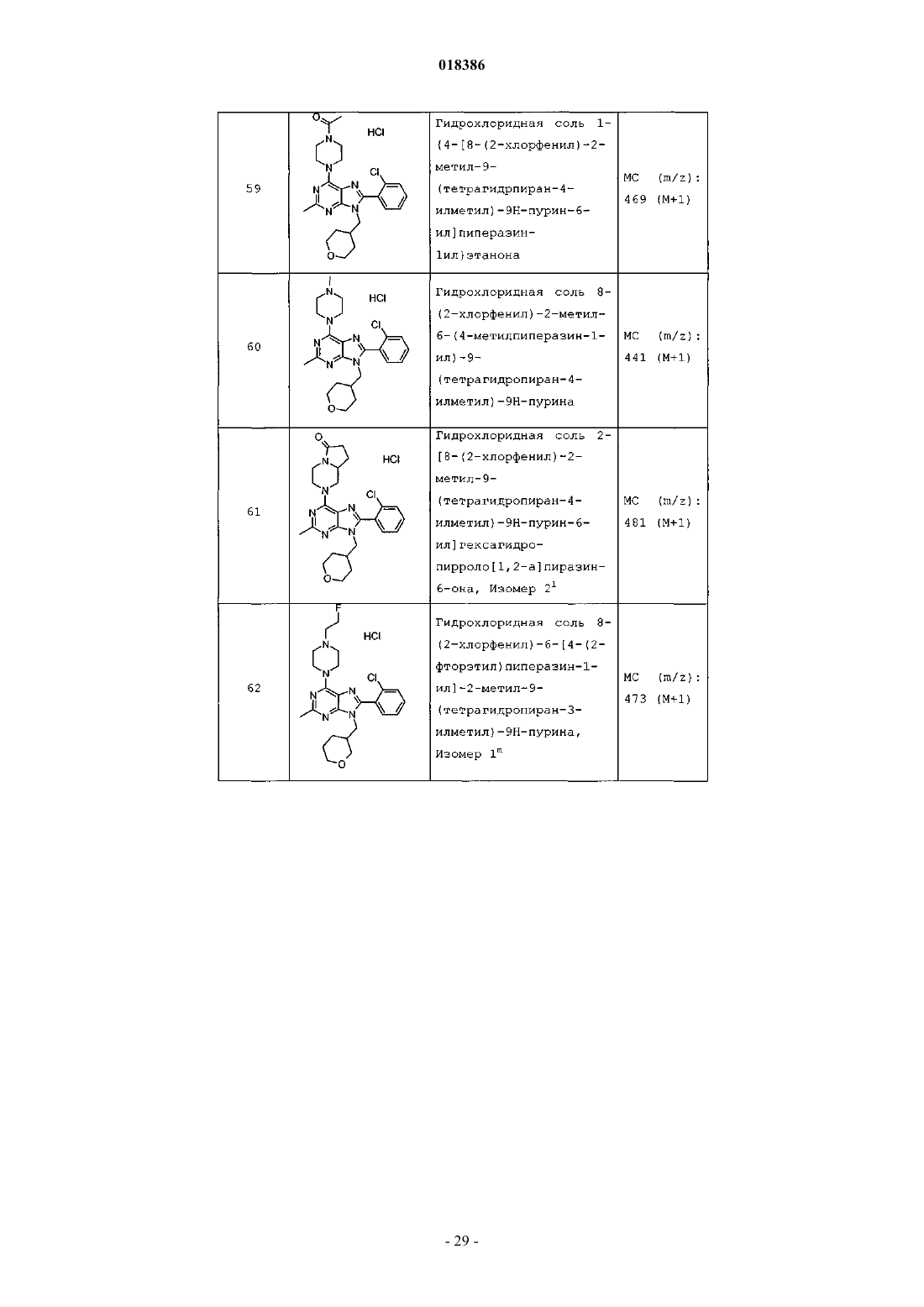
Опубликовано: 24.06.2004
Автор: Монахан Сандра Марина
МПК: C07H 19/167, A61K 31/70, A61P 11/00...
Метки: пуриновые, производные
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемые соль либо сольват, где R1 представляет собой водород или C1-C6алкил, возможно замещенный 1 или 2 заместителями, каждый из которых независимо выбран из фенила и нафтила, причем указанные фенил и нафтил возможно замещены C1-C6алкилом, C1-C6алкокси, галогено или циано; A представляет собой связь или C1-C3алкилен; R2 представляет собой (1) водород, C1-C6алкил, C3-C7циклоалкил, фенил или...
Пуриновые производные в качестве агонистов аденозиновых а1 рецепторов и способы их применения

Номер патента: 11826
Опубликовано: 30.06.2009
Авторы: Сабо Чаба, Джагтап Пракаш, Салзман Эндрю Л.
МПК: A61K 31/70
Метки: применения, рецепторов, производные, агонистов, пуриновые, качестве, способы, аденозиновых
Формула / Реферат:
1. Соединение, имеющее формулу или его фармацевтически приемлемая соль, где A представляет собой -CH2ONO2; В и С представляют собой -ОН; D представляет собой А и В представляют собой транс- в отношении друг к другу; В и С представляют собой цис- в отношении друг к другу; С и D представляют собой цис- или транс- в отношении друг к другу; R1 представляет собой тетрагидрофуранил, -C3-C8-моноциклический циклоалкил или -C8-C12-бициклический...
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Ким Сунгсуб, Ким Киоунг-Хее, Ким Сунг Хо, Ли Чанг-Сеок, Коо Ки Донг, Ким Хие Дзин, Бу Сеонг Чеол, Ким Мин-Дзунг, Хан Хее Оон, Квон Ох Хван, Лим Донгчул, Ким Геун Тае, Хонг Санг Йонг, Йео Донг-Дзун, Йим Хиеон Дзоо, Кох Дзонг Сунг, Хур Гвонг-Чеунг, Йеом Зи-Хо, Ким Дзи Янг
МПК: A61K 31/452, A61K 31/444, A61P 3/10...
Метки: способы, соединения-ингибиторы, композиции, указанные, соединения, содержащие, активного, также, ингредиента, получения, качестве, фармацевтические, дипептидилпептидазы-iv
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения

Номер патента: 12786
Опубликовано: 30.12.2009
Авторы: Ализи Мария Алессандра, Руссо Винченцо, Мангано Джорджина, Драгоне Патриция, Фурлотти Гвидо, Каццолла Никола, Поленцани Лоренцо, Колетта Изабелла
МПК: A61P 35/00, A61K 31/403, C07D 209/88...
Метки: композиция, 3-аминокарбазола, получения, фармацевтическая, содержащая, соединения, способ, указанные
Формула / Реферат:
1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы и их фармацевтически приемлемые соли. 2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным...
Предыдущий патент: Амидофеноксииндазолы в качестве ингибиторов c-met
Следующий патент: Ветроэлектростанция
Случайный патент: Дисперсия полимерной добавки для нефти, способ ее получения и применение