Ингибиторы p70 s6 киназы
Номер патента: 16445
Опубликовано: 30.05.2012
Авторы: Джозеф Саджан, Хольст Кристиан Л., Шеферд Тимоти Алан, Чжуан Цзяньпин, Дэлли Роберт Дин








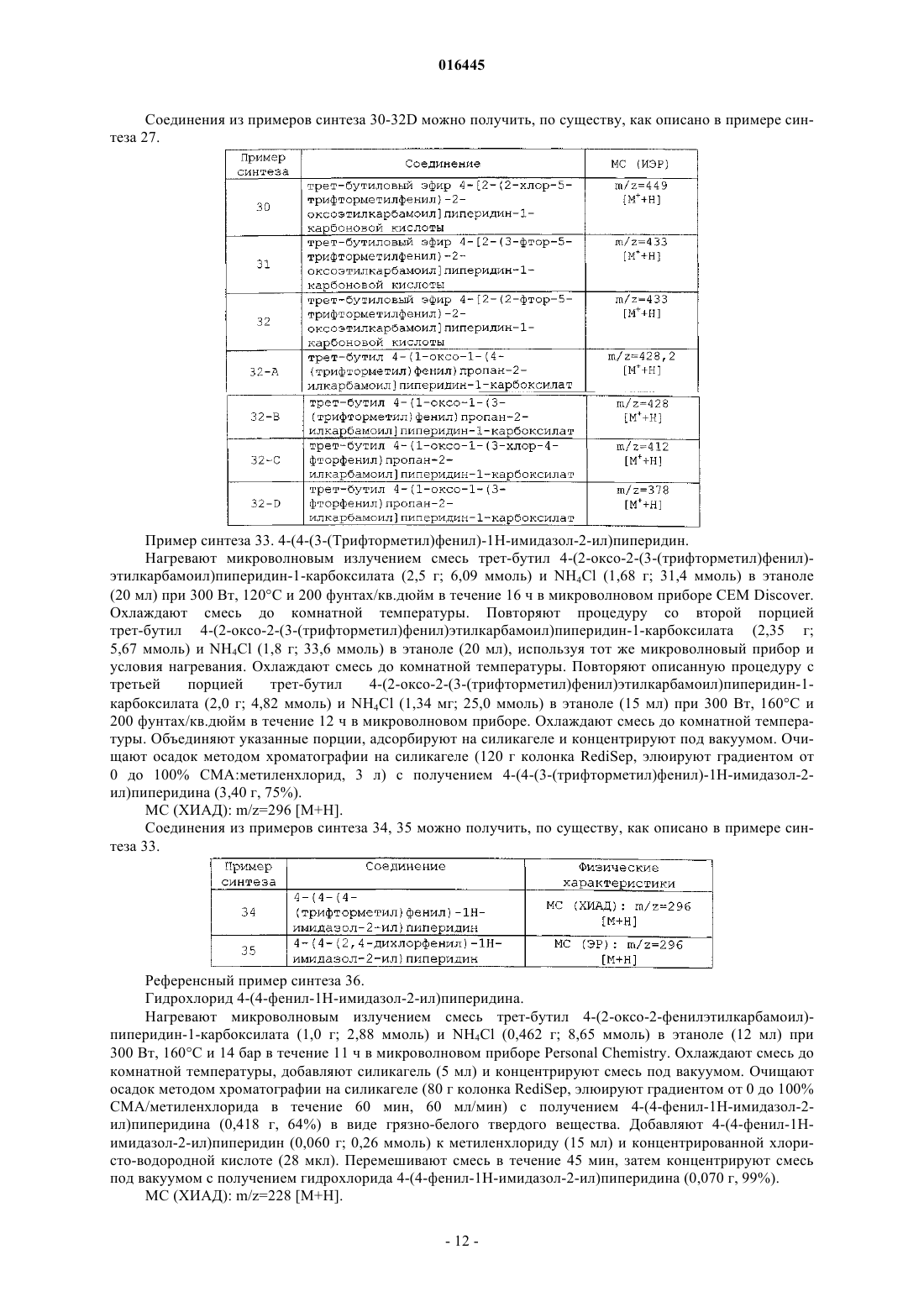














Формула / Реферат
1. Соединение формулы

где Y представляет собой CH;
Z1 и Z2 представляют собой независимо CR3 или N при условии, что Z1 и Z2, оба, не являются N;
R1 представляет собой H или CH3;
R2 представляет собой фенил, содержащий первый заместитель, выбранный из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, при этом указанный второй заместитель представляет собой галоген;
R3 представляет собой водород, галоген, C1-C4-алкил, C3-C6-циклоалкил или C2-C6-алкинил, при этом С2-C6-алкинил необязательно содержит в качестве заместителя гидроксил;
R4 и R5 представляют собой независимо водород или C1-C4-алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где Z2 представляет собой N.
3. Соединение по п.1, которое представляет собой 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-ил}-1Н-пиразоло[3,4-d]пиримидин, или его фармацевтически приемлемая соль.
4. Соединение по п.3, которое представляет собой п-толуолсульфонат 4-{4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1Н-имидазол-2-ил]пиперидин-1-ил}-1H-пиразоло[3,4-d]пиримидина.
5. Фармацевтическая композиция, включающая соединение по любому из пп.1-4 или его фармацевтически приемлемую соль, совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
6. Применение соединения по любому из пп.1-4 или его фармацевтически приемлемой соли для производства лекарственного средства для ингибирования ангиогенеза.
7. Применение соединения по любому из пп.1-4 или его фармацевтически приемлемой соли для производства лекарственного средства для лечения аденокарцином толстой кишки.
8. Способ ингибирования ангиогенеза у млекопитающего, включающий введение млекопитающему, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-4 или его фармацевтически приемлемой соли.
9. Способ лечения аденокарцином толстой кишки у млекопитающего, включающий введение млекопитающему, нуждающемуся в этом, эффективного количества соединения по любому из пп.1-4 или его фармацевтически приемлемой соли.
Текст
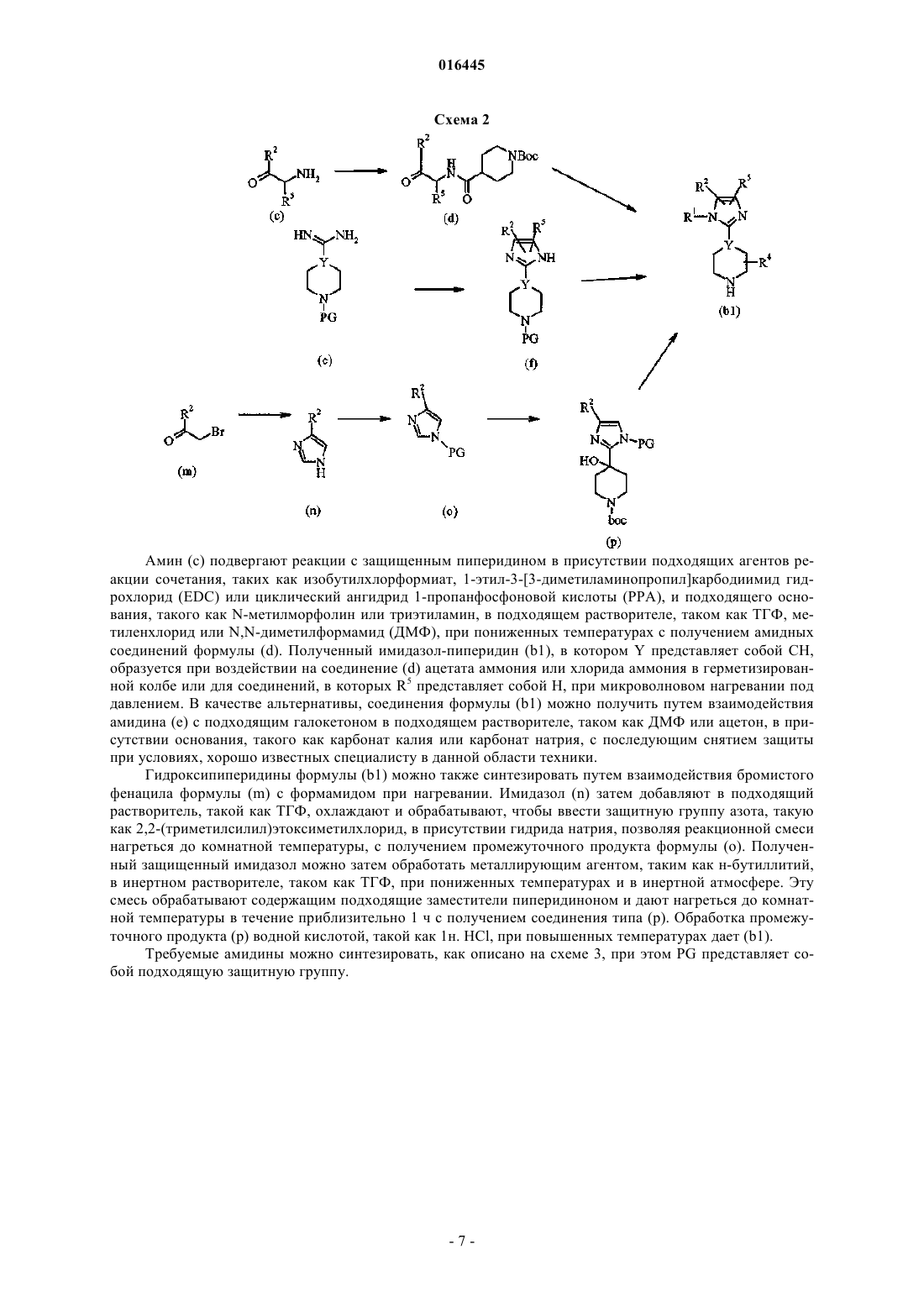
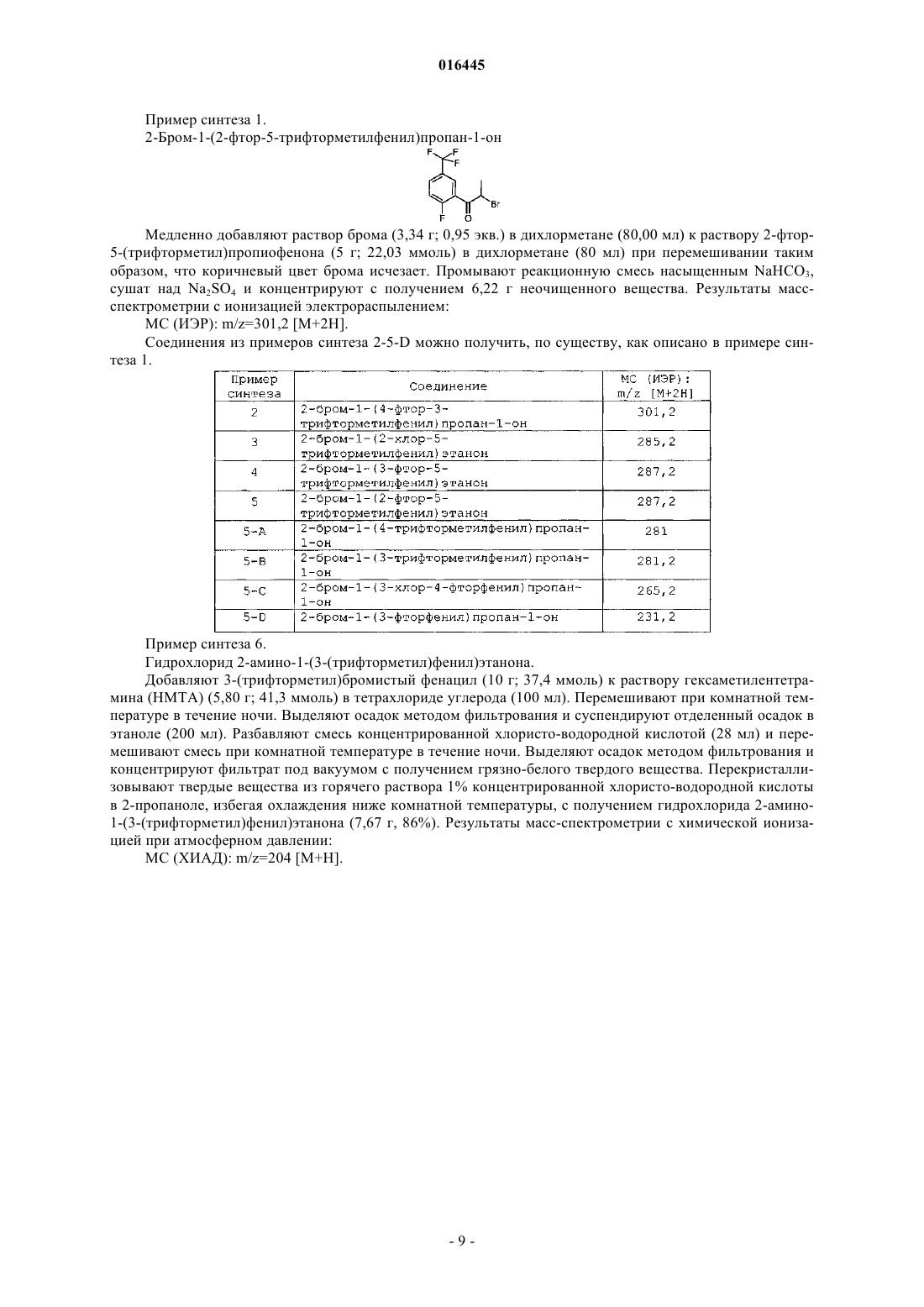
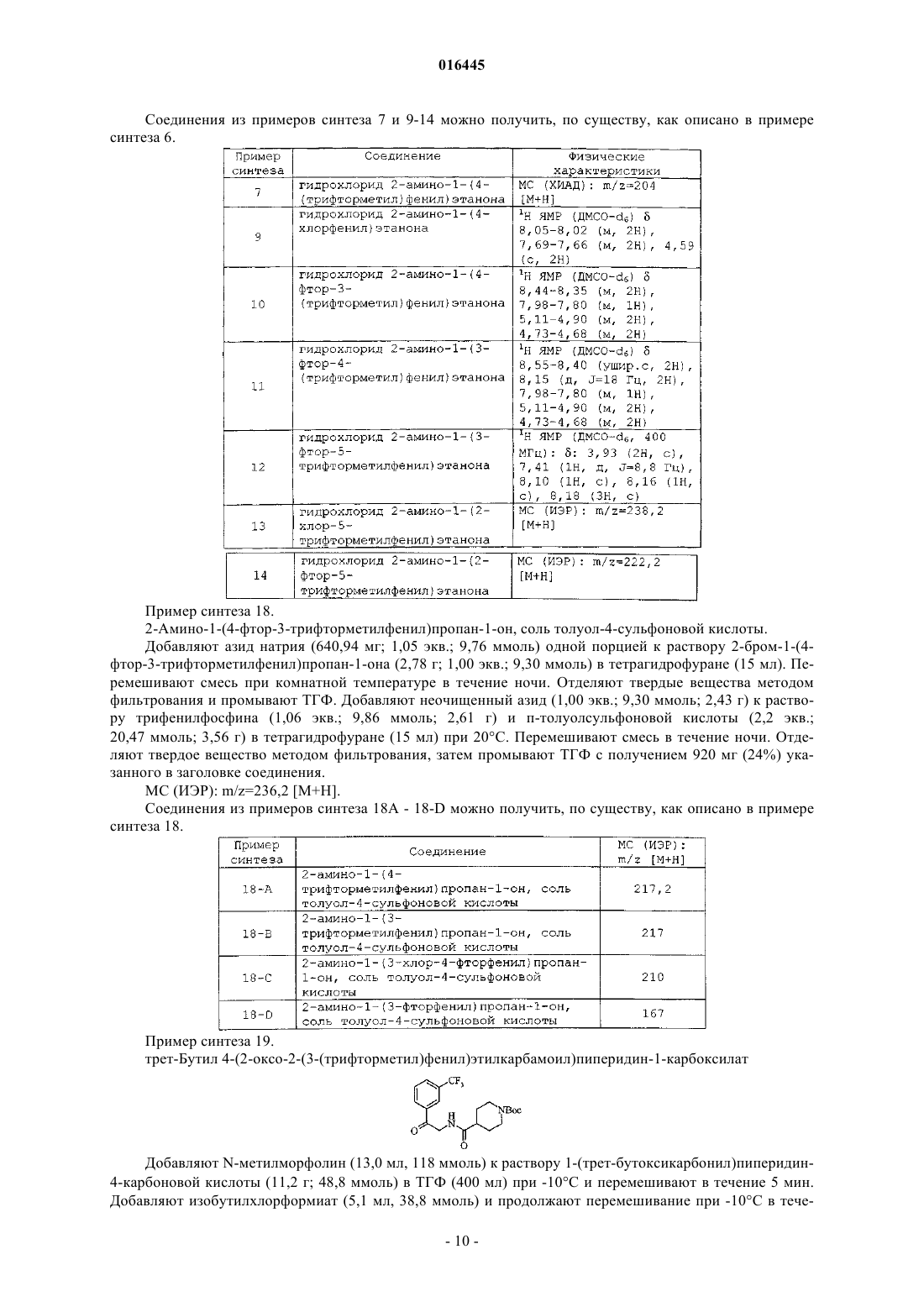
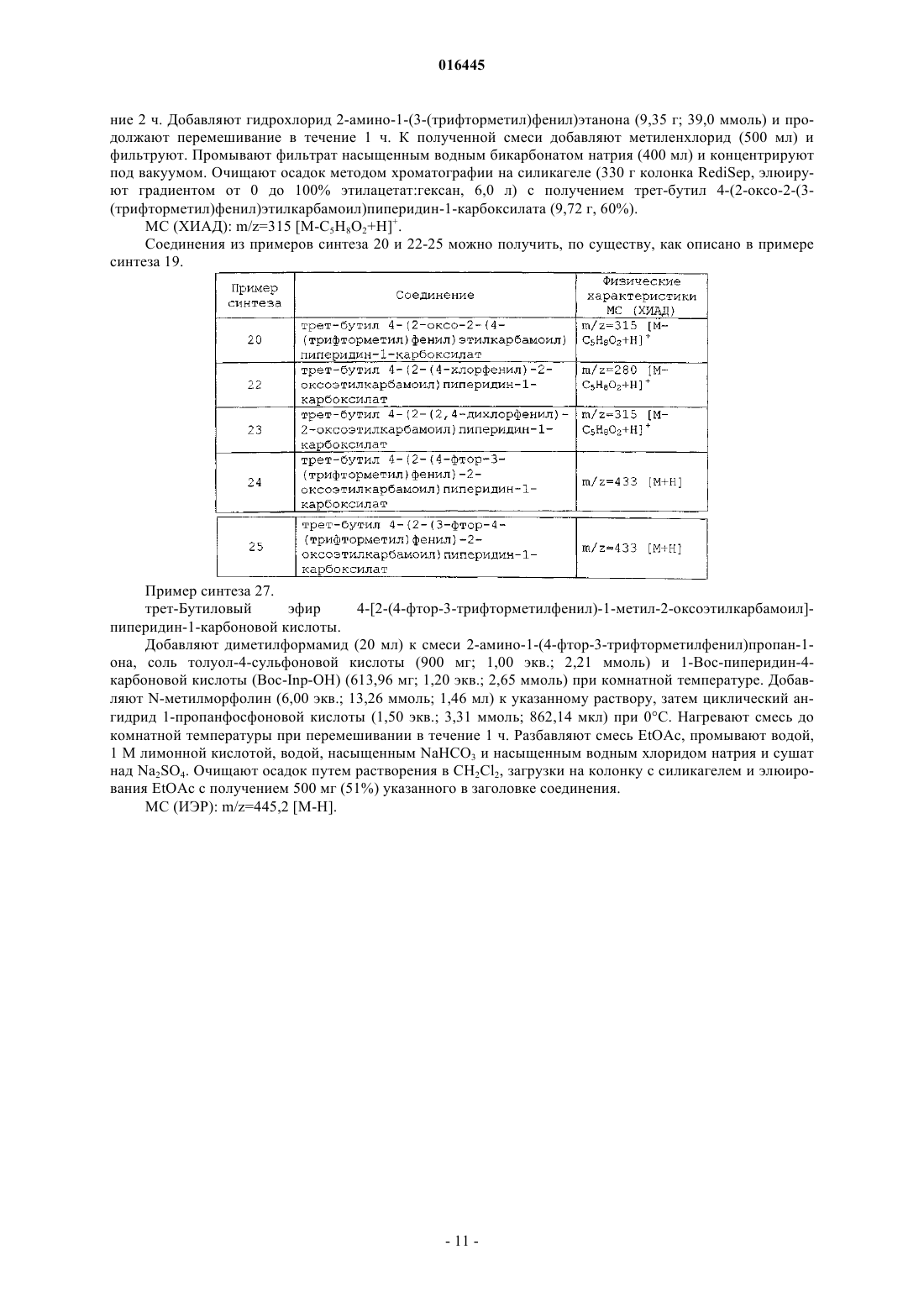
Настоящее изобретение представляет ингибиторы p70 S6 киназы формулы(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US)p70 S6 киназа представляет собой нижележащий эффектор сигнального пути фосфатидилинозит-3 киназы (PI3K)/протеинкиназы В (AKT)/мишени для рапамицина у млекопитающих (mTOR), и p70 S6 киназа обычно активирована во многих солидных опухолях человека. Активность р 70 S6 киназы регулирует биогенез рибосом, рост клеток и продвижение по клеточному циклу в ответ на митогенную стимуляцию. Поэтому подавление активности p70 S6 киназы блокирует биогенез рибосом, синтез отдельных белков, рост клеток и продвижение по клеточному циклу. Таким образом, p70 S6 киназа играет роль в пролиферации опухолевых клеток и защите клеток от апоптоза. Более того, ингибиторы р 70 S6 киназы описывают как пригодные для лечения инфекций, воспаления и образования опухолей, а также болезней и расстройств обмена веществ (WO 2005/117909, WO 2006/071819, WO 2006/046024, WO 2007/125321 иWO 2008/012635). Настоящее изобретение обеспечивает неожиданно эффективные соединения, которые ингибируют активность p70 S6 киназы. Кроме того, отдельные соединения согласно настоящему изобретению имеют высокую биодоступность. Краткое описание изобретения Настоящее изобретение обеспечивает соединения формулы IZ1 и Z2 представляют собой независимо CR3 или N при условии, что Z1 и Z2, оба, не являются N;R1 представляет собой H или CH3; первый заместитель, выбранный из галогена и трифторметила и необязательно дополнительно содержащий второй заместитель, при этом указанный второй заместитель представляет собой галоген;R4 и R5 представляют собой независимо водород или C1-C4-алкил,или фармацевтически приемлемую соль указанных соединений. Настоящее изобретение также обеспечивает способ ингибирования p70 S6 киназы у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или IA или его фармацевтически приемлемой соли. Настоящее изобретение также обеспечивает способ ингибирования ангиогенеза у млекопитающего,включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Дополнительно, настоящее изобретение также обеспечивает способ лечения аденокарцином толстой кишки у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Дополнительно, настоящее изобретение также обеспечивает способ лечения немелкоклеточного рака легкого у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также обеспечивает способ лечения мультиформной глиобластомы у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно обеспечивает способ лечения карциномы яичника у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно обеспечивает способ лечения лейкемии у млекопитающего,включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно обеспечивает способ лечения карциномы поджелудочной железы у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении,эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно обеспечивает способ лечения рака предстательной железы у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно обеспечивает способ лечения карциномы молочной железы у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли.-1 016445 Настоящее изобретение также обеспечивает способ лечения лимфангиолейомиоматоза у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Настоящее изобретение также обеспечивает фармацевтическую композицию, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, разбавителем или эксципиентом. Настоящее изобретение также обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для ингибирования p70 S6 киназы. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при ингибировании p70 S6 киназы у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для ингибирования p70S6 киназы, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение также обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для ингибирования ангиогенеза. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при ингибировании ангиогенеза у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для ингибирования ангиогенеза, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения аденокарциномы толстой кишки. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении аденокарциномы толстой кишки у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения аденокарциномы толстой кишки, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение также обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения немелкоклеточного рака легкого. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении немелкоклеточного рака легкого у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения немелкоклеточного рака легкого, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения мультиформной глиобластомы. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении мультиформной глиобластомы у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения мультиформной глиобластомы, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами,носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения карциномы яичника. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении карциномы яичника у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения карциномы яичника, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения лейкемии. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении лейкемии у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения лейкемии, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями.-2 016445 Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения карциномы поджелудочной железы. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении карциномы поджелудочной железы у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения карциномы поджелудочной железы, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения рака предстательной железы. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении рака предстательной железы у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения рака предстательной железы, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами,носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения карциномы молочной железы. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении карциномы молочной железы у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения карциномы молочной железы, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми зксципиентами,носителями или разбавителями. Настоящее изобретение дополнительно обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства для лечения лимфангиолейомиоматоза. Дополнительно, настоящее изобретение обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения при лечении лимфангиолейомиоматоза у млекопитающих. Более того, настоящее изобретение обеспечивает фармацевтическую композицию, пригодную для лечения лимфангиолейомиоматоза, включающую соединение формулы I или его фармацевтически приемлемую соль совместно с одним или более фармацевтически приемлемыми эксципиентами, носителями или разбавителями. Подробное описание изобретения Общие химические термины, используемые в формулах выше, имеют общепринятые значения. Например, термин "C1-C4-алкил" относится к прямой или разветвленной, одновалентной, насыщенной алифатической цепи, состоящей из 1-4 атомов углерода, например к метилу, этилу, пропилу, изопропилу,бутилу, изобутилу и трет-бутилу и т.п. Аналогичным образом, термин "C1-C3-алкил" включает метил,этил, изопропил и т.п. В данном описании термин "C1-C4-алкокси" относится к прямой или разветвленной алкильной цепи, состоящей из 1-4 атомов углерода, присоединенной к атому кислорода. ТипичныеC1-C4-алкоксигруппы включают метокси, этокси, пропокси, изопропокси, бутокси, трет-бутокси и т.п. В данном описании термин "галоген" относится к атому хлора, брома, йода или фтора, если не указано иначе. В данном описании термин "C3-C6-циклоалкил" означает полностью насыщенное кольцо, включающее атомы углерода и водорода, и содержит циклопропил и циклобутил. В данном описании термин "C2-C6-алкинил" описывает прямую или разветвленную алкинильную цепь, включающую от 2 до 6 атомов углерода и одну тройную связь, включая, но не ограничиваясь перечисленными, этинил, пропинил, бутинил, пентинил и т.п. Соединения согласно настоящему изобретению являются основаниями и соответствующим образом реагируют с любыми из многих органических и неорганических кислот с образованием фармацевтически приемлемых солей, и настоящее изобретение включает фармацевтически приемлемые соли соединений формулы I. Термин "фармацевтически приемлемая соль" в данном описании относится к солям соединений формулы I, которые, по существу, не токсичны для живых организмов. Такие соли включают фармацевтически приемлемые соли, перечисленные в Journal of Pharmaceutical Science, 66, 2-19 (1977), которые известны специалисту в данной области техники. Соли гидрохлорид, мезилат и тозилат (также известный как п-толуолсульфонат) являются предпочтительными солями. Соли гидрохлорид и тозилат являются наиболее предпочтительными. Некоторые соединения согласно настоящему изобретению содержат один или более хиральных центров и могут существовать во множестве стереоизомерных конфигураций. Вследствие присутствия этих хиральных центров соединения согласно настоящему изобретению встречаются в виде рацематов,смесей энантиомеров и в виде отдельных энантиомеров, а также диастереомеров и смесей диастереомеров. Все такие рацематы, энантиомеры и диастереомеры входят в объем настоящего изобретения. От-3 016445 дельные стереоизомеры и энантиомеры соединений формулы I может получить специалист в данной области техники, применяя хорошо известные методики и процедуры, такие как описанные у J. Jacques, etal., "Enantiomers, Racemates and Resolutions", John WileySons, Inc., 1981 и E.L. Eliel и S.H. Wilen,"Stereochemistry of Organic Compounds" (Wiley-Interscience 1994) и в Европейской заявке на патентEP-A-838448, опубликованной 29 апреля 1998 г. Примеры разделения включают методики перекристаллизации или хиральную хроматографию. Для специалиста в данной области техники также должно быть очевидно, что соединения формулыI, гле R1 представляет собой водород или где Z1 или Z2 представляют собой N, существуют в виде таутомеров. Хотя таутомеры различны по своей структуре, для специалиста в данной области техники должно быть очевидно, что они существуют в равновесии и легко и быстро взаимопревращаются при обычных условиях. (См., March, Advanced Organic Chemistry, третье издание, Wiley Interscience, Нью-Йорк, НьюЙорк (1985), с. 66-70 и Allinger, Organic Chemistry, второе издание, Worth Publishers, Нью-Йорк, НьюЙорк, (1976), с. 173). По этой причине представление соединения формулы I в одной таутомерной форме предполагает обе его таутомерные формы отдельно и их смеси. Аналогичным образом, использование в отношении соединения формулы I, где, например, R1 представляет собой водород, как названия 1Hимидазол, так и названия 3 Н-имидазол, предполагает обе таутомерные формы. Некоторые классы соединений формулы I являются предпочтительными ингибиторами p70 S6 киназы. В следующих пунктах описаны такие предпочтительные классы:h) R2 представляет собой фенил, содержащий первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, который представляет собой галоген;i) R2 представляет собой фенил, содержащий первый заместитель, выбранный из галогена и трифторметила, и дополнительно содержащий второй заместитель, который представляет собой галоген;j) R2 представляет собой фенил, содержащий в третьем или четвертом положении первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, который представляет собой галоген;k) R2 представляет собой фенил, содержащий в третьем или четвертом положении первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и дополнительно содержащий второй заместитель, который представляет собой галоген;l) R2 представляет собой фенил, содержащий в третьем положении первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий в четвертом положении второй заместитель, который представляет собой галоген;n) соединение формулы I представляет собой свободное основание;q) соединение формулы I представляет собой соль паратолуолсульфокислоты. Другими предпочтительными соединениями формулы I являются такие соединения, в которых Y представляет собой CH и R1 представляет собой H или CH3. Дополнительными предпочтительными соединениями формулы I являются такие соединения, в которых Y представляет собой CH; R1 представляет собой H или CH3 и R2 представляет собой фенил, содержащий первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, который представляет собой галоген. Предпочтительные соединения формулы I также включают такие соединения, в которых Y представляет собой CH и R1 представляет собой H или CH3; R2 представляет собой фенил, содержащий первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, который представляет собой галоген; R5 представляет собой H; Z1 представляет собой CR3 и Z2 представляет собой N. Также предпочтительными являются такие соединения формулы I, где Y представляет собой CH; R1 представляет собой H или CH3; R2 представляет собой фенил, содержащий первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий второй заместитель, который представляет собой галоген; R5 представляет собой H; Z1 представляет собой CH и Z2 представляет собой N.-4 016445 Кроме того, предпочтительными являются такие соединения формулы I, в которых Y представляет собой CH; R1 представляет собой H или CH3; R2 представляет собой фенил, содержащий первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и дополнительно содержащий второй заместитель, который представляет собой галоген; R5 представляет собой H; Z1 представляет собой CH и Z2 представляет собой N. Более предпочтительными соединениями формулы I являются такие, в которых Y представляет собой CH; R1 представляет собой H или CH3; R2 представляет собой фенил, содержащий в третьем положении первый заместитель, выбранный из группы, состоящей из галогена и трифторметила, и необязательно дополнительно содержащий в четвертом положении второй заместитель, который представляет собой галоген; R5 представляет собой H; Z1 представляет собой CH и Z2 представляет собой N. Кроме того, более предпочтительными соединениями формулы I являются такие, в которых Y представляет собой CH; R1 представляет собой H или СН 3; R2 представляет собой фенил, содержащий в третьем положении первый заместитель, выбранный из группы, состоящей из галогена и трифторметила,и дополнительно содержащий в четвертом положении второй заместитель, который представляет собой галоген; R5 представляет собой H; Z1 представляет собой CH и Z2 представляет собой N. В частности, предпочтительными являются такие соединения формулы I, в которых Y представляет собой CH; Z1 представляет собой CH и Z2 представляет собой N. Следующее соединение 4-4-[4-(3-(трифторметил)фенил)-1H-имидазол-2-ил]пиперидин-1-ил-1Hпиразоло[3,4-d]пиримидин или его фармацевтически приемлемая соль являются наиболее предпочтительными. Дополнительно,соединение 4-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2 ил]пиперидин-1-ил-1H-пиразоло[3,4-d]пиримидин или его фармацевтически приемлемая соль также являются наиболее предпочтительными. Соединения формулы I представляют собой ингибиторы p70 S6 киназы и, следовательно, пригодны для лечения метаболических заболеваний и расстройств, таких как ожирение, диабет, метаболический синдром, резистентность к инсулину, гипергликемия, гипераминоацидемия и гиперлипидемия, и пролиферативных расстройств, в частности мультиформной глиобластомы, колоректального рака, печеночноклеточного рака, рака легкого, рака груди, рака яичников и почечно-клеточной карциномы у млекопитающих. Кроме того, было обнаружено, что сигнальный путь p70 S6 киназы активирован в клетках доброкачественных опухолей, таких как связанные с лимфангиолейомиоматозом (LAM). См. Journal ofBiological Chemistry, 277:34, 30958-30967 (2002) и Modern Pathology. 19, 839-846 (2006). Таким образом,ингибиторы p70 S6 киназы также пригодны для лечения LAM. Ингибиторы p70 S6 киназы также пригодны для ингибирования ангиогенеза у млекопитающих. Млекопитающее, подвергаемое лечению,предпочтительно представляет собой человека. Соединения формулы I может получить специалист в данной области техники, следуя известным в данной области техники методикам и процедурам. В частности, соединения формулы I можно получить,как описано в схемах, способах и примерах, описанных ниже. Для специалиста в данной области техники должно быть очевидно, что отдельные стадии в следующих схемах можно изменять, чтобы получить соединения формулы I. Реагенты и исходные материалы легко доступны специалисту в данной области техники. Все заместители, если не указано иначе, являются такими, которые определены ранее. Некоторые заместители были исключены в следующих схемах для ясности, что никоим образом не следует рассматривать как ограничение концепции данных схем. Соединения формулы I можно получить, как показано на следующей схеме, при этом R1, R2, R4, R5,Z1, Z2 и Y такие, как описано ранее, и L представляет собой подходящую уходящую группу, такую как галоген. Содержащее заместители пиразолопиримидиновое (или пуриновое, или пирролопиримидиновое) соединение формулы (a) подвергают взаимодействию с содержащим заместители пиперидином формулы(b) с образованием соединений формулы I. Например, раствор соединения формулы (a), пиперидина (b) и подходящего основания, такого как триэтиламин, диизопропилэтиламин или N-метилморфолин, в подходящем растворителе, таком как пропанол или изопропиловый спирт, нагревают до приблизительно 70-100C с получением соединений формулы I, которые затем можно выделить и, при необходимости и желании, очистить, применяя методики, хорошо известные в данной области техники, такие как хроматография. Соединения формулы (a) доступны для приобретения или их можно синтезировать способами, известными специалисту в данной области техники. Например, соединения формулы (a), в которойZ1 представляет собой CR3 и Z2 представляет собой N, можно получить из аллопуринола в результате реакции с хлорирующим реагентом, таким как фосфорил хлорида, при нагревании до приблизительно 80-90C. Дополнительно, в соединения формулы (a) можно, при желании, ввести заместители при условиях, хорошо известных в данной области техники. Например, соединение формулы (a), в которой Z1 илиZ2 представляет собой CR3, можно фторировать при воздействии подходящего фторирующего агента,такого как [1-(хлорметил)-4-фтор-1,4-диазонийбицикло[2.2.2]октан-бис-(тетрафторборат)], в подходящем растворителе или смеси подходящих растворителей, таких как ацетонитрил и уксусная кислота. Более того, соединения формулы (a), в которой R3 представляет собой алкил или циклоалкил, синтезируют способами, известными в данной области техники. Например, дихлорпиримидин подвергают взаимодействию с циклопропанкарбальдегидом в присутствии сильного основания, такого как LDA, в растворителе, таком как ТГФ, при пониженных температурах с получением необходимого спирта. Окисление в присутствии оксида хрома (VI) при 0C в подходящем растворителе, таком как ацетон, дает требуемый кетон, который затем подвергают взаимодействию с гидратом гидразина в подходящем растворителе,таком как ТГФ, при комнатной температуре с получением пиразолопиримидинового соединения формулы (a), содержащего в качестве заместителя циклоалкил. Дополнительно, способы получения соединений, в которых R3 представляет собой алкинил, известны специалисту в данной области техники. Например, взаимодействие хлорированного соединения формулы (a) с триметилсилилацетиленом в триэтиламине и подходящем растворителе, таком как диметилформамид (ДМФ) и/или тетрагидрофуран (ТГФ),применяя тетракис-(трифенилфосфин)палладий(0) (Pd(Ph3P)4) в качестве катализатора, с получением соединения формулы (a), содержащего в качестве заместителя этинил. Необходимые пиперидиновые соединения (b) можно получить, как показано на следующей схеме,при этом R1, R2, R4, R5 и Y такие, как описано ранее, и PG представляет собой подходящую защитную группу. Амин (c) подвергают реакции с защищенным пиперидином в присутствии подходящих агентов реакции сочетания, таких как изобутилхлорформиат, 1-этил-3-[3-диметиламинопропил]карбодиимид гидрохлорид (EDC) или циклический ангидрид 1-пропанфосфоновой кислоты (РРА), и подходящего основания, такого как N-метилморфолин или триэтиламин, в подходящем растворителе, таком как ТГФ, метиленхлорид или N,N-диметилформамид (ДМФ), при пониженных температурах с получением амидных соединений формулы (d). Полученный имидазол-пиперидин (b1), в котором Y представляет собой CH,образуется при воздействии на соединение (d) ацетата аммония или хлорида аммония в герметизированной колбе или для соединений, в которых R5 представляет собой Н, при микроволновом нагревании под давлением. В качестве альтернативы, соединения формулы (b1) можно получить путем взаимодействия амидина (e) с подходящим галокетоном в подходящем растворителе, таком как ДМФ или ацетон, в присутствии основания, такого как карбонат калия или карбонат натрия, с последующим снятием защиты при условиях, хорошо известных специалисту в данной области техники. Гидроксипиперидины формулы (b1) можно также синтезировать путем взаимодействия бромистого фенацила формулы (m) с формамидом при нагревании. Имидазол (n) затем добавляют в подходящий растворитель, такой как ТГФ, охлаждают и обрабатывают, чтобы ввести защитную группу азота, такую как 2,2-(триметилсилил)этоксиметилхлорид, в присутствии гидрида натрия, позволяя реакционной смеси нагреться до комнатной температуры, с получением промежуточного продукта формулы (o). Полученный защищенный имидазол можно затем обработать металлирующим агентом, таким как н-бутиллитий,в инертном растворителе, таком как ТГФ, при пониженных температурах и в инертной атмосфере. Эту смесь обрабатывают содержащим подходящие заместители пиперидиноном и дают нагреться до комнатной температуры в течение приблизительно 1 ч с получением соединения типа (р). Обработка промежуточного продукта (p) водной кислотой, такой как 1 н. HCl, при повышенных температурах дает (b1). Требуемые амидины можно синтезировать, как описано на схеме 3, при этом PG представляет собой подходящую защитную группу. Амидин (e) можно легко получить из нитрилов. В частности, гидроксиламингидрохлорид, растворенный в подходящем растворителе, таком как вода, подвергают взаимодействию с цианопиперидином(g) в присутствии основания, такого как карбонат натрия, в подходящем растворителе, таком как метанол, и нагревают с получением гидроксикарбамимидоилпиперидина (h). Гидроксикарбамимидоилпиперидин (h) в подходящем растворителе, таком как уксусная кислота, затем гидрируют с помощью палладиевого катализатора в присутствии ангидрида уксусной кислоты под давлением с получением амидина (e). В качестве альтернативы, получают карбоксимидовую кислоту (j) путем растворения цианопиперидина (g) в подходящем растворителе, таком как метанол, охлаждения, затем проведения реакции с газообразным хлористым водородом. Амидин затем получают путем обработки карбоксимидовой кислоты (j) аммиаком в метаноле, затем насыщают полученную смесь газообразным аммиаком. Для специалиста в данной области техники должно быть очевидно, что не все заместители в соединениях формулы I будут выдерживать определенные условия реакции, используемые для синтеза указанных соединений. Эти молекулы можно ввести в удобный момент при синтезе или можно защитить и затем снять защиту при необходимости или желании. Для специалиста в данной области техники также должно быть очевидно, что защитные группы можно удалить в любой удобный момент при синтезе соединений согласно настоящему изобретению. Способы введения и удаления защитных групп азота хорошо известны в данной области техники; см., например, Greene и Wuts, Protective Groups in OrganicSynthesis, 3-е изд., John WileySons, New York, глава 7 (1999). Более того, для специалиста в данной области техники должно быть очевидно, что во многих случаях порядок, в котором вводят группы, не важен. Определенный порядок стадий, необходимый для получения соединений формулы I, зависит от конкретного синтезируемого соединения, исходного соединения и относительной неустойчивости используемых в качестве заместителей групп. Сокращения, символы и термины, используемые в примерах и анализах: Медленно добавляют раствор брома (3,34 г; 0,95 экв.) в дихлорметане (80,00 мл) к раствору 2-фтор 5-(трифторметил)пропиофенона (5 г; 22,03 ммоль) в дихлорметане (80 мл) при перемешивании таким образом, что коричневый цвет брома исчезает. Промывают реакционную смесь насыщенным NaHCO3,сушат над Na2SO4 и концентрируют с получением 6,22 г неочищенного вещества. Результаты массспектрометрии с ионизацией электрораспылением: МС (ИЭР): m/z=301,2 [M+2H]. Соединения из примеров синтеза 2-5-D можно получить, по существу, как описано в примере синтеза 1. Пример синтеза 6. Гидрохлорид 2-амино-1-(3-(трифторметил)фенил)этанона. Добавляют 3-(трифторметил)бромистый фенацил (10 г; 37,4 ммоль) к раствору гексаметилентетрамина (HMTA) (5,80 г; 41,3 ммоль) в тетрахлориде углерода (100 мл). Перемешивают при комнатной температуре в течение ночи. Выделяют осадок методом фильтрования и суспендируют отделенный осадок в этаноле (200 мл). Разбавляют смесь концентрированной хлористо-водородной кислотой (28 мл) и перемешивают смесь при комнатной температуре в течение ночи. Выделяют осадок методом фильтрования и концентрируют фильтрат под вакуумом с получением грязно-белого твердого вещества. Перекристаллизовывают твердые вещества из горячего раствора 1% концентрированной хлористо-водородной кислоты в 2-пропаноле, избегая охлаждения ниже комнатной температуры, с получением гидрохлорида 2-амино 1-(3-(трифторметил)фенил)этанона (7,67 г, 86%). Результаты масс-спектрометрии с химической ионизацией при атмосферном давлении: МС (ХИАД): m/z=204 [M+H].-9 016445 Соединения из примеров синтеза 7 и 9-14 можно получить, по существу, как описано в примере синтеза 6. Пример синтеза 18. 2-Амино-1-(4-фтор-3-трифторметилфенил)пропан-1-он, соль толуол-4-сульфоновой кислоты. Добавляют азид натрия (640,94 мг; 1,05 экв.; 9,76 ммоль) одной порцией к раствору 2-бром-1-(4 фтор-3-трифторметилфенил)пропан-1-она (2,78 г; 1,00 экв.; 9,30 ммоль) в тетрагидрофуране (15 мл). Перемешивают смесь при комнатной температуре в течение ночи. Отделяют твердые вещества методом фильтрования и промывают ТГФ. Добавляют неочищенный азид (1,00 экв.; 9,30 ммоль; 2,43 г) к раствору трифенилфосфина (1,06 экв.; 9,86 ммоль; 2,61 г) и п-толуолсульфоновой кислоты (2,2 экв.; 20,47 ммоль; 3,56 г) в тетрагидрофуране (15 мл) при 20C. Перемешивают смесь в течение ночи. Отделяют твердое вещество методом фильтрования, затем промывают ТГФ с получением 920 мг (24%) указанного в заголовке соединения. МС (ИЭР): m/z=236,2 [M+H]. Соединения из примеров синтеза 18A - 18-D можно получить, по существу, как описано в примере синтеза 18. Добавляют N-метилморфолин (13,0 мл, 118 ммоль) к раствору 1-(трет-бутоксикарбонил)пиперидин 4-карбоновой кислоты (11,2 г; 48,8 ммоль) в ТГФ (400 мл) при -10C и перемешивают в течение 5 мин. Добавляют изобутилхлорформиат (5,1 мл, 38,8 ммоль) и продолжают перемешивание при -10C в тече- 10016445 ние 2 ч. Добавляют гидрохлорид 2-амино-1-(3-(трифторметил)фенил)этанона (9,35 г; 39,0 ммоль) и продолжают перемешивание в течение 1 ч. К полученной смеси добавляют метиленхлорид (500 мл) и фильтруют. Промывают фильтрат насыщенным водным бикарбонатом натрия (400 мл) и концентрируют под вакуумом. Очищают осадок методом хроматографии на силикагеле (330 г колонка RediSep, элюируют градиентом от 0 до 100% этилацетат:гексан, 6,0 л) с получением трет-бутил 4-(2-оксо-2-(3(трифторметил)фенил)этилкарбамоил)пиперидин-1-карбоксилата (9,72 г, 60%). МС (ХИАД): m/z=315 [M-C5H8O2+H]+. Соединения из примеров синтеза 20 и 22-25 можно получить, по существу, как описано в примере синтеза 19. Пример синтеза 27. трет-Бутиловый эфир 4-[2-(4-фтор-3-трифторметилфенил)-1-метил-2-оксоэтилкарбамоил]пиперидин-1-карбоновой кислоты. Добавляют диметилформамид (20 мл) к смеси 2-амино-1-(4-фтор-3-трифторметилфенил)пропан-1 она, соль толуол-4-сульфоновой кислоты (900 мг; 1,00 экв.; 2,21 ммоль) и 1-Вос-пиперидин-4 карбоновой кислоты (Boc-Inp-OH) (613,96 мг; 1,20 экв.; 2,65 ммоль) при комнатной температуре. Добавляют N-метилморфолин (6,00 экв.; 13,26 ммоль; 1,46 мл) к указанному раствору, затем циклический ангидрид 1-пропанфосфоновой кислоты (1,50 экв.; 3,31 ммоль; 862,14 мкл) при 0C. Нагревают смесь до комнатной температуры при перемешивании в течение 1 ч. Разбавляют смесь EtOAc, промывают водой,1 М лимонной кислотой, водой, насыщенным NaHCO3 и насыщенным водным хлоридом натрия и сушат над Na2SO4. Очищают осадок путем растворения в CH2Cl2, загрузки на колонку с силикагелем и элюирования EtOAc с получением 500 мг (51%) указанного в заголовке соединения. МС (ИЭР): m/z=445,2 [M-H].- 11016445 Соединения из примеров синтеза 30-32D можно получить, по существу, как описано в примере синтеза 27. Пример синтеза 33. 4-(4-(3-(Трифторметил)фенил)-1H-имидазол-2-ил)пиперидин. Нагревают микроволновым излучением смесь трет-бутил 4-(2-оксо-2-(3-(трифторметил)фенил)этилкарбамоил)пиперидин-1-карбоксилата (2,5 г; 6,09 ммоль) и NH4Cl (1,68 г; 31,4 ммоль) в этаноле(20 мл) при 300 Вт, 120C и 200 фунтах/кв.дюйм в течение 16 ч в микроволновом приборе СЕМ Discover. Охлаждают смесь до комнатной температуры. Повторяют процедуру со второй порцией трет-бутил 4-(2-оксо-2-(3-(трифторметил)фенил)этилкарбамоил)пиперидин-1-карбоксилата (2,35 г; 5,67 ммоль) и NH4Cl (1,8 г; 33,6 ммоль) в этаноле (20 мл), используя тот же микроволновый прибор и условия нагревания. Охлаждают смесь до комнатной температуры. Повторяют описанную процедуру с третьей порцией трет-бутил 4-(2-оксо-2-(3-(трифторметил)фенил)этилкарбамоил)пиперидин-1 карбоксилата (2,0 г; 4,82 ммоль) и NH4Cl (1,34 мг; 25,0 ммоль) в этаноле (15 мл) при 300 Вт, 160C и 200 фунтах/кв.дюйм в течение 12 ч в микроволновом приборе. Охлаждают смесь до комнатной температуры. Объединяют указанные порции, адсорбируют на силикагеле и концентрируют под вакуумом. Очищают осадок методом хроматографии на силикагеле (120 г колонка RediSep, элюируют градиентом от 0 до 100% СМА:метиленхлорид, 3 л) с получением 4-(4-(3-(трифторметил)фенил)-1H-имидазол-2 ил)пиперидина (3,40 г, 75%). МС (ХИАД): m/z=296 [M+H]. Соединения из примеров синтеза 34, 35 можно получить, по существу, как описано в примере синтеза 33. Референсный пример синтеза 36. Гидрохлорид 4-(4-фенил-1H-имидазол-2-ил)пиперидина. Нагревают микроволновым излучением смесь трет-бутил 4-(2-оксо-2-фенилэтилкарбамоил)пиперидин-1-карбоксилата (1,0 г; 2,88 ммоль) и NH4Cl (0,462 г; 8,65 ммоль) в этаноле (12 мл) при 300 Вт, 160C и 14 бар в течение 11 ч в микроволновом приборе Personal Chemistry. Охлаждают смесь до комнатной температуры, добавляют силикагель (5 мл) и концентрируют смесь под вакуумом. Очищают осадок методом хроматографии на силикагеле (80 г колонка RediSep, элюируют градиентом от 0 до 100%CMA/метиленхлорида в течение 60 мин, 60 мл/мин) с получением 4-(4-фенил-1H-имидазол-2 ил)пиперидина (0,418 г, 64%) в виде грязно-белого твердого вещества. Добавляют 4-(4-фенил-1Hимидазол-2-ил)пиперидин (0,060 г; 0,26 ммоль) к метиленхлориду (15 мл) и концентрированной хлористо-водородной кислоте (28 мкл). Перемешивают смесь в течение 45 мин, затем концентрируют смесь под вакуумом с получением гидрохлорида 4-(4-фенил-1H-имидазол-2-ил)пиперидина (0,070 г, 99%). МС (ХИАД): m/z=228 [M+H].- 12016445 Соединение из примера синтеза 37 можно получить, по существу, как описано в примере синтеза 36. Пример синтеза 38. Гидрохлорид 4-(4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина. Нагревают микроволновым излучением смесь трет-бутил 4-(2-(4-фтор-3-(трифторметил)фенил)этилкарбамоил)пиперидин-1-карбоксилата (1,7 г; 3,92 ммоль) и NH4Cl (0,579 г; 10,8 ммоль) в этаноле(17 мл) при 300 Вт, 120C и 14 бар в течение 12 ч в микроволновом приборе Personal Chemistry. Охлаждают смесь до комнатной температуры и добавляют силикагель (20 г). Очищают осадок методом хроматографии на силикагеле (330 г колонка RediSep, элюируют градиентом от 0 до 100% СМА/метиленхлорид в течение 30 мин и выдерживают при 100% СМА в течение дополнительных 30 мин, 100 мл/мин) с получением 4-(4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина(600 мг, 51%). МС (ХИАД): m/z=314 [M+H]. Суспендируют 4-(4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин (250 мг; 0,80 ммоль) в метаноле (20 мл) и добавляют хлористый водород (2 М в диэтиловом эфире, 0,798 мл,0,80 ммоль). Перемешивают при комнатной температуре в течение 30 мин. Концентрируют смесь под вакуумом и добавляют воду (15 мл) и ацетонитрил (5 мл). Смесь сушат сублимацией с получением грязно-белого твердого вещества. Сушат твердое вещество под вакуумом при 100C в течение 2 ч с получением гидрохлорида 4-(4-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина в виде грязнобелого твердого вещества (220 мг, 79%). МС (ХИАД): m/z=314 [M+H]. Пример синтеза 39. Гидрохлорид 4-(4-(3-фтор-4-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина. Нагревают микроволновым излучением смесь трет-бутил 4-(2-(3-фтор-4-(трифторметил)фенил)-2 оксоэтилкарбамоил)пиперидин-1-карбоксилата (517 мг; 1,19 ммоль) и NH4Cl (350 мг; 6,54 ммоль) в этаноле (2 мл) при 300 Вт, 120C и 200 фунтах/кв.дюйм при максимальной мощности в течение 12 ч в микроволновом приборе СЕМ Discover. Охлаждают смесь до комнатной температуры и добавляют силикагель (5 г). Очищают осадок методом хроматографии на силикагеле (40 г колонка RediSep, элюируют градиентом от 0 до 100% СМА/метиленхлорид в течение 60 мин, 40 мл/мин) с получением 4-(4-(4-фтор 3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина (228 мг, 61%) в виде грязно-белого твердого вещества. МС (ХИАД): m/z=314 [M+H]. Растворяют продукт (29 мг) в метаноле (2 мл) и добавляют хлористый водород (2 М в диэтиловом эфире, 95 мкл) и перемешивают при комнатной температуре в течение 30 мин. Концентрируют смесь под вакуумом, добавляют воду (10 мл) и сушат сублимацией с получением гидрохлорида 4-(4-(3-фтор-4(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина (29 мг) в виде грязно-белого твердого вещества. МС (ХИАД): m/z=314 [M+H]. Пример синтеза 41. трет-Бутиловый эфир 4-(N-гидроксикарбамимидоил)пиперидин-1-карбоновой кислоты. Растворяют гидроксиламингидрохлорид (1,65 г; 4,99 экв.; 23,74 ммоль) в воде (5 мл) и добавляют карбонат натрия (2,53 г; 23,87 ммоль). Помещают трет-бутиловый эфир 4-цианопиперидин-1-карбоновой кислоты (1 г; 1,00 экв.; 4,76 ммоль) (Astatech) в небольшую пробирку и растворяют в метаноле (6 мл; 148,25 ммоль), затем добавляют в колбу, содержащую гидроксиламин. Нагревают смесь до кипения с обратным холодильником при перемешивании и выдерживают в течение 3 ч. Удаляют метанол выпариванием и экстрагируют водный слой этилацетатом (2100 мл). Промывают объединенные органические слои водой (1100 мл), сушат над сульфатом магния, фильтруют и концентрируют с получением белого твердого вещества. Сушат при пониженном давлении с получением 1,05 г (91%) указанного в заголовке соединения в виде белого твердого вещества. ЖХ/МС: m/z=188,2 [M-tBu, M+H]. Пример синтеза 42. трет-Бутиловый эфир 4-карбамимидоилпиперидин-1-карбоновой кислоты. Растворяют трет-бутиловый эфир 4-(N-гидроксикарбамимидоил)пиперидин-1-карбоновой кислоты(9,7 г 1,00 экв.; 39,9 ммоль) в метаноле (300 мл) и добавляют уксусную кислоту (2 экв.; 79,73 ммоль; 4,6 мл) и промытый метанолом никель Ренея (2,7 г). Нагревают реакционную смесь до 50C, затем гидрогенизируют 1 атм газообразным водородом в течение 4,5 ч. Разделяют реакционную смесь методом фильтрования через целит и концентрируют при пониженном давлении. Суспендируют твердое вещество в диэтиловом эфире, фильтруют и сушат при пониженном давлении с получением 10,39 г третбутилового эфира 4-карбамимидоилпиперидин-1-карбоновой кислоты.(15 мл; 261,77 ммоль) и ангидриде уксусной кислоты (0,5 мл; 5,29 ммоль). Гидрогенизируют при комнатной температуре в течение 7 ч при 20 фунтах/кв.дюйм. Продувают колбу азотом с получением 1,2 г пены. Растирают с ацетонитрилом и фильтруют с получением 270 мг (28%) продукта в виде белого твердого вещества. МС (ИЭР): m/z=228,0 [M+H]. Пример синтеза 44. Гидрохлорид 4-[4-(3-хлорфенил)-1H-имидазол-2-ил]пиперидина. Суспендируют трет-бутиловый эфир 4-диаминометилпиперидин-1-карбоновой кислоты, соль уксусной кислоты (685 мг; 1,00 экв.; 2,38 ммоль) в диметилформамиде (10 мл; 129,33 ммоль) и добавляют порошкообразный карбонат калия (1400 мг; 10,13 ммоль). Перемешивают 10 мин при комнатной температуре, затем 2-бром-1-(3-хлорфенил)этанон (1400 мг; 6,00 ммоль), растворенный в 4 мл ДМФ, добавляют по каплям в амидин при комнатной температуре. Через 3 ч разбавляют реакционную смесь этилацетатом и промывают 50% водным бикарбонатом натрия. Сушат органический слой над сульфатом натрия,фильтруют и концентрируют с получением неочищенного масла. Очищают, применяя хроматографию на приборе ISCO, на 40 М колонке Biotage, элюируют градиентом от DCM до 6% MeOH/DCM. Концентрируют соответствующие фракции с получением 20% выхода трет-бутилового эфира 4-[4-(3-хлорфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты. МС (ИЭР): m/z=262,0 [M+H]. Растворяют трет-бутиловый эфир 4-[4-(3-хлорфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (171 мг; 472,54 мкмоль) в дихлорметане (3 мл; 46,80 ммоль) и медленно добавляют хлористый водород (4 мл; 16,00 ммоль) (4 М HCl в диоксане) при комнатной температуре. Перемешивают раствор в течение 1 ч. Концентрируют под вакуумом (2DCM) с получением гидрохлорида 4-[4-(3-хлорфенил)-1Hимидазол-2-ил]пиперидина. МС (ИЭР): m/z=262,0 [M+H]. Соединения из примеров синтеза 45-47A можно получить, по существу, как описано в примере синтеза 44. Пример синтеза 48. 1-Бензилпиперидин-4-карбонитрил. Добавляют 4-цианопиперидин (7,19 г; 1,00 экв.; 65,269 ммоль), бензальдегид (1,05 экв.; 68,533 ммоль; 7,0 мл), затем триацетоксиборгидрид натрия (1,3 экв.; 84,85 ммоль; 18,73 г) к 2% (в объемном отношении) уксусной кислоте в тетрагидрофуране (435 мл) и перемешивают быстро до завершения. Разбавляют этилацетатом и промывают насыщенным бикарбонатом натрия, насыщенным водным хлоридом натрия, сушат над MgSO4, фильтруют, выпаривают при пониженном давлении с получением 13,0 г масла. Растворяют в эфире (300 мл), фильтруют и медленно добавляют 60 мл 1 М HCl в эфире. Охлаждают на ледяной бане. Фильтруют, ополаскивают эфиром, сушат под вакуумом и собирают 13,616 г (57,51 ммоль, 88%) 1-бензилпиперидин-4-карбонитрила. МС (ИЭР): m/z=201 [M+H]. Пример синтеза 49. Гидрохлорид метилового эфира 1-бензилпиперидин-4-карбоксимидовой кислоты. Растворяют 1-бензилпиперидин-4-карбонитрилгидрохлорид (1,00 экв.; 57,3 ммоль; 13,57 г) в 75 млMeOH и охлаждают на ледяной бане. Насыщают газообразным HCl в течение 25 мин, затем удаляют ледяную баню и перемешивают в течение 3 ч. Выпаривают при пониженном давлении с получением 17,47 г (65,0 ммоль, 113%) гидрохлорида метилового эфира 1-бензилпиперидин-4-карбоксимидовой кислоты. МС (ИЭР): m/z=233 [M+H].- 14016445 Пример синтеза 50. Дигидрохлорид 1-бензилпиперидин-4-карбоксамидина. Растворяют гидрохлорид метилового эфира 1-бензилпиперидин-4-карбоксимидовой кислоты(17,47 г; 1,00 экв.; 65 ммоль) в 2 М аммиаке в метаноле (350 мл). Насыщают газообразным аммиаком в течение 15 мин. Перемешивают в течение 18 ч и выпаривают. Выпаривают вместе с сухим метанолом. Сушат под высоким вакуумом с получением 18,29 г (63,03 ммоль, 97%) дигидрохлорида 1-бензилпиперидин-4-карбоксамидина в виде светло-желтого твердого вещества. МС (ИЭР): m/z=218 [M+H]. Пример синтеза 51. 1-Бензил-4-[5-(3-хлор-4-фторфенил)-1H-имидазол-2-ил]пиперидин. Растворяют дигидрохлорид 1-бензилпиперидин-4-карбоксамидина (2 г; 1,00 экв.; 6,89 ммоль) в диметилформамиде (90 мл). Добавляют порошкообразный карбонат калия (4 экв.; 27,56 ммоль; 3,8095 г) и нагревают до 45C. Добавляют 3-хлор-4-фторфенацилбромид (2,00 экв.; 13,782 ммоль; 3,5366 г) в 8 мл с ДМФ по каплям в течение 40 мин. Разбавляют 50 мл этилацетата, перемешивают 5 мин и фильтруют. Выпаривают и разделяют этилацетатом/насыщенным бикарбонатом натрия, промывают органический слой водой, затем насыщенным водным хлоридом натрия. Сушат над MgSO4, фильтруют и выпаривают до образования красной пены. Очищают методом флэш-хроматографии на силикагеле с 0-10%MeOH/ACN. Объединяют фракции с получением 1,9772 г (5,346 ммоль, 78%) 1-бензил-4-[5-(3-хлор-4 фторфенил)-1H-имидазол-2-ил]пиперидина. МС (ИЭР): m/z=370 [M+H]. Соединения из примеров синтеза 52-53 можно получить, по существу, как описано в примере синтеза 51.(1,00 экв.; 1,595 ммоль; 590 мг); N,N,N',N'-тетраметил-1,8-нафталиндиамин (1 экв.; 1,595 ммоль; 341,8 мг) в 1,2-дихлорэтане (12 мл) и охлаждают на ледяной бане. Добавляют 1-хлорэтилхлорформиат (3 экв.; 4,785 ммоль; 517 мкл) и нагревают до кипения с обратным холодильником в течение 1 ч. Охлаждают до комнатной температуры и фильтруют через 1-см слой из силикагеля, ополаскивают DCM. Выпаривают с получением 760 мг пены. Растворяют в MeOH и нагревают до кипения с обратным холодильником в течение 6 ч. Выпаривают с получением 556,3 мг (99%) дигидрохлорида 4-[5-(3-хлор-4-фторфенил)-1Hимидазол-2-ил]пиперидина в виде желтой пены. МС (ИЭР): m/z=280 [M+H]. Соединения из примеров синтеза 55, 56 можно получить, по существу, как описано в примере синтеза 54. Пример синтеза 57. 4-[4-(4-Фторфенил)-1H-имидазол-2-ил]пиперидин. Гидрогенизируют 1-бензил-4-[4-(4-фторфенил)-1H-имидазол-2-ил]пиперидин в 20% Pd(OH)2/C (катализатор Перлмана) (0,15 г) в 125 мл 2 В EtOH, при 30C в течение 23 ч при 60 фунтах/кв.дюйм. Фильтруют и концентрируют с получением указанного в заголовке соединения в виде масла. МС (ИЭР): m/z=246,0 [M+H]. Соединение из примера синтеза 59 можно получить, по существу, как описано в примере синтеза 57.- 15016445 Пример синтеза 60. трет-Бутил 4-(4-(3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин-1-карбоксилат. Добавляют 4-(4-(3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин (795 мг; 2,69 ммоль) к раствору CH2Cl2 (25 мл) и ТГФ (25 мл) в атмосфере азота, а затем триэтиламин (0,985 мл, 5,64 ммоль) и дитрет-бутилдикарбонат (650 мг; 2,96 ммоль). Перемешивают смесь при комнатной температуре в течение ночи. Концентрируют смесь под вакуумом и очищают осадок методом хроматографии на силикагеле(120 г колонка RediSep, элюируют градиентом от 0 до 100% этилацетат/гексаны в течение 35 мин,85 мл/мин) с получением трет-бутил 4-(4-(3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин-1 карбоксилата (550 мг, 51%) в виде грязно-белого твердого вещества. МС (ХИАД): m/z=396 [M+H]. Соединение из примера синтеза 61 можно получить, по существу, как описано в примере синтеза 60.(540 мг; 1,36 ммоль) в диэтиловый эфир (100 мл) и охлаждают до 0C. Добавляют к полученной смеси гидрид натрия (55 мг; 1,5 ммоль; 60% в минеральном масле), а затем йодистый метил (0,142 мл,2,72 ммоль). Перемешивают смесь в течение 1 ч при 0C и нагревают до комнатной температуры. Добавляют ТГФ (40 мл) и перемешивают смесь в течение 12 ч. Концентрируют смесь и очищают осадок методом хроматографии на силикагеле (120 г колонка RediSep, элюируют градиентом от 0 до 100% этилацетат:гексан в течение 40 мин, 85 мл/мин) с получением трет-бутил 4-(1-метил-4-(3(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин-1-карбоксилата (425 мг, 76%). МС (ХИАД): m/z=410 [M+H]. Соединение из примеров синтеза 63 и 63A можно получить, по существу, как описано в примере синтеза 62. Пример синтеза 64. Дигидрохлорид 4-(1-метил-4-(3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидина. Добавляют хлористый водород (4 М в 1,4-диоксане, 5 мл, 20 ммоль) к раствору трет-бутил 4-(1 метил-4-(3-(трифторметил)фенил)-1H-имидазол-2-ил)пиперидин-1-карбоксилата (400 мг; 1,10 ммоль) вCH2Cl2 (10 мл) при 0C в атмосфере азота и перемешивают в течение 2 ч. Концентрируют смесь под вакуумом с получением дигидрохлорида 4-(1-метил-4-(3-(трифторметил)фенил)-1H-имидазол-2 ил)пиперидина (435 мг, 99%). МС (ХИАД): m/z=310 [M+H]. Соединение из примера синтеза 65 можно получить, по существу, как описано в примере синтеза 64. Пример синтеза 66. трет-Бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1 карбоновой кислоты. Добавляют ацетат аммония (3,24 г; 41,63 ммоль) к раствору трет-бутил 4-(2-(4-фтор-3(трифторметил)фенил)-2-оксоэтилкарбамоил)пиперидин-1-карбоксилата (600 мг; 1,00 экв.; 1,39 ммоль) в 1-бутаноле (7 мл), а затем триэтиламин (1 экв.; 193,40 мкл). Перемешивают смесь при 160C в герметизированной пробирке в течение 2 ч. Удаляют растворитель под вакуумом путем совместного выпаривания с толуолом и CH2Cl2. Перерастворяют неочищенный материал в EtOAc, промывают водой, сушат надNa2SO4 и концентрируют. Очищают осадок методом хроматографии на силикагеле, элюируютEtOAc:гексан (6:4) с получением 280 мг (49% выход) указанного в заголовке соединения. Результаты сочетания жидкостной хроматографии и ион-распылительной тандемной масс-спектрометрии:- 16016445 ЖХ/МС: МС (ИР): m/z=314,2 [M+H]. Соединения из примеров синтеза 67, 69 и 71-73D можно получить, по существу, как описано в примере синтеза 66. Пример синтеза 74. трет-Бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1 Н-имидазол-2-ил]пиперидин-1 карбоновой кислоты. Растворяют трет-бутиловый эфир 4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2 ил]пиперидин-1-карбоновой кислоты (860 мг; 2,08 ммоль) в диэтиловом эфире (100 мл) в атмосфере N2. Охлаждают раствор до 0C на ледяной бане и добавляют гидрид натрия (1,1 экв.; 2,29 ммоль; 91,52 мг) с последующим впрыскиванием йодистого метила (2,00 экв.; 4,16 ммоль; 259,12 мкл). Перемешивают смесь в течение 1 ч при 0C и затем нагревают до комнатной температуры. Впрыскивают тетрагидрофуран (40 мл) в указанную смесь и затем перемешивают при комнатной температуре в течение ночи. Концентрируют смесь при пониженном давлении и затем растворяют осадок в EtOAc. Промывают насыщенным водным хлоридом натрия. Очищают осадок посредством хроматографии на силикагеле, элюируют смесью EtOAc:гексан (7:3) с получением 270 мг (30% выход) указанного в заголовке соединения. МС (ИР): m/z=428,2 [M+H]. Пример синтеза 74-A. трет-Бутил 4-(1,5-диметил-4-(4-(трифторметил)фенил)-1H-имидазол-2-ил]пиперидин-1-карбоксилат. Добавляют свежий порошкообразный гидроксид калия (193,45 мг; 2,93 ммоль; 4 экв.) к раствору трет-бутил 4-(5-метил-4-(4-(трифторметил)фенил)-1H-имидазол-2-ил]пиперидин-1-карбоксилата (300 мг; 0,73 ммоль) в диметилсульфоксиде (2 мл). Перемешивают смесь при комнатной температуре в течение 1 ч и добавляют одной порцией йодистый метил (156 мг; 1,5 экв.). После перемешивания в течение 3 ч,разбавляют AcOEt, промывают насыщенным водным хлоридом натрия и сушат над Na2SO4. Очищают осадок посредством хроматографии на силикагеле, элюируют EtOAc:гексан (6:4) с получением 210 мг- 17016445 Соединения из примеров синтеза 74-B - 74-F можно получить, по существу, как описано в примере синтеза 74-A. Пример синтеза 75. Гидрохлорид 4-[4-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперидина. Добавляют хлористый водород (2 мл, 12 М водный) к раствору трет-бутилового эфира 4-[4-(4-фтор 3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-карбоновой кислоты (275 мг; 665,19 мкмоль) в метаноле (10 мл). Перемешивают смесь при комнатной температуре в течение ночи и затем концентрируют и сушат с получением 210 мг (90% выход) указанного в заголовке соединения. МС (ИР): m/z=314,2 [M+H]. Соединения из примеров синтеза 76-78 и 61-83G можно получить, по существу, как описано в примере синтеза 75.- 18016445 Пример синтеза 86. 1-Бензил-4-[4-(3-хлор-4-фторфенил)-1-этил-1H-имидазол-2-ил]пиперидин. Добавляют диметилсульфоксид (0,3 М, 2,7 мл) к порошкообразному гидроксиду калия (1,5 экв.; 1,217 ммоль; 68 мг). Добавляют 1-бензил-4-[5-(3-хлор-4-фторфенил)-1H-имидазол-2-ил]пиперидин(300 мг; 1,00 экв.; 0,811 ммоль), по каплям добавляют йодэтан (1,1 экв.; 0,892 ммоль; 71 мкл) в течение 8 мин. Перемешивают реакционную смесь в течение 60 мин, затем разбавляют водой (120 мл) плюс насыщенным хлоридом натрия (25 мл) и экстрагируют четыре раза с помощью DCM. Промывают органические экстракты водой, затем насыщенным водным хлоридом натрия и сушат над MgSO4. Фильтруют и очищают на силикагеле с помощью 10% метанола/ацетонитрила с получением 262 мг (0,659 ммоль, 81%) 1-бензил-4-[4-(3-хлор-4-фторфенил)-1-этил-1H-имидазол-2-ил]пиперидина. МС (ИЭР): m/z=398 [M+H]. Соединения из примеров синтеза 87-89 можно получить, по существу, как описано в примере синтеза 86. Пример синтеза 90. Гидрохлорид 4-[4-(3-хлор-4-фторфенил)-1-этил-1H-имидазол-2-ил]пиперидина. Растворяют 1-бензил-4-[4-(3-хлор-4-фторфенил)-1-этил-1H-имидазол-2-ил]пиперидин (263,3 мг; 1,00 экв.; 0,662 ммоль) и N,N,N',N'-тетраметил-1,8-нафталиндиамин (0,05 экв.; 0,033 ммоль; 7,0 мг) в 1,2-дихлорэтане (5 мл) и охлаждают на ледяной бане. Добавляют 1-хлорэтилхлорформиат (1,2 экв.; 0,794 ммоль; 0,086 мл). Перемешивают реакционную смесь 10 мин на ледяной бане, затем нагревают до кипения с обратным холодильником в течение 20 мин и выпаривают досуха. Растворяют осадок в метаноле (5 мл), кипятят с обратным холодильником в течение 45 мин и выпаривают досуха с получением 268 мг (0,779 ммоль, 118%) гидрохлорида 4-[4-(3-хлор-4-фторфенил)-1-этил-1H-имидазол-2 ил]пиперидина. МС (ИЭР): m/z=308 [M+H]. Соединения из примеров синтеза 91-93 можно получить, по существу, как описано в примере синтеза 90.(200 мл) и уксусную кислоту (40 мл) и нагревают до 70C в течение 24 ч. За потерей исходного материала наблюдают с помощью ВЭЖХ, затем концентрируют. Добавляют две порции толуола (50 мл) и выпаривают. Фильтруют неочищенный материал через слой целита, промывая 1:1 EtOAc/CH2Cl2. В конце,концентрируют фильтрат и проводят хроматографию на колонке с силикагелем, элюируя CH2Cl2/MeOH[градиент 0-10% MeOH]. Проверяют фракции с помощью МС и объединяют фракции продукта с получением 1,931 г (58%) 4-хлор-5-фтор-7H-пирроло[2,3-d]пиримидина. МС (ИЭР): m/z=172 [M+H].- 19016445 Пример синтеза 95. 4-Хлор-1H-пиразоло[3,4-d]пиримидин. К раствору аллопуринола (20 г; 146,94 ммоль) в толуоле (205,71 мл) добавляют хлористый фосфорил (68,27 мл, 734,68 ммоль) и диизопропилэтиламин (56,38 мл, 323,26 ммоль) и нагревают смесь на 80C в течение 2 ч. Удаляют растворитель под вакуумом наполовину и вливают смесь в 2 М фосфат калия, двухосновный (734,68 мл, 1,47 моль), в воде при 4C. Перемешивают смесь в течение ночи при комнатной температуре. Отфильтровывают осадок через слой целита и затем промывают его EtOAc. Отделяют фильтрат, промывают водный слой дополнительным EtOAc, объединяют органические слои, сушат его над MgSO4, фильтруют и концентрируют под вакуумом с получением 4-хлор-1H-пиразоло[3,4d]пиримидина (16 г, 70,45% выход) в виде желтого твердого вещества. МС (ХИАД): m/z=155,l [M+H]. Пример синтеза 95-A. 4,6-Дихлорпиримидин-5-карбальдегид. Загружают ДМФ (8,9 мл, 1,3 экв.) в круглодонную колбу и охлаждают до 0C. Добавляют в реакционную смесь POCl3 (32,6 мл, 4,0 экв.) по каплям при 0C. Перемешивают реакционную массу при 0C в течение 1 ч. Загружают в реакционную массу 4,6-дигидроксипиримидин (10,0 г; 1,0 экв.) и дают ей медленно дойти до комнатной температуры. Кипятят реакционную массу с обратным холодильником в течение 4 ч и наблюдают за ходом реакции с помощью тонкослойной хроматографии (ТСХ) (10% ацетон вDCM). Концентрируют реакционную массу под вакуумом и выливают концентрированную реакционную массу на дробленый лед. Экстрагируют продукт диэтиловым эфиром и промывают насыщенным водным хлоридом натрия. Сушат органический слой над безводным сульфатом натрия и концентрируют под вакуумом с получением продукта в виде бледно-желтого твердого вещества (6,2 г, 40%). Пример синтеза 95-В. 1-(4,6-Дихлорпиримидин-5-ил)пропан-1-ол. Загружают 4,6-дихлорпиримидин-5-карбальдегид (2,5 г; 1,0 экв.) и толуол (50 мл) в круглодонную колбу. Охлаждают реакционную массу до -10C. Добавляют бромэтилмагний (3 М) в раствор ТГФ(5,1 мл, 1,1 экв.) по каплям при -10C. Дают реакционной массе медленно достичь комнатной температуры за 1 ч. Загружают охлажденный раствор хлорида аммония в реакционную массу и экстрагируют диэтиловым эфиром. Промывают эфирный слой насыщенным водным раствором хлорида натрия. Сушат эфирный слой над безводным сульфатом натрия и концентрируют при пониженном давлении с получением желаемого продукта (2,3 г, 79,3%). Пример синтеза 95-C. 1-(4,6-Дихлорпиримидин-5-ил)этанол. Соединение из примера синтеза 95-C можно получить, по существу, как описано в примере синтеза 95-B. Пример синтеза 95-D. 4-Хлор-3-йод-1H-пиразоло[3,4-d]пиримидин. Загружают 4-хлор-1H-пиразоло[3,4-d]пиримидин (6,1 г; 1,0 экв.), N-йодсукцинимид (NIS) (21,55 г; 2,0 экв.) и ДМФ (213,5 мл) в круглодонную колбу. Перемешивают реакционную массу при 50C в течение 16 ч. Наблюдают за ходом реакции с помощью ТСХ (10% ацетон в DCM). Концентрируют реакционную массу при пониженном давлении. Загружают этилацетат и промывают водой и насыщенным водным хлоридом натрия. Сушат органический слой над безводным сульфатом натрия и концентрируют органический слой при пониженном давлении с получением 4-хлор-3-йод-1H-пиразоло[3,4d]пиримидина (6,8 г, 61,43%). Пример синтеза 95-E. 4-Хлор-3-триметилсилил)этинил)-1H-пиразоло[3,4-d]пиримидин. Загружают 4-хлор-3-йод-1H-пиразоло[3,4-d]пиримидин (5,4 г; 1,0 экв.), триметилсилилацетилен(11,347 г; 6,0 экв.), CuI (1,833 г; 0,5 экв.), TEA (2,68 мл, 1,0 экв.), ДМФ (67,5 мл) и ТГФ (202,5 мл) в круглодонную колбу в атмосфере аргона. Перемешивают реакционную массу в атмосфере аргона в течение 30 мин. Загружают (PPh3)4Pd (2,225 г; 0,1 экв.) и перемешивают реакционную массу при 35C в течение 3 ч. Наблюдают за ходом реакции с помощью ТСХ (10% ацетон в DCM). Концентрируют реакционную массу при пониженном давлении. Загружают этилацетат и промывают насыщенным раствором бикарбоната натрия, водой и насыщенным водным хлоридом натрия. Сушат органический слой над безводным сульфатом натрия и концентрируют органический слой при пониженном давлении. Очищают полученное соединение методом колоночной хроматографии (силикагель 100-200 меш, DCM - ацетон) с получением желаемого продукта (1,81 г, 36,14%).- 20016445 Пример синтеза 96. Циклопропил-(4,6-дихлорпиримидин-5-ил)метанол. Добавляют медленно n-BuLi (2,37 г; 36,0 ммоль) в охлажденный раствор диизопропиламина (3,72 г; 36,0 ммоль) в 50,0 мл ТГФ при -78C в атмосфере азота. Перемешивают реакционную смесь в течение 30 мин при той же температуре, затем добавляют 4,6-дихлорпиримидин (5,0 г; 33,0 ммоль), растворяют в 15 мл ТГФ. Перемешивают полученную реакционную смесь в течение дополнительных 30 мин при-78C, затем добавляют циклопропанкарбальдегид (2,58 г; 36,8 ммоль). Дают реакционной смеси нагреться до комнатной температуры. Добавляют 50,0 мл воды и экстрагируют органическую фазу этилацетатом (350 мл), промывают насыщенным водным хлоридом натрия, затем сушат над безводным сульфатом натрия. Концентрируют при пониженном давлении под вакуумом с получением циклопропил-(4,6-дихлорпиримидин-5-ил)метанола (4,0 г, 54% выход). МС (ХИАД): m/z=220 [M+H]. Пример синтеза 97. Циклопропил-(4,6-дихлорпиримидин-5-ил)метанон. Добавляют частями оксид хрома (VI) (5,84 г; 58,4 ммоль) к циклопропил-(4,6-дихлорпиримидин-5 ил)метанолу (4,0 г; 18,2 ммоль) в 80,0 мл ацетона при 0C и перемешивают в течение 30 мин при 0C. Затем добавляют изопропиловый спирт, чтобы погасить избыточный реагент, и перемешивают в течение дополнительных 15 мин при комнатной температуре. Охлаждают до 0C и вливают в насыщенный раствор NaHCO3. Фильтруют через подложку целита, экстрагируют этилацетатом (350 мл) и промывают объединенные органические слои насыщенным водным хлоридом натрия. Сушат и концентрируют при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (2,0 г,выход 50%, 9,2 ммоль). МС (ИЭР): m/z=218 [M+H]. Соединения из следующих примеров синтеза можно получить, по существу, как описано в примере синтеза 97. Пример синтеза 98. 4-Хлор-3-циклопропил-1H-пиразоло[3,4-d]пиримидин. Медленно добавляют гидрат гидразина (2,67 г; 53,4 ммоль) в циклопропил-(4,6-дихлорпиримидин 5-ил)метанон (9,66 г; 44,5 ммоль), растворенный в 300 мл ТГФ, при комнатной температуре и перемешивают в течение 4 ч. По завершении разделяют реакционную смесь между водой и этилацетатом, собирают органический слой, промывают насыщенным водным хлоридом натрия, сушат над безводным сульфатом натрия и концентрируют под вакуумом. Очищают полученное указанное в заголовке соединение путем пропускания через небольшой слой силикагеля (60-120 меш), применяя хлороформ/метанол (97:3) в качестве элюента. МС (ИЭР): m/z=195 [M+H]. Пример синтеза 109. 4-Хлор-5-йод-7H-пирроло[2,3-d]пиримидин. Растворяли 6-хлор-7-деазапурин (10,75 г; 70 ммоль) и N-йодсукцинимид (16,8 г; 75 ммоль) в 400 мл безводного ДМФ и выдерживали при температуре окружающей среды в темноте в течение ночи. Выпаривали растворитель. Распределяли темный осадок между 500 мл этилацетата и 150 мл 10% Na2SO3. Органическую фракцию промывали 10% Na2SO3 (2100 мл), насыщенным водным раствором хлорида натрия (150 мл), высушивали над Na2SO4 и выпаривали. Кристаллизовали желтый осадок из этанола с получением 16,2 г (83%) 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидина в виде грязно-белых кристаллов. Выпаривали маточную жидкость, растворяли в толуоле и очищали с помощью флэш-хроматографии на силикагеле (74 см). Промывали колонку толуолом до тех пор, пока элюент не станет бесцветным, затем элюировали указанное в заголовке соединение 5% этилацетатом в толуоле с получением дополнительно 3,5 г продукта. Пример синтеза 110. 4-(4-Хлор-7H-пирроло[2,3-d]пиримидин-5-ил)-2-метилбут-3-ин-2-ол. Смешивали 4-хлор-5-йод-7H-пирроло[2,3-d]пиримидин (5,0 г; 1,0 экв.), 2-метил-3-бутин-2-ол(9,02 г; 6,0 экв.), TEA (1,68 г; 0,93 экв.), CuI (1,36 г; 0,4 экв.), ДМФ (62,5 мл) и ТГФ (187,5 мл) при комнатной температуре в атмосфере аргона. Перемешивали реакционную смесь при комнатной температуре в атмосфере аргона в течение 5 мин. Загружали Pd(PPh3)4 (1,03 г; 0,05 экв.) и перемешивали реакционную массу при 45C в течение 16 ч. За ходом реакции наблюдали с помощью ТСХ (65% CHCl3: 23% гексан: 12% ацетон). Концентрировали реакционную смесь под вакуумом. Загружали этилацетат и промывали водой и насыщенным водным хлоридом натрия. Органический слой высушивали над безводнымNa2SO4 и концентрировали под вакуумом. Кристаллизовали соединение в смеси 65% CHCl3:23% гексан:12% ацетон с получением желаемого соединения (3,35 г; 79,7%). Соединение из примеров синтеза 111, 112 можно получить, по существу, как описано в примере синтеза 110. Пример синтеза 113. 4-Хлор-5-(3-метил-3-(триметилсилилокси)бут-1-инил)-7H-пирроло[2,3-d]пиримидин. Смешивали 4-(4-хлор-7H-пирроло[2,3-d]пиримидин-5-ил)-2-метилбут-3-ин-2-ол (3,35 г; 1,0 экв.),имидазол (2,9 г; 3,0 экв.), TEA (2,16 г; 1,5 экв.) и диэтиловый эфир (84 мл) при комнатной температуре. Охлаждали реакционную смесь до 0C и добавляли триметилсилилхлорид (1,53 г; 1,0 экв.). Перемешивали реакционную смесь при комнатной температуре в течение 4 ч. За ходом реакции наблюдали с помощью ТСХ (5% MeOH в DCM). Вливали охлажденную деминерализованную воду и экстрагировали диэтиловым эфиром. Промывали эфирный слой насыщенным водным хлоридом натрия. Органический слой высушивали над безводным Na2SO4 и концентрировали под вакуумом с получением желаемого соединения (3,06 г, 70%). Пример синтеза 114. 4-Хлор-5-(3-(триметилсилилокси)проп)-1 инил)-7H-пирроло[2,3-d]пиримидин. Пример 1. Гидрохлорид 4-(4-(5-(2,4-дихлорфенил)-1H-имидазол-2-ил)пиперидин-1-ил)-1H-пиразоло[3,4d]пиримидина. Нагревали смесь 4-(4-(2,4-дихлорфенил)-1H-имидазол-2-ил)пиперидина (270 мг; 0,91 ммоль),4-хлор-1H-пиразоло[3,4-d] пиримидина (280 мг; 1,82 ммоль), Et3N (0,63 мл, 4,5 ммоль) и 2-пропанола(10 мл) при 90C в течение ночи в атмосфере N2. Смесь охлаждали до комнатной температуры и вливали в воду (100 мл). Экстрагировали смесь CH2Cl2 (2200 мл), объединяли органические слои и промывали водой (50 мл). Высушивали (Na2SO4) органический слой, фильтровали смесь и концентрировали фильтрат под вакуумом. Очищали осадок с помощью хроматографии на силикагеле (25 г SiO2, элюировалиCH2Cl2/СМА 4:1, 1000 мл) с получением 4-(4-(5-(2,4-дихлорфенил)-1H-имидазол-2-ил)пиперидин-1-ил)1H-пиразоло[3,4-d]пиримидина (304 мг, 80%). Добавляли хлористо-водородную кислоту (2,0 М водную,0,36 мл, 0,72 ммоль) в суспензию 4-(4-(5-(2,4-дихлорфенил)-1H-имидазол-2-ил)пиперидин-1-ил)-1Hпиразоло[3,4-d]пиримидина (300 мг, 0,72 ммоль) в метаноле (7 мл). Концентрировали смесь под вакуумом до сухого состояния. Растворяли осадок в метаноле (2 мл) и добавляли диэтиловый эфир (50 мл) с получением осадка. Осадок фильтровали, после чего промывали Et2O. Растворяли твердое вещество в метаноле (10 мл) и удаляли растворитель под вакуумом до сухого состояния с получением гидрохлорида 4-(4-(5-(2,4-дихлорфенил)-1H-имидазол-2-ил)пиперидин-1-ил)-1H-пиразоло[3,4-d]пиримидина (210 мг,65%) в виде грязно-белого твердого вещества. МС (ХИАД): m/z=414 [M+H].- 22016445 Соединения из примеров 2, 4-18, 23-28 и 31-33 Н можно получить, по существу, как описано в примере 1. Референсный пример 34. Гидрохлорид 4-4-[5-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперазин-1-ил-1Hпиразоло[3,4-d]пиримидина. Смешивали 1-[5-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперазин (195 мг; 1,00 экв.; 0,620 ммоль); 4-хлор-1H-пиразоло[3,4-d]пиримидин (1,00 экв.; 0,620 ммоль; 96 мг); изопропиловый спирт (3 мл); диизопропилэтиламин (1 мл) и нагревали в микроволновом реакторе при 80C в течение 60 мин. Выпаривали реакционную смесь и очищали на силикагеле с помощью 5% MeOH/DCM. Объединяли фракции с получением 213,2 мг (0,494 ммоль, 80%) 4-4-[5-(4-фтор-3-трифторметилфенил)-1Hимидазол-2-ил]пиперазин-1-ил-1H-пиразоло[3,4-d]пиримидина. Растворяли полученное свободное основание в DCM/MeOH и добавляли 1,0 экв. 1 М HCl в эфире. Концентрировали смесь с получением 237,2 мг гидрохлорида 4-4-[5-(4-фтор-3-трифторметилфенил)-1H-имидазол-2-ил]пиперазин-1-ил-1Hпиразоло[3,4-d]пиримидина. МС (ИЭР): m/z=443 [М+Н].- 25016445 Следующие соединения можно получить, по существу, как описано в примере 34. Пример 53-А. 4-(4-(4-(4-Фтор-3-(трифторметил)фенил)-1-метил-1H-имидазол-2-ил)пиперидин-1-ил)-3-метил-1Hпиразоло[3,4-d]пиримидин. В пробирку для микроволнового прибора помещали соль дигидрохлорида 4-[4-(4-фтор-3 трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидина (0,5 г; 1,0 экв.), 1-(4,6-дихлорпиримидин-5 ил)этанон (0,22 г; 1,0 экв.), TEA (1,2 мл, 8,0 экв.) и изопропиловый спирт (5 мл). Перемешивали реакционную смесь при 80C в течение 45 мин в микроволновом приборе. Наблюдали за ходом реакции с помощью ТСХ (10% MeOH в DCM). Охлаждали реакционную смесь до 0C и добавляли гидрат гидразина(0,07 мл, 1,2 экв.). Медленно доводили реакционную смесь до комнатной температуры. Перемешивали реакционную смесь при 80C в течение 45 мин в микроволновом приборе. Наблюдали за ходом реакции с помощью ТСХ (10% MeOH в DCM). Концентрировали реакционную массу под вакуумом. Загружали этилацетат, а затем промывали водой и насыщенным водным хлоридом натрия. Высушивали органический слой над безводным Na2SO4 и концентрировали при пониженном давлении. Очищали соединение с помощью колоночной хроматографии (силикагель 60-120 меш, DCM - метанол). Кристаллизовали продукт в диэтиловом эфире и фильтровали с получением 4-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-метил 1H-имидазол-2-ил)пиперидин-1-ил)-3-метил-1H-пиразоло[3,4-d]пиримидина (0,254 г, 50,29%). МС (М+Н): m/z=460,5. Соединение из следующего примера можно получить, по существу, как описано в примере 53-А.- 27016445 Пример 53-С. 3-Этинил-4-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-метил-1H-имидазол-2-ил)пиперидин-1-ил)-1Hпиразоло[3,4-d]пиримидин. В пробирку для микроволнового прибора загружали 4-[4-(4-фтор-3-трифторметилфенил)-1-метил 1H-имидазол-2-ил]пиперидин.2HCl (0,2 г; 1,0 экв.), 4-хлор-3-триметилсиланилэтинил-1H-пиразоло[3,4d]пиримидин (0,151 г; 1,1 экв.), диизопропилэтиламин (0,72 мл, 7,6 экв.) и изопропиловый спирт (6 мл). Перемешивали реакционную смесь при 80C в течение 45 мин в микроволновом приборе. Наблюдали за ходом реакции с помощью ТСХ (30% ацетон в DCM). Концентрировали реакционную смесь. Загружали этилацетат, а затем промывали водой и насыщенным водным хлоридом натрия. Высушивали органический слой над безводным Na2SO4 и концентрировали при пониженном давлении. Очищали соединение с помощью колоночной хроматографии (силикагель 100-200 меш, DCM - ацетон) с получением 4-4-[4-(4 фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-ил-3-триметилсиланилэтинил 1H-пиразоло[3,4-d]пиримидина (0,18 г, 60,56%). Загружали 4-4-[4-(4-фтор-3-трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидин-1-ил-3 триметилсиланилэтинил-1H-пиразоло[3,4-d]пиримидин (0,18 г; 1,0 экв.), KOH (0,057 г; 3,0 экв.), MeOH(3,7 мл) и DCM (1,85 мл) в круглодонную колбу. Перемешивали реакционную смесь при комнатной температуре в течение 40 мин. Наблюдали за ходом реакции с помощью ТСХ (30% ацетон в DCM). Концентрировали реакционную смесь под вакуумом. Загружали DCM и промывали водой и насыщенным водным хлоридом натрия. Высушивали органический слой над безводным Na2SO4 и концентрировали под вакуумом с получением 3-этинил-4-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-метил-1H-имидазол-2 ил)пиперидин-1-ил)-1H-пиразоло[3,4-d]пиримидина (0,074 г, 46,07%). ЖХ/МС = 470,4 (М+1). Следующее соединение можно получить, по существу, как описано в примере 53-С. В пробирку для микроволнового прибора загружали дигидрохлорид 4-[4-(4-фтор-3 трифторметилфенил)-1-метил-1H-имидазол-2-ил]пиперидина (0,5 г; 1,0 экв.), 4-хлор-3-(3-метил-3 триметилсиланилоксибут-1-инил)-1H-пирроло[3,4-d]пиримидин (0,47 г; 1,0 экв.), диизопропилэтиламин(2 мл, 7,6 экв.) и изопропиловый спирт (10 мл). Перемешивали реакционную смесь при 80C в течение 1 ч в микроволновом приборе. Наблюдали за ходом реакции с помощью ТСХ (10% MeOH в DCM). Концентрировали реакционную смесь под вакуумом. Загружали этилацетат и промывали водой и насыщенным водным хлоридом натрия. Высушивали органический слой над безводным Na2SO4 и концентрировали под вакуумом. Очищали полученное соединение с помощью колоночной хроматографии (силикагель 60-120 меш, DCM - MeOH). Кристаллизовали продукт в диэтиловом эфире и фильтровали с получением желаемого продукта 4-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-метил-1H-имидазол-2-ил)пиперидин-1 ил)-5-(3-метил-3-(триметилсилилокси)бут-1-инил)-7H-пирроло[2,3-d]пиримидина (0,21 г, 23%). Загружали 4-(4-(4-(4-фтор-3-(трифторметил)фенил)-1-метил-1 Н-имидазол-2-ил)пиперидин-1-ил)-5(3-метил-3-(триметилсилилокси)бут-1-инил)-7H-пирроло[2,3-d]пиримидин (0,21 г; 1,0 экв.) и ТГФ (2 мл) в круглодонную колбу. Охлаждали реакционную смесь до 0C. Добавляли по каплям фторид тетрабутиламмония (0,19 мл, 2,0 экв.). Перемешивали реакционную смесь при комнатной температуре в течение 40 мин. Наблюдали за ходом реакции с помощью ТСХ (10% MeOH в DCM). Загружали этилацетат и промывали насыщенным раствором бикарбоната натрия и насыщенным водным хлоридом натрия. Высушивали органический слой над безводным Na2SO4 и концентрировали под вакуумом. Кристаллизовали(0,11 г, 64%). МС (М+Н): m/z=474,6. Следующие соединения можно получить, по существу, как описано в примере 53-Е. 4-4-[4-(3-трифторметилфенил)-1H-имидазол-2-ил]пиперидин-1-ил-1H-пиразоло В 22-литровую четырехгорлую круглодонную колбу (оборудованную воронкой для дозирования,холодильником, скруббером, механической мешалкой и заполненную азотом) добавляли 3-трифторметилацетофенон (1500 г; 1,00 экв.; 7,97 моль) и дихлорметан (7,5 л). Перемешивали полученный прозрачный бесцветный раствор при комнатной температуре с добавлением раствора брома (1274 г; 1,00 экв.; 7,97 моль) в дихлорметане при комнатной температуре через делительную воронку в течение 4 ч. Гасили реакцию путем медленного добавления насыщенного водного NaHCO3 (2000 мл), контролируя температуру с помощью ледяной бани на уровне менее чем 25C. Разделяли фазы и промывали органический слой насыщенным водным хлоридом натрия (2000 мл), затем высушивали раствор над сульфатом натрия, фильтровали и концентрировали с получением прозрачного бесцветного масла. Очищали полученное неочищенное масло с помощью хроматографии на силикагеле (ступенчатый градиент, от 20 до 50% CH2Cl2 в гептане) с получением 2-бром-1-(3-трифторметилфенил)этанона (1667 г; 6,24 моль; 78%) в виде прозрачного, бесцветного масла. Загружали 2-бром-1-[3(трифторметил)фенил]-1-этанон (1664,7 г; 1,00 экв.; 6,23 моль) и тетрагидрофуран (7500 мл) в 12-литровую трехгорлую круглодонную колбу (оборудованную холодильником с водяным охлаждением, механической мешалкой, охлаждающей баней и заполненную азотом). Одной порцией загружали азид натрия (425,6 г; 1,05 экв.; 6,55 моль). Количественно переносили в сосуд с водой(135 мл). Перемешивали бледно-желтую суспензию при комнатной температуре в атмосфере азота. Через
МПК / Метки
МПК: A61P 35/00, A61K 31/437, C07D 487/04
Метки: киназы, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-16445-ingibitory-p70-s6-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы p70 s6 киназы</a>
Ингибиторы c – fms киназы

Номер патента: 13250
Опубликовано: 30.04.2010
Авторы: Дежарле Рене, Флорес Кристофер, Менти Карл Л., Баллентайн Шелли К., Моллой Кристофер Дж., Иллиг Карл Р., Мигалла Санатх, Чэнь Цзиньшэн, Уолл Марк Дж., Рудольф М.Джонатан, Уилсон Кен
МПК: A61K 31/4439
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединения формулы Iили их сольват, гидрат, таутомер или фармацевтически приемлемая соль, гдеА представляет собой фенил или пиридил, каждый из которых может быть замещен одним из хлора, фтора, метила, -N3, -NH2, -NH(C1-6алкил), -N(C1-6алкил)2, -S(С1-6алкил), -О(С1-6алкил) или 4-аминофенила;W представляет собой пирролил, имидазолил, изоксазолил, оксазолил, 1,2,4-триазолил или фуранил, каждый из которых может быть соединен через любой атом...
Ингибиторы киназы

Номер патента: 15189
Опубликовано: 30.06.2011
Авторы: Худзиак Кевин Джон, Мэйдер Мэри Маргарет, Блас Де Блас Хесус Андрес, Бастиан Джолие Анне, Побанс Марк Эндрю, Де Дьес Альфонсо, Ших Чуан, Ли Течао, Лопес Де Уралде-Гармениа Беатрис, Майерс Майкл Рэй, Чжун Боюй
МПК: C07D 401/14, A61K 31/44, A61K 31/497...
Метки: ингибиторы, киназы
Формула / Реферат:
1. Соединение формулы Iгде Z выбран из группы, которая включаетX выбран из группы, которая включаетR1 представляет собой С1-С7алкил, необязательно замещенный от одного до шести заместителями, выбранными из группы, которая включает галоген и C1-C4алкилгалоген; С3-С6циклоалкил, необязательно замещенный одним или двумя заместителями, выбранными из группы, которая включает C1-C4алкил и трифторметил; или триметилсилил;R2 представляет собой фенил,...
Сульфоксиминные производные как ингибиторы p38 мар киназы

Номер патента: 15497
Опубликовано: 31.08.2011
Авторы: Лохрай Видья Бхушан, Чаттерджи Абхиджит, Чакрабарти Ганес, Пател Панкадж Раманбхай, Джаин Мукул Р., Лохрай Брадж Бхушан, Шетти Шанкар Джайрам, Пател Гаутам Д.
МПК: A61P 29/00, A61K 31/427, C07D 417/04...
Метки: мар, ингибиторы, киназы, производные, сульфоксиминные
Формула / Реферат:
1. Соединения общей формулы (I)или их фармацевтически приемлемые соли,где R1, R2 могут быть одинаковыми или различными и независимо представляют собой атом водорода, необязательно замещенные группы, выбранные из линейного или разветвленного (C1-С6)алкила, (С3-С7)циклоалкила, фенила или нафтила;R3 и R4могут быть одинаковыми или различными и независимо могут быть выбраны из необязательно замещенного линейного или разветвленного (C1-С6)алкила,...
Замещенные пиразолы как ингибиторы р38 киназы

Номер патента: 5205
Опубликовано: 30.12.2004
Автор: undefined
МПК: C07D 401/04, A61P 29/00, A61K 31/415...
Метки: замещенные, ингибиторы, киназы, пиразолы
Формула / Реферат:
1. Соединение, таутомер этого соединения либо фармацевтически приемлемая соль этого соединения или таутомера, где структура упомянутого соединения соответствует формуле IB относительно R1: R1 выбирают из группы, включающей водород, гидроксил, алкил, циклоалкил, алкенил, циклоалкенил, алкинил, арил, гетероциклил, циклоалкилалкил, циклоалкенилалкил, гетероциклилалкил, галоидалкил, галоидалкенил, галоидалкинил, гидроксиалкил, гидроксиалкенил,...
Соединения и композиции, как ингибиторы киназы c-kit и pdgfr

Номер патента: 16329
Опубликовано: 30.04.2012
Авторы: Ли Сяолинь, Мольтени Валентина, Лю Сяодон
МПК: A61K 31/506, A61P 35/00, A61P 37/00...
Метки: соединения, композиции, киназы, pdgfr, c-kit, ингибиторы
Формула / Реферат:
1. Соединение формулы Iв которой R1, R2a и R2b все независимо представляют собой водород;R3 и R4 вместе с атомом углерода, к которому R3 и R4 присоединены, образуют фенил, необязательно замещенный 1-3 радикалами, независимо выбранными из группы, включающей галоген, C1-C6-алкил, галогензамещенный C1-C6-алкил и -X2S(O)0-2R5a, гдеX2 выбран из группы, включающей связь и C1-C4-алкилен;R5a независимо выбран из водорода и C1-C6-алкила;и его...
Предыдущий патент: Опорный каркас фотоэлектрической панели и наружная стена здания с такими каркасами
Следующий патент: Производные n5-(2-этоксиэтил)-n3-(2-пиридинил)-3,5-пиперидиндикарбоксамида, предназначенные для применения в качестве ингибиторов ренина
Случайный патент: Способ извлечения углеводородных соединений и устройство для извлечения углеводородов из газообразных побочных продуктов