Соединения и композиции как ингибиторы активирующей канал протеазы
Номер патента: 16199
Опубликовано: 30.03.2012
Авторы: Талли Дейвид С., Видаль Аньес, Бурсулая Бадри, Спраггон Глен, Чаттерджи Арнаб К.





























Формула / Реферат
1. Соединение формулы (1)

или его фармацевтически приемлемые соли,
в которой O-(CR2)p-R2 обозначает заместитель в любом положении кольца А;
J обозначает
, пиридил или пиперидинил;
где один или более из Z1, Z2, Z3, Z4, Z5, Z6 и Z7 обозначает гетероатом, выбранный из N, NR, О или S, а другие атомы Z1-Z7 обозначают СН при условии, что J не обозначает триазолил;
В обозначает
или (CR2)k-R5;
Y обозначает связь, -SO2-, -NHCO- или -O-(CO)-;
R1 обозначает галоген, C1-C6-алкил, CF3, OCF3, фенил, -(CR2)l-NR6R7, -(CR2)l-C(=NR)-NR6R7, -C(O)-(CR2)l-NR6R7, -(CR2)l-NR-SO2R6, -(CR2)l-NR-C(O)-R6, -(CR2)l-SO2NR6R7 или -(CR2)l-OR6;
R2 обозначает фенил или циклогексил, каждый из которых является незамещенным или замещенным заместителем, выбранным из галогена, SO2(C1-6алкила), незамещенного C1-6алкила, галогенированного C1-6алкила, незамещенного C1-6алкоксила или галогенированного C1-6алкоксила;
R3 обозначает C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил или -(CR2)l-R5;
альтернативно, NH-Y-R3 вместе образуют NH2;
R4 обозначает пиперидинил, циклогексил, фенил, или
или
;
R5 обозначает арил или R5 обозначает пиперидинил, если В обозначает (CR2)k-R5;
R, R6 и R7 независимо обозначают H или C1-C6-алкил;
k равно 2-3;
l равно 0-1;
р равно 1;
m и n независимо равны 1-2;
х равно 0-3;
при условии, что R4 обозначает пиперидинил, если NH-Y-R3 вместе образуют NH2.
2. Соединение по п.1, в котором J обозначает тиофенил, тиазолил, фенил, пиридил, индазолил или пиперидинил.
3. Соединение по п.1, в котором Y обозначает связь, -SO2- или -O-(СО)-.
4. Соединение по п.1 формулы (2)

в которой R2 обозначает незамещенный фенил, фенил, замещенный галогеном, SO2(C1-6алкилом), незамещенным галогенированным C1-6алкилом, замещенным галогенированным C1-6алкилом или C1-6алкоксилом;
J обозначает фенил;
R3 обозначает C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил или -(CR2)l-R5 или
NH-Y-R3 вместе образуют NH2;
каждый R в (CR2) обозначает Н или C1-C6-алкил;
m и n независимо равны 1-2;
р равно 1;
значения R1, R4, R5, R, Y и х определены в п.1;
или его фармацевтически приемлемые соли.
5. Соединение по п.4, в котором х равно 1-2.
6. Соединение по п.4, в котором Y обозначает SO2.
7. Соединение по п.4, в котором R2 обозначает фенил или фенил, замещенный галогеном.
8. Соединение по п.4, в котором R3 обозначает C1-C6-алкил или бензил.
9. Соединение по п.4, в котором R4 обозначает пиперидинил.
10. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из




или его фармацевтически приемлемые соли.
11. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из

или его фармацевтически приемлемые соли.
12. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из

или его фармацевтически приемлемые соли.
13. Соединение по п.1, где указанное соединение выбрано из группы, состоящей из

или его фармацевтически приемлемые соли.
14. Соединение по п.1, где указанное соединение представляет собой

или его фармацевтически приемлемые соли.
15. Соединение по п.1, где указанное соединение представляет собой

или его фармацевтически приемлемые соли.
16. Соединение по п.1, где указанное соединение представляет собой

или его фармацевтически приемлемые соли.
17. Фармацевтическая композиция, включающая соединение по любому из пп.1-16 в терапевтически эффективном количестве.
18. Соединение по любому из пп.1-16 или его фармацевтически приемлемые соли и второе терапевтическое средство, выбранное из противовоспалительного, бронхолитического, антигистаминного, противокашлевого средства, антибиотика и ДНКазы.
19. Применение соединений по любому из пп.1-16 для ингибирования активирующей канал протеазы в клетках или системе тканей или у млекопитающего, в котором указанная активирующая канал протеаза представляет собой простазин, матриптазу (MTSP-1) или трипсин.
20. Применение соединений по любому из пп.1-16 для приготовления лекарственного средства, предназначенного для лечения патологического состояния, опосредуемого активирующей канал протеазой в системе клеток или тканей или у млекопитающего, и необязательно в комбинации со вторым терапевтическим средством, в котором указанная активирующая канал протеаза представляет собой простазин, матриптазу (MTSP-1) или трипсин.
21. Применение соединений по п.20, в котором указанное патологическое состояние связано с перемещением жидкости через переносящие ионы эпителии или с накоплением слизи и мокроты на тканях органов дыхания либо с их комбинацией.
22. Применение соединений по п.20, в котором указанным патологическим состоянием является муковисцидоз, первичная цилиарная дискинезия, карцинома легких, хронический бронхит, хроническое обструктивное заболевание легких, астма или инфекция дыхательных путей.
23. Применение соединений по п.20, в котором указанное второе терапевтическое средство представляет собой противовоспалительное, бронхолитическое, антигистаминное, противокашлевое средство, антибиотик или ДНКазу и вводится до, одновременно или после соединения по любому из пп.1-16.
24. Применение соединений по п.19 или 20, где указанная активирующая канал протеаза представляет собой простазин.
25. Применение соединений по п.19 или 20, где указанная система клеток или тканей включает эпителиальные клетки бронхов.
Текст
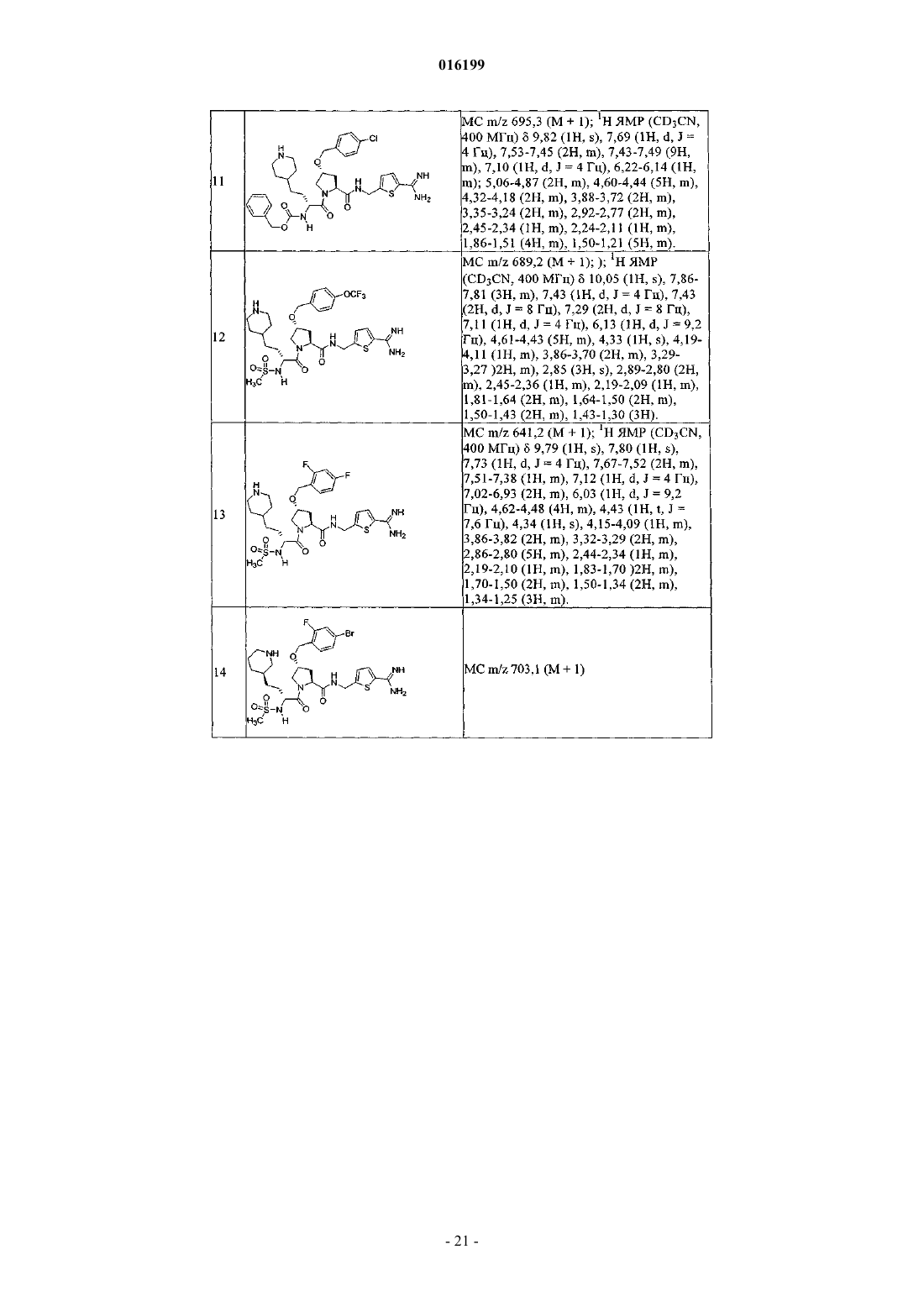
СОЕДИНЕНИЯ И КОМПОЗИЦИИ КАК ИНГИБИТОРЫ АКТИВИРУЮЩЕЙ КАНАЛ ПРОТЕАЗЫ В изобретении приведены соединения и содержащие их фармацевтические композиции, которые применимы для модулирования активирующих канал протеаз, и способы применения таких соединений для лечения, улучшения протекания или предупреждения патологического состояния,связанного с активирующей канал протеазой, включая, но не ограничиваясь только ими, простазин,PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E), TMPRSS2, TMPRSS3, TMPRSS4 016199 Перекрестная ссылка на родственные заявки Заявка на данное изобретение является родственной предварительной заявке U.S.60/891474, поданной 23 февраля 2007 г., и предварительной заявке US60/884334, поданной 10 января 2007 г., которые во всей своей полноте включены в настоящее изобретение в качестве ссылки, по заявке на изобретение испрашивается приоритет по предварительной заявке Область техники, к которой относится изобретение Настоящее изобретение в целом относится к ингибиторам активирующей канал протеазы (САР). Уровень техники Простазин является трипсиноподобной серинпротеазой, которая содержится в различных тканях млекопитающих. Он является закрепленной на мембране протеазой, которая экспрессируется на внеклеточной мембране клеток, но которая также может выделяться в жидкости организма, такой как сперма,моча и жидкость, находящаяся на поверхности дыхательных путей. Простазин (PRSS8) вместе с протеазами, такими как матриптаза, САР 2, САР 3, трипсин, PRSS22, TMPRSS11, катепсин А и нейтрофильная эластаза, могут стимулировать активность чувствительного к амилориду эпителиального натриевого канала (ENaC). Ингибирование этих ферментов может включать изменения переноса ионов в эпителии и тем самым влиять на равновесие переноса через эпителиальные мембраны. Например, полагают, что ингибирование САР в почках стимулирует диурез, тогда как ингибирование САР в дыхательных путях стимулирует выведение слизи и мокроты из легких. Поэтому ингибирование САР в почках можно использовать терапевтически для лечения гипертензии. Ингибирование САР в дыхательных путях предупреждает накопление выделений из органов дыхания, которые в противном случае могут привести к вторичным бактериальным инфекциям у пациента. Описание изобретения Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам применения таких соединений для модулирования активирующих канал протеаз (САР). Например, соединения и композиции, предлагаемые в настоящем изобретении, можно использовать для модулирования простазина, PRSS22, TMPRSS11 (например, TMPRSS11 В, TMPRSS11E), TMPRSS2, TMPRSS3,TMPRSS4 (MTSP-2), матриптазы (MTSP-1), САР 2, САР 3, трипсина, катепсина А и нейтрофильной эластазы. Одним объектом настоящего изобретения являются соединения формулы (1) или их фармацевтически приемлемые соли,в которой O-(CR2)p-R2 обозначает заместитель в любом положении кольца А;J обозначает 5-12-членное моноциклическое или конденсированное карбоциклическое кольцо, гетероциклическое кольцо, содержащее N, О и/или S, арильное или гетероарильное кольцо при условии,что J не обозначает триазолил;R1 обозначает галоген, -(CR2)l-NR6R7, -(CR2)l-NRC(=NR)-NR6R7, -(CR2)l-C(=NR)-NR6R7,-C(O)-(CR2)l-NR6R7, -(CR2)l-NR-SO2R6, -(CR2)l-NR-C(O)-R6, -(CR2)l-SO2NR6R7, или -(CR2)l-OR6, или необязательно замещенные C1-C6-алкоксигруппу, C1-C6-алкил, C2-C6-алкенил или C2-C6-алкинил или необязательно замещенное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил;R2, R4 и R5 независимо обозначают необязательно замещенное 5-12-членное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил илиR4 обозначает Н, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил или -CR= , где Р обозначает С или N и кольцо Е вместе с Р образует необязательно замещенное 5-12-членное моноциклическое или конденсированное кольцо;-1 016199 при условии, что R4 обозначает пиперидинил, если NH-Y-R3 вместе образуют NH2; и также при условии, что R5 обозначает пиперидинил, если В обозначает (CR2)k-R5. В приведенной выше формуле (1) J может обозначать тиофенил, тиазолил, фенил, пиридил, индазолил, пиперидинил или пирролидинил. В других примерах R2 может обозначать фенил или циклогексил, каждый из которых необязательно может содержать в качестве заместителей галоген, SO2(C1-C6-алкил) или необязательно замещенныйC1-C6-алкил или C1-C6-алкоксигруппу, такие как необязательно галогенированный C1-C6-алкил илиC1-C6-алкоксигруппа. В одном варианте осуществления настоящее изобретение относится к соединениям формулы (2) в которой R2 и J независимо обозначают необязательно замещенный 6-членный арил;m, n и р независимо равны 1-2. В приведенных выше формулах (1) и (2) Y может обозначать связь, SO2 или -О-(СО)-. В других примерах R1 обозначает галоген, C1-C6-алкил, CF3, OCF3, фенил, -(CR2)l-NR6R7,-(CR2)l-C(=NR)-NR6R7, -C(O)-(CR2)l-NR6R7, -(CR2)l-NR-SO2R6, -(CR2)l-NR-C(O)-R6, -(CR2)i-SO2NR6R7 или-(CR2)l-OR6, где каждый l равен 0-1; и R, R6 и R7 независимо обозначают Н или C1-C6-алкил. В приведенных выше формулах (1) и (2) R4 может обозначать необязательно замещенное 5-6 членное карбоциклическое кольцо, гетероциклическое кольцо, арил, гетероарил или -CR= , где Р обозначает С или N и кольцо Е вместе с Р образует необязательно замещенное 5-6-членное моноциклическое кольцо. Например, R4 может обозначать необязательно замещенный пиперидинил, циклогексил,фенил,или. В предпочтительных примерах R3 в формуле (2) обозначает C1-C6-алкил или необязательно замещенный бензил. В некоторых примерах Y обозначает SO2. В других примерах R4 обозначает необязательно замещенный пиперидинил. В других примерах J и R2 независимо обозначают необязательно замещенный фенил. Например, J может быть замещен с помощью 1-3 R1 (т.е. когда х равно 1-3) и R2 необязательно может быть замещен галогеном. Другим объектом настоящего изобретения являются фармацевтические композиции, включающие соединение формулы (1) или (2) и фармацевтически приемлемый инертный наполнитель. Настоящее изобретение также относится к способам модулирования активирующей канал протеазы, включающим введение в систему или млекопитающему терапевтически эффективного количества соединения, описывающегося формулой (1) или (2), или его фармацевтически приемлемых солей или фармацевтических композиций и тем самым проведение модулирования указанной активирующей канал протеазы. В одном варианте осуществления настоящее изобретение относится к способу ингибирования активирующей канал протеазы, включающему введение в систему клеток или ткани или млекопитающему терапевтически эффективного количества соединения, описывающегося формулой (1) или (2) или его фармацевтически приемлемых солей или фармацевтических композиций; где указанной активирующей канал протеазой является простазин, PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E),TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптаза (MTSP-1), CAP2, САР 3, трипсин, катепсин А или нейтрофильная эластаза, и тем самым ингибирование указанной активирующей канал протеазы. В предпочтительных примерах настоящее изобретение относится к способу ингибирования простазина. Другим объектом настоящего изобретения является способ улучшения протекания или лечения патологического состояния, опосредуемого активирующей канал протеазой, включающий введение в систему клеток или ткани или млекопитающему эффективного количества соединения, описывающегося формулой (1) или (2), или его фармацевтически приемлемых солей или фармацевтических композиций и необязательно в комбинации со вторым терапевтическим средством; где указанной активирующей канал протеазой является простазин, PRSS22, TMPRSS11 (например, TMPRSS11B, TMPRSS11E), TMPRSS2,TMPRSS3, TMPRSS4 (MTSP-2), матриптаза (MTSP-1), САР 2, САР 3, трипсин, катепсин А или нейтрофильная эластаза, и тем самым лечение указанного патологического состояния.-2 016199 Кроме того, настоящее изобретение относится к соединениям формулы (1) или (2), предназначенным для применения для лечения патологического состояния, опосредуемого активирующей канал протеазой. Настоящее изобретение также относится к применению соединения формулы (1) или (2) необязательно в комбинации со вторым терапевтическим средством для приготовления лекарственного средства,предназначенного для лечения патологического состояния, опосредуемого активирующей канал протеазой. В предпочтительных примерах соединения, предлагаемые в настоящем изобретении, можно использовать для лечения патологического состояния, опосредуемого простазином. В одном варианте осуществления второе терапевтическое средство может представлять собой противовоспалительное, бронхолитическое, антигистаминное, противокашлевое средство, антибиотик или ДНКазу и его вводят до,одновременно или после соединения формулы (1) или (2). В некоторых примерах соединения, предлагаемые в настоящем изобретении, вводят в эпителиальные клетки бронхов, предпочтительно в эпителиальные клетки бронхов человека. Примеры патологических состояний, протекание которых можно улучшать или которые можно лечить с использованием соединений, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, патологическое состояние, связанное с перемещением жидкости через переносящие ионы эпителии или с накоплением слизи и мокроты на тканях органов дыхания, или с их комбинацией. В некоторых примерах патологическим состоянием, которое можно опосредовать с использованием соединений, предлагаемых в настоящем изобретении, является муковисцидоз, первичная цилиарная дискинезия, карцинома легких, хронический бронхит, хроническое обструктивное заболевание легких, астма или инфекция дыхательных путей. Определения"Алкил" означает фрагмент и структурный элемент других групп, например галогензамещенного алкила и алкоксигруппы, и он может обладать линейной или разветвленной цепью. Необязательно замещенный алкил, алкенил или алкинил при использовании в настоящем изобретении может быть необязательно галогенирован (например, CF3) или содержащиеся в нем один или большее количество атомов углерода могут быть замещены или заменены гетероатомом, таким как NR, О или S (например,-ОСН 2 СН 2 О-, алкилтиолы, тиоалкоксигруппа, алкиламины и т.п.)."Арил" означает моноциклическое или конденсированное бициклическое ароматическое кольцо,содержащее атомы углерода. Например, арил может представлять собой фенил или нафтил. "Арилен" означает двухвалентный радикал, образованный из арильной группы."Гетероарил" при использовании в настоящем изобретении является таким, как определенный выше арил, в котором один или большее количество элементов кольца представляет собой гетероатом. Примеры гетероарилов включают, но не ограничиваются только ими, пиридил, индолил, индазолил, хиноксалинил, хинолинил, бензофуранил, бензопиранил, бензотиопиранил, бензо[1,3]диоксол, имидазолил, бензоимидазолил, пиримидинил, фуранил, оксазолил, изоксазолил, триазолил, тетразолил, пиразолил, тиенил и т.п."Карбоциклическое кольцо" при использовании в настоящем изобретении означает насыщенное или частично ненасыщенное моноциклическое, конденсированное бициклическое или мостиковое полициклическое кольцо, содержащее атомы углерода, которые необязательно могут быть замещены, например, с помощью =O. Примеры карбоциклических колец включают, но не ограничиваются только ими,циклопропил, циклобутил, циклопентил, циклогексил, циклопропилен, циклогексанон и т.п."Гетероциклическое кольцо" при использовании в настоящем изобретении является таким, как определенное выше карбоциклическое кольцо, в котором один или большее количество кольцевых атомов углерода представляет собой гетероатом. Например, гетероциклическое кольцо может содержать N, О, S,-N=, -S-, -S(O), -S(O)2- или -NR-, где R может обозначать водород, C1-C4-алкил или защитную группу. Примеры гетероциклических колец включают, но не ограничиваются только ими, морфолиновую группу, пирролидинил, пирролидинил-2-он, пиперазинил, пиперидинил, пиперидинилон, 1,4-диокса-8 азаспиро[4,5]дец-8-ил и т.п. Если не указано иное, то, когда заместитель указан как "необязательно замещенный," это означает,что заместитель представляет собой группу, которая может быть замещена одной или большим количеством групп, по отдельности и независимо выбранных из группы, включающей, например, необязательно галогенированный алкил, алкенил, алкинил, алкоксигруппу, алкиламиногруппу, алкилтиогруппу, алкинил, амидную группу, аминогруппу, включая моно- и дизамещенные аминогруппы, арил, арилоксигруппу, арилтиогруппу, карбонил, карбоциклил, цианогруппу, циклоалкил, галоген, гетероалкил,гетероалкенил, гетероалкинил, гетероарил, гетероциклил, гидроксигруппу, изоцианатную группу, изотиоцианатную группу, меркаптогруппу, нитрогруппу, О-карбамил, N-карбамил, О-тиокарбамил,N-тиокарбамил, С-амидную группу, N-амидную группу, S-сульфонамидную группу, N-сульфонамидную группу, С-карбоксигруппу, О-карбоксигруппу, пергалогеналкил, перфторалкил, силил, сульфонил, тиокарбонил, тиоцианатную группу, тригалогенметансульфонил и их производные, содержащие защитные группы. Защитные группы, которые могут образовывать соединения, защищенные по указанным выше заместителям, известны специалистам в данной области техники и описаны в литературе, в таких публи-3 016199 кациях, как Greene and Wuts, Protective Groups in Organic Synthesis, 3rd Ed., John WileySons, New York,NY, 1999, и Kocienski, Protective Groups, Thieme Verlag, New York, NY, 1994, которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Термины "совместное введение" или "комбинированное введение" и т.п. при использовании в настоящем изобретении означают введение выбранных терапевтических средств одному пациенту и включают режимы лечения, при которых выбранные терапевтические средства необязательно вводят одним и тем же путем или в одно и то же время. Термин "фармацевтическая комбинация" при использовании в настоящем изобретении означает препарат, который получен смешиванием или объединением более одного активного ингредиента и включает и фиксированные, и нефиксированные комбинации активных ингредиентов. Термин "фиксированная комбинация" означает, что активные ингредиенты, например соединение формулы (1) и дополнительное средство, оба, вводят пациенту одновременно в виде одного препарата или дозированной формы. Термин "нефиксированная комбинация" означает, что активные ингредиенты, например соединение формулы (1) дополнительное средство, оба, вводят пациенту в виде отдельных препаратов одновременно, по отдельности или последовательно без наложения специальных ограничений по времени, и при таком введении в организме пациента создаются терапевтически эффективные уровни активных ингредиентов. Последнее также относится к смешанному лечению, например с введением 3 или большего количества активных ингредиентов. Термин "терапевтически эффективное количество" означает количество соединения, предлагаемого в настоящем изобретении, которое вызывает биологический или медицинский ответ в клетке, ткани, органе, системе, у животного или человека, который необходим исследователю, ветеринару, врачу или другому клиницисту. Термин "введение" соединения, предлагаемого в настоящем изобретении, следует понимать как доставку соединения, предлагаемого в настоящем изобретении, включая пролекарство соединения, предлагаемого в настоящем изобретении, индивидууму, нуждающемуся в лечении. При использовании в настоящем изобретении термины "лечение", "лечить" означают способ облегчения протекания или ослабление болезни и/или сопровождающих ее симптомов."Простазин" также можно назвать как активирующая канал протеаза человека (hCAP); активирующая канал протеаза-1 и PRSS8, MERPOPS ID S01.159. Способы осуществления настоящего изобретения Настоящее изобретение относится к соединениям, фармацевтическим композициям и способам применения таких соединений для модулирования активирующих канал протеаз (САР). Одним объектом настоящего изобретения являются соединения формулы (1) или их фармацевтически приемлемые соли,в которой O-(CR2)p-R2 обозначает заместитель в любом положении кольца А;J обозначает 5-12-членное моноциклическое или конденсированное карбоциклическое кольцо, гетероциклическое кольцо, содержащее N, О и/или S, арильное или гетероарильное кольцо, при условии,что J не обозначает триазолил; или (CR2)k-R5; В обозначаетR1 обозначает галоген, -(CR2)l-NR6R7, -(CR2)l-NRC(=NR)-NR6R7, -(CR2)l-C(=NR)-NR6R7,-C(O)-(CR2)l-NR6R7, -(CR2)l-NR-SO2R6, -(CR2)l-NR-C(O)-R6, -(CR2)l-SO2NR6R7, или -(CR2)l-OR6, или необязательно замещенные C1-C6-алкоксигруппу, C1-C6-алкил, C2-C6-алкенил или C2-C6-алкинил, или необязательно замещенное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил;R2, R4 и R5 независимо обозначают необязательно замещенное 5-12-членное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил илиR4 обозначает Н, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил или -CR= , где Р обозначает С или N и кольцо Е вместе с Р образуют необязательно замещенное 5-12-членное моноциклическое или конденсированное кольцо;k, m, n и р независимо равны 1-6; х равно 0-4; при условии, что R4 обозначает пиперидинил, если NH-Y-R3 вместе образуют NH2; и также при условии, что R5 обозначает пиперидинил, если В обозначает (CR2)k-R5. В других вариантах осуществления настоящее изобретение относится к соединению формулы (2) в которой R2 и J независимо обозначают необязательно замещенный 6-членный арил;m, n и р независимо равны 1-2. Альтернативно, k, m, n и р в приведенных выше формулах (1) и (2) могут независимо равняться 0-6. В предпочтительных примерах k в формуле (1) равно 2-3 и J обозначает гетероарил, такой как тиофенил. В других альтернативных вариантах осуществления Y в формулах (1) и (2) может обозначать -CO-. В каждой из приведенных выше формул J также может быть выбран из группы, включающей где один или большее количество Z1, Z2, Z3, Z4, Z5, Z6 и Z7 обозначает гетероатом, выбранный из группы, включающей N, NR, О и S, где R обозначает Н или C1-C6-алкил, и другие Z1-Z7 обозначают СН. В некоторых примерах по меньшей мере два из Z1, Z2, Z3, Z4, Z5, Z6 и Z7 обозначают гетероатом, выбранный из группы, включающей N, NR, О и S, где R обозначает Н или C1-C6-алкил, и другие Z1-Z7 обозначают CH. В приведенных выше формулах (1) и (2), в которых каждый необязательно замещенный фрагмент может содержать в качестве заместителей галоген, =O, аминогруппу, гуанидинил, амидиновую группу,необязательно замещенную C1-C6-алкоксигруппу; C1-C6-алкил, C2-C6-алкенил или C2-C6-алкинил, каждый из которых необязательно может быть галогенирован или необязательно может содержать атом углерода, который может быть замещен или заменен посредством N, О или S; CO2R8, -O-(CR2)l-C(O)-R8;-(CR2)l-R8, -(CR2)l-C(O)-R8 или -(CR2)l-SO2-R8; или их комбинации, где каждый R8 обозначает Н, C1-C6 алкил или необязательно замещенное карбоциклическое кольцо, гетероциклическое кольцо, арил или гетероарил. Настоящее изобретение также относится ко всем подходящим изотопозамещенным вариантам соединений, предлагаемых в настоящем изобретении, или их фармацевтически приемлемым солям. Изотопозамещенный вариант соединения, предлагаемого в настоящем изобретении, или его фармацевтически приемлемой соли определяется как такой, в котором по меньшей мере один атом заменен на атом, обладающий таким же атомным номером, но атомной массой, отличающейся от атомной массы, обычно обнаруживаемой в природе. Примеры изотопов, которые можно включать в соединения, предлагаемые в настоящем изобретении, и их фармацевтически приемлемые соли, включают, но не ограничиваются только ими, изотопы водорода, углерода, азота и кислорода, такие как 2 Н, 3 Н, 13 С, 14 С, 15N, 17 О, 18 О, 35S,18F и 36Cl. Некоторые изотопозамещенные варианты соединений, предлагаемых в настоящем изобретении, и их фармацевтически приемлемых солей, например, такие, в которые включены радиоактивные изотопы, такие как 3 Н или 14 С, применимы в лекарственных средствах и/или для исследовании распределения в соответствующих тканях. В предпочтительных примерах изотопы 3H и 14 С можно использовать вследствие легкости их приготовления и обнаружения. В других примерах замещение изотопами, такими как 2 Н, может обеспечить некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например увеличением периода полувыведения in vivo или уменьшением необходимой дозы. Изотопозамещенные варианты соединений, предлагаемых в настоящем изобретении, или их фармацевтически приемлемые соли обычно можно получить по стандартным методикам с использованием соответствующих изотопозамещенных вариантов подходящих реагентов.-5 016199 Соединения и композиции, предлагаемые в настоящем изобретении, можно применять для модулирования активирующей канал протеазы. Примеры активирующих канал протеаз, которые можно модулировать с использованием соединений и композиций, предлагаемых в настоящем изобретении, включают, но не ограничиваются только ими, простазин, PRSS22, TMPRSS11 (например, TMPRSS11B,TMPRSS11E), TMPRSS2, TMPRSS3, TMPRSS4 (MTSP-2), матриптазу (MTSP-1), CAP2, САР 3, трипсин,катепсин А и нейтрофильную эластазу. Соединения, предлагаемые в настоящем изобретении, также могут ингибировать активность протеаз, которые стимулируют активность ионных каналов, таких как эпителиальный натриевый канал, и их можно использовать для лечения связанных с САР заболеваний. Фармакология и применение. Соединения, предлагаемые в настоящем изобретении, модулируют активность активирующей канал протеазы, в частности трипсиноподобных серинпротеаз, таких как простазин, и как таковые они применимы для лечения заболеваний или нарушений, при которых простазин, например, способствует патологии и/или симптоматике заболевания. Заболевания, опосредуемые ингибированием активирующей канал протеазы, в частности трипсиноподобной серинпротеазой, такой как простазин, включают заболевания, связанные с регулированием объемов жидкостей, проходящих через эпителиальные мембраны. Например, объем жидкости, находящейся на поверхности дыхательных путей, является ключевым регулятором мукоцилиарного клиренса и поддержания легких в здоровом состоянии. Ингибирование активирующей канал протеазы будет стимулировать накопление жидкости на слизистой стороне эпителия дыхательных путей и тем самым стимулировать удаление слизи и предупреждать накопление слизи и мокроты на тканях дыхательных путей(включая дыхательные пути легких). Такие заболевания включают заболевания органов дыхания, такие как муковисцидоз, первичная цилиарная дискинезия, хронический бронхит, хроническое обструктивное заболевание легких (ХОЗЛ), астма, инфекции дыхательных путей (острые и хронические; вирусные и бактериальные) и карцинома легких. Заболевания, опосредуемые ингибированием активирующих канал протеаз, также включают заболевания, не представляющие собой заболевания органов дыхания, которые связаны с аномальным регулированием перемещения жидкости через эпителий, возможно, включая аномальные физиологические характеристики защитных жидкостей, находящихся на их поверхности, например ксеростомию (сухость слизистой оболочки рта) или сухой кератоконъюнктивит (сухость слизистой оболочки глаз). Кроме того, регулирование ENaC посредством САР в почках можно использовать для стимулирования диуреза и тем самым вызывать гипотензивный эффект. Хроническое обструктивное заболевание легких включает хронический бронхит или связанную с ним одышку, а также осложнение гиперреактивности дыхательных путей вследствие лечения другими лекарственными средствами, в частности применения другой ингаляционной лекарственной терапии. Настоящее изобретение также относится к лечению бронхита любого типа или генеза, включая, например, острый, арахиновый, катаральный, крупозный, хронический или гнойный туберкулезный бронхит. Астма включает наследственную астму (неаллергическую) и приобретенную (аллергическую) астму, слабую, средней тяжести, тяжелую астму, бронхиальную астму, астму напряжения, профессиональную астму и астму, вызванную бактериальной инфекцией. Астма также включает патологическое состояние, называющееся "бронхит младенцев", которое наблюдается у субъектов, например, в возрасте менее 4 или 5 лет, у которых наблюдается свистящее дыхание и которым поставлен или может быть поставлен диагноз "бронхит младенцев", установившаяся категория пациентов, вызывающих большую обеспокоенность медиков, которых в настоящее время часто называют страдающими от зарождающейся астмы или ранней стадии астмы. Применимость ингибитора активирующей канал протеазы, такого как ингибитор простазина, для лечения заболевания, опосредуемого ингибированием активирующей канал протеазы, можно исследовать путем определения ингибирующего воздействия ингибитора активирующей канал протеазы по методикам, описанным ниже, и по методикам, известным в данной области техники. В соответствии с вышеизложенным настоящее изобретение также относится к способу предупреждения или лечения любого из заболеваний или нарушений, описанных выше, у субъекта, нуждающегося в таком лечении, и этот способ включает введение указанному субъекту терапевтически эффективного количества соединения формулы (1) или (2) или его фармацевтически приемлемой соли. Для любого из указанных случаев применения необходимая доза меняется в зависимости от пути введения, конкретного подвергающегося лечению патологического состояния и необходимого эффекта (см. ниже "Введение и фармацевтические композиции").-6 016199 Введение и фармацевтические композиции. Обычно соединения, предлагаемые в настоящем изобретении, вводят в терапевтически эффективных количествах посредством любого из обычных и приемлемых путей, известных в данной области техники, по отдельности или в комбинации с одним или большим количеством терапевтических средств. Ингибиторы активирующей канал протеазы, предлагаемые в настоящем изобретении, также применимы в качестве совместно применяющихся терапевтических средств, предназначенных для использования в комбинации с другим лекарственным веществом. Например, ингибитор активирующей канал протеазы можно использовать в комбинации с противовоспалительным, бронхолитическим, антигистаминным или противокашлевым средством, антибиотиком или ДНКазой. Ингибитор активирующей канал протеазы и другое терапевтическое средство могут находиться в одной или разных фармацевтических композициях. Ингибитор активирующей канал протеазы можно смешать с другим терапевтическим средством в фиксированной фармацевтической композиции или можно вводить отдельно, до, одновременно или после другого терапевтического средства. Комбинацию можно использовать, в частности, для лечения муковисцидоза или обструктивных или воспалительных заболеваний дыхательных путей, таких как указанные выше в настоящем изобретении, например, в качестве средств, усиливающих терапевтическую активность таких лекарственных средств, или в качестве средств, обеспечивающих снижение необходимой дозы или ослабление возможных побочных эффектов таких лекарственных средств. Подходящие противовоспалительные лекарственные средства включают стероиды, в частности глюкокортикостероиды, такие как будезонид, бекламетазон дипропионат, флутиказон пропионат, циклезонид или мометазонфуроат, или стероиды, описанные в WO 02/88167, WO 02/12266, WO 02/100879,WO 02/00679 (в частности, примеры 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 и 101),WO 03/35668, WO 03/48181, WO 03/62259, WO 03/64445, WO 03/72592, WO 04/39827 и WO 04/66920; нестероидные агонисты глюкокортикоидного рецептора, такие как описанные в DE 10261874,WO 00/00531, WO 02/10143, WO 03/82280, WO 03/82787, WO 03/86294, WO 03/104195, WO 03/101932,WO 04/05229, WO 04/18429, WO 04/19935 и WO 04/26248; антагонисты LTB4, такие как монтелукаст и зафирлукаст; ингибиторы PDE4, такие как циломиласт (АРИФЛО GlaxoSmithKline),РОФЛУМИЛАСТ (Byk Gulden),V-11294A (Napp), BAY19-8004 (Bayer), SCH-351591 (Schering-Plough),АРОФИЛЛИН (Almirall Prodesfarma), PD189659/PD168787 (Parke-Davis), AWD-12-281 (Asta Medica),CDC-801 (Celgene), SelCID(TM) CC-10004 (Celgene), VM554/UM565 (Vernalis), T-440 (Tanabe), KW-4490(Kyowa Hakko Kogyo), и описанные в WO 92/19594, WO 93/19749, WO 93/19750, WO 93/19751,WO 98/18796, WO 99/16766, WO 01/13953, WO 03/104204, WO 03/104205, WO 03/39544, WO 04/000814,WO 04/000839, WO 04/005258, WO 04/018450, WO 04/018451, WO 04/018457, WO 04/018465,WO 04/018431, WO 04/018449, WO 04/019944, WO 04/019945, WO 04/045607 и WO 04/037805; и антагонисты аденозинового рецептора A2B, такие как описанные в WO 02/42298, которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Подходящие бронхолитические лекарственные средства включают агонисты бета-2 адренорецептора, такие как албутерол (салбутамол), метапротеренол, тербуталин, салметерол фенотерол,прокатерол, формотерол, кармотерол, или их фармацевтически приемлемые соли; и соединения (в свободной форме или в форме соли или сольвата) формулы (1), описанные в WO 00/75114, соединение формулы соединения формулы (1), описанные в WO 04/16601 (в свободной форме или в форме соли или сольвата),и соединения, описанные в ЕР 1440966, JP 05025045, WO 93/18007, WO 99/64035, US 2002/0055651,WO 01/42193, WO 01/83462, WO 02/66422, WO 02/70490, WO 02/76933, WO 03/24439, WO 03/42160,WO 03/42164, WO 03/72539, WO 03/91204, WO 03/99764, WO 04/16578, WO 04/22547, WO 04/32921,WO 04/33412, WO 04/37768, WO 04/37773, WO 04/37807, WO 04/39762, WO 04/39766, WO 04/45618,WO 04/46083 и WO 04/80964 или их фармацевтически приемлемые соли, которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Подходящие бронхолитические лекарственные средства также включают антихолинергические или антимускариновые средства, в частности ипатропийбромид, окситропийбромид, тиотропиевые соли иCHF 4226 (Chiesi), и гликопирролат, а также описанные в ЕР 424021, US 3714357, US 5171744,WO 01/04118, WO 02/00652, WO 02/51841, WO 02/53564, WO 03/00840, WO 03/33495, WO 03/53966,WO 03/87094, WO 04/018422 и WO 04/05285, которые во всей своей полноте включены в настоящее изобретение в качестве ссылки.-7 016199 Подходящие обладающие двойным действием противовоспалительные и бронхолитические лекарственные средства включают обладающие двойным действием агонисты бета-2 адренорецептора/антагонисты мускарина, такие как раскрытые в US 2004/0167167, WO 04/74246 иWO 04/74812. Подходящие антигистаминные лекарственные средства включают цетиризингидрохлорид, ацетаминофен, клемастин фумарат, прометазин, лоратидин, деслоратидин, дифенгидрамин и фексофенадингидрохлорид, активастин, астемизол, азеластин, эбастин, эпинастин, мизоластин и тефенадин, а также раскрытые в JP 2004107299, WO 03/099807 и WO 04/026841, которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Подходящие антибиотики включают макролидные антибиотики, например тобрамицин (TOBI). Подходящие терапевтические средства типа ДНКазы включают дорназу-альфа (ПУЛЬМОЗИМ), а высокоочищенный раствор рекомбинантной дезоксирибонуклеазы I человека I (рчДНКазы), которая селективно расщепляет ДНК. Дорназу-альфа используют для лечения муковисцидоз. Другими полезными комбинациями ингибиторов активирующей канал протеазы с противовоспалительными лекарственными средствами являются комбинации с антагонистами хемокиновых рецепторов,например CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 и CCR-10, CXCR1,CXCR2, CXCR3, CXCR4, CXCR5, в особенности с антагонистами CCR-5, такими как выпускающиеся фирмой Schering-Plough антагонисты SC-351125, SCH-55700 и SCH-D, выпускающиеся фирмойTakeda антагонисты, такие как N-4-6,7-дигидро-2-(4-метилфенил)-5 Н-бензоциклогептен-8 ил]карбонил]амино]фенил]метил]тетрагидро-N,N-диметил-2 Н-пиран-4-аминийхлорид (TAK-770), и антагонисты CCR-5, описанные в US 6166037, WO 00/66558, WO 00/66559, WO 04/018425 и WO 04/026873,которые во всей своей полноте включены в настоящее изобретение в качестве ссылки. Для лечения заболевания, опосредуемого ингибированием простазина, в контексте настоящего изобретения ингибитор активирующей канал протеазы, предлагаемый в настоящем изобретении, в свободной форме или в форме фармацевтически приемлемой соли, можно вводить в виде фармацевтических композиций любым обычным путем, в частности энтерально, например перорально, например в виде таблеток или капсул или в жидкой форме, парентерально, например в виде растворов или суспензий для инъекций, или назально, например в виде аэрозоля или другой распыляемой композиции с использованием подходящего устройства введения в нос, например назального распылительного устройства, такого как известные в данной области техники, или путем ингаляции, в особенности для использования с устройством типа небулайзер. Ингибитор активирующей канал протеазы можно вводить в фармацевтической композиции вместе с фармацевтически приемлемым разбавителем или носителем, такие композиции могут представлять собой сухие порошки, таблетки, капсулы и жидкости, а также растворы для инъекции, растворы для вливания или суспензии для ингаляции, которые можно приготовить с использованием других вспомогательных ингредиентов и методик, известных в данной области техники. Дозировка ингибитора активирующей канал протеазы в свободной форме или в форме фармацевтически приемлемой соли может зависеть от различных факторов, таких как активность и длительность воздействия активного ингредиента, тяжесть подвергающегося лечению патологического состояния,путь введения, вид, пол, этническое происхождение, возраст и масса тела субъекта и/или конкретное патологическое состояние. Типичная суточная доза, например, для перорального введения теплокровному животному, в частности человеку, массой примерно 75 кг, по оценке составляет от примерно 0,7 до примерно 1400 мг, более предпочтительно от примерно 5 до примерно 200 мг. Эту дозу можно вводить, например, в виде разовой дозы или нескольких разделенных доз, составляющих, например, от 5 до 200 мг. Если композиция представляет собой аэрозольный препарат, то он может содержать в качестве пропеллента, например, гидрофторалкан (HFA), такой как HFA134a или HFA227 или их смесь, и может содержать один или большее количество сорастворителей, известных в данной области техники, таких как этанол (до 20 мас.%), и/или одно или большее количество поверхностно-активных веществ, таких как олеиновая кислота или сорбитантриолеат, и/или одно или большее количество наполнителей, таких как лактоза. Если композиция представляет собой сухой порошкообразный препарат, то она может содержать, например, ингибитор активирующей канал протеазы, обладающий частицами диаметром до 10 мкм, необязательно совместно с разбавителем или носителем, таким как лактоза, обладающим желательным распределением частиц по размерам, и соединение, которое способствует защите от ухудшения характеристик продукта вследствие влажности, например стеарат магния. Если композиция представляет собой препарат для распыления, то она может содержать, например, ингибитор активирующей канал протеазы, растворенный или суспендированный в разбавителе, содержащем воду, сорастворитель, такой как этанол или пропиленгликоль, и стабилизатор, которым может являться поверхностно-активное вещество.-8 016199 В предпочтительных вариантах осуществления настоящее изобретение относится к соединениям формулы (1) или (2) в пригодной для вдыхания форме, например в виде аэрозоля или другой распыляемой композиции или в виде вдыхаемого измельченного вещества, например в микронизированной форме. Настоящее изобретение также относится к предназначенному для вдыхания лекарственному средству, включающему соединения, предлагаемые в настоящем изобретении, в пригодной для вдыхания форме вместе с устройством для ингаляции; и устройство для ингаляции, содержащее соединения, предлагаемые в настоящем изобретении, в пригодной для вдыхания форме. Способы получения соединений, предлагаемых в настоящем изобретении. Соединения, предлагаемые в настоящем изобретении, можно получить по методикам, приведенным в примерах. В описанных реакциях реакционноспособные функциональные группы, если они необходимы в конечном продукте (например, гидроксигруппа, аминогруппа, иминогруппа, тиогруппа или карбоксигруппа), чтобы избежать их нежелательного участия в реакциях, могут быть защищены с помощью защитных групп, известных в данной области техники. В соответствии со стандартной практикой можно использовать обычные защитные группы, например, см. T.W. Greene and P.G.M. Wuts in "Protective Groups inOrganic Chemistry", John Wiley and Sons, 1991. Соединения, предлагаемые в настоящем изобретении, также можно получить в виде фармацевтически приемлемой соли присоединения с кислотой по реакции соединения в форме свободного основания с фармацевтически приемлемой неорганической или органической кислотой. Альтернативно, фармацевтически приемлемую соль присоединения с основанием соединения, предлагаемого в настоящем изобретении, можно получить по реакции соединения в форме свободной кислоты с фармацевтически приемлемым неорганическим или органическим основанием. Альтернативно, соли соединений, предлагаемых в настоящем изобретении, можно получить с использованием солей исходных веществ или промежуточных продуктов. Соединения, предлагаемые в настоящем изобретении, в форме свободной кислоты или свободного основания можно получить из соответствующей соли присоединения с основанием или соли присоединения с кислотой соответственно. Например, соединение, предлагаемое в настоящем изобретении, в форме соли присоединения с кислотой можно превратить в соответствующее свободное основание путем обработки подходящим основанием (например, раствором гидроксида аммония, гидроксидом натрия и т.п.) Соединение, предлагаемое в настоящем изобретении, в форме соли присоединения с основанием можно превратить в соответствующую свободную кислоту путем обработки подходящей кислотой (например, хлористо-водородной кислотой и т.п.). Соединения, предлагаемые в настоящем изобретении, в неокисленной форме можно получить изN-оксидов соединений, предлагаемых в настоящем изобретении, путем обработки восстановительным реагентом (например, серой, диоксидом серы, трифенилфосфином, борогидридом лития, борогидридом натрия, трихлоридом, трибромидом фосфора и т.п.) в подходящем инертном органическом растворителе(например, ацетонитриле, этаноле, водном диоксане и т.п.) при температуре от 0 до 80 С. Пролекарственные производные соединений, предлагаемых в настоящем изобретении, можно получить по методикам, известным специалисту с общей подготовкой в данной области техники (дополнительные подробности см., например, в публикации Saulnier et al. (1994), Bioorganic and MedicinalChemistry Letters, vol. 4, p. 1985). Например, подходящие пролекарства можно получить по реакции не являющегося производным соединения, предлагаемого в настоящем изобретении, с подходящим карбамилирующим реагентом (например, 1,1-ацилоксиалкилкарбанохлоридатом, паранитрофенилкарбонатом и т.п.). Защищенные производные соединений, предлагаемых в настоящем изобретении, можно получить по методикам, известным специалисту с общей подготовкой в данной области техники. Подробное описание методик, использующихся для образования защитных групп и их удаления, приведено в публикации Т.W. Greene, "Protecting Groups in Organic Chemistry", 3rd edition, John Wiley and Sons, Inc., 1999. Соединения, предлагаемые в настоящем изобретении, обычно можно получить или сформировать способом, предлагаемым в настоящем изобретении, в виде сольватов (например, гидратов). Гидраты соединений, предлагаемых в настоящем изобретении, обычно можно получить путем перекристаллизации из смеси водного/органического растворителей с использованием органических растворителей, таких как диоксан, тетрагидрофуран или метанол. Соединения, предлагаемые в настоящем изобретении, можно получить в виде отдельных стереоизомеров по реакции рацемической смеси соединения с оптически активным разделяющим реагентом с образованием пары диастереоизомерных соединений, с разделением диастереоизомеров и извлечением оптически чистых энантиомеров. Хотя разделение энантиомеров можно проводить с использованием ковалентных диастереоизомерных производных соединений, предлагаемых в настоящем изобретении,предпочтительными являются диссоциирующие комплексы (например, кристаллические диастереоизомерные соли). Диастереоизомеры обладают разными физическими характеристиками (например, температурами плавления, температурами кипения, растворимостями, реакционными способностями и т.п.) и их можно легко разделить с использованием этих различий. Диастереоизомеры можно разделить с по-9 016199 мощью хроматографии или по методикам разделения, основанных на различиях растворимостей. Затем оптически чистый энантиомер вместе с разделяющим реагентом извлекают по любой эффективной методике, которая не приводит к рацемизации. Более подробное описание методик, применимых для выделения стереоизомеров соединений из рацемической смеси, приведено в публикации Jean Jacques, AndreCollet, Samuel H. Wilen, "Enantiomers, Racemates и Resolutions", John Wiley and Sons, Inc., 1981. Можно заключить, что соединения, предлагаемые в настоящем изобретении, можно получить так,как в качестве примеров описано в примерах, и соединения формул (1) и (2) можно получить способом,который включает:(b) необязательное превращение соли соединения, предлагаемого в настоящем изобретении, в несолевую форму;(c) необязательное превращение неокисленной формы соединения, предлагаемого в настоящем изобретении, в фармацевтически приемлемый N-оксид;(e) необязательное выделение отдельного изомера соединения, предлагаемого в настоящем изобретении, из смеси изомеров;(f) необязательное превращение не являющегося производным соединения, предлагаемого в настоящем изобретении, в фармацевтически приемлемое пролекарственное производное и(g) необязательное превращение пролекарственного производного соединения, предлагаемого в настоящем изобретении, в его форму, не являющуюся производным. Хотя получение исходных веществ специально не описано, эти соединения являются известными или их можно получить по методикам, аналогичным известным в данной области техники, или так, как описано в приведенных ниже примерах. Специалист в данной области техники должен понимать, что указанные выше превращения являются только примерами методик получения соединений, предлагаемых в настоящем изобретении, и что также можно использовать другие хорошо известные методики. Настоящее изобретение дополнительно поясняется, но не ограничивается приведенными ниже промежуточными продуктами (эталонные соединения) и примерами, которые иллюстрируют получение соединений, предлагаемых в настоящем изобретении. Эталонное соединение 1 1-В: 4-Пиперидинэтанол (1-А) (5 г, 39,7 ммоль) растворяют в ТГФ (тетрагидрофуран) (120 мл). Добавляют триэтиламин (5,6 мл, 40 ммоль) и раствор охлаждают до 0 С. Добавляют Boc2O (9,59 г,44 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и неочищенный остаток растворяют в этилацетате (120 мл). Раствор промывают 0,1 н. раствором НС 1 (3100 мл) и рассолом (1100 мл), сушат над MgSO4, фильтруют и растворитель выпаривают в вакууме и получают соединение 1-В в виде прозрачного масла. 1-С: К раствору спирта 1-В (2,39 г, 10,42 ммоль) в ДХМ (дихлорметан) добавляют трихлоризоциануровую кислоту (2,66 г, 11,46 ммоль) и раствор перемешивают и выдерживают при 0 С, затем добавляют каталитическое количество TEMPO. После добавления смесь нагревают до комнатной температуры и перемешивают в течение 1 ч и затем фильтруют через целит. Органическую фазу промывают насыщенным водным раствором Na2CO3, затем 1 н. раствором HCl и рассолом. Органический слой сушат Н-ЯМР (CDCl3, 400 МГц)9,72 (1 Н, s), 4,07-4,01 (2 Н, m), 2,70-2,57 (2 Н, m), 2,35-2,31 (2 Н, m), 2,051,94 (1 Н, m), 1,64-1,46 (2 Н, m), 1,39 (9 Н, s), 1,30-1,02 (2 Н, m). 1-D: К раствору триметилового эфира Cbzфосфоноглицина (2,8 г, 8,45 ммоль) в ТГФ при -78 С добавляют 1,1,3,3-тетраметилгуанидин (1,022 мл, 8,14 ммоль). Через 10 мин добавляют альдегид 1-С(1,76 г, 7,76 ммоль). Раствор помещают в баню со льдом и выдерживают при 0 С в течение 1 ч и затем раствору дают нагреться до комнатной температуры и перемешивают в течение еще 1 ч. Раствор разбавляют с помощью EtOAc, промывают 1 М раствором NaHSO4, сушат (MgSO4) и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии на силикагеле, элюируя смесью этилацетат/гексан от 0 до 100%, и получают соединение 1-D в виде белого твердого вещества. МС (масс-спектрометрия) m/z 333,2 (М+1). 1 Н-ЯМР (CDCl3, 400 МГц)7,35-7,33 (5 Н, m), 6,63 (1H, t, J=8 Гц), 6,30 (1 Н, bs), 5,12 (2 Н, s), 4,104,04 (2 Н, m), 3,73 (3H, s), 2,67-2,62 (2 Н, m), 2,14 (2 Н, t, J=6,8 Гц), 1,63-1,46 (3H, m), 1,43 (9 Н, s), 1,14-1,06(2 Н, m). 1-Е: В реактор Парра в атмосфере азота помещают соединение 1-D (1 г, 2,31 ммоль) и МеОН(100 мл). Раствор 3 раза вакуумируют и продувают азотом, затем добавляют катализатор, (R,R)-этилDuPHOS-Rh(COD)трифлат (30 мг, 0,04 ммоль). Смесь выдерживают в атмосфере водорода под давлением, равным 60 фунт-сила/дюйм 2, при комнатной температуре в течение 24 ч. Через 24 ч превращение в соединение 1-Е завершается с ЭИ (энантиомерный избыток) 99%, растворитель удаляют в вакууме и неочищенный продукт очищают с помощью хроматографии на силикагеле (гексаны/EtOAc). 1-F: Промежуточный продукт 1-Е растворяют в МеОН. Раствор продувают азотом и добавляютPd/уголь (5 вес.%, Degussa). Смесь помещают в атмосферу водорода под давлением, равным 50 фунтсила/дюйм 2, при комнатной температуре и встряхивают в течение 24 ч. Смесь продувают азотом и фильтруют через целит. Отфильтрованный осадок промывают с помощью МеОН и объединенные органические растворы концентрируют в вакууме. Добавляют гексаны и затем смесь выпаривают для азеотропной отгонки оставшегося метанола и получают соединение 1-F в виде масла, которое используют на следующей стадии без дополнительной очистки. МС m/z 201,4 (М+1 - Boc). 1 Н-ЯМР (CDCl3, 400 МГц)4,06-3,97 (2 Н, m), 3,63 (3H, s), 3,36-3,31 (1H, m), 2,63-2,50 (2 Н, m), 1,701,61 (1H, m), 1,61-1,43 (3H, m), 1,36 (3H, s), 1,55 (6 Н, s), 1,34-1,15 (3H, m), 1,02-1,97 (2 Н, m). 1-G: Неочищенное соединение 1-F (0,6 г, 1,99 ммоль) растворяют в ТГФ (10 мл) и к раствору добавляют 2,4,6-коллидин (315 мг, 2,38 ммоль) и метансульфонилхлорид (0,170 мл, 2,19 ммоль) и перемешивают в течение 2 ч. Реакционную смесь разбавляют с помощью EtOAc (50 мл) и раствор промывают 1 М раствором NaHSO4 (225 мл), рассолом (25 мл) и сушат (MgSO4). Растворитель удаляют в вакууме и неочищенный остаток очищают с помощью флэш-хроматографии, используя градиентный режим смеси гексанов и EtOAc, и получают соединение 1-G. МС m/z 279,4 (М+1 - Boc). 1 Н-ЯМР (CDCl3, 400 МГц)5,60-5,42 (1H, m), 3,99-3,96 (3H, m), 3,68 (3H, s), 2,86 (3H, m), 2,60-2,54(232 мг, 5,55 ммоль), растворенный в воде (4 мл). Реакционную смесь перемешивают в течение 1 ч. Растворитель выпаривают и остаток разбавляют с помощью EtOAc (25 мл) и промывают 1 н. растворомNaHSO4 (25 мл) и рассолом (25 мл) и сушат (MgSO4). Растворитель удаляют в вакууме и неочищенный остаток очищают с помощью хроматографии на силикагеле (градиентный режим гексаны/EtOAc) и получают эталонное соединение 1 в виде белого твердого вещества. МС m/z 265,4 (М+1 - Boc). 1 Н-ЯМР (CDCl3, 400 МГц)8,97 (1 Н, широкий s), 5,44 (1H, d, J=8,8 Гц), 4,15-3,90 (3H, m), 2,94 (3H,s), 2,77-2,55 (2 Н, m), 1,88-1,87 (1H, m), 1,78-1,58 (3H), 1,42-1,37 (12 Н, m), 1,16-0,94 (2 Н, m). Эталонное соединение 2 Промежуточный продукт 1-Е омыляют с помощью LiOHH2O по той же методике, которая использована для получения соединения 1-Н. МС m/z 421,5 (М+1). 1 3-В: Соединение 3-А (2 г, 9,28 ммоль) объединяют с CBr4 (4,46 г, 13,47 ммоль) и трифенилфосфином (3,28 г, 12,54 ммоль) в ТГФ (0,2 М) и раствор перемешивают в течение ночи. Затем реакционную смесь фильтруют и растворитель выпаривают. При медленном добавлении неочищенной смеси к большому объему эфира осаждается значительная часть трифенилфосфиноксида. После фильтрования и концентрирования остаток очищают с помощью хроматографии (градиентный режим EtOAc:гексаны) и получают соединение 3-В. 1 Н-ЯМР (CDCl3, 400 МГц)4,05-3,99 (1 Н, m), 3,83-3,78 (1 Н, m), 3,27-3,24 (2 Н, m), 2,84-2,77 (1 Н, m),2,66-2,59 (1H, m), 1,91-1,74 (2H, m), 1,67-1,56 (1H, m), 1,42 (9H, s), 1,32-1,20 (2H, m). 3-C: Смесь соединения 3-В (1 г, 3,6 ммоль) и KCN (281 мг, 4,3 ммоль) в безводном ДМФ (20 мл) перемешивают при кипячении с обратным холодильником в течение ночи. Остаток растворяют в EtOAcMgSO4. Растворитель выпаривают и неочищенное вещество очищают с помощью хроматографии (градиентный режим EtOAc:гексаны) и получают соединение 3-С в виде масла. 1 Н-ЯМР (CDCl3, 400 МГц)3,83-3,78 (1H, m), 3,78-3,69 (1H, m), 2,87-2,73 (2 Н, m), 2,28-2,15 (2 Н, m),1,84-1,72 (2 Н, m), 1,61-1,52 (1H, m), 1,42-1,15 (11 Н, m). 3-D: К раствору соединения 3-С (750 мг, 3,34 ммоль) в ТГФ (20 мл) при -78 С добавляют ДИБАЛ(диизобутилалюминийгидрид) (1 М раствор в ТГФ, 5 мл). Этой смеси дают нагреться до комнатной температуры и ее перемешивают при комнатной температуре в течение 1 ч. Смесь охлаждают до 0 С и последовательно добавляют воду (0,2 мл), 15% водный раствор NaOH (0,2 мл) и воду (0,5 мл). После добавления MgSO4 смесь энергично перемешивают и фильтруют. Выпаривание растворителей дает соединение 3-D в виде бесцветного масла. Это соединение используют на следующей стадии без дополнительной очистки. 1 Н-ЯМР (CDCl3, 400 МГц)3,78-3,67 (1 Н, m), 3,67-3,64 (1 Н, m), 2,81-2,71 (1 Н, m), 2,71-2,50 (1 Н, m),2,24-2,09 (2 Н, m), 1,79-1,66 (2 Н, m), 1,56-1,48 (1 Н, m), 1,39-1,13 (11 Н, m). 3-Е: Это соединение получают из соединения 3-D по методикам, аналогичным тем, которые описаны для получения эталонного соединения 1-D. 3-F: Это соединение получают из соединения 3-Е по методикам, аналогичным тем, которые описаны для получения эталонного соединения 1-F. 3-G: Это соединение получают из соединения 3-F по методикам, аналогичным тем, которые описаны для получения эталонного соединения 1-G. 3-Н: Это соединение получают из соединения 3-G по методикам, аналогичным тем, которые описаны для получения эталонного соединения 1-Н. Эталонное соединение 4 4-В: Гидрохлорид этилового эфира D-гомофениланилина (5,00 г, 20,5 ммоль) и ДИЭА (8,7 мл,51,25 ммоль) растворяют в ТГФ (100 мл) и перемешивают при комнатной температуре. По каплям добавляют мезилхлорид (1,67 мл, 21,52 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 6 ч. ТГФ выпаривают и неочищенный остаток растворяют в EtOAc (100 мл), промывают водой (100 мл), 1 н. раствором HCl (2100 мл) и рассолом (100 мл) и сушат (MgSO4). Растворитель удаляют в вакууме и неочищенное вещество очищают с помощью флэш-хроматографии (гексаны:EtOAc) и- 12016199 получают этиловый эфир. 4-С: Этиловый эфир 4-В растворяют в диоксане (50 мл) и перемешивают при комнатной температуре. Добавляют LiOHH2O (1,00 мг, 24 ммоль), растворенный в воде (20 мл), и реакционную смесь перемешивают до исчезновения этилового эфира (по данным ТСХ (тонкослойная хроматография) и ЖХМС(жидкостная хроматография масс-спектрометрия. Растворитель удаляют в вакууме и неочищенное вещество подвергают распределению между EtOAc (50 мл) и 1 н. раствором HCl (50 мл). Водный слой экстрагируют с помощью EtOAc (250 мл) и объединенные органические фазы промывают 1 М растворомNaHSO4 (250 мл) и рассолом (50 мл) и сушат над MgSO4. Растворитель выпаривают и неочищенное вещество очищают с помощью флэш-хроматографии (градиентный режим EtOAc:гексаны) и получают эталонное соединение 4 в виде белого порошкообразного вещества. Эталонное соединение 5Boc-D-гомофениланилин (1,0 г, 3,58 ммоль) растворяют в ТГФ (10 мл) и к суспензии NaH (60% дисперсия в минеральном масле; 10,0 ммоль) в тетрагидрофуране (12 мл), в течение 20 мин по каплям добавляют воду (18 мкл, 0,72 ммоль), поддерживая внутреннюю температуру равной 20 С. Смесь перемешивают при той же температуре в течение 10 мин и в течение 20 мин добавляют диметилсульфат(1,05 мл, 6,44 ммоль), поддерживая температуру равной 20 С. Реакционную смесь перемешивают в течение 2 ч, затем в течение 10 мин реакцию останавливают 30% раствором гидроксида аммония (6 мл), поддерживая внутреннюю температуру равной 20 С. Перемешивание продолжают в течение еще 1 ч (для обеспечения полного разложения диметилсульфата). Смесь разбавляют с помощью EtOAc (20 мл) и водой (20 мл). Органический слой отделяют, промывают водой (10 мл), сушат (MgSO4) и выпаривают в вакууме и получают эталонное соединение 5 в виде белого твердого вещества. Эталонное соединение 6 6-В: Гидрохлорид этилового эфира D-гомофениланилина (6-А) (25,0 г, 102,5 ммоль) растворяют в 10% водном растворе EtOH (500 мл). Добавляют каталитическое количество 5% Rh/C и реакционную смесь помещают в атмосферу H2 (1000 фунт-сила/дюйм 2), перемешивают и нагревают до 50 С. Через 18 ч реакционную смесь охлаждают до комнатной температуры, устройство подачи H2 отсоединяют и давление в реакторе доводят до атмосферного. Катализатор фильтруют через целит и растворитель удаляют в вакууме и получают гидрохлорид этилового эфира D-гомоциклогексиланилина в виде белого порошкообразного вещества. 6-С: Это соединение получают из соединения 6-В по методикам, аналогичным тем, которые описаны для получения эталонного соединения 4-В. 6-D: Это соединение получают из соединения 6-С по методикам, аналогичным тем, которые описаны для получения эталонного соединения 4-С. Эталонное соединение 7 7-А: В круглодонную колбу, содержащую ТГФ (60 мл) и воду (20 мл), добавляют гидрохлорид этилового эфира D-гомоциклогексиланилина (3,83 г, 18,0 ммоль) и N-(бензилоксикарбонилокси)сукцинимид(Cbz-OSu) (4,49 г, 18,0 ммоль). Смесь перемешивают при комнатной температуре и добавляют Et3N(10,1 мл, 72,0 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение ночи. Прозрачный раствор разбавляют с помощью EtOAc (200 мл) и промывают 1 н. раствором HCl (3100 мл) и рассолом (1100 мл) и сушат над MgSO4. Растворитель выпаривают в вакууме и получают искомый продукт в виде белого твердого вещества, которое используют без дополнительной очистки.- 13016199 7-В: Это соединение получают из соединения 7-А по методикам, аналогичным тем, которые описаны для получения эталонного соединения 4-С. Эталонное соединение 8 8-В: Тонкоизмельченный KOH (19,4 г, 0,346 моль) растворяют в ДМСО (диметилсульфоксид) и перемешивают при комнатной температуре в течение 20 мин и затем охлаждают до 0 С. N-Вос-транс-4 Гидрокси-L-пролин (Вос-Нур-ОН, 8-А) (10 г, 43,3 ммоль) растворяют в ДМСО (10 мл) и добавляют к смеси и реакционную смесь перемешивают при 0 С в течение еще 10 мин. Затем добавляют 4-хлорбензилхлорид (33 г, 0,204 моль) и реакционную смесь перемешивают при 0 С в течение еще 15 мин. После этого баню со льдом удаляют и реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 4 ч. Реакционную смесь выливают в воду (300 мл) и реакционный сосуд промывают еще одной аликвотой воды (300 мл). Объединенный водный слой экстрагируют эфиром (2300 мл) и эфирный экстракт отбрасывают. Водный слой подкисляют до рН 2,3 с помощью 87% Н 3 РО 4 и затем экстрагируют эфиром (3300 мл). Объединенные эфирные экстракты промывают водой (2400 мл) и рассолом (2400 мл) и затем сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексаны (градиентный режим от 0 до 100%), и получают соединение 8-В в виде прозрачного масла. МС m/z 256,1 (М+1 - Boc). 1(2 Н, m), 2,40-2,30 (1 Н, m), 2,03-1,94 (1H, m), 1,39-1,34 (9 Н, m). 8-С: К соединению 8-В добавляют раствор ТФК (трифторуксусная кислота) в дихлорметане (50/50) и смесь перемешивают до полного удаления группы Boc. Затем реакционную смесь концентрируют в вакууме и неочищенный остаток используют на следующей стадии без дополнительной очистки. МС m/z 256,1 (М+1). 1 Н-ЯМР (CDCl3, 400 МГц)8,32 (1 Н, широкий s), 7,16-6,93 (4 Н, m), 4,41-4,12 (4 Н, m), 4,10-3,75(2 Н, m), 3,70-3,53 (1H, m), 3,51-3,30 (1H, m), 2,38-2,24 (1H, m), 2,06-1,88 (1 Н, m). 8-D: Промежуточный продукт 8-С растворяют в 200 мл раствора смеси 1,4-диоксан/Н 2 О (1:1). Добавляют NaHCO3 (17,9 г, 0,213 моль), затем Fmoc-Cl (12 г, 46,3 ммоль). Смесь перемешивают в течение ночи. Затем раствор подкисляют 1 н. раствором HCl и осадок отфильтровывают и сушат (MgSO4) и получают соединение 8-D в виде белого твердого вещества. 1 Н-ЯМР (CDCl3, 400 МГц)8,11 (1H, широкий s), 7,77-7,66 (2 Н, m), 7,58-7,52 (2 Н, m), 7,42-7,21 Для осуществления приведенной выше схемы реакций используют следующие реагенты:(с) катализатор реакции обмена Hoveyda-Grubbs (8 мол.%), N-Boc-4-метиленпиперидин (3,0 экв.),ДХМ, 40 С, 51%;- 14016199 9-А: D-Аллилглицин (5,03 г, 43,73 ммоль, 1,0 экв.) суспендируют в метаноле (70 мл) в бане с водой и льдом. В течение 10 мин по каплям добавляют тионилхлорид (9,6 мл, 131,19 ммоль, 3,0 экв.). Реакционную смесь нагревают до комнатной температуры и степень завершения реакции оценивают с помощью ЖХ/МС. Растворитель выпаривают и полученное белое твердое вещество, соединение 9-А, непосредственно используют на следующей стадии. 9-В: Гидрохлорид метилового эфира D-аллилглицина (9-А, 7,20 г, 43,73 ммоль), Et3N (18 мл,131,19 ммоль, 3,0 экв.) и ДМАП (4-диметиламинопиридин) (10 мг, каталитическое количество) растворяют в ТГФ (110 мл) и перемешивают при комнатной температуре. По каплям добавляют мезилхлорид(4,0 мл, 52,48 ммоль, 1,2 экв.) и реакционную смесь перемешивают при комнатной температуре в течение 6 ч. ТГФ выпаривают и неочищенный остаток растворяют в EtOAc (100 мл) и промывают водой (100 мл),1 н. раствором HCl (2100 мл) и рассолом (100 мл) и сушат (MgSO4). Растворитель удаляют в вакууме и неочищенное вещество очищают с помощью флэш-хроматографии (гексаны:EtOAc) и получают соединение 9-В в виде желтого масла. 9-С: К соединению 9-В (2,15 г, 10,37 ммоль, 1,0 экв.) с помощью шприца добавляют безводный дихлорметан (10 мл, 0,1 М) и в атмосфере азота катализатор реакции метатезиса Hoveyda-Grubbs 2-го поколения,[(1,3-бис-(2,4,6-триметилфенил)-2-имидазолидинилиден)дихлор(о-изопропоксифенилметилен)рутений(II)дихлорид (510 мг, 0,815 ммоль, 8 мол.%)]. С помощью шприца добавляют N-Boc-4 метилетпиперидин (6 мл, 31,11 ммоль, 3,0 экв.) и к реакционному сосуду присоединяют обратный холодильник и смесь нагревают при 40 С в течение 12 ч. После окончания реакции по данным ЖХ/МС реакционную смесь непосредственно очищают с помощью системы автоматической очистки на силикагеле(0-100% этилацетат в гексанах) и получают соединение 9-С в виде темно-зеленого масла. МС m/z 277,2 (М-Вос+1). Эталонное соединение 9: Омыление соединения 9-С проводят по методике, описанной выше для получения эталонного соединения 4. Эталонное соединение 10 Эталонное соединение 10 получают по методикам, аналогичным тем, которые описаны для получения эталонного соединения 8. Пример 1(1 мэкв./г) в ДМФ (диметилформамид) в присутствии АсОН (8 экв.). Смесь встряхивают в течение 1 ч,- 15016199 затем добавляют NaH(AcO)3 (3 экв.) и смеси дают реагировать в течение ночи. Затем смолу промывают с помощью ДМФ (2), ДХМ (2), МеОН (2) и ДХМ (2). 1-В: Аминокислоту 8-D с защитной группой Fmoc (3 экв.) добавляют к 200 мг смолы 1-А в ДМФ в присутствии HOBt (1-гидроксибензотриазол) (3,5 экв.) и ДИК (диизопропилкарбодиимид) (3,5 экв.). Смесь встряхивают в течение 3 ч. Смолу промывают с помощью ДМФ (2), ДХМ (2), МеОН (2) и ДХМ (2). 1-С: Смолу встряхивают в течение 30 мин в 20% растворе пиперидина в ДМФ и промывают с помощью ДМФ (2) и ДХМ (2). 1-D: Аминокислоту вводят в реакцию сочетания со смолой 1-С по методике, аналогичной методике 1-В. 1-Е: Раствор гидроксиламингидрохлорида (40 экв.) и ДИЭА (диизопропилэтиламин) (40 экв.) в ДМФ добавляют к смоле 1-D и смесь встряхивают в течение ночи. Смолу промывают с помощью ДМФ(2), ДХМ (2), МеОН (2) и ДХМ (2). 1-F: К раствору смолы 1-Е в ДХМ добавляют уксусный ангидрид (10 экв.). Смесь встряхивают в течение 2 ч и затем промывают с помощью ДХМ (2), ДМФ (2) и ДХМ (2). 1-G. Смолу 1-F промывают безводным ТГФ (2). Затем в атмосфере азота добавляют раствор SmI2(0,1 M в ТГФ). Смесь встряхивают в течение 2 ч и смолу промывают с помощью ДМФ (2), МеОН (2) ДМФ (2) и ДХМ (2). 1-Н: Конечное соединение 1-Н получают путем отщепления от смолы в присутствии раствора ТФК/ДХМ/H2O (45:45:10). Фильтрат концентрируют в вакууме, растворяют в ацетонитриле и очищают с помощью ВЭЖХ (высокоэффективная жидкостная хроматография) с обращенной фазой (градиентный режим H2O-ACN). После лиофилизации получают соль соединения 1-Н с ТФК в виде белого порошкообразного вещества. МС m/z 639,5 (М+1). 1 Н-ЯМР (CD3CN, 400 МГц)9,30 (1 Н, s), 7,89 (1H, s), 7,72 (1H, d, J=4 Гц), 7,36-7,26 (4 Н, m), 7,09(1H, d, J=4 Гц), 6,06 (1 Н, d, J=8 Гц), 4,60-4,41 (5 Н, m), 4,33-4,21 (1H, m), 4,11-4,05 (1H, m), 3,82-3,65 (2 Н,m), 3,29-3,27 (2 Н, m), 2,86 (3H, s), 2,86-2,76 (2 Н, m), 2,46-2,36 (1 Н, m), 2,15-2,07 (1H, m), 1,75-1,68 (2 Н,m), 1,63-1,46 (2 Н, m), 1,46-1,31 (2 Н, m), 1,31-1,27 (3H, m). Примеры 2-31. Соединения примеров 2-31 синтезируют по методикам, аналогичным тем, которые описаны для синтеза соединения примера 1. Пример 32 Реагент 32-А получают из бензиламина и смолы Pal по методикам, аналогичным тем, которые описаны для получения соединения примера 1-А. Промежуточный продукт 32-В получают из иммобилизированного соединения 32-А по методикам, аналогичным тем, которые описаны для получения соединения примера 1-В. Промежуточные продукты 32-С и 32-D получают из связанных с подложкой соединений 32-В и 32-С соответственно по методикам, аналогичным тем, которые описаны для получения со- 16016199 единений примеров 1-С и 1-D соответственно. Конечное соединение 32-Е получают путем отщепления соединения 32-D от смолы по методикам, аналогичным тем, которые описаны для получения соединения примера 1-Н. Примеры 33-54. Соединения примеров 33-54 синтезируют по методикам, аналогичным тем, которые описаны для синтеза соединения примера 32. Пример 55 55-А: К раствору гидрохлорида метилового эфира 8-С (1,6 г, 5,2 ммоль), РуВОР (бензотриазол-1 илокси-трис-(пирролидино)фосфонийгексафторфосфат) (3,79 г, 7,28 ммоль) и ДИЭА (2,7 мл, 15,6 ммоль) в ДХМ (50 мл) добавляют соединение 1-Н (1,9 г, 5,2 ммоль). Смесь перемешивают в течение ночи, затем промывают 1 М раствором NaHSO4 (250 мл), насыщенным раствором NaHCO3 (250 мл) и рассолом(150 мл). Органическую фазу сушат над MgSO4 и концентрируют в вакууме. Остаток очищают с помощью флэш-хроматографии (гексаны:EtOAc) и получают соединение 55-А в виде белого твердого вещества.MC m/z 616,2 (М+1). 55-В: Метиловый эфир 55-А (2,2 г, 3,72 ммоль) растворяют в диоксане (20 мл) и перемешивают при комнатной температуре. Добавляют LiOHH2O (467 мг, 11,12 ммоль), растворенный в воде (50 мл), и реакционную смесь перемешивают до исчезновения метилового эфира (по данным ТСХ и ЖХМС). Раствор подкисляют с помощью добавления 1 М раствора NaHSO4 и экстрагируют с помощью EtOAc (250 мл). Объединенные органические фазы промывают рассолом (50 мл) и сушат над MgSO4. Растворитель выпаривают и получают соединение 55-В в виде белого порошкообразного вещества. МС m/z 602,2 (М+1). 1 Н-ЯМР (CDCl3, 400 МГц)7,33 (2 Н, d, J=8,4 Гц ), 7,22 (2 Н, d, J=8,4 Гц ), 5,87 (1 Н, d, J=9,6 Гц ),4,43-4,57 (4 Н, m), 4,29-4,32 (1H, m), 3,95-4,17 (4 Н, m), 3,87-3,93 (1H, m), 3,60-3,64 (1H, m), 2,89 (3H, s),2,58-2,64 (2 Н, m), 2,45-2,51 (1 Н, m), 2,15-2,51 (1H, m), 1,48-1,70 (3H, m), 1,44 (9H, s), 1,22-1,35 (2H, m),0,95-1,10 (2H, m). 55-C: К раствору соединения 55-В (60 мг, 0,1 ммоль) в дихлорметане (10 мл) добавляют HATU(0,035 мл, 0,2 ммоль) и 2,5-дихлорбензиламин (23 мг, 0,13 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, затем последовательно промывают 1 М раствором NaHSO4 (10 мл), насыщенным раствором NaHCO3 (10 мл) и рассолом (10 мл). Раствор сушат над MgSO4, фильтруют, выпаривают и непосредственно используют на следующей стадии. МС m/z 659,2 (М+1 - Boc). 55-D: К раствору соединения 55-С в ДХМ медленно добавляют 50% раствор ТФК в ДХМ. Смесь перемешивают в течение 30 мин, затем растворители выпаривают и остаток растворяют в ацетонитриле и очищают с помощью ВЭЖХ с обращенной фазой. После лиофилизации растворителей получают соль соединения 55-D с ТФК в виде белого порошкообразного вещества. МС m/z 659,2 (М+1). 1 Н-ЯМР (CDCl3, 400 МГц)9,30 (1 Н, bs), 8,56 (1H, bs), 7,31 (1 Н, d, J=2 Гц ), 7,07-7,27 (6 Н, m), 5,88- 17016199 Примеры 56-70. Соединения примеров 56-70 синтезируют по методикам, аналогичным тем, которые описаны для синтеза соединения примера 55. В таблице приведены соединения формулы (1), описанные в примерах 1-70.
МПК / Метки
МПК: A61P 29/00, A61K 31/425, A61K 31/401, A61P 11/00, A61K 31/444, A61P 11/06, A61P 11/08, C07K 5/06
Метки: активирующей, ингибиторы, протеазы, композиции, канал, соединения
Код ссылки
<a href="https://eas.patents.su/30-16199-soedineniya-i-kompozicii-kak-ingibitory-aktiviruyushhejj-kanal-proteazy.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения и композиции как ингибиторы активирующей канал протеазы</a>
Новые соединения и композиции в качестве ингибиторов протеазы

Номер патента: 5369
Опубликовано: 24.02.2005
Авторы: Брайант Клиффорд М., Пэттерсон Джон У., Бьюнин Бэрри А., Крейнэк Эрика А.
МПК: A61K 31/275, C07C 271/22, A61P 11/00...
Метки: новые, качестве, композиции, протеазы, ингибиторов, соединения
Формула / Реферат:
1. Соединение формулы (I) где R1 представляет собой группу формулы (a) где X1 представляет собой -C(O)- или -CH2S(O)2-; R5 представляет собой водород, R7 представляет собой водород; R9 представляет собой (C1-6)алкил, замещенный -(O)R14 или SR14, и где R14 представляет собой (C3-6) циклоалкил(C0-6)алкил, фенил(C0-6)алкил, бифенилил(C0-6)алкил или гетеро(C5-6)арил(C0-6)алкил, где в R9 любая присутствующая алициклическая или ароматическая...
Ингибиторы вич-протеазы, содержащие их композиции, их фармацевтические применения и вещества для их синтеза

Номер патента: 8169
Опубликовано: 27.04.2007
Авторы: Мерфи Син Т., Тэтлок Джон Х., Берк Бенджамин Дж., Прасад Джосиула Вара, Болтон Гари, Кусера Дэвид Дж., Александр Терез Н., Мелник Майкл, Линтон Мария А., Скалицкий Дональд Дж., Митчелл Леннерт Дж., Уэббер Стефен Е., Барвиан Марк, Уорлэнд Стефен Т., Мачак Джефф, Кэнон-Кох Стэйси С., Джеуэлл Таня М., Бойер Фредрик Е., Вирджил Скотт С., Варни Майкл Д., Райх Зигфрид Х., Холлер Тод
МПК: A61P 31/18, A61K 31/425, C07D 207/16...
Метки: фармацевтические, содержащие, синтеза, ингибиторы, вич-протеазы, применения, вещества, композиции
Формула / Реферат:
1. Соединение формулы (I) и все его возможные стереоизомеры где R1 означает фенил, где указанный фенил замещен одним или более заместителями, независимо выбранными из C1-C6алкила, галогена или гидроксила; R2 означает C2-C6алкенильную или C2-C6алкинильную группу, которые являются линейными или разветвленными, и где указанные алкенильная или алкинильная группы являются замещенными или незамещенными одним или более атомом галогена; R2' означает Н;...
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Ким Киоунг-Хее, Йео Донг-Дзун, Бу Сеонг Чеол, Хонг Санг Йонг, Хур Гвонг-Чеунг, Ли Чанг-Сеок, Ким Мин-Дзунг, Квон Ох Хван, Йеом Зи-Хо, Ким Сунгсуб, Лим Донгчул, Ким Хие Дзин, Хан Хее Оон, Ким Сунг Хо, Ким Дзи Янг, Ким Геун Тае, Коо Ки Донг, Йим Хиеон Дзоо, Кох Дзонг Сунг
МПК: A61K 31/444, A61P 3/10, A61K 31/452...
Метки: фармацевтические, качестве, активного, способы, соединения, получения, композиции, ингредиента, соединения-ингибиторы, дипептидилпептидазы-iv, также, содержащие, указанные
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Новые соединения и композиции как ингибиторы катепсина

Номер патента: 7335
Опубликовано: 25.08.2006
Авторы: Тимм Эндрис П., Зипфель Шейла, Ли Джиайао, Линк Джон О., Грауп Майкл, Турайратнам Сукантини, Элдос Дэвид Дж.
МПК: A61P 33/06, A61P 19/02, A61K 31/16...
Метки: ингибиторы, композиции, катепсина, соединения, новые
Формула / Реферат:
1. Соединение формулы I в которой X1 является -NHC(R1)(R2)X3 или -NHX4; X2 представляет водород, фтор, -ОН, -OR4, -NHR15 или -NR17R18 и X7 является водородом, или X2 и X7, оба представляют фтор; X3 представляет циано, -С(R7)(R8)R16, -С(О)С(О)NR5R6; где R5 представляет водород, (C6-10)арил(С0-6)алкил, гетеро(C5-10)арил(С0-6)алкил; R6 представляет водород, гидрокси или (C1-6)алкил; R7 представляет водород или (C1-4)алкил и R8 представляет...
Ингибиторы протеазы вич

Номер патента: 2378
Опубликовано: 25.04.2002
Авторы: Зханг Канйин Е., Варни Майкл Д., Кобаяси Такуо, Альбицати Ким Ф., Рейх Зигфрид
МПК: C07D 217/26, A61K 31/472, A61P 37/00...
Метки: вич, ингибиторы, протеазы
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль или сольват. 2. Соединение по п.1, обладающее чистотой более 90%. 3. Соединение по п.1, обладающее чистотой, по крайней мере, 95%. 4. Соединение по п.1, обладающее чистотой, по крайней мере, 97%. 5. Соединение по п.1, обладающее чистотой, по крайней мере, 99%. 6. Фармацевтическая композиция для лечения ВИЧ инфекции, содержащая эффективное количество соединения или его соли или...
Предыдущий патент: Способ и система сбора и переработки твердых коммунальных отходов
Следующий патент: Способ склеивания слоя фторсиликонового каучука со слоем силиконового либо фторсиликонового каучука
Случайный патент: Вентилируемое курительное изделие