Пиридил- и пиримидинилзамещенные производные пиррола, тиофена и фурана в качестве ингибиторов киназ
Номер патента: 15126
Опубликовано: 30.06.2011
Авторы: Форте Барбара, Сколаро Алессандра, Кальдарелли Марина, Пиллан Антонио, Ванотти Эрмес, Чирла Алессандра, Эрмоли Антонелла, Меничинчери Мария





























Формула / Реферат
1. Соединение формулы (I)

где G представляет собой СН или атом азота;
W представляет собой атом кислорода, NR1или S(O)n;
где n равно 0;
R1 представляет собой атом водорода или C1-С6-алкил;
R2 представляет собой
атом водорода;
атом галогена;
арильную группу, где арильная группа представляет собой одновалентную ароматическую карбоциклическую группу с 6-14 атомами углерода, имеющую одно кольцо или несколько конденсированных колец, где конденсированные кольца могут быть ароматическими или неароматическими, при условии, что место присоединения находится у атома углерода ароматического кольца, и где арильная группа может быть необязательно замещена 1-3 заместителями, выбранными из группы, включающей
C1-С3-ацил,
аминоС1-С3-ацил,
C1-С6-алкил, необязательно замещенный гидроксигруппой,
C1-С3-алкокси,
арил,
арилокси,
C1-С3-алкоксикарбонил,
циано,
галоген,
нитро или гетероциклил, необязательно замещенный C1-C6-алкилом;
циклоалкильную группу, где циклоалкильная группа представляет собой одно кольцо, имеющее 3-10 атомов углерода; или
гетероциклильную группу, где гетероциклильная группа представляет собой насыщенную или ненасыщенную группу, имеющую одно кольцо или несколько конденсированных колец, от 1 до 10 атомов углерода и от 1 до 4 гетероатомов, выбранных из группы, состоящей из атомов азота, серы или кислорода, в кольце, где в системе конденсированных колец одно или несколько колец могут быть циклоалкилом, арилом или гетероарилом, при условии, что местом присоединения является гетероциклическое кольцо, и где гетероциклильная группа может быть необязательно замещена
C1-С6-алкилом,
галогеном или
C1-С3-алкоксикарбонилом;
R3 представляет собой атом водорода или C1-С6-алкил, необязательно замещенный арилом или циклоалкилом, где арил и циклоалкил являются такими, как определены выше;
R4 представляет собой атом водорода или галогена, C1-С6-алкил или С2-С6-алкенил;
R5 представляет собой атом водорода или галогена;
R6 представляет собой атом водорода или NHR7;
R7 представляет собой атом водорода или арил, где арил является таким, как определен выше;
или его фармацевтически приемлемая соль, при условии, что исключены следующие соединения:
амид 2,5-ди(пиридин-4-ил)тиофен-3-карбоновой кислоты,
метиламид 2,5-ди(пиридин-4-ил)тиофен-3-карбоновой кислоты,
амид 2,5-ди(пиридин-4-ил)-4-метилпиррол-3-карбоновой кислоты.
2. Соединение формулы (I) по п.1, отличающееся тем, что W представляет собой NR1, R1 и R3 имеют значения, указанные в п.1, и R6представляет собой NHR7, где R7 имеет значения, указанные в п.1.
3. Соединение формулы (I) по п.1 или 2, отличающееся тем, что W представляет собой NR1, где R1 имеет значения, указанные в п.1, R3 и R4представляют собой атомы водорода, R2 представляет собой арильную или гетероциклильную группу, где арил и гетероциклил имеют значения, указанные в п.1, и R6 представляет собой NH2.
4. Соединение формулы (I) по любому из предшествующих пунктов, где W представляет собой NH или R3представляет собой атом водорода.
5. Соединение формулы (I) по любому из предшествующих пунктов или его фармацевтически приемлемая соль, которое выбрано из группы, состоящей из
амида 2-фенил-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А1),
амида 2-(2-фторфенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А2),
амида 2-(3-фторфенил)-5-пиридин-4-ил-1H-пиррол-3-карбоновой кислоты (A3),
амида 2-(4-фторфенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А4),
амида 5-пиридин-4-ил-2-о-толил-1Н-пиррол-3-карбоновой кислоты (А7),
амида 5-пиридин-4-ил-2-м-толил-1Н-пиррол-3-карбоновой кислоты (А8),
амида 5-пиридин-4-ил-2-п-толил-1Н-пиррол-3-карбоновой кислоты (А9),
амида 2-(3-метоксифенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (A11),
амида 2-(4-метоксифенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А12),
амида 2-(2-нитрофенил)-5-пиридин-4-ил-1H-пиррол-3-карбоновой кислоты (А13),
амида 2-(3-нитрофенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А14),
амида 2-(2,3-диметилфенил)-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (А20),
амида 5-пиридин-4-ил-2-тиофен-3-ил-1Н-пиррол-3-карбоновой кислоты (С1),
амида 2-фуран-3-ил-5-пиридин-4-ил-1Н-пиррол-3-карбоновой кислоты (С2),
амида 5-(3-фторпиридин-4-ил)-2-фенил-1Н-пиррол-3-карбоновой кислоты (Е1),
амида 5-(3-фторпиридин-4-ил)-2-о-толил-1Н-пиррол-3-карбоновой кислоты (Е2),
амида 5-(2-аминопиримидин-4-ил)-2-фенил-1Н-пиррол-3-карбоновой кислоты (F1),
амида 5-(2-аминопиримидин-4-ил)-2-о-толил-1Н-пиррол-3-карбоновой кислоты (F2),
амида 5-(2-аминопиримидин-4-ил)-2-(2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F4),
амида 5-(2-аминопиримидин-4-ил)-2-(4-фтор-2-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F13),
амида 5-(2-аминопиримидин-4-ил)-2-(5-фтор-2-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F14),
амида 5-(2-аминопиримидин-4-ил)-2-(2,3-диметилфенил)-1Н-пиррол-3-карбоновой кислоты (F15),
амида 5-(2-аминопиримидин-4-ил)-2-(2,3-дифторфенил)-1Н-пиррол-3-карбоновой кислоты (F16),
амида 5-(2-аминопиримидин-4-ил)-2-(2,4-дифторфенил)-1Н-пиррол-3-карбоновой кислоты (F17),
амида 5-(2-аминопиримидин-4-ил)-2-(2,5-дифторфенил)-1Н-пиррол-3-карбоновой кислоты (F18),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлорфенил)-1Н-пиррол-3-карбоновой кислоты (F19),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлор-4-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F23),
амида 5-(2-аминопиримидин-4-ил)-2-(2,4-дихлорфенил)-1Н-пиррол-3-карбоновой кислоты (F26),
амида 5-(2-аминопиримидин-4-ил)-2-(2-фтор-4-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F28),
амида 5-(2-аминопиримидин-4-ил)-2-(2-фтор-3-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F30),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлор-5-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F31),
амида 5-(2-аминопиримидин-4-ил)-2-(3-хлор-2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F33),
амида 5-(2-аминопиримидин-4-ил)-2-(2,3-дихлорфенил)-1Н-пиррол-3-карбоновой кислоты (F34),
амида 5-(2-аминопиримидин-4-ил)-2-(2-фтор-3-метоксифенил)-1Н-пиррол-3-карбоновой кислоты (F35),
амида 5-(2-аминопиримидин-4-ил)-2-(4-хлор-2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F36),
амида 5-(2-аминопиримидин-4-ил)-2-(2-бромфенил)-1Н-пиррол-3-карбоновой кислоты (F38),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлор-3-метоксифенил)-1Н-пиррол-3-карбоновой кислоты (F39),
амида 5-(2-аминопиримидин-4-ил)-2-(3-метокси-2-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F40),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлор-3-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F41),
амида 5-(2-аминопиримидин-4-ил)-2-(3-бром-2-хлорфенил)-1Н-пиррол-3-карбоновой кислоты (F42),
амида 5-(2-аминопиримидин-4-ил)-2-(2-бром-3-хлорфенил)-1Н-пиррол-3-карбоновой кислоты (F43),
амида 5-(2-аминопиримидин-4-ил)-2-(2,3-дибромфенил)-1Н-пиррол-3-карбоновой кислоты (F44),
амида 5-(2-аминопиримидин-4-ил)-2-(3-бром-2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F45),
амида 5-(2-аминопиримидин-4-ил)-2-(3-бром-2-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F46),
амида 5-(2-аминопиримидин-4-ил)-2-(2-бром-3-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F47),
амида 5-(2-аминопиримидин-4-ил)-2-(4-метокси-3-метилфенил)-1Н-пиррол-3-карбоновой кислоты (F48),
амида 5-(2-аминопиримидин-4-ил)-2-(3,4-диметоксифенил)-1Н-пиррол-3-карбоновой кислоты (F49),
амида 5-(2-аминопиримидин-4-ил)-2-(2-фтор-4-метоксифенил)-1Н-пиррол-3-карбоновой кислоты амид (F50),
амида 5-(2-аминопиримидин-4-ил)-2-(2-хлор-4-метоксифенил)-1Н-пиррол-3-карбоновой кислоты (F51),
амида 5-(2-аминопиримидин-4-ил)-2-(2-бром-4-фторфенил)-1H-пиррол-3-карбоновой кислоты (F52),
амида 5-(2-аминопиримидин-4-ил)-2-(4-метокси-2-метилфенил)-1H-пиррол-3-карбоновой кислоты (F53),
амида 5-(2-аминопиримидин-4-ил)-2-тиофен-3-ил-1Н-пиррол-3-карбоновой кислоты (G1),
амида 5-(2-аминопиримидин-4-ил)-2-тиофен-2-ил-1Н-пиррол-3-карбоновой кислоты (G2),
амида 5-(2-аминопиримидин-4-ил)-2-(5-метилтиофен-2-ил)-1Н-пиррол-3-карбоновой кислоты (G3),
амида 5-(2-аминопиримидин-4-ил)-2-(5-хлортиофен-2-ил)-1Н-пиррол-3-карбоновой кислоты (G4),
амида 5-(2-амино-5-хлорпиримидин-4-ил)-2-фенил-1Н-пиррол-3-карбоновой кислоты (N1),
амида 5-(2-амино-5-бромпиримидин-4-ил)-2-фенил-1Н-пиррол-3-карбоновой кислоты (N2),
амида 5-(2-аминопиримидин-4-ил)-4-иод-2-фенил-1Н-пиррол-3-карбоновой кислоты (N3),
амида 5-(2-амино-5-хлорпиримидин-4-ил)-2-(2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (N7),
амида 5-(2-амино-5-бромпиримидин-4-ил)-2-(2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (N8),
амида 5-(2-аминопиримидин-4-ил)-2-фенилтиофен-3-карбоновой кислоты (S1),
амида 5-(2-амино-5-фторпиримидин-4-ил)-2-фенил-1Н-пиррол-3-карбоновой кислоты (V1) и
амида 5-(2-аминопиримидин-4-ил)-4-хлор-2-фенил-1Н-пиррол-3-карбоновой кислоты (Z1).
6. Соединение формулы (I) по любому из предшествующих пунктов или его фармацевтически приемлемая соль, которое представляет собой
амид 5-(2-аминопиримидин-4-ил)-2-(2,3-диметилфенил)-1Н-пиррол-3-карбоновой кислоты (F15),
амид 5-(2-аминопиримидин-4-ил)-2-(2,4-дихлорфенил)-1Н-пиррол-3-карбоновой кислоты (F26) и
амид 5-(2-аминопиримидин-4-ил)-2-(4-хлор-2-фторфенил)-1Н-пиррол-3-карбоновой кислоты (F36).
7. Способ получения соединения формулы (I) по п.1, который включает:
а) сочетание соединения формулы 1E

где R1, R4, R5, R6 и G имеют значения, указанные в п.1, и циклил представляет собой необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп, где необязательно замещенные арильные, циклоалкильные и гетероциклильные группы являются такими, как определены в п.1, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, где R3имеет значения, указанные в п.1, с получением таким образом соединения формулы (I) по п.1, где W представляет собой NR1, где R1 имеет значения, указанные в п.1, и R2 представляет собой циклил, который имеет значения, определенные выше;
b) необязательное превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
8. Способ получения соединения формулы (I) по п.1, который включает:
а) амидирование соединения формулы 5D

где R1, R5, R6, G имеют значения, указанные в п.7, циклил имеет значения, указанные в п.7, и R4 имеет значения, указанные в п.1, но не является атомом водорода, и
b) необязательное превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
9. Способ получения соединения формулы (I) по п.1, который включает:
а¢) сочетание соединения формулы 2D

где R2представляет собой атом водорода или галогена и R5, R6 и G имеют значения, указанные в п.1, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, где R3имеет значения, указанные в п.1, с получением таким образом соединения формулы (I), где W представляет собой N, R1 представляет собой атом водорода и R2 представляет собой атом водорода или атом галогена;
a'1) необязательное превращение образовавшегося соединения формулы (I), где R2 представляет собой атом галогена, в другое соединение формулы (I), где R2 представляет собой атом водорода или циклил, где циклил имеет значения, указанные в п.7, и/или
а'2) превращение образовавшегося соединения формулы (I), где R1представляет собой атом водорода, в другое соединение формулы (I), где R1 представляет собой C1-C6-алкил; и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращением соли в свободное соединение (I).
10. Способ получения соединения формулы (I) по п.1, который включает:
а') расщепление соединения формул 3А или 3В

где R3, R5, R6, X, G имеют значения, указанные в п.1, циклил имеет значения, указанные в п.7, и символ ![]() представляет собой твердый носитель, с которым связана химическая молекула, и, если необходимо, превращение образовавшегося соединения формулы (I), где W представляет собой атом азота и R2 представляет собой атом галогена или циклил, где циклил имеет значения, указанные в п.7, в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
представляет собой твердый носитель, с которым связана химическая молекула, и, если необходимо, превращение образовавшегося соединения формулы (I), где W представляет собой атом азота и R2 представляет собой атом галогена или циклил, где циклил имеет значения, указанные в п.7, в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
11. Способ получения соединения формулы (I) по п.1, который включает:
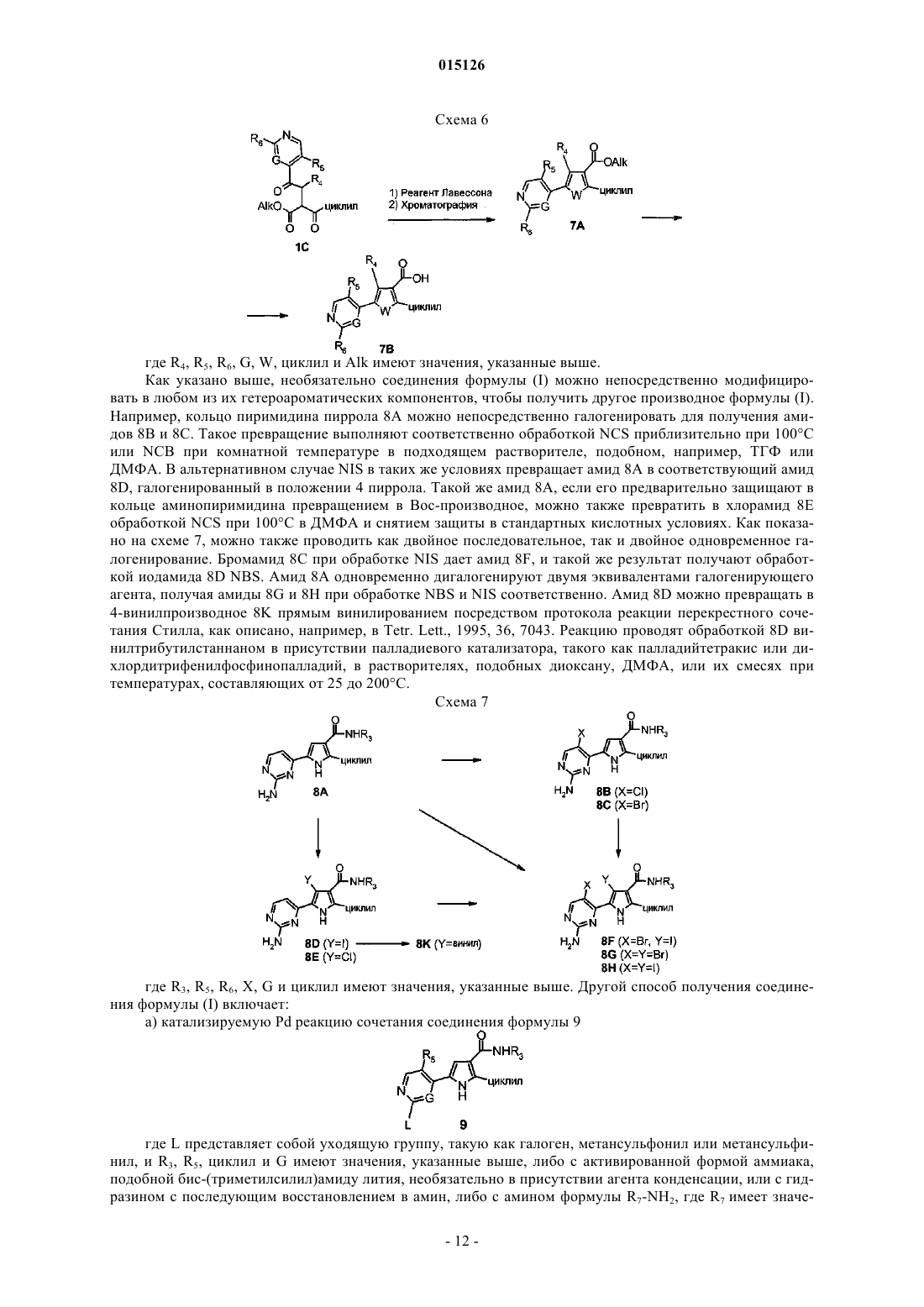
а) сочетание соединения формулы 7В

где W представляет собой атом кислорода или серы, R4, R5, R6 и G имеют значения, указанные в п.1, и циклил имеет значения, указанные в п.7, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, где R3 имеет значения, указанные в п.1, с образованием таким образом описываемого выше соединения формулы (I), где W представляет собой атом кислорода или серы и R2 представляет собой циклил, где циклил имеет значения, указанные в п.7;
b) необязательное превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
12. Способ получения соединения формулы (I) по п.1, который включает:
а) катализируемую Pd реакцию сочетания соединения формулы 9

где L представляет собой уходящую группу, такую как галоген, метансульфонил или метансульфинил, и R3, R5 и G имеют значения, указанные в п.1, и циклил имеет значения, указанные в п.7, либо с активированной формой аммиака, подобной бис-(триметилсилил)амиду лития, необязательно в присутствии агента конденсации, или с гидразином с последующим восстановлением в амин, либо с амином формулы R7-NH2, где R7 имеет значения, указанные в п.1, с получением таким образом указанного выше соединения формулы (I), где W представляет собой NR1, R1 представляет собой атом водорода и R6 представляет собой NH-R7, и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I).
13. Способ лечения клеточных пролиферативных нарушений, вызванных измененной активностью протеинкиназы, и/или связанных с такой активностью, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I) по п.1.
14. Способ по п.13 для лечения клеточных пролиферативных нарушений, вызванных измененной Cdc7-киназой и/или связанных с такой киназой.
15. Способ по п.13 или 14, где клеточным пролиферативным нарушением является рак, выбранный из карциномы, гемопоэтических опухолей лимфоидной линии; гемопоэтических опухолей миелоидной линии; опухолей мезенхимального происхождения; опухолей центральной и периферической нервной системы; меланомы, семиномы, тератокарциномы, остеосаркомы, пигментной ксеродермы, кератоксантомы, фолликулярного рака щитовидной железы и саркомы Капоши.
16. Способ по п.13 или 14, где клеточное пролиферативное нарушение выбрано из доброкачественной гиперплазии простаты, семейного аденоматоза, полипоза, нейрофиброматоза, псориаза, пролиферации клеток сосудов гладких мышц, связанной с атеросклерозом, фиброзом легких, артритом, гломерулонефритом и послехирургическим стенозом и рестенозом.
17. Способ по п.13, дополнительно включающий лечение млекопитающего, нуждающегося в этом, по схеме лечения лучевой терапией или химиотерапией в сочетании по меньшей мере с одним цитостатическим или цитотоксическим агентом.
18. Способ по п.13, где млекопитающим, нуждающимся в этом, является человек.
19. Способ ингибирования активности Cdc7-киназы, который включает контактирование указанной киназы с эффективным количеством соединения по п.1.
20. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по п.1 и по меньшей мере один фармацевтически приемлемый эксципиент, носитель и/или разбавитель.
21. Фармацевтическая композиция по п.20, дополнительно содержащая один или несколько химиотерапевтических агентов.
22. Набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль по п.1 либо его фармацевтические композиции по п.20 и один или несколько химиотерапевтических агентов в качестве комбинированного препарата для одновременного, раздельного или последовательного применения в противораковой терапии.
23. Применение соединения формулы (I) или его фармацевтически приемлемой соли по п.1 в качестве лекарственного средства.
24. Применение соединения формулы (I) или его фармацевтически приемлемой соли по п.1 при изготовлении лекарственного средства с противоопухолевой активностью.
25. Промежуточное соединение формул 1D или 1E

где G, R1, R4, R5и R6 имеют значения, указанные в п.1, циклил имеет значения, указанные в п.7, и Alk представляет собой C1-С6-алкил, при условии, что исключены следующие соединения:
метиловый эфир 2,5-ди-4-пиридинил-1H-пиррол-3-карбоновой кислоты,
метиловый эфир 4-метил-2-фенил-5-(4-пиридинил)-1Н-пиррол-3-карбоновой кислоты,
4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновая кислота, соединение с морфолином (1:1),
4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновая кислота,
этиловый эфир 4-бутил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
этиловый эфир 4-(1-метилэтил)-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
этиловый эфир 4-пропил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
2-метоксиэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
бутиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
пропиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
1,1-диметилэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
1-метилэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
метиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты,
этиловый эфир 4-метил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты и
этиловый эфир 4-этил-2,5-ди-4-пиридинил-1Н-пиррол-3-карбоновой кислоты.
Текст
Настоящее изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям, где G, W, R2, R3, R4, R5 и R6 имеют значения, указанные в описании. Следующими объектами настоящего изобретения являются способы получения соединений формулы (I) и промежуточные соединения таких способов, фармацевтические композиции, содержащие их, и способы лечения клеточных пролиферативных нарушений. Фактически соединения формулы (I) являются пригодными в терапии, при лечении заболеваний, связанных с нарушением регуляции активности протеинкиназы, подобных раку.(71)(73) Заявитель и патентовладелец: НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.Р.Л. (IT) 015126 Настоящее изобретение относится к гетеропентациклам, способу их получения, содержащим их фармацевтическим композициям и их применению в качестве терапевтических агентов, особенно при лечении рака и нарушений клеточной пролиферации. Нарушение функции протеинкиназ (РК) является отличительным признаком многочисленных заболеваний. Большая часть онкогенов и протоонкогенов принимает участие в коде для РК раковых заболеваний человека. Повышенные активности РК также участвуют во многих незлокачественных заболеваниях. В качестве основной ссылки в отношении нарушения функции или нарушения регуляции РК смотрите, например, Current Opinion in Chemical Biology, 1999, 3, 459-465. Среди нескольких протеинкиназ, известных в данной области как принимающих участие в росте раковых клеток, имеется Cdc7, эволюционно сохраненная серинтреонинкиназа, которая играет основную роль в связывании регуляции клеточного цикла с дупликацией генома, являясь существенным фактором для запуска инициации репликации ДНК (см. Montagnoli A. et al., EMBO Journal, 2002, vol. 21, No. 12,3171; Montagnoli A. et al. Cancer Research, 2004, vol. 64, 1 октября, 7110). Несколько гетероциклических соединений являются известными в данной области в качестве ингибиторов протеинкиназ. Среди них имеются, например, пирролопиразолы, описанные в WO 2002/12242; тетрагидроиндазолы, описанные в WO 2000/69846; пирролопиридины, описанные в WO 2001/98299; аминофталазиноны,описанные в WO 2003/014090, и аминоиндазолы, описанные в WO 2003/028720. Производные пиррола описаны в WO 2001/001986, 98/35944, производные тиазола описаны в WO 2002/030358 и вWO 2005/095386 заявлено, что тиофены являются ингибиторами киназ. В WO 2006/012642 описаны производные пиррола, модулирующие активность одного или нескольких стероидных ядерных рецепторов,и в WO 2003/068749 описаны производные фурана, модулирующие ваниллоидные рецепторы. Пиридилфураны и пиридилтиофены описаны в ЕР 853083 в качестве ингибиторов биосинтеза TNF и экспрессии САМ; пиридилпирролы описаны в WO 98/02430 в качестве антагонистов интерлейкина и фактора некроза опухолей; в ней описаны также кислотные и сложноэфирные производные пиррола. Производные пиперазинилфенилкарбоксамида, содержащие кольцо фурана, описаны в WO 95/04729 в качестве антагонистов рецептора 5-HT1D. В WO 2005/100342 описаны и заявлены пиримидин/пиридинзамещенные пирролы, обладающие антипролиферативными и ингибирующими Erk2-киназу активностями. В WO 2000/006085 описаны и заявлены гетероциклкарбоксамиды в качестве модуляторов рецептораCCR5. Авторы настоящего изобретения теперь обнаружили некоторые соединения, пригодные в терапии в качестве агентов против носителя заболеваний, вызванных активностью протеинкиназы с нарушенной регуляцией и/или связанных с ней и более конкретно активностью Cdc7 или Cdc7/Cdks. Другой целью настоящего изобретения является предоставление соединений, обладающих активностью, ингибирующей протеинкиназу, более конкретно активностью, ингибирующей Cdc7 или Cdc7/Cdks. В частности, настоящее изобретение относится к гетеропентациклам, которые обладают активностью,ингибирующей протеинкиназу, особенно активностью, ингибирующей Cdc7 или Cdc7/Cdks. Более конкретно соединения настоящего изобретения являются применимыми при лечении различных раковых заболеваний, включающих, но не ограничивающихся перечисленным, карциному, такую как карцинома мочевого пузыря, молочной железы, толстой кишки, почек, печени, легких, в том числе мелкоклеточного рака легких, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, простаты и кожи, включая плоскоклеточный рак; гемопоэтические опухоли лимфоидной линии, в том числе лейкоз, острый лимфолейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, ходжкинскую лимфому, неходжкинскую лимфому, лимфому "волосатых" клеток и Биркитта лимфому; гемопоэтические опухоли миелоидной линии, включающие острые и хронические миелоидные лейкозы, синдром миелодисплазии и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включающие фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включающие астроцитому, нейробластому, глиому и невриномы; другие опухоли, включающие меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши. Вследствие ключевой роли РК и, в частности, Cdc7 и Cdks, подобных Cdk2, в регуляции клеточной пролиферации указанные гетеропентациклы являются также применимыми при лечении различных клеточных пролиферативных нарушений, таких как, например, доброкачественная гиперплазия простаты,семейный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация клеток сосудов гладких мышц, связанная с атеросклерозом, фиброзом легких, артритом, гломерулонефритом и послехирургическим стенозом и рестенозом. Соединения настоящего изобретения могут быть также активными в качестве ингибиторов других протеинкиназ, таких как, например, протеинкиназа С в различных изоформах, Met, PAK-4, PAK-5, ZC-1,STLK-2, DDR-2, Aurora 1, Aurora 2, Bub-1, PLK, Chk1, Chk2, HER2, raf1, MEK1, MAPK, EGF-R, PDGF-R,FGF-R, IGF-R, VEGF-R, PI3K, weel-киназа, Src, Ab1, Akt, ILK, MK-2, IKK-2, Nek, CK2, GSK3, SULU,PKA, PKC, PDK, RET, KIT, LCK, TRKA, и поэтому являются эффективными при лечении заболеваний,связанных с другими протеинкиназами.-1 015126 Соответственно этому в первом варианте осуществления настоящее изобретение относится к соединению формулы (I)W представляет собой атом кислорода, NR1 или S(O)n; где n равно 0, 1 или 2;R1 и R3 независимо представляют собой атом водорода или необязательно замещенную группу, выбранную из алкильной, циклоалкильной, алкенильной, алкинильной, гетероциклильной, гетероциклилалкильной, арильной, арилалкильной, гетероциклилоксиалкильной и алкоксикарбонильной групп;R2 представляет собой атом водорода или галогена или необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп;R4 представляет собой атом водорода или галогена или необязательно замещенную алкильную или алкенильную группу;R5 представляет собой атом водорода или галогена;R6 представляет собой атом водорода или NHR7;R7 представляет собой атом водорода, необязательно замещенную группу, выбранную из алкильной, арильной, циклоалкильной и гетероциклильной групп, или -CO-R1, где R1 имеет значения, указанные выше; или его фармацевтически приемлемой соли при условии, что исключены следующие соединения: амид 2,5-ди(пиридин-4-ил)тиофен-3-карбоновой кислоты,метиламид 2,5-ди(пиридин-4-ил)тиофен-3-карбоновой кислоты,амид 2,5-ди(пиридин-4-ил)-4-метилпиррол-3-карбоновой кислоты,[4-метокси-3-(4-метилпиперазин-1-ил)фенил]амид 5-пиридин-4-илфуран-3-карбоновой кислоты,(1-метил-1,2,3,4-тетрагидрохинолин-7-ил)амид 5-пиридин-4-илфуран-3-карбоновой кислоты иN-[2-амино-1-(2,4-дихлорбензил)этил]-5-[2-(метиламино)пиримидин-4-ил]тиофен-3-карбоксамид. Соединения формулы (I), являющиеся объектом настоящего изобретения, можно получить посредством синтетического способа, включающего хорошо известные реакции, проводимые согласно общепринятым методикам, а также посредством очень универсального твердофазного и/или комбинаторного способа, причем все такие способы входят в объем настоящего изобретения. Настоящее изобретение относится также к фармацевтической композиции, содержащей указанное выше соединение формулы (I) и по меньшей мере один фармацевтически приемлемый эксципиент, носитель или разбавитель. Предпочтительно соединение формулы (I) отличается тем, что W представляет собой NR1, R1 и R3 независимо представляют собой атом водорода или необязательно замещенную алкильную группу и R6 представляет собой NHR7, где R7 представляет собой атом водорода или необязательно замещенную арильную группу. Более предпочтительно соединение формулы (I) отличается тем, что W представляет собой NR1; R1 представляет собой атом водорода или необязательно замещенную алкильную группу; R3 и R4 представляют собой атом водорода, R2 представляет собой необязательно замещенную арильную или гетероциклильную группу и R6 представляет собой NH2. Еще более предпочтительными являются соединения формулы (I), где W представляет собой NH или R3 представляет собой атом водорода. Конкретными, но не ограничивающимися указанными соединениями, предпочтительными соединениями формулы (I) настоящего изобретения, всякий раз, когда это является подходящим, в форме фармацевтически приемлемых солей, являются следующие соединения: амид 2-фенил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 1),амид 2-(2-фторфенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 2),амид 2-(3-фторфенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (A3),амид 2-(4-фторфенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 4),амид 5-пиридин-4-ил-2-о-толил-1 Н-пиррол-3-карбоновой кислоты (А 7),амид 5-пиридин-4-ил-2-м-толил-1 Н-пиррол-3-карбоновой кислоты (А 8),амид 5-пиридин-4-ил-2-п-толил-1 Н-пиррол-3-карбоновой кислоты (А 9),амид 2-(3-метоксифенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (A11),амид 2-(4-метоксифенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 12),амид 2-(2-нитрофенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 13),амид 2-(3-нитрофенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 14),амид 2-(2,3-диметилфенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 20),амид 5-пиридин-4-ил-2-тиофен-3-ил-1 Н-пиррол-3-карбоновой кислоты (С 1),-2 015126 амид 2-фуран-3-ил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (С 2),амид 5-(3-фторпиридин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (E1),амид 5-(3-фторпиридин-4-ил)-2-о-толил-1 Н-пиррол-3-карбоновой кислоты (Е 2),амид 5-(2-аминопиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (F1),амид 5-(2-аминопиримидин-4-ил)-2-о-толил-1 Н-пиррол-3-карбоновой кислоты (F2),амид 5-(2-аминопиримидин-4-ил)-2-(2-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (F4),амид 5-(2-аминопиримидин-4-ил)-2-(4-фтор-2-метилфенил)-1 Н-пиррол-3-карбоновой кислоты (F13),амид 5-(2-аминопиримидин-4-ил)-2-(5-фтор-2-метилфенил)-1 Н-пиррол-3-карбоновой кислоты (F14),амид 5-(2-аминопиримидин-4-ил)-2-(2,3-диметилфенил)-1 Н-пиррол-3-карбоновой кислоты (F15),амид 5-(2-аминопиримидин-4-ил)-2-(2,3-дифторфенил)-1 Н-пиррол-3-карбоновой кислоты (F16),амид 5-(2-аминопиримидин-4-ил)-2-(2,4-дифторфенил)-1 Н-пиррол-3-карбоновой кислоты (F17),амид 5-(2-аминопиримидин-4-ил)-2-(2,5-дифторфенил)-1 Н-пиррол-3-карбоновой кислоты (F18),амид 5-(2-аминопиримидин-4-ил)-2-(2-хлорфенил)-1 Н-пиррол-3-карбоновой кислоты (F19),амид 5-(2-аминопиримидин-4-ил)-2-(2-хлор-4-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (F23),амид 5-(2-аминопиримидин-4-ил)-2-(2,4-дихлорфенил)-1 Н-пиррол-3-карбоновой кислоты (F26),амид 5-(2-аминопиримидин-4-ил)-2-(2-фтор-4-метилфенил)-1 Н-пиррол-3-карбоновой кислоты (F28),амид 5-(2-аминопиримидин-4-ил)-2-(2-фтор-3-метилфенил)-1 Н-пиррол-3-карбоновой кислоты (F30),амид 5-(2-аминопиримидин-4-ил)-2-(2-хлор-5-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (F31),амид 5-(2-аминопиримидин-4-ил)-2-(3-хлор-2-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (F33),амид 5-(2-аминопиримидин-4-ил)-2-(2,3-дихлорфенил)-1 Н-пиррол-3-карбоновой кислоты (F34),амид 5-(2-аминопиримидин-4-ил)-2-(2-фтор-3-метоксифенил)-1 Н-пиррол-3-карбоновой кислоты(F53),амид 5-(2-аминопиримидин-4-ил)-2-тиофен-3-ил-1 Н-пиррол-3-карбоновой кислоты (G1),амид 5-(2-аминопиримидин-4-ил)-2-тиофен-2-ил-1 Н-пиррол-3-карбоновой кислоты (G2),амид 5-(2-аминопиримидин-4-ил)-2-(5-метилтиофен-2-ил)-1 Н-пиррол-3-карбоновой кислоты (G3),амид 5-(2-аминопиримидин-4-ил)-2-(5-хлортиофен-2-ил)-1 Н-пиррол-3-карбоновой кислоты (G4),амид 5-(2-амино-5-хлорпиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (N1),амид 5-(2-амино-5-бромпиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (N2),амид 5-(2-аминопиримидин-4-ил)-4-иод-2-фенил-1 Н-пиррол-3-карбоновой кислоты (N3),амид 5-(2-амино-5-хлорпиримидин-4-ил)-2-(2-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (N7),амид 5-(2-амино-5-бромпиримидин-4-ил)-2-(2-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (N8),амид 5-(2-аминопиримидин-4-ил)-2-фенилтиофен-3-карбоновой кислоты (S1),амид 5-(2-амино-5-фторпиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (V1) и амид 5-(2-аминопиримидин-4-ил)-4-хлор-2-фенил-1 Н-пиррол-3-карбоновой кислоты (Z1). Более предпочтительными соединениями согласно настоящему изобретению являются амид 5-(2-аминопиримидин-4-ил)-2-(2,3-диметилфенил)-1 Н-пиррол-3-карбоновой кислоты (F15),амид 5-(2-аминопиримидин-4-ил)-2-(2,4-дихлорфенил)-1 Н-пиррол-3-карбоновой кислоты (F26) и амид 5-(2-аминопиримидин-4-ил)-2-(4-хлор-2-фторфенил)-1 Н-пиррол-3-карбоновой кислоты (F36) или их фармацевтически приемлемые соли.-3 015126 Предложен также способ лечения клеточных пролиферативных нарушений, вызванных измененной активностью киназы Cdc7 и/или связанных с ней, введением млекопитающему, нуждающемуся в этом,эффективного количества указанного выше соединения формулы (I). В предпочтительном варианте осуществления описанного выше способа клеточным пролиферативным нарушением является рак. Определенные типы рака, которые можно лечить, включают карциному, плоскоклеточный рак, гемопоэтические опухоли миелоидной или лимфоидной линии, опухоли мезенхимального происхождения,опухоли центральной и периферической нервной системы, меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши. Связи гетеропентацикла являются ароматическими; нумерация (атомов кольца) указанного гетеропентацикла показана ниже В настоящем описании, если не оговорено иначе, нижеследующие термины имеют следующие значения. Арильная, циклоалкильная и гетероциклильная группы иногда будут сообща для удобства указаны как "циклил". Термин "алкил" или "Alk" относится к разветвленным или неразветвленным одновалентным насыщенным алифатическим углеводородным группам, имеющим от 1 до 6 атомов углерода. Примером такого термина являются такие группы, как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил,трет-бутил, н-пентил, н-гексил и тому подобное. "Замещенный алкил" относится к алкильной группе,имеющей от 1 до 3 заместителей, выбранных из группы, состоящей из алкокси, замещенного алкокси,ацила, ациламино, ацилокси, амино, замещенного амино, аминоацила, арила, замещенного арила, арилокси, замещенного арилокси, циано, атомов галогена, гидроксила, нитро, карбоксила, сложных эфиров карбоксила, циклоалкила, замещенного циклоалкила, гетероциклила и замещенного гетероциклила."Циклоалкил" относится к циклическим алкильным группам с 3-10 атомами углерода, имеющим одно или несколько колец и включающим в качестве примера адамантил, циклопропил, циклобутил,циклопентил, циклогексил, циклооктил и тому подобное."Замещенный циклоалкил" относится к циклоалкилу, имеющему от 1 до 5 заместителей, выбранных из группы, состоящей из оксо (=O), тиоксо (=S), алкокси, замещенного алкокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминоацила, арила, замещенного арила, арилокси, замещенного арилокси, циано, атома галогена, гидроксила, нитро, карбоксила, сложных эфиров карбоксила, циклоалкила,замещенного циклоалкила, гетероциклила и замещенного гетероциклила."Алкенил" относится к алкенильным группам, имеющим от 2 до 6 атомов углерода. Примерами таких групп являются винил, аллил, бут-3-ен-1-ил и тому подобное."Замещенный алкенил" относится к алкенильным группам, имеющим от 1 до 3 заместителей, предпочтительно 1-2 заместителя, выбранных из группы, состоящей из алкокси, замещенного алкокси, ацила,ациламино, ацилокси, амино, замещенного амина, аминоацила, арила, замещенного арила, арилокси, замещенного арилокси, циано, атома галогена, гидроксила, нитро, карбоксила, сложных эфиров карбоксила, циклоалкила, замещенного циклоалкила, гетероциклила и замещенного гетероциклила, при условии,что любой гидроксильный заместитель не присоединен к атому углерода винильной (ненасыщенной) группы."Алкинил" относится к алкинильным группам, имеющим от 2 до 6 атомов углерода. "Замещенный алкинил" относится к алкинильным группам, имеющим от 1 до 3 заместителей, предпочтительно 1-2 заместителя, выбранных из группы, состоящей из алкокси, замещенного алкокси, ацила, ациламино, ацилокси, амино, замещенного амино, аминоацила, арила, замещенного арила, арилокси, замещенного арилокси, циано, атома галогена, гидроксила, нитро, карбоксила, сложных эфиров карбоксила, циклоалкила,замещенного циклоалкила, гетероциклила и замещенного гетероциклила, при условии, что любой гидроксильный заместитель не присоединен к атому углерода ацетиленовой (ненасыщенной) группы."Ацил" относится к группам Н-С(О)-, алкил-С(О)-, замещенный алкил-С(О)-, циклоалкил-С(О)-,замещенный циклоалкил-С(О)-, арил-С(О)-, замещенный арил-С(О)-, гетероциклил-С(О)- и замещенный гетероциклил-С(О)-, где алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, арил, заме-4 015126 щенный арил, гетероциклил и замещенный гетероциклил имеют значения, указанные в настоящем описании."Ациламино" относится к группе -C(O)NR'R', где каждый R' независимо выбран из группы, состоящей из водорода, алкила, замещенного алкила, замещенного арила, циклоалкила, замещенного циклоалкила, гетероарила, замещенного гетероарила, гетероциклила, замещенного гетероциклила, и где оба R' могут быть соединены с образованием вместе с атомом азота гетероциклильного или замещенного гетероциклильного кольца, и где алкил, замещенный алкил, циклоалкил, замещенный циклоалкил, арил, замещенный арил, гетероциклил и замещенный гетероциклил имеют значения, указанные в настоящем описании."Ацилокси" относится к группам алкил-С(О)O-, замещенный алкил-С(О)О-, арил-С(О)О-, замещенный арил-С(О)О-, циклоалкил-С(O)O-, замещенный циклоалкил-С(О)O-, гетероциклил-С(О)О- и замещенный гетероциклил-С(О)O-, где алкил, замещенный алкил, циклоалкил, замещенный циклоалкил,арил, замещенный арил, гетероциклил и замещенный гетероциклил имеют значения, указанные в настоящем описании."Замещенный амино" относится к группе -NR'R', где R' имеют значения, указанные выше, при условии, что оба R' не являются водородом. Когда R' представляет собой водород и другой R' представляет собой алкил, замещенную аминогруппу иногда называют в настоящем описании алкиламино. Когда оба из R' представляют собой алкил, замещенную аминогруппу иногда называют в настоящем описании диалкиламино. Указание на монозамещенный амино означает, что любой из R', но не оба, представляет собой водород. Указание на дизамещенный амино означает, что ни один из R' не является водородом."Сложный эфир карбоксила" относится к группам -С(O)O-алкил, -С(О)О-замещенный алкил,-С(О)О-арил и -С(О)O-замещенный арил, где алкил, замещенный алкил, арил и замещенный арил имеют значения, указанные в настоящем описании."Арил" или "Ar" относится к одновалентной ароматической карбоциклической группе с 6-14 атомами углерода, имеющей одно кольцо (например, фенил) или несколько конденсированных колец (например, нафтил или антрил), где конденсированные кольца могут или не могут быть ароматическими(например, 2-бензоксазолинон, 2 Н-1,4-бензоксазин-3(4 Н)-он-7-ил и тому подобное), при условии, что место присоединения находится у атома углерода ароматического кольца. Предпочтительные арилы включают фенил и нафтил."Гетероциклил" или "гетероциклический" относится к насыщенной или ненасыщенной группе,имеющей одно кольцо или несколько конденсированных колец, от 1 до 10 атомов углерода и от 1 до 4 гетероатомов, выбранных из группы, состоящей из атомов азота, серы или кислорода, в кольце, где в системе конденсированных колец одно или несколько колец могут быть циклоалкилом, арилом или гетероарилом, при условии, что местом присоединения является гетероциклическое кольцо."Замещенный гетероциклил" относится к гетероциклильным группам, которые замещены 1-3 такими же заместителями, как указаны для замещенного циклоалкила. Примеры гетероциклилов включают, но не ограничиваются перечисленным, пиридинил, пирролил,индолил, тиенил, фурил, бензотиенил, бензофуранил, имидазолил, бензоимидазолил, пиразолил, тиазолил, бензотиазолил, изотиазолил, оксазолил, изоксазолил, пиразинил, пиримидинил, пиридазинил, изоиндолил, пуринил, хинолил, изохинолил, дигидрохинолинил, 2,3-дигидро-1 Н-индолил, хиноксалинил,бензодиоксолил, инданил, инденил, триазолил, азетидинил, индолизинил, дигидроиндолил, индазолил,хинолизинил, фталазинил, нафтилпиридинил, хиназолинил, циннолинил, птеридинил, карбазолил, карболинил, фенантридинил, акридинил, фенантролинил, феназинил, феноксазинил, фенотиазинил, имидазолидинил, имидазолинил, пиперидинил, пиперазинил, индолинил, фталимидил, 1,2,3,4-тетрагидроизохинолинил, 4,5,6,7-тетрагидробензо[b]тиофенил, тиазолидинил, морфолинил, тиоморфолинил (называемый также тиаморфолинилом), пиперидинил, пирролидинил, пирролинил, пиразолинил, пиразолидинил и тетрагидрофуранил.-5 015126 Следует указать, что при упоминании гетероциклила и замещенного гетероциклила любые атомы азота или серы, которые могут присутствовать, могут быть необязательно окислены. Из всего вышеуказанного специалисту в данной области очевидно, что любая из указанных групп или заместителей, например галогеналкил, алкокси, алкоксикарбонил, арилокси, гетероарилокси, аминоалкил, алкиламино, алкиламиноалкил, диалкиламиноалкил и тому подобное, должна истолковываться из названий групп, из которых они происходят. В этом отношении в качестве примера любая группа, которая идентифицирована как арилалкил или гетероциклоалкил, должна рассматриваться как алкильная группа, которая дополнительно замещена арилом или гетероциклом, где арил, гетероцикл и алкил имеют значения, указанные выше. Соединения формулы (I) настоящего изобретения могут иметь асимметричные атомы углерода и поэтому могут существовать в виде индивидуальных оптических изомеров, в виде рацемических смесей или в виде любой другой смеси, включающей в доминирующем количестве один из двух оптических изомеров, которые все следует рассматривать как находящиеся в пределах объема настоящего изобретения. В случаях, когда соединения могут существовать в таутомерных формах, например в виде кетоенольных таутомеров, каждая таутомерная форма рассматривается как включенная в настоящее изобретение независимо от того, находится ли она в равновесии или преимущественно в одной форме. Таким же образом, применение в качестве противоопухолевого агента всех возможных изомеров и их смесей и как метаболитов, так и фармацевтически приемлемых биопредшественников (иначе называемых фармацевтически приемлемыми пролекарствами) соединений формулы (I) также находится в пределах объема настоящего изобретения. Применяемый в настоящем описании термин "фармацевтически приемлемые соли" относится к солям нетоксичных кислот или щелочно-земельных металлов соединений формулы (I). Такие соли можно получить in situ во время последнего выделения и очистки соединений формулы (I) или отдельно реакцией основных или кислотных функциональных групп с подходящей органической или неорганической кислотой или основанием соответственно. Репрезентативные соли включают, но не ограничиваются перечисленным, следующие соли: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат,бисульфат, бутират, камфорат, камфорасульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, малонат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, пальмоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат и ундеканоат. Кроме того, основные азотсодержащие группы можно кватернизовать такими агентами, как алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -иодиды, диалкилсульфаты, подобные диметил-, диэтил-, дибутил- и диамилсульфаты, галогениды с длинной цепью, такие как децил-, лаурил-,миристил- и стеарилхлориды, -бромиды и -иодиды, аралкилгалогениды, подобные бензил- и фенетилбромидам, и другие соединения. Тем самым получают растворимые или диспергируемые в воде или масле продукты. Примеры кислот, которые можно применять для получения фармацевтически приемлемых кислотно-аддитивный солей, включают такие неорганические кислоты, как хлористо-водородная кислота, серная кислота, азотная кислота, бромисто-водородная кислота, перхлорная кислота и фосфорная кислота, и такие органические кислоты, как щавелевая кислота, малеиновая кислота, метансульфоновая кислота,янтарная кислота и лимонная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, малоновая кислота, яблочная кислота, винная кислота, бензойная кислота, коричная кислота, миндальная кислота, изетионовая кислота и салициловая кислота. Основно-аддитивные соли можно получить in situ во время последнего выделения и очистки соединений формулы (I) или отдельно реакцией фрагментов карбоновых кислот с подходящим основанием,таким как гидроксид, карбонат или бикарбонат катиона фармацевтически приемлемого металла или с аммиаком, или органическим первичным, вторичным или третичным амином. Фармацевтически приемлемые соли включают, но не ограничиваются перечисленным, катионы на основе щелочных и щелочноземельных металлов, такие как соли натрия, лития, калия, кальция, магния, алюминия и тому подобное, а также катионы аммония, четвертичного аммония и амина, включающие, но не ограничивающиеся перечисленным, катионы аммония, тетраметиламмония, тетраэтиламмония, метиламина, диметиламина, триметиламина, триэтиламина, этиламина и тому подобное. Другие репрезентативные органические амины,пригодные для получения основно-аддитивных солей, включают диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и тому подобное. Термины "фармацевтически приемлемое пролекарство" и "фармацевтически приемлемые биопредшественники", применяемые в настоящем описании, относятся к таким пролекарствам соединений настоящего изобретения, которые в пределах объема надежной медицинской оценки являются подходящими для применения в контакте с тканями человека или низших животных без чрезмерной токсичности,раздражения, аллергической реакции и тому подобного, соразмерных с приемлемым отношением польза/риск, и являются эффективными для предназначенного для них применения, а также, когда возможно,-6 015126 цвиттерионным формам соединений настоящего изобретения. Термин "пролекарство" относится к соединениям, которые быстро трансформируются in vivo с образованием активного "родительского" лекарственного средства согласно формуле (I), например, гидролизом в крови. Обсуждение описано в публикациях Т. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, vol. 14 of the A.C.S. Symposium Series,и Edward B. Roche, ea., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, обе из которых включены в настоящее описание посредством ссылки. Как указано ранее, следующей задачей настоящего изобретения является способ получения указанных выше соединений формулы (I) и их фармацевтически приемлемых солей. Соединения настоящего изобретения можно получить из легкодоступных исходных соединений с применением следующих общих способов и методик. Если не оговорено особо, исходными соединениями являются известные соединения или их можно получить из известных соединений согласно хорошо известным методикам. Должно быть понятно, что когда указываются типичные или предпочтительные условия способа (т.е. температуры, время, молярные отношения реагентов, растворители, давления реакции), можно также применять другие условия способа, если не оговорено иначе. Оптимальные условия реакции могут изменяться в зависимости от конкретных применяемых реагентов или растворителей, но такие условия могут быть определены специалистом в данной области стандартными процедурами оптимизации. Кроме того, как должно быть очевидно специалисту в данной области, могут быть необходимы общепринятые защитные группы для предотвращения участия некоторых функциональных групп в нежелательных реакциях. Подходящие защитные группы для различных функциональных групп, а также подходящие условия для защиты конкретных функциональных групп и снятия у них защиты являются хорошо известными в данной области. Например, многие защитные группы описаны в публикации T.W. Greene and P.G.M. Wuts, Protecting Groups in Organic Synthesis, Second Edition, Wiley, New York,1991 и ссылках, цитированных в ней. В частности, настоящее изобретение относится к способу, включающему: а) сочетание соединения формулы 1E где R1, R4, R5, R6 и G имеют значения, указанные выше, и циклил представляет собой необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп, которые имеют значения, указанные выше, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, где R3 имеет значения, указанные выше, с получением таким образом указанного выше соединения формулы (I), где W представляет собой NR1, где R1 имеет значения, указанные выше, и R2 представляет собой необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп;b) необязательное превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I). Сочетание для превращение 1E в требуемое соединение формулы (I) можно проводить хорошо известными протоколами получения первичного амида [например,аммониевой соли 1-гидроксибензотриазола (HOBTNH3) в присутствии либо гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI), либо тетрафторбората O-бензотриазол-1-ил-N,N,N',N'тетраметилурония (TBTU) или CDI и карбоната аммония]; превращение во вторичные амиды можно проводить сочетанием с амином формулы R3-NH2, где R3 имеет значения, указанные выше, в различных хорошо известных условиях образования амидов. Таким же образом, превращение соединения формулы (I) в соль или превращение его соли в свободное соединение (I), выполняемое согласно хорошо известным в данной области методикам, все же находится в пределах объема настоящего изобретения.-7 015126 Получение соединений формулы 1E показано на нижеследующей схеме 1. Схема 1 где R1, R4, R5, R6, G и циклил имеют значения, указанные выше, X представляет собой атом галогена, такой как атом брома или хлора, и Alk представляет собой C1-C5-алкильную группу. Соединение 1D можно получить сочетанием галогенкетона 1 А со сложным бета-кетоэфиром 1 В в присутствии подходящего основания, такого как гидрид натрия, в растворителе, подобном тетрагидрофурану (ТГФ) или диметилформамиду (ДМФА), при температурах, составляющих от -20 до 50 С, и затем выдерживанием промежуточного соединения 1 С в условиях реакции Hantzsch в присутствии ацетата аммония (когда R1 представляет собой Н) или амина формулы R1-NH2 в подходящем растворителе, таком как этанол или уксусная кислота или смесь этих двух растворителей, при температурах от комнатной до 150 С в течение периода времени от приблизительно 10 мин до 16 ч, необязательно внутри микроволновой полости. Сложный эфир 1D можно затем омылить в стандартных основных условиях с получением кислоты формулы 1E. В некоторых случаях, когда R4 является отличным от атома водорода, гидролиз сложного эфира 1D может привести к декарбоксилированному аналогу. Согласно этому предложен другой способ получения соединения формулы (I), который включает амидирование соединения формулы 5D где R1, R5, R6, G и циклил имеют значения, указанные выше, и R4 не является атомом водорода, иb) необязательно превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I). В этом случае амидирование выполняют, как описано, например, в Synthesis, 1978, 374, обработкой соединения 5D хлорсульфонилизоцианатом в подходящем растворителе, таком как ацетонитрил, дихлорметан или диэтиловый эфир, и обработкой полученного хлорсульфониламида щелочью для получения соответствующей сульфаминовой кислоты. Сульфаминовую кислоту затем гидролизуют в требуемое соединение формулы (I) в кислотной среде, такой как, например, концентрированная хлористо-водородная кислота. Соединения формул 1 А и 1 В, а также любой другой реагент способа являются известными и, если не являются коммерчески доступными per se, их можно легко получить согласно известным способам. Соединения формулы 1A можно получить галогенированием, например бромированием или хлорированием, подходящего производного гетероарилэтанона или его активированного эквивалента. Реакцию проводят общепринятыми методами, например, в присутствии брома и в подходящем растворителе, таком как водная бромисто-водородная кислота или смесь уксусной кислоты и бромисто-водородной кислоты, в течение времени, изменяющегося от приблизительно 1 ч до приблизительно 24 ч. В альтернативном случае подходящим образом активированное гетероарилпроизводное, например алкиловый простой эфир енола или силиловый простой эфир енола, можно подвергать реакции с источником галогена,например N-бромсукцинимидом (NBS), в подходящем растворителе, таком как смеси тетрагидрофуран/вода. В частности, среди подходящих галогенпроизводных формулы 1 А авторы настоящего изобретения рассматривают 2-бром-1-пиридин-4-илэтанон (коммерческий), 2-бром-1-(3-фторпиридин-4-ил)этанон(описан в WO 2005013986), 2-бром-1-пиридин-4-илпропан-1-он (коммерческий), 2-бром-1-пиримидин-4-8 015126 илэтанон (описан в WO 2005014572), 1-(2-аминопиримидин-4-ил)-2-бромэтанон (коммерческий), 2-бром 1-(2-хлорпиридин-4-ил)этанон (описан в WO 2004058762), 2-бром-1-(2-метилсульфанилпиримидин-4 ил)этанон (описан в WO 2003011838), 1-(2-амино-5-фторпиримидин-4-ил)-2-бромэтанон 1'А (схема 2.1) и 2-бром-1-(2-фениламинопиримидин-4-ил)этанон 1"А (схема 2.2). Среди подходящих производных гетероарилэтанона, подвергаемых галогенированию, авторы настоящего изобретения рассматривают, например, 1-(3-фторпиридин-4-ил)этанон (коммерческий), 1-(2 хлорпиридин-4-ил)этанон (коммерческий), 1-(пиримидин-4-ил)этанон (коммерческий), 1-[2-(метилтио)пиримидин-4-ил]этанон (коммерческий) и 4-(1-этоксивинил)-5-фторпиримидин-2-иламин iii (см. схему 2.1). Промежуточное соединение 1-(2-амино-5-фторпиримидин-4-ил)-2-бромэтанон 1'А (1 А, где R4 представляет собой Н, R5 представляет собой F, G представляет собой N, R6 представляет собой NH2, X представляет собой Br) можно получить согласно следующей схеме реакций (схема 2.1): Схема 2.1 Коммерчески доступный 2,4-дихлор-5-фторпиримидин i подвергают реакции с (1-этоксивинил)трибутилстаннаном в стандартных условиях в присутствии палладиевого катализатора (например,дихлордитрифенилфосфинопалладия) в ДМФА, получая соответствующий простой 4-виниловый эфир ii. Аминогруппу можно ввести в положение 2 непосредственной обработкой водным концентрированным аммиаком в этаноле и нагреванием при обработке микроволновым излучением iii, в то время как бромирование винилового простого эфира с получением -бромкетона (1'А) проводят с применением NBS в водных растворителях. Среди силиловых простых эфиров енола, подвергаемых галогенированию, авторы настоящего изобретения рассматривают, например, (трет-бутилдиметилсиланил)-4-[1-(трет-бутилдиметилсиланилокси)винил]пиримидин-2-илфениламин iv (описан в WO 2005014572), из которого бромкетон 1"А (1 А, гдеR4 и R5 представляют собой Н, G представляет собой N, R6 представляет собой NHAr, X представляет собой Br) можно получить согласно стадии реакции, показанной на схеме 2.2. Схема 2.2 где Ar представляет собой арильную группу, которая имеет значения, указанные выше. Галогенирование соединения формулы iv можно быстро провести N-бромсукцинимидом в водном тетрагидрофуране при комнатной температуре в течение приблизительно 20 ч. Соединения формулы 1 В,которые не являются коммерчески доступными, можно получить различными способами согласно ссылкам в литературе. Например, гомологизацию кислот с получением сложных бета-кетоэфиров можно проводить, исходя из ацилхлоридов или карбоновых кислот активацией 2,2-диметил-1,3-диоксан-4,6-дионом(кислота Мелдрума), как описано в J. Med. Chem., 2001, 44, 90, из ацилхлоридов и этилгидромалоната,как описано в J. Het. Chem., 1990, 27, 1609, или из арилэтанонов с диэтилкарбонатом, как описано в Can.J. Chem., 1992, 1323. В альтернативном случае соединение формулы (I), указанное выше, можно получить: а') сочетанием соединения формулы 2D где R2 представляет собой атом водорода или галогена, и R5, R6 и G имеют значения, указанные выше, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, где R3 имеет значения, указанные выше, с образованием таким образом указанного выше соединения формулы (I), где W представляет собой N, R1 представляет собой атом водорода и R2 представляет собой атом водорода или атом галогена;a'1) необязательно превращением образовавшегося соединения формулы (I), где R2 представляет со-9 015126 бой атом галогена, в другое соединение формулы (I), где R2 представляет собой атом водорода или необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп,которые имеют значения, указанные выше; и/или а'2) превращением образовавшегося соединения формулы (I), где R1 представляет собой атом водорода, в другое соединение формулы (I), где R1 представляет собой необязательно замещенную группу,выбранную из алкила, циклоалкила, алкенила, алкинила, гетероциклила, арила, гетероциклилоксиалкила и алкоксикарбонила; и, если необходимо, превращением соединения формулы (I) в его фармацевтически приемлемую соль или превращением соли в свободное соединение (I). Исходное соединение формулы 2D получают посредством реакции, подобной реакции Пиннера, как показано на нижеследующей схеме 3, аналогично или согласно способам, описанным в литературе (например, Il Farmaco, 1999, 54, 542 или Tetrahedron Letters, 1994, 35, 5989). Схема 3 где R5, R6, X, G, циклил и Alk имеют значения, указанные выше. Соединение 2 В можно получить обработкой галогенкетона 1 А, который имеет значения, указанные выше, со сложным цианоэфиром 2 А в присутствии подходящего основания, такого как этоксид натрия в этаноле, при температуре в пределах от комнатной до температуры флегмы в течение периодов времени от приблизительно от 1 до 16 ч. Галогенпиррол 2 С можно получить обработкой 2 В галогенводородными кислотами, такими как хлористо-водородная кислота в диоксане или бромисто-водородная кислота в уксусной кислоте, в растворителе, подобном дихлорметану или простому диэтиловому эфиру или их смесям, при температурах, составляющих от -20 С до температуры флегмы, наиболее часто при комнатной температуре. 2-Незамещенные пирролы можно получить дегалогенированием галогенпиррола 2 С,которое можно выполнить, например, гидрогенолизом с получением 4 А. В альтернативном случае образование функциональной группы в положение 2 можно выполнить превращением сложного галогенэфира 2 С с получением сложного эфира 4D. Сложные эфиры 2 С, 4 А и 4D можно затем омылить в стандартных основных условиях в соответствующие кислоты 2D. В альтернативном случае указанное выше соединение формулы (I) можно получить: а') расщеплением соединения формул 3 А или 3 В где R3, R5, R6, циклил, X, G имеют значения, указанные выше; и символ представляет собой твердый носитель, с которым связана химическая молекула,и, если необходимо, превращением образовавшегося соединения формулы (I), где W представляет собой атом азота и R2 представляет собой атом галогена или необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп, в его фармацевтически приемлемую соль или превращением соли в свободное соединение (I). Расщепление предпочтительно выполняют смесью TFA/DCM.- 10015126 Указанное выше получение позволяет избежать образования нежелательных побочных продуктов. Получение соединений 3 А и 3 В изображено на схеме 4. Кислоту 2D загружают на твердый носитель,такой как смола (например, амидная смола МВНА Ринка, предварительно расщепленная встряхиванием при комнатной температуре в 20% растворе пиперидина в ДМФА), перемешиванием при комнатной температуре в течение ночи в ДМФА в присутствии EDCI и НОВТ, получая амид 3 А, который удачно подвергают реакции образования углерод-углеродной связи, например реакции Сузуки, получая соединение 3 В. Схема 4 где R3, R5, R6, X, G и циклил имеют значения, указанные выше. Конкретное соединение формулы 1D, названное соединением 6D, где циклил имеет заместитель,можно необязательно превратить в другое соединение формулы 1D, названное соединением 7D, как представлено на схеме 5. Схема 5 где R1, R4, R5, R6, G, циклил и Alk имеют значения, указанные выше, Е представляет собой галоген,трифлатную, мезилатную или тозилатную группу и М представляет собой арил, гетероарил, гетероциклил, алкенил, алкинил или необязательно замещенные аминофрагменты. Конкретную циклильную часть, а именно подходящим образом замещенное арильное или гетероциклильное кольцо, имеющее заместитель Е, который имеет значения, указанные выше, можно подвергать реакции образования углерод-углеродной или углерод-азотной связи рядом хорошо известных из литературы способов, например протоколов Сузуки, Стилла, Соногашира или Бухвальда, при помощи которых получают требуемое соединение 7D, в котором М имеет значения, указанные выше. Кроме того, настоящее изобретение относится к способу, включающему: а) сочетание соединения формулы 7 В где W представляет собой атом кислорода или серы, R4, R5, R6 и G имеют значения, указанные выше, и циклил представляет собой необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп, которые имеют значения, указанные выше, либо с активированной формой аммиака, необязательно в присутствии агента конденсации, либо с амином формулы R3-NH2, гдеR3 имеет значения, указанные выше, с образованием таким образом описанного выше соединения формулы (I), где W представляет собой атом кислорода или серы и R2 представляет собой необязательно замещенную группу, выбранную из арильной, циклоалкильной и гетероциклильной групп;b) необязательное превращение соединения формулы (I) в другое соединение формулы (I) и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I). В необязательной стадии b), когда W представляет собой атом серы, кольцо тиофена можно окислить в соответствующие 1-оксо- или 1,1-диоксотиофены хорошо известными из литературы методиками. Сложные эфиры общей формулы 7 А, принадлежащие к рядам тиофена и фурана, можно подходящим образом получить в виде смеси из сложных кетоэфиров 1 С циклизацией, например, с применением 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфида (реагент Лавессона). После хроматографического разделения два сложных эфира можно по отдельности подвергать стандартному основному гидролизу, чтобы получить требуемые кислоты 7 В (схема 6). где R4, R5, R6, G, W, циклил и Alk имеют значения, указанные выше. Как указано выше, необязательно соединения формулы (I) можно непосредственно модифицировать в любом из их гетероароматических компонентов, чтобы получить другое производное формулы (I). Например, кольцо пиримидина пиррола 8 А можно непосредственно галогенировать для получения амидов 8 В и 8 С. Такое превращение выполняют соответственно обработкой NCS приблизительно при 100 С или NCB при комнатной температуре в подходящем растворителе, подобном, например, ТГФ или ДМФА. В альтернативном случае NIS в таких же условиях превращает амид 8 А в соответствующий амид 8D, галогенированный в положении 4 пиррола. Такой же амид 8 А, если его предварительно защищают в кольце аминопиримидина превращением в Вос-производное, можно также превратить в хлорамид 8 Е обработкой NCS при 100 С в ДМФА и снятием защиты в стандартных кислотных условиях. Как показано на схеме 7, можно также проводить как двойное последовательное, так и двойное одновременное галогенирование. Бромамид 8 С при обработке NIS дает амид 8F, и такой же результат получают обработкой иодамида 8D NBS. Амид 8 А одновременно дигалогенируют двумя эквивалентами галогенирующего агента, получая амиды 8G и 8 Н при обработке NBS и NIS соответственно. Амид 8D можно превращать в 4-винилпроизводное 8K прямым винилированием посредством протокола реакции перекрестного сочетания Стилла, как описано, например, в Tetr. Lett., 1995, 36, 7043. Реакцию проводят обработкой 8D винилтрибутилстаннаном в присутствии палладиевого катализатора, такого как палладийтетракис или дихлордитрифенилфосфинопалладий, в растворителях, подобных диоксану, ДМФА, или их смесях при температурах, составляющих от 25 до 200 С. Схема 7 где R3, R5, R6, X, G и циклил имеют значения, указанные выше. Другой способ получения соединения формулы (I) включает: а) катализируемую Pd реакцию сочетания соединения формулы 9 где L представляет собой уходящую группу, такую как галоген, метансульфонил или метансульфинил, и R3, R5, циклил и G имеют значения, указанные выше, либо с активированной формой аммиака,подобной бис-(триметилсилил)амиду лития, необязательно в присутствии агента конденсации, или с гидразином с последующим восстановлением в амин, либо с амином формулы R7-NH2, где R7 имеет значе- 12015126 ния, указанные выше, с получением таким образом указанного выше соединения формулы (I), где W представляет собой NR1, R1 представляет собой атом водорода и R6 представляет собой NH-R7; и, если необходимо, превращение соединения формулы (I) в его фармацевтически приемлемую соль или превращение соли в свободное соединение (I). Исходные соединения 9 настоящего способа показаны на схеме 8. Галогенпиридин 9, где L представляет собой галоген 9 А, можно получить, например, из 2-бром-1(2-хлорпиридин-4-ил)этанона, полученного галогенированием 1-(2-хлорпиридин-4-ил)этанона, получаемого из 2-хлор-4-цианопиридина, как описано в J. Het. Chem., 1983, 20, 533. 1-[2-(Метилтио)пиримидин 4-ил]производное 9, у которого L представляет собой CH3-S- 9B, получаемое из 2-бром-1-(2-метилсульфанилпиримидин-4-ил)этанона (описан в WO 03/011838), можно активировать превращением в соответствующий сульфоксид или сульфон 9, где L представляет собой CH3-S(O)- или CH3-S(O)2- 9D,окислением, например, оксоном. Производное галогенпиримидина 9, у которого L представляет собой галоген и R5 представляет собой фтор 9 С, можно вместо этого получить из 2-бром-1-(2-хлор-5-фторпиримидин-4-ил)этанона, полученного галогенированием 1-(2-хлор-5-фторпиримидин-4-ил)этанона, в свою очередь, полученного, как описано в WO 04/058762. Схема 8 где R3, R5, X и циклил имеют значения, указанные выше. Следующим объектом настоящего изобретения является промежуточное соединение формулы 1D или 1E где G, Alk, циклил, R1, R4, R5 и R6 имеют значения, указанные выше, при условии, что исключены следующие соединения: этиловый эфир 1-(метоксиметил)-4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; метиловый эфир 2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; метиловый эфир 4-метил-2-фенил-5-(4-пиридинил)-1 Н-пиррол-3-карбоновой кислоты; 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновая кислота, соединение с морфолином (1:1); 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновая кислота; метиловый эфир 4-(метоксиметил)-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; этиловый эфир 4-бутил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; этиловый эфир 4-(1-метилэтил)-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; этиловый эфир 4-пропил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; 2-метоксиэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; бутиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; пропиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; 1,1-диметилэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; 1-метилэтиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; 2-пропениловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; фенилметиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; метиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты; этиловый эфир 4-метил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты и этиловый эфир 4-этил-2,5-ди-4-пиридинил-1 Н-пиррол-3-карбоновой кислоты. Фармакология Соединения формулы (I) являются активными в качестве ингибиторов протеинкиназы и поэтому являются пригодными, например, для ограничения неконтролируемой пролиферации опухолевых кле- 13015126 ток. В терапии их можно применять при лечении различных опухолей, таких как опухоли, описанные выше, а также при лечении других клеточных пролиферативных нарушений, таких как псориаз, пролиферация клеток сосудов гладких мышц, связанная с атеросклерозом, послехирургическим стенозом и рестенозом, и при лечении болезни Альцгеймера. Анализ ингибирования активности Cdc7. Ингибирование активности предполагаемых ингибиторов Cdc7 и эффективность выбранных соединений определяют посредством способа анализа на основе применения методики захвата смолой Dowex. Анализ состоит из переноса меченного радиоактивным изотопом фосфатного фрагмента киназой к субстрату-акцептору. Образовавшийся 33 Р-меченый продукт отделяют от непрореагировавшего меченого вещества, переносят в сцинтилляционный коктейль и измеряют излучение света в сцинтилляционном счетчике. Анализ ингибирования активности Cdc7/Dbf4 проводят согласно следующему протоколу. Субстрат МСМ 2 трансфосфорилируют комплексом Cdc7/Dbf4 в присутствии АТФ, меченного 33-АТФ. Реакцию останавливают добавлением смолы Dowex в присутствии муравьиной кислоты. Частицы смолы Dowex захватывают непрореагировавший 33-АТФ и тащат его ко дну лунки, тогда как фосфорилированный 33 Р субстрат МСМ 2 остается в растворе. Супернатант собирают, переносят в планшетыOptiplate и степень фосфорилирования субстрата оценивают -подсчетом. Анализ ингибирования активности Cdc7/Dbf4 проводят в 96-луночном планшете согласно следующему протоколу. К каждой лунке планшета добавляют 10 мкл испытуемого соединения (10 повышающихся концентраций в диапазоне от нМ до мкМ для построения кривой доза-реакция), причем растворитель для испытуемых соединений содержал 3% ДМСО (конечная концентрация 1%); 10 мкл субстрата МСМ 2 (конечная концентрация 6 мкМ), смесь не содержащего радиоактивный изотоп АТФ (конечная концентрация 2 мкМ) и радиоактивного АТФ (молярное отношение его к не содержащему радиоактивный изотоп АТФ 1/5000); 10 мкл фермента (Cdc7/Dbf4, конечная концентрация 2 нМ), который запускает реакцию, причем буфер для реакции состоял из 50 мМ HEPES, рН 7,9, содержащего 15 мМ MgCl2, 2 мМ DTT, 3 мкМNaVO3, 2 мМ глицерофосфат и 0,2 мг/мл BSA. После инкубации в течение 60 мин при комнатной температуре реакцию останавливали добавлением к каждой лунке 150 мкл смолы Dowex в присутствии 150 мМ муравьиной кислоты. После еще 60 мин инкубации отбирали 50 мкл суспензии и переносили в 96-луночные планшеты Optiplate, содержащие 150 мкл MicroScint 40 (Packard); после 5-10 мин встряхивания планшетов считывание данных радиоактивности проводили в течение 1 мин в планшет-ридере Packard TOP-Count. Определение IC50: ингибиторы испытывали при различных концентрациях, составляющих от 0,0005 до 10 мкМ. Экспериментальные данные анализировали компьютерной программой Assay Explorer с применением четырехпараметрического логистического уравненияy=низ+(верх-низ)/(1+10logIC50-x)угол наклона,где х представляет собой логарифм концентрации ингибитора; у представляет собой ответную реакцию; у начинается внизу и достигает верха при сигмоидной форме. Соединения формулы (I) настоящего изобретения обнаруживают величины IC50 в отношенииCdc7/Dbf4 между 1 и 1000 нМ. В частности, соединения, кодированные в настоящем описании ниже как А 1, А 2, A3, А 4, А 5, А 6, А 7, А 8, А 9, А 10, А 11, А 12, А 20, С 1, С 2, E1, F1, G1, H1, L1, М 2, М 3, М 4, обнаружили величины IC50 в отношении Cdc7/Dbf4 между 1 и 100 нМ. Помимо этого, выбранные соединения характеризовали на специфичность для набора многих других киназ, среди которых имеются Cdk2A, IGF1-R, Aurora-2, AKT1, PLK1, SULU1, ERK2, CK2, GSK3,PKA, PKC, VEGFR3, PDGFR. Анализ ингибирования активности ферментов Cdk2/циклин А. Реакция киназы: 1,5 мкМ субстрат гистон H1, 2,5 мкМ АТФ (0,2 мкКи 33 Р-АТФ), 30 нг коэкспрессированных бакуловирусом ферментов Cdk:2/циклин А, 10 мкМ ингибитор в конечном объеме 100 мкл буфера (10 мМ TRIS HCl, рН 7,5, 10 мМ MgCl2, 7,5 мМ DTT) добавляли к каждой лунке 96-луночного планшета с лунками с U-образным дном. После инкубации 10 мин при 37 С реакцию останавливали 20 мкл 120 мМ ЭДТА. Захват: 100 мкл переносили из каждой лунки в планшет Multiscreen, давая тем самым возможность субстрату связываться с фосфоцеллюлозным фильтром. Планшеты затем промывали 3 раза 150 мкл/лунку PBS без Са/Mg и фильтровали с помощью системы фильтрования Multiscreen. Детекция: фильтрам давали возможность высохнуть при 37 С, затем добавляли 100 мкл/лунку сцинтиллятора и меченный 33 Р гистон H1 детектировали подсчетом радиоактивности в приборе TopCount. Результаты: получали данные анализа и выражали их как процент ингибирования относительно- 14015126 общей активности фермента (равной 100%). Все соединения, проявляющие ингибирование, больше или равное 50%, далее анализировали для изучения и определения активности (IC50), a также кинетического профиля ингибитора посредством вычисления Ki. Определение IC50: применяемый протокол был таким же, как описан выше, где ингибиторы испытывали при разных концентрациях, составляющих от 0,0045 до 10 мкМ. Экспериментальные данные анализировали компьютерной программой GraphPad Prizm с применением четырехпараметрического логистического уравнения: у=низ+(верх-низ)/(1+10logIC50-x)угол наклона,где х представляет собой логарифм концентрации ингибитора; у представляет собой ответную реакцию; у начинается внизу и достигает верха при сигмоидной форме. Вычисление Ki: изменяли концентрацию любого из АТФ и субстрата гистона H1: применяли 4, 8,12, 24, 48 мкМ для АТФ (содержащего пропорционально разведенный 33 Р-АТФ) и 0,4, 0,8, 1,2, 2,4,4,8 мкМ для гистона в отсутствие или в присутствии ингибитора при двух разных, должным образом выбранных концентрациях. Экспериментальные данные анализировали компьютерной программой "SigmaPlot" для определения Ki с применением уравнения для произвольной системы двух реагентов где А представляет собой АТФ и В представляет собой гистон H1. Соединения настоящего изобретения можно вводить либо в виде отдельных агентов, либо в альтернативном случае в комбинации с известными противораковыми лечениями, такими как схема лечения лучевой терапией или химиотерапией в сочетании с цитостатическими или цитотоксическими агентами,агентами антибиотического типа, алкилирующими агентами, антиметаболическими агентами, гормональными агентами, иммунологическими агентами, агентами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторами СОХ-2), ингибиторами металлопротеазы матрикса, ингибиторами теломеразы, ингибиторами тирозинкиназы, агентами против рецептора фактора роста, агентами противHER, агентами против EGFR, агентами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути трансдукции сигнала ras-raf, ингибиторами клеточного цикла, другими ингибиторами cdks, тубулинсвязывающими агентами, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и тому подобными. При изготовлении в виде установленной дозы такие комбинированные продукты содержат соединения настоящего изобретения в пределах диапазона доз, описанного ниже, и другой фармацевтически активный агент в пределах подходящего диапазона доз. Соединения формулы (I) можно применять последовательно с известными противораковыми агентами, когда комбинированный препарат является неподходящим. Соединения формулы (I) настоящего изобретения, подходящие для введения млекопитающему, например людям, можно вводить обычными путями и уровень дозы зависит от возраста, массы, состояний пациента и пути введения. Например, подходящая доза, адаптированная для перорального введения соединения формулы (I), может быть в диапазоне от приблизительно 10 до приблизительно 1000 мг на дозу от 1 до 10 раз в сутки. Соединения настоящего изобретения можно вводить в различных дозированных формах, например орально в форме таблеток, капсул, таблеток, покрытых сахаром или пленкой, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например внутримышечно или посредством внутривенной, и/или подоболочечной, и/или интраспинальной инъекции или инфузии. Настоящее изобретение включает также фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым эксципиентом, который может быть носителем или разбавителем. Фармацевтические композиции, содержащие соединения настоящего изобретения, обычно получают следующими общепринятыми методами и вводят в подходящей фармацевтической форме. Например, твердые оральные формы могут содержать вместе с активным соединением разбавители, например лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; лубриканты, например диоксид кремния, тальк,стеариновую кислоту, стеарат магния или стеарат кальция, и/или полиэтиленгликоли; связывающие агенты, например крахмалы, аравийскую камедь, желатин, метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезинтегрирующие агенты, например крахмал, альгиновую кислоту, альгинаты или натриевую соль гликолята крахмала; вспенивающие смеси; красители; подслащивающие агенты; увлажняющие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и в общем нетоксичные и фармакологически неактивные вещества, применяемые в фармацевтических препаратах. Указанные фармацевтические препараты можно изготовить известным способом, например посредством способов- 15015126 смешивания, гранулирования, таблетирования, покрытия сахаром или покрытия пленкой. Жидкими дисперсиями для орального введения могут быть, например, сиропы, эмульсии и суспензии. В качестве примера сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннитом и сорбитом. Суспензии и эмульсии могут содержать в качестве примеров носителей природную камедь, агар,альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например стерильную воду, оливковое масло, этилолеат, гликоли, например пропиленгликоль, и, если нужно, подходящее количество гидрохлорида лидокаина. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду или предпочтительно они могут быть в форме стерильных, водных, изотонических, солевых растворов или они могут содержать пропиленгликоль в качестве носителя. Суппозитории могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например какао-масло, полиэтиленгликоль, эфир жирной кислоты полиоксиэтиленсорбитана в качестве поверхностно-активного вещества или лецитин. С целью лучшей иллюстрации настоящего изобретения, но без намерения какого-либо ограничения его, теперь приводятся нижеследующие примеры. Примеры Для ссылки на любое указанное соединение формулы (I) настоящего изобретения, необязательно в форме фармацевтически приемлемой соли, смотрите экспериментальную часть и формулу настоящего изобретения. Что касается примеров, которые приводятся ниже, соединения настоящего изобретения синтезировали с применением способов, описанных в настоящем описании, или других способов, которые хорошо известны в данной области. Общие способы Флэш-хроматографию проводили на силикагеле (Merck grade 9395, 60 А). Когда указываются хроматографические разделения, такие разделения проводили на системе Biotage Horizon. Реакции с помощью микроволнового излучения проводили с применением Biotage/Personal Chemistry Smith Creator. ВЭЖХ проводили на колонке Waters X Terra RP 18 (4,650 мм, 3,5 мкм) с применением системы ВЭЖХ Waters 2790, снабженной детектором 996 Waters PDA и одним квадрупольным масс-спектрометром Micromass mod. ZQ, снабженным источником ионизации электрораспылением (ESI). Подвижной фазой А был буфер 5 мМ ацетат аммония (рН 5,5 со смесью уксусная кислота/ацетонитрил, 95:5) и подвижной фазой В была смесь Н 2 О/ацетонитрил (5:95). Градиент от 10 до 90% В достигали за 8 мин, 90% В поддерживали в течение 2 мин. УФ-детектирование проводили при 220 и 254 нм. Скорость потока была 1 мл/мин. Объем инъекции 10 мкл. Проводили полное сканирование, диапазон масс от 100 до 800 атомных единиц массы. Напряжение на капилляре было 2,5 кВ; температура источника была 120 С; напряжение конуса было 10 В. Времена удерживания (в.у. ВЭЖХ) указывались в минутах при 220 нм или при 254 нм. Масса указывалась как отношение m/z. Когда было необходимо, соединения очищали препаративной ВЭЖХ на колонке Waters Symmetry C18 (1950 мм, 5 мкм) с применением препаративного хроматографаWaters HPLC 600, снабженного детектором 996 Waters PDA и одним квадрупольным масс-спектрометром Micromass mod. ZMD с источником ионизации электрораспылением в режиме определения положительных ионов. Подвижной фазой была вода с 0,01% TFA и подвижной фазой В был ацетонитрил. Градиент от 10 до 90% В достигали за 8 мин, 90% В поддерживали в течение 2 мин. Скорость потока была 20 мл/мин; на колонке Waters X Terra Prep RP18 (19100 мм, 5 мкм) с применением системы Waters FractionLynx System (FL2), снабженной детектором Waters 2996 PDA в области от УФ до видимой области и одним квадрупольным масс-спектрометром Waters ZQ. Подвижной фазой А была смесь 0,05% NH4OH вH2O, рН 10/ацетонитрил, 95/5, и подвижной фазой В был ацетонитрил. Градиент от 0 до 80% В достигали за 8 мин, 100% В поддерживали в течение 2 мин. Скорость потока была 20 мл/мин. Масс-спектральные (МС) данные низкого разрешения получали на приборе с ионной ловушкойFinnigan MAT LCQ, снабженным источником образования ионов электрораспылением (ESI). Массспектры высокого разрешения (HRMS) получали на приборе Waters Q-TOF Utima, снабженным источником ионизации электрораспылением (ESI), и с применением резерпина (мол. масса 609,28065) для коррекции фиксированной массы. Если не указано иначе, 1 Н-ЯМР-спектрометрию проводили на прибореMercury VX 400, действующем при 400,45 МГц и снабженным 5-мм двойным резонансным датчиком [1H(15N-31P) IDPFG Varian]. Химические сдвиги указаны как(м.д.). В этих примерах и в других разделах аббревиатуры имеют следующие значения. АсОН = уксусная кислотаHCOONH4 = формиат аммония НОВТ = гидроксибензотриазол НОВТNH3 = аммониевая соль гидроксибензотриазола ВЭЖХ = высокоэффективная жидкостная хроматографияKOH = гидроксид калия реагент Лавессона = 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидTFA = трифторуксусная кислота ТГФ = тетрагидрофуран ксантфос = 9,9-диметил-9 Н-ксантен-4,5-диил)-бис-[дифенилфосфин] Стадия 1. Образование кольца пиррола (3). Гидробромид 2-бром-1-пиридин-4-илэтанона 1 (1,7 г, 6,2 ммоль) добавляют к смеси этилового эфира 3-оксо-3-фенилпропионовой кислоты 2 (R представляет собой Н, 1 г, 5,2 ммоль) в 100 мл сухого ТГФ и NaH (0,5 г, 13,0 ммоль) при 0 С. Раствор выдерживают при 0 С в течение 1 ч и затем перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют и остаток растворяют в 60 мл EtOH,добавляют ацетат аммония (1,4 г, 18,7 ммоль) и реакционную смесь выдерживают в течение ночи при комнатной температуре. Сырой продукт очищают флэш-хроматографией (DCM/MeOH, 98:2), получая 920 мг (60%) этилового эфира 2-фенил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты в виде твердого вещества. 1(МН+). Стадия 2. Омыление в карбоновые кислоты (4). Сложный эфир 3 (440 мг, 1,5 ммоль) в 3 мл EtOH и 3 мл 4 М водном NaOH нагревают при 100 С в течение 3 ч. Реакционную смесь охлаждают до 0 С и подкисляют конц. HCl, наблюдая осаждение продукта, который отделяют фильтрованием, промывают небольшим количеством воды и ацетона и сушат,получая 400 мг (88%) 2-фенил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты в виде твердого вещества, которое применяют в следующей стадии без дополнительной очистки. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 7,51 (м, 3 Н), 7,68 (м, 2 Н), 7,75 (д, J=2,44 Гц, 1 Н), 8,28 (д,J=6,65 Гц, 2 Н), 8,74 (д, J=6,65 Гц, 2 Н), 12,51 (с, 1 Н); МС: m/z 263 [М-Н]. Стадия 3. Конденсация с получением амидов (А 1). Кислоту 4 (380 мг, 1,44 ммоль) растворяют в 10 мл сухого ТГФ в присутствии DIEA (0,5 мл,2,90 ммоль). К раствору, охлажденному до 0 С, добавляют EDCI (414 мг, 2,16 ммоль) и НОВТNH3(330 мг, 2,16 ммоль). Реакционную смесь выдерживают в течение ночи при комнатной температуре. Растворитель удаляют, добавляют воду и суспензию экстрагируют DCM. Органический слой сушат(Na2SO4), растворитель выпаривают и сырой продукт очищают флэш-хроматографией (DCM/MeOH,95:5), получая 150 мг (40%) амида 2-фенил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты в виде светло-желтого твердого вещества. 1H-ЯМР (ДМСО-d6/400 МГц)м.д. 6,90 (ушир.с, 2 Н), 7,27 (д, J=2,56 Гц, 1 Н), 7,37 (м, 1 Н), 7,44 (м,2 Н), 7,67-7,71 (м, 4 Н), 8,53 (м, 2 Н), 11,82 (с, 1 Н); ESI (+) МС: m/z 264 (МН+). Указанную выше методику применяют для синтеза нижеследующих соединений. Пример 2, стадия 1. Этиловый эфир 2-(2-фторфенил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,11 (т, J=7,07 Гц, 3 Н), 4,09 (т, J=7,07 Гц, 2 Н), 7,28-7,34 (м, 5 Н),7,75 (дд, J=1,46, 4,63 Гц, 2 Н), 8,52 (м, 2 Н), 12,31 (с, 1 Н); ESI (+) МС: m/z 311 (МН+). Пример 2, стадия 2. 2-(2-Фторфенил)-5-пиридин-4-ил-1H-пиррол-3-карбоновая кислота. 1(ушир.с, 2 Н), 6,86 (ушир.с, 2 Н), 7,20 (д, J=7,93 Гц, 1 Н), 7,24 (д, J=2,68 Гц, 1 Н), 7,44 (м, 1 Н), 7,47 (м, 1 Н),7,67 (д, J=6,22 Гц, 2 Н), 8,51 (д, J=6,10 Гц, 2 Н), 11,72 (ушир.с, 1 Н); ESI (+) MC: m/z 419 (МН+). Обработкой кислотами (например, трифторуксусной кислотой при комнатной температуре в течение 24 ч) получают соответствующий аналог без защитной группы. Пример 8. Амид 5-пиридин-4-ил-2-(1,2,3,4-тетрагидроизохинолин-7-ил)-1 Н-пиррол-3-карбоновой кислоты (А 28). 1(+) MC: m/z 319 (МН+). Пример 9. Амид 2-(2-метил-1,2,3,4-тетрагидроизохинолин-7-ил)-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты (А 29). Восстановительным аминированием, проводимым формальдегидом и цианоборгидридом натрия в кольце тетрагидроизохинолина соединения А 28, получают указанное в заголовке соединение А 29. 1 Н-ЯМР (ДМСО-d6, 400 МГц)м.д. 2,94 (с, 3 Н), 3,09 (д, J=16,90 Гц, 1 Н), 3,69 (д, J=7,44 Гц, 1 Н),4,31 (дд, J=14,90, 6,60 Гц, 1 Н), 4,50 (д, J=14,88 Гц, 1 Н), 7,05 (ушир.с, 1 Н), 7,33 (д, J=8,05 Гц, 1 Н), 7,44 Стадия 1. Образование кольца пиррола (6). К раствору этилового эфира 3-циклогексил-3-оксопропионовой кислоты 5 (1,6 г, 8,3 ммоль) в безводном ТГФ (200 мл), охлажденному до 0 С, добавляют NaH (900 мг, 21 ммоль). Через 15 мин добавляют гидробромид 2-бром-1-пиридин-4-илэтанона 1 (3 г, 10,8 ммоль) и смесь перемешивают 5 ч при 0 С. Растворитель удаляют и остаток растворяют в EtOH (120 мл). Добавляют ацетат аммония (1,9 г,25 ммоль) и раствор перемешивают в течение ночи при комнатной температуре. После удаления растворителя остаток растворяют в EtOAc и органическую фазу промывают насыщенным водным растворомNa2CO3, затем водой, сушат (Na2SO4) и концентрируют. Сырой продукт очищают хроматографией на силикагеле (элюент: EtOAc), получая этиловый эфир 2-циклогексил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества (1,1 г, 43%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,33 (м, 1 Н), 1,30 (т, J=7,13 Гц, 3 Н), 1,70-1,88 (м, 7 Н), 3,44-3,56(м, 1 Н), 4,20 (кв., J=7,15 Гц, 2 Н), 7,06 (д, J=2,68 Гц, 1 Н), 7,71 (д, J=6,22 Гц, 2 Н), 8,50 (д, J=6,22 Гц, 2 Н),11,37 (ушир.с, 1 Н); ESI (+) МС: m/z 299 (МН+). Стадия 2. Омыление в карболовую кислоту (7). Раствор сложного эфира 6 (0,58 г, 1,95 ммоль) в 4 М водном NaOH и EtOH (1:1, 20 мл) кипятят с обратным холодильником в течение 3 ч, охлаждают во льду и подкисляют 2 н. HCl. Осадок отделяют фильтрованием, промывают небольшим количеством воды и сушат. Гидрохлорид 2-циклогексил-5 пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты получают в виде белого твердого вещества (0,55 г,90%). 1H-ЯМР (ДМСО-d6/400 МГц)м.д. 1,22-1,42 (м, 3 Н), 1,69-1,89 (м, 7 Н), 3,50-3,64 (м, 1 Н), 7,56 (д,J=2,56 Гц, 1 Н), 8,26 (д, J=6,71 Гц, 2 Н), 8,69 (д, J=6,71 Гц, 2 Н), 11,85 (с, 1 Н), 12,17 (ушир.с, 1 Н); МС: m/z 269 [М-Н]. Стадия 3. Конденсация для получения амида (Q1). Раствор кислоты 7 (0,3 г, 1 ммоль), HOBTNH3 (0,3 г, 2 ммоль), TBTU (0,64 г, 2 ммоль), DIEA (1 мл,6 ммоль) в ДМФА (4 мл) перемешивают при комнатной температуре в течение 6 ч. Реакционную смесь выливают в воду и водную фазу экстрагируют (3) EtOAc. Органическую фазу промывают 1 н. NaOH,затем водой, насыщенным раствором соли, сушат (Na2SO4) и концентрируют. Сырой продукт очищают хроматографией на силикагеле (DCM/MeOH, 12:1), получая амид 2-циклогексил-5-пиридин-4-ил-1 Нпиррол-3-карбоновой кислоты (0,12 г, 43%) в виде белого твердого вещества. 1 Стадия 1. Образование кольца пиррола (9). К раствору трет-бутилового эфира 4-(2-этоксикарбонилацетил)пиперидин-1-карбоновой кислоты 8(2,5 г, 8,3 ммоль) в безводном ТГФ (200 мл), охлажденному до 0 С, добавляют NaH (900 мг, 21 ммоль). Через 15 мин добавляют гидробромид 2-бром-1-пиридин-4-илэтанона 1 (3 г, 10,8 ммоль) и смесь перемешивают 5 ч при 0 С. Растворитель удаляют при пониженном давлении и остаток растворяют в EtOH(120 мл). Добавляют ацетат аммония (1,9 г, 25 ммоль) и раствор перемешивают в течение ночи при комнатной температуре. После удаления растворителя остаток растворяют в EtOAc и органическую фазу промывают насыщенным водным раствором Na2CO3, затем водой, сушат (Na2SO4) и концентрируют. Сырой продукт очищают хроматографией на силикагеле (элюент: EtOAc), получая трет-бутиловый эфир 4-(3-этоксикарбонил-5-пиридин-4-ил-1 Н-пиррол-2-ил)пиперидин-1-карбоновой кислоты в виде розового твердого вещества (1,55 г, 47%). 1(МН+). Стадия 2. Омыление в карбоновую кислоту (10). Раствор сложного эфира 9 (0,8 г, 2 ммоль) в 4 М водном NaOH и EtOH (1:1, 20 мл) кипятят с обратным холодильником в течение 2 ч, охлаждают во льду и подкисляют 2 н. HCl. Осадок отделяют фильтрованием, промывают небольшим количеством воды и сушат. Гидрохлорид трет-бутилового эфира 4-(3 карбокси-5-пиридин-4-ил-1 Н-пиррол-2-ил)пиперидин-1-карбоновой кислоты получают в виде белого твердого вещества (0,54 г, 66%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,43 (с, 9 Н), 1,67-1,92 (м, 4 Н), 2,79 (ушир.с, 2 Н), 3,68-3,79 (м,1 Н), 4,13 (ушир.д, J=11,58 Гц, 2 Н), 7,57 (д, J=2,56 Гц, 1 Н), 8,22 (д, J=6,83 Гц, 2 Н), 8,69 (д, J=6,83 Гц, 2 Н),11,82 (ушир.с, 1 Н); МС: m/z 370 [М-Н]. Стадия 3. Конденсация для получения защищенного амида (Q3). Раствор кислоты 10 (0,53 г, 1,4 ммоль), HOBTNH3 (0,43 г, 2,8 ммоль), TBTU (0,9 г, 2,8 ммоль),DIEA (1,4 мл) в ДМФА (4 мл) перемешивают при комнатной температуре в течение 15 ч. Реакционную смесь выливают в воду и водную фазу экстрагируют (3) EtOAc. Органическую фазу промывают 1 н.NaOH, затем водой, насыщенным раствором соли, сушат (Na2SO4) и концентрируют. При выпаривании растворителя осаждается сырой продукт. Его фильтруют, промывают EtOAc, затем Et2O. Трет-бутиловый эфир 4-(3-карбамоил-5-пиридин-4-ил-1 Н-пиррол-2-ил)пиперидин-1-карбоновой кислоты получают в виде белого твердого вещества (25 г, 47%). 1H-ЯМР (ДМСО-d6/400 МГц)м.д. 1,45 (с, 9 Н), 1,67 (ушир.д, J=12,32 Гц, 2 Н), 1,76-1,89 (м, 2 Н),2,71 (ушир.с, 2 Н), 3,81-3,91 (м, 1 Н), 4,12 (ушир.д, J=11,10 Гц, 2 Н), 6,76 (ушир.с, 1 Н), 7,18 (д, J=2,56 Гц,1 Н), 7,29 (ушир.с, 1 Н), 7,59 (д, J=6,22 Гц, 2 Н), 8,50 (д, J=6,10 Гц, 2 Н), 11,22 (ушир.с, 1 Н); ESI (+) МС: m/z 371 (МН+). Стадия 4. Удаление защитной группы для получения амида (Q2). Амид Q3 (30 мг, 0,08 ммоль) растворяют в МеОН (5 мл), добавляют 2 н. HCl (1 мл) и прозрачный раствор нагревают при 50 С при перемешивании в течение 5 ч. Осадок отделяют фильтрованием и промывают МеОН. Дигидрохлорид амида 2-пиперидин-4-ил-5-пиридин-4-ил-1 Н-пиррол-3-карбоновой кислоты получают в виде белого твердого вещества (25 мг, 90%). 1 Стадия 1. Образование кольца пиррола (12). Гидробромид бромацетилфторпиридина 11 (3,6 г, 12,0 ммоль) добавляют к смеси этилового эфира 3-оксо-3-фенилпропионовой кислоты 2 (R представляет собой Н, 2,0 г, 10,0 ммоль) в 200 мл безводного ТГФ и NaH (0,5 г, 13,0 ммоль) при 0 С. Раствор выдерживают при 0 С в течение 1 ч и затем перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют и остаток растворяют в 120 млEtOH, добавляют ацетат аммония (2,7 г, 36,0 ммоль), реакционную смесь выдерживают в течение ночи при комнатной температуре, затем нагревают при 50 С в течение 2 ч. Сырой продукт очищают флэшхроматографией (DCM/MeOH, 98:2), получая 1,88 г (60%) этилового эфира 5-(3-фторпиридин-4-ил)-2 фенил-1 Н-пиррол-3-карбоновой кислоты в виде желтого твердого вещества. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,25 (т, J=7,09 Гц, 3 Н), 4,23 (кв., J=7,09 Гц, 2 Н), 7,30-7,60 (м,7 Н), 8,40 (м, 1 Н), 8,53 (м, 1 Н), 11,80 (с, 1 Н); ESI (+) МС: m/z 311 (МН+). Стадия 2. Омыление в карбоновую кислоту (13). Сложный эфир 12 (1,8 г, 5,8 ммоль) в 10 мл EtOH и 12 мл 4 М водного NaOH нагревают при 100 С в течение 4 ч. Реакционную смесь охлаждают до 0 С и подкисляют конц. HCl, наблюдая осаждение продукта, который отделяют фильтрованием, промывают небольшим количеством воды и ацетона и сушат,получая 5-(3-фторпиридин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновую кислоту (1,7 г, 92%) в виде твердого вещества, которое применяют без дополнительной очистки. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 7,30-7,70 (м, 7 Н), 8,35 (м, 1 Н), 8,51 (м, 1 Н); МС: m/z 281 [М-Н]. Стадия 3. Конденсация для получения амидов (Е 1). Кислоту 13 (1,0 г, 3,1 ммоль) растворяют в 40 мл сухого ТГФ в присутствии DIEA (1,1 мл,6,2 ммоль). Раствор охлаждают до 0 С и добавляют EDCI (0,9 г, 4,6 ммоль) и HOBTNH3 (0,7 г,4,6 ммоль). Реакционную смесь выдерживают в течение ночи при комнатной температуре. Растворитель удаляют, добавляют воду и смесь экстрагируют DCM. Органический слой сушат (Na2SO4), растворитель выпаривают и сырой продукт очищают флэш-хроматографией (DCM/MeOH, 95:5), получая 350 мг (40%) амида 5-(3-фторпиридин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 6,86 (ушир.с, 2 Н), 7,24 (т, J=3,05 Гц, 1 Н), 7,36-7,45 (м, 3 Н), 7,65 Стадия 1. Образование кольца пиррола (15). К раствору сложного эфира 2 (R представляет собой Н, 1,34 г, 7 ммоль) в безводном ТГФ (100 мл) при 0 С добавляют NaH (0,7 г, 17,5 ммоль) в атмосфере аргона при перемешивании. Через 5 мин добавляют бромкетон 14 (2,5 г, 8,4 ммоль) и смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель выпаривают, остаток растворяют в EtOH (65 мл), добавляют ацетат аммония (1,6 г,21 ммоль) и раствор перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают досуха и остаток очищают флэш-хроматографией (EtOAc/н-гексан, 7:3). Получают этиловый эфир 5-(2-аминопиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновой кислоты (0,99 г, 3,2 ммоль, 46%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,20 (т, J=7,13 Гц, 3 Н), 4,14 (кв., J=7,07 Гц, 2H), 6,45 (с, 2 Н), 7,10(ушир. с, 1 Н); ESI (+) МС: m/z 309 (МН+). Стадия 2. Омыление в карбоновые кислоты (16). К суспензии сложного эфира 15 (3,65 г, 11,85 ммоль) в 95% EtOH (45 мл) добавляют 4 М водныйNaOH (45 мл) и смесь кипятят с обратным холодильником в течение 5 ч. Большую часть растворителя выпаривают и остаток, охлажденный на ледяной бане, подкисляют до рН 5 конц. HCl, наблюдая осаждение продукта. Осадок отделяют фильтрованием, промывают небольшим количеством холодной воды и сушат. 5-(2-Аминопиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновую кислоту, полученную в виде белого твердого вещества (3,5 г), применяют на следующей стадии без дополнительной очистки. 1H-ЯМР (ДМСО-d6/400 МГц)м.д. 7,35-7,69 (м, 6 Н), 7,76 (ушир.с, 2 Н), 8,31 (д, J=5,73 Гц, 1 Н),12,37 (ушир.с, 1 Н); МС: m/z 279 [М-Н]. Стадия 3. Конденсация для получения амидов (F1). К суспензии кислоты 16 (4 г, 14,3 ммоль) в безводном ТГФ (80 мл), DIEA (5,5 г, 42,9 ммоль) и безводного ДМФА (8 мл), охлажденной на ледяной бане, при перемешивании добавляют HOBTNH3 (3,26 г,21,4 ммоль) и EDCI (4,1 г, 21,4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем выливают в перемешиваемую смесь 1:1 воды и EtOAc. Органическую фазу промывают водой, водный слой экстрагируют EtOAc и объединенные органические слои промывают водой,сушат (Na2SO4) и концентрируют, получая указанное в заголовке соединение в виде осадка, который отделяют фильтрованием и промывают небольшим количеством холодного МеОН. Исходный раствор очищают флэш-хроматографией (DCM/MeOH/ацетон, 9:1:1), получая требуемый амид. Две порции продукта объединяют, суспендируют в МеОН и подкисляют до рН 1 1,2 5 М HCl в МеОН. Растворитель удаляют и остаток обрабатывают диэтиловым эфиром. Образовавшееся твердое вещество отделяют фильтрованием, промывают Et2O и сушат (Na2SO4), получая гидрохлорид амида 5-(2-аминопиримидин-4-ил)-2 фенил-1 Н-пиррол-3-карбоновой кислоты в виде белого твердого вещества (1,6 г, 5,1 ммоль, 43%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 7,37-7,49 (м, 4 Н), 7,49-7,52 (м, 2 Н), 7,61 (д, J=2,44 Гц, 1 Н), 7,657,71 (м, 2 Н), 8,01 (ушир.с, 3 Н), 8,31 (д, J=6,58 Гц, 1 Н), 12,28 (с, 1 Н); ESI (+) МС: m/z 280 (МН+). Указанную выше методику применяют для синтеза нижеследующих соединений. Пример 15, стадия 1. Этиловый эфир 5-(2-аминопиримидин-4-ил)-2-о-толил-1 Н-пиррол-3-карбоновой кислоты. 1 Н-ЯМР (400 МГц, ДМСО-d6)м.д. 1,07 (т, J=7,07 Гц, 3 Н), 2,14 (с, 3 Н), 4,02 (кв., J=7,07 Гц, 2 Н),6,54 (ушир.с, 2 Н), 7,04 (д, J=5,37 Гц, 1 Н), 7,22-7,37 (м, 5 Н), 8,20 (д, J=5,37 Гц, 1 Н), 12,12 (ушир.с, 1 Н); ESI(ушир.с, 2 Н), 6,89 (ушир.с, 1 Н), 7,11-7,39 (м, 5 Н), 7,64 (д, J=2,19 Гц, 1 Н), 7,75 (ушир.с, 2 Н), 8,27 (д,J=6,34 Гц, 1 Н), 12,28 (ушир.с, 1 Н); ESI (+) МС: m/z 435 (МН+). Пример 27. Амид 5-(2-аминопиримидин-4-ил)-2-(1,2,3,4-тетрагидроизохинолин-5-ил)-1 Н-пиррол-3 карбоновой кислоты (G8). Обработкой соединения G7, полученного в примере 26, кислотами, например трифторуксусной кислотой, при комнатной температуре в течение 24 ч получают соответствующий аналог G8 с удаленной защитной группой. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 2,78 (т, J=5,91 Гц, 2 Н), 4,35 (т, J=4,51 Гц, 2 Н), 6,90 (ушир.с, 1 Н),7,25 (д, J=6,58 Гц, 1 Н), 7,27-7,30 (м, 1 Н), 7,31-7,39 (м, 3 Н), 7,70 (д, J=2,44 Гц, 1 Н), 7,86 (ушир.с, 3 Н), 8,30 Стадия 1. Алкилирование кольца пиримидина (18). К раствору 2,4-дихлор-5-фторпиримидина 17 (1,2 г, 7,24 ммоль) в ДМФА (14 мл) добавляют трибутил-(1-этоксивинил)станнан (2,7 мл, 7,9 ммоль) с последующим добавлением дихлор-бис-(трифенилфосфин)палладия(II) (100 мг, 0,145 ммоль). Смесь нагревают при 70 С в течение 1 ч, охлаждают, добавляют насыщенный раствор фторида калия (водный) и смесь перемешивают при комнатной температуре в течение 18 ч. После разбавления смесью вода/диэтиловый эфир и фильтрования через целит органические фазы тщательно промывают водой и концентрируют. Сырой продукт очищают системой HorizonH-ЯМР (400 МГц, ДМСО-d6)м.д. 1,32 (т, J=6,95 Гц, 3 Н), 3,95 (кв., J=6,99 Гц, 2 Н), 4,88 (д,J=2,80 Гц, 1H), 5,20 (д, J=2,93 Гц, 1 Н), 8,90 (д, J=3,17 Гц, 1 Н); ESI (+) MC: m/z 203 (МН+). Стадия 2. Аминирование кольца пиримидина (19). Раствор простого эфира енола 18 (15,5 г, 76,73 ммоль) в абсолютном этаноле (25 мл) и 30% водном аммиаке (50 мл) при встряхивании нагревают при 100 С в течение 1,5 ч в аппарате Парра. После охлаждения этанол удаляют и соединение экстрагируют дихлорметаном. Сырой продукт очищают системой(9 г, 49,2 ммоль, 64%). 1 Н-ЯМР (400 МГц, ДМСО-d6)м.д. 1,29 (т, J=7,01 Гц, 3 Н), 3,87 (кв., J=6,95 Гц, 2 Н), 4,62 (д,J=2,44 Гц, 1 Н), 4,91 (дд, J=2,38, 0,55 Гц, 1 Н), 6,64 (ушир.с, 2 Н), 8,28 (д, J=3,54 Гц, 1 Н); ESI (+) MC: m/z 184 (МН+). Стадия 3. Бромирование для получения бромкетона (20). К раствору простого эфира енола 19 (510 мг, 2,78 ммоль) в ТГФ (25 мл) добавляют воду (1,7 мл) с последующим добавлением NBS (515 мг, 2,78 ммоль). Смесь перемешивают при комнатной температуре в течение 1,5 ч. Растворитель выпаривают, остаток тщательно перемешивают в метаноле и фильтруют. Получают 1-(2-амино-5-фторпиримидин-4-ил)-2-бромэтанон (500 мг, 77%). 1MC: m/z 235 (МН+). Стадия 4. Образование кольца пиррола (21). К раствору сложного кетоэфира 2 (192 мг, 1 ммоль) в ТГФ (5 мл), охлажденному до 0 С, при перемешивании добавляют гидрид натрия (80 мг, 2 ммоль). Через 5 мин добавляют раствор бромкетона 20(234 мг, 1 ммоль) в ДМФА (2 мл) и реакционную смесь перемешивают при 50 С в течение 8 ч. После удаления ТГФ добавляют этанол (10 мл) и ацетат аммония (240 мг, 3 ммоль) и смесь перемешивают при комнатной температуре в течение 20 ч. После удаления растворителя добавляют этилацетат, органическую фазу промывают водой и сырой продукт очищают при помощи системы Horizon, элюируя смесью н-гексан/EtOAc, 1:1. Получают этиловый эфир 5-(2-амино-5-фторпиримидин-4-ил)-2-фенил-1 Н-пиррол 3-карбоновой кислоты (50 мг, 16%).- 27015126 Стадия 5. Гидролиз в кислоты (22). К суспензии сложного эфира 21 (25 мг, 0,077 ммоль) в 95% EtOH (0,5 мл) добавляют 4 М водныйNaOH (0,5 мл) и смесь кипятят с обратным холодильником в течение 2 ч. Смесь подкисляют до рН 5 конц. HCl, наблюдая осаждение продукта. Осадок отделяют фильтрованием, промывают небольшим количеством холодной воды и сушат. 5-(2-Амино-5-фторпиримидин-4-ил)-2-фенил-1 Н-пиррол-3-карбоновую кислоту, полученную в виде белого твердого вещества (16 мг, 64%), применяют в следующей стадии без дополнительной очистки.ESI (+) MC: m/z 299 (МН+). Стадия 6. Конденсация для получения амидов (V1). К раствору кислоты 22 (16 мг, 0,054 ммоль) в ДМФА (0,5 мл) и DIEA (0,03 мл), перемешиваемому при 0 С, добавляют НОВТNH3 (13 мг, 0,08 ммоль) и EDCI (16 мг, 0,08 ммоль). Смесь перемешивают при комнатной температуре в течение 20 ч. После разбавления этилацетатом органическую фазу промывают водой, насыщ. водным раствором бикарбоната натрия, сушат над Na2SO4 и концентрируют. Сырой продукт очищают флэш-хроматографией (элюент: AcOEt/н-гексан, 9:1). Указанное в заголовке соединение получают с выходом 74%. 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 6,34 (с, 2 Н), 6,87 (ушир.с, 1 Н), 7,27 (т, J=2,80 Гц, 1 Н), 7,33-7,43 Стадия 1. Бромирование для получения бромкетона (24). К раствору силилового эфира енола 23 (1 г, 2,27 ммоль) в ТГФ (40 мл) и воде (5 мл) при комнатной температуре добавляют твердый NBS (0,43 г, 62,4 ммоль) и смесь перемешивают в течение 20 ч. После выпаривания растворителя и обработки водной части этилацетатом сырой продукт очищают флэшхроматографией (элюент: н-гексан/EtOAc, 4:1), получая 2-бром-1-(2-фениламинопиримидин-4-ил)этанон в виде желтого твердого вещества (0,27 г, 40%). 1 Н-ЯМР (ДМСО-d6/300 МГц)м.д. 4,65 (с, 2 Н), 6,7 (м, 1 Н), 6,9 (д, 1H), 7,0 (м, 2 Н), 7,4 (д, 2 Н), 8,4(д, 1 Н), 9,6 (с, 1 Н); ESI (+) МС: m/z 293 (МН+). Стадия 2. Образование кольца пиррола (25). К раствору сложного эфира 2 (150 мкл, 0,87 ммоль) в безводном ТГФ (40 мл) при 0 С добавляютNaH (50 мг, 1,2 ммоль) в атмосфере аргона с перемешиванием. Через 40 мин добавляют бромкетон 24(260 мг, 0,89 ммоль, получен, как описано в WO 2005014572) и смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель выпаривают досуха, остаток растворяют в EtOH (10 мл), добавляют ацетат аммония (343 г, 4,45 ммоль) и раствор перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают досуха и к сырому продукту добавляют EtOAc и воду и органический слой отделяют, промывают водой, сушат (Na2SO4) и концентрируют. Остаток растворяют в смесиEt2O/EtOAc/H-гексан (1:1:1) и фильтруют. Получают этиловый эфир 2-фенил-5-(2-фениламинопиримидин-4-ил)-1 Н-пиррол-3-карбоновой кислоты (120 мг, 36%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,20 (т, 3 Н), 4,14 (кв., 2 Н), 6,90-7,85 (м, 11 Н), 7,35 (д, J=5,27 Гц,1 Н), 8,46 (д, J=5,27 Гц, 1 Н), 9,45 (с, 1 Н), 12,10 (с, 1 Н); ESI (+) MC: m/z 385 (МН+). Стадия 3. Омыление в карбоновые кислоты (26). К суспензии сложного эфира 25 (120 мг, 0,31 ммоль) в 95% EtOH (3 мл) добавляют 4 М водныйNaOH (4 мл) и смесь кипятят с обратным холодильником в течение 4 ч. Большую часть растворителя выпаривают и остаток, охлажденный на ледяной бане, подкисляют до рН 5 конц. АсОН, наблюдая осаждение продукта. Осадок отделяют фильтрованием, промывают небольшим количеством холодной воды и сушат. 2-Фенил-5-(2-фениламинопиримидин-4-ил)-1 Н-пиррол-3-карбоновую кислоту, полученную в виде белого твердого вещества (100 мг), применяют на следующей стадии без дополнительных очисток.- 28015126 Стадия 3. Конденсация для получения амидов (H1). К суспензии кислоты 26 (90 мг, 0,25 ммоль) в ДМФА (3 мл) добавляют DIEA (120 мкл, 0,67 ммоль),EDCI (100 мг, 0,52 ммоль) и НОВТNH3 (79 мг, 0,52 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем ее выливают в перемешиваемую смесь 1:1 воды и EtOAc. Органическую фазу промывают водой, водный слой экстрагируют EtOAc и объединенные органические слои промывают водой, сушат (Na2SO4) и концентрируют, получая сырой продукт, который очищают флэш-хроматографией (DCM/MeOH, 96:4) с получением амида 2-фенил-5-(2-фениламинопиримидин-4 ил)-1 Н-пиррол-3-карбоновой кислоты (35 мг, 30% после двух стадий). 1 Стадия 1. Образование кольца пиррола (27). К раствору сложного эфира 2 (1,34 г, 7 ммоль) в безводном ТГФ (100 мл) при 0 С в атмосфере аргона и при перемешивании добавляют NaH (0,7 г, 17,5 ммоль). Через 5 мин добавляют бромкетон 14(2,5 г, 8,4 ммоль) и смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель выпаривают, остаток растворяют в АсОН (30 мл) и 2 М EtNH2 в ТГФ (8,7 мл, 17,5 ммоль). Смесь обрабатывают микроволновым излучением при 170 С в течение 5 мин, затем ее разбавляют EtOAc и промывают водным насыщенным раствором NaHCO3. Водный слой экстрагируют EtOAc и объединенные органические слои промывают водой, сушат (Na2SO4) и концентрируют. Остаток очищают флэш-хроматографиейESI (+) МС: m/z 337 (МН+). Стадия 2. Омыление в карбоновые кислоты (28). К суспензии сложного эфира 27 (0,7 г, 2,08 ммоль) в 95% EtOH (8 мл) добавляют 4 М водный NaOH(8 мл) и смесь перемешивают в течение 1 ч при 100 С. Растворитель удаляют в вакууме и водный остаток подкисляют конц. HCl до рН 5, наблюдая осаждение продукта. Смесь фильтруют, твердое вещество промывают небольшим количеством холодной воды и сушат, получая таким образом 0,66 г 5-(2-аминопиримидин-4-ил)-1-этил-2-фенил-1 Н-пиррол-3-карбоновой кислоты, которую применяют на следующей стадии без дополнительной очистки. МС: m/z 307 [М-Н]. Стадия 3. Конденсация для получения амидов (L1). К суспензии кислоты 28 (400 мг, 1,31 ммоль) в 10 мл ТГФ и 600 мкл DIEA (3,52 ммоль), охлажденной на ледяной бане, добавляют 336 мг EDCI (1,75 ммоль) и 267 мг HOBTNH3 (1,75 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Добавляют EtOAc и воду, слои разделяют, водный слой экстрагируют EtOAc и объединенные органические слои промывают 1 М водным NaOH и водой. Слои затем сушат (Na2SO4) и концентрируют. Остаток отделяют фильтрованием и промывают небольшим количеством холодного МеОН. Исходный раствор очищают флэш-хроматографией(DCM/MeOH/ацетон, 90:5:5), получая требуемый амид. Две порции продукта объединяют, суспендируют в МеОН и подкисляют до рН 1 1,25 М HCl в МеОН. Растворитель удаляют и остаток обрабатывают Et2O,образовавшееся твердое вещество отделяют фильтрованием, промывают Et2O и концентрируют, получая 380 мг соли с хлористо-водородной кислотой амида 5-(2-аминопиримидин-4-ил)-1-этил-2-фенил-1 Н-пиррол-3-карбоновой кислоты (1,1 ммоль, выход 83%). 1 Н-ЯМР (ДМСО-d6/400 МГц)м.д. 1,08 (т, J=6,89 Гц, 3 Н), 4,37 (кв., J=6,91 Гц, 2 Н), 6,87 (ушир.д,J=21,95 Гц, 2 Н), 7,18 (д, J=6,58 Гц, 1 Н), 7,38-7,43 (м, 2 Н), 7,50-7,55 (м, 3 Н), 7,81 (с, 1 Н), 8,00 (ушир.с, 3 Н),8,23 (д, J=6,71 Гц, 1 Н); ESI (+) МС: m/z 308 (МН+).
МПК / Метки
МПК: C07D 403/14, C07D 401/14, C07D 403/04, C07D 405/14, A61K 31/495, C07D 405/04, C07D 401/04, A61K 31/435, A61P 35/00, C07D 409/04
Метки: тиофена, качестве, киназ, пиридил, пиррола, производные, ингибиторов, пиримидинилзамещенные, фурана
Код ссылки
<a href="https://eas.patents.su/30-15126-piridil-i-pirimidinilzameshhennye-proizvodnye-pirrola-tiofena-i-furana-v-kachestve-ingibitorov-kinaz.html" rel="bookmark" title="База патентов Евразийского Союза">Пиридил- и пиримидинилзамещенные производные пиррола, тиофена и фурана в качестве ингибиторов киназ</a>
Замещённые производные пиррола и их применение в качестве ингибиторов hmg-cоa

Номер патента: 9646
Опубликовано: 28.02.2008
Авторы: Ариян Рам Чандер, Чугх Анита, Салман Мохаммад, Кумар Ятендра, Саттигери Джитендра, Раманатан Викрам Кришна
МПК: A61K 31/366, C07D 207/34, A61K 31/40...
Метки: качестве, пиррола, hmg-cоa, производные, применение, замещённые, ингибиторов
Формула / Реферат:
1. Соединения, имеющие структурную формулу I их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, полиморфы, таутомеры, или N-оксиды, где или R1 может быть С1-С6алкил, С3-С6 циклоалкил или необязательно замещенный фенил (где до трех заместителей независимо выбраны из галоидов, C1-С6алкила, циано или С1-С3перфторалкила); R2 может быть необязательно замещенный фенил (где до трех заместителей независимо выбраны из циано,...
Производные бензо[3,4] циклобута [1,2-c] пиррола и их применение в качестве ингибиторов обратного захвата серотонина

Номер патента: 3302
Опубликовано: 24.04.2003
Авторы: Пеглион Жан-Луи, Гумен Бертран, Декейн Анн, Миллан Марк, Дессанж Эме, Риве Жан-Мишель
МПК: A61P 25/24, C07D 209/58, A61K 31/403...
Метки: циклобута, производные, ингибиторов, обратного, качестве, серотонина, применение, захвата, пиррола, 1,2-c, бензо[3,4
Формула / Реферат:
1. Соединение формулы (I), имеющее цис-соединение колец в которой R1, R2 и R3, которые могут быть одинаковыми или различными, каждый независимо от других, представляет группу, выбранную из атома водорода, атома галогена, линейной или разветвленной (C1-C6)алкоксигруппы, линейной или разветвленной (C1-C6)тригалогеналкильной группы, или два из R1, R2 и R3 в смежных положениях представляют (C1-C2)алкилендиоксигруппу; R4 представляет группу,...
Производные пиразолохиназолина: способ получения и применение в качестве ингибиторов киназ

Номер патента: 10904
Опубликовано: 30.12.2008
Авторы: Вульпетти Анна, Ролетто Фульвия, Фанчелли Даниеле, Фергюсон Рон, Певарелло Паоло, Д`алессио Роберто, Квартьери Франческа, Вианелло Паола, Получчи Паоло, Браска Мария Габриелла, Панцери Акилле, Тракванди Габриелла
МПК: C07D 487/04, A61P 35/00, A61K 31/519...
Метки: получения, качестве, способ, пиразолохиназолина, киназ, производные, ингибиторов, применение
Формула / Реферат:
1. Производное пиразолохиназолина, представленное формулой (Ia) или (Ib) или его фармацевтически приемлемая соль, где R обозначает водород или группу, выбранную из амино, линейного или разветвленного C1-C6алкила, C3-C10циклоалкила, арила, арилC1-C6алкила, гетероциклила или гетероциклилC1-C6алкила; X обозначает простую связь или двухвалентный радикал, выбранный из -NR'-, -CONR'-, -NH-CO-NH-, -О- или -S-, где R' обозначает водород или группу,...
Производные аминоиндазола, активные в качестве ингибиторов киназ, способ их получения и содержащая их фармацевтическая композиция

Номер патента: 7430
Опубликовано: 27.10.2006
Авторы: Мартина Катия, Д'анелло Маттео, Вульпетти Анна, Амичи Раффаэлла, Салом Барбара
МПК: C07D 231/56, A61P 35/00, A61K 31/416...
Метки: содержащая, аминоиндазола, композиция, киназ, активные, производные, фармацевтическая, качестве, ингибиторов, способ, получения
Формула / Реферат:
1. Способ лечения заболеваний, обусловленных и/или связанных с измененной (нарушенной) активностью протеинкиназ, включающий введение нуждающемуся в этом млекопитающему эффективного количества производного аминоиндазола, представленного формулой (I) где R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С6алкила, С2-С6 алкенила или...
Птеридиноны в качестве ингибиторов киназ

Номер патента: 5287
Опубликовано: 30.12.2004
Авторы: Добрусин Эллен Майра, Тугуд Питер Лоренс, Мак Намара Деннис Джозеф, Рукасл Гордон Уильям, Крамер Джеймс Бернард, Денни Уильям Эликзэндер, Шоуолтер Хауард Дэниэл Холлис
МПК: A61K 31/505, C07D 475/00, A61P 35/00...
Метки: птеридиноны, ингибиторов, киназ, качестве
Формула / Реферат:
1. Соединение формулы и его фармацевтически приемлемые соли, эфиры, амиды и пролекарства, где R2 представляет собой (CH2)nарил и (CH2)nгетероарил, где каждая из вышеупомянутых арильных и гетероарильных групп может быть не замещена или замещена 1-5 группами-заместителями, выбранными из (а) галогена; (б) амино, алкиламино и диалкиламино; (в) C1-C10алкокси, аминоалкокси, алкиламиноалкокси и диалкиламиноалкокси; (г) гидрокси; (д) алканоила,...