3 – аминоциклопентанкарбоксамиды в качестве модуляторов хемокиновых рецепторов
Номер патента: 12649
Опубликовано: 30.12.2009
Авторы: Чжэн Чаншэн, Цао Ганьфэн, Сюэ Чу-Бяо, Гленн Джозеф, Меткаф Брайан В., Фэн Хао, Ксиа Майкл


























Формула / Реферат
1. Соединение формулы I

или его фармацевтически приемлемая соль или пролекарство,
где прерывистая линия указывает необязательную связь;
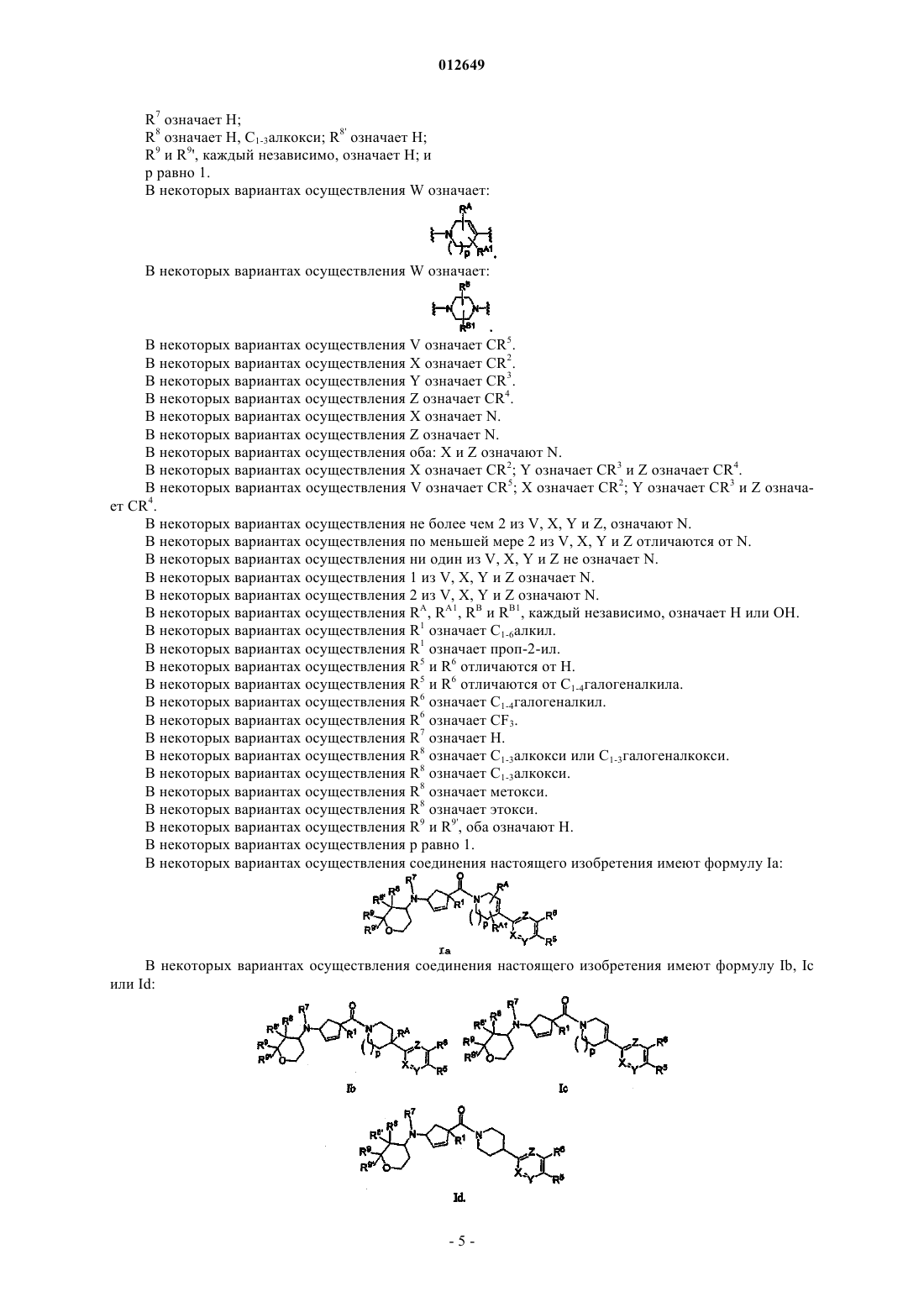
W означает

V означает N или CR5;
X означает N или CR2;
Y означает N или CR3;
Z означает N или CR4;
RA, RA1, RB или RB1, каждый независимо, означает Н, ОН;
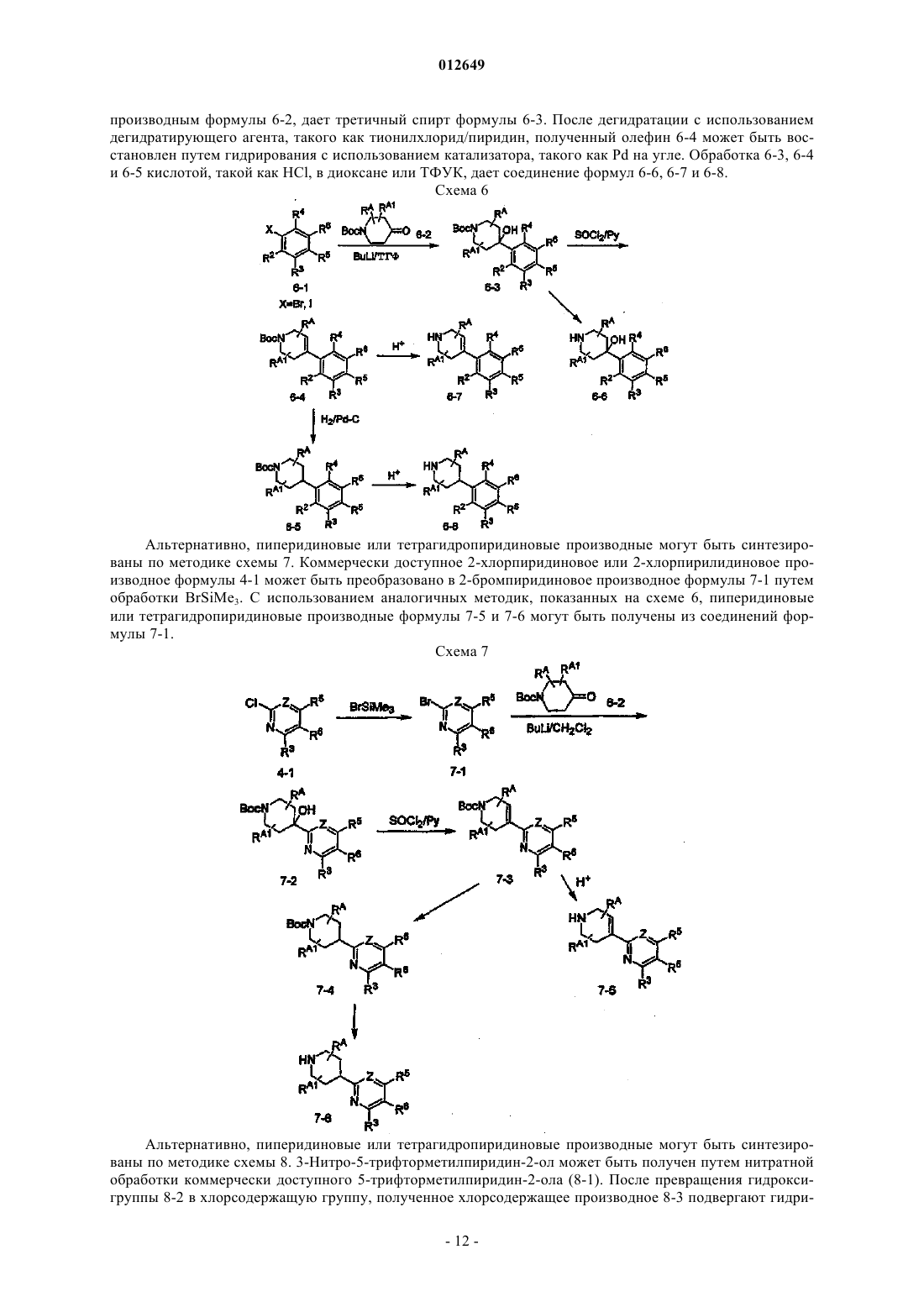
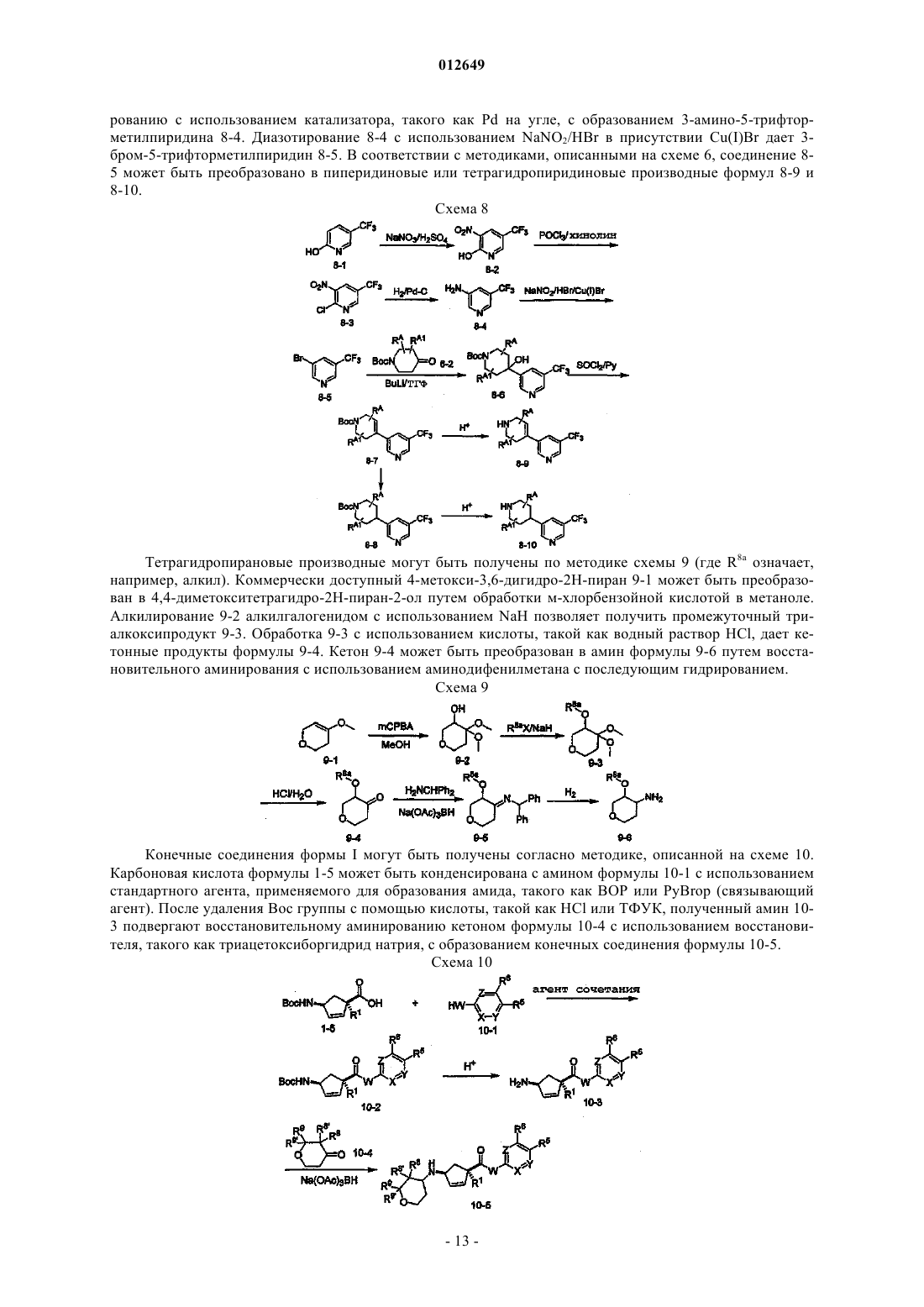
R1 означает Н, ОН, С1-6алкил, С1-6гидроксиалкил, -(С0-6алкил)-О-(C1-6алкил), тетрагидрофуранил;
R2, R3, R4, R5 и R6, каждый независимо, означает Н, С1-6алкил, C1-6галогеналкил;
R7 означает Н;
R8 означает Н, C1-3алкокси;
R8' означает Н;
R9 и R9', каждый независимо, означает Н;
р равно 1.
2. Соединение по п.1, где W означает

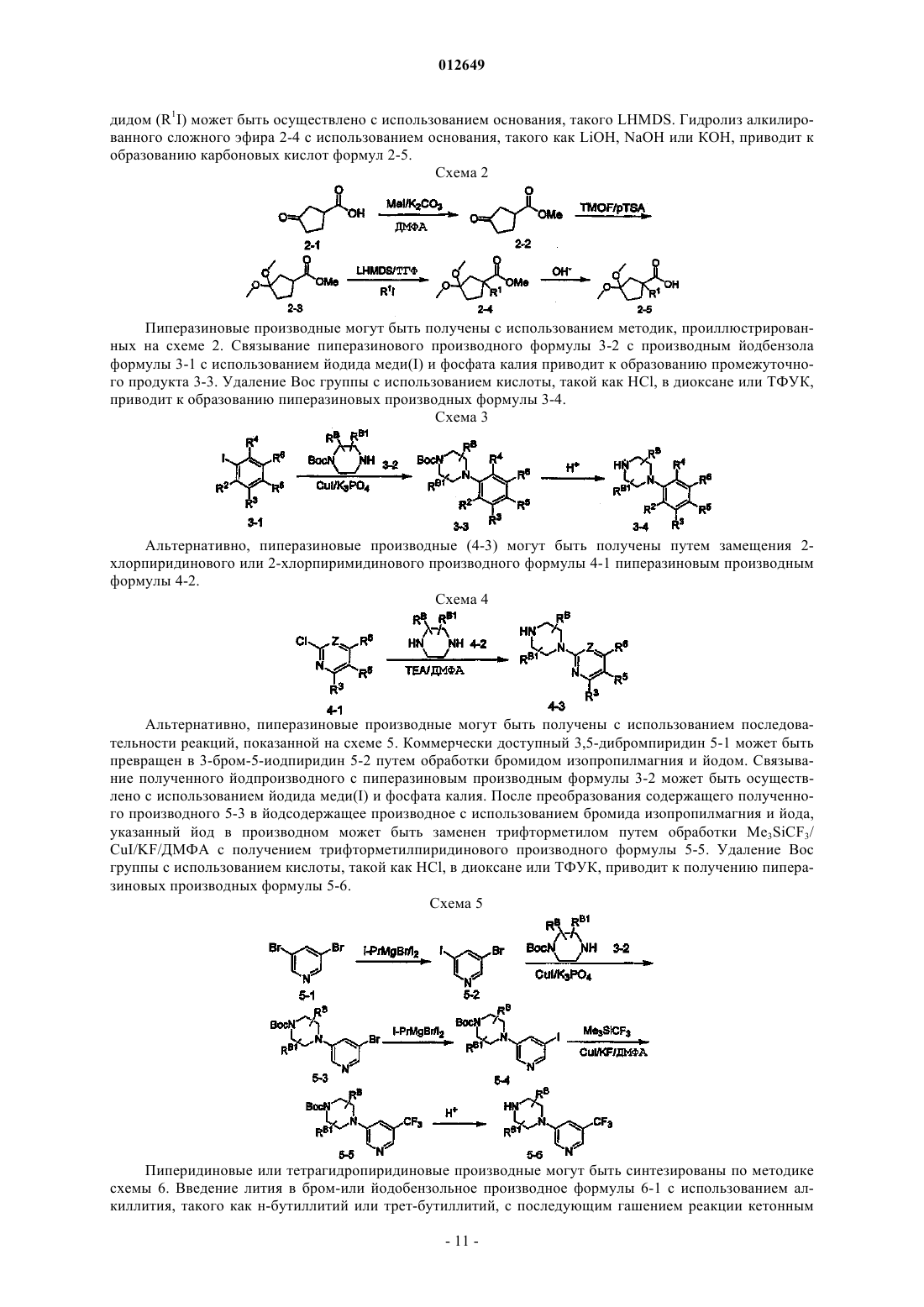
3. Соединение по п.1, где W означает

4. Соединение по п.1, где V означает CR5.
5. Соединение по п.1, где X означает CR2.
6. Соединение по п.1, где Y означает CR3.
7. Соединение по п.1, где Z означает CR4.
8. Соединение по п.1, где X означает CR2, Y означает CR3 и Z означает CR4.
9. Соединение по п.1, где V означает CR5, X означает CR2, Y означает CR3 и Z означает CR4.
10. Соединение по п.1, где R1 означает C1-6алкил.
11. Соединение по п.1, где R1 означает проп-2-ил.
12. Соединение по п.1, где один из R5 и R6 отличен от Н.
13. Соединение по п.1, где один из R5 и R6 означает С1-4галогеналкил.
14. Соединение по п.1, где R6 означает С1-4галогеналкил.
15. Соединение по п.1, где R6 означает CF3.
16. Соединение по п.1, где R8 означает метокси.
17. Соединение по п.1, где R8 означает этокси.
18. Соединение по п.1 формулы Ia

19. Соединение по п.1 формулы Ib, Ic или Id


20. Соединение по п.1 формулы Ie или If

21. Соединение по п.1 формулы Ig

22. Соединение по п.1 формулы In или Ii

23. Соединение по п.1, которое выбирают из группы, включающей
N-[(1R,3S)-3-изопропил-3-({4-[3-(трифторметил)фенил]пиперазин-1-ил}карбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
3-этокси-N-[(1R,3S)-3-изопропил-3-({4-[3-(трифторметил)фенил]пиперазин-1-ил}карбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-({4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-ил}карбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-({4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-ил}карбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2Н)-ил)карбонил]циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
1-({(1S,3R)-1-изопропил-3-[(3-метокситетрагидро-2Н-пиран-4-ил)амино]циклопентил}карбонил)-4-фенилпиперидин-4-ол;
1-({(1S,3R)-1-изопропил-3-[(3-метокситетрагидро-2Н-пиран-4-ил)амино]циклопентил}карбонил)-4-[2-(трифторметил)фенил]пиперидин-4-ол;
1-[((1S,3R)-1-изопропил-3-{[3-метокситетрагидро-2Н-пиран-4-ил]амино}циклопентил)карбонил]-4-[3-(трифторметил)фенил]пиперидин-4-ол;
1-[((1S,3R)-1-изопропил-3-{[3-метокситетрагидро-2Н-пиран-4-ил]амино}циклопентил)карбонил]-4-[4-(трифторметил)фенил]пиперидин-4-ол;
N-((1R,3S)-3-изопропил-3-{[4-[2-(трифторметил)фенил]-3,6-дигидропиридин-1(2Н)-ил]карбонил}циклопентил)-3-метокситетрагидро-2Н-пиран-4-амин;
N-((1R,3S)-3-изопропил-3-{[4-[3-(трифторметил)фенил]-3,6-дигидропиридин-1(2Н)-ил]карбонил}циклопентил)-3-метокситетрагидро-2Н-пиран-4-амин;
3-этокси-N-((1R,3S)-3-изопропил-3-{[4-[3-(трифторметил)фенил]-3,6-дигидропиридин-1(2Н)-ил]карбонил}циклопентил)тетрагидро-2Н-пиран-4-амин;
N-((1R,3S)-3-изопропил-3-{[4-(трифторметил)-3',6'-дигидро-2,4'-бипиридин-1'(2'Н)-ил]карбонил}циклопентил)-3-метокситетрагидро-2Н-пиран-4-амин;
N-((1R,3S)-3-изопропил-3-{[5-(трифторметил)-3',6'-дигидро-3,4'-бипиридин-1'(2'Н)-ил]карбонил}циклопентил)-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-({4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-ил}карбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-({4-[6-(трифторметил)пиридин-2-ил]пиперазин-1-ил}карбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-(4-[6-(трифторметил)пиримидин-4-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1R,3S)-3-изопропил-3-(4-[6-метил-4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-N-[(1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперидин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
2-[(1R,3S)-3-[(3-метокситетрагидро-2Н-пиран-4-ил)амино]-1-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]пропан-2-ол;
2-[(1R,3S)-3-[(4R)-3-метокситетрагидро-2Н-пиран-4-ил]амино-1-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]пропан-2-ол;
2-[(1S,3S)-3-[(3-метокситетрагидро-2Н-пиран-4-ил)амино]-1-(4-[6-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]пропан-2-ол;
N-[(1S,3S)-3-этил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-N-[(1R,3S)-3-этил-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
N-[(1S,3S)-3-этил-3-(4-[6-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-N-[(1R,3S)-3-метил-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-3-метокси-N-[(1R,3S)-3-(2-метоксиэтил)-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
3-метокси-N-[(1S,3S)-3-(2-метоксиэтил)-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
(4R)-N-[(1R,3S)-3-(этоксиметил)-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-N-[(1R,3S)-3-(этоксиметил)-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2Н-пиран-4-амин;
(4R)-3-метокси-N-[(1R,3S)-3-(метоксиметил)-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
(4R)-3-метокси-N-[(1R,3S)-3-(метоксиметил)-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
(4R)-3-метокси-N-[(1R,3S)-3-[(3R)-тетрагидрофуран-3-ил]-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин и
(4R)-3-метокси-N-[(1R,3S)-3-[(3R)-тетрагидрофуран-3-ил]-3-(4-[4-(трифторметил)пиримидин-2-ил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2Н-пиран-4-амин;
или его фармацевтически приемлемую соль.
24. Композиция для лечеэшя заболеваний, ассоциированных с экспрессией или активностью хемокинового рецептора, содержащая соединения по любому из пп.1-23 и фармацевтически приемлемый носитель.
25. Способ модуляции активности хемокинового рецептора, включающий приведение в контакт указанного хемокинового рецептора с соединением по любому из пп.1-23.
26. Способ по п.25, где указанный хемокиновый рецептор представляет собой CCR2 или CCR5.
27. Способ по п.25, где указанная модуляция представляет собой ингибирование.
28. Способ по п.25, где указанное соединение ингибирует и CCR2 и CCR5.
29. Способ лечения заболевания, ассоциированного с экспрессией или активностью хемокинового рецептора, у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-23.
30. Способ по п.29, где указанный хемокиновый рецептор представляет собой CCR2 или CCR5.
31. Способ по п.29, где указанное заболевание представляет собой воспалительное заболевание.
32. Способ по п.29, дополнительно включающий введение противовоспалительного средства.
33. Способ по п.32, где указанное противовоспалительное средство представляет собой антитело.
34. Способ по п.29, где указанное заболевание представляет собой иммунное расстройство.
35. Способ по п.29, где указанное заболевание представляет собой ревматоидный артрит, атеросклероз, волчанку, рассеянный склероз, невропатическую боль, реакцию отторжения трансплантата, диабет или ожирение.
36. Способ по п.29, где указанное заболевание представляет собой рак.
37. Способ по п.36, где указанный рак характеризуется наличием ассоциированных с опухолью макрофагов.
38. Способ по п.36, где указанный рак представляет собой рак молочной железы, рак яичника или множественную миелому.
39. Способ по п.29, где указанное заболевание или состояние представляет собой вирусную инфекцию.
40. Способ по п.39, где указанная вирусная инфекция представляет собой ВИЧ-инфекцию.
41. Способ леченияВИЧ-инфекции у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по любому из пп.1-23.
42. Способ по п.41, дополнительно включающий одновременное или последовательное введение по меньшей мере одного противовирусного средства.
Текст
012649 Настоящее изобретение относится к соединениям, которые модулируют активность хемокиновых рецепторов, таких как CCR2 и CCR5. В некоторых вариантах осуществления соединения настоящего изобретения модулируют как CCR2, так и CCR5. Соединения могут быть полезны, например, для лечения заболеваний, ассоциированных с экспрессией или активностью хемокинового рецептора. Миграция и транспорт лейкоцитов из кровеносных сосудов в пораженные заболеванием ткани вовлекаются в инициацию нормальных воспалительных реакций организма, направленных на борьбу с заболеванием. Процесс, известный также как рекрутинг лейкоцитов, связан с возникновением и прогрессированием угрожающего жизни воспаления, а также истощающих организм аутоиммунных заболеваний. Развивающаяся при таких заболеваниях патология связана с атакой иммунной системы организма, направленной на нормальные ткани. Соответственно, предотвращение и блокирование рекрутинга лейкоцитов в ткани-мишени при воспалительном аутоиммунном заболевании и раке будут очень эффективными подходами в терапевтической интервенции. Различные классы лейкоцитарных клеток, которые вовлекаются в клеточные иммунные реакции,включают моноциты, лимфоциты, нейтрофилы, эозинофилы и базофилы. В большинстве случаев лимфоциты представляют собой класс лимфоцитов, которые инициируют, координируют и поддерживают хронические воспалительные реакции, и в этой связи желательно препятствовать поступлению данных клеток в сайты воспаления. Лимфоциты притягивают моноциты к тканевым сайтам, которые вместе с лимфоцитами отвечают за большую часть фактических повреждений тканей, имеющих место при воспалении. Известно, что инфильтрация лимфоцитов и/или моноцитов приводит к развитию широкого круга хронических аутоиммунных заболеваний, а также реакции отторжения трансплантированных органов. Такие заболевания включают, но без ограничения, ревматоидный артрит, хронический контактный дерматит, воспалительное заболевание кишечника, волчанку, системную красную волчанку, рассеянный склероз, атеросклероз, псориаз, саркоидоз, идиопатический легочный фиброз, дерматомиозит, пемфигоид кожи и родственные заболевания (например, вызванные Pemphigus vulgaris, P. foliacious, P. erythematosis), гломерулонефриты, васкулиты, гепатит, диабет, реакцию отторжения аллотрансплантата и реакцию трансплантат против организма хозяина. Процесс, в ходе которого лейкоциты покидают кровоток, накапливаются в сайтах воспаления и приводят к началу заболевания, как считается включает по меньшей мере три стадии, которые были описаны как (1) свертывание, (2) активация/плотная адгезия и (3) миграция через эндотелий [Springer, Т.A.,Nature 346; 425-433 (1990); Lawrence and Springer, Cell 65: 859-873 (1991); Butcher, E.C., Cell 67: 10331036 (1991)]. Вторая стадия опосредована на молекулярном уровне действием хемоатрактирующих рецепторов. Хемоатрактирующие рецепторы на поверхности лейкоцитов далее связывают хемоатрактирующие хемокины, которые секретируются клетками в сайте повреждения или инфекции. Связывание рецептора активирует лейкоциты, повышает адгезивность молекул адгезии, которые вовлекаются в миграцию через эндотелий и ускоряют направленную миграцию клеток к источнику хемоатрактирующего хемокина. Хемотаксические хемокины (лейкоцитарные хемоатрактирующие/активирующие факторы), также известные как хемокины, иначе называемые интерцинами и SIS хемокинами, представляют собой группу воспалительных/иммуномодулирующих полипептидных факторов с молекулярной массой 6-15 кДа, которые высвобождаются большим числом клеток, таких как макрофаги, моноциты, эозинофилы, нейтрофилы, фибробласты, клетки эндотелия сосудов, гладкомышечные клетки и тучные клетки, в сайтах воспаления (см. обзоры: Luster, New Eng. J. Med., 338, 436-445 (1998) and Rollins, Blood, 90, 909-928 (1997. Кроме того, хемокины были описаны Oppenheim, J.J. et al., в Annu. Rev. Immunol., 9:617-648 (1991);Schall and Bacon, в Curr. Opin. Immunol., 6: 865-873 (1994); Baggiolini, M., et al., в Adv. Immunol., 55: 97179 (1994). Хемокины обладают способностью стимулировать направленную миграцию клеток в ходе процесса, известного как хемотаксис. Каждый хемокин содержит четыре цистеиновых остатка (С) и две внутренних дисульфидных связи. Хемокины могут быть сгруппированы в два подсемейства, в зависимости от того, соседствуют ли непосредственно оба цистеиновых остатка на аминоконце (СС семейство) или они разделяются одной аминокислотой (СХС семейство). Указанные различия коррелируют с организацией двух подсемейств в два отдельных генных кластера. В пределах каждого генного кластера хемокины в типичном случае демонстрируют сходство по последовательности на уровне от 25 до 60%. Хемокины класса СХС, такие как интерлейкин-8 (IL-8), нейтрофил-активирующий протеин-2 (NAP-2) и белок с активностью, стимулирующей рост меланомы (MGSA), обладают хемотаксическим действием прежде всего в отношении нейтрофилов и Т лейкоцитов, тогда как СС хемокины, такие как RANTES,MIP-1, MIP-1, моноцитарные хемотаксические белки (МСР-1, МСР-2, МСР-3, МСР-4 и МСР-5) и эотаксины (-1 и -2), оказывают хемотаксический эффект, в числе клеток других типов, на макрофаги, Т лейкоциты, эозинофилы, дендритные клетки и базофилы. Существуют также хемокины: лимфотактин-1,лимфотактин-2 (оба относятся к С хемокинам) и фракталкин (СХХХС хемокин), которые не попадают ни в одно из основных хемокиновых подсемейств. МСР-1 (также известные как MCAF (аббревиатура, применяемая для обозначения макрофагального хемотаксического и активирующего фактора) или JE) представляет собой СС хемокин, продуцируемый моноцитами/макрофагами, гладкомышечными клетками, фибробластами и клетками эндотелия сосудов,-1 012649 и вызывает миграцию клеток и клеточную адгезию моноцитов (см., например, Valente, A.J., et al., Biochemistry, 1988, 27, 4162; Matsushima, K., et al., J. Exp. Med., 1989, 169, 1485; Yoshimura, Т., et al., J. Immunol., 1989, 142, 1956; Rollins, B.J., et al., Proc. Natl, Acad. Sci. USA, 1988, 85, 3738; Rollins, В., et al.,Blood, 1991, 78, 1112; Jiang, Y., et al., J. Immunol., 1992, 148, 2423; Vaddi, K., et al., J. Immunol., 1994, 153,4721), T лимфоцитов памяти (см., например, Carr, M.W., et al., Proc. Natl. Acad. Sci. USA, 1994, 91, 3652),T лимфоцитов (см., например, Loetscher, P., et al., FASEB J., 1994, 8, 1055) и естественных клетоккиллеров (см., например, Loetscher, P., et al., J. Immunol., 1996, 156, 322; Allavena, P., et al., Eur. J. Immunol., 1994, 24, 3233), а также вовлекается в высвобождение гистамина, опосредованное базофилами (см.,например, Alam, R., et al., J. Clin. Invest., 1992, 89, 723; Bischoff, S.,C., et al., J. Exp. Med., 1992, 175, 1271;Kuna, P., et al., J. Exp. Med., 1992, 175, 489). Кроме того, имеются сообщения о высоком уровне экспрессии МСР-1 при таких заболеваниях, где накопление моноцитов/макрофагов и/или Т клеток рассматривается в качестве важного компонента для инициации или прогрессирования таких заболеваний, как атеросклероз (см., например, Hayes, I.M., et al., Arterioscler. Thromb. Vase. Biol., 1998, 18, 397; Takeya, M., et al.,Hum. Pathol., 1993, 24, 534; Yla-Herttuala, S., et al., Proc. Natl. Acad. Sci. USA, 1991, 88, 5252; Nelken, N.A.,J. Clin. Invest., 1991, 88, 1121), ревматоидный артрит (см., например, Koch, A.E., et al., J. Clin. Invest.,1992, 90, 772; Akahoshi T., et al., Arthritis Rheum., 1993, 36, 762; Robinson E., et al., Clin. Exp. Immunol.,101, 398), нефрит (см., например, Noris, M., et al., Lab. Invest., 1995, 73, 804; Wada, Т., et al., Kidney Int, . 1996, 49, 761; Gesualdo, L., et al., Kidney Int., 1997, 51, 155), нефропатия (см., например, Saitoh A., et al., J.al., J. Invest, Dermatol., 1993, 101, 127), воспалительная болезнь кишечника (см., например, Grimm, M.C.,et al., J. Leukoc. Biol., 1996, 59, 804; Reinecker, H.C., et al., Gastroenterology, 1995, 106, 40), миокардит (см.,например, Sieno, Y., et al., Chemokine, 1995, 7, 301), эндометриоз (см., например, Jolicoeur, С, et al. , Am.J. Pathol., 1998, 152, 125), внутрибрюшинная адгезия (см., например, Zeyneloglu, H., et al., Human Reproduction, 1998, 13, 1194), застойная сердечная недостаточность (см., например, Aurust, P., et al., Circulation,1998, 97, 1136), хроническая болезнь печени (см., например, Marra, F., et al., Am. J. Pathol., 1998, 152,423), вирусный менингит (см., например, Lahrtz, F., et al, Eur. J. Immunol., 1997, 27, 2484), болезнь Кавасаки (см., например, Wong, M., et al., J. Rheumatol., 1997, 24, 1179) и сепсис (см., например, Salkowski,С.A., et al., Infect. Immun., 1998, 66, 3569). Кроме того, сообщается, что антитела против МСР-1 демонстрируют ингибирующий эффект или терапевтический эффект на моделях животных с ревматоидным артритом (см, например, Schimmer, R.C., et al., J. Immunol., 1998, 160, 1466; Schrier, D.J., J. Leukoc. Biol. 1998, 63, 359; Ogata, H., et al., J. Pathol., 1997, 182, 106), с рассеянным склерозом (см., например, Karpus,W.J., et al., J. Leukoc. Biol. 1997, 62, 681), с нефритом (см., например, Lloyd, СМ., et al., J. Exp. Med., 1997,185, 1371; Wada, Т., et al., FASEB J., 1996, 10, 1418), с астмой (см., например, Gonzalo, J.-A., et al., J. Exp.Med. 1998, 188, 157; Lukacs, N.W., J. Immunol., 1997, 158, 4398), с атеросклерозом (см., например,Guzman, L.A., et al., Circulation, 1993, 88 (suppl.), 1-371), с реакцией гиперчувствительности задержанного типа (см., например, Rand, M.L., et al., Am. J. Pathol, 1996, 148, 855), с легочной гипертензией (см., например, Kimura, H., et al., Lab. Invest., 1998, 78, 571) и с внутрибрюшинной адгезией (см., например, Zeyneloglu, H., et al., Av. J. Obstet. Gynecol., 1998, 179, 438). Сообщается также, что пептидный антагонист МСР-1, МСР-1 (9-76) подавляет развитие артрита на модели мышей (см. Gong, J.-H., J. Exp. 4ed., 1997,186, 131), а также в исследованиях с использованием МСР-1-дефицитных мышей было показано, что МСР-1 необходим для рекрутинга моноцитов in vivo (см., например, Lu, В., et al., J. Exp. Med., 1998, 187,601; Gu, L., et al., Moll. Cell, 1998, 2, 275). Хроническое обструктивное заболевание легких (ХОЗЛ) входит в число наиболее частых причин смерти в западном обществе. Она определяется прогрессирующим снижением функций легких, которое лишь частично восстанавливается бронходилататорами. ХОЗЛ характеризуется хроническим воспалением дыхательных путей или альвеол, которое отличается от патологии, развивающейся при астме, и включает повышение числа нейтрофилов, макрофагов, CD8+ клеток и/или тучных клеток в стенках дыхательных путей, в альвеолярных отсеках и в гладкомышечных клетках сосудов. Считается, что цитокины, ассоциированные с ХОЗЛ, включают фактор некроза опухоли (TNF)-альфа, интерферон (IFN)-гамма,интерлейкин (IL-1)-бета, IL-6, IL-8 и МСР-1. Известно, что CCR2 является рецептором для МСР-1, и последние данные указывают на роль МСР-1 и CCR2 в ремоделировании дыхательных путей и при воспалении, осуществляемую непосредственно или через макрофаги. Таким образом, антагонисты CCR2 представляют собой перспективный подход в терапии ХОЗЛ (De Boer, W. I., Chest, 2002, 121, 209S-218S). Литературный данные указывают на то, что хемокины, такие как МСР-1 и MIP-1, привлекают моноциты и лимфоциты к сайту заболевания и опосредуют их активацию и, таким образом, как считается,тесно вовлекаются в инициацию, прогрессирование и поддержание заболеваний, в которых в существенной мере задействованы моноциты и лимфоциты, таких как атеросклероз, рестеноз, ревматоидный артрит, псориаз, астма, язвенный колит, нефрит (нефропатия), рассеянный склероз, фиброз легкого, миокар-2 012649 дит, гепатит, панкреатит, саркоидоз, болезнь Крона, эндометриоз, застойная сердечная недостаточность,вирусный менингит, церебральный инфаркт, нейропатия, болезнь Кавасаки и сепсис (см., например,Rovin, B.H.,. et al., Am. J. Kidney. Dis., 1998, 31, 1065; Lloyd, C., et al., Curr. Opin. Nephrol. Hypertens.,1998, 7, 281; Conti, P., et al., Allergy and Asthma Proc., 1998, 19, 121; Ransohoff, R.M., et al., Trends Neurisci., 1998, 21, 154; MacDermott R.P., et al., Inflammatory Bowel Diseases, 1998, 4, 54). Хемокины связываются со специфичными рецепторами на поверхности клеток, которые относятся к семейству связанных с G-белком протеинов с семью трансмембранными доменами (см. обзор в Horuk,Trends Pharm. Sci., 15, 159-165 (1994, которые называют хемокиновыми рецепторами. При связывании своих соответствующих лигандов хемокиновые рецепторы переносят внутриклеточный сигнал через ассоциированные трехмерные G-белки, что приводит, в числе других ответов, к быстрому повышению внутриклеточной концентрации кальция, к изменениям формы клеток, к повышению экспрессии молекул клеточной адгезии, к дегрануляции и к усилению миграции клеток. Гены, кодирующие рецепторы специфических хемокинов, были клонированы, в результате чего стало известно, что указанные рецепторы представляют собой связанные с G-белком рецепторы с семью трансмембранными доменами, присутствующие на поверхности различных популяций лейкоцитов. Соответственно, были идентифицированы по меньшей мере пять СХС хемокиновых рецепторов (CXCR1CXCR5) и восемь СС хемокиновых рецепторов (CCR1-CCR10). Например, IL-8 представляет собой лиганд для CXCR1 и CXCR2, MIP-1 представляет собой лиганд для CCR1 и CCR5, и МСР-1 представляет собой лиганд для CCR2A и CCR2B (ссылки см., например, Holmes, W.E., et al., Science 1991, 253, 12781280; Murphy P.M., et al., Science, 253, 1280-1283; Neote, K., et al., Cell, 1993, 72, 415-425; Charo, I.F., et al.,Proc. Natl. Acad. Sci. USA, 1994, 91, 2752-2756; Yamagami, S., et al., Biochem. Biophys. Res. Commun.,1994, 202, 1156-1162; Combadier, C., et al., the Journal of Biological Chemistry, 1995, 270, 16491-16494;Power, C.A., et al., J. Biol. Chem., 1995, 270, 19495-19500; Samson, M., et al., Biochemistry, 1996, 35, 33623367; Murphy, P.M., Annual Review of Immunology, 1994, 12, 592-633). Сообщалось, что воспаление легких и образование грануромы подавляется у CCR1-дефицитных мышей (см. Gao, J.-L., et al., J. Exp. Med.,1997, 185, 1959; Gerard, C., et al., J. Clin. Invest., 1997, 100, 2022) и рекрутинг макрофагов и образование атеросклеротических поражений снижается у CCR2-дефицитных мышей (см. Boring, L., et al., Nature,1998, 394, 894; Kuziel, W.A., et al. , Proc. Natl. Acad. Sci. USA, 1997, 94, 12053; Kurihara, Т., et al., J. Exp.Med., 1997, 186, 1757; Boring, L., et al., J. Clin. Invest., 1997, 100, 2552). Известно, что хемокиновые рецепторы выполняют функцию сорецепторов в механизме вхождения вирусов в клетку, ведущего к вирусной инфекции, такой как, например, ВИЧ инфекция. Процессы обратной транскрипции и процессинг белков представляют собой классические стадии жизненного цикла вирусов, на которые нацелено блокирующее действие антиретровирусных лекарственных агентов. Хотя существует много новых лекарственных средств, которые, как считается, блокируют вхождение вирусов в клетку, в настоящее время отсутствует агент, к которому бы ВИЧ-1 не был способен выработать резистентность. Необходимые для развития вируса множественные циклы репликации генерируют генетическое разнообразие, которое создает основу для резистентности. Комбинированная терапия, в ходе которой репликация максимально подавляется, остается краеугольным камнем в стратегии лечения с использованием ингибиторов вхождения вируса в клетку, аналогично варианту использования других агентов. Полагают, что подход, направленный, в качестве мишени воздействия, на многие стадии механизма вхождения и встраивания вируса в клетку, может оказывать синергический эффект (Starr-Spires et al., Clin.Lab. Med., 2002, 22 (3), 681). Для вхождения ВИЧ-1 в CD4(+) клетки требуется последовательное взаимодействие гликопротеинов вирусной оболочки с CD4 и сорецептором, таким как хемокиновые рецепторы CCR5 и CXCR4. Соответствующий подход, направленный на блокирование данного процесса, сопряжен с использованием малых молекул антагонистов, обладающих функцией сорецептора. Молекула ТАК-779 представляет собой один такой антагонист CCR5, действие которого препятствует инфекции ВИЧ-1. ТАК-779 ингибирует репликацию ВИЧ-1 на стадии слияния с мембраной путем блокирования взаимодействия gp120, гликопротеина поверхности вируса, с CCR5. Сайт связывания ТАК-779 на CCR5 расположен вблизи внеклеточной поверхности рецептора в полости, образованной между трансмембранными спиралями 1, 2, 3 и 7(Dragic . et al., Proc. Natl. Acad. Sci. USA, 2000, 97(10), 5639). Считается, что хемокиновые рецепторы CXCR4 и CCR5 используются как сорецепторы штаммами ВИЧ-1, тройными к Т клеткам (Х 4) и тройными к макрофагам (R5), соответственно, для вхождения в хозяйские клетки. Размножение штаммов R5 ВИЧ-1 на CD4 лимфоцитах и макрофагах требует экспрессии CCR5 сорецептора на клеточной поверхности. Индивидуумы, не имеющие CCR5 (гомозиготный генотип CCR5 Delta 32), являются фенотипически нормальными и резистентны к инфекции ВИЧ-1. Вхождение вируса в клетку может быть ингибировано природными лигандами для CXCR4 (СХС хемокинSDF-1) и CCR5 (СС хемокины RANTES, MIP-1-альфа и MIP-1-бета). Первый из них, будучи соединением непептидной природы, которое взаимодействуют с CCR5, но не с CXCR4, представляет собой производное четвертичного аммония, называемое ТАК-779, которые также обладает мощной, но варьирующей анти-ВИЧ активностью (De Clercq et al., Antivir. Chem. Cchemother. 2001, 12, Suppl. 1, 19).SCH-C (SCH 351125) представляет собой другую малую молекулу ингибитора вхождения ВИЧ-1 в-3 012649 клетку, функция которого реализуется через сорецептор CCR5. SCH-C, как оксим-пиперидиновое соединение, представляет собой специфический антагонист CCR5, по данным многих анализов связывания с рецептором и тестов на сигнальную трансдукцию. Данное соединение специфически ингибирует ВИЧ-1 инфекцию, опосредованную CCR5, в клетках астроглиомы U-87, но не оказывает эффекта на инфекциюAD101, химически близкий к SCH-C, также ингибирует вхождение вируса иммунодефицита человека типа 1 (ВИЧ-1) через человеческий CCR5. Было показано, что AD101 ингибирует поступление ВИЧ-1 через CCR5 макаки резус, тогда как SCH-C не обладает такой способностью. В число восьми остатков, которые отличаются у сорецепторов в вариантах человека и макаки, входит лишь один, метионин-198, который отвечает за нечувствительность CCR5 макаки к ингибированию под действием SCH-C. Положение 198 локализовано на трансмембранном участке (ТМ) спирали 5 в CCR5 и не относится к области ранее идентифицированного сайта связывания AD101 и SCH-C, который вовлекает остатки в ТМ спиралях 1, 2, 3 и 7. По результатам исследования аминокислотных замещений в CCR5, было высказано предположение о том, что участок CCR5 вблизи остатка 198 может влиять на конформационное состояние данного рецептора (Billick et al., 2004, J. Virol., 78(8), 4134). Идентификация соединений, которые модулируют активность хемокиновых рецепторов, вносят вклад в искомую стратегию поиска лекарственных средств, направленную на разработку фармакологических агентов для лечения заболеваний, ассоциированных с активностью хемокиновых рецепторов. Соединения по настоящему изобретению дают возможность ответить на эти и другие потребности. Настоящее изобретение относится к соединениям формулы I или к их фармацевтически приемлемым солям или их пролекарствам, где представители этого класса приведены в настоящем описании. Настоящее изобретение также относится к композициям, содержащим соединение формулы I и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу модуляции активности хемокинового рецептора,включающему взаимодействие хемокинового рецептора с соединением формулы I. Настоящее изобретение также относится к способу лечения заболевания, ассоциированного с экспрессией или активностью хемокинового рецептора у пациента, включающему введение пациенту терапевтически эффективного количества соединения формулы I. Настоящее изобретение также относится к способу лечения ВИЧ инфекции у пациента, включающему введение такому пациенту терапевтически эффективного количества соединения формулы I. Настоящее изобретение также относится к соединению, приведенному в настоящем описании, с целью применения в терапии. Настоящее изобретение также относится к соединению, приведенному в настоящем описании, с целью получения лекарственного средства, предназначенного для использования в терапии. Соединения Настоящее изобретение относится, в том числе к соединению формулы I или к его фармацевтически приемлемым солям или его пролекарствам, где прерывистая линия указывает на необязательную связь; W означаетR9 и R9', каждый независимо, означает Н; и р равно 1. В некоторых вариантах осуществления W означает: В некоторых вариантах осуществления W означает: В некоторых вариантах осуществления V означает CR5. В некоторых вариантах осуществления X означает CR2. В некоторых вариантах осуществления Y означает CR3. В некоторых вариантах осуществления Z означает CR4. В некоторых вариантах осуществления X означает N. В некоторых вариантах осуществления Z означает N. В некоторых вариантах осуществления оба: X и Z означают N. В некоторых вариантах осуществления X означает CR2; Y означает CR3 и Z означает CR4. В некоторых вариантах осуществления V означает CR5; X означает CR2; Y означает CR3 и Z означает CR4. В некоторых вариантах осуществления не более чем 2 из V, X, Y и Z, означают N. В некоторых вариантах осуществления по меньшей мере 2 из V, X, Y и Z отличаются от N. В некоторых вариантах осуществления ни один из V, X, Y и Z не означает N. В некоторых вариантах осуществления 1 из V, X, Y и Z означает N. В некоторых вариантах осуществления 2 из V, X, Y и Z означают N. В некоторых вариантах осуществления RA, RA1, RB и RB1, каждый независимо, означает Н или ОН. В некоторых вариантах осуществления R1 означает С 1-6 алкил. В некоторых вариантах осуществления R1 означает проп-2-ил. В некоторых вариантах осуществления R5 и R6 отличаются от Н. В некоторых вариантах осуществления R5 и R6 отличаются от С 1-4 галогеналкила. В некоторых вариантах осуществления R6 означает С 1-4 галогеналкил. В некоторых вариантах осуществления R6 означает CF3. В некоторых вариантах осуществления R7 означает Н. В некоторых вариантах осуществления R8 означает C1-3 алкокси или C1-3 галогеналкокси. В некоторых вариантах осуществления R8 означает C1-3 алкокси. В некоторых вариантах осуществления R8 означает метокси. В некоторых вариантах осуществления R8 означает этокси. В некоторых вариантах осуществления R9 и R9', оба означают Н. В некоторых вариантах осуществления р равно 1. В некоторых вариантах осуществления соединения настоящего изобретения имеют формулу Ia: В некоторых вариантах осуществления соединения настоящего изобретения имеют формулу Ib, Ic или Id:-5 012649 В некоторых вариантах осуществления соединения настоящего изобретения имеют формулу Ie или В некоторых вариантах осуществления соединения настоящего изобретения имеют формулу Ig: В некоторых вариантах осуществления соединения настоящего изобретения имеют формулу Ih или В разных частях настоящего описания заместители в соединениях настоящего изобретения рассматриваются при их объединении в группы или согласно определенной схеме. При этом в объем настоящего изобретения предусматривается включение каждого и любого объединения представителей таких групп и классов в любом варианте их сочетания. Например, термин C1-6 алкил относится к метилу, этилу, С 3 алкилу, С 4 алкилу, С 5 алкилу и С 6 алкилу. Для соединений по настоящему изобретению, где переменная величина появляется более чем один раз, каждая такая переменная величина может представлять собой отличающийся фрагмент, выбранный из группы Маркуша, определяющей такие переменные. Например, в том случае, когда в структуре имеются две R группы, которые одновременно присутствуют в одном и том же соединении, обе указанные R группы могут обозначать разные фрагменты, выбранные из группы Маркуша, определенной для R. Следует также понимать, что некоторые признаки настоящего изобретения, которые для простоты описания рассматриваются в контексте отдельных вариантов, могут быть реализованы в сочетании,представляющем собой уже один определенный вариант. И наоборот, различные признаки настоящего изобретения, которые для краткости описываются в контексте одного варианта его осуществления, могут быть реализованы по отдельности или в виде любых подходящих подкомбинаций. Как использовано в настоящем описании, термин алкил относится к насыщенной углеводородной группе, которая может быть линейно-цепочечной или разветвленной. Примеры алкильных групп включат метил (Me), этил (Et), пропил (например, н-пропил или изопропил), бутил (например, н-бутил, изобутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил) и т.п. Алкильная группа может содержать от 1 до примерно 20, от 2 до примерно 20, от 1 до примерно 10, от 1 до примерно 8, от 1 до примерно 6, от 1 до примерно 4 или от 1 до примерно 3 атомов углерода. Как использовано в настоящем описании, термин алкилен относится к двухвалентной алкильной группе. Как использовано в настоящем описании, термин С 2-4 алкилен относится к алкиленовой группе,включающей от 2 до 4 атомов углерода. Как использовано в настоящем описании, термин алкенил относится к любой алкильной группе,содержащей одну или несколько двойных углерод-углеродных связей. Примеры алкенильных групп включают этенил, пропенил, циклогексенил и т.п. Как использовано в настоящем описании, термин алкинил относится к алкильной группе, содержащей одну или несколько тройных углерод-углеродных связей. Примеры алкинильных групп включают этинил, пропинил и т.п. Как использовано в настоящем описании, термин галогеналкил относится к алкильной группе,содержащей один или несколько галогеновых заместителей. Примеры галогеналкильных групп включают CF3, C2F5, CHF2, CCl3, CHCl2, C2Cl5 и т.п. Как использовано в настоящем описании, термин арил относится к моноциклическим или полициклическим (например, содержащим 2, 3 или 4 конденсированных кольца) ароматическим углеводородам, таким как, например, фенил, нафтил, антраценил, фенантренил, инданил, инденил и т.п. В некото-6 012649 рых вариантах осуществления арильная группа содержит от 6 до примерно 20 атомов углерода. Как использовано в настоящем описании, карбоциклильные группы представляют собой насыщенные (то есть не содержат двойных или тройных связей) или ненасыщенные (то есть содержат одну или несколько двойных или тройных связей) циклические углеводородные фрагменты. Карбоциклильные группы могут быть моно-, поли- (например, содержащие 2, 3 или 4 конденсированных кольца) группами. Примеры карбоциклильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, 1,3-циклопентадиенил, циклогексенил, норборнил, норпинил, норкарнил, адамантил, фенил и т.п. Карбоциклильные группы могут быть ароматическими (например, арил) или неароматическими (например, циклоалкил). В некоторых вариантах осуществления карбоциклильные группы могут содержать примерно от 3 до примерно 30 атомов углерода, примерно от 3 до примерно 20 атомов углерода, примерно от 3 до примерно 10 атомов углерода или примерно от 3 до примерно 7 углеродных атомов, формирующих кольцо. Как использовано в настоящем описании, термин циклоалкил относится к неароматическим карбоциклам, включающим циклизованные алкильные, алкенильные и алкинильные группы. Цикоалкильные группы могут включать моно- или полициклические (например, содержащие 2, 3 или 4 конденсированных кольца) кольцевые системы, а также спиро-кольцевые системы. Примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклопентенил, циклогексенил, циклогексадиенил, циклогептатриенил, норборнил, норпинил, норкарнил, адамантил и т.п. В определение циклоалкила также включаются фрагменты, которые содержат одно или несколько ароматических колец, конденсированных (то есть имеющих общую связь) с циклоалкильным кольцом, например, бензопроизводные пентана, пентена, гексана и т.п. В некоторых вариантах осуществления циклоалкильные группы могут содержать примерно от 3 до примерно 10 атомов углерода, примерно от 3 до примерно 10 атомов углерода, или примерно от 3 до примерно 7 атомов углерода, формирующих кольцо. В некоторых вариантах осуществления циклоалкильная группа может содержать 0, 1, 2, 3, 4 или 5 двойных или тройных связей. В еще других вариантах осуществления один или несколько атомов углерода, формирующих кольцо, могут быть замещены в циклоалкильной группе оксогруппой или сульфидогруппой. Как использовано в настоящем описании, термин гетероциклил или гетероцикл относится к насыщенному или ненасыщенному циклическому углеводороду, где один или несколько атомов углерода, образующих кольцо в циклическом углеводороде, заменены гетероатомом, таким как О, S или N. Гетероциклильные группы могут быть ароматическими (например, представлять собой гетероарил) или неароматическими (например, представлять собой гетероциклоалкил). Гетероциклильные группы могут также соответствовать галогенированным и частично галогенированным гетероарильным группам. Гетероциклильные группы могут включать моно- или полициклические кольцевые системы (например,содержащие 2, 3 или 4 конденсированных кольца). Гетероциклильные группы могут отличаться наличием 3-14 или 3-7 атомов, образующих кольцо. В некоторых вариантах осуществления гетероциклильные группы могут содержать, в дополнение по меньшей мере к одному гетероатому, примерно от 1 до примерно 13 атомов углерода, примерно от 2 до примерно 10 атомов углерода или примерно от 2 до примерно 7 атомов углерода и могут быть присоединены через атом углерода или гетероатом. В еще других вариантах осуществления любой атом углерода или гетероатом, образующий кольцо, может быть окислен (например, может содержать оксо- или сульфидозаместитель), или атом азота может быть кватернизован. Примеры гетероциклильных групп включают морфолино, тиоморфолино, пиперазинил, тетрагидрофуранил, тетрагидротиенил, 2,3-дигидробензофурил, 1,3-бензодиоксол, бензо-1,4-диоксан, пиперидинил, пирролидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, оксазолидинил, тиазолидинил,имидазолидинил и т.п., а также любую группу, приведенную ниже применительно к гетероарилу и гетероциклоалкилу. Другие примеры гетероциклов включают пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил, феноксатинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, 3,6-дигидропиридил, 1,2,3,6-тетрагидропиридил, 1,2,5,6-тетрагидропиридил, пиперидонил, 4 пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиридил, пиримидинил, пирролидинил, пирролил, 2 Н-пирролил, пирролил, тетрагидрофуранил, тетрагидроизохинолинил,тетрагидрохинолинил, тетразолил, 6 Н-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5 тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил,ксантенил, октагидроизохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5 оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, оксазолидинил, хиназолинил, хинолинил,4 Н-хинолизинил, хиноксалинил, хинуклидинил, акридинил, азоцинил, бензимидазолил, бензофуранил,бензотиофуранил, бензотиофенил, бензоксазолил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, метилендиоксифенил, морфолинил, нафтиридинил,декагидрохинолинил, 2 Н,6 Н-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуран, фуранил, фуразанил, карбазолил, 4 аН-карбазолил, карболинил, хроманил, хроменил, циннолинил, имидазолидинил, имидазолинил, имидазолил, 1 Н-индазолил, индоленил, индолинил, индолизинил, индолил, 3 Н-индолил, изобензофуранил, изохроманил, изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил и-7 012649 изоксазолил. Другие примеры гетероциклов включают азетидин-1-ил, 2,5-дигидро-1 Н-пиррол-1-ил, пиперидин-1-ил, пиперазин-1-ил, пирролидин-1-ил, изохинол-2-ил, пиридин-1-ил, 3,6-дигидропиридин-1 ил, 2,3-дигидроиндол-1-ил, 1,3,4,9-тетрагидрокарболин-2-ил, тиено[2,3-c]пиридин-6-ил, 3,4,10,10 атетрагидро-1 Н-пиразино[1,2-а]индол-2-ил, 1,2,4,4 а, 5,6-гексагидропиразино[1,2-а]хинолин-3-ил, пиразино[1,2-а]хинолин-3-ил, диазепан-1-ил, 1,4,5,6-тетрагидро-2 Н-бензо[f]изохинолин-3-ил, 1,4,4 а,5,6,10bгексагидро-2 Н-бензо[f]изохинолин-3-ил,3,3 а,8,8 а-тетрагидро-1 Н-2-аза-циклопента[а]инден-2-ил и 2,3,4,7-тетрагидро-1 Н-азепин-1-ил и азепан-1-ил. Как использовано в настоящем описании, гетероарильные группы включают ароматический гетероцикл, содержащий по меньшей мере один гетероатом в кольце, такой как сера, кислород или азот. Гетероарильные группы включают моноциклические и полициклические системы (например, содержащие 2, 3 или 4 конденсированных кольца). Примеры гетероарильных групп включают, но без ограничения, пиридил, пиримидинил, пиразинил, пиридазинил, триазинил, фурил (фуранил), хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пиррил, оксазолил, бензофурил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, тиазолил, тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, бензотиенил, пуринил, карбазолил, бензимидазолил, индолинил и т.п. В некоторых вариантах осуществления гетероарильная группа содержит от 1 до примерно 20 атомов углерода и в других вариантах осуществления - примерно от 3 до примерно 20 атомов углерода. В некоторых вариантах осуществления гетероарильная группа содержит от 3 до примерно 14, от 3 до примерно 7 или от 5 до 6 атомов, образующих кольцо. В некоторых вариантах осуществления гетероарильная группа содержит от 1 до примерно 4, от 1 до примерно 3 или от 1 до 2 гетероатомов. Как использовано в настоящем описании, термин гетероциклоалкил относится к неароматическим гетероциклам, включающим циклизованные алкильные, алкенильные и алкинильные группы, где один или несколько атомов углерода, образующих кольцо, заменены гетероатомом, таким как О, N илиS. Примеры гетероциклоалкильных групп включают морфолино, тиоморфолино, пиперазинил, тетрагидрофуранил, тетрагидротиенил 2,3-дигидробензофурил, 1,3-бензодиоксол, бензо-1,4-диоксан, пиперидинил, пирролидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, оксазолидинил, тиазолидинил,имидазолидинил и т.п. В определение гетероциклоалкила также включаются фрагменты, которые содержат одно или несколько ароматических колец, конденсированных (то есть имеющих общую связь) с неароматическим гетероциклическим кольцом, например, фталимидильные, нафталимидильные и бензопроизводные гетероциклов, такие как индоленовые и изоиндоленовые группы. В некоторых вариантах осуществления циклоалкильные группы могут содержась от 1 до примерно 20 атомов углерода, и в других вариантах осуществления примерно от 3 до примерно 20 атомов углерода. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 3 до примерно 14, от 3 до примерно 7 или от 5 до 6 атомов, образующих кольцо. В еще других вариантах осуществления гетероциклоалкильная группа содержит от 1 до примерно 4, от 1 до примерно 3 или 1 или 2 гетероатома. В некоторых вариантах осуществления гетероциклоалкильная группа содержит от 0 до 3 двойных связей. В некоторых вариантах осуществления гетероциклоалкильная группа содержит 0-2 двойных или тройных связи. Как использовано в настоящем описании, термин спироциклил относится к 3-14-членному циклоалкилу или 3-14-членной гетероциклоалкильной группе, имеющей один общий атом с другой циклоалкильной или гетероциклоалкильной группой, к которой она присоединена. Как использовано в настоящем описании, термин галоген включает фтор, хлор, бром и йод. Как использовано в настоящем описании, термин алкокси относится к -О-алкильной группе. Пример алкоксигрупп включает метокси, этокси, пропокси (например, н-пропокси и изопропокси), третбутокси и т.п. Как использовано в настоящем описании, термин тиоалкокси относится к -S-алкильной группе. Как использовано в настоящем описании, термин галогеналкокси относится к -О-галогеналкильной группе. Примером галогеналкоксигруппы является OCF3. Как использовано в настоящем описании, термин карбоксициклилокси относится к -О-карбоциклилу. Как использовано в настоящем описании, термин циклоалкилокси относится к -О-циклоалкилу. Как использовано в настоящем описании, термин карбоциклилалкил относится к алкилу, замещенному карбоциклилом. Как использовано в настоящем описании, термин аралкил или арилалкил относится к алкильной группе, замещенной арильной группой. Как использовано в настоящем описании, термин циклоалкилалкил относится к алкильной группе, замещенной циклоалкильной группой. Как использовано в настоящем описании, термин гетероциклилалкил относится к алкильному фрагменту, замещенному гетерокарбоциклильной группой. Примеры гетероциклилалкильных групп включают гетероарилалкил (алкил, замещенный гетероарилом) и гетероциклоалкилалкил (алкил,замещенный гетероциклоалкилом). В некоторых вариантах осуществления гетероциклилалкильные группы содержат от 3 до 24 атомов углерода, в дополнение по меньшей мере к одному гетероатому, участвующему в формировании кольца.-8 012649 Как использовано в настоящем описании, термин оксо относится к =O. Приведенные в настоящем описании соединения могут быть асимметрическими (то есть могут иметь один или несколько стереоцентров). Все стереоизомеры, такие как энантиомеры и диастереомеры,включены в объем настоящего описания, если не указано иное. Соединения настоящего изобретения,которые содержат асимметрически замещенные атомы углерода, могут быть выделены в виде оптически активных или рацемических форм. Способы получения оптически активных форм из оптически активных исходных материалов известны в данной области и включают, например, разделение рацемических смесей или получение путем стереоселективного синтеза. Многие геометрические изомеры олефинов,C=N двойные связи и т.п. могут присутствовать в соединениях, приведенных в настоящем описании, и все такие стабильные изомеры охватываются объемом настоящего изобретения. Цис- и транс- геометрические изомеры соединений по настоящему изобретению описаны и могут быть выделены в виде смеси изомеров и в виде отдельных изомерных форм. Разделение рацемических смесей соединений может быть осуществлено с использованием множества известных в настоящей области методик. В качестве одного из примеров таких методик можно назвать фракционную перекристаллизацию с использованием хиральной разделяющей кислоты, представляющей собой оптически активную, солеобразующую органическую кислоту. Подходящими разделяющим агентами для фракционной перекристаллизации являются, например, оптически активные кислоты, такие как D и L формы винной кислоты, диацетилвинной кислоты, дибензоилвинной кислоты,миндальной кислоты, яблочной кислоты, молочной кислоты, или различные оптически активные камфорсульфоновые кислоты, такие как -камфорсульфоновая кислота. Другие разделяющие агенты, подходящие для использования в рамках фракционной кристаллизации, включают стереоизомерные чистые формы -метилбензиламина (например, S и R формы или диастереомерно чистые формы), 2-фенилглицинол, норэфедрин, эфедрин, N-метилэфедрин, циклогексилэтиламин, 1,2-диаминоциклогексан и т.п. Разделение рацемических смесей может быть осуществлено элюированием на колонке, содержащей оптически активный разделяющий агент (например, динитробензоилфенилглицин). Специалист в данной области может определить подходящую для такого рода элюирования композицию растворителей. Соединения настоящего изобретения также включают таутомерные формы, такие как кетоенольные таутомеры. Соединения настоящего изобретения могут также включать изотопы атомов, содержащиеся в промежуточных или конечных соединениях. Изотопы включают такие атомы, которые имеют тот же самый атомный номер, но отличаются по массовом числу. Например, изотопы водорода включают тритий и дейтерий. Фраза фармацевтически приемлемый, используемая в настоящем изобретении, относится к таким соединениям, материалам, композициям и/или дозированным формам, которые на основании весомого медицинского заключения являются подходящими для применения, включающего контакт с тканями человека и животных и не вызывают при этом выраженной токсичности, раздражения, аллергической реакции или других проблем или осложнений, не соответствующих разумному соотношению польза/риск. Настоящее изобретение также включает фармацевтически приемлемые соли приведенных в описании соединений. Как использовано в настоящем описании, фармацевтически приемлемые соли включают производные соединений настоящего изобретения, где исходное, родительское соединение было модифицировано путем превращения имевшегося фрагмента кислоты или основания в их солевую форму. Примеры фармацевтически приемлемых солей включают, но без ограничения, соли минеральной или органической кислоты и основных остатков, таких как амины, основные или органические соли кислотных остатков, такие как карбоновые кислоты, и т.п. Фармацевтически приемлемые соли соединений по настоящему изобретению включают традиционные нетоксичные соли или соли четвертичного аммония исходного соединения, образуемые, например, с помощью нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли настоящего изобретения могут быть синтезированы из соответствующего исходного соединения, которое содержит основную или кислотную группу традиционными химическими методами. Обычно такие соли могут быть получены посредством взаимодействия свободных кислотных или основных форм данных соединений со стехиометрическим количеством соответствующего основания или соли в воде или в органическом растворителе, или в смеси двух таких компонентов, при этом предпочтительны неводные среды, такие как диэтиловый эфир, этилацетат, этанол,изопропанол или ацетонитрил. Перечень подходящих солей приведен, например, в руководстве Ремингтона и в других литературных источниках (Remington's Pharmaceutical Science, 17th ed., Mack PublishingCompany, Easton, Pa., 1985, p. 1418; и Journal of Pharmaceutical Science, 66, 2 (1977, которые включены в настоящее описание во всей своей полноте в качестве ссылки. Настоящее изобретение также включат пролекарства соединений, приведенных в данном описании. Как использовано в настоящем описании, термин пролекарства относится к любым ковалентно связанным носителям, которые высвобождают активное исходное соединение при его введении в организм млекопитающего. Пролекарства могут быть получены путем модификации функциональных групп, при-9 012649 сутствующих в соединениях, таким образом, что модифицированные формы расщепляются либо в результате несложных манипуляций, либо in vivo, с образованием родительских соединений. Пролекарства включают соединения, в которых гидроксильные, амино, сульфидрильные или карбоксильные группы присоединены к любой группе таким образом, что при введении в организм млекопитающего, она расщепляется с образованием свободной гидроксильной, амино, сульфидрильной или карбоксильной группы, соответственно. Примеры пролекарств включают, но без ограничения, ацетатные, формиатные и бензоатные производные функциональных спиртовых и аминогрупп соединений настоящего изобретения. Получение и применение пролекарств описано в литературе (Т. Higuchi and V. Stella, Pro-drugs asNovel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series, и в Bioreversible Carriers in Drug Design,ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987), обе указанные работы включены в настоящее описание во всей своей полноте в качестве ссылки. Синтез Соединения по настоящему изобретению, включая их соли, гидраты и сольваты, могут быть получены с помощью известных методов органического синтеза и могут быть синтезированы по любому из многочисленных возможных путей синтеза. Реакции получения соединений настоящего изобретения могут быть осуществлены в подходящих растворителях, которые любой специалист в области органического синтеза может без труда выбрать. Подходящие растворители могут быть, по существу, не реактивными относительно исходных материалов (реагентов), относительно промежуточных продуктов или продуктов в условиях тех температур, при которых проводят реакции, например, при температурах, изменяющихся от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии, для каждой из них могут быть выбраны подходящие растворители. Получение соединений настоящего изобретения может включать введение защитных групп для различных химических групп и затем удаление защиты. Любой специалист в данной области может определить потребность в защитных группах и удалении защитных групп, а также выбрать соответствующие защитные группы. Описание химии защитных групп приводится, например, в работе Грина с соавт.(1999, которая включена в настоящее описание во всей своей полноте в качестве ссылки. Мониторинг реакций может проводиться с использованием любого приемлемого метода, известного в данной области. Например, образование продукта может выявляться спектроскопическими методами, такими как спектроскопия ядерно-магнитного резонанса (например, по 1 Н или 13 С), инфракрасная спектроскопия, спектрофотометрия (например, в УФ-видимых областях спектра) или масс-спектроскопия, а также методами хроматографии, такой как высокоэффективная жидкостная хроматография(ВЭЖХ) или тонкослойная хроматография. Примеры путей синтеза соединений по настоящему изобретению показаны ниже, на схемах 1-13, и соединения, участвующие в этих процессах, описаны в настоящем изобретении. 3-Аминопентанкарбоновые кислоты формул 1-5 могут быть получены с использованием процедуры, описанной на схеме 1. Коммерчески доступная карбоновая кислота 1-1 может быть преобразована в сложный эфир, такой как сложный метиловый эфир, путем обработки йодметаном/карбонатом калия в ДМФА. Полученный сложный эфир 1-2 может быть алкилирован галогенидом, таким как йодид (R1I), с использованием основания, такого как гексаметилдисилазид лития (LHMDS), с получением алкилированного продукта 1-3 в виде смеси цис и транс диастереомеров (в соотношении 4:1). Небольшое количество транс диастериомера может быть удалено кристаллизацией с последующим гидролизом сложного эфира до кислоты. Полученная чистая в энантиомерном отношении кислота 1-4 может быть подвергнута гидрированию с использованием катализатора, такого как Pd-C, с получением насыщенной карбоновой кислоты 1-5. Схема 1 Циклопентанкарбоновые кислоты формулы 2-5 могут быть получены с использованием методик,показанных на схеме 2. Коммерчески доступная 3-оксоциклопентанкарбоновая кислота 2-1 может быть преобразована в сложный эфир, такой как сложный метиловый эфир. Кетон полученного сложного эфира 2-2 может быть защищен путем обработки триметилортоформиатом в присутствии кислотного катализатора, такого как паратолуолсульфоновая кислота. Алкилирование полученного кеталя 2-3 алкилио- 10012649 дидом (R1I) может быть осуществлено с использованием основания, такого LHMDS. Гидролиз алкилированного сложного эфира 2-4 с использованием основания, такого как LiOH, NaOH или КОН, приводит к образованию карбоновых кислот формул 2-5. Схема 2 Пиперазиновые производные могут быть получены с использованием методик, проиллюстрированных на схеме 2. Связывание пиперазинового производного формулы 3-2 с производным йодбензола формулы 3-1 с использованием йодида меди(I) и фосфата калия приводит к образованию промежуточного продукта 3-3. Удаление Boc группы с использованием кислоты, такой как HCl, в диоксане или ТФУК,приводит к образованию пиперазиновых производных формулы 3-4. Схема 3 Альтернативно, пиперазиновые производные (4-3) могут быть получены путем замещения 2 хлорпиридинового или 2-хлорпиримидинового производного формулы 4-1 пиперазиновым производным формулы 4-2. Схема 4 Альтернативно, пиперазиновые производные могут быть получены с использованием последовательности реакций, показанной на схеме 5. Коммерчески доступный 3,5-дибромпиридин 5-1 может быть превращен в 3-бром-5-иодпиридин 5-2 путем обработки бромидом изопропилмагния и йодом. Связывание полученного йодпроизводного с пиперазиновым производным формулы 3-2 может быть осуществлено с использованием йодида меди(I) и фосфата калия. После преобразования содержащего полученного производного 5-3 в йодсодержащее производное с использованием бромида изопропилмагния и йода,указанный йод в производном может быть заменен трифторметилом путем обработки Me3SiCF3/CuI/KF/ДМФА с получением трифторметилпиридинового производного формулы 5-5. Удаление Вос группы с использованием кислоты, такой как HCl, в диоксане или ТФУК, приводит к получению пиперазиновых производных формулы 5-6. Схема 5 Пиперидиновые или тетрагидропиридиновые производные могут быть синтезированы по методике схемы 6. Введение лития в бром-или йодобензольное производное формулы 6-1 с использованием алкиллития, такого как н-бутиллитий или трет-бутиллитий, с последующим гашением реакции кетонным- 11012649 производным формулы 6-2, дает третичный спирт формулы 6-3. После дегидратации с использованием дегидратирующего агента, такого как тионилхлорид/пиридин, полученный олефин 6-4 может быть восстановлен путем гидрирования с использованием катализатора, такого как Pd на угле. Обработка 6-3, 6-4 и 6-5 кислотой, такой как HCl, в диоксане или ТФУК, дает соединение формул 6-6, 6-7 и 6-8. Схема 6 Альтернативно, пиперидиновые или тетрагидропиридиновые производные могут быть синтезированы по методике схемы 7. Коммерчески доступное 2-хлорпиридиновое или 2-хлорпирилидиновое производное формулы 4-1 может быть преобразовано в 2-бромпиридиновое производное формулы 7-1 путем обработки BrSiMe3. С использованием аналогичных методик, показанных на схеме 6, пиперидиновые или тетрагидропиридиновые производные формулы 7-5 и 7-6 могут быть получены из соединений формулы 7-1. Схема 7 Альтернативно, пиперидиновые или тетрагидропиридиновые производные могут быть синтезированы по методике схемы 8. 3-Нитро-5-трифторметилпиридин-2-ол может быть получен путем нитратной обработки коммерчески доступного 5-трифторметилпиридин-2-ола (8-1). После превращения гидроксигруппы 8-2 в хлорсодержащую группу, полученное хлорсодержащее производное 8-3 подвергают гидри- 12012649 рованию с использованием катализатора, такого как Pd на угле, с образованием 3-амино-5-трифторметилпиридина 8-4. Диазотирование 8-4 с использованием NaNO2/HBr в присутствии Cu(I)Br дает 3 бром-5-трифторметилпиридин 8-5. В соответствии с методиками, описанными на схеме 6, соединение 85 может быть преобразовано в пиперидиновые или тетрагидропиридиновые производные формул 8-9 и 8-10. Схема 8 Тетрагидропирановые производные могут быть получены по методике схемы 9 (где R8a означает,например, алкил). Коммерчески доступный 4-метокси-3,6-дигидро-2 Н-пиран 9-1 может быть преобразован в 4,4-диметокситетрагидро-2 Н-пиран-2-ол путем обработки м-хлорбензойной кислотой в метаноле. Алкилирование 9-2 алкилгалогенидом с использованием NaH позволяет получить промежуточный триалкоксипродукт 9-3. Обработка 9-3 с использованием кислоты, такой как водный раствор HCl, дает кетонные продукты формулы 9-4. Кетон 9-4 может быть преобразован в амин формулы 9-6 путем восстановительного аминирования с использованием аминодифенилметана с последующим гидрированием. Схема 9 Конечные соединения формы I могут быть получены согласно методике, описанной на схеме 10. Карбоновая кислота формулы 1-5 может быть конденсирована с амином формулы 10-1 с использованием стандартного агента, применяемого для образования амида, такого как ВОР или PyBrop (связывающий агент). После удаления Boc группы с помощью кислоты, такой как HCl или ТФУК, полученный амин 103 подвергают восстановительному аминированию кетоном формулы 10-4 с использованием восстановителя, такого как триацетоксиборгидрид натрия, с образованием конечных соединения формулы 10-5. Схема 10- 13012649 Альтернативно, соединения по настоящему изобретению могут быть обработаны в соответствии с методикой схемы 11. Связывание карбоновой кислоты формулы 2-5 с амином формулы 10-1 при использовании стандартного метода образования амида, позволяет получить амид формулы 11-1. После преобразования кеталя в кетон с использованием водного раствора кислоты, далее восстановительного аминирования полученного кетона 11-2 амином формулы 11-3 с использованием восстановителя, такого как триацетоксиборгидрид натрия, получают соединение формулы 11-4. Схема 11 Методы В некоторых вариантах осуществления соединения настоящего изобретения могут модулировать активность одного или нескольких хемокиновых рецепторов. Термин модулирует относится к способности повышать или снижать активность рецептора. Соответственно, соединения по настоящему изобретению могут быть полезны в способах модуляции хемокиновых рецепторов через взаимодействие рецептора с одним или несколькими соединениями или композициями, приведенными в настоящем описании. В некоторых вариантах осуществления соединения по настоящему изобретению могут действовать в качестве ингибиторов хемокиновых рецепторов. В других вариантах осуществления соединения по настоящему изобретению могут быть полезны для модуляции активности хемокинового рецептора у индивидуума, при наличии потребности в модуляции рецептора, путем введения модулирующего количества соединения формулы I. Хемокиновые рецепторы, с которыми соединения по настоящему изобретению связываются и/или которые они модулируют, включают любой хемокиновый рецептор. В некоторых вариантах осуществления хемокиновые рецепторы относятся к СС семейству хемокиновых рецепторов и включают, например,CCR1, CCR2, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8 и CCR10. В некоторых вариантах осуществления хемокиновый рецептор представляет собой CCR2. В некоторых вариантах осуществления хемокиновый рецептор представляет собой CCR5. В некоторых вариантах осуществления хемокиновый рецептор связывает и модулирует активность и CCR2, иCCR5. Соединения по настоящему изобретению могут действовать селективно. Термин селективно относится к соединению, которое связывает или ингибирует хемокиновый рецептор с большей аффинностью или эффективностью, соответственно, по сравнению по меньшей мере с одним другим хемокиновым рецептором. Соединения по настоящему изобретению могут обладать двойной ингибирующей или связывающей активностью для CCR2 и CCR5, иными словами, соединения по настоящему изобретению могут связывать и ингибировать как CCR2, так и CCR5 с большей аффинностью или эффективностью, соответственно, чем любые другие хемокиновые рецепторы, такие как CCR1, CCR3, CCR4, CCR6, CCR7, CCR8 иCCR10. В некоторых вариантах осуществления соединения по настоящему изобретению обладают связывающей или ингибирующей селективностью в отношении CCR2 и CCR5 в сравнении с любым другим хемокиновым рецептором. Селективность может быть по меньшей мере примерно 10-кратной, по меньшей мере примерно 20-кратной, по меньшей мере примерно 50-кратной, по меньшей мере примерно 100 кратной, по меньшей мере примерно 200-кратной, по меньшей мере примерно 500-кратной или по меньшей мере примерно 1000-кратной. Аффинность связывания и эффективность ингибирования соединения может быть измерена с использованием традиционных для данной области методик, например, в соответствии с приведенными в настоящем описании анализами. Настоящее изобретение также относится к способам лечения заболеваний или расстройств, ассоциированных с хемокиновым рецептором, у индивидуума (например, у пациента) путем введения индивидууму, нуждающемуся в таком лечении, терапевтически эффективного количества или дозы соединения по настоящему изобретению или его фармацевтической композиции. Заболевания, ассоциированные с хемокиновым рецептором, могут включать любые заболевания, расстройства или состояния, которые- 14012649 прямо или косвенно связаны с экспрессией или активностью хемокинового рецептора. Заболевание, ассоциированное с хемокиновым рецептором, может также включать любое заболевание, расстройство или состояние, которое может быть предотвращено, ослаблено или излечено путем модуляции активности хемокинового рецептора. Заболевание, ассоциированное с хемокиновым рецептором, может также включать любое заболевание, расстройство или состояние, которое характеризуется связыванием инфекционного агента, такого как вирус или вирусный белок, с хемокиновым рецептором. В некоторых вариантах осуществления заболевание, ассоциированное с хемокиновым рецептором, представляет собой CCR5 ассоциированное заболевание, такое как ВИЧ инфекция. Примеры заболеваний, расстройств и состояний, ассоциированных с хемокиновым рецептором,включают воспаление и воспалительные заболевания, иммунные расстройства, рак и вирусные инфекции. Примеры воспалительных заболевания включают заболевания, характеризующиеся наличием воспалительного компонента, такие как астма, сезонный и хронический аллергический ринит, синусит,конъюнктивит, возрастная макулярная дегенерация, пищевая аллергия, скомброидное отравление, псориаз, крапивница, зуд, экзема, воспалительная болезнь кишечника, тромботическая болезнь, средний отит, цирроз печени, болезнь сердца, болезнь Альцгеймера, сепсис, рестеноз, атеросклероз, рассеянный склероз, болезнь Крона, язвенный колит, аллергическое заболевание легких, фиброз легкого, вызванный введением лекарственных средств, хроническое обструктивное заболевание легких (ХОЗЛ), ревматоидный артрит, нефрит, язвенный колит, атопический дерматит, инсульт, острое повреждение нервов, саркоидоз, гепатит, эндометриоз, невропатическая боль, гиперчувствительная пневмония, эозинофильная пневмония, реакция гиперчувствительности задержанного типа, интерстициальная болезнь легкого(ИБЛ) (например, идиопатический легочный фиброз или ИБЛ, ассоциированная с ревматоидным артритом, системная красная волчанка, анкилозирующий спондилит, системный склероз, синдром Шегрена,полимиозит или дерматомиозит), болезни глаз (например, нейродегенерация сетчатки, хороидальная нейроваскуляризация и т.п.) и т.п. Примеры иммунных расстройств включают ревматоидный артрит,псориатический артрит, системную красную волчанку, тяжелую псевдопаралитическую миастению, начало юношеского диабета, гломерулонефрит, аутоиммунный тиреоидит, реакцию отторжения трансплантата органа, включающую отторжение реакцию аллотрансплантата и болезнь трансплантат против хозяина. Примеры рака включают рак молочной железы, рак яичника, множественную миелому и другие виды, который характеризуется инфильтрацией макрофагов (например, макрофагов, ассоциированных с опухолью, ТАМ) в опухоли или пораженные заболеванием ткани. Примеры вирусных инфекции включают инфекцию вирусом герпеса, ВИЧ инфекцию или СПИД. Как использовано в настоящем описании, термин контактирование означает объединение вместе указанных фрагментов в системе in vitro или в системе in vivo. Например, приведение в контакт хемокинового рецептора с соединением по настоящему изобретению включает введение соединения по настоящему изобретению индивидууму или пациенту, такому как человек, содержащему хемокиновый рецептор, а также, например, встраивание соединения по настоящему изобретению в образец, содержащий клеточный или очищенный препарат, включающий хемокиновый рецептор. Как использовано в настоящем описании, термины индивидуум или пациент, используемые взаимозаменяемо, относятся к любому животному, включая млекопитающих, предпочтительно мышей,крыс, других представителей грызунов, кроликов, собак, кошек, свиней, представителей крупного рогатого скота, овец, лошадей или приматов, и наиболее предпочтительно, людей. Как использовано в настоящем описании, фраза терапевтически эффективное количество относится к количеству активного соединения или фармацевтического агента, которые проявляют биологический или медицинский ответ, необходимый для данной ткани, системы, животного, индивидуума или человека, с точки зрения исследователя, ветеринара, лечащего врача или другого медицинского специалиста, который включает один или несколько из следующих указанных событий:(1) предупреждение заболевания, например предупреждение развития заболевания, состояния или расстройства у индивидуума, который предрасположен к заболеванию, состоянию или расстройству, но у которого еще не начались или не выявлены патология или симптоматика заболевания (неограничивающие примеры включают аллергическое заболевание легких, фиброз легкого, вызванный лекарственными препаратами, хроническое обструктивное заболевание легких (ХОЗЛ), реакция трансплантат против хозяина и/или реакция отторжения аллотрансплантата после трансплантации, или предупреждение развития аллергических реакций, таких как атопический дерматит или сезонный или хронический аллергический ринит);(2) подавление заболевания, например подавление развития заболевания, состояния или расстройства у индивидуума, у которого уже начались или проявлены патология или симптоматика заболевания,состояния или расстройства (т.е., остановка дальнейшего развития патологии и/или симптоматики), такое как подавление аутоиммунной реакции при аллергической заболевании легких, при фиброзе легкого,вызванном лекарственными препаратами, при хроническом обструктивном заболевании легких (ХОЗЛ),ревматоидном артрите, волчанке или псориазе, или ингибирование роста опухоли, или стабилизация вирусной нагрузки в случае вирусной инфекции; и(3) ослабление заболевания, например ослабление заболевания, состояния или расстройства у инди- 15012649 видуума, у которого имеются или проявлены патология или симптоматика заболевания, состояния или расстройства (т.е., реверсирование патологии и/или симптоматики), такое как ослабление аутоиммунной реакции при аллергическом заболевании легких, при фиброзе легкого, вызванного лекарственными препаратами, при хроническом обструктивном заболевании легких (ХОЗЛ), ревматоидном артрите, волчанке или псориазе, или уменьшение опухоли, ассоциированной с раком, или снижение вирусной нагрузки в случае вирусной инфекции. Один или несколько дополнительных фармацевтических агентов, таких как, например, антитела,противовоспалительные средства, иммуносупрессоры, химиотерапевтические средства, могут использоваться в сочетании с соединениями по настоящему изобретению для лечения заболеваний, состояний или расстройства, ассоциированных с хемокиновым рецептором. Такие агенты могут быть объединены с соединением по настоящему изобретению с получением одной дозированной формы или указанные агенты могут вводиться одновременно или последовательно в виде разных дозированных форм. Один или несколько дополнительных фармацевтических агентов, таких как, например, противовирусные средства, антитела, противовоспалительные средства, средства, способствующие секреции инсулина, и сенсибилизаторы их секреции, агенты, модулирующие уровень липидов в сыворотке крови и липидных носителей, и/или иммуносупрессоры могут использоваться в сочетании с соединениями по настоящему изобретению для лечения заболевания, состояния или расстройства, ассоциированного с хемокиновым рецептором. Указанные агенты могут быть объединены с соединениями по настоящему изобретению в виде одной или непрерывно вводимой дозированной формы или такие агенты могут вводиться одновременно или последовательно в виде отдельных дозированных форм. Подходящие антивирусные агенты для применения в сочетании с соединениями по настоящему изобретению могут включать нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы(NRTI), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы протеазы и другие противовирусные средства. Подходящие антивирусные агенты для применения в сочетании с соединениями по настоящему изобретению могут включать нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы(NRTI), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы протеазы, ингибиторы вхождения вируса в клетку, ингибиторы слияния, ингибиторы созревания и другие противовирусные средства. Примеры подходящих NRTI включают зидовудин (AZT), диданозин (ddl), залцитабин (ddC), ставудин (d4T), ламивудин (ЗТС), абакавир (1592U89), адефовира дипивоксил [bis(РОМ)-РМЕА], лобукавир(BMS-180194), ВСН-10652, эмитрицитабин [(-)-FTC], бета-L-FD4 (также называемый как бета-L-D4 С и обозначаемый как бета-L-2',3'-диклеокси-5-фторцитидин), DAPD, -)-бета-D-2,6-диаминопурина диоксолан) и лоденозин (FddA). Типичные подходящие NNRTI включают невирапин (BI-RG-587), делавирадин (ВНАР, U-90152),эфавиренц (DMP-266), PNU-142721, AG-1549, МКС-442, (1-(этоксиметил)-5-(1-метилэтил)-6-(фенилметил)-(2,4-(1 Н,3 Н)пиримидиндион) и (+)-каланолид A (NSC-675451) и В. Типичные подходящие ингибиторы протеазы включают саквинавир (Ro 31-8959), ритонавир(АВТ 538), индинавир (МК-639), нелфинавир (AG-1343), ампренавир (141W94), лазинавир (BMS-234475),DMP-450, BMS-2322623, АВТ-378 и AG-1549. Другие противовирусные агенты включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафузид,энфувиртид, С-34, циклотриазадисульфонамид CADA, РА-457 и Yissum Project No. 11607. В некоторых вариантах осуществления противовоспалительные или анальгетические агенты для применения в сочетании с соединениями по настоящему изобретению могут включать, например, опиатный агонист, ингибитор липоксигеназы, такой как ингибитор 5-липоксигеназы, ингибитор циклооксигеназы, такой как ингибитор циклооксигеназы-2, ингибитор интерлейкина, такой как ингибитор интерлейкина-1, ингибитор TNF, такой как инфликсимаб, этанерцепт или адалимумаб, антагонист NNMA, ингибитор оксида азота или ингибитор синтеза оксида азота, нестероидное противовоспалительное средство или противовоспалительный агент, подавляющий цитокин, например, такой как ацетаминофен, аспирин,кодеин, фентанил, ибупрофен, индометацин, кетодолак, морфин, напроксен, фенацетин, пироксикам,стероидный анальгетик, суфентанил, сунлиндак, тенидап и т.п. Аналогично, соединения по настоящему изобретению могут вводиться вместе со средством, облегчающим боль, с потенцирующим средством,таким как кофеин, Н 2-антагонист, симетикон, гидроксид алюминия или магния; средством, снижающим застойные явления, таким как фенилэфрин, фенилпропаноламин, псевдофедрин, оксиметазолин, эфинефрин, нафазолин, ксилометазолин, пропилгекседфин или лево-дезоксиэфедрин; противокашлевым средством, таким как кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; с диуретиком и седативным или неседативным антигистаминным средством. В некоторых вариантах осуществления фармацевтические агенты для применения в сочетании с соединениями по настоящему изобретению, могут включать, но без ограничения, (а) антагонисты VLA-4,такие как описано в US 5510332, WO95/15973, WO96/01644, WO96/06108, WO96/20216, WO96/229661,WO96/31206, WO96/4078, WO97/030941, WO97/022897, WO98/426567, WO98/53814, WO98/53817,WO98/538185, WO98/54207 и WO98/58902; (b) стероидные соединения, такие как беклометазон, метил- 16012649 преднизолон, бетарнетазон, преднизон, дексаметазон и гидрокортизон; (с) иммуносупрессоры, такие как циклоспорин, такролимус, рапарницин и другие иммуносупрессоры типа FK506; (d) антигистаминные агенты (антагонисты HI-гистамина), такие как бромофенирамин, хлорфенирамин, дексхлорфенирамин,трипролидин, клемастин, дифенгидрамин, дифенилпиралин, трипеленнамин, гидроксизин, метдилазин,прометазин, тримепразин, азатадин, ципрогептадин, антазолин, фенирамина пириларнин, астернизол,терфенадин, лоратадин, цетиризин, фексофенадин, дезеарбоэтоксилоратадин и т.п.; (е) нестероидные противоастматические средства, такие как тербуталин, метапротеренол, фенотерол, изоэтанин, албутерол, битолтерол, пирбутерол, теофиллин, натрийкромолин, атропин, бромид ипратропия, лейкотриеновые антагонисты (например, зафирлукаст, монтелукаст, пранлукаст, иралукаст, побилукаст, SKB-106 203), ингибиторы биосинтеза лейкотриена (например, зилейтон, BAY-1005); (f) нестероидные противовоспалительные средства (NSAID), такие как производные пропионовой кислоты (например, аминопрофен, беноксапрофен, буклоксиновая кислота, карпрофен, фенбуфен, фенопрофен, флупрофен, флурбипрофен, ибупрофен, индопрофен, кетопрофен, миропрофен, напркосен, оксапрозин, пирпрофен, пранопрофен, супрофен, тиапрофеновая кислота и тиоксапрофен), производные уксусной кислоты (например,индометацин, ацернетацин, алклофенак, клиданак, диклофенак, фенклофенак, фенклозиновая кислота,фентиазак, фурофенак, ибуфенак, изоксепак, окспинак, сулиндак, тиопинак, толметин, зидометацин и зомепирак), производные фенарниновой кислоты (например, флуфенарниновая кислота, меклофенаминовая кислота, рнефенаминовая кислота, нифлуминовая кислота и толфенарниновая кислота), производные бифениларбоновой кислоты (дифлунизал и флуфенизал), оксикарны (изоксикарн, пироксикам, судоксикам и теноксикам), салицилаты (ацетилсалициловая кислота, сульфасалазин) и пиразолоны (апазон,безпиперилон, фепразон, мофебутазон, оксифенбутазон, фенилбутазон); (g) ингибиторы циклооксигеназы-2 (СОХ-2); (h) ингибиторы фосфодиэстеразы типа IV (PDE-IV); (i) другие антагонисты хемокиновых рецепторов, в особенности CXCR-4, CCR1, CCR2, CCR3 и CCR5; (j) средства, снижающие уровень холестерина, такие как ингибиторы HMG-CoA редуктазы (ловастатин, сирривастатин и правастатин, флувастатин, аторвастатин и другие статины), секвестранты (холестирамин и колестипол), никотиновая кислота, производные фенофибриновой кислоты (гемфиброзил, клофибрат, фенофибрат и бензафибрат) и пробукол; (k) противовоспалительные биологические агенты, такие как агенты, используемые в анти-TNF терапии, анти-IL-1 рецептор, CTLA-4Ig, анти-CD20, и анти-VLA4 антитела; (1) антидиабетические агенты, такие как инсулин, сульфонилмочевины, бигуаниды (метформин), ингибиторы U-гликозиды (акарбоза) и орлитазоны (троглитазон и пиоглитазон); (m) препараты бета-интерферона (интерферон бета-lo.,интерферон бета-1 Р); (n) другие соединения, такие как аминосалициловые кислоты, антиметаболиты,такие как азатиоприн и 6-меркаптопурин, и цитотоксические химиотерапевтические противораковые агенты. Массовое соотношение соединения по настоящему изобретению ко второму активному ингредиенту может варьировать и зависит от эффективной дозы каждого ингредиента. Например, антагонист CCR2 и/или CCR5 может применяться в сочетании с противовоспалительным фармацевтическим агентом при лечении воспаления, метаболического заболевания, аутоиммунного заболевания, рака или вирусной инфекции с целью улучшения ответа организма на лечение, в сравнении с его ответом в случае применения одного терапевтического агента, но без усиления токсических эффектов. Дополнительные или синергические эффекты являются желательным результатом сочетания антагониста CCR2 и/или CCR5 настоящего изобретения с дополнительным агентом. Кроме того, резистентность раковых клеток к агентам, таким как дексаметазон, может реверсировать при лечении с применением антагониста CCR2 и/или CCR5 по настоящему изобретению. Фармацевтические препараты и дозированные формы При применении в качестве фармацевтических средств соединения по настоящему изобретению могут вводиться в виде фармацевтических композиций. Указанные композиции могут быть получены по способу, известному в фармацевтике, и могут вводиться с использованием множества способов, в зависимости от того, какое лечение желательно: местное или системное, а также, какая область подлежит лечению. Введение может быть местным (включая введение в глаза и в слизистые мембраны, включая интраназальную, вагинальную или ректальную доставку), через легкие (например, путем ингаляции или инсуффляции порошков или аэрозолей, включая применение небулайзера; интратекальным, интраназальным, эпидермальным и чрескожным введением), пероральное или парентеральное введение. Парентеральное введение включает внутривенную, внутриартериальную, подкожную, внутрибрюшинную,внутримышечную инъекцию или инфузию; а также внутричерепное, например интратекальное или интравентикулярное введение. Парентеральное введение может проводиться в виде однократной дозы болюса или может представлять собой, например, непрерывную перфузию, осуществляемую с помощью насоса. Фармацевтические композиции и препараты для местного введения могут включать чрескожные пластыри, мази, лосьоны, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. При этом могут быть необходимы или желательны традиционные фармацевтические носители, водные, порошковые или масляные основы, загустители и другие аналогичные компоненты. Могут быть также полезны покрытые оболочкой презервативы, перчатки и т.п. Настоящее изобретение также включает фармацевтические композиции, которые содержат в качестве активного ингредиента один или несколько соединений формулы I, указанной выше, в сочетании с- 17012649 одним или несколькими фармацевтически приемлемыми носителями. В процессе изготовления композиций по настоящему изобретению активный ингредиент обычно смешивают с эксципиентом, разбавляют эксципиентом или вводят в такой носитель с получением, например, капсулы, пакетика, бумажной формы или формы, представленной в виде другого контейнера. В том случае, когда эксципиент служит в качестве разбавителя, он может быть представлен твердым, полутвердым или жидким материалом, который действует в качестве наполнителя, носителя или среды для активного ингредиента. Так, композиции могут иметь форму таблеток, пилюль, порошков, леденцов, пакетиков, крахмальных капсул, эликсиров,суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердого материала или в жидкой среде),мазей, содержащих, например, до 10 мас.% активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъецируемых растворов и стерильных упакованных порошков. При изготовлении композиции активное соединение может быть размолото с образованием частиц соответствующего размера перед объединением его с другими ингредиентами. Если активное соединение является, по существу, нерастворимым, его можно измельчить до образования частиц с размером менее чем 200 меш. Если активное соединение является, по существу, водорастворимым, то размер частиц может быть откорректирован путем размалывания с достижением, по существу, однородного распределения его по размеру в композиции, например, с размером примерно 40 меш. Некоторые примеры подходящих эксципиентов включают лактозу, декстрозу, сахарозу, сорбит,маннит, крахмалы, аравийскую камедь, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, сироп и метилцеллюлозу. Композиции могут дополнительно включать: лубриканты, такие как тальк, стеарат магния и минеральное масло; смачивающие агенты; эмульгаторы и средства, способствующие суспендированию; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители; а также вкусовые вещества. Композиции по настоящему изобретению могут быть изготовлены таким образом, чтобы обеспечить быстрое, пролонгированное или задержанное высвобождение активного ингредиента после введения пациенту при применении для этого методов, известных в данной области. Соединения могут быть изготовлены с получением единичной дозированной формы, в которой каждая дозировка содержит примерно от 5 до примерно 1000 мг (1 г), чаще примерно от 100 до примерно 500 мг активного ингредиента. Термин единичная дозированная форма относится к физически дискретным единицам, подходящим в качестве единичных дозировок для принимающего их человека и других млекопитающих, где каждая единица содержит заданное количество активного материала, рассчитанного таким образом, чтобы обеспечивать желаемый терапевтический эффект, в сочетании с подходящим фармацевтическим эксципиентом. В некоторых вариантах осуществления соединения или композиции по настоящему изобретению содержат примерно от 5 до примерно 50 мг активного ингредиента. При этом для среднего специалиста в данной области очевидно, что в указанный диапазон входят соединения или композиции, содержащие примерно от 5 до примерно 10, примерно от 10 до примерно 15, примерно от 15 до примерно 20, примерно от 20 до примерно 25, примерно от 25 до примерно 30, примерно от 30 до примерно 35, примерно от 35 до примерно 40, примерно от 40 до примерно 45, примерно от 45 до примерно 50 мг активного ингредиента. В некоторых вариантах осуществления соединения или композиции по настоящему изобретению содержат примерно от 50 до примерно 500 мг активного ингредиента. Средний специалист в данной области понимает, что это утверждение охватывает соединения или композиции, содержащие примерно от 50 до примерно 75, примерно от 75 до примерно 100, примерно от 100 до примерно 125, примерно от 125 до примерно 150, примерно от 150 до примерно 175, примерно от 175 до примерно 200, примерно от 200 до примерно 225, примерно от 225 до примерно 250, примерно от 250 до примерно 275, примерно от 275 до примерно 300, примерно от 300 до примерно 325, примерно от 325 до примерно 350, примерно от 350 до примерно 375, примерно от 375 до примерно 400, примерно от 400 до примерно 425, примерно от 425 до примерно 450, примерно от 450 до примерно 475, примерно от 475 до примерно 500 мг активного ингредиента. В некоторых вариантах осуществления соединения или композиции по настоящему изобретению содержат примерно от 500 до примерно 1000 мг активного ингредиента. Средний специалист в данной области понимает, что это утверждение охватывает соединения или композиции, содержащие примерно от 500 до примерно 550, примерно от 550 до примерно 600, примерно от 600 до примерно 650, примерно от 650 до примерно 700, примерно от 700 до примерно 750, примерно от 750 до примерно 800, примерно от 800 до примерно 850, примерно от 850 до примерно 900, примерно от 900 до примерно 950, примерно от 950 до примерно 1000 мг активного ингредиента. Активное соединение может быть эффективным в широком диапазоне дозировок и в основном вводится в фармацевтически эффективном количестве. Следует однако понимать, что количество фактически вводимого соединения обычно определяется лечащим врачом, в соответствии с имеющимися обстоятельствами, включающими состояние, подлежащее лечению, выбранный путь введения, фактическая природа вводимого соединения, возраст, массу и реакцию конкретного пациента, тяжесть симптомов у- 18012649 пациента и т.п. Для получения твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим эксципиентом с образованием твердой предварительной композиции, содержащей гомогенную смесь соединения по настоящему изобретению. Утверждение о том, что предварительные композиции являются гомогенными, следует понимать так, что активный ингредиент обычно диспергирован равномерно по композиции, так что композиция может быть легко разделена на равные эффективные дозированные единичные формы, такие как таблетки, пилюли и капсулы. Указанную твердую предварительную композицию затем разделяют на единичные дозированные формы описанного выше типа, которые содержат, например, от 0,1 до примерно 1000 мг активного ингредиента настоящего изобретения. Таблетки или пилюли по настоящему изобретению могут быть покрыты оболочкой или иным образом изготовлены с получением дозированной формы, создающей преимущество пролонгированного действия. Например, таблетка или пилюля может включать внутренний компонент дозированной формы и внешний компонент дозированной формы, причем последний находится в форме оболочки, охватывающей первый компонент. Оба компонента могут быть разделены энтеросолюбильным слоем, который служит для того, чтобы противостоять разложению внутреннего компонента в желудке, позволяя ему проходить в интактном виде в двенадцатиперстную кишку или задержать его высвобождение. Могут использоваться различные материалы для получения таких энтеросолюбильных слоев или покрытий,включающих большое число полимерных кислот и смесей полимерных кислот с такими материалами,как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы, в состав которых соединения и композиции по настоящему изобретению могут быть включены для введения через рот или путем инъекции, включают водные растворы, сиропы с соответствующими вкусовыми добавками, водные или масляные суспензии и соответствующим образом ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло, а также эликсиры и аналогичные фармацевтические наполнители. Композиции для ингаляции или инсуффляции включают растворы или суспензии в фармацевтически приемлемых водных или органических растворителях или их смесях и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые эксципиенты, описанные выше. В некоторых вариантах осуществления композиции вводят перорально или в назальном, респираторном режиме для достижения локального или системного эффекта. Композиции могут быть распылены с помощью небулайзера, в который включаются инертные газы. Распыляемые небулайзером растворы могут вдыхаться непосредственно из небулайзера или такое распыляющее устройство может быть подсоединено к устройству возле лица или к устройству, усиливающему дыхание. Растворы, суспензии или порошковые композиции могут вводиться перорально или назально из устройств, которые доставляют композицию соответствующим способом. Количество соединения или композиции, вводимое пациенту, будет варьировать в зависимости от того, что вводится, какова цель введения - для профилактики или для лечения, а также от состояния пациента, способа введения и т.п. В случае терапевтического применения композиции могут вводиться пациенту, уже имеющему заболевание, в количестве, достаточном для излечивания или, по меньшей мере, для частичной остановки симптомов заболевания и его осложнений. При этом эффективные дозы будут зависеть от природы заболевания, подлежащего лечению, а также от заключения лечащего врача,который учитывает такие факторы, как тяжесть заболевания, масса, возраст и состояние пациента, и т.п. Композиции, вводимые пациенту, могут иметь форму фармацевтических композиций, описанных выше. Указанные композиции могут быть простерилизованы с использованием традиционных методов стерилизации или могут быть подвергнуты стерильному фильтрованию. Водные растворы могут быть упакованы для последующего непосредственного применения или могут быть лиофилизированы, и перед введением лиофилизированный препарат объединяют со стерильным водным носителем. рН препаратов данных соединений может составлять от 3 до 11, более предпочтительно от 5 до 9 и наиболее предпочтительно от 7 до 8. Следует понимать, что использование некоторых указанных эксципиентов, носителей или стабилизаторов будет приводить к образованию фармацевтических солей. Фармацевтическая дозировка соединений по настоящему изобретению будет варьировать в зависимости, например, от конкретного варианта применения, в рамках которого проводится лечение, от способа введения соединения, состояния здоровья и природы заболевания пациента, а также от заключения лечащего врача. Пропорция или концентрация соединения по настоящему изобретению в фармацевтической композиции может варьировать, в зависимости от множества факторов, включающих дозировку,химические характеристики (например, гидрофобность) и способ введения. Например, соединения по настоящему изобретению могут вводиться в водном физиологически совместимом буферном растворе,содержащем примерно от 0,1 до примерно 10% (масса/объем) соединения, в случае парентерального введения. Некоторые конкретные диапазоны дозировок включают дозы примерно от 1 мкг/мг до примерно 1 г/кг массы тела в день. В некоторых вариантах осуществления диапазон дозировок составляет примерно от 0,01 до примерно 100 мг/кг массы тела в день. Как очевидно, дозировка зависит от таким переменных,как тип и степень прогрессирования заболевания или расстройства, общее состояние здоровья конкрет- 19012649 ного больного, относительная биологическая эффективность выбранного соединения, композиция эксципиента и способ введения. Эффективные дозы могут быть определены экстраполяцией по кривой доза-ответ, полученной в исследованиях in vitro или в исследованиях на моделях тест-систем животных. Соединения по настоящему изобретению могут быть также изготовлены в сочетании с одним или несколькими дополнительными активными ингредиентами, которые могут включать любой фармацевтический агент, такой как антитела, иммуносупрессоры, противовоспалительные средства, химиотерапевтические средства, агенты, снижающие уровень липидов, агенты, повышающие уровень ЛПВП, средства,способствующие секреции инсулина или сенсибилизаторы его секреции, лекарственные средства, используемые при лечении ревматоидного артрита и т.п. Режим лечения ревматоидного артрита (РА) Пациенты с ревматоидным артритом (РА), которые подвергаются агрессивному лечению агентами,модифицирующими данное заболевание (метотрексат, противомалярийные средства, средства, содержащие золото, пеницилламин, сульфасалазин, дапсон, лефлунамид или биологические агенты), могут позволить достичь различных степеней контроля заболевания, включая полную ремиссию. Указанные клинические ответы ассоциированы с улучшением, выражаемым по стандартизованной шкале оценки активности заболевания, в частности, по критериям ACR, которые включают такие факторы, как боль,функция и число болезненных суставов, число набухших суставов, общая оценка состояния пациента,общая оценка лечащего врача, результаты лабораторного исследования для оценки степени воспаления(СРВ и СОЭ) и радиологическая оценка структурного повреждения суставов. Имеющиеся в настоящее время средства модификации заболевания (DMARD) требуют длительного введения для поддержания оптимальных результатов. Хроническое введение доз указанных агентов связано со значительной токсичностью и ухудшает защиту организма хозяина. Дополнительно, пациенты часто становятся устойчивыми к проводимой терапии, и в этом случае требуется переключение на альтернативный режим. По указанным причинам новая эффективная терапия, которая может стать заменой стандартной терапии с применением DMARD, будет клинически важным подходом. Пациенты, демонстрирующие выраженную реакцию на анти-TNF терапию (инфликсимаб, этанерцепт, адалимумаб), анти-IL-1 терапию (кинарет) или другие противоревматические средства, модифицирующие заболевание (DMARD), включают, но без ограничения, метотрексат, циклоспорин, соли золота,противомалярийные средства, пеницилламин или лефлунамид, те пациенты, у которых отмечается клиническая ремиссия заболевания, могут подвергаться лечению веществами, которые ингибируют экспрессию и/или активность CCR2, к числу которых относятся, например, нуклеиновые кислоты (в частности,антисмысловые молекулы или молекулы siPHK), белки (например, анти-CCR2 антитела), малые молекулы ингибиторов (например, приводимые в настоящем описании соединения и другие ингибиторы хемокиновых рецепторов, известные в данной области). В некоторых вариантах осуществления вещество, которое ингибирует экспрессию и/или активностьCCR2, представляют собой малую молекулу ингибитора CCR2 (или его антагониста). Антагонист CCR2 может вводиться перорально ежедневно или дважды в день в дозе, не превышающей примерно 500 мг в день. В этом случае у пациентов может быть отменена полностью проводившаяся ранее терапия или ее дозировка может быть снижена, так что они будут поддерживаться на лечении антагонистами CCR2. Лечение пациентов с использованием сочетания антагонистов CCR2 и других препаратов в рамках ранее проводившейся терапии может осуществляться в течение примерно от одного до примерно двух дней, перед отменой или снижением дозы DMARD, после чего будет продолжаться с использованием антагониста CCR2. Преимущества замены традиционных препаратов DMARD антагонистами CCR2 многочисленны. Традиционные препараты DMARD оказывают серьезные кумулятивные побочные эффекты, ограничивающие вводимые дозы, и самыми распространенными являются поражение печени и иммуносупрессорное действие. Как ожидается, антагонизм в отношении CCR2 будет улучшать профиль безопасности в течение длительного периода времени, без сопутствующего иммуносупрессорного действия, свойственного традиционным препаратам DMARD. Кроме того, известно, что период полувыведения биологических компонентов составляет обычно несколько дней или недель, что является проблемой в плане побочного действия. Ожидается, что период полувыведения биологически доступного при пероральном приеме антагониста CCR2 будет порядка нескольких часов, так что риск длительного воздействия лекарственного средства, в случае возникновения побочного действия, минимален в сравнении с биологическими агентами. Кроме того, имеющиеся в настоящее время биологически агенты (инфликсимаб, этанерцепт, адалимумаб, кинарет) обычно вводятся либо в/в, либо п/к, что требует участия врача или сам пациент должен осуществлять соответствующий самоконтроль. В иной случае может развиться инфузионная реакция или реакции в месте инъекции. Все они могут быть исключены при использовании перорально вводимого антагониста CCR2. Режим лечения диабета и инсулинрезистентности Диабет типа 2 представляет одну из ведущих причин заболеваемости и смертности в западных страна. У большинства пациентов заболевание характеризуется дисфункцией бета-клеток поджелудочной железы, сопровождаемой инсулинрезистентностью, развивающейся в печени и в периферических- 20012649 тканях. На основе первичных механизмов, ассоциированных с данным заболеванием, выделяются два основных класса пероральных терапевтических препаратов, применяемых для лечения диабета 2 типа: средства, способствующие секреции инсулина (сульфонилмочевины, такие как глибурид) и сенсибилизаторы инсулина (метформин и тиазолидиндионы, такие как розиглитазон). Было показано, что комбинированная терапия, воздействуя на оба механизма, позволяет контролировать метаболические дефекты,свойственные данному заболевания, и во многих случаях приводит к снижению потребности в экзогенном введении инсулина. Однако зачастую с течением времени инсулинрезистентность прогрессирует,что вызывает потребность в дальнейшем введении инсулина. Кроме того, как было показано, преддиабетическое состояние, называемое как метаболический синдром, характеризуется нарушенной толерантностью к глюкозе, в особенности в сочетании с ожирением. Большая часть пациентов, у которых развивается диабет 2 типа, характеризовались развитием инсулинрезистентности в сочетании с гипергликемией, которая наступала в том случае, когда данные пациенты не могли дальше поддерживать уровень гиперинсулинемии, необходимый для предупреждения нарушения глюкозного гомеостаза. Появление инсулинрезистентного компонента является высоко прогностическим фактором развития заболевания и ассоциируется с повышением риска развития диабета 2 типа, гипертензии и коронарной болезни сердца. Одним из сильнейших факторов, поддерживающих корреляцию нарушенной толерантности к глюкозе и прогрессирования от инсулинрезистентности к диабету 2 типа, является наличие центрального. ожирения. Большая часть пациентов 2 типа характеризуются ожирением, и ожирение само по себе ассоциируется с инсулинрезистентностью. Очевидно, что центральное ожирение является важнейшим фактором риска развития инсулинорезистентности, ведущей к диабету 2 типа, так что, по-видимому, висцеральный жир вовлекается в передачу сигналов, контролирующих развитие инсулинорезистентности и прогрессирование заболевания. Ожирение, в дополнение к секреции белковых факторов, индуцирует клеточный воспалительный ответ, в процессе которого макрофаги из костного мозга накапливаются в депо жировой ткани, превращаясь в макрофагов жировой ткани. Макрофаги жировой ткани накапливаются в жировой ткани пропорционально степени ожирения. Макрофаги, инфильтрующие ткань, представляют собой источник многих воспалительных цитокинов, которые, как было показано, индуцируют инсулинрезистентность в адипоцитах. Жировая ткань продуцирует МСР-1 пропорционально ожирению, и это позволяет полагать, что их сигнальная активность, осуществляемая через CCR2, также может играть важную роль в накоплении макрофагов в жировой ткани. Неизвестно, отвечает ли непосредственно взаимодействие МСР-1/CCR2 за рекрутинг моноцитов в жировую ткань, ведет ли сниженный рекрутинг макрофагов в жировую ткань непосредственно к снижению у людей продукции провоспалительных молекул и связана ли напрямую продукция провоспалительных молекул с инсулинрезистентностью. Пациенты, у которых отмечается инсулинрезистентность, либо преддиабетическая (нормогликемический уровень), либо диабетическая (гипергликемический уровень), могут подвергаться лечению веществами, которые ингибируют экспрессию и/или активность CCR2 и которые включают, например, нуклеиновые кислоты (например, антисмысловые молекулы или молекулы siPHK), белки (например, антиCCR2 антитела), малые молекулы ингибиторов (например, приводимые в настоящем описании соединения и другие известные в данной области ингибиторы хемокиновых рецепторов). В некоторых вариантах осуществления вещество, которое ингибируют экспрессию и/или активность CCR2, представляет собой малую молекулу ингибитора CCR2 (или его антагониста). Антагонист CCR2 может вводиться перорально ежедневно или дважды в день в дозе, не превышающей примерно 500 мг в день. У пациента может быть отменено введение или может быть снижена дозировка проводившейся ранее терапии, так что далее они могут поддерживаться на лечении с использованием антагонистов CCR2. Альтернативно, лечение пациентов с использованием антагониста CCR2 может применяться в дополнение к текущей терапии для повышения ее эффективности или для предупреждения дальнейшего прогрессирования инсулиновой зависимости. Преимущества замены или добавления к традиционным агентам антагонистов CCR2 многочисленны. Такие агенты могут быть полезны, например, для предотвращения прогрессирования от преддиабетического, инсулинрезистентного состояния к диабетическому состоянию. Такие агенты могут также снижать или заменять потребность в применении сенсибилизаторов инсулина, сопровождающихся свойственной им токсичностью. Такие агенты могут также снижать потребность в экзогенном введении инсулина или увеличивать период времени до возникновения такой потребности. Режим лечения атеросклероза Атеросклероз представляет собой состояние, характеризующееся отложением жировых веществ в стенках артерий. Бляшки включают такие отложения жирных веществ, как холестерин, отходы клеточного метаболизма, кальциевые и другие вещества, которые накапливаются во внутреннем выстилающем слое артерий. Бляшки могут расти и значительно снижать кровоток через артерию. Однако происходит более значительное повреждение, когда бляшка становится нестабильной и разрывается. Бляшки, которые разрываются, могут вызывать образование сгустков крови, которые, в свою очередь, могут блокировать кровоток или могут далее разрушаться и перемещаться в другие части организма. Если сгусток блокирует кровеносный сосуд, который питает сердце, он вызывает сердечный приступ. Если сгусток бло- 21012649 кирует кровеносный сосуд, который питает мозг, он вызывает мозговой удар. Атеросклероз представляет собой медленно протекающее сложное заболевание, которое обычно начинается в детском возрасте и часто прогрессирует по мере старения людей. Высокий уровень холестерина в крови является важным фактором риска коронарной болезни сердца. На основании уровня холестерина, как главного компонента бляшек, можно контролировать образование бляшек за счет снижения уровня циркулирующего холестерина или путем повышения холестерина, связанного с липопротеинами высокой плотности (ЛПВП). Уровень циркулирующего в крови холестерина может быть снижен, например, путем ингибирования его синтеза в печени или путем снижения его захвата из пищи. Такие лекарственные средства, которые действуют через указанные механизмы,могут включать лекарственные препараты, используемые для снижения высокого уровня холестерина: абсорбенты желчных кислот, ингибиторы синтеза липопротеина, ингибиторы синтеза холестерина и производные фибриновой кислоты. Уровень циркулирующего ЛПВП может быть дополнительно повышен путем введения, например, пробукола или высоких доз ниацина. Показано, что терапия, воздействующая на множественные механизмы, замедляет прогрессирование заболевания, в том числе его прогрессирование, ведущее к разрыву бляшек. Обычно атеросклероз сопровождается клеточной воспалительной реакцией, в процессе которой макрофаги из костного мозга накапливаются в жировых тяжах вдоль стенок сосудов, становясь пенными клетками. Пенные клетки представляют собой источник многочисленных воспалительных цитокинов,которые, как было показано, индуцируют прогрессирование бляшек и ферментов, которые могут способствовать дестабилизации бляшек. Пораженная атеросклеротическим бляшками ткань также продуцирует МСР-1, и можно полагать, что его активность, осуществляемая путем проведения сигнала через CCR2,также будет играть важную роль в накоплении макрофагов в виде пенных клеток в бляшках. Было показано, что CCR2-/- мыши имеют значительно сниженный уровень макрофагов в жировых полосах, образующихся в результате высокожирной диеты или генетического изменения в липидном метаболизме. Пациенты, у которых выявлено наличие высокого уровня циркулирующего в крови холестерина,низкое содержание ЛПВП или повышенный уровень в кровотоке СРБ, а также у которых различными методами визуализации показано наличие бляшек в сосудистых стенках или у которых выявлены другие показатели атеросклероза, могут подвергаться лечению веществом, которое ингибирует экспрессию и/или активность CCR2 и которое включает, например, нуклеиновые кислоты (например, антисмысловые молекулы или молекулы siPHK), белки (например, анти-CCR2 антитела), малые молекулы ингибиторов(например, соединения, приведенные в настоящем описании, и другие ингибиторы хемокиновых рецепторов, известные в данной области). В некоторых вариантах осуществления вещество, которое ингибирует экспрессию и/или активность CCR2, представляет собой малую молекулу ингибитора CCR2 (или антагониста), такое как соединение по настоящему изобретению. Антагонист CCR2 может вводиться перорально ежедневно или дважды в день в дозе, не превышающей примерно 500 мг в день. У пациента может быть отменен прием или может быть снижена дозировка текущей терапии и они могут поддерживаться на лечении с использованием антагониста CCR2. Альтернативно, лечение антагонистом CCR2 может проводиться в виде добавки к текущей терапии для усиления ее эффективности, например, для предотвращения прогрессирования роста бляшек, стабилизации бляшек, которые уже сформировались,или для индукции регрессии бляшек. Преимущества замены или добавления к традиционным агентам антагонистов CCR2 многочисленны. Такие агенты могут быть полезны, например, для предотвращения прогрессирования бляшек в направлении стадии нестабильности, ассоциированной с риском разрыва бляшек. Такие агенты могут также снижать или заменять потребность в использовании средств, модифицирующих холестерин, или лекарственных препаратов, повышающих ЛПВП, со свойственными им токсическими эффектами, которые включают, но без ограничения, гиперемию, повреждение печени и повреждение мышц, такое как миопатия. Такие агенты могут также снижать потребность или продлевать период до хирургического вмешательства, необходимого для расширения стенок сосудов, или до использования антикоагулянтов с целью ограничения повреждения, которое может вызвать разрыв бляшек. Меченые соединения и методы анализа Другой аспект настоящего изобретения относится к флуоресцентному красителю, спиновой метке,меченым тяжелым металлом или радиоактивно меченым соединениям формулы I, которые могут быть полезны не только в плане визуализации, но также при проведении количественного анализа, как in vitro,так и in vivo, для определения локализации и количественного анализа хемокинового рецептора в образцах ткани, включая ткани человека, а также для идентификации лиганда хемокинового рецептора посредством ингибирования связывания меченого соединения. Соответственно, настоящее изобретение включает тесты на хемокиновые рецепторы, которые содержат такие меченые соединения. Настоящее изобретение также включает соединения формулы I, меченные изотопами. Соединение,меченное изотопом или радиоактивной меткой, представляет собой соединение по настоящему изобретению, в котором один или несколько атомов заменены или замещены атомом, имеющим атомный номер или массовое число, отличные от атомного номера или массового числа, обычно встречающегося в природе (например, в случае природного соединения). Подходящие радионуклиды, которые могут вклю- 22012649 чаться в соединения по настоящему изобретению, включают, но без ограничения, 2 Н (также называемый как дейтерий и обозначаемый D), 3 Н (также называемый как тритий и обозначаемый Т), 11 С, 13 С, 14 С, 13N,15N, 15O, 17O, 18O, 18F, 35S, 36Cl, 82Br, 75Br, 76Br, 77Br, 123I, 124I, 125I и 131I. Радионуклид, который может включаться в радиоактивно меченое соединение по настоящему изобретению, будет определяться конкретным применением радиоактивно меченного соединения. Например, для мечения и конкурентного анализа хемокинового рецептора in vitro будут особенно полезны соединения, которые включают 3 Н, 14 С, 82Br,125 131I, I или 35S. В случае методов, направленных на радиоактивную визуализацию, наиболее полезными будут , 11C, 18F, 125I, 123I, 124I,131I,75Br,76Br, или 77Br. Следует понимать, что термины радиоактивно меченое или меченое соединение относятся к соединению, которое содержит включенный в него по меньшей мере один радионуклид. В некоторых вариантах осуществления указанный радионуклид выбирают из группы, состоящей из 3 Н, 14 С, 125I, 35S и 82Br. Подходящие способы синтеза органических соединений с включением в них радиоактивных изотопов, применимые также для соединений по настоящему изобретению, хорошо известны в данной области. Радиоактивно меченые соединения по настоящему изобретению могут использоваться в скрининганализе для идентификации/оценки соединений. В общих чертах метод состоит в том, что вновь синтезированное или идентифицированное соединение (например, исследуемое соединение) может быть проанализировано на его способность снижать связывание радиоактивно меченого соединения по настоящему изобретению с хемокиновым рецептором. Соответственно, способность исследуемых соединений конкурировать с радиоактивно меченым соединением за связывание с хемокиновым рецептором напрямую коррелирует с его связывающей аффинностью. Наборы Настоящее изобретение также включает фармацевтические наборы, применяемые, например, при лечении и профилактике заболеваний, ассоциированных с хемокинами, которые включают один или несколько контейнеров, включающих фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы I. Такие наборы могут также включать, при желании, один или несколько различных традиционных компонентов фармацевтического набора, таких как, например,контейнеры с одним или несколькими фармацевтически приемлемыми носителями, дополнительные контейнеры и другие компоненты, известные специалистам в данной области. В данный набор могут также входить инструкции либо в виде вкладышей, либо в виде ярлычков, указывающих количество компонентов, подлежащих введению, а также руководство по введению и/или руководство по смешиванию компонентов. Ниже настоящее изобретение будет описано более подробно с помощью конкретных примеров. Приведенные ниже примеры даны лишь с целью иллюстрации и никоим образом не ограничивают настоящее изобретение. Для специалистов в данной области очевидно наличие множества несущественных параметров, которые могут быть изменены или могут быть модифицированы с достижением, по существу, тех же результатов. Примеры Пример 1. Получение N-[(1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2 Н-пиран-4-амина 4,4-Диметокситетрагидро-2 Н-пиран-3-ол. К раствору 4-метокси-3,6-дигидро-2 Н-пирана (5,00 г, 43,8 ммоль) в метаноле (100 мл) при температуре 0 С добавляют по каплям раствор м-хлорпербензойной кислоты (15,1 г, 87,6 ммоль) в метаноле (15 мл). После перемешивания в течение 5 ч метанол удаляют в вакууме и белый остаток растворяют в метиленхлориде (300 мл). К раствору добавляют K2CO3. Полученный раствор перемешивают в течение 1 ч и фильтруют через целит. Фильтрат выпаривают в вакууме с получением желаемого продукта, который используют непосредственно на следующей стадии без очистки. Стадия А-2.- 23012649 3,4,4-Триметокситетрагидро-2 Н-пиран. К раствору 4,4-диметокситетрагидро-2 Н-пиран-3-ола (6,00 г, 37,0 ммоль) в ТГФ (100 мл) при температуре 0 С добавляют гидрид натрия (1,48 г, 37,0 ммоль). После перемешивания при температуре 0 С в течение 1 ч добавляют по каплям метилйодид (4,61 г, 74,0 ммоль). Реакционную смесь оставляют нагреться до температуры окружающей среды и реакцию гасят добавлением водного NH4Cl. Полученный продукт экстрагируют три раза диэтиловым эфиром. Объединенный экстракты сушат над Na2SO4 и концентрируют. Очистка флэш-хроматографией на силикагеле (10% диэтиловый эфир 60% смесь диэтиловый эфир/гексан) приводит к получению желаемого продукта. 1 3-Метокситетрагидро-2 Н-пиран-4-он. К раствору 3,4,4-триметокситетрагидро-2 Н-пирана (4 г, 20 ммоль) в смеси ТГФ/Н 2 О (60 мл/10 мл) добавляют концентрированную HCl (6 мл). После перемешивания в течение 1 ч ТГФ удаляют в вакууме. Водный раствор экстрагируют эфиром (3100 мл). Экстракты сушат и концентрируют в вакууме с получением желаемого продукта. 1 Метил-(1R,4S)-4-[(трет-бутоксикарбонил)амино]циклопент-2-ен-1-карбоксилат. К раствору (1R,4S)-4-[(трет-бутоксикарбонил) амино]циклопент-2-ен-1-карбоновой кислоты (10,0 г,44,0 ммоль) в ДМФА (25 мл) добавляют карбонат калия (6,33 г, 45,8 ммоль) и затем метилйодид (4,0 г, 64 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляют EtOAc. Раствор промывают водой четыре раза, один раз насыщенным раствором соли, сушат(MgSO4) и концентрируют. Остаток сушат в высоком вакууме в течение ночи с получением указанного в заголовке соединения (11 г, 99%). МС вычислено для C12H19NO4: (M+H)+ 242; найдено 142,1 (М-Вос-Н)+. 1 Метил-(1R,4S)-4-[(трет-Бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоксилат. К раствору 1,00 М гексаметилдизилазида лития в ТГФ (202 мл) при -78 С добавляют в течение 10 мин раствор метил-(1R,4S)-4-[(трет-бутоксикарбонил)амино]циклопент-2-ен-1-карбоксилата (22,10 г,91,59 ммоль) в ТГФ (36,2 мл). Раствор перемешивают при температуре -78 С в течение 30 мин перед добавлением одной порцией изопропилиодида (10,0 г, 100 ммоль). Затем смесь переносят в морозильную камеру при температуре -24 С и выдерживают в течение ночи. Реакцию гасят добавлением водного раствора хлорида аммония и полученный раствор экстрагируют три раза диэтиловым эфиром. Эфирные слои сушат над сульфатом натрия и выпаривают в вакууме. Остаток очищают флэш-хроматографией на силикагеле при элюировании смесью 10% этилацетат/гексан с получением указанного в заголовке соединения (20,2 г). МС вычислено для C15H25NO4: (М+Н)+ 284; найдено 184,2 (М-Вос+Н)+. Стадия В-3.(1S,4S)-4-[(трет-Бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоновая кислота. К раствору метил-(1R,4S)-4-[(трет-бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоксилата (18,42 г, 65 ммоль) в ТГФ (500 мл), метаноле (500 мл) и воде (100 мл) добавляют моногидрат гидроксида лития (5,00 г, 119 ммоль). Смесь нагревают до температуры кипения с обратным холодильником в течение ночи. Через 18 ч проводят ТСХ, которая указывает на наличие очень небольших следовых количеств исходного материала. Органические растворители удаляют в вакууме и водный слой экстрагируют диэтиловым эфиром (200 мл) для удаления непрореагировавшего исходного вещества. Вод- 24012649 ный слой подкисляют концентрированной HCl до рН 4 при охлаждении на ледяной бане. Полученный раствор экстрагируют три раза метиленхлоридом. Экстракты сушат над MgSO4 и концентрируют с получением твердого вещества (17 г). Указанное твердое вещество растворяют в горячем этилацетате (22 мл) и к раствору добавляют гексаны (550 мл). Раствор медленно охлаждают до комнатной температуры перед перенесением в морозильную камеру при температуре от -22 до -24 С. Через два дня кристаллы удаляют и жидкость выпаривают в вакууме с получением желаемого продукта в виде белого пенообразного твердого вещества (9,78 г, 56%). МС вычислено для C14H23NO4: (М+Н)+ 270; найдено 170,1 (М-Вос+Н)+. Стадия В-4.(1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновая кислота. К раствору (1S,4S)-4-[(трет-бутоксикарбонил)амино]-1-изопропилциклопент-2-ен-1-карбоновой кислоты (9,78 г, 36,3 ммоль) в этаноле (250 мл) добавляют 10% палладий на угле (550 мг). Смесь встряхивают в атмосфере водорода при давлении 55 фунт/дюйм 2 в течение ночи и фильтруют через целит. Фильтрат выпаривают в вакууме с получением указанного в заголовке соединения (9,45 г, 96%). МС вычислено для C14H25NO4: (М+Н)+ 272; найдено 172,1 (М-Вос+Н)+. Стадия C.(1S,3R)-4-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновую кислоту (0,10 г,0,37 ммоль), N-[3-трифторметил]фенилпиперазин (85 г, 0,37 ммоль), триэтиламин (0,10 мл, 0,74 ммоль) и гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (0,16 г, 0,37 ммоль) объединяют в метиленхлориде (5 мл) и перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют EtOAc и промывают насыщенным NaHCO3. Водный слой экстрагируют 3 раза EtOAc. Объединенные органические слои сушат (MgSO4), концентрируют и подвергают флэш-хроматографии (50%EtOAc/HexЕА) с получением желаемого продукта (86 мг, 53%). МС вычислено для C25H36F3N3O3:(82 мг, 0,18 ммоль) обрабатывают трифторуксусной кислотой (3 мл, 0,04 моль) в метиленхлориде (3 мл) при комнатной температуре в течение 1 ч. Смесь концентрируют с получением желаемого продукта (98 мг, 93%). МС вычислено для C20H28F3N3O: (М+Н) 384,3; найдено 384,2. Стадия Е.N-[(1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперазин-1-илкарбонил)циклопентил]-3 метокситетрагидро-2 Н-пиран-4-амин. К раствору бис(трифторацетата) (1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперазин-1 илкарбонил)циклопентанамина (140 мг, 0,23 ммоль), 3-метокситетрагидро-4 Н-пиран-4-она (90 мг, 0,69 ммоль) и триэтиламина (0,096 мл, 0,69 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (97 мг, 0,46 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляют EtOAc и промывают насыщенным Na2CO3. Водный слой экстрагируют три раза EtOAc. Объединенные органические слои сушат (MgSO4), концентрируют и очищают на силикагеле при элюировании EtOAc1% Et3N/EtOAc, получая 105 мг (92%) желаемого продукта. Продукт выделяют хиральной ВЭЖХ с получением двух основных изомеров. МС вычислено для C26H38F3N3O3: (M+H) 4 98; найдено 498,2, для обоих изомеров. 3-Этокси-4,4-диметокситетрагидро-2 Н-пиран. К раствору 4,4-диметокситетрагидро-2 Н-пиран-3-ола (2,00 г, 12 ммоль) в ТГФ (20 мл), охлажденном на ледяной бане, медленно добавляют гидрид натрия (0,60 г, 15 ммоль) и полученную взвесь перемешивают в течение 1 ч. Добавляют йодэтан (1,5 мл, 19,0 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Добавляют еще гидрид натрия (0,6 г) и йодэтан (3 мл) и перемешивание продолжают в течение ночи. Реакцию гасят добавлением воды. Полученный раствор экстрагируют два раза EtOAc и два раза диэтиловым эфиром. Объединенный экстракты сушат, концентрируют и очищают на силикагеле (20% EtOAc/гексаны) с получением 2,1 г (90%) желаемого продукта. 1 3-Этокситетрагидро-4 Н-пиран-4-он. К раствору 3-этокси-4,4-диметокситетрагидро-2 Н-пирана (2,1 г, 11 ммоль) в смеси ТГФ/вода (50 мл/10 мл) добавляют концентрированную HCl (6 мл). После перемешивания в течение 1 ч при комнатной температуре смесь разбавляют EtOAc. Органический слой отделяют и водный слой экстрагируют пять раз диэтиловым эфиром. Объединенный органически слои сушат над MgSO4, фильтруют и концентрируют с получением 1,55 г (97%) желаемого продукта. 1 Н ЯМР (CDCl3)4,30-3,50 (7 Н, м) , 2,62-2,57 (2 Н, м) , 1,30-1,20 (3 Н, т, J=5 Гц). Стадия В. 3-Этокси-N-[(1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперазин-1-илкарбонил)циклопентил]тетрагидро-2 Н-пиран-4-амин. К раствору дигидрохлорида (1R,3S)-3-изопропил-3-(4-[3-(трифторметил)фенил]пиперазин-1 илкарбонил)циклопентанамина (200 мг, 0,438 ммоль), 3-этокситетрагидро-4 Н-пиран-4-она (130 мг, 0,88 ммоль) и триэтиламина (0,18 мл, 1,3 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (180 мг, 0,88 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь разбавляют EtOAc и промывают насыщенным Na2CO3. Водный слой экстрагируют три разаEtOAc. Объединенные органические слои сушат (MgSO4), концентрируют и очищают на силикагеле(EtOAc1% Et3N/EtOAc5% Et3N/EtOAc) с получением 230 мг продукта. Указанный продукт выделяют хиральной ВЭЖХ с получением двух основных изомеров: изомера 1 (110 мг) и изомера 2 (77 мг). МС вычислено для C27H40F3N3O3: (M+H) 512; найдено 512,2. Пример 3.- 26012649 1-[4-(Трифторметил)пиридин-2-ил]пиперазин. Раствор 2-хлор-4-(трифторметил)пиридина (2,00 г, 11 ммоль), пиперазина (3 г, 30 ммоль) и триэтиламина (3,1 мл, 22 моль) в ДМФА (10 мл) нагревают при температуре 100 С в течение ночи и концентрируют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (EtOAcEtOAc/MeOH/Et3N=9/1/0,5) с получением 1,09 г (43%) чистого продукта. МС вычислено для C10H12F3N3: (M+H) 232; найдено 232,1. Стадия В. трет-Бутил[(1R,3S)-3-изопропил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил) циклопентил]карбамат. К раствору 1-[4-(трифторметил)пиридин-2-ил]пиперадина (145 мг, 0,627 ммоль), (1S,3R)-3-[(третбутоксикарбонил)амино]-1-изопропилциклопентанкарбоновой кислоты (140 мг, 0,52 ммоль) в метиленхлориде (10 мл) добавляют гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (253 мг, 0,572 ммоль) и затем триэтиламин (0,156 мл, 1,12 ммоль). После перемешивания в течение ночи реакционную смесь разбавляют EtOAc и промывают насыщенным NaHCO3. Водный слой экстрагируют три раза EtOAc. Объединенные органические слои сушат (MgSO4), концентрируют и очищают флэшхроматографией на силикагеле (20% EtOAc/тексаны 40% EtOAc/гексаны) с получением 0,15 г желаемого продукта. МС вычислено для C24H35F3N4O3: (М+Н) 485; найдено 385,2 (М-Н+Вос). Стадия С.(1R,3S)-3-Изопропил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентанамин. трет-Бутил[(1R,3S)-3-изопропил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил) циклопентил]карбамат (150 г, 0,31 ммоль) обрабатывают 4,0 М раствором HCl в 1,4-диоксане (10 мл) при комнатной температуре в течение 1 ч и концентрируют при пониженном давлении с получением продукта, который используют на следующей стадии без очистки. МС вычислено для C19H27F3N4O: (М+Н) 385; найдено 385,2. Стадия D.N-[(1R,3S)-3-Изопропил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2 Н-пиран-4-амин. К раствору дигидрохлорида (1R,3S)-3-изопропил-3-(4-[4-(трифторметил)пиридин-2-ил]пиперазин 1-илкарбонил)циклопентанамина (140 мг, 0,31 ммоль), 3-метокситетрагидро-4 Н-пиран-4-она (80 мг,0,61 ммоль) и триэтиламина (0,21 мл, 1,5 ммоль) в метиленхлориде (10 мл) добавляют триацетоксиборгидрид натрия (190 мг, 0,92 ммоль). После перемешивания в течение ночи реакционную смесь разбавляют EtOAc и промывают насыщенным Na2CO3. Водный слой экстрагируют три раза EtOAc. Объединенные органические слои сушатEt3 EtOAc) с получением 101 мг продукта. Данный продукт далее выделяют хиральной ВЭЖХ с получением изомера 1 (55 мг) и изомера 2 (37 мг). ЖХМС вычислено для C25H37F3N4O3: (M+1) 499; найдено 499,2, для обоих изомеров. Пример 4. Получение N-[(1R,3S)-3-изолропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1 илкарбонил)циклопентил]-3-метокситетрагидро-2 Н-пиран-4-амина- 27012649 3-Бром-5-йодпиридин. К раствору 3,5-дибромпиридина (48 г, 200 мл) в ТГФ (200 мл) добавляют 2 М раствор хлорида изопропилмагния в ТГФ (80 мл). После перемешивания при комнатной температуре в течение 2 ч раствор охлаждают до -78 С. Добавляют предварительно охлажденный раствор йода (51 г, 2 00 ммоль) в ТГФ(100 мл). Смесь разбавляют диэтиловым эфиром и промывают насыщенным раствором хлорида аммония, 2M раствором тиосульфата натрия и насыщенным раствором соли. Полученный органический слой сушат над MgSO4, фильтруют и концентрируют. Кристаллизация из этанола дает 33,5 г (58%) желаемого продукта. 1 трет-Бутил-4-(5-бромпиридин-3-ил)пиперазин-1-карбоксилат. Раствор 3-бром-5-йодпиридина (13,0 г, 45,8 ммоль), трет-бутилпиперазин-1-карбоксилата (8,53 г,45,8 ммоль), йодида меди(I) (0,871 г, 4,57 ммоль), K3PO4 (19,46 г, 91,68 ммоль), 1,2-этандиола (5,1 мл, 91 ммоль) в изопропиловом спирте (80 мл) нагревают в запаянной пробирке при температуре 80 С на масляной бане в течение 3 дней. После охлаждения до комнатной температуры реакционную смесь фильтруют через целит. Фильтрат концентрируют в вакууме. Остаток отбирают EtOAc и раствор промывают насыщенным NaHCO3, сушат (MgSO4) и концентрируют. Очистка флэш-хроматографией на силикагеле(20% EtOAc/гексаны 30% EtOAc/гексаны) приводит к получению 5,75 г (37%) желаемого продукта. МС вычислено для C14H20BrN3O2: (М+Н) 343; найдено 342,0, 344,0. Стадия А-3. трет-Бутил-4-(5-йодпиридин-3-ил)пиперазин-1-карбоксилат. К раствору трет-бутил-4-(бромфенил)пиперазин-1-карбоксилата (2,0 г, 5,9 ммоль) в ТГФ (20 мл) добавляют 2 М раствор хлорида изопропилмагния в ТГФ (5 мл). После перемешивания при комнатной температуре в течение 2 ч раствор охлаждают до температуры -78 С. Добавляют предварительно охлажденный раствор йода (3,0 г, 12 ммоль) в ТГФ (2 мл). После перемешивания при температуре -78 С в течение 30 мин и при комнатной температуре еще в течение 30 мин, смесь разбавляют этилацетатом, промывают насыщенным раствором хлорида аммония, 2 М раствором тиосульфата натрия и насыщенным раствором соли, сушат (MgSO4) и концентрируют. Остаток очищают флэш-хроматографией на силикагеле (20% EtOAc/гексаны 50% EtOAc/гексаны) с получением желаемого продукта (1,40 г) с 75% чистотой. МС вычислено для C15H21IN2O2: (М+Н) 390; найдено 390,0. Стадия А-4. трет-Бутил-4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-карбоксилат. Колбу с йодидом меди(I) (0,49 г, 2,6 мл) и фторидом калия (0,15 г, 2,6 мл) нагревают в пламени при осторожном встряхивании при высоком вакууме до появления зеленоватого окрашивания. Добавляют раствор трет-бутил-4-(3-йодфенил)пиперазин-1-карбоксилата (0,5 г, 1,0 мл) и (трифторметил)триметилсалана (0,37 г, 2,6 ммоль) в ДМФА (5 мл). Коричневый раствор перемешивают при комнатной температуре в течение ночи. Добавляют еще (трифторметил)триметилсилан (0,37 г). Смесь нагревают при температуре 50 С в течение ночи, разбавляют EtOAc и промывают насыщенным раствором хлорида аммония. Водный слой экстрагируют три раза EtOAc. Объединенные органические слои сушат (MgSO4), концентрируют и очищают флэш-хроматографией на силикагеле (20%-40% EtOAc/гексаны) с получением 120 мг желаемого продукта. МС вычислено для C15H20F3N3O2: (M+H) 332; найдено 332,1. Стадия А-5. 1-[5-(Трифторметил)пиридин-3-ил]пиперазин. трет-Бутил-4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-карбоксилат (0,24 г, 0,25 мл) обрабатывают 4,0 М раствором HCl в 1,4-диоксане (7 мл) при комнатной температуре в течение 1 ч и концентрируют. Остаток используют на следующей стадии без дальнейшей очистки. МС вычислено для трет-Бутил[(1R,3S)-3-изопропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-илкарбонил) циклопентил]карбамат. К раствору тригидрохлорида 1-[5-(трифторметил)пиридин-3-ил]пиперазина (0,22 г, 0,23 мл),(1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновой кислоты (0,18 г, 0,66 мл) в метиленхлориде (10 мл) добавляют гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (0,338 г, 0,764 мл) и затем триэтиламин (0,22 г, 1,6 мл). Смесь перемешивают при комнатной температуре в течение ночи и разбавляют EtOAc. Раствор промывают насыщенным NaHCO3, сушатEtOAc/гексаны 40% EtOAc/гексаны) с получением 0,19 г (61%) желаемого продукта. МС вычислено для C24H35F3N4O3: (М+Н) 485; найдено 485,2. Стадия С.(1R,3S)-3-Изопропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-илкарбонил)циклопентанамин. трет-Бутил[(1R,3S)-3-изопропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-илкарбонил) циклопентил]карбамат (190 мг, 0,14 ммоль) обрабатывают 4,0 М раствором HCl в 1,4-диоксане (5 мл) при комнатной температуре в течение 1 ч. Смесь концентрируют и очищают методом ВЭЖХ с получением 35 мг желаемого продукта. МС вычислено для C19H27F3N4O: (М+Н) 38 5; найдено 385,1. Стадия D.N-[(1R,3S)-3-изопропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-илкарбонил)циклопентил]-3-метокситетрагидро-2 Н-пиран-4-амин. К раствору трис(трифторацетата)(1R,3S)-3-изопропил-3-(4-[5-(трифторметил)пиридин-3-ил]пиперазин-1-илкарбонил)циклопентанамина (25 мг, 0,034 ммоль), 3-метокситетрагидро-4 Н-пиран-4-она (13 мг, 0,10 ммоль) и триэтиламина (0,024 мл, 0,17 ммоль) в метиленхлориде (5 мл) добавляют триацетоксиборгидрид натрия (22 мг, 0,10 ммоль). Смесь перемешивают в атмосфере N2 при комнатной температуре в течение ночи и разбавляют EtOAc. Полученный раствор промывают насыщенным NaHCO3, сушатMeOH/Et3N=9/1/0,5) с получением 14 мг желаемого продукта в виде смеси двух изомеров. Оба изомера разделяют хиральной ВЭЖХ с получением пика 1 (6,6 мг) и пика 2 (4,7 мг). ЖХМС вычислено для трет-Бутил(1R,3S)-3-изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2 Н)-ил)карбонил]циклопентилкарбамат. В высушенной колбе суспендируют (1S,3R)-3-[(трет-бутоксикарбонил)амино]-1-изопропилциклопентанкарбоновую кислоту (200 мг, 0,7 ммоль) и 4-фенил-1,2,3,6-тетрагидропиридин (160 мг, 0,81 ммоль) в метиленхлориде (4 мл) в атмосфере N2. Добавляют триэтиламин (0,22 г, 2,2 ммоль) и затем гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (0,36 г, 0,81 ммоль). Реакционную- 29012649 смесь перемешивают в течение ночи при комнатной температуре и реакцию гасят добавлением насыщенного раствора NaHCO3. Полученный раствор экстрагируют метиленхлоридом три раза. Объединенные экстракты сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией на силикагеле (градиент: 0-45% В в течение 15 мин; бутыль А=гексаны, бутыль B=EtOAc) с получением 234 мг(80%) желаемого продукта. МС вычислено для C25H36N2O3: (M+H) 413; найдено 413,2. Стадия В.(1R,3S)-3-Изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2H)-ил)карбонил]циклопентанамин. Трет-бутил(1R,3S)-3-изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2 Н)-ил)карбонил]циклопентилкарбамат (0,23 г, 0,56 ммоль) растворяют в 1,0 М растворе HCl в этиловом эфире (4 мл). После перемешивания при комнатной температуре в течение 2 ч раствор концентрируют с получением бесцветного масла (170 мг). МС вычислено для C20H28N2O: (M+H) 313; найдено 313,2. Стадия С. N-(1R,3S)-3-изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2 Н)-ил)карбонил]циклопентилтетрагидро-2 Н-пиран-4-амин. К раствору гидрохлорида (1R,3S)-3-изопропил-3-[(4-фенил-3,6-дигидропиридин-1(2 Н)-ил)карбонил]циклопентанамина (50 мг, 0,1 ммоль), 3-метокситетрагидро-4 Н-пирин-4-она (56 мг, 0,43 ммоль) и триэтиламина (0,07 мл, 0,5 ммоль) в метиленхлориде (2 мл) добавляют триацетоксиборгидрид натрия (91 мг, 0,43 ммоль). После перемешивания при комнатной температуре в течение ночи добавляют насыщенный NaHCO3. Полученный раствор экстрагируют три раза метиленхлоридом. Объединенные экстракты сушат (MgSO4), фильтруют и концентрируют. Очистка флэш-хроматографией на силикагеле (градиент: 0-15% В в течение 15 мин; бутыль А=1% NH4OH/3% MeOH/EtOAc, бутыль B=l% NH4OH/MeOH) с получением желаемого продукта. МС вычислено для C26H38N2O3: (M+H) 427; найдено 427,3. Пример 6. Получение 1-1S,3R)-1-изопропил-3-[(3-метокситетрагидро-2H-пиран-4-ил)амино] циклопентилкарбонил)-4-фенилпиперидин-4-ола.(0,42 г, 0,81 ммоль). После перемешивания в течение ночи при комнатной температуре реакцию гасят добавлением насыщенного раствора NaHCO3. Полученный раствор экстрагируют метиленхлоридом три раза. Объединенные экстракты сушат (MgSO4), фильтруют и концентрируют. Неочищенный продукт переносят на следующую стадию без очистки. МС вычислено для C25H38N2O4: (М+Н) 431; найдено 431,2. Стадия В. 1-[(1S3R)-3-Аминоизопропилциклопентил]карбонил-4-фенилпиперидин-4-ол. трет-Бутил(1R,3S)-3-[(4-гидрокси-4-фенилпиперидин-1-ил)карбонил]-3-изопропилциклопентил карбамат (0,30 г, 0,70 ммоль) растворяют в 2,0 М растворе HCl в диэтиловом эфире (10 мл). После перемешивания в течение 3,5 ч добавляют несколько капель MeOH до образования прозрачного раствора. Смесь концентрируют с получением масла. Неочищенный продукт используют в следующей реакции без очистки. МС вычислено для C20H30N2O2: (M+H) 331; найдено 331,2. Стадия С
МПК / Метки
МПК: A61P 35/00, C07D 405/12, A61K 31/496, A61P 31/18, C07D 405/14, A61K 31/453, A61K 31/4433, A61P 29/00
Метки: модуляторов, хемокиновых, качестве, аминоциклопентанкарбоксамиды, рецепторов
Код ссылки
<a href="https://eas.patents.su/30-12649-3-aminociklopentankarboksamidy-v-kachestve-modulyatorov-hemokinovyh-receptorov.html" rel="bookmark" title="База патентов Евразийского Союза">3 – аминоциклопентанкарбоксамиды в качестве модуляторов хемокиновых рецепторов</a>
3-аминоциклопентанкарбоксамиды в качестве модуляторов хемокиновых рецепторов

Номер патента: 11487
Опубликовано: 28.04.2009
Авторы: Меткаф Брайан В., Ксуе Чу-Биао, Фэн Хао, Чжэн Чаншэн, Гленн Джозеф, Ксиа Майкл, Цао Ганьфэн
МПК: A61K 31/497, A61K 31/44, A61K 31/445...
Метки: 3-аминоциклопентанкарбоксамиды, качестве, хемокиновых, рецепторов, модуляторов
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, или пролекарственная форма, где пунктирная линия обозначает необязательную связь; W представляет V представляет N или CR5; X представляет N или CR2; Y представляет N или CR3; Z представляет N или CR4; А представляет О; RA, RA1, RB и RB1, каждый независимо, представляют Н, ОН; R1 представляет C1-6алкил, C1-6гидроксиалкил, -(С0-6алкил)-О-(C1-6алкил), гетероциклил; R2, R3, R4, R5 и...
3-(4-гетероарилциклогексиламино) циклопентанкарбоксамиды в качестве модуляторов хемокиновых рецепторов

Номер патента: 11403
Опубликовано: 27.02.2009
Авторы: Фэн Хао, Меткаф Брайан, Ксиа Майкл, Гленн Джозеф, Чжэн Чаншэн, Цао Ганьфэн, Сюэ Чу-Бяо, Ананд Раджан
МПК: C07D 217/00, A61K 31/47
Метки: циклопентанкарбоксамиды, модуляторов, хемокиновых, 3-(4-гетероарилциклогексиламино, рецепторов, качестве
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль или пролекарство, где пунктирная линия означает необязательную связь; X представляет собой N, NO или CR3; R1 представляет собой C1-6 алкил, (С0-6 алкил)-O-(C1-6 алкил), (С0-6 алкил)-S-(C1-6 алкил), (С0-6 алкил)-(C3-7 циклоалкил)-(С0-6 алкил), ОН, CO2R10, гетероциклил, CN, NR10R12, NSO2R10, NCOR10, NCO2R10, NCOR10, CR11CO2R10, CR11OCOR10 или фенил; R2 представляет собой H, ОН,...
N-уреидоалкилпиперидины в качестве модуляторов активности рецепторов хемокинов

Номер патента: 3147
Опубликовано: 27.02.2003
Авторы: Сантелла Джозеф Б.III, Вэкер Дин А.К., Ким Уи Тае, Делукка Джордж В., Ко Соо С., Данкиа Джон В.
МПК: A61P 11/06, A61K 31/445, C07D 211/32...
Метки: рецепторов, активности, качестве, хемокинов, n-уреидоалкилпиперидины, модуляторов
Формула / Реферат:
1. Соединение формулы (I) или его стереоизомеры или фармацевтически приемлемые соли, где M отсутствует или выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; Q выбран из CH2, CHR5, CHR13, CR13R13 и CR5R13; J и K выбраны из CH2, CHR5, CHR6, CR6R6 и CR5R6; L представляет CHR5; при условии, что если M отсутствует, то J выбран из CH2, CHR5, CHR13 и CR5R13; Z выбран из O и S; E выбран из кольцо A представляет C3-6карбоциклический остаток, при условии,...
Производные 3-аминопирролидина в качестве модуляторов рецепторов хемокинов

Номер патента: 10027
Опубликовано: 30.06.2008
Авторы: Хань Эми Ци, Чжэн Чаншэн, CЮЭ Чу-Бяо, Хуан Тайшинг, Меткаф Брайан, Цао Ганьфэн, Робинсон Дариус Дж., Фэн Хао
МПК: C07D 207/14
Метки: производные, качестве, рецепторов, хемокинов, модуляторов, 3-аминопирролидина
Формула / Реферат:
1. Соединение формулы I его энантиомеры, диастереоизомеры, энантиомерно обогащенные смеси, его рацемические смеси, кристаллические формы, некристаллические формы, его сольваты и фармацевтически приемлемые соли, где R1 независимо выбирают из группы, состоящей из арила, гетероарила, арилкарбоксамидо, гетероарилкарбоксамидо, арилокси, арилалкокси или ариламино, и где указанные группы арила, арилалкила или гетероарила могут быть замещены 0-3...
Производные пропионамида, полезные в качестве модуляторов андрогенных рецепторов

Номер патента: 12430
Опубликовано: 30.10.2009
Авторы: Карьялайнен Арья, Вольфарт Герд, Ратилайнен Яри, Термекангас Олли, Каллио Пекка, Хухтала Пааво, Мойланен Ану
МПК: A61K 31/395, A61K 31/167, A61K 31/277...
Метки: модуляторов, качестве, производные, рецепторов, пропионамида, полезные, андрогенных
Формула / Реферат:
1. Производные пропионамида формулы (I) в которой R1 представляет (С1-С7)алкил, гидрокси(С1-С7)алкил или -СНО; R2 представляет нитро, циано или атом галогена; R3 представляет (С1-С7)алкил; R4 представляет атом водорода; X представляет О или NH; А является группой, выбранной из в которых R5, R6, R7, R8 и R9 независимо представляют атом водорода, атом галогена, нитро, циано, (C1-C7)алкил, галоген (С1-С7)алкил, циано (С1-С7)алкил,...
Предыдущий патент: Фармацевтическая композиция для чрескожной доставки активного агента (варианты)
Следующий патент: Применение [d-meala]3-[etval]4-циклоспорина для лечения инфекции гепатита с и фармацевтическая композиция, включающая [d-meala]3-[etval]4-циклоспорин
Случайный патент: Устройство для очистки дымовых газов с разделенным сборником орошающей жидкости