Хинолиновые и хиназолиновые соединения, применяемые в терапии, особенно при лечении доброкачественной гиперплазии простаты




























Формула / Реферат
1. Соединение формулы I

где R1 представляет собой C1-4 алкокси, необязательно замещенную одним или несколькими атомами фтора;
R2 представляет собой Н или C1-6 алкокси, необязательно замещенную одним или несколькими атомами фтора;
R3 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно замещен одной или несколькими группами, выбранными из галогена, C1-4 алкокси, C1-4 алкила и СF3;
R4 представляет собой 4-, 5-, 6- или 7-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно конденсирован с бензольным кольцом или с 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S,
эта циклическая система в целом необязательно замещена одной или несколькими группами, независимо выбранными из ОН, C1-4 алкила, C1-4 алкокси, галогена, CONR8R9, SO2NR8R9, (СН2)bNR8R9 и NHSO2(C1-4 алкила), и если S является членом циклической системы, он может быть замещен одним или двумя атомами кислорода;
R8 и R9 независимо представляют собой Н или C1-4 алкил или вместе с атомом N, к которому они присоединены, могут представлять собой 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S;
b равно 0, 1, 2 или 3;
X представляет собой СН или N; и
L отсутствует или представляет собой циклическую группу формулы Iа

в которой N присоединен ко второй позиции хинолинового или хиназолинового цикла;
А отсутствует или представляет собой СО или SO2;
Z представляет собой СН или N;
m равно 1 или 2, и,
кроме того, если Z представляет собой СН, оно может быть равно 0; и
n равно 1, 2 или 3, при условии, что сумма m и n равна 2, 3, 4 или 5;
или представляет собой цепь формулы Ib,

в которой N присоединен ко 2 позиции хинолинового или хиназолинового цикла;
А' и Z' имеют соответственно такие же значения, как А и Z, описанные выше;
R6 и R7 независимо представляют собой Н или C1-4 алкил; и
р равно 1, 2 или 3, и,
кроме того, если Z представляет собой СН, оно может быть равно 0;
или их фармацевтически приемлемые соли.
2. Соединение по п.1, где каждый из R1 и R2 представляет собой метоксильную группу.
3. Соединение по п.1 или 2, где R3 представляет собой 2-пиридинил или 2-пиримидинил.
4. Соединение по любому из предшествующих пунктов, где Х представляет собой N.
5. Соединение по любому из предшествующих пунктов, где L отсутствует.
6. Соединение по любому из предшествующих пунктов, где R4 включает в себя насыщенный 6-членный N-содержащий цикл, который конденсирован с бензольным или пиридиновым кольцом.
7. Соединение по п. 6, где бензольное кольцо замещено NHSО2(C1-4 алкилом).
8. Фармацевтическая композиция, включающая в себя соединение формулы I, как определено в п.1, или его фармацевтически приемлемую соль, в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем.
9. Применение соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли в качестве лекарственного препарата.
10. Применение соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли в производстве лекарственного препарата для лечения доброкачественной гиперплазии простаты.
11. Способ лечения доброкачественной гиперплазии простаты, который включает в себя введение соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, пациенту, нуждающемуся в таком лечении.
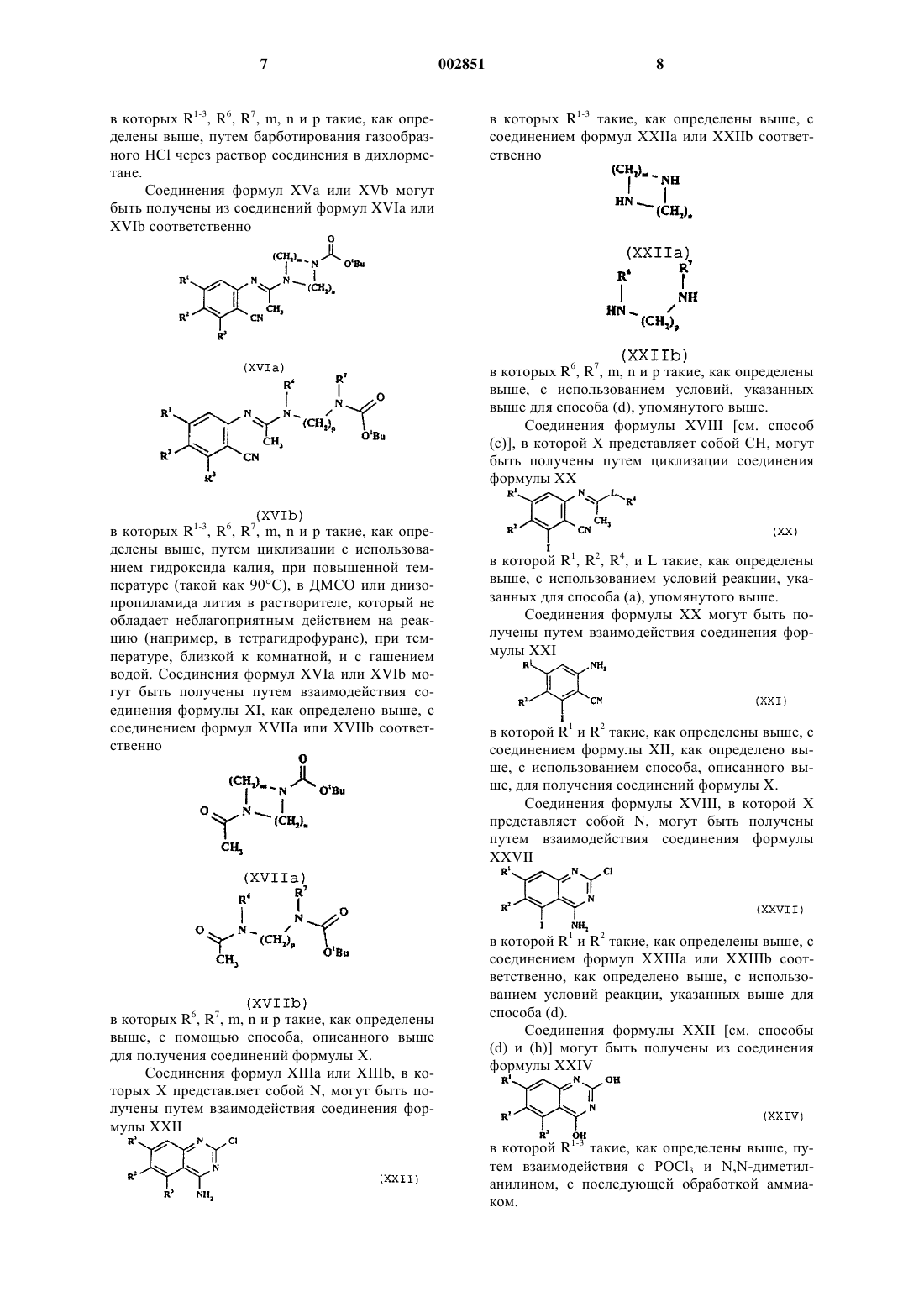
12. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя циклизацию соединения формулы X

в которой R1-4 и L такие, как определены в п.1, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
13. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XIIIa или XIIIb соответственно


в которой R1-3, R6, R7, X, m, n и р такие, как определены в п.1, с соединением формулы XIV

в которой R4 такой, как определен в п.1,
А" представляет собой СО или SO2 и
Lg представляет собой уходящую группу,
и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
14. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XVIII

в которой R1, R2, R4, X и L такие, как определены в п.1, с соединением формулы XIX,

в которой R3 такой, как определен в п.1, а М представляет собой замещенный бор, цинк или олово, в присутствии палладиевого катализатора, и,
если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
15. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XXII

в которой R1-3 такие, как определены в п.1, с соединением формулы XXIIIа или XXIIIb соответственно


в которой R4, R6, R7, А, А', Z, Z', m, n и р такие, как определены в п.1,
и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
16. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XXVIIIа или XXVIIIb соответственно


в которой R1-3, R6, R7, X, Z, Z', m, n и р такие, как определены в п.1, и Lg представляет собой уходящую группу, с соединением формулы XXIX
![]()
в которой R4a представляет собой группы, определяемые в п.1 с помощью R4, которые содержат нуклеофильный атом азота в цикле, причем этот атом азота соединен с Н,
и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
17. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, котоЁыщ включает в себя превращение соединения формулы I, в котором L представляет собой циклическую группу формулы Ia, в соответствующее соединение формулы I, в котором L представляет собой цепь формулы Ib, в которой каждый из R6 и R7 представляет собой Н, путем воздействия сильного основания, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
18. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XIIIa или XIIIb, как определено выше, с соединением формулы XXX
![]()
в которой R4 такой, как определен в п.1, и Hal представляет собой атом галогена, присоединенный к циклу, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
19. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XXII, как определено выше, с соединением формулы XXIX, как определено выше, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот.
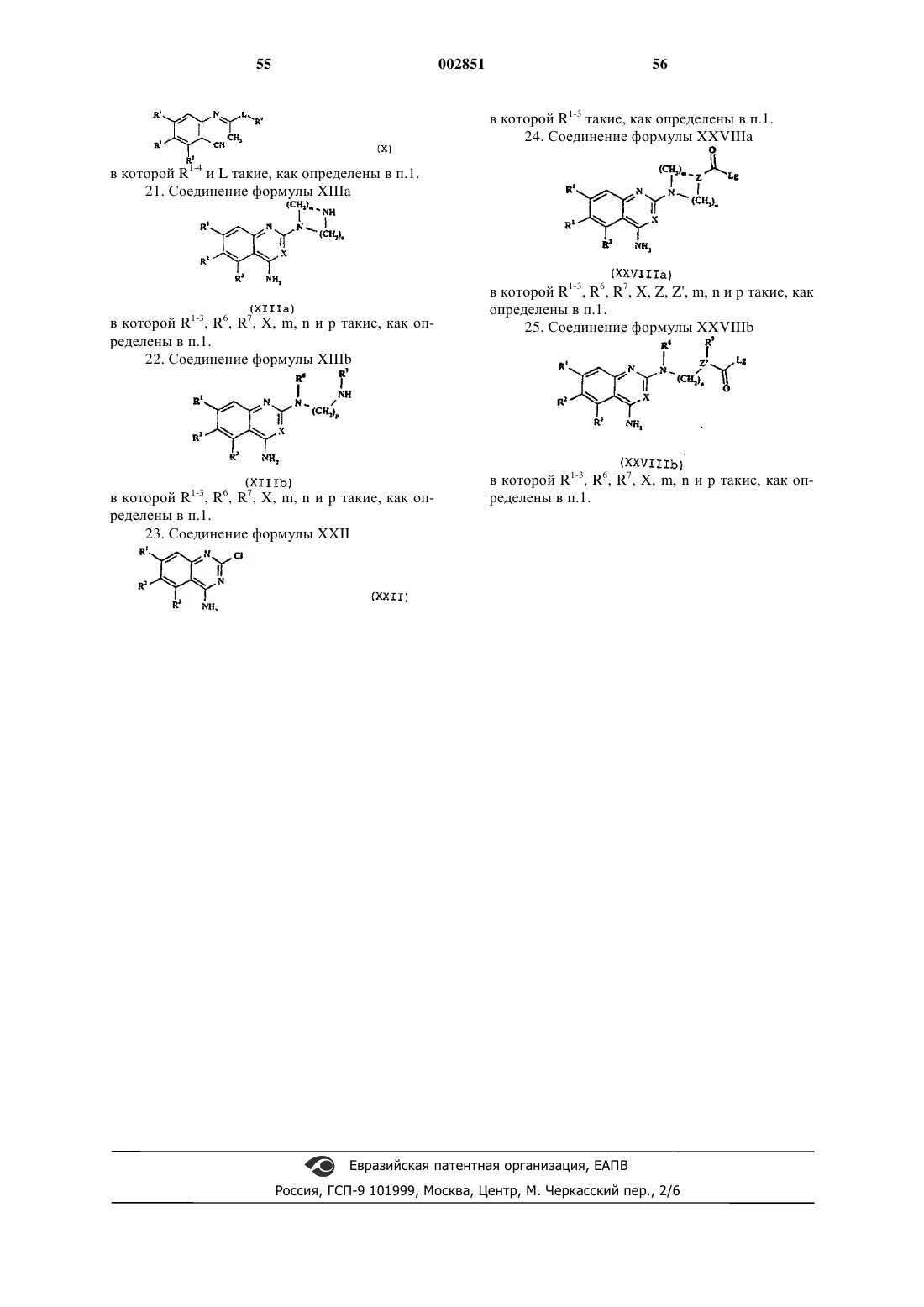
20. Соединение формулы X

в которой R1-4 и L такие, как определены в п.1.
21. Соединение формулы XIIIа

в которой R1-3, R6, R7, X, m, n и р такие, как определены в п.1.
22. Соединение формулы XIIIb

в которой R1-3, R6, R7, X, m, n и р такие, как определены в п.1.
23. Соединение формулы XXII

в которой R1-3 такие, как определены в п.1.
24. Соединение формулы XXVIIIа

в которой R1-3, R6, R7, X, Z, Z', m, n и p такие, как определены в п.1.
25. Соединение формулы XXVIIIb

в которой R1-3, R6, R7, X, m, n и р такие, как определены в п.1.
Текст
1 Данное изобретение относится к новым соединениям, применяемым в терапии, особенно при лечении доброкачественной гиперплазии простаты. В международной патентной заявке WO 89/05297 обнаружен ряд замещенных хиназолиновых соединений, которые представлены как ингибиторы секреции желудочной кислоты. В международной патентной заявке WO 97/23462 (опубликованной после даты приоритета данной заявки) обнаружены хинолиновые и хиназолиновые соединения, имеющие 5 фенильный заместитель. Эти соединения показаны при лечении доброкачественной гиперплазии простаты. В соответствии с настоящим изобретением предоставляется соединение формулы I где R1 представляет собой C1-4 алкокси, необязательно замещенную одним или несколькими атомами фтора;R2 представляет собой Н или C1-6 алкокси,необязательно замещенную одним или несколькими атомами фтора;R3 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно замещен одной или несколькими группами, выбранными из галогена, C1-4 алкокси, C1-4 алкила и СF3;R4 представляет собой 4-, 5-, 6- или 7 членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно конденсирован с бензольным кольцом или с 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S, эта циклическая система в целом необязательно замещена одной или несколькими группами, выбранными из ОН, C1-4 алкила, C1-4 алкокси, галогена,CONR8R9, SO2NR8R9, (СH2)bНR8R9 и NHSO2(C1-4 алкила), и если S является членом циклической системы, он может быть замещен одним или двумя атомами кислорода;R8 и R9 независимо представляют собой Н или C1-4 алкил или вместе с атомом N, к которому они присоединены, могут представлять собой 5- или 6-членное гетероциклическое кольцо,содержащее, по меньшей мере, один гетероатом,выбранный из N, О и S;X представляет собой СН или N; иL отсутствует или представляет собой циклическую группу формулы Iа 2 в которой N присоединен ко второй позиции хинолинового или хиназолинового цикла; А отсутствует или представляет собой СО или SO2;Z представляет собой СН или N;m равно 1 или 2, и, кроме того, если Z представляет собой СН, оно может быть равно 0; иm и n равна 2,3,4 или 5; или представляет собой цель формулы 1b в которой N присоединен ко второй позиции хинолинового или хиназолинового цикла; А' и Z' имеют соответственно такие же значения, как А и Z, описанные выше;R6 и R7 независимо представляют собой Н или C1-4 алкил; и р равно 1, 2 или 3, и, кроме того, если Z представляет собой СН, оно может быть равно 0; или их фармацевтически приемлемые соли (в этом документе упоминаются вместе как "соединения данного изобретения"). Фармацевтически приемлемые соли включают в себя аддитивные кислотные соли, такие как гидрохлориды, гидробромиды и фосфаты. Алкильные или алкоксильные группы, которые R1-4 могут представлять или включать,могут быть с прямой цепью, с разветвленной цепью, циклическими или их комбинациями. Предпочтительно R3 представляет собой ароматический цикл, например, пиридинил, пиримидинил, тиенил, фуранил или оксазолил. Гетероциклические группы,которые включает R4, могут быть насыщенными или ненасыщенными. Однако предпочтительно,чтобы цикл, присоединенный к L, или, если L отсутствует, к хинолиновому или хиназолиновому циклу, был насыщенным. Соединения данного изобретения могут быть оптически активными. В частности, они могут проявлять атропоизомеризм вокруг связи,соединяющей R3 с остатком молекулы, если заместитель R3 находится в орто-положении цикла. Данное изобретение включает в себя оптические изомеры соединений формулы I и все их диастереоизомеры. Предпочтительные группы соединений,которые могут быть упомянуты, включают в себя те, в которых:(a) R1 представляет собой метоксильную группу;(b) R2 представляет собой метоксильную группу;(d) R4 включает в себя насыщенный 6 членный N-содержащий цикл, который конденсирован с бензольным или пиридиновым цик 3 лом; например, R4 может представлять собой насыщенный 6-членный N-содержащий цикл,который конденсирован с бензольным циклом,замещенным NHSO2 (C1-4 алкил);(f) L отсутствует. В соответствии с данным изобретением также предоставляется способ получения соединения данного изобретения, который включает в себя:(а) если Х представляет собой СН, циклизацию соединения формулы X(е) если А или А' представляет СО, а R4 включает нуклеофильный атом азота в гетероциклическом кольце, присоединенном к L,взаимодействие соединения формулы XXVIIIа или XXVIIIb соответственно(b) если присутствуют А или A', a Z или Z' представляют собой N, взаимодействие соединения формулы XIIIa или XIIIb соответственно в которой R , R , R , X, m, n и р такие, как определены выше, с соединением формулы XIV в которой R4 такой, как определен выше. А" представляет СО или SO2 и Lg представляет уходящую группу;(d) если Х представляет N, взаимодействие соединения формулы XXII в которой R4a представляет группы, определенные выше с помощью R4, которые содержат нуклеофильный атом азота в цикле, причем этот атом азота соединен с Н;(f) превращение соединения формулы I, в котором L представляет собой циклическую группу формулы Iа, в соответствующее соединение формулы I, в котором L представляет собой цепь формулы Ib, в которой каждый из R6 иR7 представляют собой Н, путем воздействия сильного основания;(g) если А или А' отсутствует, a Z или Z' представляет собой N, взаимодействие соединения формулы XIIIa или XIIIb, как определено выше, с соединением формулы XXX в которой R4 такой, как определен выше, и Hal представляет собой атом галогена, присоединенный к циклу; илиh) если Х представляет собой N, L отсутствует, а R4 включает в себя нуклеофильный атом азота в гетероциклическом кольце, присоединенном к хинолиновому или хиназолиновому циклу, взаимодействие соединения формулыXXII, как определено выше, с соединением формулы XXIX, как определено выше; и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль, или наоборот. 5 В способе (а) циклизация может быть проведена в присутствии сильного основания (например, диизопропиламида лития) в растворителе, который не обладает неблагоприятным действием на реакцию (например, в тетрагидрофуране), при температуре, близкой к комнатной, и погашена водой. Как вариант, циклизация может быть проведена с использованием гидроксида калия в таком растворителе как DMSO,при повышенной температуре. Альтернативно,она может быть проведена с использованием хлорида цинка в растворителе, который не обладает неблагоприятным действием на реакцию(например, в тетрагидрофуране), при температуре кипения растворителя с обратным холодильником. В способе (b) подходящими уходящими группами являются ОН и Сl. Если соединение формулы XIV является карбоновой кислотой,реакция может быть проведена в присутствии подходящих конденсирующих агентов [например, моногидрата 1-гидроксибензотриазола,гидрохлорида 1-(3-диметиламинопропил)-3 этилкарбодиимида и 4-метилморфолина] в растворителе, который не обладает неблагоприятным действием на реакцию (например, вCH2Cl2), при температуре, близкой к комнатной. Если уходящей группой является Сl, реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в CH2Cl2), приблизительно при 0 С. В способе (с) палладиевым катализатором может быть тетракис(трифенилфосфин)палладий. М может представлять собой В(ОН)2,В(СН 2 СН 2)2, Sn(СН 2 СН 2 СН 2 СН 3)3 или ZnCl. Реакция может быть проведена в растворителе,который не обладает неблагоприятным действием на реакцию (например, если М представляет собой В(ОН)2, в смеси толуола, этанола и 1 М водного карбоната натрия) при повышенной температуре (например, при температуре кипения растворителя с обратным холодильником). Если М представляет собой ZnCl или замещенный Sn, иодид меди(I) может быть необязательно использован в качестве сокатализатора. В способе (d) реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в н-бутаноле) в присутствии основания(например, триэтиламина) при повышенной температуре (например, при 100 С). В способе (е) подходящая уходящая группа включает в себя Сl. Реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в ТГФ) в присутствии основания (например, триэтиламина) при комнатной температуре. Реакция может быть также проведена без выделения соединения формулы XXVIIIа или 6 формулы XIIIa или XIIIb с трифосгеном и соединением формулы XXIX. В этом случае уходящей группой является -Сl. Реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию(например, триэтиламина), при температуре,близкой к комнатной. В способе (f) подходящие сильные основания включают в себя диизопропиламид лития. Реакция может быть проведена в растворителе,который не обладает неблагоприятным действием на реакцию (например, в ТГФ). В способе (g) реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в смеси н-ВuОН и диметилацетамида), в присутствии основания (например, триэтиламина), при повышенной температуре (например,при 80 С). В способе (h) реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в смеси н-бутанола и диметилацетамида), в присутствии основания (например, триэтиламина), при повышенной температуре (например,при 100 С). Соединения формулы Х [см. способ (а)] могут быть получены путем взаимодействия соединения формулы XI в которой R1-3 такие, как определены выше, с комбинацией соединения формулы XII в которой R4 и L такие, как определены выше, и оксихлорида фосфора в дихлорметане при температуре кипения растворителя с обратным холодильником. Соединения формул XIIIa или XIIIb [см. способ (b)], в которых Х представляет СН, могут быть получены из соединений формул XVa или XVb соответственно 7 в которых R1-3, R6, R7, m, n и р такие, как определены выше, путем барботирования газообразного НСl через раствор соединения в дихлорметане. Соединения формул XVa или XVb могут быть получены из соединений формул XVIa или в которых R6, R7, m, n и р такие, как определены выше, с использованием условий, указанных выше для способа (d), упомянутого выше. Соединения формулы XVIII [см. способ(с)], в которой Х представляет собой СН, могут быть получены путем циклизации соединения формулы XX в которых R1-3, R6, R7, m, n и р такие, как определены выше, путем циклизации с использованием гидроксида калия, при повышенной температуре (такой как 90 С), в ДМСО или диизопропиламида лития в растворителе, который не обладает неблагоприятным действием на реакцию (например, в тетрагидрофуране), при температуре, близкой к комнатной, и с гашением водой. Соединения формул XVIa или XVIb могут быть получены путем взаимодействия соединения формулы XI, как определено выше, с соединением формул XVIIa или XVIIb соответственно в которых R6, R7, m, n и р такие, как определены выше, с помощью способа, описанного выше для получения соединений формулы Х. Соединения формул XIIIа или XIIIb, в которых Х представляет собой N, могут быть получены путем взаимодействия соединения формулы XXII в которой R1, R2, R4, и L такие, как определены выше, с использованием условий реакции, указанных для способа (а), упомянутого выше. Соединения формулы XX могут быть получены путем взаимодействия соединения формулы XXI в которой R1 и R2 такие, как определены выше, с соединением формулы XII, как определено выше, с использованием способа, описанного выше, для получения соединений формулы X. Соединения формулы XVIII, в которой Х представляет собой N, могут быть получены путем взаимодействия соединения формулы в которой R1 и R2 такие, как определены выше, с соединением формул ХХIIIа или XXIIIb соответственно, как определено выше, с использованием условий реакции, указанных выше для способа (d). Соединения формулы XXII [см. способы(d) и (h)] могут быть получены из соединения формулы XXIV 9 Соединения формулы XXIV могут быть получены из соединения формулы XXVXIX, как определено выше, с использованием условий реакции, описанных выше для способа(с). Соединения формулы XXV могут быть получены из соединений формулы XXVI в которой R1 и R2 такие, как определены выше, с использованием подходящих методов. Соединения формулы XXII могут быть также получены в соответствии со схемой 1. Схема 1XXVIIIb [см. способ (е)], в которых Lg представляет собой Сl, могут быть получены из соединений формулы XIIIа или XIIIb соответственно путем взаимодействия с трифосгеном. Эта реакция может быть проведена в растворителе, который не обладает неблагоприятным действием на реакцию (например, в CH2Cl2), в присутствии основания (например, триэтиламина), приблизительно при -10 С. Соединения формулы Х могут быть также получены путем взаимодействия соединения формулы XX с соединением формулы XIX, с использованием условий, описанных для способа (с). Соединения формул XI, XII, XIV, XVIIa,XVIIb, XIX, XXI, XXIIa, XXIIb, XXIIIa, XXIIIb,XXVI, XXIX и XXX являются или известными,или доступными с помощью известных методов,как иллюстрировано с помощью примеров. Промежуточные соединения формул X,XIIIa, XIIIb, XXII, XXVIIIa и XXVIIIb составляют следующий аспект данного изобретения. Для специалистов в данной области очевидно, что чувствительные функциональные группы могут быть защищены и защитные 10 группы удалены в процессе синтеза соединения данного изобретения. Это может быть осуществлено с помощью подходящих методов, например, как описано в 'Protective Groups in OrganicSynthesis' by T. W. Greene and P. G. M. Wuts,John Wiley and Sons Inc., 1991. Соединения данного изобретения являются полезными, потому что они обладают фармакологической активностью у животных. Особенно эти соединения являются полезными при лечении ряда заболеваний, включая гипертонию,инфаркт миокарда, нарушение эрекции у самцов, гиперлипидэмию, сердечную аритмию и доброкачественную гиперплазию простаты. Последнее заболевание представляет наибольший интерес. Таким образом, в соответствии с другим аспектом данного изобретения, предоставляется способ лечения доброкачественной гиперплазии простаты, включающий в себя введение пациенту, страдающему от такого заболевания, терапевтически эффективного количества соединения настоящего изобретения. Также предоставляется использование соединений данного изобретения в качестве лекарственных препаратов, и использование соединений данного изобретения в производстве лекарств для лечения доброкачественной гиперплазии простаты. Соединения данного изобретения могут быть введены с помощью любого подходящего способа, например, перорально, парентерально(например, внутривенно, трансдермально) или ректально. Необходимая ежедневная доза, конечно, варьируется в зависимости от конкретного используемого соединения, конкретного состояния при лечении и тяжести этого состояния. Однако в основном общая суточная доза составляет приблизительно от 0,01 до 10 мг/кг веса тела и предпочтительно приблизительно от 0,05 до 1 мг/кг, ее вводят от 1 до 4 раз в день. Пероральное введение представляет особый интерес. Соединения данного изобретения в основном вводят в виде подходящей фармацевтической композиции. Таким образом, в соответствии с другим аспектом данного изобретения,представляется фармацевтическая композиция,включающая в себя предпочтительно меньше 50 вес.% соединения данного изобретения в смеси с фармацевтически приемлемым адъювантом,разбавителем или носителем. Фармацевтическая композиция предпочтительно находится в виде единичной дозы. Такие дозы включают в себя твердые дозированные формы, например, таблетки, пилюли, капсулы, порошки, гранулы и суппозитории для перорального, парентерального или ректального введения; а также жидкие дозированные формы, например, стерильные растворы или суспензии для парентерального введения, подходящим образом ароматизированные сиропы, ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло и арахи 11 совое масло, а также эликсиры и подобные фармацевтические носители. Твердые композиции могут быть получены путем смешения активного ингредиента с фармацевтическими носителями, например, традиционными ингредиентами для таблетирования,такими как кукурузный крахмал, лактоза, сахароза, сорбит, тальк, стеариновая кислота, стеарат магния, дикальций фосфат, камеди и другие разбавители, например, вода, для образования гомогенной предварительно полученной композиции, в которой активный ингредиент однородно диспергирован таким образом, чтобы его можно было легко разделить на равнозначно эффективные единичные дозированные формы,содержащие обычно примерно от 0,1 до 500 мг активного ингредиента. Твердые дозированные формы могут быть покрыты оболочкой или другим образом приготовлены для пролонгирования действия композиции. Композиции изобретения могут также содержать соединение с ингибиторной активностью по отношению к 5 редуктазе человека[см. международную патентную заявку WO 95/28397], или соединение настоящего изобретения может быть представлено в виде фармацевтической упаковки, также содержащей соединение с ингибиторной активностью по отношению к 5 редуктазе человека в качестве комбинированного препарата для одновременного, раздельного или последовательного применения. Соединения настоящего изобретения могут быть тестированы в экспериментах, представленных ниже. Сократительные ответы простаты человека Ткань простаты разрезают на продольные полосы (приблизительно 3210 мм) и суспендируют в бане для органов при остаточном напряжении в 1 г в бикарбонатном растворе Кребса-Рингера следующего состава (мМ): NaCl(119), КСl (4,7), CaCl2 (2,5), КН 2 РO4 (1,2),MgSO4 (1,2), NaHCO3 (25), глюкоза (11), и газифицируют смесью 95%O2/5%СО 2. Этот раствор также содержит 10 мМ кокаин и 10 мМ кортикостерон. Ткани подвергают воздействию возбуждающей дозы (-)-норадреналина (100 мМ) и промывают в течение 45 мин. Изометрические сокращения получают в ответ на кумулятивные добавления (-)-норадреналина для получения контрольных кривых во всех тканях. Следующую кривую затем получают в присутствии или отсутствие антагониста (инкубируют в течение 2 ч). Значения сродства антагониста (рА 2) определяют, используя единственную концентрацию конкурирующего антагониста, рА 2=-log[А]/(DR1), где дозовое соотношение (DR) по отношению к соответствующему контролю получают с помощью единственной концентрации антагониста [А], допуская, что конкурентный антагонизм и регрессия по Шильду близки к единству. 12 Модель простатического давления и кровяного давления на анестезированной собаке. Половозрелых самцов коротконогих гончих (с весом тела 12-15 кг) анестезируют пентобарбитоном натрия (30-50 мг/кг в/в) и вставляют трахейную канюлю. Последующую анастезию поддерживают с помощью инфузии пентобарбитоном. Животных респирируют воздухом с помощью респиратора Bird Mk8 (Bird Corp.,Palm Springs, CA, USA) для поддержания уровня газов в крови в интервале рO2 90-110 мм Hg,pCO2 35-45 мм Hg, pH 7,35-7,45. Температуру тела поддерживают при 36-37,5 С, используя нагретый операционный стол. Катетеры помещают в левую бедренную артерию для регистрации кровяного давления и в левую бедренную вену для введения соединения. Сердечный пульс регистрируют через вывод II E.C.G. Лапаротомию проводят для введения канюль в обе уретры для предотвращения изменения объема жидкости в мочевом пузыре. Сердечный катетер типа 7F (с шаровой насадкой объемом 1,5 мл) вводят в мочевой пузырь через уретру. Шаровую насадку заполняют воздухом и катетер выводят, пока шарик не становится погружен в простату, что подтверждается цифровым датчиком давления. Давление в шарике регистрируют с помощью преобразователя Друка. Давление в простате и гемодинамические параметры регистрируют с помощью прибора Grass Polygraph(Grass Instruments, Quincy, Mass, U.S.A.) и данные регистрируют в непрерывном режиме с использованием микрокомпьютерной системы на основе Motorola 68000 (Motorola Inc., Temple,AZ, U.S.A.). Соединения смешивают с PEG 300 и вводят внутривенно через катетер в бедренную вену. Для получения кривых зависимости контрольная доза-ответ регистрируют ответы на введение фенилэфрина (в диапазоне 1-16 мкг/кг внутривенно в физ. растворе) (две контрольные кривые для каждого эксперимента). Соединения вводят (с учетом соединения основания) в диапазоне 10-300 мкг/кг внутривенно за 5 мин до получения кривых от фенилэфрина (получают до максимальной дозы, равной 128 мкг/кг, в присутствии тестового соединения). Вследствие 1-подобных дизритмических свойств фенилэфрина абсолютные максимальные ответные сигналы не были получены, а взяты на уровне, который на 10% превышает контрольный сигнал, полученный от 16 мкг/кг фенилэфрина. Концентрации лекарственных препаратов рассчитывают на основании соотношения молярный вес соединения/кг веса тела, таким образом позволяя проводить расчеты по схеме "псевдо рА 2" с использованием анализа по Шильду с применением дозовых соотношений, полученных по сдвигам на кривых доза фенилэфрина-ответ. Преимущество соединений данного изобретения состоит в том, что они являются более эффективными, имеют большую продолжитель 13 ность действия, более широкий диапазон активности, являются более стабильными, имеют меньше побочных эффектов или являются более селективными (в особенности они могут иметь благоприятный эффект при лечении доброкачественной гиперплазии простаты, не вызывая нежелательных сердечно-сосудистых эффектов,например, потому что они обладают способностью быть селективными антагонистами подтипов 1-адренорецепторов рецепторов простаты) или обладают другими более полезными свойствами, чем соединения предыдущего уровня техники. Изобретение иллюстрируется следующими примерами, в которых могут быть использованы следующие сокращения: ВuОН = бутанол,DMA = диметилацетамид,DMF = диметилформамид,DMPU = 1,3-диметил-3,4,5,6-тетрагидро 2(1 Н)-пиримидон,DMSO = диметилсульфоксид,EDTA = этилендиаминтетрауксусная кислота,EtOAc = этилацетат,EtOH = этанол,ч = час,МеОН = метанол,мин = минута,n-BuOH = н-бутанол,р.s.i. = фунтов на кв. дюйм,THF = тетрагидрофуран,ТСХ = тонкослойная хроматография. Промежуточное соединение 1. 1-(т-Бутилоксикарбонил)-1,4-диазепан. К раствору гомопиперазина (100 г, 1,0 моль) и триэтиламина (210 мл, 152 г, 1,5 моль) в(300 мл). Смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 18 ч, после чего CH2Cl2 упаривают при пониженном давлении. Полученный остаток распределяют между эфиром и 2N лимонной кислотой и водный слой экстрагируют эфиром (4200 мл). Водный слой подщелачивают 2N воднымNaOH и затем экстрагируют СН 2 Сl2 (4400 мл). Объединенные СН 2 Сl2 экстракты промывают Н 2 О (2 х), насыщенным солевым раствором (1 х) и высушивают над MgSO4. Упаривание при пониженном давлении сопровождается азеотропной перегонкой с CH2Cl2 (4 х), с получением указанного в заголовке соединения в виде желтого воскообразного твердого вещества (94,3 г,53%). Rf 0,25 (СН 2 Сl2/МеОН/0,88 Nнз 90/10/1,об/об). МС m/z 201 (МH+). Обнаружено: С, 58,86; Н, 10,03; N, 13,58; С 10 Н 20N2 О 2 0,05.СН 2 Сl2 соответствует С, 59,02; Н, 9,91; N, 13,70%. 14 Промежуточное соединение 2. 1-(тБутилоксикарбонил)-4-(4-морфолинкарбонил)1,4-диазепан. Раствор промежуточного соединения 1(92,0 г, 0,46 моль) и триэтиламина (96,0 мл, 69,7 г, 0,69 моль) в СН 2 Сl2 (500 мл) при 0 С обрабатывают по каплям раствором 4-морфолинкарбонил хлорида (64,0 мл, 82,0 г, 0,55 моль) в СН 2 Сl2 (100 мл) и реакцию перемешивают при комнатной температуре в атмосфере N2 в течение 18 ч. Реакционную смесь затем разбавляютCH2Cl2 (400 мл) и промывают 2N лимонной кислотой (3400 мл), насыщенным солевым раствором (1500 мл), высушивают над МgSO4 и упаривают, получая указанное в заголовке соединение в виде не совсем белого твердого вещества (141,7 г, 98%). Rf 0,80 (CH2Cl2/MeOH/ 0,88 NН 3 90/10/1, об/об). МС m/z 314 (МН+). Обнаружено: С, 57,50; Н, 8,69; N, 13,41; С 15 Н 27N3O4 соответствует С, 57,50; Н, 8,69; N,13,41%. Промежуточное соединение 3. Гидрохлорид 1-(4-морфолинкарбонил)-1,4-диазепана. Раствор промежуточного соединения 2(140,0 г, 0,44 моль) в СН 2 Сl2/МеОН (1/1, об/об,600 мл) при 0 С насыщают газообразным НСl и реакционную смесь перемешивают при комнатной температуре в атмосфере N2 в течение 18 ч,после чего реакционную смесь упаривают при пониженном давлении и суспендируют в EtOAc,получая после фильтрации белое гигроскопичное твердое вещество. Затем его очищают путем суспендирования в ацетоне, фильтрации, промывания эфиром и высушивания в вакууме при 60 С, получая указанное в заголовке соединение в виде бесцветного твердого вещества (99,0 г,90%). Rf 0,41 (CH2Cl2/MeOH/0,88 NН 3 84/14/2,об/об). МС m/z 214 (МН+) Обнаружено: С, 47,50; Н, 8,10; N, 16,55; С 10 Н 19N3O2 НСl0,2 H2O соответствует С, 47,41; Н, 8,12; N, 16,59%. Промежуточное соединение 4. 1-Ацетил-4(4-морфолинкарбонил)-1,4-диазепан. К раствору промежуточного соединения 3(50 г, 0,2 моль) и триэтиламина (42 мл, 30,5 г,0,3 моль) в СН 2 Сl2 (400 мл) при 5 С добавляют уксусный ангидрид (23 мл, 24,9 г, 0,24 моль) по каплям в течение 15 мин и реакцию затем перемешивают в течение еще 2 ч при комнатной температуре в атмосфере N2. Разбавление с помощью СН 2 Сl2 (600 мл) сопровождается промыванием насыщенным водным раствором бикарбоната натрия (2200 мл), и объединенные водные слои экстрагируют СН 2 Сl2 (1100 мл).CH2Cl2 слои объединяют и промывают насыщенным солевым раствором, высушивают над 15 насыщенным водным бикарбонатом натрия, и водный слой экстрагируют СН 2 Сl2 (5 х). Объединенные СН 2 Сl2 слои высушивают над MgSO4 и упаривают при пониженном давлении, получая желтое масло, которое затем подвергают азеотропной перегонке с CH2Cl2 (4x), получая указанное в заголовке соединение в виде желтого масла (47,1 г, 92%). Rf 0,45 (СН 2 Сl2/МеОН/ 0,88 NН 3 90/10/1, об/об). МС m/z 256 (МН+). Обнаружено: С, 52,62; Н, 8,18; N, 15,02; С 12 Н 21N3 О 30,3.CH2Cl2 соответствует С, 52,61; Н,7,75; N, 14,96%. Пример 1. 4-Амино-6,7-диметокси-2-[4-(4 морфолинкарбонил)-1,4-диазепан-1-ил]-5-(тиофен-3-ил)хинолин.a). 2-(3,4-Диметоксифенил)-4,4-диметил 2-оксазолин. Указанное в подзаголовке соединение получают из 3,4-диметоксибензойной кислоты в соответствии со способом Meyers et al., J. Org.b). 2-(3,4-Диметокси-2-иодфенил)-4,4 димeтил-2-oкcaзoлин. н-Бутиллитий (2,5 М в гексане, 8,9 мл, 22,3 ммоль) добавляют по каплям к раствору продукта стадии (а) (4,2 г, 17,8 ммоль) в сухом эфире (200 мл) при 0 С и реакцию перемешивают в атмосфере N2 в течение 2 ч. Затем по каплям добавляют раствор иода (5,46 г, 21,5 ммоль) в эфире (100 мл) и реакцию оставляют нагреваться до комнатной температуры в течение 1 ч. Реакционную смесь выливают в Н 2 О, эфирный слой отделяют, промывают насыщенным водным раствором тиосульфата натрия (1 х), затем насыщенным солевым раствором (1 х), затем высушивают над MgSO4 и упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде желтого масла (5,2 г,80%). Rf 0,60 (CH2Cl2/MeOH/0,88 NН 3 90/10/1,об/об). МС m/z 362 (МН+) .c). 3,4-Диметокси-2-иодбензонитрил. К раствору продукта стадии (b) (5,2 г, 14,4 ммоль) в пиридине (30 мл) добавляют оксихлорид фосфора (2,7 мл, 4,4 г, 28,8 ммоль) и реакцию нагревают до 85 С в течение 18 ч. Реакционную смесь охлаждают, распределяют между насыщенным водным раствором карбоната натрия (300 мл) и затем экстрагируют эфиром(2100 мл). Эфирный слой промывают 2N НСl(275 мл), затем Н 2O (1 х), высушивают над МgSO4 и упаривают при пониженном давлении,получая желтое масло. Его очищают путем суспендирования в гексане и фильтрования, получая указанное в подзаголовке соединение в виде не совсем белого твердого вещества (2,82 г,68%). Rf 0,80 (CH2Cl2/MeOH 95/5, об/об). МСd). 3,4-Диметокси-2-иод-6-нитробензонитрил. Нитрония тетрафторборат (1,73 г, 13,0 ммоль) добавляют порциями к раствору продукта стадии (с) (2,67 г, 9,2 ммоль) в ацетонитриле(40 мл) при 0 С. Реакцию перемешивают в течение 30 мин в атмосфере N2 и затем выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют EtOAc (1x). Органический слой промывают насыщенным солевым раствором (1x), высушивают над MgSO4 и упаривают при пониженном давлении, получая остаток, который суспендируют в гексане и фильтруют, получая указанное в подзаголовке соединение в виде не совсем белого твердого вещества (2,51 г, 82%). Rf 0,46 (EtOAc/гексан 1/1, об/об), МС m/z 352 (MNH4+).e). 6-Амино-3,4-диметокси-2-иодбензонитрил. К раствору продукта стадии (d) (3,50 г,0,01 моль) в СН 2 Сl2 (90 мл) добавляют раствор дитионита натрия (20,11, 0,11 моль) в Н 2 О (60 мл). К полученной смеси добавляют хлорид тетра-н-бутиламмония (1,45 г, 5,24 ммоль) и реакцию интенсивно перемешивают в течение 1,5 ч. Смесь распределяют между CH2Cl2 и H2O,органический слой отделяют, высушивают надMgSO4 и упаривают при пониженном давлении. Остаток распределяют между EtOAc и 2N НСl,затем водный слой подщелачивают 2N воднымNaOH и экстрагируют EtOAc (3 х). Объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении, получая остаток, который очищают на силикагеле,элюируя CH2Cl2, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (1,69 г, 53%), Rf 0,55 (EtOAc/гексан 1/1, об/об), МС m/z 322 (MNH4+).f). 3,4-Диметокси-2-иод-6[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]этилиденаминобензонитрил. Оксихлорид фосфора (0,6 мл, 6,08 ммоль) добавляют к раствору промежуточного соединения 4 (2,82 г, 11,0 ммоль) в CH2Cl2 (20 мл) и реакцию перемешивают в течение 20 мин при комнатной температуре. Затем добавляют продукт стадии (е) (1,68 г, 5,52 ммоль) и реакцию нагревают с обратным холодильником в течение 18 ч, после чего ее охлаждают, выливают в лед,смесь подщелачивают водным бикарбонатом натрия и продукт экстрагируют EtOAc (3 х). Объединенные органические экстракты высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH (97/3,об/об), получая указанное в подзаголовке соединение в виде бесцветного твердого веществаg). 4-Aмино-6,7-диметокси-5-иод-2-[4-(4 морфолинкарбонил)-1,4-диазепан-1-ил]хинолин. Раствор продукта стадии (f) (2,0 г, 3,7 ммоль) в смеси ТГФ (50 мл) и DMPU (10 мл) охлаждают до -78 С и обрабатывают раствором диизопропиламида лития в циклогексане (1,5 М,2,7 мл) в атмосфере N2. Реакцию нагревают до 0 С и перемешивают в течении 30 мин, после чего реакцию снова охлаждают до -78 С и добавляют еще порцию диизопропиламида лития в ТГФ (1,5 М, 2,7 мл). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 30 мин, после чего ее снова охлаждают до -78 С и обрабатывают третьей порцией диизопропиламида лития в ТГФ (1,5 М,2,0 мл). Реакцию снова нагревают до комнатной температуры и перемешивают в течение 20 мин,после чего ее гасят Н 2 О и экстрагируют EtOAc(3 х). Органический слой промывают последовательно Н 2O и насыщенным солевым раствором,высушивают над МgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(98/2, об/об). Указанное в подзаголовке соединение (1,30 г, 65%) получают в виде светлокоричневого твердого вещества. Rf -0,50h). 4-Амино-6,7-диметокси-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]-5-(тиофен-3 ил)хинолин. К раствору продукта стадии (g) (500 мг,0,92 ммоль) в смеси толуола (6 мл) и EtOH (3 мл) добавляют тиофен-3-бороновую кислоту(236 мг, 1,85 моль), тетракис(трифенилфосфин)палладий (32 мг, 0,03 ммоль) и 1 М водный раствор карбоната натрия (1 мл) и реакционную смесь нагревают с обратным холодильником в атмосфере N2 в течение 18 ч. При охлаждении реакционную смесь разбавляют Н 2O,экстрагируют EtOAc (3 х). Объединенные органические слои высушивают над МgSO4 и упаривают при пониженном давлении. Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/ 0,88 NН 3 (90/10/1, об/об), получая указанное в заголовке соединение в виде бесцветной пены 18 Указанное в заголовке соединение получают по способу примера 1(h) из соединения примера 1(g) и тиофен-2-бороновой кислоты. Неочищенный продукт очищают на силикагеле,элюируя CH2Cl2/MeOH/0,88 NН 3 (90/10/1,об/об), получая указанное в заголовке соединение (26%) в виде бесцветной пены. МС m/z 498C25H31N5O4S 0,7.CH2Cl2 соответствует С, 55,40; Н, 5,86; N, 12,57%. Пример 3. 4-Амино-6,7-диметокси-5-(2 фурил)-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]хинолин. Указанное в заголовке соединение получают по способу примера 1(h) из соединения примера 1(g) и фуран-2-бороновой кислоты(1976)]. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3C25H31N5O5 0,25.СН 2 Сl2 соответствует С, 60,29; Н, 6,31; N, 13,92%. Пример 4. 4-Амино-6,7-диметокси-5-(3 фурил)-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]хинолин. Указанное в заголовке соединение получают по способу примера 1(h) из соединения примера 1(g) и фуран-3-бороновой кислоты(1976)]. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3 19 К раствору соединения примера 1(g) (700 мг, 1,29 ммоль) в диоксане (15 мл) добавляют 2(три-н-бутилстаннил)пиридин (1,42 г, 3,88 ммоль),тетракис(трифенилфосфин)палладий(150 мг, 0,13 ммоль), иодид меди (I) (37 мг, 0,19 ммоль) и хлорид лития (271 мг, 6,5 ммоль),смесь нагревают с обратным холодильником в атмосфере N2 в течение 18 ч. При охлаждении реакционную смесь концентрируют при пониженном давлении и остаток распределяют между 2N НСl и EtOAc. Водный слой промывают тремя дополнительными порциями EtOAc и затем подщелачивают 2N водным NaOH. Продукт затем экстрагируют EtOAc (3 х), высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3(90/10/1, об/об), с получением указанного в заголовке соединения в виде бесцветного твердого вещества (210 мг, 33%). Rf 0,23a). 3,4-Диметокси-2-иодбензойная кислота. Раствор соединения примера 1(b) (115 г,0,32 моль) в смеси 3N НСl (530 мл) и EtOH (200 мл) нагревают с обратным холодильником в течение 36 ч. При охлаждении продукт фильтруют, высушивают на воздухе и затем промывают гексаном. Твердое вещество затем растворяют в CH2Cl2, высушивают над MgSO4 и упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде белого твердого вещества. Rf 0,38 (EtOAc). МС m/z 309b). Этиловый эфир 3,4-диметокси-2 иодбензойной кислоты. К суспензии продукта стадии (а) (69,3 г,0,23 моль) в CH2Cl2 при 0 С добавляют оксалил хлорид (25 мл, 0,27 моль) и DMF (0,9 мл, 11,3 ммоль) и реакцию перемешивают в течение 18 ч при комнатной температуре. Затем в реакцию добавляют EtOH (20 мл, 0,34 моль), перемешивают в течение еще 30 мин, после чего ее обрабатывают триэтиламином (78 мл, 0,56 моль). Реакционную смесь распределяют между СН 2 Сl2 и Н 2 О. Органический слой отделяют,промывают последовательно 2N НСl (3 х) и насыщенным солевым раствором, высушивают над MgSO4 и упаривают при пониженном дав 002851 20 лении, получая коричневое масло. Продукт очищают на силикагеле, элюируя CH2Cl2, с получением указанного в подзаголовке соединения в виде коричневого масла (30 г, 39%). Rf 0,73 (EtOAc). МС m/z 337 (МH+).c). Этиловый эфир 3,4-диметокси-2-иод-6 нитробензойной кислоты. Нитроний тетрафторборат (11 г, 84 ммоль) добавляют к раствору продукта стадии (b) (30 г,64 ммоль) в ацетонитриле (300 мл) при 0 С и реакцию перемешивают в течение 1,5 ч в атмосфере N2. Реакционную смесь затем разбавляют эфиром, подщелачивают 2N водным NaOH и водный слой экстрагируют эфиром (3 х). Объединенные органические слои промывают насыщенным солевым раствором, высушивают надMgSO4 и упаривают при пониженном давлении. Продукт очищают на силикагеле, элюируя гексан/EtOAc (85/15, об/об), с получением указанного в подзаголовке соединения в виде желтого твердого вещества (21,3 г, 87%), Rf 0,77d). Этиловый эфир 6-амино-3,4-диметокси 2-иодбензойной кислоты. Указанное в подзаголовке соединение получают по способу примера 1(е) из продукта стадии (с). Указанное в подзаголовке соединение (84%) получают в виде бесцветного твердого вещества. Rf 0,67 (EtOAc). MC m/z 352 (МН+).e). 2,4-Дигидрокси-6,7-диметокси-5-иодхиназолин. Цианат натрия (9 г, 0,14 моль) и трифторуксусную кислоту (11 мл, 0,14 моль) добавляют при перемешивании к раствору продукта стадии (d) (12 г, 34,2 ммоль) в СН 2 Сl2 и перемешивание продолжают в течение 18 ч. Реакционную смесь затем упаривают при пониженном давлении, добавляют Н 2O и полученное твердое вещество фильтруют, промывают водой. Суспензию твердого вещества в Н 2 О (50 мл) обрабатывают гранулированным NaOH (10 г) и смесь нагревают до 60 С в течение 30 мин, после чего реакцию охлаждают, нейтрализуют концентрированной НСl и полученное твердое вещество выделяют путем фильтрации, промывания Н 2 О и эфиром. Указанное в подзаголовке соединение получают в виде бесцветного твердого вещества (8,4 г, 71%). Rf 0,30 (EtOAc). 1f). 2,4-Дигидрокси-6,7-диметокси-5-(тиофен-3-ил)хиназолин. Указанное в подзаголовке соединение получают по способу примера 1(h) из продукта стадии (е). Указанное в подзаголовке соединение (84%) получают в виде бледно-желтого твердого вещества. Rf 0,28 (EtOAc). MC m/z 305g). 4-Амино-2-хлор-6,7-диметокси-5-(тиофен-3-ил)хиназолин. Продукт стадии (f) добавляют к смеси оксихлорида фосфора (9 мл, 96 ммоль) и N,N 21 диметаланилина (1 мл, 8 ммоль) и реакционную смесь нагревают до 110 С в течение 5 ч. При охлаждении реакционную смесь выливают в лед и распределяют между 2N НСl и эфиром. Органический слой отделяют, промывают насыщенным солевым раствором и упаривают, получая коричневое масло. Его помещают в смесь СН 2 Сl2 (100 мл) и МеОН (100 мл), охлаждают до 0 С и насыщают NН 3. Реакцию перемешивают в течение 20 ч, насыщают еще раз NH3 и перемешивают в течение еще 5 ч. Реакционную смесь упаривают досуха при пониженном давлении и остаток распределяют между EtOAc и 2N НСl. Органический слой промывают насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении. Перетирают с метанолом и фильтруют, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (255 мг, 25%). Rf 0,78 (EtOAc). MC m/z 322 (МН+).h). 4-Aмино-6,7-диметокси-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]-5-(тиофен-3 ил)хиназолин. Смесь продукта стадии (g) (220 мг, 0,68 ммоль), триэтиламина (0,24 мл, 1,7 ммоль) и промежуточного соединения 3 (250 мг, 1,0 ммоль) в н-бутаноле (50 мл) нагревают до 100 С в атмосфере N2 в течение 5 дней. После охлаждения реакционную смесь распределяют между 2N водным NaOH и EtOAc. Органический слой отделяют и промывают дополнительными порциями 2N водного NaOH (2 х), затем насыщенным солевым раствором (2 х). После высушивания над MgSO4 и упаривания при пониженном давлении, продукт перетирают с EtOAc, фильтруют и перекристаллизовывают из толуола, получая указанное в заголовке соединение в виде бесцветного твердого вещества (33 мг, 10%). Rf 0,08 (EtOAc). MC m/z 499 (МН+). 1(a). Натриевая соль 4-амино-6,7 диметокси-2-гидрокси-5-иодхиназолина. Суспензию соединения примера 1(е) (9,16 г, 30 ммоль) в СН 2 Сl2 (200 мл) обрабатывают цианатом натрия (7,9 г, 120 ммоль) и трифторуксусной кислотой (8,4 мл, 105 ммоль) по каплям при комнатной температуре в атмосфере N2 и реакцию перемешивают в течение 60 ч. Смесь затем упаривают при пониженном давлении и полученное твердое вещество суспендируют в смеси водного NaOH (20 г в 150 мл) и МеОН(200 мл) и реакцию перемешивают при комнат 002851 22 ной температуре в течение 1 ч. Полученный оранжевый раствор затем упаривают при пониженном давлении для удаления МеОН и полученную водную суспензию обрабатываютEtOAc, фильтруют и твердое вещество промывают последовательно Н 2 О (3 х), ацетоном (3 х) и эфиром, получая указанное в подзаголовке соединение в виде бледно-желтого твердого вещества (7,75 г, 69%). Rf 0,53 (CH2Cl2/MeOH/0,88DMF (1,8 мл, 23,0 ммоль) добавляют по каплям к оксихлориду фосфора (5,4 мл, 57,6 ммоль) и затем добавляют продукт стадии (а)(4,0 г, 11,5 ммоль). Полученную смесь нагревают до 90 С в течение 30 мин, после чего добавляют дополнительное количество (5 мл) оксихлорида фосфора и нагревание продолжают в течение 18 ч. Реакционную смесь охлаждают и осторожно выливают в смесь EtOAc (400 мл) и Н 2O (200 мл), смесь нейтрализуют водным бикарбонатом натрия и водный слой экстрагируютEtOAc (2 х). Объединенные органические слои объединяют, промывают насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении, получая коричневое твердое вещество. Его суспендируют в 2N водном NaOH (300 мл), добавляют диоксан (100 мл) и смесь нагревают до 90 С при интенсивном перемешивании в течение 2 мин. При охлаждении твердое вещество отделяют и собирают путем фильтрации, промывают последовательно Н 2 О и ацетоном и высушивают в вакууме при 60 С, получая указанное в подзаголовке соединение в виде не совсем белого твердого вещества (2,79 г, 66%). Rf 0,76 (EtOAc). МС m/z 366,368 (МН+).(c). 4-Aмино-6,7-диметокси-5-иод-2-[4-(4 морфолинкарбонил)-1,4-диазепан-1-ил]хиназолин. Указанное в подзаголовке соединение получают по способу примера 6(h) из продукта стадии (b). Указанное в подзаголовке соединение получают с количественным выходом в виде светло-коричневой пены. Rf 0,41 (EtOAc). МС m/z 543 (МН+).(d). 4-Aмино-6,7-диметокси-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]-5-(3-пиридил) хиназолин. К раствору продукта стадии (с) (300 мг,0,55 ммоль) в смеси THF (25 мл) и H2O (5 мл) добавляют 3-пиридилдиэтил боран (485 мг, 3,3 ммоль),тетракис(трифенилфосфин)палладий(64 мг, 0,055 ммоль) и гидроксид калия (600 мг,10,7 ммоль) и смесь нагревают с обратным холодильником в течение 18 ч в атмосфере N2. После охлаждения реакционную смесь распределяют между EtOAc и 2N водным NaOH, водный слой отделяют и экстрагируют двумя дополнительными порциями EtOAc. Объединенные органические слои промывают насыщен 23 ным солевым раствором, высушивают надMgSO4 и упаривают при пониженном давлении,получая пену. Продукт очищают на силикагеле,элюируя СН 2 Сl2/МеОН (95/5, об/об), с получением указанного в заголовке соединения в виде бесцветной пены (42 мг, 15%). Rf 0,10C24H31N7O4 0,2EtOAc 0,5 Н 2 О соответствует С,59,57; H, 6,51; N, 18,85%. Пример 8. 4-Aмино-6,7-диметокси-2-[4-(4 морфолинкарбонил)-1,4-диазепан-1-ил]-5-(2 пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 5 из соединения примера 7(с). Продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (95/5, об/об), затем перетирают с гексан/EtOAc и перекристаллизовывают из толуола, получая указанное в заголовке соединение (19%) в виде бесцветного твердого вещества. Rf 0,25 (CH2Cl2/MeOH 95/5, об/об). МС m/z 494 (MH+). 1(a). 4-Амино-6,7-диметокси-5-иод-2-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)хиназолин. Указанное в подзаголовке соединение получают по способу примера 6(h) из соединения примера 7(b) и 5,6,7,8-тетрагидро-1,6-нафтиридина [Shiozawa et al., Chem. Pharm. Bull., 32,2522 (1984)]. Указанное в подзаголовке соединение получают с количественным выходом в виде коричневой пены. Rf 0,35 (CH2Cl2/MeOH/ 0,88 NН 3 90/10/1, об/об). МС m/z 464 (МН+).(b). 4-Амино-6,7-диметокси-5-(2-пиридил)2-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)хиназолин. Указанное в заголовке соединение получают по способу примера 5 из продукта стадии(а). Продукт очищают на силикагеле, элюируяCH2Cl2/MeOH (98/2, об/об), получая указанное в заголовке соединение (30%) в виде бледножелтого твердого вещества.C23H22N6O2 0,5 Н 2 О соответствует С, 65,24; Н,5,48, N, 19,84%. Пример 10. 4-Амино-6,7-диметокси-5-(2 пиримидил)-2-(5,6,7,8-тетрагидро-1,6-нафтирид 6-ил)хиназолин. Указанное в заголовке соединение получают по способу примера 5 из соединения примера 9(а) и 2-(три-н-бутилстаннил)пиримидина[Sandosham et al., Tetrahedron, 50, 275 (1994)]. Продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН (95/5, об/об), затем распределяют между 2N НСl и EtOAc, промывают водный слой EtOAc (3 х), подщелачивают 2N воднымNaOH и экстрагируют EtOAc (3 х). Объединенные органические слои высушивают над МgSO4 и упаривают при пониженном давлении, получая указанное в заголовке соединение (21%) в виде бледно-желтого твердого вещества. Rf 0,39DMF диметилацеталь (5,82 мл, 0,044 моль) добавляют к перемешиваемому раствору 1-boc4-пиперидона [Ashwood et al., J. Chem. Soc., Perkin 1, 641 (1995)] (8,73 г, 0,044 моль) в DMF (80 мл) и реакционную смесь нагревают до 80 С в атмосфере N2 в течение 18 ч. После охлажденияDMF удаляют при пониженном давлении и остаток распределяют между EtOAc и Н 2 О. Органический слой промывают Н 2 О и насыщенным солевым раствором, затем высушивают надEtOH (150 мл), затем формамидин ацетат (3,45 г, 0,033 моль) и реакцию перемешивают при комнатной температуре в атмосфере N2 в течение 30 мин. Затем добавляют раствор продукта стадии (а) (8,43 г, 0,033 моль) в EtOH (50 мл) и реакцию нагревают с обратным холодильником в течение 18 ч, после чего смесь охлаждают и концентрируют при пониженном давлении. Остаток распределяют между EtOAc и Н 2O. Органический слой промывают насыщенным соле 25 вым раствором и высушивают над MgSO4. Очищают на силикагеле, элюируя CH2Cl2/MeOH(c). 5,6,7,8-Тетрагидро-1,3,6-триазанафталин гидрохлорид. НСl барботируют через раствор продукта стадии (b) (4,80 г, 0,020 моль) в смеси МеОН и эфира (50 мл, 1/1, об/об) при 0 С до насыщения. Затем смесь оставляют нагреваться до комнатной температуры в течение 2 ч, после чего образуется осадок. Его отделяют путем декантации супернатанта раствора, промывания эфиром (2 х) и высушивания в вакууме, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (2,85 г, 81%). Rf 0,13(d). 4-Амино-6,7-диметокси-5-иод-2(5,6,7,8-тетрагидро-1,3,6-триазанафт-6-ил)хиназолин. Указанное в подзаголовке соединение получают по способу примера 6(h) из продукта стадии (b) и соединения примера 7(b). Указанное в подзаголовке соединение (65%) получают в виде бесцветного твердого вещества. Rf 0,52(е). 4-Амино-6,7-диметокси-5-(2-пиримидил)-2-(5,6,7,8-тетрагидро-1,3,6-триазанафт-6 ил)хиназолин. Указанное в заголовке соединение получают по способу примера 5 из продукта стадии(d) и 2-(три-н-бутилстаннил)пиримидина. Продукт очищают на силикагеле, элюируя(a). 4-Амино-2-хлор-6,7-диметокси-5-(2 пиридил)хиназолин. К раствору соединения примера 7(b) (1,0 г,2,7 ммоль) в диоксане (20 мл) добавляют 2-(трин-бутилстаннил)пиридин (1,1 г, 3,0 ммоль), хлорид лития (1,5 г, 35 ммоль), тетракис(трифенилфосфин)палладия (320 мг, 0,27 ммоль) и иодид меди (I) (78 мг, 0,41 ммоль) и реакцию нагревают до 100 С в течение 2 ч. После охлаждения реакционную смесь распреде 002851 26 ляют между 2N НСl и EtOAc, водный слой дополнительно экстрагируют EtOAc (3 х), затем подщелачивают 2N водным NaOH и экстрагируют EtOAc (3 х). Объединенные органические слои высушивают над МgSO4 и упаривают при пониженном давлении, получая бледно-желтое твердое вещество. Его суспендируют в эфире и фильтруют, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества. (660 мг, 76%). Rf 0,53 (EtOAc). МС m/z 317,319 (МН+).(b). 4-Амино-2-(7-аминосульфонил-1,2,3,4 тетрагидроизохинол-2-ил)-6,7-диметокси-5-(2 пиридил)хиназолин. К раствору продукта стадии (а) (250 мг, 0,8 ммоль) в н-бутанол/DMA (3:1, об/об, 8 мл) добавляют гидрохлорид 1,2,3,4-тетрагидроизохинолин-7-сульфонамида (300 мг, 1,2 ммоль) и триэтиламин (0,33 мл, 2,4 ммоль), реакционную смесь нагревают до 100 С в атмосфере N2 в течение 18 ч. Реакцию затем охлаждают, распределяют между EtOAc и 2N водным раствором гидроксида натрия, водный слой отделяют и экстрагируют EtOAc, объединенный органический слой промывают Н 2 О, высушивают над МgSO4 и упаривают при пониженном давлении. Очищают на силикагеле,элюируяCH2Cl2/MeOH, и продукт высаживают гексаном,получая, после фильтрации и высушивания в вакууме, указанное в заголовке соединение в виде бесцветного твердого вещества (198 мг,50%). Rf 0,50 (CH2Cl2/MeOH/0,88 NН 3 84/14/2,об/об). MC m/z 493 (МН+). 1C24H24N6O4S 0,4 гексан 0,9H2O соответствует С,58,97; Н, 5,73, N, 15,64%. Пример 13. 4-Амино-6,7-диметокси-2-(2 изоиндолинил)-5-(2-пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из соединения примера 12(а) и гидрохлорида изоиндолина. Продукт очищают на силикагеле, элюируя 27 Пример 14. 4-Амино-6,7-диметокси-5-(2 пиридил)-2-(5,6,7,8-тетрагидро-1,3,6-триазанафт-6-ил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из соединения примера 12(а) и соединения примера 11(с). Продукт очищают на силикагеле, элюируяEtOAc/MeOH (95/5, об/об), затем перетирают с эфиром, получая указанное в заголовке соединение (35%) в виде бесцветного твердого вещества. Rf 0,18 (CH2Cl2/MeOH/0,88 NH3 90/10/1,об/об). МС m/z 416 (МН+). 1(40 мл) при 0 С добавляют по каплям раствор ди-(т-бутил)дикарбоната в CH2Cl2 и реакцию оставляют перемешиваться при комнатной температуре в течение 18 ч. Реакционную смесь упаривают при пониженном давлении, получая масло, которое помещают в CH2Cl2, промывают водным раствором карбоната натрия (3 х), 1N(2 х). Органический слой отделяют, высушивают над МgSO4 и упаривают, получая масло. Перетирают с гексаном, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (2,33 г, 80%). Rf 0,8 (CH2Cl2/MeOH 95/5, об/об). Т.пл. 106-108 С.(b). 7-Амино-3-т-бутилоксикарбонил 2,3,4,5-тетрагидро-1 Н,3-бензазепин. Раствор продукта стадии (а) (2,1 г, 7,2 ммоль) в смеси EtOAc (20 мл) и МеОН (20 мл) гидрогенируют над палладием на угле (5% вес/вес, 100 мг) при 345 кПа (50 psi) и комнатной температуре в течение 3 ч. После фильтрации фильтрат упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде масла (2,0 г, количественно). Rf 0,7 (СН 2 Сl2/МеОН 9/1, об/об). Обнаружено: С, 68,96; Н, 8,63; N, 10,33; С 15 Н 22N2O2 соответствует С, 68,67; Н, 8,45; N,10,68%.(c). 3-т-Бутилоксикарбонил-7-метансульфонамидо-2,3,4,5-тетрагидро-1H,3-бензазепин. Метансульфонилхлорид (0,56 мл, 7,3 ммоль) добавляют по каплям к раствору продукта стадии (b) (1,9 г, 7,2 ммоль) в пиридине(40 мл) при 0 С и полученный оранжевый раствор оставляют перемешиваться в течение 18 ч. Реакцию упаривают при пониженном давлении,получая масло, которое растворяют в CH2Cl2 и экстрагируют последовательно водным раствором бикарбоната натрия (3 х) и насыщенным солевым раствором (3 х). Органический слой отделяют, высушивают над МgSO4 и упаривают при пониженном давлении, получая масло. Продукт очищают на силикагеле, элюируяCH2Cl2/MeOH (95/5, об/об), затем перетирают с эфиром, получая указанное в подзаголовке соединение в виде белого твердого вещества (1,2 г, 49%). Rf 0,5 (CH2Cl2/MeOH 95/5, об/об). Т. пл. 153-154 С.(d). Гидрохлорид 7-метансульфонамидо 2,3,4,5-тетрагидро-1 Н,3-бензазепина. Указанное в подзаголовке соединение получают из продукта стадии (d) по способу примера 11(с). Указанное в подзаголовке соединение (71%) получают в виде бесцветного твердого вещества. Rf 0,25 (СН 2 Сl2/МеОН/0,88 NН 3 84/14/2, об/об).(e). 4-Aмино-6,7-диметокси-2-(7-метансульфонамидо-2,3,4,5-тетрагидро-1H,3-бензазепин-3-ил)-5-(2-пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из продукта стадии (d) и соединения примера 12(а). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3 (97/3/0,5, об/об) получая указанное в заголовке соединение (40%) в виде бесцветного твердого вещества. Rf 0,31(0,74 мл, 8,5 ммоль) и сразу получают тонкий белый осадок. Через 5 мин добавляют раствор карбоната натрия (1,7 г, 16,5 ммоль) в Н 2 О (20 мл), затем смесь МеОН и Н 2 О (20 мл, 1/1,об/об). Полученный прозрачный раствор перемешивают в течение 18 ч при комнатной температуре, после чего реакцию распределяют между EtOAc и 2N водным NaOH, водный слой отделяют и экстрагируют EtOAc (8x). Объединенные органические слои промывают насыщен 29 ным солевым раствором, высушивают надMgSО 4 и упаривают, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (650 мг, 70%). Rf 0,50(b). 4-Амино-6,7-диметокси-2-[7-(4-морфолинсульфонамидо)-1,2,3,4-тетрагидроизoхинолин-2-ил]-5-(2-пиридил)хиназолин. Указанное в заголовке соединение (40%) получают по способу примера 12(b) из продукта стадии (а) и соединения примера 12(а). МС m/z 563 (МН+). 1(a). 6-(т-Бутилоксикарбонил)-2-метил(5,6,7,8-тетрагидро-1,3,6-триазанафталин. Раствор этоксида натрия в EtOH получают путем добавления натрия (690 мг, 30,0 ммоль) кEtOH (75 мл) и обрабатывают соединением примера 11(а) (7,62 г, 30,0 ммоль) и гидрохлоридом ацетамида (3,12 г, 33,0 ммоль). Реакцию нагревают с обратным холодильником в течение 18 ч, после чего ее распределяют между EtOAc и водным бикарбонатом натрия. Органический слой отделяют, высушивают над МgSO4 и упаривают при пониженном давлении, получая масло. Продукт очищают на силикагеле, элюируя EtOAc/MeOH (98/2, об/об), с получением указанного в подзаголовке соединения в виде кристаллического твердого вещества (6,47 г,87%). Rf 0,31 (EtOAc/MeOH 95/5, об/об). 1(b). Гидрохлорид 2-метил-5,6,7,8-тетрагидро-1,3,6-триазанафталина. Указанное в подзаголовке соединение получают по способу примера 11(с) из продукта стадии (а). Продукт распределяют между EtOAc и 2N водным NaOH и водный слой экстрагируют несколько раз EtOAc. Объединенные органические слои высушивают над МgSO4 и упаривают при пониженном давлении, получая указанное в подзаголовке соединение (7%) в виде желтого масла, кристаллизующегося при стоянии. 1(c). 4-Амино-6,7-диметокси-2-(2-метил 5,6,7,8-тетрагидро-1,3,6-триазанафт-6-ил)-5-(2 пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из продукта стадии (b) и соединения примера 12(а). Продукт(a). 1-Тритил-3-пиперидон. Тритил хлорид (13,1 г, 47,0 ммоль) добавляют к перемешиваемой суспензии 3-пиперидон гидрохлорида (5,79 г, 42,7 ммоль) и триэтиламина (14,9 мл, 107 ммоль) в CH2Cl2 (100 мл) и реакцию перемешивают в течение 16 ч в атмосфере N2 при комнатной температуре. Полученную смесь фильтруют и фильтрат промывают последовательно Н 2 О и 5% водной лимонной кислотой, высушивают над MgSO4 и упаривают при пониженном давлении. Перетирают с пентаном, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества(b). 4-(N,N-Диметиламинометилиден)-1 тритил-3-пиперидон. Указанное в подзаголовке соединение получают по способу примера 11(а) из продукта стадии (а). Кристаллизуют из эфира, получая указанное в подзаголовке соединение (52%) в виде бесцветного твердого вещества. Rf 0,23(с). 7-Тритил-5,6,7,8-тетрагидро-1,3,7-триaзaнaфтaлин. Указанное в подзаголовке соединение получают по способу примера 11(b) из продукта стадии (b). Продукт очищают с помощью хроматографии на силикагеле,элюируя СН 2 Сl2/эфир (9/1, об/об), с получением указанного в подзаголовке соединения (51%). Rf 0,33(d). Гидрохлорид 5,6,7,8-тетрагидро-1,3,7 триазанафталина. Указанное в подзаголовке соединение получают по способу примера 11(с) из продукта стадии (с). Продукт кристаллизуют из смеси МеОН/эфир, получая указанное в подзаголовке 31 соединение (65%) в виде оранжевого гигроскопичного твердого вещества. 1(e). 4-Амино-6,7-диметокси-5-(2-пиридил)2-(5,6,7,8-тетрагидро-1,3,7-триазанафт-7-ил) хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из продукта стадии (d) и соединения примера 12(а). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(95/5, об/об), с получением указанного в заголовке соединения (66%) в виде светлокоричневого твердого вещества. Rf 0,20(40 мл) и смесь оставляют стоять в течение 72 ч. Реакционную смесь затем выливают в водную лимонную кислоту (10%, 400 мл) и экстрагируют EtOAc (2230 мл). Органический слой упаривают, получая остаток, который очищают на силикагеле, элюируя CH2Cl2/MeOH, с получением указанного в подзаголовке соединения в виде твердого вещества (3,55 г, 46%). Rf 0,03(b). Гидрохлорид 5-метансульфонамидо 1,2,3,4-тетрагидроизохинолина. Раствор продукта стадии (а) (3,50 г, 15,7 ммоль) в EtOH (205 мл) обрабатывают диоксидом платины (1,5 g) и 1N HCl (15,7 мл). Смесь гидрогенируют при 414 кПа (60 psi) в течение 16 ч, после чего реакцию фильтруют. Фильтрат упаривают при пониженном давлении и перетирают с СН 2 Сl2, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества. Этот твердый остаток после фильтрации помещают в МеОН/Н 2 О (1/2, об/об), суспензию фильтруют, промывают СН 2 Сl2 (3 х) и фильтрат упаривают, получая второй сбор указанного в подзаголовке соединения (общий выход 3,45 г, 84%). Rf 0,21 (CH2Cl2/MeOH/0,88 NН 3 90/10/1, об/об).(с). 4-Амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2 ил)-5-(2-пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из продукта стадии (b) и соединения примера 12(а). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(95/5, об/об), с получением указанного в заголовке соединения (80%) в виде светлокоричневого твердого вещества. Rf 0,21(a). 7-(4-т-Бутилоксикарбонил-1-пиперазинсульфонил)-1,2,3,4-тетрагидроизохинолин. Указанное в подзаголовке соединение получают по способу примера 16(а) из 1-тбутилоксикарбонилпиперазина и 2-трифтороацетил-1,2,3,4-тетрагидроизохинолин-7-сульфонил хлорида. Продукт очищают на силикагеле,элюируя СН 2 Сl2/МеОН (93/7, об/об), с получением указанного в подзаголовке соединения(35%) в виде бесцветного твердого вещества. Rf 0,56 (CH2Cl2/MeOH 9/1, об/об). MC m/z 382(b). 4-Амино-6,7-диметокси-2-[7-(4-тбутилоксикарбонил-1-пиперазинсульфонил)1,2,3,4-тетрагидроизохинолин-2-ил]-5-(2-пиридил)хиназолин. Указанное в подзаголовке соединение получают по способу примера 12(b) из продукта стадии (а) и соединения примера 12(а). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(96/4, об/об), с получением указанного в подзаголовке соединения (69%) в виде бесцветного твердого вещества. Rf 0,25 (CH2Cl2/MeOH 96/4,об/об).(c). Тригидрохлорид 4-амино-6,7-диметокси-2-[7-(1-пиперазинсульфонил)-1,2,3,4-тетрагидроизохинолин-2-ил]-5-(2-пиридил)хиназолина. Указанное в заголовке соединение получают по способу примера 11(с). Продукт перетирают с СН 2 Сl2, получая указанное в заголовке соединение (58%) в виде бесцветного твердого вещества. MC m/z 562 (МН+). 1(a). 5-(Трифторметансульфонато)изохинолин. Пиридин (8,35 мл, 0,10 моль) добавляют к раствору 5-гидроксиизохинолина (5,0 г, 0,034 моль) в CH2Cl2, этот раствор охлаждают до-40 С и добавляют по каплям трифторметансульфоновый ангидрид (8,47 мл, 0,052 моль). Реакцию оставляют нагреваться до комнатной температуры и перемешивают в течение 18 ч,после чего добавляют Н 2 О. Органический слой отделяют, промывают насыщенным солевым раствором, высушивают над MgSO4 и упаривают при пониженном давлении. Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH (98/2,об/об), получая указанное в подзаголовке соединение в виде твердого вещества (6,93 г,73%). Rf 0,70 (CH2Cl2/MeOH 9/1, об/об).(b). 5-(N,N-Диэтилкарбоксамидо)изохинолин. К раствору продукта стадии (а) (500 мг, 1,8 ммоль) в DMF (4 мл) добавляют ацетат палладия (12 мг, 0,054 ммоль), трифенилфосфин (28 мг, 0,11 ммоль) и диэтиламин (3,7 мл, 36 ммоль), и реакцию нагревают при 60 С в атмосфере СО в течение 20 ч. После охлаждения реакционную смесь разбавляют насыщенным солевым раствором и экстрагируют EtOAc (3 х). Объединенные органические экстракты высушивают над MgSO4 и упаривают при пониженном давлении. Продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (97/3, об/об), получая указанное в подзаголовке соединение в виде твердого вещества (220 мг, 53%). Rf 0,45(c). 5-(N,N-Диэтилкарбоксамидо)-1,2,3,4 тетрагидроизохинолин. Указанное в подзаголовке соединение получают по способу примера 19(b) из продукта стадии (b). Неочищенный продукт распределяют между CH2Cl2 и водным раствором бикарбоната натрия и водный слой экстрагируют дополнительными порциями CH2Cl2. Объединенные органические экстракты высушивают надMgSO4 и упаривают при пониженном давлении. Продукт очищают на силикагеле, элюируя(d). 5-(N,N-Диэтиламинометил)-1,2,3,4 тетрагидроизохинолин. Раствор борана в THF (1M, 18 мл, 18,0 ммоль) добавляют по каплям к раствору продукта стадии (с) (1,39 г, 6,0 ммоль) в THF (20 34 мл). Реакционную смесь нагревают с обратным холодильником в течение 18 ч в атмосфере N2,после чего реакционную смесь охлаждают, добавляют к смеси 2N HCl/MeOH (1/1, об/об, 100 мл) и перемешивают в течение 2 ч. МеОН удаляют при пониженном давлении, реакционную смесь подщелачивают до рН 10 и экстрагируют СН 2 Сl2 (3 х). Объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде масла (840 мг, 64%). МС m/z 219 (МН+). 1(е). 4-Амино-2-[5-(N,N-диэтиламинометил)-1,2,3,4-тетрагидроизохинолин-2-ил]-6,7 диметокси-5-(2-пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из продукта стадии (d) и соединения примера 12(а). Продукт очищают на силикагеле, элюируя CH2Cl2/(а). 2-Ацетил-(5-метансульфонамидо)1,2,3,4-тетрагидроизохинолин. К раствору соединения примера 19(b) (2,87 г, 10,9 ммоль) в CH2Cl2 при 0 С добавляют уксусный ангидрид (1,2 мл, 13,1 ммоль) и триэтиламин (3,4 мл, 24,0 ммоль), реакцию перемешивают при комнатной температуре в течение 16 ч, после чего реакционную смесь распределяют между EtOAc и водным раствором бикарбоната натрия, водную фазу экстрагируют дополнительными порциями EtOAc. Объединенные органические экстракты высушивают над МgSO4 и упаривают, получая масло. Его растворяют в МеОН (15 мл) и обрабатывают водным раствором карбоната натрия (7%, вес/вес, 15 мл), смесь перемешивают в течение 16 ч при комнатной температуре, после чего МеОН удаляют при пониженном давлении, рН доводят до рН 8 2N водным раствором НСl и продукт экстрагируютEtOAc (2 х). Объединенные органические экстракты высушивают над MgSO4 и упаривают,получая масло, которое очищают на силикагеле,элюируя CH2Cl2/MeOH (95/5, об/об), получая(b). 3,4-Диметокси-2-иод-6-[1-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил) этилиденамино]бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (а) и соединения примера 1(е). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(c). 3,4-Диметокси-6-[1-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)этилиденамино]-2-(2-пиримидил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (b) и 2-(три-н-бутилстаннил)пиримидина. Продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН (96/4, об/об), получая указанное в подзаголовке соединение (32%) в виде пены. Rf 0,11 (CH2Cl2/MeOH 95/5, об/об). МС m/z 507(d). 4-Амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2 ил)-5-(2-пиримидил)хинолин. Раствор хлорида цинка в THF (0,5 М, 10,6 мл, 5,3 ммоль) добавляют к раствору продукта стадии (с) (180 мг, 0,36 ммоль) в THF (5 мл) и реакционную смесь нагревают с обратным холодильником в течение 70 ч, после чего добавляют дополнительную порцию хлорида цинка вTHF (0,5 М, 3,5 мл) и нагревание продолжают с обратным холодильником в течение дополнительных 7 ч. После охлаждения реакционную смесь распределяют между CH2Cl2 и растворомEDTA в 2N водном NaOH. Органический слой промывают насыщенным солевым раствором и высушивают над MgSO4. Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3(90/10/1, об/об), получая указанное в заголовке соединение в виде бесцветного твердого вещества (38 мг, 21%). Rf 0,28 (CH2Cl2/MeOH/0,88(а). 3-Гидрокси-2-иод-4-метокси-6-нитробензонитрил. К раствору соединения примера 1(d) (10,0 г, 30 ммоль) в коллидине (100 мл) добавляют иодид лития (4,0 г, 30 ммоль) и реакцию перемешивают при комнатной температуре в течение 18 ч, что сопровождается нагреванием при 100 С в течение 1,5 ч. После охлаждения реакционную смесь распределяют между 2N воднымEtOAc. Объединенные органические слои промывают 2N НСl (2 х), высушивают над МgSО 4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH (97/3, об/об), получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (6,96 г, 73%). Rf 0,16(b). 3-Этокси-2-иод-4-метокси-6-нитробензонитрил. К раствору продукта стадии (а) (6,95 г,21,7 ммоль) и бромоэтана (1,78 мл, 23,8 ммоль) в DMF (70 мл) добавляют карбонат калия (4,49 г, 32,5 ммоль) и реакцию нагревают до 60 С в течение 18 ч. После охлаждения реакционную смесь распределяют между 2N водной НСl иEtOAc. Органический слой отделяют, промывают Н 2 О, высушивают над MgSO4 и упаривают. Неочищенный продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (97/3, об/об), получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (2,94 г, 39%). Rf 0,68 (СН 2 Сl2/МеОН 95/5, об/об). МС m/z 366(c). 6-Амино-3-этокси-2-иод-4-метоксибензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(е) из продукта стадии (b). Продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (98/2, об/об), получая указанное в подзаголовке соединение (72%) в виде бесцветного твердого вещества. Rf 0,11(d). 3-Этокси-2-иод-6-[1-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил) этилиденамино]-4-метоксибензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (с) и соединения примера 22(а). Продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН(e). 3-Этокси-6-[1-(5-метансульфонамидо 1,2,3,4-тетрагидроизохинол-2-ил)этилиденамино]-4-метокси-2-(2-пиридил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (d) и 2-(три-н-бутилстаннил)пиридина. Продукт очищают на силикагеле, элюируя 37 Раствор продукта стадии (е) (1,14 г, 2,19 ммоль) в THF (10 мл) охлаждают до -78 С и обрабатывают раствором диизопропиламида лития в циклогексане (1,5 М, 4,4 мл, 6,6 ммоль). Реакцию затем оставляют нагреваться до комнатной температуры в течение 1 ч, распределяют между EtOAc и Н 2O, и водный слой экстрагируют 3 дополнительными порциями EtOAc. Объединенные органические слои промывают насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении. Очищают на силикагеле, элюируя(а). 6-Ацетил-5,6,7,8-тетрагидро-1,6-нафтиридин. К раствору 5,6,7,8-тетрагидро-1,6-нафтиридин (4,9 г, 36,5 ммоль) в СН 2 Сl2 при 0 С добавляют по каплям триэтиламин (6,1 мл, 43,8 ммоль) и ацетилхлорид (3,11 мл, 43,8 ммоль), и реакцию оставляют нагреваться до комнатной температуры и перемешивают в течение еще 18 ч. Реакционную смесь распределяют между H2O и CH2Cl2, слои отделяют и водный слой экстрагируют еще дважды СН 2 Сl2. Объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении, получая остаток, который очищают на силикагеле, элюируя EtOAc/MeOH/0,88 NН 3 (95/5/1, об/об). Получают указанное в подзаголовке соединение(b). 3,4-Диметокси-2-иод-6-[1-(5,6,7,8 тетрагидро-1,6-нафтирид-6-ил)этилиденамино] бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (а) и соединения примера 1(е). Продукт очищают на силикагеле, элюируя EtOAc/MeOH(c). 3,4-Диметокси-2-(2-пиридил)-6-[1(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)этилиденамино]бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (b) и 2-(три-н-бутилстаннил)пиридина. Продукт очищают на силикагеле, элюируя(d). 4-Амино-6,7-диметокси-5-(2-пиридил)2-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)хинолин. Указанное в заголовке соединение получают по способу примера 23(f) из продукта стадии (с). Продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3 (96/3,5/0,5, об/об),затем перетирают с эфиром, получая указанное в заголовке соединение (22%) в виде светлокоричневого твердого вещества. Rf 0,31(a). 3-4-Диметокси-2-пиримидил-6-[1(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)этилиденамино]бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта примера 24(b) и 2-(три-н-бутилстаннил)пиримидина. Продукт очищают на силикагеле,элюируя СН 2 Сl2/МеОН (98/2, об/об), получая указанное в подзаголовке соединение в виде пены (75%). Rf 0,21 (эфир). МС m/z 415 (МН+).(b). 4-Амино-6,7-диметокси-5-(2-пиримидил)-2-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил) хинолин. Порошок гидроксида калия (72 мг, 1,29 ммоль) добавляют к раствору продукта стадии(а) (530 мг, 1,28 ммоль) в DMSO (5 мл). Реакционную смесь нагревают до 95 С в течение 45 мин. После охлаждения реакционную смесь выливают в лимонную кислоту и подщелачивают 2N водным NaOH. Продукт затем экстрагируютEtOAc (х 4). Объединенные органические слои промывают Н 2O, насыщенным солевым раствором и высушивают над MgSO4. Продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН/ 0,88 NН 3 (96/3,5/0,5, об/об). Продукт перетирают с эфиром, получая указанное в заголовке соединение в виде оранжевого твердого вещест 39 ва (91 мг, 17%). Rf 0,11 CH2Cl2/MeOH (95/5,об/об). МС m/z 415 (МН+). 1(а). Метиловый эфир 3-гидрокси-4 метоксибензойной кислоты. К суспензии 3-гидрокси-4-метоксибензойной кислоты (33,63 г, 0,2 моль) в МеОН (500 мл) добавляют концентрированную серную кислоту(25 мл) и реакционную смесь нагревают с обратным холодильником в течение 2 ч. После охлаждения реакционную смесь концентрируют при пониженном давлении до 100 мл и остаток экстрагируют EtOAc. Органический слой промывают последовательно Н 2 О (х 2), насыщенным водным бикарбонатом натрия и насыщенным солевым раствором, высушивают надMgSO4 и упаривают при пониженном давлении,получая указанное в подзаголовке соединение в виде бесцветных кристаллов (33,0 г, 91%). Rf 0,59 (EtOAc). MC m/z 183 (МН+).(b). 2,2-2-Трифторэтилтрифторметансульфонат. К раствору трифторметансульфонового ангидрида (80,4 г, 0,3 моль) в СН 2 Сl2 (50 мл) добавляют по каплям смесь 2,2,2-трифторэтанола(28,0 г, 0,28 моль) и триэтиламина (29,3 г, 0,29 моль) при -40 С в течение 45 мин. После добавления реакционную смесь нагревают до комнатной температуры, промывают последовательно H2O и насыщенным водным бикарбонатом натрия и затем высушивают над MgSO4. Получают указанное в подзаголовке соединение в виде раствора в СН 2 Сl2, который немедленно используют на следующей стадии.(с). Метиловый эфир 4-метокси-3-(2,2,2 трифторэтокси)бензойной кислоты. К раствору продукта стадии (а) (33,0 г,0,181 моль) в смеси карбоната калия (41,4 г, 0,3 моль) и DMF (100 мл) добавляют раствор продукта, полученного на стадии (b) (65,0 г, 0,28 моль). Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, после чего реакционную смесь упаривают при пониженном давлении. Остаток распределяют между эфиром и Н 2 О. Органический слой промывают насыщенным солевым раствором, высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт перетирают с гексаном, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (42,55 г, 93% с 2 стадий). Rf 0,47 (СН 2 Сl2).(d). 4-Метокси-3-(2,2,2-трифторэтокси)бензойная кислота. К раствору продукта стадии (с) (42,25 г,0,16 моль) в МеОН добавляют 2N водный NaOH(160 мл, 0,32 моль). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч и затем при 50 С в течение 1 ч. После охлаждения реакционную смесь упаривают при пониженном давлении. Остаток распределяют между EtOAc и 2N НСl. Органический слой высушивают над MgSО 4 к упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (40,4 г, 100%). Rf 0,13 (гексан/EtOAc 1/1, об/об). MC m/z 251 (МН+).(e). 4,4-Диметил-2-[4-метокси-3-(2,2,2-трифторэтокси)фенил]-2-оксазолин. К суспензии продукта стадии (d) (40,0 г,0,16 моль) в СН 2 Сl2 (200 мл) и DMF (0,5 мл) добавляют оксалилхлорид (40,6 г, 0,32 моль) при 0 С в течение 15 мин. Реакционную смесь перемешивают при 0 С в течение 30 мин, нагревают до комнатной температуре в течение 1,5 ч и упаривают при пониженном давлении. Остаток затем снова растворяют в CH2Cl2 (300 мл) и добавляют в течение 15 мин к раствору 2 амино-2-метилпропанола (17,8 г, 0,2 моль) и триэтиламина (20,2 г, 0,2 моль) в CH2Cl2 (100 мл) при 0 С. Реакционную смесь затем перемешивают при комнатной температуре в течение 30 мин, промывают последовательно 5% лимонной кислотой и разбавляют водным бикарбонатом натрия, высушивают над MgSO4 и упаривают при пониженном давлении до объема 200 мл. Тионилхлорид (21,4 г, 0,18 моль) добавляют по каплям к раствору и полученную смесь перемешивают в течение 1 ч. Продукт затем экстрагируют Н 2 О, затем 0,5N водным NaOH. Объединенные водные фазы подщелачивают 2N водным NaOH и продукт экстрагируют СН 2 Сl2(х 2). Объединенные органические фазы высушивают над MgSO4 и упаривают при пониженном давлении, получая порцию неочищенного продукта. Первичный органический экстракт затем встряхивают с 2N водным NaOH и продукт экстрагируют СН 2 Сl2 (х 3). Объединенные органические экстракты высушивают надMgSO4 и упаривают при пониженном давлении. Остаток перерастворяют в эфирном растворе НСl (150 мл), полученное белое твердое вещество фильтруют, подщелачивают еще раз 2N водным NaOH и экстрагируют CH2Cl2 (х 3). Объединенные органические экстракты высушивают над MgSO4 и упаривают при пониженном давлении, получая вторую порцию неочищенного продукта. Объединенный неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH(95/5, об/об), получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (38,8 г, 80%). Rf 0,54 (EtOAc). MC m/z 304(f). 4,4-Диметил-2-[2-иод-4-метокси-3(2,2,2-трифторэтокси)фенил]-2-оксазолин. Указанное в подзаголовке соединение получают по способу примера 1(b) из продукта 41 стадии (е). Неочищенный продукт очищают на силикагеле, элюируя EtOAc/гексан (60/40,об/об). Продукт затем перетирают с эфиром,получая указанное в подзаголовке соединение в виде оранжевого твердого вещества (53%). Rf 0,27 (эфир/гексан 1/3, об/об). MC m/z 430 (МН+).(g). 2-Иод-4-метокси-3-(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(с) из продукта стадии (f). Неочищенный продукт перетирают со смесью гексан/эфир (60/40, об/об), получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (96%). Rf 0,5(h). 2-Иод-4-метокси-6-нитро-3-(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(d) из продукта стадии (g). Неочищенное коричневое твердое вещество перетирают с эфиром, получая указанное в подзаголовке соединение (72%). Rf 0,25(i). 6-Амино-2-иод-4-метокси-3-(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(е) из продукта стадии (h). Неочищенный продукт промывают через подложку из силикагеля, получая указанное в подзаголовке соединение в виде оранжевого твердого вещества (70%). Rf 0,74j). 2-Иод-4-метокси-6-1-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]этилиденамино-3(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (i) и промежуточного соединения 4. Неочищенный продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (90/10, об/об), получая указанное в подзаголовке соединение в виде оранжевого масла, которое перекристаллизовывают из EtOAc, с получением бесцветного твердого вещества (64%). Rf 0,12 (EtOAc). МС m/z 610 (МН+).(k). 4-Метокси-6-1-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]этилиденамино-2-(2 пиридил)-3-(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (j) и 2-(три-н-бутилстаннил)пиридина. Неочищенный продукт очищают на силикагеле,элюируя CH2Cl2/MeOH (96/4, об/об), получая указанное в подзаголовке соединение в виде желтой пены (91%). Rf 0,43 (СН 2 Сl2/МеОН/0,88(l). 4-Амино-7-метокси-2-[4-(4-морфолинкарбонил)-1,4-диазепан-1-ил]-5-(2-пиридил)-6(2,2,2-трифторэтокси)хинолин. К раствору продукта стадии (k) (0,56 г, 1 ммоль) в THF (10 мл) добавляют свежеприго 002851 42 товленный диизопропиламид лития (4 мл, 2 ммоль) при -20 С. Реакционную смесь затем медленно нагревают до комнатной температуры и перемешивают в течение 20 мин, после чего ее гасят Н 2 О и выливают в EtOAc. Органический слой затем промывают 2N водным NaOH, затем насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3(90/10/1, об/об), получая указанное в заголовке соединение в виде коричневой пены (0,39 г,70%). Rf 0,37 (СН 2 Сl2/МеОН/0,88 NН 3 93/7/1,об/об). МС m/z 561 (МН+). 1(a). 2-Иод-4-метокси-6-[1-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)этилиденамин]-3(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта примера 26(i) и соединения примера 24(а). Неочищенный продукт очищают на силикагеле,элюируя EtOAc/MeOH (80/20, об/об), получая указанное в подзаголовке соединение в виде бесцветной пены (70%). Rf 0,63 (СН 2 Сl2/МеОН/ 0,88 NН 3 90/10/1, об/об). МС m/z 531 (МН+).(b). 4-Метокси-2-(2-пиримидил)-6-[1(5,6,7,8-гетрагидро-1,6-нафтирид-6-ил)этилиденамино]-3-(2,2,2-трифторэтокси)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (а) и 2-(три-н-бутилстаннил)пиримидина. Неочищенный продукт очищают на силикагеле,элюируя CH2Cl2/MeOH/0,88 NН 3 (94/6/1, об/об),получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (49%). Rf 0,29 (CH2Cl2/MeOH/0,88 NН 3 93/7/1, об/об). МС(c). 4-Амино-7-метокси-5-(2-пиримидинил)-2-(5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)6-(2,2,2-трифторэтокси)хинолин. Указанное в заголовке соединение получают по способу примера 25(b) из продукта стадии (b). Неочищенный продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН (95/5, об/об),получая указанное в заголовке соединение в виде пены (8%). Rf 0,07 (CH2Cl2/MeOH 95/5,об/об). МС m/z 483 (МН+). 1THF (15 мл) добавляют по каплям н-бутиллитий(1,6 М в гексане, 2,75 мл, 4,4 ммоль) при -78 С в атмосфере азота. Реакционную смесь перемешивают при -78 С в течение 20 мин, затем к реакционной смеси добавляют раствор хлорида цинка (1,0 М в эфире, 12 мл, 12 ммоль) и полученный раствор нагревают до комнатной температуры. Добавляют соединение примера 1(f)(1,05 г, 1,94 ммоль), затем тетракис(трифенилфосфин)палладий (200 мг). Реакционную смесь нагревают с обратным холодильником в течение 3 ч. Затем добавляют дополнительную порцию оксазолцинката, полученного как описано выше с помощью оксазола (552 мг, 8 ммоль), н-бутиллития (1,6 М в гексане, 5,5 мл,8,8 ммоль), раствор хлорида цинка (1,0 М в эфир, 24 мл, 24 ммоль), затем тетракис(трифенилфосфин)палладия (100 мг). Реакционную смесь нагревают с обратным холодильником в течение 4 ч, после чего добавляют иодид меди(I) (100 мг). Полученную смесь нагревают с обратным холодильником в течение 24 ч. Охлажденную реакционную смесь выливают вEtOAc и промывают водным раствором EDTA,подщелачивают 2N водным NaOH и отделяют органический слой, промывают насыщенным солевым раствором, высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3 (92/8/1, об/об), получая указанное в заголовке соединение (87 мг,9%). Rf 0,46 (CH2Cl2/MeOH/0,88 NН 3 93/7/1,об/об). МС m/z 483 (МН+). 1Bull., 32, 2522, (1984)] (2,73 г, 0,0184 моль) в СН 2 Сl2 (30 мл) и триэтиламину (5,1 мл, 0,0368 моль) добавляют ацетилхлорид (1,57 мл, 0,0221 моль) при 0 С. Реакционную смесь перемешивают в течение 24 ч при комнатной температуре, после чего реакционную смесь промывают последовательно насыщенным водным бикарбонатом натрия, Н 2 О, насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении, получая указанное в подзаголовке соединение (3,27 г, 93%). Rf(b). 3,4-Димeтoкcи-2-иoд-6-[1-(2-метил 5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)этилиденамино]бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (а) и соединения примера 1(е). Неочищенный продукт очищают на силикагеле, элюируя смесью EtOAc/гексан (96/4, об/об), получая указанное в подзаголовке соединение в виде пены (87%). Rf 0,42 (EtOAc). МС m/z 477 (МН+).(c). 3,4-Диметокси-6-[1-(2-метил-5,6,7,8 тетрагидро-1,6-нафтирид-6-ил)этилиденамино]2-(2-пиридил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из соединения стадии (b) и 2-(три-н-бутилстаннил)пиридина. Неочищенный продукт очищают на силикагеле,элюируя EtOAc/MeOH (97/3, об/об), получая указанное в подзаголовке соединение в виде пены (25%). Rf 0,29 (EtOAc). МС m/z 428 (МН+).(d). 4-Амино-6,7-диметокси-2-(2-метил 5,6,7,8-тетрагидро-1,6-нафтирид-6-ил)-5-(2 пиридил)хинолин. Указанное в заголовке соединение получают по способу примера 26(1) из продукта стадии (с). Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3(a). 5-Метоксиизохинолин. К раствору 5-гидроксиизохинолина (10 г,69 ммоль) в МеОН (100 мл) добавляют раствор метоксида натрия в метаноле (30% по весу, 13,8 мл, 72,4 ммоль), затем фенилтриметиламмонийхлорид (12,4 г, 72,4 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 ч, после чего ее фильтруют и фильтрат упаривают при пониженном давлении, получая масло, которое растворяют в DMF (50 мл). Реакционную смесь нагревают с обратным холодильником в течение 2 ч, после чего реакционную смесь упаривают при пониженном давлении. Полученное масло распределяют между CH2Cl2 и 1N водным NaOH. Органический слой промывают дважды 1N водным NaOH,высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя смесью(b). 5-Метокси-1,2,3,4-тетрагидроизохинолин. К раствору продукта стадии (а) (6,11 г, 384 ммоль) в EtOH (200 мл) добавляют оксид платины (0,611 г), затем концентрированную НСl(3,2 мл, 38,4 ммоль). Реакционную смесь гидрогенируют при 345 кПа (50 p.s.i.) при комнатной температуре в течение 4 ч, после чего катализатор отфильтровывают и промывают EtOH. Фильтрат упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества(c). 2-Ацетил-5-метокси-1,2,3,4-тетрагидроизохинолин. К раствору продукта стадии (b) (6,26 г,31,4 ммоль) и триэтиламина (9,6 мл, 69,0 ммоль) в CH2Cl2 (150 мл) добавляют ацетилхлорид (2,7 мл, 37,7 ммоль) при 0 С в течение 15 мин. Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, после чего раствор промывают последовательно Н 2 О и насыщенным водным раствором бикарбоната натрия,высушивают над МgSО 4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя EtOAc, получая указанное в подзаголовке соединение в виде оранжевого масла (6,07 г, 94%). Rf 0,65(d). 3,4-Диметокси-2-иод-6-[1-(5-метокси 1,2,3,4-тетрагидроизохинол-2-ил)этилиденамино]бензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (с) и соединения примера 1(е). Неочищенный продукт очищают на силикагеле, элюируя СН 2 Сl2, получая указанное в подзаголовке соединение в виде оранжевых кристаллов(e). 3,4-Диметокси-6-[1-(5-метокси-1,2,3,4 тетрагидроизохинол-2-ил)этилиденамино]-2-(2 пиридил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (d) и 2-(три-н-бутилстаннил)пиридина. Неочищенный продукт очищают на силикагеле,элюируя эфиром, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (62%). Rf 0,73 (CH2Cl2/MeOH 90/10,об/об). МС m/z 443 (МН+).(f). 4-Амино-6,7-диметокси-2-(5-метокси 1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил) хинолин. Указанное в заголовке соединение получают по способу примера 25(b) из продукта стадии (е). Неочищенный продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН (95/5, об/об),получая указанное в заголовке соединение в виде бесцветного твердого вещества (10%). Rf 0,5 (CH2Cl2/MeOH 90/10, об/об). МС m/z 443(a). 2-Aцетил-6,7-диметокси-1,2,3,4-тетрагидроизохинолин. Указанное в подзаголовке соединение получают по способу примера 30(с) из 6,7 диметокси-1,2,3,4-тетрагидроизохинолин. Неочищенный продукт очищают на силикагеле,элюируя EtOAc, получая указанное в подзаголовке соединение в виде бесцветного твердого вещества (99%). Rf 0,15 (EtOAc). 1(b). 3,4-Диметокси-6-[1-(6,7-диметокси 1,2,3,4-тетрагидроизохинол-2-ил)этилиденамино]-2-иодбензонитрил. Указанное в подзаголовке соединение получают по способу примера 1(f) из продукта стадии (а) и соединения примера 1(е). Неочищенный продукт очищают на силикагеле, элюируя СН 2 Сl2, получая указанное в подзаголовке соединение (71%). Rf 0,74 (EtOAc). МО m/z 522(c). 3,4-Диметокси-6-[1-(6,7-диметокси 1,2,3,4-гетрагидроизохинол-2-ил)этилиденамино]-2-(2-пиримидил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из продукта стадии (b) и 2-(три-н-бутилстаннил)пиримидина. Очищают на силикагеле, получая указанное в подзаголовке соединение (33%). Rf 0,38(d). 4-Амино-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2 пиримидил)хинолин. Указанное в заголовке соединение получают по способу примера 26(l) из продукта стадии (с). Неочищенный продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН/0,88 NH3(a). 4-Амино-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагидроизохинол-2-ил)-5-иодхинолин. Указанное в подзаголовке соединение получают по способу примера 1(g) из соединения примера 31(b). Неочищенный продукт очищают на силикагеле, элюируя смесью EtOAc/гексан(1/1, об/об), затем EtOAc, получая указанное в подзаголовке соединение в виде не совсем белого твердого вещества (67%). Rf 0,5 (EtOAc). МС(b). 4-Амино-6,7-диметокси-2-(6,7-диметокси-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2 пиридил)хинолин. Указанное в заголовке соединение получают по способу примера 5 из продукта стадии(а) и 2-(три-н-бутилстаннил)пиридина. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NН 3 (95/5/0,5, об/об),получая указанное в заголовке соединение(а). 6-Бензил-3,4,5,6,7,8-гексагидро-1,6 нафтиридин-2-он. К раствору 1-бензил-4-пиперидона (213 г,1,13 моль) в толуоле (700 мл) добавляют пирролидин (190 мл, 2,25 моль), реакционную смесь закрывают насадкой Дина-Старка и нагревают до 150 С в течение 18 ч. Реакционную смесь охлаждают и упаривают при пониженном давлении, затем к остатку добавляют птолуолсульфоновую кислоту (4,0 г, 0,022 моль),после чего акриламид (160 г, 2,25 моль). Реакционную смесь нагревают при быстром перемешивании до 90 С в течение 1,5 ч и затем в течение еще 2 ч при 120 С. Охлажденную смесь затем фильтруют и полученное твердое вещество промывают ацетоном, затем эфиром. Маточные жидкости объединяют, упаривают при пониженном давлении и остаток распределяют между EtOAc и H2O. Органический слой отделяют, высушивают над MgSO4 и упаривают при пониженном давлении, получая дополнительную порцию твердого вещества. Твердое веще 002851 48 ство объединяют и нагревают с обратным холодильником с 4-толуолсульфоновой кислотой (10 г, 0,056 моль) в диоксане (400 мл) в течение 18 ч. При охлаждении получают бесцветный кристаллический продукт, который фильтруют и промывают EtOAc, получая указанное в подзаголовке соединение в виде бесцветных кристаллов (176 г, 65%). Rf 0,1 (EtOAc). 1(b). 6-Бензил-2-хлор-5,6,7,8-тетрагидро 1,6-нафтиридин. К перемешиваемой суспензии продукта стадии (а) (30 г, 0,124 моль) в толуоле (400 мл) добавляют оксихлорид фосфора (57,7 мл, 0,619 моль), затем тетрахлор-1,4-бензохинон (31,98 г,0,13 моль). Реакционную смесь нагревают с обратным холодильником в атмосфере азота в течение 18 ч, после чего толуол упаривают при пониженном давлении, остаток затем подщелачивают 4N водным NaOH и продукт экстрагируют эфиром (х 3). Объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя EtOAc, получая указанное в подзаголовке соединение в виде твердого вещества (13,29 г, 41%). Rf 0,8(с). 2-Хлор-5,6,7,8-тетрагидро-1,6-нафтиридин. К перемешиваемому раствору продукта стадии (b) (13,28 г, 0,0513 моль) в толуоле (150 мл) по каплям добавляют 1-хлорэтил хлорформат (5,54 мл, 0,0513 моль) при 0 С. Реакционную смесь нагревают с обратным холодильником в течение 2 ч. После охлаждения толуол упаривают при пониженном давлении и остаток распределяют между EtOAc/H2O, органический слой промывают последовательно 1N HCl и насыщенным солевым раствором, высушивают над МgSO4 и упаривают при пониженном давлении. Полученный остаток растворяют в МеОН (150 мл) и нагревают с обратным холодильником в течение 3 ч, после чего реакционную смесь упаривают при пониженном давлении и остаток распределяют между CH2Cl2 и 2N водным NaOH, продукт экстрагируют CH2Cl2(x5). Объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/0,88 NH3(d). 2-Хлор-6-дифенилметил-5,6,7,8-тетрагидро-1,6-нафтиридин. К раствору продукта стадии (с) (1,78 г,0,01 моль) и триэтиламина (2,21 мл, 0,016 моль) в СН 2 Сl2 (20 мл) добавляют дифенилхлорметан(2,13 мл, 0,012 моль). Реакционную смесь пере 49 мешивают при комнатной температуре в течение 20 ч и упаривают при пониженном давлении. Остаток растворяют в DMA (20 мл) и нагревают до 100 С в течение 18 ч и охлаждают,раствор разбавляют CH2Cl2 и промывают последовательно насыщенным водным раствором бикарбоната натрия, Н 2O и насыщенным солевым раствором, затем высушивают над MgSO4. Упаривают при пониженном давлении, получая указанное в подзаголовке соединение в виде твердого вещества (1,01 г, 30%). Rf 0,7(е). 6-Дифенилметил-2-(4-морфолин)5,6,7,8-тетрагилро-1,6-нафтиридин. К раствору морфолина (0,62 мл, 7,17 ммоль) в THF (15 мл) добавляют этилмагнийбромид (2,4 мл, 7,17 ммоль) при 0 С, реакционную смесь перемешивают в течение 1 ч при комнатной температуре, после чего добавляют раствор продукта стадии (d) (0,8 г, 2,389 ммоль) в THF (15 мл), затем ацетилацетонат палладия(0,125 г, 0,478 ммоль) и реакционную смесь нагревают до 60 С в течение 18 ч. После охлаждения раствор распределяют между EtOAc и насыщенным водным раствором хлорида аммония. Органический слой отделяют, промывают последовательно Н 2O, насыщенным солевым раствором, высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя(f). 2-(4-Морфолин)-5,6,7,8-тетрагидро-1,6 нафтиридин. К раствору продукта стадии (е) (0,8 г, 2,08 ммоль) в MeOH/lN HCl (10/1, об/об, 33 мл) добавляют 20% гидроксида палладия на угле (0,2 г). Реакционную смесь гидрогенируют при 345 кПа (50 p.s.i.) и 50 С в течение 56 ч, после чего катализатор отфильтровывают и промывают МеОН. Полученный раствор упаривают при пониженном давлении и остаток распределяют между CH2Cl2 и насыщенным водным раствором бикарбоната натрия. Продукт экстрагируют СН 2 Сl2 (х 8), объединенные органические слои высушивают над MgSO4 и упаривают при пониженном давлении. Неочищенный продукт очищают на силикагеле, элюируя CH2Cl2/MeOH/ 0,88 NН 3 (90/10/1, об/об), получая указанное в подзаголовке соединение (0,13 г, 28%). Rf 0,4(g). 4-Амино-6,7-диметокси-2-[2-(4-морфолин)-5,6,7,8-тетрагидро-1,6-нафтирид-6-ил]5-(2-пиридил)хиназолин. Указанное в заголовке соединение получают по способу примера 12(b) из соединения примера 12(а) и продукта стадии (f). Неочищенный продукт очищают на силикагеле, элюируя(a). 3,4-Диметокси-6-[1-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)этилиденамино]-2-(2-пиридил)бензонитрил. Указанное в подзаголовке соединение получают по способу примера 5 из соединения примера 22(b) и 2-(три-н-бутилстаннил) пиридина. Продукт очищают на силикагеле,элюируя СН 2 Сl2/МеОН (95/5, об/об), получая указанное в подзаголовке соединение (45%) в виде пены. Rf 0,11 (CH2Cl2/MeOH 95/5, об/об).(b). Гидрохлорид 4-амино-6,7-диметокси 2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хинолина. Указанное в заголовке соединение получают по способу примера 23(f) из продукта стадии (а). Продукт очищают на силикагеле, элюируя СН 2 Сl2/МеОН/0,88 NН 3 (90/10/1, об/об), затем обрабатывают избытком эфирного раствора НСl, получая указанное в заголовке соединение(10%) в виде бесцветного твердого вещества. Rf 0,21 (CH2Cl2/MeOH/0,88 NН 3 93/7/1, об/об). МСC26H28ClN5O4S 0,8 СН 2 Сl2 H2O соответствует С,51,26; Н, 5,07; N, 11,15%. Пример 35. Соединение примера 28 тестировали в первом скрининге, описанном выше ("Сократительные ответы простаты человека"), и обнаружили, что оно обладает значением рА 2, равным 9,2. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I где R1 представляет собой C1-4 алкокси, необязательно замещенную одним или несколькими атомами фтора;R2 представляет собой Н или C1-6 алкокси,необязательно замещенную одним или несколькими атомами фтора;R3 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно замещен одной или несколькими группами, выбранными из галогена, C1-4 алкокси, C1-4 алкила и СF3;R4 представляет собой 4-, 5-, 6- или 7 членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно конденсирован с бензольным кольцом или с 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S,эта циклическая система в целом необязательно замещена одной или несколькими группами, независимо выбранными из ОН, C1-4 алкила, C1-4 алкокси, галогена, CONR8R9,SO2NR8R9, (СН 2)bNR8R9 и NHSO2(C1-4 алкила), и если S является членом циклической системы,он может быть замещен одним или двумя атомами кислорода;R8 и R9 независимо представляют собой Н или C1-4 алкил или вместе с атомом N, к которому они присоединены, могут представлять собой 5- или 6-членное гетероциклическое кольцо,содержащее, по меньшей мере, один гетероатом,выбранный из N, О и S;X представляет собой СН или N; иL отсутствует или представляет собой циклическую группу формулы Iа в которой N присоединен ко второй позиции хинолинового или хиназолинового цикла; А отсутствует или представляет собой СО или SO2;Z представляет собой СН или N;m равно 1 или 2, и,кроме того, если Z представляет собой СН,оно может быть равно 0; иm и n равна 2, 3, 4 или 5; или представляет собой цепь формулы Ib в которой N присоединен ко 2 позиции хинолинового или хиназолинового цикла; А' и Z' имеют соответственно такие же значения, как А и Z, описанные выше;R6 и R7 независимо представляют собой Н или C1-4 алкил; и р равно 1, 2 или 3, и,кроме того, если Z представляет собой СН,оно может быть равно 0; или их фармацевтически приемлемые соли. 2. Соединение по п.1, где каждый из R1 и 2R представляет собой метоксильную группу. 52 3. Соединение по п.1 или 2, где R3 представляет собой 2-пиридинил или 2 пиримидинил. 4. Соединение по любому из предшествующих пунктов, где Х представляет собой N. 5. Соединение по любому из предшествующих пунктов, где L отсутствует. 6. Соединение по любому из предшествующих пунктов, где R4 включает в себя насыщенный 6-членный N-содержащий цикл, который конденсирован с бензольным или пиридиновым кольцом. 7. Соединение по п.6, где бензольное кольцо замещено NHSО 2(C1-4 алкилом). 8. Фармацевтическая композиция, включающая в себя соединение формулы I, как определено в п.1, или его фармацевтически приемлемую соль, в смеси с фармацевтически приемлемым адъювантом, разбавителем или носителем. 9. Применение соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли в качестве лекарственного препарата. 10. Применение соединения формулы I,как определено в п.1, или его фармацевтически приемлемой соли в производстве лекарственного препарата для лечения доброкачественной гиперплазии простаты. 11. Способ лечения доброкачественной гиперплазии простаты, который включает в себя введение соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, пациенту, нуждающемуся в таком лечении. 12. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя циклизацию соединения формулы X в которой R1-4 и L такие, как определены в п.1,и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 13. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XIIIa или XIIIb соответственно в которой R1-3, R6, R7, X, m, n и р такие, как определены в п.1, с соединением формулы XIV в которой R4 такой, как определен в п.1,А" представляет собой СО или SO2 иLg представляет собой уходящую группу,и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 14. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы в которой R1, R2, R4, X и L такие, как определены в п.1, с соединением формулы XIX в которой R3 такой, как определен в п.1, а М представляет собой замещенный бор, цинк или олово, в присутствии палладиевого катализатора, и,если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 15. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XXII в которой R4, R6, R7, А, А', Z, Z', m, n и р такие,как определены в п.1,и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 16. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы в которой R1-3, R6, R7, X, Z, Z', m, n и р такие, как определены в п.1, и Lg представляет собой уходящую группу, с соединением формулы XXIX в которой R4a представляет собой группы, определяемые в п.1 с помощью R4, которые содержат нуклеофильный атом азота в цикле, причем этот атом азота соединен с Н,и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 17. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя превращение соединения формулы I, в котором L представляет собой циклическую группу формулы Ia, в соответствующее соединение формулы I, в котором L представляет собой цепь формулы Ib, в которой каждый из R6 и R7 представляет собой Н, путем воздействия сильного основания, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 18. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулы XIIIa или XIIIb, как определено выше, с соединением формулы XXX в которой R4 такой, как определен в п.1, и Hal представляет собой атом галогена, присоединенный к циклу, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 19. Способ получения соединения формулы I, как определено в п.1, или его фармацевтически приемлемой соли, который включает в себя взаимодействие соединения формулыXXII, как определено выше, с соединением формулы XXIX, как определено выше, и, если желательно или необходимо, превращение полученного соединения формулы I в фармацевтически приемлемую соль или наоборот. 20. Соединение формулы X
МПК / Метки
МПК: A61P 13/08, C07D 471/04, C07D 401/14, A61K 31/4709
Метки: доброкачественной, гиперплазии, особенно, простаты, применяемые, лечении, хиназолиновые, хинолиновые, терапии, соединения
Код ссылки
<a href="https://eas.patents.su/29-2851-hinolinovye-i-hinazolinovye-soedineniya-primenyaemye-v-terapii-osobenno-pri-lechenii-dobrokachestvennojj-giperplazii-prostaty.html" rel="bookmark" title="База патентов Евразийского Союза">Хинолиновые и хиназолиновые соединения, применяемые в терапии, особенно при лечении доброкачественной гиперплазии простаты</a>
Арилзамещенные пиперазины для лечения доброкачественной гиперплазии простаты

Номер патента: 1904
Опубликовано: 22.10.2001
Авторы: Мэрианофф Синтия, Рейтц Алан, Ли Ксиаобинг, Пулито Вирджиния, Жоллифф Линда, Виллани Фрэнк, Муррей Уильям, Малкахи Линда
МПК: A61P 13/08, C07D 205/08, A61K 31/40...
Метки: арилзамещенные, лечения, доброкачественной, пиперазины, простаты, гиперплазии
Формула / Реферат:
1. Соединение формулы I где А представляет собой (СН2)n, где n равно 1-6; R1 представляет собой C1-6алкил, фенил, замещенный фенил, где заместители фенила независимо выбраны из одного или нескольких из группы, состоящей из С1-5алкила, С1-5 алкоксильной группы и галогена, фенилС1-5алкила, или замещенный фенилС1-5алкил, где заместители фенила независимо выбраны из одного или нескольких из группы, состоящей из C1-5алкила, C1-5алкоксильной группы...
Способ лечения доброкачественной гиперплазии предстательной железы.

Номер патента: 946
Опубликовано: 26.06.2000
Авторы: Шипли Лиза А., Кларк Дэвид О., Джордан Уилльям Х.
МПК: A61K 31/4045, A61P 13/08
Метки: доброкачественной, способ, лечения, железы, предстательной, гиперплазии
Формула / Реферат:
1. Способ лечения доброкачественной гиперплазии предстательной железы у млекопитающих, включающий введение млекопитающему, нуждающемуся в таком лечении, эффективной дозы активного агента, отличающийся тем, что в качестве активного агента вводят агонист мелатонина. 2. Способ по п.1, отличающийся тем, что млекопитающее является человеком. 3.Способ по п.1 или 2, отличающийся тем, что агонист мелатонина выбирают из...
Хинолиновые и хиноксалиновые соединения, ингибирующие тирозинкиназы тромбоцитарного фактора роста и/или p56 lck

Номер патента: 2600
Опубликовано: 27.06.2002
Авторы: Персонс Пол Э., Спада Альфред П., Магвир Мартин П., Майерс Майкл
МПК: A61K 31/47, C07D 241/44, A61P 35/00...
Метки: ингибирующие, соединения, хинолиновые, роста, хиноксалиновые, тромбоцитарного, тирозинкиназы, фактора
Формула / Реферат:
1. Соединение формулы I где R1a представляет необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный оксагетероциклилокси или галоген; R1b представляет водород, необязательно замещенный C1-С10алкил, гидрокси, необязательно замещенный C1-С10алкокси, необязательно замещенный С3-С7циклоалкилокси, необязательно замещенный 4-7-членный...
Хинолиновые антагонисты лейкотриенов

Номер патента: 1797
Опубликовано: 27.08.2001
Авторы: Эрисон Байрон Х., Бэйлли Томас А., Балани Суреш К., Дюфресн Клод
МПК: A61P 11/06, A01N 43/42, A61K 31/47...
Метки: хинолиновые, лейкотриенов, антагонисты
Формула / Реферат:
1. Соединение формулы и его индивидуальные оптические изомеры или его фармацевтически приемлемое производное. 2. Выделенное и очищенное соединение по п.1, по существу свободное от других продуктов метаболизма монтелукаста натрия. 3. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель. 4. Способ предотвращения действия лейкотриена у млекопитающего, включающий...
Конъюгаты, используемые в лечении рака предстательной железы

Номер патента: 2066
Опубликовано: 24.12.2001
Авторы: Фенг Донг-Мей, Гарски Виктор М., Дефео-Джоунз Дебора
МПК: A61P 13/08, A61K 47/48
Метки: лечении, используемые, конъюгаты, железы, предстательной, рака
Формула / Реферат:
1. Конъюгат, применяемый в лечении рака предстательной железы, который включает в себя цитотоксический агент, связанный с олигопептидом, где олигопептид включает в себя последовательность аминокислот, которая избирательно расщепляется посредством протеолитического действия свободного специфического антигена простаты, и где средствами связывания является ковалентная связь или связывание осуществляется через химический линкер, причем указанная...