Производные транилципромина в качестве ингибиторов гистон деметилаз lsd1 и/или lsd2
Номер патента: 22459
Опубликовано: 29.01.2016
Авторы: Май Антонелло, Минуччи Саверио, Маттеви Андреа



















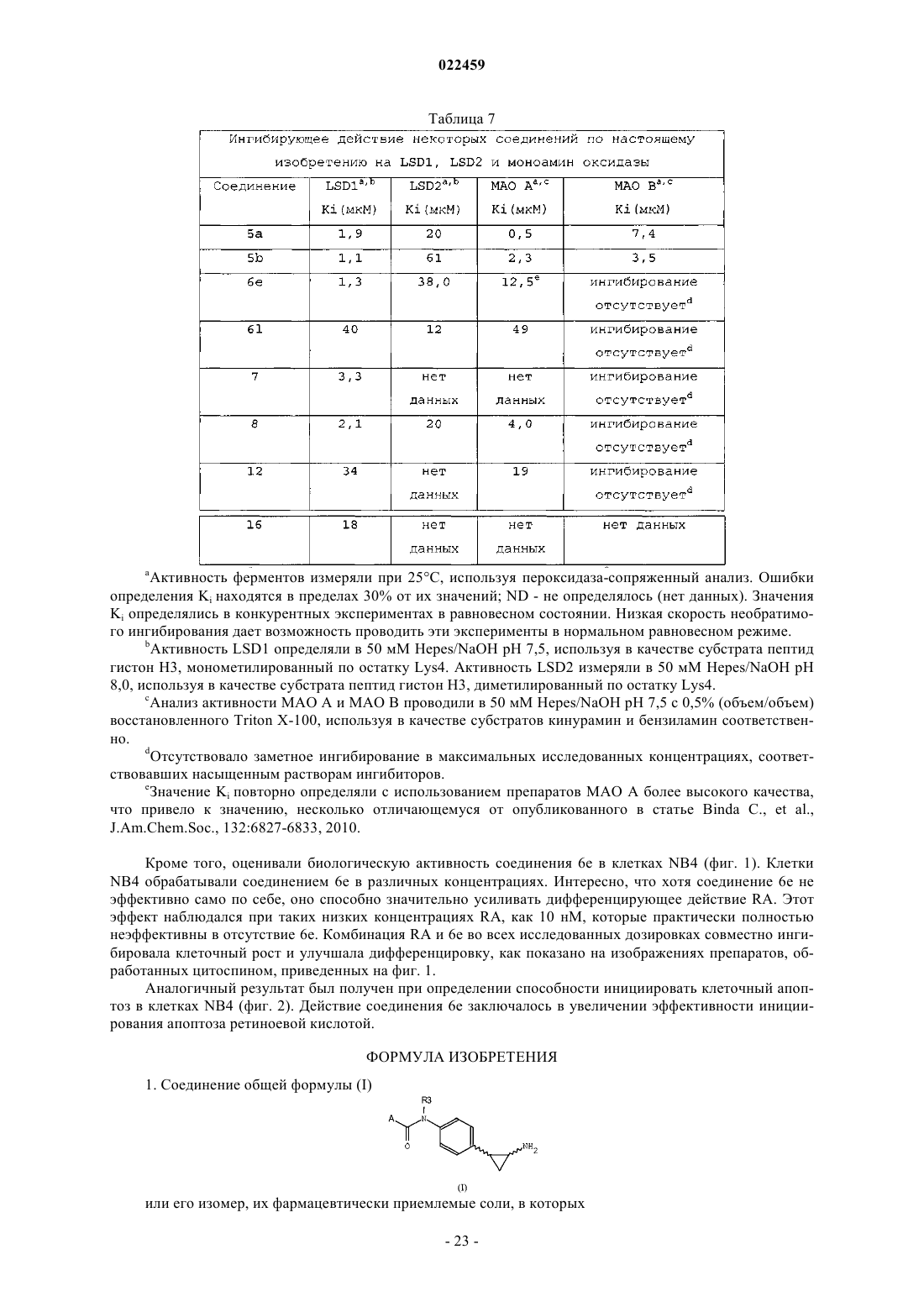


Формула / Реферат
1. Соединение общей формулы (I)

или его изомер, их фармацевтически приемлемые соли, в которых
А означает R или CH(R1)-NH-CO-R2;
R выбран из фенила, бифенила, нафтила, бензилокси, бензила, нафтилметила, HONHCO(CH2)6;
R1 выбран из изопропила, изобутила, фенила, циклогексилметила, бензила, необязательно замещенного бромом или метокси, 2-фенилэтила, 2,2'-дифенилметила, нафтилметила, индолилметила, бензотиофенилметила;
R2 выбран из бензилокси, необязательно замещенного бромом, бензиламино;
R3 представляет собой Н, метил.
2. Соединение по п.1, где
А означает R;
R3 означает Н.
3. Соединение по п.2, где
R представляет собой фенил, бифенил, нафтил, бензилокси, бензил, нафтилметил или HONHCO(CH2)6.
4. Соединение по п.1, где
А означает CH(R1)-NH-CO-R2;
R3 означает Н.
5. Соединение по п.1, где
А означает CH(R1)-NH-CO-R2;
R3 означает -CH3.
6. Соединение по любому одному из пп.1-4, где независимо или в любой комбинации
R1 означает изопропил, изобутил, фенил, циклогексилметил, бензил, необязательно замещенный бромом или метокси, 2-фенилэтил, 2,2'-дифенилметил, нафтилметил, индолилметил, бензотиофенилметил;
R2 означает бензилокси, необязательно замещенный бромом, или бензиламино.
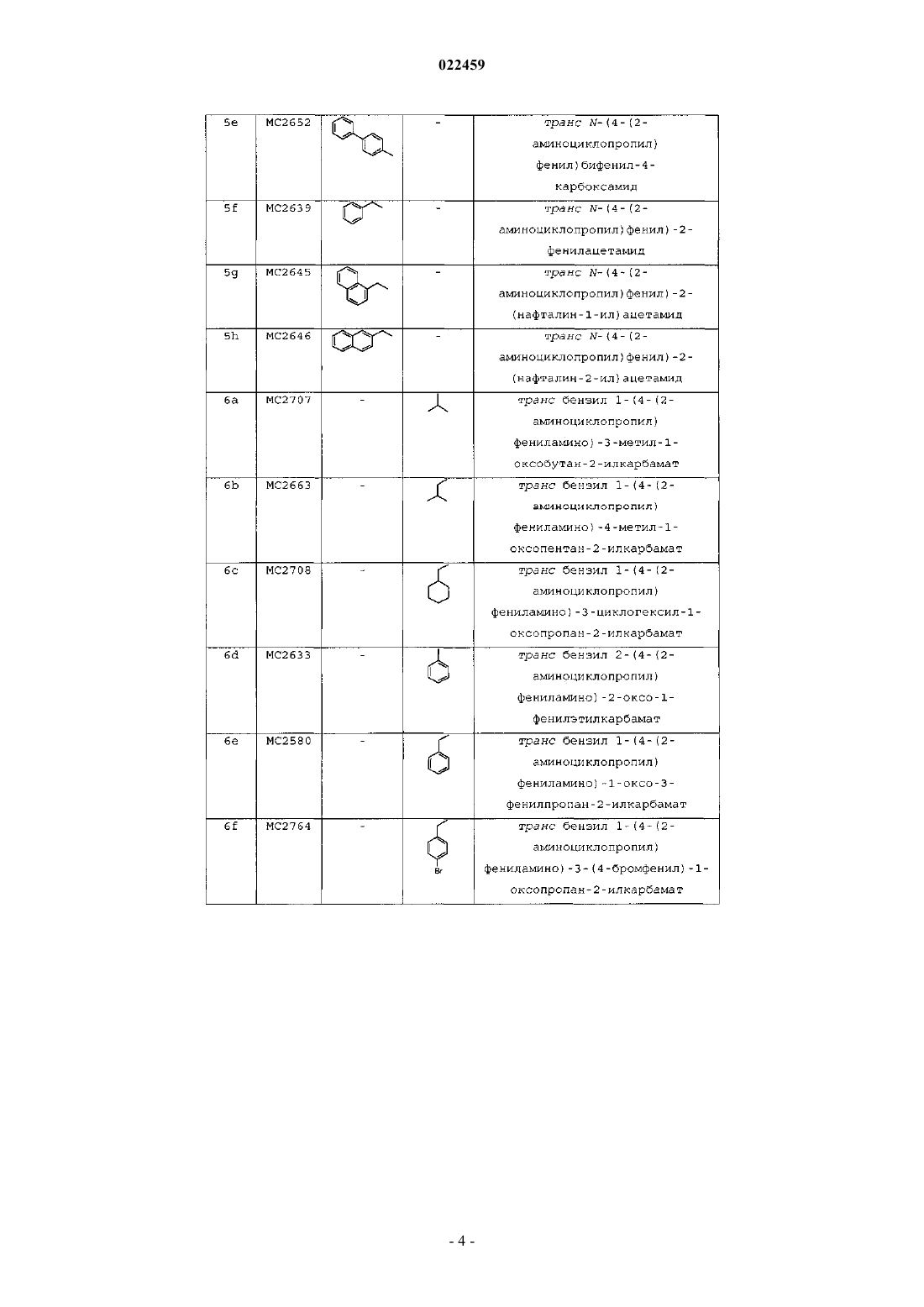
7. Соединение по п.1, входящее в следующую группу:
транс бензил-4-(2-аминоциклопропил)фенилкарбамат;
транс-N-(4-(2-аминоциклопропил)фенил)бензамид;
транс-N-(4-(2-аминоциклопропил)фенил)-1-нафтамид;
транс-N-(4-(2-аминоциклопропил)фенил)-2-нафтамид;
транс-N-(4-(2-аминоциклопропил)фенил)бифенил-4-карбоксамид;
транс-N-(4-(2-аминоциклопропил)фенил)-2-фенилацетамид;
транс-N-(4-(2-аминоциклопропил)фенил)-2-(нафталин-1-ил)ацетамид;
транс-N-(4-(2-аминоциклопропил)фенил)-2-(нафталин-2-ил)ацетамид;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-метил-1-оксобутан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-метил-1-оксопентан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-циклогексил-1-оксопропан-2-илкарбамат;
транс-бензил-2-(4-(2-аминоциклопропил)фениламино)-2-оксо-1-фенилэтилкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(4-бромфенил)-1-оксопропан-2-илкарбамат;
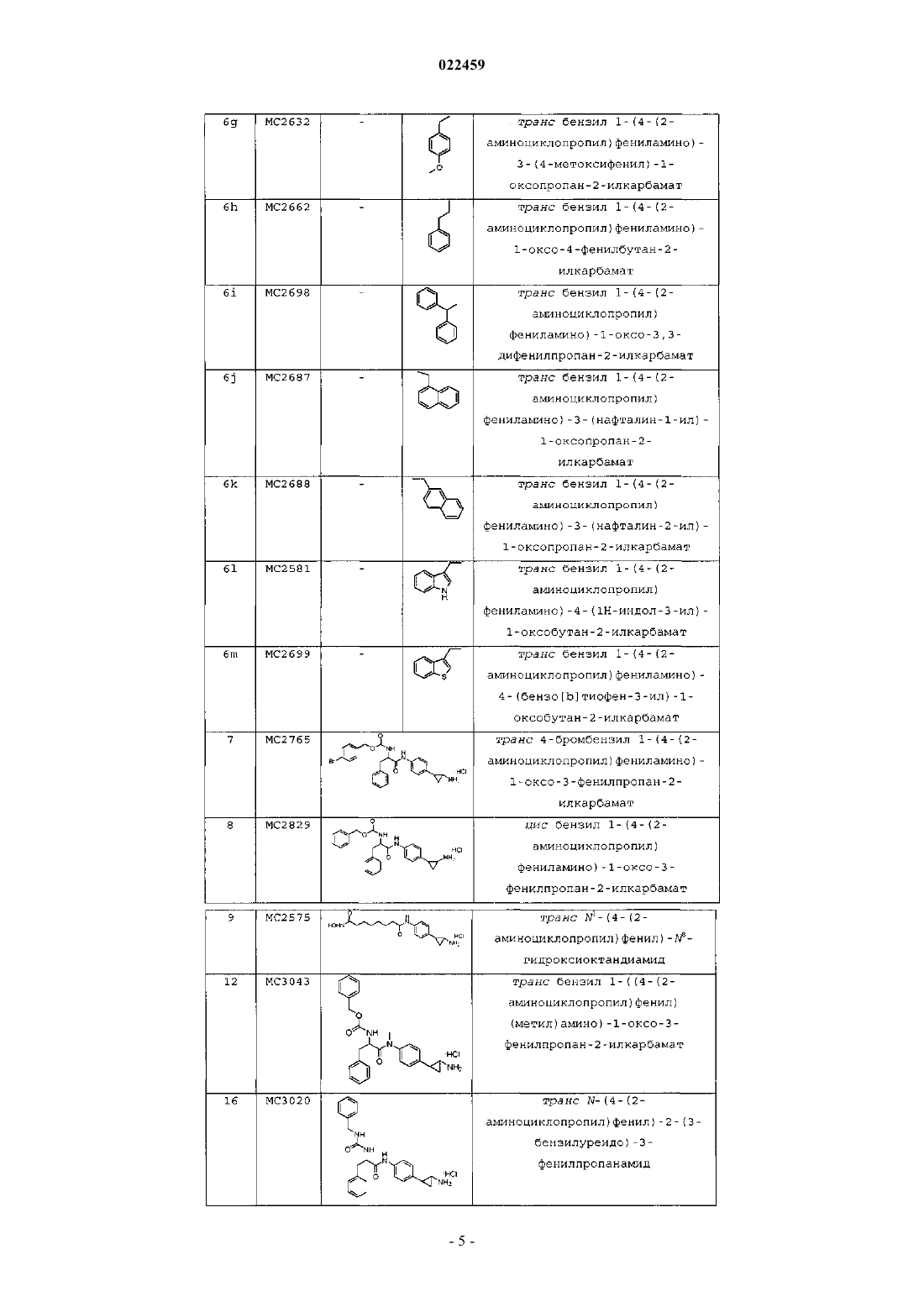
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(4-метоксифенил)-1-оксопропан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-4-фенилбутан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3,3-дифенилпропан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(нафталин-1-ил)-1-оксопропан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(нафталин-2-ил)-1-оксопропан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-(1Н-индол-3-ил)-1-оксобутан-2-илкарбамат;
транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-(бензо[b]тиофен-3-ил)-1-оксобутан-2-илкарбамат;
транс-4-бромбензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат;
цис-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат;
транс-N1-(4-(2-аминоциклопропил)фенил)-N8-гидроксиоктандиамид;
транс-бензил-1-((4-(2-аминоциклопропил)фенил)(метил)амино)-1-оксо-3-фенилпропан-2-илкарбамат;
транс-N-(4-(2-аминоциклопропил)фенил)-2-(3-бензилуреидо)-3-фенилпропанамид
или его изомер, их фармацевтически приемлемые соли.
8. Соединение по п.1 или 7, где изомер представляет собой энантиомер или диастереомер.
9. Соединение по п.8, где диастереомер представляет собой эпимер.
10. Способ получения соединения общей формулы (I) по п.1, где А представляет собой R, включающий:
(а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (III)

где R, R3 соответствуют определениям п.1 и Boc означает трет-бутоксикарбонильную защитную группу;
(b) необязательно превращение соединения формулы (III), полученного на стадии а), в другое соединение формулы (III), удаление защитной группы Boc из соединения формулы (III) с получением соединения формулы (I)

11. Способ получения соединения общей формулы (I) по п.1, где А представляет собой CH(R1)-NH-CO-R2, включающий:
а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (IV)

где R1, R2, R3 соответствуют определениям п.1 и Boc означает трет-бутоксикарбонильную защитную группу;
b) необязательно превращение соединения формулы (IV), полученного на стадии а), в другое соединение формулы (IV), удаление защитной группы Boc из соединения формулы (IV) с получением соединения формулы (I)

12. Ингибитор гистон деметилазы LSD1 и/или LSD2, являющийся соединением по любому из предшествующих пп.1-9.
13. Применение соединения по любому из пп.1-9 в терапии.
14. Применение соединения по любому из пп.1-9 в качестве проапоптотического агента.
15. Применение по п.13 в качестве лекарственного средства, предназначенного для профилактики и/или лечения заболеваний и состояний, характеризующихся нарушением регуляции транскрипции генов, дифференцировки и пролиферации клеток.
16. Применение по п.15 в качестве противоопухолевого агента.
17. Применение по п.16, где указанный противоопухолевый агент эффективен против опухоли, выбранной из группы, состоящей из нейробластомы, рака простаты, рака груди, острого миелоидного лейкоза, острого Т-клеточного лимфобластного лейкоза, рака мочевого пузыря, рака легких и колоректального рака.
18. Применение по п.15 в качестве противовирусного агента.
19. Применение по п.18, где указанный противовирусный агент эффективен против вирусной инфекции, вызванной вирусом простого герпеса.
20. Способ профилактики и/или лечения заболеваний и состояний, связанных с активностью гистон деметилазы LSD1 и/или LSD2, путем введения млекопитающему, которому необходимо такое лечение, терапевтически эффективного количества соединения общей формулы (I) по любому из пп.1-9.
21. Способ по п.20, где заболевания представляют собой опухоли или вирусные инфекции.
22. Способ по п.20 или 21, где заболевания выбраны из нейробластомы, рака простаты, рака груди, рака мочевого пузыря, рака легких, колоректального рака, острого миелоидного лейкоза, острого Т-клеточного лимфобластного лейкоза и инфекции вируса простого герпеса.
23. Фармацевтическая композиция, включающая эффективное количество одного или нескольких соединений общей формулы (I) по любому из пп.1-9, и как минимум один фармацевтически приемлемый эксципиент.
24. Фармацевтическая композиция по п.23 в виде единичной дозированной формы.
Текст
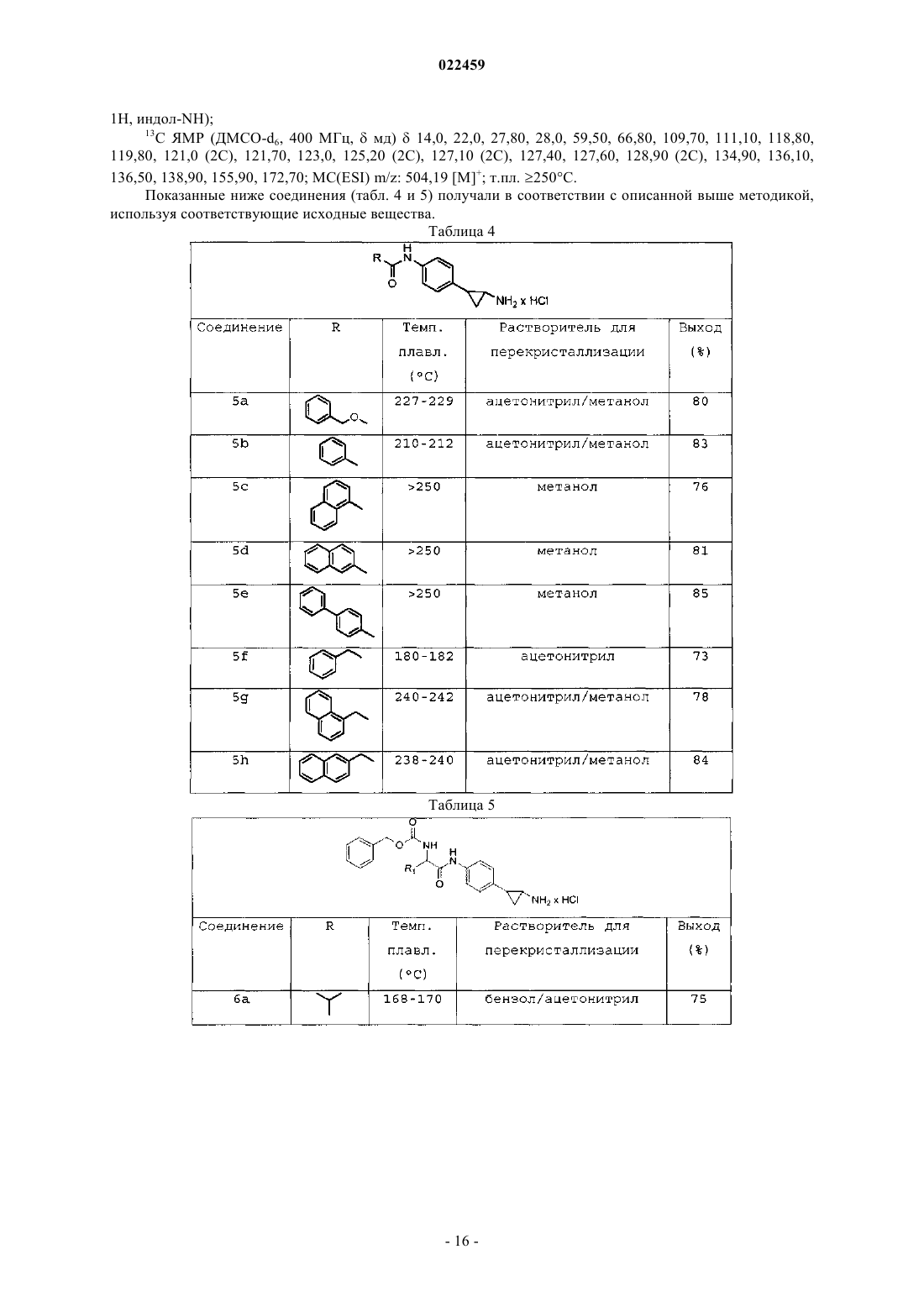
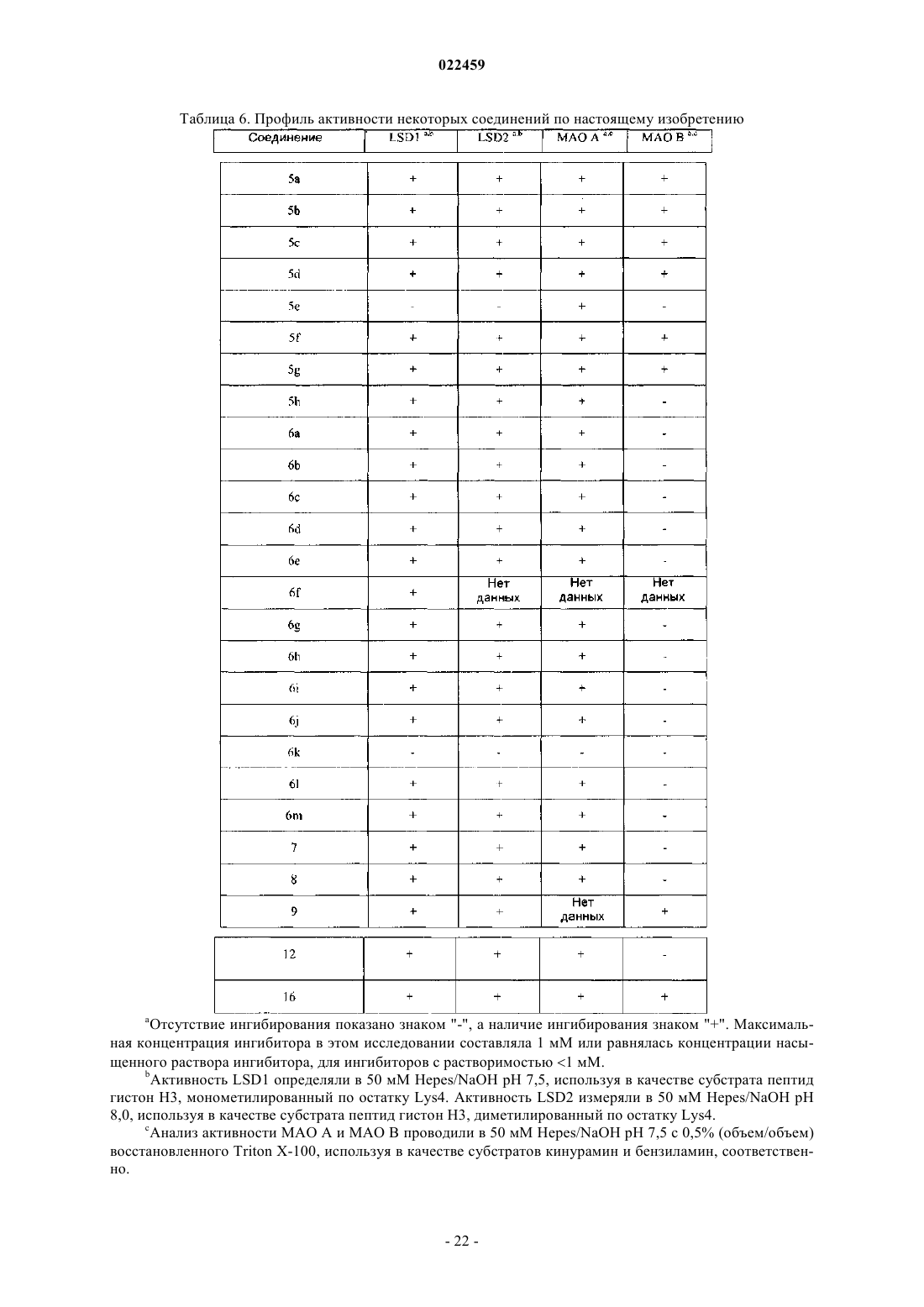
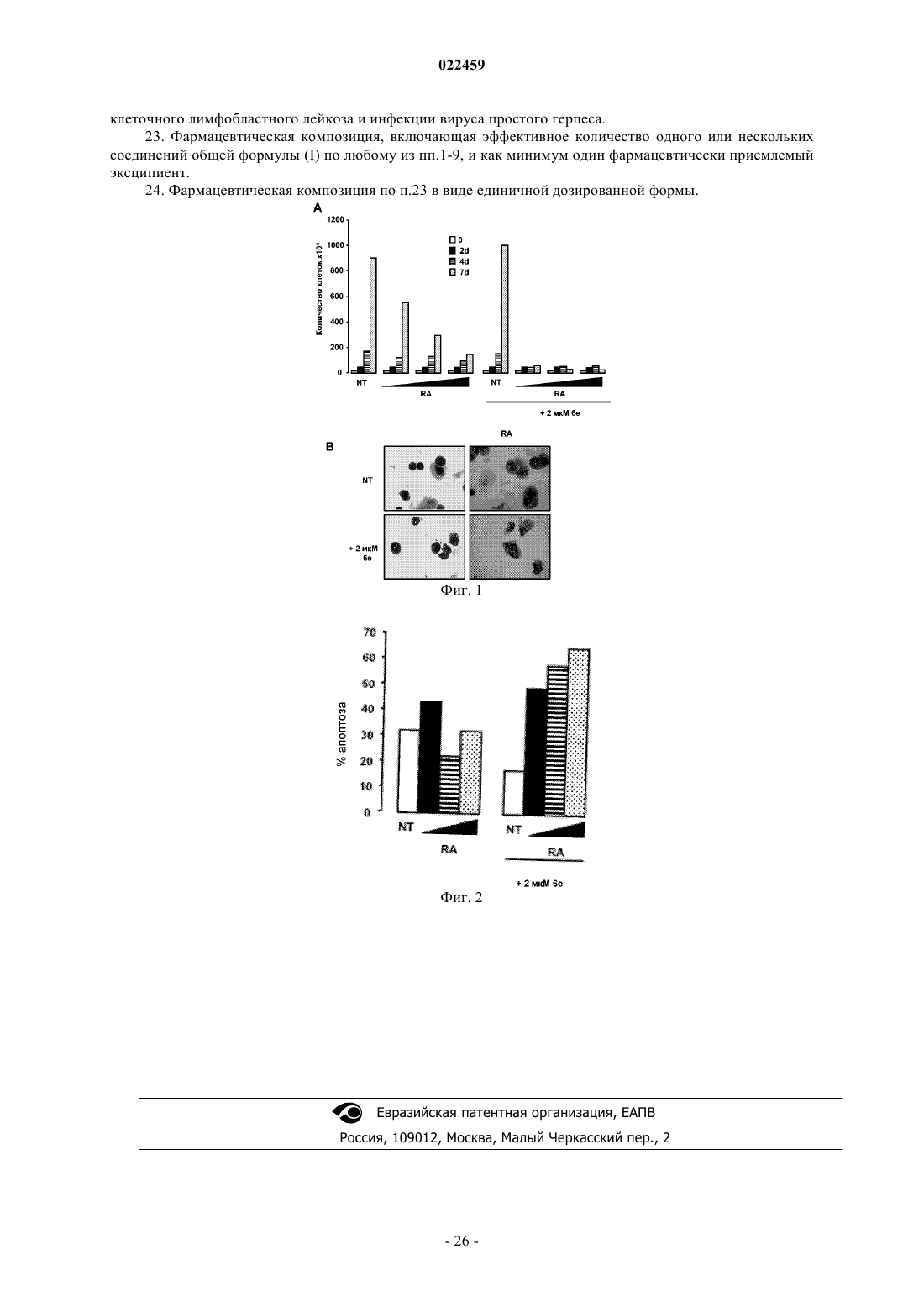
ПРОИЗВОДНЫЕ ТРАНИЛЦИПРОМИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ГИСТОН ДЕМЕТИЛАЗ LSD1 И/ИЛИ LSD2 В изобретении описаны производные транилципромина, используемые в качестве терапевтических агентов, в частности для профилактики и/или лечения заболеваний и состояний, связанных с активностью гистон деметилаз LSD1 и LSD2, например заболеваний, характеризующихся нарушением регуляции транскрипции генов, дифференцировки и пролиферации клеток, таких как опухоли и вирусные инфекции. Эти соединения соответствуют структурной формуле (I), где А иR3 определены в описании изобретения. Кроме того, изобретение относится к препаратам этих соединений, а также к содержащим их композициям и к их терапевтическому применению.(71)(73) Заявитель и патентовладелец: УНИВЕРСИТА' ДЕЛЬИ СТУДИ ДИ РОМА "ЛА САПИЕНЦА"; ФОНДАЦЬОНЕ ИЭО; УНИВЕРСИТА' ДЕЛЬИ СТУДИ ДИ ПАВИЯ; УНИВЕРСИТА' ДЕЛЬИ СТУДИ ДИ МИЛАНО (IT) Область техники, к которой относится изобретение Настоящее изобретение относится к производным транилципромина и к их применению в качестве терапевтических агентов, в частности для профилактики и/или лечения заболеваний и состояний, связанных с активностью гистон деметилаз LSD1 и LSD2, например заболеваний, характеризующихся нарушением регуляции транскрипции генов, дифференцировки и пролиферации клеток, таких как опухоли и вирусные инфекции. Кроме того, изобретение относится к получению этих соединений, а также к содержащим их композициям и их терапевтическому применению. Уровень техники Изменения в структурном и функциональном состоянии хроматина вовлечены в патогенез целого ряда заболеваний. Биохимические и ферментные процессы, которые катализируют включение и удаление пост-трансляционных модификаций нуклеосом, стали объектом исследования в качестве потенциальных мишеней для так называемой эпигенетической терапии (Urdinguio RG, Sanchez-Mut JV, EstellerM. Epigenetic mechanisms in neurological diseases: genes, syndromes and therapies. Lancet Neurol. 8:10561072, 2009). Обнаружение все возрастающего числа гистон деметилаз выдвинуло на первый план динамическую природу регулирования метилирования гистонов, т.е. ключевой модификации хроматина, которая вовлечена в эукариотический геном и регулирование генов. Гистон лизин деметилаза представляет собой очень привлекательную мишень для эпигенетических лекарственных препаратов и привлекает к себе все большее внимание. Лизин может быть моно-, ди- и триметилированным. Каждая из модификаций этой аминокислоты может определенным образом влиять на различные биологические эффекты. Последние данные исследований гистон лизин деметилаз выявили два типа ферментных механизмов(Anand R, Marmorstein R. Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 282:35425-35429, 2007). Железозависимые ферменты могут деметилировать боковые цепи лизина, находящиеся во всех трех состояниях метилирования, и к настоящему времени охарактеризованы многие деметилазы, входящие в это семейство. Напротив, реакция окисления, которая лежит в основе действия флавинзависимых гистон деметилаз, не дает этим ферментам возможности действовать на триметилированный лизин и ограничивает их действие моно- и диметилированными субстратами. В организме млекопитающих содержатся две флавоферментных деметилазы: LSD1 и LSD2. Деметилаза LSD1 явилась первой из обнаруженных гистон деметилаз и она, как правило, (но не всегда) ассоциирована с корепрессорным белком CoREST. Комплекс LSD1/CoREST способен ассоциироваться с 1/2 молекулы гистон деацетилазы (HDAC1/2), образуя мультиферментный блок, который задействован многими хроматиновыми комплексами, обычно вовлеченными в регуляцию репрессии генов (Ballas N., et al.Regulation of neuronal traits by a novel transcriptional complex. Neuron. 31:353-365, 2001). LSD1 удаляет метильные группы из моно- и диметилированного остатка Lys4 гистона H3, что является хорошо известной меткой активации гена. Этот фермент является интересной мишенью для эпигенетических лекарственных средств, как можно предположить, исходя из его избыточной экспрессии в солидных опухоляхimplications for therapy. Cancer Res 69:2065-2071, 2009), его роли в различных процессах дифференцировки (Ни X, et al. LSD1-mediated epigenetic modification is required for TALI function and hematopoiesis. Procsimplex virus 1. J Virol 83:4376-4385, 2009) и его ассоциации с HDAC1-проверенной мишени для лекарственных средств. LSD2 является позднее обнаруженной деметилазой, которая, как и LSD1, демонстрирует строгую специфичность в отношении моно- и диметилированного остатка Lys4 H3. Однако биологические свойства LSD2, которые остаются охарактеризованными лишь частично, предположительно отличаются от свойств LSD1, поскольку LSD2 не связывается с CoREST, и эта деметилаза до сих пор не была обнаружена ни в одном из белковых комплексов, включающих LSD1 (Karytinos A. et al. A novel mammalian flavin-dependent histone demethylase. J Biol Chem 284:17775-17782, 2009).LSD1 и LSD2 являются мультидоменными белками, причем в обеих молекулах присутствует сходный каталитический домен (идентичность последовательностей 45%), который структурно гомологичен моноамин оксидазам (MAOs) А и В. Транилципромин, т.е. (+)-транс-2-фенилциклопропил-1-амин В работе Gooden et al. (Bioorg. Med. Chem. Lett. 18, 3047-3051, 2008) описан путь синтеза замещенных транс-2-арилциклопропиламинов в качестве ингибиторов LSD1 и MAOs. Эффективность ингибирования МАО А и В этими соединениями более чем в 10 раз превышает эффективность ингибированияLSD1. Работа Culhane et al. (J. Am.Chem.Soc. 132, 3164-3176, 2010) относится к гидразинсодержащему ингибитору МАО фенелзину, в качестве низкомолекулярного ингибитора LSD1. В WO 2010011845 описан способ лечения вирусной инфекции организма-хозяина путем введения ингибитора белка LSD1 (молекулы, вызывающей РНК-интерференцию) и/или ингибитора моноамин оксидазы, т.е. транилципромина. ЕР 1693062 относится к применению по крайней мере одной siRNA ("короткой интерферирующей молекулы РНК") и как минимум одного антитела против LSD1, также в комбинации с ингибитором моноаминоксидазы, например транилципромином, для модулирования активности LSD1 и регулирования экспрессии генов, зависимых от андрогенных рецепторов. В WO 2010/043721, WO 2010/084160 и WO 2010/143582, WO 2011/035941, опубликованных после даты приоритета настоящей заявки, раскрыты производные фенилциклопропиламина, способные селективно ингибировать действие LSD1. Ни одно из раскрытых соединений не входит в объем настоящего изобретения. Таким образом, существует потребность в обнаружении малых молекул, которые являются эффективными и селективными ингибиторами гистон деметилаз LSD1 и/или LSD2 и применимы в профилактике или лечении заболеваний и состояний, связанных с активностью гистон деметилаз. Соединения по настоящему изобретению являются малыми молекулами, обладающими мощным ингибирующим действием на гистон деметилазы, которые применимы в лечении ряда заболеваний, при которых наблюдается нарушение регулирования транскрипции генов, дифференцировки и пролиферации клеток, например опухолей и вирусных инфекций. Описание изобретения Настоящее изобретение направлено на соединения, которые обладают способностью ингибировать гистон деметилазы LSD1 и/или LSD2 и применимы при профилактике или лечении заболеваний и состояний, связанных с активностью гистон деметилаз LSD1 и/или LSD2. Кроме того, изобретение относится к способам получения указанных соединений, содержащих их композиций и к их терапевтическому применению. В изобретении обнаружено, что производные транилципромина общей формулы (I) и их производные обладают способностью ингибировать активность гистон деметилаз. Все термины, использованные в настоящей заявке, если не указано иное, следует понимать в их обычном значении, которое известно в технике. Другие более конкретные определения некоторых терминов, использованных в настоящей заявке, приведены ниже и предполагается их применение во всем тексте заявки и формулы изобретения, если иное, явно приведенное определение, не придаст указанным терминам более широкого значения. Таким образом, объектом настоящего изобретения является соединение формулы (I)R1 выбран из изопропила, изобутила, фенила, циклогексилметила, бензила, необязательно замещенного бромом или метокси, 2-фенилэтила, 2,2'-дифенилметила, нафтилметила, индолилметила;R3 представляет собой Н, метил; а также его изомеры и его фармацевтически приемлемые соли. Формулы включают один или несколько символов для указания на все возможные конфигурации: цис, транс, (R), (S). Термин "ацилирующий агент" относится к реакционноспособному производному карбоновой кислоты, которое может обеспечить быструю реакцию сочетания кислотной группы с аминогруппой с образованием амидной связи. Примеры ацилирующих агентов включают, не ограничиваясь этим, органические ацилгалогениды, ангидриды органических кислот, карбоновые кислоты, сложные эфиры, смешанные ангидриды карбоновых-сульфоновых кислот. Термин "примерно" охватывает диапазон экспериментальных ошибок, которые обычно могут появляться при измерениях. Термин "фармацевтически приемлемые соли" относится к кислотно-аддитивным солям, образованным соединениями по настоящему изобретению и относительно нетоксичными минеральными и органическими кислотами. Эти соли можно получать in situ во время окончательного выделения и очистки со-2 022459 единений. В частности, кислотно-аддитивные соли можно получать отдельным взаимодействием соединений в очищенном виде с органической или неорганической кислотой с выделением образовавшейся при реакции соли. Образующиеся соли являются, например, гидрохлоридами, гидробромидами, сульфатами, гидросульфатами, дигидросульфатами, метансульфонатами, цитратами, оксалатами, малеатами,фумаратами, сукцинатами, трифторацетатами, 2-нафталинсульфонатами и пара-толуолсульфонатами. Настоящее изобретение относится также ко всем изомерам и их смесям. В предпочтительном варианте осуществления изобретение относится к соединению формулы (I), в котором А представляет собой R, предпочтительно фенил, бифенил, нафтил, бензилокси, бензил, нафтилметил;R3 означает Н; а также к его изомерам и их фармацевтически приемлемым солям. В другом предпочтительном варианте осуществления изобретение относится к соединению формулы (I), в которомR3 означает Н; А представляет собой CH(R1)-NH-CO-R2; предпочтительно независимо или в любых комбинациях:R1 означает изопропил, изобутил, фенил, циклогексилметил, бензил, необязательно замещенный бромом или метокси, 2-фенилэтил, 2,2'-дифенилметил, нафтилметил, индолилметил;R2 означает бензилокси, необязательно замещенный бромом, бензиламино; а также к его изомерам и их фармацевтически приемлемым солям. В другом предпочтительном варианте осуществления изобретение относится к соединению формулы (I), в которомR3 означает -CH3; А представляет собой CH(R1)-NH-CO-R2; предпочтительно независимо или в любых комбинациях;R1 означает изопропил, изобутил, фенил, индолилметил, циклогексилметил, бензил, необязательно замещенный бромом или метокси, 2-фенилэтил, 2,2'-дифенилметил, нафтилметил;R2 означает бензилокси, необязательно замещенный бромом, бензиламино; а также к его изомерам и их фармацевтически приемлемым солям. Для ознакомления с любым из конкретных соединений формулы (I) по настоящему изобретению,необязательно в форме фармацевтически приемлемой соли, смотрите приведенный ниже по тексту экспериментальный раздел. Конкретные неограничивающие примеры соединений формулы (I) показаны в приведенной ниже таблице (табл. 1). Таблица 1 Изомеры и фармацевтически приемлемые соли соединений, приведенных в табл. 1, также входят в объем настоящего изобретения. Кроме того, настоящее изобретение относится к способу получения соединений общей формулы (I),которые описаны выше, их фармацевтически приемлемых солей в соответствии со следующими способами (способ А и способ В), которые можно осуществлять способами, хорошо известными специалисту в данной области. Некоторые из подходящих методик описаны ниже и изображены на схемах, но не следует считать, что этими методиками ограничивается весь круг способов синтеза, которые подходят для получения соединений по настоящему изобретению. Эти методики приведены для получения представления о возможных путях синтеза. Представленные способы могут быть адаптированы специалистом в данной области техники в зависимости от природы соединений формулы (I), которые предполагается получить, путем выбора подходящих исходных веществ, в которых природа заместителей R, R1, R2 и R3 может подвергаться изменениям. Таким образом, объектом настоящего изобретения является способ получения соединения (Ia), соответствующего общей формуле (I), где А представляет собой R, причем указанный способ включает:(а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (III) где R1, R3 соответствуют данным выше определениям и Boc представляет собой третбутоксикарбонильную защитную группу;(b) необязательно превращение соединения формулы (III), полученного на стадии а), в другое соединение, соответствующее формуле (III), удаление защитной группы Boc из соединения формулы (III) с получением соединения формулы (Ia) На стадии (а) описываемого способа (способ А) реакцию соединения формулы (II) с ацилирующим агентом, приводящую к получению соединения формулы (III), можно проводить по различным методикам, очень хорошо известным специалистам в данной области. Например, соединение формулы (II) можно обработать подходящим ацилирующим агентом, например ацилхлоридом, в присутствии основания, с получением Вос-защищенного соединения формулы (III). Реакцию проводят в подходящем растворителе, например полярном апротонном растворителе, таком как дихлорметан, тетрагидрофуран, 1,4-диоксан,N,N'-диметилформамид или их смесях, в присутствии поглотителя протонов, например триэтиламина,N,N-диизопропилэтиламина, пиперидина, N,N-диметиланилина или пиридина, при температуре, находящейся в пределах от комнатной до температуры кипения растворителя. Предпочтительно стадию (а) осуществляют путем реакции соединения формулы (II) с ацилхлоридом в присутствии амина, например триэтиламина в дихлорметане при комнатной температуре. Перед снятием защитной группы Boc соединение формулы (III) необязательно можно превратить в другое соединение формулы (III). Например,группу NH остатка анилина можно алкилировать обработкой алкилгалогенидом в основной среде по стандартным методикам, хорошо известным специалистам в данной области. Отщепление группы Boc от соединения формулы (III) по стандартным методикам позволяет получить целевые соединения (Ia). Удаление защитной группы Boc описано в "Protective groups in Organic Chemistry" 3rd edition, T.W.Greene andP.G.Wuts, Wiley-Interscience (1999) и в "Protecting Groups", P.J.Kocienski, Georg Thieme Verlag (1994). Например, стадию (b) проводят путем добавления кислоты, например HCl или трифторуксусной кислоты, в подходящем растворителе, например полярном апротонном растворителе, например дихлорметане, тетрагидрофуране, 1,4-диоксане, N,N-диметилформамиде или их смесях, при температуре в пределах от 0C до температуры кипения. Соединения формулы (Ia) можно превратить в другие соединения формулы (Ia) с помощью любых способов синтеза, известных в технике, и/или его можно превратить в фармацевтически приемлемую соль, и/или его соли можно превратить в свободное соединение формулы (Ia). В другом варианте осуществления изобретение относится к способу получения соединения (Ib),входящего в число соединений общей формулы (I), где А представляет собой CH(R1)-NH-CO-R2, причем указанный способ включает: а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (IV) где R1, R2, R3 и Boc соответствуют данным выше определениям;b) необязательно превращение соединения формулы (IV), полученного на стадии а), в другое соединение, соответствующее формуле (IV), удаление защитной группы Boc из соединения формулы (IV) с получением соединения формулы (Ib) На стадии (а) описываемого способа (способ В) реакцию соединения формулы (II) с ацилирующим агентом, приводящую к получению соединения формулы (IV), можно проводить различными методиками, очень хорошо известными специалистам в данной области. Например, соединение формулы (II) можно обработать подходящим ацилирующим агентом, например Z-защищенной аминокислотой и основанием, необязательно в присутствии сшивающего реагента, например гексафторфосфата (бензотриазол 1-илокси)трис(диметиламино)фосфония (реагента ВОР), N,N-карбонилдиимидазола или гидрохлорида 1 этил-3-(3-диметиламинопропил)карбодиимида с получением Вос-защищенного соединения формулы(IV). Реакцию проводят в подходящих растворителях, например полярных апротонных растворителях,например дихлорметане, тетрагидрофуране, 1,4-диоксане, N,N'-диметилформамиде или их смесях в присутствии акцептора протонов, например триэтиламина, N,N-диизопропилэтиламина, пиперидина, N,Nдиметиланилина или пиридина, при температуре, находящейся в пределах от комнатной до температуры кипения растворителя. Предпочтительно стадию (а) осуществляют путем реакции соединения формулы(II) с Z-защищенной аминокислотой в присутствии амина, например триэтиламина в N,Nдиметилформамиде при комнатной температуре. Перед снятием защитной группы Boc соединение формулы (IV) необязательно можно превратить в другое соединение формулы (IV). Например, группу NH остатка анилина можно алкилировать обработкой алкилгалогенидом в основной среде по стандартным методикам, хорошо известным специалистам в данной области. Последующее отщепление группы Boc от соединения формулы (IV), которое осуществляют, как описано выше, позволяет получить целевые соединения (Ib). Соединения формулы (Ib) можно превратить в другие соединения формулы (Ib) с помощью любых способов синтеза, известных в технике, и/или его можно превратить в фармацевтически приемлемую соль, и/или его соли можно превратить в свободное соединение формулы (Ib). Упомянутые выше ацилирующий агент или Z-защищенная аминокислота имеются в продаже или их можно легко получить из известных соединений по стандартным методикам, известным специалистам в данной области. В случае, если ацилирующий агент или Z-защищенная аминокислота содержит реакционноспособную группу, такую как гидроксил, карбоксил, тиольную или аминогруппу, может оказаться необходимой защита этих групп защитными группами, например т-бутоксикарбонилом, бензилом,бензилоксикарбонилом, метилом, триметилсилилом и т.п., и на определенных стадиях синтеза снятие защиты с обратным получением свободной реакционноспособной группы. Лишенная защиты группа может быть в дальнейшем введена в реакцию, например алкилирована, ацилирована, сульфонилирована и т.п. Защита и снятие защиты функциональных групп описано в "Protective groups in Organic Chemistry" 3rd edition, T.W.Greene and P.G.Wuts, Wiley-Interscience (1999) и в "Protecting Groups", P.J.Kocienski, GeorgThieme Verlag (1994). Специалисту в данной области ясно, что если соединение формулы (I), синтезированное описанными выше способами (способ А или способ В), получено в виде смеси изомеров, разделение этой смеси на отдельные изомеры формулы (I), осуществляемое по обычным методикам, также входит в объем настоящего изобретения. Как должен понять специалист в данной области, если в ходе синтеза соединений формулы (I) некоторые функциональные группы могли бы давать нежелательные побочные реакции, эти группы необходимо защитить надлежащим образом, согласно стандартным методикам. Соответственно превращение защищенных соединений в соответствующие незащищенные соединения может осуществляться по методикам, хорошо известным специалистам в данной области. Исходные соединения формулы (II) можно получать из известных коммерчески доступных соединений по стандартным методикам, описанным в литературе и хорошо известным специалистам в данной области техники. Соединения формулы (II) можно легко получать, частично следуя описанным методи-7 022459 кам (J Am Chem Soc, 80:4015-4018, 1958; J Org Chem, 27:733-736, 1962; Bioorg Med Chem Lett, 16: 18401845, 2006). Конкретно, этил 2-(4-нитрофенил)циклопропил-1-карбоксилат получали в виде смеси цис и транс-изомеров сочетанием коммерчески доступного 4-нитростирола с этилдиазоацетатом (EDA) в присутствии хлорида меди (I) (CuCl) в сухом CHCl3 (схема 1). Два указанных изомера могут быть выделены с использованием обычных методик разделения соединений, например хроматографического разделения,перекристаллизации, а также другими способами, хорошо известными специалисту в данной области. Щелочной гидролиз сложного этилового эфира приводит к получению соответствующих карбоновых кислот, которые, в свою очередь, превращают в соответствующие т-бутоксикарбаматы реакцией с триэтиламином, дифенилфосфорилазидом, т-бутанолом и ди-т-бутилдикарбонатом в сухом бензоле. Восстановление нитрогруппы т-бутоксикарбаматов гипофосфитом натрия, палладием на угле и карбонатом калия приводит к получению соединений формулы (II). Схема 1a) EDA, CuCl, сухой CHCl3, 60C, атмосфера N2; b) 2 н KOH, EtOH, кт; с) 1)DPPA, Et3N, сухой tBuOH, сухой бензол, 80C, атмосфера N2; 2) Boc2O, сухой бензол, 80C, атмосфера N2; е) 2 н K2CO3,NaH2PO2, Pd/C, ТГФ, 60C, атмосфера N2. Было обнаружено, что соединения по настоящему изобретению являются эффективными ингибиторами LSD1 и LSD2 и демонстрируют противоопухолевое действие на лейкемические клетки при индивидуальном применении и синергический эффект со средствами против лейкемии, при применении в комбинации. Объектом настоящего изобретения является соединение формулы (I), являющееся ингибитором гистон деметилазы LSD1 и/или LSD2. Предпочтительно соединение по настоящему изобретению предназначено для применения в терапии или в качестве проапоптотического агента, также предпочтительно это соединение предназначено для применения в качестве лекарственного средства для профилактики и/или лечения заболеваний, характеризуемых нарушением регуляции транскрипции генов, дифференцировки и пролиферации клеток. В предпочтительном варианте осуществления соединение по настоящему изобретению предназначено для применения в качестве противоопухолевого агента. В еще одном варианте осуществления соединение предназначено для применения в качестве противовирусного агента. Объектом настоящего изобретения является способ профилактики и/или лечения заболеваний и состояний, связанных с активностью гистон деметилазы LSD1 и/или LSD2 при отдельных опухолях, вирусных инфекциях путем введения млекопитающему, которому необходимо такое лечение, терапевтически эффективного количества соединения общей формулы (I), которое определено выше. Предпочтительно опухоль выбрана из нейробластомы, рака простаты, рака груди, острого миелоидного лейкоза, острого Т-клеточного лимфобластного лейкоза, рака мочевого пузыря, рака легких и колоректального рака. Также предпочтительно вирусная инфекция вызвана вирусом простого герпеса (Herpes Simplex Virus). Объектом настоящего изобретения является фармацевтическая композиция, включающая одно или несколько соединений общей формулы (I), которые описаны выше, индивидуально или в комбинации с другими действующими соединениями, и как минимум один фармацевтически приемлемый эксципиент. Предпочтительно фармацевтическая композиция содержит эффективное количество соединения по настоящему изобретению, которое включено в единичную дозированную форму. Термин "эксципиент" в настоящей заявке означает любое вещество, которое само не является тера-8 022459 певтическим агентом, применяемое в качестве носителя или средства доставки терапевтического агента субъекту, или добавляемое в фармацевтическую композицию с целью улучшения ее свойств при обработке и хранении, или для получения возможности, или для облегчения изготовления дозированных форм композиции в виде дискретных единиц, таких как таблетки, капсулы, пилюли, порошки, гранулы,драже, леденцы, пастилки, эликсиры, сиропы, растворы, суспензии, эмульсии, капли, лосьоны, спреи,настойки, кремы, мази, гели, суппозитории и трансдермальные устройства для перорального, энтерального, парентерального или местного путей введения. Термин "единичные дозированные формы" относится к физически дискретным единицам, подходящим в качестве разовой дозы для людей и других млекопитающих, где каждая единица содержит заранее определенное количество действующего вещества, рассчитанного на достижение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом. Специалист в данной области техники осведомлен обо всем ассортименте таких эксципиентов, подходящих для создания фармацевтических композиций. Подходящие фармацевтически приемлемые эксципиенты хорошо известны специалисту в данной области. В качестве иллюстрации, а не ограничения эксципиенты включают разбавители, солюбилизаторы, наполнители, агглютинирующие вещества, дезинтегрирующие средства, ингибиторы дезинтеграции, ускорители абсорбции, адъюванты, связующие вещества, носители, суспендирующие/диспергирующие агенты, пленкообразующие средства/покрытия,клейкие вещества, вещества, уменьшающие липкость, смачивающие агенты, лубриканты, средства для скольжения, консерванты, сорбенты, буферные агенты, поверхностно-активные агенты, вещества, добавляемые для маскирования или противодействия неприятному вкусу или запаху, вкусоароматические средства, красители, ароматизирующие агенты, подсластители, вещества, добавляемые для улучшения внешнего вида композиции и т.п. Выбор эксципиента будет в значительной степени зависеть от такихфакторов, как конкретный путь введения, влияние эксципиента на растворимость и стабильность и природа дозированной формы. Фармацевтические композиции по настоящему изобретению могут вводиться рядом путей, включая пероральный, парентеральный, внутривенный, путем инфузии, подкожный, внутримышечный, интраперитонеальный, трансмукозальный (включая буккальный, сублингвальный, назальный, трансуретральный и ректальный), местный, трансдермальный, путем ингаляции, глазным путем (включая глазные имплантаты, имплантаты-депо и инъекции, например введение в стекловидное тело), введение через слизистые оболочки и через кожу, или с применением любого другого пути введения. Так, фармацевтические композиции по настоящему изобретению могут иметь форму твердых или жидких препаратов, растворов для инъекций или суспензий, или бутылок, вмещающих несколько доз,форму таблеток, обычных или с покрытием, таблеток, с покрытием из сахара или пленочным покрытием,капсул, облаток, гелевых капсул, пилюль, крахмальных капсул, саше, порошков, гранул, каплетов, пастилок, болюсов, драже, лекарственных кашек, паст, суппозиториев или ректальных капсул, сиропов,эликсиров, эмульсий, растворов, суспензий, кремов, мазей, линиментов, лосьонов, капель, спреев, пластырей для введения лекарственного средства через кожу или через слизистые оболочки, в которых применяются полярные растворители. Например, твердые пероральные формы, наряду с действующим компонентом, могут содержать разбавители, например карбонаты щелочно-земельных металлов, фосфат магния, лактозу, декстрозу,тростниковый сахар, сахарозу, целлюлозу, производные микрокристаллической целлюлозы, крахмалы,кукурузный крахмал или картофельный крахмал, модифицированные крахмалы и т.п.; смазывающие средства, например оксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция, и/или полиэтиленгликоли; связующие агенты, например крахмалы, гуммиарабик, желатин метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезинтегрирующие агенты, например крахмал, альгиновую кислоту, альгинаты или крахмал гликолят натрия; шипучие смеси; красящие вещества; подсластители; смачивающие агенты, например, лецитин, полисорбаты, лаурилсульфаты; и вообще любые нетоксичные и фармакологически неактивные вещества, применяемые в фармацевтических составах. Эти фармацевтические препараты могут изготавливаться известными способами, например смешиванием,гранулированием, таблетированием, нанесением сахарного покрытия или нанесением пленочного покрытия. Жидкие дисперсии для перорального введения могут представлять собой, например, сиропы,эмульсии или суспензии. В качестве носителя сиропы могут содержать, например, сахарозу или сахарозу с глицерином и/или маннитом и сорбитом. Суспензии и эмульсии могут включать, как примеры носителей, природные камеди, агар, альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечного введения, наряду с действующим соединением, могут содержать фармацевтически приемлемые носители, например стерильную воду, оливковое масло,этилолеат, гликоли, например пропиленгликоль и, если это желательно, подходящее количество лидокаина гидрохлорида. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду или предпочтительно они могут иметь форму стерильных водных изотонических солевых рас-9 022459 творов или они могут содержать пропиленгликоль в качестве носителя. Суппозитории, наряду с действующим соединением, могут содержать фармацевтически приемлемый носитель, например масло какао, полиэтиленгликоли, ПАВ на основе сложных эфиров полиоксиэтиленсорбитана и жирных кислот, салицилаты или лецитин. Ингаляционные аэрозоли могут содержать наряду с действующим соединением газ-пропеллент, например гидрофторалкан. Составы, содержащие газ-пропеллент, могут также включать другие ингредиенты, например сорастворители, стабилизаторы и, необязательно, другие эксципиенты. Составы для ингаляции, не содержащие пропеллентов, которые включают соединения по настоящему изобретению, могут иметь форму растворов или суспензий в водной, спиртовой или водно-спиртовой среде, и их можно доставлять с помощью струйных или ультразвуковых ингаляторов известного уровня техники или источников ультрадисперсного мягкого аэрозоля (soft-mist). Описанные выше компоненты фармацевтических композиций являются только иллюстративными. Другие материалы, а также методики их обработки и т.п. описаны в части 5 книги Remington's Pharmaceutical Sciences, 20th Edition, 2000, Merck Publishing Company, Easton, Pennsylvania, которая включена в настоящую заявку с помощью ссылки. Соединения формулы (I) по настоящему изобретению можно вводить в составе лекарственных форм с продолжительным высвобождением или систем доставки лекарственных средств с продолжительным высвобождением. Описание типовых материалов, обеспечивающих продолжительное высвобождение, можно также найти в материалах, включенных в Remington's Pharmaceutical Sciences. Фармацевтические композиции, содержащие соединения по настоящему изобретению, обычно получают с помощью стандартных способов и вводят в виде подходящих фармацевтических форм. Твердые пероральные композиции можно получать обычным смешиванием, заполнением или прессованием. В композициях, содержащих большие количества наполнителей, для диспергирования действующего агента операцию смешивания можно осуществлять повторно. Эти операции являются стандартными. Жидкие пероральные составы могут быть получены в форме, например, водных или масляных суспензий или растворов, эмульсий, сиропов или эликсиров, или их можно получать в виде лиофилизованных продуктов, которые перед применением необходимо регенерировать добавлением воды или подходящего носителя. Указанные жидкие препараты могут содержать стандартные добавки, например, суспендирующие агенты, такие как сорбит, сироп, метилцеллюлоза, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия или гидрированные пищевые жиры, эмульгирующие агенты, такие как лецитин, моноолеат сорбитана или гуммиарабик; неводные носители (которые могут включать пищевые масла), такие как миндальное масло, фракционированное кокосовое масло, маслянистые эфиры, например, сложные эфиры глицерина, пропиленгликоля или этилового спирта; консерванты, например, метил или пропил п-гидроксибензоат или сорбиновую кислоту и, если это желательно,стандартные вкусоароматические добавки и красители. Для парентерального введения имеется возможность получать жидкие дозированные формы, содержащие соединение и стерильный носитель. Соединение по настоящему изобретению в зависимости от выбранного носителя и концентрации можно суспендировать или растворить. Парентеральные растворы обычно получают растворением соединения в носителе, стерилизацией с помощью фильтрования,заполнением подходящих флаконов и их закупориванием. Преимущественно, имеется возможность растворить в носителе подходящие адъюванты, например местные анестетики, консерванты и буферные агенты. Для повышения устойчивости после заполнения флакона и удаления воды в вакууме композицию можно заморозить. Парентеральные суспензии получают в основном аналогичным способом, с той разницей, что соединение можно суспендировать, а не растворить в носителе, и стерилизовать добавлением оксида этилена до суспендирования в стерильном носителе. С целью облегчения равномерного распределения соединения по настоящему изобретению предпочтительно можно включать в композицию ПАВ или смачивающий агент. Соединения по настоящему изобретению можно также вводить местно. Составы для местного введения могут представлять собой, например, мази, кремы, гели, лосьоны, растворы, пасты и т.п. и/или их можно получать с применением липосом, мицелл и/или микросфер. Мази, как хорошо известно в технике получения фармацевтических составов, являются полутвердыми препаратами, которые чаще всего получают на основе вазелина или других производных нефти. Примеры мазей включают составы на маслянистой основе, например растительных маслах, животных жирах, и полутвердых углеводородах, полученных из нефти, составы на эмульгируемой основе, например гидроксистеаринсульфате, безводном ланолине и гидрофильном вазелине, составы на эмульсионной основе, например цетиловом спирте, глицерил моностеарате, ланолине и стеариновой кислоте, и составы на водорастворимой основе, полученные из полиэтиленгликолей различной молекулярной массы. Кремы, как также хорошо известно специалисту в данной области, являются вязкими жидкостями или полутвердыми эмульсиями и содержат масляную фазу, эмульгатор и водную фазу. Масляная фаза, как правило, включает вазелин и жирный спирт,например цетиловый или стеариловый спирт. Водная фаза, как правило, содержит увлажнитель. Эмульгатор, входящий в состав крема, выбирают из неионных, анионных, катионных или амфотерных ПАВ. Однофазные гели содержат органические макромолекулы, в основном равномерно распределенные в несущей жидкости, которая, как правило, является водной, но также предпочтительно содержит спирт и,необязательно, масло. Предпочтительные гелеобразующие агенты представляют собой поперечносшитые полимеры акриловой кислоты (например, "карбомерные" полимеры, такие как карбоксиполиалкилены, которые можно приобрести под торговой маркой Carbopol). Кроме этого предпочтительными являются гидрофильные полимеры, например, полиэтиленоксиды, сополимеры полиоксиэтиленполиоксипропилен и поливиниловый спирт; полимеры целлюлозы, например, гидроксипропилцеллюлоза, гидроксиэтилцеллюлоза, гидроксипропилметилцеллюлоза, фталат гидроксипропилметилцеллюлозы и метилцеллюлоза; камеди, например трагакант и ксантановая камедь; альгинат натрия и желатин. Для получения однородных гелей можно добавлять диспергирующие агенты, например спирт или глицерин,или в смеси можно диспергировать гелеобразующий агент, например, путем растирания, механического смешивания и/или перемешивания. Соединения по настоящему изобретению можно также вводить трансдермальным путем. Обычные трансдермальные составы включают традиционные водные и неводные переносчики, такие как кремы,масла, лосьоны или пасты или могут представлять собой мембраны или медицинские пластыри. В одном из вариантов осуществления соединение по настоящему изобретению наносят на чувствительный к надавливанию пластырь, который наклеивают на кожу. Это средство введения дает возможность соединению проникнуть с поверхности пластыря в организм пациента через кожу. Для обеспечения продолжительного проникновения лекарственного средства через кожу в качестве чувствительных к надавливанию клейких средств можно применять природный каучук и силикон. Соединения формулы (I) по настоящему изобретению, подходящие для введения млекопитающим,например, людям, можно вводить в виде единственного действующего агента или в комбинации с другими фармацевтически активными ингредиентами стандартными путями, и уровень дозировки зависит от ряда факторов, включая активность конкретного применяемого соединения; возраст, массу тела, общее состояние здоровья, пол и рацион питания подвергаемого лечению индивидуума; время и путь введения; скорость выведения; другие лекарственные средства, которые водились ранее; а также тяжесть конкретного подвергаемого лечению заболевания, как хорошо понятно специалисту в данной области техники. Например, дозировка соединения формулы (I), подходящая для перорального введения, может находиться в пределах от примерно 30 до 500 мг на один прием от 1 до 5 раз в день. Если применяется парентеральный путь, как правило следует вводить более низкие дозы. Так, например, для внутривенного пути введения в основном применяются дозы в диапазоне, например, от 0,5 мг до 30 мг на кг массы тела. Соединения по настоящему изобретению можно вводить в виде целого ряда дозированных лекарственных форм, например перорально в форме таблеток, таблеток, покрытых сахаром или пленкой, капсул,облаток, в виде порошка или гранул; в виде сиропов, эмульсий, растворов или суспензий в водных или неводных жидкостях, в виде жидкой эмульсии масло-в-воде или жидкой эмульсии вода-в-масле, в виде болюсов, кашицы или пасты; ректально в форме суппозиториев; парентерально, например, внутримышечно или путем внутривенной инъекции или инфузии. Предпочтительно соединения общей формулы(I) самостоятельно или в комбинации с другими действующими ингредиентами можно вводить для профилактики и/или лечения любых заболеваний, при которых необходимо ингибирование гистон деметилаз LSD1 и LSD2. Указанные заболевания включают опухоли, вирусные инфекции. Примеры Далее настоящее изобретение будет описано с помощью приведенных ниже не ограничивающих примеров, которые проиллюстрированы следующими фигурами. На фиг. 1 показаны биологические свойства соединения 6 е.(А) 6 е проявляет синергическое действие с ретиноевой кислотой (RA) при ингибировании клеточного роста. Клетки NB4 обрабатывали повышающимися концентрациями ретиноевой кислоты (10 нМ,100 нМ и 1 мкМ) в отсутствие или в присутствии соединения 6 е (2 мкМ). В показанные моменты времени подсчитывали клетки по методике исключения красителя трипанового синего. NT означает необработанные клетки (только носитель).(В) 6 е проявляет синергическое действие с ретиноевой кислотой (RA) в инициировании дифференцировки клеток NB4. Клетки NB4 обрабатывали ретиноевой кислотой (100 нМ) или носителем (NT) в отсутствие или в присутствии соединения 6 е (2 мкМ). Через 7 дней из клеток получали препараты цитоспин, помещали их на предметные стекла и окрашивали по Май-Грюнвальду-Гимзе (May GrunwaldGiemsa). На фиг. 2 показано синергическое действие с ретиноевой кислотой при инициировании апоптоза в клетках NB4. Клетки NB4 обрабатывали повышающимися концентрациями ретиноевой кислоты (10 нМ,100 нМ и 1 мкМ) или носителем в отсутствие или в присутствии соединения 6 е (2 мкМ). Апоптоз измеряли путем окрашивания пермеабилизированных клеток пропидий йодидом через 7 дней. Показаны данные типового эксперимента. 1. Химические синтезы Методики Если не указано иное, все исходные вещества имеются в коммерческой продаже или легко получаются по известным из литературы методикам, и эти вещества использовали без какой-либо очистки. Все растворители имели степень чистоты (чда, выше технической) и при необходимости их очищали и сушили стандартными способами. Концентрирование раствора после осуществления химической реакции и проведения экстракции включало использование роторного испарителя, работавшего при пониженном давлении приблизительно 20 Торр. Органические растворы сушили над безводным сульфатом натрия. Результаты элементных анализов находились в пределах 0,40% от теоретических значений. ТСХ осуществляли на силикагелевых пластинах с алюминиевой подложкой (Merck DC, AlufolienKiesegel 60 F254) и визуализацию пятен проводили УФ-светом. Спектры 1H ЯМР и 13 С ЯМР регистрировали на приборе Bruker 400 МГц. Химические сдвиги выражали в миллионных долях (м.д. единицы ). Константы спин-спинового взаимодействия выражали в Герцах (Гц) и характер расщепления сигналов описывали терминами с (синглет), уш.с (уширенный синглет), д (дублет), т (триплет), кв (квартет), квин (квинтет), м (мультиплет). Спектры EIMS (масс-спектроскопия с ионизацией электронным ударом) регистрировали на спектрометре Fisions Trio 1000; приведены данные только для молекулярных ионов (M+) и основные пики. Температуры плавления определяли на приборе для измерения температуры плавления Buchi 530, и приведенные данные являются нескорректированными. Пример 1. Получение транс и цис трет-бутил 2-(4-нитрофенил)циклопропил карбаматов: Транс-трет-бутил 2-(4-нитрофенил)циклопропил карбамат(53 ммоль, 5 мл) перемешивали при 80C в атмосфере N2 в течение 16 ч. После этого добавляли ди-третбутилдикарбонат (8 ммоль, 1,7 г) и реакционную смесь перемешивали при 80C в течение еще 2 ч. Растворитель удаляли в вакууме и остаток очищали хроматографией на силикагеле, элюируя смесью этилацетат/н-гексан 1/3 и выделяя чистый транс трет-бутил 2-(4-нитрофенил)циклопропилкарбамат в виде бледно-желтого твердого вещества. 1 Смесь транс трет-бутил 2-(4-нитрофенил)циклопропил карбамата (2,88 ммоль; 0,8 г), карбоната калия (2,04 ммоль; 0,28 г), 10% палладия на угле (0,016 г) в тетрагидрофуране (3,88 мл) и воде (3,8 мл) дегазировали в течение 5 мин, затем по каплям при энергичном перемешивании добавляли раствор гипофосфита натрия (10,96 ммоль, 1,16 г) в воде (2,32 мл). Полученную смесь перемешивали при 60C в течение 5 ч. Растворитель удаляли, остаток выливали в воду (100 мл) и экстрагировали диэтиловым эфиром (350 мл). Органические слои промывали насыщенным раствором хлорида натрия (350 мл), сушили безводным сульфатом натрия и концентрировали. Остаток хроматографировали на силикагеле, элюируя смесью этилацетат/н-гексан 1/2 и получая в качестве первого элюата трет-бутил 1-(4-аминофенил)пропан-2-ил карбамат и затем транс трет-бутил 2-(4 аминофенил)циклопропил карбамат, оба соединения в виде желтых масел. 1 К раствору транс-трет-бутил 2-(4-аминофенил)циклопропил карбамата (0,6 ммоль, 0,150 г) в сухом дихлорметане (5 мл) по каплям при внешнем охлаждении реакционного сосуда льдом добавляли триэтиламин(0,72 ммоль, 0,1 мл) и бензоилхлорид (0,6 ммоль, 0,09 мл). Полученную смесь перемешивали в течение 1 ч,затем добавляли воду (50 мл), отделяли органический слой и водный слой экстрагировали дихлорметаном(230 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (250 мл), сушили безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографической колонке с силикагелем, элюируя смесью этилацетат/н-гексан 1/3 и получая чистое соединение 1b в виде твердого вещества белого цвета. 1(д, 2 Н, ароматические протоны), 10,25 (шир.с, 1 Н, PhNHCO); 13 С ЯМР (CDCl3, 400 МГц,мд)14,40, 22,80, 28,40 (3C), 32,60, 79,50, 121,0 (2 С), 125,20 (2 С),127,50 (2 С), 128,80 (2 С), 132,10, 134,20, 134,30, 137,30, 155,60, 164,70; MC(ESI) m/z: 352,18 [М]+; т.пл.=172-174C. Показанные ниже соединения (табл. 2) получали по описанной выше методике, используя соответствующие исходные вещества. Таблица 2 Триэтиламин (2,96 ммоль, 0,41 мл) и реагент ВОР (0,89 ммоль, 0,39 г) в атмосфере N2 добавляли к раствору N-бензилоксикарбонилфенилаланина (0,74 ммоль, 0,22 г) в сухом диметилформамиде (2 мл) и перемешивали смесь в течение 0,5 ч. В атмосфере азота добавляли транс трет-бутил 2-(4 аминофенил)циклопропилкарбамат (0,81 ммоль, 0,2 г) и перемешивали полученную смесь в течение ночи. Реакционную смесь выливали в воду (50 мл) и экстрагировали этилацетатом (330 мл). Органические слои промывали насыщенным раствором хлорида натрия (350 мл), сушили безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографической колонке с силикагелем, элюируя смесью этилацетат/хлороформ 1/5 и получая чистое соединение 2 е в виде белого твердого вещества. 1(2 С), 125,20 (2 С), 125,90, 127,10 (2 С), 127,60, 127,70 (2 С), 128,60 (2 С), 128,90 (2 С), 134,90, 136,10, 136,60,137,30, 155,60, 155,90, 172,70; MC(ESI) m/z: 529,26 [М]+; т.пл.=161-163C. Приведенные ниже соединения (табл. 3) получали по описанной выше методике, используя соответствующие исходные вещества. Таблица 3 Пример 5. Получение гидрохлоридов транс-2-(4-ароил (или арилацетил или бензилоксикарбониламинофенилциклопропиламинов (5a-h); гидрохлоридов транс-4-(N-бензилоксикарбониламиноацил)аминофенилциклопропиламинов (6 а-m); гидрохлорида транс-4-бромбензил 1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан 2-илкарбамата (7); гидрохлорида цис-бензил 1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамата (8) К раствору соединения 21 (0,26 ммоль, 0,1 г) в тетрагидрофуране (2 мл) добавляли 6 н водный раствор HCl (2 мл) и перемешивали смесь в течение 12 ч при комнатной температуре. Выпавшее в осадок твердое вещество отделяли фильтрованием, промывали диэтиловым эфиром (310 мл) и сушили, получая чистое соединение 6l в виде бесцветного твердого вещества. 1 1 Н, индол-NH); 13 С ЯМР (ДМСО-d6, 400 МГц,мд)14,0, 22,0, 27,80, 28,0, 59,50, 66,80, 109,70, 111,10, 118,80,119,80, 121,0 (2 С), 121,70, 123,0, 125,20 (2 С), 127,10 (2 С), 127,40, 127,60, 128,90 (2 С), 134,90, 136,10,136,50, 138,90, 155,90, 172,70; MC(ESI) m/z: 504,19 [М]+; т.пл. 250C. Показанные ниже соединения (табл. 4 и 5) получали в соответствии с описанной выше методикой,используя соответствующие исходные вещества. Таблица 4 Стадия a: Синтез метил 8-(4-транс-(2-трет-бутоксикарбониламиноциклопропил)фениламино)-8 оксооктаноата К раствору транс-трет-бутил 2-(4-аминофенил)циклопропил карбамата (0,56 ммоль, 140 мг) в сухом дихлорметане (5 мл) по каплям при внешнем охлаждении реакционного сосуда льдом добавляли триэтиламин (0,68 ммоль, 0,09 мл) и метил 8-хлор-8-оксооктаноат (0,564 ммоль, 0,08 мл). Полученную смесь перемешивали в течение 1 ч, затем добавляли воду (50 мл), отделяли органический слой и водный слой экстрагировали дихлорметаном (230 мл). Полученный в результате органический раствор промывали насыщенным раствором хлорида натрия (350 мл), сушили безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографической колонке с силикагелем, элюируя смесью этилацетат/хлороформ 1/2 и получая чистый метил 8-(4-транс-(2-трет-бутоксикарбониламиноциклопропил)фениламино)-8-оксооктаноат в виде твердого белого вещества. 1b: Синтез 8-(4-транс-(2-трет-бутоксикарбониламиноциклопропил)фениламино)-8 оксооктановой кислоты Раствор полученного на предыдущей стадии 8-(4-транс-(2-трет-бутоксикарбониламиноциклопропил)фениламино)-8-оксооктаноата (0,53 ммоль, 220 мг) и LiOH (1,05 ммоль, 44 мг) в смеси тетрагидрофуран/вода (5 мл/5 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь гасили добавлением 2 н HCl до pH=4, затем отделяли осадок фильтрованием, промывали водой(330 мл) и сушили,получая чистую 8-(4-транс-(2-третбутоксикарбониламиноциклопропил)фениламино)-8-оксооктановую кислоту в виде твердого вещества белого цвета. 1N1-(4-транс-(2-аминоциклопропил)фенил)-N8 гидроксиоктандиамида (9) К охлаждаемому (до 0C) раствору 8-(4-транс-(2-трет-бутоксикарбониламиноциклопропил)фениламино)-8-оксооктановой кислоты (0,32 ммоль, 130 мг) в сухом тетрагидрофуране (5 мл) добавляли этилхлорформиат (0,384 ммоль, 0,04 мл) и триэтиламин (0,42 ммоль, 0,06 мл) и перемешивали смесь в течение 10 мин. Твердые вещества отделяли фильтрованием и к фильтрату добавляли О-(2 метокси-2-пропил)гидроксиламин (0,96 ммоль, 0,7 мл). Раствор перемешивали при 0C, затем добавляли 6 н раствор HCl (10 мл) и продолжали перемешивание в течение еще 12 ч. После этого осадок отделяли фильтрованием и промывали диэтиловым эфиром (310 мл), получая чистый гидрохлорид N1-(4-транс(2-аминоциклопропил)фенил)-N8-гидроксиоктандиамида (9). 1 Стадия a: синтез транс-трет-бутил 2-(4-метиламинофенил)циклопропил карбамата (10) К раствору транс-трет-бутил 2-(4-аминофенил)циклопропил карбамата (1,88 ммоль, 467 мг) в ацетонитриле (5 мл) при 0C добавляли формальдегид (1,88 ммоль, 0,052 мл), цианоборгидрид натрия (5,64 ммоль, 0,356 г) и уксусную кислоту (0,2 мл). Смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли воду (50 мл) и экстрагировали смесь этилацетатом (350 мл). Органические фазы объеди- 18022459 няли и сушили над сульфатом натрия, после чего удаляли растворитель при пониженном давлении. Оставшееся масло очищали хроматографией на силикагеле, элюируя смесью этилацетат/н-гексан 1/2 и получая указанное в заглавии соединение в виде желтого масла; выход 34%; 1(2 мл) в атмосфере N2 добавляли триэтиламин (0,61 ммоль, 0,08 мл) и РуВОР (0,18 ммоль, 0,095 г) и перемешивали смесь в течение 0,5 ч. В атмосфере N2 добавляли транс трет-бутил 2-(4 метиламинофенил)циклопропил карбамат 10 (0,15 ммоль, 0,041 г) и перемешивали смесь в течение ночи. Полученную реакционную смесь выливали в воду (30 мл) и экстрагировали этилацетатом (330 мл). Органические слои промывали насыщенным раствором хлорида натрия (330 мл), сушили безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографической колонке с силикагелем,элюируя смесью этилацетат/хлороформ 1,5 и получая чистое соединение в виде бесцветного масла с выходом 72%; 1(2 мл) и перемешивали смесь при комнатной температуре в течение 12 ч. Выпавшее в осадок твердое вещество отделяли фильтрованием, промывали диэтиловым эфиром (310 мл) и сушили, получая чистое соединение (12) в виде твердого вещества белого цвета; выход 82%; т.пл. 156-158C; для перекристаллизации использовали: бензол; 1 Стадия а: Синтез метил 2-(3-бензилуреидо)-3-фенилпропаноата (13) К раствору гидрохлорида метилового эфира фенилаланина (0,93 ммоль, 0,2 г) в тетрагидрофуране при 0C добавляли триэтиламин (1,86 ммоль, 0,26 мл) и бензилизоцианат (1,86 ммоль, 0,23 мл) и смесь перемешивали в течение 12 ч. Полученную реакционную смесь выливали в воду (30 мл) и экстрагировали этилацетатом (530 мл). Органические слои промывали насыщенным раствором хлорида натрия (330 мл), сушили безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографической колонке с силикагелем, элюируя смесью этилацетат/н-гексан 1/2 и получая чистое соединение в виде бесцветного масла, выход 95%; 1(2 С), 128,6 (2 С), 136,6, 137,9, 157,9, 171,5; MC(ESI) m/z: 312,14 [M]+. Стадия b: синтез 2-(3-бензилуреидо)-3-фенилпропановой кислоты (14) Раствор метил 2-(3-бензилуреидо)-3-фенилпропаноата (13) (2,66 ммоль, 0,83 г) и 2 н раствор гидроксида лития (5,32 ммоль, 0,22 г) в этаноле (20 мл) перемешивали в течение ночи при комнатной температуре. Реакционную смесь гасили добавлением 2 н HCl до pH=2, после чего осадок отделяли фильтрованием, промывали водой (330 мл) и сушили, получая чистую 2-(3-бензилуреидо)-3-фенилпропановую кислоту в виде беловатого твердого вещества; выход 95%, т.пл. 115-117C; вещество перекристаллизовывали из смеси циклогексан/бензол; 1H ЯМР (CDCl3, 400 МГц, ; мд)2,85-2,87 (дд, 1 Н, PhCHHCHCOO), 2,89-2,91 (дд, 1 Н,PhCHHCOO), 4,38-4,40 (м, 1 Н, PhCHHCHCOO), 6,14-6,17 (д, 1 Н, PhCHHNHCONH), 6,54-6,57 (м, 1 Н,PhCHHNHCONH), 7,18-7,30 (м, 10 Н, ароматические протоны), 12,65 (шир.с, 1 Н, СООН); 13 С ЯМР (CDCl3, 400 МГц, ; мд)36,0, 44,4, 56,8, 125,9, 126,7, 126,9 (2 С), 127,7 (2 С), 128,5 (2 С),128,6 (2 С), 136,6, 137,9, 157,6, 174,7; MC(ESI) m/z: 298,32 [M]+. Стадия с: синтез транс-трет-бутил 2-[4-[2-(3-бензилуреидо)-3-фенилпропаноил]аминофенил]циклопропил карбамата (15) К раствору 2-(3-бензилуреидо)-3-фенилпропановой кислоты (0,48 ммоль, 0,14 г) в сухом диметилформамиде (2 мл) в атмосфере N2 добавляли триэтиламин (1,92 ммоль, 0,27 мл) и PyBOP (0,57 ммоль,0,30 г) и полученную смесь перемешивали в течение 0,5 ч. В атмосфере N2 добавляли трет-бутил (2-(4 аминофенил)циклопропил)карбамат (0,52 ммоль, 0,13 г) и продолжали перемешивание в течение ночи. Реакционную смесь выливали в воду (30 мл) и экстрагировали этилацетатом (330 мл). Органические слои промывали насыщенным раствором хлорида натрия (330 мл), сушили над безводным сульфатом натрия и концентрировали. Остаток очищали на хроматографиеческой колонке с силикагелем, элюируя смесью этилацетат/н-гексан 1/1 и получая чистое соединение (15) в виде белого твердого вещества, выход 70%; т.пл. 100-102C; при перекристаллизации использовали циклогексан; 1 Н ЯМР (CDCl3, 400 МГц,мд)1,09-1,10 (м, 1 Н, CHH циклопропан), 1,18-1,19 (м, 1 Н, CHH циклопропан), 1,46 (с, 9 Н, С(CH3)3), 2,30-2,31 (м, 1 Н, PhCH циклопропан), 2,52-2,54 (м, 1 Н, CHNH циклопропан), 2,98-3,00 (дд, 1 Н, PhCHHCHCOO), 3,01-3,02 (дд, 1 Н, PhCHHCHCOO), 4,18-4,20 (м, 2 Н,PhCHHCHCOO), 4,27-4,28 (м, 1 Н, PhCHHNHCONH), 4,89 (шир.с, 1 Н, NHCOOC(CH3)3), 4,92-4,94 (д, 1 Н,PhCHHNHCONH), 6,05-6,07 (м, 1 Н, PhCHHNHCONH), 6,75-6,77 (м, 1 Н, PhCHHNHCONH), 6,90-6,94 (д,2 Н, ароматические протоны), 7,10-7,27 (м, 12 Н, ароматические протоны), 9,23 (шир.с, 1 Н, PhNHCOCH); 13 С ЯМР (CDCl3, 400 МГц, ; мд)14,40, 22,80, 28,40 (3C), 32,60, 36,90, 44,4, 59,0, 79,50, 121,0 (2 С),125,20 (2 С), 125,90, 126,7, 126,9 (2 С), 127,70 (2 С), 128,5 (2 С), 128,60 (2 С), 134,90, 136,60, 137,3, 137,9,155,6, 157,60, 172,70; MC(ESI) m/z: 528,27 [M]+. Стадия d: синтез гидрохлорида транс-N-(4-(2-аминоциклопропил)фенил)-2-(3-бензилуреидо)-3 фенилпропанамида (16) К раствору транс-трет-бутил 2-[4-[2-(3-бензилуреидо)-3-фенилпропаноил]аминофенил]циклопропил карбамата (0,30 ммоль, 0,1 г) в тетрагидрофуране (2 мл) добавляли 6 н водный растворHCl (2 мл) и перемешивали полученную смесь в течение 12 ч при комнатной температуре. Выпавшее в осадок твердое вещество фильтровали, промывали диэтиловым эфиром (310 мл) и сушили, получая чистое соединение 16 в виде твердого белого вещества, выход 82%, т.пл. 153-155C, при перекристаллизации использовали бензол; 1C ЯМР (ДМСО-d6, 400 МГц, ; мд)12,1, 20,5, 36,9, 40,3, 44,4, 59,0, 121,0 (2 С), 125,9, 125,2 (2 С),126,7, 126,9 (2 С), 127,7 (2 С), 128,5 (2 С), 128,6 (2 С), 134,9, 136,6, 137,3, 137,9, 157,6, 172,7; MC(ESI) m/z: 464,19 [M]+. 2. Биологическое тестирование Методики Человеческие рекомбинантные МАО А и МАО В экспрессировали в Pichia pastoris и очищали по опубликованной методике (Binda С. et al., Proc.Natl.Acad.Sci.USA 100: 9750-9755, 2003). При исследовании ингибирования значение Ki определяли с использованием кинурамина (МАО А) и бензиламина(МАО В) в качестве субстратов при pH 7,5 в соответствии с опубликованными методиками (Binda С. etE.Coli и очищали по описанной методике (Karytinos A. et al., J.Biol.Chem. 284:17775-17782, 2009). Человеческий рекомбинантный LSD1/CoREST экспрессировали в E.Coli в виде отдельных белков и совместно очищали, следуя ранее описанным методикам (Forneris F. et al. Trends Biochem Sci 33:181-189,2008). Исследования ферментной активности и ингибирования обеих деметилаз проводили при pH 7,5-8,0, используя метилированный пептид H3 (Forneris F. et al. J.Biol.Chem. 282: 20070-20074, 2007; Karytinos A. et al.,J.Biol.Chem. 284:17775-17782, 2009). Соединения исследовали с точки зрения их потенциального влияния на ферментную активность с помощью пероксидаза-сопряженного анализа при 25C с использованием не насыщающих концентраций субстрата. Экспериментальные значения kcat, измеренные в присутствии соединений (конечные концентрации в диапазоне от 25 до 150 мкМ в зависимости от растворимости), сравнивали со значениями, полученными в контрольном анализе, который выполняли в отсутствии тестируемого соединения, табл. 6. Активность LSD1 определяли в 50 мМ Hepes/NaOH pH 7,5, используя в качестве субстрата пептид гистон H3, монометилированный по остатку Lys4. Активность LSD2 измеряли в 50 мМ Hepes/NaOH pH 8,0, используя в качестве субстрата пептид гистон H3, диметилированный по остатку Lys4, табл. 6. Анализ активности МАО А и МАО В проводили в 50 мМ Hepes/NaOH pH 7,5 с 0,5% (объем/объем) восстановленного Triton X-100, используя в качестве субстратов кинурамин и бензиламин, соответственно,табл. 6. Клетки NB4 обрабатывали соединением 6 е в различных концентрациях (фиг. 1). Соединение 6 е и ретиноевую кислоту (RA, Sigma) растворяли в ДМСО при концентрации 1000. Клетки NB4 выращивали в среде RPMI, с добавкой 10% FBS, 100 Ед/мл пенициллина, 100 мкг/мл стрептомицина и выдерживали в увлажненном инкубаторе при 37C, в атмосфере 10% О 2 и 5% СО 2. Клетки помещали в планшеты при плотности 150000 кл/мл и обрабатывали RA (10 нМ, 100 нМ и 1 мкМ) в присутствии или отсутствии 2 мкМ соединения 6 е. К клеткам, обработанным носителем, добавляли ДМСО в конечной концентрации 0,2%. В каждый момент времени (2, 4 и 7 дней) клетки собирали, окрашивали раствором красителя трипанового синего и производили подсчет с использованием гемоцитометра. Подсчитывали только выжившие клетки. Одновременно из клеток готовили препараты цитоспин, помещали на предметные стекла и окрашивали по Май Грюнвальду-Гимзе (May Grunwald-Giemsa). Результаты Транилципромин является ковалентным ингибитором MAOs и LSDs и его связывание приводит к исчезновению поглощения флавина, связанного с белком, что легко поддается измерению (Li М., Hubalek F., Restelli N., Edmondson D.E., Mattevi A Insights into the mode of inhibition of human mitochondrialdemethylase LSD1. Biochemistry 46:4408-4416, 2007; Karytinos A., Forneris F., Profumo A., Ciossani G.,Battaglioli E., Binda C., Mattevi A. A novel mammalian flavin-dependent histone demethylase J Biol Chem 284:17775-17782, 2009). Эта особенность дает средство для быстрого и эффективного скрининга производных транилципромина по настоящему изобретению. Проводили дополнительное исследование каждого соединения, измеряя его влияние на активность ферментов, как показано в табл. 6. Значения Ki, вычисленные для отдельных соединений, приведены в табл. 7. Таблица 6. Профиль активности некоторых соединений по настоящему изобретению Отсутствие ингибирования показано знаком "-", а наличие ингибирования знаком "+". Максимальная концентрация ингибитора в этом исследовании составляла 1 мМ или равнялась концентрации насыщенного раствора ингибитора, для ингибиторов с растворимостью 1 мМ.b Активность LSD1 определяли в 50 мМ Hepes/NaOH pH 7,5, используя в качестве субстрата пептид гистон H3, монометилированный по остатку Lys4. Активность LSD2 измеряли в 50 мМ Hepes/NaOH pH 8,0, используя в качестве субстрата пептид гистон H3, диметилированный по остатку Lys4. с Анализ активности МАО А и МАО В проводили в 50 мМ Hepes/NaOH pH 7,5 с 0,5% (объем/объем) восстановленного Triton Х-100, используя в качестве субстратов кинурамин и бензиламин, соответственно. Активность ферментов измеряли при 25 С, используя пероксидаза-сопряженный анализ. Ошибки определения Ki находятся в пределах 30% от их значений; ND - не определялось (нет данных). ЗначенияKi определялись в конкурентных экспериментах в равновесном состоянии. Низкая скорость необратимого ингибирования дает возможность проводить эти эксперименты в нормальном равновесном режиме.b Активность LSD1 определяли в 50 мМ Hepes/NaOH pH 7,5, используя в качестве субстрата пептид гистон H3, монометилированный по остатку Lys4. Активность LSD2 измеряли в 50 мМ Hepes/NaOH pH 8,0, используя в качестве субстрата пептид гистон H3, диметилированный по остатку Lys4. с Анализ активности МАО А и МАО В проводили в 50 мМ Hepes/NaOH pH 7,5 с 0,5% (объем/объем) восстановленного Triton Х-100, используя в качестве субстратов кинурамин и бензиламин соответственно.d Отсутствовало заметное ингибирование в максимальных исследованных концентрациях, соответствовавших насыщенным растворам ингибиторов. е Значение Ki повторно определяли с использованием препаратов МАО А более высокого качества,что привело к значению, несколько отличающемуся от опубликованного в статье Binda С., et al.,J.Am.Chem.Soc., 132:6827-6833, 2010. Кроме того, оценивали биологическую активность соединения 6 е в клетках NB4 (фиг. 1). КлеткиNB4 обрабатывали соединением 6 е в различных концентрациях. Интересно, что хотя соединение 6 е не эффективно само по себе, оно способно значительно усиливать дифференцирующее действие RA. Этот эффект наблюдался при таких низких концентрациях RA, как 10 нМ, которые практически полностью неэффективны в отсутствие 6 е. Комбинация RA и 6 е во всех исследованных дозировках совместно ингибировала клеточный рост и улучшала дифференцировку, как показано на изображениях препаратов, обработанных цитоспином, приведенных на фиг. 1. Аналогичный результат был получен при определении способности инициировать клеточный апоптоз в клетках NB4 (фиг. 2). Действие соединения 6 е заключалось в увеличении эффективности инициирования апоптоза ретиноевой кислотой. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы (I) или его изомер, их фармацевтически приемлемые соли, в которыхR1 выбран из изопропила, изобутила, фенила, циклогексилметила, бензила, необязательно замещенного бромом или метокси, 2-фенилэтила, 2,2'-дифенилметила, нафтилметила, индолилметила, бензотиофенилметила;R3 означает -CH3. 6. Соединение по любому одному из пп.1-4, где независимо или в любой комбинацииR1 означает изопропил, изобутил, фенил, циклогексилметил, бензил, необязательно замещенный бромом или метокси, 2-фенилэтил, 2,2'-дифенилметил, нафтилметил, индолилметил, бензотиофенилметил;R2 означает бензилокси, необязательно замещенный бромом, или бензиламино. 7. Соединение по п.1, входящее в следующую группу: транс-бензил-4-(2-аминоциклопропил)фенилкарбамат; транс-N-(4-(2-аминоциклопропил)фенил)бензамид; транс-N-(4-(2-аминоциклопропил)фенил)-1-нафтамид; транс-N-(4-(2-аминоциклопропил)фенил)-2-нафтамид; транс-N-(4-(2-аминоциклопропил)фенил)бифенил-4-карбоксамид; транс-N-(4-(2-аминоциклопропил)фенил)-2-фенилацетамид; транс-N-(4-(2-аминоциклопропил)фенил)-2-(нафталин-1-ил)ацетамид; транс-N-(4-(2-аминоциклопропил)фенил)-2-(нафталин-2-ил)ацетамид; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-метил-1-оксобутан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-метил-1-оксопентан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-циклогексил-1-оксопропан-2-илкарбамат; транс-бензил-2-(4-(2-аминоциклопропил)фениламино)-2-оксо-1-фенилэтилкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(4-бромфенил)-1-оксопропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(4-метоксифенил)-1-оксопропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-4-фенилбутан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3,3-дифенилпропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(нафталин-1-ил)-1-оксопропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-3-(нафталин-2-ил)-1-оксопропан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-(1 Н-индол-3-ил)-1-оксобутан-2-илкарбамат; транс-бензил-1-(4-(2-аминоциклопропил)фениламино)-4-(бензо[b]тиофен-3-ил)-1-оксобутан-2 илкарбамат; транс-4-бромбензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат; цис-бензил-1-(4-(2-аминоциклопропил)фениламино)-1-оксо-3-фенилпропан-2-илкарбамат; транс-N1-(4-(2-аминоциклопропил)фенил)-N8-гидроксиоктандиамид; транс-бензил-1-4-(2-аминоциклопропил)фенил)(метил)амино)-1-оксо-3-фенилпропан-2-илкарбамат; транс-N-(4-(2-аминоциклопропил)фенил)-2-(3-бензилуреидо)-3-фенилпропанамид,или его изомер, их фармацевтически приемлемые соли. 8. Соединение по п.1 или 7, где изомер представляет собой энантиомер или диастереомер. 9. Соединение по п.8, где диастереомер представляет собой эпимер. 10. Способ получения соединения общей формулы (I) по п.1, где А представляет собой R, вклю- 24022459(а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (III)(b) необязательно превращение соединения формулы (III), полученного на стадии а), в другое соединение формулы (III), удаление защитной группы Boc из соединения формулы (III) с получением соединения формулы (I) 11. Способ получения соединения общей формулы (I) по п.1, где А представляет собой CH(R1)-NHCO-R2, включающий: а) взаимодействие соединения формулы (II) с ацилирующим агентом с получением соединения формулы (IV)b) необязательно превращение соединения формулы (IV), полученного на стадии а), в другое соединение формулы (IV), удаление защитной группы Boc из соединения формулы (IV) с получением соединения формулы (I) 12. Ингибитор гистон деметилазы LSD1 и/или LSD2, являющийся соединением по любому из предшествующих пп.1-9. 13. Применение соединения по любому из пп.1-9 в терапии. 14. Применение соединения по любому из пп.1-9 в качестве проапоптотического агента. 15. Применение по п.13 в качестве лекарственного средства, предназначенного для профилактики и/или лечения заболеваний и состояний, характеризующихся нарушением регуляции транскрипции генов, дифференцировки и пролиферации клеток. 16. Применение по п.15 в качестве противоопухолевого агента. 17. Применение по п.16, где указанный противоопухолевый агент эффективен против опухоли, выбранной из группы, состоящей из нейробластомы, рака простаты, рака груди, острого миелоидного лейкоза, острого Т-клеточного лимфобластного лейкоза, рака мочевого пузыря, рака легких и колоректального рака. 18. Применение по п.15 в качестве противовирусного агента. 19. Применение по п.18, где указанный противовирусный агент эффективен против вирусной инфекции, вызванной вирусом простого герпеса. 20. Способ профилактики и/или лечения заболеваний и состояний, связанных с активностью гистон деметилазы LSD1 и/или LSD2, путем введения млекопитающему, которому необходимо такое лечение,терапевтически эффективного количества соединения общей формулы (I) по любому из пп.1-9. 21. Способ по п.20, где заболевания представляют собой опухоли или вирусные инфекции. 22. Способ по п.20 или 21, где заболевания выбраны из нейробластомы, рака простаты, рака груди,рака мочевого пузыря, рака легких, колоректального рака, острого миелоидного лейкоза, острого Т- 25022459 клеточного лимфобластного лейкоза и инфекции вируса простого герпеса. 23. Фармацевтическая композиция, включающая эффективное количество одного или нескольких соединений общей формулы (I) по любому из пп.1-9, и как минимум один фармацевтически приемлемый эксципиент. 24. Фармацевтическая композиция по п.23 в виде единичной дозированной формы.
МПК / Метки
МПК: C07C 271/28, A61K 31/167, C07C 233/44, A61P 35/00, A61K 31/381, A61K 31/17, C07C 233/43, A61K 31/325, A61P 31/12, C07C 271/22, C07C 233/80, C07D 333/60, C07C 275/24
Метки: производные, гистон, ингибиторов, транилципромина, качестве, деметилаз
Код ссылки
<a href="https://eas.patents.su/27-22459-proizvodnye-tranilcipromina-v-kachestve-ingibitorov-giston-demetilaz-lsd1-i-ili-lsd2.html" rel="bookmark" title="База патентов Евразийского Союза">Производные транилципромина в качестве ингибиторов гистон деметилаз lsd1 и/или lsd2</a>
Гидроксаматы в качестве ингибиторов гистон-деацетилазы

Номер патента: 17198
Опубликовано: 30.10.2012
Авторы: Моффат Дэвид Фестус Чарльз, Дональд Элистейр Дэвид Грэм, Драммонд Алан Гастингс, Дэвидсон Алан Хорнсби, Белфилд Эндрю Джеймс, Пэтел Санджей Ратилал, Дэй Франческа Энн
МПК: A61P 25/28, A61P 19/02, A61K 31/506...
Метки: ингибиторов, качестве, гидроксаматы, гистон-деацетилазы
Формула / Реферат:
1. Соединение формулы (I) или его сольгде m равен 1;n равен 0 или 1;Q, V и W независимо обозначают -N= или -С=;В обозначает двухвалентный радикал, выбранный из (В2) и (В6)где связь, отмеченная *, связана с кольцом, содержащим Q, V и W;А обозначает циклогексильное или фенильное кольцо;-[Linker]- обозначает связь, -CH2-NH-CH2- или -NH-CH2-;Z1 обозначает радикал формулы R1R2CHNH- или R1R2CHNH-CH2-, гдеR1 обозначает карбоксильную группу (-СООН) или...
Сульфонилпроизводные в качестве ингибиторов гистон-деацетилазы

Номер патента: 7099
Опубликовано: 30.06.2006
Авторы: Ван Брандт Свен Францискус Анна, Анжибо Патрик Рене, Де Винтер Ханс Луи Йос, Ван Эмелен Кристоф, Ван Хьюсден Джимми Арнольд Вивиан, Бакс Лео Якобус Йозеф, Пилатт Изабелль Ноэлль Констанс, Вердонк Марк Густаф Селин
МПК: A61K 31/4545, A61P 35/00, C07D 207/14...
Метки: гистон-деацетилазы, качестве, ингибиторов, сульфонилпроизводные
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые соли добавления кислот и его стереохимически изомерные формы, где n равно 0, 1, 2 или 3 и, если n равно 0, то имеется в виду прямая связь; t равно 0, 1, 2, 3 или 4 и, если t равно 0, то имеется в виду прямая связь; каждый Q представляет собой азот или ; каждый Х представляет собой азот или ; каждый Y представляет собой азот или ; каждый Z представляет собой азот...
Сульфонилпроизводные в качестве новых ингибиторов гистон-деацетилазы

Номер патента: 6707
Опубликовано: 24.02.2006
Авторы: Бакс Лео Якобус Йозеф, Де Винтер Ханс Луи Йос, Пилатт Изабелль Ноэлль Констанс, Мерпул Ливен, Вердонк Марк Густаф Селин, Ван Хьюсден Джимми Арнольд Вивиан, Понселе Виржини Софи, Дяткин Алексей Борисович, Артс Янине, Ван Эмелен Кристоф, Ван Брандт Свен Францискус Анна
МПК: C07D 215/36, C07D 207/28, C07D 213/82...
Метки: гистон-деацетилазы, качестве, новых, сульфонилпроизводные, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) его N-оксидные формы, его фармацевтически приемлемые соли добавления кислот и его стереохимически изомерные формы, где n равно 0, 1, 2 или 3 и, если n равно 0, то имеется в виду прямая связь; t равно 0, 1, 2, 3 или 4, и если t равно 0, то имеется в виду прямая связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или каждый Z представляет собой азот или ...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Пейман Ануширван, Макдауэлл Роберт, Шойнеманн Карлхайнц, Гадек Томас, Бодари Сара Кэтрин, Гурвест Жан-Франсуа, Карниато Дени, Катбертсон Роберт Эндрю, Кнолле Йохен, Вилл Дэвид Вильям
МПК: C07D 239/42, A61K 31/505, A61P 19/10...
Метки: качестве, адгезии, ингибиторов, применение, костной, сульфонамидные, фармацевтическая, композиция, клеток,способ, производные, рассасывания, ткани, получения
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Сульфоксиминзамещенные анилинопиримидиновые производные в качестве cdk ингибиторов, их получение и применение в качестве лекарственных средств

Номер патента: 19230
Опубликовано: 28.02.2014
Авторы: Зимайстер Герхард, Люккинг Ульрих, Шульце Юлия, Линау Филип, Яутелат Рольф
МПК: C07D 239/47, A61K 31/505, A61P 35/00...
Метки: анилинопиримидиновые, средств, ингибиторов, получение, качестве, лекарственных, применение, производные, сульфоксиминзамещенные
Формула / Реферат:
1. Соединения общей формулы (I)в которых X представляет собой -O- или -NH-; иR1 представляет собой метильную, этильную, пропильную или изопропильную группу; иR2 и R3, независимо друг от друга, представляют собой водород, метильную или этильную группу; иR4 представляет собой C1-C6-алкильную группу или C3-C7-циклоалкильное кольцо,и их соли, диастереомеры и энантиомеры.2. Соединения, как заявлено в п.1, которые отличаются тем, что X представляет...
Предыдущий патент: 1,3,4-оксадиазол-2-карбоксамидное соединение
Следующий патент: Фармацевтические композиции в виде эмульсии с низким содержанием масла, содержащие прогестоген
Случайный патент: Дозирующий насос