Гетероароматические арилтриазольные производные в качестве ингибиторов фермента pde10a
Номер патента: 21606
Опубликовано: 30.07.2015
Авторы: Мариго Мауро, Килберн Джон Пол, Пюшль Аск, Нильсен Якоб, Келер Ян, Ланггор Мортен
























Формула / Реферат
1. Соединение, имеющее структуру I

где НЕТ-1 представляет собой гетероароматическую группу формулы II

где НЕТ-1 выбирают из группы, состоящей из имидазо[1,2-а]пиримидина, [1,2,4]триазоло[1,5-а]пиридина, имидазо[1,2-а]пиридина, имидазо[4,5-b]пиримидина, пиразоло[1,5-а]пиридина, [1,2,4]триазоло[1,5-а]пиримидина, [1,2,4]триазоло[1,5-а]пиразина и [1,2,4]триазоло[1,5-с]пиримидина,
и где НЕТ-1 может быть необязательно замещен посредством вплоть до трех заместителей R2-R4, независимо выбранных из Н; С1-С6алкила; галогена; циано, галоген (C1-C6)алкила; фенила; алкокси и С1-С6гидроксиалкила;
и где * означает место прикрепления,
Q представляет собой фенил, необязательно замещенный 1-5 заместителями, выбранными из группы, состоящей из С1-С6алкила, С1-С6алкокси и галогена, или Q представляет собой моноциклическую 5- или 6-членную гетероароматическую группу, выбранную из группы, состоящей из тиофена, фурана, тиазола, пиразола, пиридина, пиримидина и пиразина;
-L- представляет собой связующее звено, выбранное из -S-СН2-, -CH2-S-, -CH2-CH2-, -СН=СН- и -CºC-;
R1 выбирают из Н; С1-С6алкила; С1-С6алкил(С3-С8)циклоалкила; С1-С6гидроксиалкила, CH2CN, CH2C(O)NH2, С1-С6арилалкила и C1-С6алкилгетероциклоалкила;
при условии, что соединение не представляет собой
2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]-1Н-бензимидазол;
2-[[[3-(2-пиразинил)-1Н-1,2,4-триазол-5-ил]метил]тио]-1Н-бензимидазол;
2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)метил]тио]-1Н-бензимидазол;
1-этил-5-(1-пиперидинилсульфонил)-2-[[[3-(2-тиенил)-1Н-1,2,4-триазол-5-ил]тио]метил]-1Н-бензимидазол;
6-метил-2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]-1Н-бензимидазол;
2-[[[3-(3-пиридинил)-1Н-1,2,4-триазол-5-ил]метил]тио]-1Н-бензимидазол;
8-метил-2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин;
6-хлор-2-[[[3-(2-тиенил)-1Н-1,2,4-триазол-5-ил]тио]метил]имидазо[1,2-а]пиридинил;
2-[[[3-(4-пиридинил)-1Н-1,2,4-триазол-5-ил]метил]тио]-1Н-бензимидазол;
6-метил-2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин;
2-[[[3-(2-пиридинил)-1Н-1,2,4-триазол-5-ил]метил]тио]-1Н-бензимидазол;
6-хлор-2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин;
2-[[(3-фенил-1Н-1,2,4-триазол-5-ил)тио]метил]-3H-имидазо[4,5-b]пиридин или
2-[[[3-(2-фуранил)-1Н-1,2,4-триазол-5-ил]метил]тио]-1Н-бензимидазол;
и его таутомеры и фармацевтически приемлемые соли, при условии, что когда связующее звено (L) представляет собой -CH2-S-, тогда НЕТ-1 не является ни имидазо[1,2-а]пиридином, ни имидазо[1,2-а]пиримидином.
2. Соединение по п.1, где R2, R3 и R4, все, представляют собой водород.
3. Соединение по любому из пп.1-2, где по меньшей мере один из заместителей R2, R3 и R4 представляет собой С1-С6алкил, такой как метил.
4. Соединение по любому из пп.1-3, где по меньшей мере один из заместителей R2, R3 и R4 представляет собой галоген, такой как хлор или бром.
5. Соединение по п.1, где соединение выбирают из группы, состоящей из
8-метокси-5-метил-2-(5-фенил-2Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5-а]пиридина;
5-метил-2-(5-фенил-2Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5-а]пиридина;
5-метил-2-(1-метил-5-фенил-1Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5-а]пиридина;
8-метокси-5-метил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
8-метокси-5-метил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
8-метил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
5,7-диметил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиримидина;
5,8-диметил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиразина;
8-метокси-5-метил-2-[2-(5-фенил-2-пропил-2Н-[1,2,4]триазол-3-ил)этил][1,2,4]триазоло[1,5-а]пиридина;
5-метил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
8-метокси-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
этилового сложного эфира {5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}уксусной кислоты;
2-{5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}этанола;
5,8-диметил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
5,8-диметил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-с]пиримидина;
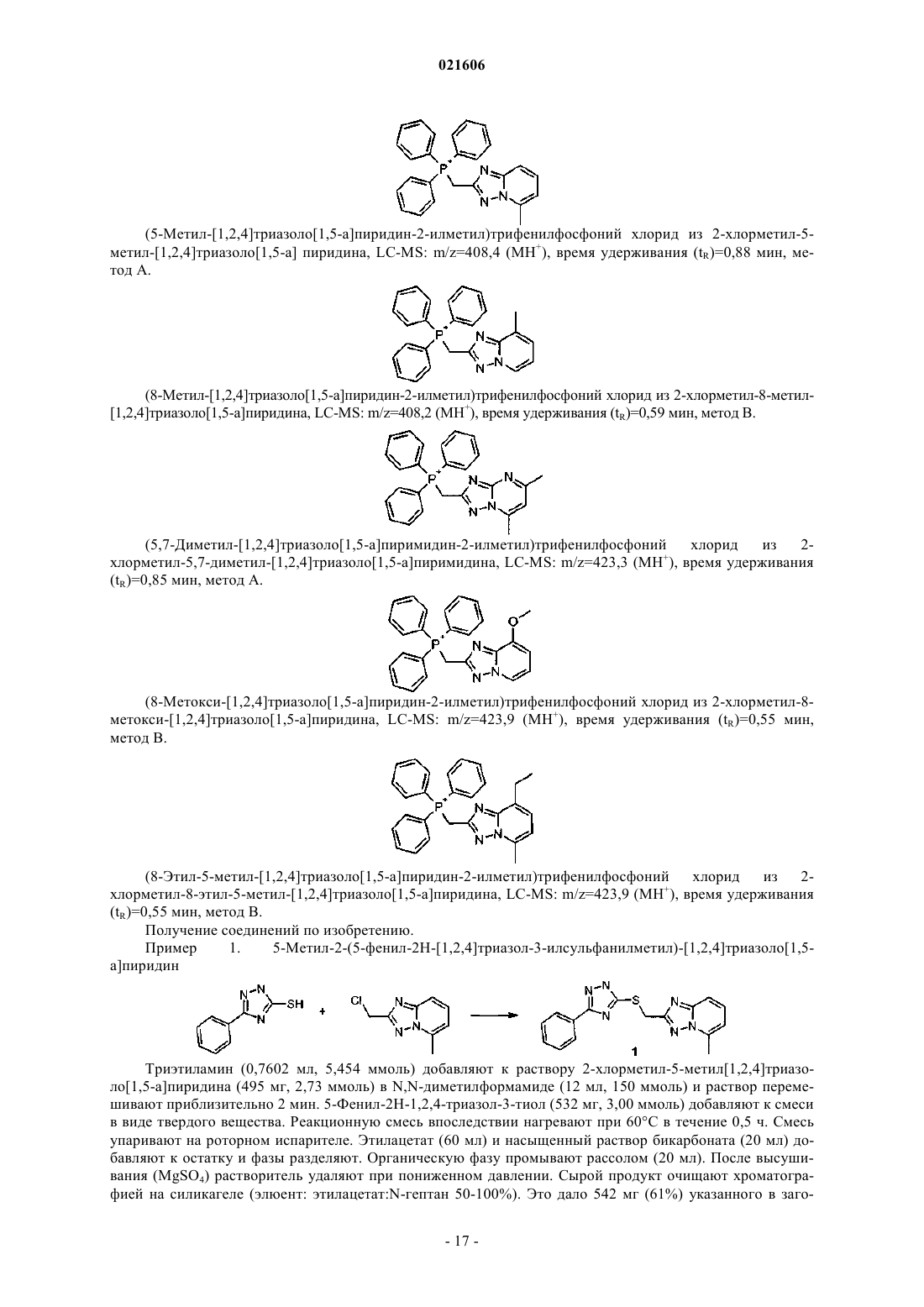
8-этил-5-метил-2-[2-(2-метил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
8-этил-5-метил-2-[2-(5-фенил-2-пропил-2Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина;
8-этил-2-[2-(2-изопропил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-5-метил-[1,2,4]триазоло[1,5-а]пиридина;
3-{5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}пропионитрила;
3-{5-[2-(8-этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}пропионитрила;
2-[2-(2-изопропил-5-фенил-2Н-[1,2,4]триазол-3-ил)этил]-8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридина;
3-{5-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}пропионитрила;
3-{2-[2-(8-этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-4-фенилимидазол-1-ил}пропиламина;
3-{5-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил}пропиламина;
2-{2-[2-(2-метоксиэтил)-5-фенил-2Н-[1,2,4]триазол-3-ил]этил}-5,8-диметил-[1,2,4]триазоло[1,5-а]пиразина и
8-метокси-2-{2-[2-(2-метоксиэтил)-5-фенил-2Н-[1,2,4]триазол-3-ил]этил}-5-метил-[1,2,4]триазоло[1,5-а]пиридина;
и их фармацевтически приемлемых солей.
6. Применение соединения по любому из пп.1-5, но без условия, в качестве лекарственного средства, которое является ингибитором фермента PDE10A.
Текст
ГЕТЕРОАРОМАТИЧЕСКИЕ АРИЛТРИАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА PDE10A Данное изобретение относится к соединениям формулы (I), где НЕТ-1 представляет собой гетероароматическую группу формулы II; где НЕТ-1 выбирают из группы, состоящей из имидазо[1,2-а]пиримидина, [1,2,4]триазоло[1,5-а]пиридина, имидазо[1,2-а]пиридина, имидазо[4,5b]пиримидина и т.д., и где НЕТ-1 может быть необязательно замещен посредством вплоть до трех заместителей R2-R4, независимо выбранных из Н; С 1-С 6 алкила; галогена; циано, галоген(C1-C6)алкила; фенила; алкокси и С 1-С 6 гидроксиалкила; и гдеозначает место прикрепления, Q представляет собой фенил, необязательно замещенный одним-пятью заместителями, выбранными из группы, состоящей из С 1-С 6 алкила, С 1-С 6 алкокси, галогена и т.д.; -L- представляет собой связующее звено, выбранное из -S-CH2-, -CH2-S-, -CH2CH2-, -СН=СН- и -СС-; R1 выбирают из Н; С 1-С 6 алкила; С 1-С 6 алкил(С 3-С 8)циклоалкила; С 1-С 6 гидроксиалкила, CH2CN, CH2C(O)NH2, C1-С 6 арилалкила и С 1-С 6 алкилгетероциклоалкила; при условии, что соединение не представляет собой 2-(3-фенил-1 Н-1,2,4-триазол-5 ил)тио]метил]-1 Н-бензимидазол; 2-3-(2-пиразинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Нбензимидазол; 2-(3-фенил-1 Н-1,2,4-триазол-5-ил)метил]тио]-1 Н-бензимидазол;1-этил-5-(1 пиперидинилсульфонил)-2-3-(2-тиенил)-1 Н-1,2,4-триазол-5-ил)тио]метил]-1 Н-бензимидазол; 6 метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]-1 Н-бензимидазол и т.д.; и их таутомерам и фармацевтически приемлемым солям, при условии, что, когда связующее звено (L) представляет собой -CH2-S-, тогда НЕТ-1 не является ни имидазо[1,2-а]пиридином, ни имидазо[1,2-а]пиримидином. Также изобретение относится к применению соединений в качестве лекарственного средства, являющегося ингибитором фермента PDE10A. Область техники, к которой относится изобретение Настоящее изобретение обеспечивает гетероароматические соединения, которые являются ингибиторами фермента PDE10A и сами по себе являются полезными для лечения нейродегенеративных и психических расстройств. Настоящее изобретение также обеспечивает фармацевтические композиции, содержащие соединения изобретения, и способы лечения расстройств с использованием соединений изобретения. Предпосылки создания изобретения В изобретении даются ссылки на различные публикации в полном объеме. Раскрытия этих публикаций, таким образом, являются включенными в эту заявку путем ссылки для более полного описания существующего уровня техники, к которому имеет отношение это изобретение. Циклические нуклеотиды циклический аденозинмонофосфат (сАМР) и циклический гуанозинмонофосфат (cGMP) действуют в качестве внутриклеточных вторичных мессенджеров, регулирующих огромное количество процессов в нейронах. Внутриклеточные сАМР и cGMP вырабатываются аденил- и гуанилциклазами и разрушаются под действием фосфодиэстераз (PDEs) циклических нуклеотидов. Внутриклеточные уровни сАМР и cGMP регулируются внутриклеточной сигнализацией, и стимуляция/подавление аденил- и гуанилциклаз в ответ на активацию GPCR (рецептор, сопряженный с Gбелком) представляет собой хорошо изученный (точно охарактеризованный) путь регулирования концентраций циклических нуклеотидов (Antoni, F.A. Front. Neuroendocrinol. 2000, 21, 103-132). Уровни сАМР и cGMP в свою очередь контролируют активность сАМР-и cGMP-зависимых киназ, а также других белков с отвечающими элементами циклических нуклеотидов, которые через последующее фосфорилирование белков и посредством других процессов регулируют важнейшие нейрональные функции,такие как синаптическая передача, нейрональная дифференциация и выживание. Существует 21 ген фосфодиэстеразы, которые могут быть подразделены на 11 семейств генов. Существует десять семейств аденилилциклаз, два семейства гуанилилциклаз и одиннадцать семейств фосфодиэстераз. PDEs представляют собой класс внутриклеточных ферментов, которые регулируют уровни сАМР и cGMP через гидролиз циклических нуклеотидов до их соответственных нуклеотидмонофосфатов. Некоторые PDEs разрушают сАМР, отчасти cGMP и отчасти и то, и другое. Большая часть PDEs имеет широкораспространенную экспрессию и имеет роли во многих тканях, хотя некоторые из них являются более специфическими в отношении тканей. Фосфодиэстераза 10 А (PDE10A) представляет собой фосфодиэстеразу с двойной специфичностью,которая может превращать как сАМР в AMP, так и cGMP в GMP (Loughney, K. et al. Gene 1999, 234, 109117; Fujishige, K. et al. Eur. J. Biochem. 1999, 266, 1118-1127 и Soderling, S. et al. Proc. Natl. Acad. Sci. 1999,96, 7071-7076). PDE10A в основном экспрессируется в нейронах в стриатуме, в прилежащем ядре и в обонятельном бугорке (Kotera, J. et al. Biochem. Biophys. Res. Comm. 1999, 261, 551-557 и Seeger, T.F. etPDE10A мыши является первым идентифицированным членом PDE10-семейства фосфодиэстеразBiochem. Biophys. Res. Comm. 1999, 261, 551-557 и Fujishige, K. et al. Eur. J. Biochem. 1999, 266, 11181127) . Существует высокая степень гомологии по видам. PDE10A локализуется у млекопитающих единственным образом в сравнении с другими PDE-семействами. мРНК для PDE10 сильно экспрессируется в семеннике и в головном мозге (Fujishige, K. et al. Eur. J. Biochem. 1999, 266, 1118-1127; Soderling, S. et al.Proc. Natl. Acad. Sci. 1999, 96, 7071-7076 и Loughney, K. et al. Gene 1999, 234, 109-117). Эти исследования показывают, что в пределах головного мозга экспрессия PDE10 является самой высокой в стриатуме(хвостатое ядро и путамен), прилежащем ядре и в обонятельном бугорке. Позднее проведен анализ картины экспрессии мРНК PDE10A в головном мозге грызунов (Seeger, T.F. et al. Abst. Soc. Neurosci. 2000,26, 345.10) и белка PDE10A (Menniti, F.S. et al. William Harvey Research Conference "Phosphodiesterase inPDE10A экспрессируется с высокими уровнями посредством срединных шипиковых нейронов(MSN) хвостатого ядра, прилежащего ядра и посредством соответствующих нейронов обонятельного бугорка. Они составляют срединную (основную) часть системы базальных ядер. Срединный шипиковый нейрон (MSN) имеет ключевую роль в кортико-ядерно-таламокортикальной петле, интегрирующей конвергентный кортикальный/таламический вход и посылающей эту интегрированную информацию обратно в кортикальный слой. MSNs экспрессируют два функциональных класса нейронов: класс D1, экспрессирующий дофаминовые рецепторы D1, и класс D2, экспрессирующий дофаминовые рецепторы D2. КлассD1 нейронов составляет часть "прямого" стриарного выводящего пути, который широко функционирует,содействуя поведенческим реакциям. Класс D2 нейронов составляет часть "непрямого" стриарного выводящего пути, который функционирует, подавляя поведенческие реакции, которые конкурируют с поведенческими реакциями, которым содействует "прямой" путь. Эти конкурирующие пути действуют подобно педали тормоза и педали акселератора в автомобиле. При самом простейшем подходе рассмотрения отсутствие/недостаток движения при болезни Паркинсона имеет место в результате чрезмерной активности "непрямого" пути, тогда как избыточное движение при заболеваниях, таких как болезнь Хан-1 021606 тингтона, отражает чрезмерную активность прямого пути. PDE10A-регуляция сАМР- и/или cGMPсигнализации в дендритном участке этих нейронов может быть вовлечена в фильтрацию кортико/таламического входа в MSN. Кроме того, PDE10A может участвовать в регуляции высвобожденияGABA (-аминомасляная кислота) в черной субстанции и в бледном шаре (Seeger, T.F. et al. Brain Research, 2003, 985, 113-126). Антагонизм дофаминового рецептора D2 точно установлен в лечении шизофрении. С 1950-х гг. антагонизм дофаминового рецептора D2 является основным направлением в лечении психозов, и все эффективные антипсихотические лекарственные средства антагонизируют рецепторы D2. Эффекты рецепторов D2, вероятно, опосредованы, в первую очередь, нейронами в стриатуме, прилежащем ядре и в обонятельном бугорке, поскольку эти зоны получают наиплотнейшие дофаминергические проекции и имеют наисильнейшую экспрессию рецепторов D2 (Konradi, С. And Heckers, S. Society of Biological Psychiatry, 2001, 50, 729-742). Агонизм дофаминового рецептора D2 приводит к снижению уровней сАМР в клетках, где он экспрессируется в результате ингибирования аденилатциклазы, и это является компонентом D2-сигнализации (Stoof, J. С; Kebabian J.W. Nature 1981, 294, 366-368 и Neve, K. A. et al. Journal ofReceptor and Signal Transduction 2004, 24, 165-205). В свою очередь, антагонизм рецептора D2 эффективно повышает уровни сАМР, и этот эффект мог быть симулирован ингибированием сАМР-расщепляющих фосфодиэстераз. Большая часть из 21 гена фосфодиэстераз экспрессируется широко; следовательно, ингибирование,вероятно, имеет побочные эффекты. Поскольку PDE10A в этом контексте имеет желательный профиль экспрессии с высокой и сравнительно специфической экспрессией в нейронах стриатума, прилежащего ядра и обонятельного бугорка, то вероятно, что ингибирование PDE10A имеет эффекты, подобные антагонизму рецептора D2, и, следовательно, имеет антипсихотические эффекты. Хотя ингибирование PDE10A, как ожидают, отчасти симулирует антагонизм рецептора D2, можно было бы предположить, что оно имеет отличный профиль. Рецептор D2 имеет компоненты сигнализации помимо сАМР (Neve, K.A. et al. Journal of Receptors and Signal Transduction 2004, 24, 165-205), в связи с чем интерференция с сАМР в результате ингибирования PDE10A может отрицательно модулировать, а не напрямую антагонизировать дофаминовую сигнализацию с помощью рецепторов D2. Это может снижать риск экстрапиримидальных побочных эффектов, которые наблюдают при сильном D2-антагонизме. С другой стороны, ингибирование PDE10A может иметь некоторые эффекты, не наблюдаемые при антагонизме рецептора D2. PDE10A также экспрессируется в стриарных нейронах, экспрессирующих рецепторы D1 (Seeger, T.F. et al. Brain Research, 2003, 985, 113-126). Поскольку агонизм рецептора D1 приводит к стимуляции аденилатциклазы и к получающемуся в результате увеличению уровней сАМР, ингибирование PDE10A, вероятно, также имеет эффекты, которые симулируют агонизм рецептора D1. В конечном итоге, ингибирование PDE10A не будет увеличивать в клетках только сАМР, но также, как можно было бы предположить, будет увеличивать уровни cGMP, поскольку PDE10A является фосфодиэстеразой с двойной специфичностью. cGMP активирует ряд белков-мишеней в клетках подобно сАМР и также взаимодействует с сАМР-сигнальными путями. В заключение необходимо отметить, что ингибированиеPDE10A, вероятно, отчасти симулирует антагонизм рецептора D2 и, следовательно, имеет антипсихотический эффект, но профиль может отличаться от профиля, наблюдаемого с классическими антагонистами рецепторов D2. Ингибитор PDE10A папаверин, как показано, является активным в нескольких антипсихотических моделях. Папаверин усиливал каталептический эффект антагониста рецептора D2 галоперидола у крыс,но не вызывал каталепсию сам по себе (WO 03/093499). Папаверин снижал гиперактивность у крыс, вызванную фенилциклидином (РСР), хотя снижение вызванной амфетамином гиперактивности было незначительным (WO 03/093499). Эти модели позволяют предположить, что ингибирование PDE10A имеет классический антипсихотический потенциал, который можно было бы ожидать по теоретическим соображениям. WO 03/093499 дополнительно раскрывает применение селективных ингибиторов PDE10 для лечения сочетанных неврологических и психических расстройств. Кроме того, ингибирование PDE10A вызывает обратное развитие подострых РСР-вызванных синдромов со сдвигом внимания у крыс (Rodeferet al. Eur. J. Neurosci. 2005, 4, 1070-1076). Эта модель позволяет предположить, что ингибированиеPDE10A может частично снимать симптомы когнитивных расстройств, сопутствующих шизофрении. Распределение PDE10A в тканях показывает, что ингибиторы PDE10A могут быть использованы для повышения уровней сАМР и/или cGMP в клетках, которые экспрессируют фермент PDE10, особенно в нейронах, которые составляют базальные ядра, и ингибиторы PDE10A настоящего изобретения, следовательно, могли бы быть полезными для лечения разнообразных сочетанных нейропсихических состояний с участием базальных ядер, таких как нейрологические и психические расстройства, шизофрения,биполярное аффективное расстройство, обсессивно-компульсивное расстройство и т.п., и могут иметь полезность, не обладая нежелательными побочными эффектами, которые имеют место в текущих методах терапии, представленных на рынке. Кроме того, недавние публикации (WO 2005/120514, WO 2005012485, Cantin et al. BioorganicMedicinal Chemistry Letters 17 (2007) 2869-2873) позволяют предположить, что ингибиторы PDE10A могут быть полезными для лечения ожирения и инсулиннезависимого сахарного диабета.-2 021606 Что касается ингибиторов PDE10A, патент ЕР 1250923 раскрывает применение селективных ингибиторов PDE10 вообще, и папаверина, в частности, для лечения некоторых неврологических и психических расстройств. Международная публикация WO 05/113517 раскрывает бензодиазепиновые стереоспецифические соединения в качестве ингибиторов фосфодиэстеразы, в особенности 2 и 4 типов фосфодиэстеразы, и предупреждение и лечение патологий, включающих в себя расстройство центральной и/или периферической нервной системы. Международная публикация WO 02/88096 раскрывает производные бензодиазепина и их применения в качестве ингибиторов фосфодиэстеразы, особенно фосфодиэстеразы 4 типа, в терапевтической области. Международная публикация WO 04/41258 раскрывает производные бензодиазепинона и их применения в качестве ингибиторов фосфодиэстеразы, в особенности фосфодиэстеразы 2 типа, в терапевтической области. Пирролодигидроизохинолины и их варианты раскрыты в качестве ингибиторов PDE10 в международных публикациях WO 05/03129 и WO 05/02579. Пиперидинил-замещенные хиназолины и изохинолины, которые служат в качестве ингибиторов PDE10, раскрыты в международной публикации WO 05/82883. Международная публикация WO 06/11040 раскрывает замещенные хиназолиновые и изохинолиновые соединения, которые служат в качестве ингибиторов PDE10. Патент US 20050182079 раскрывает замещенные тетрагидроизохинолинильные производные хиназолина и изохинолина, которые служат в качестве эффективных ингибиторов фосфодиэстеразы (PDE). В частности, патент US 20050182079 относится к вышеупомянутым соединениям, которые являются селективными ингибиторами PDE10. Аналогично, патент US 20060019975 раскрывает пиперидиновые производные хиназолина и изохинолина, которые служат в качестве эффективных ингибиторов фосфодиэстеразы (PDE). Патент US 20060019975 также относится к соединениям, которые являются селективными ингибиторами PDE10. Международная публикация WO 06/028957 раскрывает производные циннолина в качестве ингибиторов фосфодиэстеразы 10 типа для лечения психических и неврологических синдромов. Однако эти раскрытия не имеют отношения к соединениям изобретения, которые структурно не связаны ни с каким из известных ингибиторов PDE10 (Kehler, J. et al. Expert Opin. Ther. Patents 2007, 17,147-158 и Kehler, J. et al. Expert Opin. Ther. Patents 2009, 19, 1715-1725), и которые, как к настоящему времени обнаружено авторами изобретения, являются высокоактивными и селективными ингибиторами фермента PDE10A. Соединения, содержащие связующее звено -CH2-S-, и где НЕТ-1 представляет собой либо имидазо[1,2-а]пиридин, либо имидазо[1,2-а]пиримидин, раскрыты в общедоступных химических библиотеках. Эти соединения, следовательно, выведены из материалов заявки на изобретение. Соединения изобретения могут предоставить альтернативные варианты имеющимся в текущий момент на рынке методам лечения нейродегенеративных и/или психических расстройств, которые не являются эффективными у всех пациентов. Следовательно, сохраняется потребность в альтернативных способах лечения. Краткое изложение сущности изобретения Целью настоящего изобретения является предоставление соединений, которые являются селективными ингибиторами фермента PDE10A. Дополнительной целью настоящего изобретения является предоставление соединений, которые обладают такой активностью и которые имеют улучшенные растворимость, метаболическую стабильность и/или биологическую доступность в сравнении с соединениями известного уровня техники. Другой целью изобретения является обеспечение эффективного лечения, в частности длительного лечения, пациента-человека, которое не вызывает побочных эффектов, обычно сопутствующих текущим методам лечения неврологических и психических расстройств. Дополнительные цели этого изобретения станут очевидными при прочтении настоящего описания изобретения. В связи с вышеизложенным в одном аспекте настоящее изобретение относится к соединениям формулы I где НЕТ-1 представляет собой гетероароматическую группу формулы II[1,2,4]триазоло[1,5-а]пиримидина, [1,2,4]триазоло[1,5-а]пиразина и [1,2,4]триазоло[1,5-с]пиримидина, и где НЕТ-1 может быть необязательно замещен посредством вплоть до трех заместителей R2-R4, независимо выбранных из Н; С 1-С 6 алкила; галогена; циано, галоген (C1-C6) алкила; фенила; алкокси и C1-C6 гидроксиалкила; и гдеозначает место прикрепления,Q представляет собой фенил, необязательно замещенный одним-пятью заместителями, выбранными из группы, состоящей из С 1-С 6 алкила, С 1-С 6 алкокси и галогена, или Q представляет собой моноциклическую 5-членную или 6-членную гетероароматическую группу, выбранную из группы, состоящей из тиофена, фурана, тиазола, пиразола, пиридина, пиримидина и пиразина;-L- представляет собой связующее звено, выбранное из -S-СН 2-, -CH2-S-, -CH2-CH2-, -СН=СН- иR1 выбирают из Н; С 1-С 6 алкила; С 1-С 6 алкил(С 3-С 8)циклоалкила; С 1-С 6 гидроксиалкила, CH2CN,CH2C(O)NH2, С 1-С 6 арилалкила и С 1-С 6 алкилгетероциклоалкила; при условии, что соединение не представляет собой 2-(3-фенил-1H-1,2,4-триазол-5-ил)тио]метил]1 Н-бензимидазол; 2-3-(2-пиразинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 2-(3 фенил-1 Н-1,2,4-триазол-5-ил)метил]тио]-1 Н-бензимидазол; 1-этил-5-(1-пиперидинилсульфонил)-2-3(2-тиенил)-1 Н-1,2,4-триазол-5-ил)тио]метил]-1 Н-бензимидазол; 6-метил-2-(3-фенил-1 Н-1,2,4-триазол-5 ил)тио]метил]-1 Н-бензимидазол; 2-3-(3-пиридинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 8-метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин; 6-хлор-2-3-(2 тиенил)-1 Н-1,2,4-триазол-5-ил]тио]метил]имидазо[1,2-а]пиридинил; 2-3-(4-пиридинил)-1 Н-1,2,4 триазол-5-ил]метил]тио]-1 Н-бензимидазол; 6-метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил] имидазо[1,2-а]пиридин; 2-3-(2-пиридинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 6-хлор-2(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин; 2-(3-фенил-1 Н-1,2,4-триазол-5 ил)тио]метил]-3H-имидазо[4,5-b]пиридин или 2-3-(2-фуранил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Нбензимидазол; и его таутомеры и фармацевтически приемлемые соли, при условии, что, когда связующее звено (L) представляет собой -CH2-S-, тогда НЕТ-1 не является ни имидазо[1,2-а]пиридином, ни имидазо[1,2 а]пиримидином. В отдельных вариантах осуществления изобретения соединение формулы I выбирают из числа конкретных соединений, раскрытых в экспериментальном разделе в данном описании. Изобретение дополнительно обеспечивает соединение формулы I или его фармацевтически приемлемую соль для применения в качестве лекарственного средства. В другом аспекте настоящее изобретение обеспечивает применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения нейродегенеративного или психического расстройства. Подробное описание изобретения Определение заместителей. Как используют в контексте настоящего изобретения, термины "галогено-" и "галоген" используют взаимозаменяемо и относятся к фтору, хлору, брому или йоду. Термин "С 1-С 6 алкил" относится к насыщенному углеводороду с прямой или разветвленной цепью,имеющему от одного до шести атомов углерода включительно. Примеры таких групп включают метил,этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил, 2-метил-1-бутил и н-гексил, но не ограничиваются этим. Выражение "С 1-С 6 гидроксиалкил" относится к С 1-С 6 алкильной группе, которая определена выше, и которая замещена одной гидроксигруппой. Термин "галоген(C1-C6)алкил" относится к C1 С 6 алкильной группе, которая определена выше, и которая замещена посредством вплоть до трех атомов галогена, таких как трифторметил. Выражение "C1-C6 алкоксигруппа" относится к насыщенной алкоксигруппе с прямой или разветвленной цепью, имеющей от одного до шести атомов углерода включительно, с открытой валентностью на кислороде. Примеры таких групп включают метоксигруппу, этоксигруппу, н-бутоксигруппу, 2 метилпентоксигруппу и н-гексилоксигруппу, но не ограничиваются этим. Термин "С 3-С 8 циклоалкил" обычно относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу или циклооктилу. Выражение "С 1-С 6 алкил(С 3-С 8)циклоалкил" относится к С 3 С 8 циклоалкилу, определенному выше, который замещен С 1-С 6 алкилом с прямой или разветвленной цепью. Примеры таких групп включают циклопропилметил, но не ограничиваются этим. Термин "гетероциклоалкил" относится к 4-8-членному кольцу, содержащему атомы углерода и вплоть до трех атомов N, О или S, при условии, что 4-8-членное кольцо не содержит смежные атомы О или S. Открытая валентность находится либо на гетероатоме, либо на атоме углерода. Примеры таких групп включают азетидинил, оксетанил, пиперазинил, морфолинил, тиоморфолинил и [1,4]диазепанил,но не ограничиваются этим. Термин "гидроксигетероциклоалкил" относится к гетероциклоалкилу, определенному выше, который замещен одной гидроксигруппой. Термин "С 1-С 6 алкил-гетероциклоалкил" относится к гетероциклоалкилу, определенному выше, который замещен С 1-С 6 алкильной группой. Примеры таких групп включают тетрагидропиран-4-ил-метил и 2-морфолин-4-ил-этил, но не ограничиваются этим.-4 021606 Термин "арил" относится к фенильному кольцу, необязательно замещенному галогеном, С 1 С 6 алкилом, С 1-С 6 алкокси или галоген(С 1-С 6)алкилом, которые определены выше. Примеры таких групп включают фенил и 4-хлорфенил, но не ограничиваются этим. Термин "С 1-С 6 арилалкил" относится к арилу, определенному выше, который замещен С 1-С 6 алкилом с прямой или разветвленной цепью. Примеры таких групп включают бензил и 4-хлорбензил, но не ограничиваются этим. Кроме того, настоящее изобретение дополнительно обеспечивает некоторые варианты осуществления изобретения, которые описаны ниже. В одном варианте осуществления изобретения НЕТ-1 представляет собой гетероароматическую группу формулы II, содержащую 2 атома азота. В другом варианте осуществления изобретения НЕТ-1 представляет собой гетероароматическую группу формулы II, содержащую 3 атома азота. В другом варианте осуществления изобретения НЕТ-1 представляет собой гетероароматическую группу формулы II,содержащую 4 атома азота. НЕТ-1 предпочтительно выбирают из числа следующих гетероароматических групп, где означает место прикрепления: В дополнительном варианте осуществления один или несколько атомов водорода соединения формулы I были замещены дейтерием. В частности, водород был заменен дейтерием в том случае, когда R7R9 представлял собой метил или метоксигруппу. В отдельных вариантах осуществления изобретения соединение формулы I выбирают из числа следующих конкретных соединений, в форме свободного основания, в форме их одного или нескольких таутомеров или их фармацевтически приемлемых солей. Табл. 1 представляет соединения изобретения и соответствующие значения IC50, определенные таким образом, как описано в разделе "Анализ ингибирования PDE10A". Каждое из соединений составляет отдельно взятый вариант осуществления настоящего изобретения. Следует понимать, что различные аспекты, варианты осуществления, варианты реализации и отличительные признаки изобретения, упомянутые в данном описании, могут быть заявлены по отдельности или в любой комбинации, что проиллюстрировано следующими неограничивающими примерами. Таблица 1 Соединения изобретения и значения IC50 В конкретных вариантах осуществления настоящего изобретения соединения настоящего изобретения имеют значения IC50 менее чем 50 нМ, такие как, например, в диапазоне 0,2-20 нМ, в особенности в диапазоне 0,2-10 нМ, такие как, например, в диапазоне 0,2-5 нМ или в диапазоне 0,2-1 нМ. Фармацевтически приемлемые соли. Настоящее изобретение также включает в себя соли соединений, обычно фармацевтически приемлемые соли. Такие соли включают фармацевтически приемлемые соли присоединения кислоты. Соли присоединения кислоты включают соли неорганических кислот, так же как и органических кислот. Репрезентативные примеры подходящих неорганических кислот включают хлористо-водородную,бромисто-водородную, йодисто-водородную, фосфорную, серную, сульфаминовую, азотную кислоты и т.п. Репрезентативные примеры подходящих органических кислот включают муравьиную, уксусную,трихлоруксусную, трифторуксусную, пропионовую, бензойную, коричную, лимонную, фумаровую, гликолевую, итаконовую, молочную, метансульфоновую, малеиновую, яблочную, малоновую, миндальную,щавелевую, пикриновую, пировиноградную, салициловую, янтарную, метансульфоновую, этансульфоновую, винную, аскорбиновую, памовую, бисметилен-салициловую, этандисульфоновую, глюконовую,цитраконовую, аспарагиновую, стеариновую, пальмитиновую, этилендиаминтетрауксусную (EDTA),гликолевую, парааминобензойную, глутаминовую, бензолсульфоновую, паратолуолсульфоновые кислоты, теофиллинуксусные кислоты, а также 8-галогентеофиллины, например 8-бромтеофиллин, и т.п. Дополнительные примеры фармацевтически приемлемых солей присоединения неорганических или органических кислот включают фармацевтически приемлемые соли, перечисленные в публикации Berge,S.M. et al., J. Pharm. Sci. 1977, 66, 2, содержание которой включено в данное описание путем ссылки. Кроме того, соединения данного изобретения могут существовать в несольватированной, а также в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. В общем и целом, сольватированные формы рассматривают эквивалентными несольватированным формам для целей этого изобретения. Терапевтически эффективное количество. В контексте настоящего изобретения термин "терапевтически эффективное количество" соединения означает количество, достаточное для вылечивания, частичного снятия симптомов или частичного купирования клинических проявлений данного заболевания и его осложнений при терапевтическом вмешательстве, включающем в себя введение вышеупомянутого соединения. Количество, адекватное для выполнения этого, определяют как "терапевтически эффективное количество". Эффективные количества-7 021606 для каждой цели будут зависеть от тяжести заболевания или поражения, а также от массы и общего состояния субъекта. Будет понятно, что определение приемлемой дозировки может быть достигнуто с использованием обычного экспериментирования, посредством построения матрицы значений и проведения испытаний в различных точках матрицы, что полностью находится в рамках уровня средней компетентности квалифицированного врача. В контексте настоящего изобретения термин "лечение" и "лечащий" означает ведение пациента и уход за пациентом для достижения цели вылечивания состояния, такого как заболевание или расстройство. Термин, как полагают, включает полный спектр методов лечения данного состояния, которым страдает пациент, таких как, например, введение активного соединения для частичного снятия симптомов или осложнений, для отсрочивания прогрессирования заболевания, расстройства или состояния, для частичного снятия или купирования симптомов и осложнений и/или для вылечивания или устранения заболевания, расстройства или состояния, а также для предотвращения состояния, где предотвращение следует понимать как ведение пациента и уход за пациентом с целью вылечивания заболевания, состояния или расстройства и включает введение активных соединений для предотвращения начала проявления симптомов или осложнений. При этом профилактические (предотвращающие) и терапевтические (лечебные) методы лечения представляют собой два отдельных аспекта изобретения. Пациент, который подлежит лечению, предпочтительно представляет собой млекопитающее, в частности человеческое существо. Фармацевтические композиции Настоящее изобретение дополнительно обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы I и фармацевтически приемлемый носитель или разбавитель. Настоящее изобретение также предоставляет фармацевтическую композицию, содержащую терапевтически эффективное количество одного из конкретных соединений, раскрытых в экспериментальном разделе в данном описании, и фармацевтически приемлемый носитель или разбавитель. Соединения по изобретению могут быть введены по отдельности или в комбинации с фармацевтически приемлемыми носителями, разбавителями или эксципиентами либо одной дозой, либо многократно вводимыми дозами. Фармацевтические композиции согласно изобретению могут быть составлены с фармацевтически приемлемыми носителями или разбавителями, а также с любыми другими известными адъювантами и эксципиентами в соответствии с традиционными методами, такими как методы, раскрытые в Remington: The Science and Practice of Pharmacy, 19th Edition, Gennaro, Ed., Mack Publishing Co.,Easton, PA, 1995. Фармацевтические композиции могут быть особым образом составлены для введения любым подходящим путем, таким как пероральный, ректальный, назальный, пульмональный, местный (включая трансбуккальный и сублингвальный), трансдермальный, интрацистернальный, интраперитонеальный,вагинальный и парентеральный (включая подкожный, внутримышечный, интратекальный, внутривенный и интрадермальный) пути. Следует принимать во внимание, что путь будет зависеть от общего состояния и возраста субъекта, который должен пройти лечение, от природы состояния, которое должно быть вылечено, и от активного ингредиента. Фармацевтические композиции для перорального введения включают в себя тведые лекарственные формы, такие как капсулы, таблетки, драже, пилюли, пастилки для рассасывания, порошки и гранулы. В том случае, когда это приемлемо, композиции могут быть приготовлены с покрытиями, такими как кишечно-растворимые покрытия, или они могут быть составлены таким образом, чтобы обеспечивать контролируемое высвобождение активного ингредиента, такое как, например, замедленное высвобождение или пролонгированное высвобождение, в соответствии со способами, общеизвестными в данной области. Жидкие лекарственные формы для перорального введения включают растворы, эмульсии, суспензии,сиропы и эликсиры. Фармацевтические композиции для парентерального введения включают стерильные водные и неводные инъекционные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, которые должны быть ресуспендированы с получением стерильных растворов или дисперсий перед применением. Другие подходящие формы для введения включают суппозитории, спреи,мази, крема, гели, средства для ингаляции, трансдермальные пластыри и имплантаты, но не ограничиваются этим. Обычные дозировки при пероральном введении находятся в диапазоне от приблизительно 0,001 до приблизительно 100 мг/кг массы тела в день. Обычные дозировки при пероральном введении также находятся в диапазоне от приблизительно 0,01 до приблизительно 50 мг/кг массы тела в день. Обычные дозировки при пероральном введении дополнительно находятся в диапазоне от приблизительно 0,05 до приблизительно 10 мг/кг массы тела в день. Дозировки при пероральном введении обычно вводят одной или несколькими дозировками, обычно одной-тремя дозировками в день. Точная дозировка будет зависеть от частоты и способа введения, пола, возраста, веса и общего состояния субъекта, который проходит лечение, природы и тяжести состояния, которое подлежит вылечиванию, и от любых сопутствующих заболеваний, которые должны быть вылечены, и от других факторов, очевидных специалистам в данной области. Составы также могут быть представлены в единичной дозированной форме, способами, известными-8 021606 специалистам в данной области. С иллюстративными целями единичная дозированная форма для перорального введения может содержать от приблизительно 0,01 до приблизительно 1000 мг, от приблизительно 0,05 до приблизительно 500 мг или от приблизительно 0,5 мг до приблизительно 200 мг. Для парентеральных путей, таких как внутривенное, интратекальное, внутримышечное и т.п. введение, обычные дозы составляют порядка половины дозы, применяемой для перорального введения. Настоящее изобретение также предоставляет способ получения фармацевтической композиции,включающий смешение терапевтически эффективного количества соединения формулы I и по меньшей мере одного фармацевтически приемлемого носителя или разбавителя. В одном варианте осуществления настоящего изобретения соединение, используемое в вышеупомянутом способе, представляет собой одно из конкретных соединений, раскрытых в экспериментальном разделе в данном описании. Соединения по данному изобретению, как правило, используются в форме свободного вещества или в форме его фармацевтически приемлемой соли. Одним примером является кислотно-аддитивная соль соединения, имеющего полезность в форме свободного основания. В том случае, когда соединение формулы I заключает в себе свободное основание, такие соли приготавливают обычно применяемым способом путем обработки раствора или суспензии свободного основания формулы I молярным эквивалентом фармацевтически приемлемой кислоты. Репрезентативные примеры подходящих органических и неорганических кислот описаны выше. Для парентерального введения могут быть использованы растворы соединений формулы I в стерильном водном растворе, водном пропиленгликоле, водном витамине Е или сезамовом или арахисовом масле. Такие водные растворы должны быть подходящим образом забуферены, при необходимости, и жидкий разбавитель в первую очередь делают изотоническим с помощью достаточного количества солевого раствора или глюкозы. Водные растворы в особенности подходят для внутривенного, внутримышечного, подкожного и интраперитонеального введения. Соединения формулы I могут быть легко введены в известную стерильную водную среду с использованием стандартных методик, известных специалистам в данной области. Подходящие фармацевтические носители включают инертные твердые разбавители или наполнители, стерильные водные растворы и различные органические растворители. Примеры твердых носителей включают лактозу, фарфоровую глину, сахарозу, циклодекстрин, тальк, желатин, агар-агар, пектин, гуммиарабик, стеарат магния, стеариновую кислоту и низшие алкиловые простые эфиры целлюлозы. Примеры жидких носителей включают сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен и воду, но не ограничиваются этим. Подобно этому носитель или разбавитель может включать любое вещество замедленного высвобождения, известное в данной области, такое как глицерилмоностеарат или глицерилдистеарат, как таковое или смешанное с воском. Фармацевтические композиции, полученные комбинированием соединений формулы I и фармацевтически приемлемого носителя, затем хорошо подходят для введения в разнообразных лекарственных формах, подходящих для раскрытых путей введения. Составы могут быть удобным образом представлены в единичной дозированной форме способами, известными в области фармации. Составы по настоящему изобретению, подходящие для перорального введения, могут быть представлены в виде отдельных единиц, таких как капсулы или таблетки, каждая из которых содержит определенное количество активного ингредиента, и необязательно подходящий эксципиент. Кроме того, перорально усвояемые составы могут быть в форме порошка или гранул, раствора или суспензии в водной или неводной жидкости, или в форме жидкой эмульсии типа "масло-в-воде" или "вода-в-масле". Если для перорального введения используют твердый носитель, то средство может быть таблетированным, помещенным в твердую желатиновую капсулу в форме порошка или гранул, или оно может быть в форме пастилки или лекарственного леденца. Количество твердого носителя будет варьироваться широко, но будет находиться в диапазоне от приблизительно 25 мг до приблизительно 1 г на единицу дозирования. Если используют жидкий носитель, то средство может быть в форме сиропа, эмульсии,мягкой желатиновой капсулы или стерильной инъекционной жидкости, такой как водная(ый) или неводная(ый) жидкая(ий) суспензия или раствор. Фармацевтические композиции изобретения могут быть приготовлены способами, обычно применяемыми в данной области. Например, таблетки могут быть приготовлены путем смешения активного ингредиента с обычными адъювантами и/или разбавителями и путем последующего спрессовывания смеси в традиционной таблетировочной машине для изготовления таблеток. Примеры адъювантов или разбавителей включают: кукурузный крахмал, картофельный крахмал, тальк, стеарат магния, желатин,лактозу, камеди и т.п. Любые другие адъюванты или добавки, используемые для таких целей, как окрашивание, ароматизация, консервирование и т.п., могут быть использованы при условии, что они совместимы с активными ингредиентами. Лечение расстройств. Как упомянуто выше, соединения формулы I представляют собой ингибиторы фермента PDE10A и как таковые являются полезными для лечения сочетанных неврологических и психических расстройств. Изобретение, таким образом, обеспечивает соединение формулы I или его фармацевтически приемлемую соль присоединения кислоты, а также фармацевтическую композицию, содержащую такое соеди-9 021606 нение, для лечения нейродегенеративного расстройства, психического расстройства или привыкания к чрезмерному употреблению лекарственных средств/наркотиков у млекопитающих, в том числе людей; где нейродегенеративное расстройство выбирают из группы, состоящей из болезни Альцгеймера, мультиинфарктной деменции, алкогольной деменции или другой связанной с употреблением веществ деменции, деменции, сопутствующей внутричерепной опухоли или черепно-мозговой травме, деменции, сопутствующей болезни Хантингтона или болезни Паркинсона, или СПИД-ассоциированной деменции; делирии; амнестического синдрома; посттравматического стрессового расстройства; умственной отсталости; расстройства способности к обучению, например расстройства способности к чтению, расстройства способности к математике или расстройства восприятия написанного выражения; синдрома дефицита внимания с гиперактивностью; и возрастного снижения когнитивных способностей; и где психическое расстройство выбирают из группы, состоящей из шизофрении, например параноидного, дезорганизованного, кататонического, недифференцированного или остаточного типа; шизофреноформного расстройства; шизоаффективного расстройства, например бредового типа или депрессивного типа; бредового расстройства; психотического расстройства, вызванного действием веществ, например психоза, вызванного действием алкоголя, амфетамина, каннабиса, кокаина, галлюциногена, ингаляционного препарата,опиоидов, или фенциклидина; расстройства личности параноидного типа; и расстройства личности шизоидного типа; и где привыкание к чрезмерному употреблению лекарственных средств/наркотиков представляет собой привыкание к чрезмерному употреблению алкоголя, амфетамина, кокаина или опиата. Все ссылки, включая публикации, заявки на патенты и патенты, приведенные в данном описании,включены таким образом путем ссылки во всей полноте и в той же самой степени, как если бы каждая ссылка была отдельно и конкретно упомянута для того, чтобы быть включенной путем ссылки, и была изложена во всей полноте (в максимальной степени, допускаемой законом). Заголовки и подзаголовки используют в данном описании только для удобства, и они не должны быть истолкованы как ограничивающие изобретение каким-либо образом. Использование любых и всех примеров или характерного языка (включая "к примеру", "например",и "как таковой") в описании настоящего изобретения предназначено лишь для лучшей иллюстрации изобретения и не накладывает ограничение на объем изобретения, если не указано иное. Цитирование и включение патентных документов в данном описании дано только для удобства и не отражает какую-либо точку зрения на действительность, патентоспособность и/или юридическую силу. Экспериментальный раздел Получение соединений по изобретению Соединения общей формулы I по изобретению могут быть получены так, как описано в следующих реакционных схемах. Если не указано иное, в реакционных схемах и в обсуждении, которое следует,НЕТ-1, R1-R9, -L-, Z и Y являются такими же, как они определены выше. Соединения формулы I, где -L- представляет собой -S-CH2-, могут быть получены реакцией сочетания нуклеофильного соединения формулы V или Va с электрофильным соединением формулы VI, где X представляет собой уходящую группу, например Cl, Br, I, метансульфонил, 4-толуолсульфонил, как показано на схеме 1. В реакции между Va и VI алкилирование атома серы соединения Va с помощью соединения VI и замыкание кольца с образованием конденсированного бициклического триазольного кольца происходят в тех же самых реакционных условиях по методике с использованием одного реакционного сосуда без выделения промежуточных соединений. Схема 1- 10021606 Данную реакцию обычно проводят в растворителе, таком как 1-пропанол, толуол, диметилформамид (DMF) или ацетонитрил, необязательно в присутствии карбонатного основания, такого как карбонат калия, или основания на основе третичного амина, такого как триэтиламин или диизопропилэтиламин(DIPEA), при температуре, находящейся в диапазоне от приблизительно 0 до приблизительно 200C, необязательно под давлением в закрытом сосуде. Другие подходящие растворители включают бензол, хлороформ, диоксан, этилацетат, 2-пропанол и ксилол. Альтернативно, могут быть использованы смеси растворителей, такие как смесь толуола/2-пропанола. Соединения формулы V являются либо коммерчески доступными, либо могут быть получены так,как описано в литературе, см., например, публикации Brown et al. Aust. J. Chem. 1978, 31, 397-404; Yutilov et al. Khim. Geter. Soedin. 1988, 799-804; Wilde et al. Bloorg. Med. Chem. Lett. 1995, 5, 167-172; Kidwaiet al. J. Korean Chem. Soc. 2005, 49, 288-291. Соединения формулы Va могут быть получены так, как описано в международной публикации WO 96/01826 из соответствующих 1,2-диаминопиридинов по реакции с тиокарбонилдиимидазолом в подходящем растворителе, таком как хлороформ, при подходящей температуре, такой как комнатная температура или +40C. Требуемые 1,2-диаминопиридины легко получают из соответствующих коммерчески доступных 2-аминопиридинов реакцией с подходящим реагентом Nаминирования, таким как О-(мезитилсульфонил)гидроксиламин, в подходящем растворителе, таком как хлороформ, при подходящей температуре, такой как 0C или комнатная температура, см. международную публикацию WO 96/01826. 2-Галогенметил-4-(арил)-1 Н-триазолы формулы VI могут быть получены галогенированием соответствующих 2-гидроксиметил-4-(арил)-1 Н-триазолов с использованием подходящего реагента, например тионилхлорида, трихлорида фосфора или трибромида фосфора, необязательно с использованием подходящего растворителя, такого как дихлорметан, с использованием способов, общеизвестных для химиков-специалистов в данной области. Требуемые 2-гидроксиметил-4-(арил)-1 Н-имидазолы могут быть получены способами, известными в данной области (см., например, публикации: Browne, E.J.; Australian Journal of Chemistry 1971, 24(2), 393-403; Browne, E.J.; Nunn, E.E.; Polya, John B. Journal of theAnnalen der Chemie 1975, (7-8), 1264-71. Moderhack, Dietrich, Liebigs Annalen der Chemie 1984, (1), 48-65. Соединения формулы I, где -L- представляет собой -CH2-S-, могут быть получены реакцией сочетания нуклеофильного соединения формулы XII с электрофильным соединением формулы VIII, что показано на схеме 2. Схема 2 Данную реакцию обычно проводят в растворителе, таком как 1-пропанол, толуол, диметилформамид (DMF) или ацетонитрил, необязательно в присутствии карбонатного основания, такого как карбонат калия, или основания на основе третичного амина, такого как триэтиламин или диизопропилэтиламин(DIPEA), при температуре, находящейся в диапазоне от приблизительно 0 до приблизительно 200C, необязательно под давлением в закрытом сосуде. Другие подходящие растворители включают бензол, хлороформ, диоксан, этилацетат, 2-пропанол и ксилол. Альтернативно, могут быть использованы смеси растворителей, такие как смесь толуола/2-пропанола. Некоторые электрофильные соединения формулы VIII являются коммерчески доступными, и многие другие известны в данной области, см., например, патент JP 59176277. Электрофильное соединениеVIII, где X является уходящей группой, например, как Cl, Br, I, метансульфонил, 4-толуолсульфонил,также могут быть получены превращением первичного спирта соединений формулы VII в упомянутую уходящую группу способами, известными химикам-специалистам в данной области. Упомянутые способы могут быть выбраны, например, из реакционного взаимодействия соединений формулы VII с тионилхлоридом, трихлоридом фосфора, трибромидом фосфора, метансульфонилхлоридом или 4 толуолсульфонилхлоридом необязательно в присутствии подходящего растворителя, такого как дихлорметан или 1,2-дихлорэтан, и необязательно в присутствии основания, такого как триэтиламин, диизопропилэтиламин или пиридин. Альтернативно, электрофильные соединения формулы VIII могут быть полу- 11021606 чены реакционным взаимодействием коммерчески доступных ароматических аминов формулы IX с 1,3 дигалогенацетонами формулы XI, например с 1,3-дихлорацетоном, в подходящем растворителе, таком как 1,2-диметоксиэтан или этанол, при подходящей температуре, такой как комнатная температура или температура, при которой проводят кипячение с обратным холодильником. Некоторые электрофильные соединения формулы VII являются коммерчески доступными, и многие другие известны в данной области, см., например, публикации Tsuchiya, Т.; Sashida, H.J. Chem. Soc, Chem. Commun. 1980, 1109-1110;Tsuchiya, Т.; Sashida, H; Konoshita, A. Chem. Pharm. Bull. 1983, 31, 4568-4572. Альтернативно, спирты формулы VII могут быть получены реакционным взаимодействием коммерчески доступных ароматических аминов формулы IX с подходящим реагентом N-аминирования, таким как O(мезитилсульфонил)гидроксиламин, в подходящем растворителе, таком как хлороформ, при подходящей температуре, такой как 0C или комнатная температура, см. международную публикацию WO 96/01826,что дает соединения формулы X. Упомянутые соединения формулы X могут быть превращены в соединения формулы VII по реакции с метилгликолятом с последующим восстановлением метилового сложного эфира до требуемого спирта с использованием подходящего восстанавливающего агента, такого как алюмогидрид лития, в подходящем растворителе, таком как диэтиловый эфир или тетрагидрофуран, с использованием способов, известных химикам-специалистам в данной области. Соединения формулы XII являются либо коммерчески доступными, либо могут быть получены так,как описано в литературе, см., например, публикации Hoggarth, Eric. Journal of the Chemical Society 1949,1160-3. Losse, Gunter; Hessler, Willi; Barth, Alfred. Chemische Berichte 1958, 91, 150-7. Potts, K.Т.; Burton,H.R.; Roy, S.K. Journal of Organic Chemistry 1966, 31(1), 265-73. Ale, Harry L.; Piala, Joseph J.; Journal ofBarnikow, Guenter; Ebeling, Horst. Zeitschrift fur Chemie 1980, 20(2), 55-6. WO-2009087218. US2003162812. WO-200002489. Baxter, Andrew; et al. BioorganicMedicinal Chemistry Letters 2003, 13(16),2625-2628. Dhiman, A.M.; Wadodkar, K.N.; Patil, S.D. Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry 2001, 40B(7), 636-639. Соединения формулы I, где R1 не является водородом, могут быть получены алкилированием соединений формулы I, где R1 представляет собой водород, с помощью алкилгалогенида формулы XIII,что показано на схеме 3. Схема 3 Данную реакцию обычно проводят в подходящем растворителе, таком как диметилформамид, диметилацетамид или ацетонитрил, в присутствии подходящего основания, такого как карбонатное основание, например карбонат калия, или основание на основе третичного амина, например триэтиламин или диизопропилэтиламин (DIPEA), при температуре, находящейся в диапазоне от приблизительно 0 до приблизительно 100C. Соединения формулы I, где -L- представляет собой -СН=СН- или -СН 2-СН 2-, могут быть получены с помощью последовательности реакций, показанной на схеме 4. Соединения, где -L- представляет собой -СС-, могут быть получены с помощью последовательности реакций, показанной на схеме 4. Схема 4 Особым образом соединения формулы I, где -L- представляет собой -СН 2-СН 2-, могут быть получе- 12021606 ны восстановлением алкена формулы I, где -L- представляет собой -СН=СН-, гидрогенизацией с использованием катализатора на основе переходного металла, такого как палладиевый металл, вместе с источником водорода, таким как газ водород, гидрокарбонат аммония или циклогексадиен. Упомянутые алкены формулы I, где -L-представляет собой -СН=СН-, могут быть получены реакцией Виттига между фосфониевой солью формулы XIV и альдегидом формулы XV в подходящем растворителе, таком как тетрагидрофуран, в присутствии подходящего основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен. Фосфониевую соль формулы XIV легко получают реакционным взаимодействием соединений формулы VIII(см. схему 2 выше) с трифенилфосфином способами, известными химикам-специалистам в данной области. Альдегиды формулы XV легко получают окислением спиртов формулы VII (см. схему 2 выше) способами, известными химикам-специалистам в данной области, например реакционным взаимодействием спиртов формулы VII с подходящим окисляющим агентом, таким как перйодинан Десса-Мартина, в подходящем растворителе, таком как дихлорметан или 1,2-дихлорэтан. Соединения формулы I, где L имеет тройную связь (этинилен), могут быть получены реакцией сочетания между триазолилалкином формулы XVII и гетероарилгалогенидом формулы XVI или противоположной реакцией сочетания между гетероарилалкином формулы XVIII и триазолилгалогенидом формулы XIX, как показано на схеме 5. Схема 5 Реакцию обычно проводят в подходящем растворителе, таком как тетрагидрофуран, и осуществляют смешением гетероарилгалогенида с гетероарилалкином вместе с подходящим катализатором, например йодидом меди(I), с фосфиновым лигандом, например комплексом 1,1'-бис(дифенилфосфино)ферроцен-палладий(II)дихлорида и дихлорметана, и органическим основанием, таким как триэтиламин, и затем нагреванием реакционной смеси в запаянной ампуле при 120C в течение 15 мин (в условиях микроволнового излучения). Изобретение, раскрытое в данном описании, дополнительно проиллюстрировано с помощью следующих неограничивающих примеров. Общие методы Данные аналитической жидкостной хроматографии - масс-спектрометрии (LC-MS) получают с использованием одного из следующих методов. Метод А. Используют прибор РЕ Sciex API 150EX, оснащенный фотоионизацией при атмосферном давлении и хроматографической системой Shimadzu LC-8A/SLC-10A LC. Колонка: Waters Symmetry С 18 4,630 мм с размером частиц 3,5 мкм; температура колонки: 60C; система растворителей: А = вода/трифторуксусная кислота (100:0,05) и В = вода/ацетонитрил/трифторуксусная кислота (5:95:0,035); способ: линейное градиентное элюирование смесью А:В=90:10-0:100 за 2,4 мин и при скорости потока 3,3 мл/мин. Метод В. Используют прибор РЕ Sciex API 300, оснащенный фотоионизацией при атмосферном давлении и хроматографической системой Waters UPLC. Колонка: Acquity UPLC ВЕН С 18 2,150 мм с размером частиц 1,7 мкм (Waters); температура колонки: 60C; система растворителей: А = вода/трифторуксусная кислота (100:0,05) и В = вода/ацетонитрил/трифторуксусная кислота (5:95:0,035); способ: линейное градиентное элюирование смесью А:В=90:10-0:100 за 1,0 мин и при скорости потока 1,2 мл/мин. Метод С. Используют прибор РЕ Sciex API 150EX, оснащенный фотоионизацией при атмосферном давлении и хроматографической системой Shimadzu LC-8A/SLC-10A LC. Колонка: Waters Symmetry С 18 4,630 мм с размером частиц 3,5 мкм; температура колонки: 60C; система растворителей: А = вода/трифторуксусная кислота (99,95:0,05) и В = метанол/трифторуксусная кислота (99,965:0,035); способ: линейное градиентное элюирование смесью А:В=83:17-0:100 за 2,4 мин и при скорости потока 3,0 мл/мин. Очистку методом препаративной жидкостной хроматографии с масс-спектрометрическим контролем (LC-MS) проводят на приборе РЕ Sciex API 150EX с химической ионизацией при атмосферном давлении. Колонка: YMC ODS-A 5020 мм с размером частиц 5 мкм; способ: линейное градиентное элюирование смесью А:В=80:20-0:100 за 7 мин и при скорости потока 22,7 мл/мин. Сбор фракций осуществ- 13021606 ляют при масс-спектрометрическом детектировании разделенного потока. Спектры 1 Н ЯМР регистрируют при 500,13 МГц на приборе Bruker Avance AV500 или при 250,13 МГц на приборе Bruker Avance DPX250. TMS (тетраметилсилан) используют в качестве внутреннего эталонного стандарта. Значения химических сдвигов выражены в миллионных долях (м.д. = ppm). Следующие сокращения используют для ряда сигналов ЯМР: s = синглет, d = дублет, t = триплет, q = квартет, qui = квинтет, h = гептет, dd = дублет дублетов, dt = дублет триплетов, dq = дублет квартетов, tt = триплет триплетов, m = мультиплет, br s = широкий синглет и br = широкий сигнал. Сокращения даны в соответствии с руководством по стилю оформления Американского химического общества: "The ACS Styleguide - A manual for authors and editors" Janet S. Dodd, Ed. 1997, ISBN: 0841234620. Общие сведения: паратолуолсульфонилгидразид (98%) получают из авокадо. Получение промежуточных соединений 2-Метил-5-фенил-2 Н-1,2,4-триазол-3-карбальдегидN-метилгидразин (1,46 мл, 27,5 ммоль) добавляют к перемешанному раствору 2-фенилоксазол-4 она (4,03 г, 25,0 ммоль) (полученному так, как описано в публикации Yushiyuki et al. Synthesis. 2004,1359-1363) в абсолютном этаноле (12 мл) при комнатной температуре под аргоном. Экзотермическая реакция. Реакционную смесь перемешивают при комнатной температуре 1 ч. Растворитель выпаривают с получением 4,64 г сырого (2-метил-5-фенил-2 Н-1,2,4-триазол-3-ил)метанола в виде желтого/белого твердого вещества. Периодинан Десса-Мартина (11,3 г, 26,7 ммоль) добавляют одной порцией к (2-метил-5-фенил-2 Н 1,2,4-триазол-3-ил)метанолу (4,60 г, 24,3 ммоль), растворенному в метиленхлориде (72 мл) при 0C под аргоном. По истечении 2 ч реакционную смесь разбавляют дихлорметаном (DCE) (100 мл) и экстрагируют насыщенным раствором NaHCO3 (100 мл). Водную фазу отбрасывают, а органическую фазу промывают рассолом и упаривают на роторном испарителе. Сырой продукт очищают хроматографией на силикагеле (элюент: 20-50% этилацетат в гептане). Выход: 3,20 г указанного в заголовке соединения в виде твердого вещества. Н-ЯМР: (CDCl3)10,08 (с, 1 Н), 8,13 (м, 2 Н), 7,50-7,42 (м, 3H), 4,26 (с, 3H). Следующие соединения получают аналогичным образом. 2-Этил-5-фенил-2 Н-1,2,4-триазол-3-карбальдегид из этилгидразина и 2-фенилоксазол-4-она. НЯМР: (ДМСО-d6)10,01 (с, 1 Н), 8,07 (д, 2 Н), 7,50 (м, 3H), 4,59 (кв, 2 Н), 1,42 (т, 3H). 5-Фенил-2-пропил-2 Н-1,2,4-триазол-3-карбальдегид из пропилгидразина и 2-фенилоксазол-4-она. Н-ЯМР: (CDCl3)10,07 (с, 1 Н), 8,17 (д, 2 Н), 7,48 (м, 3H), 4,59 (м, 2 Н), 1,95 (м, 2 Н), 1,00 (т, 3H). 2-Изопропил-5-фенил-2 Н-1,2,4-триазол-3-карбальдегид из изопропилгидразина и 2-фенилоксазол-4 она. Н-ЯМР: (ДМСО-d6)10,02 (с, 1 Н), 8,08 (д, 2 Н), 7,50 (м, 3H), 5,37 (м, 1 Н), 1,51 (Д, 6 Н). 5-Фенил-2-(2,2,2-трифторэтил)-2 Н-1,2,4-триазол-3-карбальдегид из (2,2,2-трифторэтил)гидразина и 2-фенилоксазол-4-она. Н-ЯМР: (CDCl3)10,07 (с, 1 Н), 8,18 (д, 2 Н), 7,50 (м, 3H), 5,28 (м, 2 Н). 3-(5-Формил-3-фенил-1,2,4-триазол-1-ил)пропионитрил из 3-гидразинопропионитрила и 2 фенилоксазол-4-она. Н-ЯМР: (ДМСО-d6)10,03 (с, 1H), 8,08 (д, 2H), 7,52 (м, 3H), 4,86 (т, 2 Н), 3,20 (т,2 Н). Этиловый сложный эфир (5-формил-3-фенил-1,2,4-триазол-1-ил)уксусной кислоты из этилового сложного эфира гидразиноуксусной кислоты и 2-фенилоксазол-4-она. Н-ЯМР: (CDCl3)10,05 (с, 1 Н),8,15 (д, 2 Н), 7,50 (м, 3H), 5,38 (с, 2 Н), 4,28 (м, 2 Н), 1,32 (т, 3H). Пример 2. 2-(2-Метоксиэтил)-5-фенил-2 Н-[1,2,4]триазол-3-карбальдегид 2-Гидроксиэтилгидразин (1,60 г, 21,0 ммоль) добавляют к перемешанному раствору 2 фенилоксазол-4-она (3,08 г, 19,1 ммоль) в этаноле (9,6 мл) при комнатной температуре под аргоном. Экзотермическая реакция. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель выпаривают. Выход: 4,24 г 2-(5-гидроксиметил-3-фенил-1,2,4-триазол-1-ил)этанола. Гидрид натрия (60% в минеральном масле) (60:40, гидрид натрия: минеральное масло, 1,36 г) добавляют порциями к перемешиваемому раствору 2-(5-гидроксиметил-3-фенил-1,2,4-триазол-1 ил)этанола (2,92 г, 13,3 ммоль) в N,N-диметилформамиде (100 мл) при комнатной температуре. Реакци- 14021606 онную смесь перемешивают при комнатной температуре 5 мин под аргоном. Бензилбромид (1,35 мл, 11,3 ммоль) добавляют и раствор перемешивают при комнатной температуре под аргоном 1 ч. Реакционную смесь разлагают (реакцию останавливают) добавлением концентрированной HCl (1 мл) и затем этилацетата (200 мл) и рассола (100 мл). Фазы разделяют и органическую фазу упаривают на роторном испарителе и сырой продукт очищают хроматографией на силикагеле (элюент: 20-50% этилацетата в гептане). Выход: 1,39 г 2-(5-бензилоксиметил-3-фенил-[1,2,4]триазол-1-ил)этанола в виде масла. Гидрид натрия (60% в минеральном масле) (60:40, гидрид натрия: минеральное масло, 197,7 мг) добавляют порциями к насыщенному раствору 2-(5-бензилоксиметил-3-фенил-1,2,4-триазол-1-ил)этанола(1,39 г, 4,49 ммоль) в N,N-диметилформамиде (45 мл) при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре 5 мин под аргоном. Метилйодид (0,3077 мл, 4,942 ммоль) добавляют и раствор перемешивают при комнатной температуре под аргоном 1 ч. Добавляют дополнительный NaH (200 мг) и MeI (0,1 мл). Это добавление выполняют повторно через приблизительно 1 ч. Добавляют этилацетат (100 мл) и рассол (100 мл) и фазы разделяют. Органическую фазу промывают дополнительным рассолом, сушат (Na2SO4) и растворитель выпаривают. Сырой продукт очищают хроматографией на силикагеле (20-100% этилацетат в гептане). Выход: 1,23 г 5-бензилоксиметил-1-(2 метоксиэтил)-3-фенил-1 Н-1,2,4-отриазола в виде масла. Добавляют палладиевый катализатор на углеродном носителе Pd/C (10%) (9:1, сажа:палладий, 250 мг) к раствору 5-бензилоксиметил-1-(2-метоксиэтил)-3-фенил-1 Н-1,2,4-триазола (1,23 г, 3,80 ммоль) в метаноле (72 мл) и TFA (трифторуксусная кислота) (2 мл) при комнатной температуре. Реакционную смесь гидрогенизируют при давлении 3 бар с использованием шейкера Parr в течение ночи. Добавляют дополнительный катализатор (200 мг) и гидрогенизацию продолжают выполнять в течение ночи. Катализатор отфильтровывают и растворитель выпаривают. Сырой продукт очищают хроматографией на силикагеле (25-100% этилацетат в гептане). Выход: 0,84 г [2-(2-метоксиэтил)-5-фенил-2 Н-1,2,4-триазол-3 ил]метанола в виде масла. Периодинан Десса-Мартина (1530 мг, 3,60 ммоль) добавляют к перемешанной суспензии [2-(2 метоксиэтил)-5-фенил-2 Н-1,2,4-триазол-3-ил]метанола (840 мг, 3,6 ммоль), растворенного в метиленхлориде (36,0 мл), при комнатной температуре под аргоном. Раствор перемешивают в течение ночи. Некоторую часть твердого вещества отфильтровывают и отбрасывают. К дихлорметановому (DCM) раствору добавляют насыщенный раствор NaHCO3 (35 мл) и дополнительный DCM (35 мл). Растворитель выпаривают и сырой продукт очищают хроматографией на силикагеле (элюент: 50%-ный этилацетат в гептане). Выход: 0,76 г указанного в заголовке соединения в виде масла. Н-ЯМР: (CDCl3)10,04 (с, 1 Н), 8,13 (м,2 Н), 7,45 (м, 3H), 4,82 (т, 2 Н), 3,83 (т, 2 Н), 3,32 (с, 3H). Получение других промежуточных соединений. 2-Хлорметил-5,7-диметил-[1,2,4]триазоло[1,5-а]пиримидин К раствору 4,6-диметилпиримидин-2-иламина (25 г, 200 ммоль) в 400 мл CH2Cl2 добавляют по каплям раствор гидроксиламин-2,4,6-триметилбензолсульфоната (105 г, 488 ммоль) в 300 мл CH2Cl2 при 0C и смесь перемешивают при 0C в течение 1 ч и фильтруют. Собранное твердое вещество промываютCH2Cl2 (100 мл), что дает 2,4,6-триметилбензолсульфонат 1-амино-4,6-диметил-1 Н-пиримидин-2 илиденаммония (40 г, выход: 62%). Смесь 2,4,6-триметилбензолсульфоната 1-амино-4,6-диметил-1 Н-пиримидин-2-илиденаммония (40 г, 0,1 моль) и NaOH (10 г, 0,2 моль) в 500 мл этилового спирта перемешивают при 50-60C в течение 1 ч. После добавления метилового сложного эфира хлоруксусной кислоты (16,6 г, 0,15 моль) получающуюся в результате смесь перемешивают при кипячении с обратным холодильником в течение 4 ч. После кон- 15021606 центрирования при пониженном давлении остаток разбавляют водой (1000 мл) и экстрагируют CH2Cl2(300 мл 3). Объединенные органические слои промывают рассолом (200 мл), сушат над Na2SO4, фильтруют и концентрируют под вакуумом. Остаток очищают колоночной хроматографией на силикагелеm/z=196,9, время удерживания (tR) (мин, метод А) = 0,52. Следующие промежуточные соединения получают аналогичным образом. 7-Хлор-2-хлорметил-5,8-диметил-[1,2,4]триазоло[1,5-с]пиримидин из 6-хлор-2,5-диметилпиримидин-4-иламина, полученного, как описано в публикации Henze et al. J. Org. Chem. 1952, 17, 1320-1327. Выход 3,2%, LC-MS: m/z=231,5 (MH+), время удерживания (tR)=1,13 мин, метод С. 2-Хлорметил-5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин из 2-амино-3,6-диметилпиразина. Выход 60%, 1 Н ЯМР (500 МГц, CDCl3):7,91 (с, 1 Н), 4,87 (с, 2 Н), 2,91 (с, 3H), 2,74 (с, 3H), LC-MS: m/z=196, 9 Раствор 2-хлорметил-5,8-диметил-[1,2,4]триазоло[1,5-а]пиразина (1,351 г, 6,87 ммоль) и трифенилфосфина (1,80 г, 6,87 ммоль) в ацетонитриле (150 мл) нагревают при кипячении с обратным холодильником в течение 12 ч. Растворители удаляют в вакууме и остаток суспендируют в простом эфире, фильтруют и сушат, что дает (5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-илметил)трифенилфосфоний хлорид в виде твердого вещества грязно-белого цвета (2,412 г, 74,9%). LC-MS: m/z=423,2 ([М-Cl]+), время удерживания (tR)=0,86 мин, метод А. Следующие промежуточные соединения получают аналогичным образом.(tR)=0,55 мин, метод В. Получение соединений по изобретению. Пример 1. 5-Метил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5 а]пиридин Триэтиламин (0,7602 мл, 5,454 ммоль) добавляют к раствору 2-хлорметил-5-метил[1,2,4]триазоло[1,5-а]пиридина (495 мг, 2,73 ммоль) в N,N-диметилформамиде (12 мл, 150 ммоль) и раствор перемешивают приблизительно 2 мин. 5-Фенил-2 Н-1,2,4-триазол-3-тиол (532 мг, 3,00 ммоль) добавляют к смеси в виде твердого вещества. Реакционную смесь впоследствии нагревают при 60C в течение 0,5 ч. Смесь упаривают на роторном испарителе. Этилацетат (60 мл) и насыщенный раствор бикарбоната (20 мл) добавляют к остатку и фазы разделяют. Органическую фазу промывают рассолом (20 мл). После высушивания (MgSO4) растворитель удаляют при пониженном давлении. Сырой продукт очищают хроматографией на силикагеле (элюент: этилацетат:N-гептан 50-100%). Это дало 542 мг (61%) указанного в заго- 17021606 ловке соединения в виде твердого вещества белого цвета. LC-MS: m/z=323,1 (МН+), время удерживания(с, 2 Н), 2,67 (с, 3H). Следующее соединение получают аналогичным образом. 8-Метокси-5-метил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5 а]пиридин (2), LC-MS: m/z=353,4 (MH+), время удерживания (tR) =0,57 мин, метод В. Пример 2. 5-Метил-2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло Метилйодид (142 мг, 1,00 ммоль), растворенный в диметилформамиде (0,5 мл), добавляют к перемешанной суспензии 5-метил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5 а]пиридина (322 мг, 1,00 ммоль) и карбоната калия (276 мг, 2,0 ммоль) в N,N-диметилформамиде (3,5 мл) при комнатной температуре под аргоном. Раствор перемешивают в течение 3 ч и затем разлагают добавлением насыщенного раствора бикарбоната и экстрагируют этилацетатом. Органические фазы сушат(MgSO4) и упаривают на роторном испарителе. Сырой продукт очищают методом высокоэффективной жидкостной хроматографии (HPLC). Это дает 10 мг указанного в заголовке соединения. Жидкостная хроматография с масс-спектрометрическим контролем (LC-MS): m/z=337,1 (MH+), время удерживания(tR)=0,63 мин, метод В. 1 Н ЯМР (600 МГц, ДМСО-d6):7,96 (д, 2 Н), 7,63 (д, 1 Н), 7,60 (т, 1 Н), 7,50-7,40 (м, 3H), 7,07 (д, 1 Н),4,68 (с, 2 Н), 2, 66 (с, 3H). Следующее соединение синтезируют аналогичным образом. 5,8-Диметил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5-а]пиразин, LCMS: m/z=338,1 (MH+), время удерживания (tR)=1,38 мин, метод С. Пример 3. 5,8-Диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-илметил)трифенилфосфоний хлорид (0,490 г, 1,07 ммоль) и 2-метил-5-фенил-2 Н-[1,2,4]триазол-3-карбальдегида (0,200 г, 1,07 ммоль) в тетрагидрофуране (THF) (6,9 мл) в безводном THF (8 мл) и смесь перемешивают при комнатной температуре в атмосфере аргона в течение ночи. Добавляют дихлорметан (DCM) (50 мл) и органическую фазу экстрагируют водой (250 мл), сушат (MgSO4) и растворитель упаривают. Остатки растворяют в DCM и очищают хроматографией на силикагеле (элюент: этилацетат). Выход: 270 мг (76%) промежуточного соединения в форме смеси цис-/транс-изомеров. Это вещество (270 мг, 0,81 ммоль) растворяют в DCM (10 мл) и метиловом спирте (80 мл). Раствор фильтруют и гидрогенизируют при комнатной температуре в реакторе H-cube, пропуская через колонку с каталитическим картриджем с Pd/C, при давлении водорода 1 бар. Растворитель выпаривают и сырой продукт очищают хроматографией на силикагеле (элюент: этилацетат). Выход: 138 мг (51%) указанного в заголовке соединения в виде твердого вещества. LC-MS: m/z=334,5 (MH+), время удерживания (tR)=1,16 мин, метод С. 1 Н ЯМР (600 МГц, ДМСО-d6):8,0-7,9 (м, 3H), 7,45-7,35 (м, 3H), 3,90 (с, 3H), 3,44 (т, 2 Н), 3,35 (т,2 Н), 2,74 (с, 3H), 2,64 (с, 3H). Следующие соединения получают аналогичным образом. 8-Метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридин дигидрохлорид, LC-MS: m/z=318,9 (МН+), время удерживания (tR)=0,52 мин, метод В. 5,7-Диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиримидин,LC-MS: m/z=334,5 (МН+), время удерживания (tR)=1,05 мин, метод С. 8-Метокси-5-метил-2-[2-(5-фенил-2-пропил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридин дигидрохлорид, LC-MS: m/z=377,5 (MH+), время удерживания (tR)=0,66 мин, метод В. 8-Метокси-5-метил-2-2-[5-фенил-2-(2,2,2-трифторэтил)-2 Н-[1,2,4]триазол-3-ил]этил- 18021606[1,2,4]триазоло[1,5-а]пиридин, LC-MS: m/z=416,4 (MH+), время удерживания (tR)=0,71 мин, метод В. 8-Метокси-5-метил-2-2-[5-фенил-2-(2,2,2-трифторэтил)-2 Н-[1,2,4]триазол-3-ил]этил[1,2,4]триазоло[1,5-а]пиридин, LC-MS: m/z=319,2 (MH+), время удерживания (tR)=1,14 мин, метод С. 8-Метокси-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридин, LCMS: m/z=335,3 (MH+), время удерживания (tR)=1,09 мин, метод С. Этиловый сложный эфир 5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3 фенил-[1,2,4]триазол-1-илуксусной кислоты, LC-MS: m/z=420,6 (MH+), время удерживания (tR)=0,65 мин, метод В. 5,8-Диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридин,LC-MS: m/z=333,2 (МН+), время удерживания (tR)=1,24 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-с]пиримидин,LC-MS: m/z=334,5 (МН+), время удерживания (tR)=1,18 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-тиофен-3-ил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=340,1 (MH+), время удерживания (tR)=1,29 мин, метод С. 2-[2-(5-Фуран-2-ил-1-метил-1 Н-[1,2,4]триазоло-3-ил)этил]-5,8-диметил-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=324,1 (МН+), время удерживания (tR)=1,17 мин, метод С. 2-[(Е)-2-(5-Фуран-2-ил-1-метил-1 Н-[1,2,4]триазол-3-ил)винил]-5,8-диметил-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=322,1 (MH+), время удерживания (tR)=1,46 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-тиазол-4-ил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=341,1 (MH+), время удерживания (tR)=0,91 мин, метод С. 5,8-Диметил-2-2-[2-метил-5-(5-метилтиазол-2-ил)-2 Н-[1,2,4]триазол-3-ил]этил[1,2,4]триазоло[1,5-а]пиразин, LC-MS : m/z=355,1 (MH+), время удерживания (tR)=1,21 мин, метод С. 5,8-Диметил-2-2-[2-метил-5-(4-метилтиазол-2-ил)-2 Н-[1,2,4]триазол-3-ил]этил[1,2,4]триазоло[1,5-а]пиразин, LC-MS: m/z=355,1 (MH+), время удерживания (tR)=1,18 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-оксазол-2-ил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=325,1 (MH+), время удерживания (tR)=0,87 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-тиофен-2-ил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=340,1 (MH+), время удерживания (tR)=1,24 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-тиофен-2-ил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=340,1 (MH+), время удерживания (tR)=1,26 мин, метод С. 5,8-Диметил-2-[2-(2-метил-5-пиримидин-2-ил-2 Н-[1,2,4]-триазол-3-ил)-этил]-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: 1,8-Диазабицикло[5.4.0]ундец-7-ен (0,150 мл, 1,00 ммоль) добавляют к перемешанной суспензии 8 этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-илметил)трифенилфосфоний хлорида (472 мг, 1,00 ммоль) и 5-фенил-2-пропил-2 Н-[1,2,4]триазол-3-карбальдегида (215 мг, 1,00 ммоль) в безводном тетрагидрофуране (THF) (20 мл) и смесь перемешивают при комнатной температуре в атмосфере аргона в течение 2- 19021606 дней. Растворитель удаляют в вакууме. Остатки растворяют в дихлорметане (DCM) и очищают хроматографией на силикагеле (элюент: 0-100% этилацетата в н-гептане). Выход: 290 мг промежуточного соединения (в форме одного изомера) в виде твердого вещества белого цвета (78%). LC-MS: m/z=373,5 (MH+),время удерживания (tR)=0,93 мин, метод В. Это вещество (290 мг, 0,78 ммоль) растворяют в диметилформамиде (DMF) (7,8 мл) и добавляют паратолуолсульфонилгидразид (430 мг, 2,3 ммоль) и реакционную смесь перемешивают при 120C 8 ч под аргоном. Раствор оставляют остывать до комнатной температуры и перемешивают в течение ночи. Добавляют дополнительный паратолуолсульфонилгидразид (0,08 г) и реакционную смесь оставляют при 120C под аргоном на ночь. DMF выпаривают и остаток растворяют в этилацетате (50 мл) и экстрагируют насыщенным раствором NaHCO3 (225 мл). Органическую фазу промывают рассолом, сушат(MgSO4) и упаривают на роторном испарителе. Сырой продукт очищают хроматографией на силикагеле(элюент: 50-100% этилацетата в н-гептане). Выход: 140 мг (48%) указанного в заголовке соединения в виде твердого вещества. LC-MS: m/z=375,3 (МН+), время удерживания (tR)=1,68 мин, метод С. 1 Н ЯМР (600 МГц, CDCl3):8,08 (д, 2 Н), 7,40 (д, 2 Н), 7,34 (м, 1 Н), 7,17 (д, 1 Н), 6,70 (д, 1 Н), 4,12 (м,2 Н), 3,55 (м, 2 Н), 3,38 (м, 2 Н), 3,00 (м, 2 Н), 2,68 (с, 3H), 1,88 (м, 2 Н), 1,34 (т, 3H), 0,93 (т, 3H). Следующие соединения получают аналогичным образом. 8-Метокси-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]-триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридин, LC-MS: m/z=349,5 (MH+), время удерживания (tR)=0,59 мин, метод В. 8-Этил-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридин,LC-MS: m/z=347,1 (МН+), время удерживания (tR)=0,59 мин, метод В. 8-Этил-2-[2-(2-изопропил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-5-метил-[1,2,4]триазоло[1,5 а]пиридин, LC-MS: m/z=375,2 (МН+), время удерживания (tR)=1,67 мин, метод С. 3-5-[2-(8-Метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил пропионитрил, LC-MS: m/z=388,5 (MH+), время удерживания (tR)=1,18 мин, метод С. 3-5-[2-(8-Этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрил, LC-MS: m/z=38 6,6 (MH+), время удерживания (tR)=1,32 мин, метод С. 2-[2-(2-Изопропил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-8-метокси-5-метил-[1,2,4]триазоло[1,5 а]пиридин, LC-MS: m/z=377,4 (МН+), время удерживания (tR)=1,45 мин, метод С. 3-5-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрил, LC-MS: m/z=373,4 (МН+), время удерживания (tR)=0,56 мин, метод В. 2-2-[2-(2-Метоксиэтил)-5-фенил-2 Н-[1,2,4]триазол-3-ил]этил-5,8-диметил-[1,2,4]триазоло[1,5 а]пиразин, LC-MS: m/z=378,6 (MH+), время удерживания (tR)=1,42 мин, метод С. Температура плавления 1 М Тетрагидроалюмината лития в тетрагидрофуране (5,0 мл) добавляют к перемешанному раствору этилового сложного эфира 5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил[1,2,4]триазол-1-илуксусной кислоты (950 мг, 2,2 ммоль) в тетрагидрофуране (3 мл) при комнатной температуре. Реакция является экзотермической. Раствор перемешивают при комнатной температуре под аргоном в течение 3 ч. Раствор разбавляют тетрагидрофураном (10 мл) и разлагают путем добавления влажного Na2SO4. Раствор фильтруют через сухой фильтр, содержащий безводный Na2SO4, и упаривают на роторном испарителе. Сырой продукт очищают хроматографией на силикагеле (элюент: 0-30% метилового спирта в этилацетате). Выход: 710 мг (75%) указанного в заголовке соединения в виде прозрачного масла. LC-MS: m/z=379,4 (МН+), время удерживания (tR)=0,51 мин, метод В. Следующее соединение получают аналогичным образом. 8-Метокси-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридин, LC-MS: m/z=349,5 (MH+), время удерживания (tR)=0,59 мин, метод В. Пример 6. 3-5-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропиламин 3-5-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрил (28 мг, 0,075 ммоль) растворяют в 2 М растворе аммиака в метаноле (15 мл) и гидрогенизируют в реакторе H-cube, пропуская через колонку, заполненную никелем Ренея (водород под давлением 50 бар, комнатная температура). Растворитель выпаривают. Сырой продукт очищают хроматографией на силикагеле (элюент: 10% метиловый спирт, 10% триэтиламин, 80% этилацетат). Выход: 10 мг(35%) указанного в заголовке соединения в виде прозрачного масла. LC-MS: m/z=311,3 (MH+), время удерживания (tR)=0,81 мин, метод С. Следующее соединение получают аналогичным образом. 3-5-[2-(8-Этил-5-метил-[1,2,4]-триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1-ил пропиламин, LC-MS: m/z=390,2 (MH+), время удерживания (tR)=0,48 мин, метод В. Пример 7. 2-5-[2-(5,8-Диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илэтанол Йодтриметилсилан (92,0 мкл, 0,646 ммоль) добавляют к перемешанному раствору 2-2-[2-(2 метоксиэтил)-5-фенил-2 Н-[1,2,4]триазол-3-ил]этил-5,8-диметил-[1,2,4]триазоло[1,5-а]пиразина (61 мг,0,16 ммоль) в хлороформе (5 мл) в атмосфере аргона при комнатной температуре. Раствор перемешивают при комнатной температуре в течение 4 ч. Добавляют дополнительный йодтриметилсилан (184 мкл, 1,29 ммоль) и смесь перемешивают при комнатной температуре в течение ночи. Добавляют метиловый спирт(10 мл) и твердый сульфит натрия (0,5 г) и смесь перемешивают в течение 30 мин, фильтруют и упаривают. Сырой продукт очищают хроматографией на силикагеле (элюент: 0-5% метилового спирта в этилацетате). Выход: 27 мг (46%) указанного в заголовке соединения в виде твердого вещества. LC-MS:m/z=364,5 (MH+), время удерживания (tR)=1,30 мин, метод С. Фармакологическое тестирование Фермент PDE10A. Активный фермент PDE10A получают разными способами для применения в анализе PDE (Loughney, K. et al. Gene 1999, 234, 109-117; Fujishige, K. et al. Eur. J. Biochem. 1999, 266, 1118-1127 и Soderling,S. et al. Proc. Natl. Acad. Sci. 1999, 96, 7071-7076). PDE10A может экспрессироваться в виде непроцессированных белков или в виде процессированных белков, при условии, что они экспрессируют каталитический домен. PDE10A может быть получен в различных типах клеток, например в клетках насекомых или в клетках кишечной палочки Е. coli. Пример способа получения каталитически активного ферментаPDE10A представляет собой следующее: каталитический домен фермента PDE10A человека (аминокислоты 440-779 из последовательности с номером доступа NP 006652) амплифицирован из тотальной РНК из всего мозга человека с помощью стандартной полимеразной цепной реакции с обратной транскрипцией (RT-PCR) и клонирован в сайты BamH1 и Xho1 вектора рЕТ 28 а (Novagen). Экспрессию в клеткахE.coli проводят в соответствии со стандартными протоколами. Кратко, плазмиды экспрессии подвергают трансформации в штамм E.coli BL21(DE3), и клеточные культуры (50 мл), инокулированные клетками,оставляют расти до достижения оптической плотности OD600, равной 0,4-0,6, прежде чем индуцировать экспрессию белка посредством 0,5 мМ IPTG (изопропил-тиогалактозид). После индуцирования клетки инкубируют в течение ночи при комнатной температуре, после чего клетки собирают центрифугированием. Клетки, экспрессирующие PDE10A, повторно суспендируют в 12 мл (50 мМ Трис-HCl-рН 8,0; 1 мМMgCl2 и ингибиторы протеазы). Клетки лизируют обработкой ультразвуком и после лизирования всех клеток добавляют TritonX100 в соответствии с протоколами Novagen. Фермент PDE10A частично очищают на сефарозе Q и объединяют наиболее активные фракции. Анализ ингибирования PDE10A. Анализ PDE10A может быть выполнен, например, следующим образом: анализ проводят в 60-мкл образцах, содержащих фиксированное количество релевантного фермента PDE (достаточное для превращения 20-25% субстрата на основе циклического нуклеотида), буферный раствор (50 мМ HEPES 7,6- 21021606 на основе циклических нуклеотидов, тритий-меченый сАМР с концентрацией вплоть до конечной концентрации 5 нМ и варьирующиеся количества ингибиторов. Реакции инициируют путем добавления субстрата на основе циклического нуклеотида, и реакции оставляют протекать в течение 1 ч при комнатной температуре, прежде чем прерывают посредством смешения с 15 мкл реагента с концентрацией 8 мг/мл аналит-связывающих сцинтилляционных гранул силиката иттрия (Amersham). Гранулам дают осесть в течение 1 ч в темноте, прежде чем планшеты подвергают считыванию на гамма-счетчике Microbeta 1450(Wallac). Измеренный сигнал может быть преобразован в активность относительно неингибированных контрольных образцов (100%), и значения IC50 могут быть вычислены с использованием Xlfitрасширения EXCEL. Гиперактивность, вызванная введением фенциклидина (РСР). Используют мышей-самцов (NMRI, Charles River), имеющих вес 20-25 г. В каждой группе используют по 8 мышей, получающих испытуемое соединение (5 мг/кг) плюс фенциклидин (РСР) (2,3 мг/кг), в том числе параллельные контрольные группы, принимающие среду для испытуемого соединения плюс РСР или только инъекции со средой для лекарства. Объем вводимых инъекций составляет 10 мл/кг. Эксперимент выполняют в нормальных световых условиях в помещении со спокойной обстановкой. Испытуемое вещество вводят впрыскиванием через рот за 60 мин до инъекции РСР, которую делают подкожным введением. Сразу после инъекции РСР мышей помещают по отдельности в специально сконструированную клетку для проведения испытания (20 см 32 см). Активность измеряют с помощью источников инфракрасного света (58) и фотоэлементов, расположенных друг от друга на расстоянии 4 см. Лучи света пересекают клетку на высоте 1,8 см над уровнем дна клетки. Для регистрирования единичного импульса счета двигательной активности требуется прерывание соседних лучей света, при этом единичные импульсы счета, вызванные движениями, не связанными с линейным перемещением, не учитываются. Двигательную активность записывают с интервалами в 5 мин в течение периода времени 1 ч. Эффект лекарства вычисляют в расчете на общее число единичных импульсов в течение 1-часового периода поведенческого теста следующим образом. Среднюю двигательную активность, вызванную лечением средой для лекарства в отсутствии РСР,используют как исходный уровень. В соответствии с этим вычисляют 100% эффект РСР, который составляет общее число импульсов счета двигательной активности минус исходный уровень. Ответную реакцию в группах, принимающих испытуемое соединение, таким образом, определяют как общее число импульсов счета двигательной активности минус исходный уровень, выраженное в процентах относительно подобного результата, зарегистрированного в параллельной контрольной группе, принимающей РСР. Ответные реакции, выраженные в процентах, переводят в ингибирование, выраженное в процентах. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее структуру I где НЕТ-1 представляет собой гетероароматическую группу формулы II где НЕТ-1 выбирают из группы, состоящей из имидазо[1,2-а]пиримидина, [1,2,4]триазоло[1,5 а]пиридина, имидазо[1,2-а]пиридина, имидазо[4,5-b]пиримидина, пиразоло[1,5-а]пиридина, [1,2,4]триазоло[1,5-а]пиримидина, [1,2,4]триазоло[1,5-а]пиразина и [1,2,4]триазоло[1,5-с]пиримидина,и где НЕТ-1 может быть необязательно замещен посредством вплоть до трех заместителей R2-R4,независимо выбранных из Н; С 1-С 6 алкила; галогена; циано, галоген(C1-C6)алкила; фенила; алкокси и С 1 С 6 гидроксиалкила; и гдеозначает место прикрепления,Q представляет собой фенил, необязательно замещенный 1-5 заместителями, выбранными из группы, состоящей из С 1-С 6 алкила, С 1-С 6 алкокси и галогена, или Q представляет собой моноциклическую 5 или 6-членную гетероароматическую группу, выбранную из группы, состоящей из тиофена, фурана, тиазола, пиразола, пиридина, пиримидина и пиразина;-L- представляет собой связующее звено, выбранное из -S-СН 2-, -CH2-S-, -CH2-CH2-, -СН=СН- иCH2C(O)NH2, С 1-С 6 арилалкила и C1-С 6 алкилгетероциклоалкила; при условии, что соединение не представляет собой 2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]-1 Н-бензимидазол; 2-3-(2-пиразинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 2-(3-фенил-1 Н-1,2,4-триазол-5-ил)метил]тио]-1 Н-бензимидазол; 1-этил-5-(1-пиперидинилсульфонил)-2-3-(2-тиенил)-1 Н-1,2,4-триазол-5-ил]тио]метил]-1 Нбензимидазол; 6-метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]-1 Н-бензимидазол; 2-3-(3-пиридинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 8-метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин; 6-хлор-2-3-(2-тиенил)-1 Н-1,2,4-триазол-5-ил]тио]метил]имидазо[1,2-а]пиридинил; 2-3-(4-пиридинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 6-метил-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин; 2-3-(2-пиридинил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; 6-хлор-2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]имидазо[1,2-а]пиридин; 2-(3-фенил-1 Н-1,2,4-триазол-5-ил)тио]метил]-3H-имидазо[4,5-b]пиридин или 2-3-(2-фуранил)-1 Н-1,2,4-триазол-5-ил]метил]тио]-1 Н-бензимидазол; и его таутомеры и фармацевтически приемлемые соли, при условии, что когда связующее звено (L) представляет собой -CH2-S-, тогда НЕТ-1 не является ни имидазо[1,2-а]пиридином, ни имидазо[1,2 а]пиримидином. 2. Соединение по п.1, где R2, R3 и R4, все, представляют собой водород. 3. Соединение по любому из пп.1-2, где по меньшей мере один из заместителей R2, R3 и R4 представляет собой С 1-С 6 алкил, такой как метил. 4. Соединение по любому из пп.1-3, где по меньшей мере один из заместителей R2, R3 и R4 представляет собой галоген, такой как хлор или бром. 5. Соединение по п.1, где соединение выбирают из группы, состоящей из 8-метокси-5-метил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5 а]пиридина; 5-метил-2-(5-фенил-2 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5-а]пиридина; 5-метил-2-(1-метил-5-фенил-1 Н-[1,2,4]триазол-3-илсульфанилметил)-[1,2,4]триазоло[1,5 а]пиридина; 8-метокси-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридина; 8-метокси-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридина; 8-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина; 5,7-диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиримидина; 5,8-диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиразина; 8-метокси-5-метил-2-[2-(5-фенил-2-пропил-2 Н-[1,2,4]триазол-3-ил)этил][1,2,4]триазоло[1,5 а]пиридина; 5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина; 8-метокси-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина; этилового сложного эфира 5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3 фенил-[1,2,4]триазол-1-илуксусной кислоты; 2-5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илэтанола; 5,8-диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина; 5,8-диметил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-с]пиримидина; 8-этил-5-метил-2-[2-(2-метил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5-а]пиридина; 8-этил-5-метил-2-[2-(5-фенил-2-пропил-2 Н-[1,2,4]триазол-3-ил)этил]-[1,2,4]триазоло[1,5 а]пиридина; 8-этил-2-[2-(2-изопропил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-5-метил-[1,2,4]триазоло[1,5 а]пиридина; 3-5-[2-(8-метокси-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрила; 3-5-[2-(8-этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрила; 2-[2-(2-изопропил-5-фенил-2 Н-[1,2,4]триазол-3-ил)этил]-8-метокси-5-метил-[1,2,4]триазоло[1,5 а]пиридина; 3-5-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропионитрила; 3-2-[2-(8-этил-5-метил-[1,2,4]триазоло[1,5-а]пиридин-2-ил)этил]-4-фенилимидазол-1- 23021606 илпропиламина; 3-5-[2-(5,8-диметил-[1,2,4]триазоло[1,5-а]пиразин-2-ил)этил]-3-фенил-[1,2,4]триазол-1 илпропиламина; 2-2-[2-(2-метоксиэтил)-5-фенил-2 Н-[1,2,4]триазол-3-ил]этил-5,8-диметил-[1,2,4]триазоло[1,5 а]пиразина и 8-метокси-2-2-[2-(2-метоксиэтил)-5-фенил-2 Н-[1,2,4]триазол-3-ил]этил-5-метил-[1,2,4]триазоло[1,5-а]пиридина; и их фармацевтически приемлемых солей. 6. Применение соединения по любому из пп.1-5, но без условия, в качестве лекарственного средства, которое является ингибитором фермента PDE10A.
МПК / Метки
МПК: A61P 25/28, A61P 25/18, C07D 471/04, A61K 31/437, A61K 31/4196
Метки: pde10a, качестве, производные, гетероароматические, арилтриазольные, ингибиторов, фермента
Код ссылки
<a href="https://eas.patents.su/25-21606-geteroaromaticheskie-ariltriazolnye-proizvodnye-v-kachestve-ingibitorov-fermenta-pde10a.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероароматические арилтриазольные производные в качестве ингибиторов фермента pde10a</a>
Новые производные фенилимидазола в качестве ингибиторов фермента pde10a

Номер патента: 18880
Опубликовано: 29.11.2013
Авторы: Ланггор Мортен, Ритзен Андреас, Нильсен Якоб, Келер Ян, Фарах Мохамед М., Килберн Джон Пол
МПК: A61K 31/4184, A61K 31/4353, A61K 31/519...
Метки: фермента, pde10a, производные, новые, фенилимидазола, ингибиторов, качестве
Формула / Реферат:
1. Соединение, представленное структурной формулой Iгде НЕТ представляет собой гетероароматическую группу формулы II, содержащую от 2 до 4 атомов азотагде Y может представлять собой N или СН;Z может представлять собой N или С;НЕТ может быть необязательно замещен вплоть до трех заместителями R7-R9, независимо выбранными из Н; C1-C6-алкила; галогена; циано; галоген(C1-C6)алкила; арила; алкокси и C1-C6-гидроксиалкила;* означает точку...
Производные 2-арилимидазола в качестве ингибиторов фермента pde10a

Номер патента: 21415
Опубликовано: 30.06.2015
Авторы: Мариго Мауро, Келер Ян, Пюшль Аск, Килберн Джон Пол, Ланггор Мортен, Нильсен Якоб
МПК: A61K 31/519, A61P 25/00, C07D 471/04...
Метки: ингибиторов, 2-арилимидазола, качестве, производные, фермента, pde10a
Формула / Реферат:
1. Соединение, имеющее структуру Iгде НЕТ-1 представляет собой гетероароматическую группу, выбранную из группы, включающей [1,2,4]триазоло[1,5-а]пиридин и [1,2,4]триазоло[1,5-а]пиразин, и где НЕТ-1 необязательно может быть замещен 1-3 заместителями R7, R8 и R9, индивидуально выбираемыми из водорода, C1-C6-алкила и метокси; символ * обозначает место присоединения;Q выбран из группы, включающей фенил, тиазол, тиофен и фуран, необязательно...
Новое производное фенилимидазола в качестве ингибитора фермента pde10a, фармацевтическая композиция на его основе и его применение для лечения нейродегенеративных и психиатрических расстройств

Номер патента: 21410
Опубликовано: 30.06.2015
Авторы: Келер Ян, Ланггор Мортен, Фарах Мохамед М., Ритзен Андреас, Нильсен Якоб, Килберн Джон Пол
МПК: A61K 31/519, A61K 31/4353, A61K 31/4184...
Метки: расстройств, лечения, фармацевтическая, фермента, композиция, производное, ингибитора, фенилимидазола, психиатрических, нейродегенеративных, качестве, pde10a, основе, применение, новое
Формула / Реферат:
1. Соединение - 5,8-диметил-2-[2-(1-метил-4-фенил-1Н-имидазол-2-ил)этил][1,2,4]триазоло[1,5-а]пиразини/или его фармацевтически приемлемые кислотно-аддитивные соли.2. Применение соединения по п.1 в качестве лекарственного средства для лечения нейродегенеративного или психиатрического расстройства.3. Применение соединения по п.1 для лечения нейродегенеративного или психиатрического расстройства в комбинации с одним или несколькими...
Гетероароматические соединения хинолинов и их применение в качестве ингибиторов pde10

Номер патента: 12211
Опубликовано: 28.08.2009
Авторы: Верхоуст Патрик Роберт, Хелал Кристофер Джон, Хамфри Джон Майкл, Хувер Деннис Джей
МПК: C07D 401/14, A61K 31/14, A61P 25/18...
Метки: применение, pde10, хинолинов, гетероароматические, соединения, ингибиторов, качестве
Формула / Реферат:
1. 2-[4-(1-Метил-4-пиридин-4-ил-1Н-пиразол-3-ил)феноксиметил]хинолин или его фармацевтически приемлемая соль. 2. Соединение, выбранное из группы, состоящей из 2-[-4-(4-пиридин-4-ил-2Н-пиразол-3-ил)феноксиметил]хинолина; 2-[4-(2-метил-4-пиридин-4-ил-2Н-пиразол-3-ил)феноксиметил]хинолина и их фармацевтически приемлемых солей. 3. Фармацевтическая композиция для лечения психотических расстройств, бредовых расстройств и медикаментозного психоза;...
Бициклические гетероароматические соединения в качестве ингибиторов протеин-тирозинкиназ

Номер патента: 2455
Опубликовано: 25.04.2002
Авторы: Кокерилл Джордж Стюарт, Картер Малком Клайв, Смит Кэтрин Джейн, Лаки Карен Элизабет, Гантрип Стивен Барри
МПК: A61K 31/519, A61P 35/00, C07D 471/04...
Метки: соединения, протеин-тирозинкиназ, гетероароматические, ингибиторов, бициклические, качестве
Формула / Реферат:
1. Соединение формулы (I) или его соль или сольват; где Y представляет собой CR1, а V представляет собой N; или Y представляет собой CR1, a V представляет собой CR2; R1 представляет собой группу СН3SO2СН2СН2NНСН2-Аr-, где -Аr- выбран из фурана и тиазола, каждый из которых может быть возможно замещен одной или двумя гало, С1-4алкильной или С1-4 алкоксигруппами; R2 выбран из группы, включающей в себя водород, гало, гидрокси, С1-4алкил,...
Предыдущий патент: Непрерывный способ карбонилирования этилена
Следующий патент: Система реактора для электропорации
Случайный патент: Верхняя часть канализационного колодца