Гетероароматические соединения хинолинов и их применение в качестве ингибиторов pde10
Номер патента: 12211
Опубликовано: 28.08.2009
Авторы: Хелал Кристофер Джон, Хамфри Джон Майкл, Верхоуст Патрик Роберт, Хувер Деннис Джей




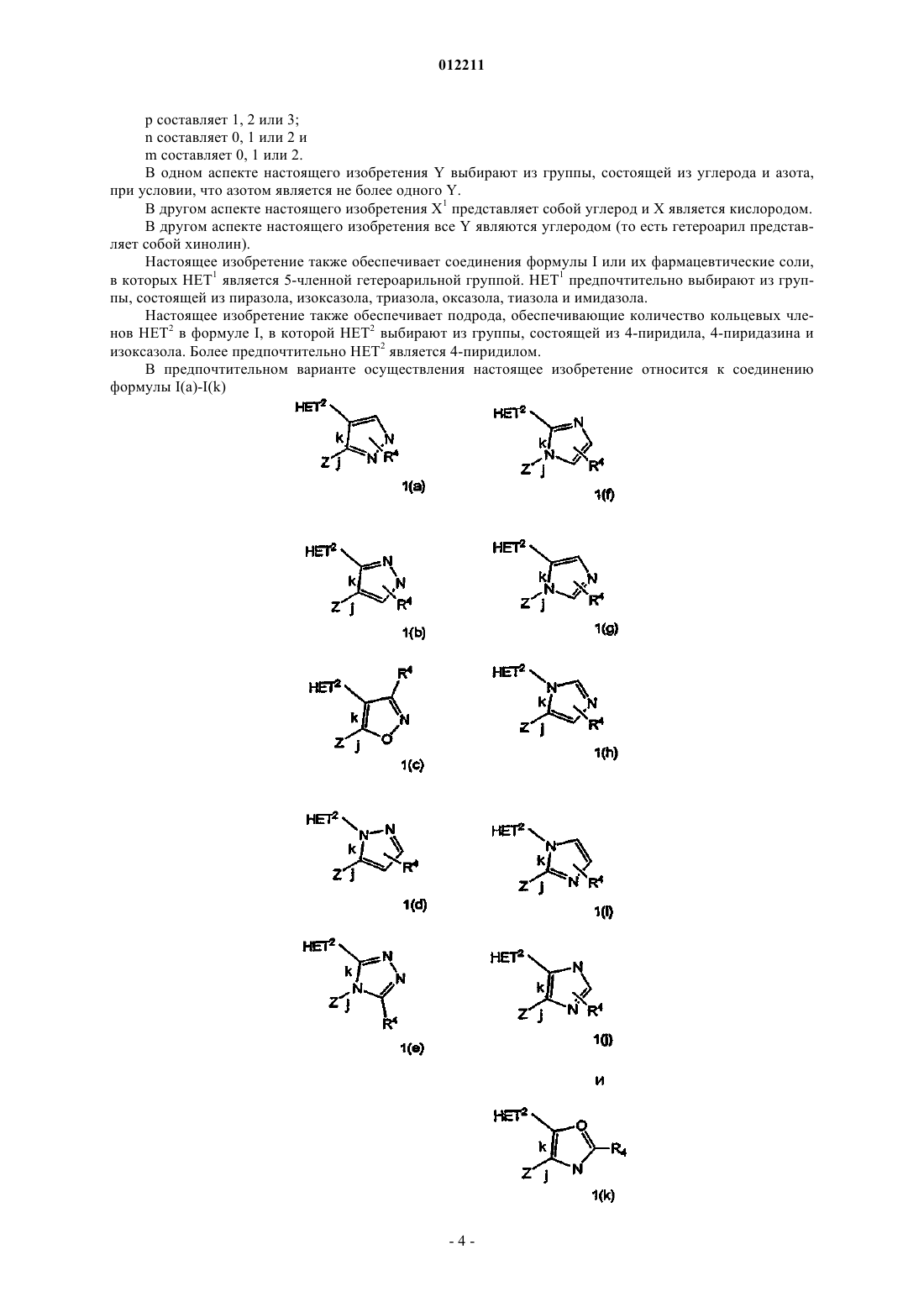



Формула / Реферат
1. 2-[4-(1-Метил-4-пиридин-4-ил-1Н-пиразол-3-ил)феноксиметил]хинолин или его фармацевтически приемлемая соль.
2. Соединение, выбранное из группы, состоящей из
2-[-4-(4-пиридин-4-ил-2Н-пиразол-3-ил)феноксиметил]хинолина;
2-[4-(2-метил-4-пиридин-4-ил-2Н-пиразол-3-ил)феноксиметил]хинолина
и их фармацевтически приемлемых солей.
3. Фармацевтическая композиция для лечения психотических расстройств, бредовых расстройств и медикаментозного психоза; тревожных расстройств, двигательных расстройств, расстройств настроения, нейродегенеративных расстройств и лекарственной зависимости, содержащая такое количество соединения хинолина по п.1 или 2, которое является эффективным для лечения указанного расстройства или состояния.
4. Способ лечения психотических расстройств, бредовых расстройств и медикаментозного психоза; тревожных расстройств, двигательных расстройств, расстройств настроения, нейродегенеративных расстройств и лекарственной зависимости, включающий введение эффективно для ингибирования PDE10 количества соединения по п.1 или 2.
Текст
012211 Область изобретения Настоящее изобретение относится к гетероароматическим соединениям, которые действуют как эффективные ингибиторы фосфодиэстеразы (PDE). Настоящее изобретение также относится к соединениям, являющимся селективными ингибиторами PDE10. Настоящее изобретение дополнительно относится к промежуточным продуктам для получения таких соединений; к фармацевтическим композициям,содержащим такие соединения; и к применению таких соединений в способах лечения некоторых заболеваний центральной нервной системы (ЦНС) или других расстройств. Настоящее изобретение также относится к способам лечения нейродегенеративных и психических расстройств, например психоза, и расстройств, симптомом которых является когнитивный дефицит. Уровень техники изобретения Фосфодиэстеразы (PDE) представляют собой класс внутриклеточных ферментов, задействованных в гидролизе нуклеотидов циклического аденозинмонофосфата (цАМФ) и циклического гуанозинмонофосфата (цГМФ) в их соответствующие нуклеотидмонофосфаты. Циклические нуклеотиды цАМФ и цГМФ, соответственно, синтезируются аденилил- и гуанилилциклазами и действуют как вторичные мессенджеры в нескольких клеточных путях метаболизма. цАМФ и цГМФ действуют в качестве внутриклеточных вторичных мессенджеров, регулирующих широкий спектр внутриклеточных процессов, особенно в нейронах центральной нервной системы. В нейронах эти процессы включают активацию цАМФ- и цГМФ-зависимых киназ и последующее фосфорилирование белков, вовлеченных в тонкую регуляцию синаптической передачи, а также в нейронную дифференциацию и выживание. На сложность передачи сигнала циклических нуклеотидов указывает молекулярное разнообразие ферментов, вовлеченных в синтез и расщепление цАМФ и цГМФ. Существуют по меньшей мере 10 семейств аденилилциклаз, 2 семейства гуанилилциклаз и 11 семейств фосфодиэстераз. Кроме того, известно, что различные типы нейронов экспрессируют множественные изоферменты (изозимы) каждого из этих классов, и существует достоверное доказательство компартментализации и специфичности функционирования различных изоферментов в пределах данного нейрона. Основным механизмом регуляции передачи сигнала циклических нуклеотидов является катаболизм циклических нуклеотидов, катализируемый фосфодиэстеразами. Существует 11 известных семействPDE, кодируемых 21 различными генами. Каждый ген обычно выдает множественные сплайс-варианты,которые вносят вклад в дополнительное разнообразие изоферментов. Семейства PDE имеют функциональные различия, основанные на субстратной специфичности циклических нуклеотидов, на механизме(механизмах) регуляции и чувствительности к ингибиторам. Кроме того, PDE дифференцированно экспрессируются во всем организме, в том числе в центральной нервной системе. Это многообразие ферментативной активности и локализаций изоферментов различных PDE в результате обуславливают различные физиологические функции. Кроме того, у соединений, способных селективно ингибировать различные семейства PDE или изоферменты, можно предположить наличие конкретных терапевтических эффектов, меньших побочных эффектов или и то, и другое. Установлено, что PDE10 представляет собой уникальное семейство, основанное на первичной аминокислотной последовательности и различной ферментативной активности. Поиск гомологов в базах данных EST среди PDE в качестве первого члена семейства PDE10 выявил мышиную PDE10A (Fujishigeet al., J. Biol. Chem. 274: 18438-18445, 1999; Loughney, K. et al., Gene 234: 109-117, 1999). Также был клонирован мышиный гомолог (Soderling, S. et al., Proc. Natl. Acad. Sci. USA 96: 7071-7076, 1999) и были идентифицированы N-концевые сплайс-варианты и крысиных, и человеческих генов (Kotera, J. et al., Biochem.Biophys. Res. Comm. 261: 551-557, 1999; Fujishige, K. et al., Eur. J. Biochem. 266: 1118-1127, 1999). Существует высокая степень межвидовой гомологии. Мышиный PDE10A1 представляет собой белок из 779 аминокислот, который гидролизует, соответственно, до АМФ и ГМФ как цАМФ, так и цГМФ. Афинность PDE10 к цАМФ (Km=0,05 мкМ) выше, чем к цГМФ (Km=3 мкМ). Вместе с тем, превышение Vmax цГМФ по сравнению с цАМФ примерно в 5 раз приводит к предположению, что PDE10 является уникальной цГМФ-азой, ингибируемой цАМФ (Fujishige et al., J. Biol. Chem. 274: 18438-18445, 1999). Семейство полипептидов PDE10 проявляет более низкую степень гомологии последовательностей по сравнению с ранее идентифицированными семействами PDE, и показано, что оно не восприимчиво к некоторым ингибиторам, известным в качестве специфических ингибиторов для других семейств PDE. Патент США 6350603 включен в качестве ссылки в настоящее изобретение. По сравнению с другими семействами PDE у млекопитающих также однозначно локализуютсяPDE10. Высокая экспрессия мРНК для PDE10 наблюдается только в яичке и мозге (Fujishige, K. et al., Eur.et al., Gene 234: 109-117, 1999). Эти первичные исследования показали, что экспрессия PDE10 в мозге наиболее высока в стриатуме (в хвостатом ядре и путамене), в прилежащем ядре и в обонятельном бугорке. Позднее был осуществлен подробный анализ паттерна экспрессии мРНК PDE10 в мозге грызуновConference 'Phosphodiesterase in Health and Disease', Porto, Portugal, Dec. 5-7, 2001). Сообщалось о различных терапевтических применениях ингибиторов PDE, включающих обструк-1 012211 тивную болезнь легких, аллергии, гипертонию, ангину, застойную сердечную недостаточность, депрессию и эректильную дисфункцию (патент WO 01/41807 А 2, включенный в настоящее описание в качестве ссылки). На основе ингибирования PDE-ассициированной активности цГМФ раскрыто применение селективного бензимидазола и родственных гетероциклических соединений в лечении ишемических состояний сердца. Патент США 5693652 включен в настоящее описание в качестве ссылки. Опубликованная патентная заявка США 2003/0032579 раскрывает способ лечения селективным ингибитором PDE10 папаверином некоторых неврологических и психических расстройств. В частности,способ относится к психотическим расстройствам, таким как шизофрения, бредовым расстройствам и к психозу, вызванному лекарственными средствами; к тревожным расстройствам, таким как паническое и обсцессивно-компульсивное расстройство; и к двигательным расстройствам, включающим болезнь Паркинсона и болезнь Хантингтона. Сущность изобретения В настоящем изобретении обеспечиваются соединения формулы I или их фармацевтические соли Каждый R1 независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, циано,С 1-С 8 алкила, С 2-С 8 алкенила, С 2-С 8 алкинила, С 1-С 8 алкокси, C1-C8 галогеналкила, С 3-С 8 циклоалкила, С 3 С 8 циклоалкил-C1-C8 алкила, 4-7-членного гетероциклоалкила, C1-C8 алкилтио, -NR3R3, -O-CF3, -S(O)n-R3,C(O)-NR3R3 и C1-C8 алкила, замещенного гетероатомом, где гетероатом выбирают из группы, состоящей из азота, кислорода и серы, и где гетероатом может быть дополнительно замещен заместителем, выбираемым из группы, состоящей из водорода, C1-C8 алкила, С 3-С 8 циклоалкила, С 2-С 8 алкенила, С 2-С 8 алкинила и C1-C8 галогеналкила; каждый R3 независимо выбирают из группы, состоящей из водорода, C1-C8 алкила, С 2-С 8 алкенила,С 2-С 8 алкинила, C1-C8 галогеналкила, С 3-С 8 циклоалкила;HET1 выбирают из группы, состоящей из моноциклического гетероарила и бициклического гетероарила, где моноциклический и бициклический гетероарил может быть необязательно замещен по меньшей мере одним R4;R4 выбирают из группы, состоящей из галогена, гидроксила, циано, C1-C8 алкила, С 2-С 8 алкенила, С 2 С 8 алкинила, C1-C8 алкокси, С 3-С 8 циклоалкила, С 3-С 8 циклоалкил-С 1-С 8 алкила, C1-C8 алкилтио и C1-C8 алкила, замещенного заместителем, выбираемым из группы, состоящей из -OR8, -NR8R8 и -SR8, где R8 независимо выбирают из группы, состоящей из водорода и С 1-С 8 алкила;HET2 представляет собой моноциклический или бициклический гетероарил, где моноциклический и бициклический гетероарил необязательно замещен по меньшей мере одним R5, при условии, что HET2 не является тетразолом;R5 независимо выбирают из группы, состоящей из галогена, гидроксила, циано, C1-C8 алкила, С 2 С 8 алкенила, С 2-С 8 алкинила, С 1-С 8 алкокси, С 3-С 8 циклоалкила, С 3-С 8 циклоалкил-C1-C8 алкила, C1-C8 алкилтио, -NR7R7 и C1-C8 галогеналкила; В 1 и В 2 представляют собой смежные атомы в Het1, которые независимо выбирают из группы, состоящей из углерода и азота; связь j является ковалентной связью между Z и В 2; связь к является ковалентной связью в Het1 между В 1 и В 2; каждый из X и X1 независимо выбирают из группы, состоящей из кислорода, серы, С(R2)2 и NR2,при условии, что по меньшей мере один из X и X1 является атомом углерода;Y выбирают из группы, состоящей из углерода и азота, при условии, если Y представляет собой углерод, он замещен R6;-2 012211 где каждый R6 независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, циано, С 1-С 8 алкила, С 2-С 8 алкенила, С 2-С 8 алкинила, C1-C8 алкокси, C1-C8 циклоалкила, С 3-С 8 циклоалкил-C1C8 алкила, С 1-С 8 алкилтио, C1-C8 галогеналкила, NR7R7, -O-CF3, -S(O)m-R7 и C(O)-NR7R7, C1-C8 алкила, замещенного гетероатомом, где гетероатом выбирают из группы, состоящей из азота, кислорода и серы, и где гетероатом может быть дополнительно замещен заместителем, выбираемым из группы, состоящей из водорода, C1-C8 алкила, С 3-С 8 циклоалкила, С 2-С 8 алкенила, С 2-С 8 алкинила и C1-C8 галогеналкила; где каждый R7 независимо выбирают из группы, состоящей из водорода и С 1-С 8 алкила; р составляет 1, 2 или 3;m составляет 0, 1 или 2. В другом варианте осуществления настоящее изобретение обеспечивает соединения формулы I или их фармацевтические соли Каждый R1 независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, циано,С 1-С 8 алкила, С 2-С 8 алкенила, С 2-С 8 алкинила, C1-C8 алкокси, C1-C8 галогеналкила, С 3-С 8 циклоалкила, С 3 С 8 циклоалкил-С 1-С 8 алкила, 4-7-членного гетероциклоалкила, C1-C8 алкилтио, -NR3R3, -O-CF3, -S(O)n-R3,C(O)-NR3R3 и C1-C8 алкила, замещенного гетероатомом, где гетероатом выбирают из группы, состоящей из азота, кислорода и серы, и где гетероатом может быть дополнительно замещен заместителем, выбираемым из группы, состоящей из водорода, С 1-С 8 алкила, С 3-С 8 циклоалкила, С 2-С 8 алкенила, С 2-С 8 алкинила и С 1-С 8 галогеналкила; каждый R3 независимо выбирают из группы, состоящей из водорода, C1-C8 алкила, С 2-С 8 алкенила,С 2-С 8 алкинила, С 1-С 8 галогеналкила, С 3-С 8 циклоалкила;HET1 выбирают из группы, состоящей из моноциклического гетероарила и бициклического гетероарила, где моноциклический и бициклический гетероарил может быть необязательно замещен по меньшей мере одним R4;HET2 представляет собой моноциклический или бициклический гетероарил, где моноциклический и бициклический гетероарил может быть необязательно замещен по меньшей мере одним R5;R5 независимо выбирают из группы, состоящей из галогена, гидроксила, циано, С 1-С 8 алкила, С 2 С 8 алкенила, С 2-С 8 алкинила, С 1-С 8 алкокси, С 3-С 8 циклоалкила, С 3-С 8 циклоалкил-C1-C8 алкила, C1-C8 алкилтио, -NR7R7 и C1-C8 галогеналкила; В 1 и В 2 представляют собой смежные атомы в Het1, которые независимо выбирают из группы, состоящей из углерода и азота; связь j является ковалентной связью между Z и В 2; связь k является связью в Het1 между В 1 и В 2; каждый из X и X1 независимо выбирают из группы, состоящей из кислорода, серы, C(R2)2 и NR2,при условии, что по меньшей мере один из X и X1 является атомом углерода;Y выбирают из группы, состоящей из углерода и азота, при условии, что, если Y представляет собой углерод, он замещен R6; где каждый R6 независимо выбирают из группы, состоящей из водорода, галогена, гидроксила, циано, C1-C8 алкила, С 2-С 8 алкенила, С 2-С 8 алкинила, C1-C8 алкокси, C1-C8 циклоалкила, С 3-С 8 циклоалкил-C1C8 алкила, C1-C8 алкилтио, C1-C8 галогеналкила, NR7R7-O-CF3, -S(O)m-R7 и C(O)-NR7R7, C1-C8 алкила, замещенного гетероатомом, где гетероатом выбирают из группы, состоящей из азота, кислорода и серы, и где гетероатом может быть дополнительно замещен заместителем, выбираемым из группы, состоящей из водорода, C1-C8 алкила, С 3-С 8 циклоалкила, С 2-С 8 алкенила, С 2-С 8 алкинила и C1-C8 галогеналкила; где каждый R7 независимо выбирают из группы, состоящей из водорода и С 1-С 8 алкила;m составляет 0, 1 или 2. В одном аспекте настоящего изобретения Y выбирают из группы, состоящей из углерода и азота,при условии, что азотом является не более одного Y. В другом аспекте настоящего изобретения X1 представляет собой углерод и X является кислородом. В другом аспекте настоящего изобретения все Y являются углеродом (то есть гетероарил представляет собой хинолин). Настоящее изобретение также обеспечивает соединения формулы I или их фармацевтические соли,в которых HET1 является 5-членной гетероарильной группой. HET1 предпочтительно выбирают из группы, состоящей из пиразола, изоксазола, триазола, оксазола, тиазола и имидазола. Настоящее изобретение также обеспечивает подрода, обеспечивающие количество кольцевых членов HET2 в формуле I, в которой HET2 выбирают из группы, состоящей из 4-пиридила, 4-пиридазина и изоксазола. Более предпочтительно HET2 является 4-пиридилом. В предпочтительном варианте осуществления настоящее изобретение относится к соединению формулы I(a)-I(k)-4 012211 где j, k, Z, HET2 и R4 соответствуют вышеупомянутым определениям. Более предпочтительно соединения формулы I имеют следующую общую структуру: Наиболее предпочтительно соединения формулы I имеют следующую общую структуру: В другом аспекте HET1 для вышеупомянутых соединений формулы I не является тетразолом. Соединения формулы I могут иметь оптические центры и поэтому могут существовать в различных энантиомерных и диастереоизомерных конфигурациях. Настоящее изобретение включает все энантиомеры, диастереоизомеры и другие стереоизомеры таких соединений формулы I, а также рацемические соединения, и рацемические смеси, и другие смеси их стереоизомеров. Фармацевтически приемлемые соли соединения формулы I включают их кислотно-аддитивные и основные соли. Подходящие кислотно-аддитивные соли представляют собой соли, образованные из кислот, которые образуют нетоксичные соли. Их примеры включают следующие соли: ацетат, адипат, аспартат, бензоат, бесилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат,эзилат, соль муравьиной кислоты, фумарат, глуцептат, глюконат, глукуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изэтионат, лактат, малат, малеат,соль малоновой кислоты, мезилат соли миндальной кислоты, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат,салицилат, сахарат, стеарат, сукцинат, сульфонат, станнат, тартрат, тозилат, трифторацетат и ксинофоат,но не ограничены вышеперечисленным. Подходящие основные соли являются солями, образованными из оснований, которые образуют нетоксичные соли. Их примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка,но не ограничены вышеперечисленным. Полусоли кислот и оснований можно образовывать, например, из солей полусульфата и полукальция. Для обзорной информации о подходящих солях см. руководство Handbook of Pharmaceutical Salts:Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002). Фармацевтически приемлемые соли соединения формулы I можно получать одним или несколькими из трех способов:(i) взаимодействием соединения формулы I с желательной кислотой или основанием;(ii) путем удаления кислотно-лабильной или щелочно-лабильной защитной группы из подходящего предшественника соединения формулы I или дециклизацией подходящего циклического предшественника, например лактона или лактама, путем использования желательной кислоты или основания или(iii) путем превращения одной соли соединения формулы I в другую соль посредством реакции с соответствующей кислотой или основанием или посредством подходящей ионообменной колонки. Все три реакции обычно осуществляют в растворе. Получаемая соль может выпадать в осадок и может быть собрана фильтрацией или может быть восстановлена путем выпаривания растворителя. Степень ионизации в получаемой соли может варьировать от полной ионизации до почти неионизированного состояния. Соединения настоящего изобретения могут существовать в диапазоне состояний твердого тела в пределах от полностью аморфного до полностью кристаллического состояния. Термин "аморфный" относится к состоянию, в котором материал не обладает дальним порядком на молекулярном уровне и, в зависимости от температуры, может проявлять физические свойства твердого тела или жидкости. Обычно такие материалы не дают отчетливой дифракционной рентгеновской картины, и более формально их описывают как жидкость, в то время как они проявляют свойства твердого тела. После нагревания свойства меняются от свойств твердого тела до свойств жидкости, что характеризуется изменением состояния, обычно второго порядка ("стеклование"). Термин "кристаллический" относится к твердой фазе, в которой материал на молекулярном уровне имеет внутреннюю структуру регулярного порядка и дает отчетливую дифракционную рентгеновскую картину с определенными пиками. Такие материалы при достаточном нагревании также проявляют свойства жидкости, но изменение от твердого состояния до жидкости характеризуется фазовым переходом, обычно первого порядка ("точка плавления"). Соединения настоящего изобретения также могут существовать в сольватных и несольватных фор-5 012211 мах. Термин "сольват" в настоящем изобретении используется для описания молекулярного комплекса,содержащего соединение настоящего изобретения и одну или несколько молекул фармацевтически приемлемого растворителя, например этанола. Если указанным растворителем является вода, употребляется термин "гидрат". В настоящее время принятой системой классификации для органических гидратов является система, которая выделяет гидраты с изолированными расположениями, канальные или ион-металлокоординированные гидраты - см. Polymorphism in Pharmaceutical Solids by K.R. Morris (Ed. H.G. Brittain, MarcelDekker, 1995). Гидраты с изолированными расположениями представляют собой гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом посредством промежуточных органических молекул. В канальных гидратах молекулы воды лежат в каналах решетчатой структуры, где они находятся рядом с другими молекулами воды. В ион-металлокоординированых гидратах молекулы воды являются связанными с ионом металла. Когда растворитель или вода тесно связаны, комплекс будет иметь четкую стехиометрию, которая не зависит от влажности. Вместе с тем, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопических соединениях, содержание воды/растворителя будет зависеть от условий влажности и высушивания. В таких случаях нормой будет являться нестехиометрия. Если соединения настоящего изобретения находятся в подходящих условиях, они также могут существовать в мезоморфном состоянии (мезоморфном или жидкокристаллическом). Мезоморфное состояние является промежуточным между истинным кристаллическим состоянием и истинным жидким состоянием (как в расплаве, так и в растворе). Мезоморфизм, возникающий в результате изменения температуры, называется "термотропным", и мезоморфизм, являющийся следствием добавления второго компонента, такого как вода или другого растворителя, называется "лиотропным". Соединения, способные образовывать лиотропные мезофазы, называют "амфифильными", и они состоят из молекул, которые обладают ионной (такой, как -COO-Na+, -COO-K+ или -SO3-Na+) или неионной (такой, как -N-N+(СН 3)3) полярной главной группой. Более подробную информацию можно найти в 4-м издании Crystails and thePolarizing Microscope by N.H. Hartshorne and A. Stuart (Edward Arnold, 1970). Далее в настоящем изобретении все ссылки на соединения формулы I включают ссылки на их соли,сольваты, многокомпонентные комплексы и жидкие кристаллы и на сольваты, многокомпонентные комплексы и жидкие кристаллы их солей. Соединения настоящего изобретения, как указано выше, включают соединения формулы I, включающие все их полиморфные формы и формы кристаллизации, их пролекарства и изомеры (включающие оптические, геометрические и таутомерные изомеры), представленные в настоящем изобретении, и меченные изотопами соединения формулы I. Как указано выше, так называемые "пролекарства" соединений формулы I также входят в объем настоящего изобретения. Таким образом, некоторые производные соединения формулы I, которые при введении в организм или на поверхность тела могут показывать отсутствие фармакологической активности или слабую активность, можно превращать, например, путем гидролитического расщепления в соединения формулы I, имеющие желаемую активность. Такие производные упоминаются как "пролекарства". Дополнительную информацию относительно использования пролекарств можно найти в изданиях Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) и в BioreversibleCarriers in Drug Design, Pergamon Press, 1987 (Ed. E.B. Roche, American Pharmaceutical Association). Пролекарства согласно настоящему изобретению, например, можно получать замещением соответствующих функциональных групп, присутствующих в соединениях формулы I, определенными группами, которые специалистам в данной области известны как "прогруппы", описанные, например, в книгеDesign of Prodrugs by H. Bundgaard (Elsevier, 1985). Некоторые примеры пролекарств согласно настоящему изобретению включают без ограничения:(i) сложный эфир карбоновой кислоты, например соединение, в котором водород функциональной группы карбоновой кислоты соединения формулы (I) замещен на (C1-C8)алкил, если соединение формулы I содержит функциональную группу карбоновой кислоты (-СООН);(ii) простой эфир, например соединение, в котором водород функциональной группы спирта соединения формулы I замещен на (С 1-С 6)алканоилоксиметил, если соединение формулы I содержит функциональную группу спирта (-ОН); и(iii) амид, например соединение, в котором, в зависимости от ситуации, один или оба водорода функциональной аминогруппы соединения формулы I замещены на (C1-С 10)алканоил, если соединение формулы I содержит первичную или вторичную функциональную аминогруппу (-NH2 или -NHR, гдеRН). Дополнительные примеры замещающих групп согласно предшествующим примерам и примеры пролекарств других типов можно найти в вышеупомянутых ссылках. Кроме того, некоторые соединения формулы I сами по себе могут действовать как пролекарства других соединений формулы I. Также в объем настоящего изобретения входят метаболиты соединений формулы I, то есть соединения, образованные in vivo после введения лекарственного средства. Некоторые примеры метаболитов-6 012211 согласно настоящему изобретению включают без ограничения:(i) если соединение формулы I содержит метильную группу, ее гидроксиметилпроизводное(ii) если соединение формулы I содержиталкокси группу, ее гидроксипроизводное (-OR-ОН);(iii) если соединение формулы I содержит третичную аминогруппу, ее вторичное аминопроизводное (-NR1R2-NHR1 или -NHR2);(iv) если соединение формулы I содержит вторичную аминогруппу, ее первичное производное(v) если соединение формулы I содержит фенильную группу, ее фенольное производное (-Ph-PhOH); и(vi) если соединение формулы I содержит амидную группу, производное ее карбоновой кислоты(vii) если соединение содержит ароматический атом азота или третичную алифатическую аминогруппу, ее N-оксидное производное. Соединения формулы I, имеющие атом азота в третичной функциональной аминогруппе, можно дополнительно замещать кислородом (т.е. N-оксидом). Соединения формулы I, содержащие один или несколько асимметричных атомов углерода, могут существовать в виде двух или более стереоизомеров. Если соединение формулы I содержит группу алкенила или алкенилена, возможно существование геометрических цис/транс (или Z/E) изомеров. Если структурные изомеры взаимопревращаемы посредством низкого энергетического барьера, может наблюдаться таутомерная изомерия ("таутомеризм"). Она может принимать форму таутомеризма протонов в соединениях формулы I содержащих, например, группу имина, кето, или оксима, или так называемого таутомеризма валентности в соединениях, которые содержат ароматическую группу. Из этого следует,что одно соединение может проявлять более одного типа изомерии. В объем настоящего изобретения входят все стереоизомеры, геометрические изомеры и таутомерные формы соединения формулы I, включающие соединения, проявляющие более одного типа изомерии,и их одна или более смеси. Также в этот объем входят кислотно-аддитивные или основные соли, в которых противоион является оптически активным, например d-лактат или l-лизин, или рацемическим, например dl-тартрат или dl-аргинин. Цис/транс изомеры можно разделять общепринятыми способами, известными специалистам в данной области, например хроматографией и фракционной кристаллизацией. Общепринятые способы получения/выделения отдельных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или расщепление рацемата (или рацемата соли или производного) с использованием, например, хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ). С другой стороны, рацемат (или рацемический предшественник) может реагировать с подходящим оптически активным соединением, например со спиртом, или, в случае, если соединение формулы I содержит кислую или основную группу, с основанием или кислотой, такой как 1-фенилэтиламин или винно-каменная кислота. Получаемую диастереоизомерную смесь можно разделять хроматографией и/или фракционной кристаллизацией и способами, известными специалисту в данной области, один или оба диастереоизомера можно превращать в соответствующий чистый энантиомер (энантиомеры). Хиральные соединения настоящего изобретения (и их хиральные предшественники) можно получать в энантиомерно-обогащенной форме, используя хроматографию, обычно ВЭЖХ, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего изопропанол от 0 до 50 об.%, обычно от 2 до 20%, и алкиламин от 0 до 5 об.%, обычно 0,1% диэтиламина. Концентрация элюата предоставляет обогащенную смесь. При кристаллизации любого рацемата возможно получение кристаллов двух различных типов. Первый тип представляет собой вышеупомянутое рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, в котором две формы кристалла образуются в эквимолярных количествах, и каждая из которых содержит единственный энантиомер. Обе кристаллические формы, присутствующие в рацемической смеси, имеют идентичные физические свойства, в то же время их физические свойства по сравнению с истинным рацематом могут иметь отличия. Рацемические смеси можно разделять общепринятыми способами, известными специалистам в данной области, например см. Stereochemistry of Organic Compounds by E.L. Eliel and S.H. Wilen (Wiley, 1994). Настоящее изобретение включает все фармацевтически приемлемые изотопно меченные соединения формулы I, в которых один или несколько атомов замещены атомами, имеющими также атомное число, но их атомная масса или массовое число отличаются от атомной массы или массового числа, преобладающего в природе. Примеры изотопов, подходящих для включения в соединения настоящего изобретения, включают,без ограничения, изотопы водорода, такие как 2 Н и 3 Н, углерода, такие как 11 С, 13 С и 14 С, хлора, такие как 36-7 012211 Некоторые изотопно меченные соединения формулы I, например соединения, включающие радиоактивный изотоп, являются полезными для исследований лекарственных препаратов и/или изучения распределения субстрата в тканях. Особенно полезными для этой цели являются радиоактивные изотопы трития, т.е. 3 Н, и углерода-14, т.е. 14 С, ввиду легкости их включения и наличия способов выявления. Определенные терапевтические преимущества может предоставлять замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, по причине их большей метаболической устойчивости, например, в результате увеличенного периода полураспада или необходимости введения меньшей дозы, и, следовательно, при некоторых обстоятельствах может быть предпочтительным. Замещение позитронно-активными изотопами, такими как 11 С, 18F, 15O и 13N, может быть полезным при позитронно-эмиссионных томографических исследованиях (PET) для исследования занятости рецепторов субстрата. В общем, путем использования соответствующего изотопно меченного реагента, вместо ранее используемого немеченного реагента, можно получать изотопно меченные соединения формулы I общепринятыми технологиями, известными специалистам в данной области, или способами, аналогичными способам, описанным в прилагаемых примерах и получениях. Фармацевтически приемлемые сольваты согласно настоящему изобретению включают сольваты, в которых растворитель кристаллизации может быть изотопно замещенным, например D2O, d6-ацетон, d6 диметилсульфоксид (ДМСО). Конкретные варианты осуществления настоящего изобретения включают соединения, ниже приведенные в качестве примеров, и их фармацевтически приемлемые соли, комплексы, сольваты, полиморфные формы, стереоизомеры, метаболиты, пролекарства и их другие производные. Настоящее изобретение также относится к фармацевтической композиции, содержащей такое количество соединения формулы I, которое эффективно ингибирует PDE10, для лечения некоторых психотических нарушений и состояний, таких как шизофрения, бредовые нарушения и психоз, вызванный лекарственными средствами; тревожных расстройств, таких как паническое обсцессивно-компульсивное расстройство; и двигательных расстройств, включающих болезнь Паркинсона и болезнь Хантингтона. В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции для лечения психотических расстройств и состояний, таких как шизофрения, бредовые нарушения и психоз, вызванный лекарственными средствами; тревожных расстройств, таких как паническое и обсцессивно-компульсивное расстройство; и двигательных расстройств, включающих болезнь Паркинсона и болезнь Хантингтона, содержащей такое количество соединения формулы I, которое эффективно для лечения указанного расстройства или состояния. Примеры психотических нарушений, которые можно лечить согласно настоящему изобретению,включают, без ограничения, шизофрению, например шизофрению параноидного, дезорганизованного,кататонического, недифференцированного или остаточного типа; шизофрениформное расстройство; шизоаффективное расстройство, например, бредового типа или депрессивного типа; бредовое расстройство; психотическое расстройство, вызванное лекарственными средствами, например алкогольный психоз,психозы, вызванные амфетамином, каннабисом, кокаином, галлюциногенами, летучими препаратами,опиатами или фенциклидином; расстройство личности параноидного типа; и расстройство личности шизоидного типа. Примеры двигательных нарушений, которые можно лечить согласно настоящему изобретению, включают, без ограничения, нарушения, выбираемые из болезни Хантингтона и дискинезии, связанной с терапией агонистами допамина, болезни Паркинсона, синдрома "беспокойных ног" и эссенциального тремора. Другие расстройства, которые можно лечить согласно настоящему изобретению, представляют собой обсцессивно-компульсивные расстройства, синдром Туретта и другие судорожные расстройства. В другом варианте осуществления настоящее изобретение относится к способу лечения тревожного расстройства или состояния у млекопитающего, содержащему введение указанному млекопитающему такого количество соединения формулы I, которое эффективно для ингибирования PDE10. Настоящее изобретение также обеспечивает способ лечения расстройства или состояния у млекопитающего, содержащий введение указанному млекопитающему такого количества соединения формулы I,которое эффективно для лечения указанного расстройства или состояния. Примеры тревожных расстройств, которые можно лечить согласно настоящему изобретению,включают, без ограничения, паническое расстройство; агорафобию; специфическую фобию; социальную фобию; обсцессивно-компульсивное расстройство; расстройство, вызванное посттравматическим стрессом; расстройство, вызванное острым стрессом; и генерализованное тревожное расстройство. Настоящее изобретение дополнительно обеспечивает способ лечения лекарственной зависимости,например алкогольной, амфетаминовой, кокаиновой или опиатной зависимости у млекопитающего,включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно для лечения лекарственной зависимости. Настоящее изобретение также обеспечивает способ лечения лекарственной зависимости, например алкогольной, амфетаминовой, кокаиновой или опиатной зависимости у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, ко-8 012211 торое эффективно ингибирует PDE10. Термин "лекарственная зависимость", используемый в настоящем изобретении, обозначает патологическое влечение к лекарственным средствам и, в общем, характеризуется мотивационными расстройствами, такими как непреодолимое влечение к приему желательного препарата и эпизоды сильного влечения к лекарственным средствам. Настоящее изобретение дополнительно обеспечивает способ лечения расстройства, содержащего в качестве симптома дефицит внимания и/или когнитивный дефицит у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно для лечения указанного расстройства. Настоящее изобретение также обеспечивает способ лечения расстройства или состояния, содержащего в качестве симптома дефицит внимания и/или когнитивный дефицит у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I,которое эффективно ингибирует PDE10. Настоящее изобретение также обеспечивает способ лечения расстройства или состояния, содержащее в качестве симптома дефицит внимания и/или когнитивный дефицит у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I,которое эффективно для лечения указанного расстройства или состояния. Термины "дефицит внимания" и/или "когнитивный дефицит", используемые в настоящем изобретении в выражении "расстройство, содержащее в качестве симптома дефицит внимания и/или когнитивный дефицит", относятся к снижению функций по одному или более когнитивным аспектам, таким как память, интеллект, или к обучаемости и логическим способностям конкретного индивидуума относительно других индивидуумов в пределах той же возрастной популяции. "Дефицит внимания и/или когнитивный дефицит у млекопитающего" также относится к снижению любых функций конкретного индивидуума по одному или более когнитивным аспектам, например, как это происходит при возрастном снижении когнитивных функций. Примерами нарушений, которые в качестве симптома включают дефицит внимания и/или когнитивный дефицит у млекопитающего, которые можно лечить согласно настоящему изобретению, являются деменция, например болезнь Альцгеймера, мультиинфарктная деменция, алкогольная деменция или другая деменция, связанная с лекарственными средствами, деменция, связанная с внутричерепными опухолями или мозговой травмой, деменция, связанная с болезнью Хантингтона или болезнью Паркинсона,или связанная со СПИДом деменция; бред; амнестическое расстройство; расстройство, вызванное посттравматическим стрессом; задержка умственного развития; расстройство обучения, например расстройство чтения, расстройство счета или расстройство письменного выражения; синдром нарушенного внимания/гиперактивности и возрастное снижение когнитивных функций. Настоящее изобретение также обеспечивает способ лечения расстройства настроения или эпизода расстройства настроения у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно для лечения указанного расстройства или эпизода. Настоящее изобретение также обеспечивает способ лечения расстройства настроения или эпизода расстройства настроения у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно ингибирует PDE10. Примеры расстройств настроения и эпизодов расстройства настроения, которые можно лечить согласно настоящему изобретению, включают, без ограничения, большой депрессивный эпизод легкого,среднего или тяжелого типа; маниакальный или смешанный эпизод настроения; гипоманический эпизод настроения; депрессивный эпизод с атипичными признаками; депрессивный эпизод с меланхолическими признаками; депрессивный эпизод с кататоническими признаками; эпизод настроения с началом в послеродовом периоде; депрессию после инсульта; большое депрессивное расстройство; дистимическое расстройство; малое депрессивное расстройство; предменструальное дисфорическое расстройство; постпсихотическое депрессивное расстройство или шизофрения; большое депрессивное расстройство, накладывающееся на психотическое расстройство, такое как бредовое расстройство или шизофрения; биполярное расстройство, содержащее биполярное расстройство I типа, биполярное расстройство II типа, циклотимическое расстройство. Настоящее изобретение дополнительно обеспечивает способ лечения нейродегенеративного расстройства или состояния у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно для лечения указанного расстройства или состояния. Настоящее изобретение дополнительно обеспечивает способ лечения нейродегенеративного расстройства или состояния у млекопитающего, включая человека, содержащий введение указанному млекопитающему такого количества соединения формулы I, которое эффективно ингибирует PDE10. Если не указано иначе, термины "нейродегенеративное расстройство или состояние", используемые в настоящем изобретении, относятся к расстройству или состоянию, которое вызвано дисфункцией и/или гибелью нейронов в центральной нервной системе. Лечению этих расстройств и состояний может спо-9 012211 собствовать введение средства, которое предотвращает дисфункцию или гибель нейронов при риске возникновения этих расстройств или состояний и/или усиливает функцию поврежденных или здоровых нейронов таким образом, что компенсируется потеря функции, вызванная дисфункцией или гибелью нейронов с повышенным риском. Используемый в настоящем изобретении термин "нейротрофное средство" относится к веществу или к средству, которые обладают некоторыми или всеми из указанных свойств. Примеры нейродегенеративных нарушений и состояний, которые можно лечить согласно настоящему изобретению, включают, без ограничения, болезнь Паркинсона; болезнь Хантингтона; деменцию,например болезнь Альцгеймера, мультиинфарктную деменцию, деменцию, связанную со СПИДом,фронтотемпоральную деменцию; нейродегенерацию, связанную с черепно-мозговой травмой; нейродегенерацию, связанную с инсультом; нейродегенерацию, связанную с инфарктом мозга; нейродегенерацию, вызванную гипогликемией; нейродегенерацию, связанную с эпилептическим припадком; нейродегенерацию, связанную с нейротоксическим воздействием; и мультисистемную атрофию. В одном варианте осуществления настоящего изобретения нейродегенеративное расстройство или состояние содержит нейродегенерацию стриарных средних дендритных нейронов у млекопитающего,включая человека. В дополнительном варианте осуществления настоящего изобретения нейродегенеративное расстройство или состояние представляет собой болезнь Хантингтона. Настоящее изобретение также обеспечивает фармацевтическую композицию для лечения психотических расстройств, бредовых расстройств и психоза, вызванного лекарственными средствами; тревожных расстройств, двигательных расстройств, расстройств настроения, нейродегенеративных расстройств,ожирения и лекарственной зависимости, содержащую такое количество соединения формулы I, которое эффективно для лечения указанного расстройства или состояния. Настоящее изобретение также обеспечивает способ лечения расстройства, выбираемого из психотических расстройств, бредовых расстройств и психоза, вызванного лекарственными средствами; тревожных расстройств, двигательных расстройств, ожирения, расстройств настроения и нейродегенеративных расстройств, содержащий введение такого количества соединения формулы I, которое эффективно для лечения указанного расстройства. Настоящее изобретение также обеспечивает способ лечения расстройств, выбираемых из группы,состоящей из деменции, болезни Альцгеймера, мультиинфарктной деменции, алкогольной деменции или другой деменции, связанной с лекарственными средствами, из деменции, связанной с внутричерепными опухолями или черепно-мозговой травмой, деменции, связанной с болезнью Хантингтона или с болезнью Паркинсона, или деменции, связанной со СПИДом; бредового расстройства; амнестического расстройства; расстройства, вызванного посттравматическим стрессом; задержки умственного развития; расстройства обучения, например расстройства чтения, расстройства счета или расстройства письменного выражения; синдрома нарушенного внимания/гиперактивности; возрастного снижения когнитивных функций, из большого депрессивного эпизода легкого, среднего или тяжелого типа; маниакального или смешанного эпизода настроения; гипоманического эпизода настроения; депрессивного эпизода с атипичными признаками; депрессивного эпизода с меланхолическими признаками; депрессивного эпизода с кататоническими признаками; эпизода настроения с началом в послеродовом периоде; из депрессии после инсульта; большого депрессивного расстройства; дистимического расстройства; малого депрессивного расстройства; предменструального дисфорического расстройства; постпсихотического депрессивного расстройства или шизофрении; большого депрессивного расстройства, накладывающегося на психотическое расстройство, содержащее бредовое расстройство или шизофрению; биполярного расстройства, содержащего биполярное расстройство I типа, биполярное расстройство II типа, циклотимическое расстройство, болезнь Паркинсона, болезнь Хантингтона; деменцию, болезнь Альцгеймера, мультиинфарктную деменцию, деменцию, связанную со СПИДом, фронтотемпоральную деменцию; нейродегенерации, связанной с черепно-мозговой травмой; нейродегенерации, связанной с инсультом; нейродегенерации, связанной с инфарктом мозга; нейродегенерации, вызванной гипогликемией; нейродегенерации,связанной с эпилептическим припадком; нейродегенерации, связанной с нейротоксическим воздействием; мультисистемной атрофии параноидального, дезорганизованного, кататонического, недифференцированного или остаточного типа; шизофрениформного расстройства; шизоаффективного расстройства бредового типа или депрессивного типа; бредового расстройства; психотического расстройства, вызванного веществами, психоза, вызванного алкоголем, амфетамином, каннабисом, кокаином, галлюциногенами, ожирением, летучими препаратами, опиатами или фенциклидином; расстройства личности параноидального типа; и расстройства личности шизоидного типа, содержащий введение такого количества соединения формулы I, которое эффективно при указанных расстройствах. Настоящее изобретение также обеспечивает способ лечения психотических расстройств, бредовых расстройств и психоза, вызванного лекарственнымт средствами; тревожных расстройств, двигательных расстройств, расстройств настроения, нейродегенеративных расстройств, ожирения и лекарственной зависимости, содержащий введение такого количества соединения формулы I, которое эффективно ингибирует PDE10. Термин "алкил", используемый в настоящем изобретении, если иначе не обозначено, включает насыщенные одновалентные углеводородные радикалы, имеющие прямые или разветвленные функцио- 10012211 нальные группы. Примеры алкилированных групп включают метил, этил, пропил, изопропил и трет-бутил, но не ограничены вышеперечисленным. Термин "алкенил", используемый в настоящем изобретении, если иначе не обозначено, включает алкильные функциональные группы, имеющие по меньшей мере одну двойную связь углерод-углерод, в которых алкил определен согласно вышеуказанному. Примеры алкенила включают, без ограничения,этенил и пропенил. Термин "алкинил", используемый в настоящем изобретении, если не обозначено иначе, включает алкильные функциональные группы, имеющие по меньшей мере одну тройную связь углерод-углерод, в которой алкил определен согласно вышеуказанному. Примеры алкинильных групп включают, без ограничения, этинил и 2-пропинил. Термин "алкокси", используемый в настоящем изобретении, если не обозначено иначе, используемый здесь единственный или в качестве части другой группы, относится к алкилу, к группам, связанным с атомом кислорода. Термин "алкилтио", используемый в настоящем изобретении, если иначе не обозначено, используемый здесь единственный или в качестве части другой группы, включает любую из вышеупомянутых алкильных групп, связанных посредством атома серы. Термин "галоген" или "гало", используемый в настоящем изобретении единственный или в качестве части другой группы, относится к хлору, брому, фтору и иоду. Термин "галогеналкил", используемый в настоящем изобретении, если не обозначено иначе, относится по меньшей мере к одной галогенгруппе, связанной с алкильной группой. Примеры галогеналкильных групп включают, без ограничения, трифторметильные, трифторэтильные, дифторметильные и фторметильные группы. Термин "циклоалкил", используемый в настоящем изобретении, если не обозначено иначе, включает неароматические насыщенные циклические алкильные функциональные группы, в которых алкил определен согласно вышеуказанному. Примеры циклоалкилов включают, без ограничения, циклопропил,циклобутил, циклопентил, циклогексил и циклогептил. Термин "арил", используемый в настоящем изобретении, если не обозначено иначе, включает органический радикал, такой как фенил, нафтил, инденил и флуоренил, получаемый путем удаления одного атома водорода из ароматического углеводорода. Термин "арил" охватывает соединенные кольцевые группы, в которых по меньшей мере одно кольцо является ароматическим. Термины "гетероциклический", "гетероциклоалкил" и подобные термины, используемые в настоящем изобретении, относятся к неароматическим циклическим группам, содержащим 1 или несколько гетероатомов, предпочтительно от 1 до 4 гетероатомов, каждый из которых предпочтительно выбирают из атомов кислорода, серы и азота. Гетероциклические группы настоящего изобретения также могут включать кольцевые системы, замещенные одной или несколькими функциональными оксогруппами. Примерами неароматических гетероциклических групп являются азиридинил, азетидинил, пирролидинил, пиперидинил, азепинил, пиперазинил, 1,2,3,6-тетрагидропиридинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, морфолино, тиоморфолино, тиоксанил, пирролинил, индолинил, 2 Н-пиранил, 4 Н-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3,1,0]гексанил, 3-азабицикло[4,1,0]гептанил, хинолизинил, хинуклидинил, 1,4-диоксаспиро[4,5]децил, 1,4-диоксаспиро[4,4]нонил, 1,4-диоксаспиро[4,3]октил и 1,4-диоксаспиро[4,2]гептил. Термин "гетероарил", используемый в настоящем изобретении, относится к ароматическим группам,содержащим 1 или несколько гетероатомов (предпочтительно кислород, серу и азот), предпочтительно от 1 до 4 гетероатомов. Мультициклическая группа, содержащая 1 или несколько гетероатомов, в которых по меньшей мере одно кольцо из группы является ароматическим, представляет собой "гетероарильную" группу. Гетероарильные группы настоящего изобретения могут также включать кольцевые системы, замещенные одной или несколькими функциональными оксогруппами. Гетероарильные группы, содержащие третичный азот, также можно дополнительно замещать кислородом (т.е. N-оксидом). Примерами гетероарильных групп являются пиридинил, пиридазинил, имидазолил, пиримидинил, пиразолил,триазолил, пиразинил, хинолил, изохинолил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, триазинил, изоиндолил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотриазолил, бензотиазолил, бензоксазолил, квиназолинил, квиноксалинил,нафтиридинил, дигидрохинолил, тетрагидрохинолил, дигидроизохинолил, тетрагидроизохинолил, бензофурил, фуропиридинил, пиролопиримидинил и азаиндолил. Для ясности, термин "гетероарил" в формулеI включает гетероарильную структуру в заместителе Z (т.е. гетероарильную структуру, содержащую Y). Если не обозначено иначе, термин "один или несколько" заместителей или "по меньшей мере один" заместитель, используемый в настоящем изобретении, относится к числу заместителей от одного до максимально возможного числа заместителей, на основании числа доступных для связи положений. Если не обозначено иначе, все полученные из углеводородов вышеупомянутые группы могут иметь примерно от 1 до примерно 20 атомов углерода (например, C1-C20 алкил, С 2-С 20 алкенил, С 3-С 20 циклоалкил, 3-20-членный гетероциклоалкил; С 6-С 20 арил, 5-20-членный гетероарил и т.д.), или примерно от 1 до- 11012211 15 атомов углерода (например, C1-C15 алкил, C2-C15 алкенил, C3-C15 циклоалкил, 3-15-членный гетероциклоалкил, C5-C15 арил, 5-15-членный гетероарил и т.д.), или примерно от 1 до 12 атомов углерода, или примерно от 1 до 8 атомов углерода, или примерно от 1 до 6 атомов углерода."Нейротоксическая интоксикация" относится к интоксикации, вызванной нейротоксином. Нейротоксином является любой химикат или вещество, которые могут вызывать гибель нейронов и, таким образом, неврологическое повреждение. Примером нейротоксина является алкоголь, который, в случае злоупотребления его беременной женщиной, может приводить к алкогольной интоксикации и неврологическому повреждению, называемому фетальным алкогольным синдромом новорожденных. Другие примеры нейротоксинов включают, без ограничения, каиновую кислоту, домоевую кислоту и акромелевую кислоту; некоторые пестициды, такие как дихлордифенилтрихлорэтан (ДДТ); некоторые инсектициды,такие как фосфорорганические вещества; летучие органические растворители, такие как гексауглеводороды (например, толуол); тяжелые металлы (например, свинец, ртуть, мышьяк и фосфор); алюминий; некоторые химикаты, используемые в качестве оружия, такие как "агент оранжевый" и "нервный газ"; и нейротоксические противоопухолевые вещества. Используемый в настоящем изобретении термин "селективный ингибитор PDE10" относится к веществу, например к органической молекуле, которая эффективно ингибирует фермент семейства PDE10,в большей степени, чем ферменты семейств PDE 1-9 или семейства PDE11. В одном варианте осуществления селективный ингибитор PDE10 представляет собой вещество, например органическую молекулу,имеющую коэффициент ингибирования PDE10 Ki, составляющий от около 1/10 или меньше от Ki вещества, которое ингибирует любой другой фермент PDE. Другими словами, вещество в той же степени ингибирует активность PDE10 при концентрации от около 1/10 или меньше от концентрации, необходимой для любого другого фермента PDE. В общем, считается, что, если вещество обладает Ki, меньшим или около 10 мкМ, предпочтительно меньшим или около 0,1 мкМ, оно эффективно ингибирует активность PDE10. Например, можно идентифицировать "селективный ингибитор PDE10" путем сравнения способности вещества ингибировать активность PDE10 с его способностью ингибировать ферменты PDE из других семейств PDE. Например, можно исследовать вещество на его способность ингибировать активностьPDE10, а также активность PDE1A, PDE1B, PDE1C, PDE2, PDE3A, PDE3B, PDE4A, PDE4B, PDE4C,PDE4D, PDE5, PDE6, PDE7, PDE8, PDE9 и PDE11. Термин "лечение", употребляемый в выражении "способ лечения расстройства", относится к изменению, облегчению или торможению прогрессирования расстройства, к которому применяют такой термин, или к одному или нескольким симптомам этого расстройства. Этот термин, используемый в настоящем изобретении, в зависимости от состояния пациента, также охватывает предотвращение расстройства, включая предотвращение начала расстройства или любых связанных с ним симптомов, а также уменьшение тяжести расстройства или любого из его симптомов до их возникновения. Используемый в настоящем изобретении термин "лечение" также относится к предотвращению рецидива расстройства. Например, используемое в настоящем изобретении выражение "лечение шизофрении или шизофрениформного или шизоаффективного расстройства" также охватывает лечение одного или нескольких симптомов (позитивных, негативных и других связанных с ним признаков) указанных нарушений, например для лечения бреда и/или галлюцинаций, связанных с этим расстройством. Другие примеры симптомов шизофрении и шизофрениформного и шизоаффективного расстройства включают дезорганизованную речь, эмоциональную тупость, афазию, ангедонию, неадекватный аффект, дисфорическое настроение(например, в форме депрессии, тревожности или гнева) и некоторые признаки когнитивной дисфункции. Термин "млекопитающее", используемый в настоящем изобретении, относится к любому члену класса "млекопитающие", включающего, без ограничения, людей, собак и кошек. Соединение настоящего изобретения можно вводить или единственным, или в комбинации с фармацевтически приемлемыми носителями, как в однократной дозе, так и в многократных дозах. Подходящие фармацевтические носители включают инертные твердые разбавители или наполнители, стерильные водные растворы и различные органические растворители. Образованные таким образом фармацевтические композиции затем можно легко вводить в разнообразных лекарственных формах, таких как таблетки, порошки, пастилки, в виде жидких препаратов, сиропов, растворов для инъекций и т.п. Эти фармацевтические композиции необязательно могут содержать дополнительные компоненты, такие как ароматизаторы, связующие вещества, наполнители и т.п. Таким образом, можно создавать рецептуру соединения настоящего изобретения для перорального, буккального, интраназального, парентерального (например, внутривенного, внутримышечного или подкожного), трансдермального (например, пластырь) или ректального введения или в форме, подходящей для введения путем ингаляции или инсуффляции. Фармацевтические композиции для перорального введения могут быть в виде, например, таблеток или капсул, изготовленных общепринятыми способами, с фармацевтически приемлемыми наполнителями, такими как связующие вещества (например, преварительно желатинизированный кукурузный крахмал, поливинилпирролидон или гидроксипропилметилцеллюлоза); наполнители (например, лактоза,микрокристаллическиая целлюлоза или фосфат кальция); лубриканты (например, стеарат магния, тальк или оксид кремния); дезинтегрирующие вещества (например, картофельный крахмал или натрия крахмал- 12012211 гликолят); или увлажняющие вещества (например, лаурил сульфат натрия). Таблетки могут быть покрыты способами, известными в данной области. Жидкие препараты для перорального введения могут быть в виде, например, растворов, сиропов или суспензии, или они могут быть представлены в виде сухого продукта для разбавления водой или другим подходящим носителем перед применением. Такие жидкие препараты можно изготовить общепринятыми способами с фармацевтически приемлемыми добавками,такими как суспендирующие вещества (например, сироп сорбитола, метилцеллюлоза или гидрогенизированные съедобные жиры); эмульгирующие вещества (например, лецитин или гуммиарабик); неводные носители (например, миндальное масло, масляные сложные эфиры или этиловый спирт) и консерванты(например, метил- или пропил-п-гидроксибензоаты или сорбиновая кислота). Композиция для буккального введения может быть в виде таблеток или пастилок, рецептура которых создается общепринятым способом. Можно создавать рецептуру соединений настоящего изобретения для парентерального введения путем инъекции, которое включает применение общепринятых технологий катетеризации или вливания. Рецептуры для инъекции могут быть представлены в монолитной лекарственной форме, например в ампулах или в мультидозных контейнерах, с добавлением консерванта. Они могут быть в виде таких форм,как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать рецептурообразующие вещества, такие как суспендирующие вещества, стабилизаторы и/или диспергирующие вещества. Альтернативно, активный компонент может находиться в виде порошка для восстановления подходящим носителем, например стерильной апирогенной водой, перед применением. Если необходим раствор продукта, его может делать путем растворения выделенного комплекса включения в воде (или в другой водной среде) в количестве, достаточном для образования раствора требуемой силы для перорального введения или парентерального введения пациентам. Можно создавать рецептуру соединений для лекарственных форм с быстрым диспергированием (fddf), которые предназначены для высвобождения активного компонента в ротовой полости. Их рецептура часто создается с использованием быстрорастворимых матриц на желатиновой основе. Эти лекарственные формы являются известными и могут применяться для доставки широкого диапазона лекарственных средств. В большинстве лекарственных форм с быстрым диспергированием желатин используют в качестве носителя или структурообразующего вещества. Обычно желатин используют для придания лекарственной форме достаточной прочности, чтобы предотвратить ломкость во время извлечения из упаковки, но при попадании в рот желатин дает возможность лекарственной форме немедленно растворяться. Альтернативно, для того же эффекта используют различные крахмалы. Рецептуру соединений настоящего изобретения можно создавать в ректальных композициях, таких как суппозитории или в виде удерживающих клизм, например, содержащих общепринятые основы для суппозиториев, такие как масло какао или другие глицериды. Соединение настоящего изобретения для интраназального введения или введения путем ингаляции удобно доставляют в виде раствора или суспензии из контейнера с пульверизатором, которые выдавливаются или нагнетаются пациентом, или представляет собой распыляемый аэрозоль из герметичного контейнера или распылителя, с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, углекислого газа или другого подходящего газа. В случае герметичного аэрозоля единицу дозы можно определять путем обеспечения клапана для доставки дозированного количества. Герметичный контейнер или распылитель могут содержать раствор или суспензию активного соединения. Можно создавать капсулы и картриджи (например, сделанные из желатина) для использования в ингаляторе или инсуффляторе, содержащие порошковую смесь соединения настоящего изобретения и подходящей порошковой основы, такой как лактоза или крахмал. Аэрозольные рецептуры для лечения вышеупомянутых состояний (например, мигрени) у среднего взрослого человека предпочтительно устроены таким образом, чтобы каждая регулируемая доза или "нажатие" аэрозоля содержала от около 20 до около 1000 мг соединения настоящего изобретения. Общая ежедневная доза с аэрозолем будет составлять в диапазоне от около 100 до около 10 мг. Введение можно осуществлять несколько раз в день, например 2, 3, 4 или 8 раз, выдавая каждый раз, например, 1, 2 или 3 дозы. Предлагаемая ежедневная доза соединения настоящего изобретения для перорального, парентерального, ректального или буккального введения у среднего взрослого человека для лечения вышеупомянутых состояний составляет от около 0,01 до около 2000 мг, предпочтительно от около 0,1 до около 200 мг активного компонента формулы I на единицу дозы, которую можно вводить, например, от 1 до 4 раз в день. Способы исследований позволяют отбирать вещество для ингибирования гидролиза циклических нуклеотидов PDE10 и PDE из других генетических семейств. Концентрация субстрата циклических нуклеотидов, используемая в исследованиях, составляет 1/3 от концентрации Km, что позволяет сравнивать значения IC50 у всех разных ферментов. Активность PDE измеряют, используя способ на основе анализа сцинтилляционного родства (SPA), как описано ранее (Fawcett et al., 2000). Эффект ингибиторов PDE выявляют путем анализа установленного количества фермента (PDE 1-11) в присутствии варьируемых концентраций вещества и низкой концентрации субстрата, таким образом, что значение IC50 приближается к значению Ki (не меченные изотопно цГМФ или цАМФ в соотношении 3:1 к меченным [3 Н] в концентрации 1/3 Km). Окончательный исследуемый объем доводится до 100 мкл тестовым буфером (50 мм- 13012211 Трис-HCl с уровнем рН 7,5, MgCl2 в количестве 8,3 ммоль, 1 мг/мл бычьего сывороточного альбумина). Реакции начинают с ферментом, инкубируемым в течение от 30 до 60 мин при температуре 30 С, для получения 30% круговорота субстрата и завершают SPA с гранулами силиката иттрия объемом 50 мкл(Amersham) (содержащие 3 ммоль соответствующего немеченого циклического нуклеотида для PDE 9 и 11). Планшеты вновь герметизируют и встряхивают в течение 20 мин, после чего позволяют осаждение гранул в течение 30 мин в темноте и затем производят подсчет на спектрофотометре TopCount для прочтения планшетов (Packard, Meriden, CT). Единицы радиоактивности можно конвертировать в процент активности от контроля без ингибирования (100%), рассчитываемого от концентрации ингибитора, и значения IC50 ингибитора можно получать с использованием расширения Fit Curve Microsoft Excel. Применяя указанное исследование, у соединений настоящего изобретения было определено значение IC50 для ингибирования активности PDE10, составляющее менее чем около 10 мкмоль. Настоящее изобретение также относится к получению соединений формулы I. Настоящее изобретение также обеспечивает способы синтеза соединений формулы I. Например, настоящее изобретение обеспечивает способ образования соединения формулы I, содержащий стадию взаимодействия с соединением формулы II с диметоксиметил-диметиламином и гидразином или замещенным гидразином (например, таким как R20-NHNH2, где R20 является алкилом). Настоящее изобретение также обеспечивает способ образования соединения формулы I, содержащий стадию взаимодействия с соединением формулы III с диметилоксалатом и гидразином формулы HET2-NHNH2. Настоящее изобретение также обеспечивает способ образования соединения формулы I, содержащий стадию взаимодействия с соединением формулы IV с диметоксиметилдиметиламином и гидразином или замещенным гидразином. Настоящее изобретение также обеспечивает способ образования соединения формулы I, содержащий стадию взаимодействия соединения формулы V в котором Q является гидроксилом или галидом.- 14012211 Подробное описание изобретения На схеме 1 показано получение соединений класса пиразолов по настоящему изобретению. Алкилирование замещенного фенола с 2-метилхлорхинолином обеспечивает получение желательного эфира. Гидролиз сложного эфира и обработка тионилхлоридом обеспечивают получение желательного хлорангидрида. Добавление гидрохлорида О,N-диметилгидроксиламина обеспечивает амид Вайнреба (Weinreb) для связывания (Weinreb et al., Tet Lett., 1981, 22 (39) 3815). Образование аниона с 4-пиколином и диизопропиламидом лития (LDA) с последующим добавлением амида Вайнреба дает кетон. Затем кетон можно обрабатывать диметоксиметилдиметиламином кипячением с обратным холодильником для образования промежуточного енаминона. Обработка разными гидразинами дает аналоги пиразола. Были получены различные соотношения этих двух изомеров. Эти изомеры разделяли посредством кристаллизации, Biotage MPLC,препаративной тонкослойной хроматографии ТСХ или препаративной ВЭЖХ. Эта схема реакции является общей для различных исходных замещенных фенолов, замещенных хинолинов и замещенных гидразинов. Схема 1 Альтернативно, соединения замещенного пиразола можно получать путем алкилирования NH пиразола. Одним из наборов условий является применение карбоната цезия в качестве основы с галоидалкилом в качестве электрофильного компонента в растворителе, таком как диметилформамид. Для некоторых реакций необходимо нагревание. Схема 2 Как показано в схеме 3, из промежуточного енаминона можно получать множество гетероциклических соединений. Пиримидины можно получать путем нагревания с замещенными формамидами в присутствии этанола и этоксида натрия. Изоксазолы получают нагреванием енаминона с гидроксиламином в метано- 15012211 ле/уксусной кислоте. В случае изоксазола образуется только один изомер. Путем нагревания с аминопиролами, аминоимидазолами или аминотриазолами можно образовывать 6-5 бициклическую систему. Схема 3 Множество 4-пиридилгетероциклических заместителей можно получать согласно схеме 4. Для получения желательного кетона метилгетероциклические соединения, такие как 3,5-диметилизоксазол и метилпиридазин, можно депротонировать диизопропиламидом лития и добавлять к амиду Вайнреба(Weinreb et al., Tet. Lett., 1981, 22 (39) 3815). Последущая обработка диметоксиметилдиметиламином и гидразином обеспечивает гетероциклические пиразолы. Пиримидины и изоксазолы также можно получать согласно описанию схемы 3. Схема 4N-Пиридилпиразолы можно получать согласно схеме 5. Исходные кетоны приготовлены алкилированием фенола, как показано на схеме 1. Обработка кетона с диметоксиметилдиметиламином с последующим добавлением 4-пиридилгидразина (см. J. Med. Chem. 2002, 45 (24) 5397) обеспечивает желательные соединения. Другие гетероциклические заместители для 4-пиридила можно получать, используя необходимый гидразин. Схема 5 Как показано нв схеме 6, 3-замещенные-N-пиридилпиразолы можно получать способами, описанными в литературе, (см. J. Med. Chem. 2004, 47, 2180). Обработка ацетофенона (полученного согласно схеме 1) метоксидом натрия и диметилоксалатом обеспечивает промежуточный сложный эфир. Добавление 4-пиридилгидразина (см. J. Med. Chem. 2002, 45 (24) 5397) обеспечивает пиразол сложным эфиром в 3-положении. Этот сложный эфир можно превращать в амиды путем гидролиза и связывания с аминами. Его можно превращать в эфиры восстановлением до спирта и алкилированием. Образование амина возможно путем образования амида, с последующим восстановлением или превращением в альдегид, с последующим восстановительным аминированием. Специалист в области органической химии может осуществлять все указанные превращения. Схема 6 Промежуточные бензилы можно получать способом, показанным на схеме 1. Бензиловый эфир можно удалять посредством обработки водородным газом с палладиевым катализатором, таким как палладий на угле или гидроксид палладия в ряде растворителей. Затем фенол можноалкилировать с использованием бензилхлорида в ацетоне, с нагреванием с карбонатом калия. Также для соединения фенола со спиртами можно применять химическую реакцию Мицунобу (Mitsunobu) (Hughes, D.L., The Mitsunobu Многие бензилгалогениды или спирты являются коммерчески доступными или известны в литературе. Общепринятые способы получения этих промежуточных продуктов специалистами в данной области представляют собой восстановление сложного эфира, кислоты или альдегида до образования спирта. Одной из общих методик является окисление положения бензила диоксидом селена для получения альдегида, который в последующем восстанавливают борогидридом натрия. Бензилгалогенид можно образовывать посредством галогенирования (см. Syn. Comm. 1995, 25 (21) 3427-3434). Схема 8 Аналоги триазола можно получать множеством способов. Один из путей показан на схеме 9. Обработка гидразида диметилформамиддиметилацеталем для образования промежуточного продукта, который впоследствии обрабатывают амином или анилином при нагревании и с добавлением уксусной кислоты, приводит к образованию 1,2,4-триазолов (см. Org. Lett, 2004, 6 (17), 2969-2971). Региоизомерные триазолы можно получать взаимным замещением функциональных групп исходных веществ. Схема 9 Другие триазольные изомеры можно получать согласно схеме 10, начиная с карбоксиамидов, и обработкой диметилформамиддиметилацеталем с последующим добавлением ароматических гидразинов. Региоизомерные триазолы можно получать взаимным замещением функциональных групп исходных веществ. Инвертированный изомер кетона можно получать согласно схеме 11. (Bunting et al., JACS, 1988,110, 4008). Исходный альдегид связан с фосфонатом для получения енанаминона. Енаминон гидролизуют для получения желательного кетона. Затем кетон можно использовать согласно схемам 1, 2 и 3 для получения желательных соединений. Схема 11 На схеме 12 показан способ синтеза 4,5-диарилоксазола. Как показано в этом примере, 4-бензилоксибензальдегид и 4-метилбензолсульфиновую кислоту нагревали с формамидом для получения замещенного формамида. Это взаимодействие известно в литературе (J. Med Chera., 2002, 45, 1697). Дегидратация формамида в реакции, опосредованной POCl3, дает изоцианат тозилметила. Этот класс соединения можно обрабатывать альдегидом и основанием для получения оксазола. В показанном случае тозилметилизоцианат обрабатывают изоникотинальдегидом и карбонатом калия. Продуктом этой реакции является оксазол, обладающий 4-бензилоксифенильной группой в 4-положении оксазольного кольца и заместителем 4-пиридил в 5-положении. Эти заместители можно просто замещать другими арильными группами путем применения различных арилальдегидов для первой и третьей стадий из последовательности. Расщепление бензилоксигруппы достигают стандартным способом каталитического гидрирования, и получаемый фенол легко алкилируется путем обработки галоидалкилом, таким как 2-(хлорметил)хинолин, и фторидом цезия в ДМФ. Способ не ограничен показанным случаем, поскольку относительные положения фенильных и пиридильных колец могут сдвигаться и указанные кольца могут содержать множество арильных групп, показывающих различные схемы замещения. Схема 12- 19012211 На схеме 13 показан способ получения 4,5-замещенных оксазолов, обладающих замещением алкильной группой во 2-положении оксазольного кольца. В показанном случае 1-(4-бензилоксифенил)-2 пиридин-4-илэтанон бромируют путем обработки бромом в уксусной кислоте согласно общепринятым способам. Получаемый -бромкетон затем обрабатывают ацетатом аммония и ацетатом натрия в уксусной кислоте, что приводит к получению метилзамещенного оксазольного кольца, согласно раскрытию в патентной литературе (WO 9513067). Метильную группу можно замещать другими алкильными группами. Например, замещение этаноата аммония, этаноата натрия и этаноевой кислоты будет приводить к замещению этильной группы. Расщепление бензилоксигруппы достигают стандартным способом каталитического гидрирования, и получаемый фенол легко алкилируется обработкой галоидалкилом, как описано выше. Способ не ограничен показанным случаем, поскольку относительные положения фенильного и пиридильного колец могут сдвигаться и указанные кольца могут содержать множество арильных групп, показывающих различные схемы замещения. Схема 13 Стадией 1 из схемы 14 является образование имина/образование гетероциклического соединения. Соединение формулы 2 А, в которой R1 является алкилом, бензилом или аллилом, конденсируют с 4-пиридинкарбоксальдегидом в растворителе, таком как толуол, и нагревают с обратным холодильником аппаратом Dean-Stark, применяемым для удаления воды, в течение примерно 40 ч. После удаления толуола неочищенный имин смешивали с тозилметилизоцианидом и основанием, таким как карбонат калия, в смеси растворителей 1,2-диметоксиэтана и метанола и нагревали с обратным холодильником в течение примерно 3 ч, чтобы получить соединение 3 А. Стадией 2 из схемы 14 является деалкилирование фенола. Если R1 является метилом, для получения 4 А деалкилирование можно проводить с трибромидом бора (BBr3) в некоординированном растворителе, таком как метиленхлорид, при температуре примерно от 20 до 40 С в течение около от 3 до 48 ч,где предпочтительным является примерно 24 ч. Если R2 является бензилом, для получения 4 А деалкилирование можно проводить в неразбавленной трифторацетиловой кислоте с анизолом при температуре примерно 75 С в течение примерно 3-48 ч, где предпочтительным является примерно 24 ч. Если R1 является аллилом, для получения 4 А деалкилирование можно проводить с палладиевым катализатором, таким как бис(трифенилфосфин)дихлорпалладия, палладия ацетат, где предпочтительным является бис(трифенилфосфин)дихлорпалладия, с восстановителем, таким как н-бутиламмония формиат, в растворителе, таком как тетрагидрофуран, 1,2-дихлорэтан, метиленхлорид илиалканол, где предпочтительным является 1,2-дихлорэтан, в диапазоне температур от примерно 20 до 75 С. Стадией 3 из схемы 14 является алкилирование фенола. Обработка 4 А основанием, таким как карбонат калия, карбонат натрия, карбонат цезия, гидрид натрия или гидрид калия, где предпочтительным является карбонат цезия или гидрид натрия, в растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан, N,N-диметилформамид, диметилацетамид, N-метилпирролидинон или диметилсульфоксид, где предпочтительными являются диметилсульфоксид или N,N-диметилформамид, при температуре от примерно 20 до 70 С, где предпочтительной является температура примерно 23 С, в течение примерно 3-48 ч,где предпочтительным является время примерно 24 ч, приводит к образованию соединения 1 А. Стадией 4 из схемы 14 является депротонирование имидазола/электрофильный захват. Обработка 3 А основанием, таким как диизопропиламид лития или 2,2,6,6-тетраметилпиперидин лития, где предпочтительным является диизопропиламид лития, в растворителе, таком как тетрагидрофуран, при температуре от примерно -78 до 0 С, где предпочтительной является температура примерно -20 С, в течение от примерно 5 до 30 мин, где примерно 10 мин предпочтительны, с последующим добавлением желательного электрофильного R3-I, приводит к образованию соединения 3 В.- 20012211 Стадией 5 из схемы 14 является деалкилирование фенола, и для получения 4 В в нем используют те же способы, описанные выше для стадии 2. Стадией 6 из схемы 14 является алкилирование фенола, и для получения 1 В в нем используют те же способы, описанные выше для стадии 3. Схема 14 Стадией 1 из схемы 15 является ацилирование амина для образования амида. Соединение 2 А, в котором R1 может быть метилом, бензилом или аллилом, для получения 5 А обрабатывают хлорангидридом или карбоксильной кислотой в присутствии связующего реактива, такого как три-н-пропилфосфиновый ангидрид или дициклогексил карбодиимид, где предпочтительным является три-н-пропилфосфиновый ангидрид, в присутствии основания, такого как гидроксид натрия, калия или карбонат натрия, триэтиламин или диизопропилэтиламин, где предпочтительным является диизопропилэтиламин, в системе растворителей, такой как вода/метиленхлорид, вода/этилацетат, этилацетат, тетрагидрофуран или метиленхлорид, где предпочтительным является этилацетат, при температуре от примерно 0 до 50 С, где предпочтительной является температура от примерно 20 до 30 С. Стадия 2 состоит из хлорирования для образования иминохлорида, реакции с амином для образования амидина, с последующей обработкой кислотой для образования имидазола. Соединение 5 А обрабатывают хлорирующим агентом, таким как PCl5/POCl3, при температуре примерно 120 С в течение примерно 4 ч. Хлорирующий агент удаляли в вакууме, и добавляли избыток 1,1-диэтокси-2-этиламина в растворителе, таком как изопропанол, и смесь перемешивали в течение примерно 5-24 ч при температуре примерно 23 С. Для получения соединения 6 А растворитель удаляли в вакууме, и добавляли концентрированную соляную кислоту и изопропанол, и в течение примерно 24 ч смесь нагревали до температуры примерно 90 С. Стадией 3 из схемы 15 является деалкилирование фенола. Если R1 является метилом, для получения 7 А можно проводить деалкилирование с трибромидом бора (BBr3) в некоординированном растворителе, таком как метиленхлорид, при температуре примерно 20-40 С в течение примерно 3-48 ч, где предпочтительным является примерно 24 ч. Если R2 является бензилом, для получения соединения 7 А можно проводить деалкилирование в неразбавленной трифторацетиловой кислоте с анизолом при температуре примерно 75 С в течение примерно 3-48 ч, где предпочтительным является примерно 24 ч. Если R1 является аллилом, для получения 7 А можно проводить деалкилирование с палладиевым катализатором, таким как дихлорпалладий бис(трифенилфосфин) ацетата палладия, где предпочтительным является дихлорпалладий бис(трифенилфосфин), с восстановителем, таким как формиат н-бутиламмония, в растворителе, таком как тетрагидрофуран, 1,2-дихлорэтан, метиленхлорид или алканол, где предпочтительным является 1,2-дихлорэтан, в диапазоне температур от примерно 20 до 75 С. Стадией 4 из схемы 15 является алкилирование фенола. Обработка 7 А основанием, таким как карбонат калия, карбонат натрия, карбонат цезия, гидрид натрия или гидрид калия, где предпочтительным является карбонат цезия, в растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан, N,N-диметилформамид, диметилацетамид, N-метилпирролидинон или диметилсульфоксид, где предпочтительным является диметилсульфоксид, при температуре от примерно 20 до 70 С, где предпочтительной является температура примерно 23 С, в течение примерно 3-48 ч, где предпочтительным является примерно 24 ч,приводит к образованию соединения 1 С. Бензальдегид хинолила можно соединять с кетоном в присутствии пиперидина нагреванием с обратным холодильником для получения желательного олефина. Обработка гидразином дает NH-пиразол. Дополнительно для получения замещенных пиразолов его можно вырабатывать обработкой гидридом натрия и электрофильным соединением, таким как метилйодид. Схема 16 Как показано в схеме 17, алкин и йодид могут быть соединены посредством связи Сонагашира(Sonagashira) и метиловый эфир становится незащищенным с трибромидом бора в дихлорметане. Алкилирование фенола с 2-хлорметилхинолином согласно вышеописанным способам обеспечивает предпоследний промежуточный продукт. Обработка триметилсилилазидом в запаянной трубке при 70-190 С,предпочтительно примерно 150 С, в течение 24-72 ч, обеспечивает желательный триазол. Схема 17 Общая схема эксперимента Органические растворы высушивали с магнием или сульфатом натрия, если не указано иначе. Комнатная температура сокращенно обозначается КТ. Система для высокоэффективной жидкостной хроматографии-масс спектрометрии ВЭЖХ-МС, система 1, состояла из колонки Zorbax Bonus-RP 4,6150 мм,1,0 мл/мин, растворителем А являлся MeCN, растворителем В являлся 0,1% водный раствор муравьиной кислоты, линейный градиент составлял от 1:9 А:В до 95:5 А:В в течение 10 мин, и применялась система ВЭЖХ Hewlett Packard 1100, снабженная диодной матрицей и масс-детекторами. В системе ВЭЖХ 2 применяли линейный градиент от 3:7 А:В до 95/5 А:В в течение 15 мин. Для индикации очистки ОФВЭЖХ применяли оборудование для препаративной ВЭЖХ Shimadzu, снабженное колонкой Х-Terra 5050 мм, растворителем А являлся ацетонитрил, растворителем В являлась вода, каждый из растворителей содержал или 0,1% трифторацетиловую кислоту ("кислые условия"), или 0,1% концентрированного гидроксида аммония ("щелочные условия"), линейный градиент составлял от 25 до 85% А:В в течение 10 мин.- 22012211 Далее настоящее изобретение иллюстрируют следующие примеры. Вместе с тем, необходимо понимать, что настоящее изобретение, приводимое полностью в настоящем описании и показанное в формуле изобретения, не ограничено подробным описанием следующих примеров. Методики эксперимента Получение 1. Метиловый сложный эфир 4-(хинолин-2-илметокси)бензойной кислоты. К раствору 2-хлорметилхинолина (2 г, 9,3 ммоль) в ацетоне (47 мл, 0,2 М) добавляли метиловый сложный эфир 4-гидроксибензойной кислоты (1,42 г, 1,0 экв.) и карбонат калия (3,86 г, 3 экв.). Смесь нагревали до 60 С в течение 16 ч в атмосфере N2, охлаждали до температуры окружающей среды и вливали в 1N гидроксид натрия (50 мл)/этилацетат (100 мл). Слои разделяли и сульфатом магния высушивали органический слой, фильтровали и концентрировали. Для получения указанного соединения в виде белого твердого вещества (1,66 г, 61%) проводили хроматографию Biotage MPLC с использованием колонки 40 М с градиентом от 5 до 30% этилацетата/гексана. 1 Н ЯМР (400 МГц, CDCl3)8,18 (д, J=8,7 Гц, 1 Н), 8,07 (д, J=8,3 Гц, 1 Н), 7,95 (М, 2 Н), 7,82 (д, J=7,9 Гц,1 Н), 7,74 (дт, J=7,1, 1,7 Гц, 1 Н), 7,62 (д, J=8,3 Гц, 1 Н), 7,55 (дт, J=7,9, 1,2 Гц, 1 Н), 7,03 (д, J=9,1, 2 Н), 5,41(с, 2 Н), 3,84 (с, 3 Н); МС: (М+Н m/z=294,2). Получение 2. 4-(Хинолин-2-илметокси)бензойная кислота. К раствору метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты (500 мг, 1,7 ммоль) в тетрагидрофуране (8,5 мл) и метаноле (3 мл) добавляли 1N NaOH (3,4 мл, 2 экв.). Смесь перемешивали при температуре окружающей среды в течение 16 ч. К смеси добавляли 50 мл солевого раствора и уровень рН регулировали 1N HCl до 3, чтобы обеспечить белый осадок, который фильтровали и высушивали для получения указанного соединения в виде белого твердого вещества (463 мг, 98%). 1 Н ЯМР (400 МГц, ДМСО)8,39 (д, J=8,3 Гц, 1 Н), 7,99 (м, 2 Н), 7,81 (М, 2 Н), 7,76 (дт, J=8,3, 1,7 Гц,1 Н), 7,64 (д, J= 8,3 Гц, 1 Н), 7,60 (дт, J=7,9, 1,3 Гц, 1 Н), 7,12 (М, 2 Н), 5,41 (с, 2 Н); МС: (М+Н m/z=280,2). Получение 3. N-Метокси-N-метил-4-(хинолин-2-илметокси)бензамид. К раствору 4-(хинолин-2-илметокси)бензойной кислоты (25,98 г, 93 ммоль) добавляли 250 мл тионилхлорида в атмосфере N2. Смесь перемешивали в течение 3 ч и удаляли избыток тионилхлорида в вакууме. Хлорангидрид растворяли в тетрагидрофуране (450 мл) и медленно добавляли триэтиламин (50 мл,4 экв.). Добавляли O,N-диметилгидроксил амина гидрохлорид (27 г, 3 экв.) и смесь перемешивали в течение 18 ч. Смесь помещали в роторный испаритель для удаления растворителя, разделяли между 1NNaOH и метиленхлоридом, разделяли, высушивали сульфатом магния, фильтровали и концентрировали. Неочищенный продукт фильтровали через силикагель, элюировали 30-70% этилацетатом/гексаном до получения указанного соединения в виде коричневого масла (26,26 г, 87%); 1 Н ЯМР (400 МГц, CDCl3)8,17 (д, J=8,7 Гц, 1 Н), 8,06 (д, J=8,3 Гц, 1 Н), 7,81 (д, J=8,3 Гц, 1 Н), 7,67m/z=323,2). Получение 4. 2-Пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанон. К раствору диизопропиламида лития (1,0 М) в тетрагидрофуране по капле добавляли 4-пиколин(7,55 мл, 5 экв.) при 0 С в атмосфере N2. Через 30 мин анион охлаждали до -78 С. В отдельной круглодонной колбе в тетрагидрофуране (77 мл, 0,2 М) растворяли N-метокси-N-метил-4-(хинолин-2-илметокси)бензамид (5,0, 15,5 ммоль) и охлаждали до -78 С в атмосфере N2. 1,2 экв. аниона 4-пиколина по капле добавляли к раствору амида. Через 45 мин добавляли еще 1 экв. аниона 4-пиколина. После дополнительных 30 мин по капле добавляли уксусную кислоту (40 мл) и медленно повышали температуру реакции до температуры окружающей среды. Твердый продукт (соль ацетата) фильтровали и разделяли между насыщенным бикарбонатом натрия и дихлорметаном. Слои разделяли, высушивали сульфатом магния,фильтровали и концентрировали для получения указанного соединения в виде бурого осадка (4,41 г, 80%). 1 Н ЯМР (400 МГц, CDCl3)8,52 (д, J=5, 8 Гц, 2 Н), 8,19 (д, J=8,7 Гц, 1 Н), 8,07 (д, J=8,7 Гц, 1 Н), 7,93(м, 2 Н), 7,82 (д, J=8,3 Гц, 1 Н), 7,75 (м, 1 Н), 7,61 (д, J=8,3 Гц, 1 Н), 7,54 (дт, J=7,9, 1,0 Гц, 1 Н), 7,23 (м, 2 Н),7,07 (м, 2 Н), 5,42 (с, 2 Н), 4,19 (с, 2 Н); МС: (М+Н m/z=355,2). Получение 5. 3-Диметиламино-2-пиридин-4-ил-1-[4-(хинолин-2-илметскси)фенил]пропенон. К 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанону (4,0 г, 11,3 ммоль) добавляли диметоксиметилдиметиламин (10 мл) и осуществляли нагревание смеси с обратным холодильником в течение 1 ч. Раствор концентрировали для получения на выходе количества указанного соединения, которое использовали как есть на следующей стадии. ЖХ/МС: КТ=1,4 мин; МС: (М+Н m/z=410,2). Пример 1. 2-[-4-(4-Пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. К раствору 3-диметиламино-2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропенона (9,57 г,27 ммоль) в метаноле добавляли гидразингидрат (3,33 г, 40,5 ммоль) и смесь нагревали с обратным холодильником в течение 1 ч. Растворитель выпаривали до получения белого твердого вещества. Твердое вещество промывали водой и этиловым эфиром. Твердое вещество перекристаллизовывали из горячего этанола/этилацетата (10 мл/г) до получения 8,34 г указанного соединения (82%). 1 Н ЯМР (400 МГц, ДМСО)8,41 (м, 3 Н), 8,16 (с, 1 Н), 7,97 (м, 2 Н), 7,86 (с, 1 Н), 7,75 (т, J=7,9 Гц,- 23012211 1 Н), 7,68 (д, J=8,3 Гц, 1 Н), 7,60 (т, J=7,5 Гц, 1 Н), 7,33 (м, 2 Н), 7,18 (м, 2 Н), 7,15 (д, J=8,3 Гц, 1 Н), 7,06 (д,J=8,3 Гц, 1 Н), 5,38 (с, 2 Н); МС: (М+Н m/z=379,2). Пример 2. 2-[4-(2-Метил-4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. К раствору 2-[-4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолина (1,72 г) в этаноле (20 мл) добавляли метилгидразин (3,5 мл, 1,5 экв.) и концентрированную серную кислоту (0,1 мл). Смесь перемешивали в течение 1 ч при температуре окружающей среды и растворитель выпаривали. Смесь разделяли между метиленхлоридом и насыщенным бикарбонатом натрия. Слои разделяли и органический слой высушивали сульфатом магния, фильтровали и концентрировали. Препаративной хроматографией ВЭЖХ получали указанное соединение (минорный изомер) в виде белого твердого вещества (0,30 г, 17%). 1 Н ЯМР (400 МГц, CDCl3)8,31 (д, J=5,4 Гц, 2 Н), 8,21 (д, J=8,7 Гц, 1 Н), 7,80 (д, J=8,3 Гц, 1 Н), 7,77(с, 2 Н), 3,69 (с, 3 Н); МС: (М+Н m/z=393,3). Пример 3. 2-[4-(1-Метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. К раствору 2-[-4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолина (1,72 г) в этаноле (20 мл) добавляли метилгидразин (3,5 мл, 1,5 экв.) и концентрированную серную кислоту (0,1 мл). Смесь перемешивали в течение 1 ч при температуре окружающей среды и растворитель выпаривали. Смесь разделяли между метиленхлоридом и насыщенным бикарбонатом натрия. Слои разделяли и органический слой высушивали сульфатом магния, фильтровали и концентрировали. Препаративной хроматографией ВЭЖХ получали указанное соединение (основной изомер) в виде прозрачного масла (0,97 г, 56%). 1 Н ЯМР (400 МГц, CDCl3)8,44 (д, J=5,0 Гц, 2 Н), 8,17 (д, J=8,7 Гц, 1 Н), 8,05 (д, J=8,3 Гц, 1 Н), 7,81(д, J=5,0 Гц, 2H), 7,00 (д, J=8,7 Гц, 2 Н), 5,38 (с, 2 Н), 3,93 (с, 3 Н); МС: (М+Н m/z=393,3). Пример 4. 2-[4-(2-Этил-4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но замещенного этилгидразином. 1 Н ЯМР (400 МГц, CDCl3)8,35 (уш.с, 2 Н), 8,23 (д, J=8,3 Гц, 1 Н), 8,08 (д, J=8,3 Гц, 1 Н), 7,85 (д,J=7,4 Гц, 1 Н), 7,83 (с, 1 Н), 7,74 (м, 2 Н), 7,57 (т, J=7,9 Гц, 1 Н), 7,21 (д, J=8,7 Гц, 2 Н), 7,14 (д, J=9,1 Гц, 2 Н),7,04 (м, 2 Н), 5,42 (c, 2H), 4,03 (кв., J=7,5 Гц, 2 Н), 1,36 (т, J=7,5 Гц, 3 Н); МС: (М+Н m/z=407,3). Пример 5. 2-[4-(1-Этил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Нпиразол-3-ил)феноксиметил]хинолина, но с замещением этилгидразином. 1 Н ЯМР (400 МГц, CDCl3)8,35 (уш.с, 2 Н), 8,19 (д, J=8,3 Гц, 1 Н), 8,07 (д, J=9,1 Гц, 1 Н), 7,82 (д,J=7,9 Гц, 1 Н), 7,73 (т, J=8,3 Гц, 1 Н), 7,67 (д, J=8,3 Гц, 2 Н), 7,62 (с, 1 Н), 7,55 (т, J=7,9 Гц, 1 Н), 7,37 (д, J=9,1 Гц,2 Н), 7,21 (уш.с, 2 Н), 7,01 (д, J=8,7 Гц, 2 Н), 5,39 (с, 2 Н), 4,24 (кв, J=7,5 Гц, 2 Н), 1,56 (т, J=7,5 Гц, 3 Н); МС:(М+Н m/z=407,3). Пример 6. Диметил-(2-4-пиридин-4-ил-3-[4-(хинолин-2-илметокси)фенил]пиразол-1-илэтил)амин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Нпиразол-3-ил)феноксиметил]хинолина, но с замещением (2-гидразиноэтил)диметиламином. 1 Н ЯМР (400 МГц, CDCl3)8,44 (дд, J=4,6, 1,7, Гц, 2 Н), 8,18 (д, J=8,3 Гц, 1 Н), 8,06 (д, J=8,3 Гц, 1 Н),7,82 (д, J=8,7 Гц, 1 Н), 7,71 (м 2 Н), 7,55 (т, J=7,1 Гц, 1 Н), 7,38 (д, J=8,7 Гц, 2 Н), 7,15 (д, J=6,2 Гц, 2 Н), 7,00 (д,J=8,7 Гц, 2 Н), 5,38 (с, 2 Н), 4,25 (т, J=6,6 Гц, 2 Н), 2,82 (т, J=6,6 Гц, 2 Н), 2,28 (с, 6 Н); МС: (М+Н m/z=450,4). Пример 7. Диметил-(2-4-пиридин-4-ил-5-[4-(хинолин-2-илметокси)фенил]пиразол-1-илэтил)амин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Нпиразол-3-ил)феноксиметил]хинолина, но с замещением (2-гидразиноэтил)диметиламином. 1(с, 2 Н), 4,05 (т, J=6,6 Гц, 2 Н), 2,66 (т, J=7,1 Гц, 2 Н), 2,10 (с, 6 Н); МС: (М+Н m/z=450,4). Пример 8. 2-4-[Пиридин-4-ил-2-(2,2,2-трифторэтил)-2H-пиразол-3-ил]феноксиметилхинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением (2,2,2-трифторэтил)гидразином. МС: (М+Н m/z=461,2). Пример 9. 2-4-[Пиридин-4-ил-1-(2,2,2-трифторэтил)-1 Н-пиразол-3-ил]феноксиметилхинолин. К раствору 2-[4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолина (26,5 г) в диметилформамиде (140 мл) добавляли 1,1,1-трифтор-2-йодоэтан (21 мл, 2,0 экв.) и карбонат цезия (68,3 г, 3 экв.) и смесь нагревали при 60 С в течение 24 ч. Смесь растворяли водой, экстрагировали 3 метиленхлоридом,высушивали сульфатом магния, фильтровали и концентрировали. Очистка посредством флэш-хроматографии с элюированием 5% метанолом/70% этилацетатом/гексанами позволила получить 20,85 г указанного соединения в виде региоизомерической смеси 8:1. Препаративной ВЭЖХ с элюированием ацетонитрилом/метанолом (98:2) на колонке Chiralpak AD со скоростью потока 430 мл/мин получали 13,4 г очищенного указанного соединения в виде свободного основания. 1 Н ЯМР (400 МГц, CDCl3)8,45 (м, 2 Н), 8,16 (д, J=8,3 Гц, 1 Н), 8,04 (д, J=8,3 Гц, 1 Н), 7,96 (с, 1 Н),- 24012211 7,79 (д, J=8,3 Гц, 1 Н), 7,69 (м, 1 Н), 7,64 (д, J=8,3 Гц, 1 Н), 7,50 (м, 1 Н), 7,36 (д, J=8,7 Гц, 2 Н), 7,14 (д, J=6,2 Гц, 2 Н), 6,98(д, J=9,1 Гц, 2 Н), 5,35 (с, 2 Н), 4,75 (кв, J=8,3 Гц, 2 Н); МС: (М+Н m/z=427,1); МС: (М+Н m/z=461,2). Пример 10. 1-4-Пиридин-4-ил-3-[4-(хинолин-2-илметокси)фенил]пиразол-1-илпропан-2-ол. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Нпиразол-3-ил)феноксиметил]хинолина, но с замещением 1-гидразинопропан-2-олом. 1 Н ЯМР (400 МГц, CDCl3)8,44 (уш.с, 2 Н), 8,20 (д, J=8,3 Гц, 1 Н), 8,08 (д, J=8,3 Гц, 1 Н), 7,83 (д,J=8,3 Гц, 1 Н), 7,75 (м 2 Н), 7,67 (д, J=8,3 Гц, 1 Н), 7,56 (т, J=8,3 Гц, 1 Н), 7,36 (д, J=8,7 Гц, 2 Н), 7,30 (м, 2 Н),7,03 (д, J=9,1 Гц, 2 Н), 5,40 (с, 2 Н), 4,29 (м, 1 Н), 4,23 (м, 1 Н), 4,02 (м, 1 Н), 1,83 (м, 1 Н), 1,28 (д, J=6,2 Гц,3 Н); МС: (М+Н m/z=437,2). Пример 11. 1-4-Пиридин-4-ил-5-[4-(хинолин-2-илметокси)фенил]пиразол-1-илпропан-2-ол. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 1-гидразинопропан-2-олом. 1(м, 2 Н), 7,75 (м 2 Н), 7,57 (т, J=6,6 Гц, 1 Н), 7,20 (д, J=9,1 Гц, 2 Н), 7,13 (д, J=8,7 Гц, 2 Н), 7,00 (дд, J=6,2, 1,7 Гц,2 Н), 5,42 (с, 2 Н), 4,17 (м, 1 Н), 3,94 (м, 2 Н), 3,86 (м, 1 Н), 1,12 (д, J=6,6 Гц, 3 Н); МС: (М+Н m/z=437,3). Пример 12. 2-[4-(2-Изопропил-4-пиридин-4-ил-2H-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением изопропилгидразином. 1 Н ЯМР (400 МГц, CDCl3)8,33 (уш.с, 2 Н), 8,24 (д, J=8,3 Гц, 1 Н), 8,08 (д, J=8,3 Гц, 1 Н), 7,86 (с,1 Н), 7,83 (м, 1 Н), 7,72 (м 2 Н), 7,58 (т, J=7,9 Гц, 1 Н), 7,20 (д, J=8,7 Гц, 2 Н), 7,15 (д, J=9,1 Гц, 2 Н), 7,04 (м,2 Н), 5,43 (с, 2 Н), 4,31 (м, 1 Н), 1,43 (д, J=6,6 Гц, 6 Н); МС: (М+Н m/z=421,2). Пример 13. 2-[4-(4-Пиридин-4-илизоксазол-5-ил)феноксиметил]хинолин. 2-Пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанон (200 мг, 0,56 ммоль) нагревали с обратным холодильником в диметоксиметилдиметиламине (1 мл) в течение 1 ч и выпаривали. Неочищенный продукт растворяли в метаноле/воде (3:1, 4 мл) и добавляли гидрохлорид гидроксиламина (43 мг, 1,1 экв.). Через 1 ч добавляли уксусную кислоту (0,016 мл) и смесь нагревали с обратным холодильником в течение 1 ч. Охлажденную до температуры окружающей среды смесь вливали в насыщенный бикарбонат натрия, экстрагировали метиленхлоридом, высушивали сульфатом магния, фильтровали и концентрировали. Для получения указанного соединения в виде бурого твердого вещества (94 мг, 45%) осуществляли хроматографию на колонке 25S Biotage MPLC с элюированием 3% метанолом/1% гидроксидом аммония/50% этилацетатом в гексанах. 1 Н ЯМР (400 МГц, CDCl3)8,59 (дд, J=6,2, 1,7 Гц, 2 Н), 8,36 (с, 1 Н), 8,20 (д, J=8,3 Гц, 1 Н), 8,07 (д,J=8,7 Гц, 1 Н), 7,82 (д, J=9,1 Гц, 1 Н), 7,73 (дт, J=7,1, 1,7 Гц, 1 Н), 7,64 (д, J=8,3 Гц, 1 Н), 7,54 (м, 3 Н), 7,28 (д,J=4,2 Гц, 2 Н), 7,05 (д, J=9,1, 2 Н), 5,40 (с, 2 Н); МС: (M+H m/z=380,2). Пример 14. 2-[4-(5-Пиридин-4-ил-пиримидин-4-ил)феноксиметил]хинолин. 2-Пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанон (200 мг) нагревали с обратным холодильником в диметоксиметилдиметиламине (1 мл) в течение 1 ч и концентрировали. Неочищенную смесь растворяли в этаноле (3 мл) и добавляли гидрохлорид формамидина (90 мг, 2 экв.). В отдельной колбе к 3 мл этанола добавляли натрий (40 мг) и перемешивали 10 мин. Раствор этоксида натрия добавляли к смеси и в течение 1 ч нагревали с обратным холодильником. Смесь концентрировали и очищали посредством хроматографии на колонке 25S Biotage MPLC с элюированием 40-100% этилацетатом/гексаном для получения указанного соединения (83 мг, 38%). 1 Н ЯМР (400 МГц, CDCl3)8,53 (м, 3 Н), 8,14 (д, J=8,7 Гц, 1 Н), 8,03 (д, J=8,3 Гц, 1 Н), 7,79 (д, J=7,9 Гц,1 Н), 7,70 (м, 1 Н), 7,58 (д, J=8,7 Гц, 1 Н), 7,50 (м, 1 Н), 7,33 (д, J=9,1 Гц, 2 Н), 7,10 (д, J=6,2, 2 Н), 6,91 (д,J=9,1 Гц, 2 Н), 5,34 (с, 2 Н), 2,77 (с, 3 Н); МС: (М+Н m/z=391,2). Пример 15. 2-[4-(2-Метил-5-пиридин-4-илпиримидин-4-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(5-пиридин-4-ил-пиримидин-4 ил)феноксиметил]хинолина, но с замещением гидрохлоридом ацетамидина. 1 Н ЯМР (400 МГц, CDCl3)9,21 (с, 1 Н), 8,63 (С, 1 Н), 8,58 (м, 2 Н), 8,17 (д, J=8,7 Гц, 1 Н), 8,04 (д,J=8,7 Гц, 1 Н), 7,81 (д, J=8,3 Гц, 1 Н), 7,70 (м, 1 Н), 7,60 (д, J=8,3 Гц, 1 Н), 7,52 (м, 1 Н), 7,37 (м, 2 Н), 7,15 (д,J=6,2, 2H), 6,93 (д, J=9,1 Гц, 2 Н), 5,35 (с, 2 Н); МС: (М+Н m/z=405,2). Пример 16. 2-[4-(2-Метил-6-пиридин-4-илпиразоло[1,5-а]пиримидин-7-ил)феноксиметил]хинолин. К раствору 3-диметиламино-2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропенона (229 мг,0,56 ммоль) в этаноле (3 мл) добавляли пиперидин (2 экв.) и 5-метил-2 Н-пиразол-3-иламин (108 мг, 2 экв.) и нагревали смесь с обратным холодильником в течение 3 ч. Смесь охлаждали до КТ, фильтровали и промывали продукт этанолом и гексаном для получения указанного соединения (96 мг, 39%). 1 Н ЯМР (400 МГц, CDCl3)8,51 (д, J=7,9 Гц, 2 Н), 8,46 (С, 1 Н), 8,30 (м, 1 Н), 8,18 (м, 1 Н), 7,89 (д,J=8,3 Гц, 1 Н), 7,78 (м, 1 Н), 7,71 (м, 1 Н), 7,60 (м, 1 Н), 7,41 (д, J=8,7, 2 Н), 7,21 (м, 2 Н), 7,07 (д, J=8,7, 2 Н),6,60 (с, 1 Н), 5,50 (с, 2 Н) 2,48 (с, 3 Н); МС: (М+Н m/z=444,2). Пример 17. 2-[4-(2-Метил-6-пиридин-4-ил-[1,2,4]-триазоло[1,5-а]пиримидин-7-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(2-метил-6-пиридин-4-илпиразоло(с, 2 Н) 2,60 (с, 3 Н); МС: (М+Н m/z=445,2). Получение 6. 2-Пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 4-метилпиридазина на 4-пиколин. 1 Н ЯМР (400 МГц, CDCl3)9,12 (д, J=5,4 Гц, 1 Н), 9,08 (д, J=8,7 Гц, 2,1 Н), 8,20 (д, J=8,3 Гц, 1 Н),8,07 (д, J=8,3 Гц, 1 Н), 7,96 (м, 2 Н), 7,83 (д, J=7,9 Гц, 1 Н), 7,76 (м, 1 Н), 7,62 (д, J=8,3 Гц, 1 Н), 7,55 (м, 1 Н),7,38 (дд, J=5,4, 2,5 Гц, 1 Н), 7,09 (м, 2 Н), 5,44 (с, 2 Н), 4,23 (с, 2 Н); МС: (M+H m/z=356,2). Получение 7. 3-Диметиламино-2-пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропенон. Указанное соединение получали, следуя процедуре получения 3-диметиламино-2-пиридин-4-ил-1[4-(хинолин-2-илметокси)фенил]пропенона, но с замещением 2-пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этаноном. ЖХ/МС: КТ=1,8 мин; МС: (М+Н m/z=411,2). Пример 18. 2-[4-(4-Пиридазин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)9,11 (с, 1 Н), 9,01 (д, J=5,0 Гц, 1 Н), 8,34 (д, J=8,7 Гц, 1 Н), 8,25 (д, J=8,7 Гц,1 Н), 7,89 (м, 2 Н), 7,81 (д, J=8,3 Гц, 1 Н), 7,79 (м, 2 Н), 7,61 (т, J=7,6 Гц, 1 Н), 7,34 (м, 1 Н), 7,31 (д, J=8,7 Гц,2 Н), 7,05 (д, J=8,7, 2H), 5,49 (с, 2 Н); МС: (М+Н m/z=380,2). Пример 19. 2-[4-(1-Метил-4-пиридазин-4-ил-1H-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1(д, J=8,3 Гц, 1 Н), 7,82 (д, J=7,9 Гц, 1 Н), 7,73 (т, J=7,1 Гц, 1 Н), 7,67 (м, 2 Н), 7,55 (т, J=7,1 Гц, 1 Н), 7,34 (д,J=9,1 Гц, 2 Н), 7,24 (м, 1 Н), 7,02 (д, J=6,6 Гц, 2 Н), 5,39 (с, 2 Н), 3,97 (с, 3 Н); МС: (М+Н m/z= 394,2). Пример 20. 2-[4-(2-Метил-4-пиридазин-4-ил-2H-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-пиридазин-4-ил-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)8,99 (д, J=2,5 Гц, 1 Н), 8,90 (д, J=5,4 Гц, 1 Н), 8,24 (д, J=8,7 Гц, 1 Н), 8,08(M+H m/z=394,2). Пример 21. 2-[4-(4-Пиримидин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолина и осуществляя необходимые химические замещения. ЖХ/МС: КТ=1,8 мин; МС: (М+Н m/z=380,2). Пример 22. 2-[4-(4-Пиридазин-3-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(4-пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметилхинолина и осуществляя необходимые химические замещения. ЖХ/МС: КТ=1,7 мин; МС: (М+Н m/z=380,2). Получение 8. 2-(3-Метилизоксазол-5-ил)-1-[4-(хинолин-2-илметокси)фенил]этанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 3,5-диметилзоксазола на 4-пиколин. ЖХ/МС: КТ=2,3 мин; МС: (М+Н m/z=359,2). Получение 9. 3-Диметиламино-2-(3-метилизоксазол-5-ил)-1-[4-(хинолин-2-илметокси)фенил]пропенон. Указанное соединение получали, следуя процедуре получения 3-диметиламино-2-пиридин-4-ил-1[4-(хинолин-2-илметокси)фенил]пропенона, но с замещением 2-(3-метилизоксазол-5-ил)-1-[4-(хинолин 2-илметокси)фенил]этаноном. ЖХ/МС: КТ=2,1 мин; МС: (М+Н m/z=414,2). Пример 23. 2-4-[4-(3-Метилизоксазол-5-ил)-2 Н-пиразол-3-ил]феноксиметилхинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(4-пиридин-4-ил-2 Н-пиразол-3 ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-(3-метилизоксазол-5-ил)-1-[4-(хинолин 2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)8,23 (д, J=8,7 Гц, 1 Н), 8,12 (д, J=8,7 Гц, 1 Н), 7,94 (с, 1 Н), 7,84 (д, J=7,1 Гц,- 26012211 1 Н), 7,74 (м, 1 Н), 7,69 (д, J=8,3 Гц, 1 Н), 7,57 (т, J=6,6 Гц, 2 Н), 7,46 (д, J=8,7 Гц, 2 Н), 7,08 (д, J=8,7 Гц, 2 Н),5,88 (с, 1 Н), 5,42 (с, 2 Н), 2,23 (с, 3 Н); МС: (M+H m/z=383,2). Пример 24. 2-4-[2-Метил-4-(3-метилизоксазол-5-ил)-2H-пиразол-3-ил]феноксиметилхинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-(3-метилизоксазол-5-ил)-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)8,25 (д, J=8,7 Гц, 1 Н), 8,12 (д, J=8,3 Гц, 1 Н), 7,89 (с, 1 Н), 7,85 (д, J=8,3 Гц,1 Н), 7,74 (м, 2 Н), 7,57 (т, J=7,1 Гц, 1 Н), 7,28 (с, 1 Н), 7,26 (д, J=10,4 Гц, 2 Н), 7,16 (д, J=8,7 Гц, 2 Н), 5,45 (с,2 Н), 3,71 (с, 3 Н), 2,16 (с, 3 Н); МС: (M+H m/z=397,2). Пример 25. 2-4-[1-Метил-4-(3-метилизоксазол-5-ил)-1H-пиразол-3-ил]феноксиметилхинолин. Указанное соединение в виде белого твердого вещества получали, следуя процедуре получения 2[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 3-диметиламино-2-(3-метилизоксазол-5-ил)-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)8,18 (д, J=8,3 Гц, 1 Н), 8,07 (д, J=8,7 Гц, 1 Н), 7,81 (д, J=7,1 Гц, 1 Н), 7,77(с, 1 Н), 7,74 (т, J=7,1 Гц, 1 Н), 7,67 (д, J=8,7 Гц, 1 Н), 7,54 (т, J=7,1 Гц, 1 Н), 7,48 (д, 8,7 Гц, 2 Н), 7,07 (д,J=8,7 Гц, 2 Н), 5,81 (с, 1 Н), 5,41 (с, 2 Н), 3,92 (с, 3 Н), 2,20 (с, 3 Н); МС: (М+Н m/z=397,2). Пример 26. 2-4-[2-Метил-5-(3-метилизоксазол-5-ил)пиримидин-4-ил]феноксиметилхинолин. Указанное соединение в виде хлористо-водородной соли получали, следуя процедуре получения 2[4-(5-пиридин-4-ил-пиримидин-4-ил)феноксиметил]хинолина, но с замещением ацетамидина гидрохлоридом и 3-диметиламино-2-(3-метилизоксазол-5-ил)-1-[4-(хинолин-2-илметокси)фенил]пропеноном. 1 Н ЯМР (400 МГц, CDCl3)8,87 (с, 1 Н), 8,18 (д, J=8,3 Гц, 1 Н), 8,06 (д, J=8,3 Гц, 1 Н), 7,82 (д, J=8,3 Гц,1 Н), 7,72 (т, J=7,1 Гц, 1 Н), 7,63 (д, J=8,7 Гц, 1 Н), 7,53 (т, J=6,6 Гц, 1 Н), 7,45 (д, J=9,1 Гц, 2 Н), 7,05 (д, J=9,1 Гц,2 Н), 5,79 (с, 1 Н), 5,40 (с, 2 Н), 2,78 (с, 3 Н), 2,23 (с, 3 Н); МС: (М+Н m/z=409,2). Получение 10. 1-[4-(Хинолин-2-илметокси)фенил]этанон. К раствору 2-хлорметилхинолина (2,5 г, 14 ммоль) в ацетоне (47 мл) добавляли 4-гидроксиацетофенон (1,92 г, 1,0 экв.) и карбонат калия (2,5 г, 2 экв.). Смесь нагревали при 60 С в течение 16 ч в атмосфере N2, охлаждали до температуры окружающей среды и вливали в 1N гидроксид натрия (50 мл)/этилацетата (100 мл). Слои разделяли и высушивали органический слой сульфатом магния, фильтровали и концентрировали. Для получения указанного соединения в виде белого твердого вещества (2,75 г, 71%) осуществляли хроматографию на колонке 40 М Biotage MPLC с использованием градиента 5-40% этилацетата/гексана. 1(с, 3 Н); МС: (М+Н m/z=278,3). Получение 11. 3-Диметиламино-1-[4-(хинолин-2-илметокси)фенил]пропенон. 1-[4-(Хинолин-2-илметокси)фенил]этанон (1,0 г, 3,61 ммоль) перемешивали в диметоксиметилдиметиламине (5 мл) и нагревали с обратным холодильником в течение 18 ч. Смесь охлаждали до КТ и получали бурый осадок. Его фильтровали и промывали этиловым эфиром для получения 840 мг (71%) указанного соединения. 1 Н ЯМР (400 МГц, CDCl3)8,39 (д, J=8,3 Гц, 1 Н), 7,97 (м, 2 Н), 7,91 (м, 2 Н), 7,84 (м, 2 Н), 7,75 (т,J=6,6 Гц, 1 Н), 7,62 (м, 3 Н), 7,05 (д, J=8,7 Гц, 2 Н), 5,77 (д, J=12,0, 1 Н), 5,40 (с, 2 Н), 3,07 (уш.с, 3 Н), 2,84(уш.с, 3 Н); МС: (М+Н m/z=333,3). Пример 27. 2-[4-(2-Пиридин-4-ил-2 Н-пиразол-3-ил)феноксиметил]хинолин. К раствору 3-диметиламино-1-[4-(хинолин-2-илметокси)фенил]пропенона (46 мг) в этаноле (0,7 мл) добавляли воду (0,7 мл), уксусную кислоту (0,05 мл) и 4-пиридилгидразин (25 мг, 1 экв.). Смесь нагревали при 100 С в течение 3 ч, охлаждали до КТ, вливали в 1N NaOH, экстрагировали хлороформом, высушивали сульфатом магния, фильтровали и концентрировали. Для получения указанного соединения в виде бурого твердого вещства (31 мг, 61%) осуществляли хроматографию на колонке 25S Biotage MPLC с элюированием 20-80% этилацетатом/гексаном. 1 Н ЯМР (400 МГц, CDCl3)8,51 (уш.с, 2 Н), 8,24 (д, J=8,7 Гц, 1 Н), 8,11 (д, J=8,7, 2 Н), 7,84 (д, J=8,3 Гц,1 Н), 7,74 (м, 2 Н), 7,69 (д, J=8,7 Гц, 1 Н), 7,58 (т, J=7,1, 1H), 7,32 (уш.с, 2 Н), 7,19 (д, J=6,6 Гц, 2 Н), 7,04 (д,J= 6,6, 2 Н), 5,40 (с, 2 Н), 6,45 (с, 1 Н), 5,42 (с, 2 Н); МС: (М+Н m/z=379,2). Пример 28. 2-[4-(3-Метил-5-пиридин-4-ил[1,2,4]триазол-4-ил)феноксиметил]хинолин. К раствору изоникотинового гидразида (1,04 г, 1,12 экв.) в ацетонитриле (30 мл) добавляли N,N-диметилацетамид диметилацеталь (1,1 экв.) и смесь нагревали при 50 С в течение 3 ч. Смесь охлаждали до температуры окружающей среды и концентрировали. Вместе с уксусной кислотой (30 мл) добавляли 4(хинолин-2-илметокси)фениламин (1,70 г) и смесь нагревали с обратным холодильником в течение 3 ч и охлаждали до температуры окружающей среды. Для получения указанного соединения в виде бурого твердого вещества (56%) смесь упаривали в роторном испарителе и очищали посредством комбиимпульсной MPLC. 1 Н ЯМР (400 МГц, CDCl3)8,51 (д, J=6,2 Гц, 2 Н), 8,24 (д, J=8,7 Гц, 1 Н), 8,08 (д, J=8,7, 1 Н), 7,85 (д,- 27012211(д, J=9,1 Гц, 2 Н), 7,12 (д, J=9,1 Гц, 2 Н), 5,43 (с, 2 Н), 2,31 (с, 3 Н); МС: (М+Н m/z=394,3). Получение 12. 4-Бензилокси-N-метокси-N-метилбензамид. Указанное соединение в виде воскового твердого вещства получали, следуя процедуре получения Nметокси-N-метил-4-(хинолин-2-илметокси)бензамида, но с замещением 4-бензилоксибензойной кислотой. МС: (М+Н m/z=272,3). Получение 13. 1-(4-Бензилоксифенил)-2-пиридин-4-илэтанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 4-бензилокси-N-метокси-N-метилбензамидом. МС: (М+Н m/z=304,2). Получение 14. 4-[3-(4-бензилоксифенил)-1-метил-1 Н-пиразол-4-ил]пиридин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 1-(4-бензилоксифенил)-2-пиридин-4-ил-этаноном. МС: (М+Н m/z=342,2). Получение 15. 4-(1-Метил-4-пиридин-4-ил-1H-пиразол-3-ил)фенол. К раствору 4-[3-(4-бензилоксифенил)-1-метил-1 Н-пиразол-4-ил]пиридина (1,28 г) в этаноле (50 мл)/ этилацетате (50 мл) в колбе Парра добавляли гидроксид палладия (500 мг). Колбу Парра подвергали давлению 40 фунтов/кв.дюйм на встряхивающем устройстве в течение 6 ч. Смесь фильтровали и концентрировали. Осуществляли хроматографию Biotage MPLC с элюированием метанолом (1-7%)/хлороформом для получения указанного соединения (860 мг, 91%). 1 Н ЯМР (400 МГц, ДМСО)9,53 (с, 1 Н), 8,39 (д, J=5,8 Гц, 2 Н), 7,15 (м, 4 Н), 6,72 (д, J=8,7 Гц, 1 Н),3,84 (с, 3 Н); МС: (М+Н m/z=252,2). Пример 29. 2-[4-(1-Метил-4-пиридин-4-ил-1H-пиразол-3-ил)феноксиметил]хиноксалин. К раствору 4-(1-метил-4-пиридин-4-ил-1H-пиразол-3-ил)фенола (50 мг) в диоксане (2 мл) добавляли трифенилфосфин (84 мг), хиноксалин-2-илметанол (48 мг) и ди-трет-бутилазадикарбоксилат (73 мг) и смесь нагревали при 60 С в течение 18 ч. Смесь вливали в 1N NaOH, экстрагировали 3 метиленхлоридом, высушивали сульфатом магния, фильтровали и концентрировали. Очисткой посредством BiotageMPLC-хроматографии получали указанное соединение (54 мг, 67%). 1 Н ЯМР (400 МГц, CDCl3)9,09 (с, 1 Н), 8,45 (д, J=6,2 Гц, 2 Н), 8,10 (м, 2 Н), 7,77 (м, 2 Н), 7,55 (с,1 Н), 7,37 (д, J=9,1 Гц, 2H), 7,10 (д, J=6,9 Гц, 2 Н), 7,01 (д, J=8,7, 2H), 5,41 (с, 2 Н), 3,94 (с, 3 Н); МС: (М+Нm/z=394,4). Пример 30. Гидрохлорид 7-хлор-2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина. Указанное соединение получали, следуя процедуре получения метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением 4-(1-метил-4-пиридин-4-ил-1H-пиразол-3 ил)фенолом и 7-хлор-2-хлорметилхинолином. 1 Н ЯМР (400 МГц, ДМСО)8,66 (д, J=6,6 Гц, 2 Н), 8,54 (с, 1 Н), 8,47 (д, J=8,3, 2 Н), 8,04 (м, 2 Н), 7,70m/z=427,1). Пример 31. Гидрохлорид 6-фтор-2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина. Указанное соединение получали, следуя процедуре получения гидрохлорида 7-хлор-2-[4-(1-метил-4 пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 2-хлорметил-6-фторхинолином. 1m/z=411,2). Получение 16. Метиловый сложный эфир хинолин-2-ила 3-фтор-4-(хинолин-2-илметокси)бензойной кислоты. Указанное соединение получали, следуя процедуре получения метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением 3-фтор-4-гидроксибензойной кислотой. МС: (М+Н m/z=439,0). Получение 17. 3-Фтор-4-(хинолин-2-илметокси)бензойная кислота. Указанное соединение получали, следуя процедуре получения 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением метиловым сложным эфиром 3-фтор-4-(хинолин-2-илметокси)бензойнокислого хинолин-2-ила. МС: (М+Н m/z=298,2). Получение 18. 3-Фтор-N-метоксил-N-метил-4-(хинолин-2-илметокси)бензамид. Указанное соединение получали, следуя процедуре получения N-метокси-N-метил-4-(хинолин-2 илметокси)бензамида, но с замещением 3-фтор-4-(хинолин-2-илметокси)бензойной кислотой. МС: (М+Н m/z=341,2). Получение 19. 1-[3-Фтор-4-(хинолин-2-илметокси)фенил]-2-пиридин-4-ил-этанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 3-фтор-N-метокси-N-метил-4-(хинолин-2-илметокси)бензамидом.- 28012211 МС: (М+Н m/z=373,1). Пример 32. 2-[2-Фтор-4-(4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(4-пиридин-4-ил-2 Н-пиразол-3 ил)феноксиметил]хинолина, но с замещением 1-[3-фтор-4-(хинолин-2-илметокси)фенил]-2-пиридин-4 илэтаноном. 1 Н ЯМР (400 МГц, CDCl3)8,47 (уш.с, 2 Н), 8,19 (д, J=8,7 Гц, 1 Н), 8,05 (д, J=8,3 Гц, 1 Н), 7,71 (м,4 Н), 7,54 (т, J=7,1 Гц, 1 Н), 7,18 (м, 3 Н), 7,07 (м, 2 Н), 5,42 (с, 2 Н); МС: (М+Н m/z=397,0). Пример 33. 2-[2-Фтор-4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Нпиразол-3-ил)феноксиметил]хинолина, но с замещением 1-[3-фтор-4-(хинолин-2-илметокси)фенил]-2-пиридин-4-илэтаноном. 1(д, J=7,9 Гц, 2 Н), 7,72 (м, 2 Н), 7,55 (м, 2 Н), 7,16 (м, 2 Н), 7,07 (м, 1 Н), 6,99 (м, 2 Н), 5,45 (с, 2 Н), 3,95 (с,3 Н); МС: (М+Н m/z=411,0). Получение 20. Метиловый сложный эфир хинолин-2-ила 2,3-дифтор-4-(хинолин-2-илметокси)бензойной кислоты. Указанное соединение получали, следуя процедуре получения метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением 2,3-дифтор-4-гидроксибензойной кислотой. МС: (М+Н m/z=457,1). Получение 21. 2,3-Дифтор-4-(хинолин-2-илметокси)бензойная кислота. Указанное соединение получали, следуя процедуре получения 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением метиловым сложным эфиром хинолин-2-ила 2,3-дифтор-4-(хинолин-2 илметокси)бензойной кислоты. МС: (М+Н m/z=316,1). Получение 22. 2,3-Дифтор-N-метокси-N-метил-4-(хинолин-2-илметокси)бензамид. Указанное соединение получали, следуя процедуре получения N-метокси-N-метил-4-(хинолин-2 илметокси)бензамида, но с замещением 2,3-дифтор-4-(хинолин-2-илметокси)бензойной кислотой. МС: (М+Н m/z=359,1). Получение 23. 1-[2,3-Дифтор-4-(хинолин-2-илметокси)фенил]-2-пиридин-4-илэтанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 2,3-дифтор-N-метокси-N-метил-4-(хинолин-2-илметокси)бензамидом. МС: (М+Н m/z=391,1). Пример 34. 2-[2,3-Дифтор-4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 1-[2,3-дифтор-4-(хинолин-2-илметокси)фенил]-2 пиридин-4-ил-этаноном. 1 Н ЯМР (400 МГц, CDCl3)8,44 (уш.с, 2 Н), 8,22 (д, J=8,7 Гц, 1 Н), 8,06 (д, J=8,7 Гц, 1 Н), 7,84 (д,J=7,9 Гц, 1 Н), 7,70 (м, 2 Н), 7,66 (с, 1 Н), 7,56 (т, J=7,9 Гц, 1 Н), 7,08 (м, 3 Н), 6,88 (м, 1 Н), 5,48 (с, 2 Н); МС:(М+Н m/z=429,1). Получение 24. Метиловый сложный эфир хинолин-2-ила 2-фтор-4-(хинолин-2-илметокси)бензойной кислоты. Указанное соединение получали, следуя процедуре получения метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением 2-фтор-4-гидроксибензойной кислотой. МС: (М+Н m/z=439,0). Получение 25. 2-Фтор-4-(хинолин-2-илметоксил)бензойная кислота. Указанное соединение получали, следуя процедуре получения 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением метиловым сложным эфиром хинолин-2-ила 2-фтор-4-(хинолин-2-илметокси) бензойной кислоты. МС: (М+Н m/z=298,2). Получение 26. 2-Фтор-N-метокси-N-метил-4-(хинолин-2-илметокси)бензамид. Указанное соединение получали, следуя процедуре получения N-метокси-N-метил-4-(хинолин-2 илметокси)бензамида, но с замещением 2-фтор-4-(хинолин-2-илметоксил)бензойной кислотой. МС: (М+Н m/z=341,2). Получение 27. 1-2-Фтор-4-(хинолин-2-илметокси)фенил-2-пиридин-4-ил-этанон. Указанное соединение получали, следуя процедуре получения 2-пиридин-4-ил-1-[4-(хинолин-2-илметокси)фенил]этанона, но с замещением 2-фтор-N-метокси-N-метил-4-(хинолин-2-илметокси)бензамидом. МС: (М+Н m/z=373,0). Пример 35. 2-[3-Фтор-4-(4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолин. Указанное соединение получали, следуя процедуре получения 2-[4-(4-пиридин-4-ил-2 Н-пиразол-3 ил)феноксиметил]хинолина, но с замещением 1-2-фтор-4-(хинолин-2-илметокси)фенил-2-пиридин-4 илэтаноном.(с, 1 Н), 7,82 (м, 1 Н), 7,74 (м, 1 Н), 7,65 (д, J=8,7 Гц, 1 Н), 7,55 (м, 1 Н), 7,25 (м, 1 Н), 7,18 (д, J=6,2 Гц, 2 Н),6,85 (д, J=10,9, 2 Н), 5,38 (с, 2 Н); МС: (М+Н m/z=397,2). Получение 28. 4-(Хинолин-2-илметокси)бензальдегид. Указанное соединение получали, следуя процедуре получения метилового сложного эфира 4-(хинолин-2-илметокси)бензойной кислоты, но с замещением 4-гидроксибензальдегидом. МС: (М+Н m/z=264,2). Получение 29. 1-Пиридин-4-ил-2-[4-(хинолин-2-илметокси)фенил]этанон. К раствору 4-пиридинкарбоксальдегида (10,8 г) в 2-пропаноле (50 мл) добавляли анилин (9,3 г). Через 15 мин фенилпиридин-4-илметиленаминовый продукт (68%) фильтровали и использовали неочищенным. К раствору имина в этаноле (35 мл) добавляли дифенилфосфит (13,1 мл) и перемешивали в течение 1 ч. Добавляли этиловый эфир (200 мл) и фильтровали дифениловый сложный эфир фениламинопиридин-4-илметилфосфиновой кислоты (5,06 г). При -40 С в атмосфере N2 перемешивали фосфиновый сложный эфир (0,98 г) в ТГФ (25 мл). После добавления раствора KOH/метанола (0,146 г/10%) добавляли 4-(хинолин-2-илметокси)бензальдегид (0,62 г). Неочищенную смесь нагревали до температуры окружающей среды в течение 1 ч и концентрировали. Неочищенный продукт перемешивали в ацетонитриле(1 мл)/1 мл концентрированной HCl в течение 1 ч, гасили насыщенным бикарбонатом натрия, экстрагировали хлороформом, высушивали сульфатом магния, фильтровали и концентрировали. Очисткой посредством комбиимпульсной MPLC получали указанное соединение. МС: (М+Н m/z=355,1). Пример 36. 2-[4-(5-Пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолин. 1-Пиридин-4-ил-2-[4-(хинолин-2-илметокси)фенил]этанон (168 мг) нагревали в диэтоксиметилдиметиламине (1 мл) с обратным холодильником в течение 2 ч. Смесь концентрировали и растворяли в метаноле (1 мл), добавляли гидрат гидразина (0,023 мл) и смесь нагревали при 65 С в течение 1 ч. Смесь концентрировали и очищали посредством комбиимпульсной MPLC-хроматографии для получения указанного соединения (90%). 1 Н ЯМР (400 МГц, CDCl3)8,37 (уш.с, 2 Н), 8,18 (д, J=8,7 Гц, 1 Н), 7,99 (д, J=8,7 Гц, 1 Н), 7,78 (д,J=8,3 Гц, 1 Н), 7,66 (м, 2 Н), 7,54 (с, 1 Н), 7,48 (м, 1 Н), 7,36 (м, 2 Н), 7,11 (д, J=7,1 Гц, 2 Н), 6,94 (д, J=8,3 Гц,2 Н), 5,29 (с, 2 Н); МС: (М+Н m/z=379,2). Пример 37. 2-[4-(1-Метил-5-пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолин. Следуя процедуре получения 2-[4-(1-метил-5-пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолина, но с замещением метилгидразином, получали указанное соединение и 2-[4-(1-метил-3-пиридин-4 ил-1 Н-пиразол-4-ил)феноксиметил]хинолин. 1 Н ЯМР (400 МГц, CDCl3)8,66 (уш.с, 2 Н), 8,17 (д, J=8,7 Гц, 1 Н), 8,05 (д, J=7,9 Гц, 1 Н), 7,81 (д,J=8,3 Гц, 1 Н), 7,70 (м, 1 Н), 7,63 (м, 2 Н), 7,53 (т, J=7,1 Гц, 1 Н), 7,21 (м, 2 Н), 7,03 (д, J=9,1 Гц, 2 Н), 6,89 (д,J=8,7 Гц, 2 Н), 5,32 (с, 2 Н), 3,80 (с, 3 Н); МС: (М+Н m/z=393,2). Пример 38. 2-[4-(1-Метил-3-пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолин. Следуя процедуре получения 2-[4-(5-пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолина, но с замещением метилгидразином, получали указанное соединение и 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-4-ил)феноксиметил]хинолин. 1 Н ЯМР (400 МГц, CDCl3)8,49 (уш.с, 2 Н), 8,20 (д, J=8,3 Гц, 1 Н), 8,07 (д, J=8,3 Гц, 1 Н), 7,83 (д,J=8,3 Гц, 1 Н), 7,74 (м, 2 Н), 7,55 (т, J=7,1 Гц, 1 Н), 7,42 (м, 2 Н), 7,38 (с, 1 Н), 7,17 (д, J=8,7 Гц, 2 Н), 7,00 (д,J=8,7 Гц, 2 Н), 5,38 (с, 2 Н), 3,95 (с, 3 Н); МС: (М+Н m/z=393,2). Пример 39. 2-Метил-1-4-пиридин-4-ил-3-[4-(хинолин-2-илметокси)фенил]пиразол-1-илпропан-2-ол. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 1-гидразино-2-метилпропан-2-олом. 1 Н ЯМР (400 МГц, CDCl3)8,47 (д, J=6,2 Гц, 2 Н), 8,19 (д, J=8,7 Гц, 1 Н), 8,07 (д, J=8,7 Гц, 1 Н), 7,82m/z=451,2). Пример 40. 2-Метил-1-4-пиридин-4-ил-5-[4-(хинолин-2-илметокси)фенил]пиразол-1-илпропан-2-ол. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением 1-гидразино-2-метилпропан-2-олом. 1 Н ЯМР (400 МГц, CDCl3)8,37 (д, J=5,8 Гц, 2 Н), 8,24 (д, J=8,3 Гц, 1 Н), 8,09 (д, J=9,1 Гц, 1 Н), 7,87(с, 1 Н), 7,85 (д, J=7,9 Гц, 1 Н), 7,76 (м, 1 Н), 7,72 (м, 1 Н), 7,17 (м, 4 Н), 7,00 (д, J=6,2 Гц, 2 Н), 5,42 (с, 2 Н),3,89 (с, 2 Н), 1,04 (с, 6 Н); МС: (М+Н m/z=451,2). Пример 41. (R)-1-4-Пиридин-4-ил-3-[4-(хинолин-2-илметокси)фенил]пиразол-1-илпропан-2-ол. Указанное соединение получали, следуя процедуре получения 2-[4-(1-метил-4-пиридин-4-ил-1 Н-пиразол-3-ил)феноксиметил]хинолина, но с замещением (R)-1-гидразинопропан-2-олом. 1 Н ЯМР (400 МГц, CDCl3)8,42 (м, 2 Н), 8,18 (д, J=8,3 Гц, 1 Н), 8,06 (д, J=8,4 Гц, 1 Н), 7,81 (д, J=8,3 Гц,1 Н), 7,73 (м, 1 Н), 7,66 (д, J=8,7 Гц, 1 Н), 7,61 (с, 1 Н), 7,54 (м, 1 Н), 7,36 (д, J=9,1 Гц, 2 Н), 7,12 (м, 2 Н), 6,99
МПК / Метки
МПК: A61P 25/18, C07D 413/14, C07D 401/14, A61K 31/14
Метки: качестве, ингибиторов, соединения, хинолинов, pde10, гетероароматические, применение
Код ссылки
<a href="https://eas.patents.su/30-12211-geteroaromaticheskie-soedineniya-hinolinov-i-ih-primenenie-v-kachestve-ingibitorov-pde10.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероароматические соединения хинолинов и их применение в качестве ингибиторов pde10</a>
Бициклические гетероароматические соединения в качестве ингибиторов протеин-тирозинкиназ

Номер патента: 2455
Опубликовано: 25.04.2002
Авторы: Кокерилл Джордж Стюарт, Гантрип Стивен Барри, Лаки Карен Элизабет, Картер Малком Клайв, Смит Кэтрин Джейн
МПК: A61P 35/00, A61K 31/519, C07D 471/04...
Метки: протеин-тирозинкиназ, соединения, гетероароматические, ингибиторов, бициклические, качестве
Формула / Реферат:
1. Соединение формулы (I) или его соль или сольват; где Y представляет собой CR1, а V представляет собой N; или Y представляет собой CR1, a V представляет собой CR2; R1 представляет собой группу СН3SO2СН2СН2NНСН2-Аr-, где -Аr- выбран из фурана и тиазола, каждый из которых может быть возможно замещен одной или двумя гало, С1-4алкильной или С1-4 алкоксигруппами; R2 выбран из группы, включающей в себя водород, гало, гидрокси, С1-4алкил,...
Азотсодержащие гетероароматические соединения в качестве ингибиторов фактора ха, фармацевтическая композиция и способ лечения

Номер патента: 3056
Опубликовано: 26.12.2002
Авторы: Хан Кви, Февиг Джон Мэттью, Росси Карен Анита, Каччиола Джозеф, Пруитт Джеймс Рассел, Кван Мими Лайфен, Орват Майкл Джеймс, Пинто Дональд Джозеф Филлип
МПК: A61P 7/02, A61K 31/41, C07D 207/34...
Метки: способ, композиция, гетероароматические, фактора, ха, азотсодержащие, фармацевтическая, ингибиторов, качестве, лечения, соединения
Формула / Реферат:
1. Соединение формулы I или его стереоизомер или фармацевтически приемлемая соль, где кольцо М содержит, кроме J, 0-3 атома N, при условии, что если М содержит 2 атома N, тогда R1b отсутствует, и если М содержит 3 атома N, тогда R1a и R1b отсутствуют; J представляет собой N или NH; D выбран из CN, C(=NR8)NR7R9, NHC(=NR8)NR7R9, NR8CH(=NR7), C(O)NR7R8 и (CR8R9)tNR7R8, при условии, что D присоединен к Е в мета- или пара-положении относительно G, Е...
Аминокислотные соединения и их применение в качестве ингибиторов nep, ace и ece

Номер патента: 5004
Опубликовано: 28.10.2004
Авторы: Беннжан Каролина, Ингимбер Никола, Ренар Пьер, Пора Эрве, Фурнье-Залуски Мари-Клод, Рок Бернар П., Скальбер Элизабет
МПК: A61P 43/00, A61K 31/33, C07D 209/20...
Метки: аминокислотные, соединения, качестве, применение, ингибиторов
Формула / Реферат:
1. Соединения формулы (I) в которой n представляет собой коэффициент, в котором 0_n_3, m представляет собой коэффициент, в котором 0_m_6, R3 и R4 совместно образуют с двумя атомами углерода, которые их несут, фенильную группу, которая незамещена или замещена от 1 до 3 одинаковыми или различными группами, выбираемыми из алкила, алкенила, алкинила, алкокси, гидрокси, алкилтио, меркапто, циано, нитро, амино, алкиламино, диалкиламино,...
Аминогетероарильные соединения в качестве ингибиторов протеинкиназ

Номер патента: 10727
Опубликовано: 30.10.2008
Авторы: Шен Хонг, Ботрос Айрини, Уолтер Эллисон, Жанг Фанг-Джи, Жанг Дженнифер, Нэмбу Митчелл Дейвид, Ли Ксяоюан, Ханау Кэтлин Элизабет, Канг Пей-Пей, Тран-Дюб Мишель, Менг Джерри Джиалун, Джиа Ли, Джонсон Джоанна, Пэйриш Мейсон Алан, (US)ЛИН Джейсон, Чу Джи Ю, Колодзей Стивен А., Фанк Ли А., Куй Джингджонг Джин, Нелсон Кристофер Джи., Бумралькар Дилип, Харрис Джи Дейвис Мл.
МПК: C07D 211/72, A61K 31/4965, C07D 401/00...
Метки: качестве, соединения, протеинкиназ, ингибиторов, аминогетероарильные
Формула / Реферат:
1. Аминогетероарильное соединение формулы 1 где Y представляет собой N или CR12; R1 выбран из C6-12арила, 5-12-членного гетероарила, C3-12циклоалкила, 3-12-членного гетероалициклила, -O(CR6R7)nR4, -C(O)R4, -C(O)OR4, -CN, -NO2, -S(O)mR4, -SO2NR4R5, -C(O)NR4R5, -NR4C(O)R5, -C(=NR6)NR4R5, C1-8алкила, C2-8алкенила и C2-8алкинила; и любой атом водорода в R1 возможно заменен одной или более чем одной группой R3; R2 представляет собой водород,...
Тиенодибензоазуленовые соединения в качестве ингибиторов фактора некроза опухоли

Номер патента: 6069
Опубликовано: 25.08.2005
Авторы: Хрвачич Бошка, Пешич Дияна, Жупанович Желько, Мерчеп Младен, Месич Милан
МПК: C07D 495/04, A61K 31/55, A61P 35/00...
Метки: соединения, опухоли, фактора, тиенодибензоазуленовые, некроза, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы I и его фармакологически приемлемые соли и сольваты где X представляет CH2 или гетероатом, выбранный из группы, включающей O, S и NR13, в котором R13 означает водород, C7-C10арилкарбонил; R1, R5, R7, R8 и R9 независимо друг от друга представляют водород; R2, R3 и R4 независимо представляют водород, фтор, хлор, бром, C1-C7алкил, галогенметил, C1-C7 алкокси или C1-C7алкилтио; R6 представляет водород, фтор, хлор, бром; R10...
Предыдущий патент: Обратные мицеллы для доставки катионов металлов, включающие в себя диглицерид и фитостерол, и способ их получения
Следующий патент: Адъювантная композиция, содержащая фосфат алюминия и 3d-mpl
Случайный патент: Способ получения катализатора полимеризации олефинов и катализаторы, полученные данным способом