Спиропиперидиновые соединения
Номер патента: 20507
Опубликовано: 28.11.2014
Авторы: Маити Пранаб, Лайнсвала Джаяна Панкадж, Амдоучи Чафик







Формула / Реферат
1. Соединение формулы

или его фармацевтически приемлемая соль;
где R1 выбран из группы, состоящей из Н, F и Cl;
R5 представляет собой Н или -СºCCH3;
X выбран из группы, состоящей из -СН2СН2-, -СН=СН и -N(R7)CH2-;
R7 выбран из группы, состоящей из Н и C1-3алкила;
Z1 выбран из группы, состоящей из -C(R3)- и -N-;
R3 выбран из группы, состоящей из Н, ОСН3 и СН3;
Z2 выбран из группы, состоящей из -S- и -О-;
Z3 выбран из группы, состоящей из -C(R4)- и -N-;
R4 выбран из группы, состоящей из Н, ОСН3 и СН3;
где по меньшей мере один из группы, состоящей из Z1 и Z3, представляет собой -C(R3)- или -C(R4)-.
2. Соединение или его соль по п.1, отличающееся тем, что R1 представляет собой Н.
3. Соединение или его соль по любому из пп.1, 2, отличающееся тем, что R5 представляет собой -СºCCH3.
4. Соединение или его соль по п.3, отличающееся тем, что соединение является S-изомером.
5. Соединение или его соль по п.1, отличающееся тем, что R1 представляет собой F, a R5 представляет собой Н.
6. Соединение по любому из пп.1-5, отличающееся тем, что Z2 представляет собой -S-.
7. Соединение или его соль по любому из пп.1-6, отличающееся тем, что Z1 представляет собой -C(R3)-.
8. Соединение или его соль по любому из пп.1-7, отличающееся тем, что R3 представляет собой Н или СН3.
9. Соединение или его соль по любому из пп.1-8, отличающееся тем, что Z3 представляет собой -C(R4)-.
10. Соединение или его соль по п.9, отличающееся тем, что R4 представляет собой Н.
11. Соединение или его соль по любому из пп.1-10, отличающееся тем, что X представляет собой -СН=СН-.
12. Соединение или его соль по любому из пп.1-10, отличающееся тем, что X представляет собой
-N(R7)CH2-.
13. Соединение или его соль по п.12, отличающееся тем, что R7 представляет собой СН3.
14. Соединение по любому из пп.1-13, отличающееся тем, что указанное соединение представляет собой фармацевтически приемлемую соль.
15. Соединение формулы

или его фармацевтически приемлемая соль.
16. Соединение формулы

или его фармацевтически приемлемая соль.
17. Фармацевтическая композиция для лечения диабета, содержащая фармацевтически приемлемый носитель и соединение по любому из пп.1-16 или его фармацевтически приемлемую соль.
18. Применение соединения по любому из пп.1-16 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения диабета.
19. Применение соединения по любому из пп.1-16 или его фармацевтически приемлемой соли для лечения диабета.
Текст
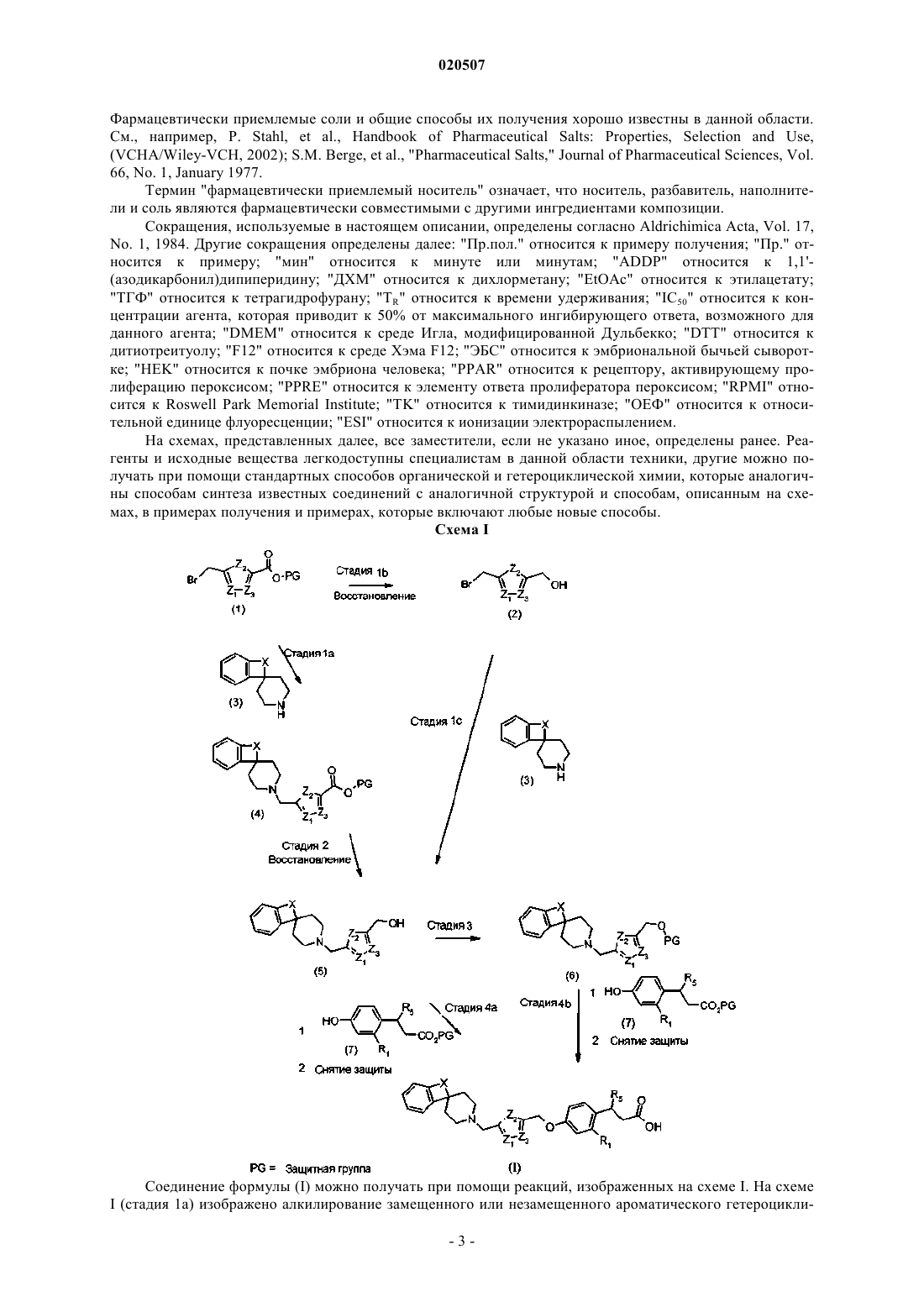
Амдоучи Чафик, Лайнсвала Джаяна Панкадж (US), Маити Пранаб (IN) Медведев В.Н. (RU) или его фармацевтически приемлемая соль, а также фармацевтическая композиция и способ лечения диабета.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Введение одобренных в настоящее время лекарственных средств для лечения диабета связывают с нежелательными побочными эффектами, которые иногда включают гипогликемию, повреждение печени,заболевания желудочно-кишечного тракта, увеличение веса или другие нежелательные эффекты, которые могут быть связаны с активностью PPAR-гамма.GPR-40 представляет собой рецептор, связанный с G-белком, который, как сообщается, главным образом экспрессируется в больших количествах в бета-клетках поджелудочной железы у грызунов, клеточных линиях инсулиномы и островках Лангерганса. Этот рецептор активируется средне- и длинноцепочечными жирными кислотами и, таким образом, этот рецептор также известен как FFAR1 (Free FattyAcid Receptor 1 - рецептор свободных жирных кислот 1). Зависимость глюкозы от секреции инсулина является важной особенностью активации GPR40, что превращает этот рецептор в отличную мишень для разработки эффективных лекарственных средств для применения для лечения диабета 2 типа (T2D). В недавно опубликованной заявке на патент США 2009/0170908 А 1 ('908) в целом предложены соединения, содержащие углеводородные спирогруппы и, как утверждается, обладающие активностью в качестве модуляторов связанного с G-белком рецептора 40 (GPR40). Соединения согласно '908 представляют собой соединения, в которых углеводородная спирогруппа не содержит гетероатомов. В противоположность этому, многие из предложенных в настоящем изобретении спиропиперидиновых соединений обеспечивают желательную селективную активацию GPR40, не вызывая поддающейся обнаружению активности PPAR. Соединения согласно настоящему изобретению являются высокоактивными активаторами GPR40. В настоящем изобретении предложен способ лечения, реализуемый через фармакологический механизм,который является уникальным по сравнению с коммерчески доступными способами лечения, а также предложены соединения, которые селективно активируют GPR40 по сравнению с PPAR-гамма. Фармакологический профиль соединений согласно настоящему изобретению в качестве селективных активаторов GPR40 может быть особенно желательным для применения для лечения T2D. Кроме того, селективная модуляция GPR40 может обеспечивать особенно желательные характеристики безопасности для применения для лечения T2D за счет отсутствия эффектов, связанных с модулированием PPAR-гамма. Настоящее изобретение направлено на соединения формулы или их фармацевтически приемлемые соли; где R1 выбран из группы, состоящей из Н, F и Cl;R4 выбран из группы, состоящей из Н, ОСН 3 и СН 3; где по меньшей мере один из группы, состоящей из Z1 и Z3, представляет собой -C(R3)- или -C(R4)-. В другом варианте реализации настоящего изобретения предложено применение соединения, заявленного в настоящем изобретении, или его фармацевтически приемлемой соли для получения лекарственного средства. Согласно другому варианту реализации лекарственное средство предназначено для лечения диабета. Дополнительный вариант реализации настоящего изобретения представляет собой применение соединения, заявленного в настоящем изобретении, или его фармацевтически приемлемой соли в качестве терапии. Дополнительный вариант реализации изобретения представляет собой соединение, заявленное в настоящем изобретении, или его фармацевтически приемлемую соль для применения для лечения диабета. Также изобретение относится к соединению, заявленному в настоящем изобретении, для применения в качестве лекарственного средства. В другом варианте реализации настоящего изобретения предложен способ лечения диабета у млекопитающего, включающий стадию введения млекопитающему соединения, заявленного в настоящем изобретении, или его фармацевтически приемлемой соли. Согласно другому варианту реализации настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение, заявленное в настоящем изобретении, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Другой вариант реализации представляет собой фармацевтическую композицию согласно настоящему изобретению, дополнительно содержащую второй фармацевтический агент. В предпочтительном варианте реализации изобретения соединения согласно настоящему изобретению селективно активируют GPR40 по сравнению с PPAR-гамма. Относительные значения IC50, харак-1 020507 теризующие активности типовых соединений в отношении PPAR, в целом, составляют более чем 10 мкМ, это подтверждает, что указанные соединения не активируют изоформы PPAR. Если R5 представляет собой -СССН 3, то соединения формулы (I) содержат хиральный центр. В настоящем изобретении описаны рацемические соединения, а также индивидуальные изомерные формы. Если R5 представляет собой -СССН 3, то S изомер, в целом, является предпочтительным. Соединения формулы (I), где R5 представляет собой -СCCH3, являются предпочтительными. Кроме того, соединения, где R5 представляет собой Н, a R1 представляет собой F, являются предпочтительными. Соединения, где R1 представляет собой Н, являются предпочтительными, если R5 представляет собой -СCCH3. Соединения, где X выбран из -СН 2 СН 2- и -СН=СН-, являются предпочтительными. Соединения, где X представляет собой -СН=СН-, являются предпочтительными. Соединения, где Z2 представляет собой -S-, являются предпочтительными. Соединения, где Z1 представляет собой -C(R3)-, являются предпочтительными. Соединения, где Z3 представляет собой -C(R4)-, являются предпочтительными. Соединения, где R3 выбран из Н и CH3, являются предпочтительными. Соединения, где R4 представляет собой Н, являются предпочтительными. Соединения, где X представляет собой -N(CH3)CH2-, являются предпочтительными. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой Н; R5 представляет собой -СCCH3; -X представляет собой -СН=СН-, Z2 представляет собой -S-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой Н; R5 представляет собой -СCCH3; -X представляет собой -СН=СН-, Z2 представляет собой -S-; Z1 представляет собой -C(R3)-; Z3 представляет собой -C(R4)-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой Н; R5 представляет собой -СCCH3; -X представляет собой -СН=СН-, Z2 представляет собой -S-; Z1 представляет собой -C(R3)-; Z3 представляет собой -СН-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой F или Cl; R5 представляет собой Н; X представляет собой -СН=СН-; Z2 представляет собой -S-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой F; R5 представляет собой Н; X представляет собой -СН=СН-; Z2 представляет собой -S-; Z1 представляет собой -C(R3)-; Z3 представляет собой -C(R4)-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой F или Cl, если R5 представляет собой Н. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой Н; R5 представляет собой -СCCH3; Z2 представляет собой -S-, а X представляет собой -СН=СН- или -N(CH3)CH2-. В другом варианте реализации настоящего изобретения предложены соединения формулы (I) или их фармацевтически приемлемые соли, где R1 представляет собой Н; R5 представляет собой -СCCH3; Z2 представляет собой -S-, а X представляет собой -СН=СН- или -N(CH3)CH2-; Z1 представляет собой-C(R3)-; Z3 представляет собой -СН-. Другим предпочтительным вариантом реализации настоящего изобретения является S-изомер соединения формулы (I), где R5 представляет собой -СCCH3. Предпочтительным вариантом реализации настоящего изобретения является соединение формулы или его фармацевтически приемлемая соль. Соединения согласно настоящему изобретению предпочтительно входят в состав фармацевтической композиции, которую вводят различными способами. Более предпочтительно указанные композиции предназначены для перорального введения. Подобные фармацевтические композиции и способы их получения хорошо известны в данной области. См., например, Remington: The Science and Practice of"Фармацевтически приемлемая соль" относится к солям соединений согласно настоящему изобретению, которые рассматривают как приемлемые для клинического и/или ветеринарного применения. Фармацевтически приемлемые соли и общие способы их получения хорошо известны в данной области. См., например, P. Stahl, et al., Handbook of Pharmaceutical Salts: Properties, Selection and Use,(VCHA/Wiley-VCH, 2002); S.M. Berge, et al., "Pharmaceutical Salts," Journal of Pharmaceutical Sciences, Vol. 66, No. 1, January 1977. Термин "фармацевтически приемлемый носитель" означает, что носитель, разбавитель, наполнители и соль являются фармацевтически совместимыми с другими ингредиентами композиции. Сокращения, используемые в настоящем описании, определены согласно Aldrichimica Acta, Vol. 17,No. 1, 1984. Другие сокращения определены далее: "Пр.пол." относится к примеру получения; "Пр." относится к примеру; "мин" относится к минуте или минутам; "ADDP" относится к 1,1'(азодикарбонил)дипиперидину; "ДХМ" относится к дихлорметану; "EtOAc" относится к этилацетату;"ТГФ" относится к тетрагидрофурану; "TR" относится к времени удерживания; "IC50" относится к концентрации агента, которая приводит к 50% от максимального ингибирующего ответа, возможного для данного агента; "DMEM" относится к среде Игла, модифицированной Дульбекко; "DTT" относится к дитиотреитуолу; "F12" относится к среде Хэма F12; "ЭБС" относится к эмбриональной бычьей сыворотке; "HEK" относится к почке эмбриона человека; "PPAR" относится к рецептору, активирующему пролиферацию пероксисом; "PPRE" относится к элементу ответа пролифератора пероксисом; "RPMI" относится к Roswell Park Memorial Institute; "TK" относится к тимидинкиназе; "ОЕФ" относится к относительной единице флуоресценции; "ESI" относится к ионизации электрораспылением. На схемах, представленных далее, все заместители, если не указано иное, определены ранее. Реагенты и исходные вещества легкодоступны специалистам в данной области техники, другие можно получать при помощи стандартных способов органической и гетероциклической химии, которые аналогичны способам синтеза известных соединений с аналогичной структурой и способам, описанным на схемах, в примерах получения и примерах, которые включают любые новые способы. Схема I Соединение формулы (I) можно получать при помощи реакций, изображенных на схеме I. На схемеI (стадия 1 а) изображено алкилирование замещенного или незамещенного ароматического гетероцикли-3 020507 ческого бромметилового соединения (1) соответствующим пиперидином (3) с получением, после восстановления сложного эфира на стадии 2, замещенного ароматического гетероциклического метилового спирта (5). К соединению (5) затем можно дополнительно проводить присоединение по гидроксилу в условиях реакции Мицунобу или алкилирование и снятие защиты с получением соединений формулы I."PG" представляет собой защитную группу, предназначенную для кислоты, такую как сложный эфир, а также предназначенную для аминогруппы, такую как карбаматы и амиды. Указанные защитные группы хорошо известны и очевидны в данной области. См., например, Greene and Wuts, Protective Groups in Organic Synthesis, см. выше. Соединение формулы (1) подвергают взаимодействию с соединением формулы (3) в условиях алкилирования (стадия 1 а). Специалисты в данной области понимают, что существует ряд способов и реагентов для алкилирования амина, происходящего в результате реакции гетероциклических бромидов и аминов. Например, взаимодействие соответствующего соединения формулы (1) с соответствующим амином или солью амина, такой как соль трифторуксусной кислоты или соль HCl, формулы (3) в присутствии основания, такого как диизопропилэтиламин или карбонат цезия, приводит к получению соединения формулы (4). Сложный эфир формулы 4 можно восстанавливать до спирта, как показано на стадии 2,с применением восстановителя, такого как гидрид диизобутилалюминия, алюмогидрид лития или боргидрид натрия, с получением соединения (5). Соединение (5) затем можно дополнительно алкилировать соединениями формулы (7) в условиях реакции Мицунобу, а затем проводить снятие защиты с получением соединений формулы (I) (стадия 4 а). Условия реакции Мицунобу хорошо известны в данной области и включают взаимодействие спирта (5) с нуклеофилом, таким как фенол (7) с применением фосфина,такого как трибутилфосфин, трифенилфосфин или триэтилфосфин, и азодикарбонила, такого как ADDP или азодикарбоксилата, такого как диэтилазодикарбоксилат (DEAD). В качестве альтернативы, можно проводить защиту соединения 5 при помощи защитной группы, такой как метилсульфонил, с получением соединения (6, стадия 3). Соединение (6) затем можно подвергать взаимодействию с (7) с применением основания, такого как карбонат цезия, а последующее снятие защиты с кислоты приводит к получению соединений формулы I. Согласно другому варианту соединение формулы (1) можно восстанавливать до бромметилгетероциклического метилового спирта (2), как показано на стадии 1b, с применением восстановителя, такого как гидрид диизобутилалюминия, или соединение (2) может быть коммерчески доступным и его можно алкилировать соответствующим амином или солью амина формулы (3, стадия 1 с) в присутствии основания, такого как диизопропилэтиламин или карбонат цезия, с получением соединения (5), а затем обрабатывать согласно представленному выше описанию с получением соединений формулы (I). На дополнительной стадии можно получать фармацевтически приемлемую соль соединения формулы (I) в результате взаимодействия соответствующей кислоты формулы (I) и соответствующего фармацевтически приемлемого основания в подходящем растворителе в стандартных условиях. Дополнительно, образование указанных солей может происходить параллельно с гидролизом сложного эфира. Получением таких солей хорошо известно и применяется в данной области техники. Схема II мещенного спиро[индолин-3,4'-пиперидина] (10), который затем можно алкилировать с получением соединений формулы (I). Например, к фенилгидразину (9) и соответствующей кислоте, такой как трифторуксусная кислота, добавляют 4-формилпиперидин с защищенным атомом азота (8). После соответствующего времени проведения реакции добавляют восстановитель, такой как боргидрид натрия, и спирт,такой как метанол, с получением целевого замещенного спиро(индолин-3,4'-замещенного пиперидина)(10). На стадии 2 атом азота индолина (10) можно алкилировать, например, путем восстановительного алкилирования с применением соответствующего альдегида и соответствующего восстановителя, такого как цианоборгидрид натрия, в соответствующей кислоте, такой как уксусная кислота, и спирте, таком как метанол. Защитную группу атома азота пиперидина можно удалять в стандартных условиях, хорошо известных в данной области, таких как условия гидрогенирования или кислые условия, с получением соединения (11). Соединение (11) можно затем алкилировать на стадии 3 с (1), описанным ранее на стадии 1 а схемы I, с получением соединения (12) и проводить восстановление на стадии 4, как описано ранее на схеме I (стадии 1 а и 2), с получением соединения (5'). Соединение (5') затем можно обрабатывать с получением соединений формулы (I), как описано на схеме I, по существу, аналогично соединению (5). Схема III На схеме III замещенную гетероциклическую кислоту восстанавливают в условиях, хорошо известных в данной области, с применением восстановителя, такого как DIBAL-H, с получением соединения(2) на стадии 1. На стадии 2 можно проводить защиту метилового спирта (2) защитной группой, такой как трет-бутилдиметилсилан, с применением основания, такого как имидазол и третбутилхлордиметилсилан, с получением (14). Соединение (14) можно алкилировать соединением (7) с применением основания, такого как карбонат калия, и снимать защиту с получением соединения (15) на стадии 3. Спиртовую группу соединения (15) можно превращать в альдегид при помощи окислителя,такого как перйодинан Десс-Мартина, с получением соединения (16) на стадии 4 или в бромид с применением условий бромирования, таких как трибромид фосфора, с получением соединения (16). Соединение (16) затем можно алкилировать соединением (3), схема I, в условиях, хорошо известных в данной области, таких как восстановительное алкилирование цианоборгидридом натрия, и после снятия защиты,можно получать соединения формулы (I). В качестве альтернативы, бромид можно алкилировать соединением (3) с применением основания, такого как карбонат цезия или N,N-диизопропилэтиламин, и проводить снятие защиты с получением соединений формулы (I). Примеры получения и примеры Следующие примеры получения и примеры дополнительно иллюстрируют изобретение и представляют типовые способы синтеза соединений формулы (I). Названия соединений согласно настоящему изобретению получены при помощи IUPACNAME ACDLABS и Symyx Draw 3.2. Пример получения 1. Бензилспиро[индолин-3,4'-пиперидин]-1'-карбоксилат Раствор фенилгидразина (1,29 г, 12,0 ммоль) и трифторуксусной кислоты (3,0 мл) в растворе толуол/ацетонитрил (49/1, 50 мл) нагревали при 35 С. Бензиловый эфир 4-формилпиперидин-1-карбоновой кислоты (2,7 г, 10,91 ммоль) растворяли в растворе толуол/ацетонитрил (49/1, 10 мл) и добавляли по каплям к смеси (WO 2005046682). Смесь перемешивали при 35 С в течение ночи. Полученный раствор охлаждали до 0 С и добавляли метанол (5 мл). Добавляли NaBH4 (0,619 г, 16,38 ммоль) медленно к раствору и смесь перемешивали в течение 45 мин. Смесь промывали водным NH4OH (6%, 25 мл) и солевым раствором (30 мл), сушили над сульфатом натрия и выпаривали насухо с получением желтого твердого вещества. Неочищенное твердое вещество перекристаллизовывали из EtOAc с получением бледножелтого твердого вещества (1,25 г, 1-я часть). Маточный раствор выпаривали и очищали путем флэшхроматографии, элюируя смесью гексан:этилацетат (8:2), с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (1,2 г, 2-я часть) с общим выходом 2,4 г (84%).ESI/MC m/z 323 (М+Н)+. Пример получения 2. Бензил-1-метилспиро[индолин-3,4'-пиперидин]-1'-карбоксилат. К охлажденному до 0 С раствору бензил-1,2-дигидро-1'Н-спиро[индол-3,4'-пиперидин]-1'карбоксилата (315 г, 977,5 ммоль), формальдегида (40% водный раствор, 138 мл, 4,9 моль) и уксусной кислоты (279 мл, 4,9 моль) в метаноле (6,0 л) добавляли цианоборгидрид натрия (184 г, 2,9 моль) по частям в течение 2 ч (наблюдали бурное выделение газов). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 6 ч. рН смеси доводили примерно до 8 при помощи 10% раствора NaHCO3 и экстрагировали в EtOAc (35,0 л). Объединенные экстракты промывали водой(22,5 л) и солевым раствором (2,5 л), сушили над сульфатом натрия и выпаривали насухо с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (310 г, 98,3%).ESI/MC m/z 337 (М+Н)+. Пример получения 3. 1-Метилспиро[индолин-3,4'-пиперидин]. К раствору бензил-1-метилспиро[индолин-3,4'-пиперидин]-1'-карбоксилата (0,85 г, 2,52 ммоль) в метаноле (50 мл) добавляли Pd(OH)2/C (10%, 0,15 г) и смесь гидрогенировали с применением баллона в течение 16 ч. Реакционную смесь фильтровали через диатомитовую землю, промывали метанолом (50 мл) и выпаривали насухо с получением указанного в заголовке соединения (0,5 г, 98%). Энантиомеры 3-(4-гидроксифенил)гекс-4-иновой кислоты разделяли путем хиральной хроматографии [колонка Chiralpak IA (250 мм 4,6 мм), мобильная фаза (А) н-гексан, мобильная фаза (В) изопропиловый спирт с 0,01% TFA; состав (85:15), расход 1,0 мл/мин, детектирование 225 нм] с получением указанного в заголовке соединения (4,2 г, 50,58%), время удерживания 7,96.ESI/MC m/z 203 (М+Н)+. Смесь энантиомеров также разделяли путем хирального разделения с применением способа, аналогичного описанному в WO 2005086661, с получением указанного в заголовке соединения. К раствору метил-4-метилтиофен-2-карбоксилата (2,7 г, 6,4 ммоль) в дихлорметане (20 мл) добавляли 48% водн. HBr (12 мл), H2SO4 (6 мл), ZnBr2 (5,4 г) и НСНО (2,2 мл, 37%) при 0-5 С. Смесь перемешивали при комнатной температуре в течение 16 ч. Раствор разбавляли водой, экстрагировали в дихлорметане, промывали раствором NaHCO3, насыщенным солевым раствором, водой, сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения в виде беловатого твердого вещества (3,5 г, 81%). Пример получения 6. Этил-2-(2-гидроксипропиламино)-2-оксоацетат К перемешиваемому раствору 1-аминопропан-2-ола (15,0 г, 199,7 ммоль) в ДХМ (150,0 мл) добавляли триэтиламин (43,30 мл, 299,56 ммоль) при 0 С. Этиловый эфир хлороксоуксусной кислоты (22,35 мл, 199,7 ммоль) добавляли по каплям к реакционной смеси при 0 С, реакционную смесь перемешивали в течение 16 ч при комнатной температуре. Смесь разбавляли водой (100 мл) при 0 С, экстрагировали в ДХМ (2100 мл), промывали водой (250 мл) и солевым раствором (50 мл), сушили над Na2SO4 и выпаривали насухо. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (3,0:7,0), с получением указанного в заголовке соединения в виде бледно-желтого геля К перемешиваемому раствору этил-2-(2-гидроксипропиламино)-2-оксоацетата (10,7 г, 61,07 ммоль) в ДХМ (100,0 мл) добавляли перйодинан Десс-Мартина (25,9 г, 61,07 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, разбавляли водой (100 мл) при 0 С и экстрагировали в ДХМ (2100 мл). Объединенные органические слои промывали водой (250 мл) и насыщенным солевым раствором (50 мл), сушили над Na2SO4 и выпаривали при пониженном давлении. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат(100,0 мл) добавляли POCl3 (4,95 мл, 53,12 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охлаждали, добавляли по частям в воду (100,0 мл) и интенсивно перемешивали. Органический слой промывали насыщенным раствором NaHCO3 (250 мл), водой(250 мл), сушили над Na2SO4 и выпаривали при пониженном давлении. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (8,0:2,0), с получением указанного в заголовке соединения (5,1 г, 61,9%) в виде желтой маслянистой жидкости.ESI/MC m/z 156,2 (М+Н)+. Пример получения 9. Этил-5-(бромметил)оксазол-2-карбоксилат. К перемешиваемому раствору этил-5-метилоксазол-2-карбоксилата (5,1 г, 32,87 ммоль) и NBS (8,19 г, 46,02 ммоль) в четыреххлористом углероде (50,0 мл) добавляли пероксид бензоила (0,79 г, 3,29 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 16 ч. Смесь охлаждали до комнатной температуры и фильтровали через диатомитовую землю. Фильтрат концентрировали в вакууме, неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетатESI/MC m/z (79Br/81Br) 234,1/236,1 [М+Н]+. Пример получения 10. Метил-3-метокситиофен-2-карбоксилат. К смеси метилового эфира 3-гидрокситиофен-2-карбоновой кислоты (5 г, 31,6 ммоль) и K2CO3 (22,2 г, 161 ммоль) в ацетоне (50 мл) добавляли CH3I (10,8 мл, 173 ммоль) по каплям при 0 С. Реакционную смесь нагревали при 55 С в течение 4 ч, охлаждали до комнатной температуры, фильтровали через диатомитовую землю и фильтрат концентрировали в вакууме. Остаток разбавляли водой (100 мл) и экстрагировали в этилацетате (2200 мл). Объединенные органические слои промывали солевым раствором (50 мл), сушили над Na2SO4 и выпаривали в вакууме с получением указанного в заголовке соединения в виде коричневого твердого вещества (5,9 г).ESI/MC m/z 173,1 (М+Н)+. Пример получения 11. Метил-5-(бромметил)-3-метокситиофен-2-карбоксилат. К раствору метилового эфира 3-метокситиофен-2-карбоновой кислоты (5 г, 29 ммоль) в безводном дихлорметане (50 мл) последовательно добавляли водную HBr (22 мл, 37%), 98% H2SO4 (11 мл), ZnBr2(8,5 г) и водный формальдегид (37%, 3,8 мл) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 14 ч. Органический слой отделяли, водный слой экстрагировали в ДХМ (2200 мл). Объединенные органические слои промывали солевым раствором (200 мл), сушили над Na2SO4 и выпаривали в вакууме. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (9,5:0,5), с получением указанного в заголовке соединения (4,4 г, 60%).(5-Бромметил-2-метокситиофен-2-ил)метанол. К раствору метилового эфира 5-бромметил-3-метокситиофен-2-карбоновой кислоты (1,4 г, 5,2 ммоль) в безводном дихлорметане (50 мл) добавляли гидрид диизобутилалюминия (1 М в гексане, 21,0 мл, 21,1 ммоль) по каплям при -78 С. После завершения добавления смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Реакцию гасили водой (15 мл) при -40 С и смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь фильтровали через диатомитовую землю. Фильтрат сушили над сульфатом натрия и концентрировали с получением указанного в заголовке соединения в виде бледно-желтой жидкости (1,2 г, 96%). 1 К раствору (5-бромметил-2-метокситиофен-2-ил)метанола (0,6 г, 2,5 ммоль) в безводном дихлорметане (10 мл) добавляли имидазол (0,516 г, 7,59 ммоль) и трет-бутилхлордиметилсилан (0,412 г, 2,75 ммоль) при 0 С. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Реакцию гасили водой и реакционную смесь концентрировали. Остаток растворяли в ДХМ, промывали водой (25 мл) и солевым раствором (25 мл), сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения (0,9 г). К раствору этилового эфира 3-(2-фтор-4-гидроксифенил)пропановой кислоты (1,03 г, 4,8 ммоль) в ацетонитриле (15 мл) добавляли карбонат калия (2,31 г, 16,8 ммоль) и (5-бромметил-3-метокситиофен-2 илметокси)-трет-бутилдиметилсилан (1,7 г, 4,8 ммоль) при комнатной температуре и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Реакционную смесь концентрировали, разбавляли водой и экстрагировали в EtOAc (230 мл). Объединенные экстракты промывали водой (15 мл) и насыщенным солевым раствором (15 мл), сушили над Na2SO4, фильтровали и концентрировали. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (9,6:0,4), с получением указанного в заголовке соединения (0,5 г, 21%). 1 Н ЯМР (400 МГц, ДМСО d6)7,51 (с, 1 Н), 7,26-7,2 (т, J=8,4 Гц, 1 Н), 6,92-6,89 (д, J=12 Гц, 1 Н),6,83-6,81 (д, J=7,6 Гц, 2 Н), 4,92 (с, 2 Н), 4,82 (с, 2 Н), 4,10-4,05 (кв, J=6,4 Гц, 2 Н), 3,82 (с, 3H), 2,86-2,82 (т,J=16 Гц, 2 Н), 2,61-2,55 (м, 2 Н), 1,21-1,17 (т, J=6,8 Гц, 3H), 0,89 (с, 9 Н), 0,13 (с, 6 Н). Пример получения 15. Этил-3-[2-фтор-4-5-(гидроксиметил)-4-метокси-2-тиенил]метокси]фенил]пропаноат. К раствору этилового эфира 3-4-[5-(трет-бутилдиметилсиланилоксиметил)-4-метокситиофен-2 илметокси]-2-фторфенилпропановой кислоты (0,5 г, 1,0 ммоль) в ТГФ (5 мл) добавляли фторид тетра-нбутиламмония (1 М раствор в ТГФ, 2 мл, 2,0 ммоль) при 0 С, реакционную смесь перемешивали в течение 30 мин при 25 С. Реакцию гасили водой и реакционную смесь выпаривали с получением остатка. Остаток экстрагировали в EtOAc, промывали водой (15 мл) и солевым раствором (15 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения(1,6 г, 4,3 ммоль) в безводном дихлорметане (10 мл), охлажденному до 0 С, добавляли перйодинан ДессМартина (2,76 г, 6,5 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Реакционную смесь разбавляли водой (15 мл), экстрагировали в этилацетате, промывали водой (15 мл), солевым раствором (15 мл), сушили над сульфатом натрия, фильтровали и выпаривали насухо. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (8,2:1,2), с получением указанного в заголовке соединения (0,8 г, 50%). Способ А. К раствору спиро[инден-1,4'-пиперидина] (1,4 г, 6,3 ммоль) в этаноле (28 мл) добавляли метил-5(бромметил)тиофен-2-карбоксилат (1,78 г, 7,6 ммоль) и диизопропиламин (3,31 мл, 19 ммоль) при комнатной температуре, реакционную смесь перемешивали в течение 16 ч при 90 С. Смесь концентрировали, разбавляли водой и экстрагировали в этилацетате (2200 мл). Объединенные экстракты промывали водой (50 мл) и насыщенным солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения (1,5 г, 58%).ESI/MC m/z 340,1 (М+Н)+. Способ В. Гидрохлорид спиро[инден-1,4'-пиперидина] (128,0 г, 560 ммоль) растворяли в этаноле (1,5 л) в атмосфере азота при 30 С. Диизопропилэтиламин (407,4 мл, 1,68 моль) добавляли по каплям при 30 С в течение 10 мин, затем перемешивали в течение 20 мин. Метил-5-(бромметил)тиофен-2-карбоксилат(50% чистота, определенная по данным ЖХМС) (300 г, 1,27 моль) добавляли при 30 С, затем перемешивали при 30 С в течение 10 мин и смесь нагревали до 60 С в течение 1 ч 45 мин. Реакцию исследовали путем ТСХ для определения присутствия исходных веществ. Реакционную смесь перемешивали в течение 13 ч при 30 С. Реакционную смесь концентрировали и растворяли в ДХМ (10 л). HCl (1 Н, 1,5 л) добавляли по каплям при постоянном перемешивании, собирали осаждавшееся белое твердое вещество. Вещество растворяли в ДХМ (1 л), перемешивали в течение 10 мин, фильтровали и сушили в вакууме Следующие соединения получали, по существу, согласно описанию способа по примеру получения 17 (способ А). Способ А. К раствору этил-5-(спиро[инден-1,4'-пиперидин]-1'-илметил)тиофен-2-карбоксилата (1,5 г, 4,4 ммоль) в дихлорметане (30 мл) добавляли DIBAL-H (11 мл, 11,06 ммоль) при -78 С. Реакционную смесь оставляли нагреваться до комнатной температуры в течение 1 ч, затем по каплям добавляли воду при 0 С. Смесь фильтровали через диатомитовую землю, экстрагировали в дихлорметане и выпаривали насухо с получением указанного в заголовке соединения (1,05 г, 76%).ESI/MC m/z 312,1 (М+Н)+. Способ В. Метил-5-(спиро[инден-1,4'-пиперидин]-1'-илметил)тиофен-2-карбоксилат (150,0 г, 446,0 ммоль) растворяли в безводном дихлорметане (3 л) в атмосфере азота при 30 С. Смесь охлаждали до -78 С иDIBAL-H (1,0 М раствор в толуоле) (1,2 л, 960 ммоль) добавляли по каплям при -78 С в атмосфере азота в течение 1 ч при постоянном перемешивании. Реакционную смесь постепенно нагревали до 0 С и перемешивали в течение 3 ч при 0 С и 3 ч при 30 С. Смесь охлаждали до -78 С и насыщенный водный раствор NH4OH (1 л) добавляли по каплям, затем по каплям добавляли ДХМ (5,0 л). Смесь нагревали до комнатной температуры и экстрагировали в ДХМ, промывали водой (2 л) и насыщенным солевым раствором (2 л), сушили над сульфатом натрия и выпаривали насухо. К остатку добавляли диэтиловый эфир(500 мл) и смесь перемешивали в течение 30 мин. Образовывавшийся осадок отфильтровывали, фильтрат выпаривали насухо и объединяли с твердым веществом с получением указанного в заголовке соединения в виде белого твердого вещества (88,0 г, 64%).ESI/MC m/z 312,1 (М+Н)+. Следующие соединения получали, по существу, согласно описанию способа по примеру получения 28 (способ А). К раствору 2-бром-4-метил-5-(спиро[инден-1,4'-пиперидин]-1'-илметил)тиазола (3,8 г, 10,12 ммоль) в ТГФ добавляли n-BuLi (0,713 г, 11,13 ммоль) при -78 С. Смесь нагревали до -45 С и перемешивали в течение 30 мин. ДМФ (1,5 мл) добавляли по каплям к смеси при -78 С, смесь медленно нагревали до комнатной температуры и перемешивали в течение 1 ч. Смесь охлаждали до -78 С, разбавляли водой,затем раствором NH4Cl. Смесь экстрагировали в этилацетате, сушили над Na2SO4 и выпаривали насухо с получением указанного в заголовке соединения (2,8 г, 85%). ESI/MC m/z 325 (М+Н)+. Следующее соединение получали, по существу, согласно описанию способа по примеру получения 39. К раствору 4-метил-5-(спиро[инден-1,4'-пиперидин]-1'-илметил)тиазол-2-карбальдегида (0,600 г, 1,8 ммоль) в ДХМ (6 мл) добавляли DIBAL-H (1 M раствор в гексане, 0,52 г, 3,6 мл, 3,6 ммоль) при -78 С,смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Смесь охлаждали до -78 С,реакцию гасили водой, фильтровали, экстрагировали в ДХМ и выпаривали насухо с получением указанного в заголовке соединения (0,500 г, 83%).ESI/MC m/z 327 (М+Н)+. Следующее соединение получали, по существу, согласно описанию способа по примеру получения 41. К охлажденному до 0 С перемешиваемому раствору [5-(спиро[инден-1,4'-пиперидин]-1'илметил)оксазол-2-ил]метанола (0,15 г, 0,51 ммоль) в ДХМ (10,0 мл) добавляли триэтиламин (0,18 мл,1,26 ммоль) и метансульфонилхлорид (0,05 мл, 0,61 ммоль). Реакционную смесь перемешивали при 0 С в течение 1 ч. Реакционную смесь промывали водой (20,0 мл) и солевым раствором (10,0 мл), экстрагировали в ДХМ, сушили над Na2SO4 и выпаривали насухо с получением неочищенного продукта (0,18 г) в виде коричневой жидкости. Следующее соединение получали, по существу, согласно описанию способа по примеру получения 43. Способ А. К раствору 1,1'-(азодикарбонил)дипиперидина (1,30 г, 5,1 ммоль) в ТГФ (5 мл) добавляли трибутилфосфин (1,21 г, 6,0 ммоль) при 0 С. Через 15 мин при 0 С добавляли раствор [5-(спиро[инден-1,4'пиперидин]-1'-илметил)-2-тиенил]метанола (0,8 г, 3,4 ммоль) в ТГФ (5 мл) и реакционную смесь перемешивали в течение 15 мин при 0 С, затем добавляли этил-(3S)-(4-гидроксифенил)гекс-4-иноат (1,07 г,3,4 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре,фильтровали, разбавляли водой и экстрагировали в этилацетате (2200 мл). Объединенные экстракты промывали водой (50 мл) и солевым раствором (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (8,5:1,5), с получением указанного в заголовке соединения (1,1 г, 61%).ESI/MC m/z 526,2 (М+Н)+. Способ В. Этил-(3S)-(4-гидроксифенил)гекс-4-иноат (30,0 г, 129,0 ммоль) растворяли в ТГФ (600 мл) в атмосфере азота и охлаждали до 0 С. DIAD (31,3 г, 154,9 ммоль) добавляли медленно при 0 С и перемешивали смесь в течение 30 мин. Раствор PPh3 (40,65 г, 154,9 ммоль) и 5-[5-(спиро[инден-1,4'-пиперидин]-1'илметил)-2-тиенил]метанола (48,26 г, 154,9 ммоль) в минимальном количестве ТГФ добавляли к реакционной смеси при 0 С, смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Растворитель выпаривали, остаток растворяли в этилацетате (300 мл), промывали водой (210 объемов) и солевым раствором (10 объемов), сушили над Na2SO4 и выпаривали насухо. Неочищенное вещество очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (9,0:1,0), с получением указанного в заголовке соединения (33,0 г, 48,6%). 1 Н ЯМР (ДМСО-d6)7,44-7,42 (д, 1 Н), 7,32-7,27 (м, 2 Н), 7,22-7,12 (м, 2 Н), 7,03-7,02 (д, 1 Н), 6,976,92 (м, 3H), 6,89-6,88 (д, 1 Н), 6,79-6,77 (д, 1 Н), 6,69-6,67 (д, 1 Н), 5,21 (с, 2 Н), 4,02-4,01 (м, 1 Н), 3,77 (с,2 Н), 2,94-2,91 (д, 2 Н), 2,68-2,62 (м, 3H), 2,41-2,36 (м, 2 Н), 2,10-2,03 (м, 2 Н), 1,77 (с, 3H), 1,39 (с, 1 Н), 1,271,14 (м, 4 Н), 1,13-1,11 (т, 3H). Следующие соединения получали, по существу, согласно описанию способа по примеру получения 45 (способ А). К перемешиваемому раствору [5-(1'Н-спиро[инден-1,4'-пиперидин]-1'-илметил)-1,3-оксазол-2 ил]метил метансульфоната (0,18 г, 0,48 ммоль) в ацетонитриле (10,0 мл) добавляли Cs2CO3 (0,39 г, 1,2 ммоль) и этил-(3S)-3-(4-гидроксифенил)гекс-4-иноат (0,13 г, 0,57 ммоль). Реакционную смесь перемешивали при 80 С в течение 1 ч и фильтровали через диатомитовую землю. Фильтрат выпаривали насухо и очищали путем хроматографии на силикагеле, элюируя смесью гексан:этилацетат (6,0:4,0), с получением указанного в заголовке соединения в виде бледно-желтой маслянистой жидкости (0,07 г, 28,2%).ESI/MC m/z 511,5 (М+1). Следующее соединение получали, по существу, согласно описанию способа по примеру получения 65.NaCNBH3 (0,273 г, 4,3 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Смесь разбавляли водой (15 мл) и экстрагировали в этилацетате. Органический слой промывали водой (10 мл) и солевым раствором (10 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Неочищенное вещество очищали путем препаративной ТСХ с получением указанного в заголовке соединения в виде бесцветной жидкости (0,2 г, 33%).ESI/MC m/z 553,2 (М+Н)+. Следующее соединение получали, по существу, согласно описанию способа по примеру получения 67. Способ А. К раствору этил-(3S)-3-[4-5-(спиро[инден-1,4'-пиперидин]-1'-илметил)-2 тиенил]метокси]фенил]гекс-4-иноата (1,1 г, 2,0 ммоль) в этаноле (6 мл) добавляли 5 М NaOH (0,83 мл,4,1 ммоль) и смесь подвергали микроволновому облучению в течение 5 мин при 90 С. Смесь концентрировали, растворяли в воде, подкисляли 2 н. HCl и экстрагировали в хлороформе (250 мл). Объединенные экстракты промывали насыщенным NaHCO3 (25 мл), NH4Cl (25 мл), водой (25 мл) и насыщенным солевым раствором (25 мл), сушили над сульфатом натрия, фильтровали и концентрировали с получением указанного в заголовке соединения в виде беловатого твердого вещества (0,746 г, 71%). 1 Н ЯМР (ДМСО d6, 400 МГц)12,2 (ушир.с, 1 Н), 7,43-7,41 (д, J=7,2 Гц, 1 Н), 7,31-7,25 (дд, J=6,8 Гц,3H), 7,21-7,13 (м, J=7,2 Гц, 2 Н), 7,02-7,01 (д, J=3,2 Гц, 1 Н), 6,96-6,94 (д, J=8,4 Гц, 1 Н), 6,92-6,90 (д, J=5,6 Гц, 1 Н), 6,88-6,87 (д, J=2,8 Гц, 1 Н) 6,77-6,76 (д, J=5,2 Гц, 1 Н) 5,19 (с, 2 Н), 3,94 (с, 1 Н), 3,76 (с, 2 Н), 2,942,901 (т, J=11,2 Гц, 2 Н), 2,59-2,57 (д, J=7,6 Гц, 2 Н), 2,41-2,36 (т, J=11,2 Гц, 2 Н), 2,08-2,03 (т, J=10,4 Гц,2 Н), 1,76 (с, 3H), 1,22 (с, 2 Н).(33,0 г, 62,7 ммоль) растворяли в этаноле (600 мл) и смесь охлаждали до 10-15 С. 5 М NaOH (5,02 г,125,5 ммоль) медленно добавляли и смесь перемешивали в течение 10 мин, затем нагревали до 80 С в течение 2 ч. Растворитель выпаривали и смесь растворяли в минимальном количестве воды. рН доводили до 7,0 при помощи 1 н. раствора HCl. Раствор экстрагировали в этилацетате (2300 мл), промывали солевым раствором, сушили над сульфатом натрия и выпаривали досуха. К неочищенному веществу добавляли метил-т-бутиловый эфир (300 мл) и 0,2% метанола, смесь перемешивали в течение 30 мин. Полученный осадок отфильтровывали и сушили в вакууме при 48 С с получением указанного в заголовке соединения в виде твердого вещества кремового цвета (24,0 г, 76%). 1 Н ЯМР (ДМСО-d6)12,217 (с, 1 Н), 7,438-7,421 (д, 1 Н), 7,330-7,271 (м, 3H), 7,228-7,126 (м, 2 Н),7,046-7,039 (д, 1 Н), 6,978-6,906 (м, 4 Н), 6,795-6,781 (д, 1 Н), 5,213 (с, 2 Н), 3,972-3,928 (м, 1 Н), 3,788(ушир.с, 2 Н), 2,945 (м, 2 Н), 2,608-2,589 (д, 2 Н), 2,422 (м, 2 Н), 2,107-2,051 (м, 2 Н), 1,778-1,772 (с, 3H),1,233-1,148 (м, 2 Н). Следующие соединения получали, по существу, согласно описанию способа по примеру 1 (способ А). Полагают, что существуют две основные причины T2D: резистентность к инсулину и прогрессирующий недостаток продуцирования соответствующих количеств инсулина бета-клетками поджелудочной железы для снижения уровня глюкозы в кровотоке. Резистентность к инсулину развивается, если инсулин, содержащийся в нормальном количестве, не способен распределять глюкозу, содержащуюся в плазме, по целевым тканям, включая скелетные мышцы и жировую ткань. Так как поджелудочная железа продуцирует большее количество инсулина для компенсации избыточного содержания глюкозы, возникающего в результате резистентности к инсулину, то бета-клетки поджелудочной железы истощаются, и дополнительная секреция инсулина становится невозможной. Со временем бета-клетки поджелудочной железы полностью разрушаются, и симптомы человека, страдающего от T2D, становятся неотличимыми от симптомов диабета 1 типа. Высокое содержание глюкозы в большом круге кровообращения является отличительным признаком диабета и в итоге может приводить к серьезным осложнениям, таким как заболевания сердца, инсульт, высокое кровяное давление, слепота, повреждения почек и нервной системы,инфекции и пародонтит. Следовательно, важно контролировать и лечить T2D настолько рано, насколько это возможно, при помощи упражнений; соответствующей диеты; пероральных противодиабетических терапий; и, наконец, инсулина. Соединения, заявленные в настоящем изобретении, обеспечивают дополнительные способы фармакологического лечения. Соединения, селективно модулирующие GPR40, могут быть особенно предпочтительными.GPR40. Информация. Результаты исследований трансгенных мышей, сверхэкспрессирующих человеческий ген GPR40 под контролем промотора инсулина II, представленные недавно Nagasumi, также подтверждают, чтоGPR40 играет важную роль в регуляции GDIS и содержания глюкозы в плазме in vivo, в частности, на моделях резистентности к инсулину грызунов. Nagasumi, K. et al., Overexpression of GPR40 in pancreaticGPR40 is activated by medium and long chain fatty acids, Journal Biological Chemistry 278: 11303-11311,2003. Эти факты также подтверждают, что разработка новых соединений, модулирующих GPR40, может быть особенно желательной для применения для лечения T2D. Первичное исследование тока кальция. Соединения, представленные в настоящем описании, исследовали по существу согласно описанию,представленному далее, и указанные соединения имеют значение EC50 в первичном исследовании тока кальция, составляющее менее 1 мкМ. Это исследование применяют для исследования соединений путем измерения увеличения внутриклеточного содержания кальция, которое возникает в результате присоединения лиганда и активацииGPR40, что, таким образом, показывает активность и эффективность агонистов GPR40. Клетки HEK293,сверхэкспрессирующие кДНК GPR40 человека, которые выдерживали в модифицированной Дульбекко среде Игла и среде F12 в соотношении 3:1, содержащих 10% ЭБС и 800 мкг/мл генетицина, при 37 С и 5% СО 2, использовали для исследования. Исследования агонистов проводили с применением набора для исследования Calcium D4 Dye (Molecular Devices) в присутствии (0,1%) или отсутствии не содержащего жирных кислот БСА в буфере для исследования (1X HBSS (сбалансированный солевой раствор Хэнка) и 20 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфокислота). Активацию рецептора измеряли в виде увеличения внутриклеточного содержания кальция с применением спектрофотометра для чтения планшетов для визуализации флуоресценции (FLIPR). Максимальное изменение флуоресценции относительно базовой линии применяли для определения ответа агониста. Значение EC50 (эффективная концентрация для половины максимального ответа) для соединения рассчитывали с применением программного обеспечения Excel Fit (версия 4; IDBS) путем построения зависимости концентрации от относительных единиц флуоресценции (ОЕФ). Эффективность в процентах рассчитывали с учетом максимального ответа, полученного для соединения по сравнению с естественным лигандом, линолевой кислотой. Исследуемое соединение по примеру 1 обладало значением EC50, составляющим 11493 нМ, и эффективностью 919%, определенной в этом же исследовании. Эти результаты также подтверждают желательную активность и эффективность соединений в качестве агонистов GPR40. Исследования глюкозозависимой секреции инсулина (GDIS). Так как известно, что активация GPR40 приводит к секреции инсулина, которая зависит от высоких концентраций глюкозы, были разработаны две различные системы исследований (клеточная линия инсулиномы и первичные островки грызунов) для дополнительной характеристики соединений, которые увеличивают внутриклеточное содержание кальция в первичном исследовании GPR40, обсуждаемом выше. Исследования GDIS проводили с применением клеточной линии инсулиномы мышей линии Min6. Клетки Min6 выдерживали в модифицированной Дульбекко среде Игла (DMEM), содержащей заменимые аминокислоты, 10% ЭБС, 50 мМ 2-меркаптоэтанола и 1% пенициллина и стрептомицина, при 37 С и 5% СО 2. В день эксперимента клетки промывали дважды 200 мкл предварительно нагретого буфера Кребса-Рингера, не содержащего глюкозу. Добавление 200 мкл предварительно нагретого буфера Кребса-Рингера, содержащего 2,5 мМ глюкозы применяли для истощения клеток, затем добавляли соединения в присутствии глюкозы в высокой концентрации (25 мМ). Планшет инкубировали при 37 С в течение 2 ч. После завершения 2 ч инкубирования надосадочную жидкость осторожно переносили на фильтрующий планшет Millipore и центрифугировали при 200 g в течение 3 мин. Содержание инсулина исследовали с применением набора Mercodia Insulin estimation. Добавление соединения по примеру 1 в концентрации 1 мкМ и 25 мМ глюкозы к клеткам Min6 приводило к статистически значимому двукратному увеличению секреции инсулина по сравнению с увеличением, возникающим при применении исключительно 25 мМ глюкозы. Исследования GDIS с применением первичных островков. Лангерганса поджелудочной железы грызунов также применяли для характеристики представленных соединений. Островки поджелудочных желез выделяли у самцов крыс линии SD (Спраг-Доули) путем усвоения коллагеназы и разделения по градиенту плотности Histopaque. Островки культивировали в течение ночи в среде RPMI-1640, содержащей GlutaMAXn (стабилизированная дипептидная форма Lглутамина (Invitrogen, номер в каталоге 61870-010, для ускорения выделения. Секрецию инсулина определяли путем 90-минутного инкубирования в буфере EBSS (сбалансированный солевой раствор Эрла) в 48-луночном планшете. Вкратце, островки сначала предварительно инкубировали в EBSS с 2,8 мМ глюкозы в течение 30 мин, а затем переносили в 48-луночный планшет (четыре островка/лунку), содержащий 150 мкл 2,8 мМ глюкозы, и инкубировали с 150 мкл EBSS, содержащего 2,8 или 11,2 мМ глюкозы в присутствии или отсутствии исследуемых соединений в течение 90 мин. Буфер удаляли из лунок после завершения инкубирования и исследовали содержание инсулина с применением набора Rat Insulin ELISA (Mercodia). В этой системе для исследования введение соединения по примеру 1 в различных концентрациях приводило к 2-4-кратному увеличению содержания инсулина по сравнению с содержанием при применении исключительно 11,2 мМ глюкозы. Исследования селективности. Исследования ,исвязывания рецептора, активирующего активацию пероксисом (PPAR) и функциональные исследования. Так как известно, что GPR40 активируется лигандами PPAR, то представленные соединения исследовали на связывание PPAR, PPAR и PPAR, а также проводили функциональные исследования для определения селективности представленных соединений к GPR40. Представленные соединения исследовали по существу согласно представленному далее описанию связывания PPAR, и в целом они имели значения связывания, составляющие более чем 1000 нМ при концентрациях исследуемого соединения, составляющих 10 мкМ, и, таким образом, показано, что они неактивны в отношении PPAR. Аффинности связывания соединений и рецепторов PPAR ,иопределяли с применением технологии сцинтилляционного анализа сближения (SPA). Биотинилированный олигонуклеотид Прямой Повтор 2 (DR2) применяли для связывания рецепторов с частиц SPA из силиката иттрия, покрытых стрептавидином. Рецепторы PPAR ,ии ретиноидов X (RXR) сверхэкспрессируются в клетках НЕК 293 и клеточные лизаты, содержащие специфические рецепторы, применяли для конкретных исследований.HEPES рН 7,8, 80 мМ KCl, 0,5 мМ MgCl2, 1 мМ DTT, 0,5% 3-[(3-холамидопропил)диметиламмоний]пропансульфокислоты (CHAPS) и 4,4% бычьей сыворотки. Клеточные лизаты инкубировали в лунках,- 20020507 содержащих одну из 11 концентраций соединения в присутствии соединений сравнения, радиоактивного меченого (0,033,8 мкКи 3 Н) двойного агониста PPAR/ для исследований рецептора альфа и дельта и радиоактивного меченого (0,037,3 мкКи 3 Н) агониста PPAR для исследования рецептора гамма, 110,3 мкг покрытых оболочкой стрептавидина частиц SPA из иттрия, 0,126 нМ HD Oligo DR2 и либо 0,3 мкгPPAR с 0,5 мкг RXR, 0,5 мкг PPAR с 0,5 мкг RXR или 1,25 мкг PPAR с 3,03 мкг RXR в буфере для связывания, описанном выше, а также 14% глицерина и 5 мкг дегидратированной в результате гидродинамического сдвига ДНК молок лососевых. Неспецифическое связывание определяли в присутствии 10000 нМ соединений сравнения, немеченого двойного агониста PPAR / для исследований рецепторов альфа и дельта и агониста PPAR для исследования рецептора гамма. Реакционную смесь для связывания(100 мкг на лунку в 96 луночном планшете [Costar 3632]) инкубировали в течение 10 ч и подсчитывали количество распадов в минуту (dpm) на Wallac Microbeta. Аффинность связывания рецептора (IC50) для соединений определяли путем построения кривой концентрация-ответ по 11 точкам с применением 4 параметрового логистического уравнения. Ki определяли по значению IC50 с применением уравнения Ченга-Прусоффа, a Kd определяли путем насыщения связывания. Для соединения по примеру 1 связывание не было детектировано ни в одном из трех исследований связывания PPAR для концентраций, не превышающих 10 мкМ. Таким образом, исследования, представленные в настоящем описании, подтверждают, что соединение по примеру 1 селективно активирует GPR40 и не вызывает нежелательную активность PPAR. Относительные значения IC50 представленных соединений, в целом, составляли более чем 10 мкМ для изоформ PPAR, что подтверждает, что соединения не вызывают активность PPAR, обеспечивая целевую активация GPR40. Функциональные исследования репортеров Gal4 PPAR, Gal4 PPAR и PPAR также применяли для исследования селективности представленных соединений. Клетки CV1, которые получали из почечной ткани африканской зеленой мартышки, трансфицировали различными рецепторами и репортерными плазмидами с применением Fugene. Для исследований Gal4 PPAR и PPAR репортерную плазмиду,содержащую пять тандемных копий элемента ответа транскрипционного белка Gal4 дрожжей, клонированных выше гена люциферазы светлячков, находящуюся под контролем основного позднего промотора аденовируса, трансфицировали совместно с плазмидой под контролем SV40, устойчиво экспрессирующей гибридный белок, содержащий ДНК-связывающий домен (DBD) Gal4, a также связывающий PPAR или PPAR лиганд. Для исследования PPAR плазмиды, кодирующие PPAR и RXR под контролем промотора цитомегаловируса (CMV), трансфицировали совместно с плазмидой, содержащей кДНК репортера люциферазы под контролем промотора TK, и элемент ответа рецептора (2 Х PPRE). Клетки трансфицировали в Т 225 см 3 колбы для клеточных культур в среду DMEM, содержащей очищенную на активированном угле 5% ЭБС. После ночи инкубирования трансфицированные клетки трипсинизировали, помещали в непрозрачные 96-луночные планшеты (15000 клеток/лунку) в среде DMEM, содержащей очищенную на активированном угле 5% ЭБС, инкубировали в течение 4 ч и подвергали воздействию от 0,17 нМ до 10 мкМ исследуемых соединений или соединения сравнения с проведением полулогарифмических разбавлений. После 24 ч инкубирования с соединениями клетки лизировали и определяли активность люциферазы, как меру активации рецептора, при помощи люминесценции. Данные вводили в четырехпараметровую логистическую модель для определения значений ЕС 50. Максимальную стимуляцию определяли в процентах от максимальной стимуляции, достигаемой при 10 мкМ соответствующего агониста PPAR сравнения. Для соединения по примеру 1 не детектировали активацию PPAR, PPAR илиPPAR при исследовании концентраций, не превышающих 10 мкМ, в исследованиях специфической совместной трансфекции PPAR/функциональных исследованиях, описанных выше. Таким образом исследование подтверждает, что представленные соединения не вызывают активности PPAR, что является желательным. Эффективность in vivo. Интраперитонеальный тест на толерантность к глюкозе (IPGTT). Для исследования способности представленных соединений активировать GPR-40 in vivo, приводящей к противодиабетической эффективности, т.е. к увеличению содержания инсулина и снижению содержания глюкозы, проводили интраперитонеальный тест на толерантность к глюкозе (IPGTT) для каждого соединения, исследуемого в IPGTT. Самцов мышей Balb/с (мыши-альбиносы) (возраст 8-9 недель) помещали в индивидуальные клетки и предоставляли доступ к традиционной пище грызунов и воде ad libitum. Животных взвешивали и произвольно распределяли по группам по массе тела и ежедневно записывали массу тела. После начала исследования животным дозировали перорально один раз день в течение трех дней состав, содержащий метилцеллюлозу и tween-80. Ночью перед 4 днем исследования IPGTT животных подвергали голоду в течение ночи в чистых клетках. Утром IPGTT (4 день) животным дозировали перорально соединение или только носитель за 60 мин перед IPGTT (глюкоза 2 г/кг i.p.). Содержание глюкозы в крови определяли по образцам крови, взятых из хвоста через 0, 3, 7, 15, 30 и 60 мин после введения глюкозы. Плазму отделяли и применяли для оценки соответствующего содержания инсулина. Данные, характеризующие профиль колебания глюкозы в крови в диапазоне от t=0 до t=60, интегрировали с получением площади под кривой(AUC) для каждого способа лечения. Снижение содержания глюкозы в процентах рассчитывали по от- 21020507 ношению данных AUC для соединений и AUC группы, которой вводили носитель. Исследуемое соединение перорально вводили в дозах 0,1, 0,3, 1,0, 3,0 или 10 мг/кг, для положительного контроля вводили дозу 10 мг/кг. Отсутствие соединения по примеру 1 или положительный контроль приводили к значительному снижению содержания глюкозы через 3 мин GTT. В противоположность этому содержание глюкозы значительно снижалось для доз соединения по примеру 1, составляющих 0,3, 1,0, 3,0 и 10 мг/кг,и положительного контроля через 7 мин и для доз соединения по примеру 1, составляющих 0,1, 0,3, 1,0 и 3,0 мг/кг, через 15, 30 и 60 мин. Положительный контроль значительно снижал содержание глюкозы через 30 и 60 мин. Значение ED50 для соединения по примеру 1, рассчитанное по данным AUC снижения содержания глюкозы, составляло 0,21 мг/кг. В этом исследовании содержание инсулина значительно повышалось для соединения по примеру 1 в дозах, составляющих 3,0 и 10,0 мг/кг, что подтверждает активацию GPR40. Результаты этого исследования показывают, что активация GPR40 соединением по примеру 1 приводит к противодиабетической эффективности in vivo. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы или его фармацевтически приемлемая соль; где R1 выбран из группы, состоящей из Н, F и Cl;R4 выбран из группы, состоящей из Н, ОСН 3 и СН 3; где по меньшей мере один из группы, состоящей из Z1 и Z3, представляет собой -C(R3)- или -C(R4)-. 2. Соединение или его соль по п.1, отличающееся тем, что R1 представляет собой Н. 3. Соединение или его соль по любому из пп.1, 2, отличающееся тем, что R5 представляет собой-СCCH3. 4. Соединение или его соль по п.3, отличающееся тем, что соединение является S-изомером. 5. Соединение или его соль по п.1, отличающееся тем, что R1 представляет собой F, a R5 представляет собой Н. 6. Соединение по любому из пп.1-5, отличающееся тем, что Z2 представляет собой -S-. 7. Соединение или его соль по любому из пп.1-6, отличающееся тем, что Z1 представляет собой-C(R3)-. 8. Соединение или его соль по любому из пп.1-7, отличающееся тем, что R3 представляет собой Н или СН 3. 9. Соединение или его соль по любому из пп.1-8, отличающееся тем, что Z3 представляет собой-C(R4)-. 10. Соединение или его соль по п.9, отличающееся тем, что R4 представляет собой Н. 11. Соединение или его соль по любому из пп.1-10, отличающееся тем, что X представляет собой-СН=СН-. 12. Соединение или его соль по любому из пп.1-10, отличающееся тем, что X представляет собой-N(R7)CH2-. 13. Соединение или его соль по п.12, отличающееся тем, что R7 представляет собой СН 3. 14. Соединение по любому из пп.1-13, отличающееся тем, что указанное соединение представляет собой фармацевтически приемлемую соль. 15. Соединение формулы или его фармацевтически приемлемая соль. или его фармацевтически приемлемая соль. 17. Фармацевтическая композиция для лечения диабета, содержащая фармацевтически приемлемый носитель и соединение по любому из пп.1-16 или его фармацевтически приемлемую соль. 18. Применение соединения по любому из пп.1-16 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения диабета. 19. Применение соединения по любому из пп.1-16 или его фармацевтически приемлемой соли для лечения диабета.
МПК / Метки
МПК: A61K 31/438, C07D 405/06, C07D 471/10, C07D 409/06, C07D 417/06, C07D 413/06, A61P 3/10
Метки: соединения, спиропиперидиновые
Код ссылки
<a href="https://eas.patents.su/24-20507-spiropiperidinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Спиропиперидиновые соединения</a>
Спиропиперидиновые соединения в качестве антагонистов рецептора orl-1

Номер патента: 20391
Опубликовано: 30.10.2014
Авторы: Лафуэнте Бланко Селия, Бенито Колладо Ана Белен, Педрегал-Терсеро Консепсьон, Мартинес-Грау Мария Анхелес, Толедо Эскрибано Мигель Анхель, Диас Буэсо Нурия, Хименес-Агуадо Альма Мария
МПК: C07D 495/20, A61P 25/00, A61K 31/438...
Метки: качестве, антагонистов, рецептора, orl-1, соединения, спиропиперидиновые
Формула / Реферат:
1. Соединение формулыгдеR1 представляет собой фтор или хлор;каждый из R2a и R2b представляет собой водород или фтор;R3 представляет собой водород, метил, гидроксиметил или (С1-С3)алкоксиметил;R4 выбран из группы, состоящей из фтора, хлора, циано, цианометила, (C1-С3)алкила, циклопропила, гидроксиметила, метокси, метоксиметила, аминокарбонилоксиметила, метиламинокарбонилоксиметила, диметиламинокарбонилоксиметила, метилкарбонила, аминокарбонила,...
Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения

Номер патента: 12786
Опубликовано: 30.12.2009
Авторы: Колетта Изабелла, Ализи Мария Алессандра, Фурлотти Гвидо, Каццолла Никола, Поленцани Лоренцо, Драгоне Патриция, Руссо Винченцо, Мангано Джорджина
МПК: C07D 209/88, A61P 35/00, A61K 31/403...
Метки: фармацевтическая, содержащая, 3-аминокарбазола, композиция, указанные, способ, получения, соединения
Формула / Реферат:
1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы и их фармацевтически приемлемые соли. 2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Данвидди Кристофер Т., Кюродо Алан Х., Юзан Андре, Перроне Марк Х., Лидли Роберт Дж.
МПК: A61P 7/02, A61K 31/715
Метки: эти, активностью, агрегации, соединения, фармкомпозиция, содержащий, лечения, тромбоцитов, сопутствующих, содержащая, применение, анти-ха, антагониста, профилактики, способ, набор, тромбообразованию, заболеваний
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Ким Геун Тае, Ким Киоунг-Хее, Кох Дзонг Сунг, Йим Хиеон Дзоо, Ли Чанг-Сеок, Йео Донг-Дзун, Ким Дзи Янг, Йеом Зи-Хо, Ким Сунгсуб, Квон Ох Хван, Хур Гвонг-Чеунг, Хан Хее Оон, Ким Мин-Дзунг, Хонг Санг Йонг, Коо Ки Донг, Ким Хие Дзин, Бу Сеонг Чеол, Лим Донгчул, Ким Сунг Хо
МПК: A61K 31/444, A61K 31/452, A61P 3/10...
Метки: способы, композиции, также, ингредиента, содержащие, фармацевтические, дипептидилпептидазы-iv, активного, соединения-ингибиторы, указанные, получения, качестве, соединения
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения

Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Вийенов Николь, Кеньяр Паскаль, Гумен Бертран, Шименти Стефано, Пеглион Жан-Луи, Толлон Катрин, Вилен Жан-Поль, Дессинже Эмея
МПК: C07D 223/16, A61P 9/00, A61K 31/55...
Метки: фармацевтические, способ, эти, получения, композиции, 1,2,4,5-тетрагидро-3н-бензазепина, соединения, содержащие
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или...
Предыдущий патент: Бициклические соединения в качестве лигандов a4b2 никотинового ацетилхолинового рецептора
Следующий патент: Выделенные антитела к sp35 и их применение
Случайный патент: Экологически безопасная обработка криогенными текучими средами в конденсированной фазе