Производные пирроло[2,3-d]пиримидина в качестве ингибиторов протеинкиназы в
Номер патента: 18512
Опубликовано: 30.08.2013
Авторы: Матусьяк Збигнев Станли, Лич Эндрью, Льюк Ричард Уилльям Артур, Моррис Джеффри Джеймс, Джонсон Пол Дейвид





















Формула / Реферат
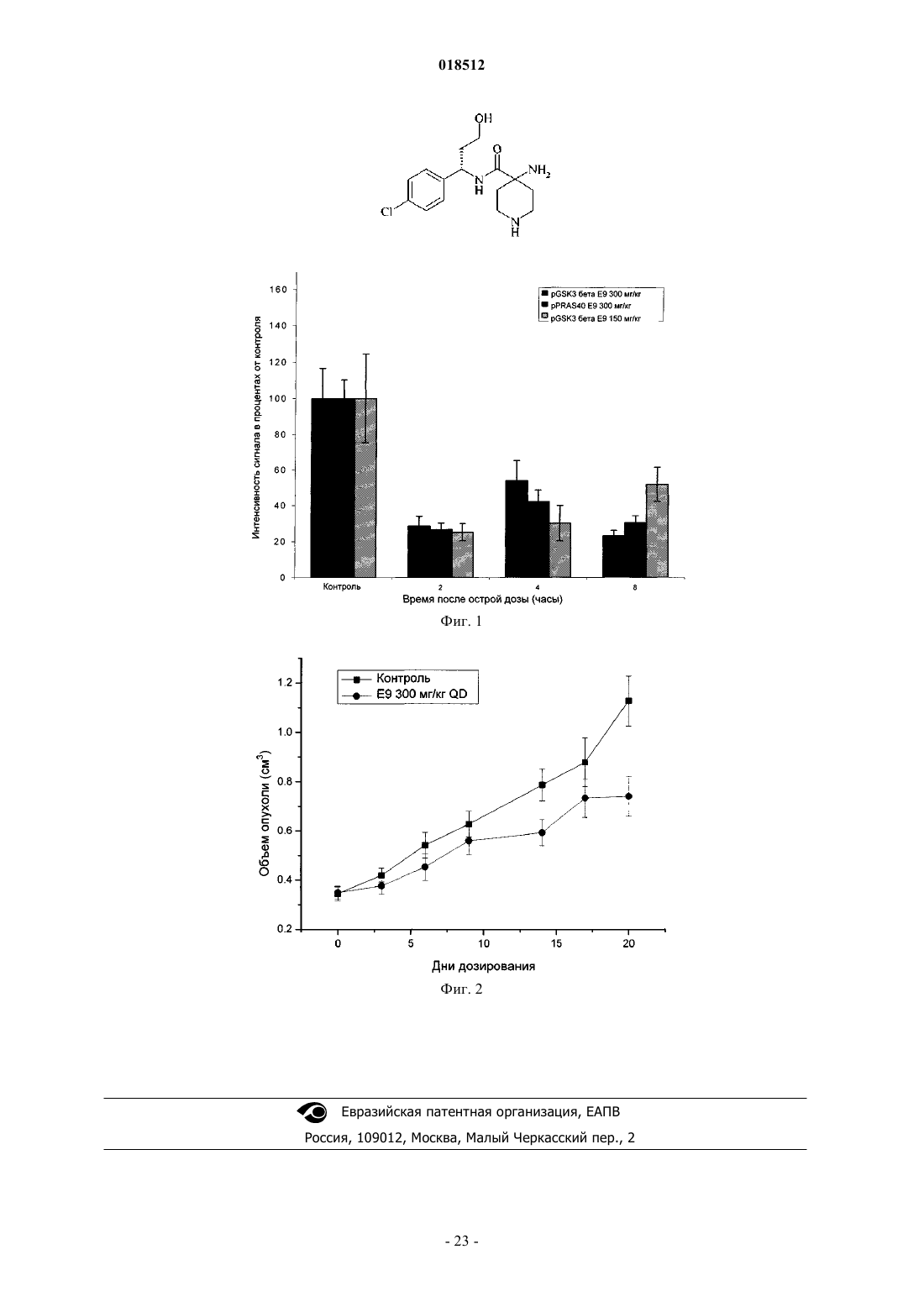
1. Соединение (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид

или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция, включающая соединение по п.1 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем или носителем.
3. Применение соединения (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида

или его фармацевтически приемлемой соли для приготовления медикамента, предназначенного для лечения рака.
4. Применение соединения, определенного в п.3, или его фармацевтически приемлемой соли для приготовления медикамента, предназначенного для лечения рака молочной железы.
5. Соединение по п.1, которое представляет собой (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид.
6. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий реакцию кислоты формулы (II) с (S)-3-амино-3-(4-хлорфенил)пропан-1-олом

где Р1 означает пригодную защитную группу;
и затем, при необходимости,
(i) удаление каких-либо защитных групп и/или
(ii) образование его фармацевтически приемлемой соли.
7. Способ по п.6, в котором Р1 означает трет-бутоксикарбонильную защитную группу.
8. Соединение (S)-трет-бутил 4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамат

9. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий реакцию (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамида с бициклическим гетероциклом формулы (V)

где L1 означает пригодную уходящую группу;
и затем, при необходимости, образование его фармацевтически приемлемой соли.
10. Способ по п.9, в котором L1 означает хлор.
11. Соединение (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамид

Текст
ПРОИЗВОДНЫЕ ПИРРОЛО[2,3-D]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ В Настоящее изобретение относится к соединению (S)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида или его фармацевтически приемлемой соли, которое может быть полезным при лечении или предупреждении заболевания или состояния, опосредованного протеинкиназой В (PKB), такого как рак. Изобретение также относится к фармацевтической композиции, включающей вышеуказанное соединение, к применению вышеуказанного соединения для лечения заболеваний, опосредованныхPKB, и к способам получения этого соединения. Джонсон Пол Дейвид, Лич Эндрью,Льюк Ричард Уилльям Артур,Матусьяк Збигнев Станли, Моррис Джеффри Джеймс (GB) Веселицкая И.А., Пивницкая Н.Н.,Кузенкова Н.В., Веселицкий М.Б.,Каксис Р.А., Комарова О.М., Белоусов Ю.В. (RU) Настоящее изобретение относится к бициклическому гетероциклу, который может быть полезным при лечении или предупреждении заболевания или медицинского состояния, опосредованного протеинкиназой В (PKB, также известна как AKT). Следовательно, такое соединение может быть полезным при лечении или предупреждении ряда различных типов рака. Изобретение также относится к фармацевтическим композициям, включающим вышеупомянутое соединение, к способам получения вышеупомянутого соединения и к способам лечения заболеваний, опосредованных PKB с применением вышеупомянутого соединения.PKB является компонентом фосфатидил 3-киназного (PI3K) сигнального пути, который играет важную роль в пролиферации и выживании клеток, в том числе в клеточных ответах на факторы роста. После присоединения фактора роста, например эпидермального фактора роста (EGF), к его рецепторной тирозинкиназе на клеточной поверхности, например EGF рецептору (EGFR), рецептор димеризуется и подвергается автофосфорилированию. Это событие автофосфорилирования позволяет 85 кДа регуляторной субъединице PI3K (р 85) взаимодействовать с рецептором либо непосредственно, либо через адаптерный белок, например связанный с рецептором фактора роста белок 2 (GRB2), и таким образом активирует 110 кДа каталитическую субъединицу PI3K (p110). После активации, p110 катализирует фосфорилирование фосфатидилинозитол-4,5-бисфосфата (PIP2) с получением фосфатидилинозитол-3,4,5 трифосфата (PIP3), молекулы вторичного мессенджера, которая прикрепляет и фосфатадилинозитолзависимую киназу 1 (PDK1), и PKB к плазматической мембране, где PDK1 фосфорилирует и активируетPKB. Известны три изоформы PKB (PKB/AKT1, PKB/AKT2 и PKB/АКТ 3), производные от трех разных генов. Активирование PKB связывают с событиями в системе клеточных сигналов, которые опосредствуют пролиферацию и выживание клеток, тогда как активирование PKB связывают с инвазией,подвижностью и инсулинопосредованными процессами метаболизма. Активированная PKB защищает клетки от апоптоза путем инактивации проапоптических факторов, например BAD, прокаспазы-9 и транскрипционного фактора forkhead (FKHR), и активации факторов транскрипции, что повышающе регулирует антиапоптозные гены, например циклический-АМР ответный элемент активирующего белка(CREB). PKB также может способствовать выживанию клеток путем инактивирования р 53 через фосфорилирование MDM2. Подобным образом, активированная PKB индуцирует пролиферацию клеток с помощью активирующих белков, вовлеченных в рост и метаболизм клеток, например, посредством пути,ведущего к активированию мишени рапамицина в клетках млекопитающих (mTOR) и через гликогенсинтаза-киназу-3 (GSK3).PKB-опосредованная стимуляция пролиферации клеток и защита от апоптоза, таким образом, содействует образованию опухолей, и при раке обычно находят генетические нарушения компонентов в пределах PI3K пути. Например, мутацию или амплификацию генов, кодирующих p110 изоформы PI3K,находят при раке молочной железы, раке кишечника, раке яичников, раке головы и шеи, и плоскоклеточном раке шейки матки, раке желудка и легких, ангиопластических олигодендроглиомах, амапластических астроцитомах, мультиформной глиобластоме и медуллобластомах. Подобным образом, мутацию,амплификацию и/или сверхэкспрессию генов, кодирующих изоформы PKB, находят при опухолях поджелудочной железы, молочной железы и яичника. Кроме того, ген, кодирующий PTEN (фосфатазу, которая играет роль, обратную PI3K, катализирующую превращение PIP3 в PIP2) инактивируется во многих типах опухолей, в том числе опухолях яичников, колоректальных опухолях, опухолях молочной железы,глиоме, меланоме, опухолях легких, лейкемиях и лимфомах; это приводит к активированию PKB/AKT. Принимая во внимание важность PI3K сигнального пути в пролиферации и выживании опухолевых клеток, какое-либо соединение, которое нарушает этот путь, в том числе PKB ингибиторы, могут быть полезны при лечении рака. Подробные обзоры PI3K сигнального пути и его вовлечения в образование опухолей обеспечены исследователями Hennessy и др., Nature Reviews/Drug Discovery (декабрь 2005) т. 4,988-1004 и Cully и др., Nature reviews/Cancer (март 2006) т. 6, 184-192. Потенциалозависимый калиевый канал, кодированный посредством гена специфических калиевых каналов сердца человека (hERG), как полагают, играет ключевую роль в реполяризации потенциала действия желудочков сердца. Изменения в его активности, вызываемые либо унаследованными мутациями генной последовательности, либо фармакологической модификацией, могут приводить к увеличению продолжительности потенциала действия. Это может привести к увеличению интервала QT, регистрируемого у человека на электрокардиограмме, и к потенциально фатальной аритмии сердца, известной как"пируэтная желудочковая тахикардия" (Vandenberg и др. (2001). Trends Pharmacol. Sci. 22, 240-246). Согласно современным нормативным рекомендациям (СРМРЛСН/539/00), in vitro исследование, изучающее действия тестируемых соединений в канале hERG, могло бы быть одним из элементов доклинической стратегии, нацеленной на прогнозирование вероятности того, что новые химические единицы будут увеличивать интервал QT, регистрируемый у человека на электрокардиограмме. По существу, устранение hERG-блокирующей активности остается важным фактором в разработке любого нового лекарственного средства. Был описан ряд соединений, которые нацелены на путь PI3K. Например в документах WO 2006/046023 и WO 2006/046024 (Astex Therapeutics Limited) описываются пуриновые, пуриноновые и деазапуриноновые соединения, которые ингибируют или модулируют активность протеинкиназы В(PKB) и протеинкиназы А (PKA). Тем не менее, до сих пор существует потребность в дальнейших улучшенных средствах, имеющих лучшую активность по отношению к PKB, и/или полезные физические свойства (например, более высокую растворимость в воде, более высокую проницаемость, и/или меньшее связывание с белком плазмы крови), и/или благоприятные профили токсичности (например, сниженную склонность к блокированию hERG), и/или благоприятные метаболические профили в сравнении с другими известными ингибиторами PKB. Заявителями было неожиданно установлено, что определенное бициклическое гетероциклическое производное особенно эффективно при ингибировании активности PKB и поэтому может быть полезным при лечении болезненных состояний, в которые вовлечена активность PKB, например рака. В соответствии с одним вариантом осуществления изобретения обеспечивается соединение или его фармацевтически приемлемая соль, которое представляет собой (S)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид. Синтез оптически активных соединений можно провести с помощью стандартных методов органической химии, хорошо известных в уровне техники, например с помощью синтеза из оптически активных исходных веществ или с помощью расщепления рацемической формы. Рацемические соединения и рацемические промежуточные соединения для их получения начерчены в данном документе в виде плоских структур, тогда как стереоспецифические соединения и стереоспецифические промежуточные соединения для их получения начерчены с соответствующим образом указанной стереохимией. Изобретение также относится ко всем без исключения таутомерным формам заявленных соединений, которые являются ингибиторами активности PKB. Подходящей фармацевтически приемлемой солью заявленного соединения является, например, кислотно-аддитивная соль соединения согласно изобретению, которое является достаточно основным, например кислотно-аддитивная соль, например, с неорганической или органической кислотой, например соляной, бромисто-водородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислотой. Следует понимать, что соединение согласно настоящему изобретению может существовать в сольватированной, например гидратированной, а также в несольватированной формах. Ясно, что настоящее изобретение охватывает все такие сольватированные формы, которые являются ингибиторами активности PKB. Соединение может вводиться в виде пролекарства, которое разлагается в теле человека или животного с получением соединения. Примеры пролекарств включают гидролизуемые in vivo сложные эфиры соединения. Различные формы пролекарств известны в уровне техники. Примеры таких пролекарственных производных см.:Pharm Bull, 32, 692 (1984). В соответствии с дополнительным аспектом изобретения обеспечивается фармацевтическая композиция, которая включает заявленное соединение или его фармацевтически приемлемую соль, как определено выше, совместно с фармацевтически приемлемым разбавителем или носителем. Композиции в соответствии с изобретением могут находиться в форме, пригодной для перорального применения (например, в виде таблеток, пастилок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей, или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельчнного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкоизмельчнного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного,подкожного или внутримышечного дозирования или в виде суппозитория для ректального дозирования). Композиции в соответствии с изобретением могут быть получены с помощью обычных способов с использованием обычных фармацевтических наполнителей, хорошо известных в уровне техники. Так,композиции, предназначенные для перорального использования, могут содержать, например, один или несколько окрашивающих, подслащивающих, ароматизирующих и/или консервирующих агентов. Заявленное соединение может вводиться теплокровному животному обычно в однократной дозе в диапазоне 5-5000 мг/м площади тела животного, то есть приблизительно 0,1-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Форма однократной дозы, такая как таблетка или капсула, должна обычно содержать, например, 1-250 мг активного ингредиента. Предпочтительно применяют суточную дозу в диапазоне 1-50 мг/кг, например 4-7 мг/кг дважды в сутки. Тем не менее, суточная доза будет непременно варьироваться в зависимости от субъекта, получающего лечение, конкретного пути введения, и серьзности болезни, подлежащей лечению. Таким образом, практикующий врач, осуществляющий лечение какого-либо отдельного пациента, может определить оптимальную дозу самостоятельно. Например, фармацевтическая композиция в соответствии с настоящим изобретением, пригодная для перорального введения, может включать 1-200 мг/мл заявленного соединения или его фармацевтически приемлемой соли (такого как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамид) в 0,5% гидроксипропилметилцеллюлозе (НРМС). Как здесь определено, термин "терапия", если специально не указано обратное, также включает"профилактику". Термины "терапевтический" и "терапевтически" следует толковать соответственно. Как здесь определено, термин "лечение" имеет свое обычное значение борьбы с заболеванием с целью полного или частичного уменьшения одного, нескольких или всех его симптомов или с целью устранения или компенсирования патологии, лежащей в его основе. Как здесь определено, термин "профилактика" имеет свое обычное значение и включает первичную профилактику для предотвращения развития заболевания, и вторичную профилактику, посредством которой пациент с уже развившимся заболеванием временно или постоянно защищается от обострения или ухудшения течения заболевания или от развития новых симптомов, связанных с заболеванием. Благодаря своей PKB ингибирующей активности ожидается, что соединение в соответствии с настоящим изобретением будет полезным для лечения заболеваний или медицинских состояний, опосредованных исключительно или частично активностью PKB, например рака. Типы рака, которые могут быть чувствительными к лечению с использованием соединения в соответствии с настоящим изобретением включают, но не ограничиваются перечисленными, рак яичников, рак шейки матки, колоректальный рак,рак молочной железы, рак поджелудочной железы, глиому, глиобластому, меланому, рак предстательной железы, лейкемию, лимфому, неходжкинскую лимфому, рак желудка, рак легких, гепатоцеллюлярный рак, рак желудка, стромальные опухоли желудочно-кишечного тракта (GIST), глиому, рак щитовидной железы, рак желчных протоков, рак эндометрия, рак почки, анапластическую крупноклеточную лимфому, острую миелоидную лейкемию (AML), множественную миелому, меланому и мезотелиому. Рак молочной железы и более конкретно люминальный рак молочной железы может быть особенно чувствительным к лечению с использованием соединений в соответствии с настоящим изобретением. Представляется, что для способов лечения рака, упоминаемых здесь, заявленное соединение должно быть введено млекопитающему, более предпочтительно человеку. Подобным образом, для применений заявленного соединения для лечения рака, упоминаемого здесь, представляется, что заявленное соединение должно быть введено млекопитающему, более предпочтительно человеку. В соответствии с другим аспектом изобретения, таким образом, обеспечивается заявленное соединение, как определено выше, или его фармацевтически приемлемая соль для применения в качестве лекарственного средства. В соответствии с дополнительным аспектом изобретения обеспечивается заявленное соединение,как определено выше, или его фармацевтически приемлемая соль для применения при лечении заболевания, опосредованного PKB. В одном варианте осуществления изобретения вышеупомянутое заболевание, опосредованное PKB, представляет собой рак. В дальнейшем варианте осуществления изобретения вышеупомянутый рак выбирают из рака яичников, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии, лимфомы, неходжкинской лимфомы, рака желудка, рака легких, гепатоцеллюлярного рака,рака желудка, стромальных опухолей желудочно-кишечного тракта (GIST), глиомы, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острой миелоидной лейкемии (AML), множественной миеломы, меланомы и мезотелиомы. В одном варианте осуществления изобретения вышеупомянутый рак выбирают из рака молочной железы, неходжкинской лимфомы, рака поджелудочной железы, гепатоцеллюлярного рака, рака желудка, рака предстательной железы и рака легких. В одном отдельном варианте осуществления вышеупомянутый рак представляет собой рак молочной железы, более предпочтительно люминальный рак молочной железы. В соответствии с дополнительным аспектом изобретения обеспечивается применение заявленного соединения, как определено выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения заболевания, опосредованного PKB. В одном варианте осуществления изобретения вышеупомянутое заболевание, опосредованное PKB, представляет собой рак. В дальнейшем варианте осуществления изобретения вышеупомянутый рак выбирают из рака яичников, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии, лимфомы, неходжкинской лимфомы, рака желудка, рака легких, гепатоцеллюлярного рака, рака желудка, стромальных опухолей желудочнокишечного тракта (GIST), глиомы, рака щитовидной железы, рака желчных протоков, рака эндометрия,рака почки, анапластической крупноклеточной лимфомы, острой миелоидной лейкемии (AML), множественной миеломы, меланомы и мезотелиомы. В одном варианте осуществления изобретения вышеупомянутый рак выбирают из рака молочной железы, неходжкинской лимфомы, рака поджелудочной желе-3 018512 зы, гепатоцеллюлярного рака, рака желудка, рака предстательной железы и рака легких. В одном отдельном варианте осуществления вышеупомянутый рак представляет собой рак молочной железы, более предпочтительно люминальный рак молочной железы. В соответствии с дополнительным аспектом изобретения обеспечивается применение заявленного соединения, как определено выше, или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения рака. В одном варианте осуществления изобретения вышеупомянутый рак выбирают из рака яичников, рака шейки матки, колоректального рака, рака молочной железы, рака поджелудочной железы, глиомы, глиобластомы, меланомы, рака предстательной железы, лейкемии,лимфомы, неходжкинской лимфомы, рака желудка, рака легких, гепатоцеллюлярного рака, рака желудка, стромальных опухолей желудочно-кишечного тракта (GIST), глиомы, рака щитовидной железы, рака желчных протоков, рака эндометрия, рака почки, анапластической крупноклеточной лимфомы, острой миелоидной лейкемии (AML), множественной миеломы, меланомы и мезотелиомы. В одном варианте осуществления изобретения вышеупомянутый рак выбирают из рака молочной железы, неходжкинской лимфомы, рака поджелудочной железы, гепатоцеллюлярного рака, рака желудка, рака предстательной железы и рака легких. В одном отдельном варианте осуществления вышеупомянутый рак представляет собой рак молочной железы, более предпочтительно люминальный рак молочной железы. Противораковое лечение, описанное выше, может применяться в виде монотерапии или дополнительно к соединению согласно изобретению можно применять обычное хирургическое лечение, лучевую терапию или химиотерапию. Такая химиотерапия может включать один или несколько следующих классов противоопухолевых средств:(i) другие антипролиферативные/противоопухолевые лекарственные средства и их комбинации, которые применяются в медицинской онкологии, такие как алкилирующие средства (например, цисплатин, оксалиплатин, карбоплатин, циклофосфамид, азотный иприт, мельфалан, хлорамбуцил, бусульфан, темозоламид и нитрозомочевины); антиметаболиты (например, гемцитабин и антифолаты, такие как фторпиримидины, подобные 5-фторурацилу и тегафуру, ралтитрексед, метотрексат, арабинозид цитозина, и гидроксимочевины); противоопухолевые антибиотики (например, антрациклины, подобные адриамицину, блеомицину, доксорубицину, дауномицину, эпирубицину, идарубицину, митомицину-C, дактиномицину и митрамицину); антимитотические средства (например, алкалоиды барвинка, подобные винкристину, винбластину, виндезину и винорелбину и таксоиды, подобные таксолу и таксотеру и ингибиторы киназы); и ингибиторы топоизомеразы (например, эпиподофиллотоксины, подобные этопозиду и тенипозиду, амсакрину, топотекану и камфотецину);(ii) цитостатические средства, такие как антиэстрогены (например, тамоксифен, фульвестрант, торемифен, ралоксифен, дролоксифен и йодоксифен), антиандрогены (например, бикалутамид, флутамид,нилутамид и ципротерон ацетат), LHRH антагонисты или LHRH агонисты (например, гозерелин, лейпрорелин и бузерелин), прогестогены (например, мегестрол ацетат), ингибиторы ароматазы (например,анастрозол, летрозол, воразол и эксеместан) и ингибиторы 5 а-редуктазы, такие как финастерид;(iii) антиинвазионные средства (например, ингибиторы семейства c-Src киназы, подобные 4-(6 хлор-2,3-метилендиоксианилино)-7-[2-(4-метилпиперазин-1-ил)этокси]-5-тетрагидропиран-4-илоксихиназолину (AZD0530; международная патентная заявка WO 01/94341) и N-(2-хлор-6-метилфенил)-2-6[4-(2-гидроксиэтил)пиперазин-1-ил]-2-метилпиримидин-4-иламинотиазол-5-карбоксамиду (дазатиниб,BMS-354825; J. Med. Chem., 2004, 47, 6658-6661), и ингибиторы металлопротеиназы, подобные маримастату, ингибиторы функции рецептора активатора плазминогена урокиназного типа или антител к гепараназе);(iv) ингибиторы действия факторов роста, например, такие ингибиторы включают антитела к фактору роста и антитела к рецепторам фактора роста (например, анти-erbB2 антитело трастузумаб [Hercepti], анти-EGFR антитело панитумумаб, анти-erbB1 антитело цетуксимаб [Erbitux, C225] и любые антитела к фактору роста или рецепторам фактора роста, описанные Stern и др. Critical reviews in oncology/haematology, 2005, т. 54, cc. 11-29); такие ингибиторы также включают ингибиторы тирозинкиназы,например ингибиторы семейства фактора роста эпидермиса (например, ингибиторы семейства EGFR тирозинкиназ, такие как N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4 амин(гефитиниб, ZD1839), N-(3-этинилфенил)-6,7-бис(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб,OSI-774) и 6-акриламидо-N-(3-хлор-4-фторфенил)-7-(3-морфолинопропокси)-хиназолин-4-амин(CI1033), ингибиторы erbB2 тирозинкиназы, такие как лапатиниб, ингибиторы семейства фактора роста гепатоцитов, ингибиторы семейства фактора роста тромбоцитов такие как иматиниб, ингибиторы сериновых/реониновых киназ (например, ингибиторы Ras/Raf пути передачи сигналов, такие как ингибиторы фарнезилтрансферазы, например, сорафениб (BAY 43-9006, ингибиторы передачи сигналов в клетках посредством MEK и/или AKT киназ, ингибиторы семейства фактора роста гепатоцитов, c-kit ингибиторы, ингибиторы abl киназы, ингибиторы киназы IGF рецептора (инсулиноподобного фактора роста); ингибиторы аурора-киназы (например, AZD1152, РН 739358, VX-680, MLN8054, R763, МР 235, МР 529, VX528 и АХ 39459) и ингибиторы циклинзависимой киназы, такие как CDK2 и/или CDK4 ингибиторы;(v) антиангиогенные средства, такие как те, которые ингибируют действия фактора роста эндотелия сосудов [например, антитело к фактору роста эндотелиальных клеток сосудов бевацизумаб (Avastin) и ингибиторы тирозинкиназы рецептора VEGF, такой как 4-(4-бром-2-фторанилино)-6-метокси-7-(1 метилпиперидин-4-илметокси)хиназолин (ZD6474; пример 2 в заявке WO 01/32651), 4-(4-фтор-2 метилиндол-5-илокси)-6-метокси-7-(3-пирролидин-1-илпропокси)хиназолин (AZD2171; пример 240 в заявке WO 00/47212), ваталаниб (PTK787; WO 98/35985) и SU11248 (сунитиниб; WO 01/60814), соединения, такие как те, что раскрыты в международных патентных заявках WO 97/22596, WO 97/30035, WO 97/32856 и WO 98/13354, и соединения, которые действуют по другим механизмам (например, линомид,ингибиторы действия интегрина v3 и ангиостатин)];(vi) вещества, повреждающие сосуды, такие как комбрестатин А 4, и соединения, раскрытые в международных патентных заявках WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 и(vii) антисмысловая терапия, например, такая, которая направлена на мишени, перечисленные выше, такие как ISIS 2503, антисмысловая терапия на основе гена ras;(viii) подходы генной терапии, включая, например, подходы на основе замены аберрантных генов,такие как способы аберрации р 53 или аберрации BRCA1 или BRCA2, GDEPT (пролекарственная терапия, направленная на ген фермента) подходы с использованием деаминазы цитозина, тимидинкиназы или бактериальной нитроредуктазы и подходы на основе повышения устойчивости пациента к химиотерапии или радиотерапии, такие как генная терапия резистентности ко многим лекарственным средствам; и(ix) иммунотерапевтические подходы, включая, например подходы повышения иммуногенности опухолевых клеток пациента в условиях ex vivo и in vivo, такие как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или фактор стимуляции колоний гранулоцитов-макрофагов, подходы снижения анэргии Т-клеток, подходы с использованием трансфектированных иммунных клеток, таких как цитокинтрансфектированные дендритные клетки, подходы с использованием цитокин-трансфектированных линий опухолевых клеток и подходы с использованием антиидиотипичных антител. Соединение согласно изобретению или его соль можно получить с помощью любого способа, известного в качестве подходящего для получения таких соединений или структурно родственных соединений. Функциональные группы могут быть защищены, и защита может быть снята с использованием обычных способов. Примеры защитных групп, таких как защитные группы для аминогруппы и карбоновой кислоты (а также средств образования защиты и последующего ее снятия), см. T.W. Greene и P.G.M.Wuts, "Protective Groups in Organic Synthesis", Second Edition, John WileySons, New York, 1991. Определенные способа синтеза заявленного соединения представляют собой дополнительную характерную особенность изобретения. Таким образом, в соответствии с дополнительным аспектом изобретения обеспечивается способ получения заявленного соединения или его фармацевтически приемлемой соли, который включает:(а) реакцию кислоты формулы (II) с (S)-3-амино-3-(4-хлорфенил)пропан-1-олом где P1 означает пригодную защитную группу; или(b) реакцию (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамида с бициклическим гетероциклом формулы (V) где L1 означает пригодную уходящую группу; и затем, при необходимости:(ii) образование его фармацевтически приемлемой соли. Характерные условия реакций для способов (а) и (b), упомянутых ранее, являются следующими: Способ (а) - кислоты формулы (II) и амины, такие как (S)-3-амино-3-(4-хлорфенил)пропан-1-ол, могут реагировать вместе в присутствии подходящего реагента сочетания, например гексафторфосфата O(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), и подходящего основания, напримерN,N'-диизопропилэтиламина (DIPEA), в подходящем растворителе, например диметилацетамиде (DMA),и при подходящей температуре, например от 50 до 70C, более подходяще при приблизительно 60C; Способ (b) - карбоксамиды, такие как (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамид, и гетероциклы формулы (V), могут реагировать вместе в присутствии подходящего основания, например N,N'-диизопропилэтиламина (DIPEA), в подходящем растворителе, например бутан-1-оле, и при подходящей температуре, например от 50 до 70C, более подходяще при приблизительно 60C. Соединения формулы (II) могут быть получены в соответствии со схемой 1. Схема 1 где Р 1 означает пригодную защитную группу, например трет-бутоксикарбонил, L1 означает пригодную уходящую группу, например хлор. Соединения,такие как(S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4 карбоксамид, могут быть получены в соответствии со схемой 2. Схема 2 где Р 1 и Р 2 означают пригодные защитные группы, например трет-бутоксикарбонил. Соединения, такие как (S)-3-амино-3-(4-хлорфенил)пропан-1-ол, и соединения формул (V) и (VIII) доступны для приобретения, известны в литературе, получаются с помощью стандартных способов, известных в уровне техники, или могут получаться в соответствии со способами, описанными здесь. Следующие примеры приведены в целях иллюстрации и не предназначены для ограничения объема этой заявки. Каждое иллюстрируемое соединение представляет специфический и независимый аспект изобретения. Все исходные вещества являются доступными для приобретения соединениями, или они известны в литературе, или они получаются с помощью стандартных способов, известных в уровне техники. Как правило, применительно к следующим примерам(i) температуры приведены в градусах Цельсия (C); операции проводили при комнатной температуре или температуре окружающей среды, то есть при температуре в диапазоне от 18 до 25C;(ii) органические растворы сушили над безводным сульфатом магния или безводным сульфатом натрия; упаривание растворителя проводили с использованием роторного испарителя при пониженном давлении (600-4000 Па; 4,5-30 мм Hg) с температурой бани вплоть до 60C;(iii) хроматография подразумевает флэш-хроматографию на силикагеле; тонкослойную хроматографию (ТСХ) проводили на силикагелевых пластинках;(iv) в общем, протекание реакций контролировали с помощью ТСХ и/или аналитической ЖХ-МС, а время реакций, если приведено, дано исключительно в иллюстративных целях;(v) конечные продукты имели удовлетворительный спектр протонного ядерного магнитного резонанса (ЯМР) и/или масс-спектральные данные;(vi) выходы приведены исключительно в иллюстративных целях и не являются непременно теми,которые можно получить с помощью старательного усовершенствования способа; процесс получения повторяли, если требовалось большее количество вещества;(vii) ЯМР данные, когда приведены, находятся в виде значениядля главных отличительных протонов, приведенных в миллионных долях (м.д.) относительно тетраметилсилана (ТМС) в качестве внутреннего стандарта, определенного при 500 МГц с использованием пердейтеродиметилсульфоксида(ДМСО-d6) в качестве растворителя, если не указано иное; использовались следующие сокращения: s,синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; bs, широкий синглет;(viii) химические символы имеют их обычные значения; использовали единицы и символы СИ;(ix) масс-спектр (МС) и данные ЖХ-МС были генерированы на системе ЖХ-МС, где ВЭЖХ компонент включал обычно или оборудование Agilent 1100, Waters Alliance HT (27902795) или насос HP 1100 и диодную матрицу с автосамплером СТС и работали на колонке Phenomenex Gemini C18 5 мкм,502 мм (или подобной) с элюированием либо кислотным элюентом (например, с использованием градиента между 0-95% воды/в ацетонитриле с 5% 1%-ной муравьиной кислоты в 50:50 смеси вода:ацетонитрил (об./об., или основный элюент (например, с использованием градиента между 0-95% воды/в ацетонитриле с 5% 0,1%-ного 880 аммиака в ацетонитрильной смеси); а МС компонент включал обычно масс-спектрометр Waters ZQ, сканирующий при подходящем диапазоне масс. Были получены хроматограммы и сняты интенсивности основных пиков МС с положительным и отрицательным режимом электрораспыления (ESI), и полного УФ-поглощения при 220-300 нм, и приведены значения m/z; как правило, представлены только ионы, которые показывают массу, согласующуюся с исходным соединением, и, если не указано иное, приведенное значение (M+H)+ соответствует режиму определения положительных ионов, а (M-H)- соответствует режиму определения отрицательных ионов;(х) если не определено иное, соединения, содержащие асимметрично замещенный атом углерода и/или атом серы не разделяли;(xi) какие-либо микроволновые реакции проводили либо в микроволновой печи Biotage OptimizerEXP, либо в микроволновой печи СЕМ Explorer;(xii) препаративную высокоэффективную жидкостную хроматографию (ВЭЖХ) выполняли на приборе от "Gilson instrument" с использованием следующих условий: Колонка: кремнеземная C18 с обращенной фазой, например Waters "Xbridge", кремнезем 5 мкм,19100 мм или 30100 мм, с использованием смеси растворителей с уменьшающейся полярностью в качестве элюента (убывающее отношение растворителя А - растворителя В),растворитель А: вода с 1% гидроксидом аммония,растворитель В: ацетонитрил,скорость потока: 28 мл/мин или 61 мл/мин,градиент: приспособленный специально для каждого соединения, обычно продолжительностью 710 мин,длина волны: 254 нм. СокращенияCAS - химическая реферативная служба ДХМ - дихлорметанLDA - диизопропиламид лития ЖХСД - жидкостная хроматография среднего давленияSCX - катионообменная (сильный катион) СЖХ - сверхкритическая жидкостная хроматография ТВМЕ - т-бутилметиловый эфирTEA - триэтиламин ТФУ - трифторуксусная кислота ТГФ - тетрагидрофуран. Пример 9. (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4 ил)пиперидин-4-карбоксамид (Е 9).(20 мл) добавляли HCl (4 М в диоксане) (3,00 мл, 12,00 ммоль). Получающуюся суспензию перемешивали при 20C в течение 16 ч. Реакционную смесь фильтровали через ПТФЭ чашечный фильтр и сырое твердое вещество очищали с помощью препаративной ВЭЖХ (колонка Waters XTerra C18, кремнезем 5 мкм,диаметр 19 мм, длина 100 мм), используя смеси с уменьшающейся полярностью воды (содержащей 1% ТФУ) и MeCN в качестве элюентов. Фракции, содержащие целевое соединение, очищали с помощью ионообменной хроматографии, используя SCX колонку. Целевой продукт элюировали из колонки с использованием 7 М NH3/MeOH и чистые фракции упаривали досуха с получением (S)-4-амино-N-(1-(4 хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (0,200 г, 19,4%) в виде белого твердого вещества. 1 К (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамиду (промежуточное соединение 49) (1 г, 3,21 ммоль) и 4-хлор-7 Н-пирроло[2,3-d]пиримидину (0,493 г, 3,21 ммоль) в бутан-1 оле (15 мл) добавляли N-этилдиизопропиламин (1,676 мл, 9,62 ммоль). Получающийся раствор перемешивали при 60C в течение 18 ч. Реакционную смесь разбавляли EtOAc (50 мл) и последовательно промывали водой (25 мл) и насыщенным раствором соли (25 мл). Органический слой сушили над MgSO4,фильтровали и упаривали с получением сырого продукта. Сырой продукт очищали с помощью флэшхроматографии на кремнеземе, элюирование градиентом 0-6% MeOH аммиака в ДХМ. Чистые фракции упаривали досуха с получением (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамида (842 мг) в виде белой пены. (S)-4-амино-N-(1-(4 хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид перемешивали в этилацетате (7 мл) в течение 18 ч. Твердое вещество собирали путем фильтрования, промывали небольшим количеством этилацетата и сушили в вакуумном шкафу при 55C в течение 18 ч с получением (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (0,585 г, 42,5%) в виде белого твердого вещества, m/z (ES+) (M+H)+ = 429; ВЭЖХ К 4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоновой кислоте (промежуточное соединение 1) (4 г, 11,07 ммоль) и DIPEA (5,80 мл, 33,20 ммоль) в DMA (40 мл) одной порцией добавляли (S)-3-амино-3-(4-хлорфенил)пропан-1-ол (промежуточное соединение 47)(2,055 г, 11,07 ммоль). Добавляли HATU (4,63 г, 12,18 ммоль) и получающийся раствор перемешивали при 20C в течение 24 ч. Реакционную смесь упаривали досуха, затем разбавляли EtOAc (300 мл), и последовательно промывали водой (50 мл) и насыщенным раствором соли (50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением сырого продукта. Сырой продукт очищали с помощью флэш-хроматографии на кремнеземе, элюирование градиентом 2-6% MeOH аммиака в ДХМ. Чистые фракции упаривали досуха и растирали с диоксаном (40 мл) с получением (S)-трет-бутил 4-(1-(4 хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамата (промежуточное соединение 22) (4,82 г, 82%) в виде белого твердого вещества. (S)-трет-бутил 4-(1(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамат (промежуточное соединение 22) (4,82 г, 82%) суспендировали в диоксане (40,0 мл) и добавляли 4 М хлороводород в диоксане (7,69 мл, 221,36 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 2 ч. Сырой продукт очищали с помощью ионообменной хроматографии,используя SCX колонку. Целевой продукт элюировали из колонки с использованием 3,5 М NH3/MeOH и чистые фракции упаривали досуха. Сырой продукт очищали с помощью препаративной ВЭЖХ (колонкаWaters XBridge Prep C18 OBD, кремнезем 5 мкм, диаметр 19 мм, длина 100 мм), используя смеси с уменьшающейся полярностью воды (содержащей 1% NH3) и MeCN в качестве элюентов. Фракции, содержащие целевое соединение, упаривали досуха с получением (S)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (1,200 г, 25,3%) в виде белого твердого вещества, m/z (ES+) (M+H)+ = 429; ВЭЖХ tR = 1,67 мин. 1 К суспензии трет-бутил 4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-илкарбамата (промежуточное соединение 36) (175 мг, 0,33 ммоль) в дихлорметане (7 мл) под аргоном добавляли ТФУ (0,7 мл). Получающийся раствор перемешивали при 20C в течение 16 ч. Растворители удаляли в вакууме и реакционную смесь очищали с помощью препаративной ВЭЖХ, используя колонку с обращенной фазой Waters X-Bridge (С-18, кремнезем 5 мкм, диаметр 19 мм, длина 100 мм) и смеси с уменьшающейся полярностью воды (содержащей 0,2% карбонат аммония) и ацетонитрила в качестве элюента. Чистые фракции упаривали досуха с получением 4-амино-N-(1-(4 хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (99 мг,69,8%) в виде белого порошка. 1 К (R)-трет-бутил 4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамату (промежуточное соединение 42) (0,160 г, 0,30 ммоль) в ДХМ (3 мл) добавляли HCl (4 М в диоксане) (0,378 мл, 1,51 ммоль). Получающуюся суспензию перемешивали при 20C в течение 3 ч. Реакционную смесь упаривали. Сырой продукт очищали с помощью препаративной ВЭЖХ (колонка Waters XTerra C18, кремнезем 5 мкм, диаметр 19 мм, длина 100 мм), используя смеси с уменьшающейся полярностью воды (содержащей 1% аммиака) и MeCN в качестве элюентов. Фракции,содержащие целевое соединение, упаривали досуха с получением (R)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (0,051 г, 39,3%) в виде белого твердого вещества. 1CH3CN-H2O (6 л) добавляли NaHCO3(181 г) и далее 4-хлор-7 Н-пирроло[2,3-d]пиримидин (72,7 г). Смесь нагревали с обратным холодильником под азотом в течение 24 ч и затем экстрагировали EtOAc (1 л 4). Водный слой концентрировали и добавляли MeOH (1,5 л). Смесь встряхивали в течение 30 мин при 45C и фильтровали. Фильтрат вновь концентрировали и растворяли в H2O (300 мл). До момента достижения значения pH, равным 4.5, добавляли 6 н. HCl (ок. 80 мл). Смесь фильтровали и кек сушили в вакууме с получением сырого продукта, который дополнительно очищали с помощью гель-хроматографии на кремнеземе (элюирование MeOH: ДХМ=1:3) с получением 4-[(2-метилпропан-2-ил)оксикарбониламино]1-(3,5,7-триазабицикло[4.3.0]нона-2,4,8,10-тетраен-2-ил)пиперидин-4-карбоновой кислоты в виде бледно-серого твердого вещества (105 г, 63%). 1 К (S)-3-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропановой кислоте (1 г, 3,34 ммоль) и карбонату калия (0,922 г, 6,67 ммоль) в ДМФА (15 мл) одной порцией добавляли йодметан (1,038 мл, 16,68 ммоль). Получающуюся суспензию перемешивали при 80C в течение 24 ч. Реакционную смесь концентрировали и разбавляли EtOAc (50 мл) с водой (50 мл). Органический слой сушили над MgSO4, фильтро- 10018512(1,340 г, 128%) в виде оранжевого твердого вещества. 1 К (S)-метил 3-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропаноату (промежуточное соединение 19) (1,05 г, 3,35 ммоль) в ДХМ (20 мл) при 20C одной порцией добавляли соляную кислоту (4,0 М в 1,4-диоксане) (4,18 мл, 16,73 ммоль). Получающийся раствор перемешивали при 20C в течение 5 ч. Реакционную смесь упаривали с получением (S)-метил 3-амино-3-(4-хлорфенил)пропаноата (гидрохлорида) (0,850 г, 102%) в виде белого твердого вещества. 1 К 4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоновой кислоте (промежуточное соединение 1) (1 г, 2,77 ммоль) и N,N-диизопропилэтиламину (1,006 мл, 6,09 ммоль) в NMP (10 мл) при 20C одной порцией добавляли гексафторфосфат О-(7-азабензотриазол-1-ил)N,N,N'N'-тетраметилурония (1,157 г, 3.04 ммоль). Получающийся раствор перемешивали при 20C в течение 5 мин. К раствору затем добавляли (S)-метил 3-амино-3-(4-хлорфенил)пропаноат (гидрохлорид)(промежуточное соединение 20) (0,692 г, 2,77 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли EtOAc (100 мл) и последовательно промывали водой (250 мл) и раствором соли (50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением сырого продукта. Сырой продукт очищали с помощью флэш-хроматографии на кремнеземе, элюирование градиентом 0-5% MeOH в ДХМ. Чистые фракции упаривали досуха с получением (S)-метил 3-(4(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамидо)-3-(4 хлорфенил)пропаноата (1,27 г, 82%) в виде белого твердого вещества. 1(70 мл), охлажденному до 0C, под азотом по каплям добавляли LiAlH4 (2,280 мл, 2,28 ммоль). Получающийся раствор перемешивали при 20C в течение 1 ч. Реакционную смесь гасили гидроксидом натрия (2 М) (2 мл) и водой (1 мл). Раствор фильтровали и разбавляли EtOAc (200 мл) и последовательно промывали водой (100 мл), водой (100 мл) и насыщенным раствором соли (100 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением (S)-трет-бутил 4-(1-(4-хлорфенил)-3 гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамата (1,04 г,86%) в виде белого твердого вещества. M/z (ESI+) (M+H)+ = 529; ВЭЖХ tR = 2,00 мин. Промежуточное соединение 29. 3-Амино-3-(4-хлорфенил)пропан-1-ол. К перемешиваемой суспензии 3-амино-3-(4-хлорфенил)пропионовой кислоты (2,50 г, 12,52 ммоль) в ТГФ (75 мл) при 0C на протяжении 20 мин под азотом по каплям добавляли борантетрагидрофурановый комплекс (94,0 мл, 93,92 ммоль). Получающуюся суспензию перемешивали при 0C в течение 30 мин, затем при 22C в течение 5 ч. Реакционную смесь порциями добавляли к метанолу(этот прим повторяли трижды). Остаток растворяли в ДХМ (200 мл) и промывали 1 н. NaOH (150 мл). Водный слой экстрагировали ДХМ (5100 мл) и экстракты объединяли с органическим слоем. Объединенную органику промывали насыщенным раствором соли (2150 мл), сушили над MgSO4 и концентрировали с получением белого полутвердого вещества. Сырой продукт очищали с помощью флэшхроматографии на кремнеземе, элюирование градиентом 5-7% (10:1 MeOH/конц. NH3 (водн. в ДХМ. Чистые фракции упаривали досуха с получением 3-амино-3-(4-хлорфенил)пропан-1-ола (1,320 г, 56,8%) в виде белого твердого вещества. 1 К 3-амино-3-(4-хлорфенил)пропан-1-олу (промежуточное соединение 29) (0,500 г, 2,69 ммоль) в ДХМ (30 мл) при 22C добавляли ди-трет-бутилдикарбонат (0,705 г, 3,23 ммоль). Получающийся раствор перемешивали при 22C в течение 2 ч. Смесь концентрировали и остаток очищали с помощью флэшхроматографии на кремнеземе, элюирование градиентом 0-4% (10:1 MeOH/конц. NH3 (водн. в ДХМ. Чистые фракции упаривали досуха с получением трет-бутил 1-(4-хлорфенил)-3-гидроксипропилкарбамата (0,759 г, 99%) в виде белого твердого вещества. 1 К 4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоновой кислоте (промежуточное соединение 1) (72,3 мг, 0,20 ммоль), гидрохлориду сложного метилового эфира 3-амино-3-(4-хлорфенил)пропионовой кислоты (50 мг, 0,20 ммоль) и N,N-диизопропилэтиламину (0,104 мл, 0,60 ммоль) в N-метил-2-пирролидиноне (1,5 мл) при 20C под аргоном одной порцией добавляли гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N'N'-тетраметилурония (84 мг, 0,22 ммоль). Получающийся раствор перемешивали в течение 5 ч. Реакционную смесь очищали с помощью препаративной ВЭЖХ с использованием колонки с обращенной фазой Waters X-Bridge (С-18, кремнезем 5 мкм, диаметр 19 мм, длина 100 мм, скорость потока 40 мл/мин) и смеси с уменьшающейся полярностью воды (содержащей 0,2% карбоната аммония) и ацетонитрила в качестве элюента. Фракции, содержащие целевое соединение, упаривали досуха с получением метил 3-(4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамидо)-3-(4-хлорфенил)пропаноата (71,0 мг, 63,8%) в виде кристаллического твердого вещества. 1 К перемешиваемой суспензии метил 3-(4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамидо)-3-(4-хлорфенил)пропаноата (промежуточное соединение 35) (1,0 г, 1,80 ммоль), растворенного в EtOH (400 мл) под аргоном одной порцией добавляли борогидрид натрия (1,019 г, 26,93 ммоль). Получающуюся суспензию перемешивали при 20C в течение 70 ч. Несколько капель воды добавляли с целью погасить избыток гидрида и смесь перемешивали в течение 1 ч. Смесь упаривали досуха и остаток распределяли между CH2Cl2 и водой, насыщенной NaCl. Органическую фазу сушили и концентрировали с обеспечением сырого продукта в виде белых кристаллов. Сырой продукт очищали с помощью флэш-хроматографии на силикагеле, элюирование 0-10% метанолом в дихлорметане. Чистые фракции упаривали досуха с получением трет-бутил 4-(1-(4-хлорфенил)-3 гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамата (286 мг,30,1%) в виде белого твердого вещества. 1(876 мг, 6,34 ммоль) в ДМФА (15 мл) одной порцией добавляли йодметан (0,987 мл, 15,85 ммоль). Получающуюся суспензию перемешивали при 80C в течение 24 ч. Реакционную смесь концентрировали и разбавляли EtOAc (25 мл) с водой (25 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением (R)-метил 3-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропаноата (990 мг, 100%) в виде оранжевого твердого вещества. 1 К (R)-метил 3-(трет-бутоксикарбониламино)-3-(4-хлорфенил)пропаноату (промежуточное соединение 39) (990 мг, 3,16 ммоль) в ДХМ (20 мл) при 20C одной порцией добавляли соляную кислоту (4,0 М в диоксане) (7,89 мл, 31,55 ммоль). Получающийся раствор перемешивали при 20C в течение 3 ч. Реакционную смесь упаривали с получением (R)-метил 3-амино-3-(4-хлорфенил)пропаноата (гидрохлорид) (777 мг, 98%) в виде желтого твердого вещества. 1 К 4-(трет-бутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоновой кислоте (промежуточное соединение 1) (0,5 г, 1,38 ммоль) и DIPEA (0,503 мл, 3,04 ммоль) в NMP (5 мл) при 20C одной порцией добавляли гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилурония (0,579 г, 1,52 ммоль). Получающийся раствор перемешивали при 20C в течение 5 мин. К раствору затем добавляли (R)-метил 3-амино-3-(4-хлорфенил)пропаноат (гидрохлорид) (промежуточное соединение 40) (0,346 г, 1,38 ммоль) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь разбавляли EtOAc (100 мл) и последовательно промывали водой (50 мл), водой (50 мл) и водой (50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением сырого продукта. Сырой продукт очищали с помощью флэш-хроматографии на кремнеземе, элюирование градиентом 0-5% MeOH в ДХМ. Чистые фракции упаривали досуха с получением (R)-метил 3-(4-(третбутоксикарбониламино)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамидо)-3-(4-хлорфенил)пропаноата (0,800 г, 100%) в виде белого твердого вещества. 1(40 мл), охлажденному до 0C, под азотом по каплям добавляли LiAlH4 (1,398 мл, 1,40 ммоль). Получающийся раствор перемешивали при 20C в течение 3 ч. Реакционную смесь гасили водой (1 мл) и нейтрализовали 2 М HCl. Реакционную смесь упаривали досуха и повторно растворяли в ДХМ (100 мл) и промывали водой (100 мл). Органический слой сушили над MgSO4, фильтровали и упаривали с получением сырого продукта. Сырой продукт очищали с помощью флэш-хроматографии на кремнеземе, элюирование градиентом 0-10% MeOH в ДХМ. Чистые фракции объединяли и упаривали с получением (R)трет-бутил 4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-илкарбамата (160 мг, 21,63%) виде твердого вещества. 1 Н ЯМР (399,9 МГц, ДМСО-d6)1.37-1.41 (9 Н, s), 1.86-1.92 (2H, m), 1.94 (2H, d), 2.00 (2H, d), 3.38 К перемешиваемой суспензии (S)-3-амино-3-(4-хлорфенил)пропановой кислоты (10 г, 50,09 ммоль) в ТГФ (500 мл) при 0C на протяжении 45 мин под азотом по каплям добавляли борантетрагидрофурановый комплекс (376 мл, 375,69 ммоль). Получающуюся суспензию перемешивали при 0C в течение 30 мин, затем при 22C в течение 5 ч. Реакционную смесь порциями добавляли к метанолу(500 мл). Раствор перемешивали при комнатной температуре в течение трех дней. Смесь концентрировали, повторно растворяли в метаноле (250 мл) и повторно концентрировали (этот прим повторяли трижды). Остаток растворяли в ДХМ (75 мл) и промывали 1 н. NaOH (50 мл). Водный слой экстрагировали ДХМ (3100 мл) и экстракты объединяли с органическим слоем. Объединенную органику промывали насыщенным раствором соли (50 мл), сушили над MgSO4 и концентрировали с получением сырого продукта. Сырой продукт очищали с помощью флэш-хроматографии на кремнеземе, элюирование градиентом 2-6% MeOH/аммиака в ДХМ. Чистые фракции упаривали досуха с получением (S)-3-амино-3-(4-хлорфенил)пропан-1-ола (5,76 г,61,9%) в виде белого твердого вещества. 1(промежуточное соединение 70) (2,022 г, 5,87 ммоль) и DIPEA (3,08 мл, 17,61 ммоль) в DMA (20 мл) одной порцией добавляли (S)-3-амино-3-(4-хлорфенил)пропан-1-ол (промежуточное соединение 47) (1,09 г,5,87 ммоль). Добавляли гексафторфосфат О-(7-азабензотриазолил)-N,N,N',N'-тетраметилурония (2,456 г,6,46 ммоль), и получающийся раствор перемешивали при 20C в течение 24 ч. Реакционную смесь упаривали досуха, затем разбавляли EtOAc (300 мл) и последовательно промывали водой (50 мл) и насыщенным раствором соли (50 мл). Органический слой сушили над MgSO4, фильтровали и упаривали, затем растирали с диэтиловым эфиром с получением сырого (S)-трет-бутил 4-(трет-бутоксикарбониламино)-4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)пиперидин-1-карбоксилата (4,66 г,155%) в виде белого твердого вещества. 1 К (S)-трет-бутил 4-(трет-бутоксикарбониламино)-4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил) пиперидин-1-карбоксилату (промежуточное соединение 48) (3 г, 5,86 ммоль) в диоксане (30 мл) добавляли хлороводород 4 М в диоксане (11,72 мл, 46,87 ммоль). Получающийся раствор перемешивали при 20C в течение 2 ч. Реакционную смесь растворяли в метаноле и очищали с помощью ионообменной хроматографии, используя SCX колонку. Целевой продукт элюировали из колонки с использованием 3,5 н. аммиака/MeOH и чистые фракции упаривали досуха с получением (S)-4-амино-N-(1-(4-хлорфенил)3-гидроксипропил)пиперидин-4-карбоксамида (1,970 г, 108%) в виде бесцветной смолы. Метил 4-(трет-бутоксикарбониламино)пиперидин-4-карбоксилат (WO 2008075109) (10 г, 38,71 ммоль) растворяли в ДХМ (194 мл). К раствору порциями добавляли ди-трет-бутилдикарбонат (10,67 мл,46,46 ммоль). После добавления реакционную смесь перемешивали при 25C в течение 1 ч. Сырой продукт промывали насыщенным бикарбонатом натрия (350 мл) и органические слои сушили над MgSO4 и упаривали досуха с получением 1-трет-бутил 4-метил-4-(трет-бутоксикарбониламино)пиперидин-1,4 дикарбоксилата (13,6 г, 98%) в виде белого твердого вещества. 1 К 1-трет-бутил 4-метил-4-(трет-бутоксикарбониламино)пиперидин-1,4-дикарбоксилату (промежуточное соединение 69) (13,88 г, 38,72 ммоль) в воде (21,51 мл), ТГФ (86 мл) и метаноле (86 мл) добавляли моногидрат гидроксида лития (8,13 г, 193,62 ммоль). Получающийся раствор перемешивали при 20C в течение 1 дня. Реакционную смесь разбавляли EtOAc (75 мл) и промывали водой (75 мл). Значение pH водной фазы устанавливали на 5 с помощью 1 М раствора лимонной кислоты, затем экстрагировалиEtOAc (3200 мл). Органические экстракты промывали насыщенным раствором соли (50 мл), затем сушили над MgSO4, фильтровали и упаривали с получением целевого продукта 1-(трет-бутоксикарбонил)4-(трет-бутоксикарбониламино)пиперидин-4-карбоновой кислоты (10,36 г, 78%) в виде мелкозернистого твердого вещества. 1(2H, широкий), 3.62-3.65 (2H, d), 7.16 (обмен), 12.36 (обмен). Биологическое действие заявленного соединения может быть исследовано, как изложено ниже. Исследование фосфорилирования GSK-3 аденокарциномы молочной железы человекаMDA-MB-468 в условиях in vitro В этом исследовании определяли способность тестируемых соединений ингибировать фосфорилирование остатка серина-9 в киназе-3 гликоген-синтазы (GSK-3) в качестве суррогатного маркера активности PKB (Akt) в клетках, что оценивали с помощью технологии Acumen Explorer Fluorescent PlateReader. Клеточную линию аденокарциномы молочной железы человека MDA-MB-468 (LGC Promochem,Teddington, Middlesex, UK,каталога НТВ-132) поддерживали общепринятым способом в ростовой среде Игла, модифицированной Дульбекко (DMEM; Invitrogen Limited, Paisley, UKкаталога 11966025), содержащей 10% инактивированную нагреванием фетальную телячью сыворотку (ФТС; Sigma,Poole, Dorset, UK,каталога F0392) и 1% L-глутамин (Gibco,каталога 25030-024) при температуре 37C с 5% CO2 вплоть до слияния 70-90%. Для исследования фосфорилирования клетки отсоединяли от культивируемой колбы, используя Трипсин-EDTA (Invitrogen Limited,каталога 25300-062), и высевали в лунки черного планшета с прозрачным дном Corning Costar Polystyrene на 96 лунок (Fisher Scientific UK, Loughborough, Leicestershire,UK;каталога 3904 и DPS-130-020K) при плотности 5000 клеток на лунку в 100 мкл полной ростовой среды. Клетки инкубировали в течение ночи при 37C с 5% CO2, что давало возможность им прилипнуть. В день 2 клетки обрабатывали тестируемыми соединениями и инкубировали в течение 2 ч при 37C с 5% CO2. Тестируемые соединения приготавливали в виде 10 мМ маточных растворов в ДМСО и дозировали непосредственно до необходимых концентраций в тестируемые лунки, используя неконтактную(акустическое распределение множества капелек 2,5 нл непосредственно в исследуемые лунки) ECHO технологию дозирования (Labcyte Inc. Sunnyvale, California, USA). Каждый планшет содержал контрольные лунки без тестируемого соединения. Затем в каждую лунку добавляли 20 мкл фиксирующего буфера (фосфатно-солевой буфер (ФСБ),содержащий 10% формальдегид; Sigma;каталога F1635) для получения конечных концентраций в лунке 1,6%. После этого планшеты инкубировали в течение 30 мин при комнатной температуре перед удалением фиксажа. Каждую лунку один раз промывали 250 мкл ФСБ и затем в каждую лунку добавляли 50 мкл ФСБ. Затем ФСБ отсасывали и клетки пермеализировали и блокировали путем инкубирования каждой лунки с 50 мкл пермеализирующего/блокирующего буфера (ФСБ, содержащий 0,5% Твин 20(Sigma;каталога Р 5927) и 5% Marvel Milk Powder (Andrews Pharmacy Ltd., Macclesfield, Cheshire, UK;каталога АРС 100199) в течение 1 ч при комнатной температуре до окрашивания. После удаления пермеализирующего/блокирующего буфера в каждую лунку добавляли 50 мкл первичного антифосфо-GSK-3 антитела (Cell Signalling Technology (New England Biolabs (Uk) Ltd.), Hitchin,Hertfordshire, UK;каталога 9336, разведенного 1:400 в блокирующем буфере (ФСБ, содержащий 5%Marvel и 0,05% Твин/Полисорбат 20) и инкубировали в течение ночи при 4C. Каждую лунку промывали три раза в 250 мкл промывочного буфера (ФСБ, содержащий 0,05% полисорбат 20) и клетки инкубировали в течение 1 ч при комнатной температуре с 50 мкл вторичного флюоресцентно-меченого антикроличьего Alexa Fluor 488 антитела (Molecular Probes, Invitrogen Limited, каталога А 11008), разведенного 1:750 в блокирующем буфере. Планшеты промывали три раза в 250 мкл промывочного буфера и хранили содержащими 50 мкл ФСБ при 4C до дальнейших исследований. Планшеты анализировали, используя планшет-ридер Acumen Explorer для количественного определения флуоресцентного сигнала, который характеризует количество фосфорилированного-GSK-3. Активные соединения вызывают снижение фосфо-GSK-3 фосфорилирования относительно максимума(недозированного) контроля для каждого исследования, который измеряли по количеству фосфорилированных объектов на лунку, и это предоставляет возможность оценить потенциал ингибиторов PKB (Akt).IC50 расчет - IC50 представляет собой концентрацию соединения, необходимую для получения 50% действия по сравнению с уровнем активности, оказываемой соединением, между максимумом (без соединения) и минимумом (избыточный уровень соединения) ответной реакции относительно контрольных данных. Значения IC50 определяли путем подгонки скорректированного фона, данных исследования ответной реакции для модели 4-параметрической логистической подгонки кривой соответствия уравнению с минимальным ответом, установленным равным нулю. Это осуществляли с помощью собственного разработанного алгоритма с Origin графическим программным обеспечением (OriginLab Corporation, Northampton, MA, USA). Соединения-примеры изобретения тестировали в вышеупомянутом исследовании и вычисляли их средние значения IC50. Эти значения представлены в табл. G. Исследование AKT1 киназы в условиях in vitro В этом исследовании детектировали ингибиторы активности AKT1 (PKBa) киназы с использованием прибора Caliper LabChip LC3000. Исследование по сдвигу пятна с инкубированием вне чипа на Caliper включает использование микрожидкостного чипа для измерения превращения флуоресцентномеченого пептида в фосфорилированный продукт посредством соответствующей киназы. Необратимую ферментативную реакцию проводят в микротитрационных планшетах и затем гасят. Получающиеся остановленные растворы серийно "протягивают" через капилляр в чип, где пептидный субстрат и фосфорилированный продукт разделяется с помощью электрофореза. Затем их детектируют с помощью лазерно-индуцированной флуоресценции. Субстрат и продукт разделяют на два пика путем наложения высокого электрического поля и непосредственно детектируют с использованием флуоресценции. Сигнатура флуоресцентного сигнала показывает степень реакции. Растворитель для ЕСНО-дозирования представлял собой 100% ДМСО. Мастер-планшет приготовляли исходя из 40 мкл 10 мМ маточного раствора из своего первичного запаса жидкостей в 1 квадранте 384-луночного планшета Labcyte. 1:100 разбавление приготовляли из 1 квадранта во 2 квадранте путем извлечения 0,4 мкл и добавления их к 39,6 мкл ДМСО. Последующие 1:100 разбавления приготовляли в 3 квадранте из разбавлений 2 квадранта и в квадранте 4 из разбавлений 3 квадранта. Для получения диапазона доз, который был необходим для данного исследования из каждого квадранта мастер-планшета многократно дозировали 2,5 нл капель с использованием технологии ЕСНОдозирования (Labcyte Inc. Sunnyvale, California, USA). Обычно используемый диапазон доз был следующим: 100, 30, 10, 3, 1, 0,3, 0,1, 0,03, 0,01, 0,003, 0,001, 0,0001 мкМ. Каждую лунку заполняли диметилсульфоксидом (ДМСО) до общего объема 120 нл так, что когда добавляли смесь фермента и субстрата,конечная концентрация ДМСО составляла 1%. ДМСО добавляли к лункам с максимальным контролем в количестве 120 нл, лунки с минимальным контролем были обработаны 120 нл соединения при концентрации, которая ингибирует активность фермента на 100%. После добавления соединения или контроля к аналитическому планшету, добавляли 6 мкл пептидной смеси, содержащей 3 мкМ субстрат (5-FAM-GRPRTSSFAEG-CONH2; CRB) и 40 мкМ АТФ в киназном основном буфере (100 мкМ Hepes pH 7.5, 0,015% Brij-35) и 6 мкл ферментной смеси, содержащей 8 нМ AKT1/PKBa активный фермент (Upstate Biotechnology, кат.14-276), 8 мМ DTT и 20 мМ MgCl2 в киназном основном буфере. Все буферы приготовляли из воды с сопротивлением 18 МОм. Планшеты запечатывали и инкубировали при комнатной температуре в течение 50 мин. Реакцию останавливали путем добавления 10 мкл останавливающего буфера (100 мМ Hepes pH 7.5, 0,015% раствор Brij-35, 0,1% покрывающий реактив 3, 40 мМ EDTA, 5% ДМСО) к каждой лунке (прим. планшеты могут быть заморожены после остановки реакции и прочитаны позднее). Планшеты затем анализировали, используя систему поиска новых лекарств Caliper LabChip LC3000 (Caliper Life Sciences, 1 Wellfield, Preston Brook,Runcorn, WA7 3AZ), с использованием следующих условий разделения: - 1,8 фунта на кв.дюйм, - 500 входное напряжение, - 1700 выходное напряжение, время введения образца 0,2 с, время начала введения образца 30 с и конечная задержка 120 с. Интегрирование субстрата и пиков продукта проводили с использованием программного обеспечения Caliper LabChip, a кривые IC50 рассчитывали с использованием программного обеспечения Origin (OriginLab Corporation, Northampton, MA, USA). Соединенияпримеры в соответствии с изобретением тестировали в исследовании фермента AKT1 в условиях in vitro,и полученные средние значения IC50 сводили в табл. G. Таблица GhERG анализ Культура клеток. Описанные hERG-экспрессирующие клетки яичника китайского хомячка K1 (СНО) (Persson, Carlsson, Duker,Jacobson, 2005) выращивали до полуконфлюэнтного состояния при 37C во влажной атмосфере (5% CO2) в F-12 Ham среде, содержащей L-глутамин, 10% фетальной телячьей сыворотки (ФТС) и 0,6 мг/мл гигромицина (все от Sigma-Aldrich). Перед использованием монослой промывали с использованием предварительно нагретой (37C) 3 мл аликвоты версена 1:5,000 (Invitrogen). После аспирации этого раствора колбу инкубировали при 37C в инкубаторе с дополнительными 2 мл версена 1:5,000 в течение 6 мин. Клетки затем отделяли от дна колбы путем легкого постукивания и затем к колбе добавляли 10 мл физиологического раствора, забуференного фосфатом Дульбекко, содержащего кальций (0,9 мМ) и магний (0,5 мМ) (ФСБ; Invitrogen), и аспирировали в 15 мл центрифужную пробирку перед центрифугированием (50 g, в течение 4 мин). Получающийся супернатант отбрасывали и осадок осторожно ресуспендировали в 3 мл ФСБ. Аликвоту 0,5 мл суспензии клеток удаляли и ряд жизнеспособных клеток определяли (на основе эксклюзии трипанового синего) в автоматическом планшет-ридере (Cedex; Innovatis) с целью отрегулировать с помощью ФСБ объем ресуспендированных клеток с получением требуемой конечной концентрации клеток. Последняя является концентрация клеток в данной точке исследования,которая приведена при ссылке на этот параметр. Клетки CHO-Kv1.5, которые использовали для корректирования смещения напряжения на Ion Works HT, хранили и приготовляли для использования таким же образом. Электрофизиология. Принципы и функционирование этого устройства описано ранее (Schroeder, Neagle, Trezise,Worley, 2003). Вкратце, технология основывается на 384-луночном планшете (PatchPlate), в котором регистрацию предпринимают в каждой лунке путем использования всасывания для расположения и фиксации клетки в небольшом отверстии, разделенном двумя отдельными камерами с жидкостями. После запечатывания раствор в нижней части PatchPlate загружают в камеру, которая содержит амфотерицин В. Это делает участок клеточной мембраны, которая покрывает отверстие у каждой лунки, проницаемым и, в сущности, позволяет записывать перфорированный, фиксированный потенциал (петч-кламп) в целой клетке. Для Р-теста использовали прибор Ion Works HT от Essen Instrument. Прибор не обладал свойством подогревать растворы, поэтому работу проводили при комнатной температуре (21C), как изложено ниже. Резервуар в положении "Буфер" загружали 4 мл ФСБ, а в положении "Клетки" вносили суспензию клеток CHO-hERG, описанную выше. 96-Луночный планшет (с V-подобным дном, Greiner Bio-one), содержащий подлежащие тестированию соединения (при 3-кратной вышеуказанной их конечной тестируемой концентрации), помещали в положение "Планшет 1" и PatchPlate зажимали в терминале PatchPlate. Планшет с каждым соединением выставляли в 12 колонках таким образом, чтобы обеспечить построение десяти 8-точковых кривых зависимости действия от концентрации; оставшиеся на планшете две колонки заполняли наполнителем (конечная концентрация 0,33% ДМСО), для определения исходного уровня исследования и надмаксимальной блокирующей концентрацией цизаприда (конечная концентрация 10 мкМ) для определения 100% уровня ингибирования. Потом через жидкостную головку (Fголовка) Ion Works HT прибавляли 3,5 мкл PBS в каждую лунку PatchPlate, и его нижнюю часть обрызгивали "внутренним" раствором следующего состава (в мМ): K-глюконат 100, KCl40, MgCl2 3,2,- 18018512EGTA 3 и HEPES 5 (все производства Sigma-Aldrich) (pH 7,25-7,30 с помощью 10 М KOH). После примирования и дебарботирования электронную головку (Е-головку) перемещали вокруг PatchPlate, выполняя тест на проницаемость (то есть прикладывали напряжение для определения того, является ли отверстие в каждой клетке открытым). После этого через F-головку вводили 3,5 мкл суспензии клеток, описанной выше, в каждую лунку PatchPlate, и клеткам давали 200 с для реагирования и закрытия отверстия в каждой лунке. После этого Е-головку перемещали вокруг PatchPlate для определения сопротивления закрытию, полученного в каждой лунке. Далее раствор на нижней части PatchPlate загружали в раствор "доступа", который имел следующий состав (в мМ): KCl 140, EGTA 1, MgCl2 1 и HEPES 20 (рН 7.25-7.30 с использованием 10 М KOH) плюс 100 мг/мл амфотерицина В (Sigma-Aldrich). После выдерживания в течение 9 мин для осуществления перфорации участка Е-головку перемещали вокруг 48 лунокPatchPlate за раз для получения измерений тока hERG перед действием соединения. Через F-головку затем добавляли 3,5 мкл раствора из каждой лунки планшета с соединением к 4 лункам на PatchPlate(конечная концентрация ДМСО составляла 0,33% в каждой ячейке). Это достигали путем перемещения из наиболее разбавленной к наиболее концентрированной ячейки планшета с соединением для минимизирования влияния какого-либо переноса соединения. Через приблизительно 3,5 мин инкубирования Еголовку затем передвигали вокруг всех 384-лунок PatchPlate для получения измерений тока hERG после действия соединения. Таким способом получали некумулятивные дозазависимые кривые при условии, что критерий исходного контроля достигали в достаточном проценте лунок (см. ниже), действие каждой концентрации исследуемого соединения рассчитывали на основании записей от 1 до 4 лунок с клетками. Токи hERG до и после введения соединения вызывали при помощи одиночного импульса напряжения, состоящего из 20 с периода, поддерживаемом на уровне -70 мВ, 160 мс шага до -60 мВ (с получением оценки утечки), 100 мс шага обратно к -70 мВ, 1 с шага до +40 мВ, 2 с шага до -30 мВ и в заключение 500 мс шага до -70 мВ. В интервале между импульсами напряжения, снятыми до и после введения соединения, потенциал мембраны не фиксировали. Токи корректировали, вычитая токи утечки, полученные на основе оценки тока, вызываемого во время шага +10 мВ в начале протокола импульса напряжения. Любые сдвиги напряжения в Ion Works HT корректировали одним из двух путей. При определении активности соединения к клеткам CHO-Kv1.5 прилаживали деполяризующее линейно изменяющееся напряжение и отмечали напряжение, при котором наблюдалась точка перегиба в следе тока (то есть точку, в которой активирование каналов было видно из линейно изменяющегося протокола). Напряжение,при котором это происходило, предварительно определяли с использованием таких же команд управления напряжением в обычном электрофизиологическом тесте и, как установлено, было равным -15 мВ(данные не показаны); таким образом, потенциал сдвига мог быть введен в программное обеспечение IonWorks HT с использованием этого значения в виде контрольной точки. При определении основных электрофизиологических свойств hERG любой сдвиг корректировали путем определения потенциала реверсии hERG следового тока в Ion Works HT, сравнивая его с таковым, установленным в обычном электрофизиологическом тесте (-82 мВ) и затем осуществляя необходимый ввод значения сдвига в программное обеспечение Ion Works HT. Сигнал тока был дискретный с частотой 2,5 кГц. Магнитуду hERG тока до и после сканирования измеряли автоматически из вычтенных следов утечки с помощью программного обеспечения Ion Works HT, беря 40 мс среднего тока во время начального периода, поддерживаемого на уровне -70 мВ (исходный ток), и вычитая результат из ответного пика следового тока. Критериями приемлемости для вызываемых токов в каждой лунке являлись сопротивление закрытию перед сканированием 60 МОм, амплитуда следового hERG тока перед сканированием 150 пА; сопротивление закрытия после сканирования 60 МОм. Степень ингибирования hERG тока определяли путем деления hERG тока после сканирования на соответствующий hERG ток перед сканированием для каждой лунки. СсылкиSchroeder, K., Neagle, В., Trezise, D.J.,Worley, J. (2003). Ionworks HT: a new high-throughput electrophysiology measurement platform. J. Biomol. Screen., 8, 50-64. Результаты hERG анализа. Соединение-пример 9, (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамид, испытывали вплоть до 100 мкМ в соответствии с методикой, описанной выше, и среднее hERG значение IC50 для 177 мкМ получали путем экстраполяции кривой. Эксперименты в условиях in vivo. Фармакодинамический анализ PKB субстратных белков GSK3 и PRAS40 в ответ на (S)-4-амино-N(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид. 2,5106 U87-MG клеток (АТСС номер НТВ-14) + 50% матригеля инъецировали п/к (подкожно) в- 19018512 бок голой мыши. Когда опухоли достигали объема около 0,5 см 3, давали п/о (перорально, принудительно) острую дозу 150 или 300 мг/кг (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Нпирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (Е 9). Животных умерщвляли в заданный момент времени и опухоли вырезали и быстро замораживали в жидком азоте.Ex-vivo опухолевые лизаты приготовляли в 10% тритон-Х-100 Трис-лизирующем буфере, содержавшем используемые ингибиторы протеазы и фосфатазы, используя методику "FastPrep 24" (МР Biomedicals, матрикс 6910-500). Концентрации белка оценивали по БСА калибровочной кривой с использованием набора Pierce BCA (23225). pGSK3 измеряли с помощью вестерн-блоттинга, a pPRAS40 с помощью "сэндвич"-метода иммуноферментного твердофазного анализа - ELISA (Biosource KHO0421). Для вестерн-блоттинга эквивалентные количества белка разделяли с помощью 4-12% градиента заранее приготовленного Bis-Tris полиакриламида (Invitrogen NP0323), переносили на Hybond С Extra нитроцеллюлозные мембраны (Invitrogen LC2001) и инкубировали с первичной антисывороткой (pGSK3ser9, Cell Signaling Technology 9336; весь GSK3, BD Transduction Labs 610202) и впоследствии либо с конъюгированными с пероксидазой хрена антикроличьими IgG (Cell Signaling Technology 7074), либо с антимышиными IgG (Cell Signalling Technology 7076). Иммунореактивные белки детектировали с помощью усиленной хемилюминесценции (34076 Pierce Supersignal Dura) и полосы количественно определяли с помощью ChemiGenius II (Syngene). Нанеснные на график значения показывают ингибирование pGSK3, выраженное в процентах, по сравнению с контролями, содержащими наполнитель, следуя нормировки общего pGSK3. Для ELISA (энзим связанный иммуносорбентный анализ), эквивалентные количества белков добавляли к 96-луночному планшету, предварительно покрытому "захватывающим" pPRAS40 специфическим моноклональным антителом. Затем добавляли "обнаруживающее" антитело, специфическое дляpPRAS40 (Thr 246) и далее HRP меченное антикроличье IgG антитело. Исследование затем проводили в количественной форме с использованием стабилизированного ТМВ (тетраметилбензидин) при 450 нм на планшет-ридере. Интенсивность окраски является пропорциональной концентрации pPRAS40 (Thr 246). Результаты показаны на фиг. 1. Изучение MCF-7 противоопухолевого действия с использованием (S)-4-амино-N-(1-(4-хлорфенил)3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (Е 9). Мышей с тяжелым комбинированным иммунодефицитом (SCID) получали от Charles River Laboratories. Мышей обеспечивали местом обитания и содержали в специфических, непатогенных условиях. Для in vivo имплантации клетки получали из колбы с культурой Т 225 ткани путем 2-5-минутной обработки последней 3 трипсином (Invitrogen) в EDTA растворе с последующим суспендированием в основной среде и троекратным промыванием в забуференном фосфатом физиологическом растворе (Invitrogen). Для инъекции использовали исключительно суспензии из отдельных клеток с более чем 90% жизнеспособностью, как установлено с помощью эксклюзии трипановым синим. MCF-7 клетки опухоли молочной железы (АТСС номер НТВ-22) (5106 клеток + 50% Matrigel) инъецировали подкожно в левый бок животного в объеме 0,1 мл. Перед имплантацией MCF-7 клеток SCID мышей анестезировали и осуществляли имплантацию пеллет с 0,5 мг эстрогена в течение 21 дня. Когда средний размер опухоли достигал 0.3 см 3, мышей рандомизировали в контрольную и лечебную группы. Лечебная группа получала 300 мг/кг соединения Е 9, солюбилизированного в наполнителе, состоящем из 10% (об./об.) ДМСО,25% (мас./об.) Kleptose в воде, путем перорального введения, принудительно. Контрольная группа получала один наполнитель, один раз в день путем перорального введения, принудительно. Объем опухоли(измеренный штангенциркулем) регистрировали через определенные интервалы времени проведения исследования. Мышей умерщвляли с помощью СО 2. Объем опухоли рассчитывали, беря длину, являющуюся наибольшим диаметром от края до края опухоли, и ширину, являющуюся соответствующим перпендикулярным диаметром, с использованием формулы (длинаширина)(длинаширина)(/6). Ингибирование роста с самого начала лечения оценивали с помощью сравнения различий объема опухоли между контрольной и лечебной группами. Поскольку вариация данных среднего объема опухоли возрастает пропорционально объему (и является, вследствие этого, непропорциональной между группами),данные log-преобразовывали для устранения какой-либо зависимости от размера перед статистической оценкой. Статистическую достоверность оценивали с использованием двухвыборочного t теста с использованием одностороннего критерия. Результаты показаны на фиг. 2. Прогнозирование дозы для человека. Прогнозирование дозы для человека для клинических исследований требует оценки параметров фармакокинетики у человека (ФК), важных для определения T1/2 элиминации и формы и амплитуды концентрации в плазме в зависимости от времени при конкретной дозе. Эти параметры элиминации включают клиренс, объем распределения в стационарном состоянии (Vss), константу скорости всасывания(Ka), бионакопление (F) и периодичность дозирования. Прогнозирование дозы для человека также требует некоторых фармакологических данных о том, каким образом доза сопоставляется с эффективностью перидин-4-карбоксамида (Е 9), клиренс для человека прогнозировали исходя из данных собственного клиренса (Clint), определенного у гепатоцитов человека с поправкой на in vivo клиренс путем включения"хорошоперемешиваемой" модели (Riley, McGinnityAustin, Drug MetabolismDisposition 2005 33(9) 1304-1311). Незначительные различия наблюдали в связывании белка плазмы крови с соединением у крысы, собаки и человека; следовательно, наблюдаемые у крысы и собаки значения Vss корректировали с помощью фактора Fu, человек/Fu (крыса или собака) (где Fu = фракция, не связанная в плазме). С использованием этого подхода Vss человека прогнозировали на уровне 3,3 л/кг. Фракцию, абсорбировавшуюся (Fabs) у людей, принимали в качестве средней между крысой и человеком. Этот Fabs параметр затем был подогнан под бионакопление (F) с помощью поправки на печеночную экстракцию. Скорость абсорбции (Ka) полагали обеспечивающей значение Tmax для Е 9 равным 1 ч, причем дано относительно установившееся значение, которое наблюдалось доклинически, и должно в равной степени приводить к установившемуся значению Cmax для использования в подсчете границ, что касается исследований безопасности. Предпочтительная частота дозирования препарата, как предполагалось, составляет дважды в сутки (BID). Определив форму и величину концентрации в плазме человека в зависимости от кривой времени,на заключительной стадии доза должна была быть установлена так, чтобы в плазме были достигнуты такие концентрации, которые, вероятно, обеспечат эффективность. В ФД исследованиях на мышах ингибирование pGSK (суррогатная концевая точка противоопухолевой активности) наблюдалась в то время,когда концентрации в свободной плазме 1-кратно превышали концентрацию IC50 для каждого соединения, определяемую в клеточном исследовании AKT ингибирующей активности, скорректированную с учетом связывания вследствие присутствия фетальной телячьей сыворотки при исследовании. Следовательно, ФК модель человека допускает, что свободная экспозиция Е 9 для обеспечения клинической эффективности должна превышать свободную PKB IC50. Суммируя все эти данные вместе, можно предположить, что эффективная для человека доза (S)-4 амино-Н-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4 карбоксамида (Е 9) будет составлять около 7 мг/кг два раза в день, а период полувыведения будет равным приблизительно 6 ч. Перечень фигур Фиг. 1 - уровни GSK3 и PRAS40 фосфорилирования у образцов, взятых из U87-MG опухолей, выращенных в голых мышах, после острой дозы 150 или 300 мг/кг (S)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамида (Е 9), n=5 в определенный момент времени. Фиг. 2 - результаты MCF-7 противоопухолевого исследования на SCID мышах с использованием дозы 300 мг/кг (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4 ил)пиперидин-4-карбоксамида (Е 9), вводимой один раз в день (QD). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение или его фармацевтически приемлемая соль. 2. Фармацевтическая композиция, включающая соединение по п.1 или его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым разбавителем или носителем. 3. Применение соединения (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-карбоксамида или его фармацевтически приемлемой соли для приготовления медикамента, предназначенного для лечения рака. 4. Применение соединения, определенного в п.3, или его фармацевтически приемлемой соли для приготовления медикамента, предназначенного для лечения рака молочной железы. 5. Соединение по п.1, которое представляет собой (S)-4-амино-N-(1-(4-хлорфенил)-3 гидроксипропил)-1-(7 Н-пирроло[2,3-d]пиримидин-4-ил)пиперидин-4-карбоксамид. 6. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий реакцию кислоты формулы (II) с (S)-3-амино-3-(4-хлорфенил)пропан-1-олом где Р 1 означает пригодную защитную группу; и затем, при необходимости,(i) удаление каких-либо защитных групп и/или(ii) образование его фармацевтически приемлемой соли. 7. Способ по п.6, в котором Р 1 означает трет-бутоксикарбонильную защитную группу. 8. Соединение (S)-трет-бутил 4-(1-(4-хлорфенил)-3-гидроксипропилкарбамоил)-1-(7 Н-пирроло[2,3d]пиримидин-4-ил)пиперидин-4-илкарбамат 9. Способ получения соединения по п.1 или его фармацевтически приемлемой соли, включающий реакцию (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамида с бициклическим гетероциклом формулы (V) где L1 означает пригодную уходящую группу; и затем, при необходимости, образование его фармацевтически приемлемой соли. 10. Способ по п.9, в котором L1 означает хлор. 11. Соединение (S)-4-амино-N-(1-(4-хлорфенил)-3-гидроксипропил)пиперидин-4-карбоксамид
МПК / Метки
МПК: C07D 487/04, A61P 35/00, A61K 31/519
Метки: качестве, протеинкиназы, ингибиторов, производные, пирроло[2,3-d]пиримидина
Код ссылки
<a href="https://eas.patents.su/24-18512-proizvodnye-pirrolo23-dpirimidina-v-kachestve-ingibitorov-proteinkinazy-v.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пирроло[2,3-d]пиримидина в качестве ингибиторов протеинкиназы в</a>
Оптическое разделение (1-бензил-4-метилпиперидин-3-ил)метиламина и его применение для получения производных пирроло 2,3-пиримидина в качестве ингибиторов протеинкиназы

Номер патента: 12666
Опубликовано: 30.12.2009
Авторы: Кехер Кристиан, Манчхоф Майкл Джон, Уилкокс Гленн Эрнест, Врис Тон, Флэнэган Марк Эдвард
МПК: A61K 31/505, A61P 37/06, C07D 487/04...
Метки: 1-бензил-4-метилпиперидин-3-ил)метиламина, пирроло, ингибиторов, производных, применение, протеинкиназы, разделение, качестве, получения, оптическое, 2,3-пиримидина
Формула / Реферат:
1. Способ получения соединения формулы или его фармацевтически приемлемой соли, в котором R1 представляет собой группу формулы где у равен 0, 1 или 2; R4 выбран из группы, состоящей из водорода, (C1-С6)алкила, (C1-С6)алкилсульфонила, (C2-С6)алкенила, (C2-С6)алкинила, в которой группы алкила, алкенила и алкинила необязательно замещены дейтерием, гидрокси, амино, трифторметилом, (C1-С4)алкокси, (C1-С6)ацилокси, (C1-С6)алкиламино,...
Замещенные амидные производные в качестве ингибиторов протеинкиназы

Номер патента: 13231
Опубликовано: 30.04.2010
Авторы: Кси Нинг, Грасеффа Рассел, Беллон Стивен, Букер Шон, Д`анжело Ноел, Янг Кевин, Чокитте Дебора, Хйраи Сатоко, Ровето Филип, Ла Дэниэл, Д`амико Дерин К, Сигмунд Аарон К., Лиу Лонгбин, Ким Тэ-Сеонг, Арманж Жан-Кристоф, Боер Дэвид, Норман Марк Х., Джермейн Джули, Потейшман Майкл, Боезио Алессандро, Феллоус Ингрид М., Ли Мэтью, Домингуез Селия
МПК: C07D 401/12, A61K 31/435, C07D 401/14...
Метки: производные, качестве, протеинкиназы, ингибиторов, амидные, замещенные
Формула / Реферат:
1. Соединение формулы Iего энантиомеры, диастереомеры, соли, сольваты и N-оксиды,где R представляет собойТ выбран из фенила, 5-6-членного гетероарила или 5-6-членного гетероциклила;Z выбран из N или CR7;Z1 выбран из N или CR7;W представляет собой замещенный или незамещенный фенил, замещенный или незамещенный бензоморфолинил, замещенный или незамещенный 6-членный гетероарил, содержащий атом азота; C3-7 циклоалкил, C1-6-алкил и C1-6-алкинил;X...
Соединения пирроло[2,3-d]пиримидина в качестве иммунодепрессантов

Номер патента: 6153
Опубликовано: 27.10.2005
Авторы: Флэнэган Марк Эдвард, Манчхоф Майкл Джон, Блуменкопф Тодд Эндрю
МПК: A61P 17/06, C07D 487/04, A61K 31/505...
Метки: качестве, иммунодепрессантов, соединения, пирроло[2,3-d]пиримидина
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль; где R1 представляет собой группу формулы где R4 представляет собой (C1-C6)алкил; R5 представляет собой пиперидинил, который должен быть замещен группами в количестве от одной до пяти, состоящими из (C1-C6)алкила, циано(C1-C6)алкила, [(циано(C1-C6)алкил)((C1-C6)алкил)амино]ацила, гидрокси(C1-C6)алкилацила, нитро(C1-C6)алкилацила, (C1-C6)алкилSO2HN(C1-C6)алкилацила,...
Производные 6-пиридинил-4-пиримидона в качестве ингибиторов тау-протеинкиназы 1

Номер патента: 13004
Опубликовано: 26.02.2010
Авторы: Фукунага Кендзи, Ватанабе Казутоси, Хики Синсуке, Кохара Тосиюки, Уехара Фумиаки, Йокосима Сатоси
МПК: A61P 25/28, A61K 31/505, A61K 31/506...
Метки: ингибиторов, тау-протеинкиназы, качестве, производные, 6-пиридинил-4-пиримидона
Формула / Реферат:
1. Соединение, представленное формулой (I), его оптически активный изомер или его фармацевтически приемлемая сольгде в указанной формуле каждый символ имеет значения, указанные ниже:R1 представляет метильную группу;R2 представляет атом водорода;R3 представляет атом фтора, замещающий положение 3 пиридинового кольца;q равен 1;R5 представляет группу, представленную следующей формулой (II):где А представляет (С6-С10)-арил или гетероцикл;R6 могут...
Производные пиримидина и их применение в терапии в качестве ингибиторов киназы csbp/rk/p38

Номер патента: 12875
Опубликовано: 30.12.2009
Авторы: Ливия Стефано, Купер Энтони Уилльям Джеймс, Каллахан Джеймс Фрэнсис, Вань Цзехун, Невинс Нейса, Нортон Бет А., Боэм Джеффри С.
МПК: A61P 29/00, A61K 31/5365, A61K 31/519...
Метки: качестве, применение, пиримидина, киназы, терапии, производные, ингибиторов
Формула / Реферат:
1. Соединение формул в которых G3 представляет CH2; G4 представляет CH; R1 представляет C(Z)N(R10')(CR10R20)vRb, C(Z)O(CR10R20)vRb, N(R10')C(Z)(CR10R20)vRb, N(R10')C(Z)N(R10')(CR10R20)vRb или N(R10')OC(Z)(CR10R20)vRb; R1' независимо выбран в каждом случае из атома галогена, C1-4алкила, замещенного атомом галогена C1-4алкила, циано, нитро, (CR10R20)v'NRdRd', (CR10R20)v'C(О)R12, SR5, S(O)R5, S(O)2R5 или (CR10R20)v'OR13; Rb представляет атом...
Предыдущий патент: Система для распределения жидкости
Следующий патент: Способ получения 1,2-пропандиола
Случайный патент: Применение n-(4-((3-(2-амино-4-пиримидинил)-2-пиридинил)окси)фенил)-4-(4-метил-2-тиенил)-1-фталазинамина для лечения резистентного к антимитотическому агенту рака