Новые производные фенантридина в качестве антагонистов брадикинина
Номер патента: 15419
Опубликовано: 31.08.2011
Авторы: Сентирмай Эва, Бозо Эва, Фаркаш Шандор, Ваго Иштван, Шмидт Эва, Ваштаг Моника, Елеш Янош, Хорнок Каталин, Беке Дьюла, Цира Габор







Формула / Реферат
1. Антагонисты В1 рецептора брадикинина - производные фенантридина формулы (I)

где R1является атомом водорода или С1-С4 алкильной группой;
R2 выбран из (1) атома водорода при условии, что R1 и R2не могут быть одновременно атомом водорода; (2) -(CH2)n-NRaRb; (3) -(CH2)n-CO-NRaRb; (4) -(CH2)m-X-Q; (5) -CHRc-NRaRb; или
R1 и R2вместе с атомом азота, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо при необходимости замещено -CO-NRaRb, C1-C4 алкилом, 4-(4,5-дигидро-1H-имидазол-2-ил)бензилом или 4-(1,4,5,6-тетрагидропиримидин-2-ил)бензилом;
R3, R4, R5, R6и R7 являются независимо один от другого атомом водорода, атомом галогена, трифторметилом, С1-С4 алкилом, С1-С4 алкокси- или ацетильной группой;
n является целым числом от 1 до 4;
Ra и Rb являются атомом водорода, при необходимости замещенным С1-С4алкильной группой, или Ra, Rb и атом азота, к которому они присоединены, вместе образуют насыщенное, частично ненасыщенное или ароматическое 4-7-членное кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо при необходимости замещено 1-пиперидинильной, 2-пиперидинильной, 4-пиперидинильной, 2-пиридильной или 4-пиридильной группой;
Rc является метильной, гидроксиметильной, бензильной или фенильной группой;
m является целым числом от 0 до 6;
X является одинарной связью, О или S;
Q является фенильной группой, при необходимости замещенной [1,4']-бипиперидинил-1'-ильной, 4,5-дигидро-1H-имидазол-2-ильной, -(CH2)n-NH-(C=NH)-NH2или -(CH2)m-(C=NH)-NH2группой; или 4-пиперидинильной группой, при необходимости замещенной 4-пиперидинильной группой; или С5-С7 циклоалкильной группой, не обязательно замещенной -(CH2)m-NRaRb группой,
и их оптические антиподы, или рацематы, и/или соли, и/или гидраты, и/или сольваты.
2. Соединения по п.1, выбранные из группы из
гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-8-фтор-5,6-дигидрофенантридин-6-ил]-N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}ацетамида,
гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}ацетамида,
2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[2-(4-пиридин-4-ил-пиперазин-1-ил)этил]ацетамида,
транс-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[4-(2-пирролидин-1-ил-этил)циклогексил]ацетамида,
2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-1-{4-[4-(4,5-дигидро-1Н-имидазол-2-ил)бензил]пиперидин-1-ил}этанона,
2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-1-{4-[4-(1,4,5,6-тетрагидропиримидин-2-ил)бензил]пиперидин-1-ил}этанона,
N-(4-[1,4']-пиперидинил-1'-ил-фенил)-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида,
2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[3-(4-пиридин-4-ил-пиперазин-1-ил)пропил]ацетамида,
гидрохлорида N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}-2-[5-(толуол-4-сульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида,
гидрохлорида N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}-2-[5-(2,4,6-триметилбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида,
гидрохлорида N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}-2-[5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида,
гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]-N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}ацетамида,
гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]-N-{2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил}ацетамида,
гидрохлорида 2-[8-ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-{2-[4-(4,5-дигидро-1Н-имидазол-2-ил)фенил]этил}ацетамида,
N-[2-(4-карбамимидоилфенил)этил]-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида или
транс-2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]-N-[4-(2-пирролидин-1-илэтил)циклогексил]ацетамида.
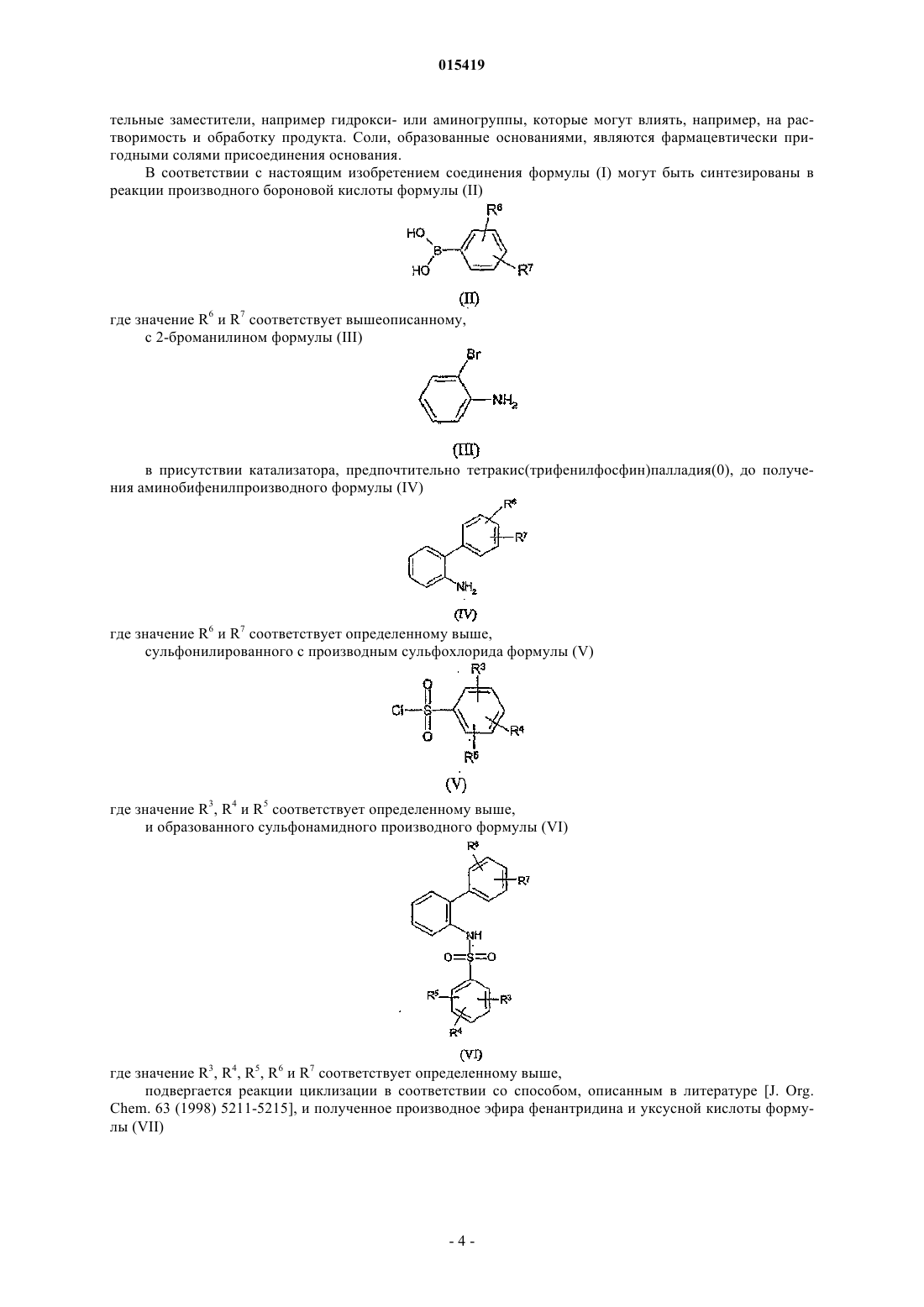
3. Способ приготовления соединений формулы (I) по п.1, включающий реакцию производного бороновой кислоты формулы (II)

где значение R6 и R7соответствует вышеописанному,
с 2-броманилином формулы (III)

в присутствии катализатора, предпочтительно тетракис(трифенилфосфин)палладия(0), до получения аминобифенил производного формулы (IV)

где значение R6 и R7соответствует определенному выше,
сульфонилированного с производным сульфохлорида формулы (V)

где значение R3, R4и R5 соответствует определенному выше,
и образованного сульфонамидного производного формулы (VI)

где значение R3, R4, R5, R6и R7 соответствует определенному выше,
подвергается реакции циклизации и полученное производное эфира фенантридина и уксусной кислоты формулы (VII)

где значение R3, R4, R5, R6и R7 соответствует определенному выше, a R является С1-С4 алкильной группой,
гидролизуется в присутствии основания до получения фенантридинового производного уксусной кислоты формулы (VIII)

где значение R3, R4, R5, R6и R7 соответствует определенному выше,
затем последнее реагирует с аминовым производным формулы (IX)

где значение R1 и R2соответствует вышеописанному.
4. Способ по п.1, включающий трансформацию соединений из формулы (I) в другие соединения формулы (I) путем введения новых заместителей и/или модификаций, или удаления существующих заместителей, и/или образования соли, и/или высвобождения соединения из солей.
5. Соединение формулы (IX), выбранное из группы дигидрохлорид транс-4-(2-пирролидин-1-ил-этил)циклогексиламина, 4-[4-(4,5-дигидро-1Н-имидазол-2-ил)бензил]пиперидина, 2-(4-пиперидин-4-илметилфенил)-1,4,5,6-тетрагидропиримидина.
6. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов и одного или более фармацевтически пригодных наполнителей.
7. Применение соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов для производства медикамента для профилактики и/или лечения состояния, требующего ингибирования рецептора брадикинина.
8. Применение по п.7, где рецептором брадикинина является В1 рецептор брадикинина.
9. Способ лечения и/или профилактики состояния, требующего ингибирования рецептора брадикинина, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов.
10. Способ по п.9, где рецептором брадикинина является В1 рецептор брадикинина.
Текст
НОВЫЕ ПРОИЗВОДНЫЕ ФЕНАНТРИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ БРАДИКИНИНА Настоящее изобретение относится к новым производным фенантридина формулы (I) где R1 является атомом водорода или С 1-С 4 алкильной группой; R2 выбран из (1) атома водорода с условием, что R1 и R2 не могут быть одновременно атомом водорода; (2) -(CH2)n-NRaRb, (3)-(CH2)n-CO-NRaRb, (4) -(CH2)m-X-Q, (5) -(CHRc-NRaRb; или R1 и R2 вместе с атомом азота, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо не обязательно замещено -CO-NRaRb,C1-C4 алкилом, 4-(4,5-дигидро-1H-имидазол-2-ил)бензилом или 4-(1,4,5,6-тетрагидропиримидин 2-ил)бензилом; R3, R4, R5, R6 и R7 являются независимо один от другого атомом водорода, атомом галогена, трифторметилом, С 1-С 4 алкилом, С 1-С 4 алкокси- или ацетильной группой; n является целым числом от 1 до 4; Ra и Rb являются атомом водорода, не обязательно замещенными С 1-С 4 алкильной группой, или Ra, Rb и атом азота, к которому они присоединены, вместе образуют насыщенное, частично ненасыщенное или ароматическое 4-7-членное кольцо, включающее 13 гетероатома, выбранных из О, S и N; где указанное кольцо не обязательно замещено 1 пиперидинильной, 2-пиперидинильной, 4-пиперидинильной, 2-пиридильной или 4-пиридильной группой; Rc является метильной, гидроксиметильной, бензильной или фенильной группой; m является целым числом от 0 до 6; X является одинарной связью, О или S; Q является фенильной группой, не обязательно замещенной [1,4']-бипиперидинил-1'-ильной, 4,5-дигидро-1H-имидазол 2-ильной, -(CH2)n-NH-(C=NH)-NH2 или -(CH2)m-(C=NH)-NH2 группой; или 4-пиперидинильной группой, не обязательно замещенной 4-пиперидинильной группой; или С 5-С 7 циклоалкильной группой, не обязательно замещенной -(CH2)m-NRaRb группой, и их оптическим антиподам, или рацематам, и/или солям, и/или гидратам, и/или сольватам; к процессам их производства, к фармакологическим композициям, включающим их, и к их применению в лечении или профилактике болезненных и воспалительных процессов. 015419 Область техники, к которой относится изобретение Настоящее изобретение относится к новым производным фенантридина формулы (I) и их оптическим антиподам, или рацематам, и/или солям, и/или гидратам, и/или сольватам, пригодным для лечения или профилактики болезненных и воспалительных процессов. Настоящее изобретение также относится к процессам производства соединений формулы (I) и к фармакологическим композициям, содержащим их. Уровень техники Кинины являются эндогенными пептидами, образующимися в плазме и периферических тканях в ответ на повреждение ткани или инфекцию, сопровождающиеся каталитическим расщеплением кининогенов калликреиновыми ферментами. Кинины играют важную роль в патофизиологических процессах,сопровождающих боль и воспаление. Их биологическая роль опосредована двумя связанными с Gбелком мембранными рецепторами, обозначаемыми как В 1 и В 2. Как В 1, так и В 2 рецепторы были клонированы [Biochem. Biophys. Res. Commun., 184 (1992) 260-268 и J. Biol. Chem., 269 (1994) 21583-21586],и механизмы, регулирующие их экспрессию, самоподдержание и сигнальную функцию, интенсивно исследуются [Pharmacol. Rev., 57 (2005) 27-77]. Первая группа кининов, брадикинин (BK) и каллидин (LysBK) предпочтительно действует посредством стимуляции постоянно экспрессируемых и быстро десенсибилизирующихся В 2 рецепторов, которые широко распространены во многих тканях. С другой стороны, их активные карбоксипептидазные метаболиты, вторая группа кининов, desArg9BK (DABK) и LysdesArg9BK (LysDABK) активируют индуцибельные и недесенсибилизирующиеся В 1-рецепторы, которые редко экспрессируются в непатологических условиях. Обычно В 1 рецепторы быстро появляются после повреждения различной природы (травма тканей, инфекция и т.д.). Поэтому позитивная регуляция В 1-рецептора кажется частью генерализованного ответа, который включает локальную совместную экспрессию (в результате позитивной регуляции) ферментов, рецепторов, аутокоидов, цитокинов и хемокинов, которые, как известно, играют роль в раннем и позднем ответе тканей на различные типы повреждения. На животных моделях было продемонстрировано, что имеется переключение преобладания функции от В 2 на В 1 при хронических воспалительных состояниях. В то время как В 2 рецептор вовлечен в острую фазу воспалительного и болевого ответа, В 1 рецептор вовлекается в хроническую фазу данного ответа. Вовлечение кининовых рецепторов в воспаление и передачу сигнала боли было подтверждено результатами исследования на мышах, не имеющих В 1 рецепторы брадикинина. В 1-Рецептордефицитные мыши отличались от мышей дикого типа по сенсорным функциям, проявлению повышенного анальгетического порога на повреждающие химические и тепловые стимулы и по резкому уменьшению накопления полиморфно-ядерных лейкоцитов в местах воспаления [PNAS, 97 (2000) 8140-8145 иNeuropharmacology 41 (2201) 006-1012]. Далее наиболее интересной находкой у В 1-рецептордефицитных мышей было прямое доказательство роли центральных кининовых рецепторов в ноцицепции, позволяющее предположить, что гипоальгезия, наблюдаемая у В 1-рецептор-нокаутных мышей, частично обусловлена сниженной центральной сенсибилизацией спинного мозга. Однако за исключением вышеуказанных изменений, В 1-нокаутные мыши были явно нормальными, без каких-либо заметных патологических изменений. Помимо доказательств основной экспрессии В 1 рецепторов на периферии, все больше и больше доказательств демонстрируют, что В 1 рецепторы постоянно экспрессируются центрально в некоторых нейрональных элементах, включая спинной мозг, а также некоторые высшие структуры. Функция этих рецепторов неясна, но они вовлечены в передачу сигнала боли и гипералгезию. Таким образом, считается, что антагонисты В 1 рецептора пригодны для облегчения боли не только в периферических участках,но также возможно, обладают более широким спектром анальгезирующих эффектов, если они также блокируют центральные В 1 рецепторы [NeuroReport 11 (2000) 4003-4005; NeuroReport, 12 (2001) 23112313; Neuroscience 107 (2001) 665-673 и Neuroscience Letters 294 (2000) 175-178]. На основе научных данных рецепторы брадикинина вовлечены в опосредование боли и гиперальгезии несколькими путями. Антагонисты В 1 рецепторов могут иметь различные способы действия. Они оказывают (I) непрямые (периферические) эффекты на ноцицепторы посредством ингибирования высвобождения других альгогенных медиаторов (простагландинов, цитокинов и оксида азота) из клеток,иных чем сенсорные нейроны (макрофаги, фибробласты или эндотелиальные клетки); (2) прямые (периферические) эффекты на ноцицепторы, экспрессирующие В 1 рецепторы (постоянно) или при индукции, и (3) центральные эффекты на боль, возникающую в поверхностном спинном роге спинного мозга. Таким образом, активные при пероральном приеме непептидные антагонисты рецепторов брадикинина могут быть потенциальными терапевтическими агентами в лечении хронической воспалительной боли. Раскрытие изобретения Мы обнаружили класс производных фенантридина, которые обладают высокой аффинностью к В 1 рецепторам брадикинина и избирательностью (селективностью), не реагируя с В 2 рецепторами брадикинина. Избирательность особенно важна, поскольку нежелательные побочные эффекты соединений вы-1 015419 ражены гораздо меньше. Настоящее изобретение относится к новым производным фенантридина формулы (I) где R1 является атомом водорода или С 1-С 4 алкильной группой;R2 выбран из (1) атома водорода при условии, что R1 и R2 не могут быть одновременно атомом водорода; (2) -(CH2)n-NRaRb, (3) -(CH2)n-CO-NRaRb, (4) -(CH2)m-X-Q, (5) -CHRc-NRaRb; илиR1 и R2 вместе с атомом азота, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо не обязательно замещено -CO-NRaRb, C1-C4 алкилом, 4-(4,5-дигидро-1 Н-имидазол-2-ил)бензилом или 4-(1,4,5,6 тетрагидропиримидин-2-ил)бензилом;R3, R4, R5, R6 и R7 являются независимо один от другого атомом водорода, атомом галогена, трифторметилом, С 1-С 4 алкилом, С 1-С 4 алкокси- или ацетильной группой;n является целым числом от 1 до 4;Ra и Rb являются атомом водорода, не обязательно замещенным С 1-С 4 алкильной группой, или Ra,bR и атом азота, к которому они присоединены, вместе образуют насыщенное, частично ненасыщенное или ароматическое 4-7-членное кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо не обязательно замещено 1-пиперидинильной, 2-пиперидинильной, 4-пиперидинильной,2-пиридильной или 4-пиридильной группой;Rc является метильной, гидроксиметильной, бензильной или фенильной группой;m является целым числом от 0 до 6;Q является фенильной группой, при необходимости замещенной [1,4']-бипиперидинил-1'-ильной,4,5-дигидро-1H-имидазол-2-ильной, -(CH2)n-NH-(C=NH)-NH2 или -(СН 2)m-(C=NH)-NH2 группой; или 4 пиперидинильной группой, не обязательно замещенной 4-пиперидинильной группой; или С 5-С 7 циклоалкильной группой, не обязательно замещенной -(CH2)m-NRaRb группой,и их оптическим антиподам, или рацематам, и/или солям, и/или гидратам, и/или сольватам. Изобретение также относится к фармацевтическим композициям, включающим соединения формулы (I) или их оптические антиподы, или рацематы, или соли, или гидраты, или сольваты в качестве активного ингредиента. Далее объектами настоящего изобретения являются синтез соединений формулы (I) и химическое или фармацевтическое производство медикаментов, включающих данные соединения, а также способы лечения данными соединениями, что означает введение лечимым млекопитающим, включая человека,эффективного количества/количеств соединений формулы (I) настоящего изобретения как таковых или в качестве медикамента. Осуществление изобретения Настоящее изобретение относится к новым антагонистам В 1 рецептора брадикинина - производным фенантридина формулы (I) где R1 является атомом водорода или С 1-С 4 алкильной группой;R2 выбран из (1) атома водорода при условии, что R1 и R2 не могут быть одновременно атомом водорода; (2) -(CH2)n-NRaRb, (3) -(CH2)n-CO-NRaRb, (4) -(CH2)m-X-Q, (5) -CHRc-NRaRb; илиR1 и R2 вместе с атомом азота, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо при необходимости замещеноR3, R4, R5, R6 и R7 являются независимо один от другого атомом водорода, атомом галогена, трифторметилом, С 1-С 4 алкилом, С 1-С 4 алкокси- или ацетильной группой;n является целым числом от 1 до 4;Ra и Rb являются атомом водорода, при необходимости замещенными С 1-С 4 алкильной группой,или Ra, Rb и атом азота, к которому они присоединены, вместе образуют насыщенное, частично ненасыщенное или ароматическое 4-7-членное кольцо, включающее 1-3 гетероатома, выбранных из из О, S и N; где указанное кольцо при необходимости замещено 1-пиперидинильной, 2-пиперидинильной, 4 пиперидинильной, 2-пиридильной или 4-пиридильной группой;Rc является метильной, гидроксиметильной, бензильной или фенильной группой;m является целым числом от 0 до 6;Q является фенильной группой, при необходимости замещенной [1,4']-бипиперидинил-1'-ильной,4,5-дигидро-1 Н-имидазол-2-ильной, -(CH2)n-NH-(C=NH)-NH2 или -(CH2)m-(C=NH)-NH2 группой; или 4 пиперидинильной группой, не обязательно замещенной 4-пиперидинильной группой; или С 5-С 7 циклоалкильной группой, не обязательно замещенной -(CH2)m-NRaRb группой,и их оптическим антиподам, или рацематам, и/или солям, и/или гидратам, и/или сольватам. Изобретение также относится к фармацевтическим композициям, включающим соединения формулы (I) или их оптические антиподы, или рацематы, или соли, или гидраты, или сольваты в качестве активного ингредиента. Далее объектами настоящего изобретения являются синтез соединений формулы (I) и химическое или фармацевтическое производство медикаментов, включающих данные соединения, а также способы лечения данными соединениями, что означает введение лечимым млекопитающим, включая человека,эффективного количества/количеств соединений формулы (I) настоящего изобретения как таковых или в качестве медикамента. Термин галогеновый заместитель обозначает атомы фтора, хлора, брома или йода. Термин С 1-С 4 алкильная группа, использованный в представленном описании, обозначает метильные, этильные, пропильные и изопропильные и различные бутильные группы. Эти С 1-С 4 алкильные группы могут быть в С 1-С 4 алкоксильных группах и С 1-С 4 алкоксикарбонильных группах. 4-7-Членное гетероциклическое кольцо в значении R1 и R2 может быть, например, приперидином,пирролидином, пиперазином, гомопиперазином, морфолином, тиоморфолином и тому подобным. С 1-С 4 Алкильная группа в значении R1 и R2 может быть замещена, например, 4-пиперидинильной,1-пирролидинильной или пиперазинильной группой. Насыщенное, частично ненасыщенное или ароматическое 4-7 членное кольцо в значении R1 и R2 может быть, например, пиперидином, пирролидином, пиперазином, гомопиперазином, морфолином,тиоморфолином и тому подобным. Изобретение относится также к солям соединений формулы (I), образованными с кислотами или основаниями. Как органические, так и неорганические кислоты могут применяться для образования солей присоединения кислот. Подходящими неорганическими кислотами могут быть, например, соляная кислота,серная кислота и фосфорная кислота. Представителями одновалентных органических кислот могут быть,например, муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, и различные бутановые кислоты, валериановые кислоты и каприновые кислоты. Представителями двухвалентных органических кислот могут быть, например, щавелевая кислота, малоновая кислота, малеиновая кислота, фумаровая кислота и янтарная кислота. Могут применяться также другие органические кислоты, такие как гидроксикислоты, например лимонная кислота, винная кислота, или ароматические карбоновые кислоты, например бензойная кислота или салициловая кислота, а также алифатические и ароматические сульфоновые кислоты, например метансульфоновая кислота и п-толуолсульфоновая кислота. Особо ценной группой солей присоединения кислоты являются те, в которых кислый компонент сам по себе не имеет лечебного эффекта в применяемой дозе или не оказывает нежелательного влияния на эффект активного ингредиента. Эти соли присоединения кислоты являются фармацевтически пригодными солями присоединения кислоты. Причиной, по которой соли присоединения кислоты, не принадлежащие к фармацевтически пригодным солям присоединения кислоты, применяются в настоящем изобретении,является то, что в данном случае они могут иметь преимущество при очистке и выделении желаемых соединений. Среди солей, образованных с основаниями, особо важными являются соли, образованные щелочными металлами, например натрием, калием, щелочно-земельными металлами, например кальцием и магнием, а также аммиаком или органическими аминами. Последние основания могут иметь дополни-3 015419 тельные заместители, например гидрокси- или аминогруппы, которые могут влиять, например, на растворимость и обработку продукта. Соли, образованные основаниями, являются фармацевтически пригодными солями присоединения основания. В соответствии с настоящим изобретением соединения формулы (I) могут быть синтезированы в реакции производного бороновой кислоты формулы (II) в присутствии катализатора, предпочтительно тетракис(трифенилфосфин)палладия(0), до получения аминобифенилпроизводного формулы (IV) где значение R6 и R7 соответствует определенному выше,сульфонилированного с производным сульфохлорида формулы (V) где значение R3, R4 и R5 соответствует определенному выше,и образованного сульфонамидного производного формулы (VI)Chem. 63 (1998) 5211-5215], и полученное производное эфира фенантридина и уксусной кислоты формулы (VII) где значение R3, R4, R5, R6 и R7 соответствует определенному выше, a R является С 1-С 4 алкильной группой,гидролизуется в присутствии основания до получения фенантридинового производного уксусной кислоты формулы (VIII) где значение R3, R4, R5, R6 и R7 соответствует определенному выше,затем последнее реагирует с аминовым производным формулы (IX) где значение R1 и R2 соответствует вышеописанному,и полученное производное фенантридина формулы (I) в данном случае может быть преобразовано в другое соединение формулы (I) путем введения новых заместителей и/или модификации или удаления существующих, и/или образования солей, и/или высвобождения соединения из солей. Реакцию сульфонилирования предпочтительно выполняют в надлежащем растворителе, предпочтительно в присутствии основания. Реакции контролируют с помощью тонкослойной хроматографии. Необходимое время реакции составляет 6-20 ч. Обработку реакционной смеси можно осуществлять различными способами.A) Реакционную смесь концентрируют, и продукт выделяют кристаллизацией или экстракцией. Если сырой продукт является недостаточно чистым, для его очистки можно применить колоночную хроматографию. Колоночную хроматографию проводят или в нормальной фазе, с применением Кизельгеля 60 в качестве адсорбента и различных систем растворителей, например, н-гексана/этилацетата, хлороформа/метанола, дихлорметана/этилацетата или хлороформа/ацетона в качестве элюента, или в обращенной фазе, с применением колонки типа YMC-Pack ODS-AQ (производства YMC) и ацетонитрила/воды/трифторуксусной кислоты в качестве элюента.B) Реакционную смесь наливают в воду со льдом, а продукт выделяют фильтрацией или экстракцией. Сырой продукт кристаллизуют или очищают колоночной хроматографией, как описано выше. Структуру продуктов определяют с помощью ИК, ЯМР и масс-спектрометрии. Образование амидной связи предпочтительно осуществляют путем приготовления активного производного карбоновой кислоты формулы (VIII), которая реагирует с амином формулы (IX) предпочтительно в присутствии основания. Преобразование карбоновой кислоты в активное производное может быть проведено in situ во время образования амидной связи в подходящем растворителе (например, диметилформамиде, ацетонитриле, хлорированных углеводородах или углеводородах). Активные производные могут быть хлоридными кислотами (например, приготовленными из карбоновой кислоты и тионилхлорида), смешанными ангидридами (например, приготовленными из карбоновой кислоты и изобутилхлорформиата в присутствии основания, например триэтиламина), активными эфирами (например, приготовленными из карбоновой кислоты и гидроксибензтриазола (HOBt) и дициклогексилкарбодиимидом (DCC) или O-бензотриазол-1-5 015419 ил-N,N,N',N'-тетраметилуроний-гексафторфосфатом (HBTU) в присутствии основания, например триэтиламина), азидами кислот (например, приготовленными из гидразида карбоновой кислоты). Активные производные могут быть приготовлены при температуре в диапазоне от 0 С до комнатной температуры. Подходящий амин формулы (IX) добавляют в качестве основания или соли, образованной с неорганической кислотой, к полученному таким образом раствору или суспензии в присутствии основания, например триэтиламина, необходимого для высвобождения амина. Реакции конденсации сопровождаются тонкослойной хроматографией. Необходимое время реакции составляет 6-20 ч. Обработка реакционной смеси может осуществляться различными способами. Когда реакционная смесь является суспензией, осадок отделяют фильтрацией, промывают водой и/или органическим растворителем и проводят перекристаллизацию из подходящего растворителя до получения чистого продукта. Если кристаллизация не дает чистый продукт, то может применяться колоночная хроматография для его очистки. Колоночную хроматографию проводят или в нормальной фазе, с применением Кизельгеля 60 в качестве адсорбента и различных систем растворителей, например толуола/метанола, хлороформа/метанола или толуола/ацетона в качестве элюента, или в обращенной фазе с применением колонки типа YMC-Pack ODS-AQ (производства YMC) и ацетонитрила/воды/трифторуксусной кислоты в качестве элюента. Если реакционная смесь является раствором в конце реакции образования амидной связи, его концентрируют, а осадок кристаллизуют или экстрагируют с подходящим органическим растворителем и в данном случае очищают колоночной хроматографией, как описано выше. Структуры продуктов определяют с помощью ИК, ЯМР и масс-спектрометрии. Полученные амидные производные формулы (I) независимо от способа приготовления в данном случае могут быть преобразованы в другое соединение формулы (I) ведением дополнительных заместителей, и/или их модификацией, и/или удалением существующих, и/или образованием солей с кислотами,и/или высвобождением бензамидных производных формулы (I) из полученных солей присоединения кислот путем обработки основанием, и/или свободные сульфонамидные производные формулы (I) могут быть преобразованы в соль путем обработки основанием. Бороновые кислоты формулы (II) и сульфонилхлориды формулы (V) являются коммерчески доступными. Большинство аминов формулы (IX) может быть синтезировано различными известными способами. Синтез некоторых новых аминов формулы (IX) описан в примерах. Следуя этим процедурам,можно также приготовить другие амины формулы (IX). Соединения настоящего изобретения, а также их фармацевтически пригодные соли, или гидраты,или сольваты могут применяться как таковые, или в подходящей форме фармацевтических композиций. Эти композиции (лекарства) могут быть в твердой, жидкой или полужидкой форме и могут быть добавлены фармацевтические адъюванты и вспомогательные материалы, которые обычно применяются на практике, такие как носители, наполнители, разбавители, стабилизаторы, увлажняющие агенты или эмульгаторы, агенты, влияющие на рН и осмотическое давление, вкусовые и ароматизирующие, а также поддерживающие и обеспечивающие рецептуру добавок. Дозировка, требуемая для проявления лечебного эффекта, может варьировать в широких пределах и быть приспособлена к индивидуальным требованиям в каждом частном случае, в зависимости от стадии заболевания, состояния и веса тела пациента, которому назначено лечение, а также чувствительности пациента к активному ингредиенту, способу применения и числу ежедневных приемов. Действительная доза применяемого активного ингредиента может безопасно быть определена лечащим врачом, сведущим в данной области техники, знающим пациента, которому назначено лечение. Фармацевтические композиции, включающие активный ингредиент в соответствии с настоящим изобретением, обычно включают от 0,01 до 100 мг активного ингредиента в отдельной форме дозирования. Конечно, возможно, что количество активного ингредиента в некоторых композициях может выходить за верхний или нижний пределы, указанные выше. Твердыми формами фармацевтических композиций могут быть, например, таблетки, драже, капсулы, пилюли или ампулы с лиофилизированным порошком, пригодные для приготовления инъекций. Жидкими композициями являются композиции для введения и инфузии, жидкие медикаменты, упакованные жидкости и капли. Полужидкими композициями могут быть мази, бальзамы, кремы, микстуры для встряхивания и суппозитории. Для простого введения подходит фармацевтическая композиция, включающая единицы дозирования, содержащие количество активного ингредиента, предназначенного для применения единовременно,или несколько раз, или его половину, третью или четвертую часть. Такими единицами дозирования являются, например, таблетки, которые могут быть снабжены бороздками, дающими половину или четвертую часть таблетки, чтобы точно вводить требуемое количество активного ингредиента. Таблетки могут быть покрыты кислото-растворимым слоем, чтобы достичь высвобождения содержимого активного ингредиента после прохождения через желудок. Такие таблетки являются таблетками с энтеросолюбильным покрытием. Подобный эффект может достигаться также путем инкапсулирования активного ингредиента. Фармацевтические композиции для перорального применения могут включать, например, лактозу или крахмал в качестве наполнителей, натрия карбоксиметилцеллюлозу, метилцеллюлозу, поливинил-6 015419 пирролидон или крахмальную пасту в качестве связующих или гранулирующих агентов. Картофельный крахмал или микрокристаллическая целлюлоза добавляются как дезинтегрирующие агенты, но ультраамилопектин или формальдегид-казеин также могут применяться. Тальк, коллоидная кремниевая кислота, стеарин, стеарат кальция или магния могут применяться в качестве антиадгезивных средств и любрикантов. Таблетки могут быть произведены, например, влажным гранулированием с последующим прессованием. Смешанные активные ингредиенты и наполнители, так же, как и в данном случае часть дезинтегрантов, гранулируют в водном, спиртовом или водно-спиртовом растворе связующих агентов в подходящем оборудовании, а затем высушивают гранулят. В высушенный гранулят добавляют другие дезинтегранты, любриканты и антиадгезивные агенты и прессуют смесь в таблетки. В данном случае таблетки делают с бороздкой, разделяющей на две части, для облегчения применения. Таблетки могут быть сделаны непосредственно из смеси активного ингредиента и подходящих вспомогательных веществ путем прессования. В данном случае таблетки могут быть покрыты с применением добавок, обычно используемых в фармацевтической практике, например стабилизаторов, вкусовых веществ, красящих агентов, таких как сахар, производные целлюлозы (метил- или этилцеллюлоза,натрия карбоксиметилцеллюлоза и т.д.), поливинилпирролидон, фосфат кальция, карбонат кальция, пищевые красители, пищевые добавки, ароматические агенты, пигменты из оксида железа и т.д. В случае капсул смесью активного ингредиента и вспомогательных веществ наполняют капсулы. Жидкие пероральные композиции, например суспензии, сиропы, эликсиры могут быть приготовлены с применением воды, гликолей, масел, спиртов, красящих и вкусовых агентов. Для ректального применения композиция составляется в виде суппозиториев или клистиров. Суппозиторий может включать помимо активного ингредиента носитель, так называемый адепс для суппозитория. Носителями могут быть растительные масла, такие как гидрогенированные растительные масла,триглицериды С 12-С 18 жирных кислот (предпочтительно носители под торговым наименованием Witepsol). Активный ингредиент гомогенно смешивают с расплавленным адепсом для суппозитория и отливают суппозитории. Для парентерального применения композицию составляют в виде раствора для инъекций. Для производства раствора для инъекций активные ингредиенты растворяют в дистиллированной воде и/или различных органических растворителях, таких как гликоль-эфиры, в данном случае в присутствии солюбилизаторов, например полиоксиэтиленсорбитана-монолаурата, -моноолеата, или моностеарата (Твин 20,Твин 60, Твин 80). Раствор для инъекций может также включать различные вспомогательные вещества,такие как консервирующие агенты, например этилендиаминтетраацетат, а также агенты, регулирующие рН и буферы, и в отдельном случае местные анестетики, например лидокаин. Раствор для инъекций, содержащий активный ингредиент изобретения, фильтруют перед розливом в ампулы и стерилизуют после розлива. Если активный ингредиент является гигроскопичным, он может затем быть стабилизирован лиофилизацией. Антагонисты В 1 рецептора брадикинина описаны, например, в следующих международных заявках на изобретение: WO 200075107, WO 02076964, WO 04054584, WO 02099388, WO 05004810. Применение Соединения настоящего изобретения являются антагонистами рецептора брадикинина, в частности избирательными антагонистами В 1 рецепторов брадикинина, соответственно, они пригодны для лечения или профилактики болезненных и воспалительных процессов. Соединения могут быть эффективными в лечении боли, включая, например, хроническую боль, в частности боль, вызванную воспалением, гиперальгезию, боль в костях и связках (остеоартрит), синдром повторяющихся движений, мышечнофасциальную боль (повреждение мышц, фибромиалгия), висцеральную боль (язвенный колит, панкреатит, цистит, увеит), периоперативную боль (общая хирургия, гинекология), постоперативную боль (постхирургический болевой синдром), посттравматическую боль (например, растяжения или перелом),невропатологическую боль (постгерпетическая невралгия, повреждение нерва, фантомная боль лимба,мононейропатия, полинейропатия), зубную боль, и боль при онкологии. Далее для лечения боли, связанной с ангиной, менструацией, диабетической васкулопатией, посткапиллярной резистентностью или симптомами диабета, связанными с инсулитом (например, гипергликемией, нарушением диуреза, протеинурией, и повышенной экскрецией нитритов и калликреина с мочой), диабетической гиперальгезией. Далее соединения могут применяться для лечения ангиоэдемы, атеросклероза, септического шока, например, в качестве антигиповолемических и/или антигипотензивных агентов, и сепсиса. Они могут применяться в качестве релаксантов гладких мышц для лечения спазма желудочно-кишечного тракта или матки. Далее соединения данного изобретения могут дополнительно применяться для лечения воспалительных заболеваний кожи, таких как псориаз и эдема, и повреждений кожи, включая ожоги и солнечные ожоги (УФ-эритема и боль). Соединения могут применяться для лечения воспалительной боли различного происхождения (например, ревматоидного артрита, ревматического заболевания, теносиновита, заболевания печени, синдрома раздраженного кишечника, воспалительного заболевания кишечника, болезни Крона, нефрита, аллергического ринита, вазомоторного ринита, увеита, гингивита), аллергий. Такие со-7 015419 единения могут применяться для лечения заболевания воздухоносных путей, например хронического обструктивного заболевания легких, респираторного дистресс-синдрома взрослых, бронхита, пневмонии,астмы. Они могут применяться для контроля, ограничения или обращения гиперактивности воздухоносных путей при астме, лечения эндогенной астмы и экзогенной астмы, включая аллергическую астму(атоническую или неатопическую), профессиональную астму, астму, отягощенную бактериальной или вирусной инфекцией, другие неаллергические астмы, синдром одышки у детей, а также бронхоконстрикцию физического напряжения. Они могут быть эффективными против пневмокониоза, включая алюминоз, антракоз, асбестоз, халикоз, филоз, сидероз., силикоз, табакоз и биссиноз. Дополнительно они могут быть эффективными при некоторых неврологических заболеваниях, например рассеянном склерозе, болезни Альцгеймера, эпилепсии, отеке головного мозга, головной боли, включая гистаминовую головную боль, мигрень, включая профилактику и применение в остром периоде, а также закрытую травму головы. Биологическая оценка. Оценка антагонистической мощности в отношении В 1 и В 2 рецепторов in vitro посредством измерения концентрации ионов кальция в цитозоле с флуориметром для сканирования микропланшетов, в клетках, экспрессирующих рекомбинантные человеческие В 1 и В 2 рецепторы. Культура клеток. Клетки яичников китайского хомячка (СНО), экспрессирующие рекомбинантный человеческий В 1(СНО-В 1, Euroscreen) или В 2 (СНО-В 2, Perkin-Elmer) рецепторы культивировали в Дульбекко модифицированной среде Игла (DMEM), содержащей 10% эмбриональную телячью сыворотку (FCS), 100 ед./мл пенициллина, 0,1 мг/мл стрептомицина, 0,25 мкг/мл амфотерицина В, 1% минимальной эссенциальной среды Игла (MEM), раствор заменимых аминокислот, 600 мкг/мл G418, 1% пируват (для В 2 клеточной линии). Клетки хранили при 37 С в увлажненном инкубаторе в атмосфере 5%СО 2/95% воздуха и пересеивали 1:4 три раза в неделю. Клетки вносили на планшет по 1,5-2,5104 клеток/лунку в стандартный 96-луночный планшет; измерение концентрации ионов кальция в цитозоле ([Ca2+]i) проводили 1-2 дня спустя после помещения клеток на планшет. Флуоресцентное измерение концентрации кальция в цитозоле. Измерение [Ca2+]i проводили на CHO-В 1 и CHO-В 2 клетках, стабильно экспрессирующих человеческие В 1 и В 2 рецепторы соответственно. Клетки выращивали в стандартных 96-луночных микропланшетах и перед измерением нагружали флуоресцентной [Са 2+]-чувствительной краской, флуо-4/АМ (2 мкМ): после удаления культуральной среды добавляли к клеткам краситель (растворенный в буфере для анализа: 145 мМ NaCl, 5 мМ KCl, 2 мМ MgCl2, 2 мМ CaCl2, 10 мМ HEPES, 20 мМ D-глюкоза, 2 мМ пробенецид, 100 млк/лунку) и инкубировали клетки при 37 С в увлажненном инкубаторе в атмосфере 5% СО 2/95% воздуха в течение 40-120 мин. Для остановки нагрузки красителем клетки отмывали дважды буфером для анализа. После отмывания различные концентрации анализируемых соединений (разведенных в экстрацеллюлярной среде от ДМСО маточного раствора, конечная концентрация ДМСО была 0,1%) или буфер добавляли в каждую лунку, в зависимости от экспериментальной установки. После инкубации при 37 С в течение 20-25 мин измеряли базовую линию и вызванные агонистом изменения[Ca2+]i от колонки к колонке с флуориметром для сканирования микропланшетов (Fluoroskan Ascent,Labsystems). Возбуждение и детекцию эмиссии проводили со дна планшетов. Использованные фильтры для Fluo-4: фильтр возбуждения - 485 нм, фильтр эмиссии - 538 нм. В целом процесс измерения проводили при 37 С и контролировали с обычным программным обеспечением. Ингибирующую мощность анализируемых соединений оценивали путем измерения уменьшения вызванного агонистом повышения[Ca2+]i в присутствии различных концентраций соединений. Агонистами были LysDABK для СНО-В 1 и брадикинин для СНО-В 2 клеток. Агонисты вносили в концентрации EC80, значения EC80 были получены из ежедневно определяемых кривых ответа на дозу. Данные флуоресценции выражали в виде F/F (данные флуоресценции, нормализованные по отношению к базовой линии). Все обработки на отдельном планшете измеряли во многих лунках. Данные ото всех лунок с одной и той же обработки усредняли и среднее значение использовали для анализа. Ингибирующую мощность соединения в отдельной точке концентрации выражали как процент ингибирования контрольного ответа на агонист. Сигмоидальные кривые концентрации/ингибирования подгоняли к данным (полученным по крайней мере по трем независимым экспериментам), и определяли значения IC50 как концентрацию, вызывающую половину от максимального ингибирования, вызываемого соединением. В табл. 1 перечислены наиболее эффективные соединения данного изобретения и некоторые из исследованных контрольных соединений, оцененных в данном анализе.+ IC500,5 мкМIC50 между 0,1 и 0,5 мкМIC500,1 мкм Контрольными соединениями были следующие: 70002460: 4-2-[(2,2-дифенилэтил)амино]-5-[4-/(4-метил-этил-1-пиперазинил)карбонил/-1-пиперидинил]сульфонилбензоилморфолин (WO 200075107),70003770: (+)-N-[1-(4-аминометилбензил)-2-оксо-2-пирролидин-1-ил-этил]-3-(нафтален-2-сульфониламино)-3-фенилпропионамид (WO 02076964),70004287: 2-[1-(3,4-дихлорбензилсульфонил)-3-оксо-1,2,3,4-тетрагидрохиноксалин-2(R)-ил]-N-2[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамид (WO 04054584),70004387:-N-[4-(1,4'-бипиперидин)-1'-ил-фенил]-N'-[2,3-дигидро-5-(4-метил-фенил)-2-оксо-1 пропил-1H-1,4-бензодиазепин-3-ил]мочевина (WO 02099388). Анализы связывания с рецепторами 1. Связывание с рекомбинантным человеческим рецептором В 1 к брадикинину. Анализы связывания проводили на человеческих рекомбинантных рецепторах-1 к брадикинину(экспрессируемых на СНО клетках) в соответствии с Перечнем технических данных Euroscreen (Кат.ES-091). 20 мкг белка/пробирку инкубировали с [3,4-пролил-3,4-3H(N)]-[Des-Arg10]-каллиндином в качестве радиолиганда. Неспецифическое связывание определяли в присутствии 10 мкМ Lys-des-Arg9 брадикинина. Конечный объем инкубации составил 250 мкл. Образцы инкубировали в течение 15 мин при 25 С, а затем быстро фильтровали через GF/B фильтры, предварительно промытые в течение по крайней мере 1 ч 0,5% PEI. Радиоактивность определяли жидкостной сцинтилляционной спектроскопией. В табл. 2 перечислены наиболее эффективные соединения из данного изобретения и некоторые из исследованных контрольных соединений, оцененных в данном анализе.+ Ki0,5 мкМKi между 0,1 и 0,5 мкМKi0,1 мкм Контрольными соединениями были следующие: 70002460: 4-2-[(2,2-дифенилэтил)амино]-5-[4-/(4-метилэтил-1-пиперазинил)карбонил/-1-пиперидинил]сульфонилбензоилморфолин (WO 200075107),70003770: (+)-N-[1-(4-аминометилбензил)-2-оксо-2-пирролидин-1-ил-этил]-3-(нафтален-2-сульфониламино)-3-фенилпропионамид (WO 02076964),70004287: 2-[1-(3,4-дихлорбензилсульфонил)-3-оксо-1,2,3,4-тетрагидрохиноксалин-2(R)-ил]-N-2[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамид (WO 04054584),70004387:(+)-N-[4-(1,4'-бипиперидин)-1'-ил-фенил]-N'-[2,3-дигидро-5-(4-метилфенил)-2-оксо-1 пропил-1H-1,4-бензодиазепин-3-ил]мочевина (WO 02099388). 2. Связывание с рекомбинантным человеческим рецептором В 2 к брадикинину. Анализы связывания проводили на человеческих рекомбинантных рецепторах-2 к брадикинину(экспрессируемых на СНО клетках) в соответствии с Перечнем технических данных Euroscreen (Кат. .:RBHB2M) с минимальными изменениями. 8,4 мкг белка/пробирку инкубировали с [2,3-пролил-3,43 Н(N)]-брадикинином в качестве радиолиганда. Неспецифическое связывание определяли в присутствии 5 мкМ брадикинина. Конечный объем инкубации составил 200 мкл. Образцы инкубировали в течение 90 мин при +4 С, а затем быстро фильтровали через GF/B фильтры, предварительно промытые в течение по крайней мере 1 ч 0,5% PEI. Радиоактивность определяли жидкостной сцинтилляционной спектроскопией. Соединения проявляли высокую аффинность и избирательность (боле чем 50-кратную) для человеческих B1-рецепторов по сравнению с человеческими В 2-рецепторами в соответствии как с функциональным анализом, так и с анализом связывания. Синтез соединений и фармацевтических композиций в соответствии с изобретением проиллюстрирован следующими неограничивающими примерами. Ссылочный пример 1. Дигидрохлорид 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина. а) 4-[2-(1,3-Диоксо-1,3-дигидроизоиндол-2-ил-этил]бензонитрил. Раствор 4-(2-гидроксиэтил)бензонитрила [Helv. Chim. Acta 64 (1981) 1688-1703] (4,49 г, 30,5 ммоль), фталимида (4,94 г, 33,55 ммоль), трифенилфосфина (8,8 г, 33.55 ммоль) и диметилформамида(100 мл) перемешивали под аргоном при 0 С в течение 20 мин, затем добавляли по каплям диэтилазидокарбоксилат (7,59 мл, 48,8 мл) при 0 С. Так называемую реакционную смесь перемешивали при комнатной температуре в течение ночи, затем вливали в ледяную воду (740 мл). Осажденный продукт отделяли фильтрацией, промывали водой и высушивали. Сырой продукт отделяли перекристаллизацией из 2-пропанола до получения 7,83 г (93%) названного соединения в виде желтого твердого вещества.b) 2-2-[4-(4,5-Дигидро-1 Н-имидазол-2-ил)фенил]этилизоиндол-1,3-дион. Сухой хлористый водород пропускали через охлажденный на льду раствор 4-[2-(1,3-диоксо-1,3 дигидроизоиндол-2-ил)этил]бензонитрила (7,83 г, 28,3 ммоль) в этаноле (400 мл) в течение 3 ч, затем полученную таким образом смесь хранили при 8 С в течение ночи. Реакционную смесь концентрировали под вакуумом, осадок растворяли в сухом этаноле (400 мл), добавляли этилендиамин (2,0 мл, 29,7 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение ночи. Смесь концентрировали под вакуумом, осадок разделяли между дихлорметаном (400 мл) и концентрированным гидроксидом- 10015419 аммония (400 мл), сепарировали фазы и экстрагировали водную фазу дихлорметаном (2200 мл). Объединенные органические слои высушивали над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт отделяли перекристаллизацией из 2-пропанола до получения 5,58 г (62%) названного соединения в качестве белого твердого вещества.c) Дигидрохлорид 2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этиламина. Смесь 2-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилизоиндол-1,3-диона (5,58 г, 17,47 ммоль), этанола (140 мл) и гидразина гидрата (98%, 6,57 мл, 135,4 ммоль) перемешивали при комнатной температуре в течение 2 ч, затем концентрировали под вакуумом. Осадок разделяли между дихлорметаном (250 мл) и 1 н. гидроксидом натрия (250 мл), фазы сепарировали и экстрагировали водную фазу дихлорметаном (6250 мл). Объединенные органические слои высушивали на сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт растворяли в метаноле (15 мл), рН раствора доводили до 5 добавлением метанольного раствора хлористого водорода, затем перемешивали смесь при комнатной температуре в течение 1 ч. После добавления диэтилового эфира (200 мл) суспензию перемешивали при 0 С в течение 2 ч, осажденные кристаллы отделяли фильтрацией, промывали диэтиловым эфиром и высушивали до получения 4,11 г (90%) названного соединения в виде белого твердого вещества. Ссылочный пример 2. Тригидрохлорид (3-[1,4']бипиперидинил-1'-ил)пропиламина. а) трет-Бутиловый эфир 3-[1,4']бипиперидинил-1'-ил-пропил)карбаминовой кислоты. Смесь 4-пиперидиндинопиперидина (Aldrich) (2,0 г, 11,88 ммоль), (3-бромопропил)карбаминовой кислоты трет-бутилового эфира [Eur. J. Med. Chem. Chim. Ther. 37 (2002) 573-584] (3,96 г, 16,63 ммоль),диметилформамида (130 мл) и калия карбоната (1,64 г, 11,88 ммоль) перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Осадок растворяли в воде (150 мл), экстрагировали с дихлорметаном (3150 мл), объединенные органические слои промывали рассолом (150 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт подвергали колоночной хроматографии с применением Кизельгеля 60 (0,040-0,063 мм) (Merck) в качестве адсорбента и хлороформа:метанола:NH4OH = 10:1:0,1 в качестве элюента до получения 2,27 г (59%) названного соединения в виде масла.b) Тригидрохлорид 3-[1,4']бипепиридинил-1'-ил)пропиламина. Смесь трет-бутилового эфира (3-[1,4']бипиперидинил-1'-ил-пропил)карбаминовой кислоты (2,15 г,6,6 ммоль), сухого диоксана (40 мл) и 6,5 н. хлористого водорода в диоксане (22 мл) перемешивали при комнатной температуре в течение ночи, затем разбавляли диэтиловым эфиром и перемешивали при 0 С в течение 1 ч. Очищенные кристаллы отделяли фильтрацией, промывали диэтиловым эфиром и высушивали до получения 2,03 г (92%) названного соединения в виде твердого вещества бежевого цвета. Ссылочный пример 3. Дигидрохлорид транс-4-(2-пирролидин-1-ил-этил)циклогексиламина. а) Транс-2-1-[4-(N-трет-бутоксикарбонил)амино]циклогексилэтанол. Раствор транс-2-1-[4-(N-трет-бутоксикарбонил)амино]циклогексилуксусной кислоты метилового эфира [J. Med. Chem. 43 (2000) 1878-1885] (28,5 г, 105,2 ммоль) в сухом тетрагидрофуране (500 мл) охлаждали до -2 С, добавляли порциями лития алюминия гидрид (5,4 г, 142 ммоль) и перемешивали смесь при -2 С в течение 60 мин. Реакционную смесь охлаждали до -10 С и гасили этилацетатом (15 мл), затем в смесь медленно добавляли рассол (43 мл) при 0 С. Осажденные соли фильтровали и отмывали этилацетатом. Фильтрат концентрировали под вакуумом. Осадок отделяли перекристаллизацией из диизопропилового эфира (100 мл) до получения 23,7 г (93%) названного соединения в виде белого порошка.(15 г, 62 ммоль) и триэтиламина (10,5 мл, 75 ммоль) в сухом дихлорметане (150 мл) добавляли по каплям метансульфонилхлорид (5,7 мл, 73,4 ммоль) в дихлорметане (25 мл) при 0 С. После перемешивания в течение 30 мин при 0 С раствор экстрагировали трехкратно водой. Органический раствор высушивали трехкратно над сульфатом натрия и концентрировали под вакуумом до получения 13,0 г (65%) названного соединения.c) трет-Бутиловый эфир транс-[4-(2-пирролидин-1-ил-этил)циклогексил]карбаминовой кислоты. Смесь метансульфоновой кислоты транс-2-(4-трет-бутоксикарбониламиноциклогексил)этилового эфира (3,2 г, 10 ммоль), калия карбоната (1,4 г, 10 моль) и пирролидина (1,25 мл, 15 ммоль) в ацетонитриле (40 мл) перемешивали при 60 С в течение 2 ч. Смесь охлаждали до комнатной температуры и вливали в воду (200 мл). Осажденные белые кристаллы отделяли фильтрацией и промывали водой до получения 1,9 г (64%) названного соединения.d) транс-4-(2-Пирролидин-1-ил-этил)циклогексиламин дигидрохлорид. Названное соединение готовили из транс-[4-(2-пирролидин-1-ил-этил)циклогексил]карбаминовой кислоты трет-бутилового эфира в соответствии со способом, описанным в ссылочном примере 2b. Ссылочный пример 4. 4-[4-(4,5-Дигидро-1 Н-имидазол-2-ил)бензил]пиперидин.- 11015419 подвергали дистилляции под вакуумом до получения 52,2 г (97%) названного соединения.b) 4-(1-Бензилпиперидин-4-илиденметил)бензонитрил. К перемешиваемой под аргоном смеси N-бензил-4-пиперидона (Aldrich) (26,0 г, 0,137 моль) и диэтилового эфира (4-цианобензил)фосфониевой кислоты (36,6 г, 0,1445 моль) в диметилформамиде (260 мл) добавляли гидрид натрия (60%, 7,7 г, 0,195 моль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем по каплям добавляли этанол (10 мл) и полученную таким образом смесь выливали в воду (300 мл) и экстрагировали с диэтиловым эфиром (3300 мл). Органический слой высушивали над сульфатом натрия и концентрировали. Осадок очищали колоночной хроматографией с применением Кизельгеля 60 (0,040-0,063 мм) (Merck) в качестве адсорбента, и n-гексана: этилацетата = 2:1 в качестве элюента до получения 34,65 г (87%) названного соединения в виде масла.c) Этиловый эфир 4-(1-бензилпиперидин-4-илиденметил)бензимидной кислоты. Смесь 4-(1-бензилпиперидин-4-илиденметил)бензонитрила (14 г, 48,6 ммоль), хлороформа (10 мл) и 6 М хлористого водорода в этаноле (300 мл) перемешивали при комнатной температуре в течение 48 ч. Раствор концентрировали под вакуумом, оставшееся твердое вещество последовательно растворяли в этаноле (400 мл) и концентрировали под вакуумом до получения 16,4 г (92,4%) названного соединения,которое применяли на следующих этапах без дальнейшей очистки.d) 1-Бензил-4-[4-(4,5-дигидро-1 Н-имидазол-2-ил)бензилиден]пиперидин. К раствору 4-(1-бензилпиперидин-4-илиденметил)бензимидной кислоты этилового эфира (6,8 г,18,3 ммоль) в этаноле (250 мл) добавляли этилендиамин (2,45 мл, 36,6 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Осажденное твердое вещество отделяли фильтрацией и фильтрат концентрировали под вакуумом. Осадок последовательно растворяли в этаноле (100 мл) и концентрировали под вакуумом. Неочищенный продукт очищали путем хроматографии с испарительной колонной с применением Кизельгеля 60 (0,015-0,040 мм) в качестве адсорбента (Merck) и хлороформа:метанола:NH4OH = 9:2:0,1 в качестве элюента до получения 2,7 г (44,6%) названного соединения. е) 4-[4-(4,5-Дигидро-1 Н-имидазол-2-ил)-бензил]-пиперидин. К перемешиваемому раствору 1-бензил-4-[4,5-дигидро-1 Н-имидазол-2-ил)бензилиден]пиперидина(0,1 г, 0,3 ммоль) в этаноле (20 мл), добавляли муравьинокислый аммоний 90,19 г, 3 ммоль) и 10% Pd/C(20 мг) в смесь, нагреваемую с обратным холодильником в течение 8 ч. Катализатор отделяли фильтрацией и концентрировали фильтрат под вакуумом. Остающийся неочищенный материал очищали колоночной хроматографией с применением окиси алюминия (150 меш, Aldrich) в качестве адсорбента и 10% метанола в хлороформе в качестве элюента до получения 68 мг (92%) названного соединения. Ссылочный пример 5. 2-(4-Пиперидин-4-илметилфенил)-1,4,5,6-тетрагидропиримидин.a) 2-[4-(1-Бензилпиперидин-4-илиденметил)фенил]-1,4,5,6-тетрагидропиримидин. Названное соединение готовили из этилового эфира 4-(1-бензилпиперидин-4-илиденметил)бензимидной кислоты и 1,2-диаминопропана в соответствии со способом, описанным в ссылочном примере 4d.b) 2-(4-Пиперидин-4-илметилфенил)-1,4,5,6-тетрагидропиримидин. Названное соединение готовили из 2-[4-(1-бензилпиперидин-4-илиденметил)фенил]-1,4,5,6 тетрагидропиримидина в соответствии со способом, описанном в ссылочном примере 4 е. Пример 1. Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-8-фтор-5,6-дигидрофенантридин-6-ил]N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида. а) 4'-Фторобифенил-2-иламин. К перемешиваемому раствору 2-броманилина (1,72 г, 10,0 ммоль) в диглиме (30 мл) добавляли тетракис(трифенилфосфин)палладий(0) (0,13 г, 0,3 ммоль) и 2,0 М водный раствор карбоната натрия (15 мл,30,0 ммоль). В отдельном флаконе растворяли 4-фторфенилбороновую кислоту (2,23 г, 16,0 ммоль) в этаноле (8 мл) и смесь, содержащую 2-броманилин, добавляли к этому раствору бороновой кислоты. Полученную коричневую реакционную смесь нагревали до 80 С в течение 6 ч, затем охлаждали, разбавляли этилацетатом и промывали насыщенным раствором хлорида аммония. Органический слой затем высушивали над сульфатом натрия, фильтровали и концентрировали. Осадок очищали путем хроматографии с испарительной колонной с применением Кизельгеля 60 (0,015-0,040 мм) в качестве адсорбента (Merck) и n-гексан:этилацетата=2:1 в качестве элюента до получения, после перекристаллизации из этанола, 1,8 гb) 3,4-Дихлор-N-(4'-фторбифенил-2-ил)бензилсульфонамид. К перемешиваемому раствору 4'-фторбифенил-2-иламина (2,17 г, 11,6 ммоль) и 4 диметиламинопиридина (13 мг, 0,11 ммоль) в пиридине (20 мл) добавляли 3,4-дихлорбензилсульфонилхлорид (4,23 г, 17,4 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали. Осадок разделяли между хлороформом (50 мл) и водой (50 мл). Водный слой отделяли и экстрагировали хлороформом (250 мл). Объединенные органические экстракты промывали насыщенным водным раствором бикарбоната натрия (20 мл) и рассолом,высушивали над сульфатом натрия, фильтровали и концентрировали. Продукт кристаллизовали из 2 пропанола до получения 3,58 г (78%) названного соединения в виде белых кристаллов.c) Метиловый эфир [5-(3,4-дихлорбензилсульфонил)-8-фторо-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Ацетат натрия (370 мг, 4,5 ммоль) высушивали и 100 мл колбе с двумя горловинами при 150 С под вакуумом в течение 2 ч, затем добавляли ацетат палладия (100,8 мг, 0,45 ммоль), меди(II) ацетат гидрат(90,0 мг, 0,45 ммоль), 3,4-дихлор-N-(4'-фторобифенил-2-ил)бензилсульфонамид (3,58 г, 9 ммоль), метилакрилат (2,32 г, 27 ммоль), 4 молекулярные сита (3,6 г) и диметилформамид (45 мл) и полученную смесь перемешивали при 120 С в течение 16 ч. После охлаждения реакционную смесь концентрировали,затем разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром (230 мл), высушивали над сульфатом натрия, фильтровали и концентрировали. Осадок очищали путем хроматографии с испарительной колонной с применением Кизельгеля 60 (0,015-0,040 мм) в качестве адсорбента (Merck) и н-гексана: этилацетата=4:1 в качестве элюента до получения 1,91 г (44%) названного соединения в виде белых кристаллов.d) [5-(3,4-Дихлорбензилсульфонил)-8-фторо-5,6-дигидрофенантридин-6-ил]уксусная кислота. К перемешиваемому раствору [5-(3,4-дихлорбензилсульфонил)-8-фторо-5,6-дигидрофенантридин 6-ил]уксусной кислоты метилового эфира (0,48 г, 1 ммоль) в 1:1 смеси тетрагидрофурана и воды (30 мл) добавляли лития гидроксид моногидрат (105 мг, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали. Осадок растворяли в смеси этилацетата (25 мл) и воды (25 мл) и подкисляли 1 н. раствором гидрохлорида. Водную фазу сепарировали и экстрагировали этилацетатом (225 мл). Объединенные органические экстракты высушивали над сульфатом натрия, фильтровали и концентрировали до получения 0,46 г (98%) названного соединения в виде белых кристаллов. е) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-8-фторо-5,6-дигидрофенантридин-6-ил]-N-2-[4(4,4-дигидро-1 Н-имидазол-2-ил)фенил]этил)ацетамид. Раствор [5-(3,4-дихлорбензилсульфонил)-8-фторо-5,6-дигидрофенантридин-6-ил]уксусной кислоты(210 мг, 0,45 ммоль), триэтиламина (0,07 мл, 1,5 ммоль) и гидрохлорида N-(3-диметиламинопропил)-N'этилкарбодиимида (0,19 мг, 0,5 ммоль) в сухом диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 5 мин перед добавлением гидрохлорида 2-[4-(4,5-дигидро-1 Н-имидазо-2-ил)фенил]этиламина (ссылочный пример 2) (131 мг, 0,5 ммоль). Доводили рН реакционной смеси до 8 путем добавления триэтиламина и перемешивали полученную таким образом смесь при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Осадок очищали путем препаративной тонкослойной хроматографии (дихлорметан:метанол:NH4OH=5:1:0.1) до получения 382 мг (75%) названного соединения в виде белых кристаллов. MS (EI) 638,1 (MH+). Пример 2. Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-2-[4(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида.a) N-Бифенил-2-ил-3,4-дихлорбензилсульфонамид. Названное соединение готовили из 2-аминобифенила (Aldrich) в соответствии со способом, описанным в примере 1b.b) Метиловый эфир [5-3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из N-бифенил-2-ил-3,4-дихлорбензилсульфонамида в соответствии со способом, описанным в примере 1 с.c) [5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанном в примере 1d.d) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-2-[4-(4,5 дигидро-1 Н-имидазол-2-ил)фенил]этил)ацетамида. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина (ссылочный пример 2) в соответствии со способом, описанным в примере 1 е. MS (EI) 620,5 (МН+). Пример 3. 2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[2-(4-пиридин-4 ил-пиперазин-1-ил)этил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 2-(4-пиридин-4-ил-пиперазин-1-ил)этиламин (ссылочный пример 1),в соответствии со способом, описанным в примере 1 е. MS (EI) 637,3 (МН+). Пример 4. транс-2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[4-(2-пиролидин-1-ил-этил)циклогексил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и дигидрохлорида транс-4-(2-пирролидин-1-ил-этил)циклогексиламина- 13015419 Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 4-[4-(4,5-дигидро-1 Н-имидазол-2-ил)бензил]пиперидина (ссылочный пример 6) в соответствии со способом, описанном в примере 1e. MS (EI) 674,3 (МН+). Пример 6. 2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-1-4-[4-(1,4,5,6 тетрагидропиримидин-2-ил)бензил]пиперидин-1-илэтанон. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 2-(4-пиперидин-4-илметилфенил)-1,4,5,6-тетрагидропиримидина(ссылочный пример 7) в соответствии со способом, описанным в примере 1e. MS (EI) 688,3 (МН+). Пример 7. N-[2-(4-Цианофенил)этил]-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин 6-ил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 4-(2-аминоэтил)бензонитрила [J. Am. Chem. Soc. 125 (2003) 75167517] в соответствии со способом, описанным в примере 1e. MS (EI) 577,2 (МН+). Пример 8. N-(4-[1,4']Бипиперидинил-1'-ил-фенил)-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 4-[1,4']бипиперидинил-1'-ил-фениламина [J. Med. Chem. 46 (2003) 1803-1806] в соответствии со способом, описанным в примере 1e. MS (EI) 690,3 (МН+). Пример 9. 2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[3-(4-пиридин-4 ил-пиперазин-1-ил)пропил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и 2-(4-пиперидин-4-ил-пиперазин-1-ил)пропиламин (ссылочный пример 8) в соответствии со способом, описанным в примере 1e. MS (EI) 651,2 (МН+). Пример 10. N-(3-[1,4']Бипиперидинил-1'-ил-пропил)-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и тригидрохлорида (3-[1,4']бипиперидинил-1'-ил)пропиламина (ссылочный пример 3) в соответствии со способом, описанном в примере 1e. MS (EI) 655,68 (МН+). Пример 11. 2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[2-(4-пиридин-2 ил-пиперазин-1-ил)этил]ацетамид. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]уксусной кислоты (пример 2 с) и тетрагидрохлорида 2-(4-пиридин-2-ил-пиперазин-1-ил)этиламина(ссылочный пример 5) в соответствии со способом, описанным в примере 1e. MS (EI) 637,3 (МН+). Пример 12. Гидрохлорид 2-Г 5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N[(пиперидин-4-илметил)карбамоил]метилацетамида. а) Метиловый эфир 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноуксусной кислоты. Раствор [5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты (пример 2 с) (225 мг, 0,5 ммоль), триэтиламина (0,14 мл, 1 ммоль) и HBTU [О-бензотриазол-1-ил-N,N,N',N'тетраметилуроний гексафторфосфат] (208 мг, 0,55 ммоль) в сухом диметилформамиде (5 мл) перемешивали при комнатной температуре в течение 5 мин перед добавлением глицинметилового эфира гидрохлорида (63 мг, 0,5 ммоль). Доводили рН реакционной смеси до 8 путем добавления триэтиламина, полученную таким образом смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Осадок очищали препаративной тонкослойной хроматографией (дихлорметан:метанол:NH4OH=15:1:0.1) до получения 248 мг (95%) названного соединения в виде белых кристаллов.b) 2-[5-(3,4-Дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноуксусная кислота. Названное соединение готовили из метилового эфира 2-[5-(3,4-дихлорбензилсульфонил)-5,6 дигидрофенантридин-6-ил]ацетиламиноуксусной кислоты в соответствии со способом, описанном в примере 1d. с) трет-Бутиловый эфир 4-[(2-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноацетиламино)метил]пиперидин-1-карбоновой кислоты. Названное соединение готовили из 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]ацетиламиноуксусной кислоты и трет-бутилового эфира 4-аминометилпиперидин-1-карбоновой кислоты в соответствии со способом, описанном в примере 1 е.d) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[(пиперидин 4-илметил)карбамоил]метилацетамида. К раствору трет-бутилового эфира 4-[(2-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноацетиламино)метил]пиперидин-1-карбоновой кислоты (140 мг, 0,2 ммоль) в этилацетате (5 мл) добавляли 2,4 М хлористый водород в этилацетате (4 мл, 9,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи, затем разбавляли диэтиловым- 14015419 эфиром (25 мл). Осажденный продукт отделяли фильтрацией и промывали диэтиловым эфиром до получения 127 мг (87%) названного соединения в виде белых кристаллов. MS (EI) 602,2 (МН+). Пример 13. Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[(2 пиперидин-4-ил-этилкарбамоил)метил]ацетамида.a) трет-Бутиловый эфир 4-[2-(2-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноацетиламино)этил]пиперидин-1-карбоновой кислоты. Названное соединение готовили из 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]ацетиламиноуксусной кислоты (пример 12b) и трет-бутилового эфира 4-(2-аминоэтил)пиперидин-1 карбоновой кислоты в соответствии со способом, описанным в примере 1 е.b) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[(2-пиперидин-4-ил-этилкарбамоил)метил]ацетамида. Названное соединение готовили из трет-бутилового эфира 4-[2-(2-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетиламиноацетиламино)этил]пиперидин-1 карбоновой кислоты в соответствии со способом, описанным в примере 12d. MS (EI) 616,2 (МН+). Пример 14. Гидрохлорид N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этил)-2-[5-(толуол-4 сульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида. а) N-Бифенил-2-ил-4-метил-бензилсульфонамид Названное соединение готовили из 2-аминобифенила (Aldrich) и 4-метилбензилсульфонилхлорида в соответствии со способом, описанном в примере 1b.b) Метиловый эфир [5-(толуол-4-сульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из N-бифенил-2-ил-4-метилбензилсульфонамида в соответствии со способом, описанным в примере 1 с.c) [5-(Толуол-4-сульфонил)-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(толуол-4-сульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d.d) Гидрохлорид N-2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил-2-[5-(толуол-4-сульфонил)5,6-дигидрофенантридин-6-ил]адетамида. Названное соединение готовили изN-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этил-2-[5-(2,4,6 триметилбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида. а) N-Бифенил-2-ил-2,4,4-метилбензилсульфонамид. Названное соединение готовили из 2-аминобифенила (Aldrich) и 2,4,6-триметилбензилсульфонилхлорида в соответствии со способом, описанным в примере 1b.b) Метиловый эфир [5-(2,4,6-триметилбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из N-бифенил-2-ил-2,4,6-триметилбензилсульфонамида в соответствии со способом, описанным в примере 1 с.c) [5-(2,4,6-Триметилбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(2,4,6-триметилбензилсульфонил)-5,6 дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d.d) N-2-[4-(4,5-Дигидро-1 Н-имидазол-2-ил)фенил]этил-2-[5-(2,4,6-триметилбензилсульфонил)-5,6 дигидрофенантридин-6-ил]ацетамид. Названное соединение готовили из [5-(2,4,6-триметилбензилсульфонил)-5,6-дигидрофенантридин 6-ил]уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина (ссылочный пример 2) в соответствии со способом, описанным в примере 1 е. Пример 16. Гидрохлорид N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этил-2-[5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида. а) N-Бифенил-2-ил-4-метоксибензилсульфонамид. Названное соединение готовили из 2-аминобифенила (Aldrich) и 4-метоксибензилсульфонилхлорида в соответствии со способом, описанным в примере 1b.b) Метиловый эфир [5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из N-бифенил-2-ил-4-метоксибензилсульфонамида в соответствии со способом, описанным в примере 1 с. с) [5-(4-Метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d.d) Гидрохлорид N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этил-2-[5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида. Названное соединение готовили из [5-(4-метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]- 15015419 уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина (ссылочный пример 2) в соответствии со способом, описанным в примере 1e. MS (EI) 581,6 (MH+). Пример 17. Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин 6-ил]-N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида.a) 2'-Метоксибифенил-2-иламин. Названное соединение готовили из 2-метоксифенилбороновой кислоты в соответствии со способом,описанным в примере 1 а.b) 3,4-Дихлор-N-(2'-метоксибифенил-2-ил)бензилсульфонамид. Названное соединение готовили из 2'-метоксибифенил-2-иламина в соответствии со способом, описанным в примере 1b.c) Метиловый эфир [5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из 3,4-дихлор-N-(2'-метоксибифенил-2-ил)бензилсульфонамида в соответствии со способом, описанным в примере 1 с.d) [5-(3,4-Дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(3,4-дихлорбензилсульфонил)-10-метокси 5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d. е) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]-N2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамида. Данное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина (ссылочный пример 2) в соответствии со способом, описанным в примере 1e. Пример 18. Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин 6-ил]-N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида.a) 2',4'-Дифторбифенил-2-иламин. Названное соединение готовили из 2,4-дифторфенилбороновой кислоты в соответствии со способом, описанным в примере 1a.b) 3,4-Дихлор-N-(2',4'-дифторбифенил-2-ил)бензилсульфонамид. Названное соединение готовили из 2',4'-дифторбифенил-2-иламина в соответствии со способом,описанным в примере 1b.c) Метиловый эфир [5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из 3,4-дихлор-N-(2',4'-дифторбифенил-2-ил)бензилсульфонамида в соответствии со способом, описанным в примере 1 с.d) [5-(3,4-Дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(3,4-дихлорбензилсульфонил)-8,10 дифтор-5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d.e) Гидрохлорид 2-[5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]-N2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида. Данное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламина (ссылочный пример 2) в соответствии со способом, описанным в примере 1e. MS (EI) 656,1a) 4'-Ацетилбифенил-2-иламин. Названное соединение готовили из 4-ацетилфенилбороновой кислоты в соответствии со способом,описанным в примере 1 а.b) N-(4'-Ацетилбифенил-2-ил)-3,4-дихлорбензилсульфонамид. Названное соединение готовили из 4'-ацетилбифенил-2-иламина в соответствии со способом, описанным в примере 1b.c) Метиловый эфир [8-ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусной кислоты. Названное соединение готовили из N-(4'-ацетилбифенил-2-ил)-3,4-дихлорбензилсульфонамида в соответствии со способом, описанным в примере 1c.d) [8-Ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [8-ацетил-5-(3,4-дихлорбензилсульфонил)5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d.e) Гидрохлорид 2-[8-ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-2[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамида. Данное соединение готовили из [8-ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенан- 16015419 тридин-6-ил]уксусной кислоты и дигидрохлорида 2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этиламин(ссылочный пример 2) в соответствии со способом, описанным в примере 1e. MS (EI) 662,3 (МН+). Пример 20. N-[2-(Карбамоилфенил)этил]-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамид. Раствор N-[2-(4-цианофенил)этил]-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]ацетамида (пример 7) (200 мг, 0,35 ммоль) в 9 М хлористом водороде в этаноле (10 мл) оставляли на ночь при комнатной температуре, затем смесь концентрировали под вакуумом. Осадок растворяли в этаноле (10 мл) и добавляли карбонат аммония (335 мг, 3,5 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем концентрировали под вакуумом. Осадок очищали препаративной тонкослойной хроматографией (дихлорметан:метанол:NH4OH=5:1:0,1) до получения 113 мг(10 мл) перемешивали при комнатной температуре в течение 5 мин перед добавлением дигидрохлорида транс-4-(2-пирролидин-1-ил-этил)циклогексиламин (ссылочный пример 4) (0,09 г, 0,34 ммоль). Доводили рН реакционной смеси до 8 путем добавления триэтиламина, полученную таким образом смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали под вакуумом. Осадок обрабатывали насыщенным раствором гидрокарбоната натрия (30 мл), осажденные кристаллы отделяли фильтрацией, промывали водой и высушивали. Неочищенный продукт очищали колоночной хроматографией с применением Кизельгеля 60 (0,040-0,063 мм) (Merck) в качестве адсорбента и хлороформа: метанола:NH4OH=9:1:0,1 в качестве элюента. Продукт кристаллизовали диэтиловым эфиром до получения 0,135 г (60%) названного соединения. MS (EI) 657,2 (МН+). Пример 22. транс-2-[5-(3,4-Дихлорбензилсульфонил)-8,10-диметокси-5,6-дигидрофенантридин-6 ил]-N-[4-(2-пирролидин-1-илэтил)циклогексил]ацетамид.a) 2',4'-Диметоксибифенил-2-иламин. Названное соединение готовили из 2,4-диметоксифенилбороновой кислоты в соответствии со способом, описанным в примере 1 а.b) 3,4-Дихлор-N-(2',4'-диметоксибифенил-2-ил)бензилсульфонамид. Названное соединение готовили из 2',4'-диметоксибифенил-2-иламина в соответствии со способом,описанным в примере 1b.c) Метиловый эфир [5-(3,4-дихлорбензилсульфонил)-8,10-диметокси-5,6-дигидрофенантридин-6 ил]уксусной кислоты. Названное соединение готовили из 3,4-дихлор-N-(2',4'-диметоксибифенил-2-ил)бензилсульфонамида в соответствии со способом, описанным в примере 1 с.d) [5-(3,4-Дихлорбензилсульфонил)-8,10-диметокси-5,6-дигидрофенантридин-6-ил]уксусная кислота. Названное соединение готовили из метилового эфира [5-(3,4-дихлорбензилсульфонил)-8,10 диметокси-5,6-дигидрофенантридин-6-ил]уксусной кислоты в соответствии со способом, описанным в примере 1d. е) Гидрохлорид 2-[5-(3,4-Дихлорбензилсульфонил)-8,10-диметокси-5,6-дигидрофенантридин-6-ил]N-2-[4-(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида. Названное соединение готовили из [5-(3,4-дихлорбензилсульфонил)-8,10-диметокси-5,6-дигидрофенантридин-6-ил]уксусной кислоты и дигидрохлорида транс-4-(2-пирролидин-1-ил-этил)циклогексиламин (ссылочный пример 4) в соответствии со способом, описанным в примере 21. MS (EI) 687,2 (МН+). Пример 23. Приготовление фармацевтических композиций.a) Таблетки. 0,01-50% активного ингредиента из формулы (I), 15-50% лактозы, 15-50% картофельного крахмала,5-15% поливинилпирролидона, 1-5% талька, 0,01-3% магния стеарата, 1-3% коллоидного диоксида кремния и 2-7% ультра-амилопектина смешивали, затем гранулировали влажным гранулированием и прессовали в таблетки.b) Драже, покрытые пленкой таблетки. Таблетки, приготовленные способом, описанным выше, покрывали слоем, включающим пленку,растворяющуюся в кишечнике или в желудке, или сахаром и тальком. Драже полировали смесью пчелиного воска и воска карнаубы.c) Капсулы. 0,01-50% активного ингредиента из формулы (I), 1-5% натрия лаурилсульфата, 15-50% крахмала,15-50% лактозы, 1-3% коллоидного диоксида кремния и 0,01-3% магния стеарата смешивали, смесь пропускали через сито и заполняли в капсулы из твердого желатина.- 17015419 Ингредиенты: 0,01-15% активного ингредиента из формулы (I), 0,1-2% гидроксида натрия, 0,1-3% лимонной кислоты, 0,05-0.2% нипагина (натрия метил-4-гидроксибензоата), 0,005-0,02% нипазола, 0,010,5% карбопола (полиакриловой кислоты), 0,1-5% 96% этанола, 0,1-15 вкусового агента, 20-70% сорбитола (70% водного раствора) и 30-50% дистиллированной воды. К раствору нипагина и лимонной кислоты в 20 мл дистиллированной воды добавляли карбопол маленькими порциями при энергичном смешивании и оставляли раствор на 10-12 ч. Затем добавляли гидроксид натрия в 1 мл дистиллированной воды, водный раствор сорбитола и, наконец, малиновый вкусовой агент в этаноле при перемешивании. К этому носителю добавляли активный ингредиент маленькими порциями и суспендировали с погруженным гомогенизатором. Окончательно суспензию доводили до желаемого конечного объема дистиллированной водой, пропускали сироп суспензии через перемалывающее коллоиды оборудование.e) Суппозитории. Для каждого суппозитория тщательно смешивали 0,01-15% активного ингредиента из формулы (I) и 1-20% лактозы, затем 50-95% адепса для суппозиториев (например, Witepsol 4) плавили, охлаждали до 35 С и перемешивали со смесью активного ингредиента и лактозы в гомогенизаторе. Полученную смесь отливали в охлаждаемые формы.f) Композиции из лиофилизированного порошка для ампул. Готовили 5% раствор маннитола или лактозы на бидистиллированной воде для инъекций и раствор фильтровали так, чтобы получить стерильный раствор. Также готовили 0,01-5% раствор активного ингредиента из формулы (I) на бидистиллированной воде для инъекций и этот раствор фильтровали так,чтобы получить стерильный раствор. Эти два раствора смешивали в асептических условиях, разливали порциями по 1 мл в ампулы, содержимое ампул лиофилизировали и ампулы запаивали под азотом. Содержимое ампул растворяли в стерильной воде или 0,9% (физиологическом) стерильном водном растворе хлорида натрия перед применением. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Антагонисты В 1 рецептора брадикинина - производные фенантридина формулы (I) где R1 является атомом водорода или С 1-С 4 алкильной группой;R2 выбран из (1) атома водорода при условии, что R1 и R2 не могут быть одновременно атомом водорода; (2) -(CH2)n-NRaRb; (3) -(CH2)n-CO-NRaRb; (4) -(CH2)m-X-Q; (5) -CHRc-NRaRb; илиR1 и R2 вместе с атомом азота, к которому они присоединены, образуют 4-7-членное гетероциклическое кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо при необходимости замещено -CO-NRaRb, C1-C4 алкилом, 4-(4,5-дигидро-1H-имидазол-2-ил)бензилом или 4-(1,4,5,6 тетрагидропиримидин-2-ил)бензилом;R3, R4, R5, R6 и R7 являются независимо один от другого атомом водорода, атомом галогена, трифторметилом, С 1-С 4 алкилом, С 1-С 4 алкокси- или ацетильной группой;n является целым числом от 1 до 4;Ra и Rb являются атомом водорода, при необходимости замещенным С 1-С 4 алкильной группой, илиR , R и атом азота, к которому они присоединены, вместе образуют насыщенное, частично ненасыщенное или ароматическое 4-7-членное кольцо, включающее 1-3 гетероатома, выбранных из О, S и N; где указанное кольцо при необходимости замещено 1-пиперидинильной, 2-пиперидинильной, 4 пиперидинильной, 2-пиридильной или 4-пиридильной группой;Rc является метильной, гидроксиметильной, бензильной или фенильной группой;m является целым числом от 0 до 6;Q является фенильной группой, при необходимости замещенной [1,4']-бипиперидинил-1'-ильной,4,5-дигидро-1H-имидазол-2-ильной, -(CH2)n-NH-(C=NH)-NH2 или -(CH2)m-(C=NH)-NH2 группой; или 4 пиперидинильной группой, при необходимости замещенной 4-пиперидинильной группой; или С 5-С 7 циклоалкильной группой, не обязательно замещенной -(CH2)m-NRaRb группой,- 18015419 и их оптические антиподы, или рацематы, и/или соли, и/или гидраты, и/или сольваты. 2. Соединения по п.1, выбранные из группы из гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-8-фтор-5,6-дигидрофенантридин-6-ил]-N-2-[4(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамида,гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-2-[4-(4,5 дигидро-1H-имидазол-2-ил)фенил]этилацетамида,2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[2-(4-пиридин-4-ил-пиперазин-1-ил)этил]ацетамида,транс-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[4-(2-пирролидин-1-илэтил)циклогексил]ацетамида,2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-1-4-[4-(4,5-дигидро-1 Н-имидазол-2-ил)бензил]пиперидин-1-илэтанона,2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-1-4-[4-(1,4,5,6-тетрагидропиримидин-2-ил)бензил]пиперидин-1-илэтанона,N-(4-[1,4']-пиперидинил-1'-ил-фенил)-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин 6-ил]ацетамида,2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-[3-(4-пиридин-4-ил-пиперазин-1-ил)пропил]ацетамида,гидрохлорида N-2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил-2-[5-(толуол-4-сульфонил)-5,6 дигидрофенантридин-6-ил]ацетамида,гидрохлоридаN-2-[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этил-2-[5-(4 метоксибензилсульфонил)-5,6-дигидрофенантридин-6-ил]ацетамида,гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]-N-2[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамида,гидрохлорида 2-[5-(3,4-дихлорбензилсульфонил)-8,10-дифтор-5,6-дигидрофенантридин-6-ил]-N-2[4-(4,5-дигидро-1H-имидазол-2-ил)фенил]этилацетамида,гидрохлорида 2-[8-ацетил-5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6-ил]-N-2-[4(4,5-дигидро-1 Н-имидазол-2-ил)фенил]этилацетамида,N-[2-(4-карбамимидоилфенил)этил]-2-[5-(3,4-дихлорбензилсульфонил)-5,6-дигидрофенантридин-6 ил]ацетамида или транс-2-[5-(3,4-дихлорбензилсульфонил)-10-метокси-5,6-дигидрофенантридин-6-ил]-N-[4-(2-пирролидин-1-илэтил)циклогексил]ацетамида. 3. Способ приготовления соединений формулы (I) по п.1, включающий реакцию производного бороновой кислоты формулы (II) в присутствии катализатора, предпочтительно тетракис(трифенилфосфин)палладия(0), до получения аминобифенил производного формулы (IV) где значение R6 и R7 соответствует определенному выше,- 19015419 сульфонилированного с производным сульфохлорида формулы (V) где значение R3, R4 и R5 соответствует определенному выше,и образованного сульфонамидного производного формулы (VI) где значение R3, R4, R5, R6 и R7 соответствует определенному выше,подвергается реакции циклизации и полученное производное эфира фенантридина и уксусной кислоты формулы (VII) где значение R3, R4, R5, R6 и R7 соответствует определенному выше, a R является С 1-С 4 алкильной группой,гидролизуется в присутствии основания до получения фенантридинового производного уксусной кислоты формулы (VIII) где значение R3, R4, R5, R6 и R7 соответствует определенному выше,затем последнее реагирует с аминовым производным формулы (IX) где значение R1 и R2 соответствует вышеописанному. 4. Способ по п.1, включающий трансформацию соединений из формулы (I) в другие соединения формулы (I) путем введения новых заместителей и/или модификаций, или удаления существующих заместителей, и/или образования соли, и/или высвобождения соединения из солей. 5. Соединение формулы (IX), выбранное из группы дигидрохлорид транс-4-(2-пирролидин-1-илэтил)циклогексиламина, 4-[4-(4,5-дигидро-1 Н-имидазол-2-ил)бензил]пиперидина, 2-(4-пиперидин-4 илметилфенил)-1,4,5,6-тетрагидропиримидина. 6. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов и одного или более фармацевтически пригодных наполнителей. 7. Применение соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов для производства медикамента для профилактики и/или лечения состояния, требующего ингибирования рецептора брадикинина. 8. Применение по п.7, где рецептором брадикинина является В 1 рецептор брадикинина. 9. Способ лечения и/или профилактики состояния, требующего ингибирования рецептора брадикинина, включающий введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I) по п.1, или его оптических антиподов, или рацематов, или фармацевтически пригодных солей, или гидратов, или сольватов. 10. Способ по п.9, где рецептором брадикинина является В 1 рецептор брадикинина.
МПК / Метки
МПК: C07D 401/10, A61K 31/473, C07D 221/12, C07D 401/14, C07D 401/12, A61P 25/02, C07D 295/12, C07D 213/74
Метки: фенантридина, новые, антагонистов, качестве, брадикинина, производные
Код ссылки
<a href="https://eas.patents.su/23-15419-novye-proizvodnye-fenantridina-v-kachestve-antagonistov-bradikinina.html" rel="bookmark" title="База патентов Евразийского Союза">Новые производные фенантридина в качестве антагонистов брадикинина</a>
Производные n-бифенилметиламиноциклоалканкарбоксамида с заместителем на метиле, в качестве антагонистов брадикинина

Номер патента: 7297
Опубликовано: 25.08.2006
Авторы: Бок Марк Дж., Су Дай-Си, Вуд Майкл Р., Вэй Дженни Миу-Чун, Фенг Донг-Мей, Кудук Скотт Д., Энтони Невилл Дж.
МПК: C07D 213/82, C07D 239/00, C07C 237/24...
Метки: заместителем, антагонистов, качестве, брадикинина, n-бифенилметиламиноциклоалканкарбоксамида, производные, метиле
Формула / Реферат:
1. Соединение формулы I(3) и его фармацевтически приемлемые соли где m равно 0 или 1, R6a обозначает 2-метил-2Н-тетразол-5-ил, 3-метил-1,2,4-оксадиазол-5-ил, CO2Ra или C(O)NHORa, где Ra означает С1-4алкил; R6b означает водород, фтор или хлор; R3b означает С1-4алкил; R5 выбран из С1-4алкила, необязательно замещенного 1-5 атомами галогена или цианогруппой, С3-6циклоалкила, изоксазолила, пиримидинила и пиридинила (и его N-оксида), необязательно...
Новые ацилгуанидиновые производные в качестве ингибиторов резорбции костной ткани и антагонистов витронектиновых рецепторов

Номер патента: 2921
Опубликовано: 31.10.2002
Авторы: Брайполь Герхард, Пейман Ануширван, Карниато Дени, Макдауэлл Роберт, Шойнеманн Карл-Хайнц, Кнолле Йохен, Феррара Наполиэйн, Гурве Жан-Франсуа, Катбертсон Роберт Эндрю, Бодари Сара Кэтрин, Гейдек Томас
МПК: C07C 279/22, A61K 31/505, A61P 19/10...
Метки: ацилгуанидиновые, производные, костной, антагонистов, рецепторов, качестве, ткани, ингибиторов, резорбции, витронектиновых, новые
Формула / Реферат:
1. Соединение формулы I где радикалы R1 и R2 вместе представляют насыщенную или ненасыщенную двухвалентную (С2-С3)-алкиленовую группу; R4 представляет собой водород или (C1-C6)-алкил; R5 представляет собой фенил или бензил; R6 представляет собой водород; А представляет собой СН2 или О; m равно 1, 2 или 3; n равно 0 или 1; во всех его стереоизомерных формах и их смесях в любых соотношениях, и его физиологически переносимые соли, и его...
О-замещенные производные дибензилмочевины в качестве антагонистов рецепторов trpv1

Номер патента: 15152
Опубликовано: 30.06.2011
Авторы: Павани Мария Джованна, Бореа Пьер Андреа, Джеппетти Пьеранджело, Фруттароло Франческа, Баральди Пьер Джованни, Тревизани Марчелло
МПК: A61K 31/17, A61P 29/00, C07C 275/24...
Метки: антагонистов, производные, рецепторов, дибензилмочевины, качестве, trpv1, о-замещенные
Формула / Реферат:
1. Соединения формулы (I)где R выбран из галогена, алкила, алкокси, арила и гетероарила;R1 выбран из 2-гидроксиэтила, 2,3-дигидроксипропила, 3-гидроксипропила, 2,2-дигидроксиэтила, 3,3-дигидроксипропила, 1,3-диоксоланэтила, 1,3-диоксанметила, 1,3-диоксоланметила, 1,3-диоксанэтила, 3-фтор-2-гидроксипропила, 3-карбокси-2-гидроксипропила, 3-хлор-2-гидроксипропила, 2-гидроксипропила, 2-гидроксипропен-2-ила, морфолиноэтила, пиперазиноэтила,...
Производные тетрагидрохинолина в качестве антагонистов глицина

Номер патента: 3276
Опубликовано: 24.04.2003
Автор: Ди Фабио Романо
МПК: A61P 25/28, A61K 31/4709, C07D 401/04...
Метки: тетрагидрохинолина, производные, качестве, антагонистов, глицина
Формула / Реферат:
1. Соединение формулы (I) или его соль, или нетоксичные метаболически лабильные сложные эфиры, где Y представляет собой атом углерода, Z представляет собой группу CH, которая связана с группой Y посредством двойной связи, а X представляет собой CH или Z представляет собой метилен или NR11, a X представляет собой атом углерода, связанный с группой Y посредством двойной связи, A представляет собой C1-2алкиленовую цепь, которая может быть...
Производные (тио) карбамоилциклогексана в качестве антагонистов d3/d2 рецептора

Номер патента: 9022
Опубликовано: 26.10.2007
Авторы: Ласловски Иштван, Саги Каталин, Агаине Чонгор Эва, Кишш Бела, Ваго Иштван, Ногради Каталин, Ласи Юдит, Дьертьян Иштван, Галамбош Янош
МПК: A61P 25/00, C07D 243/08, A61K 31/495...
Метки: карбамоилциклогексана, антагонистов, производные, качестве, тио, рецептора
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 независимо представляют собой заместитель, выбранный из водорода, C1-6алкила, С2-7алкенила, арила, циклоалкила, ароила, или R1 и R2 могут образовывать гетероциклическое кольцо со смежным атомом азота; X представляет собой атом кислорода или серы; n является целым числом от 1 до 2, и/или его геометрические изомеры, и/или стереоизомеры, и/или диастереомеры, и/или соли, и/или гидраты, и/или сольваты. 2....
Предыдущий патент: Получение прегабалина и родственных соединений
Следующий патент: Способ получения r-энантиомера n-карбамоилметил-4-фенил-2-пирролидона
Случайный патент: Способ удаления газообразного компонента из текучей среды