Замещенные производные индола для лечения иммунологических расстройств
Номер патента: 23238
Опубликовано: 31.05.2016
Авторы: Каммертэн Карен, Ван Эйс Морис, Кук Найджел Грэхэм, Рамос Рита
















Формула / Реферат
1. Соединение формулы (I) или его фармацевтически приемлемая соль или гидрат:

где X обозначает СН или N;
R обозначает H или PO3H2;
R1 обозначает H или С1-4алкил;
R2 обозначает H или С1-4алкил;
R3 обозначает H или С1-4алкил и
R4 и R5, независимо друг от друга, обозначают H или С1-4алкил или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу.
2. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль или гидрат, где
X обозначает СН;
R обозначает Н;
R1 обозначает Н;
R2 обозначает H или С1-4алкил;
R3 обозначает H или С1-4алкил и
R4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу.
3. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль или гидрат, где
X обозначает N;
R обозначает PO3H2;
R1 обозначает Н;
R2 обозначает Н или С1-4алкил;
R3 обозначает H и
R4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу.
4. Соединение по п.1, которое представляет собой соединение формулы (III)

или его фармацевтически приемлемую соль или гидрат.
5. Соединение по п.1 или 4, которое представляет собой моногидрат моно[3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1-илметилового] эфира фосфорной кислоты.
6. Соединение по любому из пп.1 или 2, которое представляет собой 3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-1-гидроксиметил-4-(7-метил-1Н-индол-3-ил)пиррол-2,5-дион или его фармацевтически приемлемую соль.
7. Соединение по любому из пп.1 или 3, которое представляет собой моно{3-(1Н-индол-3-ил)-4-[2-(4-метилпиперазин-1-ил)хиназолин-4-ил]-2,5-диоксо-2,5-дигидропиррол-1-илметиловый} эфир фосфорной кислоты или его фармацевтически приемлемую соль.
8. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или гидрата для изготовления лекарственного средства для лечения расстройств или заболеваний, где указанное лечение может быть показано в случае острого или хронического отторжения органа или ткани алло- или ксенотрансплантатов, и реакции "трансплантат против хозяина".
9. Способ лечения острого или хронического отторжения органа или ткани алло- или ксенотрансплантатов и реакции "трансплантат против хозяина" у нуждающегося в этом субъекта, включающий введение указанному субъекту эффективного количества соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или гидрата.
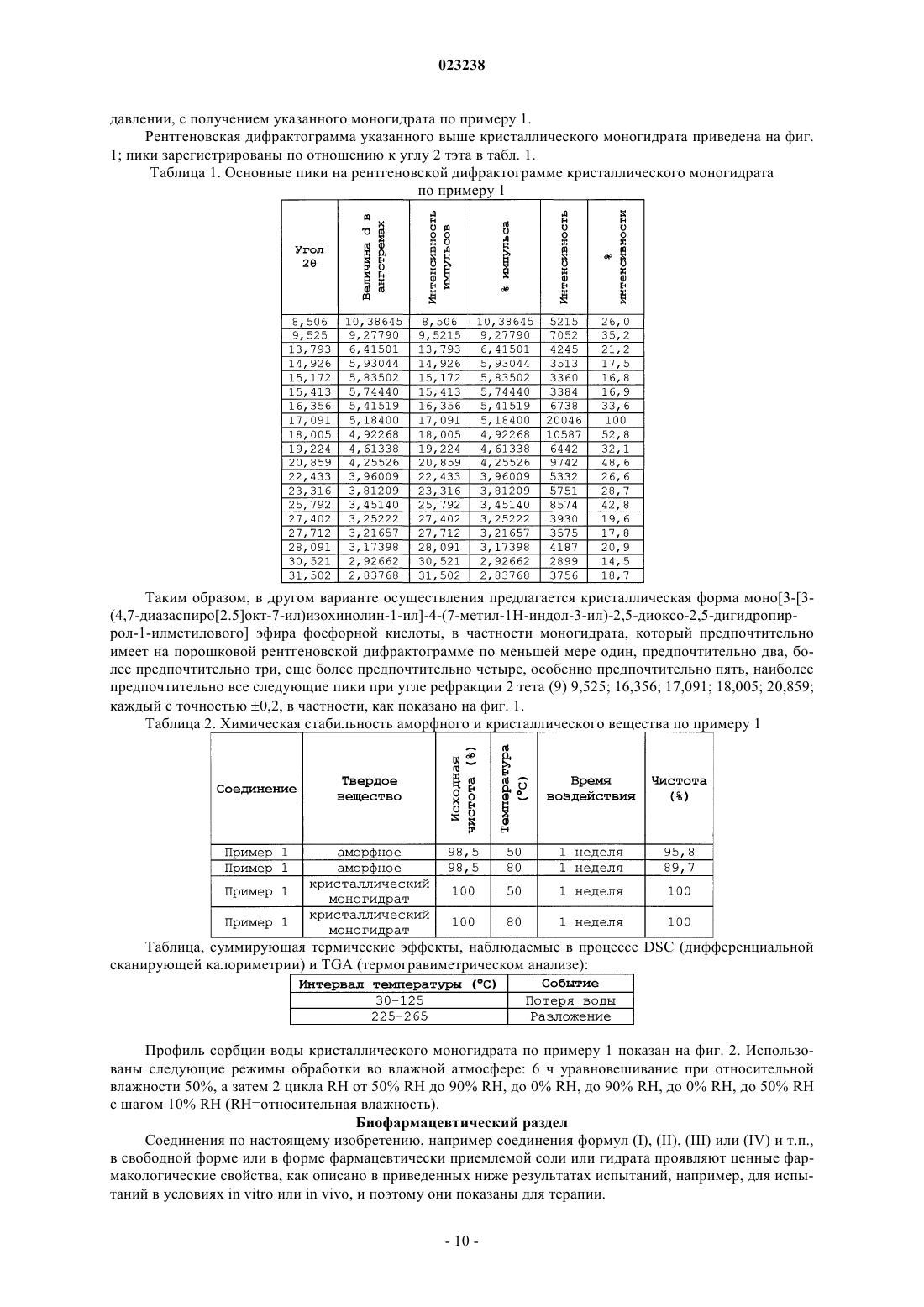
10. Кристаллическая форма моно[3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1-илметилового] эфира фосфорной кислоты, которая имеет на порошковой рентгеновской дифрактограмме следующие пики при угле рефракции 2 тета (θ): 9,525; 16,356; 17,091; 18,005; 20,859; каждый с точностью ±0,2.
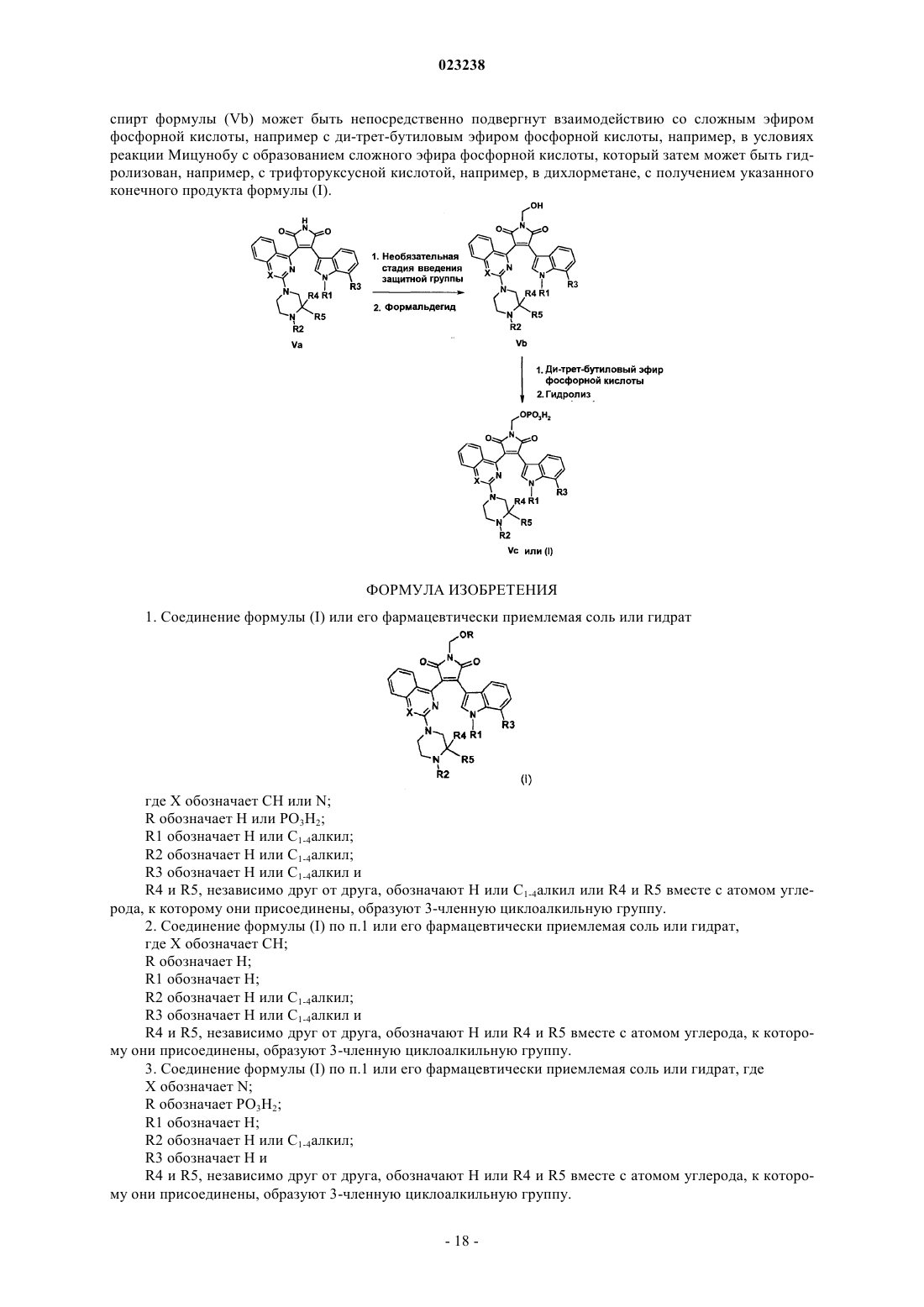
11. Способ получения соединения формулы (I) по п.1

где X обозначает СН или N;
R обозначает H или PO3H2;
R1 обозначает H или С1-4алкил;
R2 обозначает H или С1-4алкил;
R3 обозначает Н или С1-4алкил и
R4 и R5, независимо друг от друга, обозначают H или C1-4алкил или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу,
включающий:
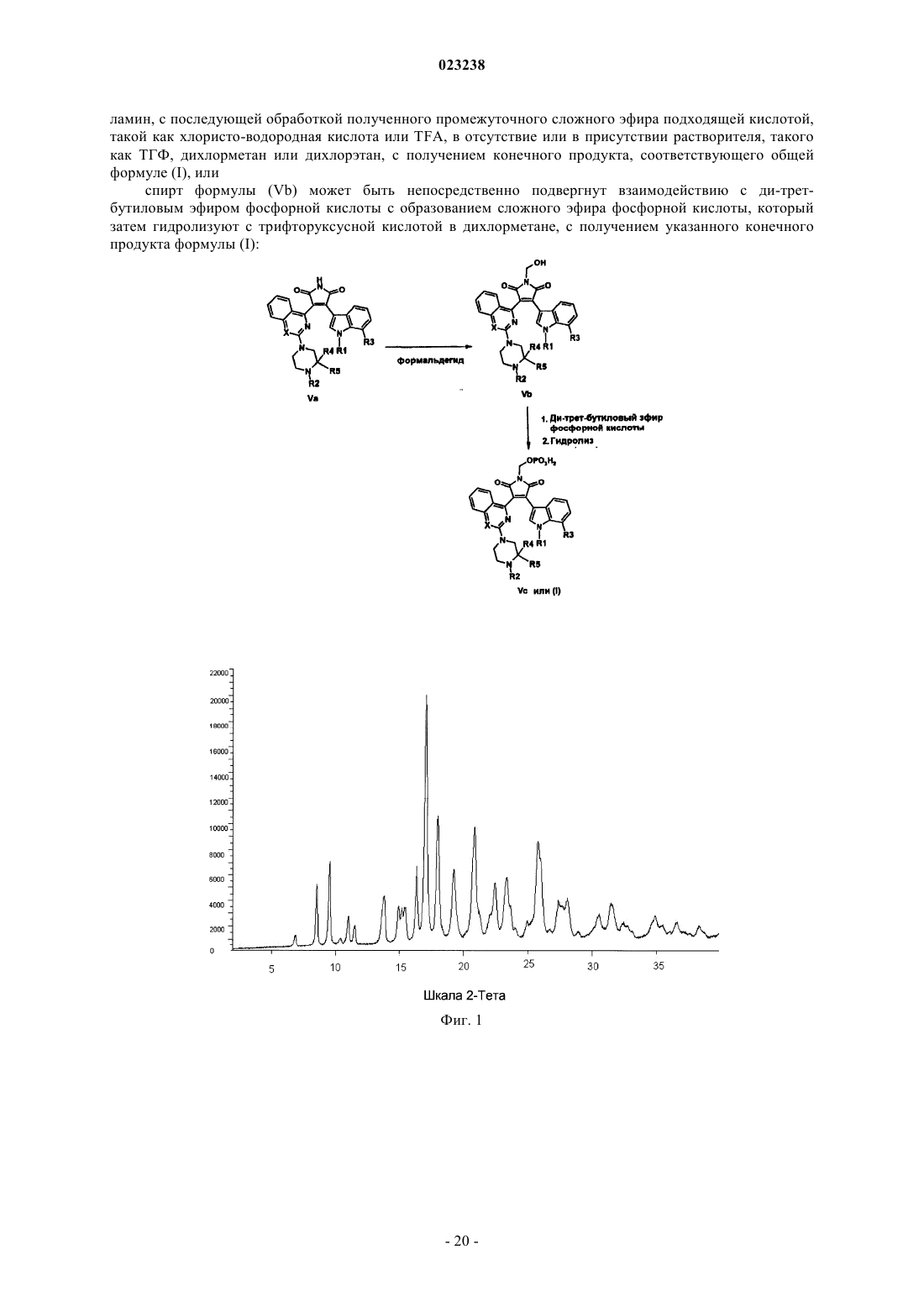
(а) обработку малеимида формулы (Va) формальдегидом в присутствии или в отсутствие растворителя и/или основания, такого как карбонат калия, с получением спирта формулы (Vb), где R=H; причем для соединений, где R1 и/или R2 представляют собой атом водорода, до указанной реакции малеимид формулы (Va) обрабатывают ди-трет-бутилдикарбонатом в присутствии или в отсутствие растворителя, такого как ТГФ или дихлорметан, и/или основания, такого как триэтиламин, с получением малеимида формулы (Va), содержащего трет-бутоксикарбонильные группы вместо атомов водорода у R1 и/или R2;
(b) обработку спирта формулы (Vb), включающую
обработку спирта формулы (Vb) трихлорацетонитрилом в присутствии основания, такого как DBU или триметиламин, с получением реакционноспособного сложного эфира, с последующей обработкой ди-трет-бутиловым эфиром фосфорной кислоты в присутствии основания, в присутствии основания, такого как DBU или триметиламин, с последующей обработкой полученного промежуточного сложного эфира подходящей кислотой, такой как хлористо-водородная кислота или TFA, в отсутствие или в присутствии растворителя, такого как ТГФ, дихлорметан или дихлорэтан, с получением конечного продукта, соответствующего общей формуле (I), или
спирт формулы (Vb) может быть непосредственно подвергнут взаимодействию с ди-трет-бутиловым эфиром фосфорной кислоты с образованием сложного эфира фосфорной кислоты, который затем гидролизуют с трифторуксусной кислотой в дихлорметане, с получением указанного конечного продукта формулы (I)

Текст
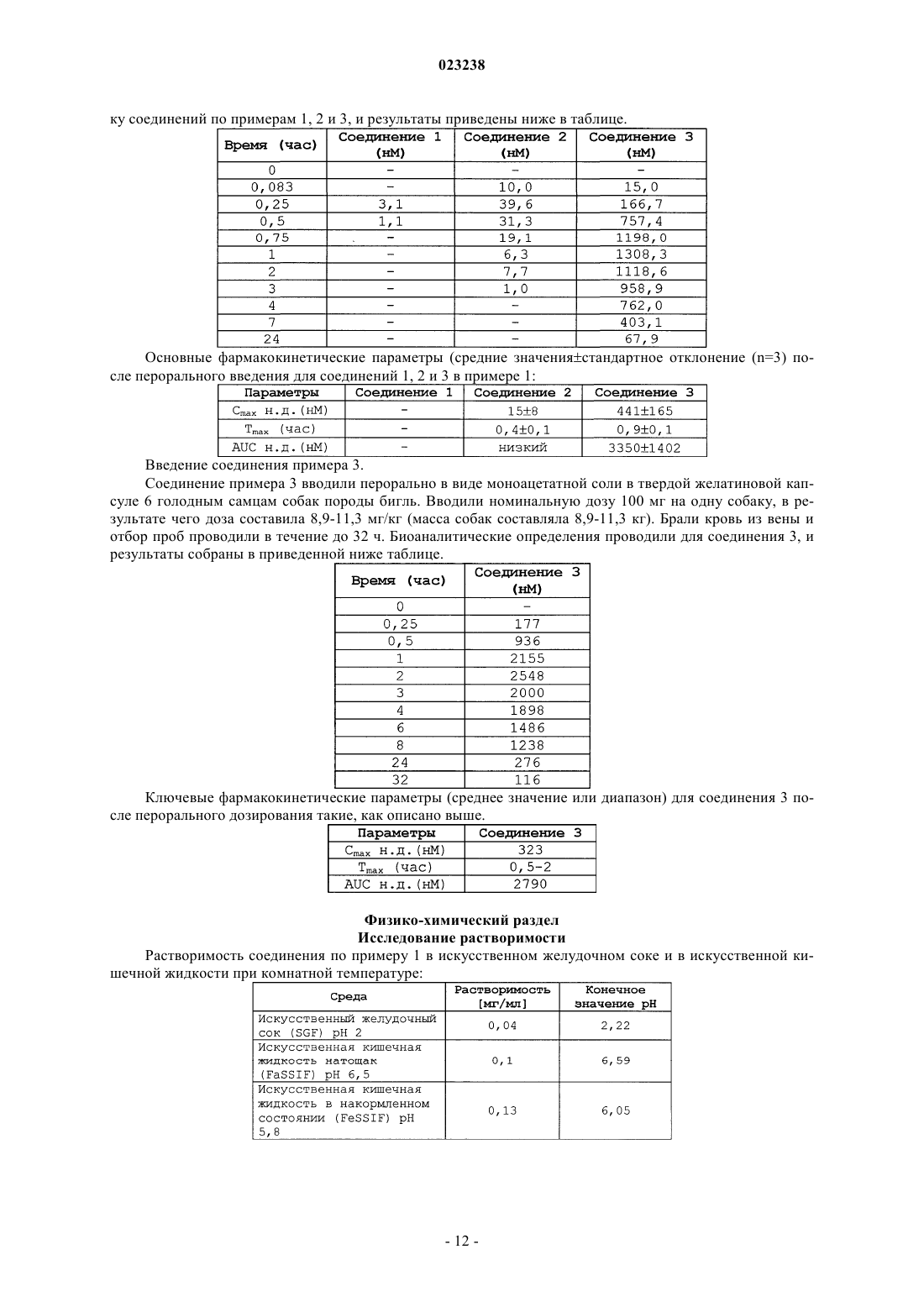
ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДОЛА ДЛЯ ЛЕЧЕНИЯ ИММУНОЛОГИЧЕСКИХ РАССТРОЙСТВ Настоящее изобретение относится к замещенным производным индола, к способам их получения,их применению в качестве фармацевтических средств и к содержащим их фармацевтическим композициям. Настоящее изобретение относится к замещенным производным индола, к способам их получения,их применению в качестве фармацевтических средств и к содержащим их фармацевтическим композициям. Уровень техники Низкая биодоступность лекарственных средств часто является ограничивающим фактором для фармацевтически эффективных ингредиентов. Указанная проблема решается в настоящее время, в частности, в области производных индола, путем преобразования соответствующего родительского лекарственного средства в его производное, которое, как неожиданно оказалось, оказывает положительное воздействие, по сравнению с его родительскими соединениями. Сущность изобретения Более конкретно, настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или гидратуX обозначает СН или N;R3 обозначает Н, С 1-4 алкил; CN; атом галогена или ОН; иR4 и R5, независимо друг от друга, обозначают H или C1-4 алкил или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют (3-6)-членную циклоалкильную группу. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или гидрату,где X обозначает СН;R4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют (3-6)-членную циклоалкильную группу. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или гидрату,гдеR4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют (3-6)-членную циклоалкильную группу. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или гидрату,где X обозначает N;R4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют (3-6)-членную циклоалкильную группу. В другом варианте осуществления настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или гидрату,гдеR4 и R5, независимо друг от друга, обозначают Н или C1-4 алкил. В другом варианте осуществления настоящее изобретение относится к соединению формулы (II) или его фармацевтически приемлемой соли. В другом варианте осуществления настоящее изобретение относится к соединению формулы (III) или его фармацевтически приемлемой соли или гидрату. В другом варианте осуществления настоящее изобретение относится к соединению формулы (IV) или его фармацевтически приемлемой соли. Подробное описание фигур На фиг. 1 приведена рентгеновская дифрактограмма кристаллического моногидрата моно[3-[3-(4,7 диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1 илметилового] эфира фосфорной кислоты (моногидрат по примеру 1). На фиг. 2 приведен профиль абсорбции воды для кристаллического моногидрата по примеру 1[уравновешивание в течение 6 ч при 50% относительной влажности с последующими 2 циклами при изменении относительной влажности от 50% RH до 90% RH, до 0% RH, до 90% RH, до 0% RH, до 50% RH с шагом 10% RH]. (RH=относительная влажность) Предшествующий уровень техникиN. Fotouhi et al. (EP 1224181) описывают замещенные производные пиррола, где химическая модификация указанного пиррольного кольца состоит из большого числа переменных, а также может содержать гидроксиметиленовые или метиленфосфатные группы. Определения Как использовано в данном описании, термин "атом галогена" (или галоген) означает атом фтора,брома, хлора или йода, в частности атом фтора, хлора. Как использовано в данном описании, термин "алкил" относится к полностью насыщенному разветвленному или неразветвленному углеводородному остатку, имеющему вплоть до 4 атомов углерода. Иллюстративные примеры алкила включают, но, не ограничиваясь ими, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил и т.п. Как использовано в данном описании, термин "алкокси" относится к группе алкил-О-, где алкил является таким, как определено выше в данном описании. Иллюстративные примеры алкокси включают,но, не ограничиваясь ими, метокси, этокси, пропокси, 2-пропокси, бутокси, трет-бутокси, пентилокси,гексилокси, циклопропилокси, циклогексилокси и т.п. Как правило, алкоксигруппы содержат 1-4 атома углерода. Как использовано в данном описании, термин "циклоалкил" относится к насыщенной или ненасыщенной моноциклической углеводородной группе, содержащей 3-6 атомов углерода, в частности, 3-5 атомов углерода, в особенности, 3-4 или 3 атома углерода. Как использовано в данном описании, термин "соль" или "соли" относится к кислотно-аддитивной соли или основно-аддитивной соли соединения по настоящему изобретению. "Соли" включают, в частности, "фармацевтически приемлемые соли". Термин "фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства соединений по настоящему изобретению и которые, как правило, не являются биологически или иным образом нежелательными. Во многих случаях соединения по настоящему изобретению способны образовывать кислотные и/или основные соли благодаря присутствию амино и/или карбоксильных групп или сходных с ними групп. Фармацевтически приемлемые кислотно-аддитивные соли могут быть образованы с неорганическими кислотами и органическими кислотами; например, такие соли, как ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид, хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изетионат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат,сукцинат, субсалицилат, тартрат, тозилат и трифторацетат. Неорганические кислоты, из которых могут быть получены соли, включают, например, хлористоводородную кислоту, бромисто-водородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту и т.п. Органические кислоты, из которых могут быть получены соли, включают, например, уксусную кислоту, пропионовую кислоту, гликолевую кислоту, щавелевую кислоту, малеиновую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, лимонную кислоту, бензойную кислоту, миндальную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, толуолсульфоновую кислоту, сульфосалициловую кислоту и т.п. Фармацевтически приемлемые основно-аддитивные соли могут быть образованы с неорганическими и органическими основаниями. Неорганические основания, из которых могут быть получены соли, включают, например, соли аммония и металлов из групп I-XII периодической таблицы. В некоторых вариантах осуществления соли получают из натрия, калия, аммония, кальция, магния, железа, серебра, цинка и меди; в частности, пригодные для использования соли включают соли аммония, калия, натрия, кальция и магния. Органические основания, из которых могут быть получены соли, включают, например, первичные,вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т.п. Некоторые органические амины включают изопропиламин, бензатин, холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, основной или кислотной группы обычными химическими способами. Как правило,подобные соли могут быть получены взаимодействием свободной кислотной формы указанных соединений со стехиометрическим количеством подходящего основания (такого как гидроксид, карбонат, бикарбонат Na, Ca, Mg, K и т.п.), или взаимодействием формы свободного основания указанных соединений со стехиометрическим количеством соответствующей кислоты. Подобные реакции, как правило, осуществляют в воде или в органическом растворителе, или в смеси воды и органического растворителя. Как правило, желательно использовать, если это возможно, неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечень дополнительных подходящих солей можно найти, например, в "Remington's Pharmaceutical Sciences", 20th d., Mack Publishing Company, Easton, Pa., (1985); и в"Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002). Любая формула, приведенная в данном описании, представляет не содержащие изотопы формы, а также меченные изотопами формы соединений. Содержащие радиоактивные метки соединения имеют структуры, представленные формулами в данном описании, за исключением того, что один или более атомов заменены атомами, имеющими выбранную атомную массу или массовое число. Примеры изотопов, которые могут быть введены в соединения по настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2 Н, 3 Н, 11 С, 13 С, 14 С, 15N,18F, 31P, 32P, 35S, 36Cl, 125I соответственно. Настоящее изобретение включает различные меченные изотопами соединения, как указано в данном описании, например соединения, в которые включены радиоактивные изотопы, такие как 3 Н и 14 С, или соединения, в которых присутствуют нерадиоактивные изотопы,такие как 2 Н и 13 С. Такие меченные изотопами соединения (содержащие 14 С) пригодны для проведения метаболических исследований, исследования кинетики реакций (например, содержащие 2 Н или 3 Н), для обнаружения или для таких методов визуализации, как позитронно-эмиссионная томография (PET) или однофотонная эмиссионная компьютерная томография (SPECT), включая распределение в тканях лекарственных средств или субстратов, или для радиотерапии пациентов. В частности, меченное атомами 18F соединение наиболее пригодно для проведения PET или SPECT исследований. Меченные изотопами со-3 023238 единения по настоящему изобретению и их пролекарства можно, в общем случае, получить, осуществлением методик, описанных на схемах или в примерах и препаративных примерах, как описано ниже, путем замены не меченного изотопом реагента на легкодоступный меченный изотопом реагент. Кроме того, замещение более тяжелыми изотопами, в частности дейтерием (т.е. 2 Н, или D), может предоставлять определенные терапевтические преимущества, которые являются результатом большей метаболической стабильности, например, увеличенным периодом полураспада в условиях in vivo, или снижением требований дозировки, или увеличением широты терапевтического действия. Следует понимать, что дейтерий в данном контексте рассматривается в качестве заместителя соединения по настоящему изобретению. Концентрацию такого более тяжелого изотопа, в частности, дейтерия, можно определить с помощью коэффициента изотопного обогащения. Термин "коэффициент изотопного обогащения" в данном описании означает отношение между содержанием изотопа и природной распространенностью указанного изотопа. Если заместителем в соединении по настоящему изобретению является дейтерий, то подобное соединение имеет коэффициент изотопного обогащения для каждого указанного атома дейтерия, равный по меньшей мере 3500 (включение 52,5% дейтерия в каждом указанном атоме дейтерия), по меньшей мере 4000 (включение 60% дейтерия), по меньшей мере 4500 (включение 67,5% дейтерия), по меньшей мере 5000 (включение 75% дейтерия), по меньшей мере 5500 (включение 82,5% дейтерия), по меньшей мере 6000 (включение 90% дейтерия), по меньшей мере 6333,3 (включение 95% дейтерия), по меньшей мере 6466,7 (включение 97% дейтерия), по меньшей мере 6600 (включение 99% дейтерия) или по меньшей мере 6633,3 (включение 99,5% дейтерия). Меченные изотопами соединения по настоящему изобретению могут быть получены обычными способами, известными специалистам из данной области техники, или способами, аналогично описанным в прилагаемых примерах и препаративных примерах, с использованием соответствующих меченных изотопами реагентов вместо ранее использованных не содержащих метку реагентов. Фармацевтически приемлемые сольваты согласно настоящему изобретению включают такие сольваты, в которых растворитель кристаллизации может быть изотопнозамещенным, например, D2O, d6 ацетон, d6-ДМСО. Соединения по настоящему изобретению, например, соединения формулы (I), которые содержат группы, способные играть роль доноров и/или акцепторов водородных связей, могут быть способны образовывать сокристаллы с подходящими для образования сокристаллов агентами. Указанные сокристаллы могут быть получены из соединений формулы (I) известными методиками получения сокристаллов. Подобные методики включают помол, нагревание, совместную сублимацию, совместное плавление или контакт в растворе соединений формулы (I) с образующими сокристаллы агентами в условиях кристаллизации и выделение образовавшихся сокристаллов. Подходящие образующие сокристаллы агенты включают такие агенты, которые описаны в WO 2004/078163. Таким образом, в настоящем изобретении предлагаются также сокристаллы, содержащие соединение формулы (I). Как использовано в данном описании, термин "фармацевтически приемлемый носитель" включает любой и все растворители, дисперсионные среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные агенты, противогрибковые агенты), изотонические агенты, замедляющие абсорбцию агенты, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие вещества, эксципиенты, дезинтегранты, лубриканты, подсластители,ароматизаторы, красители и т.п. и их комбинации, которые известны специалистам из данной области техники (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329). За исключением тех случаев, когда какой-либо носитель несовместим с активным ингредиентом, предполагается, что он может быть использован в терапевтических или фармацевтических композициях. Термин "терапевтически эффективное количество" соединения по настоящему изобретению относится к такому количеству соединения по изобретению, которое способно вызывать у субъекта биологический или терапевтический ответ, например, снижение или ингибирование ферментативной или белковой активности или облегчение симптомов, облегчение состояний, замедление или задержку развития заболевания или предотвращение заболевания и т.д. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при его введении субъекту эффективно (1) для, по меньшей мере, частичного облегчения, ингибирования, предотвращения и/или облегчения состояния, или расстройства, или заболевания, которое (i) опосредуется протеинкиназой С, или (ii) связано с активностью протеинкиназы С, или(iii) определяется активностью (нормальной или аномальной) протеинкиназы С; или (2) для снижения или ингибирования активности протеинкиназы С; или (3) для снижения или ингибирования экспрессии протеинкиназы С. В другом, неограничивающем варианте осуществления термин "терапевтически эффективное количество" относится к количеству соединения по настоящему изобретению, которое при его введении в клетку или ткань, или внеклеточный биологический материал, или среду эффективно, по меньшей мере, для частичного уменьшения или ингибирования активности протеинкиназы С; или, по меньшей мере, для частичного снижения или ингибирования экспрессии протеинкиназы С. Как использовано в данном описании, термин "субъект" относится к животному. Как правило, жи-4 023238 вотным является млекопитающее. Субъект также относится, например, к приматам (в частности, людям,мужчинам или женщинам), коровам, овцам, козам, лошадям, собакам, кошкам, кроликам, крысам, мышам, рыбам, птицам и т.п. В некоторых вариантах осуществления субъектом является примат. Наконец,в других вариантах осуществления субъектом является человек. Как использовано в данном описании, термин "ингибировать" или "ингибирование" означает ослабление или подавление данного состояния, симптома или расстройства, или заболевания, или значительное снижение исходного уровня биологической активности или процесса. Как использовано в данном описании, термин "лечение" или "терапия" любого заболевания или расстройства в одном варианте осуществления относится к облегчению заболевания или расстройства(т.е. замедлению или остановке, или ослаблению развития заболевания, или, по меньшей мере, одного из его клинических симптомов). В другом варианте осуществления "лечить", "лечение" или "терапия" относится к облегчению или снижению интенсивности симптомов, по меньшей мере, одного физического параметра, включая такие физические параметры, которые пациент не может различить. В еще одном варианте осуществления "лечить", "лечение" или "терапия" относится к изменению заболевания или расстройства как физически (например, путем стабилизации различимого симптома), физиологически (например, путем стабилизации физического параметра), так и физически и физиологически одновременно. Наконец, в еще одном варианте осуществления "лечить", "лечение" или "терапия" относится к предупреждению или задержке начала, или развития, или прогрессирования заболевания или расстройства. Как использовано в данном описании, выражение субъект, "нуждающийся в" лечении, является таким субъектом, если такое лечение приведет к биологическому, медицинскому улучшению или к улучшению качества жизни. Как использовано в данном описании, в единственном числе и подобные термины, используемые в контексте настоящего изобретения (особенно в контексте формулы изобретения) следует рассматривать как охватывающие как единственное, так и множественное число, если в настоящем описании не указано иное или явно не противоречит контексту. Все описанные в настоящем описании способы могут быть выполнены в любом подходящем порядке, если в настоящем описании не указано иное или иным образом явно не противоречит контексту. Использование приведенного любого и всех примеров или поясняющих формулировок (например, "такой как") предназначено только для лучшего освещения настоящего изобретения и не накладывает ограничений на объем настоящего изобретения и его формулу. Способы получения Соединения по изобретению могут быть получены указанными ниже способами, например, путем преобразования малеимида формулы (Va) в спирт формулы (Vb), например, формальдегидом в присутствии или в отсутствие растворителя или основания, такого как карбонат калия, и необязательного перед проведением указанной реакции введения защитных групп, например трет-бутоксикарбонильных групп,в соответствии с принятыми в данной области реакциями, например, в случае, когда в соединении формулы (Va) имеются свободные и реакционноспособные аминогруппы, где переменные X, R, R1, R2, R3,R4 и R5 имеют значения, указанные для формулы (I). Спирт формулы (Vb) необязательно может быть преобразован в реакционноспособный сложный эфир, например, с трихлорметилацетонитрилом и подходящим основанием, например DBU, а затем сложный эфир может быть подвергнут взаимодействию с подходящим фосфорилирующим агентом, например ди-трет-бутиловым эфиром фосфорной кислоты в присутствии или в отсутствие подходящего растворителя, например, апротонного растворителя, такого как ацетонитрил, и затем может быть подвергнут гидролизу, например, с трифторуксусной кислотой, например, в дихлорметане или 1,2 дихлорэтане, с получением конечного продукта (Vc). Альтернативно, спирт формулы (Vb) можно подвергнуть взаимодействию непосредственно со сложным эфиром фосфорной кислоты, например, ди-трет-бутиловым эфиром фосфорной кислоты, например, в условиях реакции Мицунобу, с получением указанного эфира фосфорной кислоты, который затем может быть подвергнут гидролизу, например, с трифторуксусной кислотой, в частности, в дихлорметане, с получением конечного продукта (Vc). Экспериментальная часть Получение исходных соединений специально не описано, и указанные соединения известны или могут быть получены по аналогии со способами, известными из данной области техники, или как описано ниже. Следующие примеры иллюстрируют настоящее изобретение и никоем образом его не ограничивают. Сокращения: Химическую номенклатуру всех соединений формулируют с помощью AutoNom. ЯМР спектры регистрируют на спектрометре Bruker Avance DPX 400 при комнатной температуре. Используемые методы ЖХ-МС ЖХ метод 1 (Rt(1: Время удерживания (Rt) получают на системе Acquity UPLC, сопряженной с масс-спектрометромWaters ZQ 2000 с использованием колонки Waters ВЕН С 18 1,7 мкм, 2,150 мм (скорость потока=0,7 мл/мин, детектирование 240-350 нм; DAD) путем градиентного элюирования (растворитель А: вода+0,1% муравьиной кислоты, растворитель В: ацетонитрил; t=0 мин: 99% А, 1% В; t=1 мин 98% А, 2% В; t=2,25 мин 1% А, 99% В; t=4,5 мин 0% А, 100% В). ЖХ метод 2 (Rt(2: Время удерживания (Rt) получают на системе Agilent HPLC с колонкой Ascentis Express C18 2,7 мкм, 302,1 мм (Supelco) путем градиентного элюирования (Н 2 О+0,05% муравьиной кислоты+3,75 мМ ацетата аммония)/(CH3CN+0,04% муравьиной кислоты) от 90/10 до 5/95 в течение 1,7 мин в токе растворителя 1,2 мл/мин и затем 5/95 в течение 0,7 мин в токе растворителя 1,4 мл/мин и при температуре термостата 40 С. Метод обнаружения УФ 214-350 нм - МС. Способ очистки Препаративная ВЭЖХ с обращенной фазой на приборе компании Gilson Заполненная колонка SunFire C18 OBD 5 мкм, 30100 мм на установке Waters, H2O+0,l% TFA и ацетонитрил+0,1% TFA в качестве подвижной фазы. Метод обнаружения УФ 220-400 нм.(1,90 г, 2,42 ммоль) в 1,2-дихлорэтане (50 мл) добавляют TFA (8,27 г, 72,5 ммоль) в атмосфере аргона при 0 С. Реакционную смесь перемешивают в течение 3,5 ч в атмосфере аргона при 0 С до тех пор, покаUPLC-MC не показывает, что произошло полное преобразование исходных веществ. Реакционную смесь разбавляют 1,2-дихлорэтаном (50 мл) и концентрируют при пониженном давлении, с получением неочищенного продукта в виде твердого вещества красного цвета. Неочищенный продукт реакции растворяют в МеОН и медленно концентрируют при пониженном давлении до тех пор, пока не начнется кристаллизация. Добавляют пентан, и твердые вещества отфильтровывают и промывают Et2O. Дальнейшую очистку проводят суспендированием неочищенного продукта в ДМСО, с последующей обработкой ультразвуком в течение 30 мин. Твердые вещества отфильтровывают, промывают Et2O и сушат в глубоком вакууме (1 мм рт.ст.), с получением указанного в заголовке соединения в виде твердого вещества темно-красного цвета. 1 К раствору трет-бутилового эфира 7-1-[1-гидроксиметил-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо 2,5-дигидро-1 Н-пиррол-3-ил]изохинолин-3-ил-4,7-диазаспиро[2.5]октан-4-карбоновой кислоты (7,30 г,12,30 ммоль) в ацетонитриле (60 мл) добавляют по каплям раствор DBU (0,374 г, 0,371 мл, 2,46 ммоль) в трихлорацетонитриле (17,8 г, 12,3 мл, 123 ммоль) при комнатной температуре в атмосфере аргона. Реакционную смесь перемешивают в течение 5 ч при комнатной температуре до тех пор, пока ТСХ (SiO2,EtOAc/циклогексан 6:4) не укажет на полное преобразование. Реакционную смесь упаривают досуха при пониженном давлении и остаток суспендируют в ацетонитриле (60 мл). Добавляют ди-трет-бутиловый эфир фосфорной кислоты (3,36 г, 15,99 ммоль) и реакционную смесь перемешивают в течение приблизительно 3,5 ч при комнатной температуре в атмосфере аргона до тех пор, пока ТСХ (SiO2,EtOAc/циклогексан 6:4) не покажет, что реакция завершена. Реакционную смесь концентрируют при пониженном давлении и остаток распределяют между этилацетатом и водой. Слои разделяют и органическую фазу промывают водой (5 раз). Органическую фазу сушат над Na2SO4 и концентрируют при пониженном давлении с получением твердого вещества красного цвета. Неочищенный продукт очищают FCC(система Biotage SP4, SiO2, циклогексан/EtOAc, 20:80), с получением указанного в заголовке соединения в виде твердого вещества красного цвета. 1 Н-ЯМР (400 МГц, ДМСО-d6): 12,03 (с, 1 Н), 8,08 (д, 1 Н), 7,70-7,65 (м, 2 Н), 7,47 (т, 1 Н), 7,11 (с, 1 Н),7,11-7,07 (м, 1 Н), 6,77 (д, 1 Н), 6,44 (т, 1 Н), 5,97 (д, 1 Н), 5,39 (д, 1 Н), 3,49-3,08 (м, 6 Н), 2,39 (с, 3 Н), 1,45 (с,18 Н), 1,41 (с, 9 Н) , 0,87-0,56 (м, 4 Н). 31 Р-ЯМР (162 МГц, ДМСО-d6): -12,1. ЖХ-МС: [М+1]+=786,4; Rt(1)=2,53 мин, Rt(2) = l,62 мин. К раствору трет-бутилового эфира 7-1-[4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо-2,5-дигидро-1 Нпиррол-3-ил]изохинолин-3-ил-4,7-диазаспиро[2.5]октан-4-карбоновой кислоты (3,00 г, 5,32 ммоль) в МеОН (25 мл) добавляют 37%-ный водный раствор формальдегида (9,5 г, 8,72 мл, 117 ммоль) в атмосфере аргона при комнатной температуре. Реакционную смесь нагревают до 85 С и перемешивают в течение 4 ч. Реакционную смесь при непрерывном перемешивании охлаждают до комнатной температуры и фильтруют. Твердое вещество промывают ледяной водой и сушат в глубоком вакууме (1 мм рт.ст.), с получением указанного в заголовке соединения в виде темно-красных кристаллов. 1(5,0 г, 10,8 ммоль) в ТГФ (50 мл) в атмосфере аргона при комнатной температуре. Реакционную смесь перемешивают в течение 16 ч и концентрируют при пониженном давлении. Остаток распределяют между насыщенным водным раствором NH4Cl и CH2Cl2. Слои разделяют и водный слой экстрагируютCH2Cl2. Органические слои объединяют и промывают насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении, с получением указанного в заголовке соединения в виде твердого вещества оранжевого цвета. 1 К раствору трет-бутилового эфира 7-1-[1-гидроксиметил-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо 2,5-дигидро-1 Н-пиррол-3-ил]изохинолин-3-ил-4,7-диазаспиро[2.5]октан-4-карбоновой кислоты (500 мг,0,842 ммоль) в 1,2-дихлорэтане (5 мл) добавляют TFA (1,44 г, 0,973 мл, 12,7 ммоль) в атмосфере аргона при 0 С. Реакционную смесь перемешивают в течение 1 ч в атмосфере аргона при 0 С и добавляют дополнительное количество TFA (0,768 г, 0,52 мл, 6,74 ммоль). Перемешивание продолжают в течение 1,5 ч при 0 С. Реакционную смесь упаривают досуха при пониженном давлении и неочищенный продукт кристаллизуют из МеОН, с получением указанного в заголовке соединения в виде твердого вещества красного цвета (TFA соль). 1 Синтез указанного в заголовке соединения описан как пример 69 в WO 03082859. Пример 4. Моно 3-(1H-индол-3-ил)-4-[2-(4-метилпиперазин-1-ил)хиназолин-4-ил]-2,5-диоксо-2,5 дигидропиррол-1-илметиловыйэфир фосфорной кислоты В атмосфере аргона ди-трет-бутиловый эфир хлорметилового эфира фосфорной кислоты (1,24 г,4,81 ммоль) и Cs2CO3 (3,14 г, 9,63 ммоль) добавляют к раствору 3-(1 Н-индол-3-ил)-4-[2-(4 метилпиперазин-1-ил)хиназолин-4-ил]пиррол-2,5-диона (2,0 г, 4,01 ммоль) в ацетоне (40 мл). Реакционную смесь перемешивают в атмосфере аргона в течение 16 ч при 50 С и затем концентрируют при пониженном давлении. Остаток распределяют между EtOAc и насыщенным водным раствором NH4Cl, слои разделяют и органический слой сушат над безводным Na2SO4. Концентрирование при пониженном давлении дает неочищенный продукт в виде красной пены. Неочищенный продукт очищают ВЭЖХ с обращенной фазой на установке Gilson, как описано выше. После концентрирования нужных фракций в вакууме, получают твердое вещество красного цвета. UPLC-MC показывает, что произошло частичное расщепление трет-бутилового эфира. Полученную таким образом смесь (2 80 мг) растворяют в смеси 1,2 дихлорэтана (4 мл) и ацетонитрила (2,0 мл). Добавляют TFA (145 мг, 98 мкл, 1,27 ммоль) и полученный раствор перемешивают в течение 3 ч в атмосфере аргона при 0 С до тех пор, пока UPLC-MC не покажет,что исходные вещества полностью израсходованы. Реакционную смесь разбавляют 1,2-дихлорэтаном(4,0 мл) и концентрируют при пониженном давлении с получением неочищенного продукта в виде твердого вещества красного цвета. Неочищенный продукт реакции растворяют в МеОН (3 мл) и медленно концентрируют при пониженном давлении до тех пор, пока не начнется кристаллизация. Кристаллы отфильтровывают и промывают Et2O и пентаном, с получением указанного в заголовке соединения в виде твердого вещества оранжевого цвета. 1H-ЯМР (400 МГц, ДМСО-d6): 12,27 (с, 1 Н), 8,21 (с, 1 Н), 7,72-7,66 (м, 2 Н), 7,58 (д, 1 Н) , 7,40 (д, 1 Н),7,14 (т, 1 Н), 7,02 (т, 1 Н), 6,65 (т, 1 Н), 6,24 (д, 1 Н), 5,34 (д, 2 Н), 4,14-3,72 (шир.с, 4 Н), 3,00-2,73 (шир.с,4 Н), 2,60 (с, 3 Н). 31 Р-ЯМР (162 МГц, ДМСО-d6): -2,7. ЖХ-МС: [М]+=548,6; Rt(1) = 1,72 мин, Rt(2)=0,73 мин. Получение кристаллического вещества соединения по примеру 1: 2 г моно[3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо-2, 5-дигидропиррол-1-илметилового] эфира фосфорной кислоты (пример 1) диспергируют в смеси 800 мл этанола и 200 мл воды. Полученную суспензию перемешивают при комнатной температуре в течение 3 дней. Затем указанную суспензию фильтруют через стеклянный фильтр и полученные кристаллы сушат в токе воздуха при нормальном атмосферном давлении. К 1 г полученных кристаллов добавляют 4 мл раствора 80% этанола/20% воды (об./об.) и полученную смесь упаривают досуха при атмосферном давлении, с получением указанного моногидрата по примеру 1. Рентгеновская дифрактограмма указанного выше кристаллического моногидрата приведена на фиг. 1; пики зарегистрированы по отношению к углу 2 тэта в табл. 1. Таблица 1. Основные пики на рентгеновской дифрактограмме кристаллического моногидрата по примеру 1 Таким образом, в другом варианте осуществления предлагается кристаллическая форма моно[3-[3(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1-илметилового] эфира фосфорной кислоты, в частности моногидрата, который предпочтительно имеет на порошковой рентгеновской дифрактограмме по меньшей мере один, предпочтительно два, более предпочтительно три, еще более предпочтительно четыре, особенно предпочтительно пять, наиболее предпочтительно все следующие пики при угле рефракции 2 тета (9) 9,525; 16,356; 17,091; 18,005; 20,859; каждый с точностью 0,2, в частности, как показано на фиг. 1. Таблица 2. Химическая стабильность аморфного и кристаллического вещества по примеру 1 Профиль сорбции воды кристаллического моногидрата по примеру 1 показан на фиг. 2. Использованы следующие режимы обработки во влажной атмосфере: 6 ч уравновешивание при относительной влажности 50%, а затем 2 цикла RH от 50% RH до 90% RH, до 0% RH, до 90% RH, до 0% RH, до 50% RH с шагом 10% RH (RH=относительная влажность). Биофармацевтический раздел Соединения по настоящему изобретению, например соединения формул (I), (II), (III) или (IV) и т.п.,в свободной форме или в форме фармацевтически приемлемой соли или гидрата проявляют ценные фармакологические свойства, как описано в приведенных ниже результатах испытаний, например, для испытаний в условиях in vitro или in vivo, и поэтому они показаны для терапии.A. In vitro 1. Исследования альфа-протеинкиназы С и тета-протеинкиназы С Соединения по настоящему изобретению были исследованы на их активность по отношению к различным изотипам протеинкиназ (РКС) в соответствии с приведенной ниже методикой. Все анализы проводили на 384-луночных планшетах для микротитрования. Каждый используемый для анализа планшет содержал серию 8-точечных последовательных разведений для 40 тестируемых соединений, а также две серии 16-точечных последовательных разведений стауроспорина, используемого в качестве эталонного соединения, плюс 16 верхних и 16 нижних контролей. Стадии манипулирования с жидкими образцами и инкубирования жидких образцов проводили на автоматизированной станции, оснащенной блоком Innovadyne Nanodrop Express. Планшеты для анализа готовили добавлением 50 нл на лунку раствора соединения в 90%-ном ДМСО. Реакции киназы инициировали путем ступенчатого добавления 4,5 мкл на лунку 2 раствора пептид/АТФ и 4,5 мкл на лунку 2 раствора фермента. Конечная концентрация реагентов в процессе реакции киназ составила: 50 мМ HEPES, рН 7,5; 1 мМ DTT, 0,02% Tween 20, 0,02% BSA, 0,6% ДМСО, 10 мМ бета-глицерофосфата и 10 мкМ ортованадата. В качестве пептидного субстрата при исследовании РКС-альфа и РКС-тета использовали Dy4 95-X5-ME-Mpr-RFARKGSLRQKNV-COOH. Оба фермента представляют собой полноразмерные рекомбинантные белки человека, экспрессированные в клетках насекомых (Invitrogen AG, Basel, Швейцария). Другие компоненты были подобраны специально для анализа соответствующих киназы: РКС-альфа: 12 пМ фермента, 17 мкМ АТФ, 1 мкМ пептидного субстрата,7 мМ MgCl2, 0,2 мМ CaCl2. РКС-тета: 29 пМ фермента, 70 мкМ АТФ, 1 мкМ пептидного субстрата, 7 мМMgCl2, 0,2 мМ CaCl2. Реакционные смеси киназы инкубировали при 30 С в течение 60 мин, а затем прерывали реакции добавлением 16 мкл на лунку стоп-раствора (100 мМ HEPES, рН 7,5; 5% ДМСО, 0,1% реагента для нанесения покрытия Caliper, 10 мМ EDTA и 0,015% Brij35). Планшеты с прерванными реакциями киназы переносили в рабочие станции Caliper LC3000 для считывания. Фосфорилированные и нефосфорилированные пептиды разделяли, используя технологию микрофлюидного микроанализа по сдвигу пятна фирмы Caliper, и активность киназы рассчитывали из количества образовавшегося фосфопептида. Приведенные выше и ниже в данном описании результаты испытаний могут поддерживать концепцию пролекарства соединения по настоящему изобретению. 2. Исследование пролиферации клеток костного мозга (ВМ) Клетки костного мозга мышей линии СВА (2,5104 клеток на лунку в микропланшетах с плоским дном для культивирования тканей) инкубировали в 100 мкл среды RPMI, содержащей 10% FCS, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина (Gibco BRL, Basel, Switzerland), 50 мкМ 2 меркаптоэтанола (Fluka, Buchs, Switzerland), кондиционированную среду WEHI-3 (7,5% об./об.) и кондиционированную среду L929 (3% об./об.), в качестве источника факторов роста, и серийно разведенные соединения. Проводили две серии по семь циклов трехкратного разведения тестируемых соединений. После четырех дней инкубирования добавляли 1 мкКи 3 Н-тимидина. Клетки собирали после дополнительного пятичасового инкубационного периода, и включенный 3 Н-тимидин определяли в соответствии со стандартными методиками. Кондиционированные среды готовили следующим образом. КлеткиWEHI-3 (АТСС TIB68) и клетки L929 (АТСС CCL 1) выращивали в среде RPMI до начала слияния в течение 4 дней и одной недели, соответственно. Клетки собирали, вновь суспендировали в тех же колбах с культуральной средой С, содержащей 1% FCS (Schreier and Tess 1981) для клеток WEHI-3, и среду RPMI для клеток L929 и инкубировали в течение 2 дней (WEHI-3) или одной недели (L929). Супернатант собирали, фильтровали через поры 0,2 мкм и хранили в виде аликвот при -80 С. Культуры без тестируемых соединений и без WEHI-3 и супернатант L929 использовали в качестве нижних контрольных значений. Низкие контрольные значения вычитали из всех значений. Верхние контрольные значения без какихлибо образцов принимали за 100% пролиферации. Рассчитывали процент ингибирования образцами и определяли концентрации, необходимые для 50%-ного ингибирования (величины IC50). В. In vivo Введение соединения по примеру 1. Однократную дозу соединения по примеру 1 (3,0 мг/кг) вводили перорально 3 самцам собак породы бигль. Соединение 1 дозировали в виде водной суспензии кристаллической формы моногидрата с метилцеллюлозой (0,5%):Tween 80 (1%) (90:10). Кровь брали из вены через регулярные промежутки времени и образцы анализировали в течение вплоть до 24 ч. С течением времени проводили количественную оцен- 11023238 ку соединений по примерам 1, 2 и 3, и результаты приведены ниже в таблице. Основные фармакокинетические параметры (средние значениястандартное отклонение (n=3) после перорального введения для соединений 1, 2 и 3 в примере 1: Введение соединения примера 3. Соединение примера 3 вводили перорально в виде моноацетатной соли в твердой желатиновой капсуле 6 голодным самцам собак породы бигль. Вводили номинальную дозу 100 мг на одну собаку, в результате чего доза составила 8,9-11,3 мг/кг (масса собак составляла 8,9-11,3 кг). Брали кровь из вены и отбор проб проводили в течение до 32 ч. Биоаналитические определения проводили для соединения 3, и результаты собраны в приведенной ниже таблице. Ключевые фармакокинетические параметры (среднее значение или диапазон) для соединения 3 после перорального дозирования такие, как описано выше. Физико-химический раздел Исследование растворимости Растворимость соединения по примеру 1 в искусственном желудочном соке и в искусственной кишечной жидкости при комнатной температуре: Растворимость соединения по примеру 3 (свободная форма/форма ацетатной соли) в искусственной желудочном соке: Исследование на стабильность Стабильность соединения по примеру 1 в искусственном желудочном соке и в искусственной кишечной жидкости при 37 С: Стабильность соединения по примеру 3 (свободная форма/форма ацетатной соли) в искусственном желудочном соке и в искусственной кишечной жидкости при 37 С: Полезность Соединения по настоящему изобретению обычно пригодны для предотвращения или лечения расстройств или заболеваний, где важную роль играют протеинкиназы С (РКС) или медиаторы других киназ, например заболеваний или расстройств, опосредованных Т-лимфоцитами, В-лимфоцитами, тучными клетками, эозинофилами или кардиомиоцитами и, следовательно, как правило, имеют показания в случае острого или хронического отторжения органа или ткани алло- или ксенотрансплантата, реакции "трансплантат против хозяина", реакции "хозяин против трансплантата", при атеросклерозе, инфаркте головного мозга, окклюзии сосудов, вызванной повреждением сосудов, таким как ангиопластика, рестенозе,фиброзе (в особенности легочном фиброзе, а также других видах фиброза, таких как почечный фиброз),ангиогенезе, гипертензии, сердечной недостаточности, хроническом обструктивном заболевании легких,заболеваниях ЦНС, таких как болезнь Альцгеймера или боковой амиотрофический склероз, раке, инфекционных заболеваниях, таких как СПИД, септическом шоке или респираторном дистресс-синдроме взрослых, при ишемическом/реперфузионном повреждении, например инфаркте миокарда, инсульте,ишемии кишечника, почечной недостаточности или геморрагическом шоке или травматическом шоке. Соединения по настоящему изобретению также пригодны для лечения и/или предотвращения острых или хронических воспалительных заболеваний или расстройств или аутоиммунных заболеваний,например, саркоидоза, пневмосклероза, идиопатической интерстициальной пневмонии, обструктивных заболеваний дыхательных путей, в том числе таких состояний, как астма, наследственная бронхиальная астма, приобретенная бронхиальная астма, астма, вызванная пыльцой, в частности, хроническая или застарелая астма (например, поздняя астма и гиперчувствительность верхних дыхательных путей), бронхит, включая бронхиальную астму, инфантильная астма, ревматоидный артрит, остеоартрит, системная красная волчанка, волчаночный нефротический синдром, тиреоидит Хашимото, рассеянный склероз,тяжелая псевдопаралитическая миастения, сахарный диабет I типа и связанные с ним осложнения, сахарный диабет II типа у взрослых, увеит, нефротический синдром, стероид-зависимой нефроз и стероидрезистентный нефротический синдром, ладонно-подошвенный пустулез, аллергический энцефаломиелит, гломерулонефрит, псориаз, псориатический артрит, атопическая экзема (атопический дерматит),аллергический контактный дерматит, контактный дерматит вследствие раздражения и другие экзематозные дерматиты, себорейный дерматит, красный плоский лишай, пузырчатка, буллезный пемфигоид,врожденный буллезный эпидермолиз, крапивница, ангионевротический отек, васкулиты, эритемы, кожные эозинофилии, угри, гнездная алопеция, эозинофильный фасциит, атеросклероз, конъюнктивит, кератоконъюнктивит, кератит, весенний конъюнктивит, увеит связанный с болезнью Бехчета, герпетический кератит, коническая роговица, синдром Шегрена, дистрофия роговицы эпителиальная, кератолейкома,атрофический мукосинехиальный буллезный дерматит, язва Мурена, склерит, эндокринная офтальмопатия, сильное внутриглазное воспаление, воспаление слизистой оболочки или кровеносных сосудов, такое как лейкотриен В 4-опосредованные заболевания, язвы желудка, сосудистые повреждения, вызванные ишемической болезнью и тромбозом, гипертрофия сердца, ишемическая болезнь кишечника, воспалительные болезни кишечника (например, болезнь Крона или язвенный колит), некротический энтероколит, почечные заболевания, включая интерстициальный нефрит, синдром Гудпасчера, гемолитический уремический синдром и диабетическую нефропатию, нервные заболевания, выбранные из дерматомиозита, синдрома Гийена-Барре, синдрома Меньера и радикулопатии, коллагеновые болезни, включая склеродермию, гранулематоз Вегенера и синдром Шегрена, хронические аутоиммунные заболевания печени, включая аутоиммунный гепатит, первичный билиарный цирроз печени и склерозирующий холангит), частичная резекция печени, острый некроз печени (например, некроз, вызванный токсинами,вирусным гепатитом, шоком или гипоксией), цирроз печени, молниеносный гепатит, пустулезный псориаз, болезнь Бехчета, активный хронический гепатит, синдром Эванса, поллиноз, идиопатический гипопаратиреоз, болезнь Аддисона, аутоиммунный атрофический гастрит, волчаночный гепатит, тубулоинтерстициальное воспаление почек, мембранозный гломерулонефрит или ревматический полиартрит. Соединения по настоящему изобретению могут быть также пригодны для лечения опухолей, например рака молочной железы, рака мочеполовой системы, рака легких, желудочно-кишечного рака,плоскоклеточного рака, меланомы, рака яичников, рака поджелудочной железы, нейробластомы, рака головы и/или шеи или рака мочевого пузыря, или в более широком смысле рака почек, головного мозга или желудка, в частности (i) опухоли молочной железы; плоскоклеточные опухоли, такие как плоскоклеточная опухоль головы и/или шеи, или опухоли ротовой полости; опухоли легкого, например, мелкоклеточный или немелкоклеточный рак легкого; желудочно-кишечные опухоли, например опухоли толстого кишечника; или опухоли мочеполовой системы, например опухоль предстательной железы (в особенности гормонорезистентная опухоль простаты); или (ii) пролиферативное заболевание, которое трудно поддается лечению другими химиотерапевтическими средствами; или (iii) опухоль, которая устойчива к лечению другими химиотерапевтическими средствами вследствие множественной лекарственной устойчивости. Соединения могут быть также полезны для лечения опухолей крови и лимфатической системы (например, болезни Ходжкина, неходжкинской лимфомы, лимфома Беркитта, СПИД-ассоциированных лимфом, злокачественных иммунопролиферативных заболеваний, множественной миеломы и злокачественных новообразований плазматических клеток, лимфолейкоза, острого или хронического миелоидного лейкоза, острого или хронического лимфолейкоза, моноцитарного лейкоза, других лейкозов указанных типов клеток, для лечения лейкемии клеток неопределенного типа, других неуточненных злокачественных новообразований лимфоидной, кроветворной ткани и родственных тканей, например, диффузной крупноклеточной лимфомы, Т-клеточной лимфомы или кожной Т-клеточной лимфомы). Миелоидный рак включает, в частности, острый или хронический миелоидный лейкоз. Когда речь идет об опухоли, опухолевом заболевании, злокачественной опухоли или раке, то альтернативно или в дополнение, подразумеваются также метастазы в исходном органе или ткани и/или в любом другом месте независимо от расположения опухоли и/или метастазов. Предпочтительно соединения по настоящему изобретению особенно пригодны для предотвращения и/или лечения заболевания или расстройства, опосредованного Т-лимфоцитами, такого как острое или хроническое отторжение органа или ткани алло- или ксенотрансплантата, реакция "трансплантат против хозяина", реакция "хозяин против трансплантата", рассеянный склероз, псориаз или ревматоидный артрит. Низкая биодоступность лекарственных средств очень часто является ограничивающим фактором для фармацевтически эффективных ингредиентов. Кроме того, биодоступность может зависеть от видов животных. Например, препарат, который хорошо всасывается у мыши, крысы или собаки и т.п., может не превратиться в соединение с нужной биодоступностью для людей. В настоящем изобретении предлагается пролекарство соединения формулы (I), которое обеспечивает благоприятную биодоступность родительскому(им) соединению(ям), в частности, для людей. Например, как указано в экспериментальной части (раздел В, in vivo), соединение по примеру 1 преобразовывают, например, в соединение по примеру 3, которое можно обнаружить в крови в качестве основного компонента сразу после введения (например, примерно через час) и, следовательно, демонстрирует эффективное и удобное преобразование в родительское соединение. Для указанных выше применений требуемая доза, конечно, будет меняться в зависимости от способа введения, конкретного состояния, подлежащего лечению, и от желаемого эффекта. Как правило, удовлетворительные системные результаты должны быть получены при ежедневных дозах от примерно 0,02 до 25 мг на один кг массы тела. Назначенная суточная доза для более крупных млекопитающих, например, людей, как правило, может быть в пределах примерно от 0,2 мг до примерно 2 г, и ее удобно вводить, например, в виде разделенных общих доз вплоть до четырех раз в день или в форме, обеспечивающей замедленное высвобождение. Пригодные единичные лекарственные формы для перорального введения обычно содержат приблизительно от 0,1 до 500 мг активного ингредиента. Соединения по изобретению можно вводить любым обычным способом, в частности парентерально, например, в форме растворов или суспензий для инъекций, энтерально, в частности, перорально, например в форме таблеток или капсул, местно, например в виде лосьонов, гелей, мазей или кремов, или интраназально или форме суппозиториев. Примером местного введения может быть, например, нанесение на кожу. Другой формой местного введения может быть введение в глаз. Фармацевтические композиции, содержащие соединение по настоящему изобретению в сочетании по меньшей мере с одним фармацевтически приемлемым носителем или разбавителем, могут быть изготовлены обычным способом путем смешивания с фармацевтически приемлемым носителем или разбавителем. Соединения по изобретению можно вводить в свободной форме или в форме фармацевтически приемлемой соли или в форме гидрата, например, как указано выше. Подобные соли или гидраты могут быть получены обычным способом и, как правило, могут проявлять тот же уровень активности, как и соединения в свободной форме. В соответствии с вышеизложенным, в настоящем изобретении предлагаются также:(1) соединение по настоящему изобретению или его фармацевтически приемлемая соль или гидрат для применения в качестве фармацевтического средства;(2) соединение по настоящему изобретению или его фармацевтически приемлемая соль или гидрат для применения в качестве ингибитора РКС, например для применения в любом из приведенных выше конкретных показаний;(3) фармацевтическая композиция, например, для применения в любых приведенных выше показаниях, содержащая соединение по настоящему изобретению или его фармацевтически приемлемую соль или гидрат, вместе с одним или более фармацевтически приемлемыми разбавителями или носителями;(4) способ лечения или предупреждения заболевания или состояния, в котором определенную роль играет активация РКС или в котором РКС принимают участие, например, для лечения любого из приведенных выше конкретных показаний у нуждающегося в этом субъекта, при этом способ включает введение указанному субъекту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли или гидрата;(5) применение соединения по настоящему изобретению или его фармацевтически приемлемой соли или гидрата для изготовления лекарственного средства, предназначенного для лечения или предупреждения заболевания или состояния, в котором определенную роль играет активация РКС или в котором РКС принимают участие, например, для лечения указанных выше состояний. Комбинации Соединения по настоящему изобретению можно вводить в виде единственного активного ингредиента или в сочетании, например, в качестве вспомогательного лекарственного средства, с другими лекарственными средствами, в частности, в схемах лечения с использованием иммуносупрессивных или им- 15023238 муномодулирующих средств, или с другими противовоспалительными агентами, например, для лечения или предупреждения острого или хронического отторжения алло- или ксенотрансплантатов или для лечения воспалительных или аутоиммунных расстройств, в комбинации с химиотерапевтическим агентом или антибактериальным средством, в частности с противовирусным агентом, таким как, например, антиретровирусный агент или антибиотик. Например, соединения по настоящему изобретению могут использоваться в комбинации с ингибитором кальциневрина, в частности, циклоспорином A, ISA247 или FK 506; ингибитором mTOR, в частности, рапамицином, 40-О-(2-гидроксиэтил)рапамицином, CCI779, АВТ 578, TAFA-93, AP23573,АР 23464, АР 23841, биолимусом-7 или биолимусом-9; аскомицином, обладающим иммуносупрессивными свойствами, в частности, АВТ-281, ASM981 и т.д.; кортикостероидами, циклофосфамидом; азатиопреном; метотрексатом, лефлуномидом; мизорибином; микофенольной кислотой или ее солью; микофенолятом мофетила; 15-дезоксиспергуалином или его иммуносупрессивным гомологом, аналогом или производным; ингибитором РКС, например, как раскрыто в WO 02/38561 или WO 03/82859, в частности,например, соединением по примеру 56 или 70; агонистом или модулятором S1P рецептора, в частности,FTY720, необязательно фосфорилированным, или его аналогом, например, 2-амино-2-[4-(3 бензилоксифенилтио)-2-хлорфенил]этил-1,3-пропандиолом, необязательно фосфорилированным, или 14-[1-(4-циклогексил-3-трифторметилбензилоксиимино)этил]-2-этилбензилазетидин-3-карбоновой кислотой или ее фармацевтически приемлемыми солями; с иммуносупрессивными моноклональными антителами, например, моноклональными антителами против рецепторов лейкоцитов, в частности, МНС,CD2, CD3, CD4, CD7, CD8, CD25, CD28, CD40, CD45, CD52, CD58, CD80, CD86 или их лигандам; с другими иммуномодулирующими соединениями, в частности, рекомбинантной связывающей молекулой,включающей, по меньшей мере, часть внеклеточного домена CTLA4 или его мутант, например, по меньшей мере, внеклеточную часть CTLA4 или его мутант, присоединенную к отличной от CTLA4 белковой последовательности, в частности CTLA41g (например, имеющей обозначение АТСС 68629) или его мутант, например LEA29Y; ингибиторами адгезивных молекул, например, антагонистами LFA-1, антагонистами ICAM-1 или -3, антагонистами VCAM-4 или антагонистами VLA-4, в частности, натализумабом (ANTEGREN); или с антителами против хемокина или антителами против рецептора хемокина,или низкомолекулярными антагонистами рецептора хемокина, в частности, антителами против МСР-1. Соединение по настоящему изобретению может также применяться в комбинации с другими антипролиферативными агентами. Подобные антипролиферативные агенты включают, но, не ограничиваясь ими:(iii) ингибиторы топоизомеразы I, например топотекан, иринотекан, 9-нитрокамптотецин и макромолекулярный конъюгат камптотецина PNU-166148 (соединение A1 в WO99/17804);(iv) ингибиторы топоизомеразы II, например, антрациклины доксорубицина (в том числе липосомальные составы, например, CAELYX), эпирубицин, идарубицин и неморубицин, антрахиноны митоксантрона и лозоксантрона, и этопозид подофиллотоксинов и тенипозид;(v) вещества, активные по отношению к микротрубочкам, например таксаны паклитаксел и доцетаксел, алкалоиды винка, например винбластин, в частности сульфат винбластина, винкристин, в частности сульфат винкристина, и винорелбин, дискодермолид и эпотилоны, такие как эпотилон В и D;(xii) противоопухолевые антиметаболиты, например 5-фторурацил, тегафур, капецитабин, кладрибин, цитарабин, флударабина фосфат, фторуридин, гемцитабин, 6-меркаптопурин, гидроксимочевина,метотрексат, эдатрексат и соли подобных соединений и, кроме того, ZD 1694 (RALTITREXED),LY231514 (ALIMTA), LY264618 (LOMOTREXOL) HOGT719;(xiv) соединения, снижающие активность протеинкиназы и другие антиангиогенные препараты, например, (i) соединения, которые снижают активность фактора роста эндотелия сосудов (VEGF), (Ь) эпидермального фактора роста (EGF), c-Src, протеинкиназы С, фактора роста тромбоцитов (PDGF), Bcr-Ab1 тирозинкиназы, c-kit, Flt-3 и рецептора инсулиноподобного фактора роста I (IGF-IR) и циклинзависимых киназ (CDK), (ii) иматиниб, мидостаурин, Iressa (ZD1839), CGP 75166, ваталаниб, ZD6474, GW2016,CHIR-200131, CEP-7055/CEP-5214, СР-547632 и KRN-633; (iii) талидомид (THALOMID), целекоксиб(xxi) статины. Структуру активных агентов, определенных закодированными номерами, типовыми названиями и фирменными наименованиями, можно узнать из последнего издания стандартного справочника "MerckIndex" или из баз данных, например, Общества содействия внедрению изобретений (например, IMSWorld Publications). В соответствии с вышеизложенным в настоящем изобретении также предлагаются:(6) указанный выше способ, включающий совместное введение, например, одновременно или последовательно, терапевтически эффективного количества а) соединения формулы (I) или его фармацевтически приемлемой соли или гидрата и b) второго лекарственное средства, причем указанное второе лекарственное средство применяется, например, для любого из приведенных выше конкретных показаний;(7) комбинации, например, наборы реагентов, содержащие терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли или гидрата, и второе лекарственное средство, причем указанным вторым лекарственным средством является, например, описанное выше лекарственное средство. В том случае, когда соединение по настоящему изобретению вводят в сочетании с другим иммуносупрессивным/иммуномодулирующим, противовоспалительным и противоопухолевым средством, например описанным выше средством, то дозировки совместно вводимого лекарственного средства или агента будут, конечно, меняться в зависимости от типа совместно используемого лекарственного средства или агента, или конкретного используемого лекарственного средства или агента, а также от состояния, подлежащего лечению, и т.п. В другом варианте осуществления предлагается способ получения соединения формулы (I): где X обозначает СН или N, R обозначает H или РО 3 Н 2; R1 обозначает H или С 1-4 алкил; R2 обозначает H или С 1-4 алкил; R3 обозначает Н, С 1-4 алкил, CN, атом галогена или ОН; и R4 и R5, независимо друг от друга, обозначают H или С 1-4 алкил; или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют (3-6)-членную циклоалкильную группу,включающий:(a) необязательно для соединений, где R1 и/или R2 представляют собой атом водорода, обработку малеимида формулы (Va), например, ди-трет-бутилдикарбонатом в присутствии или в отсутствие растворителя, такого как ТГФ или дихлорметан, и/или основания, такого как триэтиламин, с получением малеимида формулы (Va), содержащего трет-бутоксикарбонильные группы вместо атомов водорода у R1 и/или R2, где это необходимо;(b) обработку необязательно защищенного малеимида формулы (Va), например, формальдегидом в присутствии или в отсутствие растворителя и/или основания, такого как карбонат калия, с получением спирта формулы (Vb), где R=H;(c) необязательно обработку спирта формулы (Vb), например, трихлорацетонитрилом, как правило,в присутствии основания, например DBU или триметиламина, с получением реакционноспособного сложного эфира, с последующей обработкой фосфорилирующим агентом, например сложным эфиром фосфорной кислоты, в частности ди-трет-бутиловым эфиром фосфорной кислоты, обычно в присутствии основания, например DBU или триметиламина, с последующей обработкой полученного промежуточного сложного эфира подходящей кислотой, в частности хлористо-водородной кислотой или TFA, в отсутствие или в присутствии растворителя, такого как ТГФ, дихлорметан, дихлорэтан или т.п., с получением конечного продукта, соответствующего общей формуле (I), или в качестве альтернативной стадии (с) спирт формулы (Vb) может быть непосредственно подвергнут взаимодействию со сложным эфиром фосфорной кислоты, например с ди-трет-бутиловым эфиром фосфорной кислоты, например, в условиях реакции Мицунобу с образованием сложного эфира фосфорной кислоты, который затем может быть гидролизован, например, с трифторуксусной кислотой, например, в дихлорметане, с получением указанного конечного продукта формулы (I). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) или его фармацевтически приемлемая соль или гидрат где X обозначает СН или N;R4 и R5, независимо друг от друга, обозначают H или С 1-4 алкил или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу. 2. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль или гидрат,где X обозначает СН;R4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу. 3. Соединение формулы (I) по п.1 или его фармацевтически приемлемая соль или гидрат, гдеR4 и R5, независимо друг от друга, обозначают Н или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу. 4. Соединение по п.1, которое представляет собой соединение формулы (III) или его фармацевтически приемлемую соль или гидрат. 5. Соединение по п.1 или 4, которое представляет собой моногидрат моно[3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил-1 Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1-илметилового] эфира фосфорной кислоты. 6. Соединение по любому из пп.1 или 2, которое представляет собой 3-[3-(4,7-диазаспиро[2.5]окт-7 ил)изохинолин-1-ил]-1-гидроксиметил-4-(7-метил-1 Н-индол-3-ил)пиррол-2,5-дион или его фармацевтически приемлемую соль. 7. Соединение по любому из пп.1 или 3, которое представляет собой моно 3-(1 Н-индол-3-ил)-4-[2(4-метилпиперазин-1-ил)хиназолин-4-ил]-2,5-диоксо-2,5-дигидропиррол-1-илметиловый эфир фосфорной кислоты или его фармацевтически приемлемую соль. 8. Применение соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или гидрата для изготовления лекарственного средства для лечения расстройств или заболеваний, где указанное лечение может быть показано в случае острого или хронического отторжения органа или ткани алло- или ксенотрансплантатов, и реакции "трансплантат против хозяина". 9. Способ лечения острого или хронического отторжения органа или ткани алло- или ксенотрансплантатов и реакции "трансплантат против хозяина" у нуждающегося в этом субъекта, включающий введение указанному субъекту эффективного количества соединения по любому из пп.1-7 или его фармацевтически приемлемой соли или гидрата. 10. Кристаллическая форма моно[3-[3-(4,7-диазаспиро[2.5]окт-7-ил)изохинолин-1-ил]-4-(7-метил 1 Н-индол-3-ил)-2,5-диоксо-2,5-дигидропиррол-1-илметилового] эфира фосфорной кислоты, которая имеет на порошковой рентгеновской дифрактограмме следующие пики при угле рефракции 2 тета : 9,525; 16,356; 17,091; 18,005; 20,859; каждый с точностью 0,2. 11. Способ получения соединения формулы (I) по п.1 где X обозначает СН или N;R4 и R5, независимо друг от друга, обозначают H или C1-4 алкил или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют 3-членную циклоалкильную группу,включающий:(а) обработку малеимида формулы (Va) формальдегидом в присутствии или в отсутствие растворителя и/или основания, такого как карбонат калия, с получением спирта формулы (Vb), где R=H; причем для соединений, где R1 и/или R2 представляют собой атом водорода, до указанной реакции малеимид формулы (Va) обрабатывают ди-трет-бутилдикарбонатом в присутствии или в отсутствие растворителя,такого как ТГФ или дихлорметан, и/или основания, такого как триэтиламин, с получением малеимида формулы (Va), содержащего трет-бутоксикарбонильные группы вместо атомов водорода у R1 и/или R2;(b) обработку спирта формулы (Vb), включающую обработку спирта формулы (Vb) трихлорацетонитрилом в присутствии основания, такого как DBU или триметиламин, с получением реакционноспособного сложного эфира, с последующей обработкой ди-трет-бутиловым эфиром фосфорной кислоты в присутствии основания, такого как DBU или тримети- 19023238 ламин, с последующей обработкой полученного промежуточного сложного эфира подходящей кислотой,такой как хлористо-водородная кислота или TFA, в отсутствие или в присутствии растворителя, такого как ТГФ, дихлорметан или дихлорэтан, с получением конечного продукта, соответствующего общей формуле (I), или спирт формулы (Vb) может быть непосредственно подвергнут взаимодействию с ди-третбутиловым эфиром фосфорной кислоты с образованием сложного эфира фосфорной кислоты, который затем гидролизуют с трифторуксусной кислотой в дихлорметане, с получением указанного конечного продукта формулы (I):
МПК / Метки
МПК: C07D 401/14, A61P 37/00, A61K 31/496, C07F 9/141
Метки: индола, расстройств, лечения, производные, иммунологических, замещенные
Код ссылки
<a href="https://eas.patents.su/22-23238-zameshhennye-proizvodnye-indola-dlya-lecheniya-immunologicheskih-rasstrojjstv.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные производные индола для лечения иммунологических расстройств</a>
Производные индола, пригодные для лечения расстройств цнс

Номер патента: 6315
Опубликовано: 27.10.2005
Авторы: Банг-Андерсен Бенни, Келер Ян, Андерсен Ким, Феллинг Якоб
МПК: A61K 31/4045, C07D 401/14, A61P 25/06...
Метки: расстройств, цнс, индола, производные, пригодные, лечения
Формула / Реферат:
1. Замещенное производное индола формулы I где (a) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет собой CO, CS, SO или SO2 и Y4 представляет собой CH2; (b) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет собой CH2 и Y4 представляет собой CO, CS, SO или SO2; или (c) один из Y1 и Y2 представляет собой N, который связан с Y4, а другой из Y1 и Y2 представляет...
Производные индола и их применение для лечения психических и неврологических расстройств

Номер патента: 6104
Опубликовано: 25.08.2005
Авторы: Роттлендер Марио, Крог-Енсен Кристиан, Андерсен Ким, Мольтсен Айнер Кнуд, Миккельсен Иван, Руланд Томас
МПК: A61K 31/497, C07D 401/12
Метки: психических, лечения, неврологических, применение, производные, расстройств, индола
Формула / Реферат:
1. Соединение, представленное общей формулой I где A представляет собой O или S; n равно 2, 3, 4, 5, 6, 7, 8, 9 или 10; m равно 2 или 3; W представляет собой N, C или CH; Q представляет собой N, C или CH; и пунктирная линия представляет возможную связь; R1 представляет собой водород, C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-8-циклоалкил-C1-6-алкил, арил-C1-6-алкил или ацил; R2, R3, R4, R5 и R6 независимо представляют собой водород, галоген,...
Замещенные производные арила и гетероарила в качестве модуляторов метаболизма и для профилактики и лечения связанных с ним расстройств

Номер патента: 11671
Опубликовано: 28.04.2009
Авторы: Шин Янг-Дзун, Джоунз Роберт М., Брюс Марк А., Леманн Юэрг, Чои Цзинь Сунь Кэролайн, Фьораванти Беатриц, Сэмпл Грэем, Сюн Ифэн, Рен Альберт С.
МПК: A61K 31/506, A61P 3/10, C07D 401/04...
Метки: гетероарила, арила, ним, расстройств, качестве, замещенные, профилактики, модуляторов, лечения, производные, связанных, метаболизма
Формула / Реферат:
1. Соединение, выбранное из соединений формулы (I) и их фармацевтически приемлемых солей, сольватов, гидратов и N-оксидов: где A1 и А2 оба являются -СН2СН2- и каждый A1 и A2 необязательно замещен 1, 2, 3 или 4 метильными группами; D является CR1R2 или NR2, где R1 выбирают из Н, C1-6алкила, C1-6алкокси, галогена и гидроксила; Е является N или CR3, где R3 является Н или C1-6алкилом; является одинарной связью; K отсутствует, является...
Производные индол-2-она для лечения расстройств центральной нервной системы, желудочно-кишечных расстройств и сердечно-сосудистых расстройств

Номер патента: 10865
Опубликовано: 30.12.2008
Авторы: Шимиг Дьюла, Гиглер Габор, Мезеи Тибор, Капиллерне Дежёфи Рита, Баркоци Йожеф, Гачальи Иштван, Вольк Балаж, Левелеки Чилла, Харсинг Ласло Габор, Мориц Кристина, Леваи Дьёрдь, Паллаги Каталин, Сенаши Габор, Сираи Нора, Эдьед Андраш
МПК: A61P 25/00, C07D 401/06, A61K 31/404...
Метки: системы, сердечно-сосудистых, нервной, желудочно-кишечных, индол-2-она, производные, расстройств, лечения, центральной
Формула / Реферат:
1. 3,3-Дизамещенные производные индол-2-она общей формулы (I) где R1 и R2 независимо представляют собой водород, галоген, алкил, имеющий в своем составе от 1 до 7 атомов углерода, или сульфамоил; R3 представляет собой водород или алкил с прямой или разветвленной цепью, имеющий в своем составе от 1 до 7 атомов углерода; R4 означает алкил, имеющий в своем составе от 1 до 7 атомов углерода; R5 является водородом и R6 означает фенил, необязательно...
Новые замещенные производные индола в качестве модуляторов гамма-секретазы

Номер патента: 23045
Опубликовано: 29.04.2016
Авторы: Бертело Дидье Жан-Клод, Велтер Адриана Ингрид, Минн Гарретт Берлонд, Питерс Серж Мария Алоисиус, Бисхофф Франсуа Пол, Гейсен Хенрикус Якобус Мария
МПК: C07D 487/04, A61P 25/28, A61K 31/5383...
Метки: качестве, индола, замещенные, gamma;-секретазы, производные, новые, модуляторов
Формула / Реферат:
1. Соединение формулы (I)его таутомер или стереоизомерная форма, гдеR3 выбран из группы, состоящей из водорода, С1-4алкилокси, циано и Het1;А1 представляет собой CR4a или N, где R4a представляет собой водород, галоген или С1-4алкилокси, необязательно замещенный одним или более заместителями, выбранными из группы, состоящей из С1-4алкилокси и галогена;А2 представляет собой CR4b или N, где R4b представляет собой водород, галоген, С1-4алкилокси...
Предыдущий патент: Производные гетероциклических аминов
Следующий патент: Соединения в качестве ингибиторов диацилглицерин ацилтрансферазы
Случайный патент: Применение пептидных соединений тро и фармацевтических композиций для лечения анемии