Антагонисты рецептора 5-ht2b
Номер патента: 20967
Опубликовано: 31.03.2015
Авторы: Тюринг Йоханнес Вильхельмус Йохн Ф., Вер Донк Люк Аугюст Лаурентиус

















Формула / Реферат
1. Соединение формулы (I)

или его стереохимически изомерная форма, где
R представляет собой водород или фтор,
или его аддитивная соль, или сольват.
2. Соединение по п.1, где R представляет собой фтор и указанное соединение представляет собой рацемическую смесь, или его аддитивная соль, или сольват.
3. Соединение по п.1, где R представляет собой фтор и указанное соединение обладает оптическим вращением [α]=-14,4° (c=0,3, MeOH, l=598 нм; 20°C), или его аддитивная соль, или сольват.
4. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп.1-3 и фармацевтически приемлемый носитель.
5. Способ получения фармацевтической композиции по п.4, отличающийся тем, что фармацевтически приемлемый носитель тщательно смешивают с терапевтически эффективным количеством соединения по любому из пп.1-3.
6. Применение соединения по любому из пп.1-3 для лечения или предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки.
7. Способ лечения или предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-3.
Текст
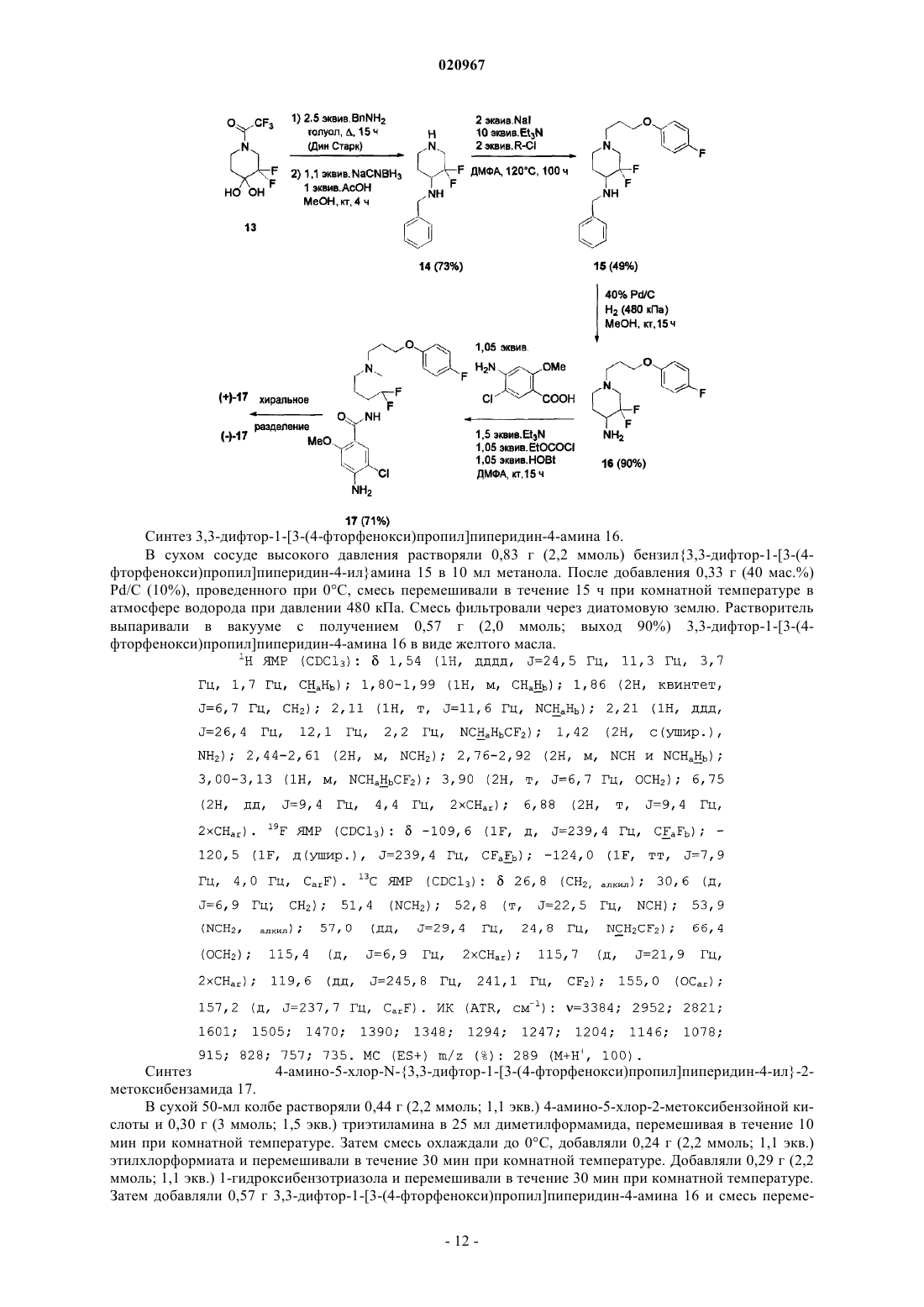
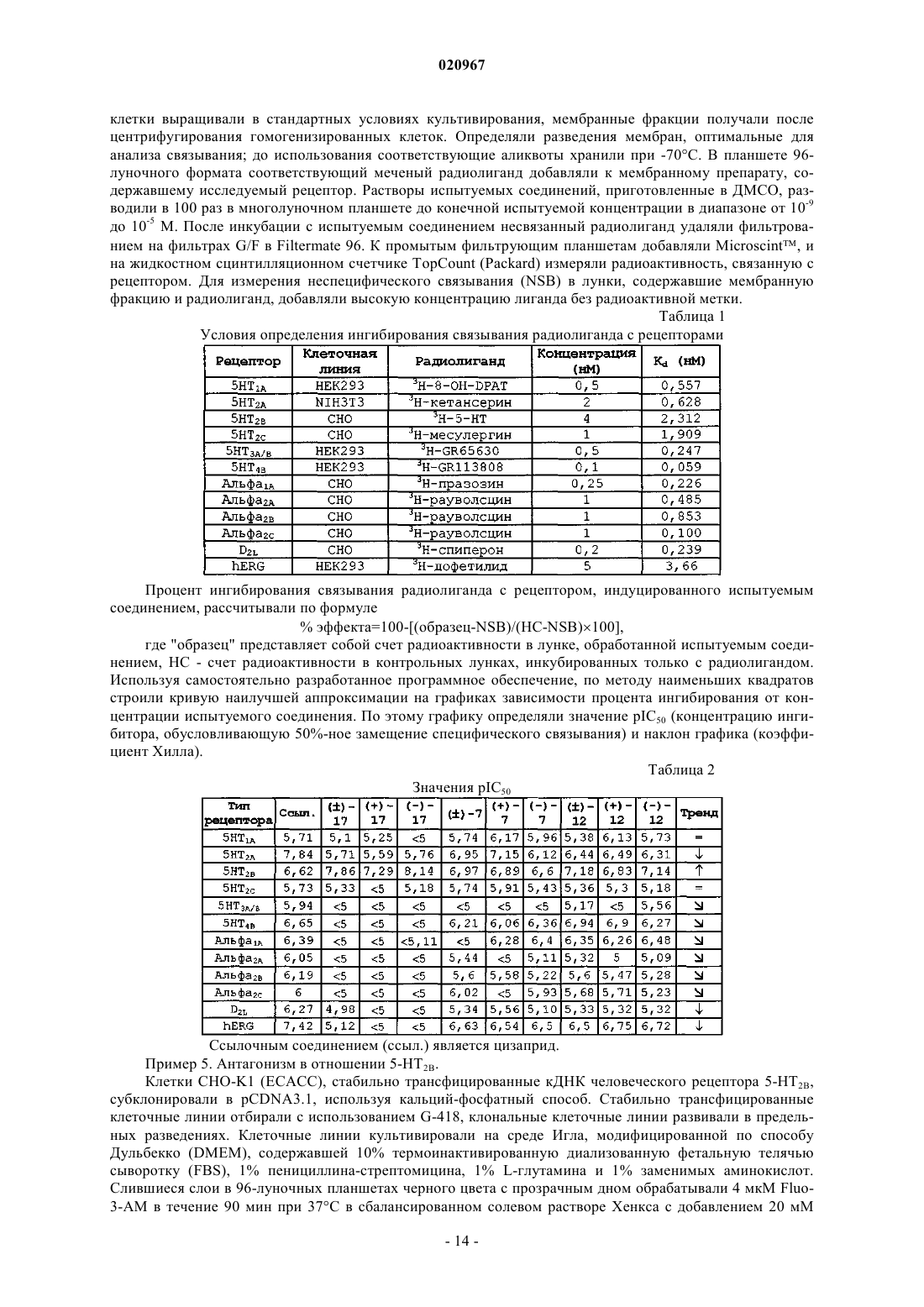
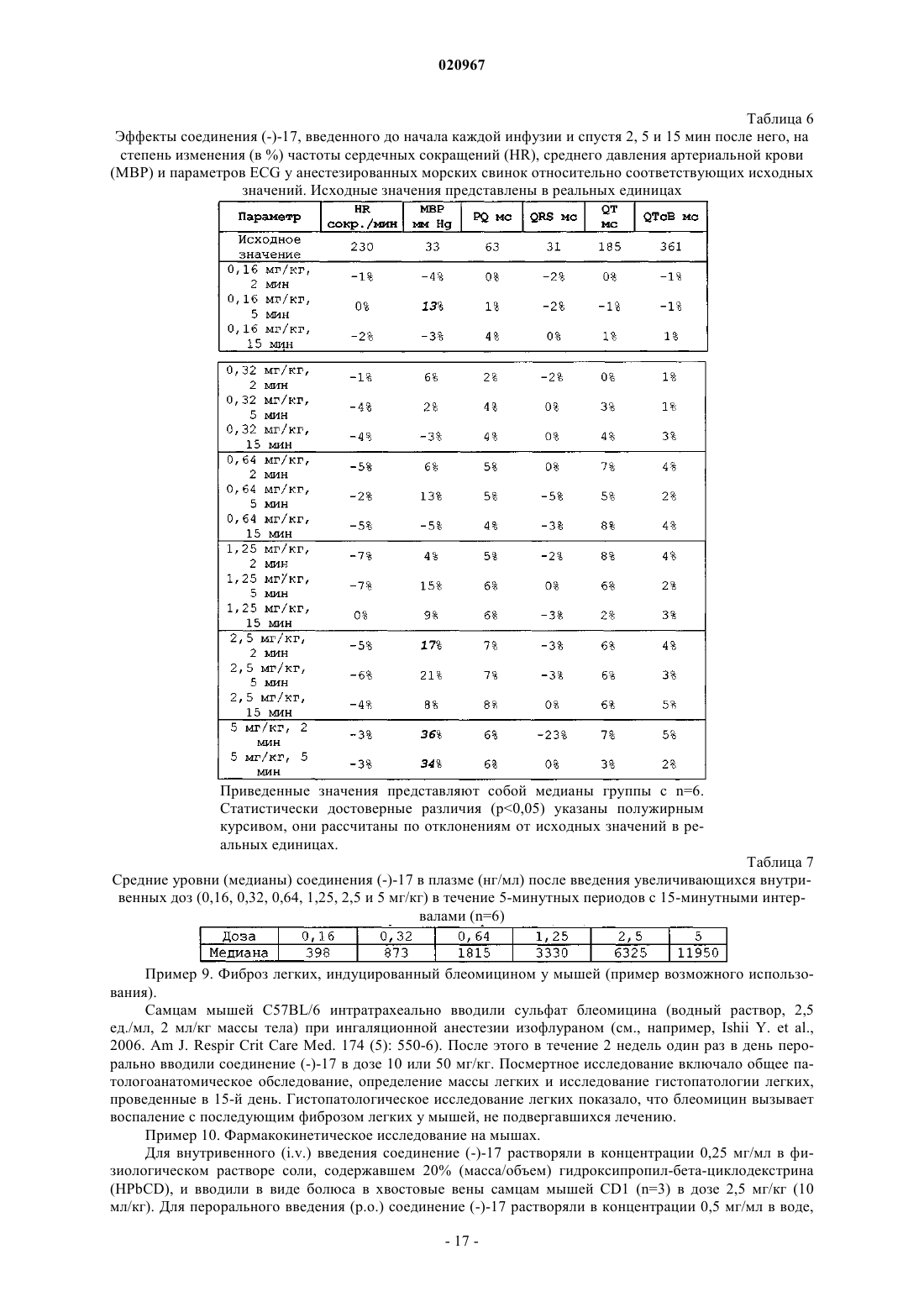
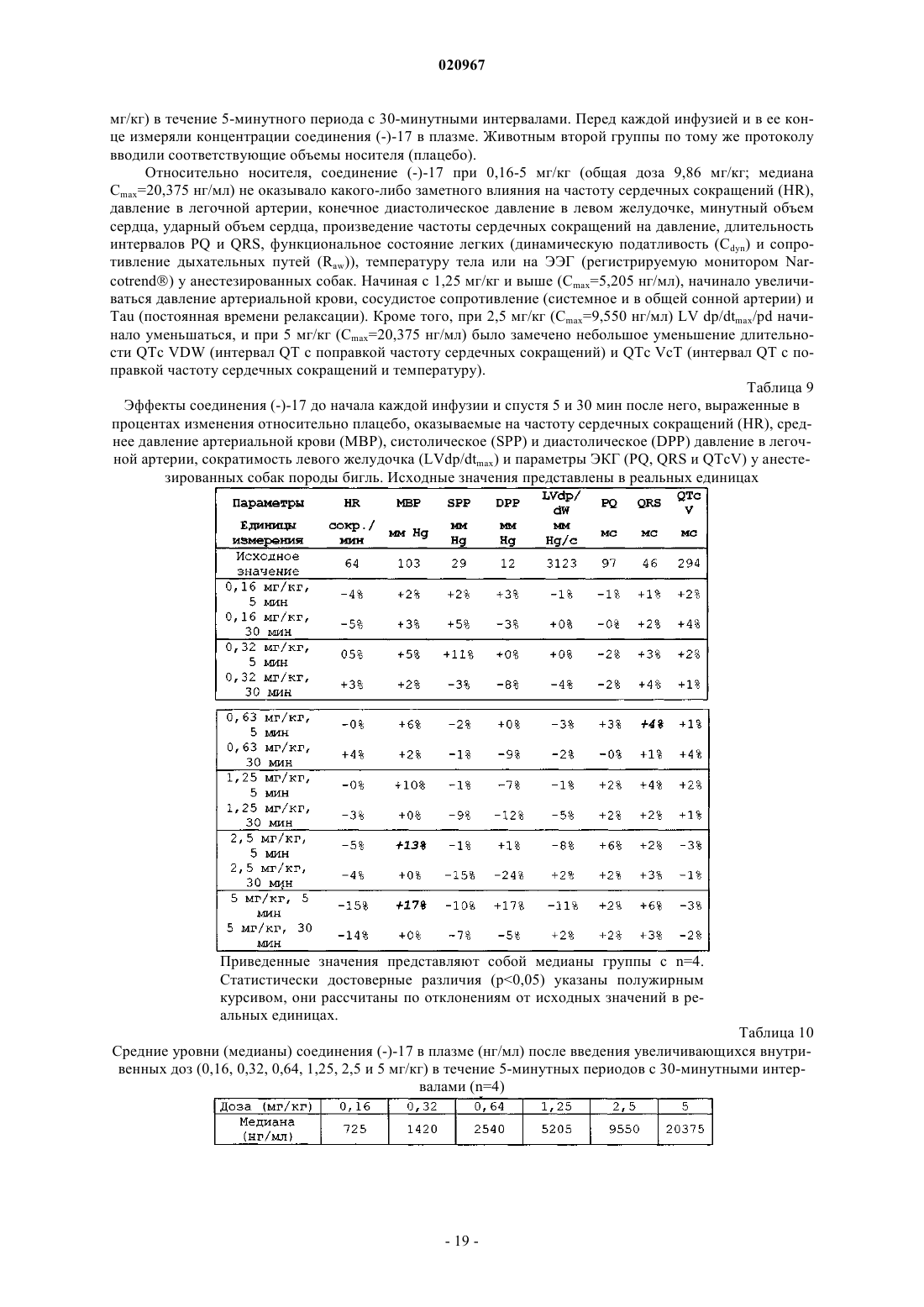
Настоящее изобретение относится к новым фторированным производным пиперидина,обладающим антагонистической активностью в отношении рецептора 5-HT2B, к фармацевтическим композициям, содержащим эти соединения, и к их применению в качестве лекарственных средств для лечения и предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки. Тюринг Йоханнес Вильхельмус Йохн Ф., Вер Донк Люк Аугюст Лаурентиус(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) Область техники, к которой относится изобретение Настоящее изобретение относится к новым фторированным производным пиперидина, обладающим антагонистической активностью в отношении рецептора 5-НТ 2 В, к фармацевтическим композициям,содержащим эти соединения, и к их применению в качестве лекарственного средства. Предпосылки создания изобретения Цизаприд представляет собой агонист рецептора 5-НТ 4, применяемый в качестве гастропрокинетического лекарственного средства. Он также в значительной степени взаимодействует с некоторыми другими рецепторами, такими как 5-НТ 2 А и 5-НТ 2 с; D2L; 5-HT3A/B, Альфа 1 А, Альфа 2 А, Альфа 2 В и Альфа 2 С. В 2000 году, вследствие появления сообщений о внезапных сердечных аритмиях, его удалили с некоторых рынков. В основе этого побочного эффекта находится индуцированное этим лекарственным средством удлинение интервала QT по механизму блокады калиевых каналов hERG (ген специфических калиевых каналов сердца человека, ген hERG). Один из известных фармакофоров блокаторов каналов hERG содержит гидрофильный и гидрофобный фрагменты, связанные со средней частью, содержащей основный атом азота. При физиологическом pH основный азот протонируется и вовлекается в -катионное взаимодействие с остатками Tyr 652 внутри поры в канале hERG. Для уменьшения значения pKa пиперидинового атома азота (и, тем самым, для уменьшения вероятности блокады канала hERG) были получены производные цизаприда, в которых 3-метоксипиперидин заменен 3-фторпиперидином и 3,3 дифторпиперидином. Сущность изобретения Настоящее изобретение относится к соединению формулы (I) или к его стереохимически изомерной форме, гдеR представляет собой водород или фтор,или к его аддитивной соли, или сольвату. Иллюстрацией настоящего изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и любое описанное выше соединение. Иллюстрацией настоящего изобретения является фармацевтическая композиция, полученная посредством смешивания любого описанного выше соединения и фармацевтически приемлемого носителя. Иллюстрацией настоящего изобретения является способ получения фармацевтической композиции, включающий смешивание любого описанного выше соединения и фармацевтически приемлемого носителя. Примерами настоящего изобретения являются способы лечения расстройств, опосредуемых рецепторами 5-HT2B, включающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Кроме того, примерами настоящего изобретения являются способы ингибирования рецептора 5HT2B, включающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Примером настоящего изобретения является способ лечения расстройства, выбранного из группы,состоящей из легочной артериальной гипертензии, фиброза легких, синдрома раздраженной толстой кишки, сердечно-сосудистых расстройств, таких как хроническая болезнь сердца, застойная сердечная недостаточность и гипертензия, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или любой из фармацевтических композиций, описанных выше. Другим примером настоящего изобретения является любое из соединений, описанных выше, предназначенных для лечения легочной артериальной гипертензии, фиброза легких, синдрома раздраженной толстой кишки, сердечно-сосудистых расстройств, таких как хроническая болезнь сердца, застойная сердечная недостаточность и гипертензия, у субъекта, нуждающегося в этом. Подробное описание изобретения Настоящее изобретение направлено на соединения формулы (I), как определено в настоящем описании выше, и их фармацевтически приемлемые соли. Соединения формулы (I) являются селективными антагонистами рецептора 5-HT2B. В варианте осуществления настоящего изобретения R представляет собой фтор и указанное соединение, или его аддитивная соль, или сольват представляют собой рацемическую смесь. В другом варианте осуществления настоящего изобретения R представляет собой фтор и указанное соединение, или его аддитивная соль, или сольват обладают оптическим вращением []=+14,1 (c=0,3,MeOH, =598 нм; 20C). В другом варианте осуществления настоящего изобретения R представляет собой фтор и указанное соединение, или его аддитивная соль, или сольват обладают оптическим вращением []=-14,4 (c=0,3,-1 020967MeOH, =598 нм; 20C). В другом варианте осуществления настоящего изобретения R представляет собой водород и заместители в положении 3 и 4 пиперидинового фрагмента указанного соединения, или его аддитивной соли,или сольвата имеют цис-ориентацию. В другом варианте осуществления настоящего изобретения R представляет собой водород, заместители в положении 3 и 4 пиперидинового фрагмента имеют цис-ориентацию и указанное соединение, или его аддитивная соль, или сольват обладают оптическим вращением []=+39,8 (c=0,2, MeOH, =598 нм; 20C). В другом варианте осуществления настоящего изобретения R представляет собой водород, заместители в положении 3 и 4 пиперидинового фрагмента имеют цис-ориентацию и указанное соединение, или его аддитивная соль, или сольват обладают оптическим вращением []=-45,5 (c=0,2, MeOH, =598 нм; 20C). В другом варианте осуществления настоящего изобретения R представляет собой водород, и заместители в положении 3 и 4 пиперидинового фрагмента указанного соединения, или его аддитивной соли,или сольвата имеют транс-ориентацию. В другом варианте осуществления настоящего изобретения R представляет собой водород, заместители в положении 3 и 4 пиперидинового фрагмента имеют транс-ориентацию и указанное соединение,или его аддитивная соль, или сольват обладают оптическим вращением []=+19,2 (c=0,4, MeOH, =598 нм; 20C). В другом варианте осуществления настоящего изобретения R представляет собой водород, заместители в положении 3 и 4 пиперидинового фрагмента имеют транс-ориентацию и указанное соединение,или его аддитивная соль, или сольват обладают оптическим вращением []=-22,8 (c=0,3, MeOH, =598 нм; 20C). Как и ожидалось, фторированные производные цизаприда являются, по существу, менее сильными блокаторами каналов hERG, чем цизаприд, и, таким образом, значительно уменьшается вероятность того, что они будут вызывать удлинение интервала QT. Однако оказалось неожиданным, что сродство к рецепторам изменяется различными путями, результатом чего являются соединения с более избирательным профилем. Достоверно уменьшается сродство к рецепторам 5-HT2A и D2L и наблюдается тенденция к некоторому уменьшению сродства к рецепторам 5-HT3A/B, 5-HT4B, Альфа 1 А, Альфа 2 А, Альфа 2 В и Альфа 2 С. Единственным исключением является сродство к рецептору 5-HT2B, которое достоверно увеличивается. Антагонисты рецептора 5-HT2B показаны для лечения или предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки. Легочная артериальная гипертензия может быть идиопатической, семейной или связанной с другими заболеваниями, таким как инфекция ВИЧ, или она может быть связанной с применением определенных лекарственных средств. Она также может быть связанной с заболеваниями сердца или легких, таких как хроническое обструктивное заболевание легких (COPD), интерстициальное заболевание легких или хроническое воздействие пребывания на большой высоте. Фиброз легких характеризуется хроническим воспалением и прогрессирующим фиброзом альвеолярных стенок, с постоянно прогрессирующей одышкой, конечным результатом чего является смерть от кислородной недостаточности или недостаточности правого сердца. Синдром раздраженной толстой кишки является хроническим невоспалительным заболеванием, характеризующимся болью в животе, изменением ритма дефекации с диареей или запором, или и с тем, и с другим, но без патологических изменений. Он представляет собой распространенное расстройство с психофизиологической основой. Антагонисты рецептора 5-HT2B можно также применять для лечения сердечнососудистых расстройств, таких как хроническая (ишемическая) болезнь сердца, застойная сердечная недостаточность и гипертензия. Определения. Термин "субъект", используемый в настоящем описании, относится к животному (предпочтительно к млекопитающем, наиболее предпочтительно к человеку), которое является или являлось объектом лечения, наблюдения или эксперимента. Термин "терапевтически эффективное количество", используемый в настоящем описании, означает количество активного соединения или фармацевтического средства, способствующее биологическому или медицинскому ответу тканевой системы, животного или человека, который желателен исследователю, ветеринару, врачу или другому клиницисту и который включает облегчение симптомов заболевания или расстройства, лечение которых они осуществляют. Термин "композиция", используемый в настоящем описании, относится к продукту, содержащему указанные ингредиенты в указанных количествах, а также к любому продукту, получаемому (прямо или косвенно) в результате комбинирования указанных ингредиентов в указанных количествах. Следует понимать, что некоторые из соединений согласно формуле (I) или их аддитивные соли,гидраты или сольваты могут содержать один или более центров хиральности и существовать в виде стереоизомерных форм. Термин "стереоизомерные формы", используемый выше или ниже в настоящем описании, опреде-2 020967 ляет все возможные стереоизомерные формы, которыми могут обладать соединения согласно формуле(I) и их аддитивные соли. Если не отмечено или не указано иное, химическое обозначение соединений означает смесь всех возможных стереохимически изомерных форм, причем такие смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры, а также каждую из индивидуальных изомерных форм согласно формуле (I) и их соли, сольваты, по существу, свободные, т.е. связанные менее чем с 10% других изомеров (предпочтительно менее чем с 5%, в частности менее чем с 2%, наиболее предпочтительно менее чем с 1%). В тех случаях, когда соединения согласно настоящему изобретению имеют по меньшей мере один хиральный центр, они соответственно могут существовать в виде энантиомеров. В тех случаях, когда указанные соединения обладают двумя или более хиральными центрами, они, кроме того, могут существовать в виде диастереомеров. Следует понимать, что такие изомеры и их смеси включены в объем настоящего изобретения. Предпочтительно в тех случаях, когда указанное соединение присутствует в виде энантиомера, данный энантиомер присутствует в энантиомерном избытке, большем или равном примерно 80% (более предпочтительно в энантиомерном избытке, большем или равном примерно 90%, еще более предпочтительно в энантиомерном, большем или равном примерно 95%, еще более предпочтительно в энантиомерном избытке, большем или равном примерно 98%, наиболее предпочтительно в энантиомерном избытке, большем или равном примерно 99%). Аналогичным образом, в тех случаях, когда указанное соединение присутствует в виде диастереомера, данный диастереомер присутствует в диастереомерном избытке, большем или равном примерно 80% (более предпочтительно в диастереомерном избытке, большем или равном примерно 90%, еще более предпочтительно в диастереомерном избытке, большем или равном примерно 95%, еще более предпочтительно в диастереомерном избытке, большем или равном примерно 98%, наиболее предпочтительно в диастереомерном избытке, большем или равном примерно 99%). Кроме того, некоторые из кристаллических форм соединений согласно настоящему изобретению могут существовать в виде полиморфов и как таковые должны быть включены в объем настоящего изобретения. В дополнение, некоторые из соединений согласно настоящему изобретению могут образовывать сольваты с водой (т.е. гидраты) и обычными органическими растворителями, и такие сольваты также должны быть охвачены объемом настоящего изобретения. Соли соединений согласно настоящему изобретению, предназначенные для применения в медицине, относятся к нетоксичным "фармацевтически приемлемым солям". Однако для получения соединений согласно настоящему изобретению или их фармацевтически приемлемых солей можно использовать и другие соли. Подходящие фармацевтически приемлемые соли указанных соединений включают кислотно-аддитивные соли, которые могут быть образованы, например, посредством смешивания раствора указанного соединения с раствором фармацевтически приемлемой кислоты, такой как хлористо-водородная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота,бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, в тех случаях, когда соединения согласно настоящему изобретению несут кислотный фрагмент, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов (например,соли натрия или калия), соли щелочно-земельных металлов (например, соли кальция или магния) и соли,образованные подходящими органическими лигандами (например, четвертичные аммонийные соли). Примеры кислот, которые можно использовать при получении фармацевтически приемлемых солей, включают, но не ограничиваются ими, следующие: уксусную кислоту, 2,2-дихлоруксусную кислоту,ацетилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, Lаспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорсульфоновую кислоту, каприновую кислоту, капроновую кислоту,каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, этан-1,2 дисульфоновую кислоту, этансульфоновую кислоту, 2-гидроксиэтансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту,D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, бета-оксоглутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, бромисто-водородную кислоту, хлористо-водородную кислоту, (+)-L-молочную кислоту, -DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, -DL-миндальную кислоту, меглумин, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси 2-нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту, оротовую кислоту,щавелевую кислоту, пальмитиновую кислоту, памовую кислоту, фосфорную кислоту, Lпироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту,стеариновую кислоту, янтарную кислоту, серную кислоту, дубильную кислоту, (+)-L-винную кислоту,тиоциановую кислоту, п-толуолсульфоновую кислоту, трифторметилсульфоновую кислоту и ундециленовую кислоту. Некоторые из соединений согласно формуле (I) могут также существовать в таутомерной форме. Хотя в формуле, представленной выше, такие формы явно не указаны, они также должны быть включен-3 020967 ными в объем настоящего изобретения. Фармацевтические композиции. Настоящее изобретение также предоставляет композиции для предупреждения или лечения заболеваний, при которых полезно ингибирование легочной артериальной гипертензии или фиброза легких. Указанные композиции содержат терапевтически эффективное количество соединения согласно формуле (I) и фармацевтически приемлемый носитель или разбавитель. Хотя активный ингредиент можно вводить отдельно, однако предпочтительно предоставлять его в виде фармацевтической композиции. Соответственно, настоящее изобретение дополнительно предоставляет фармацевтическую композицию, содержащую соединение согласно настоящему изобретению совместно с фармацевтически приемлемым носителем или разбавителем. Носитель или разбавитель должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами композиции и невредным для его реципиентов. Фармацевтические композиции согласно настоящему изобретению можно получать любыми способами, хорошо известными в фармации. Терапевтически эффективное количество конкретного соединения (в форме основания или в форме аддитивной соли), используемого в качестве активного ингредиента, объединяют в тщательно перемешиваемой смеси с фармацевтически приемлемым носителем, который может иметь разнообразные формы в зависимости от формы препарата, желаемой для введения. Желательно, чтобы фармацевтические композиции были в стандартной лекарственной форме, подходящей предпочтительно для системного введения (такого как пероральное, чрескожное или парентеральное введение) или для местного применения (такого как ингаляция или инсуффляция). Например, при получении композиций в пероральной дозированной форме можно использовать любые обычные фармацевтические среды, такие как, например, вода, гликоли, масла, спирты и т.п. (в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы), или твердые носители, такие как крахмалы, сахара, каолин, лубриканты, связующие, дезинтегранты и т.п. (в случае порошков, пилюль, капсул и таблеток). Таблетки и капсулы, вследствие легкости их введения, представляют собой наиболее удобную форму пероральной единичной дозы, для которой, как очевидно, используют твердые фармацевтические носители. Носитель, используемый для парентеральных композиций, обычно будет содержать стерильную воду (хотя бы в большей части), но возможно включение и других ингредиентов, например средств, улучшающих растворимость. Например, можно готовить инъецируемые растворы, в которых носитель содержит раствор соли, раствор глюкозы или смесь раствора соли и раствора глюкозы. Можно также готовить инъецируемые суспензии, в которых можно использовать подходящие жидкие носители,суспендирующие средства и т.п. В композициях, подходящих для чрескожного введения, носитель, необязательно, содержит средство, повышающее проницаемость, и/или подходящее смачивающее средство, необязательно, в комбинации с подходящими дополнительными компонентами любой природы, добавляемыми в малых пропорциях, причем эти дополнительные компоненты не вызывают каких-либо значительных неблагоприятных эффектов на коже. Указанные дополнительные компоненты могут облегчать нанесение желательных композиций на кожу и/или могут служить вспомогательными средствами при их изготовлении. Такие композиции можно вводить разнообразными путями, например в виде трансдермального пластыря, средства точечной обработки или мази. Особенно выгодно составлять вышеуказанные фармацевтические композиции в стандартной лекарственной форме, удобной для введения и для обеспечения однородности дозирования. Термин "стандартная лекарственная форма", используемый в настоящем описании и в пунктах формулы настоящего изобретения, относится к физически дискретным единицам, используемым в качестве единичных доз,причем каждая единица содержит заданное количество активного ингредиента, рассчитанное на создание желаемого терапевтического эффекта, в сочетании с требующимся фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая делимые таблетки с насечками и таблетки, покрытые оболочками), капсулы, пилюли, расфасованные в пакеты порошки, суппозитории, облатки, инъецируемые растворы или суспензии, порошки для ингаляции, мерные чайные ложки, мерные столовые ложки и т.п. и их разделяемые многозвенные упаковки. Точные дозы и частота введения зависят от применяемого конкретного соединения формулы (I),конкретного состояния, лечение которого осуществляют, тяжести состояния, лечение которого осуществляют, возраста, массы тела, пола, степени расстройства и общего физического состояния конкретного пациента, а также от других лекарственных средств, которые индивид может принимать, как хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшать или увеличивать в зависимости от реакции субъекта, лечение которого осуществляют, и/или в зависимости от его оценки лечащим врачом, назначающим соединения согласно настоящему изобретению. В зависимости от способа введения фармацевтическая композиция будет содержать активный ингредиент в количестве, составляющем от 0,05 до 99 мас.% (предпочтительно от 0,1 до 70 мас.%, более предпочтительно от 0,1 до 50 мас.%), и фармацевтически приемлемый носитель в количестве, составляющем от 1 до 99,95 мас.% (предпочтительно от 30 до 99,9 мас.%, более предпочтительно от 50 до 99,9 мас.%), причем все процентные доли даны в расчете на общую массу композиции. Соединения согласно настоящему изобретению можно применять для системного введения (такого как пероральное, чрескожное или парентеральное введение) или для местного нанесения (такого как при ингаляции, введении интраназального спрея, закапывании глазных капель или нанесении крема, использовании геля, шампуня и т.п.). Предпочтительно указанные соединения вводят перорально. Точная доза и частота введения зависят от применяемого конкретного соединения формулы (I), конкретного состояния,лечение которого осуществляют, тяжести состояния, лечение которого осуществляют, возраста, массы тела, пола, степени расстройства и общего физического состояния конкретного пациента, а также от других лекарственных средств, которые индивид может принимать, как хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное суточное количество можно уменьшать или увеличивать в зависимости от реакции субъекта, лечение которого осуществляют, и/или в зависимости от его оценки лечащим врачом, назначающим соединения согласно настоящему изобретению. Количество соединения формулы (I), которое можно вносить в комбинацию с материалом носителя для получения стандартной лекарственной формы, будет разным в зависимости от заболевания, лечение которого осуществляют, биологического вида млекопитающего и конкретного способа введения. Однако в качестве общего руководства можно указать, что предпочтительно подходящие единичные дозы соединений согласно настоящему изобретению могут содержать, например, от 0,1 до примерно 1000 мг активного соединения. Предпочтительная единичная доза составляет от 1 мг до примерно 500 мг. Более предпочтительная единичная доза составляет от 1 мг до примерно 300 мг. Еще более предпочтительная единичная доза составляет от 1 мг до примерно 100 мг. Такие единичные дозы можно вводить более одного раза в день (например, 2, 3, 4, 5 или 6 раз в день, но предпочтительно их вводить 1 или 2 раза в день,так что общая доза для взрослого человека с массой тела 70 кг находится в диапазоне от 0,001 до примерно 15 мг на 1 кг массы тела субъекта в расчете на одно введение). Предпочтительная доза составляет от 0,01 до примерно 1,5 мг на один килограмм массы тела в расчете на одно введение, и такую терапию можно продолжать в течение нескольких недель или месяцев (в некоторых случаях, в течение нескольких лет). Однако следует понимать, что конкретный уровень дозы для любого конкретного пациента будет зависеть от многих факторов, включая активность конкретного применяемого соединения, возраст,массу тела, общее состояние здоровья, пол и характер диеты индивида, лечение которого осуществляют,времени и пути введения, скорости выведения, других лекарственных средств, которые вводили ранее, и тяжести конкретного заболевания, лечение которого осуществляют, как хорошо понятно специалистам в данной области. Конкретная доза может составлять от 1 до примерно 100 мг в виде таблетки или от 1 мг до примерно 300 мг, принимаемых один раз в день или неоднократно в течение дня, или в виде одной капсулы или таблетки с пролонгированным высвобождением, принимаемой один раз в день и содержащей пропорционально большее количество активного ингредиента. Эффект пролонгированного высвобождения может быть получен с использованием материалов капсул, которые растворяются при разных значенияхpH, капсул, которые медленно высвобождают под действием осмотического давления или с использованием других средств регулируемого высвобождения. В некоторых случаях может потребоваться применение доз, находящихся за пределами указанных диапазонов, как будет понятно специалистам в данной области. Кроме того, очевидно, что клиницист или лечащий врач будет знать, как и когда начинать, прерывать, изменять и заканчивать терапию в зависимости от реакции индивидуального пациента. Следующие примеры предназначены для иллюстрации настоящего изобретения без ограничения его объема. Экспериментальная часть. Примеры синтеза. Далее в настоящем описании термин "Т.пл." означает точку плавления, "ТГФ" означает тетрагидрофуран, "ДМФА" означает диметилформамид, "DCM" означает дихлорметан, "EtOAc" означает этилацетат, "АсОН" означает уксусную кислоту, "MeOH" означает метанол, "рац" означает рацемический,"Et2O" означает простой диэтиловый эфир, "DMAP" означает диметиламинопиридин, "ДМСО" означает диметилсульфоксид, "hex" означает гексаны и "ТФУК" означает трифторуксусную кислоту, "DEA" означает диэтиламин. Синтез цис-1-Вос-3-фтор-4-гидроксипиперидина 3. К раствору 0,5 г (2,30 ммоль) N-Boc-3-фтор-4-пиперидинона 2 (J. Med. Chem. 1999, 42, 2087-2104) в 10 мл сухого ТГФ добавляли по каплям 2,8 мл (2,76 ммоль) 1 М раствора Li-селектрида в ТГФ в атмосфере N2 при 0C. Раствор перемешивали при 0C в течение 4 ч, затем при 0C добавляли 10 мл 2 МNaOH и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь экстрагировали Et2O, сушили над MgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенную смесь подвергали флэш-хроматографии на силикагеле (hex/EtOAc/Et3N 1:1:0,1) с получением 0,27 г(56%) чистого цис-1-Вос-3-фтор-4-гидроксипиперидина 3 в виде бесцветного масла, которое затвердевало при стоянии при -20C (морозильная камера). Т.пл. 48C. Синтез транс-4-азидо-1-Вос-3-фторпиперидина 4. К раствору 0,60 г (2,74 ммоль) цис-1-Вос-3-фтор-4-гидроксипиперидина 3 в 15 мл DCM при комнатной температуре добавляли 0,42 г (4,11 ммоль) триэтиламина и 37 мг (0,3 ммоль) 4-(N,Nдиметиламино)пиридина (DMAP). Затем при комнатной температуре в атмосфере сухого воздуха (трубка с CaCl2) добавляли раствор 0,57 г (3,01 ммоль) п-толуолсульфонилхлорида в 2 мл DCM. После перемешивания в течение 15 ч при комнатной температуре полученный раствор выливали в насыщенный раствор соли (20 мл) и экстрагировали DCM (325 мл). После сушки над MgSO4, фильтрования и выпаривания растворителя неочищенную смесь использовали непосредственно на следующей стадии без дальнейшей очистки. Полученный тозилат растворяли в 5 мл сухого ДМСО и добавляли 0,36 г (5,48 ммоль)NaN3. Смесь в течение 15 ч перемешивали при 90C в атмосфере N2. После охлаждения смесь выливали в насыщенный раствор соли (10 мл) и экстрагировали EtOAc. Объединенный экстракт промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и выпаривали в вакууме. Транс-4-азидо-1 Вос-3-фторпиперидин 4 получали в виде бесцветного масла с выходом 92% из 4-гидроксипиперидина 3; он был достаточно чистым для дальнейшего использования. Синтез транс-4-амино-1-Вос-3-фторпиперидина 5. К раствору 0,59 г (2,42 ммоль) транс-4-азидо-1-Вос-3-фторпиперидина 4 в 10 мл MeOH добавляли 0,61 г (9,67 ммоль) формиата аммония и 0,25 г (0,24 ммоль Pd) 10%-ного Pd на углероде. Реакционную смесь в течение 5 ч перемешивали при 50C в атмосфере N2. После охлаждения смесь фильтровали через диатомовую землю и выпаривали при пониженном давлении. Затем неочищенную смесь подвергали флэш-хроматографии на силикагеле (5% Et3N в EtOAc, короткая колонка) с получением 0,42 г (80%) транс-4-амино-1-Вос-3-фторпиперидина 5 в виде масла. Синтез транс-трет-бутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3-фторпиперидин-1 карбоксилата 6. К раствору 0,41 г (2,02 ммоль) 4-амино-5-хлор-2-метоксибензойной кислоты в 10 мл сухого ДМФА при комнатной температуре в атмосфере N2 добавляли 0,29 г (2,89 ммоль) триэтиламина. После перемешивания в течение 10 мин при комнатной температуре по каплям добавляли раствор 0,22 г (2,02 ммоль) этилхлорформиата в 2 мл ДМФА и перемешивание продолжали в течение 30 мин, поддерживая комнатную температуру (охлаждая на водяной бане с комнатной температурой). Затем при комнатной температуре одной порцией добавляли 0,27 г (2,02 ммоль) твердого гидроксибензотриазола и раствор перемешивали в течение 30 мин. После этого при комнатной температуре по каплям добавляли раствор 0,42 г (1,93 ммоль) амина 5 в 3 мл ДМФА и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем смесь выливали в 20 мл насыщенного раствора соли и экстрагировали EtOAc (325 мл). Объединенную органическую фракцию промывали насыщенным раствором соли, сушили над MgSO4,фильтровали и выпаривали при пониженном давлении. Неочищенную смесь подвергали флэшхроматографии на силикагеле (hex/EtOAc/Et3N 1:1:0,1) с получением 0,72 г (93%) чистого транс-третбутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3-фторпиперидин-1-карбоксилата 6 в виде твердого вещества. Синтез транс-4-амино-5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2 метоксибензамида 7. К раствору 0,14 г (0,34 ммоль) трет-бутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3 фторпиперидин-1-карбоксилата 6 в 5 мл DCM добавляли 0,39 г (3,4 ммоль) трифторуксусной кислоты при 0C в атмосфере сухого воздуха (трубка с CaCl2). После перемешивания в течение 5 ч при 0C смесь выпаривали при пониженном давлении. Маслянистый остаток растворяли в 10 мл сухого простого диэтилового эфира, охлаждали до 0C и выделяли образовавшуюся кристаллическую соль с ТФУК (на фильтре или декантируя Et2O). После сушки и дальнейшего выпаривания белую кристаллическую соль 4-амино-5-хлор-N-(3-фторпиперидин-4-ил)-2-метоксибензамида с ТФУК растворяли в 5 мл сухого ДМФА. К раствору добавляли 0,17 г (1,70 ммоль) триэтиламина, 55 мг (0,34 ммоль) йодида натрия и затем 65 мг (0,34 ммоль) 3-(4-фторфенокси)пропил-1-хлорида при комнатной температуре в атмосфере сухого воздуха. Смесь нагревали до 110-120C в течение 4 ч. После охлаждения смесь разбавляли 25 млEtOAc, выливали в насыщенный раствор соли (25 мл) и экстрагировали EtOAc (325 мл). Объединенную органическую фазу промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенную смесь подвергали градиентной флэш-хроматографии на силикагеле (от EtOAc/hex/Et3N 3:2:0,1 до 1% Et3N в EtOAc) с получением 85 мг (55%) транс-4-амино 5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2-метоксибензамида 7 в виде светложелтого твердого вещества. Т.пл. 125C. Возможна перекристаллизация из EtOAc/EtOH. Хиральное разделение транс-4-амино-5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4 ил-2-метоксибензамида 7. Соединение 7 разделяли на его энантиомеры сверхкритической флюидной хроматографией. Количество: 80 мг (нагрузка: 10 мг/3,00 мл) Условия: Колонка: OD 20250 мм (I) Подвижная фаза: 37% MeOH (с 0,2% изо-PrNH2), выдержка 9,00 мин Параметры: поток 50 мл/мин Температура колонки 40C Давление сопла 10 МПа Тип инъекции: многослойные инъекции (8) Способ сбора: сбор с использованием стандартного детектирования пика Пик 1 элюировался при 5 мин 20 с, давая левовращающий энантиомер (-)-7[]=-22,8 (c=0,3, MeOH, =598 нм; 20C). Пик 2 элюировался при 7 мин 30 с, давая правовращающий энантиомер (+)-7 Синтез цис-N-(1-Вос-3-фторпиперидин-4-ил)амина 10. Указанное соединение получали, как описано в литературных ссылках: 1) J. Med. Chem. 1999, 42, 2087-2104, и 2) WO 2007071965. Синтез цис-трет-бутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3-фторпиперидин-1 карбоксилата 11. К раствору 0,97 г (4,82 ммоль) 4-амино-5-хлор-2-метоксибензойной кислоты в 25 мл сухого ДМФА в атмосфере N2 при комнатной температуре добавляли 0,70 г (6,88 ммоль) триэтиламина. После перемешивания при комнатной температуре в течение 10 мин добавляли по каплям раствор 0,52 г (4,82 ммоль) этилхлорформиата в 1 мл ДМФА при комнатной температуре и перемешивание продолжали в течение 30 мин. Затем при комнатной температуре одной порцией добавляли 0,65 г (4,82 ммоль) твердого гидроксибензотриазола и раствор перемешивали в течение 30 мин. Затем при комнатной температуре по каплям добавляли раствор 1,0 г (4,59 ммоль) амина 10 в 3 мл ДМФА и реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем смесь выливали в 100 мл насыщенного раствора соли и экстрагировали EtOAc (430 мл). Объединенную органическую фракцию промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенную смесь подвергали флэш-хроматографии на силикагеле (hex/EtOAc/Et3N 1:1:0,1; Rr=0,01) с получением 1,25 г (68%) чистого трет-бутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3-фторпиперидин-1 карбоксилата 11 в виде твердого вещества. Т.пл. 198-199C. Синтез цис-4-амино-5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2 метоксибензамида 12. К раствору 1,00 г (2,49 ммоль) трет-бутил-4-(4-амино-5-хлор-2-метоксибензоиламино)-3 фторпиперидин-1-карбоксилата 11 в 10 мл DCM в атмосфере сухого воздуха (трубка с CaCl2) при 0C добавляли 2,83 г (24,9 ммоль) трифторуксусной кислоты. После перемешивания в течение 4 ч при 0C смесь выпаривали при пониженном давлении. Маслянистый остаток растворяли в 25 мл сухого диэтилового эфира, охлаждали до 0C и выделяли образовавшуюся кристаллическую соль с ТФУК (на фильтре или декантируя Et2O). После сушки и дополнительного выпаривания в вакууме получали 0,78 г соли 4-9 020967 амино-5-хлор-N-(3-фторпиперидин-4-ил)-2-метоксибензамида с ТФУК в виде белого твердого вещества. К раствору 0,78 г полученной соли в 10 мл ДМФА при комнатной температуре в атмосфере сухого воздуха добавляли 1,26 г (12,45 ммоль) триэтиламина, 0,37 г (2,49 ммоль) йодида натрия и затем 0,47 г (2,49 ммоль) 3-(4-фторфенокси)пропил-1-хлорида. Смесь нагревали до 120C в течение 2 ч. После охлаждения смесь разбавляли 25 мл EtOAc, выливали в насыщенный раствор соли (25 мл) и экстрагировали EtOAc(325 мл). Объединенную органическую фазу промывали насыщенным раствором соли, сушили надMgSO4, фильтровали и выпаривали при пониженном давлении. Неочищенную смесь подвергали градиентной флэш-хроматографии на силикагеле (от EtOAc/hex/Et3N 3:1:0,1 до 1% Et3N в EtOAc) с получением 49% 4-амино-5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2-метоксибензамида 12 в виде светло-желтого твердого вещества. Т.пл. 137C. Возможна перекристаллизация из EtOAc/EtOH. Хиральное разделение цис-4-амино-5-хлор-N-3-фтор-1-[3-(4-фторфенокси)пропил]пиперидин-4 ил-2-метоксибензамида 12. Соединение 12 разделяли на его энантиомеры сверхкритической флюидной хроматографией. Количество: 152 мг (нагрузка: 8,5 мг/1,250 мл) Условия: Колонка: OJ 20250 мм (I) Подвижная фаза: 19% MeOH (с 0,2% изо-PrNH2), выдержка 14,00 мин Параметры: поток 50 мл/мин Температура колонки 40C Давление сопла: 10 МПа Тип инъекции: многослойные инъекции (18) Способ сбора: сбор с использованием стандартного детектирования пиков. Пик 1 элюировался при 10 мин 20 с, давая левовращающий энантиомер (-)-12[]=-45,5 (c=0,2, MeOH, =598 нм; 20C). Пик 2 элюировался при 11 мин 40 с, давая правовращающий энантиомер (+)-12(8,0 ммоль) 3,3-дифтор-4,4-дигидрокси-1 трифторацетилпиперидина 13 (J. Org. Chem. 2010, 75, 929-932) и 2,15 г (20,0 ммоль; 2,5 экв.) бензиламина в 50 мл толуола. Смесь кипятили с обратным холодильником и ловушкой Дина Старка в течение 15 ч. После охлаждения до комнатной температуры растворитель удаляли в вакууме. Полученное в результате масло растворяли в 25 мл абсолютного метанола и при комнатной температуре медленно добавляли 0,56 г (8,8 ммоль; 1,1 экв.) цианоборгидрида натрия и 0,48 г (8,0 ммоль; 1 экв.) уксусной кислоты. Раствор перемешивали в течение 4 ч при комнатной температуре. После удаления растворителя в вакууме неочищенное масло повторно растворяли в 50 мл дихлорметана, выливали в 50 мл насыщенного водного раствора NaHCO3 и затем экстрагировали дихлорметаном (350 мл). Объединенные органические слои промывали насыщенным раствором соли и сушили над MgSO4. Отфильтровывали твердые вещества и выпаривали растворитель с получением неочищенного масла, которое очищали флэш-хроматографией Синтез бензил 3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-иламина 15. В 100-мл колбе при 120C в течение 30 ч перемешивали смесь 1,22 г (5,4 ммоль) бензил(3,3 дифторпиперидин-4-ил)амина 14, 0,81 г (5,4 ммоль; 1 экв.) йодида натрия, 2,73 г (27,0 ммоль; 5 экв.) триэтиламина и 1,05 г (5,4 ммоль; 1 экв.) 1-(3-хлорпропокси)-4-фторбензола в 70 мл ДМФА. Добавляли другую порцию триэтиламина (2,73 г; 27,0 ммоль; 5 экв.) и смесь перемешивали при 120C в течение 16 ч. Затем добавляли 1,05 г (5,4 ммоль; 1 экв.) 1-(3-хлорпропокси)-4-фторбензола и смесь перемешивали при 120C в течение 54 ч до завершения реакции. Растворитель удаляли в вакууме и неочищенное масло повторно растворяли в 100 мл EtOAc, промывали насыщенным раствором соли и сушили над MgSO4. Отфильтровывали твердые вещества и выпаривали растворитель с получением неочищенного масла, которое очищали флэш-хроматографией (гексан/EtOAc 1:1, Rf=0,19-0,38) (EtOAc, Rf=0,03), с получением 1,00 г (2,6 ммоль; выход 49%) бензил 3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-иламина 15 в виде масла коричневого цвета. Синтез 3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-амина 16. В сухом сосуде высокого давления растворяли 0,83 г (2,2 ммоль) бензил 3,3-дифтор-1-[3-(4 фторфенокси)пропил]пиперидин-4-иламина 15 в 10 мл метанола. После добавления 0,33 г (40 мас.%)Pd/C (10%), проведенного при 0C, смесь перемешивали в течение 15 ч при комнатной температуре в атмосфере водорода при давлении 480 кПа. Смесь фильтровали через диатомовую землю. Растворитель выпаривали в вакууме с получением 0,57 г (2,0 ммоль; выход 90%) 3,3-дифтор-1-[3-(4 фторфенокси)пропил]пиперидин-4-амина 16 в виде желтого масла. Синтез 4-амино-5-хлор-N-3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2 метоксибензамида 17. В сухой 50-мл колбе растворяли 0,44 г (2,2 ммоль; 1,1 экв.) 4-амино-5-хлор-2-метоксибензойной кислоты и 0,30 г (3 ммоль; 1,5 экв.) триэтиламина в 25 мл диметилформамида, перемешивая в течение 10 мин при комнатной температуре. Затем смесь охлаждали до 0C, добавляли 0,24 г (2,2 ммоль; 1,1 экв.) этилхлорформиата и перемешивали в течение 30 мин при комнатной температуре. Добавляли 0,29 г (2,2 ммоль; 1,1 экв.) 1-гидроксибензотриазола и перемешивали в течение 30 мин при комнатной температуре. Затем добавляли 0,57 г 3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-амина 16 и смесь переме- 12020967 шивали при комнатной температуре в течение 15 ч. Выпаривали в вакууме растворитель, неочищенное масло повторно растворяли в EtOAc, выливали в 50 мл насыщенного раствора соли и экстрагировалиEtOAc (450 мл). Органические фазы промывали насыщенным раствором соли и сушили над MgSO4. Отфильтровывали твердые вещества и выпаривали растворитель с получением неочищенного масла, которое очищали флэш-хроматографией (гексан/EtOAc 3:7, Rf=0,35), с получением 0,66 г (1,4 ммоль; выход 71%) чистого 4-амино-5-хлор-N-3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4-ил-2 метоксибензамида 17 в виде белых кристаллов. Т.пл. 125,8C (гексан/EtOH 1:1). Хиральное разделение 4-амино-5-хлор-N-3,3-дифтор-1-[3-(4-фторфенокси)пропил]пиперидин-4 ил-2-метоксибензамида 17. 170 мг соединения 17 разделяли на его энантиомеры сверхкритической флюидной хроматографией на приборе Berger Multigram SFC (Mettler, Toledo Co., Ltd) с 5-мкм колонкой IC 250 мм 50 мм. Подвижная фаза: сверхкритический CO2:MeOH с 0,05% DEA (75:25) при 160 мл/мин Температура колонки: 38C Давление сопла: 30 МПа Температура сопла: 60C Температура испарителя: 20C Температура триммера: 25C Длина волны: 220 нм. Пик 1 элюировался при 7,4 мин, давая правовращающий энантиомер (+)-17 е.е.%=100%; []=+14,1 (c=0,3, MeOH, =598 нм; 20C). Пик 2 элюировался при 8,5 мин, давая левовращающий энантиомер (-)-17 е.е.%=98,6%; []=-14,4 (c=0,3, MeOH, =598 нм; 20C). Фармакологические примеры. Пример 4. Связывание с рецепторами. Для определения сродства испытуемых соединений к конкретному рецептору использовали анализ конкурентного связывания радиолигандов. К инкубационной смеси с мембранной фракцией, содержавшей соответствующий рецептор, и с фиксированной низкой концентрацией (нМ) радиолиганда добавляли различные концентрации немеченого испытуемого соединения. Во время инкубации радиолиганд связывался с рецептором, но это связывание ингибировалось немеченым испытуемым соединением пропорционально его аффинности связывания и концентрации. Были установлены клеточные линии, которые стабильно экспрессировали человеческий вариант исследуемого рецептора после трансфекции соответствующей кДНК (табл. 1). Трансфицированные клетки выращивали в стандартных условиях культивирования, мембранные фракции получали после центрифугирования гомогенизированных клеток. Определяли разведения мембран, оптимальные для анализа связывания; до использования соответствующие аликвоты хранили при -70C. В планшете 96 луночного формата соответствующий меченый радиолиганд добавляли к мембранному препарату, содержавшему исследуемый рецептор. Растворы испытуемых соединений, приготовленные в ДМСО, разводили в 100 раз в многолуночном планшете до конечной испытуемой концентрации в диапазоне от 10-9 до 10-5 М. После инкубации с испытуемым соединением несвязанный радиолиганд удаляли фильтрованием на фильтрах G/F в Filtermate 96. К промытым фильтрующим планшетам добавляли Microscint, и на жидкостном сцинтилляционном счетчике TopCount (Packard) измеряли радиоактивность, связанную с рецептором. Для измерения неспецифического связывания (NSB) в лунки, содержавшие мембранную фракцию и радиолиганд, добавляли высокую концентрацию лиганда без радиоактивной метки. Таблица 1 Условия определения ингибирования связывания радиолиганда с рецепторами Процент ингибирования связывания радиолиганда с рецептором, индуцированного испытуемым соединением, рассчитывали по формуле% эффекта=100-[(образец-NSB)/(HC-NSB)100],где "образец" представляет собой счет радиоактивности в лунке, обработанной испытуемым соединением, НС - счет радиоактивности в контрольных лунках, инкубированных только с радиолигандом. Используя самостоятельно разработанное программное обеспечение, по методу наименьших квадратов строили кривую наилучшей аппроксимации на графиках зависимости процента ингибирования от концентрации испытуемого соединения. По этому графику определяли значение pIC50 (концентрацию ингибитора, обусловливающую 50%-ное замещение специфического связывания) и наклон графика (коэффициент Хилла). Таблица 2 Значения pIC50 Ссылочным соединением (ссыл.) является цизаприд. Пример 5. Антагонизм в отношении 5-HT2B. Клетки CHO-K1 (ЕСАСС), стабильно трансфицированные кДНК человеческого рецептора 5-HT2B,субклонировали в pCDNA3.1, используя кальций-фосфатный способ. Стабильно трансфицированные клеточные линии отбирали с использованием G-418, клональные клеточные линии развивали в предельных разведениях. Клеточные линии культивировали на среде Игла, модифицированной по способу Дульбекко (DMEM), содержавшей 10% термоинактивированную диализованную фетальную телячью сыворотку (FBS), 1% пенициллина-стрептомицина, 1% L-глутамина и 1% заменимых аминокислот. Слившиеся слои в 96-луночных планшетах черного цвета с прозрачным дном обрабатывали 4 мкМ Fluo3-AM в течение 90 мин при 37C в сбалансированном солевом растворе Хенкса с добавлением 20 мМHEPES и 2,5 мМ пробенецида. К промытым клеткам добавляли испытуемое соединение и регистрировали максимальную флуоресценцию, являвшуюся реакцией на 0,1 нМ серотонина, с использованием флуориметрического планшетного анализатора изображений (FLIPR) для детектирования изменений внутриклеточных уровней кальция. Максимальную флуоресценцию, зарегистрированную в присутствии испытуемого соединения, выражали в процентах от максимальной флуоресценции, обусловленной реакцией на агонист (0,1 нМ серотонина). Значение IC50 определяли посредством нелинейного регрессионного анализа кривых зависимости реакции от концентрации, построенных по средним значениям, используя аппроксимацию по уравнению Хилла (Y=D+[(A-D)/(1+(C/C50)nH)], где Y - специфическая реакция, D - минимальная специфическая реакция (без добавления испытуемых соединений или серотонина), А - максимальная специфическая реакция (0,1 нМ серотонина, без испытуемых соединений), С - концентрация испытуемого соединения, С 50=IC50, и nH - коэффициент наклона) (SigmaPlot 4,0, SPSS Inc.). Эффективную константу диссоциации(KB=IC50/(1+(A/EC50A, где А - концентрация серотонина и ЕС 50 А представляет собой значение ЕС 50 для серотонина в этом исследовании) (Porter et al. (1999), Br. J. Pharmacol, 128: 13-20). Таблица 3 Антагонизм в отношении 5-HT2B Пример 6. Использование аппарата PatchXpress с hERG-трасфицированными клетками HEK293. Эксперименты проводили с клетками HEK293, стабильно экспрессирующими калиевый каналhERG. Клетки выращивали при 37C и 5% CO2 в культуральных сосудах на среде MEM с добавлением 10% термоинактивированной фетальной телячьей сыворотки, 1%-ного раствора L-глутаминапенициллина-стрептомицина, 1% заменимых аминокислот (100), 1% пирувата натрия (100 мМ) и 0,8% генетицина (50 мг/мл). Перед использованием клетки субкультивировали в среде MEM в отсутствие 5 млL-глутамина-пенициллина-стрептомицина. Для использования в автоматизированной системе локальной фиксации потенциала ("пэтч-кламп") PatchXpress 7000A (Axon Instruments) клетки собирали для получения суспензии единичных клеток. Внеклеточный раствор содержал 150 мМ NaCl, 4 мМ KCl, 1 мМ MgCl2, 1,8 мМ CaCl2, 10 мМEGTA, 4 мМ АТР-Mg2, 2 мМ MgCl2, 0,5 мМ CaCl2 (pH 7,2 с KOH). Эксперименты по методике "пэтч-кламп" проводили в режиме фиксации потенциала, записывая токи с цельной клетки в автоматизированном исследовании с локальной фиксацией потенциала, используя систему PatchXpress 7000A (Axon Instruments). Зарегистрированные токи усиливали и оцифровывали усилителем Multiclamp, сохраняли и анализировали с помощью программного пакета DataXpress системы PatchXpress и Igor 5,0 (Wavemetrics). Исходный потенциал составлял -80 мВ. Ток hERG (K+-селективный выходящий ток) определяли как максимальный следовой ток при -40 мВ после 2 секунд деполяризации до +60 мВ. Продолжительность циклической пульсации составляла 15 с. Перед каждым испытательным импульсом задавали короткий импульс (0,5 с) от исходного потенциала до -60 мВ для определения (линейного) тока утечки. После создания конфигурации с цельной клеткой и некоторого периода стабильности следовал 5 минутный период с нанесенным плацебо (водный ДМСО в качестве контроля), после которого применяли возрастающие концентрации тестируемого вещества (10-7 М, 310-7 М и 310-6 М). Каждую концентрацию тестируемого вещества наносили дважды. Эффект каждой концентрации определяли спустя 5 мин в виде среднего тока трех последовательных импульсов напряжения. Для определения степени блокирования остаточный ток сравнивали с током, зарегистрированным при предварительной обработке с плацебо. Данные представляли в виде степени блокирования (в %) при концентрациях, указанных в табл. 4. Значения в скобках относятся к степени блокирования при нанесении плацебо. Таблица 4 Степень блокирования (в %) канала hERG Пример 7. Легочная артериальная гипертензия, индуцированная монокроталином у крыс. Соединение (-)-17 испытывали при легочной артериальной гипертензии, индуцированной монокроталином у крыс (см., например, Stenmark et al., 2009, Am J. Physiol Lung Cell Mol Physiol 297, L1013L1032). Измерения включали определение среднего давления артериальной крови и давления в правом желудочке in vivo, расчет отношения массы правого желудочка к суммарной массе левого желудочка и перегородки как показателя гипертрофии правого желудочка, определение времени ускорения легочного артериального кровотока и гистологическую оценку мускуляризации легочных артерий. Монокроталин растворяли в 1 н. HCl и затем в дистиллированной воде, pH доводили до 7,4, добавляя NaOH. Единичную дозу монокроталина (60 мг/кг) вводили подкожно в нулевой день трем группам самцов крыс Sprague Dawley. Тестируемое соединение (-)-17 растворяли в 20%-ном гидроксипропилбета-циклодекстрине с NaOH, HCl и маннитом в апирогенной воде и вводили перорально через зонд(10 мл/кг) один раз в день с первого дня в течение 21 дня в дозах 10 и 50 мг/кг. Концентрацию соединения (-)-17 измеряли через 2 ч (что приблизительно соответствует величине Cmax после перорального введения крысе) после последнего введения в 21-й день. Животным третьей группы по тому же протоколу вводили соответствующие объемы 20%-ного гидроксипропил-бета-циклодекстрина в качестве носителя(плацебо). Трехнедельное пероральное введение 10 или 50 мг соединения (-)-17 один раз в день (средняя концентрация в плазме, спустя 2 ч после введения дозы в 21-день составляла 80 и 1000 нг/мл соответственно) не приводило к токсичным эффектам и не влияло на среднее давление артериальной крови (MAP),но понижало давление в правом желудочке (RVP), уменьшало гипертрофию правого желудочка (правый желудочек/(левый желудочек+перегородка); RV/(LV+S и увеличивало время ускорения легочного артериального кровотока (РААТ) (табл. 5). Обработка монокроталином достоверно увеличивала среднюю толщину стенок малых легочных артерий, и трехнедельное лечение соединением (-)-17 в пероральной дозе 50 мг/кг или 10 мг уменьшало степень этого утолщения (Р=0,0539 и 0,005 соответственно). Таблица 5 Приведенные данные представляют собой средние значения ( стандартное отклонение), измеренные в 21-й день.р 0,05 по сравнению с плацебо. Пример 8. Сердечно-сосудистые эффекты у анестезированных морских свинок. Самок морских свинок анестезировали пентобарбиталом натрия (66 мг/кг внутрибрюшинно с последующей непрерывной внутривенной инфузией в дозе 6 мг/ч) и подготавливали к измерениям поверхностной электрокардиограммы (ECG), частоты сердечных сокращений и среднего давления артериальной крови (см. De Clerck et al., Fundam. Clin. Pharm.; 2002; 16: 125-140). Соединение (-)-17 растворяли в 20%-ном гидроксипропил-бета-циклодекстрине с NaOH, HCl и маннитом в апирогенной воде и внутривенно вводили (0,5 мл/кг) в увеличивающихся дозах (0,16, 0,32, 0,64, 1,25, 2,5 и 5 мг/кг) в течение 5 минутного периода с 15-минутными интервалами. В конце каждой инфузии измеряли концентрацию соединения (-)-17 в плазме. Животным второй группы по тому же протоколу вводили соответствующие объемы носителя. Относительно носителя, соединение (-)-17 в дозах от 0,16 до 5 мг/кг (общая доза 9,87 мг/кг;Cmax=11,950 нг/мл) не оказывало существенного влияния на частоту сердечных сокращений, на длительность интервалов PQ, QRS, QT и QTcB или на морфологию ECG у анестезированных морских свинок(табл. 6). Начиная с 2,5 мг/кг и выше (Cmax=6,325 нг/мл; табл. 7), начинало увеличиваться среднее давление артериальной крови (табл. 6). Ссылочное соединение дофетилид (0,02 мг/кг внутривенно в течение 1 мин), введенное через 15 мин после начала последней инфузии носителя, уменьшало частоту сердечных сокращение и удлиняло интервалы QT и QTcB. Таблица 6 Эффекты соединения (-)-17, введенного до начала каждой инфузии и спустя 2, 5 и 15 мин после него, на степень изменения (в %) частоты сердечных сокращений (HR), среднего давления артериальной крови(MBP) и параметров ECG у анестезированных морских свинок относительно соответствующих исходных значений. Исходные значения представлены в реальных единицах Приведенные значения представляют собой медианы группы с n=6. Статистически достоверные различия (р 0,05) указаны полужирным курсивом, они рассчитаны по отклонениям от исходных значений в реальных единицах. Таблица 7 Средние уровни (медианы) соединения (-)-17 в плазме (нг/мл) после введения увеличивающихся внутривенных доз (0,16, 0,32, 0,64, 1,25, 2,5 и 5 мг/кг) в течение 5-минутных периодов с 15-минутными интервалами (n=6) Пример 9. Фиброз легких, индуцированный блеомицином у мышей (пример возможного использования). Самцам мышей C57BL/6 интратрахеально вводили сульфат блеомицина (водный раствор, 2,5 ед./мл, 2 мл/кг массы тела) при ингаляционной анестезии изофлураном (см., например, Ishii Y. et al.,2006. Am J. Respir Crit Care Med. 174 (5): 550-6). После этого в течение 2 недель один раз в день перорально вводили соединение (-)-17 в дозе 10 или 50 мг/кг. Посмертное исследование включало общее патологоанатомическое обследование, определение массы легких и исследование гистопатологии легких,проведенные в 15-й день. Гистопатологическое исследование легких показало, что блеомицин вызывает воспаление с последующим фиброзом легких у мышей, не подвергавшихся лечению. Пример 10. Фармакокинетическое исследование на мышах. Для внутривенного (i.v.) введения соединение (-)-17 растворяли в концентрации 0,25 мг/мл в физиологическом растворе соли, содержавшем 20% (масса/объем) гидроксипропил-бета-циклодекстрина(HPbCD), и вводили в виде болюса в хвостовые вены самцам мышей CD1 (n=3) в дозе 2,5 мг/кг (10 мл/кг). Для перорального введения (р.о.) соединение (-)-17 растворяли в концентрации 0,5 мг/мл в воде,- 17020967 содержавшей 20% (масса/объем) HPbCD, и вводили через зонд самцам мышей CD1 (n=3) в дозе 10 мг/кг(20 мл/кг). Образцы крови отбирали из подкожной вены задней конечности через определенные интервалы времени до 24 ч после введения дозы. Плазму получали центрифугированием и до анализа хранили при -20C. Анализ проводили жидкостной хроматографией (ЖХ) с тандемным масс-спектрометрическим детектированием (МС/МС) в режиме положительных ионов. Соединение (-)-17 элюировали на колонке с обращенной фазой градиентом ацетонитрила в воде, содержавшей 0,1% (об./об.) муравьиной кислоты. Во время анализа образцы плазмы (20 мкл) размораживали и из них удаляли белок (добавляя 200 мкл ацетонитрила и центрифугируя). Аликвоты супернатанта инъецировали колонку с обращенной фазой типа UPLC и анализировали масс-спектрометрией с электрораспылением. Стандарты для калибровки и контроля качества, анализ которых проводили перед исследованием образцов и после него, готовили в плазме крови мыши одновременно с исследованием образцов. Точность (внутриотраслевая точность,оцениваемая с независимыми образцами, применяемыми для контроля качества) составляла 85% и 115% от номинального значения во всем диапазоне концентраций. Некомпартментный фармакокинетический анализ кривых зависимости концентрации в плазме от времени проводили, используя WinNonLin для оценки клиренса из плазмы (CLp), объема распределения в стационарном состоянии (Vss), периода полувыведения в конечной фазе (t1/2) и пероральной биодоступности (F); результаты сведены в табл. 8. Пример 11. Фармакокинетическое исследование на крысах. Для внутривенного (i.v.) введения соединение (-)-17 растворяли в концентрации 1 мг/мл в физиологическом растворе соли, содержавшем 20% (масса/объем) гидроксипропил-бета-циклодекстрина(HPbCD), и вводили в виде болюса в подкожную вены задней конечности самцу крысы Sprague Dawley(n=1) в дозе 2,5 мг/кг (2,5 мл/кг). Для перорального введения (р.о.) соединение (-)-17 растворяли в концентрации 1 мг/мл в воде, содержавшей 20% (мас./об.) HPbCD, и вводили через зонд самцам крыс Sprague Dawley (n=3) в дозе 10 мг/кг (10 мл/кг). Образцы крови отбирали из хвостовой вены через определенные интервалы времени до 24 ч после введения дозы. Плазму получали центрифугированием и до анализа хранили при -20C. Анализ проводили посредством жидкостной хроматографии (ЖХ) с тандемным масс-спектрометрическим детектированием (МС/МС) в режиме положительных ионов. Соединение(-)-17 элюировали на колонке с обращенной фазой градиентом ацетонитрила в воде, содержавшей 0,1%(об./об.) муравьиной кислоты. Во время анализа образцы плазмы (50 мкл) размораживали и из них удаляли белок (добавляя по меньшей мере три объема ацетонитрила и центрифугируя). Аликвоты супернатанта инъецировали на колонку с обращенной фазой типа UPLC и анализировали масс-спектрометрией с электрораспылением. Стандарты для калибровки и контроля качества, анализ которых проводили перед исследованием образцов и после него, готовили в плазме крови крысы одновременно с исследованием образцов. Точность (внутриотраслевая точность, оцениваемая с независимыми образцами, применяемыми для контроля качества) составляла 85% и 115% от номинального значения во всем диапазоне концентраций. Некомпартментный фармакокинетический анализ кривых зависимости концентрации в плазме от времени проводили, используя WinNonLin для оценки клиренса из плазмы (CLp), объема распределения в стационарном состоянии (Vss), периода полуэлиминации в конечной фазе (t1/2) и пероральной биодоступности (F); результаты сведены в табл. 8. Таблица 8 Параметры некомпартментной фармакокинетики для соединения (-)-17, полученные у мышей и крыс после внутривенного и перорального введения соединения (-)-17-ААА (свободного основания) Пример 12. Кардиогемодинамические, кардиоэлектрофизиологические, электроэнцефалографические и пульмонореспираторные эффекты у анестезированных собак (породы бигль) при искусственной вентиляции. Животных анестезировали смесью 0,015 мг/кг внутривенного скополамина и 0,075 мг/кг внутривенного лофентанила и вызывали релаксацию сукцинилхолином (5 мг/кг, внутривенно) с последующей внутривенной инфузией этомидата (1,5 мг/кг/ч) и небольшими дополнительными дозами фентанила(0,025 мг/кг, внутривенно), вводимыми с 60-минутными интервалами. Животных подключали к искусственной вентиляции легких и подготавливали к измерениям поверхностной ЭКГ, давления крови в аорте,легочной артерии и в левом желудочке, кровотока в сонной артерии, монофазного потенциала действия,температуры тела, газов в крови и ЭЭГ (см. Van Deuren et al., J. Pharmacol Toxicol Methods; 2009; 60: 1123). Соединение (-)-17 растворяли в 20%-ном гидроксипропилциклодекстрине с NaOH, HCl и маннитом в апирогенной воде и вводили внутривенно (1 мл/кг) в возрастающих дозах (0,16, 0,32, 0,63, 1,25, 2,5 и 5 мг/кг) в течение 5-минутного периода с 30-минутными интервалами. Перед каждой инфузией и в ее конце измеряли концентрации соединения (-)-17 в плазме. Животным второй группы по тому же протоколу вводили соответствующие объемы носителя (плацебо). Относительно носителя, соединение (-)-17 при 0,16-5 мг/кг (общая доза 9,86 мг/кг; медианаCmax=20,375 нг/мл) не оказывало какого-либо заметного влияния на частоту сердечных сокращений (HR),давление в легочной артерии, конечное диастолическое давление в левом желудочке, минутный объем сердца, ударный объем сердца, произведение частоты сердечных сокращений на давление, длительность интервалов PQ и QRS, функциональное состояние легких (динамическую податливость (Cdyn) и сопротивление дыхательных путей (Raw, температуру тела или на ЭЭГ (регистрируемую монитором Narcotrend) у анестезированных собак. Начиная с 1,25 мг/кг и выше (Cmax=5,205 нг/мл), начинало увеличиваться давление артериальной крови, сосудистое сопротивление (системное и в общей сонной артерии) иTau (постоянная времени релаксации). Кроме того, при 2,5 мг/кг (Cmax=9,550 нг/мл) LV dp/dtmax/pd начинало уменьшаться, и при 5 мг/кг (Cmax=20,375 нг/мл) было замечено небольшое уменьшение длительности QTc VDW (интервал QT с поправкой частоту сердечных сокращений) и QTc VcT (интервал QT с поправкой частоту сердечных сокращений и температуру). Таблица 9 Эффекты соединения (-)-17 до начала каждой инфузии и спустя 5 и 30 мин после него, выраженные в процентах изменения относительно плацебо, оказываемые на частоту сердечных сокращений (HR), среднее давление артериальной крови (МВР), систолическое (SPP) и диастолическое (DPP) давление в легочной артерии, сократимость левого желудочка (LVdp/dtmax) и параметры ЭКГ (PQ, QRS и QTcV) у анестезированных собак породы бигль. Исходные значения представлены в реальных единицах Приведенные значения представляют собой медианы группы с n=4. Статистически достоверные различия (р 0,05) указаны полужирным курсивом, они рассчитаны по отклонениям от исходных значений в реальных единицах. Таблица 10 Средние уровни (медианы) соединения (-)-17 в плазме (нг/мл) после введения увеличивающихся внутривенных доз (0,16, 0,32, 0,64, 1,25, 2,5 и 5 мг/кг) в течение 5-минутных периодов с 30-минутными интервалами (n=4) или его стереохимически изомерная форма, гдеR представляет собой водород или фтор,или его аддитивная соль, или сольват. 2. Соединение по п.1, где R представляет собой фтор и указанное соединение представляет собой рацемическую смесь, или его аддитивная соль, или сольват. 3. Соединение по п.1, где R представляет собой фтор и указанное соединение обладает оптическим вращением []=-14,4 (c=0,3, MeOH, =598 нм; 20C), или его аддитивная соль, или сольват. 4. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по любому из пп.1-3 и фармацевтически приемлемый носитель. 5. Способ получения фармацевтической композиции по п.4, отличающийся тем, что фармацевтически приемлемый носитель тщательно смешивают с терапевтически эффективным количеством соединения по любому из пп.1-3. 6. Применение соединения по любому из пп.1-3 для лечения или предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки. 7. Способ лечения или предупреждения легочной артериальной гипертензии, фиброза легких или синдрома раздраженной толстой кишки, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп.1-3.
МПК / Метки
МПК: A61K 31/4468, A61P 11/00, C07D 211/58
Метки: рецептора, 5-ht2b, антагонисты
Код ссылки
<a href="https://eas.patents.su/21-20967-antagonisty-receptora-5-ht2b.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты рецептора 5-ht2b</a>
Антагонисты рецептора 5-нт7

Номер патента: 16787
Опубликовано: 30.07.2012
Авторы: Ваттон Мария Энн, Галлахер Питер Таддеуш, Бадеску Валентина О., Филла Сандра Энн
МПК: A61K 31/497, A61P 25/00, C07D 401/14...
Метки: антагонисты, 5-нт7, рецептора
Формула / Реферат:
1. Соединение формулыгде А представляет собой -С(Н)= или -N=;R1 представляет собой заместитель, выбранный из группы, состоящей из i) водорода, ii) метила, iii) этила, iv) гидроксиметила, v) гидроксиэтила, vi) фенила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, vii) бензила, необязательно содержащего в качестве заместителей от 1 до 3 фторгрупп, и viii) пиридила;R2 представляет собой водород, метил или этил;R3...
Антагонисты рецептора il-8

Номер патента: 1436
Опубликовано: 26.02.2001
Авторы: Херцберг Роберт Филип, Вебер Дэниел Франк, Юревич Энтони Джозеф, Ратледж Мельвин Кларенс Мл., Виддаусон Кетрин Луиза
МПК: A61P 11/06, A61K 31/17
Метки: рецептора, антагонисты
Формула / Реферат:
1. Способ лечения болезненного состояния, опосредованного хемокином, где хемокин связывается у млекопитающих, нуждающихся в таком лечении, с IL-8 а- или b-рецептором, включающий введение млекопитающему эффективного количества соединения формулы где Х является кислородом или серой; R является любой функциональной группой, имеющей ионизируемый водород и рКа, равный 10 или менее; R1 независимо выбирают из водорода; галогена; нитро; циано;...
Бензоксазиноновые антагонисты рецептора допамина d4.

Номер патента: 1486
Опубликовано: 23.04.2001
Авторы: Беллиотти Томес, Вайс Лауренс Дейвид, Вустров Дейвид Юрген
МПК: C07D 265/36, A61K 31/535
Метки: бензоксазиноновые, допамина, антагонисты, рецептора
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 означают независимо водород или алкил с числом углеродных атомов от 1 до 6, X означает С, N или СН, при этом в случае X=С штрихованная линия означает связь, а в других случаях отсутствует; R3 означает фенил, нафтил, гетероарил, замещенный фенил, замещенный нафтил или замещенный гетероарил, при этом каждый заместитель независимо выбран из группы, включающей галоген, алкоксигруппу с числом углеродных...
Антагонисты рецептора интегрина

Номер патента: 2822
Опубликовано: 31.10.2002
Авторы: Аскью Бен С., Хант Сесилия, Смит Гарри Р., Пэйтан Майкл А., Мейсснер Роберт С., Хартман Джордж Д., Даггэн Марк Е., Халщенко Василь, Хатчинсон Джон Х., Ванг Дзиабинг, Коулман Пол Дж.
МПК: A61P 19/00, C07D 471/04, A61K 31/4375...
Метки: рецептора, антагонисты, интегрина
Формула / Реферат:
1. Соединение формулы где Х выбирают из группы, состоящей из Y выбирают из группы, состоящей из (СН2)m, (СН2)m-S-(СН2)n и (СН2)m-NR4-(СН2)n, где каждый атом углерода метиленовой группы (СН2) в группировке Y в отличие от R4 может быть замещен одним или двумя заместителями R3, при условии, что когда Y обозначает -(СН2)m-NR4-(СН2)n- и n=1, то заместитель R3 на атоме углерода метиленовой группы в -(СН2)m-, соседнем с атомом азота, не может...
Антагонисты рецептора гистамина-3

Номер патента: 13602
Опубликовано: 30.06.2010
Авторы: Чандрасекаран Рамалакшми Йегна, Вагер Тревис Т., Батлер Тодд Уилльям
МПК: A61K 31/165, C07D 207/04, C07C 237/24...
Метки: рецептора, антагонисты, гистамина-3
Формула / Реферат:
1. Соединение формулы Iили его фармацевтически приемлемая соль,где R1и R2, каждый независимо, выбран из группы, включающейводород;С1-С8-алкил, необязательно замещенный 1-4 галогенами;С1-С4-алкильную группу, необязательно замещенную заместителем, выбранным из группы, состоящей из ОН, одного до четырех С1-С4-алкила, С3-С7-циклоалкила, С1-С4-диалкиламино, С6-С14-арила, необязательно замещенного галогеном и необязательно замещенного группой...
Предыдущий патент: Способ получения и очистки солей солифенацина
Следующий патент: Ингаляторное лекарственное средство
Случайный патент: Прибор для измерения и прогнозирования шероховатости дорожных покрытий и способ его использования