Соединение, способ его получения и применение
Номер патента: 18326
Опубликовано: 30.07.2013
Авторы: Хоупис Иоаннес Николаос, Вертс Йохан Эрвин Эдмонд, Лан Иоланд Лидия, Лейс Карина, Стокбрукс Сигрид Карл Мария, Дикенс Йюлиус В.Й.












Формула / Реферат
1. Соединение формулы (XI)

его N-оксид, аддитивная соль или стереохимически изомерная форма.
2. Способ получения соединения по п.1, включающий взаимодействие промежуточного продукта формулы (IX) с промежуточным продуктом формулы (X) в присутствии подходящего растворителя

3. Применение соединения по п.1 в способе получения соединения формулы (XVIII), включающем:
а) взаимодействие промежуточного продукта формулы (VIII) с промежуточным продуктом формулы (XI) в присутствии подходящего растворителя

b) взаимодействие промежуточного продукта формулы (I) с промежуточным продуктом формулы (II) в подходящем растворителе с последующим восстановлением и образованием соли с получением промежуточного продукта формулы (XIII)

с) превращение промежуточного продукта формулы (XIII) нейтрализацией и гидролизом с помощью основания и подкислением соляной кислотой в промежуточный продукт формулы (XVII) и

d) взаимодействие промежуточного продукта формулы (XVII) с О-(тетрагидро-2Н-пиран-2-ил)гидроксиламином в присутствии соответствующих конденсирующих реагентов

4. Применение по п.3, в котором количество воды в промежуточном продукте (XVII) составляет от 15 до 25% об./об.
Текст
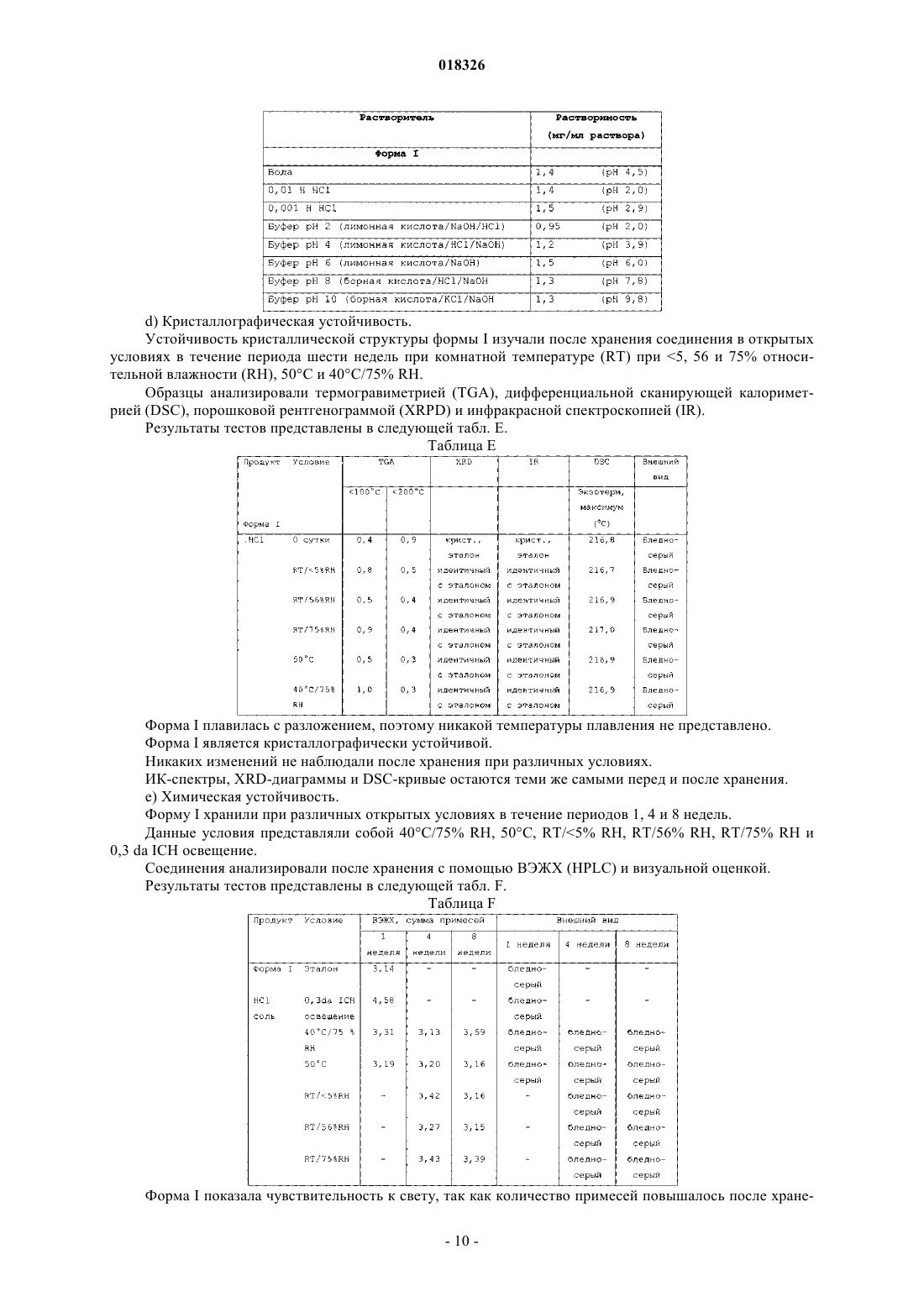
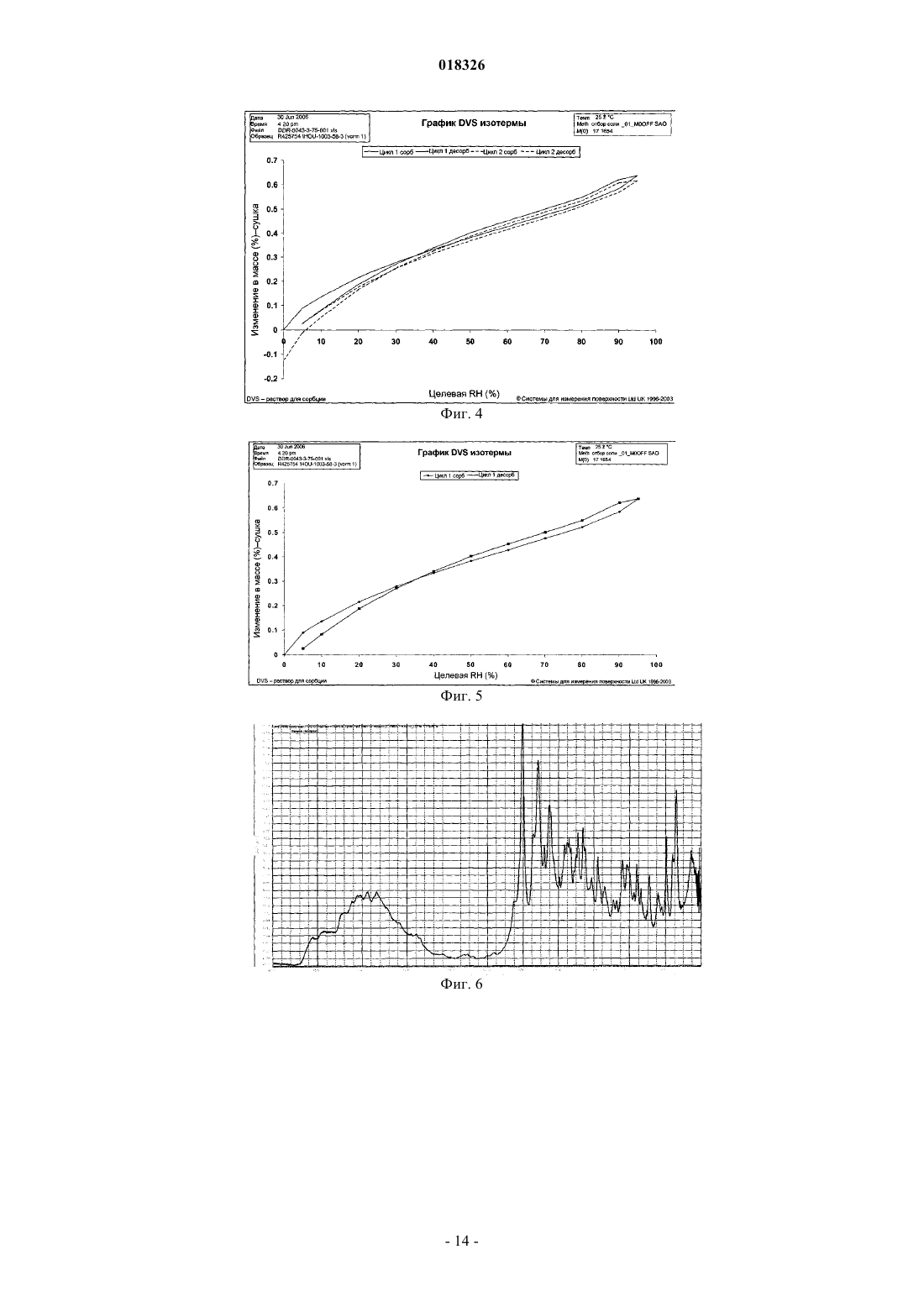
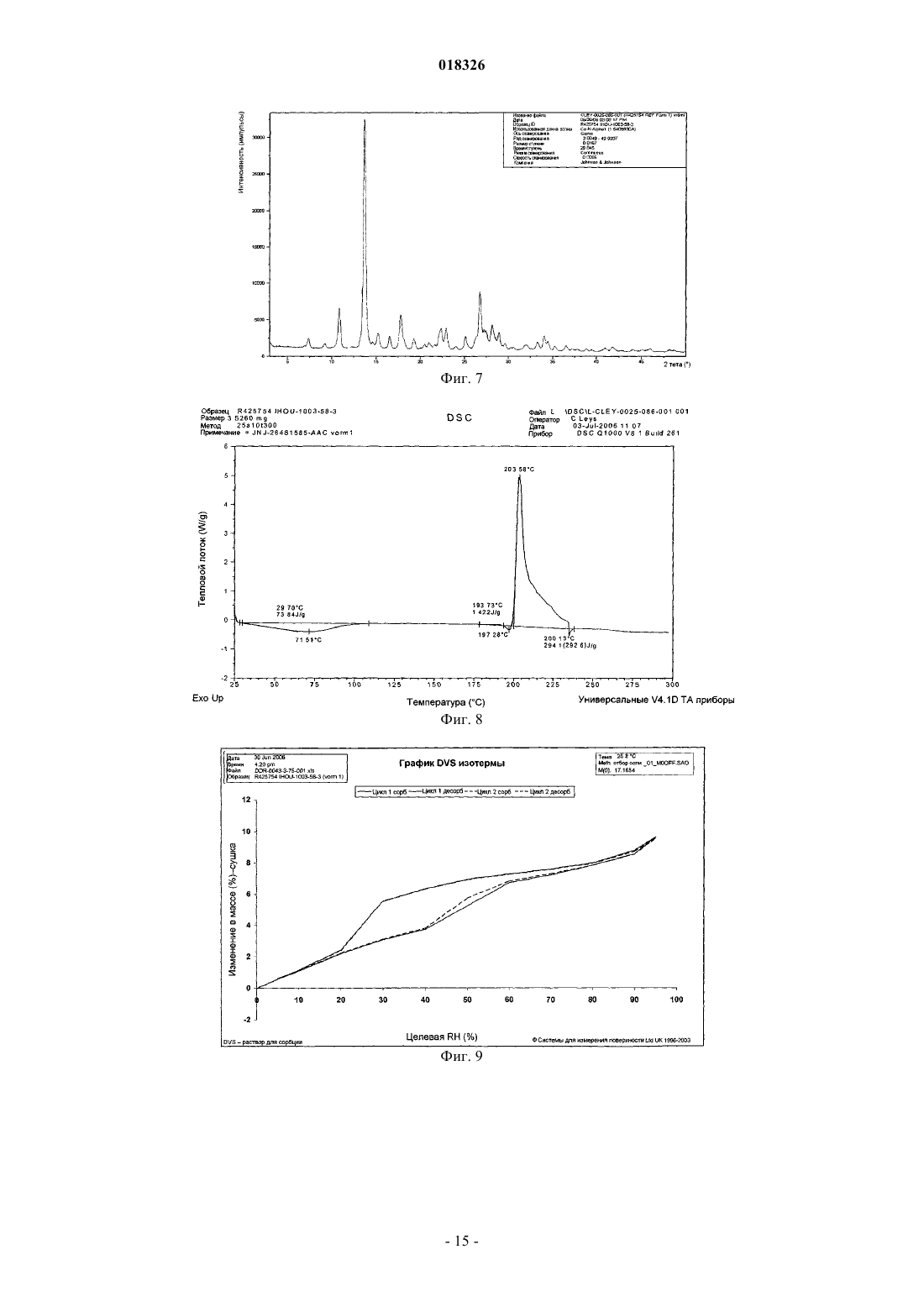
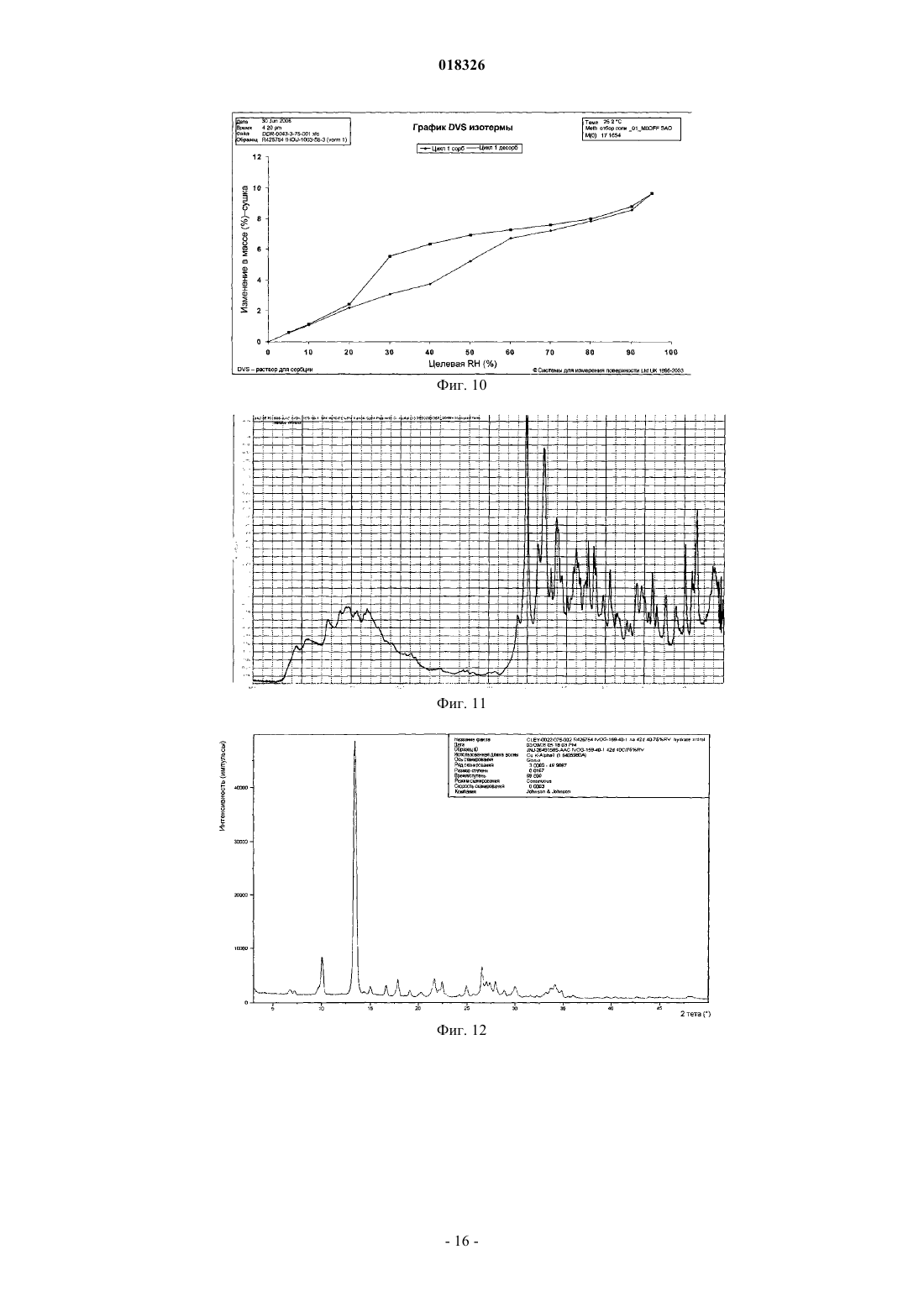
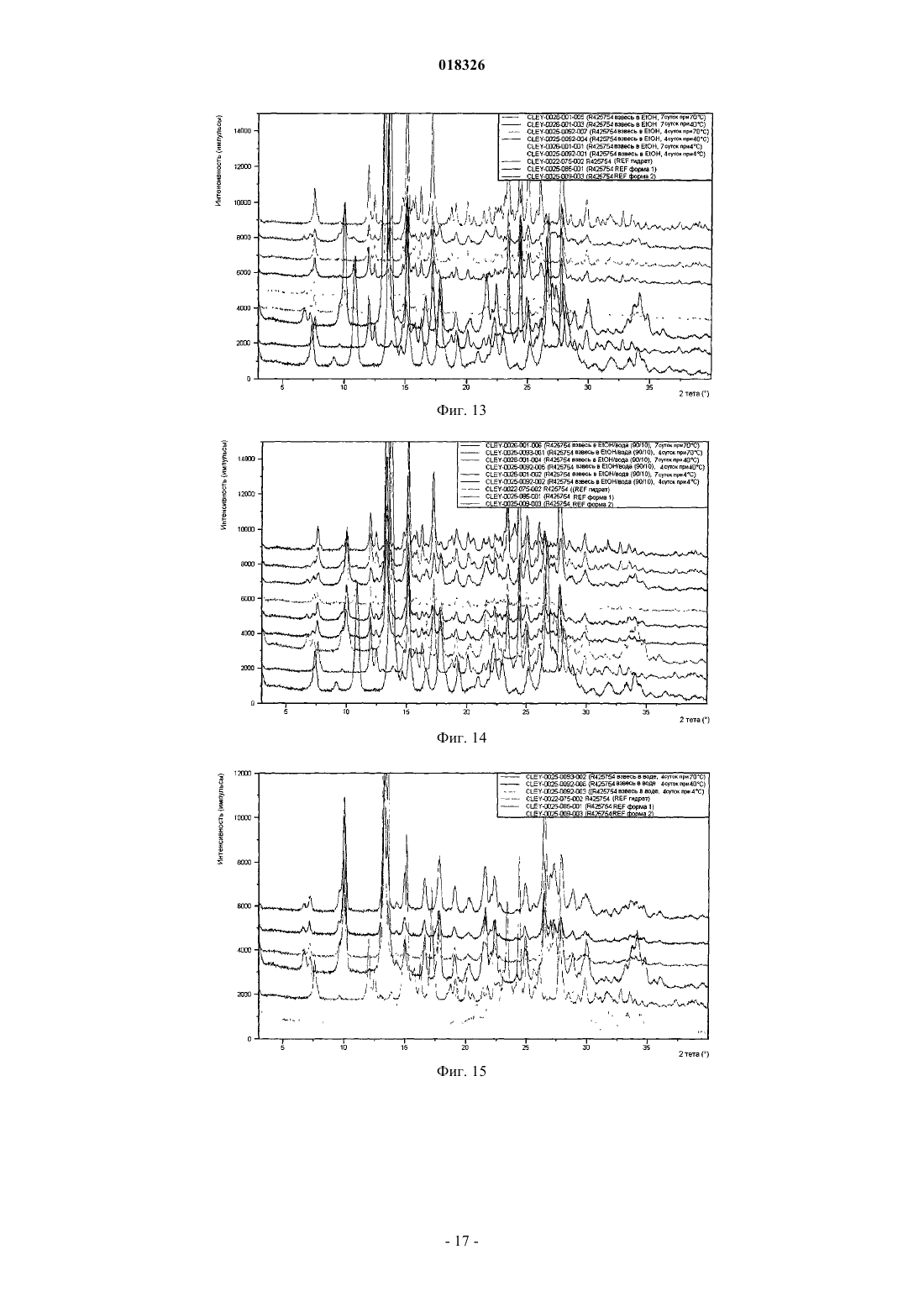
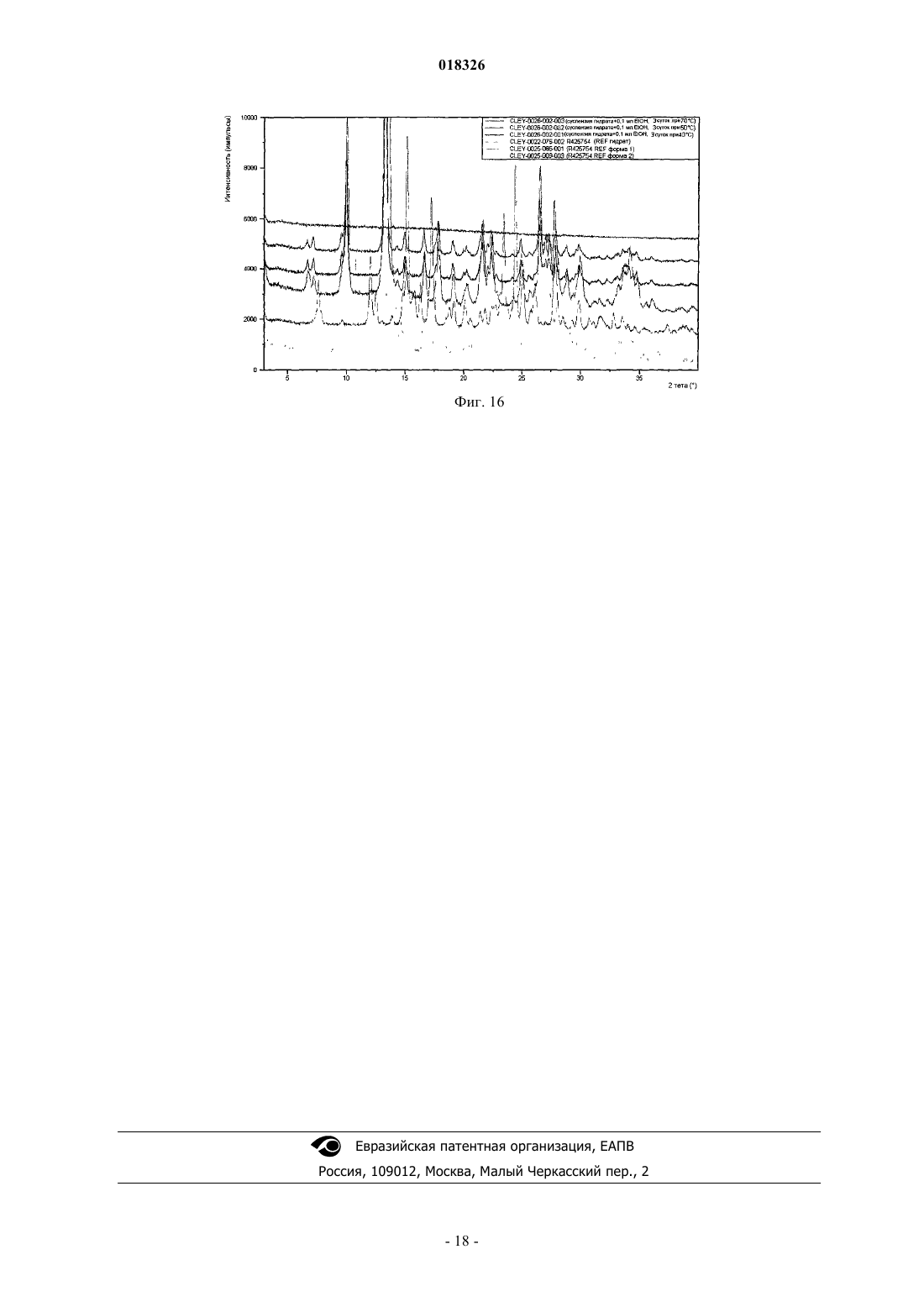
СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ Дикенс Йюлиус В.Й., Хоупис Иоаннес Николаос, Лан Иоланд Лидия, Лейс Карина, Стокбрукс Сигрид Карл Мария, Вертс Йохан Эрвин Эдмонд Изобретение также относится к способу получения соединения формулы (XI) и к применению данного соединения в способе получения моногидрохлорида соединения формулы (XVIII)(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) Область техники, к которой относится изобретение Данное изобретение относится к соединению формулы (XI) Изобретение также относится к способу получения соединения формулы (XI) и к применению данного соединения в способе получения моногидрохлорида соединения формулы (XVIII) которое может быть использовано для получения моно-HCl солей соли соединения JNJ-26481585,ингибитора гистондеацетилаз. Уровень техники Многие фармацевтические вещества могут существовать в разных физических формах, например в аморфной форме, в одной или нескольких кристаллических формах (например, безводные или сольватированные формы), в форме смеси различных кристаллических форм, или как смесь аморфной формы и кристаллической форм(ы). Аморфная форма представляет собой форму, в которой отсутствует трехмерная протяженная упорядоченная структура. В аморфной форме положения одной молекулы относительно другой являются в основном беспорядочными, т.е. без регулярного расположения молекул в форме решетки. Аморфные и неупорядоченные продукты часто имеют улучшенные свойства, но создание и стабилизация данного состояния может представлять различной кристаллической симметрии и/или параметрам ячейки. Полиморфы отличаются друг от друга по их физико-химическим параметрам, но не по их химическому составу. Полиморфизм обычно труден для контроля и представляет проблему для галенистов. Псевдополиморфы, также называемые сольватами, являются особым случаем твердых кристаллических форм, в которых присутствуют либо стехиометрические, либо нестехиометрические количества молекул растворителя или включены в решетчатую структуру соединения. Водный сольват также называется гидратом. Химия твердого состояния представляет интерес для фармацевтической промышленности и особенно для отраслей, вовлеченных в разработку подходящих лекарственных форм. Например, превращения твердого состояния могут серьезно влиять на устойчивость фармацевтических средств (срок годности). Метастабильная фармацевтическая твердая форма может переходить в кристаллическую структуру(например, из аморфной в кристаллическую) или сольват/десольват в ответ на изменения условий в окружающей среде или в течение времени. Различные кристаллические формы или аморфная форма того же самого лекарственного средства могут иметь существенные различия в таких фармацевтически важных свойствах, как скорости растворения, термодинамическая растворимость и биодоступность. Скорость растворения активного ингредиента в желудочном соке пациента может иметь терапевтические последствия, так как она задает верхний предел скорости, при которой перорально введенный активный ингредиент может достигать кровотока пациента. Скорость растворения, следовательно, принимается во внимание при изготовлении твердых лекарственных форм и жидких лекарственных средств, таких как сиропы и эликсиры. Аналогично, различные кристаллы или аморфная форма могут иметь разные свойства в отношении обработки, такие как гигроскопичность, текучесть, компактность, и тому подобное, которые могут влиять на их пригодность в качестве активных фармацевтических средств для коммерческого производства. В процессе клинической разработки лекарственных средств, если полиморфная форма не остается постоянной, то точная используемая или изучаемая лекарственная форма может быть несопоставима от одной партии с другой. Таким образом, желательно иметь способы для получения соединения с выбранной полиморфной формой высокой чистоты, когда данное соединение используют в клинических испытаниях или коммерческих продуктах, так как наличие примесей может давать нежелательные токсикологические эффекты. Некоторые полиморфные формы могут проявлять повышенную термодинамическую устойчивость или их можно легко производить с высокой степенью чистоты в больших количествах, и,таким образом, эти формы являются более подходящими для включения в фармацевтические препараты. Данное соединение представляет собой ингибитор гистондеацетилазы (HDAC).WO 2006/010750, опубликованная 2 февраля 2006, раскрывает аморфную форму соли JNJ-26481585.C2HF3O2 и ди-HCl соли и способы их получения. Синтез соли JNJ-26481585. C2HF3O2, первоначально описанной в WO 9721701, представлен на схеме 1. Здесь, на стадии 1 промежуточные продукты формулы (III) получали взаимодействием промежуточного продукта формулы (I) с карбоксальдегидом формулы (II) в присутствии тетрагидробората натрия в метаноле. На стадии 2 промежуточные продукты формулы (IV) получали взаимодействием промежуточного продукта формулы (III) с гидроксидом натрия в этаноле. На стадии 3 промежуточные продукты формулы (V) получали взаимодействием промежуточного продукта формулы (IV) с O-(тетрагидро-2 Н-пиран-2-ил)гидроксиламином в присутствии соответствующих реагентов, таких как N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамин, моногидрохлорид(EDC) и 1-гидрокси-1 Н-бензотриазол (НОВТ). Реакцию проводили в смеси дихлорметана и тетрагидрофурана. На стадии 4 C2HF3O2 соль гидроксамовой кислоты формулы (VI) получали взаимодействием промежуточного продукта формулы (V) с трифторуксусной кислотой. Указанную реакцию проводили в метаноле. Схема 1 В альтернативном случае соль JNJ-26481585. 2HCl, первоначально описанную в WO 97/21701, получали взаимодействием промежуточного продукта формулы (III) с гидроксиламином в присутствии гидроксида натрия. Указанную реакцию проводили в метаноле, дальнейшее превращение в ди-HCl соль проводили в этаноле. Способ, описанный в WO 2006/010750, не подходит для промышленного производства вследствие низких выходов и высокого количества примесей на различных стадиях способа, который, следовательно, требует несколько хроматографических стадий. Очистка соединений с применением хроматографии является дорогой и неблагоприятной для окружающей среды из-за необходимости применения растворителя и специализированного оборудования, требуемого для проведения промышленной хроматографии. Проблема, решаемая данным изобретением, состоит в том, чтобы предложить новые кристаллические формы моно-HCl соли и гидрата моно-HCl соли соединения JNJ-26481585. Другой аспект данного изобретения представляет собой способ, в котором получены новая кристаллическая HCl соль и гидратная форма HCl соли с высоким выходом и высокой чистоты. Полезные свойства данных HCl форм состоят в исключительных физико-химических свойствах, включающих их негигроскопичную природу и химическую устойчивость, обеспечивающую длительный срок годности данного соединения. Описание фигур Фиг. 1 представляет собой изображение инфракрасного (ИК/IR) спектра формы I. Фиг. 2 представляет собой изображение порошковой рентгенограммы (XPRD) формы I. Фиг. 3 представляет собой кривую дифференциальной сканирующей калориметрии (DSC) для формы I. Фиг. 4 представляет собой изменение массы формы I в качестве функции относительной влажности. Фиг. 5 представляет собой изображение кривой адсорбция-десорбция (ADS/DES) формы I. Фиг. 6 представляет собой изображение инфракрасного ИК (IR) спектра формы II. Фиг. 7 представляет собой изображение XPRD формы II. Фиг. 8 представляет собой кривую DSC формы II. Фиг. 9 представляет собой изменение массы формы II в качестве функции относительной влажности. Фиг. 10 представляет собой изображение кривой ADS/DES формы II. Фиг. 11 представляет собой изображение инфракрасного ИК (IR) спектра гидратной формы. Фиг. 12 представляет собой изображение XPRD гидратной формы. Фиг. 13 представляет собой наложение диаграмм XPRD при исследовании конверсии взвеси формыI и формы II в этаноле при разных температурах. Фиг. 14 представляет собой наложение диаграмм XPRD при исследовании конверсии взвеси формыI и формы II в смеси этанол/вода (90/10, об./об. %) при разных температурах. Фиг. 15 представляет собой наложение диаграмм XPRD при исследовании конверсии взвеси формыI и формы II в воде при разных температурах. Фиг. 16 представляет собой наложение диаграмм XPRD при исследовании конверсии взвеси гидрата в этаноле при разных температурах. Описание изобретения Получение промежуточных продуктов А. Получение промежуточного продукта формулы (I) а) Промежуточный продукт формулы (XI) может быть получен взаимодействием промежуточного продукта формулы (IX) с промежуточным продуктом формулы (X) в присутствии подходящего растворителя, таком как полярный или неполярный апротонный углеводородный растворитель, например толуол, метиленхлорид, изопропилацетат, этилацетат, тетрагидрофуран и тому подобное. В данном способе могут быть использованы другие ароматические или алифатические альдегиды. Данную реакцию можно также проводить в протонных растворителях, например метаноле, этаноле, изопропаноле и тому подобное. Реакцию можно проводить при температуре от 25 до 60 С, предпочтительно при температуре 45 С. Более высокие температуры не рекомендуются из-за потенциальной неустойчивости промежуточного продукта формулы (X).b) Промежуточный продукт формулы (VIII) может быть получен превращением промежуточного продукта формулы (VII) в присутствии подходящего окислителя, такого как мета-хлорпероксибензойная кислота (МСРВА), в подходящем растворителе, таком как полярный или неполярный апротонный углеводородный растворитель, например толуол, метиленхлорид, изопропилацетат, этилацетат, тетрагидрофуран и тому подобное. Реакцию можно проводить при температуре от -20 до 40 С, предпочтительно при температуре от 0 до 5 С, предпочтительнее при 0 С. При более высоких температурах метахлорпероксибензойная кислота не устойчива и промежуточный продукт формулы (XIII) может разлагаться. Полное превращение промежуточного продукта (VII) в промежуточный продукт (VIII) может быть обеспечено добавлением соответствующего количества МСРВА. Таким образом, количество МСРВА предпочтительно составляет 1 эквивалента. с) Промежуточный продукт формулы (I) может быть получен взаимодействием промежуточного продукта формулы (VIII) с промежуточным продуктом формулы (XI) в присутствии подходящего растворителя, такого как полярный или неполярный апротонный углеводородный растворитель или их смесь, например толуол, метиленхлорид, изопропилацетат, тетрагидрофуран, смесь диизопропилэтиламина или других третичных аминных оснований и этилацетата и тому подобное. Реакцию можно проводить при температуре от -20 до 40 С, предпочтительно при температуре от 0 до 5 С, предпочтительнее при 0 С с нагреванием вплоть до 25 С. Данный синтез с временной защитой аминопиперидина формулы (IX) с помощью пнитробензальдегида формулы (X) с образованием промежуточного продукта формулы (XI) создает возможность для предпочтительной реакции более замещенного кольцевого азота. Если данная защита не осуществлена, то образуются большие количества димера (А) и изомера (В), так как оба азота промежуточного продукта формулы (IX) будут реагировать с промежуточным продуктом формулы (VIII). Нагревание реакционной смеси в продолжение ночи обеспечивает полное взаимодействие промежуточного продукта формулы (XI) до промежуточного продукта формулы (I) и полное превращение любого оставшегося промежуточного продукта формулы (IX) в димер (А), который вместе с оставшейся МСРВА может быть легко удален при последующей кислотной обработке. Вариант осуществления данного изобретения включает промежуточный продукт формулы (XI). В. Получение промежуточного продукта формулы (XIII) а) Промежуточный продукт формулы (XII) может быть получен взаимодействием промежуточного продукта формулы (I) с промежуточным продуктом формулы (II) в подходящем растворителе. Реакцию можно проводить при температуре от 50 до 150 С, предпочтительно при температуре 110 С (нагревание с обратным холодильником при температуре кипения толуола). Для продолжения данной реакции требуется азеотропное удаление воды. В качестве растворителя можно использовать полярный или неполярный апротонный углеводородный растворитель, такой как толуол, изопропилацетат и тому подобное. Данные растворители хорошо образуют азеотроп с водой.b) Промежуточный продукт формулы (XII) обрабатывают тетраборатом натрия в подходящем растворителе, таком как полярный или неполярный апротонный и протонный углеводородный растворители и их смеси, например толуол, изопропилацетат, этанол, метанол, изопропанол и тому подобное. Восстановление тетраборатом натрия может происходить при температуре от 0 до 50 С, предпочтительно при 10 С. В течение восстановления предпочитают низкую температуру, чтобы избежать образования примесей с большей степенью восстановления.c) Затем получение соли осуществляют с фумаровой кислотой в смеси ацетон/этанол с 5% об./об. воды, с образованием промежуточного продукта (XIII). Вариант осуществления данного изобретения включает соль фумарата формулы (XIII). С. Получение промежуточного продукта формулы (XVIII). В первой попытке найти улучшенный способ синтеза для получения JNJ-26481585, промежуточный продукт формулы (III) подвергали взаимодействию с О-(тетрагидро-2 Н-пиран-2-ил)гидроксиламином, в присутствии основания и растворителя без конденсирующего реагента. Данная попытка была неуспешной. Во второй попытке стремились защитить группировки амино и гидроксамовой кислоты кислотолабильными защитными группами, чтобы повлиять на самопроизвольное снятие защиты-образование соли. Поэтому промежуточный продукт формулы (XIII) превращали в свободное основание с получением промежуточного продукта формулы (III) и затем превращали в промежуточный продукт формулы (XIV),где R представляет собой третичный бутил, бензил или фульвенил, с последующим гидролизом с помощью NaOH в этаноле и выделением промежуточного продукта формулы (XV) путем подкисления и кри-4 018326 сталлизации непосредственно из реакционной смеси. Конденсация промежуточного продукта формулы (XV) с O-(тетрагидро-2 Н-пиран-2-ил)гидроксиламином при стандартных условиях аминокислотной конденсации (EDC, НОВТ, триэтиламин, тетрагидрофуран) давала промежуточный продукт формулы (XVI) с отличным выходом. Реакция конденсации для промежуточных продуктов формулы (XV), где R представляет собой фульвенил, вызывает некоторое отщепление фульвенильной группы из-за триэтиламина, необходимого для оптимальной конденсации. Затем были предприняты попытки снятия защиты с промежуточного продукта формулы (XVI), гдеR представляет собой третичный бутил, при разных условиях (растворители: этанол, этилацетат, толуол,ацетон, метилизобутилкетон, диметилформамид; кислоты: этансульфоновая кислота, метансульфоновая кислота, хлороводородная кислота, трифторуксусная кислота). К сожалению, как при температуре окружающей среды (2-3 ч), так и при 50 С (10 мин) единственные наблюдаемые продукты представляли собой продукты от расщепления индольной группировки. Гидрогенолиз промежуточного продукта формулы (XVI), где R представляет собой бензил, был осуществлен в атмосфере водорода и в присутствии соответствующего катализатора, такого как, например, палладий на угле, и был признан неуспешным из-за конкурирующего разрыва (вплоть до 20%) N-O связи в продукте с гидроксамовой кислотой. С другой стороны, отщепление фульвенильной группы от промежуточного продукта формулы (XVI), где R представляет собой фульвенил, называемого в данной работе как промежуточный продукт формулы (XVI-a), в мягких условиях и улавливание фульвенового побочного продукта использованием тиолсиликагеля было успешным. Соответствующий свободный амин формулы (V) может дать соединение формулы (XIX) в аналогичных условиях (1,05 эквивалентов хлороводородной кислоты, этанол, 70 С), как описано ниже. а) Наконец, свободное основание промежуточного продукта (XIII) может быть получено нейтрализацией с помощью водного гидроксида натрия и экстракцией в метилтетрагидрофуран. Органический слой, содержащий свободное основание, затем подвергали основному гидролизу с 3 мольными эквивалентами гидроксида натрия в воде при кипячении с обратным холодильником. Натриевую соль в водном слое затем отделяли от метилтетрагидрофуранового слоя и подкисляли 5 мольными эквивалентами HCl при 10 С. Промежуточный продукт (XVII) может иметь различное содержание воды. Сразу после сушки содержание воды составляет 0,7%. Когда образец оставляли на 24 ч при атмосферных условиях, содержание воды повышалось и устанавливалось при 8%, что представляет 2 моля воды.b) Влияние воды в промежуточном продукте (XVII) является решающим, так как ниже приведенная реакция конденсации требует присутствия определенного количества воды для успешного прохождения. Количество воды в промежуточном продукте (XVII) предпочтительно находится в интервале от 15 до 25% об./об., наиболее предпочтительно равно примерно 16% об./об. Промежуточные продукты формулы (XVIII) могут быть получены взаимодействием промежуточного продукта формулы (XVII) с O-(тетрагидро-2 Н-пиран-2-ил)гидроксиламином в присутствии соответствующих реагентов, таких как N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамин, моногидро-5 018326 хлорид (EDC) в качестве конденсирующего реагента. Реакция может быть проведена в полярных или неполярных апротонных и протонных углеводородных растворителях и их смесях, например, таких как метилтетрагидрофуран, диметилформамид (ДМФА/DMF), дихлорметан (DCM), толуол, изопропанол,этанол, ацетонитрил, этилацетат, изопропанолацетат, их смесь, и смесь одного или нескольких разных растворителей с водой, предпочтительно смесь этилацетата и этанола, предпочтительнее смесь этилацетата, этанола и воды. Температура в течение реакции может принимать значения в интервале 10-40 С,предпочтительно комнатную температуру. Реакция является быстрой и полной, когда присутствует достаточно воды. Реакция проходит медленнее при сухих условиях, присутствует больше примесей и скорость реакции по направлению к продукту снижается. с) Промежуточный продукт формулы (XVIII) может быть растворен в растворителе, таком как диметилформамид или диметилацетамид, предпочтительно диметилацетамид. Добавление со-растворителя предоставляет продукту возможность кристаллизоваться. Могут быть использованы со-растворители,такие как ацетон, метилизобутилкетон или метилэтилкетон, предпочтительно метилизобутилкетон. Очистка может быть проведена при температуре в интервале 25-90 С, предпочтительно в интервале от 50 до 70 С. Время кристаллизации не должно быть больше чем 5 ч. При более высоких температурах или более продолжительных периодах кристаллизации выход конечного продукта снижается. Перекристаллизация может быть проведена в растворителе, таком как этанол, в присутствии со-растворителя, такого как метилэтилкетон, при температуре от 50 до 70 С, предпочтительно при 70 С. Экспериментальная часть Следующие примеры предназначены для иллюстрации данного изобретения и не для его ограничения. Пример 1. Получение промежуточного продукта (I)a) 4-Пиперидинметанамин (2,6 моль) и этилацетат (5,2 л) загружали в инертный реактор (20 л) и нагревали вплоть до 45 С. 4-Нитробензальдегид (2,7 моль) добавляли и реакционную смесь перемешивали в течение 2 ч при 45 С. Реакционную смесь охлаждали при 0 С и добавляли диизопропилэтиламин (6,6 моль), получая раствор 1.b) Этиловый эфир 2-(метилтио)-5-пиримидинкарбоновой кислоты (2,7 моль) и этилацетат (2,6 л) помещали в инертный реактор и охлаждали до 0 С. Раствор мета-хлорфпероксибензойной кислоты (1,2 моль) в этилацетате (2,6 л) добавляли в течение 1 ч при температуре от 0 до 5 С. Реакционную смесь перемешивали в течение 30 мин при 0 С, получая раствор 2.c) Раствор 2 добавляли к раствору 1 в течение 1 ч при температуре от 0 до 5 С. Реакционную смесь оставляли на ночь при комнатной температуре. Смесь подкисляли до рН 2 раствором 640 мл концентрированной хлороводородной кислоты в 10 л воды. Водный слой отбирали и промывали 1 л этилацетата. Водный слой собирали и к нему добавляли 1 л дихлорметана. Смесь подщелачивали до рН 10 с помощью 450 мл 50% гидроксида натрия. Смесь перемешивали в течение 30 мин при комнатной температуре. Органический слой собирали, получая фракцию 1. Водный слой затем экстрагировали 2 л дихлорметана и органический слой собирали, получая фракцию 2. Фракцию 1 и 2 объединяли и дихлорметан выпаривали с получением 511,25 г (1,93 моль) промежуточного продукта (I) (выход 74%). Пример 2. Получение промежуточного продукта (XIII)a) Промежуточный продукт (I) (0,97 моль) и толуол (4,5 л) добавляли в инертный реактор (20 л). 1-Метил-1 Н-индол-3-карбоксальдегид (0,97 моль) добавляли в реакционную смесь при комнатной температуре. Реакционную смесь нагревали при температуре кипения с обратным холодильником, кипятили с обратным холодильником в течение ночи и охлаждали до комнатной температуры. Добавляли этанол (1,5 л) с примесью метанола, получая раствор 3.b) Тетрагидроборат натрия (55,2 г) и толуол (1,5 л) добавляли в инертный реактор (20 л). Смесь доводили до 10 С при непрерывном перемешивании. К смеси добавляли раствор 3 в течение 1 ч при температуре 10 С. Смесь перемешивали в течение 1 ч при температуре 10 С. Реакционную смесь доводили до комнатной температуры. Добавляли ацетон (8,79 моль) в течение 30 мин. Реакционную смесь перемешивали в течение 4 ч. В реакционную смесь закапывали воду (5,1 л) в течение 15 мин. Реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Водный слой не учитывали, и органический слой промывали два раза раствором 300 г бикарбоната натрия в 4,1 л воды. Органический слой фильтровали над сульфатом магния и выпаривали с получением фракции 3 (397 г остатка после выпаривания). С) Этанол (5 л) с примесью 2% метилэтилкетона добавляли к фракции 3 при комнатной температуре. Концентрированный ацетон (5 л) и 0,5 л воды добавляли при комнатной температуре и смесь затем нагревали до 50 С. Готовили смесь фумаровой кислоты (0,97 моль), этанола (1,4 л) с добавкой 2% метилэтилкетона, ацетона (1,4 л) и 140 мл воды с получением раствора 4. Раствор 4 добавляли к реакционной смеси в течение 2 ч при температуре 50 С. Реакционную смесь перемешивали в течение 2 ч при 50 С,охлаждали в течение 4 ч при комнатной температуре и перемешивали в течение ночи при комнатной температуре. Осадок собирали и затем промывали с помощью 1,4 л этанола с примесью 2% метилэтилкетона, 1,4 л концентрированного ацетона и 140 мл воды. Осадок сушили в продолжение ночи при 50 С,получая 371 г (0,7 моль) промежуточного продукта (XIII) (выход 73%). Пример 3. Получение промежуточного продукта (XVII) Четырехгорлую колбу (2 л) загружали промежуточным продуктом (XIII) (100 г; 191,0 ммоль). Добавляли воду (3 л/моль чистого лимитирующего реагента; 573,0 мл) и 2-метилтетрагидрофуран (2,2 л/моль чистого лимитирующего реагента; 420,2 мл). После перемешивания добавляли 50% гидроксид натрия (2,5 моль/моль чистого лимитирующего реагента; 477,5 ммоль; 25,13 мл). Затем реакционную смесь перемешивали в течение 40-60 мин при комнатной температуре, после чего реакционную смесь оставляли для отстаивания. Верхний органический слой собирали и промывали водой (2 л/моль чистого лимитирующего реагента; 382,0 мл). Воду (1,5 л/моль чистого лимитирующего реагента; 286,5 мл) и гидроксид натрия (3 моль/моль чистого лимитирующего реагента; 573,0 ммоль; 45,84 г) добавляли к органическому слою. Реакционную смесь нагревали до 80 С и перемешивали в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры и собирали нижний водный слой. Добавляли изопропиловый спирт (90 мл; 1,177 моль) и смесь охлаждали до 10 С на ледяной бане. Реакционную смесь подкисляли концентрированной хлороводородной кислотой (5 моль/моль чистого лимитирующего реагента; 954,9 ммоль; 100,9 г) до рН 1 (рН 13,8: темно-зеленый раствор; рН 7,5: темно-красный раствор; рН 4,7: розовый раствор, белый осадок). Реакционную смесь перемешивали в течение 4 ч при 10 С. Белый осадок фильтровали, промывали 4 раза водой и сушили в вакууме при 40 С, получая 84 г промежуточного продукта (XVII) (выход: 97%). Пример 4. Получение промежуточного продукта (XVIII)a) В четырехгорлую колбу (1 л) помещали 0,093 моль промежуточного продукта (XVII) и добавляли 220 мл этилацетата. Реакционную смесь перемешивали и к ней добавляли 5 мл воды, получая раствор 5. В 250-мл колбу помещали 0,122 моль N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамин моногидрохлорида (EDC) в 130 мл этанола и реакционную смесь перемешивали, получая раствор 6. О(Тетрагидро-2 Н-пиран-2-ил)гидроксиламин (0,123 моль) добавляли к раствору 5 и воронку для добавления промывали 26 мл этилацетата. Сразу после этого 200 мл раствора 6 добавляли в реакционную смесь,содержащую раствор 5, в течение 1 ч 30 мин (реакционная смесь становилась гомогенной, когда было добавлено 90% раствора 6, затем требуемый продукт кристаллизовался). Реакционную смесь перемешивали в течение 5 ч при комнатной температуре. Осадок фильтровали и промывали 55 мл этилацетата и сушили в вакууме при 50 С в течение 16 ч, получая 35,7 г (0,07 моль) промежуточного продукта (XVIII)b) В четырехгорлую колбу (1 л) помещали 0,073 моль промежуточного продукта (XVIII) в атмосфере азота. Добавляли N,N-диметилацетамид (377 мл) и 377 мл метилизобутилкетона и смесь нагревали до 70 С. Реакционную смесь перемешивали в течение 5 ч при 70 С, затем охлаждали в течение 1 ч при 25 С и затем перемешивали в течение другого часа при 25 С. Осадок фильтровали и затем промывали 94 мл смеси N,N-диметилацетамида и метилизобутилкетона, потом 150 мл метилизобутилкетона во взвешенной промывочной воде и затем 150 мл метилизобутилкетона в замененной промывочной воде. Осадок(XVIII) (выход: 89%). Пример 5. Получение кристаллической формы I HCl соли соединения JNJ-26481585a) В инертную четырехгорлую колбу (0,5 л) помещали 0,03 моль очищенного промежуточного продукта (XVIII). Добавляли этанол (300 мл) (обычное содержание воды равно 0,07% (мас./мас Реакционную смесь перемешивали и нагревали до 57-60 С. Вносили как затравку 30 мг формы I промежуточного продукта (XVIII) JNJ-26481585 HCl соли. В реакционную смесь при 57 С добавляли концентрированную хлороводородную кислоту (0,05 мол.%) и реакционную смесь перемешивали в течение 16 ч. Осадок фильтровали при 50 С и промывали 3 раза 20 мл этанола, получая 10 г кристаллической формы I JNJ26481585 HCl соли.b) В инертную четырехгорлую колбу (50 мл) помещали 2,6 г кристаллической формы I JNJ26481585 HCl соли, полученной на стадии а). Добавляли этанол (20 мл). Реакционную смесь перемешивали в азоте и темноте и нагревали до 50 С. Реакционную смесь перемешивали в течение 12 ч при 50 С,охлаждали при 40 С в течение 1 ч и фильтровали. Осадок промывали один раз 20 мл этанола и два раза 20 мл ацетона. Затем продукт сушили при 50 С в вакууме в течение 16 ч с получением 2 г (80%) очищенной кристаллической формы I JNJ-26481585 HCl соли. Пример 6. Превращение смеси полиморфа I и II при использовании процедуры суспендирования. а) Получение взвесей. Примерно 25 мг формы I и примерно 25 мг формы II взвешивали в сосуде. Примерно 0,2 мл этанола добавляли и сосуд закрывали. Готовили три сосуда и каждый сосуд хранили в течение 4 суток при различной температуре, при 4 С (холодильник), 40 и 70 С. Данную процедуру повторяли для взвесей в смеси этанол/вода (90/10, % об./об.) и в воде. Взвеси хранили в течение 4 суток и 7 суток при различных температурах. После хранения сосуд открывали и образец сушили распределением нескольких мг взвеси на бумажном фильтре.b) Аналитические способы (порошковая XRD). Все полученные фракции анализировали, применяя порошковую XRD. Анализы порошковой рентгенограммы (XPRD) проводили на Philips X"PertPRO MPD дифрактометре PW3050/60 с генератором PW3040. Прибор был снабжен Cu LFF трубкой для рентгеновских лучейPW3373/00. Соединение распределяли на держателе образца с нулевым фоном. Параметры приборов Результаты, полученные в исследованиях превращений взвесей через 4 и 7 суток хранения в этаноле, собраны в следующей табл. А. Таблица АXRD диаграмма сольватированной формы сравнима с XRD диаграммой гидрата. Результаты, полученные в исследованиях превращений взвесей через 4 и 7 суток хранения в смеси этанол/вода (90/10, об./об.%), собраны в следующей табл. В. Таблица В Растворитель, присутствующий во взвеси, полностью испарился. Через 4 суток хранения к смеси снова добавляли 0,2 мл растворителя. Результаты, полученные в исследованиях превращений взвесей через 4 суток хранения в воде, собраны в следующей табл. С. Таблица С Данные гидратные образцы (за исключением взвесей в воде) хранили в течение 3 суток с 0,1 мл этанола при разных температурах, 40, 50 и 70 С. Гидратные образцы, хранившиеся в течение 3 суток при 40 и 50 С, оставались гидратом. Гидратный образец, хранившийся в течение 3 суток при 70 С, был полностью сжижен (масло). Пример 7. Устойчивость формы I. а) Информация о соединении. Графическая формула:b) Исследования адсорбция/десорбция. Адсорбцию и десорбцию воды при 25 С в различных условиях относительной влажности исследовали на 17 мг формы I. Регистрировали изменение массы как функцию относительной влажности. Результаты изображены на фиг. 4. Фракция формы I адсорбирует вплоть до 0,6% воды при высокой относительной влажности, она не показала гигроскопического поведения и оставалась кристаллической в течение теста. с) Растворимость. Водные растворимости формы I определяли в растворителях с различными рН. Избыток растворенного вещества приводили в равновесие растворителем при 20 С в течение 24 ч. После удаления нерастворенного соединения концентрацию в растворе определяли, используя ИК-спектрометрию. Растворимости перечислены в следующей табл. D. Таблица Dd) Кристаллографическая устойчивость. Устойчивость кристаллической структуры формы I изучали после хранения соединения в открытых условиях в течение периода шести недель при комнатной температуре (RT) при 5, 56 и 75% относительной влажности (RH), 50 С и 40 С/75% RH. Образцы анализировали термогравиметрией (TGA), дифференциальной сканирующей калориметрией (DSC), порошковой рентгенограммой (XRPD) и инфракрасной спектроскопией (IR). Результаты тестов представлены в следующей табл. Е. Таблица Е Форма I плавилась с разложением, поэтому никакой температуры плавления не представлено. Форма I является кристаллографически устойчивой. Никаких изменений не наблюдали после хранения при различных условиях. ИК-спектры, XRD-диаграммы и DSC-кривые остаются теми же самыми перед и после хранения. е) Химическая устойчивость. Форму I хранили при различных открытых условиях в течение периодов 1, 4 и 8 недель. Данные условия представляли собой 40 С/75% RH, 50C, RT/5% RH, RT/56% RH, RT/75% RH и 0,3 da ICH освещение. Соединения анализировали после хранения с помощью ВЭЖХ (HPLC) и визуальной оценкой. Результаты тестов представлены в следующей табл. F. Таблица F Форма I показала чувствительность к свету, так как количество примесей повышалось после хране- 10018326b) Исследование адсорбция/десорбция. Адсорбцию и десорбцию воды при 25 С в различных условиях относительной влажности исследовали на примерно 24 мг формы II. Регистрировали изменение массы как функцию относительной влажности. Результаты изображены на фиг. 9. В течение начальной стадии сушки регистрируют для формы II 1,67% потери массы. Полученный высушенный продукт был гигроскопичным. Он адсорбировал вплоть до 9,6% воды при высокой относительной влажности. Продукт высыхал полностью в течение цикла десорбции и оставался кристаллическим в течение теста.d) Кристаллографическая устойчивость. Устойчивость кристаллической структуры формы II изучали после хранения соединения в открытых условиях в течение периода шести недель при комнатной температуре (RT) при 5, 56 и 75% относительной влажности (RH), 50 С и 40 С/75% RH. Образцы анализировали термогравиметрией (TGA), дифференциальной сканирующей калориметрией (DSC), порошковой рентгенограммой (XRPD) и инфракрасной спектроскопией (IR). Результаты тестов представлены в следующей табл. G. Таблица GRef: идентичный с эталоном Форма II плавилась с разложением, поэтому никакой температуры плавления не представлено. Дополнительный эндотермический сигнал в DSC-кривой был обусловлен испарением растворителя. Форма II является кристаллографически неустойчивой. Наблюдали изменения после хранения при различных условиях влажности. ИК-спектры и XRD-диаграммы отличаются от исходного продукта после хранения при RT/56% RH,RT/75% RH и 40 С/75% RH состоянии. Изменения после хранения при RT/56% RH, RT/75% RH и 40C/75% RH состоянии были обусловлены поглощением воды. е) Химическая устойчивость Форму II хранили при различных открытых условиях в течение периодов 1, 4 и 8 недель. Данные условия представляли собой 40 С/75% RH, 50C, RT/5% RH, RT/56% RH, RT/75% RH и 0,3 da ICH освещение. Соединения анализировали после хранения с помощью ВЭЖХ (HPLC) и визуальной оценкой. Результаты тестов представлены в следующей табл. Н. Таблица Н Исследование химической устойчивости R425754 предоставило следующие наблюдения.R425754 показала чувствительность к свету, так как количество примесей повышалось после хранения в 0,3 da ICH условиях освещения. Также наблюдается изменение цвета от белого до оранжево-коричневого после хранения при 0,3 daICH освещении и от белого до розового после хранения в условиях влажности RT/56% RH, RT/75% RH и 40 С/75% RH и повышенной температуре в 50 С. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (XI) его N-оксид, аддитивная соль или стереохимически изомерная форма. 2. Способ получения соединения по п.1, включающий взаимодействие промежуточного продукта формулы (IX) с промежуточным продуктом формулы (X) в присутствии подходящего растворителя 3. Применение соединения по п.1 в способе получения соединения формулы (XVIII), включающем: а) взаимодействие промежуточного продукта формулы (VIII) с промежуточным продуктом формулы (XI) в присутствии подходящего растворителяb) взаимодействие промежуточного продукта формулы (I) с промежуточным продуктом формулы(II) в подходящем растворителе с последующим восстановлением и образованием соли с получением промежуточного продукта формулы (XIII) с) превращение промежуточного продукта формулы (XIII) нейтрализацией и гидролизом с помощью основания и подкислением соляной кислотой в промежуточный продукт формулы (XVII) иd) взаимодействие промежуточного продукта формулы (XVII) с О-(тетрагидро-2 Н-пиран-2-ил)гидроксиламином в присутствии соответствующих конденсирующих реагентов 4. Применение по п.3, в котором количество воды в промежуточном продукте (XVII) составляет от 15 до 25% об./об.
МПК / Метки
МПК: A61K 31/506, C07D 211/26, C07D 401/14, A61P 35/00
Метки: способ, применение, соединение, получения
Код ссылки
<a href="https://eas.patents.su/19-18326-soedinenie-sposob-ego-polucheniya-i-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Соединение, способ его получения и применение</a>
Циклическое пептидное соединение и его применение, бициклопептидное соединение, пептидное соединение и его применение, фармацевтическая композиция

Номер патента: 2819
Опубликовано: 31.10.2002
Авторы: Кондон Стефен М., Мориз Изабелль
МПК: A61K 38/12, C07K 7/64, A61P 19/00...
Метки: композиция, фармацевтическая, применение, соединение, бициклопептидное, пептидное, циклическое
Формула / Реферат:
1. Циклическое пептидное соединение формулы X-A10-A11-A12-A13-A14-A15-A16-A17-A18-A19-A20-A21-A22-A23-A24-A25-A26-A27-Y, или его фармацевтически приемлемая соль, или пролекарственный предшественник, где Х выбирают из группы, состоящей из (a) R1a-Ao-A1-A2-A3-A4-A5-A6-A7-A8-A9-, (b) R1a-А2-А3-А4-А5-А6-А7-А8-А9-, (c) R1b-А3-А4-А5-А6-А7-А8-А9-, (d) R1a-A4-A5-A6-A7-A8-A9-, (e) R1a-A5-A6-A7-A8-A9-, (f) R1a-А6-А7-А8-А9-, (g) R1a-A7-A8-A9-, (h)...
Железо-декстрановое соединение для применения в качестве компонента терапевтической композиции для профилактики или лечения дефицита железа, способ получения указанного железо-декстранового соединенияи применение указанного соединения для изготовления терапевтической композиции для парентерального введения

Номер патента: 3427
Опубликовано: 24.04.2003
Авторы: Андреасен Ханс Берг, Христенсен Ларс
МПК: A61K 47/48, A61P 7/06
Метки: железа, применение, компонента, соединенияи, терапевтической, введения, получения, дефицита, железо-декстрановое, применения, лечения, способ, изготовления, парентерального, железо-декстранового, соединения, качестве, соединение, композиции, указанного, профилактики
Формула / Реферат:
1. Соединение железо-декстран для применения в производстве терапевтической композиции для профилактики или лечения дефицита железа у животного или человека путем парентерального введения, содержащее гидрированный декстран со среднемассовой молекулярной массой (СМММ) от 700 до 1400 Да, предпочтительно около 1000 Да, среднечисловой молекулярной массой (СЧММ) от 400 до 1400 Да, где 90 мас.% декстрана имеет молекулярные массы менее 2700 Да, а СМММ...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Систи Николас Дж., Чандер Мадхави С., Свинделл Чарльз С.
МПК: C07D 305/14
Метки: соединения, получения, паклитаксела, промежуточное, способ, промежуточного, соединение
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Способ получения стробилуринового фунгицида, соединение и композиция

Номер патента: 13635
Опубликовано: 30.06.2010
Авторы: Васс Джек, Уиттон Алан Джон, Бойд Эван Кэмпбелл
МПК: C07C 59/64, C07D 239/52
Метки: соединение, композиция, получения, стробилуринового, фунгицида, способ
Формула / Реферат:
1. Способ получения соединения формулы (I)который включает по выбору(а) взаимодействие соединения формулы (II)с 2-цианофенолом или его солью в присутствии от 0,1 до 2 мол.% 1,4-диазабицикло[2.2.2]октана или(b) взаимодействие соединения формулы (III)с соединением формулы (IV)в присутствии от 0,1 до 2 мол.% 1,4-диазабицикло[2.2.2]октана;где W представляет собой метил-(Е)-2-(3-метокси)акрилатную группу С(СО2СН3)=СНОСН3, или...
Промежуточное соединение и способ получения обогащенных β-аномером 21-дезокси-21, 21-дифтор-d-рибофуранозилнуклеозидов

Номер патента: 11558
Опубликовано: 28.04.2009
Авторы: Панда Биджан Кумар, Бхатт Дипендра, Маикап Голак Чандра
МПК: C07H 13/08, C07H 19/073
Метки: получения, соединение, 21-дезокси-21, обогащенных, промежуточное, beta;-аномером, 21-дифтор-d-рибофуранозилнуклеозидов, способ
Формула / Реферат:
1. Соединение формулы (I) где Р представляет собой водород или гидроксизащитную группу. 2. Соединение формулы (I) по п.1, где защитную группу Р выбирают из формила, 2-хлорацетила, бензила, дифенилметила, трифенилметила, 4-нитробензила, феноксикарбонила, третичного бутила, метоксиметила, тетрагидропиранила, аллила, тетрагидротиенила, 2-метоксиэтоксиметила, метоксиацетила, феноксиацетила, изобутирила, этоксикарбонила, бензилоксикарбонила, мезила,...
Предыдущий патент: Растения brassica oleracea с устойчивостью к albugo candida и их получение
Следующий патент: Способ химической обработки поверхностей полупроводников и устройство для его выполнения
Случайный патент: Способ стабилизации водного раствора производного пиразолоакридона