Феноксиэтилпиперидиновые соединения














Формула / Реферат
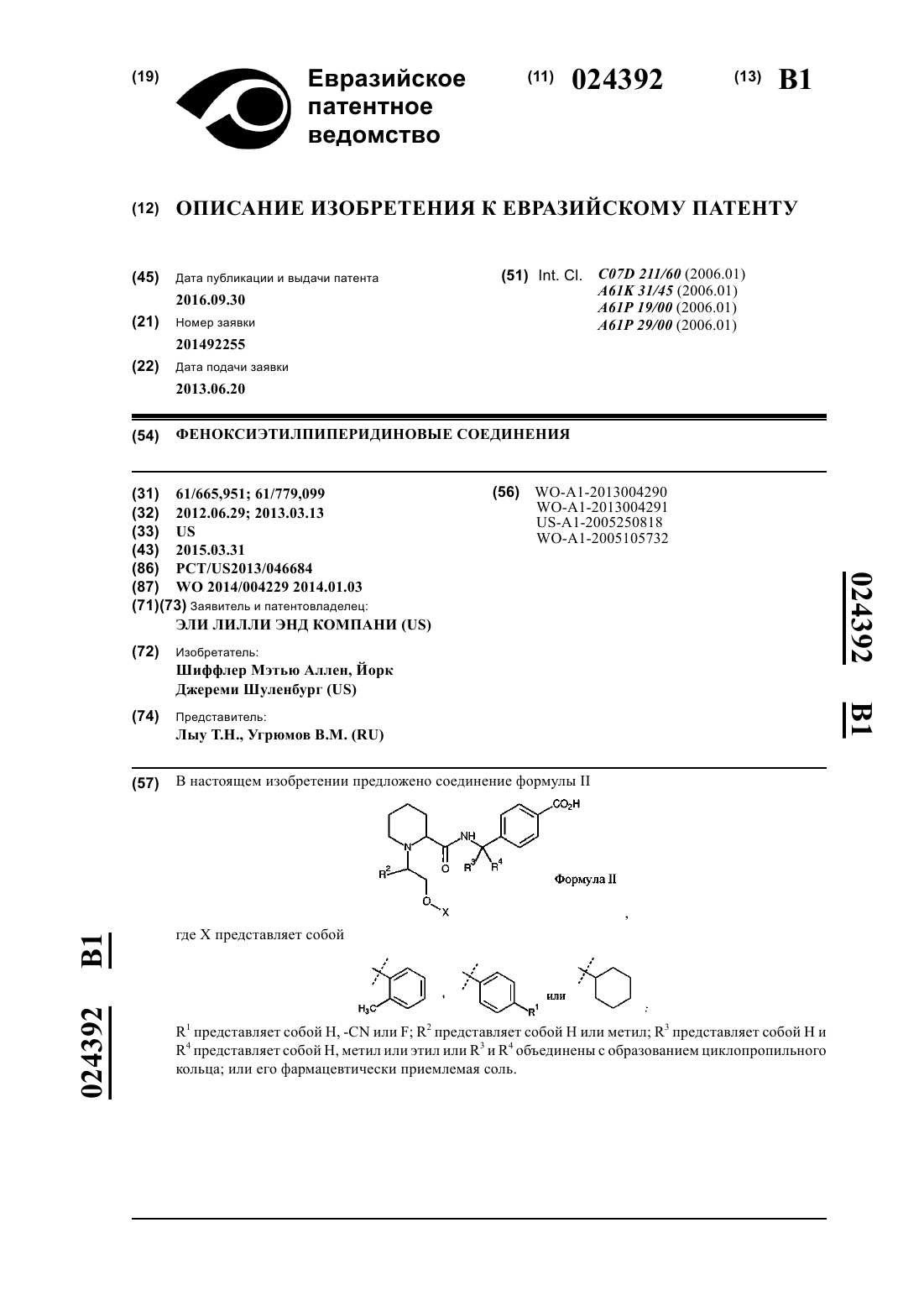
1. Соединение формулы

где X представляет собой

R1 представляет собой H, -CN или F;
R2 представляет собой H или метил;
R3 представляет собой H и
R4 представляет собой H, метил или этил или
R3 и R4 объединены с образованием циклопропильного кольца;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что R2 представляет собой Н.
3. Соединение по п.1 или 2, отличающееся тем, что R3 представляет собой H и R4 представляет собой метил.
4. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой

5. Соединение по п.1, представляющее собой

или его фармацевтически приемлемая соль.
6. Соединение по п.5, представляющее собой

или его фармацевтически приемлемая соль.
7. Гидрохлоридная соль соединения по п.6, представляющая собой

Текст
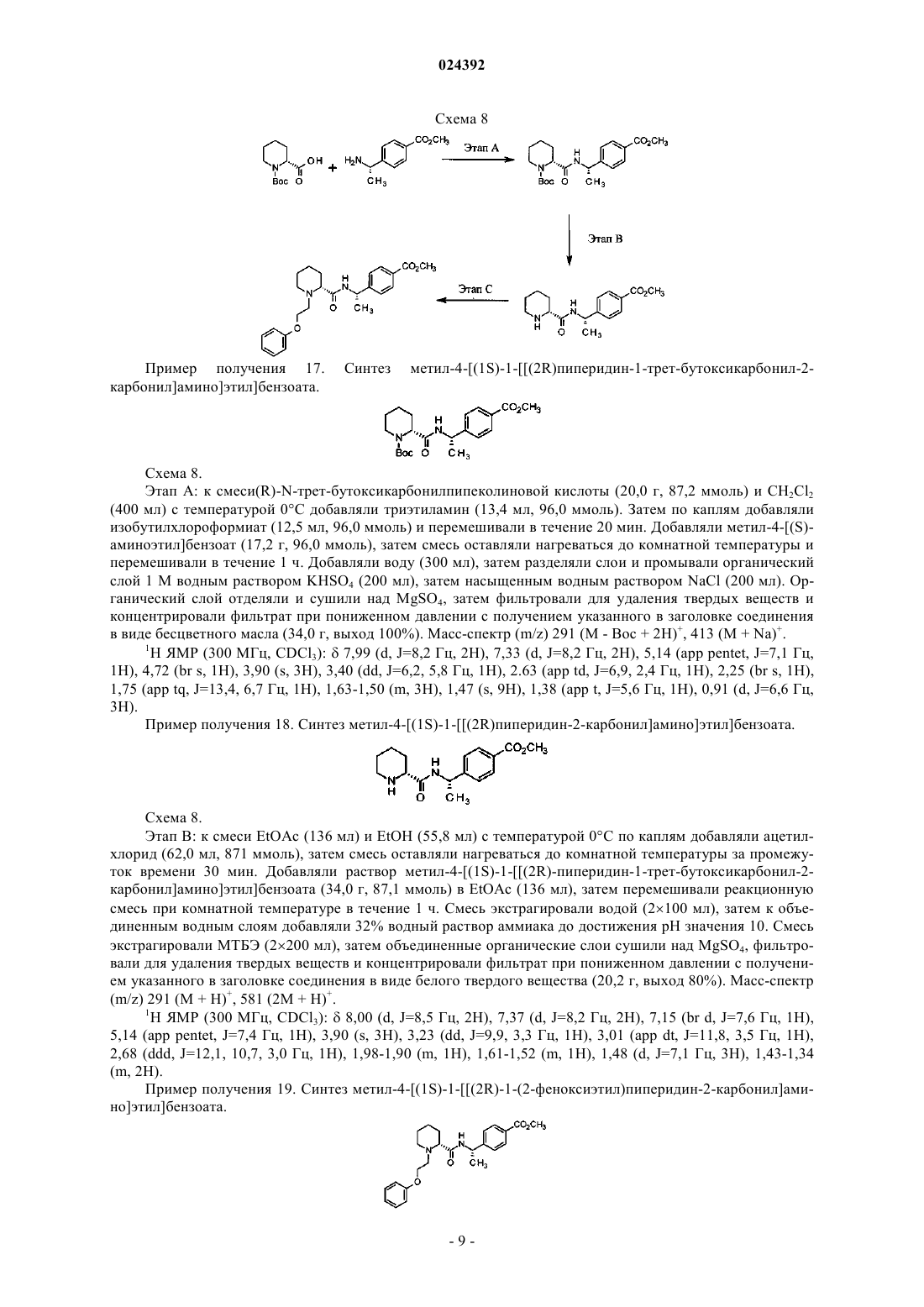
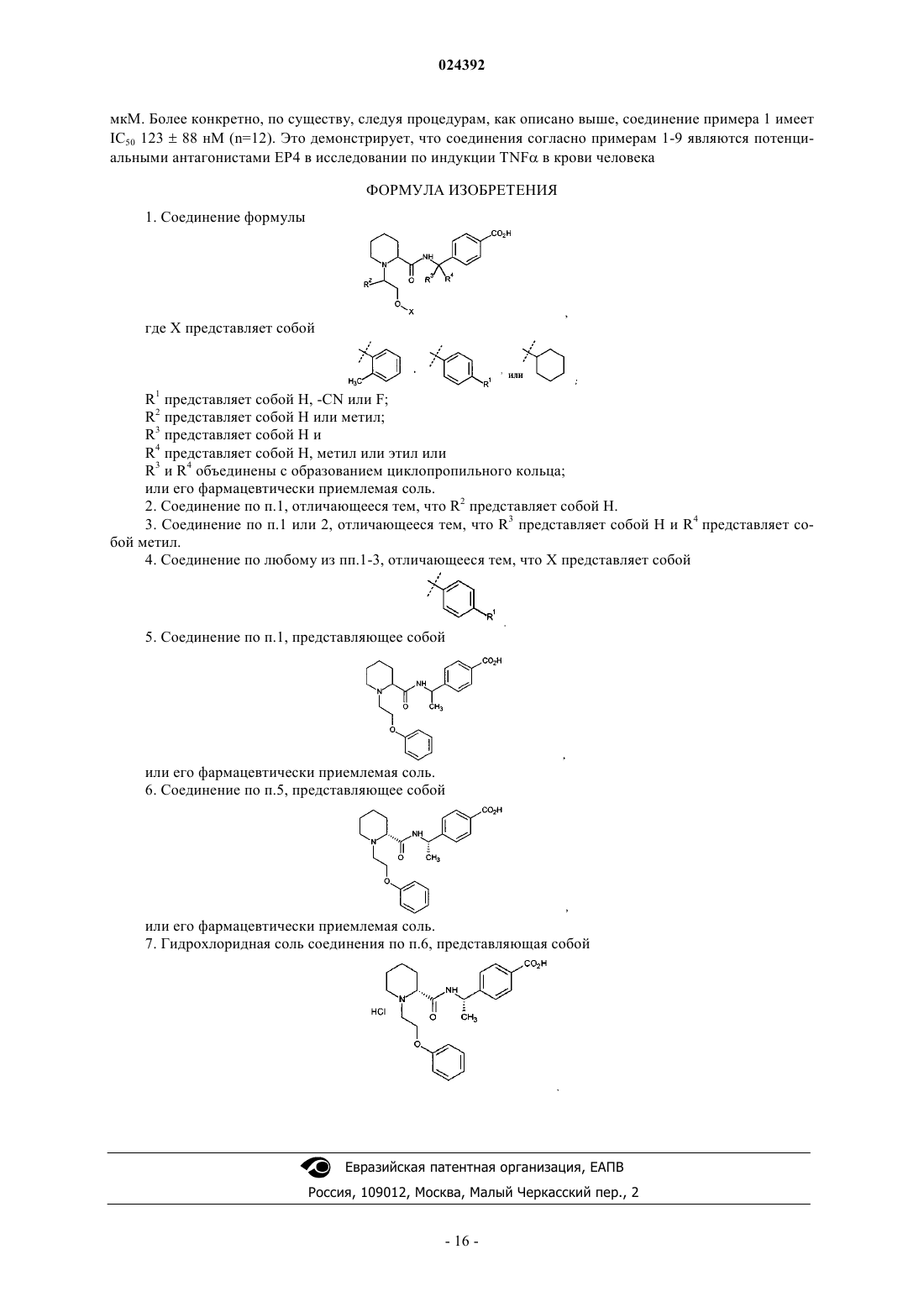
В настоящем изобретении предложено соединение формулы II Шиффлер Мэтью Аллен, Йорк Джереми Шуленбург (US) Лыу Т.Н., Угрюмов В.М. (RU)R4 представляет собой H, метил или этил или R3 и R4 объединены с образованием циклопропильного кольца; или его фармацевтически приемлемая соль.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Настоящее изобретение относится к новым феноксиэтилпиперидиновым соединениям, к фармацевтическим композициям, содержащим указанные соединения, к способам применения указанных соединений для лечения физиологических расстройств и к промежуточным соединениям и способам, подходящим для синтеза указанных соединений. Настоящее изобретение относится к области лечения воспалительных состояний, таких как артрит,включая остеоартрит и ревматоидный артрит, и также включая боли, связанные с этими состояниями Артрит поражает миллионы пациентов в одних только Соединенных Штатах и является одной из основных причин инвалидности. Лечение часто включает НПВС (нестероидные противовоспалительные средства) или ингибиторы циклооксигеназы-2 (ЦОГ-2, СОХ-2), которые могут быть причиной нежелательных побочных эффектов со стороны сердечно-сосудистой системы и/или желудочно-кишечного тракта. По этой причине для пациентов, которые имеют плохой профиль риска сердечно-сосудистых заболеваний, таких как гипертония, применение НПВП или ингибиторов ЦОГ-2 может быть исключено. Таким образом, существует необходимость в альтернативном лечении остеоартрита и ревматоидного артрита,предпочтительно без побочных эффектов, присущих имеющимся в настоящее время способам лечения. Были идентифицированы следующие четыре подтипа рецепторов простагландина: E2 (ПГЕ 2, PGE2).EP1, ЕР 2, EP3 и ЕР 4. С помощью моделей ревматоидного артрита и остеоартрита у грызунов было обнаружено, что ЕР 4 является основным рецептором, вовлеченным в возникновение боли, связанной с воспалением суставов (см., например, J. Pharmacol. Exp. Ther., 325. 425 (2008. Следовательно, селективный антагонист ЕР 4 может быть подходящим для лечения артрита, включая боли при артрите. Кроме того,было высказано предположение, что поскольку антагонизм ЕР 4 не мешает биосинтезу простаноидов,таких как PGI2 и TxA2, селективный антагонист ЕР 4 возможно не будет обладать потенциальными побочными эффектами в отношении сердечно-сосудистой системы, наблюдаемыми в случае НПВС и ингибиторов ЦОГ-2. (см., например, BioorganicMedicinal Chemistry Letters, 21, 484 (2011. В WO 2013/004290 предложены производные циклических аминов в качестве антагонистов рецепторов ЕР 4. В US 2005/0250818 предложены некоторые ортозамещенные арил- и гетероариламидные соединения, которые являются селективными антагонистами рецептора ЕР 4 с анальгетической активностью. Кроме того, в WO 2011/102149 предложены некоторые соединения, которые являются селективными антагонистами ЕР 4, подходящими для лечения заболеваний, связанных с IL-23. Согласно настоящему изобретению предложены новые соединения, которые являются селективными ингибиторами ЕР 4 по сравнению с ЕР 1, ЕР 2 и EP3. Кроме того, согласно настоящему изобретению предложены новые соединения, обладающие потенциалом в отношении снижения побочных эффектов со стороны сердечно-сосудистой системы или желудочно-кишечного тракта по сравнению с традиционными НПВП. Соответственно, согласно настоящему изобретению предложено соединение формулы IIR3 и R4 объединены с образованием циклопропильного кольца; или его фармацевтически приемлемая соль. Согласно настоящему изобретению также предложено соединение формулы I или его фармацевтически приемлемая соль. Согласно настоящему изобретению также предложен способ лечения артрита у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения фор-1 024392 мулы I или формулы II, или их фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ лечения остеоартрита у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I или формулы II, или их фармацевтически приемлемой соли. Кроме того, согласно настоящему изобретению также предложен способ лечения ревматоидного артрита у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I или формулы II, или их фармацевтически приемлемой соли. Согласно настоящему изобретению также предложен способ лечения боли, связанной с артритом, у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I или формулы II, или их фармацевтически приемлемой соли. Согласно настоящему изобретению дополнительно предложен способ лечения боли, связанной с остеоартритом или ревматоидным артритом, у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения формулы I или формулы II, или их фармацевтически приемлемой соли. Кроме того, согласно настоящему изобретению предложено соединение формулы I или формулы II,или его фармацевтически приемлемая соль, для применения в терапии, в частности для лечения остеоартрита. Кроме того, согласно настоящему изобретению предложено соединение формулы I или формулы II, или его фармацевтически приемлемая соль, для применения для лечения ревматоидного артрита. Согласно настоящему изобретению также предложено соединение или его фармацевтически приемлемая соль для применения для лечения боли, связанной с остеоартритом или ревматоидным артритом. Более того, согласно настоящему изобретению предложено применение соединения формулы I или формулы II,или его фармацевтически приемлемой соли для получения лекарственного средства для лечения остеоартрита. Согласно настоящему изобретению предложено применение соединения формулы I или формулы II, или его фармацевтически приемлемой соли, для получения лекарственного средства для лечения ревматоидного артрита. Согласно настоящему изобретению также предложено применение соединения формулы I или формулы II, или его фармацевтически приемлемой соли, для получения лекарственного средства для лечения боли, связанной с остеоартритом или ревматоидным артритом. Согласно настоящему изобретению дополнительно предложена фармацевтическая композиция, содержащая соединение формулы I или формулы II, или его фармацевтически приемлемую соль, в комбинации с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами. В конкретном варианте реализации настоящего изобретения, композиция дополнительно содержит один или более других терапевтических агентов. Данное изобретение также охватывает новые промежуточные соединения и способы синтеза соединений формулы I или формулы II, или их фармацевтически приемлемой соли. Кроме того, настоящее изобретение включает способ лечения воспалительных состояний, таких как артрит, включая остеоартрит и ревматоидный артрит, у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества соединения антагониста провоспалительного простагландина, такого как антагонист ЕР 4, в комбинации с эффективным количеством модулятора рецепторов липоксина или резолвина, такого как модулятор BLT-1, BLT-2, ALX/FPR1, GPR32, CysLT1,CysLT2 или ChemR23. Дополнительный аспект настоящего изобретения включает способ лечения воспалительных заболеваний, таких как артрит, включая остеоартрит и ревматоидный артрит, у пациента, включающий введение пациенту, нуждающемуся в таком лечении, эффективного количества ингибитора синтазы провоспалительного простагландина, такого как ингибитор mPGES-1, в комбинации с эффективным количеством модулятора рецепторов липоксина или резолвина, такого как модулятор BLT-1, BLT-2, ALX/FPR1,GPR32, CysLT1, CysLT2 или ChemR23. В настоящем документе термины "лечение" или "лечить" включают подавление, сдерживание, замедление, прекращение или обращение развития или тяжести существующего симптома или расстройства. В настоящем документе термин "пациент" относится к млекопитающему, такому как мышь, морская свинка, крыса, собака или человек. Это понятно, что предпочтительным пациентом является человек. В настоящем документе термин "эффективное количество" относится к количеству или дозе соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, которое при однократном или многократном введении дозы пациенту обеспечивает необходимый эффект у пациента при диагностике или лечении. Эффективное количество может быть легко определено практикующим диагностом, являясь специалистом в данной области, посредством применения известных методик и наблюдения результатов,полученных в аналогичных условиях. При определении эффективного количества для пациента практикующие диагносты рассматривают ряд факторов, включая, но не ограничиваясь ими: вид млекопитающего; его размер, возраст и общее состояние здоровья; поражение конкретным заболеванием или расстройством; степень поражения или степень тяжести заболевания или расстройства; реакцию индивидуального пациента; конкретное вводимое соединение; способ введения; характеристики биодоступности вводимого препарата; выбранную схему приема; применение сопутствующего лекарственного средства и другие важные обстоятельства. Соединения формулы I или формулы II, или их фармацевтически приемлемые соли, являются в целом эффективными в широком диапазоне доз. Например, ежедневные дозы приема обычно находятся в диапазоне от примерно 0,01 до примерно 50 мг/кг массы тела. В некоторых случаях уровни доз ниже нижнего предела вышеуказанного диапазона могут быть более чем достаточными, тогда как в других случаях, более высокие дозы вс ещ можно применять с приемлемыми побочными эффектами, и, следовательно, доза выше диапазона не ограничивает объем настоящего изобретения в любом случае. Соединения согласно настоящему изобретению предпочтительно изготавливают в виде фармацевтических композиций для введения любым способом, который делает соединение биодоступным. Наиболее предпочтительно такие композиции являются композициями для перорального введения. Такие фармацевтические композиции и способы их получения хорошо известны в данной области. См., например, Remington: The Science and Practice of Pharmacy (D.B. Troy, Editor, 21st Edition, Lippincott, WilliamsWilkins, 2006). Соединения формулы I и формулы II подходят для лечения согласно настоящему изобретению, но некоторые группы, заместители и конфигурации являются предпочтительными для соединений формулыI и формулы II. Следующие параграфы описывают такие предпочтительные группы, заместители и конфигурации. Следует понимать, что эти предпочтения применимы как к способам лечения, так и к новым соединениям согласно настоящему изобретению. Предпочтительным является то, что R1 представляет собой H, R2 представляет собой H, R3 представляет собой H и X представляет собой Также предпочтительным является то, что если R3 представляет собой H, то R4 представляет собой метил. Также предпочтительным является то, что X представляет собой и ее фармацевтически приемлемая соль являются особенно предпочтительными. 4-[(1S)-1-(2R)-1-(2-феноксиэтил)пиперидин-2-карбонил]амино]этил]бензойной кислоты гидрохлорид также является особенно предпочтительным. В настоящем документе "кПа изб." относится к килопаскалям избыточного давления; "Boc" относится к трет-бутоксикарбонильной защищающей группе; "DMEM" относится к модифицированной по способу Дульбекко среде Игла; "ACN" относится к ацетонитрилу; "ДМСО" относится к диметилсульфоксиду; "DMF" относится к N,N-диметилформамиду; "EtOH" относится к этанолу; "ТГФ" относится к тетрагидрофурану; "MeOH" относится к метанолу; "EtOAc" относится к этилацетату; "Et2O" относится к диэтиловому эфиру; "МТБЭ" относится к трет-бутилметиловому эфиру; "ВОР" относится к бензотриазол-1-ил-окситрис(диметиламино)фосфония гексафторфосфату; "NaHMDS" относится к натрия бис(триметилсилил)амиду; "PGE2" относится к простагландину E2; "FBS" относится к фетальной бычьей сыворотке; "IBMX" относится к 3-изобутил-1-метилксантину; "MES" относится к 2-(Nморфолино)этансульфоновой кислоте; "HEPES" относится к 2-[4-(2-гидроксиэтил)пиперазин-1 ил]этансульфоновой кислоте; "HTRF" относится к технологии гомогенной флуоресценции с временным разрешением; "HEK" относится к мезонефросу человека; "HBSS" относится к сбалансированному солевому раствору Хенкса; "EC50" относится к концентрации агента, которая позволяет получить 80% от максимальной эффективности, возможной для данного агента; и "IC50" относится к концентрации агента,которая позволяет получить 50% от максимального ингибирующего эффекта, возможного для данного агента. Фармацевтически приемлемые соли и общая методология их получения хорошо известны в данной области. См., например, Gould P.L., "Salt selection for basic drags', International Journal of Pharmaceutics, 33: 201-217 (1986); Bastin R.J., et al. "Salt Selection and Optimization Procedures for Pharmaceutical New Chemical Entities", Organic Process Research and Development, 4: 427-435 (2000) и Berge S.M., et al., "Pharmaceutical Salts", Journal of Pharmaceutical Sciences, 66: 1-19, (1977). Специалистам в области синтеза будет понятно, что соединения формулы I и формулы II, легко могут быть преобразованы и могут быть изолированы в виде фармацевтически приемлемых солей, таких как гидрохлориды, с применением методов и условий, хорошо известных специалистам в данной области. Кроме того, специалистам в области синтеза будет понятно, что соединения формулы I и формулы II, легко могут быть преобразованы и могут быть изолированы в виде соответствующего свободного основания или свободной кислоты из соответствующей фармацевтически приемлемой соли. В настоящем изобретении рассмотрены все индивидуальные энантиомеры или диастереомеры, а также смеси энантиомеров и диастереомеров указанных соединений, включая рацематы. Индивидуальные изомеры, энантиомеры или диастереомеры могут быть выделены или разделены специалистами в данной области в любой удобной точке синтеза соединений согласно настоящему изобретению, способами, такими как способ селективной кристаллизации или хиральной хроматографии (см., например, J.S.H. Wilen, "Stereochemistry of Organic Compounds", Wiley-Interscience, 1994). Соединения согласно настоящему изобретению или их фармацевтически приемлемые соли могут быть получены посредством различных процедур, известных в данной области, некоторые из которых проиллюстрированы на схемах, способах получения и примерах, приведенных ниже. Конкретные этапы синтеза для каждого из описанных путей могут быть скомбинированы по-разному или в сочетании с этапами из разных схем, для получения соединения формулы I или его фармацевтически приемлемой соли. Продукты каждого этапа на схемах, приведенных ниже, могут быть извлечены с помощью традиционных методов, включая экстракцию, выпаривание, осаждение, хроматографию, фильтрование, растирание и кристаллизацию. Реагенты и исходные материалы легко доступны специалистам в данной области. Все заместители, если не указано иное, определены заранее. Следует понимать, что не предполагается, что объем настоящего изобретения ограничен данными схемами, способами получения и примерами. Схема 1 Схема 1. Этап А: к перемешанному раствору (-)-1-(4-бромфенил)этиламина (1,00 г, 5,0 ммоль) в дихлорметане (10 мл) при 0C добавляли ди-трет-бутилдикарбонат (1,09 г, 5,0 ммоль). Реакционную смесь оставляли нагреваться при комнатной температуре, затем перемешивали в течение двух часов. К перемешанной смеси добавляли 1 М водный раствор хлористо-водородной кислоты (25 мл), после чего добавляли Et2O(25 мл). Слои разделяли, и экстрагировали водный слой Et2O (225 мл). Органические слои объединяли,промывали насыщенным водным раствором NaCl (25 мл), высушивали органический слой над MgSO4,фильтровали для удаления твердых частиц и концентрировали фильтрат при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (1,50 г, выход 99%). Масс-спектр (m/z) (79Br/81Br) 244/246 (M + 2H - t-Bu)+, 322/324 (M + Na)+. 1H ЯМР (400 МГц, CDCl3):7,46-7,43 (m, 2H), 7,19-7,15 (m, 2H), 4,81-4,65 (m, 1H), 1,48-1,36 (m,12H). Следующее соединение получали, по существу, согласно примеру получения 1, используя 1-(4 бромфенил)циклопропанамин вместо (-)-1-(4-бромфенил)этиламина Схема 1. Этап В: в автоклав от Parr с механическим перемешиванием добавляли Pd(OAc)2 (120 мг, 0,53 ммоль), 1,Г-бис(дифенилфосфино)ферроцен (355 мг, 0,64 ммоль), (S)-N-трет-бутоксикарбонил-1-(4 бромфенил)этиламин (1,50 г, 5,0 ммоль), безводный CH3CN (45 мл), безводный СH3OH (30 мл) и триэтиламин (1,9 мл, 13,63 ммоль). Сосуд закрывали и нагнетали давление с применением монооксида углерода до 724 кПа изб. Сосуд нагревали до 85C и перемешивали смесь на протяжении ночи. Реакционный сосуд продували (Осторожно - ядовитый газ) и перемещали смесь в круглодонную колбу, промываяCH3OH. Смесь концентрировали при пониженном давлении до получения оранжевого осадка. Добавляли воду (50 мл), далее экстрагировали EtOAc (250 мл). Объединенную органическую фазу промывали насыщенным водным раствором NaCl (25 мл), затем разделяли слои, высушивали органическую фазу надMgSO4, фильтровали для удаления твердых частиц и концентрировали фильтрат при пониженном давлении до получения неочищенного продукта. Продукт очищали флэш-хроматографией на силикагеле, проводя элюирование градиентом от 0 до 60% EtOAc/гексан. Фракции, содержащие необходимый продукт,концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (1,00 г, выход 72%). Масс-спектр (m/z) 224 (М + 2 Н - t-Buf, 302 (М + Na)+, 581 (2M + Na)+. 1H ЯМР (400 МГц, ДМСО-d6):7,89 (d, J = 8,4 Гц, 2 Н), 7,41 (d, J = 8,2 Гц, 2 Н), 4,64 (dq, J = 7,4, 6,8 Гц, 1H), 3,82 (s, 3H), 1,34 (br s, 9H), 1,28 (d, J = 7,2 Гц, 3H). Следующее соединение получали, по существу, согласно примеру получения 3, используя третбутил-N-[1-(4-бромфенил)циклопропил]карбамат вместо Схема 1. Этап С: к метил-(S)-4-(1-трет-бутоксикарбониламиноэтил)бензоату (1,00 г, 3,58 ммоль), добавляли хлороводород (4M в диоксане, 5 мл, 20 ммоль) и перемешивали полученную смесь при комнатной температуре в течение одного часа. Смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (750 мг, выход 97%). 1H ЯМР (400 МГц, ДМСО-d6):8,57 (br s, 3H), 7,99 (d, J= 8,4 Гц, 2 Н), 7,65 (d, J = 8,4 Гц, 2 Н), 4,47 (q,J= 6,7 Гц, 1 Н), 3,84 (s, 3H), 1,50 (d, J= 6,8 Гц, 3H). Следующее соединение получали по существу согласно примеру получения 5, используя метил-4[1-(трет-бутоксикарбониламино)циклопропил]бензоат вместо метил-(S)-4-(1-трет-бутоксикарбониламиноэтил)бензоата: Схема 2. Этап А: 4-фторфенол растворяли (5,5 г, 49,1 ммоль) в ацетонитриле (49 мл) и обрабатывали раствор 2-бром-1,1-диметоксиэтаном (11,6 мл, 98,1 ммоль) и K2CO3 (16,95 г, 122,7 ммоль). Раствор нагревали с обратным холодильником при перемешивании в течение пяти дней. Смесь фильтровали и концентрировали фильтрат при пониженном давлении. Полученный неочищенный материал очищали с помощью хроматографии с силикагелем, проводя элюирование градиентом от 0 до 50% EtOAc/гексан. Фракции,содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (5,85 г, выход 60%). Масс-спектр (m/z) 218 (М +(1 М раствор в ТГФ, 21,0 мл, 21,0 ммоль) и перемешивали раствор при комнатной температуре в течение 5 мин. Добавляли 2-бром-1,1-диметоксиэтан (2,26 мл, 19,1 ммоль), затем перемешивали смесь при комнатной температуре в атмосфере азота в течение трех дней. Смесь разбавляли EtOAc (250 мл) и промывали насыщенным водным раствором NaCl (2250 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали фильтрат при пониженном давлении. Полученный неочищенный материал очищали с помощью хроматографии с силикагелем, проводя элюирование градиентом от 0 до 10%EtOAc/гексан. Фракции, содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бледно желтого масла (1,10 г, выход 31%). 1 Схема 4. Этап А: 1-(2,2-диметоксиэтокси)-4-фторбензен растворяли (1,00 г, 4,99 ммоль) в хлороформе (5,0 мл) и обрабатывали смесь трифторуксусной кислотой (0,755 мл, 9,99 ммоль). Смесь перемешивали при комнатной температуре в течение двух дней, затем нагревали до 65C и перемешивали в течение 4 ч. Смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла примерно 70% чистоты, что определяли с помощью 1H ЯМР-анализа (550 мг,нескорректированный выход 71%). 1 Схема 5. Этап А: смесь (2,2-диметоксиэтокси)циклогексана и воды (30 мл) подкисляли до pH 1,0 серной кислотой (9,0 М водный раствор) и присоединяли смесь к колонке для молекулярной перегонки. Понижали давление до 26,7 кПа и нагревали смесь до 100C в течение 1 ч. Смесь охлаждали до комнатной температуры, затем водный слой экстрагировали МТБЭ (275 мл). Объединенные органические слои промывали насыщенным водным раствором NaHCO3 (75 мл) и насыщенным водным раствором NaCl (75 мл). Органическую фазу сушили над MgSO4, фильтровали и концентрировали фильтрат при пониженном давлении с получением указанного в заголовке соединения (634 мг, выход 51%). 1 Схема 6. Этап А: метил-(R)-пиперидин-2-карбоксилат (5,00 г, 34,9 ммоль) растворяли в DMF (87 мл) и обрабатывали K2CO3 (14,48 г, 104,8 ммоль) и Р-бромфенетолом (7,16 г, 34,9 ммоль). Смесь перемешивали в течение ночи при 100C. Смесь охлаждали до комнатной температуры и добавляли EtOAc (250 мл). Органическую фазу промывали водой (4100 мл) и насыщенным водным раствором NaCl (100 мл). Органическую фазу сушили над K2CO3, фильтровали для удаления твердых веществ и концентрировали фильтрат при пониженном давлении до получения желтого масла. Полученный неочищенный материал очищали с помощью флэш-хроматографии с силикагелем, проводя элюирование градиентом от 20 до 100%EtOAc/гексан. Фракции, содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла со степенью чистоты примерно 90% согласно 1H ЯМР анализу (6,50 г, выход 64%). Масс-спектр (m/z) 264 (М + Н)+. 1 Схема 6. Этап В: метил-(R)-1-(2-феноксиэтил)пиперидин-2-карбоксилат (6,50 г, 22,2 ммоль) растворяли в ТГФ (11,1 мл) при комнатной температуре и добавляли NaOH (5 М водный раствор, 8,89 мл, 44,4 ммоль) и нагревали до 65C в течение ночи с перемешиванием. Добавляли хлороводород (5 М водный раствор) до достижения pH водной фазы значения 1,0. Водную фазу промывали CH2Cl2 (375 мл). Водную фазу концентрировали при пониженном давлении до получения белого твердого вещества. Твердое вещество растирали с EtOH (50 мл), фильтровали для удаления суспендированных солей и концентрировали фильтрат при пониженном давлении с получением указанного в заголовке соединения в виде белого твердогоH ЯМР (400 МГц, CD3OD):7,31 (dd, J = 8,8, 7,5 Гц, 2H), 7,02-6,97 (m, 3H), 4,47 (ddd, система связывания типа АВ, J= 11,5, 7,1, 3,0 Гц, 1H), 4,38 (ddd, система связывания типа АВ, J=11,8, 6,3, 3,1 Гц, 1H),4,15 (dd, J=11,2, 2,8 Гц, 1H), 3,80 (d, J=12,2 Гц, 1H), 3,73-3,68 (m, 2H), 3,29 (арр td, J=13,2, 3,8 Гц, 1H), 2,35 Схема 7. Этап А: (R)-1-(2-феноксиэтил)пиперидинкарбоновой кислоты гидрохлорид (750 мг, 2,62 ммоль) и метил-(S)-4-(1-аминоэтил)бензоата гидрохлорид (566 мг, 2,62 ммоль) растворяли в DMF (5,25 мл) при комнатной температуре. Добавляли триэтиламин (1,65 мл, 11,81 ммоль), затем ВОР (1,51 г, 3,41 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч, затем разбавляли EtOAc (25 мл). Смесь промывали насыщенным водным раствором LiCl (225 мл). Органический слой сушили над MgSO4,фильтровали для удаления твердых веществ и концентрировали при пониженном давлении. Полученное желто-оранжевое масло очищали с помощью флэш-хроматографии с силикагелем, проводя элюирование градиентом от 0% до 100% EtOAc/гексан. Фракции, содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (930 мг, выход 86%). Масс-спектр (m/z) 411 (М + Н)+. 1(m, 3H), 1,36 (d, J=7,0 Гц, 3H), 1,27-1,18 (m, 1H). Следующие соединения получали, по существу, согласно примеру получения 13, используя подходящие соли аммония вместо метил-(S)-4-(1-аминоэтил)бензоата гидрохлорида:(400 мл) с температурой 0C добавляли триэтиламин (13,4 мл, 96,0 ммоль). Затем по каплям добавляли изобутилхлороформиат (12,5 мл, 96,0 ммоль) и перемешивали в течение 20 мин. Добавляли метил-4-[(S)аминоэтил]бензоат (17,2 г, 96,0 ммоль), затем смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Добавляли воду (300 мл), затем разделяли слои и промывали органический слой 1 М водным раствором KHSO4 (200 мл), затем насыщенным водным раствором NaCl (200 мл). Органический слой отделяли и сушили над MgSO4, затем фильтровали для удаления твердых веществ и концентрировали фильтрат при пониженном давлении с получением указанного в заголовке соединения в виде бесцветного масла (34,0 г, выход 100%). Масс-спектр (m/z) 291 (M - Boc + 2H)+, 413 (М + Na)+. 1 Схема 8. Этап В: к смеси EtOAc (136 мл) и EtOH (55,8 мл) с температурой 0C по каплям добавляли ацетилхлорид (62,0 мл, 871 ммоль), затем смесь оставляли нагреваться до комнатной температуры за промежуток времени 30 мин. Добавляли раствор метил-4-[(1S)-1-(2R)-пиперидин-1-трет-бутоксикарбонил-2 карбонил]амино]этил]бензоата (34,0 г, 87,1 ммоль) в EtOAc (136 мл), затем перемешивали реакционную смесь при комнатной температуре в течение 1 ч. Смесь экстрагировали водой (2100 мл), затем к объединенным водным слоям добавляли 32% водный раствор аммиака до достижения pH значения 10. Смесь экстрагировали МТБЭ (2200 мл), затем объединенные органические слои сушили над MgSO4, фильтровали для удаления твердых веществ и концентрировали фильтрат при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (20,2 г, выход 80%). Масс-спектр Схема 8. Этап С: к суспензии силикагеля (100 г) в CH2Cl2 (705 мл) при комнатной температуре по каплям добавляли раствор NaIO4 (35,0 г, 161,9 ммоль) в воде (235 мл). Смесь перемешивали в течение 30 мин, затем добавляли 1,2-дигидрокси-3-феноксипропан (21,5 г, 121,4 ммоль) и перемешивали смесь в течение дополнительных 30 мин. Смесь фильтровали для удаления твердых веществ и слои фильтрата разделяли. Органический слой сушили над MgSO4 и фильтровали для удаления твердых веществ. К фильтрату добавляли метил-4-[(1S)-1-(2R)-пиперидин-2-карбонил]амино]этил]бензоат (23,5 г, 80,9 ммоль), затем маленькими порциями добавляли триацетоксиборогидрид натрия (35,7 г, 161,9 ммоль). Перемешивали в течение одного часа при комнатной температуре, затем добавляли 32% водный раствор аммиака до достижения pH значения 10. Слои разделяли, и органическую фазу сушили над MgSO4. Фильтровали для удаления твердых веществ, затем концентрировали фильтрат при пониженном давлении до получения неочищенного материала. Материал растворяли в EtOAc (300 мл) и фильтровали через слой силикагеля(30 г). Фильтрат концентрировали при пониженном давлении до получения 36 г материала. Добавляли МТБЭ (180 мл) и нагревали до 50C. В процессе достижения температуры 50C, в течение 15 мин добавляли гексан (360 мл), затем перемешивали в течение одного часа. Смесь оставляли нагреваться до комнатной температуры, затем отделяли твердые вещества посредством фильтрования и сушили при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества(7,80 г, 19,98 ммоль) обрабатывали хлористо-водородной кислотой (4 М раствор в 1,4-диоксане, 25,0 мл,99,9 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 1 ч. Смесь концентрировали при пониженном давлении с получением целевого соединения в виде белого твердого вещества (6,0 г, выход 92%). Масс-спектр (m/z) 291 (М + Н)+, 581 (2 М + Н)+, 603 (2 М + Na)+. Пример получения 21. Синтез метил-4-[(1S)-1-(2R)-1-(2-(4-фторфенокси)этил)пиперидин-2 карбонил]амино]этил]бензоата.(650 мг, 1,99 ммоль) и 2-(4-фторфенокси)ацетальдегида (337 мг, 2,19 ммоль) в DCE (9,9 мл) перемешивали при комнатной температуре в течение 30 мин. Добавляли уксусную кислоту (0,113 мл, 1,99 ммоль) и триацетоксиборогидрид натрия (590 мг, 2,78 ммоль) перемешивали при комнатной температуре в течение трех дней. Реакцию останавливали насыщенным водным раствором NaHCO3 (25 мл) и экстрагировали водный слой EtOAc (225 мл). Объединенные органические слои промывали насыщенным водным раствором NaCl (25 мл), затем органическую фазу сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученное масло очищали с помощью флэш-хроматографии с силикагелем,проводя элюирование градиентом от 0 до 100% EtOAc/гексан. Фракции, содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (600 мг, выход 70%). Масс-спектр (m/z) 429 (М + Н)+, 451 (М + Na)+. Следующие соединения получали, по существу, согласно примеру получения 21, используя подходящие альдегиды вместо 2-(4-фторфенокси)ацетальдегида:(150 мг, 0,46 ммоль), 1-фенокси-2-пропанона (69 мкл, 0,50 ммоль), DCE (2,3 мл), уксусной кислоты (26 мкл, 0,46 ммоль) и триацетоксиборогидрида натрия (136 мг, 0,64 ммоль) перемешивали при 65C в течение двух дней. Реакцию останавливали насыщенным водным раствором NaHCO3 (75 мл) и экстрагировали водный слой EtOAc (75 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении. Неочищенный материал подвергали хроматографии с обращенной фазой на силикагеле C18, проводя элюирование 0,1% TFA с градиентом от 5 до 50% ACN/вода. Фракции, содержащие необходимый продукт, концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде бледно-желтого масла в смеси 2:1 диастереомеров (24 мг, выход 10%). Масс-спектр (m/z) 425 (M + H)+. Схема 11NaOH (1 M водный раствор, 4,5 мл, 4,5 ммоль), затем перемешивали полученную смесь при комнатной температуре в течение трех дней. Реакционную смесь концентрировали при пониженном давлении до получения липкого твердого вещества. Добавляли хлороводород (4 М раствор в диоксане, 2 мл, 8 ммоль) и интенсивно перемешивали в течение 10 мин. Суспендированные твердые вещества удаляли посредством фильтрования и концентрировали фильтрат при пониженном давлении до получения белого твердого вещества Твердое вещество растирали в кипящем диэтиловом эфире (25 мл) и суспендированные твердые вещества удаляли посредством фильтрования с получением указанного в заголовке соединения(650 мг, выход 66%) в виде белого твердого вещества. Масс-спектр (m/z): 397 (М + Н)+. 1(m, 1H), 3,37-3,18 (m, 2H), 2,15 (d, J=13,5 Гц, 1H), 1,82-1,66 (m, 4H), 1,50-1,43 (m, 1H), 1,39 (d, J=7,2 Гц,3H). Следующие соединения получали, по существу, согласно способу, представленному в примере 1,используя подходящие метиловые эфиры вместо метил-4-[(1S)-1-(2R)-1-(2-феноксиэтил)пиперидин-2 карбонил]амино]этил]бензоата: Схема 12. Этап А: метил-4-[(1S)-1-(2R)-1-(1-метил-2-феноксиэтил)пиперидиниум-2-карбонил]амино]этил]бензоата трифторацетат (24 мг, 0,045 ммоль) растворяли в ТГФ (226 мкл) и обрабатывали смесь метанолом (226 мкл) и гидроксидом натрия (1 н. водный раствор, 170 мкл, 0,17 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, затем концентрировали при пониженном давлении до получения липкого твердого вещества. Неочищенный материал подвергали хроматографии с обращенной фазой на силикагеле C18, проводя элюирование 0,1% TFA с градиентом от 5 до 50% ACN/вода с получением двух отдельных фракций, каждая из которых содержала отдельный диастереомер продукта. Каждую фракцию концентрировали при пониженном давлении, каждую растворяли в минимальном объеме метанола, каждую растирали с диэтиловым эфиром (5 мл) и концентрировали каждую при пониженном давлении с получением изомера 1 (3,0 мг, выход 13%) и изомера 2 (1,1 мг, выход 5%) указанного в заголовке соединения в виде белых твердых веществ. Пример 9 а: Основной изомер (изомер 1). Масс-спектр (m/z): 411 (М + Н)+. 1H ЯМР (ДМСО-d6)9,75 (br s), 9,30 (d, J=7,4 Гц, 1H), 7,82 (d, J=7,6 Гц, 2H), 7,43 (d, J=7,6 Гц, 2H),7,33-7,25 (m, 2H), 7,04-6,95 (m, 3H), 5,03 (app p, J=6,8 Гц, 1H), 4,35-4,24 (m, 2H), 4,07 (dd, J=12,1, 3,5 Гц,1H), 3,74-3,65 (m, 1H), 3,52 (br d, J=12,5 Гц, 1H), 3,11-2,99 (m, 1H), 2,15 (br d, J=12,4 Гц, 1H), 1,89-1,71 (m,4H), 1,52-1,45 (m, 1H), 1,40 (d, J=6,8 Гц, 3H), 1,33 (d, J=6,8 Гц, 3H). Пример 9b: Второстепенный изомер (изомер 2). Масс-спектр (m/z): 411 (М + Н)+. Специалистам в данной области следует понимать, что соли HCl из примеров 1-9 легко преобразовать в соответствующие свободные основания, используя условия, хорошо известные в данной области. Тест на связывание с человеческими рецепторами EP1, ЕР 2, EP3 и ЕР 4 in vitro Мембраны hEP1 и hEP4 получали из рекомбинантных клеток HEK293, стабильно экспрессирующих человеческие EP1 (Genbank accession number AY275470) или ЕР 4 (Genbank accession number AY429109) рецепторы. Мембраны hEP2 и hEP3 получали из клеток HEK293, временно трансфицированных плазмидами рецепторов ЕР 2 (Genbank accession number AY275471) или EP3 (isoform VI: Genbank accession number AY429108). Замороженные клеточные пеллеты гомогенизировали в гомогенизационном буфере, используя тефлоновый/стеклянный гомогенизатор. Протеин мембраны распределяли и быстро замораживали с использованием сухого льда для хранения при -80C. Гомогенизационный буфер содержал 10 мМ трис-HCl, pH 7,4, 250 мМ сахарозы, 1 мМ ЭДТА, 0,3 мМ индометацина и с добавлением Complete с ЭДТА, полученного от Roche Molecular Biochemicals (Каталожный номер 1 697 498). Значения Kd для [3H]-PGE2, связывающегося с каждым рецептором, определяли посредством метода связывания до насыщения или метода конкуренции гомологов. Соединения тестировали с использованием 96-луночного микропланшета, используя серии трехкратных разведений для получения 10 точечной кривой. Разбавленное соединение инкубировали с мембранами, содержащими 20 мкг на лунку ЕР 1, 10 мкг на лунку ЕР 2, 1 мкг на лунку EP3 или от 10 до 20 мг на лунку ЕР 4 в течение 90 мин при 25C в присутствии от 0,3 до 0,5 нМ [3H]-PGE2 (PerkinElmer, от 118 до 180 Ci/ммоль). Реакцию связывания проводили в 200 мкл MES буфера (10 мМ MES pH 6,0 с KOH, 10 мМ MgCl2 и 1 мМ ЭДТА) с использованием 96-луночных планшетов с глубокими лунками на 0,5 мл из полистирола. Неспецифическое связывание подсчитывали путем сравнения связывания в присутствии и отсутствие 2 мкМ PGE2. Мембраны собирали посредством фильтрования (TomTek коллектор), промывали 4 раза холодным буфером (10 мМMES pH 6,0 с KOH, 10 мМ MgCl2), высушивали в печи при 60C и определяли радиоактивность как число импульсов в минуту (СРМ) с использованием детектора TopCount. Процент специфического связывания рассчитывали как процент связывания в отсутствие любого ингибитора, с поправкой на связывание в присутствии 2 мкМ PGE2. Данные анализировали с использованием следующего 4-параметрического нелинейного логистического уравнения (ABase уравнение 205): y = (A+B-A)/(1+C/x)D, где y = % специфического ингибирования, A = нижнее значение кривой; B = верхнее значение кривой; C = относительная IC50 = концентрация, вызывающая 50% ингибирование, основываясь на изменении данных от верхнего до нижнего значения; D = коэффициент Хилла = наклон кривой. Расчет Ki из IC50 оценивали как(Ki = IC50/(1 + [L]/Kd), где [L] представляет собой концентрацию лиганда). Соединения согласно примерам 1-9 настоящего документа тестировали, в сущности, как описано выше и показали значение Ki дляhEP4 ниже, чем примерно 1 мкМ. Таблица 1. Тест на связывание примера 1 с человеческими рецепторами ЕР 1, ЕР 2, EP3 и ЕР 4 in vitro Более конкретно, по существу, следуя процедурам, как описано выше, данные в табл. 1 показывали,что соединение примера 1 связывается с hEP4 при низких наномолярных концентрациях. Данные, приведенные в табл. 1, также демонстрируют, что соединение примера 1 связывается с hEP4 сильнее, чем сhEP1, hEP2 и hEP3, показывая селективность в отношении рецептора hEP4. Функциональная антагонистическая активность по отношению к ЕР 4 человека in vitro Анализы проводили в рекомбинантных клетках HEK293, стабильно экспрессирующих рецептор ЕР 4 человека. Клеточные линии поддерживали культивированием в среде DMEM с высоким содержанием глюкозы и пиридоксина гидрохлорида (Invitrogen) с добавлением 10% фетальной бычьей сыворотки(FBS), 1 мМ пирувата натрия, 10 мМ HEPES, 500 мкг/мл генетицина и 2 мМ L-глутамина. Конфлюентные культуры выращивали при 37 С в атмосфере, содержащей 5% CO2. Клетки собирали, используя 2,5% Трипсин-ЭДТА, суспендированный в сублимационной среде (FBS с 6% ДМСО) при концентрации 107 клеток/мл, и аликвоты хранили в жидком азоте. Непосредственно перед анализом клетки оттаивали в среде DMEM, центрифугировали и ресуспендировали в цАМФ (сАМР) буфере. Ингибирование PGE2-стимулированного образования цАМФ (сАМР) антагонистами ЕР 4 измеряли с помощью HTRF (Cisbio каталожный номер 62 АМ 4 РЕВ). Аликвоту, эквивалентную 4000 клеток, инкубировали с 50 мкл буфера для анализа цАМФ, содержащего PGE2 в концентрации, заданной для получения EC80 (0,188 нМ PGE2 от Sigma, каталожный номер Р 5640-10 мг) и антагонисты ЕР 4 при комнатной температуре в течение 20 мин. Буфер для анализа цАМФ содержал 500 мл HBSS, 0,1% BSA, 20 мМHEPES и 200 мкМ IBMX (Sigma I5879). CJ-042794 (4-(1S)-1-[(5-хлор-2-[(4-фторфенил)окси]фенилкарбонил)амино]этилбензойную кислоту) использовали как положительный контроль. Для определения количества цАМФ коньюгат цАМФ-d2 и анти-цАМФ-криптат в буфере для лизиса инкубировали с клетками, обработанными при комнатной температуре в течение 1 ч. Сигнал HTRF фиксировали с помощью спектрофотометра для считывания планшетов EnVision (Perkin-Elmer), чтобы вычислить соотношение флуоресценции при 665 нм к флуоресценции при 620 нм. Исходные данные преобразовы- 14024392 вали в количество цАМФ (пмоль/лунка) с помощью стандартной кривой цАМФ, полученной для каждого эксперимента. Данные анализировали с использованием следующего 4-параметрического нелинейного логистического уравнения (ABase уравнение 205): y = (A+B-A)/(1+C/x)D, где y = % специфического ингибирования, A = нижнее значение кривой; B = верхнее значение кривой; C = относительнаяIC50 = концентрация, вызывающая 50% ингибирование, основываясь на изменении данных от верхнего до нижнего значения; D = Коэффициент Хилла = наклон кривой. По существу, следуя процедурам, как описано выше, соединения согласно примерам 1-9 настоящего документа, тестировали, в сущности, как описано выше и показали значение IC50 ниже, чем примерно 1 мкМ. Более конкретно, по существу, следуя процедурам, как описано выше, пример 1 имеет IC50 6,92,5 нМ (n=5), определенную относительно ЕР 4 человека. Это демонстрирует, что соединения согласно примерам 1-9 являются потенциальными антагонистами ЕР 4 человека in vitro. Функциональная антагонистическая активность по отношению к ЕР 4 крыс in vitro кДНК ЕР 4 крыс (Genebank Accession NM03276) клонировали в 3,1 вектор пкДНК (pcDNA) и затем трансфицировали в клетки HEK293 для экспрессии рецептора. Наращивали количество стабильного клона ЕР 4 крыс и затем замораживали как банк клеток для будущего скрининга соединений. Для тестирования соединений антагонистов в клетках rEP4, замороженные клетки оттаивали и затем ресуспендировали в буфере для анализа цАМФ (сАМР). Буфер для анализа цАМФ готовили с использованием HBSS без добавления фенолового красного (Hyclone, SH30268) с добавлением 20 мМ HEPES (Hyclone, SH30237),0,1% BSA (Gibco, 15260) и 125 мкМ IBMX (Sigma, 15879). Клетки помещали в 96-луночные плоскодонные планшеты с половинным объемом лунок из полистирола (Costar 3694). Соединения последовательно разводили ДМСО, чтобы получить 10-точечные концентрационные кривые. Затем разбавленные соединения добавляли в буфер для анализа цАМФ, содержащий PGE2 (Cayman 14010, в концентрации, заданной для получения EC80) в соотношении ДМСО/буфер 1/100. Клетки обрабатывали соединениями в присутствии PGE2 (концентрация EC80) в течение 30 мин при комнатной температуре. Количество цАМФ,произведенного из клеток, подсчитывали с применением набора для анализа HTRF assay kit (Cisbio 62AM4PEC). Планшеты считывали с помощью спектрофотометра для считывания планшетов EnVision(Perkin-Elmer), используя протокол, оптимизированный для HTRF. IC50 рассчитывали с помощью нелинейной регрессии программы Graphpad Prism (v. 4), сигмоидальной кривой дозовой зависимости. По существу, следуя процедурам, как описано выше, соединения согласно примерам 1-9 настоящего документа тестировали, в сущности, как описано выше и показали значение IC50 ниже, чем примерно 1 мкМ. Более конкретно, по существу, следуя процедурам, как описано выше, соединение примера 1 имеет IC50 15 нМ, определенную относительно ЕР 4 крыс. Это демонстрирует, что соединения согласно примерам 1-9 являются потенциальными антагонистами ЕР 4 крыс in vitro. Антагонистическая активность в цельной крови человека in vitro Ингибирующие эффекты PGE2 на LPS-индуцированной продукции TNF (фактора некроза опухолей) из макрофагов/моноцитов, как полагают, опосредованы рецепторами ЕР 4 (См Murase, A., et al., LifeSciences, 82:226-232 (2008. Способность соединения примера 1 к обратному ингибирующему действиюPGE2 на ЛПС-индуцированную продукцию TNF в цельной крови человека является индикатором функциональной активности. Кровь собирали от нормальных доноров-добровольцев в вакуумные пробирки с гепарином натрия. Доноры не принимали НПВП или целекоксиб в течение 48 ч или глюкокортикоиды в течение двух недель до сдачи крови. Все пробирки/доноры объединяли в 50 мл конические центрифужные пробиркиFalcon и распределяли в 96-луночные планшеты для культивирования тканей по 98 мкл в лунку (Falcon 3072). Соединения разводили в ДМСО до конечного разведения в 100 раз и добавляли к крови по 1 мкл на лунку в трех повторностях, чтобы получить 7-точечные концентрационные кривые. Кровь предварительно обрабатывали соединениями при 37 С, в 5% увлажненной CO2 атмосфере, в течение 30 мин, после чего добавляли раствор 1 мг/мл липополисахарида (LPS) (Sigma 0111: В 4) в 0,2 мг/мл бычьего сывороточного альбумина (BSA)/PBS по 1 мкл в лунку, как с добавлением, так и без добавления 1 мМ PGE2(Cayman 14010) с получением конечной концентрации LPS 10 мкг/мл, как с добавлением, так и без добавления 10 нМ PGE2. Планшеты инкубировали в течение 20-24 ч при 37C в 5% увлажненной CO2 атмосфере. Планшеты центрифугировали при 1800g в течение 10 мин при 22C в центрифуге Eppendorf 5810R. Плазму удаляли с клеточного слоя и переносили в полипропиленовые планшеты с V-дном. Количество TNF в 2 мкл плазмы определяли с помощью коммерчески доступного иммуноферментного анализа (RD Systems DY210) с использованием планшетов Immulon 4 НВХ (Thermo 3855) и субстрата с 3,3',5,5'-тетраметилбифенил-4,4'-диамином (KPL 50-76-03). Планшеты считывали при A450-A650 с помощью спектрофотометра для считывания планшетов (Molecular Devices Versamax), используя программное обеспечение SOFTmaxPRO (v. 4,3.1). IC50 рассчитывали с помощью нелинейной регрессии программы Graphpad Prism (v. 4), сигмоидальной кривой дозовой зависимости. Результаты представили в виде геометрического значениястандартное отклонение; n = количество независимых определений. По существу, следуя процедурам, как описано выше, соединения согласно примерам 1-9 настоящего документа тестировали, в сущности, как описано выше и показали значение IC50 ниже, чем примерно 1IC50 12388 нМ (n=12). Это демонстрирует, что соединения согласно примерам 1-9 являются потенциальными антагонистами ЕР 4 в исследовании по индукции TNF в крови человека ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулыR4 представляет собой H, метил или этил илиR3 и R4 объединены с образованием циклопропильного кольца; или его фармацевтически приемлемая соль. 2. Соединение по п.1, отличающееся тем, что R2 представляет собой Н. 3. Соединение по п.1 или 2, отличающееся тем, что R3 представляет собой H и R4 представляет собой метил. 4. Соединение по любому из пп.1-3, отличающееся тем, что X представляет собой или его фармацевтически приемлемая соль. 6. Соединение по п.5, представляющее собой или его фармацевтически приемлемая соль. 7. Гидрохлоридная соль соединения по п.6, представляющая собой
МПК / Метки
МПК: A61K 31/45, C07D 211/60, A61P 29/00, A61P 19/00
Метки: соединения, феноксиэтилпиперидиновые
Код ссылки
<a href="https://eas.patents.su/17-24392-fenoksietilpiperidinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Феноксиэтилпиперидиновые соединения</a>
Соединения 3-аминокарбазола, фармацевтическая композиция, содержащая указанные соединения, и способ их получения

Номер патента: 12786
Опубликовано: 30.12.2009
Авторы: Колетта Изабелла, Драгоне Патриция, Мангано Джорджина, Ализи Мария Алессандра, Руссо Винченцо, Каццолла Никола, Поленцани Лоренцо, Фурлотти Гвидо
МПК: A61P 35/00, A61K 31/403, C07D 209/88...
Метки: композиция, соединения, указанные, способ, фармацевтическая, получения, 3-аминокарбазола, содержащая
Формула / Реферат:
1. Соединение 3-аминокарбазола, отличающееся тем, что его выбирают из группы, включающей соединения из таблицы и их фармацевтически приемлемые соли. 2. Фармацевтическая композиция, отличающаяся тем, что она содержит терапевтически эффективную дозу соединения 3-аминокарбазола, выбранного из группы, включающей соединения из таблицы по п.1 или их фармацевтически приемлемую соль, вместе по меньшей мере с одним фармацевтически приемлемым инертным...
Пуриновые соединения

Номер патента: 18386
Опубликовано: 30.07.2013
Авторы: Гвидетти Росселла, Эстлз Питер Чарльз, Холлиншед Шон Патрик, Тидуэлл Майкл Вэйд
МПК: C07D 473/34, A61K 31/52, A61P 25/04...
Метки: соединения, пуриновые
Формула / Реферат:
1. Соединение формулыгде R1 выбран из Н, F, Cl, C1-C2-алкила, CF3, циклопропила, ОСН3, OCF3 и CN;R2 выбран из тетрагидрофуранила, тетрагидропиранила, метилового эфира азетидин-1-карбоновой кислоты и тетрагидротиофен-1,1-диоксида;R3 представляет собой Н или совместно с R4 образует конденсированный пирролидин-2-он;R4 выбран из C1-C2-алкила, C1-С2-фторалкила, циклопропила и СОСН3;R5 выбран из Н, СН3 и CF3;n равно 0 или 1;X1 и X3 независимо выбраны...
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Йеом Зи-Хо, Ким Мин-Дзунг, Хонг Санг Йонг, Хан Хее Оон, Хур Гвонг-Чеунг, Ким Дзи Янг, Ким Киоунг-Хее, Бу Сеонг Чеол, Кох Дзонг Сунг, Ким Сунг Хо, Йео Донг-Дзун, Ким Геун Тае, Ли Чанг-Сеок, Йим Хиеон Дзоо, Лим Донгчул, Ким Хие Дзин, Ким Сунгсуб, Квон Ох Хван, Коо Ки Донг
МПК: A61P 3/10, A61K 31/444, A61K 31/452...
Метки: активного, указанные, ингредиента, содержащие, также, способы, дипептидилпептидазы-iv, качестве, фармацевтические, соединения-ингибиторы, соединения, композиции, получения
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Соединения, ингибирующие ферменты гистондеацетилазы, и фармацевтическая композиция, содержащая данные соединения

Номер патента: 22964
Опубликовано: 31.03.2016
Авторы: Шмидт Дарби, Балоглу Эркан, Гхош Шомир, Лобера Мерседес
МПК: A61K 31/497
Метки: соединения, ферменты, данные, гистондеацетилазы, композиция, ингибирующие, содержащая, фармацевтическая
Формула / Реферат:
1. Соединение формулы (I-а)где R1 представляет собой галоген(C1-C2)алкил, где указанный галоген(C1-C2)алкил содержит по меньшей мере 2 атома фтора;А представляет собой необязательно замещенный (С3-С6)циклоалкил, фенил или 5-6-членный гетероарил, где указанный циклоалкил, фенил или гетероарил необязательно замещен 1-3 группами, независимо выбранными из (C1-C4)алкила, галогена, циано и (C1-C4)алкокси;Z представляет собой -C(=O)NRX-; -NRXC(=O)-,...
Хиральные бифосфиновые соединения, комплексы на их основе и способ получения хирального бифосфинового соединения

Номер патента: 1377
Опубликовано: 26.02.2001
Авторы: Россен Кай, Воланте Ральф П., Пай Филип
МПК: C07F 9/50, C07C 25/22, C07B 53/00...
Метки: получения, хирального, комплексы, соединения, бифосфиновые, хиральные, способ, основе, бифосфинового
Формула / Реферат:
1. Хиральные бифосфины формулы где R представляет C1-4-алкил, С3-6-циклоалкил или арил, незамещенный или замещенный -F, -СН3, -СF3 или СН3О-; и Х1 и Х2 связывают два R2Р-замещенные бензола и независимо образуют 2-4-членную связывающую группу, состоящую из 2-4 атомов углерода и одного незамещенного или замещенного гетероатома, выбранных из О, S, SO, SO2 или 2. Соединение по п.1, в котором число атомов в связи X1 является таким же, как и...
Предыдущий патент: Производные бензамида для ингибирования активности abl1, abl2 и bcr-abl1
Следующий патент: Синергетическая гербицидная композиция
Случайный патент: Рабочее колесо шламового насоса