Способ получения эхинокандиновых соединений
Номер патента: 7964
Опубликовано: 27.02.2007
Авторы: Боффелли Филипп, Эльтер Мишель, Дру Серж, Лемэтр Ги, Ферру Дидье, Бруйар Аньес, Паладино Жозеф, Колладан Колетт







Формула / Реферат
1. Способ получения соединений формулы (I)

в которой R представляет собой линейную или разветвленную, или циклическую цепь, содержащую до 30 атомов углерода, необязательно, содержащую один или несколько гетероатомов и один или несколько гетероциклов, включающий в себя следующие стадии:
а) соединение формулы (II)

подвергают действию кислоты формулы R-СО2Н, указанная кислота находится, если это приемлемо, в выделенной или невыделенной активированной форме, с получением соединения формулы (III)

b) если это приемлемо, соединение формулы (III) подвергают реакции алкилирования спиртом по положению 5, под действием спирта формулы Alk-ОН, в присутствии PPTS, Alk представляет собой алкильный радикал, содержащий 1-4 атома углерода, с получением соединения формулы (III')

с) соединение формулы (III) или (III') подвергают реакции дегидратации, с получением соединения формулы (IV)

d) соединение формулы (IV) подвергают реакции восстановительного аминирования под действием этилендиамина в присутствии восстановительного агента, такого как NаВН3СN, в присутствии кислоты Льюиса, или NaBH(OCOR'), где OCOR' представляет собой Boc-L-Pro, Bzl-L-Pro или любую другую оптически активную аминокислоту, а также любую другую хиральную или нехиральную карбоновую кислоту, с получением соединения формулы (I), как определено выше, содержащего преимущественно один из активных изомеров,
затем указанное соединение формулы (I) подвергают, если это приемлемо, и в любом приемлемом порядке, одной или нескольким из следующих далее операций:
хроматографированию;
кристаллизации;
действию основания;
преобразованию в форму соли.
2. Способ по п.1, в котором соединения формул (I), (III), (III') или (IV) содержат радикал R, представляющий собой следующие группы:

3. Способ по п.1, в котором соединения формулы (I), (III), (III'), (IV) содержат радикал R, представляющий собой следующую группу:

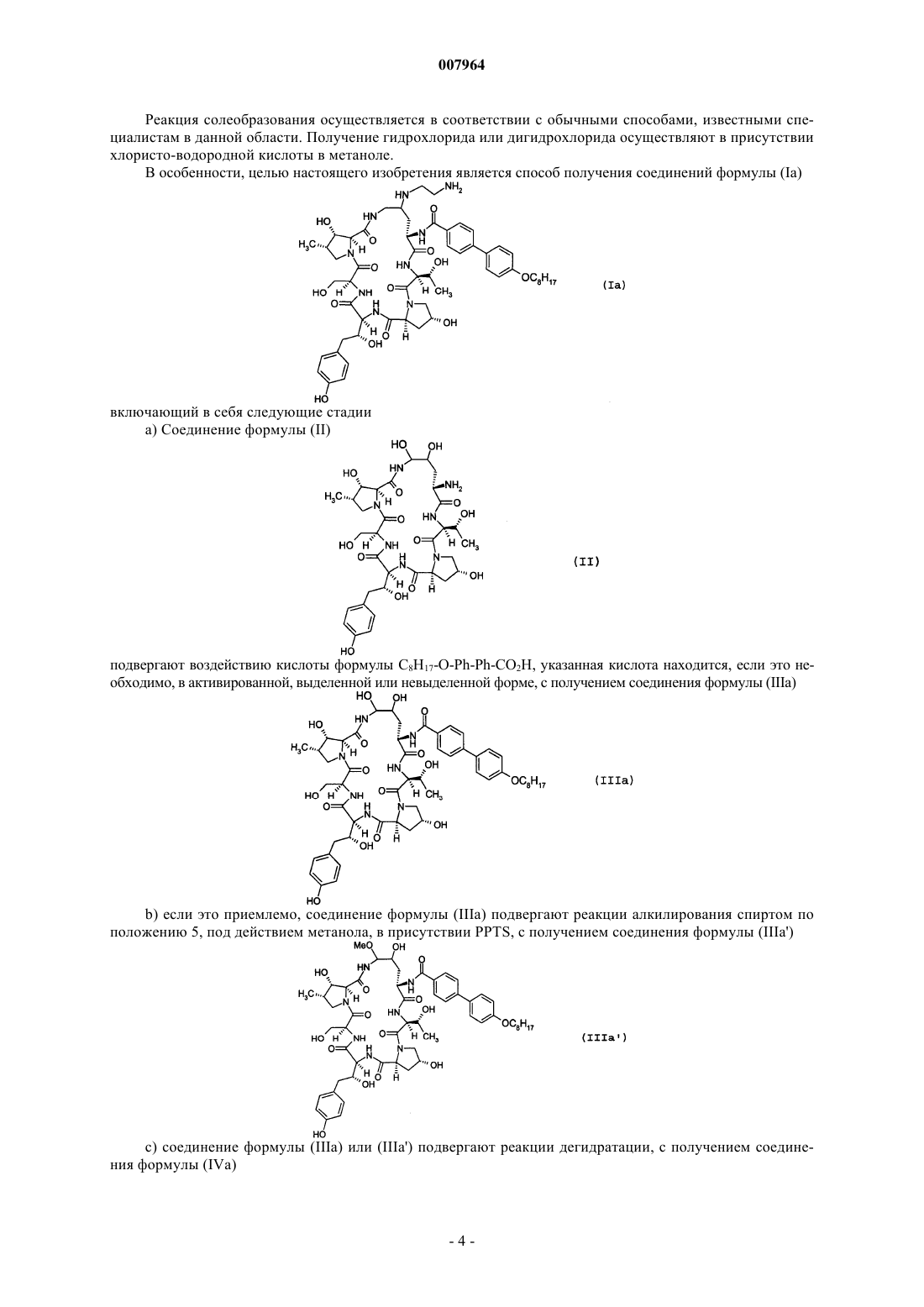
4. Способ получения соединений формулы (Iа)

включающий в себя следующие стадии:
а) соединение формулы (II)

подвергают действию кислоты формулы С8Н17-О-Рh-Рh-СО2Н, указанная кислота находится, если это приемлемо, в активированной, выделенной или невыделенной форме, с получением соединения формулы (IIIа)

b) если это приемлемо, соединение формулы (IIIа) подвергают реакции алкилирования спиртом по положению 5, под действием метанола, в присутствии PPTS, с получением соединения формулы (IIIа')

с) соединение формулы (IIIа) или (IIIа') подвергают реакции дегидратации, с получением соединения формулы (IVa)

d) соединение формулы (IVa) подвергают реакции восстановительного аминирования под действием этилендиамина, в присутствии восстановительного агента, такого как NaBH3CN, связанного с TiCl4, NaBH(Boc-L-Pro)3 или NaBH(Bzl-L-Pro)3, с получением соединения формулы (Iа), как определено выше, преимущественно один из активных изомеров, указанное соединение формулы (Iа) подвергают одной или нескольким из следующих далее операций, в приемлемом порядке:
хроматографираванию;
кристаллизации;
действию основания;
преобразованию в форму соли под действием хлористо-водородной кислоты.
5. Способ по п.4, отличающийся тем, что соединение формулы (Iа) последовательно подвергается следующим операциям:
a) очистке хроматографией на силикагеле, затем хроматографией с обращенной фазой с использованием смеси органических растворителей, воды и трифторуксусной кислоты с получением соли трифторуксусной кислоты соединения формулы (Iа);
b) действию основания, например, действию водного раствора бикарбоната натрия, с получением соединения формулы (Iа) в форме основания;
c) преобразованию в форму соли под действием хлористо-водородной кислоты, с получением соответствующей соли, а именно, дигидрохлорида соединения формулы (Iа).
6. Способ по любому из пп.1-5, отличающийся тем, что реакцию активирования кислоты осуществляют в присутствии пентафторфенола, N-гидроксисукцинимида или, необязательно, N-гидроксибензотриазола.
7. Способ по любому из пп.1-5, отличающийся тем, что реакцию ацилирования в присутствии выделенной активированной кислоты осуществляют в присутствии диизопропилэтиламина.
8. Способ по любому из пп.1-5, отличающийся тем, что реакцию активирования, а затем реакцию ацилирования в присутствии невыделенной активированной кислоты осуществляют в присутствии N-метилпирролидона и DMTMM.
9. Способ по любому из пп.1-5, отличающийся тем, что реакцию дегидратации осуществляют в присутствии AIBB и, если это приемлемо, МgI2.
10. Способ по любому из пп.1-7, отличающийся тем, что реакцию дегидратации осуществляют в присутствии HBr-АсОН и MgI2.
11. Способ по пп.9 и 10, отличающийся тем, что продукт, получаемый от дегидратации, очищают кристаллизацией из смеси ДМФ/ацетон или ДМФ/AcOEt.
12. Способ по любому из пп.1-5, отличающийся тем, что реакцию восстановительного аминирования осуществляют в присутствии восстановительного агента, выбранного из NаВН3СN, связанного с TiCl4, NaBH(Boc-L-Pro)3 и NaBH(Bzl-L-Pro)3.
13. Способ получения соединения формулы (III) или (IIIа) ацилированием соединений формулы (II), как определено в любом из пп.1-4 и 6-8.
14. Способ получения соединения формулы (IV) или (IVa) дегидратацией соединений формулы (III), (III'), (IIIa) или (III'а), как определено в любом из пп.1-4 и 9-11.
15. Способ получения соединения формулы (I) или (Iа) восстановительным аминированием соединений формулы (IV) или (IVa), как определено в любом из пп.1-4 и 12.
16. Дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-[[4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в 4S или 4R форме или в форме смеси стереоизомеров.
17. Дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-[[4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в форме активного изомера А, который может быть получен способом, описанным в пп.1-15.
18. Применение дигидрохлорида 1-[4-[(2-аминоэтил)амино]-N2-[[4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в форме 4R или 4S изомера или в виде смеси указанных стереоизомеров в качестве лекарственного средства.
19. Применение дигидрохлорида 1-[4-[(2-аминоэтил)амино]-N2-[[4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-cepинэхинокандина В в виде активного изомера А в качестве противогрибкового лекарственного средства.
20. Фармацевтическая композиция, содержащая дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-[[4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В, как определено в п.16 или 17, и фармацевтически приемлемый носитель.
21. Промежуточные соединения формул (III') или (III'а), как определено в пп.1 и 6.
22. Промежуточные соединения формул (IV), R является таким, как определено в люсюь из пп.1-2, или (IV'a), где R представляет собой Ph-Ph-OC8H17

Текст
007964 Целью настоящего изобретения является способ получения эхинокандиновых соединений, промежуточных соединений и полученных из них продуктов, и их применение в качестве противогрибкового лекарственного средства. Из литературы известно множество соединений, имеющих противогрибковую активность. В частности, могут быть рассмотрены эхинокандиновые соединения, как описано в заявке WO99/29716. Целью настоящего изобретения является создание нового способа получения эхинокандиновых соединений и для получения их новых солей. Эхинокандин может присутствовать в форме двух эпимеров, называемых изомер А и изомер В, соответствующих R и S конфигурациям заместителя в положении 4. Одной из целей настоящего изобретения является достижение, в ходе реализации способа, количественного преобладания одного из двух изомеров, то есть, по меньшей мере, более 50% изомера, называемого форма А, соответствующего фармакологически активному соединению формулы (I), что не является случаем, соответствующим способу,раскрытому в заявке WO99/29716, в ходе которого активный изомер А, получаемый в ходе реализации способа, перед очисткой на хроматографической колонке, находится в количественном меньшинстве. Еще одной целью настоящего изобретения является исключение операций очистки промежуточных продуктов с помощью хроматографии и получения кристаллизованных продуктов, что делает возможным значительное повышение выходов. Следовательно, целью настоящего изобретения является способ получения соединений формулы (I) в которой R представляет собой линейную или разветвленную, или циклическую цепь, содержащую до 30 атомов углерода, необязательно, содержащую один или несколько гетероатомов и один или несколько гетероциклов, включающий в себя следующие стадии а) Соединение формулы (II) подвергают действию кислоты формулы R-CO2H, где R является таким, как определено выше, указанная кислота находится, если приемлемо, в выделенной или невыделенной активированной форме, с получением соединения формулы (III)b) если приемлемо, соединение формулы (III) подвергают реакции алкилирования спиртом по по-1 007964 ложению 5 действием спирта формулы Alk-OH, в присутствии PPTS, Alk представляет собой алкильный радикал, содержащий 1-4 атома углерода, с получением соединения формулы (III') с) соединение формулы (III) или (III') подвергают реакции дегидратации, с получением соединения формулы (IV)d) соединение формулы (IV) подвергают реакции восстановительного аминирования под действием этилендиамина в присутствии восстановительного агента, такого как NаВН 3 СN, в присутствии кислоты Льюиса, или NaBH(OCOR'), где OCOR' представляет собой Boc-L-Pro, Bzl-L-Pro или любую другую оптически активную аминокислоту, а также любую другую хиральную или нехиральную карбоновую кислоту, с получением соединения формулы (I), как определено выше, содержащего количественное большинство одного из активных изомеров,затем указанное соединение формулы (I) подвергают, если необходимо, и в любом приемлемом порядке, одной или нескольким из следующих далее операций: очистке хроматографией; очистке кристаллизацией; действию основания; преобразованию в форму соли. Предпочтительно целью настоящего изобретения является способ, определенный ранее, в котором соединения формул (I), (III), (III') или (IV) содержат радикал R, представляющий собой следующие группы: Предпочтительно целью настоящего изобретения является способ, определенный ранее, в котором соединения формул (I), (III), (III') или (IV) содержат радикал R, представляющий собой следующую группу: Соединение формулы (II) получают в соответствии со способом, описанным в заявке WO99/29716 страница 18 (Получение 2, "ядро дезоксимулундокандина"). Активирование кислоты формулы R-СО 2 Н осуществляют в соответствии со стандартными методами, известными специалистам в данной области (Journet M. et al., J. Org. Chem. (1999) , 64, 2411-2417;Pochlauer P. et al. Tetrahedron 54 (1998) 3489-3494; Kunushima et al. Tetrahedron (1999) 55 13159-13170). В частности, пентафторфенол используют для получения активированного сложного пентафторфенолового эфира, который выделяют перед использованием в реакции ацилирования амина. Другой способ пред-2 007964 ставляет собой использование N-гидроксисукцинимида (необязательно, N-гидроксибензотриазола), с получением активированного сложного(необязательно,Nгидроксибензотриазолового) эфира, который также выделяют. Стадия ацилирования, которая следует далее, для получения соединения формулы (III), осуществляется в присутствии основания, в соответствии со способами, известными специалистам в данной области, и иллюстрируется в примерах, описываемых далее. Также возможно осуществление ацилирования без выделения активного сложного эфира и без добавления основания, проводя реакцию в присутствии хлорида 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-мил-морфолиния (DMTMM). Эта последняя альтернатива обладает преимуществом исключения стадии выделения и, следовательно, повышения выходов. Стадия дегидратации соединения формулы (III), с получением соединения формулы (IV), может осуществляться следующим образом. Стадия дегидратации осуществляется в присутствии галогеноводородной кислоты, такой как НВr, в присутствии MgI2. В этом случае не является необходимым проходить через промежуточное соединение формулы (III'). В качестве одного из вариантов, может быть желательным алкилировать гидроксильную группу в положении 5, перед переходом к стадии дегидратации. Например, метилирование может осуществляться с помощью метанола, в присутствии PPTS (паратолуолсульфоната пиридиния), с получением соединения формулы (III'), где Alk представляет собой метил. Наконец, эта реакция дегидратации может также осуществляться в присутствии альфа-ацетокси изобутирил бромида (AIBB) в диоксане. В этом последнем случае, в реакцию может быть введено либо соединение формулы (III'), либо непосредственно соединение формулы (III), в присутствии MgI2. Предпочтительно соединение, полученное после реакции дегидратации, затем очищают кристаллизацией из смеси ДМФ (диметилформамид)/MeCN (ацетонитрил). Также могут использоваться смеси следующих растворителей: ДМФ/ацетон и ДМФ/AcOEt (этилацетат). Реакции восстановительного аминирования соединений формулы (IV) с получением эхинокандина формулы (I), осуществляют в соответствии со способами, описанными в литературе и известными специалистам в данной области (Yamada K. et al. J. Chem. Soc. Perkin Trans I (1983) 265-270; Hutchins R. et al.NaBH3CN, проводя сочетание с TiCl4 или любой другой кислотой Льюиса, или в присутствииNaBH(OCOR'), где OCOR' представляет собой Boc-L-Pro, Bzl-L-Pro или любую другую оптически активную аминокислоту, а также, любую другую хиральную или нехиральную карбоновую кислоту. Сочетание восстановительного реагента NaBH3CN с кислотой Льюиса, в частности, с TiCl4, является существенным для селективности и, следовательно, для получения смеси изомера А и изомера В, в которой активный изомер А находится в количественном большинстве. Как правило, получают отношение изомера А, большее чем 70%, и в частности, находящееся в пределах между 80 и 85%. Похожая ситуация происходит и с другими реагентами, рассмотренными выше. В частности, для соединения формулы (Iа), на этой стадии, получают отношение изомер А/изомер В, равное 80-85%/20-15%, соответственно, в то время как способ, описанный в WO 97/29716, приводит к получению смеси, в которой изомер А находится в количественном меньшинстве или в равном отношении (рацемическая смесь А и В). Во время стадии восстановительного аминирования, перед восстановлением, может быть, если необходимо, выделен промежуточный продукт реакции, и, следовательно, он представляет собой цель настоящего изобретения. Он представляет собой имидазолидин формулы (IV) или (IV'а), где R представляет собой группу -Ph-Ph-O-C8H17. Может быть желательной очистка полученного продукта или продуктов, с помощью хроматографии или перекристаллизации. Эта операция осуществляется в соответствии с обычными способами, известными специалистам в данной области. Когда полученный продукт, в частности, находится, после хроматографии, в форме соли трифторуксусной кислоты, желательно разрушение этой соли под действием основания, с получением соединения формулы (I) в форме основания.-3 007964 Реакция солеобразования осуществляется в соответствии с обычными способами, известными специалистам в данной области. Получение гидрохлорида или дигидрохлорида осуществляют в присутствии хлористо-водородной кислоты в метаноле. В особенности, целью настоящего изобретения является способ получения соединений формулы (Iа) включающий в себя следующие стадии а) Соединение формулы (II) подвергают воздействию кислоты формулы C8H17-O-Ph-Ph-CO2H, указанная кислота находится, если это необходимо, в активированной, выделенной или невыделенной форме, с получением соединения формулы (IIIа)b) если это приемлемо, соединение формулы (IIIа) подвергают реакции алкилирования спиртом по положению 5, под действием метанола, в присутствии PPTS, с получением соединения формулы (IIIа') с) соединение формулы (IIIа) или (IIIа') подвергают реакции дегидратации, с получением соединения формулы (IVa)d) соединение формулы (IVa) подвергают реакции восстановительного аминирования действием этилендиамина, в присутствии восстановительного агента, такого как NaBH3CN, связанного с TiCl4,NaBH(Boc-L-Pro)3 или NaBH(Bzl-L-Pro)3, с получением соединения формулы (Iа), как определено выше,содержащего количественное большинство одного из активных изомеров, указанное соединение формулы (Iа) подвергают одной или нескольким из следующих далее операций, в приемлемом порядке: хроматографии; кристаллизации; воздействию основания; преобразованию в форму соли под действием хлористо-водородной кислоты. Соединение формулы (Iа) может находиться в форме смеси двух эпимеров (асимметричный атом углерода в положении 4), которые называют изомер А или изомер В. Соединение формулы (I), полученное в соответствии со способом, определенным выше, делает возможным получение смеси изомер А/изомер В, в соотношении приблизительно 70-85%/30-15%. Стадии очистки/кристаллизации и преобразования в форму соли, которые осуществляют после реакции восстановительного аминирования, дают возможность для селективного получения активного эпимера А соединения формулы (Iа). Предпочтительно соединение формулы (Iа) очищают с помощью хроматографии на силикагеле, затем с помощью хроматографии с обращенной фазой, используя смеси органических растворителей, воды и трифторуксусной кислоты, для получения соли трифторуксусной кислоты соединения формулы (Iа). Затем эту соль подвергают действию основания, например, действию водного раствора бикарбоната натрия, с получением соединения формулы (Iа) в форме основания. Затем полученное основание преобразуют в форму соли действием хлористо-водородной кислоты, с получением соответствующей соли, а именно, дигидрохлорида соединения формулы (Iа). По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что соединение формулы (Iа) подвергают последовательно следующим операциям:a) очистке хроматографией на силикагеле, затем хроматографии с обращенной фазой, с использованием смеси органических растворителей, воды и трифторуксусной кислоты, с получением соли трифторуксусной кислоты соединения формулы (Iа);b) действию основания, например, действию водного раствора бикарбоната натрия, с получением соединения формулы (Iа) в форме основания;c) преобразованию в форму соли под действием хлористо-водородной кислоты, с получением соответствующей соли, а именно, дигидрохлорида соединения формулы (Iа). По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакция активирования кислоты осуществляется в присутствии пентафторфенола, N-гидроксисукцинимида или, необязательно, N-гидроксибензотриазола. По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакция ацилирования в присутствии выделенной активированной кислоты осуществляется в присутствии диизопропилэтиламина. По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакцию активирования, а затем реакцию ацилирования в присутствии выделенной активированной кислоты осуществляют в присутствии N-метилпирролидона и DMTMM. По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакция дегидратации осуществляется в присутствии AIBB и, если это приемлемо, MgI2. По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакция дегидратации осуществляется в присутствии НВr-АсОН и MgI2. По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что продукт, получаемый после дегидратации, очищают кристаллизацией из смеси диметилформамид (ДМФ)/ацетонитрил, ДМФ/ацетон или ДМФ/AcOEt.-5 007964 По этой причине совершенно конкретной целью настоящего изобретения является способ, определенный ранее, отличающийся тем, что реакцию восстановительного аминирования осуществляют в присутствии восстановительного агента, выбранного из NaBH3CN, связанного с TiCl4/NaH(Boc-L-Pro)3 иNaH(Bzl-L-Pro)3. По этой причине целью настоящего изобретения является также дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-Lтреонин]-5-L-серин эхинокандина В в 4S или 4R форме, или в форме смеси этих двух стереоизомеров. Способ, описанный ранее, дает возможность для получения изомера А соединения формулы (I) или(Iа) в количественном большинстве, до разделения изомеров с помощью хроматографии. По этой причине, целью настоящего изобретения также является дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-Lтреонин]-5-L-серин эхинокандина В в форме активного изомера А, полученного с помощью способа, как описано ранее. Целью настоящего изобретения является также дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В в 4S или 4R форме, или в форме смеси этих двух стереоизомеров, в качестве лекарственного средства и, в частности, в качестве, противогрибкового лекарственного средства. Наконец, целью настоящего изобретения является фармацевтические композиции, содержащие дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]-карбонил]-L-орнитин]4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В в 4S или 4R форме, или в форме смеси этих двух стереоизомеров, и фармацевтически приемлемый носитель. 1-[4-[(2-Aминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-cepин эхинокандин В получают в заявке WO 99/29716 в форме основания или соли трифторуксусной кислоты (пример 14). Преимущество дигидрохлорида заключается в лучшей стабильности и растворимости в воде. Кроме того, дигидрохлорид представляет собой предпочтительную фармацевтически приемлемую соль, которая представляет собой цель настоящего изобретения. Пример 1. Дигидрохлорид 1-[4-[(2-аминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В.I) Первая стадия. Ацилирование соединения формулы (IIа) и получение соединения формулы (III). 1-[(4R,5R)-4,5-Дигидрокси-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4 гидроксифенил)-L-треонин]-5-L-cepин эхинокандин В. Способ 1. C помощью активированного сложного эфира выделенного пентафторфенола. Стадия 1. Получение активированного сложного эфира. Пентафторфенил 4'-октилокси-бифенил-4-карбоксилат. 100 г 4-октилоксибифенил-4' карбоновой кислоты и 62 г пентафторфенола вводят в 1 л дихлорметана. Перемешивание осуществляют в течение 15 мин и добавляют 69,6 г N,N'дициклогексилкарбодиимида в виде раствора 500 мл дихлорметана, в течение 15 мин. Темно-бежевую суспензию перемешивают в течение 20 ч при температуре окружающей среды. Дициклогексилмочевину фильтруют и фильтрат концентрируют. Дистилляцию до получения постоянного объема осуществляют с регулярным добавлением этанола до получения температуры паров 74 С. Реакционную среду охлаждают до температуры окружающей среды с последующим перемешиванием, в течение 1 ч, фильтрованием, а затем промывкой этанолом. После сушки получают 145,7 г 5 целевого продукта в форме кристаллов. Выход: 96,5%. ЯМР (CDCl3): 8,23-7,72 (АА'ВВ') 4 Н; 7,01-7,60 (АА'ВВ') 4 Н; 4,02 (т) 2 Н; 1,82 (квинт.) 2 Н; 1,48(квинт.) 2 Н; 1,32 (м) 8 Н; 0,89 (т) 3 Н. Стадия 2. Ацилирование. Вводят 23,2 г соединения формулы (II) ("ядра деоксимулундокандина", полученного в соответствии с получением 2 WO 99/29716) и 14,1 г сложного эфира, полученного выше, в 60 мл ДМФ. В суспензию вводят 6,7 мл диизопропилэтиламина и перемешивание осуществляют в течение 24 ч, в атмосфере азота,при температуре окружающей среды. Гомогенную реакционную среду выливают в 1 л воды, в течение 10 мин, при этом осуществляют острожное перемешивание, суспензию перемешивают в течение 2 ч, фильтруют, и твердый продукт промывают водой. Твердый продукт сушат в вакууме при комнатной температуре, затем нагревают с обратным холодильником в 100 мл метиленхлорида, в атмосфере азота, при этом перемешивают в течение 2 ч (40 С), с последующим охлаждением до температуры окружающей среды в течение 1 ч, перемешиванием в течение 1 ч, фильтрованием твердого продукта и промывкой его метиленхлоридом 3 раза и сушкой в вакууме при комнатной температуре. Получают 26,9 г экстрагированного продукта в форме бежевого твердого продукта. Выход: 91,5%. ТСХ: Rf: 0,13 пластинка с силикагелем; проявление УФ 254 нм; элюент: СН 2 Сl2-МеОН-вода: 86-13-1. ЯМР: (ДМСО). Треонин: 8,16 (1 Н), 4,85 (1 Н), 4,41 (1 Н), 1,13 (3 Н); -гидроксипролин: 4,42 (1 Н), 1,92-2,28 (2 Н), 4,44(2 Н), 1,25 (2 Н), 1,31 (2 Н), 1,27 (2 Н), 0,86 (3 Н). Способ 2. C помощью активированного сложного эфира выделенного HOSu. Стадия 1. Синтез активированного сложного эфира 2,5-диоксопирролидин 4'-октилокси-бифенил-4 карбоксилата. Вводят 9,3 г октилоксибифениловой кислоты, 93 мл дихлорметана, 3,8 г N-гидроксисукцинимида,6,3 г EDC и осуществляют перемешивание в течение 3 ч при температуре окружающей среды. Добавляют 6 мл воды, осуществляют перемешивание в течение 10 мин с последующим декантированием и повторным экстрагированием 45 мл дихлорметана. Органическую фазу промывают водой (3 раза по 45 мл) и сушат над сульфатом натрия. После высушивания в вакууме получают 12,05 г ожидаемого продукта в форме кристаллов. Выход: 99,9%. ЯМР CDCl3: 8,2-7,65 (АА'ВВ') 4 Н; 7,6-6,97 (АА'ВВ') 4 Н; 4,02 (т, 2 Н); 2,93 (шир.) 4 Н; 1,83 (квинт. 2 Н); 1,45 (м) 2 Н; 1,3 (м) 8 Н; 0,9 (т) 3 Н. Стадия 2. Ацилирование. 1,43 г сложного сукцинимидного эфира, как получено выше, растворяют в 6 мл ДМФ. Вводят 2,42 г"ядра диоксимулункандина" (полученного в соответствии с Получением 2, WO 99/29716) и 0,66 мл диизопропилэтиламина. Раствор перемешивают в течение 18 ч, при температуре окружающей среды. Вводят 35 мл воды и перемешивание осуществляют в течение 2 ч при температуре окружающей среды. После фильтрования твердый продукт извлекают в 30 мл воды, при перемешивании в течение 2 ч в реакционной емкости, с последующим фильтрованием и промывкой водой. Твердый продукт сушат в вакууме,при температуре окружающей среды и получают 2,75 г ожидаемого продукта в форме бежевого твердого продукта. Выход: 98,2%. Способ 3. Синтез путем непосредственного активирования с помощью DMTMM. 2,8 г диоксимулундокандина (полученного в соответствии с Получением 2, WO 99/29716) растворяют в 8,3 мл N-метилпиролидона. Добавляют 1,12 г октилоксибифениловой кислоты и 0,95 г 4-(4,6 диметокси-1,3,5-триазин-2-ил)-4-Ме-морфолиний хлорида (DMTMM). После перемешивания в течение 24 ч при температуре окружающей среды реакционную среду выливают в 133 мл воды, при перемешивании. Перемешивание осуществляют в течение 20 мин, твердый продукт фильтруют и промывают 3 раза водой (3 раза по 7 мл). После сушки в вакууме, при 40 С, получают 2,66 г ожидаемого продукта 4, в форме бежевого твердого продукта. Выход: 73,5%.II) Вторая стадия. Дегидратация (получение соединения формулы (IVa. 1-[N2-[4'-(Oктилокси)-[1,1'-бифенил]-4-ил]карбонил]-4-оксо-L-орнитин]-4-[4-(4-гидроксифенил)-Lтреонин]-5-L-серин-эхинокандин В. Способ 1. Через соединение формулы (III'а). Стадия 1. Алкилирование. 1-[(4R,5R)-4-Гидрокси,5-метокси-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандин В (III'а). 283 г продукта IIIа, полученного на первой стадии, растворяют в 5,67 л метанола. 22,7 г птолуолсульфоната пиридиния добавляют за 1 раз при перемешивании и перемешивание осуществляют в течение 6 ч, при нагревании с обратным холодильником (42 С), и в течение 12 ч при температуре окружающей среды. Концентрирование осуществляют в вакууме до двух остаточных объемов, затем добавляют 1,2 л воды. Суспензию перемешивают в течение 18 ч. Твердый продукт фильтруют и дважды промывают водой. После сушки в печи, в вакууме, при температуре окружающей среды, получают 270,5 г ожидаемого продукта в форме бежевого порошка. Выход: 94,2%. ЯМР: (ДМСО). Треонин: 8,11 (1 Н), 4,84 (1 Н), 4,41 (1 Н), 1,11 (3 Н); -гидроксипролин: 4,40 (1 Н), 1,91-2,27 (2 Н), 4,42-7 007964 7 ч. Добавляют 4,1 л насыщенного водного раствора бикарбоната натрия (NaHCO3), в течение 15 мин. Перемешивание осуществляют в течение 15 мин и диоксан дистиллируют в вакууме, при температуре,которая не превышает 30 С. Добавляют 3,2 л воды, и перемешивание осуществляют в течение 15 ч, при температуре окружающей среды. Твердый продукт фильтруют и дважды промывают водой. После сушки в печи, в вакууме, получают 251,2 г ожидаемого продукта в форме твердого продукта бледного коричневато-желтого цвета. Выход: 97,5%. Продукт кристаллизуют в соответствии с получением, описанным ниже. Способ 2. Начиная с продукта (IIIа), полученного на стадии а), под действием АIВВ/МgI2/диоксана. 23,26 г безводного йодида магния суспендируют в 500 мл диоксана, и осуществляют перемешивание в течение 30 мин. Вводят 12,25 мл -ацетоксиизобутил бромида (AIBB), и перемешивают суспензию бледного коричневато-желтого цвета при температуре окружающей среды, в течение 45 мин. Раствор 50 г продукта (IIIа), полученного на первой стадии, растворенный в 400 мл, диоксана добавляют в течение 1 ч, и капельную воронку промывают 25 мл диоксана. Суспензию перемешивают в течение 19 ч, при температуре окружающей среды. Раствор 5 г бикарбоната натрия, растворенного в 50 мл воды, вводят в течение 30 мин (рН 5-6, к концу добавления). Перемешивание осуществляют в течение 2 ч, с последующей дистилляцией в вакууме до остаточного объема 250 мл, при внутренней температуре, меньшей, чем 35 С. Восстанавливают атмосферное давление азота и добавляют 200 мл диметилформамида с последующей дистилляцией в вакууме до остаточного объема 250 мл и этот раствор выливают при температуре окружающей среды в 2,8 л воды. После промывки ДМФ перемешивание осуществляют в течение 1 ч. Твердый продукт фильтруют и промывают водой. Твердый продукт сушат в течение 24 ч в вакууме, при 30 С. Получают 46 г ожидаемого продукта в форме бежевого твердого продукта. Выход: 98,6%. Затем продукт кристаллизуют в соответствии со способом, описанным ниже. Способ 3. Начиная с продукта IIIа, полученного на первой стадии, под действием HBrAcOH/MgI2/MEK. 10 г продукта, полученного на стадии а), растворяют в 180 мл метилэтилкетона (МЕК). Вводят 4,65 г безводного йодида магния и 10 мл МЕК. Осуществляют перемешивание в течение 35 мин при температуре окружающей среды, с последующим охлаждением до 20 С. Вводят в АсОН 3 мл 33% раствора НВr. Суспензию перемешивают в течение 4 ч при 20 С. Добавляют 10 мл насыщенного водного раствора NaHCO3 (бикарбоната натрия). Перемешивание осуществляют в течение 1 ч при 20 С, (растворение). Раствор выливают в 700 мл воды в течение 2 ч, при этом дистиллируя МЕК в вакууме при 40 С. Дистилляцию осуществляют до остаточного объема 430 мл. Добавляют 100 мл воды, и дистилляцию продолжают до остаточного объема 430 мл. Операцию повторяют дважды. Суспензию доводят до температуры окружающей среды, и перемешивают в течение ночи. Твердый продукт фильтруют и промывают 3 раза 50 мл воды. После сушки в вакууме, при температуре окружающей среды, получают 9,3 г ожидаемого продукта в форме бежевого порошка. Продукт очищают путем кристаллизации, в соответствии со способом,описанным ниже. Выход: 94,5%, растворители не учитываются. Кристаллизация продукта формулы (IVa), полученного выше (способ 1, 2 или 3). 10 г 1-[N2-[4'-(октилокси)-[1,1'-бифенил]-4-ил]карбонил]-4-оксо-L-орнитин]-4-[4-(4-гидроксифенил)L-треонин]-5-L-серин эхинокандина В (полученного выше, способ 1, 2 или 3) растворяют в течение 1 ч при перемешивании и при температуре окружающей среды, в 30 мл диметилформамида (ДМФ). Вводят 51 мг инициатора (кристаллов продукта, полученного в соответствии со способами 1, 2 или 3), и осуществляют перемешивание в течение 2 ч. Затем равномерно вводят 67 мл ацетонитрила, в течение 2 ч. После окончания введения осуществляют перемешивание в течение 19 ч, с последующим фильтрованием и промывкой, 3 раза по 10 мл ацетонитрила. После сушки в вакууме, при температуре окружающей среды,получают 7,10 г ожидаемого продукта в форме белых кристаллов. Выход: 71%. ТСХ: Rf=0,28. Силикагель 60F254, УФ 254 нм Подвижная фаза: СН 2 Сl2/МеОН/вода 86/13/1 ЯМР: (ДМСО). Треонин: 7,94 (1 Н), 4,50 (1 Н), 4,25 (1 Н), 1,18 (3 Н); -гидроксипролин: 4,39 (1 Н), 1,93-2,20 (2 Н), 4,40III) Третья стадия. Восстановительное аминирование соединения формулы (IVa) и получение соединения формулы (Iа). 1-[4-[(2-аминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4 гидроксифенил)-L-треонин]-5-L-серинэхинокандин В. Способ 1. 8 г продукта формулы (IVa), полученного в соответствии со стадией II, и 2,52 мл этилендиамина растворяют в 80 мл тетрагидрофурана (ТГФ), при этом перемешивая в течение от 30 мин до 1 ч, в атмосфере азота и при температуре окружающей среды. Раствор 0,36 г тетрахлорида титана, растворенного в 80 мл ТГФ, добавляют в течение 30 мин. Перемешивание осуществляют в течение 1 ч, с последующим охлаждением до 5 С. Добавляют 3,2 мл ледяной уксусной кислоты, растворенной в 16 мл ТГФ. Перемешивание осуществляют в течение 1 ч при 5 С с последующим охлаждением до 0 С, и раствор 1,04 г цианоборгидрида натрия, растворенного в 24 мл ТГФ, добавляют в течение 15 мин. Реакционную среду оставляют с повышением температуры до 5 С, в течение 30 мин и перемешивают в течение 30 мин при этой температуре. Затем оставляют с повышением температуры до температуры окружающей среды, при этом перемешивая в течение 3 ч, с последующими дистилляцией в вакууме до 40 мл объема, добавлением раствора 7,2 г бикарбоната натрия, растворенного в 80 мл воды, дистиллированием в вакууме до остаточного объема 80 мл, затем добавляют 32 мл метанола и 128 мл этилацетата. Перемешивание осуществляют в течение 10 мин, с последующим декантированием, повторным экстрагированием водной фазы смесью этилацетата/метанола и дистилляцией всех органических фаз в вакууме до остаточного объема 40 мл. Вводят 40 мл ДМФ, и продолжают дистилляцию до остаточного объема 60 мл. Этот раствор выливают в 320 мл воды в течение 20 мин и перемешивают в течение 15 ч при температуре окружающей среды. Если это необходимо, рН доводят до 9-9,5 с помощью разбавленного раствора соды с последующим фильтрованием и промывкой водным раствором 1,2 г бикарбоната натрия (рН 9-9,5). После сушки твердого продукта в сушильном шкафу в вакууме при 30 С в течение 18 ч получают 8,55 г ожидаемого неочищенного основания в форме белого твердого продукта. Выход: 100%. Отношение изомеров А/В=80/20. Способ 2. 10 г продукта формулы (IV), полученного на стадии II, вводят при перемешивании и в атмосфере азота, в 600 мл ТГФ. Добавляют 20 г 3 молекулярных сит, затем 3,16 мл этилендиамина и, наконец,110,4 г три-бензил-L-пролинборгидрида натрия. Гомогенный раствор перемешивают при температуре окружающей среды в течение 6 ч, с последующим фильтрованием молекулярных сит и промывкой ТГФ. Фильтрат дистиллируют досуха в вакууме при внутренней температуре, меньшей, чем 40 С. 600 мл насыщенного водного раствора бикарбоната натрия медленно добавляют на смолу, полученную во время перемешивания. К суспензии добавляют 20 г фильтрационного реагента Hyflosupercel Kieselgehr и осуществляют перемешивание в течение 16 ч, при температуре окружающей среды, с последующим фильтрованием и промывкой водой. Лепешку растворяют путем пропускания через 100 мл метанола четыре раза. Метанольный фильтрат концентрируют досуха в вакууме, при температуре, не превышающей 40 С. Получают 21,25 г ожидаемого продукта. Способ 3. Выделение промежуточного продукта реакции восстановительного аминирования (соединение формулы IVа). 1-[4-[N,N'-Имидазолидин]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4 гидроксифенил)-L-треонин]-5-L-серин эхинокандин В. Суспензию продукта формулы (IVa), полученного на стадии II, в 40 мл дихлорметана и 0,32 мл этилендиамина перемешивают в течение 18 ч. Добавляют 80 мл простого диэтилового эфира, и осуществляют перемешивание в течение 5 ч. Твердый продукт фильтруют и промывают несколько раз диэтиловым эфиром. После сушки в сушильном шкафу в вакууме в течение 15 ч при температуре окружающей среды получают 0,76 г ожидаемого соединения в форме белого твердого продукта. Это промежуточное соединение затем подвергают реакции восстановления, в соответствии с предыдущими способами 1 или 2. Это выделенное соединение является новым и представляет собой часть настоящего изобретения. Выход: 73%.IV) Четвертая стадия. Очистка с помощью хроматографии соединения формулы (Iа), полученного ранее (в соответствии со способами 1, 2 или 3), и разделение двух изомеров А и В (А или В соответствуют стереоизомерам R-9 007964 или S в положении 4). Ди-трифторацетат 1-[4-[(2-аминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-Lорнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В. Колонку Prochrom LC 200 кондиционируют с помощью 2,5 г силикагеля Merck 11763, используя в качестве подвижной фазы смесь метиленхлорид (43)-ацетонитрил (50)-метанол (7)-вода (5)трифторуксусная кислота (1). 24 г соединения, полученного выше (стадия III, способ 1), растворяют в 126 мл смеси, состоящей из ацетонитрила (50)-метанола (7)-воды (5)-ТФУ (1). Раствор фильтруют и добавляют 86 мл метиленхлорида. Этот раствор инжектируют в колонку. Элюирование осуществляют при объемном расходе 76 л/ч и при давлении 14 бар. Детектирование осуществляют при 280 нм, с последующей дистилляцией собранных фракций в вакууме при внешней температуре, меньшей, чем 40 С. Получают 36,75 г продукта в форме твердой дисоли ТФУ. Отношение изомеров А/В=99,3/0,7.V) Пятая стадия. Очистка с помощью хроматографии дитрифторацетата (устранение всех других солей). Колонку Prochrom LC 50 кондиционируют с помощью 300 г обращенной фазы силикагеля DaisogelSP 120 n5/1502, используя в качестве подвижной фазы смесь вода (90)-ацетонитрил (10)трифторуксусная кислота (0,1). 36,75 г соли, полученной выше, при первом процессе хроматографии,растворяют в смеси, содержащей 57,3 мл воды (90)-ацетонитрила (10)-трифторуксусной кислоты (0,1) и 26,8 мл чистого ацетонитрила. Раствор фильтруют, и этот раствор вводят в колонку. Элюирование осуществляют с помощью воды (90)-ацетонитрила (10)-трифторуксусной кислоты (0,1), в первом случае,для удаления всех минеральных солей, затем с помощью воды (50)-ацетонитрила (50)-трифторуксусной кислоты (0,1), для удаления продукта. Собранные фракции дистиллируют в вакууме при внешней температуре, меньшей, чем 40 С. Полученный прозрачный водный раствор лиофилизируют. Получают 15,59 г ожидаемого продукта в форме пушистой белой твердой дисоли ТФУ. Отношение изомеров А/В=99,4/0,6. Суммарный выход после двух процессов хроматографии: 53,8%. Выход аминовосстановления: 28,7%.VI) Шестая стадия. Регенерация основания. 1-[4-[(2-Aминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4 гидроксифенил)-L-треонин]-5-L-серин эхинокандин В. 14,37 г соединения, полученного выше, в форме лиофилизированной ди-трифторацетатной соли,растворяют в 143,7 мл воды. Перемешивание осуществляют в течение 15 мин в атмосфере азота и добавляют 57,5 мл насыщенного водного раствора бикарбоната натрия в течение 15 мин при температуре окружающей среды. Перемешивание осуществляют в течение 17 ч. Добавляют 430 мл смеси этилацетат/метанол 8/2, и осуществляют перемешивание в течение 15 мин. Верхнюю органическую фазу декантируют, и водную фазу повторно экстрагируют 288 мл смеси этилацетат (8)-метанол (2). Органические фазы объединяют, осторожно декантируют и дистиллируют досуха в вакууме при температуре, не превышающей 35 С. К сухому экстракту добавляют 143,7 мл воды, и суспензию перемешивают в течение 30 мин, с последующим фильтрованием и промывкой водой, до тех пор, пока фторидов больше не останется. После сушки твердого продукта в вакууме при 40 С получают 10,91 г ожидаемого продукта в форме белого порошка. Выход: 91,6%. Микроанализ: (вода: 8,8%)VII) Седьмая стадия. Преобразования в форму соли (хлористо-водородной кислоты). Дигидрохлорид 1-[4-[(2-аминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В. 10,53 г соединения, полученного на предыдущей стадии, растворяют в 263 мл метанола, при этом перемешивая в течение 30 мин при температуре окружающей среды с последующим фильтрованием и промывкой, дважды по 31,6 мл метанола. Добавляют 2,1 мл 36% хлористо-водородной кислоты при перемешивании в атмосфере азота при температуре окружающей среды. Перемешивание осуществляют в течение 30 мин, с последующей дистилляцией досуха в вакууме при температуре, не превышающей 35 С. Сухой экстракт извлекают 105,3 мл диизопропилоксида. Полученную суспензию перемешивают в течение 2 ч с последующим фильтрованием и промывкой, дважды по 21,1 мл диизопропилоксида. После- 10007964 сушки твердого продукта в печи в вакууме, при 40 С, получают 10,52 г ожидаемого продукта в форме белого порошка. Выход: 93,7%. Микроанализ: (вода: 5,75%)VIII) Восьмая стадия. Кристаллизация соли хлористо-водородной кислоты. Дигидрохлорид 1-[4-[(2-аминоил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серин эхинокандина В. 2,75 г соединения, полученного в соответствии со стадией VII, растворяют в смеси 235,4 мл ацетонитрила и 14,8 мл деминерализованной воды, при этом нагревая с обратным холодильником (77,4 С). Перемешивание осуществляют в течение 15 мин при 77 С, затем реакционную среду охлаждают до+50 С в течение 45 мин. Инициирование осуществляют с помощью 41,25 мг кристаллизованного соединения с последующим равномерным охлаждением до 20 С в течение 30 мин и перемешиванием в течение 2 ч 15 мин, давая возможность для развития кристаллизации. Суспензию концентрируют в вакууме до остаточного объема 55 мл, при этом позволяя температуре расти от 5 до 20 С. Повторно устанавливают нормальное давление азота с последующим охлаждением до 0 С, в течение 20 мин перемешиванием при 0 С в течение 17 ч, фильтрованием в атмосфере азота и промывкой 11 мл ацетонитрила. После сушки кристаллизованного твердого продукта в сушильном шкафу, в вакууме и при 40 С в течение 24 ч получают 2,29 г ожидаемого продукта в форме белого кристаллического твердого продукта. Выход: 83,3%. ВЭЖХ: Rt=5,8 (изомер A); Rt=7,1 (изомер В).(м) 2 Н; 1,44 (м) 2 Н; 1,22-1,40 (м) 8 Н; 1,13 (д, j=6) 3 Н; 0,99 (д, j=6,5) 3 Н; 0,88 (т, j=7). Пример 2. Фармацевтическая композиция. Изготавливают таблетки, содержащие Продукт примера 1 150 мг Наполнитель, s.q.f 1 г Составляющие наполнителя: крахмал, тальк, стеарат магния. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединений формулы (I) в которой R представляет собой линейную или разветвленную, или циклическую цепь, содержащую до 30 атомов углерода, необязательно, содержащую один или несколько гетероатомов и один или несколько подвергают действию кислоты формулы R-СО 2 Н, указанная кислота находится, если это приемлемо, в выделенной или невыделенной активированной форме, с получением соединения формулы (III)b) если это приемлемо, соединение формулы (III) подвергают реакции алкилирования спиртом по положению 5, под действием спирта формулы Alk-ОН, в присутствии PPTS, Alk представляет собой алкильный радикал, содержащий 1-4 атома углерода, с получением соединения формулы (III') с) соединение формулы (III) или (III') подвергают реакции дегидратации, с получением соединения формулы (IV)d) соединение формулы (IV) подвергают реакции восстановительного аминирования под действием этилендиамина в присутствии восстановительного агента, такого как NаВН 3 СN, в присутствии кислоты Льюиса,или NaBH(OCOR'), где OCOR' представляет собой Boc-L-Pro, Bzl-L-Pro или любую другую оптически активную аминокислоту, а также любую другую хиральную или нехиральную карбоновую кислоту, с получением соединения формулы (I), как определено выше, содержащего преимущественно один из активных изомеров,затем указанное соединение формулы (I) подвергают, если это приемлемо, и в любом приемлемом- 12007964 порядке, одной или нескольким из следующих далее операций: хроматографированию; кристаллизации; действию основания; преобразованию в форму соли. 2. Способ по п.1, в котором соединения формул (I), (III), (III') или (IV) содержат радикал R, представляющий собой следующие группы: 3. Способ по п.1, в котором соединения формулы (I), (III), (III'), (IV) содержат радикал R, представляющий собой следующую группу: 4. Способ получения соединений формулы (Iа) подвергают действию кислоты формулы С 8 Н 17-О-Рh-Рh-СО 2 Н, указанная кислота находится, если это приемлемо, в активированной, выделенной или невыделенной форме, с получением соединения формулы (IIIа)b) если это приемлемо, соединение формулы (IIIа) подвергают реакции алкилирования спиртом по положению 5, под действием метанола, в присутствии PPTS, с получением соединения формулы (IIIа') с) соединение формулы (IIIа) или (IIIа') подвергают реакции дегидратации, с получением соединения формулы (IVa)d) соединение формулы (IVa) подвергают реакции восстановительного аминирования под действием этилендиамина, в присутствии восстановительного агента, такого как NaBH3CN, связанного с TiCl4,NaBH(Boc-L-Pro)3 или NaBH(Bzl-L-Pro)3, с получением соединения формулы (Iа), как определено выше,преимущественно один из активных изомеров, указанное соединение формулы (Iа) подвергают одной или нескольким из следующих далее операций, в приемлемом порядке: хроматографираванию; кристаллизации; действию основания; преобразованию в форму соли под действием хлористо-водородной кислоты. 5. Способ по п.4, отличающийся тем, что соединение формулы (Iа) последовательно подвергается следующим операциям:a) очистке хроматографией на силикагеле, затем хроматографией с обращенной фазой с использованием смеси органических растворителей, воды и трифторуксусной кислоты с получением соли трифторуксусной кислоты соединения формулы (Iа);b) действию основания, например, действию водного раствора бикарбоната натрия, с получением соединения формулы (Iа) в форме основания;c) преобразованию в форму соли под действием хлористо-водородной кислоты, с получением соответствующей соли, а именно, дигидрохлорида соединения формулы (Iа). 6. Способ по любому из пп.1-5, отличающийся тем, что реакцию активирования кислоты осуществляют в присутствии пентафторфенола, N-гидроксисукцинимида или, необязательно, Nгидроксибензотриазола. 7. Способ по любому из пп.1-5, отличающийся тем, что реакцию ацилирования в присутствии выделенной активированной кислоты осуществляют в присутствии диизопропилэтиламина.- 14007964 8. Способ по любому из пп.1-5, отличающийся тем, что реакцию активирования, а затем реакцию ацилирования в присутствии невыделенной активированной кислоты осуществляют в присутствии Nметилпирролидона и DMTMM. 9. Способ по любому из пп.1-5, отличающийся тем, что реакцию дегидратации осуществляют в присутствии AIBB и, если это приемлемо, МgI2. 10. Способ по любому из пп.1-7, отличающийся тем, что реакцию дегидратации осуществляют в присутствии HBr-АсОН и MgI2. 11. Способ по пп.9 и 10, отличающийся тем, что продукт, получаемый от дегидратации, очищают кристаллизацией из смеси ДМФ/ацетон или ДМФ/AcOEt. 12. Способ по любому из пп.1-5, отличающийся тем, что реакцию восстановительного аминирования осуществляют в присутствии восстановительного агента, выбранного из NаВН 3 СN, связанного сTiCl4, NaBH(Boc-L-Pro)3 и NaBH(Bzl-L-Pro)3. 13. Способ получения соединения формулы (III) или (IIIа) ацилированием соединений формулы (II),как определено в любом из пп.1-4 и 6-8. 14. Способ получения соединения формулы (IV) или (IVa) дегидратацией соединений формулы(III), (III'), (IIIa) или (III'а), как определено в любом из пп.1-4 и 9-11. 15. Способ получения соединения формулы (I) или (Iа) восстановительным аминированием соединений формулы (IV) или (IVa), как определено в любом из пп.1-4 и 12. 16. Дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-Lорнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в 4S или 4R форме или в форме смеси стереоизомеров. 17. Дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4-ил]карбонил]-Lорнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в форме активного изомера А,который может быть получен способом, описанным в пп.1-15. 18. Применение дигидрохлорида 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4 ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-серинэхинокандина В в форме 4R или 4S изомера или в виде смеси указанных стереоизомеров в качестве лекарственного средства. 19. Применение дигидрохлорида 1-[4-[(2-аминоэтил)амино]-N2-4'-(октилокси)[1,1'-бифенил]-4 ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-L-cepинэхинокандина В в виде активного изомера А в качестве противогрибкового лекарственного средства. 20. Фармацевтическая композиция, содержащая дигидрохлорид 1-[4-[(2-аминоэтил)амино]-N2-4'(октилокси)[1,1'-бифенил]-4-ил]карбонил]-L-орнитин]-4-[4-(4-гидроксифенил)-L-треонин]-5-Lсеринэхинокандина В, как определено в п.16 или 17, и фармацевтически приемлемый носитель. 21. Промежуточные соединения формул (III') или (III'а), как определено в пп.1 и 6. 22. Промежуточные соединения формул (IV), R является таким, как определено в любом из пп.1-2,или (IV'a), где R представляет собой Ph-Ph-OC8H17
МПК / Метки
МПК: A61P 31/10, C07K 7/56, A61K 38/12
Метки: эхинокандиновых, способ, получения, соединений
Код ссылки
<a href="https://eas.patents.su/16-7964-sposob-polucheniya-ehinokandinovyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения эхинокандиновых соединений</a>
Способ получения промежуточных соединений, применимых для получения противораковых соединений

Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Норрис Тимоти, Лехнер Ричард Шелтон, Сантафьянос Динос Пол
МПК: C07D 239/94
Метки: соединений, способ, противораковых, промежуточных, получения, применимых
Формула / Реферат:
1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше, тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются...
Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений

Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Леветт Филип Чарльз, Негри Джоанна Тереза, Вуд Альберт Шо, Девриз Кейт Майкл
МПК: C07D 401/14, C07D 487/04, C07D 401/12...
Метки: соединений, получения, промежуточных, способ, пиридилсульфонильных, пиразоло[4,3-d]пиримидин-7-он-3
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом; R1 (который может быть...
(1r, 2s, 4r)-(-)-2-[n,n-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло [2,2,1]гептан высокой степени чистоты, его фармацевтически приемлемые кислотные аддитивные соли, способ получения этих соединений и их применение, а также лекарственные средства, содержащие одно или более из этих соединений

Номер патента: 2164
Опубликовано: 24.12.2001
Авторы: Краснаи Дьёрдь, Сабо Тибор, Мезеи Тибор, Порч-Маккаи Марта, Надь Калман, Будаи Золтан, Лукач Дьюла, Суладьи Янош, Шимиг Дьюла, Немет Норберт
МПК: A61K 31/13, A61P 25/04, C07C 217/12...
Метки: также, одно, чистоты, соли, 2,2,1]гептан, более, лекарственные, применение, средства, этих, соединений, получения, приемлемые, кислотные, высокой, фармацевтически, способ, содержащие, аддитивные, 4r)-(-)-2-[n,n-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло, степени
Формула / Реферат:
1. (1R,2S,4R)-(-)-2-[N,N-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло-[2,2,1]гептан по формуле и его фармацевтически приемлемые кислотные аддитивные соли, отличающиеся тем, что они содержат не более 0,2% (1R,3S,4R)-3-[2-N,N-(диметиламиноэтил)]-1,7,7-триметилбицикло[2,2,1]гептан-2-она по формуле или его фармацевтически приемлемой кислотной аддитивной соли. 2....
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Крок Вероник, Колладан Колетт, Ларкин Джон Патрик, Руссель Патрик
МПК: C07D 487/04
Метки: соединений, октагидро-6, способ, получения, производные, применение, кислоты, диазепин-1-карбоновой, 10-диоксо-6н-пиридазино, активных, 1,2-а, терапевтически, 1,2
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Способ получения противораковых соединений

Номер патента: 5892
Опубликовано: 30.06.2005
Авторы: Сантафьянос Динос Пол, Норрис Тимоти, Лехнер Ричард Шелтон
МПК: C07D 239/94
Метки: способ, соединений, противораковых, получения
Формула / Реферат:
1. Способ получения соединения формулы 9 или его фармацевтически приемлемых соли или сольвата, в которых R6 является C1-C10-алкилом или -(CH2)mO(CH2)nCH3; R7 представляет собой C1-C10-алкил или -(C1-C6-алкил)(C6-C10-арил); каждый m, независимо, является целым числом от 1 до 6, а n является целым числом от 0 до 3; R8, R9 и R10, каждый независимо, выбираются из H, C1-C10-алкила и -OR11; каждый R11, независимо, представляет собой C1-C10-алкил; R15...
Предыдущий патент: Способ снижения концентрации альдегидов в смеси, содержащей циклогексанон и один или несколько альдегидов
Следующий патент: Способ получения комбретастатинов
Случайный патент: Нерастворимые инсулинсодержащие композиции, способы их получения, суспензионный препарат, способ лечения диабета и гипергликемии