Производные n-[фенил(пиперидин-2-ил)метил]бензамида и их применение в терапии
Номер патента: 7225
Опубликовано: 25.08.2006
Авторы: Севрен Мирей, Вероник Коринн, Марабу Бенуа, Эстенн-Буту Женевьев, Роже Пьер, Мага Паскаль, Даргазанли Жихад, Медеско Флоранс














Формула / Реферат
1. Соединение в форме чистого оптического изомера (1R, 2R) или (1S, 2S) либо в форме трео-диастереоизомера, соответствующее общей формуле (I)

где А представляет собой
или группу общей формулы N-R1, где R1 представляет собой или атом водорода, или линейную или разветвленную (С1-С7)алкильную группу, возможно замещенную одним или более чем одним атомом фтора, или (С4-С7)циклоалкильную группу, или (С3-С7)циклоалкил(С1-С3)алкильную группу, или фенил(С1-С3)алкильную группу, возможно замещенную одной или двумя гидроксильными или метоксигруппами, или (С2-С4)алкенильную группу, или (С2-С4)алкинильную группу,
или группу общей формулы N+(O-)R1, где R1 является таким, как определено выше,
или альтернативно группу общей формулы N+(R')R1, где R' представляет собой линейную или разветвленную (С1-С7)алкильную группу и R1 является таким, как определено выше,
Х представляет собой атом водорода или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, линейных или разветвленных (С1-С4)алкильных и (С1-С4)алкоксигрупп,
R2 представляет собой или атом водорода, или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, (С1-С4)алкильной или (С1-С4)алкоксигрупп или аминогрупп общей формулы NR3R4, где R3 и R4, каждый независимо друг от друга, представляeт собой атом водорода или (С1-С4)алкильную группу или образуeт вместе с несущим их атомом азота пирролидиновое, пиперидиновое или морфолиновое кольцо или фенильную группу, возможно замещенную атомом или группой, как определено для символа Х выше,
в форме свободного основания или соли присоединения кислоты.
2. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию (1S, 2S) и тем, что R2 представляет собой атом галогена или трифторметильные группы в количестве один(на) или более.
3. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию (1R, 2R), и тем, что R2 представляет собой атом галогена и аминогруппу общей формулы NR3R4, как определено в п.1.
4. Лекарственное средство, отличающееся тем, что оно состоит из соединения по любому из пп.1-3.
5. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение по любому из пп.1-3 в комбинации с эксципиентом.
Текст
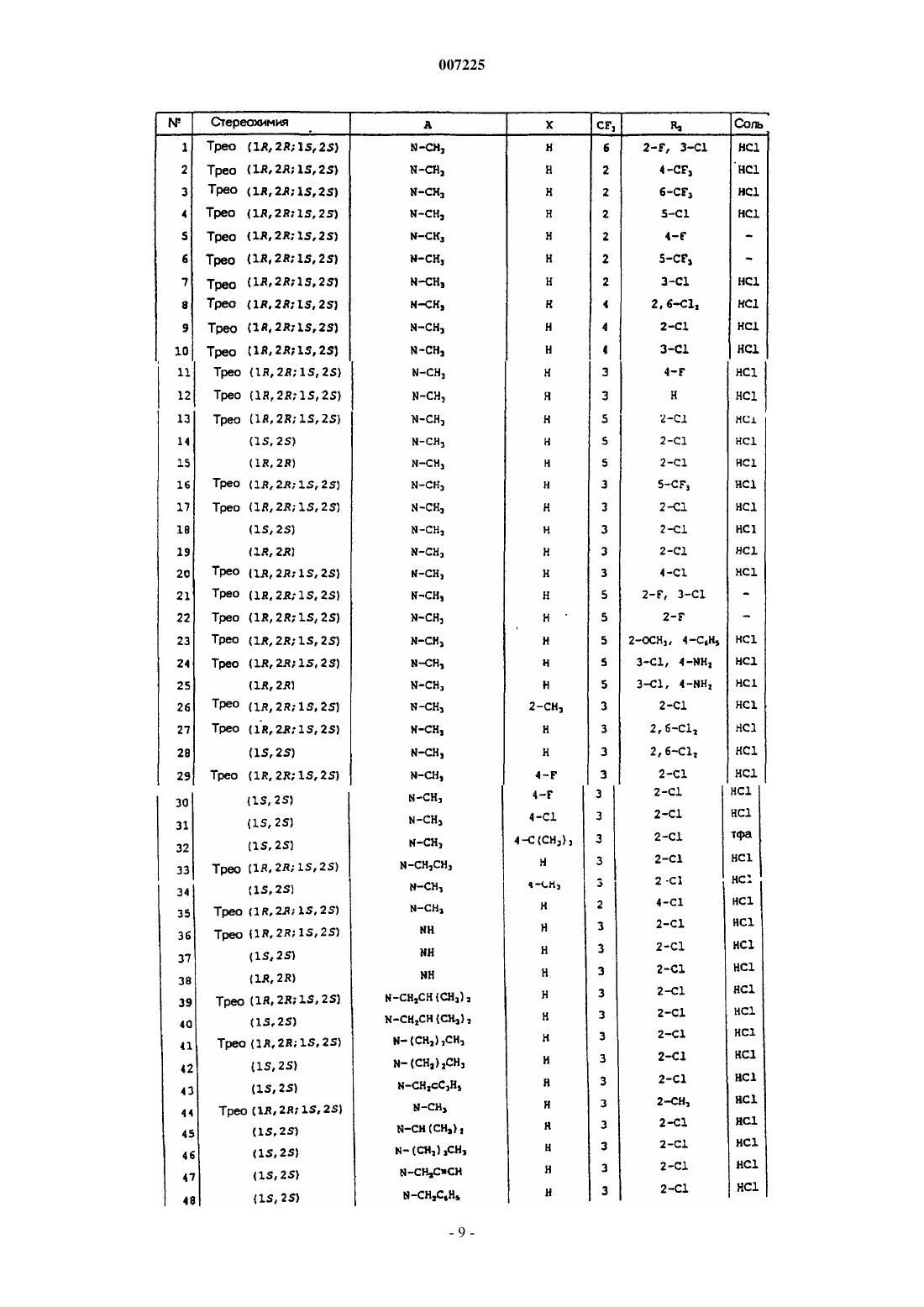
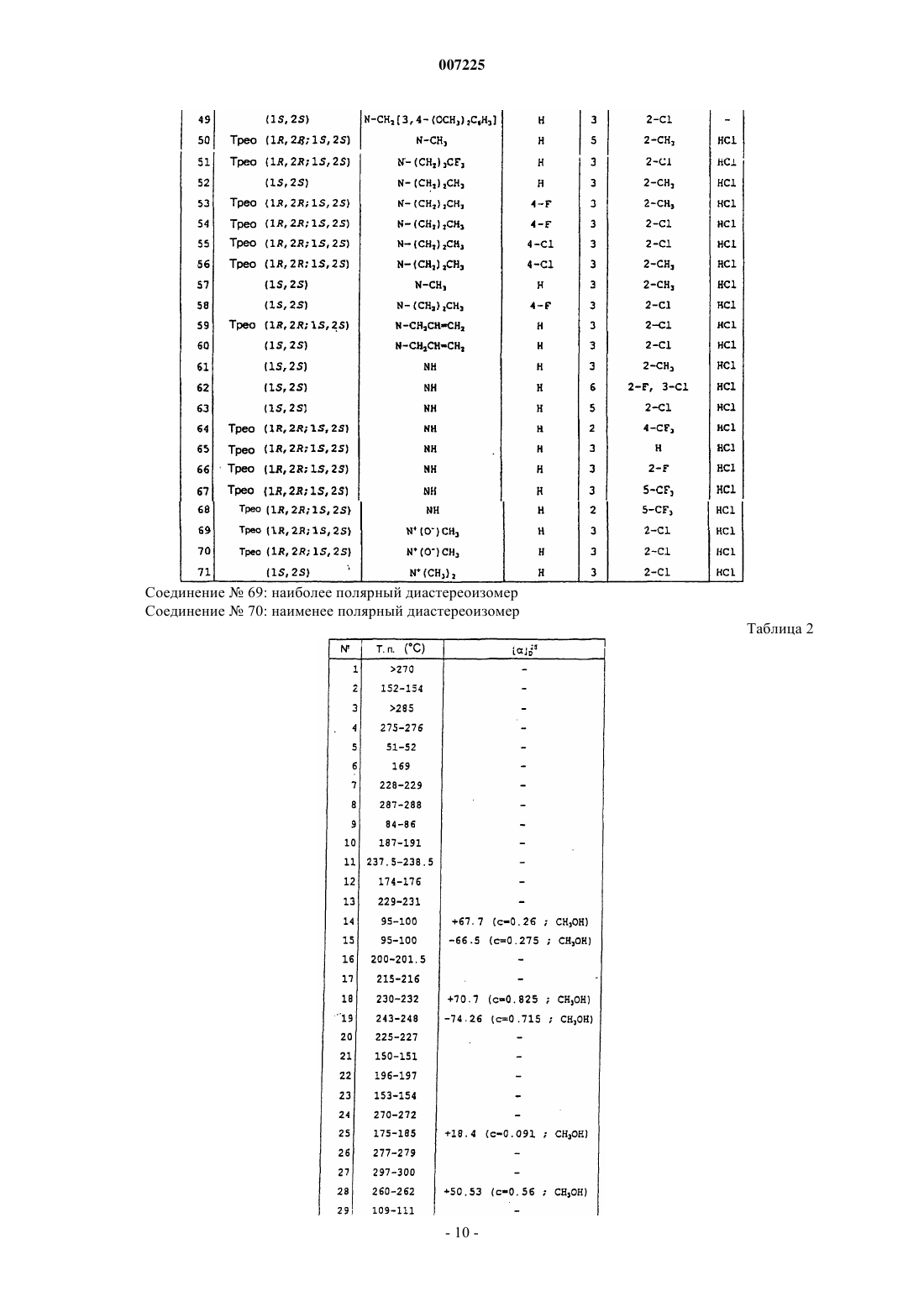
007225 Соединения по данному изобретению соответствуют общей формуле (I) где А представляет собой или группу общей формулы N-R1, где R1 представляет собой или атом водорода, или линейную или разветвленную (С 1-С 7)алкильную группу, возможно замещенную одним или более чем одним атомом фтора, или (С 4-С 7)циклоалкильную группу, или (С 3-С 7)циклоалкил(С 1-С 3)алкильную группу, или фенил(С 1-С 3)алкильную группу, возможно замещенную одной или двумя гидроксильными или метоксигруппами, или (С 2-С 4)алкенильную группу, или (C2-С 4)алкинильную группу,или группу общей формулы N+(O-)R1, где R1 является таким, как определено выше,или альтернативно группу общей формулы N+(R')R1, где R' представляет собой линейную или разветвленную (С 1-С 7)алкильную группу и R1 является таким, как определено выше,Х представляет собой атом водорода или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, линейных или разветвленных (С 1-С 4)алкильных и (С 1-С 4)алкоксигрупп,R2 представляет собой или атом водорода, или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, (С 1-С 4)алкильной или (С 1-С 4)алкоксигрупп или аминогрупп общей формулы NR3R4, где R3 и R4, каждый независимо друг от друга представляет собой атом водорода или(С 1-С 4)алкильную группу или образует вместе с несущим их атомом азота пирролидиновое, пиперидиновое или морфолиновое кольцо или фенильную группу, возможно замещенную атомом или группой, как определено для символа Х выше. Соединения общей формулы (I) могут существовать в форме трео-рацемата (1R, 2R; 1S, 2S) или в форме энантиомеров (1R, 2R) или (1S, 2S); они могут существовать в форме свободных оснований или солей присоединения кислот. Соединения, имеющие структуру, аналогичную структуре соединений по данному изобретению,описаны в патенте US 5254569 в качестве обезболивающих, мочегонных, противосудорожных, анестезирующих, успокаивающих средств, агентов, защищающих головной мозг, на основании механизма действия на опиатные рецепторы. Другие соединения, имеющие аналогичную структуру, описаны в заявке на патент ЕР 0499995 в качестве 5-НТ 3(5-гидрокситриптамин)антагонистов, которые полезны при лечении психических расстройств, неврологических заболеваний, желудочных синдромов, тошноты и рвоты. Соединения по данному изобретению проявляют особую активность в качестве специфических ингибиторов транспортеров глицина glyt1 и/или glyt2. Соединения, предпочтительные в качестве ингибиторов glyt1 транспортера, имеют конфигурацию(1S, 2S), где R2 представляет собой атомы водорода или трифторметильные группы в количестве один(на) или более, в то время как соединения, предпочтительные в качестве ингибиторов glyt2 транспортера, имеют конфигурацию (1R, 2R), где R2 представляет собой атом водорода и аминогруппу общей формулы NR3R4. Соединения общей формулы (I), где А представляет собой группу общей формулы N-R1, где R1 является отличным от атома водорода, могут быть получены способом, изображенным на следующей ниже Диамин общей формулы (II), где R1 и Х такие, как определено выше (R1 является отличным от атома водорода), соединяют с активированной кислотой или хлорангидридом общей формулы (III), где Y представляет собой уходящую группу, такую как атом галогена, и R2 является таким, как определено выше, способами, известными специалистам в данной области техники. Диамин общей формулы (II) может быть получен способом, изображенным на следующей ниже схеме 2. Проводят взаимодействие амида Вейнреба формулы (IV) с производным фениллития общей формулы (V), где Х является таким, как определено выше, в эфирном растворителе, таком как диэтиловый эфир, при температуре от -30 С до комнатной температуры; получают кетон общей формулы (VI), который восстанавливают до спирта общей формулы (VII) с трео-конфигурацией, применяя такой восстанавливающий агент, как K-Selectride или L-Selectride (три-втор-бутилборгидрид калия или лития), в эфирном растворителе, таком как тетрагидрофуран, при температуре от -78 С до комнатной. Карбамат общей формулы (VII) затем может быть восстановлен до трео-N-метиламиноспирта общей формулы (VIII) под действием смешанного гидрида, такого как литийалюминийгидрид, в эфирном растворителе, таком как тетрагидрофуран, при температуре от комнатной до температурой дефлегмации. трео-Спирт общей формулы (VIII) затем превращают в промежуточное трео-соединение общей формулы (II), где R1 представляет собой метильную группу, в две стадии: прежде всего, спиртовую функциональную группу превращают в уходящую группу, например метансульфонатную группу, под действием метилсульфонилхлорида, в хлорированном растворителе, таком как дихлорметан, и в присутствии основания, такого как триэтиламин, при температуре от 0 С до комнатной, и затем проводят взаимодействие уходящей группы со сжиженным аммиаком при -50 С, в спирте, таком как этанол, в закрытой среде, такой как автоклав, при температуре от -50 С до комнатной. Также можно удалить защиту карбамата общей формулы (VII) при помощи сильного основания, такого как гидроксид калия, в спирте, таком как метанол, для получения трео-аминоспирта общей формулы (IX), и затем осуществить N-алкилирование при помощи галогенированного производного формулыR1Z, где R1 является таким, как определено выше, но отличным от атома водорода, и Z представляет собой атом галогена, в присутствии основания, такого как карбонат калия, в полярном растворителе, таком как N,N-диметилформамид, при температуре от комнатной до 100 С. Спирт общей формулы (X), полученный таким образом, затем обрабатывают так, как описано для спирта общей формулы (VIII). Другой вариантный способ, изображенный на следующей ниже cхеме 3, можно использовать в случае, когда R1 представляет собой метильную группу, а Х представляет собой атом водорода. Оксим пиридина формулы (XI) кватернизируют, например, воздействуя метилтрифторметансульфонатом в эфирном растворителе, таком как диэтиловый эфир, при комнатной температуре. Получаемую таким образом соль пиридиния формулы (XII) затем подвергают гидрированию в атмосфере водорода, в присутствии катализатора, такого как оксид платины, в смеси спирта и водного раствора кислоты, такой как этанол и 1 н соляная кислота. Получают диамин общей формулы (II), где R1 представляет собой метильную группу и Х представляет собой атом водорода, в форме смеси двух диастереоизомеров трео/эритро 9/1. Можно получить его соль, например, с щавелевой кислотой и затем очистить путем перекристаллизации образовавшегося оксалата из смеси спирта и эфирного растворителя, такой как метанол и диэтиловый эфир, так, чтобы получить чистый трео-диастереоизомер (1R, 2R; 1S, 2S). Схема 3-2 007225 Соединения общей формулы (I), где А представляет собой группу общей формулы NR1, причем R1 представляет собой атом водорода, могут быть получены способом, изображенным на следующей cхеме 4. Схема 4 Начиная с амина общей формулы (XIII), где Х является таким, как определено выше, осуществляют связывание с активированной кислотой или хлорангидридом общей формулы (III), как описано выше, в соответствии со способами, известными специалистам в данной области техники, для получения соединения общей формулы (XIV). В заключение осуществляют гидрирование последнего соединения, например, водородом в присутствии катализатора, такого как 5%-ная платина на углероде, в кислотном растворителе, таком как ледяная уксусная кислота, так чтобы в заключение получить соединение общей формулы (I), где R1 представляет собой атом водорода. Другой способ состоит, в соответствии со схемой 2, в использовании соединения общей формулы(I), где либо R1 представляет собой возможно замещенную фенилметильную группу, с удалением защиты атома азота пиперидинового кольца, например, окисляющим агентом или кислотой Льюиса, такой как трибромид бора, или путем гидрогенолиза, либо R1 представляет собой алкенильную группу, предпочтительно аллильную группу, с последующим удалением защиты комплексом Pd0, с получением соединения общей формулы (I), где R1 представляет собой атом водорода. Соединения общей формулы (I), где А представляет собой группу общей формулы N+(O-)R1, могут быть получены из соединений общей формулы (I), где А представляет собой группу общей формулы NR1, где R1 является таким, как описано выше, путем взаимодействия с окисляющим агентом, например 3 хлорпербензойной кислотой, в хлорированном растворителе, таком как дихлорметан, при температуре от 0 С до комнатной температуры. Соединения общей формулы (I), где А представляет собой группу общей формулы N+(R')R1, могут быть получены из соединений общей формулы (I), где А представляет собой группу общей формулы NR1, путем взаимодействия с алкилгалогенидом общей формулы R'-Z, где R' является таким, как определено выше, и Z представляет собой атом галогена, в полярном растворителе, таком как ацетонитрил, при температуре от комнатной до 100 С. Кроме того, хиральные соединения общей формулы (I), соответствующие энантиомерам (1R, 2R) или (1S, 2S) трео-диастереоизомера, также могут быть получены путем разделения рацемических соединений высокоэффективной жидкостной хроматографией (HPLC) на хиральной колонке или путем разделения рацемического амина общей формулы (II) с использованием хиральной кислоты, такой как винная кислота, камфорсульфоновая кислота, дибензоилвинная кислота, N-ацетиллейцин, путем фракционированной и избирательной перекристаллизации диастереоизомерной соли из растворителя спиртового типа или путем энантиоселективного синтеза в соответствии со cхемой 2, с использованием хирального амида Вейнреба общей формулы (IV). Рацемический или хиральный амид Вейнреба формулы (IV) может быть получен способом, аналогичным описанному в Eur. J. Med. Chem., 35, (2000), 979-988 и J. Med. Chem., 41, (1998), 591-601. Фениллитиевое соединение общей формулы (V), где Х представляет собой атом водорода, имеется в продаже. Его замещенные производные могут быть получены способом, аналогичным описанному в Tetra. Lett.,57, 33, (1996), 5905-5908, способом, аналогичным описанному в заявке на патент ЕР 0366006. Амин общей формулы (IX), где Х представляет собой атом водорода, может быть получен в хиральной цепочке в соответствии со способом, описанным в патенте US 2928835. Наконец, амин общей формулы (XIII) может быть получен способом, аналогичным описанному в Chem. Pharm. Bull., 32,12, (1984), 4893-4906 иSynthesis, (1976), 593-595. Кислоты и хлорангидриды общей формулы (III) имеются в продаже, за исключением 4-амино-3 хлор-5-трифторметилбензойной кислоты. Ее можно приготовить хлорированием 4-амино-5-трифторметилбензойной кислоты сульфурилхлоридом в хлорированном растворителе, таком как хлороформ,способом, аналогичным описанному в Arzneim. Forsch., 34, 11 а, (1984), 1668-1679. Следующие ниже примеры иллюстрируют получение некоторых соединений по данному изобретению. Элементные микроанализы, IR (инфракрасные) и NMR (ядерного магнитного резонанса) спектры и HPLC на хиральной колонке подтверждают структуры и энантиомерную чистоту полученных соединений.-3 007225 Номера, указанные в скобках в заголовках примеров, соответствуют номерам в 1-й колонке таблицы, приведенной ниже. В названиях соединений тире "-" образует часть слова, а тире служит только для разделения в конце строки; при отсутствии разделения он опускается и не должен замещаться ни нормальным тире, ни пробелом. Пример 1 (соединение 33). трео-2-Хлор-N-[(1-этилпиперидин-2-ил)фенилметил]-3-трифторметилбензамида гидрохлорид 1:1. 1.1. 1,1-Диметилэтил-2-бензоилпиперидин-1-карбоксилат. 8,0 г (29,4 ммоль) 1,1-диметилэтил-2-(N-метокси-N-метилкарбамоил)пиперидин-1-карбоксилата в 100 мл безводного диэтилового эфира помещают в круглодонную колбу на 250 мл в атмосфере аргона,среду охлаждают до -25 С, по каплям добавляют 16 мл (29,4 ммоль) 1,8 М раствора фениллития в смеси циклогексана и диэтилового эфира 70/30 и перемешивание продолжают в течение 2 ч. После гидролиза насыщенным водным раствором хлорида натрия водную фазу отделяют, экстрагируют этилацетатом, органическую фазу сушат над сульфатом натрия, фильтруют, и фильтрат концентрируют при пониженном давлении, и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью этилацетата и циклогексана. Получают 2 г белого твердого вещества. 1.2. 1,1-Диметилэтил-трео-[гидрокси(фенил)метил]пиперидин-1-карбоксилат. 2,0 г (6,9 ммоль) 1,1-диметилэтил-2-бензоилпиперидин-1-карбоксилата в 30 мл безводного диэтилового эфира помещают в круглодонную колбу на 250 мл в атмосфере аргона, раствор охлаждают до -78 С,по каплям добавляют 20,7 мл (20,7 ммоль) 1 М раствора три-втор-бутилборгидрида лития в диэтиловом эфире и перемешивание продолжают в течение 3 ч. Смесь гидролизуют 16 мл воды и 16 мл 35%-ного водного раствора перекиси водорода и смеси оставляют возвращаться к комнатной температуре при перемешивании в течение 2 ч. Смесь разбавляют водой и этилацетатом, водную фазу отделяют и экстрагируют этилацетатом. После промывания объединенных органических фаз, высушивания над сульфатом натрия и выпаривания растворителя при пониженном давлении остаток очищают хроматографией на колонке с силикагелем,элюируя смесью этилацетата и циклогексана. Получают 2,0 г маслянистого продукта. 1.3. трео-Фенил(пиперидин-2-ил)метанол. Раствор 2,0 г (6,9 ммоль) 1,1-диметилэтил-трео-[гидрокси(фенил)метил]пиперидин-1-карбоксилата в 40 мл метанола помещают в круглодонную колбу на 250 мл, добавляют водный раствор гидроксида калия, приготовленный из 2,0 г гранул гидроксида калия и 20 мл воды, и смесь нагревают с обратным холодильником в течение 2 ч. Смесь охлаждают, растворитель выпаривают при пониженном давлении, добавляют воду и эту смесь несколько раз экстрагируют дихлорметаном. После промывания объединенных органических фаз,высушивания над сульфатом магния, фильтрации и выпаривания растворителя при пониженном давлении получают 1,0 г белого твердого вещества. Температура плавления: 172-174 С. 1.4. трео-(1-Этилпиперидин-2-ил)фенилметанол. Раствор 1 г (5,2 ммоль) трео-фенил(пиперидин-2-ил)метанола в 30 мл безводного N,N-диметилформамида помещают в круглодонную колбу на 100 мл, добавляют 0,39 мл (5,2 ммоль) бромэтана и 0,8 г(5,8 ммоль) карбоната калия и смесь нагревают при 80 С в течение 2 ч. Смесь охлаждают до комнатной температуры, гидролизуют путем добавления воды и несколько раз экстрагируют этилацетатом. После промывания объединенной органической фазы водой и затем насыщенным водным раствором хлорида натрия,высушивания над сульфатом магния, фильтрации и выпаривания растворителя при пониженном давлении остаток очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола. Получают 0,8 г маслянистого соединения. 1.5. трео-(1-Этилпиперидин-2-ил)фенилметанамин. 0,8 г (3,65 ммоль) трео-(1-этилпиперидин-2-ил)фенилметанола и 0,48 мл (3,65 ммоль) триэтиламина в 20 мл безводного дихлорметана помещают в круглодонную колбу на 100 мл в атмосфере аргона, смесь охлаждают до 0 С, добавляют 0,28 мл (3,63 ммоль) метансульфонилхлорида, и смесь оставляют медленно возвращаться к комнатной температуре в течение 2 ч, и ее концентрируют при пониженном давлении. Сжиженный аммиак вводят в автоклав, оборудованный магнитной мешалкой и охлажденный до-50 С, и добавляют раствор метансульфоната, предварительно приготовленный в 10 мл абсолютного этанола, автоклав закрывают и перемешивание продолжают в течение 48 ч. Смесь переносят в круглодонную колбу, концентрируют при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола. Получают 0,3 г маслянистого соединения, которое используют как таковое, на следующей стадии. 1.6. трео-2-Хлор-N-[(1-этилпиперидин-2-ил)]фенилметил]-3-трифторметилбензамида гидрохлорид 1:1. 0,3 г (1,37 ммоль) 2-хлор-3-трифторметилбензойной кислоты, 0,26 г (1,37 ммоль) 1-[3-(диметиламино)пропил]-3-этилкарбодиимида и раствор 0,19 г (1,37 ммоль) 1-гидроксибензотриазола в 10 мл ди-4 007225 хлорметана помещают в круглодонную колбу на 50 мл и смесь перемешивают при комнатной температуре в течение 30 мин. Добавляют раствор 0,3 г (1,37 ммоль) трео-(1-этилпиперидин-2-ил)фенилметанамина в нескольких миллилитрах дихлорметана и перемешивание продолжают в течение 5 ч. Смесь гидролизуют водой и несколько раз экстрагируют дихлорметаном. После промывания органических фаз водой и затем 1 н водным раствором гидроксида натрия, высушивания над сульфатом магния, фильтрации и выпаривания растворителя при пониженном давлении остаток очищают хроматографией на колонке с силикагелем,элюируя смесью дихлорметана и метанола. Получают 0,25 г маслянистого продукта. Этот продукт растворяют в нескольких миллилитрах пропан-2-ола, добавляют 5,9 мл 0,1 н раствора соляной кислоты в пропан-2-оле и смесь концентрируют при пониженном давлении для уменьшения объема растворителя. После растирания в конечном итоге выделяют 0,15 г гидрохлорида в форме белого твердого вещества. Температура плавления: 230-232 С. Пример 2 (соединение 18). 2-Хлор-N-[(1S)-[(2S)-1-метилпиперидин-2-ил)фенилметил]-3-трифторметилбензамида гидрохлорид 1:1. 2.1. 1,1-Диметилэтил-(25)-2-бензоилпиперидин-1-карбоксилат. 11,8 г (43,3 ммоль) 1,1-диметилэтил-(2S)-2-(N-метокси-N-метилкарбамоил)пиперидин-1-карбоксилата в 100 мл безводного диэтилового эфира помещают в круглодонную колбу на 500 мл в атмосфере азота, среду охлаждают до -23 С, по каплям добавляют 21,6 мл (43,2 ммоль) 1,8 М раствора фениллития в смеси циклогексана и диэтилового эфира 70/30 и перемешивание продолжают в течение 3 ч при комнатной температуре. После гидролиза насыщенным водным раствором хлорида натрия водную фазу отделяют и экстрагируют этилацетатом. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью этилацетата и циклогексана. Получают 4,55 г твердого продукта. Температура плавления: 123-125 С.[]D25=-25,4 С (с=2,22; CH2Cl2) ее (энантиомерный избыток)=97,2%. 2.2. 1,1-Диметилэтил-(1S)-2-[(2S)-гидрокси(фенил)метил]пиперидин-1-карбоксилат. 4,68 г (16,2 ммоль) 1,1-диметилэтил-(2S)-2-бензоилпиперидин-1-карбоксилата в 170 мл безводного тетрагидрофурана помещают в круглодонную колбу на 500 мл в атмосфере азота, раствор охлаждают до-78 С, по каплям добавляют 48,5 мл (48,5 ммоль) 1 М раствора L-Selectride (три-втop-бутилборгидрида лития) в тетрагидрофуране и смесь перемешивают при комнатной температуре в течение 5 ч. Смесь медленно гидролизуют в охлажденном состоянии при помощи 34 мл воды и 34 мл 35%-ного водного раствора перекиси водорода и смесь оставляют возвращаться к комнатной температуре при перемешивании в течение 2 ч. Эту смесь разбавляют водой и этилацетатом, водную фазу отделяют и экстрагируют этилацетатом. После промывания объединенных органических фаз, высушивания над сульфатом натрия, фильтрации и выпаривания остаток очищают хроматографией на колонке с силикагелем, элюируя смесью этилацетата и циклогексана. Получают 4,49 г бледно-желтого масла.[]D25=+63,75 С (с=0,8; CH2Cl2) ее=97,8%. 2.3. (1S)-[(2S)-(1-Метилпиперидин-2-ил)]фенилметанол. 2,96 г (78,1 ммоль) литийалюминийгидрида в 50 мл безводного тетрагидрофурана помещают в двугорлую колбу на 200 мл в атмосфере азота, смесь нагревают с обратным холодильником, добавляют 4,49 г(15,4 ммоль) раствора 1,1-диметилэтил-(1S)-2-[(2S)-гидрокси(фенил)метил]пиперидин-1-карбоксилата в 35 мл тетрагидрофурана и нагревание смеси с обратным холодильником поддерживают в течение 3,5 ч. Смесь охлаждают, медленно гидролизуют 0,1 М раствором калий-натрий тартрата и оставляют перемешиваться в течение ночи. Смесь фильтруют, осадок промывают тетрагидрофураном и фильтрат затем концентрируют при пониженном давлении. Получают 2,95 г бесцветного маслянистого продукта. 2.4. (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанамин. 2,95 г (14,4 ммоль) (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанола и 2 мл (14,4 ммоль) триэтиламина в 70 мл безводного дихлорметана помещают в круглодонную колбу на 250 мл в атмосфере азота,среду охлаждают до 0 С, добавляют 1,1 мл (14,4 ммоль) метансульфонилхлорида и смеси оставляют медленно возвращаться к комнатной температуре в течение 2 ч, затем ее концентрируют при пониженном давлении.-5 007225 Сжиженный аммиак вводят в автоклав, оборудованный магнитной мешалкой и охлажденный до-50 С, добавляют раствор неочищенного метансульфоната, предварительно приготовленный в 30 мл абсолютного этанола, автоклав закрывают и перемешивание продолжают в течение 48 ч. Смесь переносят в круглодонную колбу и амин выделяют в форме маслянистого продукта, который используют как таковой на следующей стадии. 2.5. 2-Хлор-N-[(1S)-[(2S)-1-метилпиперидин-2-ил)]фенилметил]-3-трифторметилбензамида гидрохлорид 1:1. Используя процедуру, описанную в п.1.6, начиная с 1 г (4,9 ммоль) 2-хлор-3-трифторметилбензойной кислоты, 0,9 г (4,9 ммоль) 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида, 0,66 г(4,6 ммоль) 1-гидроксибензотриазола и 1 г (4,9 ммоль) (1S)-[(2S)-(1-метилпиперидин-2-ил)]фенилметанамина, получают 0,45 г продукта в форме свободного основания после очистки хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола. Продукт растворяют в нескольких миллилитрах пропан-2-ола, добавляют 10,9 мл 0,1 н раствора соляной кислоты в пропан-2-оле и смесь концентрируют при пониженном давлении для уменьшения объема растворителя. После растирания в конечном итоге выделяют 0,37 г гидрохлорида в форме белого твердого вещества. Температура плавления: 230-232 С.[]D25=+70,3 С (с=0,825; СН 3 ОН) ее 99%. Пример 3 (соединение 24). трео-4-Амино-3-хлор-N-[(1-метилпиперидин-2-ил)фенилметил]-5 трифторметилбензамида гидрохлорид 1:1. 3.1. 2-(Бензилоксииминофенилметил)-1-метилпиридиния трифторметансульфонат. 17,4 мл (120 ммоль) метилтрифторметансульфоната по каплям добавляют при 0 С к суспензии 35 г(120 ммоль) фенил(пиридин-2-ил)метанона O-бензилоксима в 200 мл диэтилового эфира и смесь перемешивают при комнатной температуре в течение 3 ч. Образовавшийся осадок отделяют путем фильтрации и сушат при пониженном давлении. Получают 49 г продукта, который используют как таковой на следующей стадии. 3.2. трео-(1-Метилпиперидин-2-ил)фенилметанамина этандиоат 2:1. 14,8 г (31,89 ммоль) 2-(бензилоксииминофенилметил)-1-метилпиридиния трифторметансульфоната и 0,74 г оксида платины в 50 мл этанола и 50 мл 1 н соляной кислоты помещают в сосуд Парра и проводят гидрирование в течение 5 ч. Этанол выпаривают при пониженном давлении, остаток экстрагируют дихлорметаном, водную фазу отделяют, к ней добавляют раствор аммиака и экстрагируют дихлорметаном. После промывания объединенных органических фаз, высушивания над сульфатом натрия, фильтрации и выпаривания растворителя при пониженном давлении получают 6,7 г маслянистого продукта, содержащего 10% эритродиастереоизомера. Этандиоат готовят путем растворения полученных 6,7 г основания в метаноле, при действии 2 эквивалентов этандионовой кислоты, растворенной в минимальном количестве метанола. Полученную соль очищают перекристаллизацией из смеси метанола и диэтилового эфира. В конечном итоге выделяют 4,7 г чистого этандиоата трео-диастереоизомера. Температура плавления: 156-159 С. 3.3. 4-Амино-3-хлор-5-трифторметилбензойная кислота. 7,8 г (40 ммоль) 4-амино-5-трифторметилбензойной кислоты в 80 мл хлороформа помещают в круглодонную колбу на 500 мл в присутствии 9,97 мл (50 ммоль) сульфурилхлорида и смесь перемешивают при кипячении с обратным холодильником в течение ночи. Растворитель выпаривают при пониженном давлении, остаток переносят в воду и водный раствор аммиака и смесь экстрагируют дихлорметаном. Водную фазу подкисляют, образующийся остаток отделяют путем фильтрации и сушат при пониженном давлении. Получают 9 г продукта. Температура плавления: 229-235 С. 3.4. трео-4-Амино-3-хлор-N-[(1-метилпиперидин-2-ил)фенилметил]-5-трифторметилбензамида гидрохлорид 1:1. 0,52 г (2,15 ммоль) 4-амино-3-хлор-5-трифторметилбензойной кислоты, 0,37 г (1,96 ммоль) 1-[3(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида, 0,26 г (1,96 ммоль) 1-гидроксибензотриазола в 5 мл 1,2-дихлорэтана помещают в круглодонную колбу на 100 мл и эту смесь перемешивают при комнатной температуре в течение 10 мин. Добавляют раствор 0,4 г (1,96 ммоль) трео-(1-метилпиперидин-2 ил)фенилметанамина в 5 мл 1,2-дихлорэтана и эту смесь оставляют перемешиваться в течение 12 ч. Смесь гидролизуют водой, добавляют гранулы гидроксида калия до получения щелочного значения рН и смесь экстрагируют дихлорметаном. Органическую фазу промывают водой, сушат над сульфатом натрия, фильтруют, растворитель выпаривают при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола. Выделяют 0,4 г соединения в форме основания.-6 007225 Его растворяют в нескольких миллилитрах пропан-2-ола, добавляют 9,4 мл 0,1 н раствора соляной кислоты в пропан-2-оле и растворитель выпаривают при пониженном давлении. Остаток собирают и сушат в вакууме. Получают 0,285 г твердого продукта. Температура плавления: 270-272 С. Пример 4 (соединение 25). 4-Амино-3-хлор-N-[(1R)-[(2R)-1-метилпиперидин-2-ил]фенилметил]5-трифторметилбензамида гидрохлорид 1:1. 4.1. (1R)-[(2R)-(1-Метилпиперидин-2-ил)]фенилметанамин. Раствор 80 г (390 ммоль) трео-(1-метилпиперидин-2-ил)фенилметанамина в 300 мл метанола и раствор 68 г (390 ммоль) N-ацетил-D-лейцина в 450 мл метанола помещают в круглодонную колбу на 4 л. Полученный раствор концентрируют при пониженном давлении и остаток перекристаллизовывают из 1100 мл пропан-2-ола. Получают 72 г соли (1R)-[(2R)-(1-метилпиперидин-2-ил)]фенилметанамина. Перекристаллизацию повторяют 3 раза и в конечном итоге получают 15 г соли (1R)-[(2R)-(1-метилпиперидин-2-ил)]фенилметанамина. Температура плавления: 171,5 С.[]D25=-11 (с=1; СН 3 ОН) ее 99%. 4.2. 4-Амино-3-хлор-N-[(1R)-[(2R)-1-метилпиперидин-2-ил]фенилметил]-5-трифторметилбензамида гидрохлорид 1:1. Используя способ, описанный в п.3.4 выше, начиная с 1,04 г (4,37 ммоль) 4-амино-3-хлор-5-трифторметилбензойной кислоты, 0,46 г (3,97 ммоль) 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида, 0,53 г (3,97 ммоль) 1-гидроксибензотриазола и 1,5 г (3,97 ммоль) (1R)-[(2R)-метилпиперидин 2-ил]фенилметанамина, получают 1,12 г продукта в форме основания. Гидрохлорид указанного продукта получают путем добавления 28,2 мл 0,1 н раствора соляной кислоты в пропан-2-оле к раствору 1,12 г основания в нескольких миллилитрах пропан-2-ола. Растворитель выпаривают при пониженном давлении, полученное твердое вещество собирают и сушат при пониженном давлении. В конечном итоге выделяют 0,9 г гидрохлорида в форме белого твердого вещества. Температура плавления: 175-185 С.[]D25=+18,4 (с=0,091; СН 3 ОН) ее=97,8%. Пример 5 (соединение 36). трео-2-Хлор-N-[фенил(пиперидин-2-ил)метил]-3-трифторметилбензамида гидрохлорид 1:1. 5.1. 2-Хлор-N-[фенил(пиридин-2-ил)метил]-3-трифторметилбензамид. 1,61 г (7,16 ммоль) 2-хлор-3-трифторметилбензойной кислоты, 1,4 г (7,28 ммоль) 1-[3-(диметиламино)пропил]-3-этилкарбодиимида гидрохлорида, 0,218 г (1,79 ммоль) раствора 4-диметиламинопиридина в 60 мл дихлорметана помещают в круглодонную колбу на 250 мл, смесь перемешивают в течение 15 мин,добавляют 1,1 г (5,97 ммоль) раствора фенил(пиридин-2-ил)метанамина в 60 мл дихлорметана и смесь перемешивают при комнатной температуре в течение 24 ч. Смесь гидролизуют путем добавления воды, добавляют 35%-ный водный раствор гидроксида натрия, органическую фазу отделяют, ее промывают водой и затем насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и растворитель выпаривают при пониженном давлении. Остаток очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана и метанола, и в конечном итоге выделяют 1,34 г продукта в форме желтого масла, которое кристаллизуется и как таковое используется на следующей стадии. 5.2. трео-2-Хлор-N-[фенил(пиперидин-2-ил)метил]-3-трифторметилбензамида гидрохлорид 1:1. Раствор 4,17 г (10 ммоль) 2-хлор-N-[фенил(пиперидин-2-ил)метил]-3-трифторметилбензамида в 43 мл ледяной уксусной кислоты помещают в сосуд Парра, добавляют 0,1 г 5%-ного палладия на углероде и проводят гидрирование при давлении 0,35 МПа при 50 С в течение 3 ч. После возвращения к комнатной температуре катализатор удаляют фильтрацией, фильтрат концентрируют при пониженном давлении, остаток переносят в воду и этилацетат, добавляют концентрированный гидроксид натрия и эту смесь экстрагируют несколько раз этилацетатом. Органическую фазу промывают водой и затем насыщенным водным раствором хлорида натрия, ее сушат над сульфатом натрия,фильтруют и растворитель выпаривают при пониженном давлении. Остаток очищают двумя последовательными хроматографическими разделениями на колонке с силикагелем, элюируя смесью дихлорметана и метанола от 100/0 до 95/5, для отделения непрореагировавшего исходного вещества. Выделяют 0,8 г (менее полярного) трео-диастереоизомера. Для получения гидрохлорида его растворяют в нескольких миллилитрах пропан-2-ола и добавляют к ним 20 мл раствора 0,1 н соляной кислоты в пропан-2-оле. Растворитель частично выпаривают при пониженном давлении, путем растирания получают белое твердое вещество, его собирают путем фильтрации и сушат при пониженном давлении. В конечном итоге получают 0,6 г гидрохлорида. Температура плавления: 234-235 С.-7 007225 Пример 6 (соединение 37). 2-Хлор-N-[(S)-фенил-[(2S)-пиперидин-2-ил]метил]-3-(трифторметил) бензамида гидрохлорид 1:1. Раствор 8,36 г (3 экв.) 1,3-диметилбарбитуровой кислоты в 100 мл безводного дихлорметана помещают в двугорлую колбу на 500 мл, оборудованную магнитной мешалкой, с циркуляцией аргона и холодильником. Добавляют 0,2 г (0,01 экв.) тетракис(трифенилфосфин)палладия и реакционную среду нагревают до 35 С. Добавляют раствор 7,8 г (19,18 ммоль) N-[(S)-[(2S)-1-аллилпиперидин-2-ил](фенил)метил]-2-хлор 3-(трифторметил)бензамида (полученного способом, аналогичным способу примера 1) и за развитием реакции наблюдают при помощи тонкослойной хроматографии. Добавляют 100 мл насыщенного раствора гидрокарбоната натрия, среду после отстаивания отделяют и водную фазу дважды экстрагируют 100 мл дихлорметана, объединенные органические фазы промывают 100 мл воды и затем 100 мл насыщенного раствора хлорида натрия. Их сушат над сульфатом натрия, фильтруют и растворитель выпаривают при пониженном давлении. Получают 10,15 г бежевого твердого вещества, которое очищают хроматографией на колонке с силикагелем, элюируя смесью дихлорметана, содержащей 0,4% 33%-ного раствора аммиака. Выделяют 4,8 г белесоватого твердого вещества. Твердое вещество растворяют в 50 мл пропан-2 ола, и добавляют 125 мл 0,1 н соляной кислоты в пропан-2-оле, и смесь концентрируют при пониженном давлении для уменьшения объема растворителя. После растирания выделяют 4,33 г гидрохлорида в форме белых кристаллов. Температура плавления: 223-225 С.[]D25=+80,7 С (с=0,5; СН 3 ОН) ее 98%. Пример 7 (соединения 69 и 79). 2-Хлор-N-1-метил-1-оксидопиперидин-2-ил](фенил)метил]3-трифторметилбензамид. 0,54 г (1,3 ммоль) трео-2-хлор-N-[(1-метилпиперидин-2-ил)фенилметил]-3-трифторметилбензамида в 20 мл безводного дихлорметана при 0 С помещают в круглодонную колбу на 50 мл, оборудованную магнитной мешалкой, добавляют раствор 0,28 г (1,2 экв.) 3-хлорпербензойной кислоты в 5 мл дихлорметана и смесь оставляют возвращаться к комнатной температуре при перемешивании в течение 12 ч. Добавляют 30 мл воды, среду отделяют после отстаивания и водную фазу дважды экстрагируют 30 мл дихлорметана, объединенные фазы промывают 100 мл воды и затем 100 мл насыщенного раствора хлорида натрия. Органическую фазу сушат над сульфатом натрия, растворители удаляют при пониженном давлении и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 90/10 дихлорметана и метанола в течение 40 мин. Выделяют 0,15 г первого изомера N-оксида (температура плавления: 100-102 С) и 0,03 г второго изомера N-оксида (температура плавления: 126-128 С). Пример 8 (соединение 71). (2S)-2[(1S)-2-Хлор-3-(трифторметил)бензоил]амино](фенил)метил]1,1-диметилпиперидиния йодид. Раствор 0,15 г (0,36 ммоль) 2-хлор-N-[(1S)-[(2S)-1-метилпиперидин-2-ил]фенилметил]-3-трифторметилбензамида в 20 мл ацетонитрила помещают в двугорлую колбу на 50 мл, оборудованную магнитной мешалкой, с циркуляцией аргона и холодильником, добавляют 0,5 мл йодметана и среду нагревают при 80 С в течение 2 ч. Реакционную среду концентрируют наполовину, осаждается аммониевая соль, ее фильтруют и сушат при пониженном давлении. Выделяют 0,17 г желтого твердого вещества. Температура плавления: 121-123 С. В следующей ниже табл. 1 изображены химические структуры некоторых соединений по данному изобретению. В колонке "A" cC3H5 обозначает циклопропильную группу. В "СF3" колонке указано положение СF3 группы в общей формуле (I). В "R2" колонке С 6 Н 6 обозначает фенильную группу. В колонке "соль" "-" обозначает соединение в форме основания, "HCl" обозначает гидрохлорид и "тфа" обозначает трифторацетат. В табл. 2 показаны физические свойства, температуры плавления и значения оптического вращения для некоторых соединений. Таблица 1 Соединение 69: наиболее полярный диастереоизомер Соединение 70: наименее полярный диастереоизомер Таблица 2 Соединения по данному изобретению были подвергнуты ряду фармакологических испытаний, которые продемонстрировали их важность в качестве веществ, обладающих терапевтической активностью. Исследование транспорта глицина в SK-N-MC клетках,экспрессирующих нативный транспортер glyt1 человека Захват [14C]глицина изучают в SK-N-MC клетках (нейроэпителиальные клетки человека), экспрессирующих нативный транспортер glyt1 человека, путем измерения радиоактивности, инкорпорированной в присутствии или в отсутствиe тестируемого соединения. Клетки выращивают в монослое в течение 48 ч в планшетах, предварительно обработанных 0,02%-ным фибронектином. В день эксперимента культуральную среду удаляют, а клетки промывают буфером Кребса-HEPES ([4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) с рН 7,4. После предварительной инкубации в течение 10 мин при 37 С в присутствии или буфера (контрольная партия), или тестируемого соединения в различных концентрациях, или 10 мМ глицина (определение неспецифического захвата) добавляют 10 мкМ [14 С]глицина (специфиче- 11007225 ская активность 112 мКи/ммоль). Инкубацию продолжают в течение 10 мин при 37 С и реакцию останавливают путем 2 промывок буфером Кребса-HEPES при рН 7,4. Затем после добавления 100 мкл жидкого сцинтиллятора и перемешивания в течение 1 ч в клетках оценивают инкорпорированную радиоактивность. Подсчет осуществляют на счетчике Microbeta Tri-lux. Эффективность соединения определяют по IC50, представляющей собой концентрацию соединения, при которой на 50% уменьшается специфический захват глицина, определяемый по разнице радиоактивности, инкорпорированной в контрольной партии и партии, которая получала 10 мМ глицина. Соединения по данному изобретению в этом тесте имели значения IC50 примерно от 0,0001 до 10 мкМ.Ex vivo исследование ингибиторной активности соединения в отношении захвата [14 С]глицина в гомогенате коры головного мозга мышей Увеличивающиеся дозы исследуемого соединения вводят перорально (готовят растиранием молекул тестируемого соединения в ступке в растворе Tween/Methocel в концентрации 0,5% в дистиллированной воде) или интраперитонеально (растворение молекул тестируемого соединения в физиологическом солевом растворе или приготовление путем растирания в ступке в растворе Tween/Methocel в концентрации 0,5% в воде в соответствии с растворимостью молекул) самцам мышей Iffa Credo OF1 массой от 20 до 25 г в день эксперимента. Контрольную группу обрабатывают носителем. Дозы в мг/кг, путь введения и время обработки определяют в зависимости от исследуемых молекул. После гуманного умерщвления животных путем декапитации в заданный момент времени после введения кору головного мозга каждого животного быстро перемещают на лед, взвешивают и хранят при 4 С или замораживают при -80 С (в обоих случаях образцы хранят в течение максимум 1 суток). Каждый образец гомогенизируют в буфере Кребса-HEPES при рН 7,4 в количестве 10 мл/г ткани. 20 мкл каждого гомогената инкубируют в течение 10 мин при комнатной температуре в присутствии 10 мМ L-аланина и буфера. Неспецифический захват определяют путем добавления к контрольной группе 10 мМ глицина. Реакцию останавливают путем фильтрации в вакууме и оставшуюся радиоактивность оценивают при помощи сцинтилляции в твердом теле путем подсчета на счетчике Microbeta Tri-lux. Ингибитор захвата [14 С]глицина будет уменьшать количество радиоактивного лиганда, инкорпорированного в каждом гомогенате. Активность соединения оценивают по его ED50, дозе, которая на 50% ингибирует захват [14 С]глицина по сравнению с контрольной группой. Наиболее сильнодействующие соединения по данному изобретению в данном испытании имелиED50 от 0,1 до 5 мг/кг при интраперитонеальном или пероральном пути введения. Исследование транспорта глицина в гомогенате спинного мозга мышей Захват [14 С]глицина транспортером glyt2 исследуют в гомогенате спинного мозга мышей путем измерения радиоактивности, инкорпорированной в присутствии или в отсутствие исследуемого соединения. После гуманного умерщвления животных (самцы мышей Iffa Credo OF1 массой от 20 до 25 г в день эксперимента) спинной мозг каждого животного быстро извлекают, взвешивают и хранят на льду. Образцы гомогенизируют в буфере Кребса-HEPES ([4-(2-гидроксиэтил)пиперазин-1-этансульфоновая кислота) при рН 7,4 в количестве 25 мл/г ткани. 50 мкл гомогената предварительно инкубируют в течение 10 мин при 25 С в присутствии буфера Кребса-HEPES, рН 7,4 и различных концентраций исследуемого соединения или 10 мМ глицина для определения неспецифического связывания. Затем добавляют [14 С]глицин (специфическая активность=112 мКи/ммоль) в течение 10 мин при 25 С в конечной концентрации 10 мкМ. Реакцию останавливают путем фильтрации в вакууме и радиоактивность оценивают при помощи сцинтилляции в твердом теле путем подсчета в счетчике Microbeta Tri-lux. Эффективность соединения определяют по концентрации IC50, которая способна на 50% уменьшить специфическое захват глицина, определенный по разнице радиоактивности, инкорпорированной в контрольной партии и партии, которая получала 10 мМ глицина. Соединения по данному изобретению в этом испытании продемонстрировали значения IC50 примерно от 0,0001 до 10 мкМ.Ex vivo исследование ингибиторной активности соединения в отношении захвата [14 С]глицина в гомогенате спинного мозга мышей Увеличивающиеся дозы исследуемого соединения вводят перорально (готовят путем растирания тестируемого соединения в ступке в растворе Tween/Methocel в концентрации 0,5% в дистиллированной воде) или интраперитонеально (тестируемое соединение, растворенное в физиологическом солевом растворе или растертое в ступке в растворе Tween/Methocel в концентрации 0,5% в дистиллированной воде) самцам мышей Iffa Credo OF1 массой от 20 до 25 г в день эксперимента. Контрольную группу обрабатывают носителем. Дозы в мг/кг, путь введения, время обработки и время гуманного умерщвления определяют в зависимости от исследуемого соединения. После гуманного умерщвления животных путем декапитации в заданный момент времени после введения образцы спинного мозга быстро извлекают, взвешивают и помещают в стеклянные сцинтилляционные колбы, помещают на измельченный лед или замораживают при -80 С (в обоих случаях образцы хранят в течение максимум 1 суток). Каждый образец гомогенизируют в буфере Кребса-HEPES при рН- 12007225 7,4 в количестве 25 мл/г ткани. 50 мкл каждого гомогената инкубируют в течение 10 мин при комнатной температуре в присутствии буфера. Неспецифический захват определяют путем добавления 10 мМ глицина к контрольной группе. Реакцию останавливают путем фильтрации в вакууме и радиоактивность оценивают при помощи сцинтилляции в твердом теле с использованием счетчика Microbeta Tri-lux. Ингибирование захвата [14 С]глицина уменьшает количество радиоактивного лиганда, инкорпорированного в каждом гомогенате. Активность соединения оценивают по его ED50, дозе, которая ингибирует на 50% захват [14 С]глицина по сравнению с контрольной группой. Наилучшие соединения по данному изобретению в соответствии с этим тестом имели ED50 от 1 до 20 мг/кг при интраперитонеальном или при пероральном пути введения. Результаты испытаний, проведенных с соединениями по данному соединению, имеющими конфигурацию (1S, 2S), и их трео-рацематами, имеющими конфигурацию (1R, 2R; 1S, 2S) в общей формуле (I),где R2 представляет собой один или более чем один атом галогена или трифторметильные группы, показывают, что они являются ингибиторами транспортера глицина glyt1, который находится в головном мозге, причем это происходит и in vitro, и ex vivo. Эти результаты означают, что соединения по данному изобретению можно применять для лечения расстройств поведения, связанных со слабоумием, психозов, в частности шизофрении (неполная форма и продуктивная форма) и острых и хронических экстрапирамидных симптомов, вызванных нейролептическими средствами, для лечения различных форм тревоги, приступов паники, фобий, обсессивно-компульсивных расстройств, для лечения различных форм депрессии, включая психотическую депрессию, для лечения расстройств, являющихся следствием злоупотребления алкоголем или прекращения употребления алкоголя, расстройств сексуального поведения, расстройств приема пищи и для лечения мигрени. Результаты испытаний, проведенных с соединениями по данному изобретению, имеющими конфигурацию (1R, 2R), и их рацематами, имеющими конфигурацию (1R, 2R; 1S, 2S) в общей формуле (I), гдеR2 представляет собой не только атом галогена, но и аминогруппу NR3R4, демонстрируют, что эти соединения представляют собой ингибиторы транспортера глицина glyt2, который преимущественно находится в спинном мозге, причем это происходит как in vitro, так и ех vivo. Эти результаты означают, что соединения по данному изобретению можно применять для лечения болезненных мышечных сокращений в ревматологии и при острой патологии спинного мозга, для лечения спастических сокращений медуллярного или церебрального происхождения, для симптоматического лечения острой или подострой боли с интенсивностью от слабой до средней, для лечения сильной и/или хронической боли, нейрогенной боли и упорной боли, для лечения болезни Паркинсона и паркинсонических симптомов нейродегенеративного происхождения или вызванных нейролептическими средствами,для лечения первичной или вторичной генерализованной эпилепсии, частичной эпилепсии с простой или сложной симптоматикой, смешанных форм и других эпилептических синдромов в качестве дополнения к другому эпилептическому лечению или в монотерапии, для лечения апноэ во сне, и для нейропротекции. Соответственно объектом настоящего изобретения также являются фармацевтические композиции,содержащие эффективную дозу по меньшей мере одного соединения по данному изобретению в форме фармацевтически приемлемого основания или соли, или сольвата и в форме смеси, когда это приемлемо,с подходящими эксципиентами. Указанные эксципиенты выбирают в соответствии с фармацевтической лекарственной формой и желаемым способом введения. Фармацевтические композиции по данному изобретению, таким образом, могут быть предназначены для перорального, сублингвального, подкожного, внутримышечного, внутривенного, местного, интратрахеального, интраназального, чрескожного, ректального или внутриглазного введения. Стандартные формы для введения могут представлять собой, например, таблетки, желатиновые капсулы, гранулы, порошки, пероральные или инъекционные растворы или суспензии, пластыри или суппозитории. Для местного введения могут быть предусмотрены мази, лосьоны и коллирии. Указанные стандартные формы содержат дозы, которые позволяют ежесуточно вводить от 0,01 до 20 мг активного ингредиента на килограмм массы организма, в соответствии с галеновой формой. Для изготовления таблеток к активному ингредиенту, микронизированному или немикронизированому, добавляют фармацевтический носитель, который может состоять из разбавителей, таких как, например, лактоза, микрокристаллическая целлюлоза, крахмал, и вспомогательных веществ для изготовления препарата, таких как связующие вещества (поливинилпирролидон, гидроксипропилметилцеллюлоза и подобные им), агенты, увеличивающие текучесть, такие как диоксид кремния, смазывающие вещества,такие как стеарат магния, стеариновая кислота, трибегенат глицерина, стеарилфумарат натрия. Также могут быть добавлены увлажняющие агенты или поверхностно-активные вещества, такие как лаурилсульфат натрия. Способами изготовления могут быть прямое прессование, сухое гранулирование, влажное гранулирование или горячее плавление. Таблетки могут быть непокрытыми, покрытыми оболочкой, например сахарозой, или покрытыми различными полимерами или другими подходящими веществами. Они могут быть предназначены для- 13007225 того, чтобы обеспечить быстрое, замедленное или продолжительное высвобождение активного ингредиента при помощи полимерных матриц или специфических полимеров, используемых в оболочке. Для приготовления желатиновых капсул активный ингредиент смешивают с сухими (простая смесь,сухое или влажное гранулирование или горячее плавление), жидкими или полутвердыми фармацевтическими носителями. Желатиновые капсулы могут быть твердыми или мягкими, покрытыми пленкой или иными, так чтобы обладать быстрым, продолжительным или замедленным действием (например, для энтеральной формы). Композиция в форме сиропа или эликсира или для введения в форме капель может содержать активный ингредиент вместе с подсластителем, предпочтительно бескалорийным, метилпарабеном или пропилпарабеном в качестве антисептика, корригентом и красителем. Диспергируемый в воде порошок и гранулы могут содержать активный ингредиент в форме смеси с диспергирующими агентами, или увлажняющими агентами, или диспергирующими веществами, такими как поливинилпирролидон, и с подсластителями и корригентами вкуса. Для ректального введения применяют суппозитории, которые изготавливают со связывающими веществами, плавящимися при ректальной температуре, например с какао-маслом или полиэтиленгликолями. Для парентерального введения применяют водные суспензии, изотонические солевые растворы или стерильные растворы для инъекции, содержащие фармакологически совместимые диспергирующие агенты и/или увлажняющие агенты, например пропиленгликоль или бутиленгликоль. Активный ингредиент также можно изготовить в форме микрокапсул, возможно с одним или более чем одним носителем, или добавками, или, альтернативно, с полимерной матрицей, или с циклодекстрином (пластыри, формы с длительным высвобождением). Композиции для местного введения по данному изобретению содержат среду, совместимую с кожей. Они могут быть представлены, в частности, в форме водных, спиртовых или водно-спиртовых растворов, гелей, эмульсий вода в масле или масло в воде, имеющих вид крема или геля, микроэмульсий,аэрозолей или, альтернативно, в форме дисперсий везикул, содержащих ионные и/или неионные липиды. Эти галеновые формы готовят способами, обычными для рассматриваемой области. В заключение, фармацевтические композиции по данному изобретению могут содержать кроме соединения общей формулы (I) другие активные ингредиенты, которые могут быть пригодны в лечении указанных выше расстройств и заболеваний. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение в форме чистого оптического изомера (1R, 2R) или (1S, 2S) либо в форме треодиастереоизомера, соответствующее общей формуле (I) где А представляет собой или группу общей формулы N-R1, где R1 представляет собой или атом водорода, или линейную или разветвленную (С 1-С 7)алкильную группу, возможно замещенную одним или более чем одним атомом фтора, или (С 4-С 7)циклоалкильную группу, или (С 3-С 7)циклоалкил(С 1-С 3)алкильную группу, или фенил(С 1-С 3)алкильную группу, возможно замещенную одной или двумя гидроксильными или метоксигруппами, или (С 2-С 4)алкенильную группу, или (С 2-С 4)алкинильную группу,или группу общей формулы N+(O-)R1, где R1 является таким, как определено выше,или альтернативно группу общей формулы N+(R')R1, где R' представляет собой линейную или разветвленную (С 1-С 7)алкильную группу и R1 является таким, как определено выше,Х представляет собой атом водорода или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, линейных или разветвленных (С 1-С 4)алкильных и (С 1-С 4)алкоксигрупп,R2 представляет собой или атом водорода, или один или более чем один заместитель, выбранный из атомов галогена и трифторметила, (С 1-С 4)алкильной или (С 1-С 4)алкоксигрупп или аминогрупп общей формулы NR3R4, где R3 и R4, каждый независимо друг от друга, представляeт собой атом водорода или(С 1-С 4)алкильную группу или образуeт вместе с несущим их атомом азота пирролидиновое, пиперидиновое или морфолиновое кольцо или фенильную группу, возможно замещенную атомом или группой, как определено для символа Х выше,в форме свободного основания или соли присоединения кислоты.- 14007225 2. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию (1S, 2S) и тем, что R2 представляет собой атом галогена или трифторметильные группы в количестве один(на) или более. 3. Соединение по п.1, отличающееся тем, что оно имеет конфигурацию (1R, 2R), и тем, что R2 представляет собой атом галогена и аминогруппу общей формулы NR3R4, как определено в п.1. 4. Лекарственное средство, отличающееся тем, что оно состоит из соединения по любому из пп.1-3. 5. Фармацевтическая композиция, отличающаяся тем, что она содержит соединение по любому из пп.1-3 в комбинации с эксципиентом.
МПК / Метки
МПК: A61K 31/445, C07D 211/26, A61P 25/00
Метки: терапии, n-[фенил(пиперидин-2-ил)метил]бензамида, производные, применение
Код ссылки
<a href="https://eas.patents.su/16-7225-proizvodnye-n-fenilpiperidin-2-ilmetilbenzamida-i-ih-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные n-[фенил(пиперидин-2-ил)метил]бензамида и их применение в терапии</a>
Аминированные производные дигидро-1,3,5-триазина и их применение в терапии

Номер патента: 5378
Опубликовано: 24.02.2005
Авторы: Доар Лилиан, Кергоа Мишлин, Мезанжо Дидье, Муане Жерар, Краво Даниель
МПК: C07D 251/10, A61K 31/53, A61P 3/10...
Метки: применение, производные, дигидро-1,3,5-триазина, аминированные, терапии
Формула / Реферат:
1. Соединение общей формулы (I) где R1, R2, R3 и R4 выбираются независимо друг от друга из групп H, (C1-C20)алкил, замещенный или не замещенный галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, (C3-C8)циклоалкилом, (C2-C20)алкенил, замещенный или не замещенный галогеном, (C1-C5)алкилом, (C1-C5)алкоксилом, (C2-C20)алкинил, замещенный или не замещенный галогеном, (C1-C5)алкилом, алкоксилом, (C3-C8)циклоалкил, замещенный или не замещенный...
(r)-n-[[4-[[(2-метилфениламино)карбонил]амино]фенил]ацетил]-l-пропил-3- метил- бета-аланин и его применение в качестве ингибитора клеточной адгезии, опосредуемой интегрином альфа4бета1

Номер патента: 2988
Опубликовано: 26.12.2002
Авторы: Ли Вен-Чернг, Джилл Алан
МПК: A61P 11/06, C07D 207/16, A61K 31/40...
Метки: применение, адгезии, альфа4бета1, интегрином, метил, клеточной, опосредуемой, r)-n-[[4-[[(2-метилфениламино)карбонил]амино]фенил]ацетил]-l-пропил-3, бета-аланин, ингибитора, качестве
Формула / Реферат:
1. (R)-N-[[4-[[(2-метилфениламино)карбонил]амино]фенил]ацетил]-L-пролил-3-метил-b-аланин, ингибирующий клеточную адгезию или его фармацевтически приемлемое производное, пролекарство или соль. 2. Соединение по п.1, в котором пролекарство представляет собой сложный эфир соединения. 3. Соединение по п.2, представляющее собой сложный эфир, полученный путем взаимодействия...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Макколи Джеймс, Крокер Луис
МПК: C07D 265/32, A61P 25/28, A61K 31/5375...
Метки: метил)морфолина, антагониста, трифторметил, качестве, триазоло, 4-фтор, 2-(r, рецептора, 5-оксо-1h, тахикинина, 3,5-бис, полиморфная, 4h-1, этокси, форма, фенил
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Новые замещенные 4-фенил-4-[1н-имидазол-2-ил] пиперидиновые производные и их применение в качестве селективных непептидных агонистов дельта-опиоидов

Номер патента: 6507
Опубликовано: 29.12.2005
Авторы: Фернандес-Гадеа Франсиско Хавьер, Янссенс Франс Эдуард, Гомес-Санчес Антонио, Ленартс Йозеф Элизабет, Мерт Тео Франс
МПК: A61P 25/04, A61K 31/445, C07D 401/04...
Метки: агонистов, производные, применение, дельта-опиоидов, замещенные, непептидных, селективных, качестве, пиперидиновые, 4-фенил-4-[1н-имидазол-2-ил, новые
Формула / Реферат:
1. Соединение формулы (I) где A=B представляет собой двухвалентный радикал с p-связью; X представляет собой ковалентную связь, -CH2- или CH2CH2-; R1 представляет собой водород, гидрокси, алкилокси, алкилкарбонилокси, Ar-окси, Het-окси, Ar-карбонилокси, Het-карбонилокси, Ar-алкилокси, Het-алкилокси, алкил, полигалогеналкил, алкилоксиалкил, Ar-алкил, Het-алкил, Ar, Het, тио, алкилтио, Ar-тио, Het-тио или NR9R10, где R9 и R10, каждый независимо,...
Производные бензамида и фармацевтическая композиция на их основе

Номер патента: 3245
Опубликовано: 27.02.2003
Авторы: Андерскевич Ральф, Йенневайн Ханс Михаель, Меаде Кристофер Джон Монтэгю, Бирке Франц, Шромм Курт, Рент Эрнст-Отто
МПК: A61P 11/06, C07C 257/18, A61K 31/155...
Метки: композиция, основе, фармацевтическая, бензамида, производные
Формула / Реферат:
1. Производные бензамидина общей формулы (1) в которой A означает -OCH2CH2O-, -CH2O-, -CH2CH2CH2O-; R1 означает алкил с 1-6 атомами углерода с разветвленной или прямой цепью, алкенил с 3-6 атомами углерода с разветвленной или прямой цепью, предпочтительно аллил; R2 означает водород, алкил с 1-6 атомами углерода с разветвленной или прямой цепью, алкенил с 3-6 атомами углерода с разветвленной или прямой цепью, предпочтительно аллил; R3 и R4,...
Предыдущий патент: Производные 3-замещенных 4-пиримидонов
Следующий патент: Производные имидазохинолина
Случайный патент: Ингибиторы akt (протеинкиназы в)