Некоторые производные 1-(d-циклопропилглицинил)-4-(пиперидин-4-ил) пиперазина в качестве ингибиторов серинпротеазы фактора ха







Формула / Реферат
1. Соединение формулы (I)

в которой R представляет собой атом водорода или атом фтора, или его фармацевтически приемлемая соль.
2. Соединение по п.1, в котором R представляет собой атом водорода.
3. Соединение по п.2, выбранное из
1-(4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина и его гидрохлоридных, фумаратных и малеатных аддитивных солей кислоты.
4. Соединение по п.3, выбранное из дигидрохлоридных, дифумаратных и дималеатных аддитивных солей кислоты в кристаллической форме.
5. Соединение по п.4, представляющее собой дифумарат 1-(4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина в кристаллической форме.
6. Соединение по п.1, в котором R представляет собой атом фтора.
7. Соединение по п. 6, выбранное из
1-(3-фтор-4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина и его гидрохлоридных аддитивных солей кислоты.
8. Фармацевтическая композиция, содержащая соединение по одному любому из пп.1-7 вместе с фармацевтически приемлемым разбавителем или носителем.
9. Фармацевтическая композиция по п.8, приспособленная для перорального введения.
10. Способ получения соединения по одному любому из пп.1-7, включающий:
(а) взаимодействие соединения формулы (II)

или его соли с соединением формулы (III)

или его реакционноспособным производным; или
(b) взаимодействие соединения формулы (IV)

или его соли с соединением формулы (V)

или его реакционноспособным производным;
и последующее образование фармацевтически приемлемой соли, если требуется фармацевтически приемлемая соль.
11. Соединение формулы (II)

или его соль.
12. Соединение формулы (V)

13. Соединение по одному любому из пп.1-7, предназначенное для использования в терапии.
14. Применение соединения по одному любому из пп.1-7 для производства лекарственного препарата, предназначенного для лечения тромботического заболевания.
15. Применение по п.14, в котором лекарственный препарат предназначен для перорального лечения тромботического заболевания.
16. Способ лечения тромботического заболевания у субъекта, нуждающегося в таком лечении, включающий введение эффективного количества соединения по п.1.
17. Способ по п.16, в котором соединение вводят субъекту перорально.
Текст
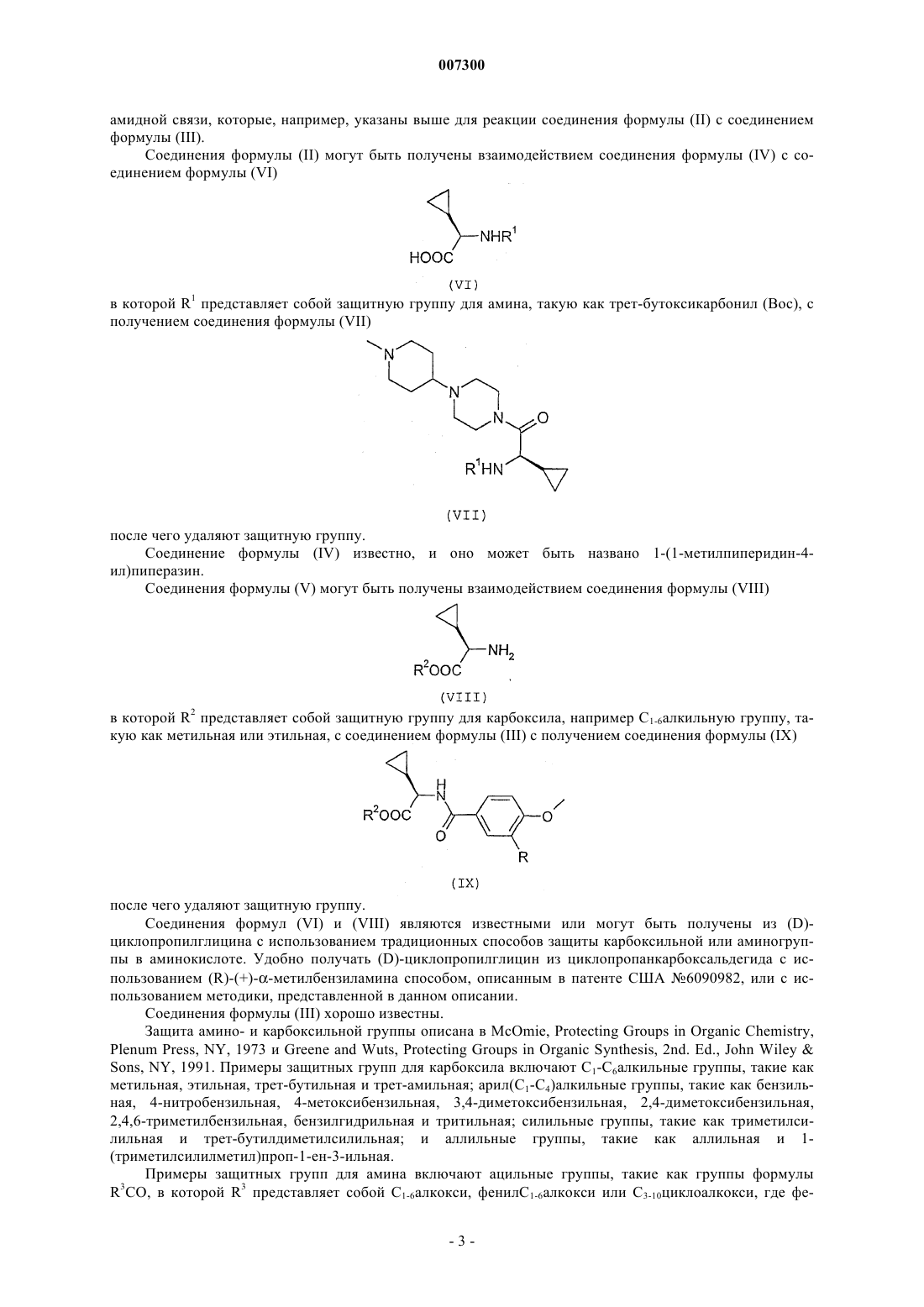
007300 Настоящее изобретение относится к соединениям, применимым в качестве лекарственных препаратов, к содержащим соединения фармацевтическим композициям, к способу получения соединений, к промежуточным продуктам, полезным при получении соединений, и к применению соединений в качестве лекарственных препаратов. Сердечно-сосудистые заболевания продолжают представлять большую проблему для здоровья людей во всем мире и являются основной причиной серьезных заболеваний и смерти. Одно из направлений исследований, осуществляемых исследователями в области новых способов лечения сердечно-сосудистого заболевания, основано на гипотезе о том, что ингибитор серинпротеазы,являющейся фактором Ха, может быть применен при лечении тромботического заболевания в качестве антикоагулянта. Ингибиторы фактора Ха известны. Так например, в WO 01/96323 раскрыты некоторые соединения,содержащие ароматическую группу, глициновый остаток, несущий циклическую группу, и 4 замещенную пиперазинильную группу. Циклическая группа может представлять собой циклоалкильную группу, такую как циклопентильная или циклогексильная, но предпочтение отдается соединениям, в которых циклическая группа является фенильной группой. Неожиданно было найдено, что при выборе в соединениях WO 01/96323 в качестве ароматической группы 4-метоксифенильной или 3-фтор-4-метоксифенильной группы, в качестве глицинового остатка циклопропилглициновой группы и в качестве 4-замещенной пиперазинильной группы (1 метилпиперидин-4-ил)пиперазинильной группы могут быть получены соединения, которые являются селективными ингибиторами фактора Ха и имеют особенно выгодные свойства. Соответственно настоящее изобретение относится к соединению формулы (I) в которой R представляет собой атом водорода или атом фтора, или к его фармацевтически приемлемой соли. Было найдено, что соединения формулы (I) являются сильнодействующими и селективными ингибиторами серинпротеазы, являющейся фактором Ха, имеют высокую противосвертывающую активность в плазме человека, оказывают эффективное действие на плазму при пероральном введении млекопитающим и обладают особенно выгодными фармакологическими и токсикологическими профилями активности. Соединения формулы (I) могут быть также представлены химическими названиями: 1-(4 метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазин и 1-(3-фтор-4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазин. Следует принять во внимание, что соединения формулы (I) содержат центр асимметрии, имеющий(D)-конфигурацию. Поэтому соединения могут существовать и быть выделены в смеси с соответствующим (L) изомером, такой как рацемическая смесь, или по отдельности. Предпочтительно выделяют соединения, по существу не содержащие (L) изомеров. Следует также принять во внимание, что соединения формулы (I) или их фармацевтически приемлемые соли могут быть выделены в форме сольватов и, что соответственно любой такой сольват включен в объем настоящего изобретения. Примеры фармацевтически приемлемых солей включают гидрохлоридные, фумаратные и малеатные аддитивные соли кислоты. Необходимо учитывать, что соединения формулы (I) содержат два основных атома азота и поэтому могут образовывать аддитивные соли одно- и двухосновной кислоты. Одной группой соединений формулы (I) является такая группа, в которой R представляет собой атом водорода. Конкретные соединения, в которых R является атомом водорода, представляют собой: 1-(4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил) пиперазин и его гидрохлоридные, фумаратные и малеатные аддитивные соли кислоты, в особенности дигидрохлоридные, дифумаратные и дималеатные аддитивные соли кислоты.-1 007300 Получены кристаллические дигидрохлоридные, дифумаратные и дималеатные аддитивные соли кислоты, которые охарактеризованы дифференциальной сканирующей калориметрией (ДСК): дигидрохлорид: разлагается перед плавлением; дифумарат: 222,8 С (начало), 225,4 С (пик); и дималеат: 195,8 С (начало), 202,0 С (пик). Особенного упоминания заслуживает дифумарат 1-(4-метоксибензоил-D-циклопропилглицинил)-4(1-метилпиперидин-4-ил)пиперазина в кристаллической форме. Другой группой соединений формулы (I) является такая группа, в которой R представляет собой атом фтора. Конкретные соединения, в которых R является атомом фтора, представляют собой 1-(3-фтор-4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазин и гидрохлоридные аддитивные соли кислоты. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть получены способом,включающим:(а) взаимодействие соединения формулы (II) или его соли (такой как тригидрохлорид) с соединением формулы (III) или его реакционноспособным производным; или(b) взаимодействие соединения формулы (IV) или его соли (такой как дигидробромид) с соединением формулы (V) или его реакционноспособным производным; и последующее образование фармацевтически приемлемой соли, если требуется фармацевтически приемлемая соль. Реакция между соединением формулы (II) и соединением формулы (III) обычно может быть осуществлена с использованием реагентов и условий реакций, традиционно применяемых для образования амидной связи. Реакцию обычно осуществляют в присутствии реагента на основе бензотриазола, такого как 1-гидроксибензотриазол или 1-гидрокси-7-азабензотриазол, и дегидратирующего агента, такого как дициклогексилкарбодиимид или 1-(3-диметиламинопропил)-3-этилкарбодиимид, в инертном органическом растворителе, таком как диметилформамид и/или метиленхлорид. Реакцию обычно проводят при температуре от 0 до 50 С, предпочтительно при комнатной температуре. Если используют соль соединения формулы (II), реакцию обычно осуществляют при дополнительном присутствии основания, такого как триэтиламин. В данной области известны другие подходящие реагенты и растворители, например галогенангидрид кислоты, такой как п-анизоилхлорид, в присутствии основания, такого как триэтиламин. Реакция между соединением формулы (IV) и соединением формулы (V) обычно может быть осуществлена с использованием реагентов и условий реакций, традиционно применяемых для образования-2 007300 амидной связи, которые, например, указаны выше для реакции соединения формулы (II) с соединением формулы (III). Соединения формулы (II) могут быть получены взаимодействием соединения формулы (IV) с соединением формулы (VI) в которой R1 представляет собой защитную группу для амина, такую как трет-бутоксикарбонил (Воc), с получением соединения формулы (VII) после чего удаляют защитную группу. Соединение формулы (IV) известно, и оно может быть названо 1-(1-метилпиперидин-4 ил)пиперазин. Соединения формулы (V) могут быть получены взаимодействием соединения формулы (VIII) в которой R2 представляет собой защитную группу для карбоксила, например C1-6 алкильную группу, такую как метильная или этильная, с соединением формулы (III) с получением соединения формулы (IX) после чего удаляют защитную группу. Соединения формул (VI) и (VIII) являются известными или могут быть получены из (D)циклопропилглицина с использованием традиционных способов защиты карбоксильной или аминогруппы в аминокислоте. Удобно получать (D)-циклопропилглицин из циклопропанкарбоксальдегида с использованием (R)-(+)метилбензиламина способом, описанным в патенте США 6090982, или с использованием методики, представленной в данном описании. Соединения формулы (III) хорошо известны. Защита амино- и карбоксильной группы описана в McOmie, Protecting Groups in Organic Chemistry,Plenum Press, NY, 1973 и Greene and Wuts, Protecting Groups in Organic Synthesis, 2nd. Ed., John WileySons, NY, 1991. Примеры защитных групп для карбоксила включают C1-С 6 алкильные группы, такие как метильная, этильная, трет-бутильная и трет-амильная; арил(C1-C4)алкильные группы, такие как бензильная, 4-нитробензильная, 4-метоксибензильная, 3,4-диметоксибензильная, 2,4-диметоксибензильная,2,4,6-триметилбензильная, бензилгидрильная и тритильная; силильные группы, такие как триметилсилильная и трет-бутилдиметилсилильная; и аллильные группы, такие как аллильная и 1(триметилсилилметил)проп-1-ен-3-ильная. Примеры защитных групп для амина включают ацильные группы, такие как группы формулыR3CO, в которой R3 представляет собой С 1-6 алкокси, фенилС 1-6 алкокси или С 3-10 циклоалкокси, где фе-3 007300 нильная группа может быть необязательно замещена, например, одним или двумя заместителями, выбранными из галогена, С 1-4 алкила и С 1-4 алкокси. Предпочтительные защитные группы для амина включают бензилоксикарбонил (CBz) и третбутоксикарбонил (Вос). Полагается, что некоторые из указанных в данном описании промежуточных продуктов, например соединения формул (II) и (V), являются новыми и, соответственно, рассматриваются в качестве дополнительных аспектов изобретения. Соединения изобретения могут быть введены любым подходящим путем, например в желудочнокишечный тракт (например, ректально или перорально), нос, легкие, мускулатуру или сосудистую сеть или транедермально. Соединения могут быть введены в любой форме, подходящей для введения, например в виде таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, аэрозолей, суппозиториев, гелей, эмульсий, пластырей и т.д. Такие композиции могут содержать компоненты, общепринятые в фармацевтических препаратах, например разбавители, носители, модификаторы рН, подсластители, наполнители и дополнительные активирующие агенты. Если требуется парентеральное введение,композиции должны быть стерильными и находиться в форме раствора или суспензии, подходящих для инъекции или вливания. Такие композиции образуют еще один аспект изобретения. С точки зрения данного аспекта изобретения предлагается фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. В соответствии с другим аспектом настоящего изобретения предлагается соединение формулы (I) или его фармацевтически приемлемая соль для использования в терапии. В соответствии с другим аспектом настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного препарата,предназначенного для лечения тромботического заболевания. В соответствии с другим аспектом настоящее изобретение относится к способу лечения тромботического заболевания у субъекта, нуждающегося в таком лечении, включающему введение эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Субъектом может быть человек или отличное от человека животное, такое как отличное от человека млекопитающее, например кошка, собака, лошадь, корова или овца. Тромботическое заболевание может представлять собой, например, венозный тромбоз, эмболию легких, артериальный тромбоз, ишемию миокарда, инфаркт миокарда или церебральный тромбоз. Соединения могут быть также использованы в соответствии со способом изобретения при лечении резкого закрытия сосуда, связанного с тромболитической терапией и рестенозом, например после транслюминальной пластической операции на коронарных сосудах или обходного сосудистого шунтирования коронарных или периферических артерий, и при сохранении сосуда больного в раскрытом состоянии при продолжительном гемодиализе больных. Доза соединения формулы (I) будет зависеть от природы и тяжести заболевания, подвергаемого лечению, способа введения лекарственного средства, массы тела и специфических особенностей субъекта. Однако, обычно лекарственное средство вводят в количестве в диапазоне от 0,01 до 100 мкмоль/кг массы тела. Использованный в данном описании термин лечение включает профилактическое применение. Термин эффективное количество относится к количеству соединения формулы (I), которое является эффективным для уменьшения или ингибирования развития симптомов подвергаемого лечению тромботического заболевания. Соединение в соответствии с изобретением может быть введено одно или в комбинации с антикоагулянтом, имеющим другой путь действия, или с тромболитическим средством. Данное изобретение иллюстрируют следующие примеры. Использованные сокращения соответствуют номенклатуре IUPAC-IUB. В описании используются следующие сокращения: Вос (третичный бутилоксикарбонил), Выч. (вычислено), DMSO (диметилсульфоксид, полностью дейтерированный, если для ЯМР), EDCI (гидрохлорид (1-(3-диметиламинопропил)-3 этилкарбодиимида), ES-MS (масс-спектр с ионизацией электрораспылением), HOBt (1 гидроксибензотриазол), ВЭЖХ (высокоэффективная жидкостная хроматография с tr в качестве времени удерживания), МеОН (метанол), ЯМР (ядерный магнитный резонанс), TFA (трифторуксусная кислота). Пример 1. 1-(4-Метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазинA. D-Циклопропилглицин Аминокислоту получали удобным путем из циклопропанкарбоксальдегида с использованием R-(+)-метилбензиламина и методики патента США 6090982 или с использованием методики, представленной в данном описании.(300 мл) и 1N NaOH (480 мл, 0,48 моль), охлажденный на бане со льдом до 0-5 С. Медленно добавляли дитрет-бутилдикарбонат (105 г, 0,48 моль) и продолжали перемешивание при комнатной температуре в-4 007300 течение 0,5 ч. Раствор концентрировали в вакууме примерно до 500 мл, охлаждали на бане со смесью льда и воды, покрывали слоем этилацетата (500 мл) и подкисляли разбавленным водным растворомKHSO4 до рН 2-3. Водную фазу экстрагировали этилацетатом (500 мл) и экстракцию повторяли до исчезновения продукта. Этилацетатные экстракты объединяли, промывали водой (0,5 л), насыщенным раствором соли (0,5 л), сушили над Na2SO4, фильтровали и концентрировали с получением белого твердого вещества (78 г, 90,6%).Boc-D-циклопропилглицин (216,0 г, 1,0 моль) и 1-(1-метилпиперидин-4-ил)пиперазин (192 г, 1,05 моль) суспендировали в водном растворе CH2Cl2 (3,2 л) в атмосфере N2. Затем смесь охлаждали на бане со льдом до 0-5 С. К полученной смеси добавляли моногидрат 1-гидроксибензотриазола (HOBt) (149 г,1,1 моль) и диизопропилэтиламин (136 г, 1,05 моль), после чего медленно добавляли гидрохлорид 1-[3(диметиламино)пропил]-3-этилкарбодиимида (EDCl) (211 г, 1,1 моль), поддерживая температуру при 05 С в течение 1 ч. Реакционной смеси давали возможность нагреться в течение ночи до комнатной температуры. Затем реакционную смесь гасили добавлением насыщенного (насыщ.) водного раствораNaHCO3 (3 л) и экстрагировали метиленхлоридом (2 л). Разделяли слои. Органический слой опять промывали насыщ. NаНСО 3 (3 л), насыщенным раствором соли (2 л), сушили над MgSO4, фильтровали и концентрировали с получением неочищенного продукта в виде вязкого масла (415 г, 109%), который использовали непосредственно. 1 Н ЯМР (ДМС-d6), : 6,91 (д, 1 Н), 3,97 (т, 1 Н), 3,41 (ушир.с, 4 Н), 2,74 (д, 2 Н), 2,40 (ушир.с, 4 Н), 2,10D. Тригидрохлорид 1-(D-циклопропилглицинил)-4- (1-метилпиперидин-4-ил)пиперазина 1-(Boc-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазин (неочищенный, 415 г, 1,0 моль) растворяли в безводном метаноле (1,5 л). К полученному раствору при 0 С добавляли раствор HClМеОН (380 г/1,5 л, 10,4 моль). Реакционную смесь перемешивали и медленно нагревали до комнатной температуры в течение 2-х ч. Затем при перемешивании добавляли этилацетат (2 л) . Перемешивание продолжали в течение 1 ч при 0-5 С, и продукт выпадал в виде белого порошка, который фильтровали и сушили в вакууме при 45 С с получением указанного в заголовке соединения в виде белого твердого вещества (357 г, 91,7%). 1E. 1-(4-Метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазин Тригидрохлорид 1-(D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина (300 г, 0,77 моль) суспендировали в безводном СН 2 Сl2 (3 л) в атмосфере N2. Затем смесь охлаждали на бане со льдом до 0-5 С. Медленно добавляли триэтиламин (450 мл, 3,23 моль), поддерживая температуру при 0-5 С,затем медленно добавляли п-анизоилхлорид (142 г, 0,83 моль), снова поддерживая температуру при 05 С. Реакционной смеси давали возможность нагреться до комнатной температуры в течение 2-х ч. Затем реакционную смесь гасили добавлением насыщенного раствора NaHCO3 (1 л) и разделяли слои. После этого водный слой экстрагировали СН 2 Сl2 (2 л). Органические слои объединяли, промывали насыщенным раствором соли (1 л), сушили над MgSO4, фильтровали и концентрировали с получением указанного в заголовке соединения (326 г, 102%). 1(315 г, 0,76 моль) в 95% этаноле (4,2 л), нагретому до 65 С, добавляли раствор фумаровой кислоты (177 г, 1,52 моль) в горячем этаноле (при 65 С, 2,8 л). Конечный прозрачный раствор перемешивали при 65 С и медленно охлаждали до комнатной температуры (в течение 2-х ч), а затем до 0-5 С. Белые кристаллы отфильтровывали, промывали 95% этанолом (1 л) и сушили в вакууме при 45 С с получением указанной в заголовке соли (448 г, 91,2%), т.пл.= 205-207 С. 1 Н ЯМР (ДМС-d6), : 11,35 (с, 1 Н), 8,58 (д, 1 Н), 7,86 (д, 2 Н), 6,96 (д, 2 Н), 6,55 (с, 4 Н), 4,39 (т, 1 Н),3,79 (с, 3H), 3,45 (с, 4 Н), 3,20 (д, 2 Н), 2,62 (т, 2 Н), 2,55 (с, 3H), 2,42 (м, 6 Н), 1,80 (д, 2 Н), 1,62 (д, 2 Н), 1,28ES-MS, m/z: 415,5 (M+l)+. Анализ C23H34N4O31,25 HCl1,0 Н 2O: Вычислено: С 57,78; Н 7,85; N 11,72; Сl 9,30; Найдено: С 57,79; Н 7,93; N 11,74; Сl 9,64. Аналитическая ВЭЖХ (Xterra RP18, 4,6 х 150 см, смесь 10% ацетонитрил/вода (0,1% TFA) - смесь 50% ацетонитрил/вода (0,1% TFA) в течение 40 мин), 1 мл/мин: 99%, tr = 10,66 мин. Пример 2. 1-[(3-Фтор-4-метоксибензоил)-D-циклопропилглицинил]-4-(1-метилпиперидин-4 ил)пиперазин К перемешиваемой суспензии тригидрохлорида 1-D-циклопропилглицинил-4-(1-метилпиперидин 4-ил)пиперазина (1,5 г, 3,85 ммоль) в дихлорметане (30 мл) добавляли триэтиламин (1,36 г, 13,5 ммоль),после чего добавляли 3-фтор-4-метоксибензойную кислоту (0,622 г, 3,66 ммоль), HOBt (0,573 г, 4,24 ммоль) и EDCl (0,813 г, 4,24 ммоль). После перемешивания в течение ночи смесь распределяли между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Затем органическую фазу опять промывали насыщенным водным раствором бикарбоната натрия, после этого насыщенным раствором соли, затем сушили MgSO4, фильтровали и концентрировали в вакууме. Затем остаток растворяли в дихлорметане и очищали хроматографией на силикагеле, осуществляя градиентное элюирование смесью 012% 2N аммиак/метанол в дихлорметане. Содержащие чистый продукт фракции объединяли и концентрировали в вакууме с получением 1,03 г (61%) указанного в заголовке соединения.ES-MS, m/z: 433,3 (M+l)+. Анализ C23H33FN4O31,6 HCl0,5 Н 2O: Вычислено: С 55,26; Н 7,18; N 11,21; F 3,80; Сl 11,35; Найдено: С 55,31; Н 7,24; N 11,38; F 3,64; Сl 11,36. Аналитическая ВЭЖХ (Xterra RP18, 4,6 х 150 см, смесь 10% ацетонитрил/вода (0,1% TFA) - смесь 50% ацетонитрил/вода (0,1% TFA) в течение 40 мин.), 1 мл/мин: 98%, tr = 8,067 мин. Анализы ингибирования фермента Способность испытуемого соединения ингибировать фактор Ха может быть оценена одним или несколькими следующими анализами ингибирования фермента или другими стандартными анализами, известными специалистам в данной области. Анализ ингибирования фермента Человеческий фактор Ха и человеческий тромбин приобретали у Enzyme Research Laboratories(South Bend, Indiana, USA). Другие протеазы получали из других коммерческих источников. Хромогенные пара-нитроанилидные субстраты для пептидазы приобретали у Midwest Biotech (Fishers, Indiana,USA). Способность к связыванию человеческого фактора Ха измеряли в виде кажущейся константы ассоциации Касса (Kass), полученной из кинетики ингибирования, как описано ранее в а' b' c' d. Значение кажущейся константы по Кассу получали с использованием автоматических (BioMek - 1000) разбавлений ингибиторов (определения по Кассу осуществляли в виде трех одинаковых опытов, проводимых при каждых четырех-восьми концентрациях ингибитора) в 96-луночных планшетах, и скорости гидролиза хромогенного субстрата определяли при 405 нм с использованием считывающего устройства Thermomax отMolecular Devices (San Francisco). Для ингибирования фактора Ха протокол анализа был следующим: 50 мкл буфера (0,06 М tris, 0,3 М NaCl, pH 7,4); 25 мкл раствора испытуемого ингибитора (в МеОН); 25 мкл человеческого фактора Ха (32 нМ в 0,03 М tris, 0,15 М NaCl, 1 мг/мл HSA); и, наконец, 150 мклBzIleGluGlyArgpNA (0,3 мМ в воде), добавленного в течение 2-х мин для начала гидролиза. Конечная концентрация [фактора Ха] составляла 3,2 нМ. Концентрацию свободного фактора [Ха] и концентрацию-6 007300 связанного фактора [Ха] определяли по линейным стандартным кривым на том же планшетном считывающем устройстве, используя компьютерное обеспечение SoftmaxPro, для каждой концентрации ингибитора и вычисляли значение кажущейся константы ассоциации по Кассу для каждой концентрации ингибитора, которая давала ингибирование гидролиза, равное значению между 20 и 80% относительно контрольного образца (3,2 нМ фактор Ха) : кажущаяся константа ассоциации по Кассу = [E:I] / [Ef] [If] =[Eb]/[Ef] [I-Ib], где [E:I] и [Eb] обозначают концентрацию фактора Ха, связанного с ингибитором, [Ef] обозначает концентрацию свободного фактора Ха, [If] и [I-Ib] обозначают исходную концентрацию ингибитора. Полученные таким образом значения кажущейся константы ассоциации по Кассу приблизительно представляют собой обратные значения Ki для соответствующих ингибиторов [1/каж. по Кассу = прибл. Ki]. Отклонения средних значений кажущейся константы по Кассу, определенные при выбранной концентрации субстрата, составляли +/- 15%. Km анализируемой системы равна 0,347 +/-0,031 мМ [число измерений n= 4]; и Vmax равна 13,11 +/- 0,76 мкМ/мин. Значения по Кассу определяли с тромбином и другими протеазами с использованием такого же протокола и следующих концентраций фермента и субстрата: 5,9 нМ тромбина с 0,2 мМ BzPheValArgpNA; 1,2 нМ фактора ХIа с 0,4 мМ пиpoGluProArgpNA; 10 нМ фактора XIIa с 0,2 мМ HDProPheArgpNA; 3,4 нМ плазмина с 0,5 мМ HDValLeuLyspNA; 1,2 нМ nt-PA с 0,8 мМ HDIleProArgpNA; 0,4 нМ урокиназы с 0,4 мМ пироGluGlyArgpNA; 3 нМ аРС с 0,174 мМ пиpoGluProArgpNA; 1,9 нМ калликреина плазмы с D-ProPheArgpNA; и 1,4 нМ бычьего трипсина с 0,18 мМ BzPheValArgpNA. Список литературыPharmacokinetic Properties of Modified C-3 Side Chain Derivatives. J. Med. Chem., 43, 649-663 (2000). Было найдено, что приведенные в данном описании в качестве примеров соединения формулы (I) показывают в анализе ингибирования фермента значения константы по Кассу, равные примерно от 14 х 106 до 35 х 106 л/моль. Способность испытуемого соединения продлевать неполное время тромбопластина (протромбиновое время) может быть оценена в следующих протоколах испытаний. Протокол определения неполного времени тромбопластина (протромбина) Венозную кровь собирали в пробирки с 3,2% (0,109 М) тринатрийцитратом при соотношении 1 объем антикоагулянта к девяти объемам крови. Клетки крови отделяли центрифугированием при 700 g в течение 10 мин с получением плазмы, которую замораживали при 70 С до возникновения необходимости в ее использовании. Для осуществления испытания 100 мкл плазмы отбирали пипеткой в стеклянную пробирку, добавляли 1 мкл испытуемого соединения в ДМСО и давали нагреться в течение более двух минут до 37 С. Добавляли 100 мкл теплого (37 С) реагента Манчестера (тромбопластин ткани) (Helena Biosciences, UK) и давали возможность установиться равновесию в течение двух минут. Для инициирования свертывания крови добавляли 100 мкл теплого (37 С) 25 мМ раствора хлорида кальция. Пробирку три раза наклоняли под углом 90 каждые пять секунд для смешивания реагентов и регистрировали время образования сгустков. Данные, полученные в результате ряда наблюдений и использования различных концентраций испытуемого соединения, анализировали с помощью программы статистического анализа SAS и СТ 2(концентрация, необходимая для удвоения времени свертывания) для каждого полученного соединения. Было найдено, что соединения изобретения значительно продлевают неполное время тромбопластина (протромбиновое время).-7 007300 Альтернативные протоколы протромбинового времени и АРТТ Определения коагуляции. Значения протромбинового времени и АРТТ определяли в человеческой плазме с использованием прибора STA (Stago). BioPT представляет специальный анализ неплазматического свертывания, инициированный человеческим тканевым фактором (Innovin). Возможное связывание с альбумином или липидом оценивали сравнением действий BioPT в присутствии/отсутствии 30 мг/мл человеческого альбумина (HSA) и 1 мг/мл фосфатидилхолина (PC) . Ингибиторы доставлялись в 50% водно-метанольном носителе. Анализ АРТТ 75 мкл плазмы Citrol Baxter-Dade, нормальная человеческая плазма с цитратом 25 мкл испытуемого раствора 75 мкл Actin Baxter-Dade, активированный цефалопластин инкубировали 2 мин при 37 С 75 мкл СаСl2 (0,02 М) Анализ РТ (ПВ) 75 мкл плазмы 25 мкл испытуемого раствора 75 мкл физиологического раствора инкубировали 1 мин при 37 С. 75 мкл Innovin Baxter-Dade, рекомбинантный человеческий тканевый фактор Дополнительные выгодные свойства соединений формулы (I) могут быть наглядно показаны определением их фармакодинамических (PD) и фармакокинетических (РК) свойств в лабораторных испытаниях на таких видах животных, как крысы и собаки, с пероральным дозированием голодным и накормленным животным. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) в которой R представляет собой атом водорода или атом фтора, или его фармацевтически приемлемая соль. 2. Соединение по п.1, в котором R представляет собой атом водорода. 3. Соединение по п.2, выбранное из 1-(4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина и его гидрохлоридных, фумаратных и малеатных аддитивных солей кислоты. 4. Соединение по п.3, выбранное из дигидрохлоридных, дифумаратных и дималеатных аддитивных солей кислоты в кристаллической форме. 5. Соединение по п.4, представляющее собой дифумарат 1-(4-метоксибензоил-Dциклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина в кристаллической форме. 6. Соединение по п.1, в котором R представляет собой атом фтора. 7. Соединение по п. 6, выбранное из 1-(3-фтор-4-метоксибензоил-D-циклопропилглицинил)-4-(1-метилпиперидин-4-ил)пиперазина и его гидрохлоридных аддитивных солей кислоты. 8. Фармацевтическая композиция, содержащая соединение по одному любому из пп.1-7 вместе с фармацевтически приемлемым разбавителем или носителем. 9. Фармацевтическая композиция по п.8, приспособленная для перорального введения. 10. Способ получения соединения по одному любому из пп.1-7, включающий:(а) взаимодействие соединения формулы (II) или его соли с соединением формулы (III) или его реакционноспособным производным; или(b) взаимодействие соединения формулы (IV) или его соли с соединением формулы (V) или его реакционноспособным производным; и последующее образование фармацевтически приемлемой соли, если требуется фармацевтически приемлемая соль. 11. Соединение формулы (II) 13. Соединение по одному любому из пп.1-7, предназначенное для использования в терапии. 14. Применение соединения по одному любому из пп.1-7 для производства лекарственного препарата, предназначенного для лечения тромботического заболевания. 15. Применение по п.14, в котором лекарственный препарат предназначен для перорального лечения тромботического заболевания. 16. Способ лечения тромботического заболевания у субъекта, нуждающегося в таком лечении,включающий введение эффективного количества соединения по п.1. 17. Способ по п.16, в котором соединение вводят субъекту перорально. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: A61K 31/495, C07D 211/58, A61P 7/02
Метки: фактора, качестве, ингибиторов, серинпротеазы, 1-(d-циклопропилглицинил)-4-(пиперидин-4-ил, производные, пиперазина, некоторые
Код ссылки
<a href="https://eas.patents.su/10-7300-nekotorye-proizvodnye-1-d-ciklopropilglicinil-4-piperidin-4-il-piperazina-v-kachestve-ingibitorov-serinproteazy-faktora-ha.html" rel="bookmark" title="База патентов Евразийского Союза">Некоторые производные 1-(d-циклопропилглицинил)-4-(пиперидин-4-ил) пиперазина в качестве ингибиторов серинпротеазы фактора ха</a>
Производные индазола и их использование в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продуцирования фактора некроза опухоли (фно)

Номер патента: 2113
Опубликовано: 24.12.2001
Автор: Марфат Энтони
МПК: A61P 11/06, A61K 31/415, C07D 401/12...
Метки: фосфодиэстеразы, типа, производные, ингибиторов, фдэ, использование, фактора, индазола, некроза, качестве, опухоли, фно, продуцирования
Формула / Реферат:
1. Соединение формулы (I) или их фармацевтически приемлемые соли, в которых R является Н, C1-C9 алкилом, -(СН2)m (5-10 членным гетероциклилом), где m равно от 0 до 2, или (Z1)b(Z2)с(С6-С10 арилом), где b и с независимо равны от 0 до 1, Z1 является C1-С6 алкиленом или C2-C8 алкениленом и Z2 является О, S, SO2 или NR12; и где указанные R группы необязательно замещены от 1 до 3 заместителями, независимо выбранными из группы, включающей галоген,...
Производные замешенных индазолов, их применение в качестве ингибиторов фосфодиэстеразы (фдэ) типа iv и продукции фактора некроза опухолей (фно)

Номер патента: 2272
Опубликовано: 28.02.2002
Автор: Марфэт Энтони
МПК: C07D 231/56, A61K 31/415, A61P 35/00...
Метки: индазолов, фно, замешенных, фосфодиэстеразы, фактора, производные, типа, применение, ингибиторов, качестве, опухолей, продукции, фдэ, некроза
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R представляет собой водород, -(СН2)n(С3-С7циклоалкил), где n равно 0, или фенил, возможно замещенный 1-3 галогено; R1 представляет собой С1-С7алкил; R2 выбран из группы, состоящей из где пунктирные линии в формулах (Iа) и (Iб) представляют простую связь; m равно от 0 до 1; R3 представляет собой циано или CH2CN; R4 представляет собой Н, CO2R14, C(Y)NR17R14, CN,...
Тиенодибензоазуленовые соединения в качестве ингибиторов фактора некроза опухоли

Номер патента: 6069
Опубликовано: 25.08.2005
Авторы: Жупанович Желько, Мерчеп Младен, Месич Милан, Пешич Дияна, Хрвачич Бошка
МПК: A61P 35/00, A61K 31/55, C07D 495/04...
Метки: опухоли, некроза, фактора, качестве, соединения, ингибиторов, тиенодибензоазуленовые
Формула / Реферат:
1. Соединение формулы I и его фармакологически приемлемые соли и сольваты где X представляет CH2 или гетероатом, выбранный из группы, включающей O, S и NR13, в котором R13 означает водород, C7-C10арилкарбонил; R1, R5, R7, R8 и R9 независимо друг от друга представляют водород; R2, R3 и R4 независимо представляют водород, фтор, хлор, бром, C1-C7алкил, галогенметил, C1-C7 алкокси или C1-C7алкилтио; R6 представляет водород, фтор, хлор, бром; R10...
Азотсодержащие гетеробициклы в качестве ингибиторов фактора xa

Номер патента: 4515
Опубликовано: 29.04.2004
Авторы: Пруитт Джеймс Р., Лэм Патрик Йук Сун, Хан Кви, Кван Мими, Пинто Дональд Дж.П., Каччола Джозеф, Кларк Чарльз Г., Февиг Джон М., Росси Карен А.
МПК: A61P 7/02, A61K 31/415, C07D 471/04...
Метки: гетеробициклы, фактора, азотсодержащие, качестве, ингибиторов
Формула / Реферат:
1. Соединение общей формулы содержащее различные конденсированные кольца F, в указанном соединение фрагмент формулы представлен одной из следующих структур где Z является N, CR1a или NR3; L является N или C, при условии, что, когда L является N, Z является N или CR1a и что, когда L является C, Z является NR3; где цикл F выбран из где соединения вышеуказанных формул замещены 0-2 R3; G выбран из A выбран из одной из следующих...
Новые миметики гуанидина в качестве ингибиторов фактора ха

Номер патента: 3210
Опубликовано: 27.02.2003
Авторы: Пинто Дональд Джозеф Филлип, Кларк Чарльз Дж., Лэм Патрик Й., Домингез Селия, Февиг Джон Мэттью, Кван Мими Лайфен, Ли Ренуа, Хан Кви, Пруитт Джеймс Расселл
МПК: A61K 31/4725, C07D 401/04, A61P 7/02...
Метки: миметики, гуанидина, новые, качестве, фактора, ингибиторов
Формула / Реферат:
1. Соединение формулы I или его стереоизомер или фармацевтически приемлемая соль, где кольцо D выбрано из -CH2N=CH-, -CH2CH2N=CH-, 5-6-членной ароматической системы, содержащей 0-2 гетероатома, выбранных из группы N, O и S; кольцо D замещено 0-2 R, при условии, что, когда кольцо D является незамещенным, оно содержит, по меньшей мере, один гетероатом; кольцо E содержит 0-2 атома N и замещено 0-1 R; R выбран из группы, включающей Cl, F, Br, I,...
Предыдущий патент: Твердая диспергируемая во рту фармацевтическая композиция агомелатина