Способ получения периндоприла, его аналогов и солей с использованием 2,5-диоксо-оксазолидиновых промежуточных соединений





Формула / Реферат
1. Способ получения соединения формулы (IV)

либо его сложного эфира или соли, включающий 1) взаимодействие соединения формулы (I)

где Ra представляет собой C1-4 алкил, Rb представляет собой С1-4 алкил, a Rc представляет собой C1-6 алкил с соединением формулы Х2С=O (где каждый Х независимо представляет собой уходящую группу) с образованием соединения формулы (II)

где Ra, Rb и Rc - такие же, как определено выше; и
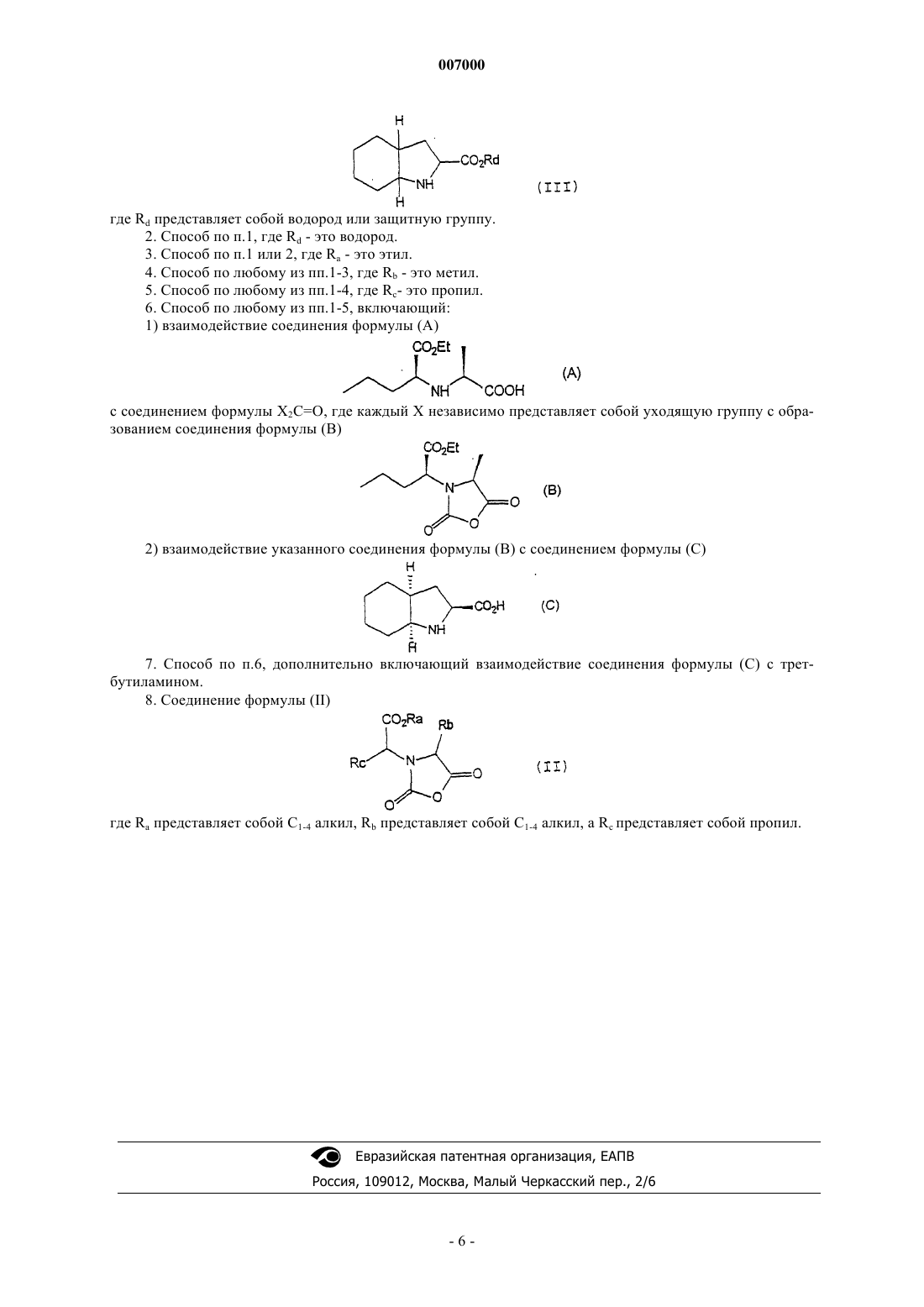
2) взаимодействие указанного соединения формулы (II) с соединением формулы (III)

где Rd представляет собой водород или защитную группу.
2. Способ по п.1, где Rd - это водород.
3. Способ по п.1 или 2, где Ra - это этил.
4. Способ по любому из пп.1-3, где Rb - это метил.
5. Способ по любому из пп.1-4, где Rc- это пропил.
6. Способ по любому из пп.1-5, включающий:
1) взаимодействие соединения формулы (А)

с соединением формулы Х2С=O, где каждый Х независимо представляет собой уходящую группу с образованием соединения формулы (В)

2) взаимодействие указанного соединения формулы (В) с соединением формулы (С)

7. Способ по п.6, дополнительно включающий взаимодействие соединения формулы (С) с трет-бутиламином.
8. Соединение формулы (II)

где Ra представляет собой C1-4 алкил, Rb представляет собой C1-4 алкил, a Rc представляет собой пропил.
Текст
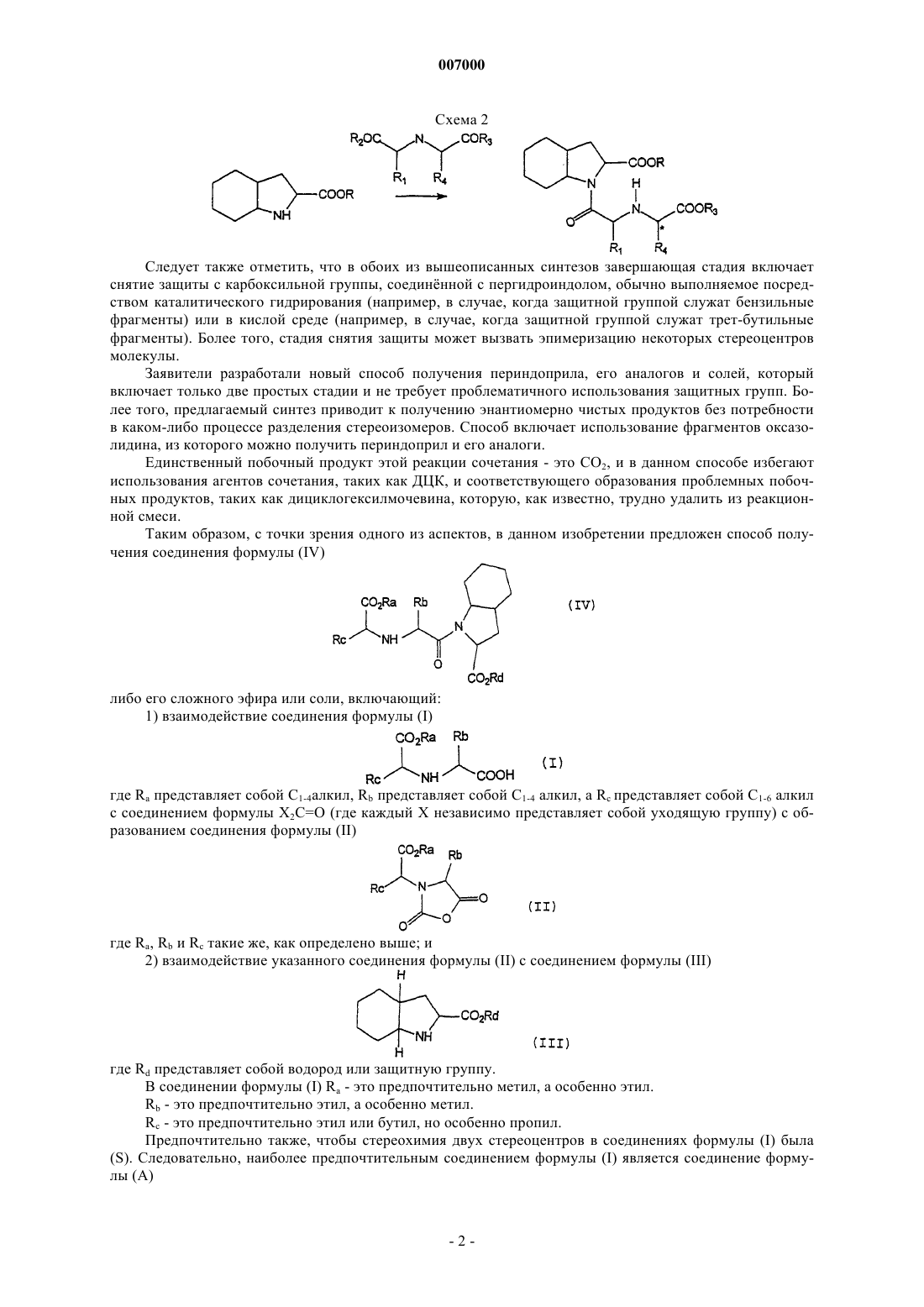
007000 В данном изобретении предложен новый способ получения периндоприла-1-этилового эфира (2S,3aS, 7aS)-1-[(S)-N-[(S)-1-карбоксибутил]аланил]гексагидро-2-индолин-карбоновой кислоты, а также его аналогов и солей, в частности трет-бутиламиновых солей. Периндоприл и его трет-бутиламиновую соль - периндоприла эрбумин используют в качестве ингибиторов ангиотензин-превращающего фермента (АПФ). Периндоприл действует как пролекарство дикислотного периндоприлата в его активной форме. Вслед за пероральным примом периндоприл быстро абсорбируется и в значительной степени метаболизируется, главным образом в печени, до периндоприлата и неактивных метаболитов, включая глюкурониды. Периндоприл используют при лечении гипертонии и сердечной недостаточности, так как ингибиторы ангиотензин-превращающего фермента ингибируют превращение ангиотензина-I в ангиотензин-II. Они являются антигипертензивными средствами, действующими как вазодилататоры, и снижают периферическое сопротивление; они оказывают благотворное воздействие на дисфункцию левого желудочка и снижают протеинурию, связанную с заболеваниями почек. Другие сферы возможной терапии - это инфаркт миокарда и диабетическая нефропатия, хотя сообщают о побочных эффектах, включая гипотонию, кожную сыпь, ангионевротический отек, кашель, нарушения вкусовых ощущений, нарушения почечной функции, и гиперкалиемию. Периндоприл был впервые синтезирован в соответствии со способом, описанным в ЕР-А-0049658. В настоящее время периндоприл традиционно получают в соответствии со способом, подробно описанным ниже. Первый способ - это четырхстадийный способ, начинающийся с пергидроиндолкарбоновой кислоты, которую необходимо прежде всего, до реакции, защитить. Затем, как показано на схеме 1, получают боковую цепь со стороны N путм сочетания соответствующим образом защищнной пергидроиндолкарбоновой кислоты с реакционно-способным производным энантиомерно чистой аминокислоты, такой как аланин. Остальную боковую цепь строят путм восстановительного аминирования, традиционно совершаемого с использованием гидрида металла, такого как цианоборогидрид натрия. Затем проводят снятие защиты. Схема 1 Вместе со стадиями защиты карбоксильной группы и снятия защиты этот маршрут синтеза периндоприла включает четыре стадии, а стадия восстановительного аминирования ведт к образованию двух возможных стереоизомеров, которые необходимо разделить. Следовательно, для того, чтобы получить энантиомерно чистое лекарство, необходимо провести трудную процедуру отделения, как только получен чистый периндоприл. Альтернативный метод получения периндоприла отражн на схеме 2 и включает сочетание предварительно образованной боковой цепи с фрагментами соответствующим образом защищнной пергидроиндолкарбоновой кислоты, такими как дициклогексилкарбодиимид (ДЦК). Этот метод требует трх стадий, опять же вместе со стадиями защиты и снятия защиты. Следует также отметить, что в обоих из вышеописанных синтезов завершающая стадия включает снятие защиты с карбоксильной группы, соединнной с пергидроиндолом, обычно выполняемое посредством каталитического гидрирования (например, в случае, когда защитной группой служат бензильные фрагменты) или в кислой среде (например, в случае, когда защитной группой служат трет-бутильные фрагменты). Более того, стадия снятия защиты может вызвать эпимеризацию некоторых стереоцентров молекулы. Заявители разработали новый способ получения периндоприла, его аналогов и солей, который включает только две простых стадии и не требует проблематичного использования защитных групп. Более того, предлагаемый синтез приводит к получению энантиомерно чистых продуктов без потребности в каком-либо процессе разделения стереоизомеров. Способ включает использование фрагментов оксазолидина, из которого можно получить периндоприл и его аналоги. Единственный побочный продукт этой реакции сочетания - это СO2, и в данном способе избегают использования агентов сочетания, таких как ДЦК, и соответствующего образования проблемных побочных продуктов, таких как дициклогексилмочевина, которую, как известно, трудно удалить из реакционной смеси. Таким образом, с точки зрения одного из аспектов, в данном изобретении предложен способ получения соединения формулы (IV) либо его сложного эфира или соли, включающий: 1) взаимодействие соединения формулы (I) где Ra представляет собой С 1-4 алкил, Rb представляет собой С 1-4 алкил, a Rc представляет собой C1-6 алкил с соединением формулы Х 2 С=O (где каждый Х независимо представляет собой уходящую группу) с образованием соединения формулы (II) где Ra, Rb и Rc такие же, как определено выше; и 2) взаимодействие указанного соединения формулы (II) с соединением формулы (III) где Rd представляет собой водород или защитную группу. В соединении формулы (I) Ra - это предпочтительно метил, а особенно этил.Rc - это предпочтительно этил или бутил, но особенно пропил. Предпочтительно также, чтобы стереохимия двух стереоцентров в соединениях формулы (I) была(S). Следовательно, наиболее предпочтительным соединением формулы (I) является соединение формулы (А) Соединения формулы (I) можно получить известными специалистам методами. Например, соединение формулы (А) можно получить реакцией возможно защищнного аланина с подходящим образом функционализированным сложным эфиром пентановой кислоты. Альтернативно соединения формулы (I) можно получить так, как показано на схеме реакции ниже: где Ra, Rb и Rc такие же, как определено выше, a Re совместно с атомом кислорода, к которому он присоединн, образует уходящую группу, например -ОSО 2 СF3. Предпочтения для Ra, Rb и Rc такие же, как определено выше. Соединение формулы (VI) можно получить из D-молочной кислоты традиционными способами. Стереохимия соединения формулы (V) предпочтительно (S), чтобы была возможность получения соединения формулы (А). Снятие защиты карбоксильной группы выполняют гидрированием. Эта реакция представляет собой дополнительный аспект данного изобретения. Соединение формулы (I) реагирует с соединением, способным к введению карбонильной группы, с тем чтобы дать оксазолидину образоваться. Всестороннее обсуждение синтеза оксазолидинов (называемых также N-карбоксиангидридами аминокислот) можно найти в книге "N-карбоксиангидриды -аминокислот и соответствующие гетероциклы, синтез, свойства, пептидный синтез, полимеризация" Ганса Рутгера Крихельдорфа ("Aminoacid-N-carboxy anhydrides and related heterocycles, syntheses, properties, peptide synthesis, polymerization" by Hans Rytger Kricheldorf) (Springer-Verlag, Berlin, 1987), которая включена сюда посредством ссылки. Таким образом, оксазолидиновое кольцо можно построить методом Фукса-Фартинга (FuchsFarthing method), как это описано здесь. Метод Фукса-Фартинга включает прямую реакцию свободных аминокислот с фосгеном, причм реакция протекает через образование промежуточного соединения - N-хлороформил аминокислоты, которая превращается в ангидрид в присутствии хлористо-водородной кислоты. Подходящие соединения, способные к введению карбонильной группы, имеют формулу Х 2 С=O. Каждый Х может независимо быть любой из пригодных уходящих групп, которые хорошо известны специалистам. Таким образом, каждый Х должен обладать способностью к замещению на отдельные нуклеофильные пары присутствующие на атомах кислорода и азота соединения (I). Х может, например, быть галогеном, тозилатом, мезилатом, алкоксигруппой, алкилтио- или имидазолилгруппой. Вообще, в случаях, когда Х образует сложноэфирную или тиоэфирную связь с СОсоставляющей, пригодное для введения карбонила соединение приведт, например, к (Сl3 СО)-. В предпочтительном случае осуществления изобретения обе группы Х одинаковы, а в ещ одном предпочтительном случае осуществления изобретения обе группы Х представляют собой галоген, предпочтительно хлор. В этом случае соединение формулы Х 2 С=O является, конечно, фосгеном (Cl2C=O). Так как фосген опасен при работе, может быть предпочтительным использовать его менее активную форму - трифосген ССl3 О)2 СО). В другом предпочтительном случае осуществления изобретения Х 2 С=O - это N,N'-карбонилдиимидазол. Нуклеофильные атомы азота и кислорода в соединении формулы (I) атакуют электрофильные фрагменты Х 2 С=O, благоприятствуя осуществлению 5-эндо-тригциклизации (5-endo-trig cyclisation). В зависимости от природы Х 2 СО группы эту реакцию можно проводить в различных растворителях. В частности, большинство инертных растворителей с низкой точкой кипения пригодно в качестве реакционной среды, например тетрагидрофуран, диоксан, дихлорметан. Когда в качестве вводящего карбонил агента выступает трифосген или фосген, специалист-химик примет во внимание, что необходим тщательный контроль над реакцией, чтобы избежать потенциальной опасности.-3 007000 Например, реакцию можно проводить в смеси вода/дихлорметан в присутствии моногидрофосфата натрия. Перед выделением соединения формулы (II) может потребоваться нейтрализовать любые остатки агента, вводящего карбонил, например, добавлением основания, такого как пиридин. Соединение формулы (II) можно затем выделить, используя стандартные методики выделения и промывочные фазы. Можно добиться такой степени превращения до соединения (II), что выход будет составлять более чем 70%, например 80% без ущерба для стереохимии. В предпочтительном случае осуществления изобретения соединение формулы (II) имеет формулу (В) Соединения формулы (II), особенно соединение формулы (В) является новым и представляет собой следующий аспект данного изобретения. Следовательно, с точки зрения следующего аспекта, в изобретении предложено соединение формулы (II), как описано здесь выше. Затем соединение (II) приводят во взаимодействие с соединением формулы (III). Предпочтительно,чтобы в соединениях формулы (III) Rd был атомом водорода, однако он может представлять собой и защитную группу, такую как бензил. Соединения формулы (III) широко представлены в литературе, а их получение описано, между прочим, в ЕР-А-0037231. Эта реакция может протекать в подходящем органическом растворителе, например в дихлорметане в присутствии слабого основания, например триэтиламина. Предпочтительно соединение формулы (III) имеет формулу (С) т.е. (2S, 3aS, 7 аS)-2-карбоксипергидроиндол. Продукт реакции соединений формул (В) и (С) - это, конечно, периндоприл, который можно очистить традиционными методиками или кристаллизовать непосредственно в виде соли, например соли трет-бутиламина. После реакций соединений (II) и (III), и если необходимо снятия защиты с пергидроиндолкарбоксильной кислоты, можно проводить выделение продукта. Для этого к реакционной смеси можно добавить воду и охладить смесь до 15 С. рН смеси можно довести до приблизительно 4,2 добавлением кислоты, например хлористоводородной кислоты, а водную фазу экстрагировать дихлорметаном. Затем можно высушить органический экстракт при пониженном давлении и температуре ниже 40 С, при этом получится масло. Реакцию между соединениями (II) и (III) можно провести, не выделяя соединение формулы (II) из исходной среды. Его можно превратить в соль трет-бутиламина (например, в периндоприла эрбумин) просто путм контакта масла в подходящем растворителе с трет-бутиламином. После выделения можно изолировать соль с выходом более 70%. Периндоприл или его производные, полученные в соответствии с предлагаемым способом, можно использовать при показаниях, описанных в разделе уровень техники данной заявки, и при показаниях,известных специалистам. Периндоприл или его производные могут быть изготовлены в виде части фармацевтической композиции и назначены любым из стандартных путей, таких как пероральный, через слизистые или в виде инъекций. Далее изобретение будет проиллюстрировано на примерах, не ограничивающих его. Пример 1. 2,5-диоксо-3-[1-(S)-этоксикарбонилбутил]-4-(S)-метилоксазолидин Приготовили раствор 28,7 г гидрофосфата натрия в 200 мл воды и нагрели до 30-35 С. После полного растворения смесь охладили до комнатной температуры и добавили к ней 160 мл дихлорметана. К хорошо перемешиваемой смеси добавляли 20 г N-[1-(S)-этоксикарбонилбутил]-(S)-аланина и охлаждали полученную смесь до 15 С. Медленно добавляли раствор 10,9 г трифосгена в 40 мл дихлорметана в течение 30 мин, поддерживая температуру ниже 20 С. После добавления трифосгена смесь перемешивали в течение 30 мин и добавляли 0,1 мл пиридина,чтобы разрушить остаточный фосген. После перемешивания в течение ещ 1 ч, или до того момента, как фосген разрушался окончательно, фазы разделяли и промывали органическую фазу сначала 100 мл 2NHCl, а затем 100 мл воды. Органическую фазу фильтровали и выпаривали растворитель при пониженном давлении. Было получено 18,85 г бледно-жлтого масла.-4 007000 Анализ: 95% (2,5-диоксо-3-[1-(S)-этоксикарбонилбутил]-4-(S)-метилоксазолидина) Выход: 80% Пример 2. трет-Бутиламин (2S, 3 аS, 7 аS)-1-2-[1-этоксикарбонил)-(S)-бутиламино]-(S)-пропионилоктагидроиндол-2-карбоксилат (периндоприла эрбумин) 20 г (2S, 3 аS, 7 аS)-2-карбоксиоктагидроиндола суспендировали в 150 мл дихлорметана при 25 С и добавляли 16,5 мл триэтиламина. Медленно добавляли раствор 27,5 г 95%-ного 2,5-диоксо-3-[1-(S)этоксикарбонилбутил]-4-(S)-метилоксазолидина (пример 1) в 40 мл дихлорметана в течение 3 ч и перемешивали смесь ещ в течение 1 ч. Добавляли 150 мл воды и охлаждали двухфазную смесь до 15 С. Доводили рН до значения 4,2 путм добавления 2N хлористо-водородной кислоты (потребовалось 52 мл). Отделяли органическую фазу и экстрагировали водную фазу 100 мл дихлорметана. Соединяли и фильтровали органические экстракты,а растворитель выпаривали при пониженном давлении, поддерживая температуру ниже 40 С, чтобы получить масло. К маслу добавляли 100 мл ацетонитрила и удаляли растворитель в вакууме. Получившееся масло растворяли в 300 мл ацетонитрила и нагревали раствор до 35 С. Медленно добавляли раствор 12,5 мл трет-бутиламина в 50 мл ацетонитрила в течение 30 мин и перемешивали получившуюся смесь при 40 С в течение 1 ч. Смесь охлаждали и перемешивали при 5 С в течение ещ 1 ч. Получившийся осадок отфильтровывали и дважды промывали 50 мл ацетонитрила. Получили 78,27 г влажного белого осадка (42,34 г сухого продукта при расчте на потерю при высушивании). 70,27 г влажного осадка суспендировали в 160 мл ацетонитрила и добавляли 4,75 г воды. Смесь перемешивали при 40 С в течение 1 ч, после чего охлаждали и перемешивали при 5 С в течение 1 ч. Осадок отфильтровывали и дважды промывали 50 мл ацетонитрила. После высушивания получили 44,52 г периндоприла эрбумина в виде белого порошка. Выход: 81%. Пример 3. трет-Бутиламин (2S,3aS,7 аS)-1-2-[(1-этоксикарбонил)-(S)-бутиламино]-(S)-пропионилоктагидроиндол-2-карбоксилат (периндоприла эрбумин) К охлажднной суспензии 5 г N-1-(S) этоксикарбонилбутил-(S)-аланина в 71 мл дихлорметана добавляли 4,47 г N,N'-карбонилдиимидазола. Смесь охлаждали при 0 С и перемешивали в течение 1 ч. К вышеуказанному раствору при -5 С добавляли 5,05 г (2s, 3aS, 7aS)-2-карбоксиоктагидроиндола и перемешивали эту смесь при -5 С в течение 1 ч. Дихлорметан выпаривали и добавляли к водной фазе 71 мл воды, после чего добавляли 11,6 мл 6N хлористо-водородной кислоты. Водный раствор насыщали хлоридом натрия и экстрагировали 120 мл дихлорметана. Растворитель выпаривали. Получившееся масло растворяли в 80 мл этилацетата. Добавляли 1,78 г трет-бутиламина при перемешивании при комнатной температуре. Чтобы способствовать растворению, смесь нагревали; а затем охлаждали до 20 С. Получившийся осадок фильтровали и промывали этилацетатом. Выход 80%, чистота 99%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (IV) либо его сложного эфира или соли, включающий 1) взаимодействие соединения формулы (I) где Ra представляет собой C1-4 алкил, Rb представляет собой С 1-4 алкил, a Rc представляет собой C1-6 алкил с соединением формулы Х 2 С=O (где каждый Х независимо представляет собой уходящую группу) с образованием соединения формулы (II) где Ra, Rb и Rc - такие же, как определено выше; и 2) взаимодействие указанного соединения формулы (II) с соединением формулы (III) где Rd представляет собой водород или защитную группу. 2. Способ по п.1, где Rd - это водород. 3. Способ по п.1 или 2, где Ra - это этил. 4. Способ по любому из пп.1-3, где Rb - это метил. 5. Способ по любому из пп.1-4, где Rc- это пропил. 6. Способ по любому из пп.1-5, включающий: 1) взаимодействие соединения формулы (А) с соединением формулы Х 2 С=O, где каждый Х независимо представляет собой уходящую группу с образованием соединения формулы (В) 2) взаимодействие указанного соединения формулы (В) с соединением формулы (С) 7. Способ по п.6, дополнительно включающий взаимодействие соединения формулы (С) с третбутиламином. 8. Соединение формулы (II)
МПК / Метки
МПК: C07D 209/42, C07D 263/44
Метки: аналогов, использованием, периндоприла, способ, промежуточных, солей, соединений, 2,5-диоксо-оксазолидиновых, получения
Код ссылки
<a href="https://eas.patents.su/7-7000-sposob-polucheniya-perindoprila-ego-analogov-i-solejj-s-ispolzovaniem-25-diokso-oksazolidinovyh-promezhutochnyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения периндоприла, его аналогов и солей с использованием 2,5-диоксо-оксазолидиновых промежуточных соединений</a>
Способ получения промежуточных соединений, применимых для получения противораковых соединений

Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Лехнер Ричард Шелтон, Сантафьянос Динос Пол, Норрис Тимоти
МПК: C07D 239/94
Метки: промежуточных, применимых, получения, противораковых, соединений, способ
Формула / Реферат:
1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше, тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются...
Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений

Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Леветт Филип Чарльз, Девриз Кейт Майкл, Вуд Альберт Шо, Негри Джоанна Тереза
МПК: C07D 487/04, C07D 401/14, C07D 401/12...
Метки: соединений, промежуточных, пиразоло[4,3-d]пиримидин-7-он-3, способ, получения, пиридилсульфонильных
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом; R1 (который может быть...
Способ получения циклопропанкарбоновых кислот и их промежуточных соединений

Номер патента: 683
Опубликовано: 28.02.2000
Авторы: Клемменсен Пер Дауселль, Колинн-Андерсен Ханс, Винкельманн Иб
МПК: C07D 307/93, C07C 61/35
Метки: циклопропанкарбоновых, получения, способ, промежуточных, кислот, соединений
Формула / Реферат:
1. Способ получения соединений общей формулы I где R' представляет Н, а два атома водорода циклопропанового кольца находятся в цис-положении по отношению друг к другу, включающий взаимодействие между соединением общей формулы II и соединением СF3-CClХ2, где Х представляет атом галогена, в частности атом хлора или брома, в инертной среде в присутствии Zn и при подходящей температуре от 0 до 150шС, предпочтительно от 20 до 100шС, в...
Получение промежуточных соединений эндотелина реакцией ассиметрического сопряженного присоединения с использованием хиральной добавки.

Номер патента: 2056
Опубликовано: 24.12.2001
Авторы: Тилльер Ричард Д., Ксу Фенг, Щаен Дэвид М.
МПК: A61K 31/44, C07B 37/02, C07D 213/55...
Метки: промежуточных, реакцией, эндотелина, использованием, сопряженного, хиральной, получение, ассиметрического, соединений, присоединения, добавки
Формула / Реферат:
1. Способ получения соединения формулы I где представляет а) 5- или 6-членный гетероциклил, содержащий одну, две или три двойные связи, по крайней мере одну двойную связь, и 1, 2 или 3 гетероатома, выбранных из О, N и S, незамещенный или замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из ОН, CO2R4, Br, Cl, F, I, СF3, N(R5)2, C1-C8 алкокси, C1-C8 алкила, C2-C8 алкенила, C2-C8 алкинила или C3-C8 циклоалкила,...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Руссель Патрик, Крок Вероник, Ларкин Джон Патрик, Колладан Колетт
МПК: C07D 487/04
Метки: получения, способ, соединений, применение, диазепин-1-карбоновой, 1,2-а, 1,2, активных, октагидро-6, производные, кислоты, 10-диоксо-6н-пиридазино, терапевтически
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...